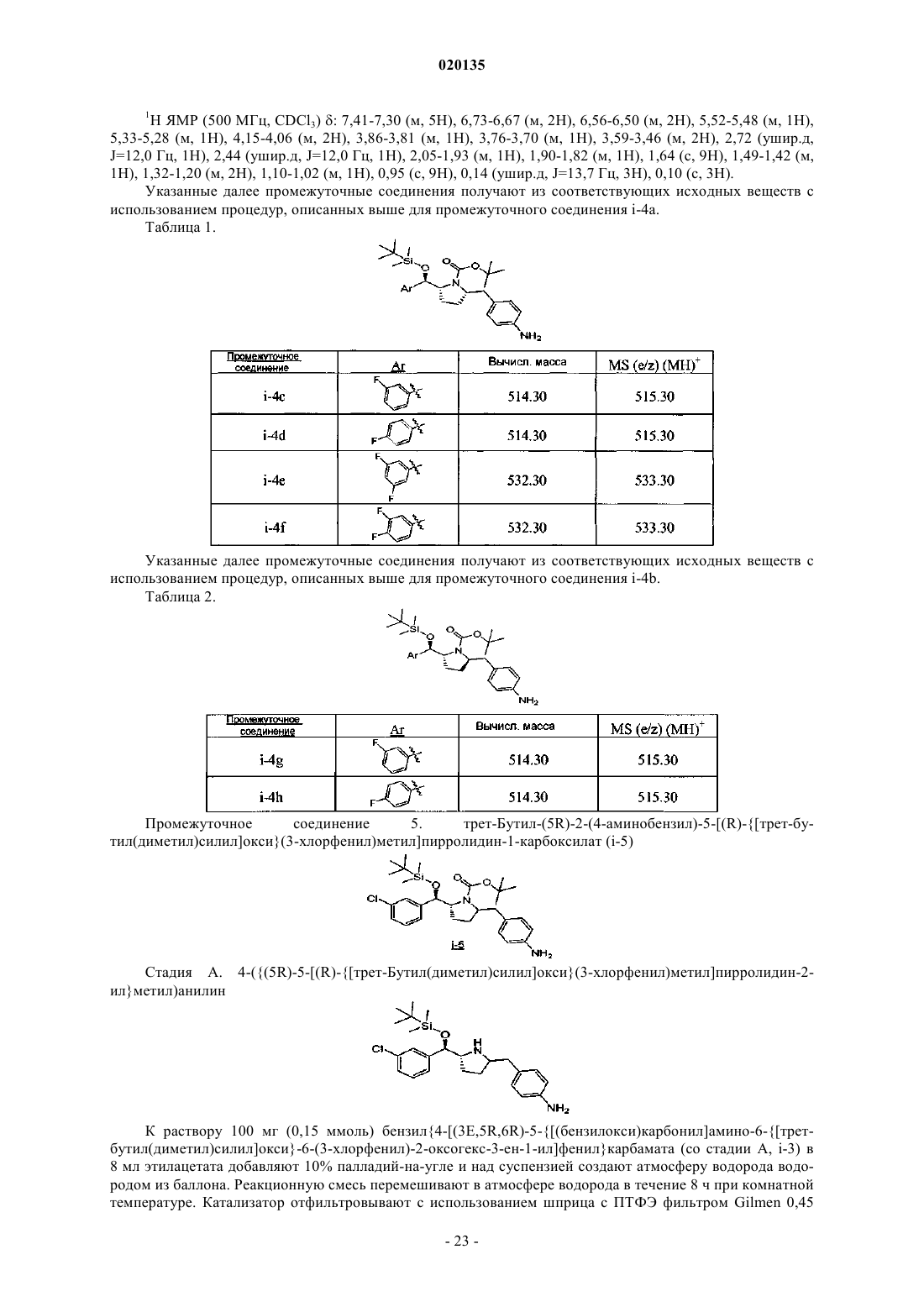
Гидроксиметилпирролидины в качестве агонистов адренергических рецепторов бета 3
Номер патента: 20135
Опубликовано: 29.08.2014
Авторы: Шэнь Дун-Мин, Мойз Крис Р., Ха Соокхее Николь, Ли Бин, Кар Нам Фунг, Копка Ихор Е., Чжу Чэн, Бергер Ричард, Гобле Стефен Д., Чан Лехуа, Мориэлло Грегори Дж., Ван Липин, Эдмондсон Скотт Д.





























Формула / Реферат
1. Соединение формулы Iа

или его фармацевтически приемлемая соль, или его стереоизомер, или фармацевтически приемлемая соль его стереоизомера,
в которой
n равен 0, 1 или 2;
Y выбирают из группы, состоящей из:
(1) (C1-C5)алкандиила и
(2) связи;
Z выбирают из группы, состоящей из тиазолила, пиридила, дигидропиридила, 1,2,4-триазолила, 1,2,3-триазолила, пиримидинила, дигидропиримидинила, тетрагидропиримидинила, пиридазинила, дигидропиридазинила, пирролидинила, пиразолила

R3 выбирают из группы, состоящей из:
(1) (C1-C6)алкила,
(2) оксо и
(3)-NH2.
2. Соединение по п.1, где Y представляет собой метилен, -СН(СН3)- или связь.
3. Соединение формулы Iа

или его фармацевтически приемлемая соль, или его стереоизомер, или фармацевтически приемлемая соль его стереоизомера,
в которой
n равен 0, 1 или 2,
Y выбирают из группы, состоящей из метилена, -СН(СН3)- и связи,
Z выбирают из группы, состоящей из тиазолила, пиридила, дигидропиридила, 1,2,4-триазолила, пиримидинила, дигидропиримидинила, пиридазинила, дигидропиридазинила, пиразолила

и R3 выбирают из группы, состоящей из:
(1) метила,
(2) оксо и
(3) -NH2.
4. Соединение, выбранное из группы, состоящей из



или его фармацевтически приемлемая соль, или его фармацевтически приемлемый стереоизомер, или фармацевтически приемлемая соль его стереоизомера.
5. Соединение по п.4, выбранное из группы, состоящей из

или его фармацевтически приемлемая соль, или его фармацевтически приемлемый стереоизомер, или фармацевтически приемлемая соль его стереоизомера.
6. Фармацевтическая композиция для лечения или предупреждения заболевания или расстройства, опосредуемого активацией адренорецептора β3, включающая соединение по п.1 и фармацевтически приемлемый носитель.
7. Способ лечения или предупреждения заболевания или расстройства, опосредуемого активацией адренорецептора β3, включающий введение пациенту, нуждающемуся в этом, терапевтически эффективного количества соединения по п.1.
8. Способ по п.7, где заболевание или расстройство выбирают из группы, состоящей из: (1) повышенной активности мочевого пузыря, (2) недержания мочи, (3) безотлагательности мочеиспускания с недержанием мочи и (4) безотлагательности мочеиспускания.
9. Применение соединения по п.1 при получении лекарственного средства для лечения или предупреждения заболевания или расстройства, опосредуемого активацией адренорецептора β3.
10. Соединение формулы

или его фармацевтически приемлемая соль, или его фармацевтически приемлемый стереоизомер, или фармацевтически приемлемая соль его стереоизомера.
11. Соединение формулы

или его фармацевтически приемлемая соль, или его фармацевтически приемлемый стереоизомер, или фармацевтически приемлемая соль его стереоизомера.
Текст
ГИДРОКСИМЕТИЛПИРРОЛИДИНЫ В КАЧЕСТВЕ АГОНИСТОВ АДРЕНЕРГИЧЕСКИХ РЕЦЕПТОРОВ БЕТА 3 Настоящее изобретение относится к соединениям формулы (Iа), фармацевтическим композициям с указанными соединениями и способам их применения при лечении или предупреждении заболеваний, опосредуемых активацией адренорецептора 3.(71)(73) Заявитель и патентовладелец: МЕРК ШАРП ЭНД ДОМЭ КОРП. Предпосылки создания изобретения Функцией нижнего отдела мочевых путей является накопление и периодическое выделение мочи. Для этого требуется гармоническое сочетание накопления и рефлексов мочеиспускания, в которые вовлекается ряд афферентных и эфферентных нервных трактов, ведущих к модуляции центральных и периферических нейроэффекторных механизмов, и как результат - координированная регуляция симпатических и парасимпатических компонентов автономной нервной системы, а также соматических моторных трактов. Они непосредственно регулируют сократительное состояние мочевого пузыря (сжиматель) и гладкой мышцы мочевого пузыря и поперечно-полосатой мышцы уретрального сфинктера. В гладкой мышце сжимателя различных видов, в том числе, человека, крысы, морской свинки, кролика, хорька, собаки, кошки, свиньи и примата, не являющегося человеком, присутствуют адренергические рецепторы (AR). Однако фармакологические исследования показывают существование заметных видовых различий в подтипах рецепторов, опосредующих релаксацию изолированного сжимателя; у кошек и морских свинок преобладает 1AR, у кролика преобладает 2AR, и 3AR вносит вклад или преобладает в сжимателе собаки, крысы, хорька, свиньи, обезьяны и человека. Экспрессия подтипов AR в сжимателе человека и крысы проверена различными методами, и присутствие 3AR подтверждено с использованием гибридизации in situ и/или полимеразной цепной реакции с обратной транскрипцией (ОТ-ПЦР, RT-PCR). Количественные ПЦР-анализы в реальном времени мРНК 1AR, 2AR и 3AR в ткани мочевого пузыря больных, подвергающихся радикальной цистэктомии, показывают преобладание мРНК 3AR(97%, сравните с 1,5% мРНК 1AR и 1,4% мРНК 2AR). Более того, экспрессия мРНК 3AR равнозначна в контрольных и закупоренных мочевых пузырях людей. Такие данные предполагают, что обструкция выхода из мочевого пузыря не приводит к даун-регуляции 3AR или к изменению релаксации сжимателя, опосредуемой 3AR. Также сравнивали реактивность 3AR в полосках мочевого пузыря, полученных во время цистэктомии или энтероцистопластики от пациентов, решивших иметь нормальную функцию мочевого пузыря, и от пациентов с гипорефлексией или гиперрефлексией сжимателя. Не наблюдали различий между степенью или силой релаксации, опосредуемой агонистом 3AR, что согласуется с концепцией, что активация 3AR является эффективным путем релаксации сжимателя в здоровом и болезненном состояниях. Официальное свидетельство в поддержку важной роли 3AR в накоплении мочи следует из исследований in vivo. После внутривенного введения крысам селективный агонист 3AR грызуна CL316243 снижает давление в мочевом пузыре и в цистометрических исследованиях повышает емкость мочевого пузыря, что ведет к пролонгации интервала между мочеиспусканиями без повышения остаточного объема мочи. Мочевой пузырь с повышенной активностью (ОАВ) характеризуется симптомами безотлагательности мочеиспускания с недержанием мочи или без него, как правило, связанными с частотой и никтурией. Распространенность ОАВ в Соединенных Штатах и Европе оценивается в 16-17% как среди женщин, так и среди мужчин в возрасте старше 18 лет. Мочевой пузырь с повышенной активностью наиболее часто классифицируется как идиопатический, но также может являться вторичным при неврологическом состоянии, обструкции выхода из мочевого пузыря и в других случаях. Из патофизиологической перспективы комплекс симптомов мочевого пузыря с повышенной активностью, когда связан с недержанием мочи, предполагает повышенную активность сжимателя. Показано, что безотлагательность мочеиспускания с недержанием мочи или без него отрицательно влияет как на социальное, так и на медицинское здоровье и представляет существенную нагрузку в смысле ежегодных расходов на медико-санитарную помощь. Более того, существующая лекарственная терапия против безотлагательности (с недержанием или без него) является субоптимальной, так как многие пациенты или не показывают адекватной реакции на существующее лечение и/или не способны переносить существующее лечение (например, сухость во рту, связанная с антихолинергической терапией). Поэтому существует потребность в новых хорошо переносимых способах лечения, которые эффективно лечат от частоты мочеиспускания, безотлагательности и недержания, или в виде монотерапии, или в сочетании с доступными видами терапии. Ожидается,что средства, которые релаксируют гладкую мышцу мочевого пузыря, такие как агонисты 3AR, будут эффективными для лечения таких расстройств мочеиспускания. Сущность изобретения Настоящее изобретение относится к новым агонистам 3AR, фармацевтическим композициям, содержащим их, а также к способам лечения или профилактики опосредуемых через 3AR расстройств с использованием таких новых соединений. Описание изобретения Настоящее изобретение относится к соединению структурной формулы 1 а или его фармацевтически приемлемой соли или его стереоизомеру или фармацевтически приемлемой соли его стереоизомера,где(3) -NH2. В одном воплощении соединения формулы 1 а представляют собой соединения, где n равен 0, 1 или 2. В одном подмножестве данного воплощения n равен 0. В другом подмножестве n равен 1. В еще одном подмножестве n равен 2. В одном воплощении соединения формулы 1 а представляют собой соединения, где Y представляет собой метилен, -CH(CH3)- или связь. В одном их подмножестве Y представляет собой метилен или-CH(CH3)-. В другом их подмножестве Y представляет собой связь. В другом воплощении соединения формулы 1 а представляют собой соединения, где Z выбирают из группы, состоящей из тиазолила, пиридила, дигидропиридила, 1,2,4-триазолила, 1,2,3-триазолила, пиримидинила, дигидропиримидинила, тетрагидропиримидинила, пиридазинила, дигидропиридазинила, пирролидинила, пиразолила В другом воплощении соединения формулы 1 а представляют собой соединения, где Z выбирают из группы, состоящей из тиазолила, пиридила, дигидропиридила, 1,2,4-триазолила, 1,2,3-триазолила, пиримидинила, дигидропиримидинила, тетрагидропиримидинила, пиридазинила, дигидропиридазинила, пирролидинила и пиразолила. В другом воплощении соединения формулы 1 а представляют собой соединения, где Z выбирают из группы, состоящей из В другом воплощении соединения формулы 1 а представляют собой соединения, где R3 выбирают из группы, состоящей из:(3) -NH2. В одном подмножестве данного воплощения R3 представляет собой метил или этил. В другом подмножестве R3 представляет собой оксо. В другом подмножестве R3 представляет собой -NH2. В другом воплощении соединения формулы 1 а представляют собой соединения со специфической стереоконфигурацией при указанном хиральном центре В другом воплощении соединения формулы 1 а представляют собой соединения со специфической стереоконфигурацией при указанных хиральных центрах, причем хиральный центр, помеченный звездочкой, представляет собой R или S В одном подмножестве конфигурация при хиральном центре, помеченном звездочкой, представляет собой S. Используемый в данном описании термин алкил предназначен для обозначения как разветвленных, так и линейных насыщенных алифатических углеводородных групп с определенным числом атомов углерода, например метила (Me), этила (Et), н-пропила (Pr), н-бутила (Bu), н-пентила, н-гексила и их изомеров, таких как изопропил (изо-Pr), 1-изобутил (изо-бут), втор-бутил (втор-Bu), трет-бутил (третBu), изопентил, изогексил и т.п. Термин циклоалкил обозначает моноциклический насыщенный карбоцикл с определенным числом атомов углерода, например 3, 4, 5 или 6 атомами углерода. Примеры циклоалкила включают циклопропил, циклобутил, циклопентил и циклогексил. Термин алкандиил относится к линейному или разветвленному двухвалентному углеводородному радикалу с определенным числом атомов углерода. Термины алкендиил и алкиндиил относятся к линейным или разветвленным ненасыщенным двухвалентным углеводородным радикалам. Алкендиил характеризуется двойной углерод-углеродной связью, и алкиндиил характеризуется тройной углерод-углеродной связью. Примеры алкандиила включают, но не ограничиваются перечисленным, метилен (-CH2-), этилен (-СН 2 СН 2-), 1,1 этандиил (-СН(CH3)-), 1,2-пропандиил (-СН(СН 3)СН 2-), 2-метил-1,1-пропандиил (-СН[С(CH3)2])-); примеры алкендиила включают, но не ограничиваются перечисленным, 1,1-этендиил (-С(=СН 2)-), 1,2-этендиил(-СН=СН-) и 2-пропен-1,1-диил (-СН(СН=СН 2)-); примеры алкиндиила включают, но не ограничиваются перечисленным, 1,2-этиндиил (-СС-) и 3-бутин-1,1-диил (-СН(СН 2 ССН)-). Примером галогензамещенного алкандиила является -С(CH3)(F)-. Термин необязательно замещенный означает незамещенный или замещенный, и, следовательно,общие структурные формулы, описанные в данном описании, охватывают соединения, содержащие определенный необязательный заместитель, а также соединения, которые не содержат необязательный заместитель. Каждая переменная определяется независимо каждый раз, когда имеет место в определениях общих структурных формул. Термины гало или галоген предназначены для включения фтора, хлора, брома и иода, если не указано иное. Предпочтительными являются фтор и хлор. Термины карбоцикл или карбоциклический относятся к насыщенным, частично ненасыщенным и ароматическим циклам, имеющим в цикле только атомы углерода. Примеры включают, но не ограничиваются перечисленным, циклопентил, циклогексил, циклогептил, циклооктил, циклопентенил, циклогексенил, циклогексадиенил и фенил. Термин арил относится к ароматическому карбоциклу. В рамках определения Z термин бензольный цикл, конденсированный с (С 5-С 10)-карбоциклом включает, но не ограничивается перечисленным, нафтил, дигидронафтил, тетрагидронафтил, инданил, инденил, бензоциклогептен, тетрагидробензоциклогептен и т.п.; предпочтительно бензол конденсирован с (C5-C6)карбоциклом. Такой конденсированный цикл может присоединяться к остальной части молекулы через атом углерода в любом цикле. Термины гетероцикл или гетероциклический относятся к насыщенным, частично ненасыщенным и ароматическим циклам, имеющим по меньшей мере один циклический гетероатом и по меньшей мере один циклический атом углерода; гетероцикл может присоединяться к остальной части молекулы через циклический атом углерода или циклический атом азота. Термины гетероарил или гетероарильный относятся к ароматическому гетероциклу. В рамках определения Z термин 5- или 6 членный гетероцикл с 1-4 гетероатомами, выбранными из атомов кислорода, серы и азота включает, но не ограничивается перечисленным, пирролил, тиенил, фуранил, имидазолил, пиразолил, оксазолил, изоксазолил, тиазолил, изотиазолил, триазолил, тетразолил, оксадиазолил, тиадиазолил, пирролидинил, тетрагидрофуранил, пиридинил, дигидропиридинил, тетрагидропиридинил, пиримидинил, дигидропиримидинил, тетрагидропиримидинил, пиразинил, дигидропиразинил, тетрагидропиразинил, пиридазинил, дигидропиридазинил, тетрагидропиридазинил, пиперидинил, пиперазинил, морфолинил, пиранил, дигидропиранил, тетрагидропиранил и т.п. В случае термина (R3)n, а также любых других подобных обозначений, когда n равен 0, тогда R3 представляет собой водород; когда n больше 1, тогда при каждом появлении R3 независимо выбирают из всех подходящих или указанных для R3 групп. Оптические изомеры - диастереомеры - геометрические изомеры - таутомеры Соединения, описанные в данном описании, могут содержать асимметрический центр и, таким образом, могут существовать в виде энантиомеров. Когда соединения по изобретению обладают двумя или большим числом асимметрических центров, они также могут существовать в виде диастереомеров. Когда в формулах в изобретении связи хирального углерода отображаются в виде прямых линий, это понимается как то, что в формулах заключаются обе конфигурации хирального углерода (R) и (S), и, следова-3 020135 тельно, оба энантиомера и их смеси. Настоящее изобретение включает все такие возможные стереоизомеры как, по существу, чистые расщепленные энантиомеры, их рацемические смеси, а также смеси диастереомеров. Приведенная выше формула 1 а показана без отличительной стереохимии в некоторых положениях. Настоящее изобретение включает все стереоизомеры соединений формулы 1 а и их фармацевтически приемлемые соли. Диастереомерные пары энантиомеров можно разделить, например, фракционной кристаллизацией из подходящего растворителя и полученную таким образом пару энантиомеров можно разделить на отдельные стереоизомеры обычными способами, например, с использованием оптически активной кислоты или основания в качестве расщепляющего агента или ВЭЖХ на хиральной колонке. Также любой энантиомер или диастереомер соединения общей формулы 1 а может быть получен стереоспецифическим синтезом с использованием оптически чистых исходных материалов или реагентов известной конфигурации. Когда соединения, описанные в данном описании, содержат олефиновые двойные связи, если не указано иное, такие двойные связи означают включение как Е, так и Z геометрических изомеров. Некоторые из соединений, описанных в данном описании, могут существовать с различными точками присоединения водорода, и такие соединения называют таутомерами. Например, соединения,включающие карбонильные группы -СН 2 С(О)- (кетогруппы), могут претерпевать таутомеризм с образованием гидроксилсодержащих групп -СН=С(ОН)- (енольные формы). Как кетоформы, так и енольные формы по отдельности, а также их смеси включены в объем настоящего изобретения. Соли Термин фармацевтически приемлемые соли относится к солям, полученным из фармацевтически приемлемых нетоксичных оснований или кислот. Когда соединение по настоящему изобретению представляет собой кислоту, его соответствующую соль можно обычно получить с фармацевтически приемлемым нетоксичным основанием, в том числе, неорганическими основаниями и органическими основаниями. Соли, полученные с такими неорганическими основаниями, включают соли алюминия, аммония,кальция, меди (одно- и двухвалентной), железа (3), железа (2), лития, магния, марганца (двух- и трехвалентного), калия, натрия, цинка и подобные соли. Предпочтительными являются соли аммония, кальция,магния, калия и натрия. Соли, полученные с фармацевтически приемлемыми нетоксичными органическими основаниями, включают соли первичных, вторичных и третичных аминов, полученных как из встречающихся в природе, так и синтетических источников. Фармацевтически приемлемые органические нетоксичные основания, с которыми можно получить соли, включают, например, аргинин, бетаин, кофеин, холин, N,N'-дибензилэтилендиамин, диэтиламин, 2-диэтиламиноэтанол, 2-диметиламиноэтанол, этаноламин, этилендиамин, N-этилморфолин, N-этилпиперидин, глюкамин, глюкозамин, гистидин, гидрабамин, изопропиламин, дициклогексиламин, лизин, метилглюкамин, морфолин, пиперазин, пиперидин,полиаминовые смолы, прокаин, пурины, теобромин, триэтиламин, триметиламин, трипропиламин, трометамин и т.п. Когда соединение по настоящему изобретению представляет собой основание, его соответствующую соль можно обычно получить с фармацевтически приемлемой нетоксичной неорганической и органической кислотой. Такие кислоты включают, например, уксусную, бензолсульфоновую, бензойную,камфорсульфоновую, лимонную, этансульфоновую, фумаровую, глюконовую, глутаминовую, бромоводородную, хлороводородную, изэтионовую, молочную, малеиновую, яблочную, миндальную, метансульфоновую, слизевую, азотную, памовую, пантотеновую, фосфорную, янтарную, серную, винную, птолуолсульфоновую кислоту и подобные кислоты. Предпочтительными являются лимонная, бромоводородная, хлороводородная, малеиновая, фосфорная, серная и винная кислоты. Сольваты Настоящее изобретение включает в свой объем сольваты соединений формулы 1 а. Используемый в данном описании термин сольват относится к комплексу переменной стехиометрии, образованному растворенными веществом (т.е. соединением формулы 1a) или его фармацевтически приемлемой солью и растворителем, что не влияет на биологическую активность растворенного вещества. Примеры растворителей включают, но не ограничиваются перечисленным, воду, этанол и уксусную кислоту. Когда растворителем является вода, сольват известен как гидрат; гидраты включают, но не ограничиваются перечисленным, геми-, моно-, сескви-, ди- и тригидраты. Пролекарства Настоящее изобретение включает в свой объем применение пролекарств соединений по данному изобретению. Вообще, такие пролекарства будут представлять собой функциональные производные соединений по данному изобретению, которые могут легко превращаться in vivo в требуемое соединение. Таким образом, в способах лечения по настоящему изобретению термин введение будет охватывать лечение различных описанных состояний соединением формулы 1 а или соединением, которое может не являться соединением формулы 1 а, но которое превращается в соединение формулы 1a in vivo после введения пациенту. Обычные процедуры отбора и получения подходящих пролекарственных производных описаны, например, в Design of Prodrugs, ed. H. Bundgaard, Elsevier, 1985. Использование Соединения по настоящему изобретению являются сильными агонистами адренорецептора 3 и как таковые применимы при лечении или предупреждении заболеваний, расстройств или состояний, опосредуемых активацией адренорецептора 3. Таким образом, один аспект настоящего изобретения относится к способу лечения, регуляции или предупреждения таких заболеваний, расстройств или состояний у млекопитающего, который включает введение такому млекопитающему терапевтически эффективного количества соединения формулы 1 а. Термин млекопитающее включает человека и животных, не относящихся к человеку, таких как собаки и кошки и т.п. Заболевания, расстройства или состояния, в случае которых соединения по настоящему изобретению применимы для лечения или предупреждения, включают, но не ограничиваются перечисленным, (1) мочевой пузырь с повышенной активностью, (2) недержание мочи, (3) безотлагательность с недержанием мочи, (4) безотлагательность мочеиспускания, (5) сахарный диабет, (6) гипергликемию, (7) ожирение, (8) гиперлипидемию, (9) гипертриглицеридемию,(10) гиперхолестеринемию, (11) атеросклероз коронарной, мозговой и периферических артерий, (12) желудочно-кишечные расстройства, включающие пептическую язву, эзофагит, гастрит и дуоденит (в том числе, вызываемый Н. pylori), кишечные изъязвления (в том числе, воспалительное заболевание кишечника, неспецифический язвенный колит, болезнь Крона и проктит) и желудочно-кишечные изъязвления,(13) нейрогенное воспаление дыхательных путей, включая кашель, астму, (14) депрессию, (15) заболевания предстательной железы, такие как доброкачественная гиперплазия предстательной железы, (16) синдром раздраженной толстой кишки и другие расстройства, нуждающиеся в пониженной подвижности кишок, (17) диабетическую ретинопатию, (18) преждевременные роды и (19) повышенное внутриглазное давление и глаукому. Любой подходящий способ введения можно использовать для предоставления млекопитающему, в особенности, человеку, эффективной дозировки соединения по настоящему изобретению. Например,можно использовать пероральный, ректальный, местный, парентеральный, глазной, легочный, назальный и подобные пути. Лекарственные формы включают таблетки, пастилки, дисперсии, суспензии, растворы,капсулы, кремы, мази, аэрозоли и т.п. Предпочтительно соединения формулы 1 а вводят перорально. Эффективная дозировка активного ингредиента может изменяться в зависимости от определенного используемого соединения, способа введения, состояния, которое лечат, и тяжести состояния, которое лечат. Такая дозировка может быть легко установлена специалистом в данной области техники. При лечении мочевого пузыря с повышенной активностью (ОАВ) одними соединениями по настоящему изобретению или в сочетании с другими средствами против ОАВ, как правило, удовлетворительные результаты получают, когда соединения по настоящему изобретению вводят в суточной дозировке от 0,01 до примерно 100 мг на кг массы тела животного, предпочтительно даваемой в однократной дозе или раздельных дозах два-шесть раз в сутки, или в форме с отсроченным высвобождением. В случае 70-кг взрослого человека общая суточная доза, как правило, будет составлять от примерно 0,7 до примерно 3500 мг, или специфичнее, от примерно 0,7 до примерно 2000 мг. Такая схема приема может быть отрегулирована для получения оптимального терапевтического отклика. При лечении одного ожирения или в сочетании с диабетом и/или гипергликемией, как правило,удовлетворительные результаты получают, когда соединения по настоящему изобретению вводят в суточной дозировке от 0,01 до примерно 100 мг на кг массы тела животного, предпочтительно даваемой в однократной дозе или раздельных дозах два-шесть раз в сутки, или в форме с отсроченным высвобождением. В случае 70-кг взрослого человека общая суточная доза, как правило, будет составлять от примерно 0,7 до примерно 3500 мг. Такая схема приема может быть отрегулирована для получения оптимального терапевтического отклика. При лечении сахарного диабета и/или гипергликемии, а также других заболеваний или расстройств,для которых применимы соединения формулы 1 а, как правило, удовлетворительные результаты получают, когда соединения по настоящему изобретению вводят в суточной дозировке от 0,001 до примерно 100 мг на кг массы тела животного, предпочтительно даваемой в однократной дозе или раздельных дозах два-шесть раз в сутки, или в форме с отсроченным высвобождением. В случае 70-кг взрослого человека общая суточная доза, как правило, будет составлять от примерно 0,07 до примерно 350 мг. Такая схема приема может быть отрегулирована для получения оптимального терапевтического отклика. В одном воплощении соединение по настоящему изобретению используют для получения лекарственного средства для лечения или предупреждения заболевания или расстройства, опосредуемого активацией адренорецептора 3. Другой аспект настоящего изобретения относится к фармацевтическим композициям, включающим соединение формулы 1 а и фармацевтически приемлемый носитель. Фармацевтические композиции по настоящему изобретению включают соединение формулы 1 а или его фармацевтически приемлемую соль как активный ингредиент и также могут содержать фармацевтически приемлемый носитель и, необязательно, другие терапевтические ингредиенты. Термин фармацевтически приемлемые соли относится к солям, полученным с фармацевтически приемлемыми нетоксичными основаниями или кислотами, включая неорганические основания или кислоты и органические основания или кислоты.(офтальмического), легочного (назальной или трансбуккальной ингаляции) или назального введения,хотя наиболее подходящий путь в любом данном случае будет зависеть от характера и тяжести состояний, которые лечат, и природы активного ингредиента. Они обычно могут предоставляться в стандартной лекарственной форме, и их получают любым из способов, хорошо известных в области фармации. При практическом использовании соединения формулы 1 а в качестве активного ингредиента можно объединять при равномерном перемешивании с фармацевтическим носителем согласно обычным методам получения фармацевтических препаратов. Носитель может принимать самые разные формы, в зависимости от формы препарата, нужного для введения, например, перорального или парентерального(включая внутривенное). При получении композиций для пероральной лекарственной формы можно использовать любую обычную фармацевтическую среду, такую как, например, вода, гликоли, масла, спирты, корригенты, консерванты, красители и т.п. для жидких пероральных препаратов, таких как, например, суспензии, эликсиры и растворы; или такие носители, как крахмалы, сахара, микрокристаллическая целлюлоза, наполнители, агенты грануляции, смазывающие вещества, связующие вещества, вещества,способствующие рассыпанию, и т.п. в случае пероральных твердых препаратов, таких как, например,порошки, твердые и мягкие капсулы и таблетки, причем твердые пероральные препараты предпочтительнее жидких препаратов. Из-за легкости введения таблетки и капсулы представляют наиболее выгодную пероральную лекарственную форму, и в таком случае, разумеется, используют твердые фармацевтические носители. При необходимости на таблетки может быть нанесено покрытие стандартными водными и неводными методами. Такие композиции и препараты должны содержать по меньшей мере 0,1% активного соединения. Процентное содержание активного соединения в таких композициях может, конечно, изменяться и обычно может составлять от примерно 2 до примерно 60 мас.% на единицу. Количество активного соединения в таких терапевтически применимых композициях является таким, что будет получаться эффективная дозировка. Активные соединения также можно вводить интраназально, в виде, например,жидких капель или спрея. Таблетки, пилюли, капсулы и т.п. также могут содержать связующее вещество, такое как трагакантовая смола, аравийская камедь, кукурузный крахмал или желатин; эксципиенты, такие как дикальцийфосфат; вещество, способствующее рассыпанию, такое как кукурузный крахмал, картофельный крахмал,альгиновая кислота; смазывающее вещество, такое как стеарат магния; и подслащивающее средство, такое как сахароза, лактоза или сахарин. Когда стандартная лекарственная форма представляет собой капсулу, она может содержать кроме материалов указанных выше типов жидкий носитель, такой как жирное масло. Могут присутствовать различные другие материалы в качестве покрытий или модифицирующие физическую форму стандартной лекарственной формы. Например, таблетки могут быть покрыты шеллаком, сахаром или тем и другим. Сироп или эликсир могут содержать, кроме активного ингредиента, сахарозу как подслащивающее вещество, метил- и пропилпарабены как консерванты, краситель и корригент, такие как вишневая или апельсиновая отдушка. Соединения формулы 1 а также можно вводить парентерально. Растворы или суспензии указанных активных соединений можно получить в воде, соответствующим образом смешанной с поверхностно-активным веществом, таким как гидроксипропилцеллюлоза. Дисперсии также можно получить в глицерине, жидких полиэтиленгликолях и их смесях в маслах. При обычных условиях хранения и применения такие препараты содержат консервант для предотвращения роста микроорганизмов. Фармацевтические формы, подходящие для применения в качестве инъекций, включают стерильные водные растворы или дисперсии и стерильные порошки для приготовления стерильных растворов или дисперсий для инъекций для немедленного применения. Во всех случаях форма должна быть стерильной и должна быть текучей до такой степени, чтобы могла легко применяться с помощью шприца. Она должна быть устойчива в условиях получения и хранения и должна быть защищена от загрязняющего действия микроорганизмов, таких как бактерии и грибы. Носитель может представлять собой растворитель или дисперсионную среду, содержащую, например, воду, этанол, полиол (например, глицерин,пропиленгликоль и жидкий полиэтиленгликоль), их подходящие смеси и растительные масла. Соединения формулы 1 а можно использовать в сочетании с другими лекарственными средствами,которые используют при лечении/предупреждении/подавлении или облегчении заболеваний или состояний, для которых применяют соединения формулы 1 а. Такие другие лекарственные средства можно вводить способом и в количестве, обычно используемыми для них, одновременно или последовательно с соединением формулы 1 а. Когда соединение формулы 1 а используют одновременно с одним или несколькими другими лекарственными средствами, предпочтительна фармацевтическая стандартная лекарственная форма, содержащая такие другие лекарственные средства в дополнение к соединению формулы 1 а. Соответственно, фармацевтические композиции по настоящему изобретению включают композиции,которые кроме соединения формулы 1 а также содержат один или несколько других активных ингредиен-6 020135 тов. Примеры других активных ингредиентов, которые можно комбинировать с соединением формулы 1 а, вводимые или раздельно, или в единых фармацевтических композициях, включают, но не ограничиваются перечисленным:(а) лекарственные средства против повышенной активности мочевого пузыря, включающие (i) антагонисты мускариновых рецепторов (например, толтеродин, оксибутинин, в том числе, S-оксибутинин,гиосциамин, пропантелин, пропиверин, троспий, включая хлорид троспия, солифенацин, дарифенацин,имидафенацин, фезотеродин, темиверин, SVT-40776, 202405 от GlaxoSmithKline, TD6301, RBX9841,DDP200, PLD179 и другие антихолинергетики; см., например, US 5382600, US 3176019, US 3480626, US 4564621, US 5096890, US 6017927, US 6174896, US 5036098, US 5932607, US 6713464, US 6858650 и DD 106643. См. также US 6103747, US 6630162, US 6770295, US 6911217, US 5164190, US 5601839, US 5834010, US 6743441, WO 2002000652, WO 200400414853; как будет понятно специалистам в данной области техники, такие лекарственные средства можно вводить перорально или местно в стандартных формах или формах с отсроченным высвобождением, таких как формы с отсроченным высвобождением толтеродина, с отсроченным высвобождением оксибутинина и трансдермального оксибутинина), (ii) антагонисты NK-1 или NK-2 (например, апрепитант, цизолиртин, соединения, раскрытые в WO 2005/073191, WO 2005/032464, и другие описанные антагонисты NK-1), (iii) антагонисты альфаадренергических рецепторов (например, альфузозин, доксазозин, празозин, тамсулозин, теразозин и другие), (iv) средства, открывающие калиевые каналы (например, кромакалим, пинацидил и другие), (v) ванилоиды и другие модуляторы афферентных нервов - агонисты и антагонисты (например, капсаицин,резинифератоксин и другие), (vi) агонисты дофаминовых рецепторов D1 (например, перголинд), (vii) ингибиторы повторного поглощения серотонина и/или норэпинефрина (например, дулоксетин), (viii) ингибиторы нервно-мышечного соединения высвобождения ацетилхолина (например, ботулинический токсин), (ix) блокаторы кальциевых каналов (например, дилитазем, нифедипин, верапамил и другие), (х) ингибиторы синтеза простагландина (например, флурбипрофен), (xi) антагонисты рецепторов гаммааминомасляной кислоты (например, баклофен), (xii) вагинальные эстрогенные препараты, (xiii) селективные ингибиторы повторного поглощения норэпинефрина, (xiv) агонисты 5-НТ 2 С, (xv) потенциалозависимый блокатор натриевых каналов, (xvi) антагонисты пуринергических рецепторов Р 2 Х (например,антагонисты Р 2 Х 1 или Р 2 Х 3), (xvii) ингибиторы PAR2, (xviii) ингибиторы фосфодиэстеразы (например,ингибиторы PDE1, PDE4 и PDE5) и (xix) АТФ-чувствительные вещества, открывающие калиевые каналы;(b) сенсибилизаторы инсулина, в том числе (i) агонисты PPAR, такие как глитазоны (например,троглитазон, пиоглитазон, энглитазон, МСС-555, BRL49653 и т.п.), и соединения, раскрытые в WO 97/27857, 97/28115, 97/28137 и 97/27847; (ii) бигуаниды, такие как метформин и фенформин;(c) инсулин или миметики инсулина;(d) сульфонилмочевины, такие как толбутамид и глипизид;(f) средства, снижающие уровень холестерина, такие как (i) ингибиторы HMG-CoA-редуктазы (ловастатин, симвастатин и правастатин, флувастатин, аторвастатин и другие статины), (ii) вещества, усиливающие секрецию (холестирамин, колестипол и диалкиламиноалкильные производные сшитого декстрана), (ii) никотиниловый спирт, никотиниловая кислота или ее соль, (iii) агонисты рецепторов пролифератора-активатора а, такие как производные фенофибриновой кислоты (гемфиброзил, клофибрат, фенофибрат и бензафибрат), (iv) ингибиторы поглощения холестерина, например, бета-ситостерин и эзетимид, и ингибиторы ацил-СоА (холестерин-ацетилтрансферазы), например метиламид, (v) пробукол, (vi) витамин Е и (vii) тиромиметики;(g) агонисты PPAR, такие как раскрытые в WO 97/28149;(h) соединения против ожирения, такие как фенфлурамин, дексфенфлурамин, фентермин, сибутрамин, орлистат и другие агонисты адренергических рецепторов 3;(i) средства, модифицирующие пищевое поведение, такие как антагонисты нейропептидов Y (например, нейропептида Y5), такие как раскрытые в WO 97/19682, WO 97/20820, WO 97/20821, WO 97/20822 и WO 97/20823;(j) агонисты PPAR, такие как описанные в WO 97/36579, Glaxo;(k) агонисты PPAR , такие как описанные в WO 97/10813; и(l) ингибиторы повторного поглощения серотонина, такие как флуоксетин и сертралин. В одном воплощении соединение по настоящему изобретению и второе активное средство, описанное выше, используют при получении лекарственного средства для лечения или предупреждения заболевания или расстройства, опосредуемого активацией адренорецептора S3. Соединения формулы 1 а по настоящему изобретению можно получить согласно процедурам, показанным на следующих далее схемах и в примерах, с использованием соответствующих материалов, и также приведенным в качестве примеров в следующих далее конкретных примерах. Кроме того, используя процедуры, описанные в данном описании, специалист в данной области техники может легко получить другие соединения по настоящему изобретению, заявляемые в данном описании. Однако соедине-7 020135 ния, приведенные в примерах, не должны рассматриваться как образующие единственный вид, который рассматривается как изобретение. Примеры также иллюстрируют детали получения соединений по настоящему изобретению. Специалисты в данной области техники легко поймут, что для получения указанных соединений можно использовать различные изменения условий и процессов описанных далее процедур получения. Соединения по настоящему изобретению, как правило, выделяют в форме их фармацевтически приемлемых солей, таких как соли, описанные в данном описании выше. Свободные основания амины, соответствующие выделенным солям, можно получить нейтрализацией подходящим основанием, таким как водный раствор гидрокарбоната натрия, карбоната натрия, гидроксида натрия и гидроксида калия, и экстрагированием свободного амина органическим растворителем с последующим выпариванием. Свободное основание амин, выделенное таким способом, затем можно превратить в другую фармацевтически приемлемую соль растворением в органическом растворителе с последующим добавлением соответствующей кислоты и последующими выпариванием, осаждением или кристаллизацией. Все температуры приводятся в градусах по Цельсию, если не указано иное. Масс-спектры (МС) измеряют масс-спектроскопией с ионизацией электронным распылением. Различные хроматографические методы можно использовать при получении соединений. Такие методы включают, но не ограничиваются перечисленным, высокоэффективную жидкостную хроматографию (ВЭЖХ), включая ВЭЖХ с нормальной, обращенной фазой и хиральной фазой; жидкостную хроматографию среднего давления (ЖХСД), сверхкритическую жидкостную хроматографию; препаративную тонкослойную хроматографию (преп. ТСХ); флэш-хроматографию на силикагеле или на силикагеле с обращенной фазой; ионообменную хроматографию и радиальную хроматографию. Все температуры приводятся в градусах по Цельсию, если не указано иное. Выражение стандартные условия реакции пептидного сочетания означает сочетание карбоновой кислоты с амином с использованием кислотного активирующего агента, такого как EDC, DCC и ВОР, в инертном растворителе, таком как дихлорметан, в присутствии катализатора, такого как НОВТ и НОАТ. Использование защитных групп для функциональных групп амина и карбоновой кислоты для облегчения нужного взаимодействия и минимизации нежелательных взаимодействий хорошо описано в документах. Условия, требуемые для удаления защитных групп, описаны в известных руководствах, таких как GreeneT. and Wuts P.G.M., Protective Groups in Organic Synthesis, John WileySons, Inc., New York. NY, 1991. Группы MOZ и ВОС являются обычно используемыми защитными группами в органическом синтезе, и условия их удаления известны специалистам в данной области техники. Например, MOZ можно удалить каталитическим гидрированием в присутствии благородного металла или его оксида, такого как платина на активированном угле, в протонном растворителе, таком как метанол или этанол. В случаях, когда каталитическое гидрирование противопоказано из-за присутствия других потенциально реакционноспособных функциональных групп, удаления групп MOZ также можно достичь обработкой раствором трифторуксусной кислоты, хлороводородной кислоты или газообразным хлороводородом в растворителе,таком как дихлорметан, метанол или этилацетат. Удаление защитных групп ВОС осуществляют с помощью сильной кислоты, такой как трифторуксусная кислота, хлороводородная кислота, или газообразным хлороводородом в растворителе, таком как дихлорметан, метанол или этилацетат. В заявке термины, приведенные далее, имеют указанные значения, если не отмечается иное. Термин Значение Ас - Ацил (СН 3 С(О)-) Водн. - ВодныйC - Градус по Цельсию Вычисл. или выч-но - Вычислено Целит - Диатомовая земля целитHATU - Гексафторфосфат О-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония НОАс - Уксусная кислота НОАТ - 1-Гидрокси-7-азабензотриазол НОВТ - 1-Гидроксибензотриазол ВЭЖХ - Высокоэффективная жидкостная хроматография ЖХ/МС или ЖХ-Масс - Жидкостная хроматография - масс-спектр л - Литр(ы) М - Молярный(е)Me - Метил МеОН - Метанол МФ (MF) - Молекулярная формула мин - Минута(ы) мг - Миллиграмм(ы) мл - Миллилитр(ы) ммоль - Миллимоль(и)MOZ (Moz) п-Метоксибензилоксикарбонил Т.пл. - Температура плавления МС - Масс-спектр нМ - НаномолярныйSCF CO2S - Жидкий диоксид углерода в сверхкритическом состоянииTf - Трифлат или трифторметансульфонат ТФК - Трифторуксусная кислота ТГФ - Тетрагидрофуран ТСХ - Тонкослойная хроматографияTMS - Триметилсилил Приведенные ниже схемы реакций иллюстрируют способы, используемые при синтезе соединений формулы 1 а по настоящему изобретению. Все заместители имеют значения, указанные выше, если не указано иное. Синтез новых соединений формулы 1 а, которые являются предметом настоящего изобретения, можно осуществлять одним или несколькими схожими путями. По схеме I коммерчески доступный I-1 обрабатывают 1-2 М раствором винилсодержащего реактива Гриньяра или в безводном ТГФ или в эфире при температуре 0C и нагревают до комнатной температуры в течение периода времени от 1 до 4 ч. Реакцию обычно осуществляют в инертном органическом растворителе, таком как ТГФ, и в атмосфере инертного газа, такого как азот. Продукт реакции представляет собой аллиловый спирт структурной формулы I-2. Конверсии I-2 в I-3 можно достигнуть, подбирая нужный силилзащитный агент, такой как трет-бутилдиметилхлорид, и слабое органическое основание, такое как имидазол, и смешивая их при комнатной температуре от 4 до 16 ч. Окисление двойной связи посредством барботирования газа озона в течение периода времени до появления устойчивого синего цвета и последующее восстановление озонида посредством добавления избытка метилсульфида дают альдегид I4. Затем I-4 обрабатывают или R-(+)-, или S-(-)-2-метил-2-пропансульфинамидом в присутствии кислоты Льюиса, такой как сульфат меди или тетрахлорид титана, которая также действует как осушитель. Взаимодействие обычно осуществляют в инертном органическом растворителе, таком как дихлорметан, при температуре от комнатной до 40C в течение 6-36 ч и продукт реакции представляет собой сульфинамид структурной формулы I-5. Как и I-1, I-5 обрабатывают винилсодержащим реактивом Гриньяра в схожих условиях и в течение схожего времени и получают аллилсульфинамид I-6. Для того чтобы селективно удалить сульфинамид, I-6 обрабатывают безводным 4 М раствором HCl в диоксане в течение не более 15 мин. Затем реакционную смесь разбавляют толуолом, концентрируют досуха и получают I-7. Наконец, I7 превращают в I-8 обработкой бензилхлорформиатом в присутствии безводного органического основания, такого как триэтиламин или диизопропилэтиламин, в инертном органическом растворителе, таком как DCM, при 0C с нагреванием до комнатной температуры в течение периода времени от 1 до 3 ч. С другой стороны, альдегид I-4 можно получить так, как показано на схеме II. Обработка I-3 тетраоксидом осмия в присутствии N-оксида N-метилморфолина дает диол I-9. Взаимодействие обычно осуществляют в смеси воды и ацетона и смесь тщательно обрабатывают для удаления токсичного тетраоксида осмия перед концентрированием раствора. Затем остаток I-9 растворяют в смеси ацетон/вода (8:1) и обрабатывают периодатом натрия в течение периода времени от 8 до 24 ч при комнатной температуре, и получают альдегид I-4 как на схеме I. Затем его доводят до конечного нужного промежуточного соединения I-8 с использованием тех же процедур, какие описаны на схеме I. Схема II Схема III описывает синтез пирролидинового каркаса с использованием защищенного CBz аллиламида I-8, описанного на схемах I и II. Виниловое соединение I-8 можно ввести в реакцию метатезиса олефинов с промежуточным винилкетоном I-10 с использованием соответствующих катализаторов, применимых при метатезисе олефинов, известных специалистам в данной области техники. Подходящие катализаторы для получения соединения структурной формулы I-11 включают, но не ограничиваются перечисленным, катализаторы как Grabbs, так и Zhan и типа, известного как Grabbs-II и Zhan I или II. Гидрирование такого промежуточного соединения I-11 посредством обработки в атмосфере водорода с катализатором 10% палладием-на-угле в растворителе, таком как этилацетат или этанол, в течение 2-16 ч приводит к гидрированию олефина вместе с удалением защитных CBz-групп в дополнение к замыканию цикла через внутримолекулярное образование имина между свободным амином и кетоном и восстановлению имина с образованием пирролидинового цикла общей структуры I-12. В зависимости от выбора растворителя галогенозаместители арила могут или остаться, или быть удаленными в то же время, в зависимости от предпочтительного конечного промежуточного соединения. Селективную защиту пирролидина Boc можно выполнить путем добавления к I-12 одного эквивалента трет-бутилдикарбоната (Вос 2 О) в присутствии безводного органического основания, такого как триэтиламин (TEA). Взаимодействие обычно осуществляют в инертном органическом растворителе, таком как ТГФ, и в инертной атмосфере, такой как азот, и получают продукт структурной формулы I-13. В зависимости от выбора амида, сульфонамида или мочевины, I-13 можно превратить в каждое указанное соединение, используя соответствующий способ, известный специалистам в данной области техники для получения таких нужных соединений. В случае сульфонамидов I-13 можно обработать нужным сульфонилхлоридом, содержащим R6, в присутствии подходящего основания, такого как пиридин. Используемый в данном описании R6 выбирают из:(1) водорода,(2) (C1-C10)-алкила, необязательно замещенного 1-5 группами, независимо выбранными из галогена,-ORa, -CO2Ra -CONRaRb,(3) фенила, необязательно замещенного 1-3 группами, независимо выбранными из галогена, (C1C5)-алкила, необязательно замещенного 1-5 атомами галогена, и -ORa, и(4) 5- или 6-членного гетероцикла с 1-4 гетероатомами, выбранными из атомов кислорода, серы и азота, и где указанный гетероцикл необязательно орто-конденсирован с бензольным циклом и необязательно замещен 1-3 группами, независимо выбранными из галогена, (C1-C5)-алкила, необязательно замещенного 1-5 атомами галогена, и -ORa. Взаимодействие обычно осуществляют в инертном органическом растворителе, таком как ДМФА,при температуре от комнатной до 80C в течение периода 12-24 ч, и продукт реакции представляет собой сульфонамид структурной формулы I-14. В случае амидов I-13 можно обработать нужным ацетилхлори- 10020135 дом, содержащим R6, в присутствии подходящего основания, такого как TEA или DIEA. Взаимодействие обычно осуществляют в инертном органическом растворителе, таком как ДМФА, при комнатной температуре в течение периода 12-24 ч, и продукт реакции представляет собой амид структурной формулы I15. Наконец, мочевину можно получить, обрабатывая I-13 CDI или фосгеном в присутствии амина, содержащего R6, в течение периода времени от 1 до 24 ч при комнатной температуре и получить мочевину структурной формулы I-16. Удаление Boc и силилзащитной групп из I-14, I-15 и I-16 одновременно посредством обработки 6 М HCl в водном метаноле при комнатной температуре в течение периода 12-24 ч дает конечные нужные продукты - амид, сульфонамид и мочевину, содержащие R6, показанные в общих структурных формулах I-17, I-18 и I-19. Можно включить дополнительные стадии удаления защитных групп, если для облегчения протекания химических процессов необходимы защитные группы для группы R6, известные специалистам в данной области техники. Такие защитные группы могут включать тритильные группы, третбутилкарбаматные группы или другие группы, подходящие для защиты гетероциклических соединений или функциональных групп, присоединенных к группе R6, таких как аминогруппы, гидроксильные группы, карбоксильные группы, известных специалистам в данной области техники. Схема III Альтернативный путь, показанный на схеме IV, описывает синтез, который дает как цис-, так и транс-пирролидины, которые разделяют перед получением конечного соединения. Исходный 2-аминоарилпропан-1,3-диол (I-20) сначала защищают как полуацеталь с использованием ацетона в подходящем растворителе, таком как толуол, в присутствии кислоты, такой как птолуолсульфоновая кислота, и затем защищают амин обработкой трет-бутилдикарбонатом, и получают промежуточное соединение I-21. С использованием стандартных условий окисления по Сверну свободный первичный гидроксил превращают в альдегид I-22. Затем реакцией Виттига с (трифенилфосфоранилиден)ацетальдегидом альдегид удлиняют на два атома углерода, и полученную двойную связь восстанавливают гидрированием с палладием-на-угле, и получают I-23. Затем полученное промежуточное соединение подвергают второй реакции Виттига с бромидом (4-нитробензил)трифенилфосфония, и получают промежуточное соединение I-24, что создает возможность циклизации соединения через присоединение по Михаэлю по двойной связи после удаления защитной группы. Защита аминогруппы третбутилдикарбонатом содействует очистке и отделению субстрата для получения как цис-, так и трансизомеров I-25 а и I-25b соответственно. Гидрирование нитрогруппы до свободного амина дает нужные промежуточные соединения I-26 а и I-26b. Промежуточные соединения I-26 а и I-26b можно использовать для стандартных амидных сочетаний с использованием EDC, однако может потребоваться селективная защита гидроксильной группы перед обработкой или ацил- или сульфонилхлоридами или фосгеном, когда идет превращение в мочевину. Схема V отражает в общих чертах синтез пирролидинового каркаса, который связывает пути на схемах I, III и IV, и дает пирролидиновый каркас с диастереоселективностью для пирролидина цис 2S,5R. Реакцию Виттига используют для превращения альдегида I-22 со схемы IV в виниловый аналог I-27 через обработку бромидом метилтрифенилфосфония. После манипуляции с защитной группой, видной на промежуточном соединении I-28, схема становится конвергентной схеме III через промежуточное соединение I-8. Затем с использованием процедур, схожих с процедурами, описанными на схеме III, можно получить промежуточное соединение I-11. Оптимизация гидрирования за счет введения хлороводородной кислоты и показателя разбавления до концентрации 0,15-0,30 М дает преимущественно пирролидиновый каркас I-13 а цис 2S,5R. Схема V Схема VI отражает в общих чертах способ синтеза промежуточного ацетилена методами химии альдолей для установления хиральности как гидроксильной группы, так и левой части пирролидина. Из указанной схемы следует, что такой промежуточный ацетилен можно использовать для синтеза как цис-,так и транс-пирролидинов. Коммерчески доступный I-29 сначала обрабатывают триметилацетилхлоридом в присутствии слабого органического основания, такого как триэтиламин, при -25C в течение 2 ч. Последовательное добавление к смеси безводного хлорида лития и/или (S)-(-)-4-бензил-, или (S)-(-)-4-фенил-2 оксазолидинона и последующее постепенное нагревание до комнатной температуры в течение периода времени от 12 до 24 ч дают имид I-30. Взаимодействие обычно осуществляют в инертном органическом растворителе, таком как ТГФ, в инертной атмосфере, такой как азот. Спирт I-32 получают согласно процедурам, описанным в литературе (см. Evans et al., J. Am. Chem. Soc, 2002, 124, 392-394). Например, обработка I-30 безводным хлоридом магния, триэтиламином и соответствующим альдегидом I-31, таким как 3-хлорбензальдегид или бензальдегид, и хлортриметилсиланом при комнатной температуре в течение периода 72 ч дает триметилсилиловый эфир альдольного продукта I-32. Взаимодействие обычно осуществляют в инертном органическом растворителе, таком как этилацетат, в инертной атмосфере, такой как азот. Обработка промежуточного соединения триметилсилилового эфира смесью трифторуксусной кислоты и метанола дает нужный спирт I-32. Гидролиз имида I-32 достигается обработкой пероксидом лития при 0C в течение 15-18 ч. Затем надкислоту восстанавливают водным раствором сульфита натрия, и получают карбоновую кислоту I-33. Взаимодействие обычно осу- 12020135 ществляют в смеси инертного органического растворителя, такого как ТГФ, и воды в инертной атмосфере, такой как азот. Конверсии I-33 в I-34 можно достигнуть выбором нужного силилсодержащего защитного агента, такого как трет-бутилдиметилсилилтрифторметансульфонат, и его реакцией в присутствии слабого органического основания, такого как DBU, при 0C в течение периода от 12 до 16 ч. Затем I-34 можно обработать дифенилфосфорилазидом в присутствии слабого органического основания, такого как триэтиламин, в течение 6 ч при комнатной температуре. Добавление соответствующего спирта, такого как 4-метоксибензиловый спирт, с нагреванием при 100C в течение периода от 12 до 16 ч дает соответствующий карбамат I-35. Взаимодействие обычно осуществляют в инертном органическом растворителе,подобном толуолу, в инертной атмосфере, такой как азот. Материал образует основу, на которой можно синтезировать пирролидиновый каркас. Схема VI Схема VII отражает в общих чертах использование I-35 для конверсии пирролидинового каркаса. Пирролидин получают замыканием цикла через внутримолекулярное восстановительное аминирование с образованием как цис-, так и транс-пирролидина. Разделение цис- и транс-пирролидина с последующим восстановлением нитрогруппы до амина дает конечный нужный пирролинанилин, используемый для синтеза аналогов. Алкин I-35 можно ввести во взаимодействие в реакции типа перекрестного сочетания по Сонагашира с соответствующим коммерчески доступным арилгалогенидом I-36 с образованием I-37 с использованием соответствующих условий реакции, известных специалистам в данной области техники. Условия реакции могут включать использование катализаторов,таких как тетракис(трифенилфосфин)палладий(0), с иодидом меди(I) или без него в присутствии органического основания, такого как триэтиламин, или ацетат палладия(II), с органическим основанием, таким как ацетат тетрабутиламмония, в органическом растворителе, таком как ацетонитрил или ДМФА, в инертной атмосфере, такой как азот. Кетон I-38 можно получить взаимодействием алкина I-37 с пирролидином при температуре 80C в растворителе, таком как ДМФА, в течение периода от 3 до 6 ч. Последующая обработка 10% водным раствором уксусной кислоты в течение периода 15-60 мин при комнатной температуре дает кетон I-38. Защитную группу в карбамате I-38 можно удалить с использованием соответствующих условий реакции, известных специалистам в данной области техники, и получить соответствующий амин,который затем подвергают внутримолекулярному замыканию цикла с кетоном, и получить имин I-39. Условия реакции могут включать трифторуксусную кислоту в органическом растворителе, таком как дихлорметан, и хлороводородную кислоту в органическом растворителе, таком как эфир. Восстановления имина I-39 можно достигнуть обработкой цианоборгидридом натрия в органическом растворителе,таком как метанол, при температуре 0C в инертной атмосфере, такой как азот, в течение периода 18-24 ч. Это дает промежуточные соединения цис-пирролидин (I-40 а) и транс-пирролидин (I-40b), которые можно разделить хроматографией на силикагеле. I-40 а является основным диастереомером, образовавшимся в реакции, и он является первым диастереомером, который элюируется из колонки. Защиты атома азота пирролидина I-40 а или I-40b группой Boc достигают обработкой трет-бутилдикарбонатом в присутствии слабого органического основания, такого как триэтиламин. Взаимодействие обычно осуществляют в органическом растворителе, таком как дихлорметан, в инертной атмосфере, такой как азот, и получают продукт реакции структурной формулы I-41 а или I-41b. Гидрирование промежуточного соединения I-41 а или I-41b путем обработки с 10% палладием-на-угле в присутствии хлороводорода при давлении водорода от 103,4 до 344,7 кПа (15-50 ф/д 2) в растворителе, таком как этилацетат или этанол, в течение периода времени 8-12 ч дает I-42 а или I-42b. В зависимости от выбора условий галогенозаместители X в это время могут или оставаться, или удаляться, в зависимости от предпочтительного конечного промежуточного соединения. В некоторых случаях порядок осуществления реакционных схем может изменяться для облегчения взаимодействия или для того, чтобы избежать нежелательных продуктов реакции. Приведенные далее примеры даются для того, чтобы изобретение можно было понять более полно. Приведенные примеры являются только пояснительными и не должны рассматриваться как ограничивающие изобретение каким-либо образом. Промежуточное соединение 1. Бензил[3-(2-оксобут-3-ен-1-ил)фенил]карбамат (i-1)DIEA (28,5 мл, 155 ммоль), и полученный раствор охлаждают до 0C и помещают в атмосферу азота. Затем к такому охлажденному раствору добавляют бензилхлорформиат (21,1 мл, 148 ммоль) и полученную смесь перемешивают в течение ночи, позволяя нагреваться до комнатной температуры. Реакционную смесь промывают 1 М HCl, водой и затем солевым раствором. Органический слой сушат над сульфатом натрия, фильтруют и концентрируют в вакууме. В дополнительной очистке нет необходимости, и материал (44 г, 99%) используют как он есть на следующей стадии реакции. ЖХ-МС: m/z (ES) 314 К раствору 44,0 г (140 ммоль) этил(3-[бензилокси)карбонил]аминофенил)ацетата (со стадии А) в ТГФ, этаноле и воде (1:1:1, 1500 мл) добавляют твердый LiOH (16,8 г, 700 ммоль) и полученный раствор греют при 60C на масляной бане в течение 3 ч. Смесь охлаждают до комнатной температуры в течение ночи и затем постепенно добавляют 40 мл концентрированной HCl, поддерживая температуру ниже 25C, до тех пор, пока рН раствора не станет примерно 2-3. Экстрагируют смесь этилацетатом (3750 мл), затем экстракты объединяют, и органическую фазу промывают водой и затем солевым раствором. Органическую фазу сушат над сульфатом натрия, фильтруют и концентрируют в вакууме. Указанное в заголовке соединение (24,7 г, 87%) используют на следующей стадии реакции без дополнительной очистки. ЖХ-МС: m/z (ES) 28 6 (МН)+, 308 (MNa)+. К суспензии 24,7 г (87 ммоль) (3-[бензилокси)карбонил]аминофенил)уксусной кислоты (со стадии В) в 200 мл дихлорметана добавляют триэтиламин (30,2 мл, 173 ммоль), что приводит к некоторому выделению тепла (+5C), и суспензия становится раствором. После охлаждения в течение 10 мин к раствору добавляют HOBt (13,2 г, 87 ммоль) и гидрохлорид N,О-диметилгидроксиламина (8,5 г, 87 ммоль) и затем EDC (16,6 г, 87 ммоль) и полученную смесь перемешивают при комнатной температуре в течение ночи в атмосфере азота. Раствор переносят в делительную воронку и промывают 1 М HCl, что вызывает образование эмульсии. Добавляют метанол для разрушения эмульсии и водную фазу отделяют. Органическую фазу сушат над сульфатом натрия, фильтруют и концентрируют в вакууме. Перекристаллизация остатка из 1000 мл 70% гексана в этилацетате (нагревание до температуры образования флегмы и последующее охлаждение до комнатной температуры в течение ночи) дает указанное в заголовке соединение(21 г, 74%) в виде белого твердого вещества. ЖХ-МС: m/z (ES) 329 (МН)+. Стадия D. Бензил[3-(2-оксобут-3-ен-1-ил)фенил]карбамат (i-1) К охлажденному до 0C на бане со смесью воды и льда раствору 15 г (45,7 ммоль) бензил(3-2[метокси(метил)амино]-2-оксоэтилфенил)карбамата (со стадии С) в 1000 мл безводного ТГФ в атмосфере азота добавляют по каплям через канюлю 1,0 М раствор винилмагнийбромида (100 мл в ТГФ, 100 ммоль) и полученный раствор перемешивают в течение 1 ч при 0C. Реакцию гасят постепенным добавлением 500 мл 1 М HCl, поддерживая температуру ниже 5C, и смесь перемешивают в течение 30 мин. Затем смесь экстрагируют этилацетатом, и объединенные органические слои промывают водой, а затем солевым раствором. Затем органические слои сушат над сульфатом натрия, фильтруют и концентрируют в вакууме. Остаток очищают флэш-хроматографией на Biotage 75M с элюированием смесью 30% этилацетата в гексане, и получают указанное в заголовке соединение (11 г, 78%) в виде светло-желтого твердого вещества. ЖХ-МС: m/z (ES) 310 (МН)+, 332 (MNa)+; 1 К охлажденному раствору 3-хлорбензальдегида (22,5 г, 160 моль) в 100 мл безводного ТГФ в инертной атмосфере азота постепенно через шприц добавляют 1,6 М раствор винилмагнийхлорида в ТГФ (100 мл, 160 ммоль), и раствор перемешивают в течение трех часов, оставляя нагреваться до комнатной температуры. Реакцию гасят насыщенным раствором хлорида аммония и органический слой отделяют, смесь экстрагируют этилацетатом (2200 мл) и органические слои объединяют, сушат над сульфатом магния, фильтруют и концентрируют в вакууме. Очистка ЖХСД Horizon с колонкой с силикагелем 40 М+ с использованием элюирования с градиентом смесями 0-40% этилацетата в гексане дает указанное в заголовке соединение (22,4 г, 44%). m/z (ES) 168, 170 (м, М+2)+, 190, 192 (MNa, MNa+2)+. 1 Н ЯМР (500 МГц, CDCl3) : 7,38 (с, 1 Н), 7,35-7,22 (м, 3H), 5,90 (ддд, J=7,3, 10,0, 17,4 Гц, 1 Н), 5,38 К раствору 22,4 г (133 ммоль) 1-(3-хлорфенил)проп-2-ен-1-ола (со стадии А) в 90 мл безводного ДМФА добавляют трет-бутилдиметилсилилхлорид (20,0 г, 133 ммоль) и имидазол (18,1 г, 266 ммоль) и полученный раствор перемешивают в течение ночи в атмосфере азота при комнатной температуре. Смесь промывают водой и экстрагируют этилацетатом. Органический слой отделяют, сушат над сульфатом магния, фильтруют и концентрируют в вакууме. Остаток очищают колоночной флэшхроматографией на силикагеле с элюированием с градиентом смесями 0-15% этилацетата в гексане и(со стадии В) в дихлорметане, охлажденному до -78C на бане сухой лед/ацетон, барботируют газ озон до тех пор, пока раствор не будет сохранять светло-синий цвет. Затем через раствор барботируют газ азот до тех пор, пока он не станет вновь прозрачным. К раствору добавляют метилсульфид, и полученную смесь перемешивают в течение ночи при комнатной температуре. Материал концентрируют в вакууме, остаток очищают ЖХСД Horizon на колонке с силикагелем 40 М+ с элюированием с градиентом смесями 0-50% этилацетата в гексане, и получают продукт реакции (3,57 г, 89%). Стадия D. N-[(1E)-2-[трет-Бутил(диметил)силил]окси-2-(3-хлорфенил)этилиден]-2-метилпропан 2-сульфинамид К раствору 3,0 г (10,6 ммоль) [трет-бутил(диметил)силил]окси(3-хлорфенил)ацетальдегида (со стадии С) и 1,3 г (10,6 ммоль) (R или S)-2-метил-2-пропансульфинамида в 50 мл безводного дихлорметана добавляют сульфат меди(II) (3,4 г, 21,2 ммоль) и полученную смесь перемешивают при комнатной температуре в атмосфере азота в течение 16 ч. Реакционную смесь промывают водой и экстрагируют дихлорметаном. Органический слой сушат над сульфатом магния, фильтруют и концентрируют в вакууме. Остаток очищают ЖХСД Horizon на колонке с силикагелем 40 М+ с элюированием элюирующей системой с градиентом смесями 0-25% этилацетата в гексане и получают указанное в заголовке соединениеN-[(1E)-2-[трет-бутил(диметил)силил]окси-2-(3 хлорфенил)этилиден]-2-метилпропан-2-сульфинамида (со стадии D) в 20 мл безводного ТГФ, охлажденному до 0C, в атмосфере азота добавляют через шприц 1,6 М раствор винилмагнийхлорида в ТГФ (3,90 мл, 6,2 ммоль) и полученную смесь перемешивают в течение 1 ч. Смесь нагревают до комнатной температуры и перемешивают еще в течение часа. Реакцию гасят насыщенным раствором хлорида аммония и смесь экстрагируют этилацетатом. Объединенные органические слои сушат над сульфатом магния,фильтруют и концентрируют в вакууме. Остаток очищают ЖХСД Horizon на колонке с силикагелем 40 М+ с элюированием элюирующей системой с градиентом смесями 0-35% этилацетата в гексане и получают все четыре диастереомера в виде отдельных изомеров. По данным ЯМР четыре полученных продукта являются диастереомерами друг друга. Изомеры метят по мере их элюирования из колонки на силикагеле. Изомер, который элюирован первым, называют изомером 1, затем следуют изомеры 2, 3 и последний называют изомером 4.N-1-трет-бутил(диметил)силил]окси(3 хлорфенил)метил]проп-2-ен-1-ил-2-метилпропан-2-сульфинамида (со стадии Е) добавляют 5 мл безводного 4 М раствора HCl в диоксане, и раствор перемешивают в течение 15 мин при комнатной температуре. Реакционную смесь концентрируют досуха, и добавляют толуол (25 мл) для азеотропного удаления избытка HCl. Затем остаток растворяют в безводном дихлорметане, создают инертную атмосферу азота,раствор охлаждают до 0C на бане со смесью воды со льдом, и затем постепенно через шприц добавляют бензилхлорформиат (0,32 мл, 2,22 ммоль) и затем диизопропилэтиламин (1,19 мл, 6,66 ммоль) и полученный раствор перемешивают в течение 2 ч при 0C. Раствор концентрируют в вакууме досуха, остаток очищают на препаративных пластинках (4 1000 мкм) с элюированием смесью 20% этилацетата в гексане и получают указанное в заголовке соединение (703 мг, 71%). m/z (ES) 446, 448 (м, М+2)+, 468, 470(д, J=4,2 Гц, 1 Н), 3,88-3,82 (м, 1 Н), 3,33 (д, J=9,4 Гц, 1 Н), 1,19 (с, 9 Н), 0,94 (с, 9 Н), 0,09 (с, 3H), -0,14 (с,3H). Промежуточные соединения различной стереохимии, родственные описанным выше, можно получить из соответствующих исходных веществ с использованием процедуры, описанной выше. 1 К раствору бензил[3-(2-оксобут-3-ен-1-ил)фенил]карбамата (i-1) (820 мг, 2,80 ммоль) и 1R)-1-[R)[трет-бутил(диметил)силил]окси)(3-хлорфенил)метил]проп-2-ен-1-илкарбамата (i-2) (500 мг, 1,12 ммоль) в 7 мл безводного дихлорметана добавляют катализатор Zhan I (740 мг, 1,12 ммоль) и полученный зеленый раствор греют при 40C в течение ночи в атмосфере азота. Реакционную смесь концентрируют досуха, остаток очищают на препаративных пластинках (41000 мкм) с элюированием смесью 40% этилацетата в гексане, и получают указанное в заголовке соединение (348 мг, 50%). m/z (ES) 713, 715 (М,М+2)+, 735, 737 (MNa, MNa+2)+. Стадия В. 4-5R)-5-[(R)-[трет-Бутил(диметил)силил]окси(фенил)метил]пирролидин-2 илметил)анилин К раствору 328 мг (0,46 ммоль) бензил 4-[(3 Е,5R,6R)-5-[(бензилокси)карбонил]амино-6-[третбутил(диметил)силил]окси-6-(3-хлорфенил)-2-оксогекс-3-ен-1-ил]фенилкарбамата (со стадии А) в 25 мл этанола добавляют 10% палладий-на-угле, и суспензию помещают в атмосферу водорода при подаче водорода из баллона. Реакционную смесь перемешивают в атмосфере водорода в течение 1 ч при комнатной температуре. ТСХ показывает, что реакция прошла полностью. Катализатор отфильтровывают с использованием шприца с фильтром из ПТФЭ, 0,45 мкм, Gilmen и промывают этанолом (45 мл). Фильтрат концентрируют в вакууме досуха, остаток очищают на препаративной пластинке (31000 мкм) с элюированием смесью 5% метанола в дихлорметане, и получают указанное в заголовке соединение (121 мг, 66%). m/z (ES) 397 (МН)+. Стадия С. трет-Бутил-(5R)-2-(4-аминобензил)-5-[(R)-[трет-бутил(диметил)силил]окси(фенил)метил]пирролидин-1-карбоксилат (i-3) К раствору 121 мг(0,315 ммоль) 4-5R)-5-[(R)-[трет-бутил(диметил)силил]окси(фенил)метил]пирролидин-2-илметил)анилина (со стадии В) в 5 мл безводного ТГФ добавляют трет-бутилкарбонат (69 мг, 0,315 ммоль), а затем TEA (44 мкл, 0,315 ммоль) и полученный раствор перемешивают при комнатной температуре в атмосфере азота в течение ночи. Реакционную смесь наносят непосредственно на препаративную пластинку (1500 мкм) и элюируют смесью 30% этилацетата в гексане, и получают указанное в заголовке соединение (100 мг, 64%). m/z (ES) 497 (МН)+, 397 (М-Вос)+. 1 Н ЯМР (500 МГц, CDCl3) : 7,40-7,30 (м, 5 Н), 6,75-6,68 (м, 2 Н), 6,56-6,51 (м, 2 Н), 5,52-5,48 (м, 1 Н),5,32-5,28 (м, 1 Н), 4,16-4,06 (м, 2 Н), 3,88-3,82 (м, 1 Н), 3,76-3,70 (м, 1 Н), 3,55-3,48 (м, 2 Н), 2,74 (ушир.д,J=11,8 Гц, 1 Н), 2,44 (ушир.д, J=11,8 Гц, 1 Н), 2,05-1,94 (м, 1 Н), 1,90-1,82 (м, 1 Н), 1,60 (с, 9 Н), 1,50-1,42 (м,1 Н), 1,32-1,22 (м, 2 Н), 1,10-1,02 (м, 1 Н), 0,95 (с, 9 Н), 0,08 (с, 3H), -0,15 (с, 3H). Разделение промежуточных соединений 4 а и 4b. трет-Бутил-(2S,5R)-2-(4-аминобензил)-5-[(R)-[трет-бутил(диметил)силил]окси(фенил)метил]пирролидин-1-карбоксилат (i-4a) трет-Бутил-(2R,5R)-2-(4-аминобензил)-5-[(R)-[трет-бутил(диметил)силил]окси(фенил)метил]пирролидин-1-карбоксилат (i-4b)(трет-бутил-(5R)-2-(4-аминобензил)-5-[(R)-[третбутил(диметил)силил]окси(фенил)метил]пирролидин-1-карбоксилат (смесь цис и транс, 4:1) растворяют в метаноле и очищают методом SFC (сверхкритическая), Berger Multigram, с использованием элюента смеси 30% метанола:60% диоксида углерода, и два стереоизомера разделяют. Первый изомер, элюируемый из колонки, называют минорным изомером 1 и второй изомер называют основным изомером 2.(ушир.д, J=13,8 Гц, 3H), 0,09 (с, 3H). Элюируется 7,78 мин на SFC, изомер 1. Синтез промежуточного соединения 4 а и промежуточного соединения 4b. трет-Бутил-(2S,5R)-2-(4-аминобензил)-5-[(R)-[трет-бутил(диметил)силил]окси(фенил)метил]пирролидин-1-карбоксилат (i-4a); трет-бутил-(2R,5R)-2-(4-аминобензил)-5-[(R)-[трет-бутил(диметил)силил]окси(фенил)метил]пир- 18020135 К раствору 69,0 г (615 ммоль) 5-гексиновой кислоты и 214 мл (1540 ммоль) триэтиламина в 1,0 л безводного тетрагидрофурана при -25C в атмосфере азота в течение 20 мин добавляют 83,0 мл (677 ммоль) триметилацетилхлорида. После добавления образуется белый осадок и полученную суспензию перемешивают в течение 2 ч. Затем последовательно добавляют 28,7 г (677 ммоль) безводного хлорида лития и 100,0 г (615,0 ммоль) (4S)-4-фенил-1,3-оксазолидин-2-она и смесь постепенно нагревают до температуры окружающей среды в течение 12 ч. Все летучие вещества удаляют в вакууме, и остаток разбавляют водой (1 л) и экстрагируют этилацетатом (3 300 мл). Объединенные органические слои промывают солевым раствором (250 мл), сушат над сульфатом магния, фильтруют и концентрируют в вакууме. Сырой остаток очищают хроматографией на силикагеле с элюированием с градиентом смесями 5-50% этилацетата в гексане, и получают указанное в заголовке соединение в виде бесцветного твердого вещества (135 г, 85,4%). 1 Н ЯМР (500 МГц, CDCl3) :7,40-7,37 (м, 2 Н), 7,36-7,32 (м, 1 Н), 7,31-7,28 (м, 2 Н),5,42 (дд, J=8,9, 3,7 Гц, 1 Н), 4,69 (т, J=8,9 Гц, 1 Н), 4,28 (дд, J=9,2, 3,7 Гц, 1 Н), 3,13-3,02 (м, 2 Н), 2,24-2,21 К раствору 56,8 г (221 ммоль) (4S)-3-гекс-5-иноил-4-фенил-1,3-оксазолидин-2-она с описанной выше стадии А в 265 мл безводного этилацетата при перемешивании при температуре окружающей среды в атмосфере азота добавляют 6,31 г (66,2 ммоль) безводного хлорида магния, 61,5 мл (442 ммоль) триэтиламина, 26,9 мл (265 ммоль) бензальдегида и 42,3 мл (331 ммоль) хлортриметилсилана и полученную смесь перемешивают в течение 72 ч. Неоднородную реакционную смесь фильтруют через 300-мл слой силикагеля с элюированием еще 1 л этилацетата. Фильтрат упаривают в вакууме досуха и остаток суспендируют в 265 мл метанола и 10 мл трифторуксусной кислоты. Полученную смесь перемешивают при температуре окружающей среды в атмосфере азота в течение 5 ч, на протяжении которых реакционная смесь становится однородной. Затем все летучие вещества удаляют в вакууме, остаток очищают хроматографией на силикагеле с элюированием с градиентом смесями 5-15% этилацетата в гексане, и получают указанное в заголовке соединение в виде белого твердого вещества (65,0 г, 81,2%). 1 Н ЯМР (500 МГц, CDCl3) :7,30-7,28 (м, 8 Н), 7,09-7,07 (м, 2 Н), 5,42 (дд, J=8,7, 3,7 Гц, 1 Н), 4,764,72 (м, 1 Н), 4,72-4,67 (м, 1 Н), 4,65 (т, J=8,7 Гц, 1 Н), 4,18 (дд, J=8,7, 3,7 Гц, 1 Н), 3,05 (д, J=7,8 Гц, 1 Н),2,24 (тд, J=7,1, 2,5 Гц, 2 Н), 2,00-1,93 (м, 2 Н), 1,67-1,61 (м, 1 Н). ЖХ-МС: m/z (ES) 346,1 (МН-Н 2 О)+, 386,0 К раствору 65,0 г (179 ммоль) (4S)-3-(2R)-2-[(S)-гидрокси(фенил)метил]гекс-5-иноил-4-фенил 1,3-оксазолидин-2-она с описанной выше стадии В в 1050 мл смеси безводного тетрагидрофурана с водой, 20 : 1, при перемешивании при 0C в атмосфере азота добавляют 77,0 мл (894 ммоль) 35% водного раствора пероксида водорода со скоростью, достаточно медленной для того, чтобы поддерживать внутреннюю температуру ниже 3C. Затем добавляют 395 мл (395 ммоль) 1,0 М водного раствора гидроксида лития со скоростью, достаточно медленной для того, чтобы поддерживать внутреннюю температуру реакционной смеси ниже 5C, и полученную смесь перемешивают в течение 3 ч при 0C. Реакцию гасят 755 мл (984 ммоль) 1,3 М водного раствора сульфита натрия со скоростью, достаточно медленной для того, чтобы поддерживать внутреннюю температуру смеси ниже 5C. Все летучие вещества удаляют в вакууме и оставшуюся водную фазу экстрагируют этилацетатом (3200 мл). Затем водную фазу охлаждают до 0C и подкисляют 6 М соляной кислотой до тех пор, пока не достигнут рН 3. Затем водную фазу экстрагируют этилацетатом (3300 мл) и объединенные органические слои промывают солевым раствором (100 мл), сушат над сульфатом магния, фильтруют и упаривают в вакууме. Остаток очищают хроматографией на силикагеле с элюированием с градиентом смесями 5-10% этилацетата и 3% уксусной кислоты в гексане, и получают указанное в заголовке соединение в виде бесцветной смолы (32,0 г, 82,0%). 1 Н ЯМР (500 МГц, CDCl3):7,39-7,28 (м, 5 Н), 4,85 (д, J=8,2, 1 Н), 3,03-2,97 (м, 1 Н), 2,29-2,15 (м,2 Н), 1,97 (т, J=2,5 Гц, 1 Н), 1,93-1,82 (м, 1 Н), 1,62-1,55 (м, 1 Н). ЖХ-МС: m/z (ES) 201,0 (МН-Н 2 О)+. Стадия D. (2R)-2-[(S)-[трет-Бутил(диметил)силил]окси(фенил)метил]гекс-5-иновая кислота К раствору 32,0 г (147 ммоль) (2R)-2-[(S)-гидрокси(фенил)метил]гекс-5-иновой кислоты с описанной выше стадии С в 500 мл безводного ацетонитрила при перемешивании при температуре окружающей среды в атмосфере азота добавляют 77,0 мл (513 ммоль) 1,8-диазабицикло[5.4.0]ундец-7-ена и затем 66,3 г (440 ммоль) трет-бутилдиметилсилилхлорида в три приема в течение 10 мин. Реакционную смесь перемешивают в течение 4 ч и затем упаривают в вакууме для удаления всех летучих веществ. Остаток разбавляют 300 мл дихлорметана и 100 мл воды. К смеси добавляют 1,0 М водного раствора соляную кислоту до тех пор, пока не достигнут рН 3. Фазы разделяют, и водную фазу экстрагируют дихлорметаном (2100 мл). Объединенные органические слои промывают водой (50 мл) и солевым раствором (50 мл) и затем сушат над сульфатом магния. После фильтрации и упаривания в вакууме остаток растворяют в 350 мл метанола и добавляют 350 мл (280 ммоль) 0,8 М водного раствора карбоната калия. Полученную смесь перемешивают в течение 1,5 ч и затем упаривают в вакууме для удаления всех летучих веществ. Остаток разбавляют 300 мл дихлорметана и водную фазу подкисляют 5,0 М водного раствора соляной кислотой до тех пор, пока не достигнут рН 3. Фазы разделяют и водную фазу экстрагируют дихлорметаном (2100 мл). Объединенные органические слои промывают водой (50 мл) и солевым раствором (50 мл), затем сушат над сульфатом магния, фильтруют и упаривают в вакууме. Остаток очищают хроматографией на силикагеле с элюированием с градиентом смесями 3-15% этилацетата в гексане и получают указанное в заголовке соединение в виде бесцветного твердого вещества (42,3 г, 86,6%). 1 К раствору 40,0 г (120 ммоль) (2R)-2-[(S)-[трет-бутил(диметил)силил]окси(фенил)метил]гекс-5 иновой кислоты с описанной выше стадии D и 33,5 мл (241 ммоль) триэтиламина в 400 мл безводного толуола при температуре окружающей среды в атмосфере азота добавляют 37,5 мл (132 ммоль) дифенилфосфорилазида. Смесь перемешивают в течение 5 ч и затем добавляют 37,5 мл (301 ммоль) 4 метоксибензилового спирта. Полученную смесь греют при 105C в течение 16 ч, охлаждают до температуры окружающей среды и затем разбавляют 250 мл насыщенного водного раствора бикарбоната натрия. Фазы разделяют, и водную фазу экстрагируют этилацетатом (2150 мл). Объединенные органические слои промывают водой (100 мл) и солевым раствором (100 мл), затем сушат над сульфатом магния,фильтруют и упаривают в вакууме. Сырой остаток очищают хроматографией на силикагеле с элюированием с градиентом смесями 3-10% этилацетата в гексане, и получают указанное в заголовке соединение в виде бесцветного масла (50,9 г, 90,5%). 1 К раствору ацетилена (со стадии Е, 40 г, 80 ммоль) и 4-иоднитробензола (21,8 г, 88 ммоль) в безводном ДМФА (500 мл) добавляют триэтиламин (111 мл, 797 ммоль). Добавляют Pd(dppf)Cl2 (1,95 г, 2,39 ммоль) и иодид меди(I) (910 мг, 4,78 ммоль) и смесь обезгаживают азотом (барботирование 15 мин), и полученный раствор перемешивают при комнатной температуре в течение 5 ч. Смесь выливают в воду(1200 мл) и экстрагируют EtOAc (3300 мл). Затем объединенные органические слои промывают водой(2500 мл) и насыщенным раствором NaCl (200 мл), сушат над сульфатом магния, фильтруют и упаривают в вакууме. Остаток очищают ЖХСД (Horizon Biotage 2 Flash 65i) с элюированием с градиентом смесями 0-30% этилацетата в гексане, и получают 41 г (84%) темно-красного масла. 1 Н ЯМР (500 МГц, CDCl3) : 8,11-8,04 (м, 2 Н), 7,94-8,01 (м, 1 Н), 7,38-7,21 (м, 8 Н), 6,87 (д, J=8,4 Гц,2 Н), 4,98 (с, 2 Н), 4,77-4,59 (м, 2 Н), 4,00-3,95 (м, 3H), 3,81 (с, 3H), 2,56 (т, J=7,1 Гц, Н = 2 Н), 2,00-1,95 (м,1 Н), 1,66-1,61 (м, 1 Н), 0,93 (с, 9 Н), 0,10 (с, 3H), -0,10 (с, 3H). ЖХ-МС: m/z (ES) 589,3 (МН)+, 611,2 (MNa)+. Стадия G. 4-Метоксибензил-[(1R)-1-[(R)-[трет-бутил(диметил)силил]окси(фенил)метил]-5-(4 нитрофенил)-4-оксопентил]карбамат К раствору нитрофенилацетилена (со стадии F, 41 г, 65,5 ммоль) в ДМФА (40 мл) добавляют пирролидин (14 мл, 196,5 ммоль) и полученную смесь греют при 80C в течение 3 ч. Смесь охлаждают до комнатной температуры, добавляют 10% раствор уксусной кислоты в воде (110 мл) и полученный раствор перемешивают при комнатной температуре еще в течение 3 ч. Смесь выливают в воду (300 мл) и экстрагируют EtOAc (3250 мл); объединенные EtOAc слои промывают водой (2250 мл) и насыщенным раствором NaCl (100 мл), сушат над MgSO4, фильтруют и упаривают. Остаток очищают Horizon Flash 75 с элюированием с градиентом с повышением от 100% гексана до смеси 50% EtOAc в гексане и получают 34 г (81%) темно-оранжевого масла. 1 Н ЯМР (500 МГц, CDCl3): 8,17-8,14 (м, 2 Н), 7,32-7,23 (м, 9 Н), 6,87 (д, J=8,4 Гц, 2 Н), 4,96 (д, J=12,2 Гц, 1 Н), 4,90 (д, J=12,1 Гц, 1 Н), 4,72 (д, J=3 Гц, 1 Н), 4,16-4,13 (м, 1 Н), 3,81 (с, 3H), 3,71-3,77 (м, 2 Н), 2,652,52 (м, 2 Н), 1,97-1,92 (м, 1 Н), 1,72-1,60 (м, 1 Н), 0,93 (с, 9 Н), 0,05 (с, 3H), -0,13 (с, 3H). Стадия Н. К раствору кетоамина с защитной группой MOZ (со стадии G, 34 г, 56 ммоль) в DCM (350 мл) добавляют ТФК (256 мл) и полученную смесь перемешивают при комнатной температуре в течение 1,5 ч. Раствор упаривают в вакууме, и остаток обрабатывают DCM и насыщенным раствором NaHCO3. Органический слой сушат над MgSO4, фильтруют и упаривают. Остаток растворяют в МеОН (750 мл) и охлаждают до 0C на бане со смесью воды со льдом. Затем добавляют цианоборгидрид натрия (21,2 г, 337 ммоль), и полученную смесь перемешивают в течение ночи, позволяя нагреваться до комнатной температуры. Реакцию гасят, добавляя воду, и органические летучие вещества удаляют в вакууме. Затем водный слой экстрагируют EtOAc ( 2), и объединенные EtOAc слои промывают насыщенным растворомNaCl, сушат над MgSO4, фильтруют и упаривают в вакууме. Остаток очищают колоночной хроматографией на силикагеле (элюент: градиент с повышением от 100% гексана до смеси 35% EtOAc в гексане), и получают 16,4 г(2R,5S)-2-[(R)-[трет-бутил(диметил)силил]окси(фенил)метил]-5-(4-нитробензил)пирролидина (12 г, 42,5 ммоль) в безводном ТГФ добавляют Вос-ангидрид (9,3 г, 42,5 ммоль), а затем TEA (17,76 мл, 127,4 ммоль) и полученный раствор перемешивают при комнатной температуре в атмосфере азота в течение 2 ч. Смесь промывают водой (100 мл) и экстрагируют этилацетатом (2200 мл). Органическую фазу сушат над сульфатом натрия, фильтруют и концентрируют в вакууме. Остаток очищают ЖХСД Horizon Biotage (колонка с силикагелем 65i) с элюированием с градиентом смесями 2075% этилацетата в гексане, и получают нужный продукт. ЖХ-МС: m/z (ES) 527,3 (МН)+, 549,2 (MNa)+. Стадия J. трет-Бутил-(2R,5R)-2-[(R)-[трет-бутил(диметил)силил]окси(фенил)метил]-5-(4-нитробензил)пирролидин-1-карбоксилат Указанное в заголовке соединение получают таким же способом, как на стадии I, но заменяя цисизомер пирролидина на транс-изомер (2R,5R)-2-[(R)-[трет-бутил (диметил)силил]окси(фенил)метил]-5(4-нитробензил)пирролидина. ЖХ-МС: m/z (ES) 527,3 (МН)+, 549,2 (MNa)+. Стадия K. трет-Бутил-(2S,5R)-2-(4-аминобензил)-5-[(R)-[трет-бутил(диметил)силил]окси(фенил)метил]пирролидин-1-карбоксилат (i-4a) В 500-мл качалочную колбу Парра загружают 10% Pd/C (4,75 г) и к нему добавляют 100 мл метанола, чтобы он закрыл катализатор. Затем к суспензии добавляют раствор промежуточного нитросоединения со стадии I (8,5 г, 18,5 ммоль) в метаноле (80 мл) и затем 15,4 мл 1,0 М раствора хлороводорода в метаноле. В реакционном сосуде создают давление водорода 344,7 кПа (50 ф/д 2) и смесь перемешивают в течение ночи. Берут аликвоту и анализируют ЖХ-МС, которая показывает, что реакция завершена. Катализатор отфильтровывают с использованием целита, который промывают метанолом (2100 мл). Фильтрат концентрируют досуха, и продукт реакции очищают ЖХСД Horizon (колонка с силикагелем 65i) с элюированием с градиентом, поднимающимся от 0% до 30% этилацетата в гексане, и получают указанное в заголовке соединение (6,2 г, 72%). m/z (ES) 497 (МН)+, 397 (М-Вос)+. 1 Указанное в заголовке соединение получают таким же способом, как на стадии K, но заменяя цисизомер пирролидина на транс-изомер трет-бутил-(2R,5R)-2-[(R)-[трет-бутил(диметил)силил]окси(фенил)метил]-5-(4-нитробензил)пирролидин-1-карбоксилата. m/z (ES) 497 (МН)+, 397 (МВос)+. Н ЯМР (500 МГц, CDCl3) : 7,41-7,30 (м, 5 Н), 6,73-6,67 (м, 2 Н), 6,56-6,50 (м, 2 Н), 5,52-5,48 (м, 1 Н),5,33-5,28 (м, 1 Н), 4,15-4,06 (м, 2 Н), 3,86-3,81 (м, 1 Н), 3,76-3,70 (м, 1 Н), 3,59-3,46 (м, 2 Н), 2,72 (ушир.д,J=12,0 Гц, 1 Н), 2,44 (ушир.д, J=12,0 Гц, 1 Н), 2,05-1,93 (м, 1 Н), 1,90-1,82 (м, 1 Н), 1,64 (с, 9 Н), 1,49-1,42 (м,1 Н), 1,32-1,20 (м, 2 Н), 1,10-1,02 (м, 1 Н), 0,95 (с, 9 Н), 0,14 (ушир.д, J=13,7 Гц, 3H), 0,10 (с, 3H). Указанные далее промежуточные соединения получают из соответствующих исходных веществ с использованием процедур, описанных выше для промежуточного соединения i-4a. Таблица 1. Указанные далее промежуточные соединения получают из соответствующих исходных веществ с использованием процедур, описанных выше для промежуточного соединения i-4b. Таблица 2. К раствору 100 мг (0,15 ммоль) бензил 4-[(3E,5R,6R)-5-[(бензилокси)карбонил]амино-6-[третбутил(диметил)силил]окси-6-(3-хлорфенил)-2-оксогекс-3-ен-1-ил]фенилкарбамата (со стадии А, i-3) в 8 мл этилацетата добавляют 10% палладий-на-угле и над суспензией создают атмосферу водорода водородом из баллона. Реакционную смесь перемешивают в атмосфере водорода в течение 8 ч при комнатной температуре. Катализатор отфильтровывают с использованием шприца с ПТФЭ фильтром Gilmen 0,45 мкм и промывают его этилацетатом (42 мл). Фильтрат концентрируют в вакууме досуха, остаток очищают на препаративной пластинке (1000 мкм) с элюированием смесью 5% метанола в дихлорметане, и получают указанное в заголовке соединение (33 мг, 51%). m/z (ES) 430, 432 (М, М+2)+. Стадия В. трет-Бутил-(5R)-2-(4-аминобензил)-5-[(R)-[трет-бутил(диметил)силил]окси(3 хлорфенил)метил]пирролидинкарбоксилат (i-5) К раствору 33 мг (0,07 ммоль) 4-5R)-5-[(R)-[трет-бутил(диметил)силил]окси(3 хлорфенил)метил]пирролидин-2-илметил)анилина (со стадии А) в 1 мл безводного ТГФ добавляют трет-бутилкарбонат (15,3 мг, 0,07 ммоль), а затем TEA (13 мкл, 0,07 ммоль) и полученную смесь перемешивают при комнатной температуре в атмосфере азота в течение ночи. Реакционную смесь помещают непосредственно на препаративную пластинку (500 мкм) и элюируют смесью 30% этилацетата в гексане,и получают указанное в заголовке соединение (25 мг, 78%). m/z (ES) 530, 532 (М, М+2)+, 430, 432 (МВос, М-Вос+2)+. Промежуточное соединение 6. 4-4-[4-(Трифторметил)фенил]-1,3-тиазол-2-илбензолсульфонилхлорид (i-6) Промежуточное соединение 6 можно получить согласно процедурам, описанным в литературе, например, в Ikemoto et al., Tetrahedron, 2003, 59, 1317-1325. Промежуточное соединение 7. 2-Метил-5,6-дигидро-4 Н-циклопента[][1,3]тиазол-4-карбоновая кислота (i-7) К раствору этил-2-оксоциклопентан-2-карбоксилата (56 г, 359 ммоль) в хлороформе (500 мл), охлажденному до 0C, добавляют бром (18,5 мл, 359 ммоль) в течение 20 мин. По завершении добавления смеси позволяют нагреваться до комнатной температуры и перемешивают в течение ночи. Газ азот барботируют через смесь в течение 90 мин для удаления большей части HBr. Смесь промывают водой (500 мл), насыщенным раствором NaHCO3 (200 мл), сушат над MgSO4, фильтруют и упаривают. Остаток растворяют в EtOH (500 мл), добавляют тиоацетамид (26,9 г, 359 ммоль), смесь перемешивают при комнатной температуре в течение 1 ч и затем кипятят с обратным холодильником в течение ночи. Смесь охлаждают и упаривают, остаток обрабатывают DCM и насыщенным раствором NaHCO3, органический слой промывают насыщенным раствором NaCl, сушат над MgSO4, фильтруют и упаривают. Остаток очищают ЖХСД (Biotage Horizon: 2 FLASH 65i), элюент: 100% гексана (450 мл), градиент с повышением от 100% гексана до смеси 25% этилацетата в гексане (1400 мл), затем смесь 25% этилацетата в гексане, и получают указанное в заголовке соединение (32 г, 42%) в виде темного масла. 1 Н ЯМР (500 МГц, CDCl3) : 4,22 (кв., J=7,0 Гц, 2 Н), 3,96 (м, 1 Н), 3,04 (м, 1 Н), 2,88 (м, 1 Н), 2,76 (м,2 Н), 2,70 (с, 3H), 1,30 (т, J=7,0 Гц, 3H). Стадия В. 2-Метил-5,6-дигидро-4 Н-циклопента[][1,3]тиазол-4-карбоновая кислота (i-7) К раствору 31,5 г (149 ммоль) этил-2-метил-5,6-дигидро-4 Н-циклопента[][1,3]тиазол-4 карбоксилата (со стадии А) в ТГФ (450 мл) и метаноле (100 мл) добавляют раствор гидроксида лития(149 мл 1 М раствора, 149 ммоль) и полученную смесь перемешивают при комнатной температуре в течение 3 ч. Органические летучие вещества удаляют выпариванием, водный остаток экстрагируют Et2O(2250 мл) и подкисляют до рН 3, добавляя 1 М HCl (170 мл), и насыщают твердым NaCl. Смесь экстрагируют DCM (3250 мл), объединенные DCM слои сушат над MgSO4, фильтруют и упаривают. Остаток растирают в ацетонитриле, твердое вещество отфильтровывают и сушат, и получают указанное в заголовке соединение (7,1 г, 26%) в виде не совсем белого твердого вещества. 1H ЯМР (500 МГц, CDCl3) : 11,75 (ушир.с, 1 Н), 4,02 (м, 1 Н), 3,00 (м, 1 Н), 2,90-2,66 (м, 6 Н). Указанное в заголовке соединение получают таким же способом, как промежуточное соединение (i-7),заменяя на стадии А тиоацетамид на тиомочевину. 1 Н ЯМР (500 МГц, CDCl3) : 5,30 (ушир.с, 2 Н), 4,21 (кв.,J=7,0, 2 Н), 3,81 (м, 1 Н), 2,91 (м, 1 Н), 2,78 (м, 1 Н), 2,66 (м, 2 Н), 1,30 (т, J=7,0, 3H). Стадия В. Этил-2-[(трет-бутоксикарбонил)амино]-5,6-дигидро-4 Н-циклопента[][1,3]тиазол-4 карбоксилат К раствору 230 мг (1,08 ммоль) этил-2-амино-5,6-дигидро-4 Н-циклопента[][1,3]тиазол-4 карбоксилата (со стадии А) в дихлорметане (5 мл) добавляют ди-трет-бутилдикарбонат (236 мг, 1,08 ммоль), триэтиламин (0,15 мл, 1,08 ммоль) и DMAP (13 мг, 0,11 ммоль) и полученную смесь перемешивают при комнатной температуре в течение 2 ч. Смесь промывают 1 н HCl (10 мл), насыщенным раствором NaCl (5 мл), сушат над MgSO4, фильтруют и упаривают. Остаток очищают ЖХСД (Biotage Horizon:(м, 1 Н), 2,86 (м, 1 Н), 2,76 (м, 2 Н), 1,55 (с, 9 Н), 1,23 (т, J=7,1 Гц, 3H). Стадия С. 2-[(трет-Бутоксикарбонил)амино]-5,6-дигидро-4 Н-циклопента[][1,3]тиазол-4 карбоновая кислота (i-8) Указанное в заголовке соединение получают из этил-2-[(трет-бутоксикарбонил)амино]-5,6-дигидро 4 Н-циклопента[][1,3]тиазол-4-карбоксилата (со стадии В) с использованием процедуры, аналогичной процедуре для стадии В при получении промежуточного соединения (i-7). 1 Указанное в заголовке соединение получают с использованием процедур, аналогичных процедурам получения промежуточного соединения (i-7), заменяя на стадии А тиоацетамид на 4-фтортиобензамид. 1 Н ЯМР (500 МГц, ДМСО-d6) : 7,90 (м, 2 Н), 7,29 (т, J=8,7, 2 Н), 3,81 (м, 1 Н), 2,99 (м, 1 Н), 2,86 (м,1 Н), 2,70-2,58 (м, 2 Н). Промежуточное соединение 10. 2-Метил-4,5,6,7-тетрагидро-1,3-бензотиазол-4-карбоновая кислота(40 мл), охлажденному до 0C, добавляют по каплям бром (4,5 мл, 88 ммоль) в течение 15 мин. По завершении добавления смеси позволяют нагреваться до комнатной температуры 90 мин. Смесь разбавляют EtOAc (100 мл) и промывают насыщенным раствором NaHCO3, насыщенным раствором NaCl, сушат над MgSO4, фильтруют и упаривают. Остаток растворяют в этаноле (100 мл) и добавляют тиоацетамид(6,6 г, 88 ммоль). Смесь перемешивают при комнатной температуре в течение 1 ч и затем кипятят с обратным холодильником в течение ночи. Смесь упаривают и остаток обрабатывают насыщенным раство- 25020135 ром NaHCO3 и DCM. Органический слой сушат над MgSO4, фильтруют и упаривают. Остаток очищают ЖХСД (Biotage Horizon: FLASH 65i), элюент: 100% гексана (500 мл), градиент смеси 0-25% EtOAc в гексане (1200 мл), затем смесь 25% EtOAc в гексане (1200 мл) и получают указанное в заголовке соединение(с, 3H), 2,18 (м, 1 Н), 2,11-1,95 (м, 2 Н), 1,85 (м, 1 Н), 1,29 (т, J=7,1, 3H). Стадия В. 2-Метил-4,5,6,7-тетрагидро-1,3-бензотиазол-4-карбоновая кислота (i-10) Указанное в заголовке соединение получают из этил-2-метил-4,5,6,7-тетрагидро-1,3-бензотиазол-4 карбоксилата (со стадии А) согласно процедуре, описанной для стадии В при получении промежуточного соединения (i-7). 1 Н ЯМР (500 МГц, CDCl3) : 9,26 (ушир.с, 1 Н), 3,81 (кв., J=7,3 и 5,9, 1 Н), 2,75 (м, 2 Н), 2,68 (с, 3H),2,24 (м, 1 Н), 2,18-2,01 (м, 2 Н), 1,82 (м, 1 Н). Промежуточное соединение 11. 2-[(трет-Бутоксикарбонил)амино]-4,5,6,7-тетрагидро-1,3 бензотиазол-4-карбоновая кислота (i-11) Указанное в заголовке соединение получают согласно процедурам, описанным для стадии А при получении промежуточного соединения (i-10), заменяя тиоацетамид на тиомочевину. 1 Н ЯМР (500 МГц, ДМСО-d6) : 9,28 (ушир.с, 2 Н), 4,11 (кв., J=7,3, 2H), 3,71 (т, J=5,0, 1 Н), 2,57-2,39(м, 2 Н), 1,90 (м, 2 Н), 1,78 (м, 1 Н), 1,59 (м, 1 Н), 1,17 (т, J=7,3, 3H). Стадия В. 2-[(трет-Бутоксикарбонил)амино]-4,5,6,7-тетрагидро-1,3-бензотиазол-4-карбоновая кислота (i-11) Указанное в заголовке соединение получают из этил-2-амино-4,5,6,7-тетрагидро-1,3-бензотиазол-4 карбоксилата (со стадии А) согласно процедурам, описанным для стадий В и С при получении промежуточного соединения (i-8). 1 Указанное в заголовке соединение получают согласно процедуре, описанной в литературе, в Journal(20,9 г, 89,1 ммоль) в CH2Cl2 (150 мл) добавляют (трифенилфосфоранилиден)ацетальдегид (27,1 г, 89,1 ммоль), и полученную смесь перемешивают при температуре окружающей среды в течение 40 ч. После удаления 1/3 растворителя добавляют достаточно гексана и полученное твердое вещество отфильтровывают. Флэш-хроматография на системе Biotage Horizon (силикагель, градиент смеси 0-20% этилацетата в гексане, затем смесь 20% этилацетата в гексане) дает 16,3 г (72%) названного в заголовке соединения вPd/C и полученную суспензию перемешивают в атмосфере водорода из баллона при температуре окружающей среды в течение 24 ч. Твердое вещество отфильтровывают на целите и фильтрат концентрируют в вакууме. Остаток очищают флэш-хроматографией на системе Biotage Horizon (силикагель, градиент смеси 0-20% этилацетата в гексане, затем смесь 20% этилацетата в гексане), и получают 11,5 г (58%) названного в заголовке соединения в виде бесцветного масла. ЖХ/МС 356,3 (М+23). Стадия С. трет-Бутил-(4R,5R)-2,2-диметил-4-[(3E)-4-(4-нитрофенил)бут-3-ен-1-ил]-5-фенил-1,3 оксазолидин-3-карбоксилат и трет-бутил-(4R,5R)-2,2-диметил-4-[(3Z)-4-(4-нитрофенил)бут-3-ен-1-ил]-5 фенил-1,3-оксазолидин-3-карбоксилат К раствору трет-бутил-(4R,5R)-2,2-диметил-4-(3-оксопропил)-5-фенил-1,3-оксазолидин-3 карбоксилата со стадии В (10,0 г, 30,0 ммоль) в CH2Cl2 (200 мл) добавляют бромид (4 нитробензил)трифенилфосфония (21,5 г, 45,0 ммоль), а затем Et3N (8,36 мл, 60,0 ммоль). Красную реакционную смесь перемешивают при температуре окружающей среды в течение 48 ч. В реакционную смесь вливают гексан (200 мл) и отфильтровывают твердое вещество. Флэш-хроматография на системе К раствору полученной выше на стадии С смеси цис/транс (7,86 г, 17,4 ммоль) в этилацетате (100 мл) добавляют 50 мл 2 н HCl и полученную смесь перемешивают при температуре окружающей среды в течение 2 ч и затем при нагревании при 45C в течение 3 ч. Летучие вещества удаляют при пониженном давлении. Полученное белое твердое вещество растворяют в N,N-диметилформамиде (100 мл) и добавляют 15,1 мл (86,7 ммоль) изо-Pr2Net. Реакционную смесь перемешивают при температуре окружающей среды в течение 7 ч. Затем добавляют ди-трет-бутилдикарбонат (4,55 г, 20,8 ммоль) и реакционную смесь перемешивают при температуре окружающей среды в течение ночи. Добавляют воду (200 мл), и смесь экстрагируют этилацетатом (200 мл 3). Объединенные органические слои сушат над Na2SO4, фильтруют и концентрируют при пониженном давлении. Остаток очищают флэш-хроматографией на системе Biotage Horizon (силикагель, градиент смеси 0-30% этилацетата в гексане), и получают названные в заголовке соединения - 1,61 г (22%) трет-бутил-(2R,5S)-2-[(R)-гидрокси(фенил)метил]-5-(4-нитробензил)пирролидин-1-карбоксилата (цис) и 3,9 г (54%) трет-бутил-(2R,5R)-2-[(R)-гидрокси(фенил)метил]-5(4-нитробензил)пирролидин-1-карбоксилата (транс). ЖХ/МС 435,4 (М+23). Стадия Е. трет-Бутил-(2S,5R)-2-(4-аминобензил)-5-[(R)-гидрокси(фенил)метил]пирролидин-1 карбоксилат (i-13a) К раствору полученного выше на стадии D (цис)трет-бутил-(2R,5S)-2-[(R)-гидрокси(фенил)метил]5-(4-нитробензил)пирролидин-1-карбоксилата (1,51 г, 3,66 ммоль) в этаноле (20 мл) добавляют 0,15 г 10% Pd/C и полученную суспензию перемешивают в атмосфере водорода из баллона при температуре окружающей среды в течение 5 ч. Фильтрация через целит и удаление растворителя дают 1,40 г (100%) названного в заголовке соединения в виде белой пены, которое используют без дополнительной очистки. ЖХ/МС 405,3 (М+23).F. трет-Бутил-(2R,5R)-2-(4-аминобензил)-5-[(R)-гидрокси(фенил)метил]пирролидин-1 карбоксилат (i-13b) К раствору полученного выше на стадии(транс)трет-бутил-(2R,5R)-2-[(R)гидрокси(фенил)метил]-5-(4-нитробензил)пирролидин-1-карбоксилата (3,90 г, 9,46 ммоль) в этаноле (40 мл) добавляют 0,4 г 10% Pd/C и полученную суспензию перемешивают в атмосфере водорода из баллона при температуре окружающей среды в течение 6 ч. Твердое вещество отфильтровывают на целите. После удаления растворителя флэш-хроматография на системе Biotage Horizon (силикагель, градиент смеси 030% этилацетата в гексане, затем смесь 30% этилацетата в гексане) дает 2,30 г (64%) названного в заголовке соединения в виде белой пены. ЖХ/МС 4 05,3 (М+23). Промежуточное соединение 14. К раствору 28 мг (0,24 ммоль) L-пролина в N,N-диметилформамиде (3 мл) при температуре окружающей среды добавляют 51 мг (0,24 ммоль) 2-бромбензотиазола, 100 мг (0,72 ммоль) карбоната калия и 6 мг (0,03 ммоль) иодида меди. Реакционную смесь перемешивают при 100C в течение ночи. Затем смесь фильтруют и фильтрат очищают ВЭЖХ с обращенной фазой (ТМС Pro-Рас С 18; градиент смеси 060% 0,1% трифторуксусной кислоты в ацетонитриле/0,1% трифторуксусной кислоты в воде). Чистые фракции лиофилизуют в течение ночи и получают 35 мг (60%) названного в заголовке соединения в виде светло-коричневого твердого вещества. 1 Н ЯМР (ДМСО-d6):7,78 (д, J=8,0 Гц, 1 Н), 7,45 (д, J=8,0 Гц, 1 Н), 7,28 (т, J=7,8 Гц, 1 Н), 7,08 (т,J=7,8 Гц, 1 Н), 4,48 (д, J=7,3 Гц, 1 Н), 3,52-3,61 (м, 2 Н), 2,37 (м, 1 Н), 2,01-2,11 (м, 3H), ЖХ/МС 249,3(М+1). Промежуточные соединения 15-22. Перечисленные далее промежуточные соединения N-замещенные L-пролины получают из соответствующих исходных веществ с использованием процедур, описанных выше, и процедур, известных в технике. Таблица 3 К раствору 3-хлор-6-оксопиридазин-1(6 Н)-ил)уксусной кислоты (1 г, 5,30 ммоль, ChemBridge) в 40 мл метанола добавляют 100 мг 10% палладия-на-угле, и полученную суспензию помещают в атмосферу водорода и энергично перемешивают в течение 4 ч при комнатной температуре. Катализатор отфильтровывают в шприце на ПФТЭ фильтре Gilmen 0,45 мкм, и фильтрат концентрируют в вакууме. Остаток используют без дополнительной очистки. 1 Н ЯМР (D2O):9,07 (дд, J=1,6, 3,9 Гц, 1 Н), 7,59 (дд, J=4,1, 9,4 Гц, 1 Н), 7,14 (дд, J=1,6, 9,3 Гц, 1H),4,97 (с, 2 Н), ЖХ/МС 155,09 (М+1). Промежуточное соединение 24. Получение 2-бром-5,6-дигидро-4 Н-циклопента[d][1,3]тиазол-4 карбоновой кислоты (i-24) К раствору трет-бутилнитрита (4,2 мл, 35,3 ммоль) и бромида меди(II) (6,3 г, 28,3 ммоль) в ацетонитриле (100 мл) добавляют по частям этил-2-амино-5,6-дигидро-4 Н-циклопента[][1,3]тиазол-4 карбоксилат (5 г, 23,6 ммоль). По завершении добавления смесь перемешивают при комнатной температуре в течение 3 ч. Смесь выливают в 2 М HCl (600 мл) и экстрагируют EtOAc (3200 мл), объединенныеEtOAc слои промывают 1 М HCl (500 мл), водой (250 мл), насыщенным раствором NaHCO3 (200 мл) и насыщенным раствором NaCl (150 мл), сушат над MgSO4, фильтруют и упаривают. Остаток очищают ЖХСД (Biotage Horizon: FLASH 40M), элюент: 100% гексана (100 мл), градиент с повышением от 100% гексана до смеси 25% EtOAc в гексане (750 мл), затем смесь 25% EtOAc в гексане (700 мл) и получают 1,87 г (29%) светло-оранжевого масла. 1 Н ЯМР (500 МГц CDCl3) : 4,22 (кв., J=7,1, 2 Н), 4,00 (м, 1 Н), 3,04-3,11 (м, 1 Н), 2,88-2,94 (м, 1 Н),2,77 (кв., J=7,3, 2 Н), 1,31 (т, J=7,1, 3H). Стадия В. 2-бром-5,6-дигидро-4 Н-циклопента[d][1,3]тиазол-4-карбоновая кислота Продукт со стадии А (4,92 г, 17,82 ммоль) растворяют в метаноле (20 мл) и раствор добавляют по каплям к смеси 5 н раствора NaOH (4,25 мл, 21,25 ммоль), воды (16 мл) и метанола (30 мл). По завершении добавления смесь перемешивают в течение 2 ч. Метанол удаляют выпариванием и рН оставшейся водной части доводят до 2,5 концентрированной HCl. Смесь насыщают твердым NaCl и экстрагируютEtOAc ( 3); объединенные EtOAc слои промывают насыщенным раствором NaCl, сушат над Na2SO4 и обрабатывают активированным углем в течение ночи. Профильтрованную смесь упаривают. Остаток растирают в EtOAc, твердое вещество отфильтровывают, и получают 1,94 г нужного продукта. Маточные жидкости упаривают и очищают ЖХСД с использованием градиента, повышающегося от 100% гексана до 100% EtOAc в гексане, и получают дополнительные 0,82 г названного в заголовке продукта (всего 2,76 г, 62%) в виде не совсем белого твердого вещества. 1 К раствору этил-2-аминотиазол-4-ацетата (8 г, 43 ммоль) в DCM (75 мл) добавляют ди-третбутилдикарбонат (20,63 г, 95 ммоль), основание Хюнига (16,51 мл, 95 ммоль) и DMAP (1,57 г, 12,89 ммоль) и полученную смесь перемешивают при комнатной температуре в течение ночи. Смесь промывают водой, насыщенным раствором NaCl, сушат над MgSO4, фильтруют и упаривают. Остаток очищают ЖХСД с использованием градиента, поднимающегося от 100% гексана до смеси 50% EtOAc в гексане, и получают указанное в заголовке соединение (12,5 г, 75%) в виде белого твердого вещества. 1H ЯМР (500 МГц CDCl3): 7,02 (с, 1 Н), 4,17 (кв., J=7,3, 2 Н), 3,73 (с, 2 Н), 1,51 (с, 18 Н), 1,25 (т, J=7,3, 3H).
МПК / Метки
МПК: C07D 487/04, C07D 401/12, C07D 417/12, C07D 403/12, A61K 31/4178, C07D 471/04, A61P 13/00, A61K 31/506
Метки: качестве, рецепторов, гидроксиметилпирролидины, адренергических, beta, агонистов
Код ссылки
<a href="https://eas.patents.su/30-20135-gidroksimetilpirrolidiny-v-kachestve-agonistov-adrenergicheskih-receptorov-beta-3.html" rel="bookmark" title="База патентов Евразийского Союза">Гидроксиметилпирролидины в качестве агонистов адренергических рецепторов бета 3</a>
Производные диазаспиро[5.5]ундекана в качестве антагонистов мускариновых рецепторов и агонистов бета-адренорецепторов для применения в лечении легочных расстройств

Номер патента: 18711
Опубликовано: 30.10.2013
Авторы: Алькараз Лилиан, Стокс Майкл Джон, Листер Эндрю Стюарт, Киндон Николас Дэвид, Роббинс Эндрю Джеймс, Джонсон Тимоти, Бэйли Эндрю, Теобальд Барри Джон, Булл Ричард Джеймс
МПК: A61P 11/08, A61P 11/06, A61K 31/357...
Метки: рецепторов, диазаспиро[5.5]ундекана, антагонистов, производные, агонистов, лечении, легочных, мускариновых, расстройств, качестве, бета-адренорецепторов, применения
Формула / Реферат:
1. Соединение, выбранное издитрифторацетата (R)-4-гидрокси-7-(1-гидрокси-2-(4-(2-(4-(2-метилтиазол-4-карбонил)-1-окса-4,9-диазаспиро[5.5]ундекан-9-ил)этокси)бензиламино)этил)бензо[d]тиазол-2(3Н)-она;дитрифторацетата (R)-4-гидрокси-7-(1-гидрокси-2-(9-(4-(2-метилтиазол-4-карбонил)-1-окса-4,9-диазаспиро[5.5]ундекан-9-ил)нониламино)этил)бензо[d]тиазол-2(3Н)-она;дигидрохлорида...
Тетрагидробензазепиновые производные в качестве агонистов дофаминовых рецепторов d1

Номер патента: 4745
Опубликовано: 26.08.2004
Автор: Тилбрук Гари
МПК: C07D 223/16, A61K 31/55, A61P 25/16...
Метки: дофаминовых, агонистов, производные, качестве, тетрагидробензазепиновые, рецепторов
Формула / Реферат:
1. Соединение-агонист дофаминовых рецепторов D1, общей формулы I где R1 представляет собой галоген; R2 представляет собой водород, метил или низший алкенил с 3-5 атомами углерода; R3 и R4 вместе образуют фурановое, дигидрофурановое, тиофеновое, дигидротиофеновое, циклопентановое или циклогексановое кольцо и R5 представляет собой водород; или R4 и R5 вместе образуют фурановое, дигидрофурановое, тиофеновое, дигидротиофеновое, циклопентановое или...
Соединения бензимидазолкарбоксамида в качестве агонистов рецепторов 5-нт4

Номер патента: 13240
Опубликовано: 30.04.2010
Авторы: Чой Сеок-Ки, Маркесс Дэниэл, Дзян Лань, Физакерли Кирстен М., Лонг Дэниэл Д., Фазери Пол Р., Гендрон Роланд, Маккиннелл Роберт Мюррей, Далзил Шон М.
МПК: C07D 401/14, A61P 1/00, A61K 31/4545...
Метки: качестве, бензимидазолкарбоксамида, соединения, рецепторов, агонистов, 5-нт4
Формула / Реферат:
1. Соединение формулы (I)где R1 является С3-5алкилом, необязательно замещенным -ОН; иX выбирают из:(a) -С(О)OR2, где R2 представляет собой С1-4алкил или -(CH2)n-фенил, где n равно 0 или 1;(b) -C(O)R3, где R3 выбирают изфенила, необязательно замещенного 1, 2 или 3 заместителями, выбранными из С1-4алкила, галогена, С1-4 алкокси, -CF3, -OCF3, -OCHF2 и -CN,С1-5алкила,С4-5циклоалкила и-(CH2)m-А, где m равно 0 или 1 и А выбирают из амино, фуранила,...
Замещенные бензопираны в качестве селективных агонистов бета-рецептора эстрогена

Номер патента: 7382
Опубликовано: 27.10.2006
Авторы: Норман Брайан Херст, Кришнан Венкатеш Гари, Лугар Чарльз Уиллис III, Пфайфер Ланс Аллен, Нойбауэр Блейк Ли, Ричардсон Тимоти Айво, Додж Джеффри Алан
МПК: C07D 311/60, A61P 35/00
Метки: замещенные, селективных, качестве, эстрогена, бензопираны, бета-рецептора, агонистов
Формула / Реферат:
1. Соединение формулы (I) где R1, R2 и R3, каждый независимо, является -Н, C1-С6 алкилом, -ОН, C1-С6 алкокси, галогеном или -СFз; R5 является водородом или C1-С6 алкилом; Y1, Y2 и Y3, каждый независимо, является -Н или C1-С6 алкилом и G является -СН2-, -СН2-СН2- или -СН2-СН2-СН2-; или его фармацевтически приемлемые соли. 2. Соединение по п.1, в котором G является -СН2-. 3. Соединение по любому из пп.1 или 2, в котором Y2 и Y3 оба являются -Н....
Производные 3-ароилиндола и их применение в качестве агонистов рецепторов cb2

Номер патента: 5854
Опубликовано: 30.06.2005
Авторы: Гийомон Кароль, Барт Франсис, Васс Фабьенн, Верне Клод, Ринальди Мирей, Конжи Кристиан
МПК: A61P 43/00, A61K 31/404, C07D 209/12...
Метки: рецепторов, применение, агонистов, качестве, производные, 3-ароилиндола
Формула / Реферат:
1. Соединение формулы где Ar представляет собой a) фенил, моно-, ди- или тризамещенный одной или более чем одной группой, выбранной из атома галогена, (C1-C4)алкила, трифторметила, амино, нитро, гидроксила, (C1-C4)алкокси, (C1-C4)алкилсульфанила или (C1-C4)алкилсульфонила; b) нафтил, который не замещен или замещен единожды или дважды атомом галогена, (C1-C4)алкилом или трифторметилом; A представляет собой C2-C6алкиленовый радикал; Y...
Предыдущий патент: Носители никорандила с повышенной стабильностью
Следующий патент: Производные пиколинамида в качестве ингибиторов киназы
Случайный патент: Бортовая штора для кузовов грузовых автомобилей и прицепов