Пирролопиримидины в качестве ингибиторов cdk4/6
Номер патента: 22355
Опубликовано: 30.12.2015
Авторы: Ван Япин, Луццио Майкл, Лагу Бхарат, Лэвэлл Джулиан, Чо Ён Шинь, Джиралдес Джон Уилльям, Перес Лоренс Блас, Брейн Кристофер Томас, Ян Фань

























Формула / Реферат
1. Соединение формулы (I)

в которой R1 обозначает С3-С7-алкил; С4-С7-циклоалкил, необязательно содержащий один заместитель, выбранный из группы, включающей C1-С6-алкил и ОН; фенил, необязательно содержащий один заместитель, выбранный из группы, включающей C1-С6-алкил, С(СН3)2CN и ОН; пиперидинил, необязательно замещенный одним циклопропилом или C1-С6-алкилом; тетрагидропиранил, необязательно замещенный одним циклопропилом или C1-C6-алкилом; или бицикло[2.2.1]гептанил;
А обозначает СН или N;
R11 обозначает водород или С1-С4-алкил;
L обозначает связь или С(О);
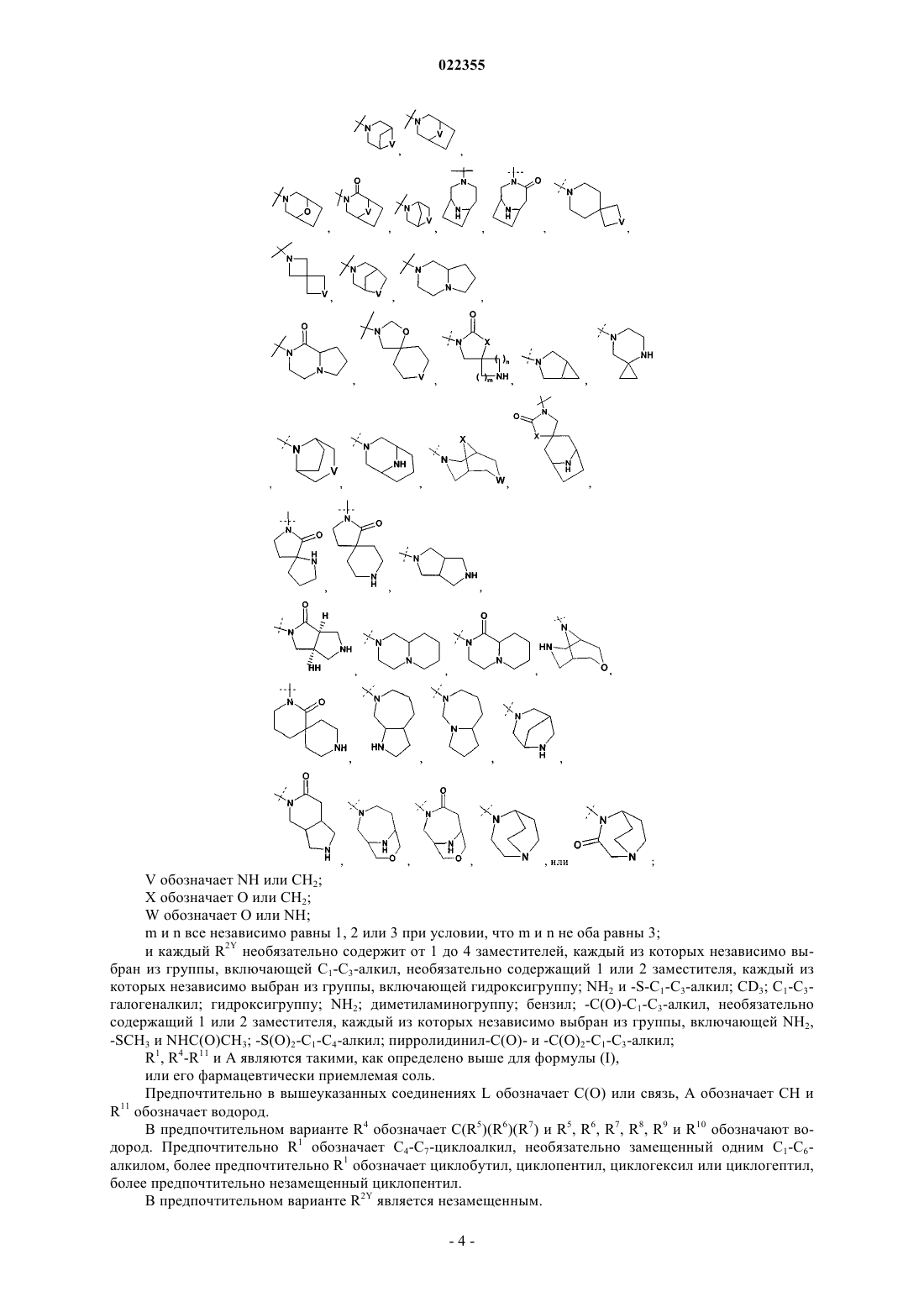
R2Y обозначает



V обозначает NH или СН2;
X обозначает О или СН2;
W обозначает О или NH;
m и n все независимо равны 1, 2 или 3 при условии, что m и n не оба равны 3;
каждый R2Y необязательно содержит от 1 до 4 заместителей, каждый из которых независимо выбран из группы, включающей С1-С3-алкил, необязательно содержащий 1 или 2 заместителя, каждый из которых независимо выбран из группы, включающей гидроксигруппу; NH2 и -S-C1-С3-алкил; CD3; галоген; оксогруппу; C1-С3-галогеналкил; гидроксигруппу; NH2; диметиламиногруппу; бензил; -С(О)-С1-С3-алкил, необязательно содержащий 1 или 2 заместителя, каждый из которых независимо выбран из группы, включающей NH2; -SCH3 и NHC(О)СН3; -S(O)2-С1-С4-алкил; пирролидинил-С(О)- и -С(О)2-C1-C3-алкил;
R4 обозначает водород, дейтерий или C(R5)(R6)(R7);
R5, R6, R7, R8, R9 и R10 все независимо обозначают Н или дейтерий,
или его фармацевтически приемлемая соль.
2. Соединение по п.1 формулы (I-B)

в которой L обозначает связь или С(O);
R2Y обозначает


V обозначает NH или СН2;
X обозначает О или СН2;
W обозначает О или NH;
m и n все независимо равны 1, 2 или 3 при условии, что m и n не оба равны 3;
каждый R2Y необязательно содержит от 1 до 4 заместителей, каждый из которых независимо выбран из группы, включающей C1-С3-алкил, необязательно содержащий 1 или 2 заместителя, каждый из которых независимо выбран из группы, включающей гидроксигруппу; NH2 и -S-C1-С3-алкил; CD3; C1-С3-галогеналкил; гидроксигруппу; NH2; диметиламиногруппу; бензил; -C(O)-C1-C3-алкил, необязательно содержащий 1 или 2 заместителя, каждый из которых независимо выбран из группы, включающей NH2;
-SCH3 и NHC(О)СН3; -S(О)2-С1-С4-алкил; пирролидинил-С(О)- и -С(О)2-С1-С3-алкил,
или его фармацевтически приемлемая соль.
3. Соединение по любому из предыдущих пунктов, в котором L обозначает С(О), или его фармацевтически приемлемая соль.
4. Соединение по п.1 или 2, в котором L обозначает связь, или его фармацевтически приемлемая соль.
5. Соединение по любому из предыдущих пунктов, в котором А обозначает СН и R11 обозначает водород, или его фармацевтически приемлемая соль.
6. Соединение по любому из предыдущих пунктов, в котором R4 обозначает С(R5)(R6)(R7) и R5, R6, R7, R8, R9 и R10 обозначают водород, или его фармацевтически приемлемая соль.
7. Соединение по любому из предыдущих пунктов, в котором R1 обозначает С4-С7-циклоалкил, необязательно замещенный одним C1-С6-алкилом, или его фармацевтически приемлемая соль.
8. Соединение по любому из предыдущих пунктов, в котором R1 обозначает циклобутил, циклопентил, циклогексил или циклогептил, или его фармацевтически приемлемая соль.
9. Соединение по любому из предыдущих пунктов, в котором R1 обозначает незамещенный циклопентил, или его фармацевтически приемлемая соль.
10. Соединение по любому из предыдущих пунктов, в котором R2Y является незамещенным, или его фармацевтически приемлемая соль.
11. Соединение по любому из пп.1-9, в котором R2Y обозначает

или

любой из которых необязательно замещен одним C1-C3-алкилом,
или его фармацевтически приемлемая соль.
12. Соединение по любому из пп.1-9, в котором R2Y обозначает

необязательно замещенный одним C1-C3-алкилом,
или его фармацевтически приемлемая соль.
13. Соединение по п.1, выбранное из группы, включающей
7-циклопентил-2-(5-(9-гидрокси-1,5,7-триметил-3,7-диазабицикло[3.3.1]нонан-3-карбонил)пиридин-2-иламино)-N,N-диметил-7Н-пирроло[2,3-d]пиримидин-6-карбоксамид;
2-(5-(2,6-диазаспиро[3.3]гептан-2-карбонил)пиридин-2-иламино)-7-циклопентил-N,N-диметил-7H-пирроло[2,3-d]пиримидин-6-карбоксамид;
диметиламид 7-(4-трет-бутилфенил)-2-[5-(3,8-диазабицикло[3.2.1]октан-3-карбонил)пиридин-2-иламино]-7Н-пирроло[2,3-d]пиримидин-6-карбоновой кислоты;
диметиламид 7-циклопентил-2-[5-(4-оксо-3,9-диазабицикло[4.2.1]нон-3-ил)пиридин-2-иламино]-7Н-пирроло[2,3-d]пиримидин-6-карбоновой кислоты;
диметиламид 7-циклопентил-2-[5-((1R,6S)-4-оксо-3,9-диазабицикло[4.2.1]нон-3-ил)пиридин-2-иламино]-7Н-пирроло[2,3-d]пиримидин-6-карбоновой кислоты;
диметиламид 7-циклопентил-2-[5-(3,8-диазабицикло[3.2.1]октан-3-карбонил)пиридин-2-иламино]-7Н-пирроло[2,3-d]пиримидин-6-карбоновой кислоты;
диметиламид 7-циклогептил-2-[5-(2,5-диазабицикло[2.2.1]гептан-2-карбонил)пиридин-2-иламино]-7Н-пирроло[2,3-d]пиримидин-6-карбоновой кислоты;
диметиламид 7-циклопентил-2-[5-(3,8-диазабицикло[3.2.1]октан-8-карбонил)пиридин-2-иламино]-7Н-пирроло[2,3-d]пиримидин-6-карбоновой кислоты;
2-(5-(2,7-диазаспиро[3.5]нонан-7-карбонил)пиридин-2-иламино)-7-циклогептил-N,N-диметил-7Н-пирроло[2,3-d]пиримидин-6-карбоксамид;
диметиламид 7-циклопентил-2-[5-(8-метил-3,8-диазабицикло[3.2.1]октан-3-карбонил)пиридин-2-иламино]-7Н-пирроло[2,3-d]пиримидин-6-карбоновой кислоты;
метиламид 7-циклопентил-2-[5-((S,S)-2,5-диазабицикло[2.2.1]гептан-2-карбонил)пиридин-2-иламино]-7Н-пирроло[2,3-d]пиримидин-6-карбоновой кислоты;
диметиламид 7-(3-трет-бутилфенил)-2-[5-(3,8-диазабицикло[3.2.1]октан-3-карбонил)пиридин-2-иламино]-7Н-пирроло[2,3-d]пиримидин-6-карбоновой кислоты;
2-(5-((1R,5S)-3-окса-7,9-диазабицикло[3.3.1]нонан-9-карбонил)пиридин-2-иламино)-7-циклопентил-N,N-диметил-7Н-пирроло[2,3-d]пиримидин-6-карбоксамид;
диметиламид 7-[4-(цианодиметилметил)фенил]-2-[5-(3,8-диазабицикло[3.2.1]октан-8-карбонил)пиридин-2-иламино]-7Н-пирроло[2,3-d]пиримидин-6-карбоновой кислоты;
диметиламид 7-циклопентил-2-[5-((1S,6R)-3,9-диазабицикло[4.2.1]нонан-3-карбонил)пиридин-2-иламино]-7Н-пирроло[2,3-d]пиримидин-6-карбоновой кислоты;
диметиламид 7-циклопентил-2-[5-((1R,6S)-3,9-диазабицикло[4.2.1]нонан-3-карбонил)пиридин-2-иламино]-7Н-пирроло[2,3-d]пиримидин-6-карбоновой кислоты;
диметиламид 7-циклопентил-2-[5-(3,6-диазабицикло[3.2.1]октан-3-карбонил)пиридин-2-иламино]-7Н-пирроло[2,3-d]пиримидин-6-карбоновой кислоты;
диметиламид 7-циклопентил-2-[5-(гексагидропирроло[1,2-а]пиразин-2-карбонил)пиридин-2-иламино]-7Н-пирроло[2,3-d]пиримидин-6-карбоновой кислоты;
7-циклопентил-N,N-диметил-2-(5-(5'-оксо-8-азаспиро[бицикло[3.2.1]октан-3,3'-пирролидин]-1'-ил)пиридин-2-иламино)-7Н-пирроло[2,3-d]пиримидин-6-карбоксамид;
диметиламид 7-циклопентил-2-[5-(1-оксогексагидропирроло[1,2-а]пиразин-2-ил)пиридин-2-иламино]-7Н-пирроло[2,3-d]пиримидин-6-карбоновой кислоты;
7-циклопентил-N,N-диметил-2-(5-(2-оксо-1-окса-3,8-диазаспиро[4.5]декан-3-ил)пиридин-2-иламино)-7Н-пирроло[2,3-d]пиримидин-6-карбоксамид;
диметиламид 7-циклопентил-2-[5-((1S,6R)-4-оксо-3,9-диазабицикло[4.2.1]нон-3-ил)пиридин-2-иламино]-7Н-пирроло[2,3-d]пиримидин-6-карбоновой кислоты;
диметиламид 2-[5-(4-оксо-3,9-диазабицикло[4.2.1]нон-3-ил)пиридин-2-иламино]-7-фенил-7Н-пирроло[2,3-d]пиримидин-6-карбоновой кислоты;
7-циклогексил-N,N-диметил-2-(5-(4-оксо-3,9-диазабицикло[4.2.1]нонан-3-ил)пиридин-2-иламино)-7Н-пирроло[2,3-d]пиримидин-6-карбоксамид,
или его фармацевтически приемлемая соль.
14. Соединение по п.1, выбранное из группы, включающей
диметиламид 7-циклопентил-2-[5-(3,8-диазабицикло[3.2.1]октан-3-карбонил)пиридин-2-иламино]-7Н-пирроло[2,3-d]пиримидин-6-карбоновой кислоты;
диметиламид 7-циклопентил-2-[5-((S,S)-2,5-диазабицикло[2.2.1]гептан-2-карбонил)пиридин-2-иламино]-7Н-пирроло[2,3-d]пиримидин-6-карбоновой кислоты;
диметиламид 7-циклопентил-2-[5-((1R,6S)-4-оксо-3,9-диазабицикло[4.2.1]нон-3-ил)пиридин-2-иламино]-7Н-пирроло[2,3-d]пиримидин-6-карбоновой кислоты;
диметиламид 7-циклопентил-2-[5-((1S,5S)-3,6-диазабицикло[3.2.1]октан-3-карбонил)пиридин-2-иламино]-7Н-пирроло[2,3-d]пиримидин-6-карбоновой кислоты;
диметиламид 7-циклобутил-2-[5-((1S,6R)-4-оксо-3,9-диазабицикло[4.2.1]нон-3-ил)пиридин-2-иламино]-7Н-пирроло[2,3-d]пиримидин-6-карбоновой кислоты;
диметиламид 7-циклогексил-2-[5-((1R,6S)-4-оксо-3,9-диазабицикло[4.2.1]нон-3-ил)пиридин-2-иламино]-7Н-пирроло[2,3-d]пиримидин-6-карбоновой кислоты;
диметиламид 7-циклопентил-2-[5-(3,8-диазабицикло[3.2.1]октан-3-карбонил)-6-метилпиридин-2-иламино]-7Н-пирроло[2,3-d]пиримидин-6-карбоновой кислоты;
диметиламид 7-циклопентил-2-[5-(3,8-диазабицикло[3.2.1]октан-3-карбонил)-4-метилпиридин-2-иламино]-7Н-пирроло[2,3-d]пиримидин-6-карбоновой кислоты;
диметиламид 7-циклопентил-2-[5-(3,9-диазабицикло[3.3.1]нонан-3-карбонил)пиридин-2-иламино]-7Н-пирроло[2,3-d]пиримидин-6-карбоновой кислоты;
7-циклопентил-N,N-диметил-2-(5-(3-оксо-1,4-диазабицикло[3.2.2]нонан-4-ил)пиридин-2-иламино)-7Н-пирроло[2,3-d]пиримидин-6-карбоксамид;
7-циклопентил-N,N-диметил-2-(5-((1R,6S)-9-метил-4-оксо-3,9-диазабицикло[4.2.1]нонан-3-ил)пиридин-2-иламино)-7Н-пирроло[2,3-d]пиримидин-6-карбоксамид,
или его фармацевтически приемлемая соль.
15. Соединение по п.1, выбранное из группы, включающей
7-циклопентил-N,N-диметил-2-(5-((3aS,6aR)-1-оксагексагидропирроло[3,4-с]пиррол-2(1Н)-ил)пиридин-2-иламино)-7Н-пирроло[2,3-d]пиримидин-6-карбоксамид;
7-циклопентил-N,N-диметил-2-(5-((3aR,6aS)-1-оксагексагидропирроло[3,4-с]пиррол-2(1Н)-ил)пиридин-2-иламино)-7Н-пирроло[2,3-d]пиримидин-6-карбоксамид;
7-циклогептил-N,N-диметил-2-(5-(цис-6-оксотетрагидро-1Н-пирроло[3,4-с]пиридин-5(6Н,7Н,7аН)-ил)пиридин-2-иламино)-7Н-пирроло[2,3-d]пиримидин-6-карбоксамид;
7-циклопентил-N,N-диметил-2-(5-(цис-6-оксотетрагидро-1Н-пирроло[3,4-с]пиридин-5(6Н,7Н,7аН)-ил)пиридин-2-иламино)-7Н-пирроло[2,3-d]пиримидин-6-карбоксамид;
7-циклопентил-N,N-диметил-2-(5-(цис-6-оксотетрагидро-1Н-пирроло[3,4-с]пиридин-5(6Н,7Н,7аН)-ил)пиридин-2-иламино)-7Н-пирроло[2,3-d]пиримидин-6-карбоксамид;
7-циклопентил-N,N-диметил-2-(5-((3aR,6aS)-5-метил-1-оксагексагидропирроло[3,4-с]пиррол-2(1Н)-ил)пиридин-2-иламино)-7Н-пирроло[2,3-d]пиримидин-6-карбоксамид,
или его фармацевтически приемлемая соль.
16. Соединение по п.1, выбранное из группы, включающей
7-циклопентил-N,N-диметил-2-(5-((1R,3r,5S)-2'-оксо-8-азаспиро[бицикло[3.2.1]октан-3,5'-оксазолидин]-3'-ил)пиридин-2-иламино)-7Н-пирроло[2,3-d]пиримидин-6-карбоксамид;
7-циклопентил-N,N-диметил-2-(5-(2-оксо-1-окса-3,8-диазаспиро[4.6]ундекан-3-ил)пиридин-2-иламино)-7Н-пирроло[2,3-d]пиримидин-6-карбоксамид;
7-циклопентил-N,N-диметил-2-(5-(2-оксо-1-окса-3,7-диазаспиро[4.5]декан-3-ил)пиридин-2-иламино)-7Н-пирроло[2,3-d]пиримидин-6-карбоксамид;
диметиламид 7-циклопентил-2-[5-((S)-2-оксо-1-окса-3,7-диазаспиро[4.5]дец-3-ил)пиридин-2-иламино]-7Н-пирроло[2,3-d]пиримидин-6-карбоновой кислоты;
диметиламид 7-циклопентил-2-[5-((R)-2-оксо-1-окса-3,7-диазаспиро[4.5]дец-3-ил)пиридин-2-иламино]-7Н-пирроло[2,3-d]пиримидин-6-карбоновой кислоты,
или его фармацевтически приемлемая соль.
17. Фармацевтическая композиция для лечения рака, содержащая соединение по любому из предыдущих пунктов или его фармацевтически приемлемую соль и фармацевтически приемлемый носитель или инертный наполнитель.
18. Соединение по п.1, которое представляет собой 7-циклопентил-N,N-диметил-2-(5-((1R,6S)-9-метил-4-оксо-3,9-диазабицикло[4.2.1]нонан-3-ил)пиридин-2-иламино)-7Н-пирроло[2,3-d]пиримидин-6-карбоксамид, описывающийся следующей формулой:

или его фармацевтически приемлемая соль.
19. Фармацевтическая композиция для лечения рака, содержащая соединение по п.18 или его фармацевтически приемлемую соль и фармацевтически приемлемый носитель или инертный наполнитель.
20. Способ лечения рака у пациента, нуждающегося в его лечении, включающий введение пациенту соединения по п.18 или его фармацевтически приемлемой соли в эффективном количестве, где рак выбран из группы, включающей лимфому из клеток зоны мантии, множественную миелому, рак молочной железы, плоскоклеточный рак пищевода, меланому, немелкоклеточный рак легких, рак толстой кишки и рак поджелудочной железы.
21. Соединение по п.1, которое представляет собой диметиламид 7-циклопентил-2-[5-((1R,6S)-4-оксо-3,9-диазабицикло[4.2.1]нон-3-ил)пиридин-2-иламино]-7Н-пирроло[2,3-d]пиримидин-6-карбоновой кислоты, описывающийся следующей формулой:

или его фармацевтически приемлемая соль.
22. Соединение по п.1, которое представляет собой метиламид 7-циклопентил-2-[5-((1R,6S)-4-оксо-3,9-диазабицикло[4.2.1]нон-3-ил)пиридин-2-иламино]-7Н-пирроло[2,3-d]пиримидин-6-карбоновой кислоты, описывающийся следующей формулой:

или его фармацевтически приемлемая соль.
23. Соединение по п.1, которое представляет собой метиламид 7-циклопентил-2-[5-((1R,6S)-9-метил-4-оксо-3,9-диазабицикло[4.2.1]нон-3-ил)пиридин-2-иламино]-7Н-пирроло[2,3-d]пиримидин-6-карбоновой кислоты, описывающийся следующей формулой:

или его фармацевтически приемлемая соль.
24. Соединение по п.1, которое представляет собой диметиламид 7-циклопентил-2-[5-(3,8-диазабицикло[3.2.1]октан-3-карбонил)пиридин-2-иламино]-7Н-пирроло[2,3-d]пиримидин-6-карбоновой кислоты, описывающийся следующей формулой:

или его фармацевтически приемлемая соль.
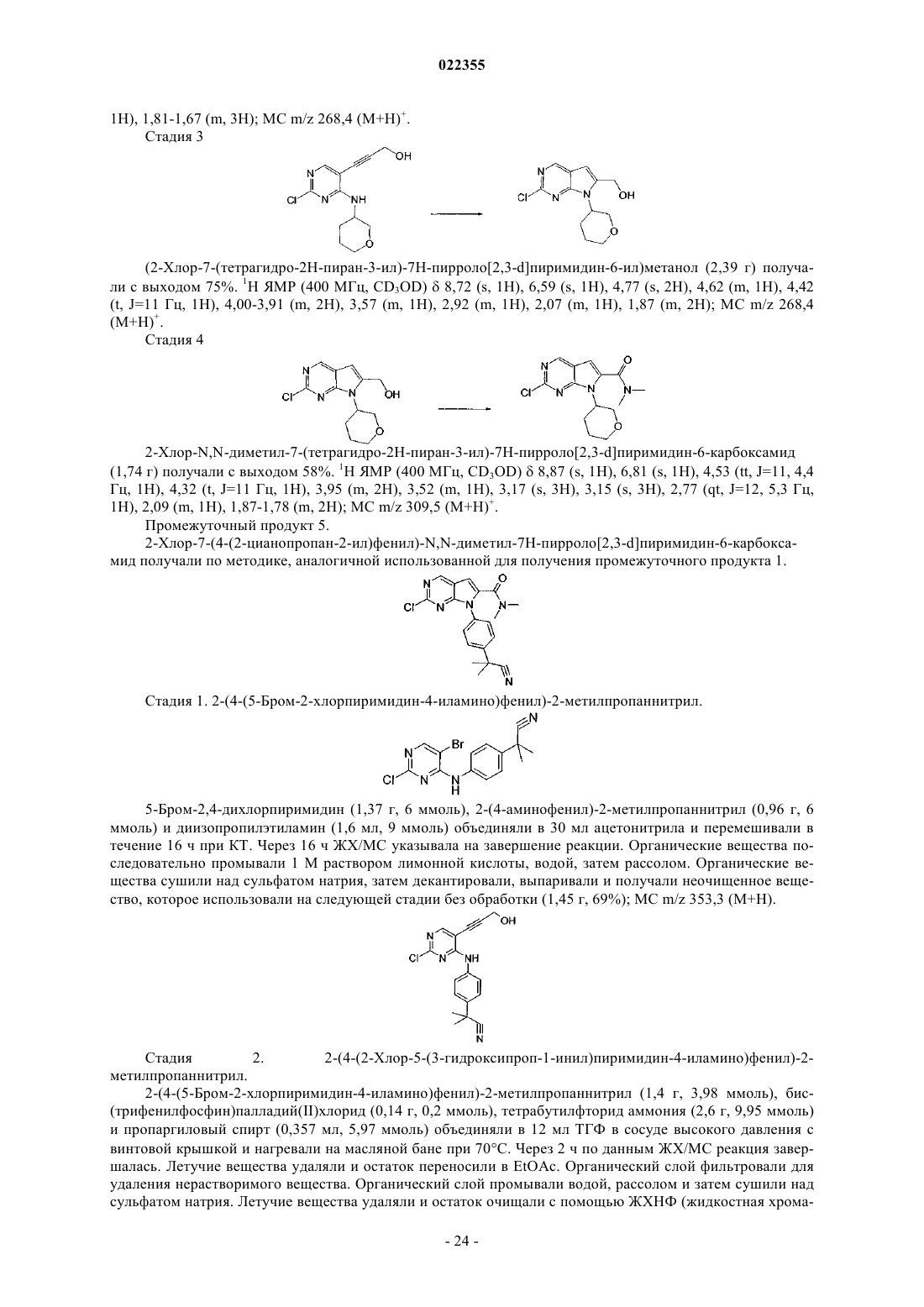
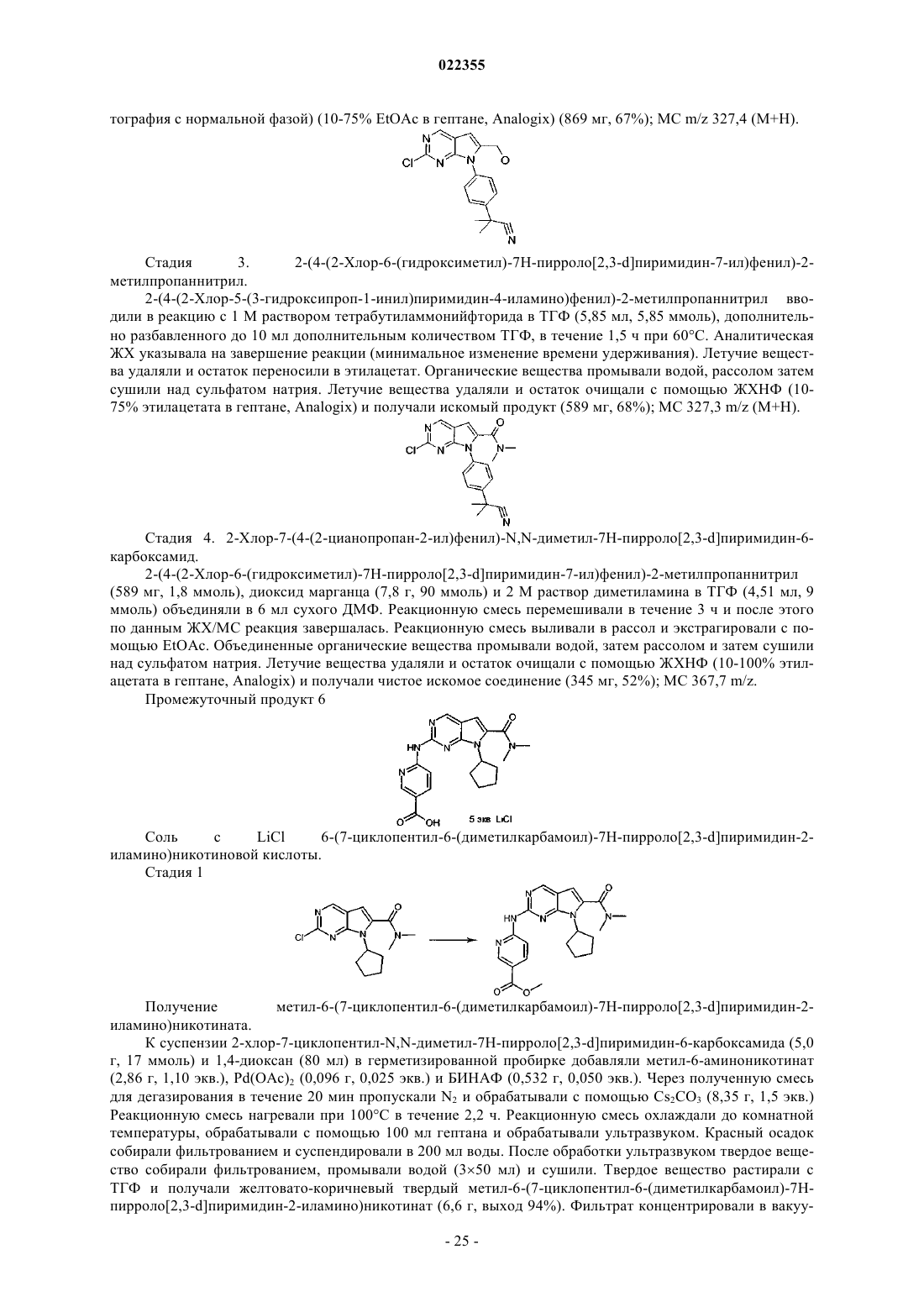
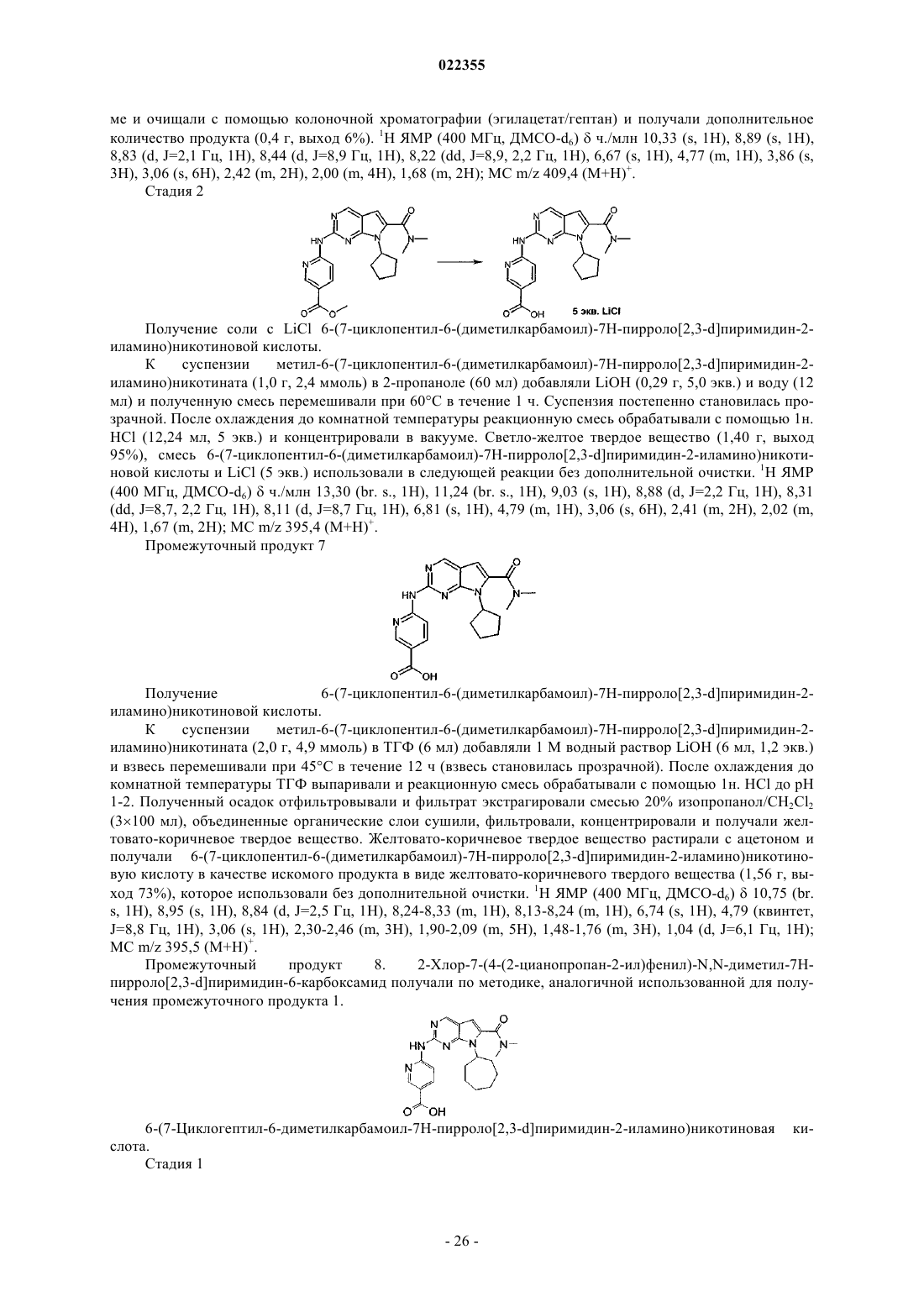
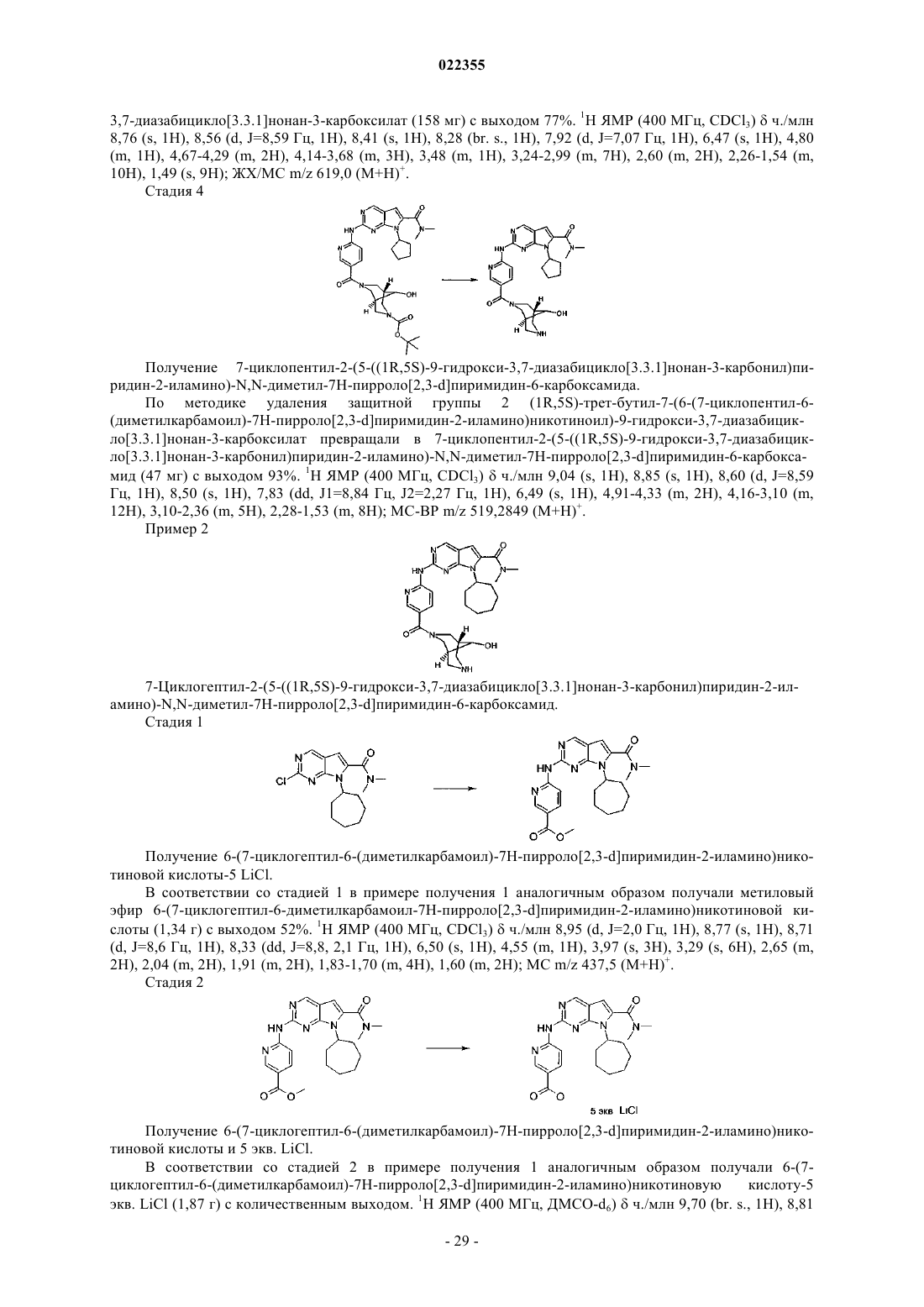
Текст
в которой R1, R2Y, R4, R8-R11, А и L определены в изобретении к их фармацевтически приемлемым солям. Изобретение также относится к фармацевтическим композициям, содержащим указанное соединение и фармацевтически приемлемый носитель или инертный наполнитель. Изобретение также относится к способам лечения рака, включающим введение пациенту соединения формулы(I) или его фармацевтически приемлемой соли в эффективном количестве, где рак выбран из группы, включающей лимфому из клеток зоны мантии, множественную миелому, рак молочной железы, плоскоклеточный рак пищевода, меланому, немелкоклеточный рак легких, рак толстой кишки и рак поджелудочной железы. Область техники, к которой относится изобретение Настоящее изобретение относится к новым пирролопиримидинам и содержащим их фармацевтическим композициям, точнее, к пирролопиримидинам и содержащим их фармацевтическим композициям,которые являются ингибиторами CDK4/6. Настоящее изобретение также относится к применению этих соединений и композиций для лечения гиперпролиферативных нарушений, таких как рак. Уровень техники Протекание клеточного цикла млекопитающих является строго регулируемым процессом, в котором переход через разные фазы протекает строго регулируемым образом и проходит через множество определенных контрольных точек. Белок ретинобластомы (pRb) является белком контрольной точки для перехода из фазы G1 в фазу S. pRb ассоциируется с семейством факторов транскрипции E2F для предупреждения их активности при отсутствии соответствующих стимулов роста (см. Ortega et al., Biochimica(11), 1770-1783). При стимулировании митогеном покоящиеся клетки начинают переходить в фазу S с помощью вновь синтезированных D-циклинов, которые являются активаторами циклинзавимимых киназ 4 и 6 (CDK4/6). После связывания с циклинами CDK4/6 дезактивирует белок pRb путем фосфорилирования. Фосфорилирование pRb приводит к высвобождению E2F, предназначенных для прямой транскрипции генов, необходимых для фазы S. Для полной дезактивации pRb необходимо фосфорилирование обеими циклинами D-CDK4/6 и E-CDK2. Показано, что фосфорилирование посредством CDK4/6 по специфическим сайтам pRb (Ser780, Ser795) необходимо для фосфорилирования циклинами E-CDK2 (см.Lundberg et al., Molecular and Cellular Biology 1998; 18 (2):753-761). В дополнение к D-циклинам активность CDK4/6 регулируется с помощью р 16, кодируемого геном INK4a, который ингибирует активность киназы (см. Kamb et al., Science 1994; 264 (5157):436-440). Белки CIP/KIP, которые являются ингибиторами циклина E-CDK2, также связываются с комплексом циклин D-CDK4/6, и это приводит к дополнительной дезактивации CDK2 путем изоляции белков CIP/KIP от их мишени (см. Sherr et al., GenesDevelopment 1999; 13 (12):1501-1512). Поэтому циклин D-CDK4/6 является ключевым ферментным комплексом, который регулирует переход от фазы G1 к фазе S. При раке путь D-циклин-CDK4/6-INK4a-pRb повсеместно нарушается для обеспечения пролиферации клеток. В большинстве случаев (80%) при раке сохраняется функциональный pRb и используются различные механизмы для повышения активности киназы CDK4/6 (см. Ortega et al., Biochimica et Biophysica Acta-Reviews on Cancer 2002; 1602 (1):73-87; Shapiro, Journal of Clinical Oncology 2006; 24(11):1770-1783. При лимфоме из клеток зоны мантии (MCL) циклин D1 подвергается транслокации в промотор IgH (t11:14), что приводит к конститутивной экспрессии белка, приводящего к активацииCDK4/6 (см. Amin, et al., Archives of PathologyLaboratory Medicine 2003; 127 (4):424-431; Oudat, et al.,Modern Pathology 2001; 14 (1):175A). Эта транслокация наблюдается в 90% случаев MCL и считается патогномоничной для заболевания. D-циклин также подвергается транслокации в 20% множественных миелом (см. Bergsagel et al. Immunological Reviews 2003; 194 (1):96-104). В дополнение к транслокации содержание D-циклина также можно увеличить с помощью амплификации или сверхэкспрессии и примерами этого могут служить плоскоклеточный рак пищевода, при котором является значительной амплификация циклина D1 (см. Jiang, et al., Cancer Research 1992; 52(10):2980-2983), и рак молочной железы, при котором является частой сверхэкспрессия циклина D1 (см.Arnold et ah, Journal of Clinical Oncology 2005). Активность киназы CDK4/6 также можно повысить с помощью амплификации самого гена CDK4 и совместной амплификации генов CDK4 и MDM2 и это наблюдается почти во всех случаях дедифференцированных липосарком (см. Sirvent, et al., American Journalof Surgical Pathology 2007; 31 (10): 1476-1489). Генетический ингибитор CDK4/6 также часто инактивируется при раке для обеспечения активации CDK4/6 и примеры такого рака включают немелкоклеточный рак легких, меланому и рак поджелудочной железы (Brambilla, et ah, Journal of Pathology 1999; 188(4):351-360; Cowgill et ah, American Journal of Surgery 2003; 186 (3):279-286; Gazzeri, et ah, Oncogene 1998; 16 (4):497-504; Kamb et ah, Science 1994; 264 (5157):436-440; Ortega et ah, Biochimica et Biophysica ActaReviews on Cancer 2002; 1602 (1):73-87). В дополнение к этим генетическим дефектам, непосредственно связанным с путем D-циклинCDK4/6-INK4a-pRb, активность киназ CDK4/6 также может повыситься онкогенными аберрациями митогенных путей, что увеличивает экспрессию D-циклина. Примеры включают амплификации EGFR при немелкоклеточном раке легких (NSCLC), активацию мутаций K-Ras при раке поджелудочной железы,мутации V600E B-Raf при меланоме и инактивацию PTEN при раке толстой кишки (см. Dailey, et al., CytokineGrowth Factor Reviews 2005; 16 (2):233-247; Engelman, Nature Reviews Cancer 2009; 9 (8)-550562; Garcia-Echeverria, Purinergic Signalling 2009; 5 (1):117-125, Gray-Schopfer et ah, Cancer and MetastasisReviews 2005; 24 (1):165-183, John, et ah, Oncogene 2009; 28:S14-S23, Sharma, et ah, Nature Reviews Cancer 2007; 7 (3):169-181). Все это показывает, что в большом количестве новообразований у человека усиленная пролиферация клеток обеспечивается путем повышения активности CDK4/6 и малая молекула-ингибитор этих киназ может оказаться эффективным средством лечения этих заболеваний. Ингибиторы разных CDK являются известными и поданы заявки на патенты, посвященные таким ингибиторам (см., например, WO2007/140222). Поэтому были предприняты усилия по синтезу соединений, которые ингибируют активностьCDK4/6 и в данной области техники раскрыт целый ряд таких соединений. Однако, поскольку существует целый ряд патологических ответов, которые опосредуются с помощью CDK4/6, постоянно необходимы ингибиторы CDK4/6, которые можно использовать для лечения различных патологических состояний, включая рак. Краткое изложение сущности изобретения Настоящее изобретение относится к новым пирролопиримидинам формулы в которой R1, R2Y, R4, R8-R11, A и L определены в настоящем изобретении, и к их солям, включая их фармацевтически приемлемые соли. Соединения, предлагаемые в настоящем изобретении, являются ингибиторами CDK4/6 и могут быть применимы для лечения заболеваний и нарушений, опосредуемых с помощью CDK4/6, таких как рак, включая лимфому из клеток зоны мантии, липосаркому, немелкоклеточный рак легких, меланому,плоскоклеточный рак пищевода и рак молочной железы. Настоящее изобретение также относится к фармацевтическим композициям, содержащим соединение, предлагаемое в настоящем изобретении. Настоящее изобретение также относится к способам ингибирования активности CDK4/6 и к лечению связанных с ним нарушений с использованием соединения, предлагаемого в настоящем изобретении, или фармацевтической композиции, содержащей соединение, предлагаемое в настоящем изобретении. Подробное описание изобретения Новые пирролопиримидины представляют собой соединения формулы в которой R1 обозначает С 3-С 7-алкил; С 4-С 7-циклоалкил, необязательно содержащий один заместитель, выбранный из группы, включающей C1-С 6-алкил и ОН; фенил, необязательно содержащий один заместитель, выбранный из группы, включающей C1-С 6-алкил, C(CH3)2CN и ОН; пиперидинил, необязательно замещенный одним циклопропилом или С 1-С 6-алкилом; тетрагидропиранил, необязательно замещенный одним циклопропилом или C1-С 6-алкилом; или бицикло[2.2.1]гептанил; А обозначает СН или N;R11 обозначает водород или С 1-С 4-алкил;L обозначает связь или С(О);m и n все независимо равны 1, 2 или 3 при условии, что m и n не оба равны 3; каждый R2Y необязательно содержит от 1 до 4 заместителей, каждый из которых независимо выбран из группы, включающей: C1-С 3-алкил, необязательно содержащий 1 или 2 заместителя, каждый из которых независимо выбран из группы, включающей гидроксигруппу; NH2 и -S-C1-С 3-алкил; CD3; галоген; оксогруппу; C1-С 3 галогеналкил; гидроксигруппу; NH2; диметиламиногруппу; бензил; -С(О)-С 1-С 3-алкил, необязательно содержащий 1 или 2 заместителя, каждый из которых независимо выбран из группы, включающей NH2,-SCH3 и NHC(O)CH3; -S(О)2-С 1-С 4-алкил; пирролидинил-С(О)- и -С(О)2-С 1-С 3-алкил;R5, R6, R7, R8, R9 и R10 все независимо обозначают Н или дейтерий,или его фармацевтически приемлемая соль. Одним вариантом осуществления настоящего изобретения является соединение формулы (I-B) в которой L обозначает связь или С(О);m и n все независимо равны 1, 2 или 3 при условии, что m и n не оба равны 3; и каждый R2Y необязательно содержит от 1 до 4 заместителей, каждый из которых независимо выбран из группы, включающей C1-С 3-алкил, необязательно содержащий 1 или 2 заместителя, каждый из которых независимо выбран из группы, включающей гидроксигруппу; NH2 и -S-C1-С 3-алкил; CD3; С 1-С 3 галогеналкил; гидроксигруппу; NH2; диметиламиногруппу; бензил; -С(О)-С 1-С 3-алкил, необязательно содержащий 1 или 2 заместителя, каждый из которых независимо выбран из группы, включающей NH2,-SCH3 и NHC(O)CH3; -S(O)2-C1-С 4-алкил; пирролидинил-С(О)- и -С(О)2-С 1-С 3-алкил;R1, R4-R11 и А являются такими, как определено выше для формулы (I),или его фармацевтически приемлемая соль. Предпочтительно в вышеуказанных соединениях L обозначает С(О) или связь, А обозначает СН иR11 обозначает водород. В предпочтительном варианте R4 обозначает C(R5)(R6)(R7) и R5, R6, R7, R8, R9 и R10 обозначают водород. Предпочтительно R1 обозначает С 4-С 7-циклоалкил, необязательно замещенный одним C1-С 6 алкилом, более предпочтительно R1 обозначает циклобутил, циклопентил, циклогексил или циклогептил,более предпочтительно незамещенный циклопентил. В предпочтительном варианте R2Y является незамещенным.-4 022355 В другом предпочтительном варианте R2Y обозначает любой из которых необязательно замещен одним C1-С 3-алкилом. В еще одном предпочтительном варианте R2Y обозначает необязательно замещенный одним C1-С 3-алкилом. Конкретные соединения, предлагаемые в настоящем изобретении, включают Термины и определения. При использовании в настоящем изобретении термин "алкил" означает полностью насыщенный разветвленный или неразветвленный углеводородный фрагмент, содержащий от 1 до 6 атомов углерода(С 1-С 6-алкил), от 3 до 7 атомов углерода (С 3-С 7-алкил), от 1 до 4 атомов углерода (С 1-С 4-алкил) или от 1 до 3 атомов углерода (C1-С 3-алкил). Типичные примеры алкилов включают, но не ограничиваются только ими, метил, этил, н-пропил, изопропил, н-бутил, изобутил, втор-бутил, трет-бутил, н-пентил, 1 метилбутил, 2-метилбутил, 3-метилбутил, 3,3-диметилпропил, гексил, 2-метилпентил и т.п. При использовании в настоящем изобретении термин "циклоалкил" означает неароматические карбоциклические кольца, которые частично или полностью гидрированы и могут представлять собой единичное кольцо, бициклическое кольцо, трициклическое кольцо или спирановое кольцо, если не указано иное, циклоалкил означает циклические углеводородные группы, содержащие 3-14 атомов углерода. Циклоалкильные группы также означают циклические углеводородные группы, содержащие от 3 до 9 кольцевых атомов углерода, или от 4 до 7 кольцевых атомов углерода. Типичные моноциклические углеводородные группы включают, но не ограничиваются только ими, циклопропил, циклобутил, циклопентил, циклопентенил, циклогексил, циклогексенил, циклогептил и т.п. Типичные бициклические углеводородные группы включают борнил, индил, гексагидроиндил, тетрагидронафтил, декагидронафтил, бицикло[2.1.1]гексил, бицикло[2.2.1]гептил, бицикло[2.2.1]гептенил, 6,6-диметилбицикло[3.1.1]гептил,2,6,6-триметилбицикло[3.1.1]гептил, бицикло[2.2.2]октил и т.п. Типичные трициклическое углеводородные группы включают адамантил и т.п."Дейтерий", или "D", или "d" означает изотоп водорода, ядро которого содержит один протон и один нейтрон. Если указано, что в конкретном положении находится дейтерий, следует понимать, что содержание дейтерия в этом положении больше, чем природное содержание дейтерия (обычно 0,015%). Если не указано иное, то, если указано, что в положении находится "D" или "дейтерий", следует понимать, что в положении находится дейтерий при содержании, превышающем природное содержание дейтерия. При использовании в настоящем изобретении термин "галоген" означает фтор, хлор, бром и йод. При использовании в настоящем изобретении термин "галогеналкил" означает алкильную группу, в которой по меньшей мере один атом водорода, присоединенный к атому углерода алкильной группы,заменен на галоген, предпочтительно фтор. Примеры галогеналкильных групп включают моно-, ди- и трифторметил, моно-, ди- и трихлорметил, моно-, ди-, три-, тетра- и пентафторэтил и моно-, ди-, три-,тетра- и пентахлорэтил. При использовании в настоящем изобретении термин "необязательно замещенный" показывает, что группа, такая как алкил, циклоалкил или конкретная группа R, может быть незамещенной или содержит один или большее количество заместителей, определенных в настоящем изобретении. Термин "замещенная" применительно к группе показывает, что атом водорода, присоединенный к атому группы, является замещенным. Следует понимать, что термин "замещенный" включает косвенное указание на то, что такое замещение соответствует допустимой валентности замещенного атома и заместителя и что замещение приводит к стабильному соединению (т.е. оно не подвергается самопроизвольному превращению,например, путем перегруппировки, циклизации или отщепления). В некоторых вариантах осуществления один атом может содержать более одного заместителя, если такое замещение соответствует допустимой валентности атома. Подходящие заместители определены в настоящем изобретении для каждой замещенной или необязательно замещенной группы. Термин "соединения, предлагаемые в настоящем изобретении" (если специально не указано иное) означает соединения формулы (I) и их соли, включая фармацевтически приемлемые соли соединений, а также все стереоизомеры (включая диастереоизомеры и энантиомеры), таутомеры и изотопно меченые соединения. Сольваты и гидраты обычно считаются фармацевтическими композициями соединений,предлагаемых в настоящем изобретении. При использовании в настоящем изобретении обозначения и условия, использующиеся в этих методиках, схемах и примерах, согласуются с использующимися в современной научной литературе, например в Journal of the American Chemical Society или the Journal of Biological Chemistry. Если не указано иное, то все исходные вещества получали от коммерческих поставщиков и использовали без дополнительной очистки. В примерах и описании могут использоваться следующие аббревиатуры: АЦН - ацетонитрил,АсОН - уксусная кислота,BnBr - бензилбромид,boc - трет-бутоксикарбонил,С - градусы Цельсия,КДИ - карбонилдиимидазол,КСК - камфорсульфоновая кислота,CS2CO3 - карбонат цезия,Да - Дальтоны,ДИБАГ, ДИБАГ-Н - диизобутилалюминийгидрид,ДИПЭА - диизопропилэтиламин,ДИПК - N,N'-диизопропилкарбодиимид,ДМФ - N,N-диметилформамид,ДМИ - 1,3-диметил-2-имидазолидинон,ПДМ - перйодинан Десса-Мартина,ДЦК - N,N-дициклогексилкарбодиимид,ДХЭ - дихлорэтан,ДХМ - дихлорметан,ДМАП - 4-диметиламинопиридин,ДМСО - диметилсульфоксид,EtOAc - этилацетат,EtOH - этанол,экв. - эквиваленты,g - газообразное вещество,ч - час,ГАТУ - О-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилуронийгексафторфосфат,ГМФА - гексаметилфосфорамид,HCl - хлористо-водородная кислота,imid. - имидазол,К - градусы Кельвина,KHMDS - гексаметилдисилилазид калия,K2CO3 - карбонат калия,ДАЛ - диизопропиламид лития,LiBH4 - борогидрид лития,LHMDS - гексаметилдисилазид лития,ЖХ - жидкостная хроматография,ЖХ/МС - жидкостная хроматография-масс-спектроскопия,М - молярная концентрация,MeCN - ацетонитрил,МеОН - метанол,MgSO4 - сульфат магния,МГц - мегагерц,Мин - минута,NaBH4 - борогидрид натрия,N - нормальная концентрация,ЯМР - ядерный магнитный резонанс,Pd/C - палладий на угле,ПЭГ(750) - О-(2-аминоэтил)-О'-метилполиэтиленгликоль 750; NH2(CH2CH2O)nCH3; CAS [8050664-5]; Fluka 07964; средняя молекулярная масса=750,ПС - полистирол,Ру - пиридин,ч./млн - частей на миллион,ОФ - обращенная фаза,КТ - комнатная температура,Rt - время удерживания,s - твердое вещество,ТБС - трет-бутилдиметилсилил,ТМС - триметилсилил,ТБАФ - тетрабутиламмонийфторид,TBTU - О-бензотриазол-1-ил-N,N,N'N'-тетраметилуронийтетрафторборат,ТСХ - тонкослойная хроматография,- 10022355 ТЭА - триэтиламин,ТФК - трифторуксусная кислота,ТГФ - тетрагидрофуран,ч - час,мин - минута,m/z - отношение массы к заряду,МС - масс-спектроскопия,ЯМР - ядерный магнитный резонанс. Специалист в данной области техники должен понимать, что можно получить соли, включая фармацевтически приемлемые соли соединений формулы (I). При использовании в настоящем изобретении термины "соль" или "соли" означает соль присоединения с кислотой или основанием соединения, предлагаемого в настоящем изобретении. Термин "фармацевтически приемлемые соли" означает соли, которые сохраняют биологическую эффективность и характеристики соединений, предлагаемых в настоящем изобретении, и которые обычно не являются нежелательными в биологическом или другом отношении. Поэтому настоящее изобретение также относится к солям, предпочтительно фармацевтически приемлемым солям соединений формулы (I). Фармацевтически приемлемые соли присоединения с кислотами, которые можно получить с неорганическими кислотами и органическими кислотами, включают, например, ацетат, аспартат, бензоат,безилат, бромид/гидробромид, бикарбонат/карбонат, бисульфат/сульфат, камфорсульфонат, хлорид/гидрохлорид, хлортеофиллонат, цитрат, этандисульфонат, фумарат, глюцептат, глюконат, глюкуронат, гиппурат, гидройодид/йодид, изетионат, лактат, лактобионат, лаурилсульфат, малат, малеат, малонат, манделат, мезилат, метилсульфат, нафтоат, напзилат, никотинат, нитрат, октадеканоат, олеат, оксалат, пальмитат, памоат, фосфат/гидрофосфат/дигидрофосфат, полигалактуронат, пропионат, стеарат,сукцинат, сульфосалицилат, тартрат, тозилат и трифторацетат. Неорганические кислоты, из которых можно получить соли, включают, например, хлористоводородную кислоту, бромисто-водородную кислоту, серную кислоту, азотную кислоту, фосфорную кислоту и т.п. Органические кислоты, из которых можно получить соли, включают, например, уксусную кислоту,пропионовую кислоту, гликолевую кислоту, щавелевую кислоту, малеиновую кислоту, малоновую кислоту, янтарную кислоту, фумаровую кислоту, винную кислоту, лимонную кислоту, бензойную кислоту,миндальную кислоту, метансульфоновую кислоту, этансуль фоновую кислоту, толуолсульфоновую кислоту, сульфосалициловую кислоту и т.п. Фармацевтически приемлемые соли присоединения с основаниями можно получить с неорганическими и органическими основаниями. Неорганические основания, из которых можно образовать соли, включают, например, соли аммония и металлов групп I-XII Периодической системы. В некоторых вариантах осуществления солями являются соли натрия, калия, аммония, кальция, магния, железа, серебра, цинка и меди; особенно подходящие соли включают соли аммония, калия, натрия, кальция и магния. Органические основания, из которых можно образовать соли, включают, например, первичные,вторичные и третичные амины, замещенные амины, включая природные замещенные амины, циклические амины, основные ионообменные смолы и т.п. Некоторые органические амины включают изопропиламин, бензатин, холин, диэтаноламин, диэтиламин, лизин, меглумин, пиперазин и трометамин. Фармацевтически приемлемые соли, предлагаемые в настоящем изобретении, можно синтезировать из исходного соединения, основного или кислотного фрагмента по обычным химическим методикам. Обычно такие соли можно получить по реакции свободных кислотных форм этих соединений со стехиометрическим количеством соответствующего основания (такого как гидроксид, карбонат, бикарбонат и т.п. Na, Ca, Mg или K) или по реакции свободных основных форм этих соединений со стехиометрическим количеством соответствующей кислоты. Такие реакции обычно проводят в воде, или в органическом растворителе, или в их смеси. Обычно, если это возможно, предпочтительными являются неводные среды, такие как эфир, этилацетат, этанол, изопропанол или ацетонитрил. Перечни дополнительных подходящих солей приведены, например, в публикациях "Remington's Pharmaceutical Sciences", 20th ed., MackUse" by Stahl and Wermuth (Wiley-VCH, Weinheim, Germany, 2002). Соединения формулы (I) могут содержать один или большее количество асимметричных центров(также называющихся хиральными центрами) и поэтому могут существовать в виде отдельных энантиомеров, диастереоизомеров или других стереоизомерных форм, или в виде их смесей. Хиральные центры,такие как хиральные атомы углерода, также могут содержаться в заместителе, таком как алкильная группа. Если стереохимическая конфигурация хирального центра, содержащегося в формуле (I) или в любой химической структуре, приведенной в настоящем изобретении, не указана, то подразумевается, что структура описывает любой стереоизомер или их смеси. Таким образом, соединения формулы (I), содержащие один или большее количество хиральных центров, можно использовать в виде рацемических смесей, энантиомерно обогащенных смесей или в виде энантиомерно чистых отдельных стереоизомеров. При использовании в настоящем изобретении термин "изомеры" означает разные соединения, которые обладают одной и той же молекулярной формулой. Кроме того, при использовании в настоящем изобретении термин "оптический изомер" или "стереоизомер" означает любую из разных стереоизомерных конфигураций, которые могут существовать для данного соединения, предлагаемого в настоящем изобретении, и включает геометрические изомеры. Следует понимать, что заместитель может быть присоединен к атому углерода, являющемуся хиральным центром. Поэтому настоящее изобретение включает энантиомеры, диастереоизомеры и рацематы соединения. "Энантиомеры" представляют собой пару стереоизомеров, которые являются зеркальными изображениями, не налагающимися друг на друга. Смесь двух энантиомеров состава 1:1 называется "рацемической" смесью. Этот термин используется для обозначения рацемической смеси, если это является подходящим. "Диастереоизомеры" являются стереоизомерами, которые содержат не менее двух асимметрических атомов, но которые не являются зеркальными изображениями друг друга. Абсолютную стереохимическую конфигурацию описывают с помощью R-S системы Кана-Ингольда-Прелога. Если соединение является чистым энантиомером, то стереохимическую конфигурацию каждого хирального атома углерода можно описать как R или S. Разделенные соединения, абсолютная конфигурация которых не установлена, можно описать с помощью символов (+) или (-) в зависимости от направления, в котором они вращают плоскость поляризации света при длине волны линии D натрия (право- и левовращающие). Некоторые соединения, описанные в настоящем изобретении, содержат один или большее количество асимметрических центров и поэтому могут образовывать энантиомеры, диастереоизомеры и другие стереоизомерные формы, абсолютную стереохимическую конфигурацию которых можно обозначить как (R)-или (S)-. В объем настоящего изобретения входят все такие возможные изомеры, включая рацемические смеси, оптически чистые формы и смеси промежуточного состава. Оптически активные (R)- и (S)-изомеры можно получить с помощью хиральных синтонов или хиральных реагентов или выделить по обычным методикам. Любые полученные рацематы конечных продуктов или промежуточных продуктов можно разделить на оптические антиподы по известным методикам, например путем разделения солей их диастереоизомеров, полученных с оптически активной кислотой или основанием, выделения оптически активного кислотного или основного соединения. В частности, таким образом можно использовать фрагмент основания, чтобы разделить соединения, предлагаемые в настоящем изобретении, на оптические антиподы, например, с помощью фракционной кристаллизации соли, образованной с оптически активной кислотой, например винной кислотой, дибензоилвинной кислотой, диацетилвинной кислотой, ди-О,О'-п-толуилвинной кислотой, миндальной кислотой, яблочной кислотой или камфор-10-сульфоновой кислотой. Рацемические продукты также можно разделить с помощью хиральной хроматографии, например высокоэффективной жидкостной хроматографии (ВЭЖХ) с использованием хирального сорбента. Если соединение, предлагаемое в настоящем изобретении, содержит двойную связь, то заместители могут находиться в Е- или Z-конфигурации. Если соединение представляет собой дизамещенный циклоалкил, то заместители циклоалкила могут находиться в цис- или транс-конфигурации. Предполагается,что в объем настоящего изобретения входят все таутомерные формы. Любой асимметрический атом (например, углерода и т.п.) соединения (соединений), предлагаемого в настоящем изобретении, может находиться в рацемической или энантиомерно обогащенной форме,например в (R)-, (S)- или (R,S)-конфигурации. В некоторых вариантах осуществления каждый асимметрический атом характеризуется энантиомерным избытком в (R)- или (S)-конфигурации, составляющим не менее 50%, энантиомерным избытком, составляющим не менее 60%, энантиомерным избытком, составляющим не менее 70%, энантиомерным избытком, составляющим не менее 80%, энантиомерным избытком, составляющим не менее 90%, энантиомерным избытком, составляющим не менее 95%, или энантиомерным избытком, составляющим не менее 99%. Заместители атомов, образующих кратную связь, если это возможно, могут находиться в цис-(Z)- или транс-(Е)-форме. С использованием различий физико-химических характеристик компонентов любые полученные смеси изомеров можно разделить на чистые или в основном чистые геометрические или оптические изомеры, диастереоизомеры, рацематы, например, с помощью хроматографии и/или фракционной кристаллизации. Замещение более тяжелыми изотопами, предпочтительно дейтерием (т.е. Н или D), может обеспечить некоторые терапевтические преимущества, обусловленные их более высокой метаболической стабильностью, например, увеличенной длительностью полувыведения in vivo, или возможностью использования меньших доз, или улучшением терапевтического индекса. Следует понимать, что в этом контексте дейтерий рассматривается в качестве заместителя в соединениях, предлагаемых в настоящем изобретении. Концентрацию такого более тяжелого изотопа, в частности дейтерия, можно определить с помощью коэффициента изотопного обогащения. Термин "коэффициент изотопного обогащения" при использовании в настоящем изобретении означает отношение содержания изотопа к содержанию конкретного изотопа в природе. Если заместитель в соединении, предлагаемом в настоящем изобретении, означает дейтерий, то такое соединение характеризуется коэффициентом изотопного обогащения для каждого обозначенного атома дейтерия, равным не менее 3500 (содержание дейтерия для каждого обозначенного атома дейтерия равно 52,5%), не менее 4000 (содержание дейтерия равно 60%), не менее 4500 (содержание дейтерия равно 67,5%), не менее 5000 (содержание дейтерия равно 75%), не менее 5500 (содержание дейтерия равно 82,5%), не менее 6000 (содержание дейтерия равно 90%), не менее 6333,3 (содержание дейтерия равно 95%), не менее 6466,7 (содержание дейтерия равно 97%), не менее 6600 (содержание дейтерия равно 99%) или не менее 6633,3 (содержание дейтерия равно 99,5%). В дополнение к приведенным в настоящем изобретении содержащим дейтерий соединениям подразумевается, что любая формула, приведенная в настоящем изобретении, также характеризует немеченые формы, а также изотопно меченые формы соединений. Изотопно меченые соединения обладают структурами, изображенными формулами, приведенными в настоящем изобретении, с тем отличием, что один или большее количество атомов заменены атомом, обладающим указанной атомной массой или массовым числом. Примеры изотопов, которые можно включить в соединения, предлагаемые в настоящем изобретении, включают изотопы водорода, углерода, азота, кислорода, фосфора, фтора и хлора, такие как 2 Н, 3 Н, 11 С, 13 С, 14 С, 15N, 18F 31P, 32 Р, 35S, 36C1, 125I соответственно. В объем настоящего изобретения входят различные изотопно меченые соединения, предлагаемые в настоящем изобретении, например, в которые включены радиоактивные изотопы, такие как 3 Н, 13 С и 14 С. Такие изотопно меченые соединения применимы для изучения метаболизма (содержащие 14 С), исследования кинетики реакций (содержащие,например, 2 Н или 3 Н), в методологиях детектирования и визуализации, таких как позитронная эмиссионная томография (ПЭТ) или однофотонная эмиссионная компьютерная томография (ОФЭКТ), включая исследование распределения лекарственного средства или субстрата в тканях или лучевую терапию пациентов. В частности, содержащее 18F или меченое им соединение может быть особенно предпочтительным для исследований с помощью ПЭТ или ОФЭКТ. Изотопно меченые соединения, предлагаемые в настоящем изобретении, и их пролекарства обычно можно получить по методикам, раскрытым в схемах или в примерах и синтезах, описанных ниже, путем замены не содержащего изотопа реагента на легко доступный изотопно меченый реагент. Изотопно-обогащенные соединения, предлагаемые в настоящем изобретении, обычно можно получить по стандартным методикам, известным специалистам в данной области техники, или по методикам,аналогичным описанным в прилагаемых примерах и синтезах, путем использования соответствующего изотопно меченого реагента вместо использовавшегося ранее немеченого реагента. Кроме того, соединения, предлагаемые в настоящемизобретении, включая их соли, также можно получить в форме их гидратов или с включением других растворителей, использовавшихся для их кристаллизации. Соединения, предлагаемые в настоящем изобретении, по своей природе или полученной структуре могут образовывать сольваты с фармацевтически приемлемыми растворителями (включая воду); поэтому в объем настоящего изобретения входят сольватированные и несольватированные формы. Термин "сольват" означает молекулярный комплекс соединения, предлагаемого в настоящем изобретении (включая его фармацевтически приемлемые соли) с одной или большим количеством молекул растворителя. Такими молекулами растворителя являются обычно использующиеся в фармацевтике, для которых известно, что они нетоксичны для реципиента, например вода, этанол и т.п. Термин "гидрат" означает комплекс, в котором молекулой растворителя является вода. Кроме того, фармацевтически приемлемые сольваты в контексте настоящего изобретения включают такие, в которых растворитель, использованный при кристаллизации, может быть изотопно замещенным, например D2O, d6-ацетон, d6 ДМСО. Настоящее изобретение также относится к пролекарствам соединений, предлагаемых в настоящем изобретении, которые in vivo превращаются в соединения, предлагаемые в настоящем изобретении. Пролекарство представляет собой активное или неактивное соединение, которое после введения субъекту химически изменяется вследствие физиологического воздействия in vivo, такого как гидролиз, метаболизм и т.п., с превращением в соединение, предлагаемое в настоящем изобретении. Методики приготовления и применения пролекарств и их применимость хорошо известны специалистам в данной области техники. Пролекарства можно по характеру разделить на две неэксклюзивные категории, пролекарствабиологические предшественники и пролекарства-носители; см. публикацию The Practice of MedicinalChemistry, Ch. 31-32 (Ed. Wermuth, Academic Press, San Diego, Calif., 2001). Обычно пролекарствабиологические предшественники являются соединениями, которые неактивны или обладают низкой активностью по сравнению с активным лекарственным соединением, содержат одну или большее количество защитных групп и превращаются в активную форму вследствие метаболизма или сольволиза. И активная форма лекарства, и любой высвободившийся продукт метаболизма должны обладать приемлемо низкой токсичностью. Пролекарства-носители являются лекарственными соединениями, которые содержат фрагментпереносчик, например, которые улучшают всасывание или локализуют высвобождение в центре (центрах) воздействия. Для такого пролекарства-носителя предпочтительно, чтобы связь между лекарственным фрагментом и фрагментом-переносчиком была ковалентной, пролекарство было неактивным или менее активным, чем лекарственное соединение, и любой высвобождающийся фрагмент-переносчик являлся в приемлемой степени нетоксичным. В случае пролекарств, для которых фрагмент-переносчик предназначен для усиления всасывания, высвобождение фрагмента-переносчика обычно должно быть быстрым. В других случаях предпочтительно использовать фрагмент, который обеспечивает медленное высвобождение, например некоторые полимеры или другие фрагменты, такие как циклодекстрины. Пролекарства-носители, например, можно использовать для улучшения одной или большего количества следующих характеристик: увеличения липофильности, увеличения длительности фармакологического воздействия, улучшения специфичности на участке воздействия, уменьшения токсичности и побочных реакций и/или улучшения характеристик лекарственного препарата (например, стабильности, растворимости в воде, подавления нежелательных органолептических или физико-химических характеристик). Например, липофильность можно увеличить путем этерификации (а) гидроксигрупп липофильными карбоновыми кислотами (например, карбоновой кислотой, содержащей по меньшей мере один липофильный фрагмент) или (b) карбоксигрупп липофильными спиртами (например, спиртом, содержащим по меньшей мере один липофильный фрагмент, например алифатическими спиртами). Типичными пролекарствами являются, например, эфиры свободных карбоновых кислот с Sацилпроизводными тиолов и О-ацилпроизводными спиртов или фенолов, где ацил обладает значением,определенным в настоящем изобретении. Подходящими пролекарствами часто являются фармацевтически приемлемые сложные эфиры, которые посредством гидролиза в физиологических условиях превращаются в исходную карбоновую кислоту, например низш. алкиловые сложные эфиры, циклоалкиловые сложные эфиры, низш. алкениловые сложные эфиры, бензиловые сложные эфиры, моно- или дизамещенные низш. алкиловые сложные эфиры, такие как -(амино, моно- или ди-низш.алкиламино-, карбокси-, низш. алкоксикарбонил)-низш. алкиловые сложные эфиры, -(низш. алканоилокси-, низш. алкоксикарбонил- или ди-низш. алкиламинокарбонил)-низш. алкиловые сложные эфиры, такие как пивалоилоксиметиловый эфир и т.п., обычно применяющиеся в данной области техники. Кроме того, амины маскируют путем образования арилкарбонилоксиметилпроизводных, которые in vivo расщепляются эстеразами с высвобождением свободного лекарственного средства и формальдегида (Bundgaard, J. Med. Chem. 2503 (1989. Кроме того, лекарственые средства, содержащие кислую группу NH, такую как имидазольную, имидную, индольную и т.п., маскируют с помощью N-ацилоксиметильных групп (Bundgaard, Design of Prodrugs, Elsevier (1985. Гидроксигруппы маскируют путем образования сложных и простых эфиров. В ЕР 039051 (Sloan and Little) раскрыты пролекарства, являющиеся основаниями Манниха гидроксамовых кислот, их получение и применение. Композиции. Другим объектом настоящего изобретения является фармацевтическая композиция, содержащая соединение формулы (I) или его фармацевтически приемлемую соль и фармацевтически приемлемый носитель. Фармацевтическую композицию можно приготовить для конкретных путей введения, таких как пероральное введение, парентеральное введение и ректальное введение и т.п. Кроме того, фармацевтические композиции, предлагаемые в настоящем изобретении, можно приготовить в твердой форме (включая без наложения ограничений капсулы, таблетки, пилюли, гранулы, порошки или суппозитории) или в жидкой форме (включая без наложения ограничений растворы, суспензии или эмульсии). Фармацевтические композиции можно подвергнуть обычной фармацевтической обработке, такой как стерилизация,и/или они могут содержать обычные инертные разбавители, смазывающие агенты или буферные агенты,а также вспомогательные вещества, такие как консерванты, стабилизаторы, смачивающие агенты, эмульгаторы и буферные агенты и т.п. Обычно фармацевтические композиции являются таблетками или капсулами из желатина, включающими активный ингредиент, а также а) разбавители, например лактозу, декстрозу, сахарозу, маннит,сорбит, целлюлозу и/или глицин; b) смазывающие агенты, например диоксид кремния, тальк, стеариновую кислоту, ее магниевую или кальциевую соль и/или полиэтиленгликоль; для таблеток также c) связующие, например алюмосиликат магния, крахмальную пасту, желатин, трагакантовую камедь, метилцеллюлозу, натриевую соль карбоксиметилцеллюлозы и/или поливинилпирролидон; при необходимостиd) разрыхлители, например крахмалы, агар, альгиновую кислоту или ее натриевую соль, или шипучие смеси; и/или е) абсорбенты, красители, ароматизаторы и подсластители. На таблетки по методикам, известным в данной области техники, можно нанести пленочное покрытие или энтеросолюбильное покрытие. Композиции, подходящие для перорального введения, содержат соединение, предлагаемое в настоящем изобретении, в эффективном количестве в виде таблеток, лепешек, водных или масляных суспензий, диспергирующихся порошков или гранул, эмульсий, твердых или мягких капсул или сиропов, или эликсиров. Композиции, предназначенные для перорального применения, получают по любой методике, известной в данной области техники для приготовления фармацевтических композиций, и такие композиции могут содержать один или большее количество агентов, выбранных из группы, включающей подсластители, ароматизаторы, красители и консерванты, чтобы получить фармацевтически привлекательные и обладающие приятным вкусом препараты. Таблетки могут содержать активный ингредиент в смеси с нетоксичными фармацевтически приемлемыми инертными наполнителями, которые являются подходящими для изготовления таблеток. Этими инертными наполнителями являются, например, инертные разбавители, такие как карбонат кальция, карбонат натрия, лактоза, фосфат кальция или фосфат натрия; гранулирующие и разрыхляющие агенты, например кукуруз- 14022355 ный крахмал или альгиновая кислота; связующие агенты, например крахмал, желатин или камедь акации; и смазывающие агенты, например стеарат магния, стеариновая кислота или тальк. Таблетки не содержат покрытия или на них по известным методикам наносят покрытие для задержки распада и всасывания в желудочно-кишечном тракте, что обеспечивает непрерывное воздействие в течение более длительного периода времени. Например, можно использовать такое замедляющее вещество, как глицерилмоностеарат или глицерилдистеарат. Препараты для перорального применения также могут представлять собой капсулы из твердого желатина, в которых активный ингредиент смешан с инертным твердым разбавителем, например карбонатом кальция, фосфатом кальция или каолином, или капсулы из мягкого желатина, в которых активный ингредиент смешан с водой или масляной средой, например арахисовым маслом, жидким парафином или оливковым маслом. Некоторые композиции для инъекций представляют собой изотонические водные растворы или суспензии, а суппозитории предпочтительно готовят из эмульсий или суспензий жиров. Указанные композиции можно стерилизовать и/или прибавлять к ним вспомогательные вещества, такие как консервирующие, смачивающие или эмульгирующие агенты, стимуляторы растворения, соли для регулирования осмотического давления и/или буферные агенты. Кроме того, они также могут содержать другие терапевтически ценные вещества. Указанные композиции получают по обычным технологиям смешивания,гранулирования или нанесения покрытий и они содержат примерно 0,1-75% или содержат примерно 150% активного ингредиента. Композиции, подходящие для чрескожного введения, содержат эффективное количество соединения, предлагаемого в настоящем изобретении, с носителем. Носители, подходящие для чрескожного введения, включают впитывающиеся фармакологически приемлемые растворители, способствующие проникновению через кожу реципиента. Например, устройства для чрескожного введения представляют собой повязку, включающую защитный слой, резервуар, содержащий соединение необязательно с носителями, необязательно барьер, регулирующий доставку соединения через кожу реципиента с заданной скоростью в течение пролонгированного периода времени, и средства закрепления устройства на коже. Композиции, подходящие для местного введения, например, на кожу или в глаза, включают водные растворы, суспензии, мази, кремы, гели или распыляемые композиции, например, для подачи в виде аэрозоля и т.п. Такие устройства местного действия являются особенно подходящими для воздействия на кожу, например для лечения рака кожи, например для использования профилактических средств в солнцезащитных кремах, лосьонах, аэрозольных препаратах и т.п. Поэтому они являются особенно подходящими для использования в средствах местного действия, включая косметические, хорошо известных в данной области техники. Они могут содержать солюбилизаторы, стабилизаторы, агенты, регулирующие тоничность, буферные агенты и консерванты. При использовании в настоящем изобретении местное введение также может представлять собой ингаляционное или назальное введение. Обычно его можно провести в виде сухого порошка (по отдельности или в виде смеси, например сухой смеси с лактозой, в виде смесей с компонентами, например с фосфолипидами) с помощью ингалятора для сухих порошков или в виде аэрозольного спрея, подаваемого из находящегося по давлением контейнера, с помощью насоса, распыляющего устройства, атомизатора или устройства типа небулайзер с использованием или без использования подходящего пропеллента. Настоящее изобретение также относится к безводным фармацевтическим композициям и дозированным формам, включающим соединения, предлагаемые в настоящем изобретении, в качестве активных ингредиентов, поскольку вода может облегчать разложение некоторых соединений. Безводные фармацевтические композиции и дозированные формы, предлагаемые в настоящем изобретении, можно получить с использованием безводных или обладающих низкой влажностью ингредиентов в условиях низкой влажности. Безводную фармацевтическую композицию можно готовить и хранить так, чтобы она оставалась безводной. В соответствии с этим безводные композиции упаковывают в материалы, для которых известно, что они защищают от воздействия воды, гак чтобы их было можно включить в подходящие наборы препаратов. Примеры подходящих упаковочных средств включают, но не ограничиваются только ими, герметично запаиваемую фольгу, пластмассы, контейнеры для разовых доз (например, флаконы), блистерные упаковки и ленточные упаковки. Настоящее изобретение также относится к фармацевтическим композициям и дозированным формам, которые включают один или большее количество агентов, которые регулируют скорость, с которой будет разлагаться соединение, предлагаемое в настоящем изобретении в качестве активного ингредиента. Такие агенты, которые в настоящем изобретении называют "стабилизаторами", включают, но не ограничиваются только ими, антиоксиданты, такие как аскорбиновая кислота, буферные агенты, регулирующие рН, и солевые буферные агенты и т.п. В одном из вариантов настоящее изобретение относится к фармацевтической композиции для лечения рака, содержащей соединение 7-циклопентил-N,N-диметил-2-(5-1R,6S)-9-метил-4-оксо-3,9 диазабицикло[4.2.1]нонан-3-ил)пиридин-2-иламино)-7 Н-пирроло[2,3-d]пиримидин-6-карбоксамид, описывающейся следующей формулой: или его фармацевтически приемлемую соль и фармацевтически приемлемый носитель или инертный наполнитель. Способы применения. Соединения, предлагаемые в настоящем изобретении, являются ингибиторами CDK4/6 и поэтому могут быть применимы для лечения заболеваний, при которых основная патология (по меньшей мере,частично) опосредуется с помощью CDK4/6. Такие заболевания включают рак и другие заболевания, при которых происходит нарушение пролиферации, апоптоза или дифференциации клеток. Таким образом, соединения, предлагаемые в настоящем изобретении, могут быть применимы для лечения опухолей RB+ve (протеинположительная ретинобластома), включая опухоли, содержащие мутации в Ras, Raf, рецепторах факторов роста или в которых сверхэкспресируются рецепторы факторов роста. Соединения, предлагаемые в настоящем изобретении, также могут быть применимы для лечения опухолей с амплификацией генов CDK4 и CDK6, а также, опухолей, в которых сверхэкспресируются партнеры циклинов циклинзависимых киназ. Соединения, предлагаемые в настоящем изобретении, также могут быть применимы для лечения опухолей RB-ve. Соединения, предлагаемые в настоящем изобретении, также могут быть применимы для лечения опухолей с генетическими аберрациями, которые активируют активность киназы CDK4/6. Они включают, но не ограничиваются только ими, раковые заболевания с транслокациями D-циклина, такие как лимфома из клеток зоны мантии и множественная миелома, амплификациями D-циклина, такие как рак молочной железы и плоскоклеточный рак пищевода, амплификациями CDK4, такие как липосаркома,амплификациями или сверхэкспресированиями CDK6, такие как Т-клеточная лимфома, и инактивацией р 16, такие как меланома, немелкоклеточный рак легких и рак поджелудочной железы. Для использования для лечения также можно рассмотреть соединения, предлагаемые в настоящем изобретении, могут быть применимы для лечения раковых заболеваний, которые содержат генетические аберрации в находящихся в обратном направлении регуляторах D-циклинов, где дефекты приводят к увеличению содержания D-циклинов. Они включают, но не ограничиваются только ими, острый миелолейкоз с активацией FLT3, раковые заболевания молочной железы со сверхэкспресированием Her2/neu,зависимостью от ER или с тройным негативным фенотипом, раковые заболевания толстой кишки с активирующими мутациями пути PI3K или WNT, меланомы с активирующими мутациями пути MAPK, немелкоклеточный рак легких с активирующими аберрациями пути EGFR и раковые заболевания поджелудочной железы с активирующими аберрациями пути MAPK, включая мутации K-ras. Примеры раковых заболеваний, которые можно лечить соединением, предлагаемым в настоящем изобретении, включают, но не ограничиваются только ими, лимфому из клеток зоны мантии, множественную миелому, рак молочной железы, плоскоклеточный рак пищевода, меланому, немелкоклеточный рак легких, рак толстой кишки и рак поджелудочной железы. Одна группа раковых заболеваний включает раковые заболевания молочной железы человека (например, первичные опухоли молочной железы, рак молочной железы без поражения лимфатических узлов, инвазивные аденокарциномы протоков молочной железы, неэндометриоидные раковые заболевания молочной железы); и раковые заболевания эндометрия. Другая подгруппа раковых заболеваний, при которых особое терапевтическое преимущество могут предоставить соединения, обладающие ингибирующей активностью по отношению к CDK4/6, включают мультиформную глиобластому, Т-клеточный ОЛЛ(острый лимфоцитарный лейкоз), саркомы, семейную меланому и меланому. Способы лечения, предлагаемые в настоящем изобретении, включают введение нуждающемуся в нем субъекту соединения формулы (I) или его фармацевтически приемлемой соли в терапевтически эффективном количестве. Отдельные варианты осуществления настоящего изобретения включают способы лечения любого из указанных выше нарушений или заболеваний путем введения нуждающемуся в нем субъекту соединения формулы (I) или его фармацевтически приемлемой соли в эффективном количестве. В одном из вариантов способ лечения рака у пациента, нуждающегося в его лечении, включает вве- 16022355 дение пациенту соединения 7-циклопентил-N,N-диметил-2-(5-1R,6S)-9-метил-4-оксо-3,9 диазабицикло[4.2.1]нонан-3-ил)пиридин-2-иламино)-7 Н-пирроло[2,3-d]пиримидин-6-карбоксамида, описывающегося следующей формулой: или его фармацевтически приемлемой соли в эффективном количестве, где рак выбран из группы,включающей лимфому из клеток зоны мантии, множественную миелому, рак молочной железы, плоскоклеточный рак пищевода, меланому, немелкоклеточный рак легких, рак толстой кишки и рак поджелудочной железы. Термин "терапевтически эффективное количество" соединения, предлагаемого в настоящем изобретении, означает количество соединения, предлагаемого в настоящем изобретении, которое приводит к биологической или медицинской реакции субъекта, например уменьшению или ингибированию активности фермента или белка, или облегчает симптомы, облегчает патологические состояния, замедляет или задерживает прогрессирование заболевания или предупреждает заболевание и т.п. В одном неограничивающем варианте осуществления термин "терапевтически эффективное количество" означает количество соединения, предлагаемого в настоящем изобретении, которое при введении субъекту эффективно (1), по меньшей мере, для частичного ослабления, подавления, предупреждения и/или облегчения протекания патологического состояния или нарушения, или заболевания, опосредуемого с помощью CDK4/6, или (ii) связанного с активностью CDK4/6, или (iii) характеризующегося активностью (нормальной или аномальной) CDK4/6; или (2) для уменьшения или ингибирования активности CDK4/6; или (3) для уменьшения или ингибирования экспрессии CDK4/6. В другом неограничивающем варианте осуществления термин "терапевтически эффективное количество" означает количество соединения, предлагаемого в настоящем изобретении, которое при введении в клетку или ткань или в неклеточный биологический материал или среду эффективно, по меньшей мере, для частичного уменьшения или ингибирования активности CDK4/6; или, по меньшей мере, для частичного уменьшения или ингибирования экспрессииCDK4/6. Значение термина "терапевтически эффективное количество", проиллюстрированное для приведенного выше варианта осуществления CDK4/6, таким же образом относится к любым другим соответствующим белкам/пептидам/ферментам. При использовании в настоящем изобретении термин "субъект" означает животное. Обычно животным является млекопитающее. Субъект также означает, например, приматов (например, людей, мужчин и женщин), коров, овец, коз, лошадей, собак, кошек, кроликов, крыс, мышей, рыб, птиц и т.п. В некоторых вариантах осуществления субъектом является примат. В других вариантах осуществления субъектом является человек. При использовании в настоящем изобретении термин "лечить" или "лечение" любого заболевания или нарушения в одном варианте осуществления означает улучшение протекания заболевания или нарушения (т.е. замедление или остановку или ослабление развития заболевания или по меньшей мере одного из его клинических симптомов). В другом варианте осуществления "лечить" или "лечение" означает улучшение по меньшей мере одного физического параметра, включая те, которые могут не ощущаться пациентом. В еще одном варианте осуществления "лечить" или "лечение" означает изменение протекания заболевания или нарушения, физическое (например, стабилизацию проявляющегося симптома), или физиологическое (например, стабилизацию физикального параметра), или и то, и другое. В еще одном варианте осуществления "лечить" или "лечение" означает предупреждение, или задержку начала, или развития, или прогрессирования заболевания или нарушения. При использовании в настоящем изобретении субъект "нуждается в" лечении, если лечение окажет на такой субъект благоприятное биологическое или медицинское воздействие или улучшит качество его жизни. Фармацевтическая композиция или комбинация, предлагаемая в настоящем изобретении, может содержать в разовой дозе примерно 1-1000 мг активных ингредиентов для субъекта массой примерно 50-70 кг или примерно 1-500 мг, или примерно 1-250 мг, или примерно 1-150 мг, или примерно 0,5-100 мг, или примерно 1-50 мг активных ингредиентов. Терапевтически эффективная доза соединения, фармацевтической композиции или их комбинаций зависит от вида субъекта, массы тела, возраста и индивидуального состояния, нарушения или заболевания, подвергающегося лечению, и его тяжести. Врач, клиницист или ветеринар с общей подготовкой должен легко определить эффективное количество каждого из активных ингредиентов, необходимое для предупреждения, лечения или подавления прогрессирования нарушения или заболевания. Указанные выше характеристики доз можно определить с помощью провидимых in vitro и in vivo исследований предпочтительно с использованием млекопитающих, например мышей, крыс, собак, обезьян, или изолированных органов, тканей и их препаратов. Соединения, предлагаемые в настоящем изобретении, можно использовать in vitro в виде растворов, например предпочтительно водных растворов, иin vivo энтерально, парентерально, предпочтительно внутривенно, например, в виде суспензии или водного раствора. Доза in vitro может находиться в диапазоне примерно от 10-3 до 10-9 М. Терапевтически эффективное количество in vivo в зависимости от пути введения может находиться в диапазоне примерно 0,1-500 мг/кг, предпочтительно примерно 1-100 мг/кг. Одним вариантом осуществления настоящего изобретения является способ модулирования активности CDK4/6 у субъекта, включающий введение субъекту соединения формулы (I) или его фармацевтически приемлемой соли в терапевтически эффективном количестве. Другим вариантом осуществления настоящего изобретения является способ лечения нарушения или заболевания, опосредуемого с помощью CDK4/6 у нуждающегося в нем субъекта, включающий введение субъекту соединения формулы (I) или его фармацевтически приемлемой соли в терапевтически эффективном количестве. Другим вариантом осуществления настоящего изобретения является способ лечения нарушения или заболевания, опосредуемого с помощью CDK4/6, у нуждающегося в лечении субъекта, включающий введение соединения формулы (I) или его фармацевтически приемлемой соли в терапевтически эффективном количестве, в котором нарушение или заболевание выбрано из группы, включающей карциномы с генетическими аберрациями, которые активируют активность киназы CDK4/6. Они включают, но не ограничиваются только ими, раковые заболевания с транслокациями D-циклина, такие как лимфома из клеток зоны мантии и множественная миелома, амплификациями D-циклина, такие как рак молочной железы и плоскоклеточный рак пищевода, амплификациями CDK4, такие как липосаркома, амплификациями или сверхэкспресированиями CDK6, такие как Т-клеточная лимфома, и инактивацией р 16, такие как меланома, немелкоклеточный рак легких и рак поджелудочной железы. Другим вариантом осуществления настоящего изобретения является применение соединения формулы (I) или его фармацевтически приемлемой соли для лечения нарушения или заболевания, опосредуемого с помощью CDK4. Другим вариантом осуществления настоящего изобретения является применение соединения формулы (I) или его фармацевтически приемлемой соли для лечения у субъекта нарушения или заболевания,опосредуемого с помощью CDK4, в котором нарушение или заболевание выбрано из группы, включающей карциномы с генетическими аберрациями, которые активируют активность киназы CDK4/6. Они включают, но не ограничиваются только ими, раковые заболевания с транслокациями D-циклина, такие как лимфома из клеток зоны мантии и множественная миелома, амплификациями D-циклина, такие как рак молочной железы и плоскоклеточный рак пищевода, амплификациями CDK4, такие как липосаркома, амплификациями или сверхэкспресированиями CDK6, такие как Т-клеточная лимфома, и инактивацией р 16, такие как меланома, немелкоклеточный рак легких и рак поджелудочной железы. Другим вариантом осуществления настоящего изобретения является соединение формулы (I) или его фармацевтически приемлемая соль, предназначенное для применения в качестве лекарственного средства. Другим вариантом осуществления настоящего изобретения является применение соединения формулы (I) или его фармацевтически приемлемой соли для приготовления лекарственного средства, предназначенного для лечения нарушения или заболевания, опосредуемого с помощью CDK4/6, в котором нарушение или заболевание выбрано из группы, включающей карциномы с генетическими аберрациями,которые активируют активность киназы CDK4/6. Они включают, но не ограничиваются только ими, раковые заболевания с транслокациями D-циклина, такие как лимфома из клеток зоны мантии и множественная миелома, амплификациями D-циклина, такие как рак молочной железы и плоскоклеточный рак пищевода, амплификациями CDK4, такие как липосаркома, амплификациями или сверхэкспресированиями CDK6, такие как Т-клеточная лимфома, и инактивацией р 16, такие как меланома, немелкоклеточный рак легких и рак поджелудочной железы. Общие методики синтеза и промежуточные продукты Общая методика N-C сочетания 1. В подходящем сосуде для проведения реакции в подходящем растворителе (таком как, но не ограничиваясь только им, диоксан) объединяли соединения структуры 1 (1 экв.) и соединения общей структуры 2 (1 экв.). К этому полученному раствору добавляли ацетат палладия(II) (0,1 экв.), лиганд, такой как БИНАФ (2,2'-бис-(дифенилфосфино)-1,1'-бинафтил), XPhos или XantPhos (0,15 экв.), и карбонат цезия(1,5 экв.). Затем через реакционную смесь осторожно пропускали азот (примерно от 5 до 10 мин). Затем полученную реакционную смесь нагревали на масляной бане или в микроволновой печи при температуре, равной примерно от 100 до 130 С, в течение подходящего периода времени, пока анализ с помощью ТСХ или ВЭЖХ МС не показывал, что реакция завершалась. Реакционную смесь удаляли от источника тепла и ей давали охладиться. Затем смесь обрабатывали путем добавления подходящего растворителя,такого как дихлорметан или этилацетат. Нерастворимые вещества удаляли фильтрованием и органический фильтрат экстрагировали водой. Водную фазу подвергали обратной экстракции. Органические фазы объединяли, сушили над сульфатом натрия или магния, фильтровали, концентрировали и получали остаток. Неочищенный остаток очищали с помощью хроматографии на силикагеле с использованием подходящей подвижной фазы, что давало искомый промежуточный продукт или соединение формулы(I). Методика восстановления нитрогруппы 1. В подходящий сосуд для проведения реакции помещали соединение структуры 3. Для растворения соединения структуры 3 использовали подходящий растворитель, такой как метанол, этилацетат, тетрагидрофуран или смеси этих растворителей. К этой полученной смеси и в потоке азота добавляли катализатор, такой как палладий на угле или гидроксид палладия (содержание металла на угле или подходящей подложке составляет от 5 до 20%) от 5 до 10 мол.% в пересчете на содержание структуры 3. Затем полученную смесь продували и перемешивали в атмосфере водорода. После того как по данным ТСХ или ЖХ/МС все исходное вещество превращалось в продукт, сосуд для проведения реакции отсоединяли от источника водорода и продували азотом для удаления оставшегося водорода. Реакционную смесь фильтровали через слой целита в потоке азота и промывали дополнительным количеством растворителя, такого как дихлорметан или метанол. Фильтраты объединяли, концентрировали и получали остаток. Остаток сушили в вакууме до постоянной массы. Полученное вещество использовали без обработки в следующей реакции или очищали с помощью перекристаллизации или хроматографии на силикагеле и затем использовали в следующей реакции. Методики получения амидов формулы (I). Приведенные ниже общие методики использовали для сочетания карбоновых кислот структуры 5 с аминами с получением соответствующих амидов структуры 6. Общая методика получения амида 1. К раствору карбоновой кислоты (1,01 ммоль) в ДМФ (5 мл) добавляли О-(бензотриазол-1-ил)N,N,N',N'-тетраметилуронийгексафторфосфат (HBTU, 580 мг, 1,53 ммоль, 1,5 экв.) и N,Nдиизопропилэтиламин (0,55 мл, 3,0 экв.) и полученную смесь перемешивали при комнатной температуре в течение примерно 5 мин. К полученной смеси добавляли амин (1,18 ммоль, 1,1 экв.). Полученную смесь перемешивали при комнатной температуре в течение подходящего периода времени, необходимого для завершения реакции по данным ТСХ или ЖХ/МС, и затем разбавляли этилацетатом и последовательно промывали с помощью 0,5 М HCl, водой, сушили над Na2SO4, фильтровали и концентрировали. Искомое вещество сразу использовали на следующей стадии без дополнительной очистки или очищали с помощью хроматографии на силикагеле с использованием подходящей подвижной фазы и затем сразу использовали в следующей реакции. Общая методика получения амида 2. К раствору карбоновой кислоты (в виде соли с 5 экв. LiCl) (1 ммоль, 1 экв.) в ДМФ (5 мл) добавляли O-(бензотриазол-1-ил)-N,N',N',N'-тетраметилуронийгексафторфосфат (HBTU, 580 мг, 1,53 ммоль, 1,5 экв.) и N,N-диизопропилэтиламин (0,55 мл, 3,0 экв.) и полученную смесь перемешивали при комнатной температуре в течение примерно 5 мин. К полученной смеси добавляли амин (1,18 ммоль, 1,1 экв.). Смесь перемешивали при комнатной температуре в течение подходящего периода времени, необходимого для завершения реакции по данным ТСХ или ЖХ/МС, и затем разбавляли этилацетатом и последовательно промывали с помощью 0,5 М HCl, водой, сушили над Na2SO4, фильтровали и концентрировали. Искомое вещество использовали сразу на следующей стадии без дополнительной очистки или очищали с помощью хроматографии на силикагеле с использованием подходящей подвижной фазы и затем сразу использовали в следующей реакции. Общая методика получения амида 3. К раствору карбоновой кислоты (1 экв., которая содержала 5 экв. LiCl) в 1 мл ДМА (диметилацетамид) или ДМФ добавляли О-(бензотриазол-1-ил)-N,N,N',N'-тетраметилуронийтетрафторборат (TBTU, 1 экв.) и полученный раствор перемешивали в течение примерно 10 мин. Затем добавляли еще 3 мл дихлорметана (конечная концентрация 0,03 М), амин (1 экв.) и ДИПЭА (4 экв.) и перемешивали при КТ,пока по данным ТСХ или ЖХ/МС реакция не завершалась. Реакционную смесь разбавляли дихлорметаном, промывали водой и затем рассолом. Объединенные водные слои подвергали обратной экстракции дихлорметаном. Объединенные органические слои сушили над сульфатом натрия или сульфатом магния,затем фильтровали, затем концентрировали и в заключение очищали с помощью колоночной хроматографии на силикагеле с использованием подходящей подвижной фазы и затем сразу использовали в следующей реакции. Общая методика получения амида 4. К суспензии/раствору смеси карбоновой кислоты (с 5 экв. LiCl) (1 экв.) в ДМА/ДХМ (1:4, 0,07 М) добавляли O-(бензотриазол-1-ил)-N,N,N',N'-тетраметилуронийтетрафторборат (TBTU, 1,5 экв., общая методика В 1) или О-(бензотриазол-1-ил)-N,N,N',N '-тетраметилуронийгексафторфосфат (HBTU, 1,5 экв.,общая методика В 2) и полученную смесь перемешивали при комнатной температуре в течение 5 мин. Реакционную смесь обрабатывали раствором амина (1 экв.) в смеси ДМА/ДХМ (1:4, 0,07 М) или суспензией соли с HCl амина (1,0 экв.) и бикарбоната натрия (1,5 экв.) в смеси ДМА/ДХМ (1:4, 0,07 М). Полученную смесь обрабатывали N,N-диизопропилэтиламином (4,0 экв.) и перемешивали при комнатной температуре в течение 1 ч. Реакционную смесь разбавляли этилацетатом, промывали водой, сушили надNa2SO4, фильтровали и концентрировали. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле с использованием подходящей подвижной фазы и затем использовали в следующей реакции. Общие методики удаления защитных групп из промежуточных продуктов. Из промежуточных продуктов, которые содержали защитные группы, необходимые для синтеза соединений формулы (I), эти защитные группы удаляли по стандартным методикам, описанным в публикации "Protective Groups in Organic Synthesis, 3rd Edition, by Green and Wutts". Методика удаления защитной группы 1. Удаление ВОС с помощью HCl. При перемешивании к раствору, содержащему защитную группу ВОС амина-соединения формулы(I) (1 ммоль) в дихлорметане (4 мл) или другом подходящем растворителе, при комнатной температуре добавляли 4 М раствор HCl в диоксане (2,54 мл, 10,2 ммоль, 10 экв.). Реакционную смесь перемешивали при комнатной температуре, пока по данным ЖХ/МС или ТСХ не израсходовалось все исходное вещество. Затем реакционную смесь фильтровали и промывали растворителем, таким как дихлорметан, этилацетат или диэтиловый эфир. Остаток собирали и переносили в воду, подщелачивали с помощью 1 МNaOH и экстрагировали дихлорметаном или смесью 20% изопропиловый спирт-дихлорметан. Объединенные органические слои сушили над Na2SO4, фильтровали, концентрировали при пониженном давлении и получали остаток. Неочищенный остаток очищали с помощью хроматографии на силикагеле с использованием подходящей подвижной фазы и затем сразу использовали в следующей реакции. Методика удаления защитной группы 2. Удаление ВОС с помощью трифторуксусной кислоты. При перемешивании к холодному 0 С раствору, содержащему защитную группу ВОС аминасоединения формулы (I) (1 ммоль) в дихлорметане (4 мл) или другом подходящем растворителе, добавляли 4 мл безводной трифторуксусной кислоты. Реакционную смесь перемешивали при 0 С, ей давали нагреться до комнатной температуры и перемешивали, пока по данным ЖХ/МС или ТСХ не израсходовалось все исходное вещество. Затем реакционную смесь концентрировали в вакууме и получали вязкий остаток. Остаток экстрагировали смесью дихлорметана и насыщенного раствора бикарбоната натрия. Водный раствор подвергали обратной экстракции дихлорметаном. В случае очень полярных аминов в качестве растворителя для экстракции органических веществ использовали смесь 20% изопропиловый спирт-дихлорметан. Органические фракции объединяли и сушили над Na2SO4, фильтровали, концентрировали при пониженном давлении и получали остаток. Объединенные органические слои сушили над сульфатом натрия или магния, фильтровали, концентрировали при пониженном давлении и получали остаток. Неочищенный остаток очищали с помощью хроматографии на силикагеле с использованием подходящей подвижной фазы и затем сразу использовали в следующей реакции. Методика удаления защитной группы 3. Удаление ВОС с помощью трифторуксусной кислоты. Раствор содержащего защитную группу ВОС амина-соединения формулы (I) в CH2Cl2 (0,1 М) при комнатной температуре обрабатывали таким же объемом трифторуксусной кислоты и перемешивали при комнатной температуре в течение 1,0 ч. Реакционную смесь концентрировали в вакууме и обрабатывали порциями 7 н. NH3 в МеОН до нейтральной реакции смеси. Полученную смесь концентрировали и получали вязкий остаток. Затем полученный остаток очищали с помощью препаративной ВЭЖХ или хроматографии на силикагеле с использованием 7 н. NH3 в МеОН/CH2Cl2 и затем сразу использовали в следующей реакции. Общая методика восстановительного аминирования. Соединение формулы (1) (1 экв.), содержащее первичную или вторичную аминогруппу, вместе с избытком (от 3 до 8 экв.) альдегида (например, формальдегида, 37% водный раствор) или кетона (например, ацетона) объединяли в подходящем растворителе, таком как тетрагидрофуран, и перемешивали при КТ в течение 1 ч над сульфатом магния (1 экв.). Затем одной порцией добавляли триацетоксиборогидрид натрия (2 экв.) и реакционную смесь перемешивали, пока по данным ТСХ или ЖХ/МС не израсходовалось все исходное вещество. Затем реакцию останавливали и переносили в этилацетат, нейтрализовывали насыщенным раствором Na2CO3 и промывали рассолом. Водные слои подвергали обратной экстракции этилацетатом. Органические слои объединяли и сушили над Na2SO4. В случае очень полярных аминов в качестве растворителя для экстракции использовали раствор 20% изопропанол-хлороформ. Летучие вещества удаляли и полученный остаток очищали с помощью хроматографии на силикагеле с использованием подходящей подвижной фазы, что давало искомый конечный продукт. Методики получения промежуточных продуктов 2-хлор-7R1-N,N-диметил-7 Н-пирроло[2,3d]пиримидин-6-карбоксамидов. К раствору 5-бром-2,4-дихлорпиримидина (13,41 г, 58,8 ммоль) в EtOAc (100 мл) добавляли ДИПЭА (13 мл, 1,3 экв.), затем циклогептанамин (8,6 мл, 1,1 экв.) и полученную смесь перемешивали при комнатной температуре в течение 5 дней. Реакционную смесь разбавляли с помощью EtOAc (350 мл),промывали водой (100 мл), рассолом (2100 мл), сушили над Na2SO4, фильтровали и концентрировали. Неочищенный продукт (18,1 г) использовали в следующей реакции без очистки. 1 Н ЯМР (400 МГц,CD3OD)8,10 (s, 1 Н), 4,17 (септет, J=4,6 Гц, 1 Н), 1,94 (т, 2 Н), 1,77-1,51 (т, 10 Гц); МС m/z 305,3 (М+Н)+.(200 мл) добавляли пропаргиловый спирт (4,5 мл, 1,4 экв.) и раствор тетрабутиламмонийфторид в ТГФ (1 М, 130 мл, 2,3 экв.) и полученную коричневую смесь обрабатывали потоком азота в течение 15 мин(пропускали через раствор). Затем смесь обрабатывали бис-(трифенилфосфин)палладий(II)хлоридом(2,09 г, 0,054 экв.) и кипятили с обратным холодильником в течение 5 ч. После охлаждения реакционную смесь фильтровали через слой целита (промывали с помощью EtOAc 350 мл). Фильтрат концентрировали для удаления ТГФ, дополнительно разбавляли с помощью EtOAc (полный объем 250 мл), промывали насыщенным раствором NaHCO3 (2150 мл) и водой (150 мл), сушили над Na2SO4, фильтровали и концентрировали. Растирание остатка с ацетоном и СН 2 С 12 давало 3-(2-хлор-4(циклогептиламино)пиримидин-5-ил)проп-2-ин-1-ол (8,65 г) с выходом 56%. 1 Н ЯМР (400 МГц, CD3OD)7,98 (s, 1 Н), 4,45 (s, 2 Н), 4,19 (септет, J=4,5 Гц, 1 Н), 1,95 (m, 2 Н), 1,77-1,52 (m, 10 Гц); МС m/z 280,4 Получение (2-хлор-7-циклогептил-7 Н-пирроло[2,3-d]пиримидин-6-ил)метанола. К суспензии 3-(2-хлор-4-(циклогептиламино)пиримидин-5-ил)проп-2-ин-1-ола (8,64 г, 30,9 ммоль) в ТГФ (200 мл) добавляли раствор ТБАФ в ТГФ (1 М, 66 мл, 2,1 экв.) и полученную смесь нагревали при 63 С в течение 5 ч. Реакционную смесь концентрировали для удаления ТГФ, разбавляли с помощьюEtOAc (350 мл), промывали водой (140 мл) и рассолом (140 мл), сушили над Na2SO4, фильтровали и концентрировали. Растирание остатка с iPrOH, CH2Cl2 и MeCN с последующей очисткой маточного раствора с помощью колоночной хроматографии (EtOAc/гептан от 20 до 100%) давало (2-хлор-7-циклогептил-7 Нпирроло[2,3-d]пиримидин-6-ил)метанол (6,24 г) с выходом 72%. 1 Н ЯМР (400 МГц, CDCl3)8,69 (s, 1 Н),6,45 (s, 1 Н), 4,83 (s, 2 Н), 4,61 (m, 1H), 2,54 (m, 2 Н), 1,98-1,86 (m, 4 Н), 1,73-1,55 (m, 6 Н); МС m/z 280,4 Получение 2-хлор-7-циклогептил-N,N-диметил-7 Н-пирроло[2,3-d]пиримидин-6-карбоксамида. К раствору (2-хлор-7-циклогептил-7 Н-пирроло[2,3-d]пиримидин-6-ил)метанол (6,24 г, 22,3 ммоль) в ДМФ (100 мл) добавляли раствор диметиламин в ТГФ (2 М, 46 мл, 4,1 экв.) и цианид натрия (1,04 г, 0,95 экв.) и полученную смесь перемешивали при комнатной температуре в течение 4 мин. Реакционную смесь обрабатывали четырьмя порциями диоксида марганца (100,4 г, 45 экв.) в течение 1 ч и перемешивали в течение еще 1 ч. Реакционную смесь фильтровали через слой целита (промывали с помощьюEtOAc 600 мл). Фильтрат промывали водой (200 мл). Водную фазу экстрагировали с помощью EtOAc(2150 мл). Объединенные органические вещества промывали с помощью 4% водного раствора NaCl(2250 мл), сушили над Na2SO4, фильтровали и концентрировали в вакууме. Растирание остатка с MeCN с последующей очисткой маточного раствора с помощью колоночной хроматографии (EtOAc/гептан от 30 до 100%) давало 2-хлор-7-циклогептил-N,N-диметил-7 Н-пирроло[2,3-d]пиримидин-6-карбоксамид(M+H)+. Промежуточный продукт 2. 2-Хлор-7-циклогексил-N,N-диметил-7 Н-пирроло[2,3-d]пиримидин-6-карбоксамид получали по методике, аналогично использованной для получения промежуточного продукта 1.(s, 1 Н); МС m/z 266,5 (М+Н)+. Стадия 4. Диметиламид 2-хлор-7-циклогексил-7 Н-пирроло[2,3-d]пиримидин-6-карбоновой кислоты (0,85 г,выход 64%): 1 Н ЯМР (400 МГц, ДМСО-d6)ч./млн 1,22 (br. s., 1 Н), 1,36 (d, J=13,6 Гц, 2 Н), 1,66 (br. s.,1 Н), 1,85 (d, J=10,6 Гц, 4 Н), 2,30 (dd, J=12,4, 3,3 Гц, 2 Н), 3,04 (d, J=17,2 Гц, 6 Н), 4,34 (br. s., 1 Н), 6,80 (s,1 Н), 8,97 (s, 1 Н); МС m/z 307,3 (М+Н)+. Промежуточный продукт 3. 2-Хлор-7-циклопентил-N,N-диметил-7 Н-пирроло[2,3-d]пиримидин-6-карбоксамид получали по методике, аналогичной использованной для получения промежуточного продукта 1, и он является описанным в литературе соединением (см. WO 2010/020675) Промежуточный продукт 4. 2-Хлор-N,N-диметил-7-(тетрагидро-2 Н-пиран-3-ил)-7 Н-пирроло[2,3-d]пиримидин-6-карбоксамид получали по методике, аналогичной использованной для получения промежуточного продукта 1.(1,74 г) получали с выходом 58%. 1 Н ЯМР (400 МГц, CD3OD)8,87 (s, 1 Н), 6,81 (s, 1 Н), 4,53 (tt, J=11, 4,4 Гц, 1 Н), 4,32 (t, J=11 Гц, 1 Н), 3,95 (m, 2 Н), 3,52 (m, 1 Н), 3,17 (s, 3 Н), 3,15 (s, 3 Н), 2,77 (qt, J=12, 5,3 Гц,1 Н), 2,09 (m, 1 Н), 1,87-1,78 (m, 2 Н); МС m/z 309,5 (М+Н)+. Промежуточный продукт 5. 2-Хлор-7-(4-(2-цианопропан-2-ил)фенил)-N,N-диметил-7 Н-пирроло[2,3-d]пиримидин-6-карбоксамид получали по методике, аналогичной использованной для получения промежуточного продукта 1. 5-Бром-2,4-дихлорпиримидин (1,37 г, 6 ммоль), 2-(4-аминофенил)-2-метилпропаннитрил (0,96 г, 6 ммоль) и диизопропилэтиламин (1,6 мл, 9 ммоль) объединяли в 30 мл ацетонитрила и перемешивали в течение 16 ч при КТ. Через 16 ч ЖХ/МС указывала на завершение реакции. Органические вещества последовательно промывали 1 М раствором лимонной кислоты, водой, затем рассолом. Органические вещества сушили над сульфатом натрия, затем декантировали, выпаривали и получали неочищенное вещество, которое использовали на следующей стадии без обработки (1,45 г, 69%); МС m/z 353,3 (М+Н). Стадия 2. 2-(4-(2-Хлор-5-(3-гидроксипроп-1-инил)пиримидин-4-иламино)фенил)-2 метилпропаннитрил. 2-(4-(5-Бром-2-хлорпиримидин-4-иламино)фенил)-2-метилпропаннитрил (1,4 г, 3,98 ммоль), бис(трифенилфосфин)палладий(II)хлорид (0,14 г, 0,2 ммоль), тетрабутилфторид аммония (2,6 г, 9,95 ммоль) и пропаргиловый спирт (0,357 мл, 5,97 ммоль) объединяли в 12 мл ТГФ в сосуде высокого давления с винтовой крышкой и нагревали на масляной бане при 70 С. Через 2 ч по данным ЖХ/МС реакция завершалась. Летучие вещества удаляли и остаток переносили в EtOAc. Органический слой фильтровали для удаления нерастворимого вещества. Органический слой промывали водой, рассолом и затем сушили над сульфатом натрия. Летучие вещества удаляли и остаток очищали с помощью ЖХНФ (жидкостная хрома- 24022355 Стадия 3. 2-(4-(2-Хлор-6-(гидроксиметил)-7 Н-пирроло[2,3-d]пиримидин-7-ил)фенил)-2 метилпропаннитрил. 2-(4-(2-Хлор-5-(3-гидроксипроп-1-инил)пиримидин-4-иламино)фенил)-2-метилпропаннитрил вводили в реакцию с 1 М раствором тетрабутиламмонийфторида в ТГФ (5,85 мл, 5,85 ммоль), дополнительно разбавленного до 10 мл дополнительным количеством ТГФ, в течение 1,5 ч при 60 С. Аналитическая ЖХ указывала на завершение реакции (минимальное изменение времени удерживания). Летучие вещества удаляли и остаток переносили в этилацетат. Органические вещества промывали водой, рассолом затем сушили над сульфатом натрия. Летучие вещества удаляли и остаток очищали с помощью ЖХНФ (1075% этилацетата в гептане, Analogix) и получали искомый продукт (589 мг, 68%); МС 327,3 m/z (M+H).(589 мг, 1,8 ммоль), диоксид марганца (7,8 г, 90 ммоль) и 2 М раствор диметиламина в ТГФ (4,51 мл, 9 ммоль) объединяли в 6 мл сухого ДМФ. Реакционную смесь перемешивали в течение 3 ч и после этого по данным ЖХ/МС реакция завершалась. Реакционную смесь выливали в рассол и экстрагировали с помощью EtOAc. Объединенные органические вещества промывали водой, затем рассолом и затем сушили над сульфатом натрия. Летучие вещества удаляли и остаток очищали с помощью ЖХНФ (10-100% этилацетата в гептане, Analogix) и получали чистое искомое соединение (345 мг, 52%); МС 367,7 m/z. Промежуточный продукт 6 Получение метил-6-(7-циклопентил-6-(диметилкарбамоил)-7 Н-пирроло[2,3-d]пиримидин-2 иламино)никотината. К суспензии 2-хлор-7-циклопентил-N,N-диметил-7H-пирроло[2,3-d]пиримидин-6-карбоксамида (5,0 г, 17 ммоль) и 1,4-диоксан (80 мл) в герметизированной пробирке добавляли метил-6-аминоникотинат(2,86 г, 1,10 экв.), Pd(OAc)2 (0,096 г, 0,025 экв.) и БИНАФ (0,532 г, 0,050 экв.). Через полученную смесь для дегазирования в течение 20 мин пропускали N2 и обрабатывали с помощью Cs2CO3 (8,35 г, 1,5 экв.) Реакционную смесь нагревали при 100 С в течение 2,2 ч. Реакционную смесь охлаждали до комнатной температуры, обрабатывали с помощью 100 мл гептана и обрабатывали ультразвуком. Красный осадок собирали фильтрованием и суспендировали в 200 мл воды. После обработки ультразвуком твердое вещество собирали фильтрованием, промывали водой (350 мл) и сушили. Твердое вещество растирали с ТГФ и получали желтовато-коричневый твердый метил-6-(7-циклопентил-6-(диметилкарбамоил)-7Hпирроло[2,3-d]пиримидин-2-иламино)никотинат (6,6 г, выход 94%). Фильтрат концентрировали в вакуу- 25022355 ме и очищали с помощью колоночной хроматографии (эгилацетат/гептан) и получали дополнительное количество продукта (0,4 г, выход 6%). 1 Н ЯМР (400 МГц, ДМСО-d6)ч./млн 10,33 (s, 1 Н), 8,89 (s, 1 Н),8,83 (d, J=2,1 Гц, 1 Н), 8,44 (d, J=8,9 Гц, 1 Н), 8,22 (dd, J=8,9, 2,2 Гц, 1 Н), 6,67 (s, 1H), 4,77 (m, 1H), 3,86 (s,3 Н), 3,06 (s, 6 Н), 2,42 (m, 2 Н), 2,00 (m, 4 Н), 1,68 (m, 2 Н); МС m/z 409,4 (М+Н)+. Стадия 2 Получение соли с LiCl 6-(7-циклопентил-6-(диметилкарбамоил)-7H-пирроло[2,3-d]пиримидин-2 иламино)никотиновой кислоты. К суспензии метил-6-(7-циклопентил-6-(диметилкарбамоил)-7H-пирроло[2,3-d]пиримидин-2 иламино)никотината (1,0 г, 2,4 ммоль) в 2-пропаноле (60 мл) добавляли LiOH (0,29 г, 5,0 экв.) и воду (12 мл) и полученную смесь перемешивали при 60 С в течение 1 ч. Суспензия постепенно становилась прозрачной. После охлаждения до комнатной температуры реакционную смесь обрабатывали с помощью 1 н.HCl (12,24 мл, 5 экв.) и концентрировали в вакууме. Светло-желтое твердое вещество (1,40 г, выход 95%), смесь 6-(7-циклопентил-6-(диметилкарбамоил)-7H-пирроло[2,3-d]пиримидин-2-иламино)никотиновой кислоты и LiCl (5 экв.) использовали в следующей реакции без дополнительной очистки. 1 Н ЯМР Получение 6-(7-циклопентил-6-(диметилкарбамоил)-7 Н-пирроло[2,3-d]пиримидин-2 иламино)никотиновой кислоты. К суспензии метил-6-(7-циклопентил-6-(диметилкарбамоил)-7H-пирроло[2,3-d]пиримидин-2 иламино)никотината (2,0 г, 4,9 ммоль) в ТГФ (6 мл) добавляли 1 М водный раствор LiOH (6 мл, 1,2 экв.) и взвесь перемешивали при 45 С в течение 12 ч (взвесь становилась прозрачной). После охлаждения до комнатной температуры ТГФ выпаривали и реакционную смесь обрабатывали с помощью 1 н. HCl до рН 1-2. Полученный осадок отфильтровывали и фильтрат экстрагировали смесью 20% изопропанол/CH2Cl2(3100 мл), объединенные органические слои сушили, фильтровали, концентрировали и получали желтовато-коричневое твердое вещество. Желтовато-коричневое твердое вещество растирали с ацетоном и получали 6-(7-циклопентил-6-(диметилкарбамоил)-7 Н-пирроло[2,3-d]пиримидин-2-иламино)никотиновую кислоту в качестве искомого продукта в виде желтовато-коричневого твердого вещества (1,56 г, выход 73%), которое использовали без дополнительной очистки. 1 Н ЯМР (400 МГц, ДМСО-d6)10,75 (br.s, 1H), 8,95 (s, 1H), 8,84 (d, J=2,5 Гц, 1 Н), 8,24-8,33 (m, 1 Н), 8,13-8,24 (m, 1 Н), 6,74 (s, 1H), 4,79 (квинтет,J=8,8 Гц, 1 Н), 3,06 (s, 1 Н), 2,30-2,46 (m, 3 Н), 1,90-2,09 (m, 5 Н), 1,48-1,76 (m, 3 Н), 1,04 (d, J=6,1 Гц, 1 Н); МС m/z 395,5 (М+Н)+. Промежуточный продукт 8. 2-Хлор-7-(4-(2-цианопропан-2-ил)фенил)-N,N-диметил-7 Нпирроло[2,3-d]пиримидин-6-карбоксамид получали по методике, аналогичной использованной для получения промежуточного продукта 1. Через смесь диметиламида 2-хлор-7-циклогептил-7 Н-пирроло[2,3-d]пиримидин-6-карбоновой кислоты (517 мг, 1,2 ммоль, 1,0 экв.), метилового эфира 6-аминоникотиновой кислоты (221 мг, 1,5 ммоль,1,2 экв.), CS2CO3 (591 мг, 1,8 ммоль, 1,5 экв.) и БИНАФ (38 мг, 0,06 ммоль, 0,05 экв.) с помощью пипетки в течение 3 мин пропускали N2. Добавляли Pd(OAc)2 (14 мг, 0,06 ммоль, 0,05 экв.) и колбу герметизировали и в течение 3 ч перемешивали на масляной бане при нагревании до 130 С. Смесь фильтровали через слой целита и промывали с помощью EtOAc. Органический слой промывали водой, затем рассолом и органические слои объединяли, сушили над Na2SO4. Растворитель выпаривали и коричневое твердое вещество растирали с ацетонитрилом, фильтровали, промывали с помощью АЦН и сушили в высоком вакууме и получали искомое соединение в виде светло-розового твердого вещества (236 мг, 0,53 ммоль,выход 44%). 1H ЯМР (400 МГц, хлороформ-d)ч./млн 8,84-8,90 (m, 1 Н), 8,69 (s, 1 Н), 8,61 (d, J=9,09 Гц,1 Н), 8,51 (br. s., 1 Н), 8,20-8,26 (m, 1 Н), 6,40 (s, 1 Н), 4,40-4,53 (m, 1 Н), 3,87 (s, 3 Н), 3,10 (s, 6 Н), 2,48-2,64 Получение 6-(7-циклогептил-6-диметилкарбамоил-7 Н-пирроло[2,3-d]пиримидин-2 иламино)никотиновой кислоты. К раствору метилового эфира 6-(7-циклогептил-6-диметилкарбамоил-7 Н-пирроло[2,3-d]пиримидин 2-иламино)никотиновой кислоты (146 мг, 0,33 ммоль, 1,0 экв.) в ТГФ (1,5 мл) добавляли LiOH (94,7 мг,4,0 ммоль, 12 экв.) в 1,5 мл воды. Смесь перемешивали при комнатной температуре в течение 48 ч. Реакционную смесь охлаждали до 0 С в бане со льдом и подкисляли с помощью 1 н. HCl до рН около 1-2. Твердый осадок отфильтровывали и промывали и получали искомое соединение в виде светло-розового твердого вещества (52 мг, 0,124 ммоль, 37%) и использовали без обработки; альтернативно, его можно было экстрагировать раствором 20% изопропанола в дихлорметане) 1 Н ЯМР (400 МГц, ДМСО-d6)ч./млн 12,99 (br. s., 1 Н), 10,38 (br. s., 1 Н), 8,87 (s, 1 Н), 8,81 (d, J=2,01 Гц, 1 Н), 8,53 (d, J=9,03 Гц, 1 Н), 8,19 6-(7-Циклогептил-6-(диметилкарбамоил)-7 Н-пирроло[2,3-d]пиримидин-2-иламино)никотиновую кислоту и 5 экв. LiCl получали по методике, аналогичной использованной для получения промежуточного продукта 6. 1 Н ЯМР (400 МГц, ДМСО-d6)ч./млн 9,70 (br. s., 1H), 8,81 (s, 1H), 8,71 (s, 1H), 8,42 (d,J=8,53 Гц, 1 Н), 8,10 (dd, J1=8,53, J2=2,01 Гц, 1 Н), 6,63 (s, 1H), 4,41 (m, 1H), 3,07 (d, 6 Н), 2,70-2,52 (m, 2 Н),1,99-1,39 (m, 10 Н); МС m/z 422,9 (М+Н)+. Получение метил-6-(7-циклопентил-6-(диметилкарбамоил)-7H-пирроло[2,3-d]пиримидин-2 иламино)никотината. По общей методике N-C сочетания 1 2-хлор-7-циклопентил-N,N-диметил-7H-пирроло[2,3d]пиримидин-6-карбоксамид (5,0 г, 17 ммоль) объединяли с метил-6-аминоникотинатом (2,86 г, 1,10 экв.), что давало метил-6-(7-циклопентил-6-(диметилкарбамоил)-7H-пирроло[2,3-d]пиримидин-2 иламино)никотинат (6,6 г) с выходом 94%. 1H ЯМР (400 МГц, ДМСО-d6)ч./млн 10,33 (s, 1 Н), 8,89 (s,1 Н), 8,83 (d, J=2,1 Гц, 1 Н), 8,44 (d, J=8,9 Гц, 1 Н), 8,22 (dd, J=8,9, 2,2 Гц, 1 Н), 6,67 (s, 1 Н), 4,77 (m, 1 Н),3,86 (s, 3 Н), 3,06 (s, 6 Н), 2,42 (m, 2 Н), 2,00 (m, 4 Н), 1,68 (m, 2 Н); МС m/z 409,4 (М+Н)+. Стадия 2 Получение 6-(7-циклопентил-6-(диметилкарбамоил)-7H-пирроло[2,3-d]пиримидин-2-иламино)никотиновой кислоты и 5 экв. LiCl. К суспензии метил-6-(7-циклопентил-6-(диметилкарбамоил)-7H-пирроло[2,3-d]пиримидин-2 иламино)никотината (1,0 г, 2,4 ммоль) в 2-пропаноле (60 мл) добавляли LiOH (0,29 г, 5,0 экв.) и воду (12 мл) и полученную смесь перемешивали при 60 С в течение 1 ч. После охлаждения до комнатной температуры реакционную смесь обрабатывали с помощью 1 н. HCl (12,24 мл, 5 экв.) и концентрировали в вакууме,что давало 6-(7-циклопентил-6-(диметилкарбамоил)-7H-пирроло[2,3-d]пиримидин-2 иламино)никотиновую кислоту в виде светло-желтого твердого вещества (1,40 г) с выходом 95% в виде соли с LiCl (5 экв.) и ее использовали в следующей реакции без дополнительной очистки. 1 Н ЯМР (400 МГц, ДМСО-d6)ч./млн 13,30 (br. s., 1 Н), 11,24 (br. s., 1 Н), 9,03 (s, 1 Н), 8,88 (d, J=2,2 Гц, 1 Н), 8,31 (dd,J=8,7, 2,2 Гц, 1 Н), 8,11 (d, J=8,7 Гц, 1 Н), 6,81 (s, 1 Н), 4,79 (m, 1 Н), 3,06 (s, 6 Н), 2,41 (m, 2 Н), 2,02 (m, 4 Н),1,67 (m, 2 Н); МС m/z 395,4 (М+Н)+. Стадия 3 По общей методике получения амида 1 6-(7-циклопентил-6-(диметилкарбамоил)-7 Н-пирроло[2,3d]пиримидин-2-иламино)никотиновую кислоту с 5 экв. LiCl объединяли с (1R,5S)-трет-бутил-9 гидрокси-3,7-диазабицикло[3.3.1]нонан-3-карбоксилатом,что давало Получение 7-циклопентил-2-(5-1R,5S)-9-гидрокси-3,7-диазабицикло[3.3.1]нонан-3-карбонил)пиридин-2-иламино)-N,N-диметил-7 Н-пирроло[2,3-d]пиримидин-6-карбоксамида. По методике удаления защитной группы 2 (1R,5S)-трет-бутил-7-(6-(7-циклопентил-6(диметилкарбамоил)-7 Н-пирроло[2,3-d]пиримидин-2-иламино)никотиноил)-9-гидрокси-3,7-диазабицикло[3.3.1]нонан-3-карбоксилат превращали в 7-циклопентил-2-(5-1R,5S)-9-гидрокси-3,7-диазабицикло[3.3.1]нонан-3-карбонил)пиридин-2-иламино)-N,N-диметил-7 Н-пирроло[2,3-d]пиримидин-6-карбоксамид (47 мг) с выходом 93%. 1 Н ЯМР (400 МГц, CDCl3)ч./млн 9,04 (s, 1 Н), 8,85 (s, 1 Н), 8,60 (d, J=8,59 Гц, 1 Н), 8,50 (s, 1 Н), 7,83 (dd, J1=8,84 Гц, J2=2,27 Гц, 1 Н), 6,49 (s, 1 Н), 4,91-4,33 (m, 2 Н), 4,16-3,10 (m,12 Н), 3,10-2,36 (m, 5 Н), 2,28-1,53 (m, 8 Н); МС-ВР m/z 519,2849 (М+Н)+. Пример 2 Получение 6-(7-циклогептил-6-(диметилкарбамоил)-7 Н-пирроло[2,3-d]пиримидин-2-иламино)никотиновой кислоты-5 LiCl. В соответствии со стадией 1 в примере получения 1 аналогичным образом получали метиловый эфир 6-(7-циклогептил-6-диметилкарбамоил-7 Н-пирроло[2,3-d]пиримидин-2-иламино)никотиновой кислоты (1,34 г) с выходом 52%. 1H ЯМР (400 МГц, CDCl3)ч./млн 8,95 (d, J=2,0 Гц, 1 Н), 8,77 (s, 1H), 8,71 Получение 6-(7-циклогептил-6-(диметилкарбамоил)-7 Н-пирроло[2,3-d]пиримидин-2-иламино)никотиновой кислоты и 5 экв. LiCl. В соответствии со стадией 2 в примере получения 1 аналогичным образом получали 6-(7 циклогептил-6-(диметилкарбамоил)-7 Н-пирроло[2,3-d]пиримидин-2-иламино)никотиновую кислоту-5 экв. LiCl (1,87 г) с количественным выходом. 1 Н ЯМР (400 МГц, ДМСО-d6)ч./млн 9,70 (br. s., 1 Н), 8,81
МПК / Метки
МПК: C07B 59/00, C07D 519/00, A61K 31/519, C07D 487/04, A61P 35/00
Метки: качестве, пирролопиримидины, ингибиторов
Код ссылки
<a href="https://eas.patents.su/30-22355-pirrolopirimidiny-v-kachestve-ingibitorov-cdk4-6.html" rel="bookmark" title="База патентов Евразийского Союза">Пирролопиримидины в качестве ингибиторов cdk4/6</a>
Аналоги гетероциклил пиразолопиримидина в качестве ингибиторов jak

Номер патента: 22120
Опубликовано: 30.11.2015
Авторы: Дагостин Клаудио, Буссар Сириль, Бэль Кэсрин, Рэтклифф Эндру, Оксенфорд Сэлли, Рэмзден Найджель, Пайтн Нелли, Хэррисон Ричард Джон
МПК: A61K 31/505, A61P 35/00, A61P 37/00...
Метки: гетероциклил, аналоги, пиразолопиримидина, качестве, ингибиторов
Формула / Реферат:
1. Соединение формулы (I)или его фармацевтически приемлемая соль, гдеR означает Н или F;кольцо А представляет собой 5-членный ароматический гетероцикл, в котором Z1, Z2 и Z3 независимо выбраны из группы, включающей C(R1), N, N(R1), О и S, при условии, что по меньшей мере один из Z1, Z2, Z3 является N;каждый R1 независимо представляет собой Н, галоген; CN; C(O)OR2; OR2; C(O)R2; C(O)N(R2R2a); S(O)2N(R2R2a); S(O)N(R2R2a); S(O)2R2; S(O)R2;...
Гетероариламины-производные пиримидина и пиридазина в качестве ингибиторов гликогенсинтаза-киназы 3-бета (ингибиторов gskз)

Номер патента: 10859
Опубликовано: 30.12.2008
Авторы: Ван Акен Кун Жанн Альфонс, Фрейн Эдди Жан Эдгар, Койманс Люсьен Мария Хенрикус, Эмбрехтс Вернер Констант Йохан, Жанссен Поль Адриан Ян, Дильс Гастон Станислас Марселла, Леви Паулус Йоаннес, Виллемс Марк, Лав Кристофер Джон, Винкерс Хендрик Мартен, Бейнстерс Петер Якобус Йоханнес Антониус, Хэрес Ян, Де Жонж Марк Рене
МПК: A61K 31/435, A61K 31/505, A61K 31/50...
Метки: пиридазина, 3-бета, ингибиторов, gskз, качестве, пиримидина, гликогенсинтаза-киназы, гетероариламины-производные
Формула / Реферат:
1. Соединение формулы его N-оксид, фармацевтически приемлемая аддитивная соль, четвертичный амин и стереохимически изомерная форма, где кольцо А является R1 обозначает водород; X обозначает -O-; -O-C1-6алкил- или прямую связь; Z обозначает прямую связь, -С(=O)- или -NR1-C1-6алкил-; R обозначает водород или R20; R3 обозначает водород; галоген; циано; полигалогенС1-6алкил; R4 обозначает моноциклический, насыщенный или частично насыщенный или...
Гетероариламины в качестве ингибиторов гликогенсинтаза-киназы 3-бета (ингибиторов gsk3)

Номер патента: 7298
Опубликовано: 25.08.2006
Авторы: Леви Паулус Йоаннес, Винкерс Хендрик Мартен, Койманс Люсьен Мария Хенрикус, Хэрес Ян, Виллемс Марк, Бейнстерс Петер Якобус Йоханнес Антониус, Фрейн Эдди Жан Эдгар, Дильс Гастон Станислас Марселла, Эмбрехтс Вернер Констант Йохан, Жанссен Поль Адриан Ян, Ван Акен Кун Жанн Альфонс, Лав Кристофер Джон, Де Жонж Марк Рене
МПК: A61K 31/505, A61K 31/435, A61K 31/50...
Метки: гликогенсинтаза-киназы, качестве, 3-бета, gsk3, гетероариламины, ингибиторов
Формула / Реферат:
1. Соединение формулы его N-оксид, фармацевтически приемлемая аддитивная соль, четвертичный амин и стереохимически изомерная форма, где R1 обозначает водород; X обозначает -O-; -О-С1-4алкил- или прямую связь; Z обозначает прямую связь, -NH-, -С1-4алкил-NH- или -С(=O)-; R2 обозначает водород или фенил; R3 обозначает водород, С1-4алкил, полигалогенC1-6алкил или циано; R4 обозначает моноциклический или бициклический насыщенный гетероцикл;...
Пирролопиримидины и их применение

Номер патента: 19094
Опубликовано: 30.01.2014
Авторы: Хоу Ин, Мортенсон Пол, Врона Войцех, Лю Ипинь, Ли Йю, Конгрив Майлс Стьюарт, Дагостин Клаудио, Вудхед Стивен, Сун Му, Хэ Гуо, Лагу Бхарат, Брукс Клинтон А., Смит Трой, Брейн Кристофер Томас, Безонг Гильберт, Хауард Стивен
МПК: C07D 487/04, A61K 31/519, A61P 35/00...
Метки: применение, пирролопиримидины
Формула / Реферат:
1. Соединение формулы Iили его фармацевтически приемлемая соль, в которойX обозначает CR9;R1 обозначает CONR5R6, где R5 и R6 оба обозначают метил;R2 обозначает циклопентил;L обозначает связь, -СН2-, -СН2СН2-, -СН2СН2СН2-, С(О) или C(O)NH;Y является частью следующей группы:где m и n равны 1 или 2;Y и W обозначают N;где может содержаться 0-3 R8 и R8 обозначает C1-C8-алкил, оксогруппу, галоген или два или более R8 могут образовать мостиковую...
Новые производные имидазолонов в качестве лекарственных средств, способ их получения, фармацевтические композиции и применение в качестве ингибиторов протеинкиназ, в частности cdc7

Номер патента: 18496
Опубликовано: 30.08.2013
Авторы: Штайнметц Анке, Ронан Батист, Леруа Венсан, Консейер Эмманюэль, Бак Эрик, Леталлек Жан-Филипп
МПК: A61K 31/437, C07D 471/04, A61P 35/00...
Метки: качестве, частности, протеинкиназ, имидазолонов, производные, средств, применение, лекарственных, способ, композиции, новые, получения, ингибиторов, фармацевтические
Формула / Реферат:
1. Соединения формулы (I)в которой X-Y обозначает NH-C(S), N=C-NR7R8, N=C-SR, N=C-R или N=C-OR;R1 обозначает атом водорода, радикал циклоалкил или радикал алкил, гетероциклоалкил, арил или гетероарил, причем все эти радикалы необязательно замещены;R, идентичный или отличающийся от R1, выбран из значений R1;R2 обозначает атом водорода, атом галогена или радикал алкил;R3 обозначает атом водорода, атом галогена, радикал гидроксил или радикал алкил...
Предыдущий патент: Терапевтическое применение конъюгатов интерферон-пэг
Следующий патент: Способ управления средами в микрофлюидных системах с обратной связью
Случайный патент: Замещенные трициклические соединения.