Гидроксаматы в качестве ингибиторов гистон-деацетилазы
Номер патента: 17198
Опубликовано: 30.10.2012
Авторы: Дэвидсон Алан Хорнсби, Белфилд Эндрю Джеймс, Дональд Элистейр Дэвид Грэм, Моффат Дэвид Фестус Чарльз, Пэтел Санджей Ратилал, Драммонд Алан Гастингс, Дэй Франческа Энн

















Формула / Реферат
1. Соединение формулы (I) или его соль

где m равен 1;
n равен 0 или 1;
Q, V и W независимо обозначают -N= или -С=;
В обозначает двухвалентный радикал, выбранный из (В2) и (В6)

где связь, отмеченная *, связана с кольцом, содержащим Q, V и W;
А обозначает циклогексильное или фенильное кольцо;
-[Linker]- обозначает связь, -CH2-NH-CH2- или -NH-CH2-;
Z1 обозначает радикал формулы R1R2CHNH- или R1R2CHNH-CH2-, где
R1 обозначает карбоксильную группу (-СООН) или сложноэфирную группу, выбранную из группы сложноэфирных групп формулы -(C=O)OR9, где R9 представляет собой R7R8CH-, где R7 и R8 вместе с атомом углерода, к которому они присоединены, образуют моноциклическое карбоциклическое кольцо из 3-7 атомов в кольце;
R2 обозначает боковую цепь природной или неприродной α-аминокислоты, причем боковая цепь неприродной α-аминокислоты выбрана из фенильной, циклогексилметильной, циклогексильной, пиридин-3-илметильной, трет-бутоксиметильной, трет-бутильной, 1-бензилтио-1-метилэтильной, 1-метилтио-1-метилэтильной, 1-меркапто-1-метилэтильной и фенилэтильной.
2. Соединение по п.1, отличающееся тем, что Q обозначает -С= и V и W, каждый, обозначают -N=.
3. Соединение по п.1 или 2, отличающееся тем, что R9 обозначает циклопентил.
4. Соединение по любому из предыдущих пунктов, отличающееся тем, что R2 обозначает циклогексилметил, пиридин-3-илметил, втор-бутил, трет-бутил, 1-бензилтио-1-метилэтил, 1-метилтио-1-метилэтил, 1-меркапто-1-метилэтил или фенилэтил.
5. Соединение по любому из пп.1-3, отличающееся тем, что R2 обозначает фенил, бензил, изобутил, циклогексил или трет-бутоксиметил.
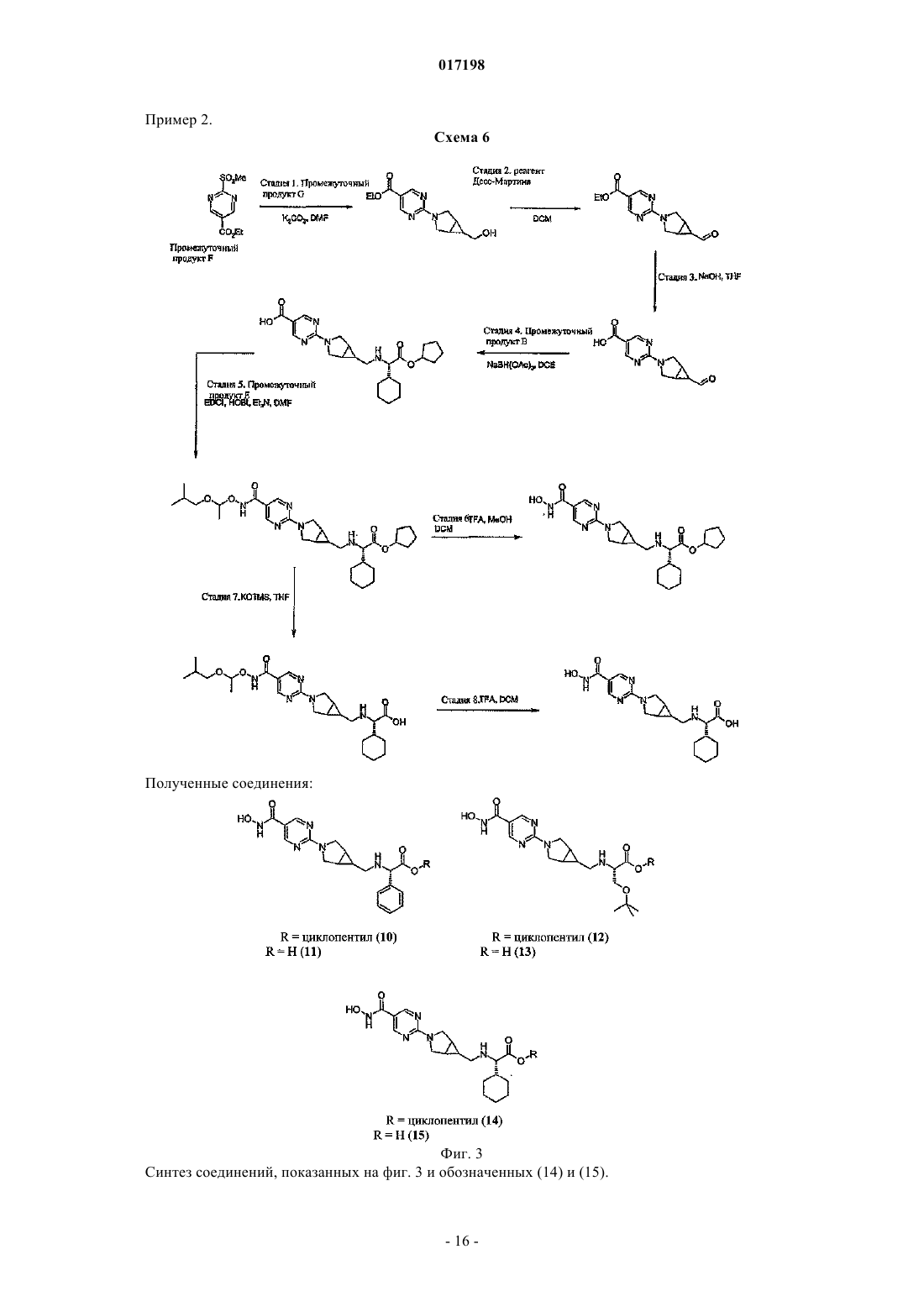
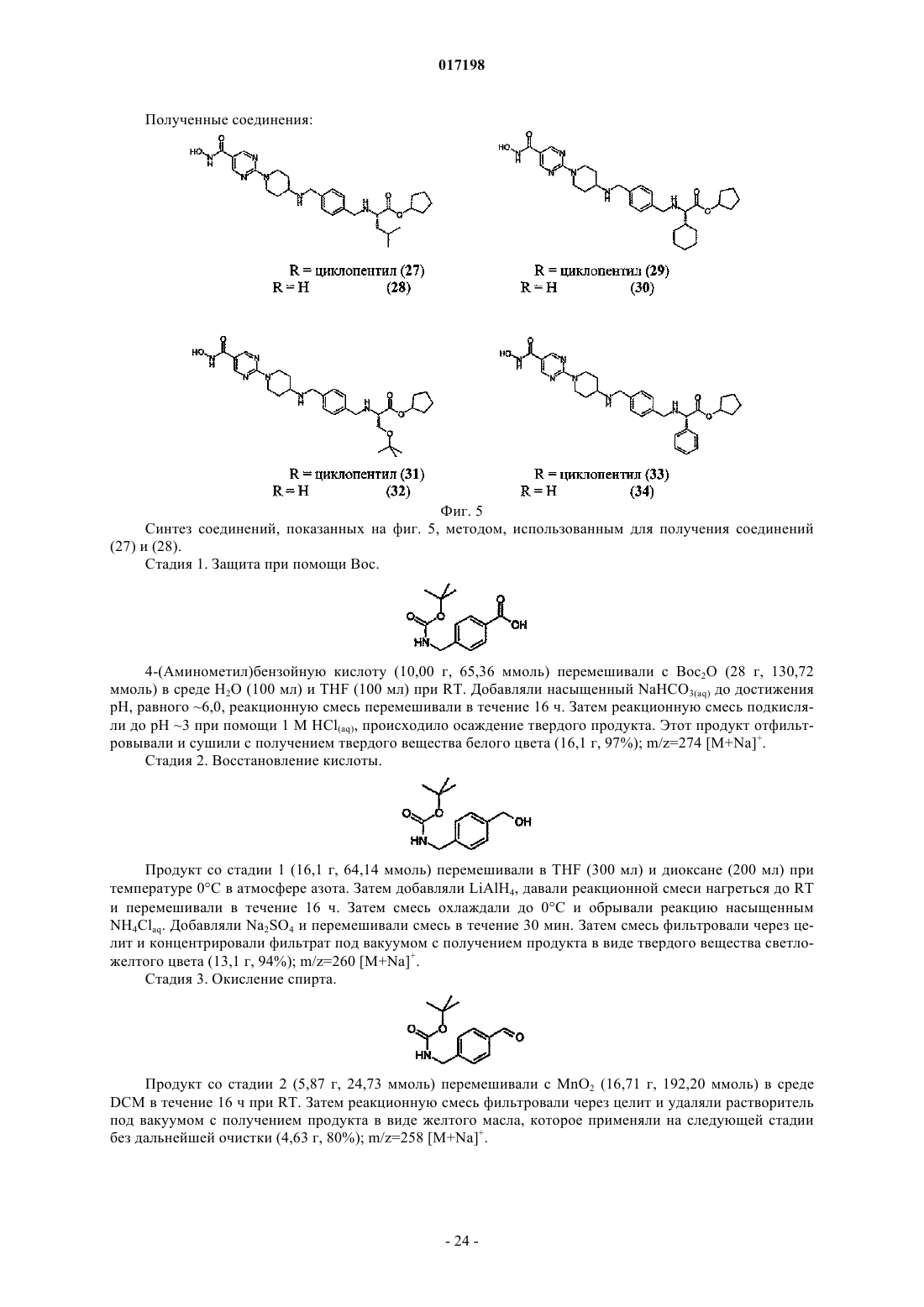
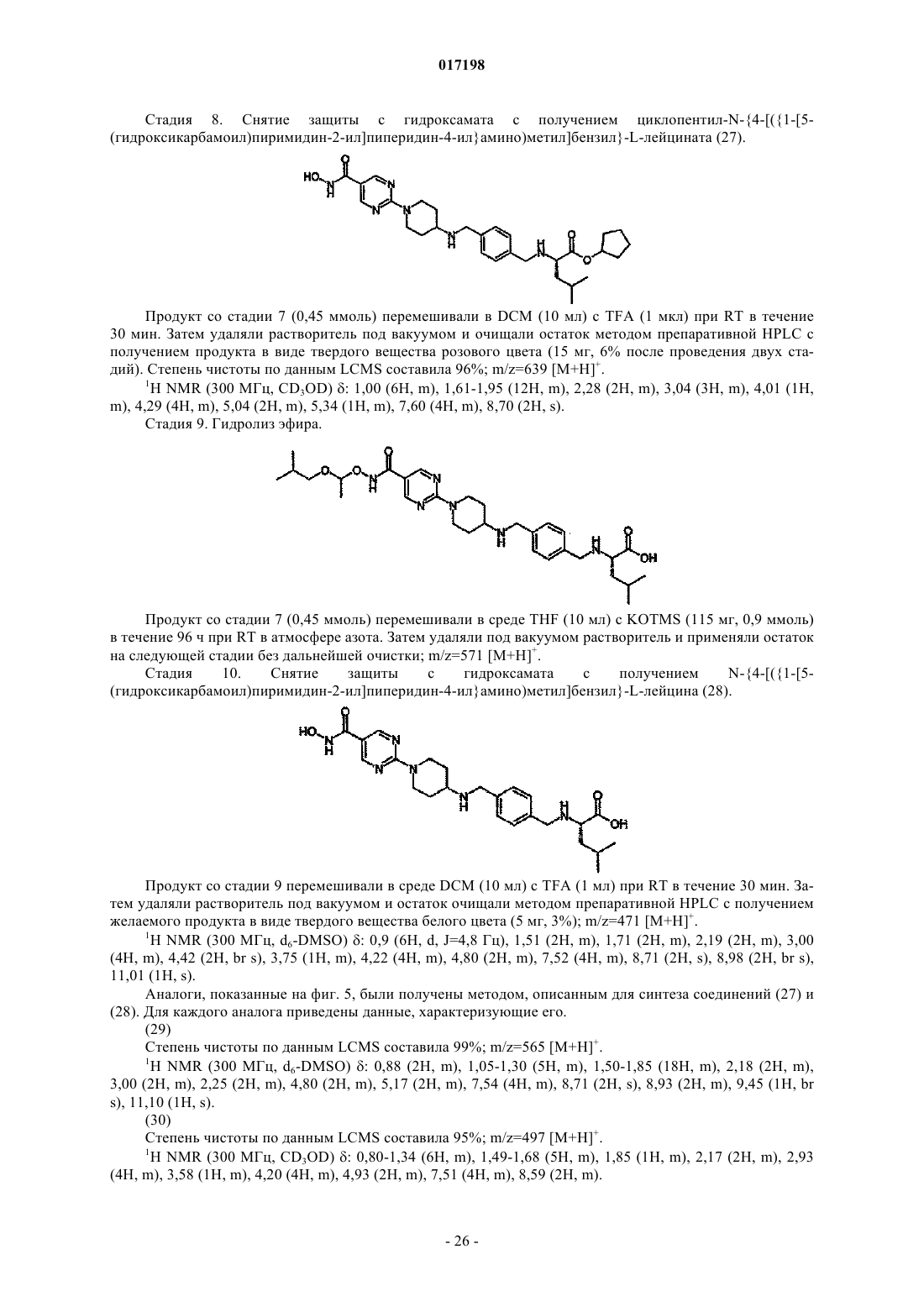
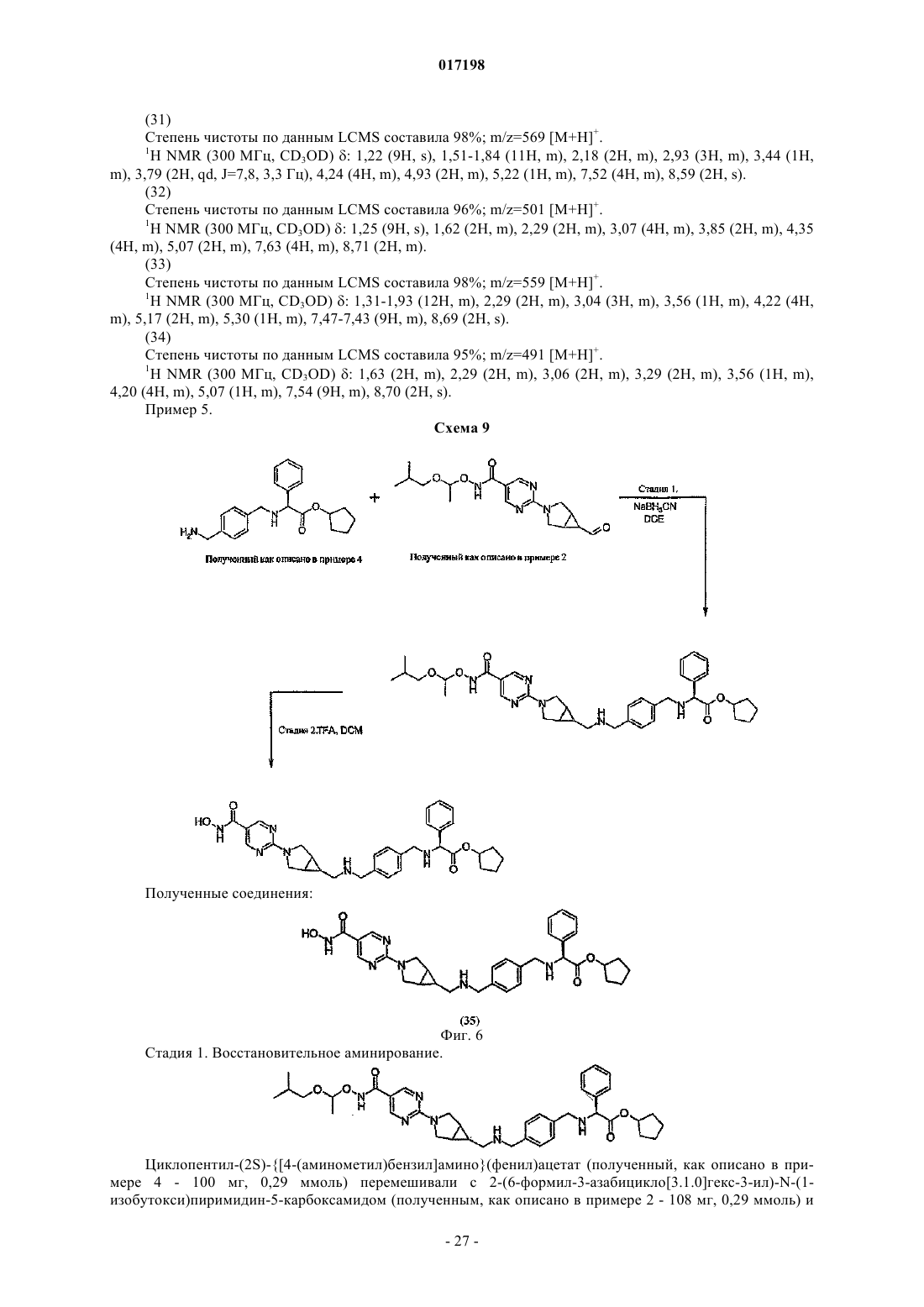
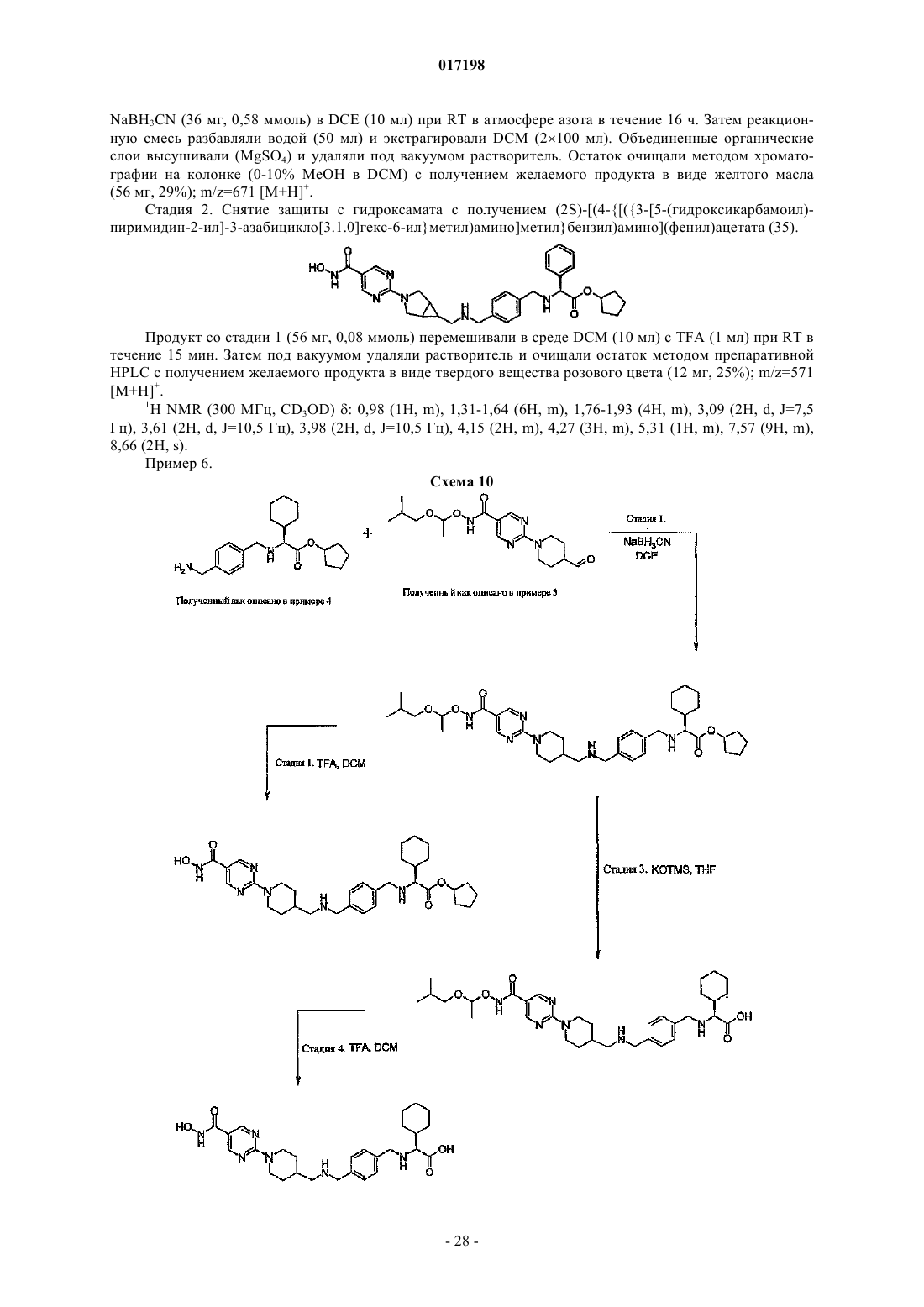
6. Соединение по п.1, отличающееся тем, что оно имеет любую из приведенных ниже структурных формул:

в которых R9 представляет собой циклопентил,
или его соль.
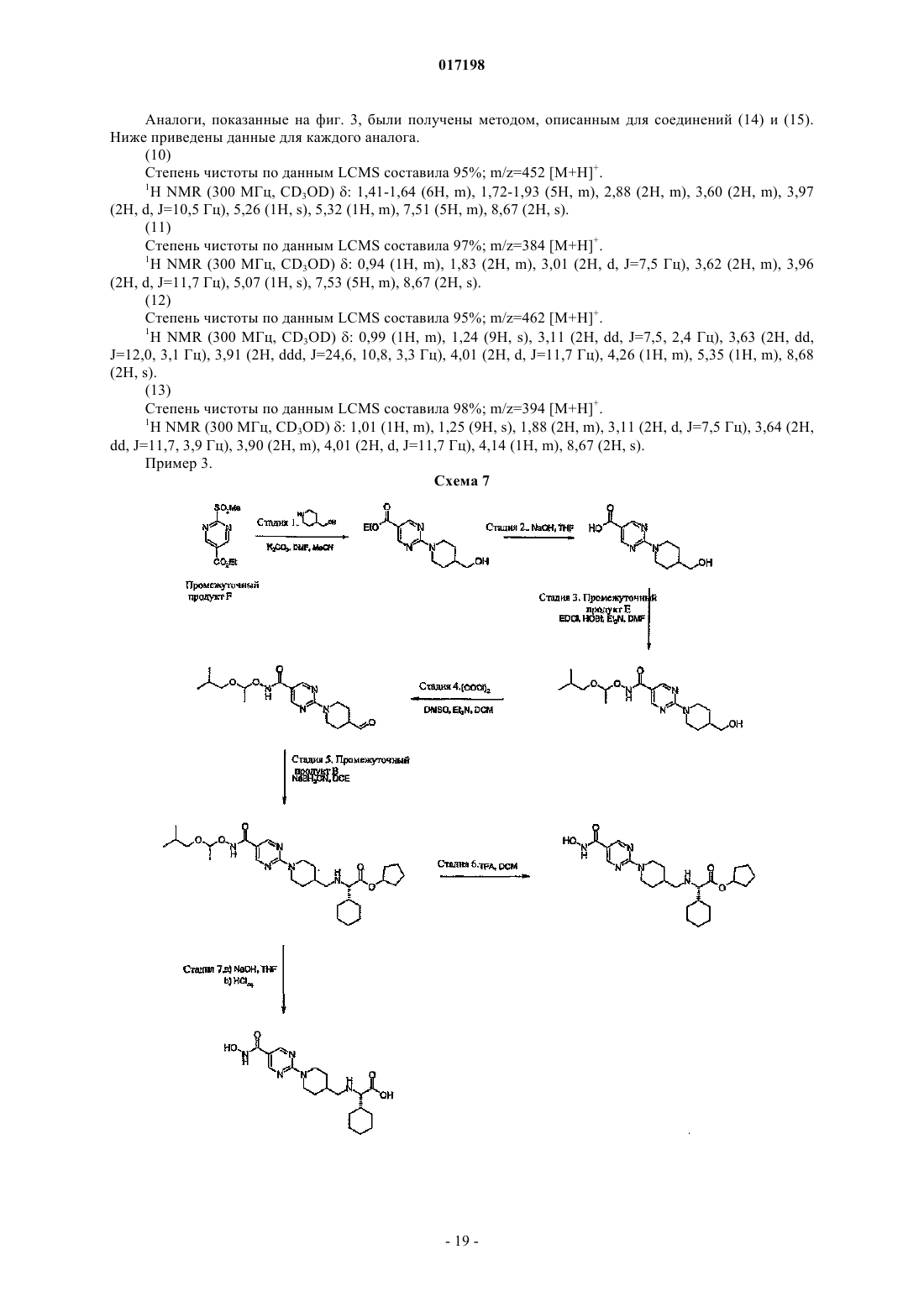
7. Соединение по п.1, отличающееся тем, что оно имеет любую из приведенных ниже структурных формул:

в которых R9 представляет собой циклопентил,
или его соль.
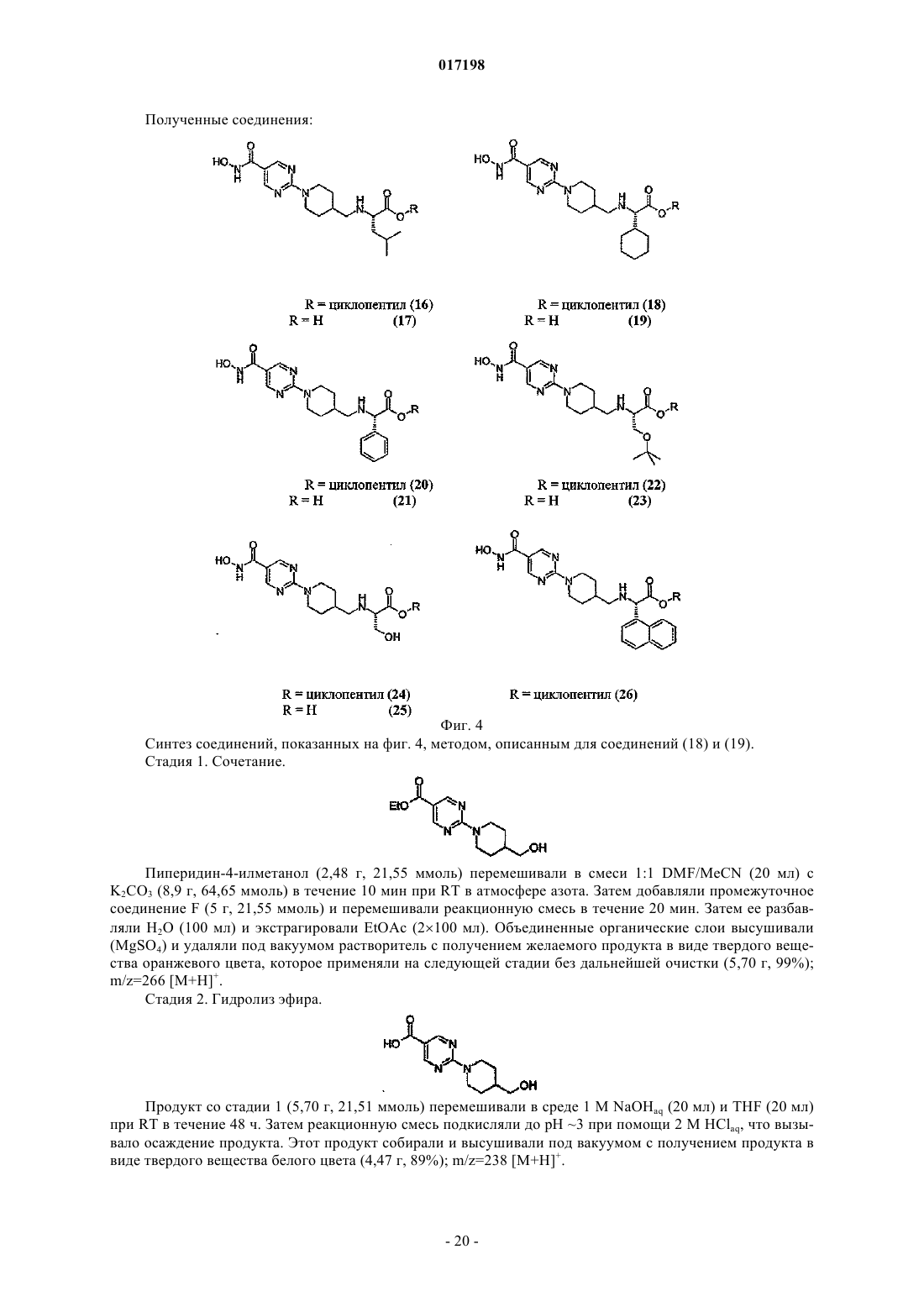
8. Соединение по п.1, отличающееся тем, что оно имеет любую из приведенных ниже структурных формул:

в которых R9 представляет собой циклопентил,
или его соль.
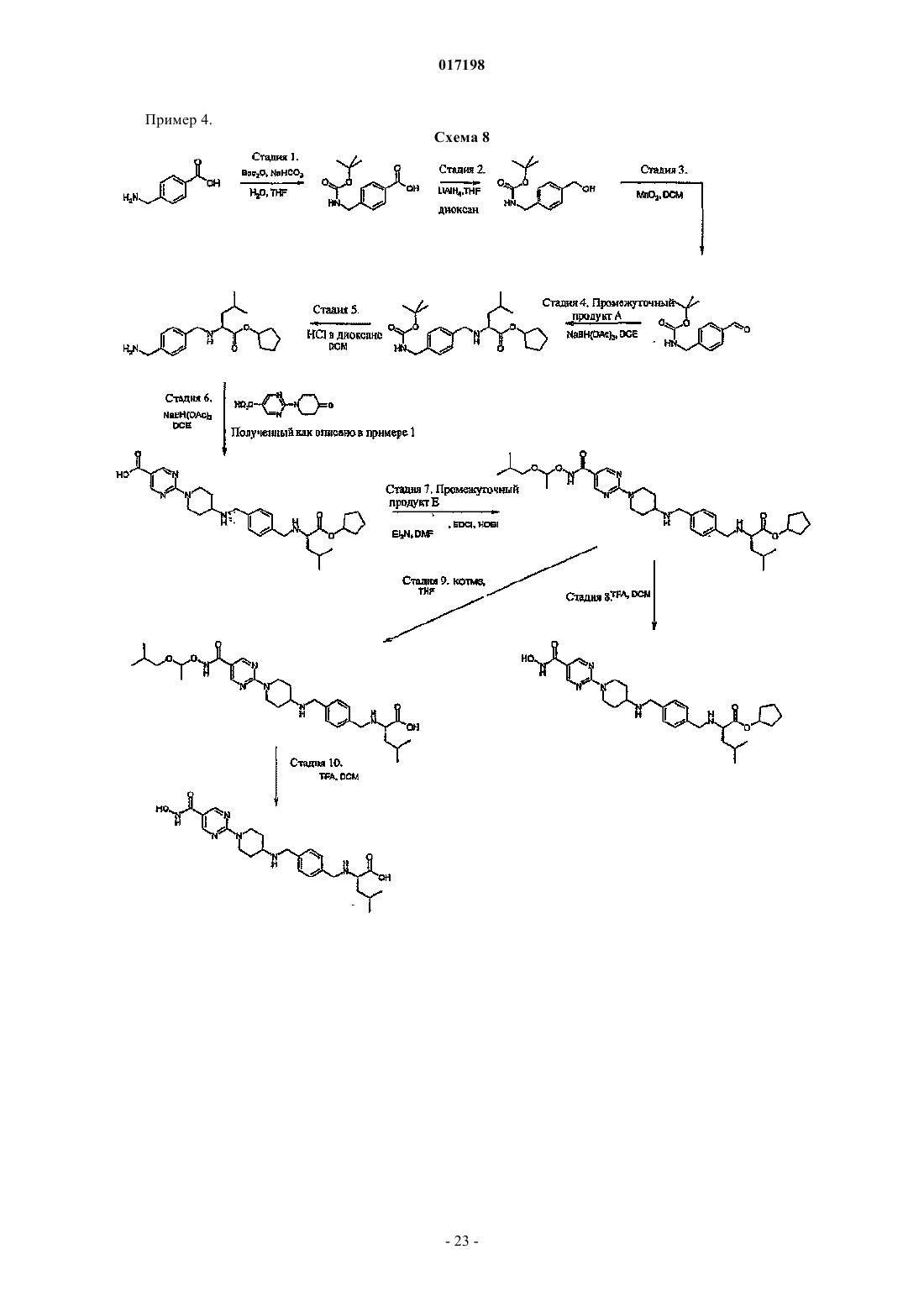
9. Соединение по п.1, отличающееся тем, что оно имеет любую из приведенных ниже структурных формул:

в которых R9 представляет собой циклопентил,
или его соль.
10. Соединение по п.1, отличающееся тем, что оно имеет любую из приведенных ниже структурных формул:

или его соль.
11. Соединение по п.1, отличающееся тем, что оно имеет любую из приведенных ниже структурных формул:

в которых R9 представляет собой циклопентил,
или его соль.
12. Соединение по п.1, отличающееся тем, что оно имеет любую из приведенных ниже структурных формул:

в которых R9 представляет собой циклопентил,
или его соль.
13. Фармацевтическая композиция, содержащая соединение по любому из предыдущих пунктов вместе с фармацевтически приемлемым носителем.
14. Применение соединения по любому из пп.1-13 для изготовления лекарственного средства для лечения болезни, связанной с пролиферацией клеток, полиглютаминовой болезни, нейродегенеративной болезни, аутоиммунной болезни, воспалительного заболевания, отторжения трансплантата органа, диабета, гематологических нарушений или инфекции, причем лекарственное средство включает эффективное количество указанного соединения.
15. Применение по п.14, предназначенное для лечения пролиферации раковых клеток, болезни Хантингтона или болезни Альцгеймера.
16. Применение по п.15, предназначенное для лечения ревматоидного артрита.
Текст
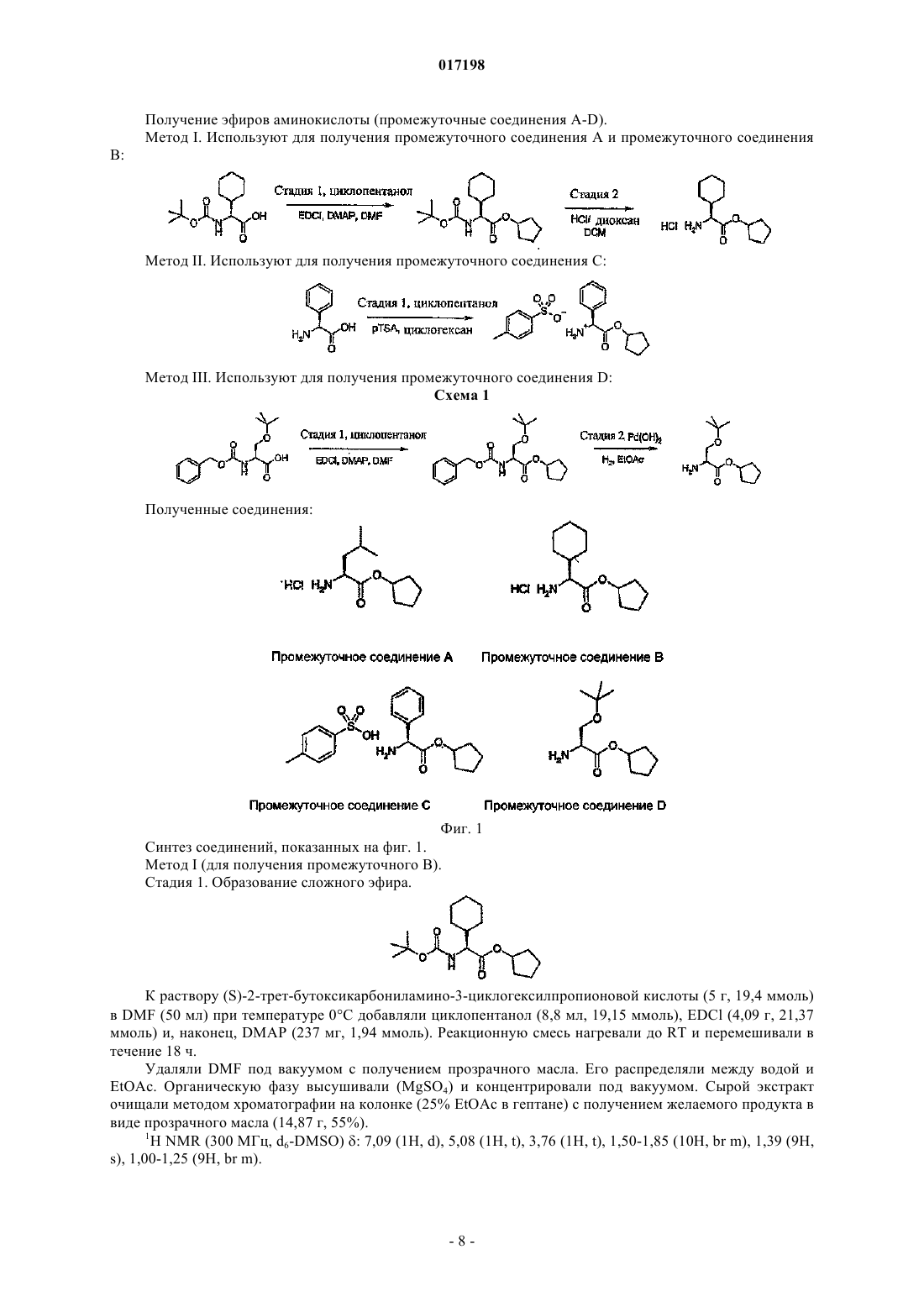
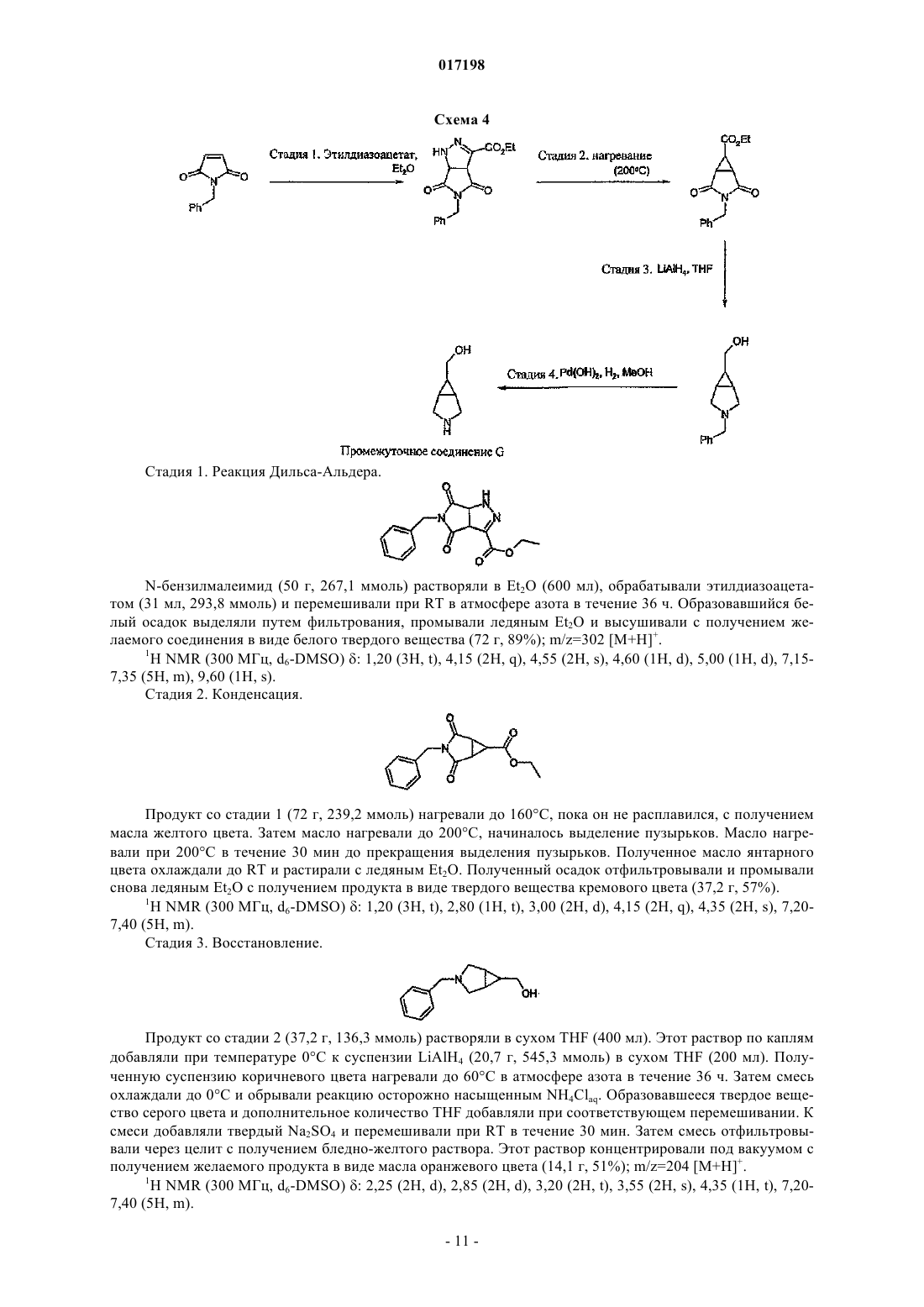
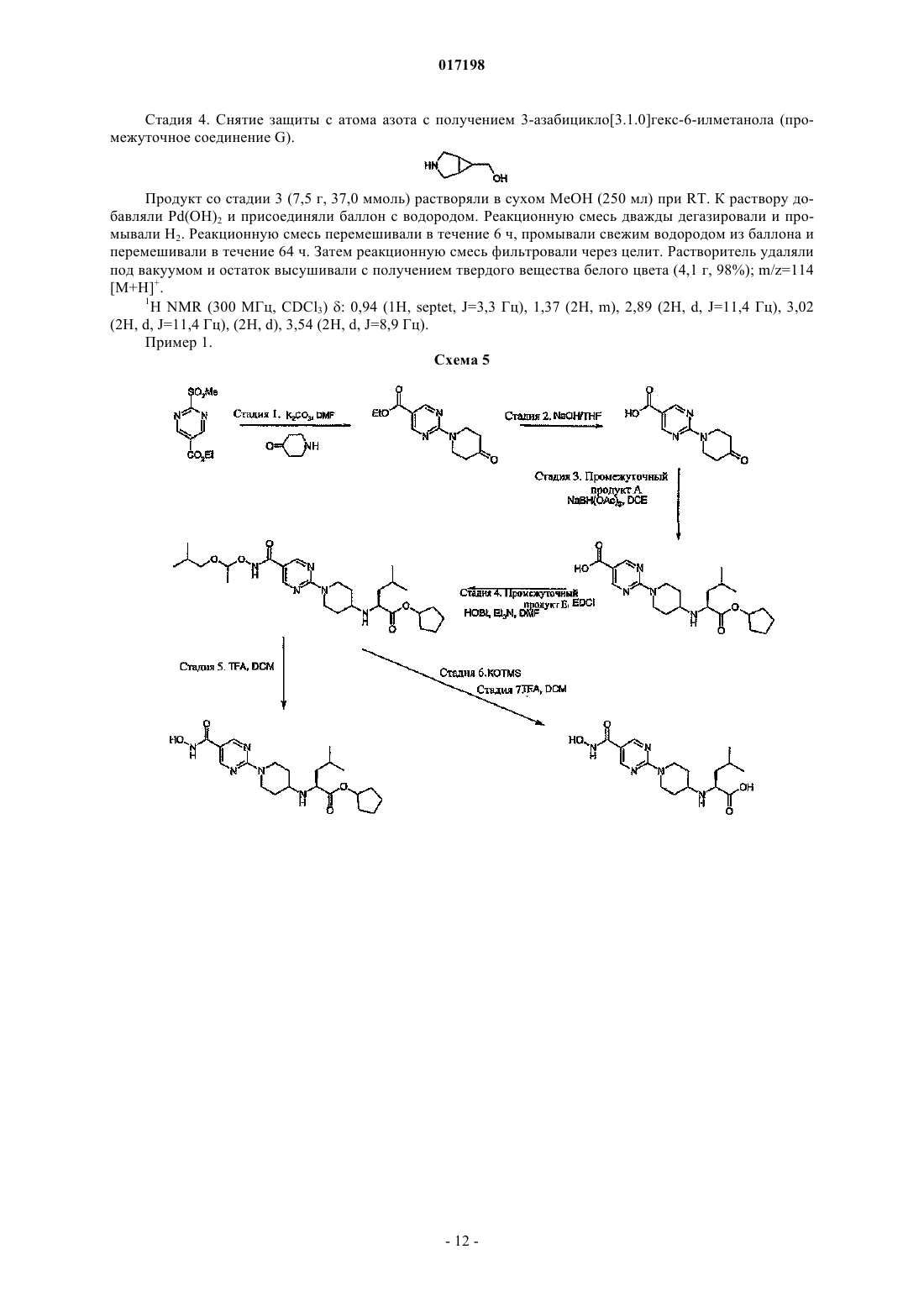
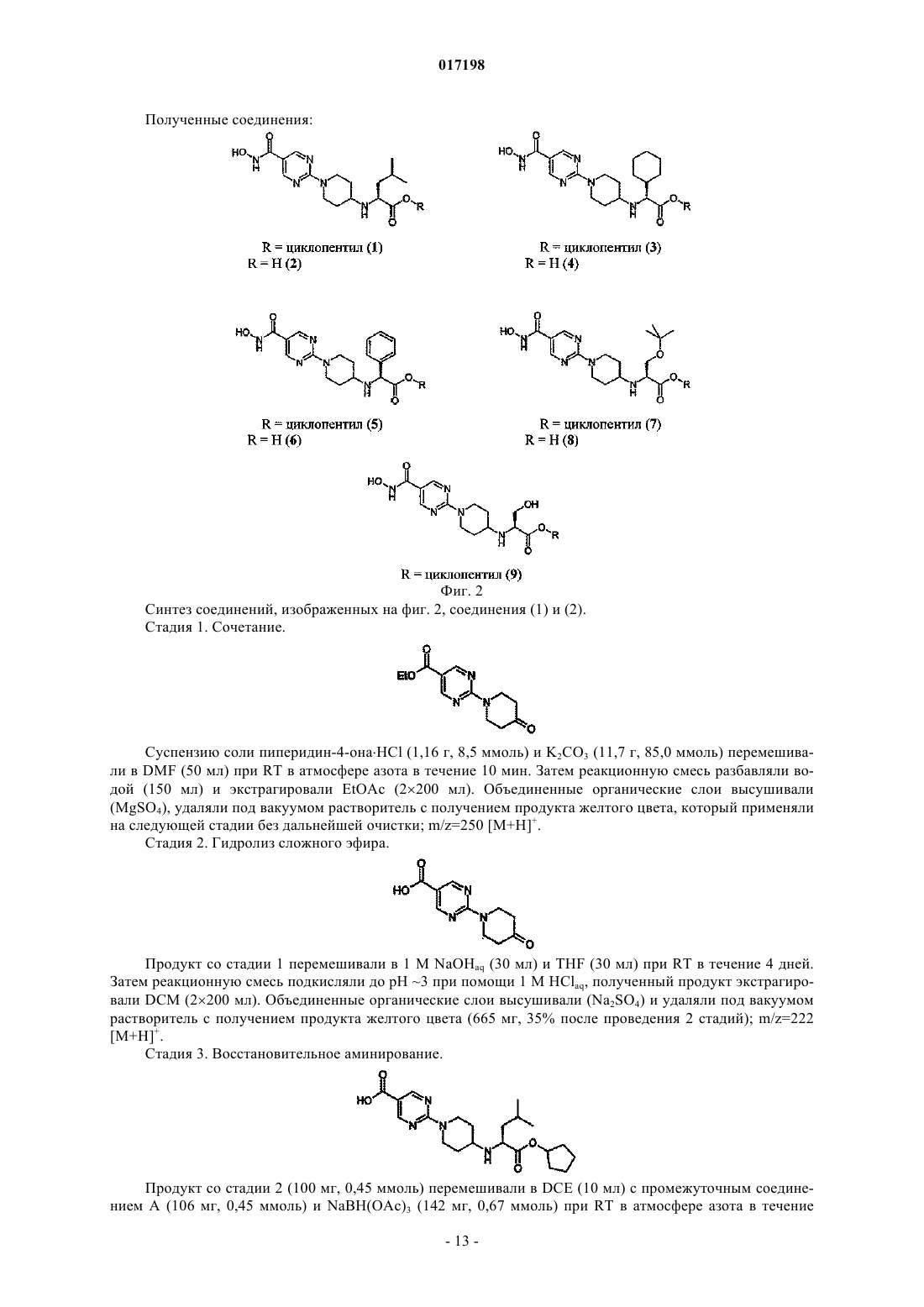
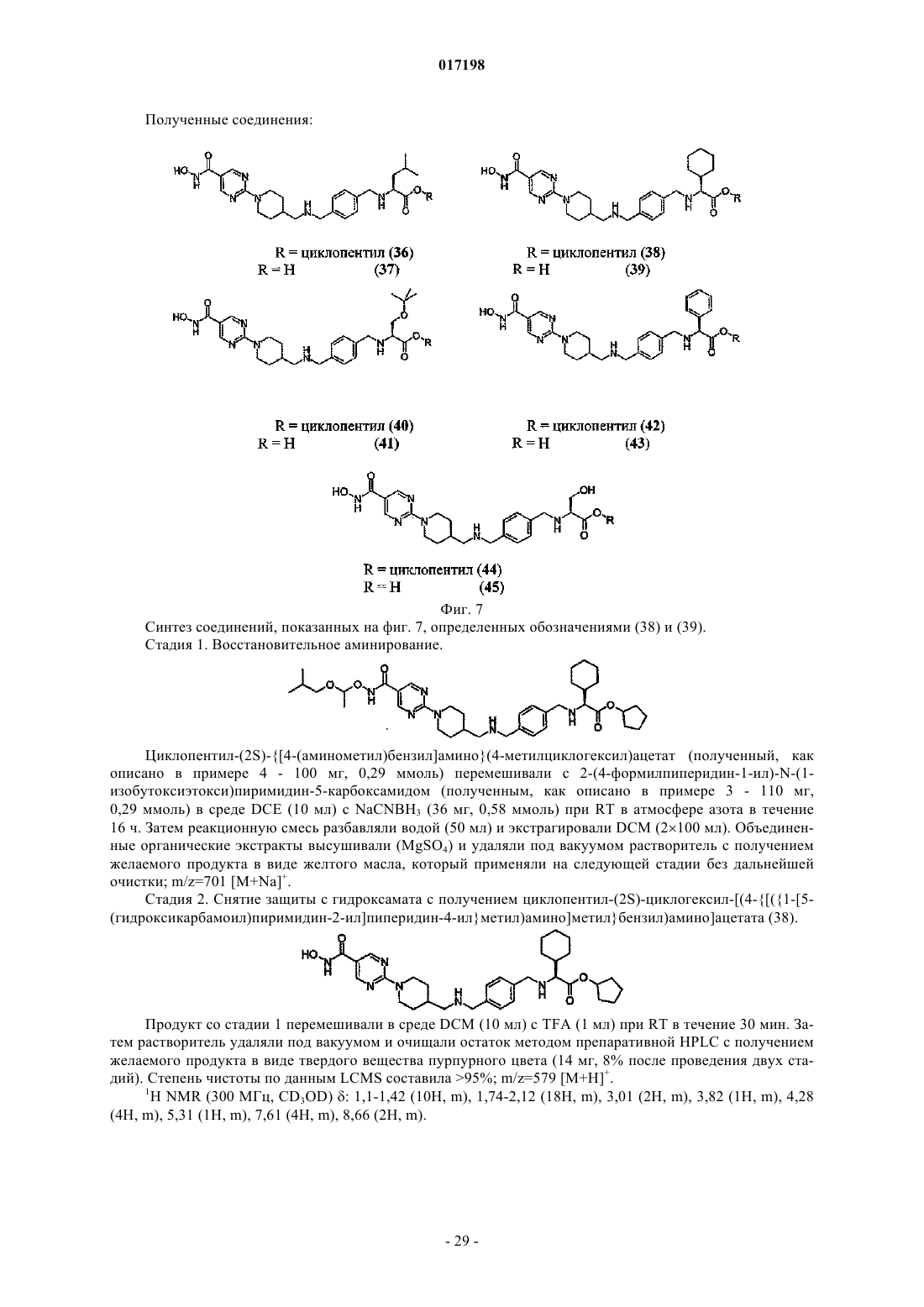
Дональд Элистейр Дэвид Грэм,Дэвидсон Алан Хорнсби, Драммонд Алан Гастингс (GB)W; А обозначает циклогексильное или фенильное кольцо; -[Linker]- обозначает связь, -CH2NH-CH2- или -NH-CH2-; Z1 обозначает радикал формулы R1R2CHNH- или R1R2CHNH-СН 2-,где R1 обозначает карбоксильную группу (-СООН) или сложноэфирную группу, выбранную из группы сложноэфирных групп формулы -(C=O)OR9, где R9 представляет собой R7R8CH-, гдеR7 и R8 вместе с атомом углерода, к которому они присоединены, образуют моноциклическое карбоциклическое кольцо из 3-7 атомов в кольце; R2 обозначает боковую цепь природной или неприродной альфа-аминокислоты, причем боковая цепь неприродной альфа-аминокислоты выбрана из фенильной, циклогексилметильной, циклогексильной, пиридин-3-илметильной, третбутоксиметильной, трет-бутильной, 1-бензилтио-1-метилэтильной, 1-метилтио-1-метилэтильной,1-меркапто-1-метилэтильной и фенилэтильной. Соединения изобретения представляют собой ингибиторы гистон-деацетилазы и пригодны для лечения болезней, связанных с пролиферацией клеток, включая рак. Моффат Дэвид Фестус Чарльз, Дэй Франческа Энн, Пэтел Санджей Ратилал, Белфилд Эндрю Джеймс,017198 Область техники, к которой относится изобретение Данное изобретение относится к соединениям, которые ингибируют членов семейства гистондеацетилаз, являющихся ферментами, и к их применению для лечения болезней, связанных с пролиферацией клеток, включая различные виды рака, полиглютаминовые болезни, например болезнь Хантингтона, нейродегенеративные болезни, например болезнь Альцгеймера, аутоиммунные болезни, например ревматоидный артрит и отторжение трансплантатов различных органов, диабет, гематологические заболевания, воспалительные болезни, сердечно-сосудистые болезни, атеросклероз и остаточные воспалительные явления, вызванные инфекцией. Сведения о предшествующем уровне техники В эукариотных клетках ДНК "набита" гистонами с образованием хроматина. Около 150 пар оснований ДНК дважды окружают октамер гистонов (каждые два гистона 2 А, 2 В, 3 и 4) с образованием нуклеосомы, основной единицы хроматина. Упорядоченная структура хроматина нуждается в модификации для осуществления транскрипции ассоциированных генов. Регуляция транскрипции представляет собой ключевой момент дифференциации пролиферации и апоптоза и, следовательно, жестко контролируется. Контролирование изменений в структуре хроматина (и, следовательно, транскрипции) опосредовано ковалентной модификацией гистонов, наиболее выраженной в N-концевых участках. Ковалентная модификация (например, метилирование, ацетилирование, фосфорилирование и убиквинирование) боковых цепей аминокислот опосредована ферментивно (обзор ковалентной модификации гистонов и их роли в регуляции транскрипции можно найти в Berger S.L. 2001, Oncogene 20, 3007-3013; см. Grunstein M. 1997,Nature 389, 349-352; Wolffe А.Р. 1996, Science 272, 371-372; и Wande P.A. et al., 1997, Trends Biochem. Sci. 22, 128-132, там описан обзор методов ацетилирования гистонов и транскрипции). Ацетилирование гистонов связано с такими участками хроматина, которые являются транскрипционно активными, в то время как нуклеосомы с низким уровнем ацетилирования обычно не активны в процессе транскрипции. Статус ацетилирования гистонов контролируется двумя классами ферментов с противоположными активностями: гистон-ацетилтрансферазами (HATs) и гистон-деацетилазами(HDACs). Полагают, что в трансформированных клетках неподходящая экспрессия HDACs приводит к сайленсингу опухоль-супрессорных генов (обзор возможной роли HDACs в генезисе опухолей см. GrayS.G. и Teh В.Т. 2001, Curr. Mol. Med, 1, 401-129). Ингибиторы ферментов HDAC описаны в литературе,показано, что они индуцируют транскрипционную реактивацию некоторых генов, что приводит к ингибированию пролиферации раковых клеток, индукции апоптоза и ингибированию роста опухолей у животных (обзор см. Kelly W.K. et al., 2002, Expert Opin. Investig Drugs, 11, 1695-1713). Эти факторы позволяют предположить, что ингибиторы HDAC имеют терапевтический потенциал при лечении пролиферативных заболеваний, таких как рак (Kramer O.H. et al., 2001, Trends Endocrinol. 12, 294-300, Vigushin D.M.and Coombes R.C. 2002, Anticancer Drugs 13, 1-13). Кроме того, в других работах утверждается, что аберрантная активность HDAC или ацетилирование гистонов вовлечены в развитие следующих заболеваний и расстройств: полиглютаминовых болезней, например болезни Хантингтона (Hughes R.Е. 2002, Curr Biol. 12, R141-R143; McCampbell A. et al.,2001, Proc. Soc. Natl. Acad. Sci. 98, 15179-15184; Hockly E. et al. 2003 Proc. Soc. Natl. Acad. Sci. 100, 20412046), других нейродегенеративных болезней, например болезни Альцгеймера (Hempen В. and Brion J.P. 1996, J. Neuropathol. Exp. Neurol. 55, 964-972), аутоиммунных болезней и отторжения трансплантатов различных органов (Skov S. et al., 2003, Blood, 101 1430-1438; Mishra N. et al., 2003, J. Clin. Invest 111,539-552), диабета (Mosley A.L. And Ozcan S. 2003, J. Biol. Chem. 278, 19660-19666) и осложнений при диабете, инфекций (включая протозоальные инфекции (Darkin-Rattray S.J. et al., 1996, Proc. Soc. Natl.Acad. Sci. 93, 13143-13147 и гематологических нарушений, включая талассемию (Witt O. et al., 2003,Blood, 101, 2001-2007). Наблюдения, описанные в этих источниках, позволяют предположить, что ингибирование HDAC будет приводить к терапевтическому эффекту при лечении этих и других родственных болезней. Были предложены многие виды ингибиторов HDAC, несколько таких соединений в настоящее время проходят клинические испытания при лечении рака. Например, такие соединения описаны в следующих патентных источниках: US 5369108 и WO 01/18171; WO 03/076400; US 4254220; WO 03/076401;WO 05/013958; WO 05/018578; WO 05/028447; WO 05/019174; WO 05/02690; WO 05/004861. Многие из ингибиторов HDAC, известных из уровня техники, имеют структуру шаблона, которая может быть представлена формулой (А): где кольцо А представляет собой карбоциклическую или гетероциклическую кольцевую систему с возможными заместителями R и [Linker] является линкерным радикалом различного типа. Гидроксамат-1 017198 ная группа, действующая как группа, связывающая металл, взаимодействующая с ионам металла в активном сайте фермента HDAC, который расположен в основании "кармана", содержащего ион металла,причем радикал [Linker] простирается вглубь этого кармана, соединяя А с группой гидроксамовой кислоты, связывающей металл. В уровне техники и в некоторых случаях в данном изобретении кольцо или кольцевая система А иногда называется "главной группой" ингибитора. В международной заявке PCT/GB 2006/001779 описан и заявлен новый класс ингибиторов HDAC,структура которых соответствует обобщенной формуле шаблона (А). Этот новый класс состоит из соединений формулы (В) и их солей, N-окисей, гидратов и сольватов: где связь, отмеченная , связана с кольцом, содержащим Q, V и W через [Linker1], а связь, отмеченная , связана с А через [Linker2], А обозначает возможно замещенную моно-, би- или трициклическую карбоциклическую или гетероциклическую кольцевую систему и -[Linker1]- и -[Linker2]- независимо обозначают связь или двухвалентный линкерный радикал. Сущность изобретения Данное изобретение основано на установлении того факта, что введение эфирной группы аминокислоты в молекулярный шаблон (В) ингибитора HDAC и некоторые структурно подобные шаблоны облегчает проникновение агента через клеточную мембрану и при этом приводит к тому, что внутриклеточная активность карбоксилэстеразы вызывает гидролиз сложного эфира с высвобождением родительской кислоты. Будучи заряженной, кислота не легко транспортируется из клетки, где она аккумулируется с увеличением внутриклеточной концентрации активного ингибитора HDAC. Это приводит к увеличению активности и продолжительности действия. Следовательно, изобретение предусматривает класс соединений, которые представляют собой конъюгаты альфа-аминокислот структур (В) и некоторые родственные структуры. Сложный эфир альфа-аминокислоты является субстратом для внутриклеточной карбоксилэстеразы (называемой здесь также "эстеразным мотивом"). Такие конъюгаты и соответствующие деэтерифицированные родительские кислоты могут применяться при лечении таких болезней, как рак, которые выигрывают от внутриклеточного ингибирования HDAC. Сведения, подтверждающие возможность осуществления изобретения Согласно данному изобретению предусмотрено соединение формулы (I) или его соль где связь, отмеченная , связана с кольцом, содержащим Q, V и W; А обозначает циклогексильное или фенильное кольцо;Z1 обозначает радикал формулы R1R2CHNH- или R1R2CHNH-CH2-, где R1 обозначает карбоксильную группу (-СООН) или сложноэфирную группу, выбранную из группы сложноэфирных групп формулы -(C=O)OR9, где R9 представляет собой R7R8CH-, где R7 и R8 вместе с атомом углерода, к которому они присоединены, образуют моноциклическое карбоциклическое кольцо из 3-7 атомов в кольце; иR2 обозначает боковую цепь природной или неприродной альфа-аминокислоты, причем боковая-2 017198 цепь неприродной альфа-аминокислоты выбрана из фенильной, циклогексилметильной, циклогексильной, пиридин-3-илметильной, трет-бутоксиметильной, трет-бутильной, 1-бензилтио-1-метилэтильной, 1 метилтио-1-метилэтильной, 1-меркапто-1-метилэтильной и фенилэтильной. Хотя вышеприведенное определение включает соединения с высоким молекулярным весом, предпочтительно в соответствии с общими принципами медицинской химии, чтобы соединения, которых касается данное изобретение, имели молекулярный вес не более 600. Согласно другому аспекту данное изобретение предусматривает применение соединения формулы(I), описанного выше, или его соли для получения композиции для ингибирования активности гистондеацетилазы. Соединения, которых касается данное изобретение, могут быть применены для ингибирования активности гистон-деацетилазы ex vivo или in vivo. Согласно одному аспекту данного изобретения соединения по изобретению могут быть применены для получения композиции для лечения болезни, связанной с пролиферацией клеток, например пролиферацией раковых клеток, и аутоиммунных болезней. Согласно другому аспекту настоящее изобретение предусматривает способ лечения указанных выше видов болезней, который включает введение субъекту, страдающему от такой болезни, эффективного количества соединения формулы (I), определение которого дано выше. Терминология. Используемый в данном изобретении термин "Ca-Cb-алкил", где а и b обозначают целые числа, относится к линейному или разветвленному алкильному радикалу, содержащему от "а" до "b" атомов углерода. Таким образом, например, когда a равен 1 и b равен 6, этот термин включает метил, этил, н-пропил,изопропил, н-бутил, изобутил, втор, бутил, трет, бутил, н-пентил и н-гексил. Применяемый термин "двухвалентный Ca-Cb-алкиленовый радикал", где а и b обозначают целые числа, относится к насыщенной углеводородной цепи, содержащей от а до b атомов углерода и две ненасыщенные валентности. Термин "Ca-Cb-алкенил", где а и b обозначают целые числа, относится к линейной или разветвленной цепи алкенильного фрагмента, содержащего от а до b атомов углерода, по меньшей мере одну двойную связь E- или Z-стереохимии, где это возможно. Этот термин включает, например, винил, аллил, 1- и 2-бутенил и 2-метил-2-пропенил. Применяемый термин "двухвалентный Ca-Cb-алкениленовый радикал" обозначает углеводородную цепь, содержащую от а до b атомов углерода, по меньшей мере одну двойную связь и две ненасыщенные валентности. Используемый в данном изобретении термин "Ca-Cb-алкинил", где а и b обозначают целые числа,относится к линейным или разветвленным углеводородным группам, содержащим от а до b атомов углерода и, кроме этого, одну тройную связь. Этот термин включает, например, этинил 1-пропинил, 1- и 2 бутинил, 2-метинил-2-пропинил, 2-пентинил, 3-пентинил, 4-пентинил, 2-гексинил, 3-гексинил, 4 гексинил и 5-гексинил. Термин "двухвалентный Ca-Cb-алкиниленовый радикал", где а и b обозначают целые числа, относится к двухвалентной углеводородной цепи, содержащей от а до b атомов углерода,по меньшей мере одну тройную связь. Применяемый термин "карбоциклический" относится к моно-, би- или трициклическому радикалу,содержащему до 16 атомов в кольце, все из которых являются атомами углерода, этот термин включает арил и циклогексил. Термин "циклоалкил" относится к моноциклическому насыщенному карбоциклическому радикалу,содержащему 3-8 атомов углерода, и включает, например, циклопропил, циклобутил, циклопентил, циклогексил, циклогептил и циклооктил. Не имеющий узаконенного определения термин "арил" относится к моно-, би- или трициклическому карбоциклическому ароматическому радикалу и включает радикалы, содержащие два моноциклических карбоциклических ароматических кольца, которые непосредственно связаны ковалентной связью. Примером таких радикалов являются фенил, бифенил и нафтил. Не имеющий узаконенного определения термин "гетероарил" относится к моно-, би- или трициклическому ароматическому радикалу, содержащему один или более гетероатомов, выбранных из S, N и O,и включает радикалы, содержащие два таких моноциклических кольца или одно такое моноциклическое кольцо и одно моноциклическое арильное кольцо, которые непосредственно соединены ковалентной связью. Примерами таких радикалов являются тиенил, бензтиенил, фурил, бензофурил, пирролил, имидазолил, бензимидазолил, тиазолил, изотиазолил, бензизотиазолил, пиразолил, оксазолили, бензоксазолил, изоксазолил, бензизоксазолил, изотиазолил, триазолил, бензтриазолил, тиадиазолил, оксадиазолил,пиридинил, пиридазинил, пиримидинил, пиразинил, триазинил, индолил и индазолил. Применяемый в данном изобретении, не имеющий общепринятого определения термин "гетероциклил" или "гетероциклический" включает "гетероарил", определение которого дано выше, и в случае его неароматического значения относится к моно-, би- или трициклическому неароматическому радикалу, содержащему один или более гетероатомов, выбранных из S, N и O, и к группам, состоящим из моноциклического неароматического радикала, содержащего один или более таких гетероатомов, который-3 017198 ковалентно соединен с другим таким радикалом или с моноциклическим карбоциклическим радикалом. Примерами таких радикалов являются пирролил, фуранил, тиенил, пиперидинил, имидазолил, оксазолил,изоксазолил, тиазолил, тиадиазолил, пиразолил, пиридинил, пирролидинил, пиримидинил, морфолинил,пиперазинил, индолил, бензфуранил, пиранил, изоксазолил, бензимидазолил, метилендиоксифенил, этилендиоксифенил, малеимидо- и сукцинимидная группы. Если иное не оговаривается в контексте данного описания, термин "замещенный" в применении к любой группе означает радикал, замещенный совместимыми заместителями в количестве до четырех,каждый из которых, независимо, может представлять собой, например, C1-C6-алкил, C1-C6-алкокси, гидрокси, гидрокси-C1-C6-алкил, меркапто, меркапто-C1-C6-алкил, C1-C6-алкилтио, фенил, галоид (включая фтор, бром и хлор), трифторметил, трифторметилокси, нитро, нитрил (-CN), оксо, -СООН, -COORA,-CORA, -SO2RA, -CONH2, -SO2NH2, -CONHRA, -SO2NHRA, -CONRARB, -SO2NRARB, -NH2, -NHRA, -NRARB-OCONH2, -OCONHRA, -OCONRARB, -NHCORA, -NHCOORA, -NRBCOORA, -NHSO2ORA, -NRBSO2OH,-NRBSO2ORA, -NHCONH2, -NRACONH2, -NHCONHRB, -NRACONHRB, -NHCONRARB или -NRACONRARB,где RA и RB независимо обозначают C1-C6-алкил, C3-C6-циклоалкил, фенил или моноциклический гетероарил, содержащий 5 или 6 атомов в кольце, или RA и RB, будучи присоединенными к одному и тому же атому азота, образуют циклическую аминогруппу (например, морфолинил, пиперидинил, пиперазинил,тетрагидропирролил). "Возможный заместитель" может быть одной из указанных выше замещающих групп. Используемый термин "заместитель у атома азота" означает заместитель у атома азота, который выбран из числа следующих: амино-C1-6 алкил, например аминоэтил, C1-3 алкиламино-С 1-6 алкил, C1-3 диалкиламино-C1-6 алкил, гидрокси-C1-6 алкил, например гидроксиэтил, C1-3 алкокси-C1-6 алкил, например метоксиэтил, меркаптоC1-3 алкил, C1-3 алкилмеркапто-C1-6 алкил, карбоксамидо-C1-6 алкил, например CH2CONH2, аминосульфонил-C1-6 алкил, например CH2SO2NH2, C1-3 алкиламиносульфонил-C1-6 алкил, например CH2SO2NMe,C1-3 диалкиламиносульфонил-C1-6 алкил, например CH2SO2NMe2, C1-6 алканоил, C1-6 алкилсульфонил, аминосульфонил (-SO2NH2), C1-6 алкиламиносульфонил, например -SO2NHMe, С 1-6 диалкиламиносульфонил,например -SO2NMe2, возможно замещенный фениламиносульфонил, карбоксамидо (-CONH2),C1-6 алкиламинокарбонил, C1-6 диалкиламинокарбонил, морфолинил-C1-6 алкил, имидазолил-C1-6 алкил,триазолил-C1-6 алкил или моноциклический гетероциклоалкил-C1-6 алкил, возможно замещенный в имидазолильном, триазолильном или гетероциклильном кольце, например пиперидинил-C1-6 алкил, пиперазинил-C1-6 алкил или 4-(C1-6 алкил)пиперазинил-C1-6 алкил. Применяемый термин "соль" включает соли присоединения к основанию, соли присоединения к кислоте и четвертичные соли. Соединения согласно изобретению, которые являются кислыми, могут образовывать соли, включая фармацевтически приемлемые соли, с основаниями, такими как гидроокиси щелочных металлов, например гидроокиси натрия и калия; гидроокиси щелочно-земельных металлов, например гидроокиси кальция, бария и магния; с органическими основаниями, например N-метил-Dглютамином, холин-трис-(гидроксиметил)аминометаном, L-аргинином, L-лизином, N-этилпиперидином,дибензиламином и т.п. Те соединения (I), которые являются основаниями, могут образовывать соли,включая фармацевтически приемлемые соли, с неорганическими кислотами, например с гидрогалоидными кислотами, такими как соляная или бромисто-водородная кислоты, серная кислота, азотная кислота или фосфорная кислота и т.п., и с органическими кислотами, например с уксусной, винной, янтарной,фумаровой,малеиновой,малевой,салициловой,лимонной,метансульфокислотой,птолуолсульфокислотой, бензойной, бензолсульфокислотой, глутаминовой, молочной и миндальной кислотами и т.п. Соединения по изобретению, которые содержат один или более имеющихся или потенциальных хиральных центров благодаря наличию асимметричных атомов углерода, могут существовать в виде ряда энантиомеров или диастереомеров с R- или S-стереохимией у каждого хирального центра. Изобретение охватывает все такие энантиомеры или диастереомеры и их смеси. В соединениях по изобретению, в любой совместимой комбинации, с учетом того, что соединения предпочтительно имеют молекулярный вес менее 600. Гидроксаматная группа -C(=O)NHOH. В соединениях по изобретению гидроксаматная группа действует как группа, связывающая металл,взаимодействуя с ионом металла в активном сайте фермента HDAC, который расположен у основания кармана в структуре фермента. Кольцо, содержащее Q, V u W. Каждая группа из Q, V и W может представлять собой -C= или по меньшей мере одна из Q, V и W может быть -N=, или Q может быть -С= и V, и W, каждая, могут быть -N=. Предпочтительным является случай, когда Q обозначает -С=, V и W, каждая, являются -N=, и HONHC(=O)- присоединен в 5 положении полученного пиримидин-2-ильного радикала.-4 017198 Кольцо А. Кольцевые радикалы могут представлять собой циклогексильное или фенильное кольцо. Радикал [Linker].-[Linker]- служит для соединения двухвалентного радикала В с кольцом А, если оно содержится. Таким образом, он может быть выбран из следующих примеров: связь, -CH2-NH-CH2- или -NH-CH2-. Группа Z1. Группа Z1 обозначает радикал формулы R1R2CHNH- или R1R2CHNH-CH2-, где R1 обозначает карбоксильную группу (-СООН) или сложноэфирную группу, выбранную из группы сложноэфирных групп формулы -(C=O)OR9, где R9 представляет собой R7R8CH-, где R7 и R8 вместе с атомом углерода, к которому они присоединены, образуют моноциклическое карбоциклическое кольцо из 3-7 атомов в кольце; иR2 обозначает боковую цепь природной или неприродной альфа-аминокислоты, причем боковая цепь неприродной альфа-аминокислоты выбрана из фенильной, циклогексилметильной, циклогексильной,пиридин-3-илметильной, трет-бутоксиметильной, трет-бутильной, 1-бензилтио-1-метилэтильной, 1 метилтио-1-метилэтильной, 1-меркапто-1-метилэтильной и фенилэтильной. Эфирная группа R1 может быть такой, которая в соединении по изобретению способна гидролизоваться под действием одного или нескольких внутриклеточных ферментов карбоксиэстераз с получением карбоксильной группы. Внутриклеточные ферменты карбоксиэстеразы, способные гидролизовать сложноэфирную группу соединения по изобретению до соответствующей кислоты, включают три известных изотипа ферментов человека:hCE-1, hCE-2 и hCE-3. Хотя они считаются основными ферментами, другие ферменты, такие как бифенилгидролаза (ВРН), также могут играть роль в гидролизе сложного эфира. В общем, если карбоксиэстераза гидролизует эфирную группу свободной аминокислоты до родительской кислоты, она вследствие Nкарбонильной зависимости hCE-2 и hCE-3, описанной выше, также гидролизует сложноэфирный мотив,ковалентно сопряженный с ингибитором HDAC. Следовательно, данные клеточного анализа расщепившейся клетки и/или анализа изолированной карбоксиэстеразы, которые описаны в данном изобретении,представляют собой прямой, быстрый и простой первый метод сканирования для эфиров, которые обладают требуемым профилем гидролиза. Эфирные мотивы, выбранные таким образом, можно повторно подвергать анализу тем же методом анализа карбоксиэстеразы в случае сопряжения с ингибитором по выбранному методу сопряжения для того, чтобы подтвердить, что соединение все еще является карбоксиэстеразным субстратом. Известно, что макрофаги играют ключевую роль в развитии воспалительных заболеваний вследствие высвобождения цитокинов, в частности TNF и IL-1 (van Roon et al., Arthritis and Rheumatism, 2003,1229-1238). В случае ревматоидного артрита они вносят основной вклад в сохранение воспаления суставов и разрушения суставов. Макрофаги вовлечены также в рост опухолей и их развитие (Naldini andCarraro, Curr. Drug Targets Inflamm. Allergy, 2005, 3-8). Следовательно, агенты, которые селективно нацелены на пролиферацию клеток макрофага, могут иметь значение при лечении рака и аутоиммунных болезней. Виды конкретных клеток, как ожидается, приведут к сниженным побочным эффектам. Изобретатели обнаружили способ таргетирования ингибиторов на макрофаги, который основан на наблюдении,что путь, которым эстеразный мотив связан с ингибитором, решает, гидролизуется ли он и, следовательно, аккумулируется ли он в различных видах клеток или нет. Конкретно, было установлено, что макрофаги содержат человеческую карбоксилазу hCE-1, в то время как другие виды клеток ее не содержат. В общей формуле (I), когда азот эстеразного мотива R1CH(R2)NH- не связан непосредственно с карбонилом(-С(=О)-), эфир будет гидролизоваться только hCE-1 и поэтому ингибиторы будут накапливаться только в макрофагах. В случае соединений по изобретению, которые должны вводиться системно, предпочтительными являются эфиры с небольшой скоростью расщепления карбоксилэстеразы, так как они менее восприимчивы к досистемному метаболизму. Их способность достигать целевую целостность ткани поэтому увеличивается, и эфир может быть превращен внутри клеток целевой ткани в кислоту. Однако в случае локального введения, когда эфир или непосредственно наносится на целевую ткань, или направляется туда,например, путем ингаляции, часто желательно, чтобы эфир характеризовался быстрым расщеплением эстеразы для минимизации системного действия и последующих нежелательных побочных эффектов. В соединениях по изобретению, если атом углерода, смежный с альфа-атомом углерода эфира альфааминокислоты, является монозамещенным, т.е. R2 обозначает CH2Rz (Rz является монозаместителем),эфиры имеют тенденцию к более быстрому расщеплению, чем в случае, когда этот атом углерода является ди- или тризамещенным, как в случае, когда R2 обозначает, например, фенил или циклогексил. Следует также отметить, что преимущества от наличия эфирного мотива аминокислоты, описанные выше (легкое попадание в клетку, гидролиз карбоксилэстеразы внутри клетки и аккумулирование активного продукта гидролиза, карбоновой кислоты), лучше реализуются, когда связь между эфирным мотивом аминокислоты и кольцами В и/или А не является субстратом для активности пептидазы внутри клетки, что может привести к высвобождению аминокислоты из молекулы. Конечно, стабильность к внутриклеточным пептидазам легко определяется путем инкубирования соединения с компонентами разрушенных клеток и анализа такого расщепления. Для соединений по изобретению, которые должны вводиться системно, предпочтительными явля-5 017198 ются сложные эфиры с небольшой скоростью расщепления эстеразой, так как они менее восприимчивы к досистемному метаболизму. Их способность достигать целевую ткань возрастает и эфир может превращаться внутри клеток целевой ткани в кислоту. Однако в случае локального применения, когда эфир или непосредственно наносится на целевую ткань, или направляется к ней, например при ингаляции, часто бывает желательно, чтобы эфир характеризовался высокой скоростью расщепления эстеразы для минимизации системной выдержки и последующих нежелательных побочных эффектов. Как указано выше, соединения, которых касается данное изобретение, представляют собой ингибиторы HDAC и, следовательно, могут найти применение при лечении болезней, связанных с пролиферацией клеток, таких как рак, у людей и других млекопитающих. Следует отметить, что величины конкретной дозы для любого пациента будут зависеть от различных факторов, включая активность конкретного применяемого соединения, возраст, вес, общее состояние здоровья, пол, питание, время введения, метод введения, скорость выделения, комбинацию лекарств и степень серьезности конкретного заболевания, подвергающегося лечению. Оптимальная величина дозы и частота приема определяются в ходе клинических испытаний. Соединения, которых касается изобретение, могут быть приготовлены для введения любым способом в соответствии с их фармакокинетическими свойствами. Вводимые перорально композиции могут быть в виде таблеток, капсул, порошков, гранул, леденцов, жидких или гелеобразных препаратов, таких как оральные, топические или стерильные парентеральные растворы или суспензии. Таблетки и капсулы для орального введения могут быть в виде стандартных доз и могут содержать обычные эксципиенты, такие как связующие, например сироп, смола акации, желатин, сорбит, трагакант или поливинилпирролидон; наполнители, например лактоза, сахар, маисовый крахмал, фосфат кальция,сорбит или глицин; смазочный агент для получения таблеток, например стеарат магния, тальк; полиэтиленгликоль или двуокись кремния; дезинтегранты, например картофельный крахмал или приемлемые смачивающие агенты, такие как лаурилсульфат натрия. Таблетки могут содержать покрытие, наносимое методами, хорошо известными в обычной фармацевтической практике. Жидкие оральные препараты могут быть, например, в виде водных или масляных суспензий, растворов, эмульсий, сиропов или эликсиров или могут быть представлены в виде сухого продукта для восстановления водой или другим подходящим носителем перед применением. Такие жидкие препараты могут содержать обычные добавки,такие как суспендирующие агенты, например сорбит, сироп, метилцеллюлоза, сироп глюкозы, желатин,гидрированные съедобные жиры; эмульгирующие агенты, например лецитин, моноолеат сорбитана или смола акации; неводные носители (которые могут включать съедобные масла), например миндальное масло, фракционированное кокосовое масло, маслянистые эфиры, такие как глицерин, пропиленгликоль или этиловый спирт; консерванты, например метил- или пропил-п-гидроксибензоат или сорбиновая кислота, и, если желательно, ароматизирующие или красящие агенты. Для топического применения путем ингаляции можно ввести лекарство в аэрозоль для доставки,например, при помощи струйных распылителей, работающих под давлением, или ультразвуковых распылителей либо предпочтительно при помощи дозированных ингаляторов с пропеллентами или путем введения не содержащих пропелленты микронизированных порошков, например капсул или других систем для доставки "сухих порошков". В таких составах для ингаляции могут содержаться эксципиенты,такие как, например, пропелленты (например, Frigen в случае дозируемых аэрозолей), поверхностноактивные вещества, эмульгаторы, стабилизаторы, консерванты, ароматизаторы и наполнители (например, лактоза в случае порошковых ингаляторов). Для ингаляции имеется большое количество устройств,при помощи которых можно получить аэрозоли с оптимальным размером частиц и ввести их с применением методов ингаляции, подходящих для пациента. Помимо применения адаптеров (спейсеров, экспандеров) и грушеобразных контейнеров (например, Nebulator, Volumatic и автоматических устройств,образующих спрей (Autohaler для дозируемых аэрозолей, в частности, в случае порошковых ингаляторов, доступен ряд технических растворов (например, Diskhaler, Rotadisk, Turboholer или ингаляторы, описанные, например, в заявке EP0505321). При топическом нанесении на кожу лекарство может быть в составе крема, лосьона или мази. Составы кремов или мазей, которые можно применять для введения лекарства, хорошо известны из уровня техники, например они описаны в обычных учебниках по фармацевтике, таких как British Pharmacopeia. При топическом введении в глаз лекарство может быть введено в состав раствора или суспензии в подходящем стерильном водном или неводном носителе. В состав могут быть также включены добавки,например буферы, такие как метабисульфит натрия или динатрийэдетат; консерванты, включая бактерицидные и фунгицидные агенты, такие как фенилацетат рутин или нитратацетат ртути, бензалконийхлорид или хлоргексидин, и загущающие агенты, такие как гипромеллоза. Активный ингредиент может быть также введен парентерально в стерильной среде. В зависимости от носителя и применяемой концентрации лекарство может быть или суспендировано, или растворено в носителе. В носителе предпочтительно также растворять адъюванты, такие как локальный анестетик,консервант и буферные агенты.-6 017198 Синтез. Существуют многие подходы к синтезу соединений формулы (I), которых касается данное изобретение, но все они основаны на известных химических принципах, известных специалисту в области органической химии. Так, соединения формулы (I) могут быть синтезированы в соответствии со способами, описанными в общепринятой литературе и хорошо известными специалисту. Обычные литературные источники представляют собой "Advanced organic chemistry", 4th Edition (Wiley), J. Mach, "Comprehensive(Pergamon), A.R. Katrizky, обзорные статьи, такие как содержащиеся в "Synthesis", "Acc. Chem. Res.","Chem. Rev.", или первичные литературные источники, обнаруживаемые в результате поиска online или во вторичных источниках, таких как "Chemical Abstracts " или "Beilstein". Способы синтеза, используемые при получении соединений в примерах, описанных ниже, могут быть адаптированы для получения аналогичных соединений. Сокращения.RT=комнатная температура; ч=час(ы). Следующие ниже примеры иллюстрируют получение конкретных соединений по изобретению, а также ингибирующие свойства в отношении HDAC.-7 017198 Получение эфиров аминокислоты (промежуточные соединения А-D). Метод I. Используют для получения промежуточного соединения А и промежуточного соединения В: Метод II. Используют для получения промежуточного соединения С: Метод III. Используют для получения промежуточного соединения D: Схема 1 К раствору (S)-2-трет-бутоксикарбониламино-3-циклогексилпропионовой кислоты (5 г, 19,4 ммоль) в DMF (50 мл) при температуре 0C добавляли циклопентанол (8,8 мл, 19,15 ммоль), EDCl (4,09 г, 21,37 ммоль) и, наконец, DMAP (237 мг, 1,94 ммоль). Реакционную смесь нагревали до RT и перемешивали в течение 18 ч. Удаляли DMF под вакуумом с получением прозрачного масла. Его распределяли между водой иEtOAc. Органическую фазу высушивали (MgSO4) и концентрировали под вакуумом. Сырой экстракт очищали методом хроматографии на колонке (25% EtOAc в гептане) с получением желаемого продукта в виде прозрачного масла (14,87 г, 55%). 1 Продукт со стадии 1 (14,87 г, 45,69 ммоль) растворяли в DCM (100 мл) и обрабатывали 4 МHCl/диоксана (22,8 мл, 91,38 ммоль), реакционную смесь перемешивали при RT в течение 24 ч. Сырую смесь концентрировали при пониженном давлении с получением оранжевого масла. Его растирали сEt2O с получением белого остатка. Остаток затем промывали Et2O с получением целевого продукта в виде белого порошка (7,78 г, 65%). 1 Н NMR (300 МГц, d6-DMSO) : 8,45 (3H, br m), 5,22 (1 Н, t), 3,28 (1H, d), 1,95-1,50 (10H, br m), 1,300,90 (9H, br m). Метод II. Стадия 1. Образование эфира с получением (1S)-2-(циклопентилокси)-2-оксо-1-фенилэтанаминий 4-метилбензосульфоната (промежуточное соединение С).(29,84 мл, 331 ммоль) и п-толуолсульфокислоту (6,92 г, 36,4 ммоль). Устанавливали ловушку ДинаСтарка и смесь нагревали до 135C для полного растворения. Через 12 ч смесь охлаждали до RT, что приводило к осаждению белого твердого продукта. Этот продукт отфильтровывали и промывали EtOAc перед сушкой при пониженном давлении с получением целевого продукта в виде порошка белого цвета К раствору (S)-бензилоксикарбониламино-3-трет-бутоксипропионовой кислоты (25 г, 84,65 ммоль) в DMF (250 мл) при температуре 0C добавляли циклопентанол (15,36 мл, 169,3 ммоль), EDCl (17,85 г,93,11 ммоль) и затем DMAP (1,03 г, 8,46 ммоль). Реакционную смесь нагревали до RT и перемешивали в течение 18 ч. Удаляли DMF под вакуумом, получая масло желтого цвета. Его распределяли между водой иEtOAc. Органическую фазу высушивали (MgSO4) и концентрировали под вакуумом. Сырой экстракт очищали методом хроматографии (25% EtOAc в гептане) с получением целевого продукта в виде прозрачного масла. Его использовали на следующей стадии без анализа. Стадия 2. Снятие защиты Cbz с получением O-трет-бутил-L-серината (промежуточное соединение Продукт со стадии 1 растворяли в EtOAc (150 мл) обрабатывали Pd(OH)2 (10 мол.%) и перемешивали в атмосфере водорода в течение 32 ч. После завершения реакции катализатор удаляли путем фильтрования через целит и концентрировали фильтрат под вакуумом, получая целевой продукт в виде прозрач-9 017198 ного масла (15,96 г, 82% в результате двух стадий). 1 Н NMR (300 МГц, d6-DMSO) : 5,17 (1H, t), 3,45 (1H, m), 3,34 (2 Н, q), 1,90-1,50 (9 Н, br m), 1,08 (9 Н,s). Получение O-(1-изобутоксиэтил)гидроксиламина (промежуточное соединение Е). Схема 2 Промежуточное соединение Е было получено по методу, описанному в заявке WO 01/60785. 1 Стадия 1. Восстановление по атому хлора. Этил-4-хлор-2-метилтио-5-пиримидин-карбоксилат (12,5 г, 53,88 ммоль) и порошок Zn (14,1 г,215,52 ммоль) соединяли и добавляли бензол (60 мл) и 3 М NH4Cl (140 мл). Суспензию энергично перемешивали и нагревали до 80C в течение 30 ч. Реакционную смесь фильтровали через целит и промывали EtOAc (200 мл). Фильтрат концентрировали под вакуумом до объема, равного примерно 50 мл, и затем распределяли между H2O (400 мл) и EtOAc (400 мл). Затем водный слой экстрагировали EtOAc (250 мл). Объединенные органические слои высушивали (MgSO4) и концентрировали под вакуумом с получением темного масла. Его очищали методом хроматографии на колонке (чистый гептан и затем 1:1:1 смесь гептан/CH2Cl2/Et2O). Целевой продукт получали в виде бесцветного масла (13 г, 61%); m/z=199 [М+Н]+. 1 Н NMR (300 МГц, d6-DMSO) : 1,30 (3H, t), 2,60 (3H, s), 4,35 (2 Н, q), 9,0 (2H, s). Стадия 2. Окисление сульфида с получением этил-2-(метилсульфонил)пиримидин-5-карбоксилата К перемешиваемому раствору продукта со стадии 1 (13 г, 47,59 ммоль) в сухом THF (250 мл) медленно в течение 30 мин добавляли раствор mCPBA (47,59 г, 275,76 ммоль) в THF (150 мл) в атмосфере азота при температуре 0C. Реакционной смеси давали нагреться до RT и перемешивали в течение 2 ч. Реакционную смесь концентрировали под вакуумом до объема, равного примерно 100 мл, и смесь полученный продукт/бензойная кислота абсорбировали на силикагеле. Очистку проводили методом хроматографии на колонке (вначале чистый гептан, затем смесь 1:5:3 CH2Cl2/гептан/ Et2O и затем 1:1:1 смесьCH2Cl2/гептан/Et2O). Целевое соединение получали в виде белого твердого вещества (10 г, 66%); m/z=231N-бензилмалеимид (50 г, 267,1 ммоль) растворяли в Et2O (600 мл), обрабатывали этилдиазоацетатом (31 мл, 293,8 ммоль) и перемешивали при RT в атмосфере азота в течение 36 ч. Образовавшийся белый осадок выделяли путем фильтрования, промывали ледяным Et2O и высушивали с получением желаемого соединения в виде белого твердого вещества (72 г, 89%); m/z=302 [М+Н]+. 1 Н NMR (300 МГц, d6-DMSO) : 1,20 (3H, t), 4,15 (2 Н, q), 4,55 (2 Н, s), 4,60 (1H, d), 5,00 (1H, d), 7,157,35 (5 Н, m), 9,60 (1H, s). Стадия 2. Конденсация. Продукт со стадии 1 (72 г, 239,2 ммоль) нагревали до 160C, пока он не расплавился, с получением масла желтого цвета. Затем масло нагревали до 200C, начиналось выделение пузырьков. Масло нагревали при 200C в течение 30 мин до прекращения выделения пузырьков. Полученное масло янтарного цвета охлаждали до RT и растирали с ледяным Et2O. Полученный осадок отфильтровывали и промывали снова ледяным Et2O с получением продукта в виде твердого вещества кремового цвета (37,2 г, 57%). 1 Н NMR (300 МГц, d6-DMSO) : 1,20 (3H, t), 2,80 (1H, t), 3,00 (2 Н, d), 4,15 (2 Н, q), 4,35 (2 Н, s), 7,207,40 (5 Н, m). Стадия 3. Восстановление. Продукт со стадии 2 (37,2 г, 136,3 ммоль) растворяли в сухом THF (400 мл). Этот раствор по каплям добавляли при температуре 0C к суспензии LiAlH4 (20,7 г, 545,3 ммоль) в сухом THF (200 мл). Полученную суспензию коричневого цвета нагревали до 60C в атмосфере азота в течение 36 ч. Затем смесь охлаждали до 0C и обрывали реакцию осторожно насыщенным NH4Claq. Образовавшееся твердое вещество серого цвета и дополнительное количество THF добавляли при соответствующем перемешивании. К смеси добавляли твердый Na2SO4 и перемешивали при RT в течение 30 мин. Затем смесь отфильтровывали через целит с получением бледно-желтого раствора. Этот раствор концентрировали под вакуумом с получением желаемого продукта в виде масла оранжевого цвета (14,1 г, 51%); m/z=204 [М+Н]+. 1 Н NMR (300 МГц, d6-DMSO) : 2,25 (2 Н, d), 2,85 (2 Н, d), 3,20 (2 Н, t), 3,55 (2 Н, s), 4,35 (1H, t), 7,207,40 (5 Н, m). Продукт со стадии 3 (7,5 г, 37,0 ммоль) растворяли в сухом MeOH (250 мл) при RT. К раствору добавляли Pd(OH)2 и присоединяли баллон с водородом. Реакционную смесь дважды дегазировали и промывали H2. Реакционную смесь перемешивали в течение 6 ч, промывали свежим водородом из баллона и перемешивали в течение 64 ч. Затем реакционную смесь фильтровали через целит. Растворитель удаляли под вакуумом и остаток высушивали с получением твердого вещества белого цвета (4,1 г, 98%); m/z=114 Суспензию соли пиперидин-4-онаHCl (1,16 г, 8,5 ммоль) и K2CO3 (11,7 г, 85,0 ммоль) перемешивали в DMF (50 мл) при RT в атмосфере азота в течение 10 мин. Затем реакционную смесь разбавляли водой (150 мл) и экстрагировали EtOAc (2200 мл). Объединенные органические слои высушивали(MgSO4), удаляли под вакуумом растворитель с получением продукта желтого цвета, который применяли на следующей стадии без дальнейшей очистки; m/z=250 [М+Н]+. Стадия 2. Гидролиз сложного эфира. Продукт со стадии 1 перемешивали в 1 М NaOHaq (30 мл) и THF (30 мл) при RT в течение 4 дней. Затем реакционную смесь подкисляли до pH 3 при помощи 1 М HClaq, полученный продукт экстрагировали DCM (2200 мл). Объединенные органические слои высушивали (Na2SO4) и удаляли под вакуумом растворитель с получением продукта желтого цвета (665 мг, 35% после проведения 2 стадий); m/z=222 Продукт со стадии 2 (100 мг, 0,45 ммоль) перемешивали в DCE (10 мл) с промежуточным соединением А (106 мг, 0,45 ммоль) и NaBH(OAc)3 (142 мг, 0,67 ммоль) при RT в атмосфере азота в течение- 13017198 3 дней. Затем реакционную смесь разбавляли водой (50 мл) и экстрагировали DCM (2100 мл). Объединенные органические слои высушивали (Na2SO4) и удаляли растворитель под вакуумом с получением продукта в виде твердого вещества желтого цвета, которое использовали на следующей стадии без дальнейшей очистки; m/z=405 [М+Н]+. Стадия 4. Образование защищенного гидроксамата. Продукт со стадии 3 (182 мг, 0,45 ммоль) перемешивали в DMF (10 мл) с EDCl (103 мг, 0,54 ммоль),HOBt (73 мг, 0,54 ммоль), Et3N (314 мкл, 2,25 ммоль) и с промежуточным соединением E (310 мкл, 2,25 ммоль) в течение 16 ч при RT в атмосфере азота. Затем реакционную смесь разбавляли водой (50 мл) и экстрагировали DCM (2100 мл). Объединенные органические слои высушивали (MgSO4) и удаляли под вакуумом растворитель. Остаток очищали путем хроматографии на колонке (0-15% MeOH в DCM) с получением продукта в виде желтого масла (110 мг, 47%); m/z=520 [М+Н]+. Стадия 5. Снятие защиты с получением циклопентил-N-1-[5-(гидроксикарбамоил)пиримидин-2 ил]пиперидин-4-ил-L-лейцина (1). Продукт со стадии 4 (110 мг, 0,21 ммоль) перемешивали в DCM (20 мл) с TFA (0,5 мл) в течение 1 ч при RT. Затем растворитель удаляли под вакуумом и остаток очищали методом препаративной HPLC с получением продукта в виде твердого вещества пурпурного цвета (12 мг, 11%). Чистота по данным Продукт со стадии 4 (182 мг, 0,45 ммоль) перемешивали в THF (10 мл) с KOTMS (115 мг, 0,9 ммоль) в течение 4 дней при RT атмосфере азота. Затем растворитель удаляли под вакуумом и остаток применяли на следующей стадии без дальнейшей очистки; m/z=452 [М+Н]+. Стадия 7. Снятие защиты с гидроксамата с получением N-1-[5-(гидроксикарбамоил)пиримидин-2 ил]пиперидин-4-ил-L-лейцина (2). Продукт со стадии 6 (0,45 ммоль) перемешивали в DCM (10 мл) с TFA (1 мл) в течение 30 мин приRT. Затем растворитель удаляли под вакуумом и остаток очищали методом препаративной HPLC с получением целевого продукта в виде твердого вещества розового цвета (7 мг, 5% после проведения двух стадий). Чистота по данным LCMS составила 95%; m/z=352 [М+Н]+, не растворим в NMR растворителях. Аналоги, показанные на фиг. 2, были получены методом, описанным для соединений (1) и (2). Для каждого аналога приведены данные.(3) Степень чистоты по данным LCMS составила 97%; m/z=440 [М+Н]+. 1 Н NMR (300 МГц, CD3OD) : 1,30-1,61 (9 Н, m), 1,78-1,90 (4 Н, m), 2,23 (2 Н, m), 2,90 (2 Н, m), 5,00(4) Степень чистоты по данным LCMS составила 98%; m/z=378 [М+Н]+, не растворяется в NMR растворителях.(5) Степень чистоты по данным LCMS составила 97%; m/z=446 [М+Н]+. 1 Н NMR (300 МГц, CD3OD) : 0,83-1,82 (24 Н, m), 2,02 (2 Н, m), 2,94 (2 Н, m), 4,86 (1H, m), 5,23 (1H,m), 8,58 (2H, s).(6) Степень чистоты по данным LCMS составила 95%; m/z=372 [М+Н]+. 1(7) Степень чистоты по данным LCMS составила 98%; m/z=450 [М+Н]+. 1 Н NMR (300 МГц, CD3OD) : 1,25 (9 Н, s), 1,58-1,77 (8 Н, br m), 1,96 (2 Н, m), 2,22 (2 Н, m), 3,02 (2 Н,m), 3,55 (1H, m), 3,87 (1H, dd, J=10,8, 2,7 Гц), 3,99 (1 Н, dd, J=10,8, 2,7 Гц), 4,45 (1H, m), 5,03 (2 Н, m), 5,35(8) Степень чистоты по данным LCMS составила 98%; m/z=382 [М+Н]+. 1(9) Степень чистоты по данным LCMS составила 95%; m/z=394 [М+Н]+. 1 Н NMR (300 МГц, CD3OD) : 1,52-1,98 (11H, m), 2,10 (2 Н, m), 2,87 (3H, m), 3,44 (1H, m), 3,95 (2 Н,m), 4,18 (1H, m), 5,25 (1H, m), 8,58 (2 Н, m).K2CO3 (5,96 г, 43,1 ммоль) при RT в атмосфере азота в течение 10 мин. Затем добавляли промежуточное соединение F (2,00 г, 8,62 ммоль) и перемешивали смесь еще в течение 20 мин. Затем реакционную смесь разбавляли водой (100 мл) и экстрагировали EtOAc (2100 мл). Объединенные органические слои высушивали (MgSO4) и удаляли растворитель под вакуумом с получением твердого продукта светло-желтого цвета, который применяли на следующей стадии без дальнейшей очистки (1,8 г, 78%); m/z=264 [М+Н]+. Стадия 2. Окисление спирта. Продукт со стадии 1 (1,80 г, 6,84 ммоль) перемешивали в DCM (50 мл) при 0C и добавляли периодинан Десс-Мартина (3,50 г, 8,22 ммоль). Реакционной смеси давали нагреться до RT и перемешивали в течение 4 ч. Затем реакцию обрывали 1:1 смесью насыщенного NaHCO3(aq) и насыщенного Na2S2O4(aq),полученную смесь перемешивали в течение 20 мин. Затем смесь экстрагировали DCM (2100 мл), объединенные органические экстракты сушили (MgSO4) и удаляли под вакуумом растворитель. Остаток очищали методом хроматографии на колонке (0-10% MeOH в DCM) с получением продукта в виде желтого масла (1,35 г, 72%); m/z=262 [М+Н]+. Стадия 3. Гидролиз эфира. Продукт со стадии 2 (1,35 г, 5,17 ммоль) перемешивали в 1 М NaOH(aq) (20 мл) и THF (20 мл) приRT в течение 3 ч. Затем реакционную смесь подкисляли до pH 3 при помощи 1 М HCl(aq), что вызывало осаждение твердого продукта белого цвета. Этот твердый продукт отфильтровывали, высушивали и сохраняли. Затем фильтрат экстрагировали DCM (2100 мл), объединенные органические слои высушивали (Na2SO4), растворитель удаляли под вакуумом с получением твердого вещества светло-желтого цвета,которое соединяли с предварительно полученным твердым продуктом (900 мг, 82%); m/z=236 [М+Н]+. Стадия 4. Восстановительное аминирование. Продукт со стадии 3 (1,75 мг, 0,75 ммоль) перемешивали в DCE (10 мл) с промежуточным соединением В (196 мг, 0,75 ммоль) и NaBH(OAc)3 (222 мг, 1,05 ммоль) при RT в атмосфере азота в течение 16 ч. Затем реакционную смесь разбавляли водой (50 мл) и экстрагировали Et2O. Объединенные органические слои высушивали (Na2SO4) и удаляли растворитель под вакуумом с получением желаемого продукта в виде желтого масла, который применяли на следующей стадии без дальнейшей очистки (171 мг, 52%); Продукт со стадии 4 (171 мг, 0,39 ммоль) перемешивали в DMF (10 мл) с промежуточным соединением Е (539 мкл, 3,9 ммоль), EDCl (90 мг, 0,47 ммоль), HOBt (63 мг, 0,47 ммоль) и Et3N (543 мкл, 3,9 ммоль) при RT в атмосфере азота в течение 16 ч. Затем реакционную смесь разбавляли водой (50 мл) и экстрагировали DCM (2100 мл). Объединенные органические слои высушивали (MgSO4) и удаляли под вакуумом растворитель. Остаток очищали методом хроматографии на колонке (0-10% MeOH в DCM) с получением целевого продукта в виде бесцветного масла (91 мг, 42%); m/z=558 [М+Н]+. Стадия 6. Снятие защиты с гидроксамата с получением циклопентил-(2S)-циклогексил-[(3-[5(гидроксикарбамоил)пиримидин-2-ил]-3-азабицикло[3.1.0]-гекс-6-илметил)амино]ацетата (14). Продукт со стадии 5 (91 мг, 0,163 ммоль) перемешивали в смеси 1:1 MeOH/DCM (4 мл) с TFA (2 мл) при RT в течение 1 ч. Затем растворитель удаляли под вакуумом и остаток очищали методом препаративной HPLC с получением белого твердого вещества (15 мг, 20%). Степень чистоты по данным LCMS составила 98%; m/z=588 [М+Н]+. 1 Н NMR (300 МГц, CD3OD) : 0,92-1,38 (6 Н, m), 1,73-1,95 (16 Н, m), 3,09 (2 Н, m), 3,621 (2 Н, d,J=11,4 Гц), 3,91 (1H, d, J=3,6 Гц), 4,00 (2 Н, d, J=11,4 Гц), 5,11 (1H, m), 8,67 (2 Н, s). Стадия 7. Гидролиз сложного эфира. Продукт со стадии 5 (161 мг, 0,29 ммоль) перемешивали в среде THF (10 мл) с KOTMS (74 мг, 0,58 ммоль) при RT в атмосфере азота в течение 48 ч. Затем добавляли снова KOTMS (112 мг, 0,87 ммоль) и перемешивали реакционную смесь при температуре 50C в течение 48 ч. Затем под вакуумом удаляли растворитель, и остаток применяли на следующей стадии без дальнейшей очистки; m/z=490 [М+Н]+. Стадия 8. Снятие защиты с гидроксамата с получением (2S)-циклогексил-[(3-[5(гидроксикарбамоил)пиримидин-2-ил]-3-азабицикло[3.1.0]гекс-6-илметил)амино]уксусной кислоты Продукт со стадии 7 (0,29 ммоль) перемешивали в DCM (10 мл) с TFA (1 мл) при RT в течение 30 мин. Затем растворитель удаляли под вакуумом и остаток очищали методом препаративной HPLC с получением целевого продукта в виде твердого вещества белого цвета (13 мг, 12%). Степень чистоты по данным LCMS составила 99%; m/z=390 [М+Н]+. 1 Н NMR (300 МГц, CD3OD) : 0,96 (1H, m), 1,10-1,47 (6 Н, m), 1,72-1,87 (6 Н, m), 1,99 (1H, m), 2,99 (1 Н,m), 3,18 (1H, m), 3,63 (2 Н, dd, J=11,7, 3,3 Гц), 3,85 (1H, d, J=3,3 Гц), 3,99 (2 Н, d, J=11,7 Гц), 8,67 (2H, s).- 18017198 Аналоги, показанные на фиг. 3, были получены методом, описанным для соединений (14) и (15). Ниже приведены данные для каждого аналога.(10) Степень чистоты по данным LCMS составила 95%; m/z=452 [М+Н]+. 1 Н NMR (300 МГц, CD3OD) : 1,41-1,64 (6 Н, m), 1,72-1,93 (5 Н, m), 2,88 (2 Н, m), 3,60 (2 Н, m), 3,97(11) Степень чистоты по данным LCMS составила 97%; m/z=384 [М+Н]+. 1 Н NMR (300 МГц, CD3OD) : 0,94 (1 Н, m), 1,83 (2 Н, m), 3,01 (2 Н, d, J=7,5 Гц), 3,62 (2 Н, m), 3,96(12) Степень чистоты по данным LCMS составила 95%; m/z=462 [М+Н]+. 1 Н NMR (300 МГц, CD3OD) : 0,99 (1H, m), 1,24 (9 Н, s), 3,11 (2 Н, dd, J=7,5, 2,4 Гц), 3,63 (2 Н, dd,J=12,0, 3,1 Гц), 3,91 (2 Н, ddd, J=24,6, 10,8, 3,3 Гц), 4,01 (2 Н, d, J=11,7 Гц), 4,26 (1H, m), 5,35 (1 Н, m), 8,68(13) Степень чистоты по данным LCMS составила 98%; m/z=394 [М+Н]+. 1 Н NMR (300 МГц, CD3OD) : 1,01 (1 Н, m), 1,25 (9 Н, s), 1,88 (2 Н, m), 3,11 (2 Н, d, J=7,5 Гц), 3,64 (2 Н,dd, J=11,7, 3,9 Гц), 3,90 (2 Н, m), 4,01 (2 Н, d, J=11,7 Гц), 4,14 (1H, m), 8,67 (2 Н, s). Пример 3. Схема 7 Фиг. 4 Синтез соединений, показанных на фиг. 4, методом, описанным для соединений (18) и (19). Стадия 1. Сочетание.K2CO3 (8,9 г, 64,65 ммоль) в течение 10 мин при RT в атмосфере азота. Затем добавляли промежуточное соединение F (5 г, 21,55 ммоль) и перемешивали реакционную смесь в течение 20 мин. Затем ее разбавляли H2O (100 мл) и экстрагировали EtOAc (2100 мл). Объединенные органические слои высушивали(MgSO4) и удаляли под вакуумом растворитель с получением желаемого продукта в виде твердого вещества оранжевого цвета, которое применяли на следующей стадии без дальнейшей очистки (5,70 г, 99%); Продукт со стадии 1 (5,70 г, 21,51 ммоль) перемешивали в среде 1 М NaOHaq (20 мл) и THF (20 мл) при RT в течение 48 ч. Затем реакционную смесь подкисляли до pH 3 при помощи 2 М HClaq, что вызывало осаждение продукта. Этот продукт собирали и высушивали под вакуумом с получением продукта в виде твердого вещества белого цвета (4,47 г, 89%); m/z=238 [М+Н]+. Продукт со стадии 2 (4,47 г, 18,86 ммоль) перемешивали в среде DMF (50 мл) с промежуточным соединением Е (13,00 мл, 94,30 ммоль), EDCl (4,33 г, 22,60 ммоль), HOBt (3,05 г, 22,60 ммоль) и Et3N (13,10 мл, 94,30 ммоль) при RT в атмосфере азота в течение 48 ч. Затем реакционную смесь разбавляли водой(200 мл) и экстрагировали при помощи DCM (2200 мл). Объединенные органические слои высушивали(MgSO4) и удаляли под вакуумом растворитель. Остаток очищали методом хроматографии на колонке(0-15% MeOH в DCM) с получением желаемого продукта в виде масла желтого цвета (5,12 г, 77%); Раствор (COCl)2 (253 мкл, 2,90 ммоль) в DCM (50 мл) перемешивали в атмосфере азота и охлаждали до внутренней температуры, равной -70C. Затем медленно добавляли DMSO (363 мкл, 5,11 ммоль), поддерживая температуру, равной -70C. Когда добавление было завершено, медленно добавляли раствор продукта со стадии 3 (1,00 г, 2,84 ммоль) в DCM (50 мл), опять-таки поддерживая внутреннюю температуру, равной -70C. После окончания прибавления медленно добавляли Et3N (1,70 мл, 12,21 ммоль), снова поддерживая внутреннюю температуру, равной -70C. Реакционной смеси давали нагреться до RT и затем под вакуумом удаляли растворитель. Затем остаток очищали методом хроматографии на колонке(0-10% MeOH в DCM) с получением конечного продукта в виде бесцветного масла (890 мг, 89%); Продукт со стадии 4 (100 мг, 0,28 ммоль) перемешивали в среде DCE (10 мл) с промежуточным соединением В (73 мг, 0,28 ммоль) и NaBH3CN (35 мг, 0,56 ммоль) при RT в атмосфере азота в течение 16 ч. Затем реакционную смесь разбавляли водой (50 мл) и экстрагировали DCM (2100 мл). Объединенные органические слои высушивали (MgSO4) и удаляли растворитель под вакуумом. Остаток очищали методом хроматографии на колонке (0-10% MeOH в DCM) с получением конечного продукта в виде бесцветного масла (145 мг, 92%); m/z=560 [М+Н]+. Стадия 6. Снятие защиты с гидроксамата с получением циклопентил-(2S)-циклогексил-[(1-[5(гидроксикарбамоил)пиримидин-2-ил]пиперидин-4-илметил)амино]ацетата (18). Продукт со стадии 5 (145 мг, 0,26 ммоль) перемешивали в среде DCM (10 мл) с TFA (0,5 мл) при RT в течение 10 мин. Затем под вакуумом удаляли растворитель и остаток очищали методом препаративнойHPLC с получением желаемого продукта в виде твердого вещества светло-пурпурного цвета (11 мг, 9%). Степень чистоты по данным LCMS составила 95%; m/z=460 [М+Н]+. 1 Н NMR (300 МГц, CD3OD) : 1,02-1,37 (8 Н, m), 1,73-1,94 (19 Н, m), 3,02 (3H, m), 3,90 (1H, m), 5,37 Продукт со стадии 5 (78 мг, 0,14 ммоль) перемешивали в среде 1 М NaOHaq (10 мл) и THF (10 мл) в течение 4 дней при температуре 40C. Затем реакционную смесь охлаждали до RT и подкисляли до pH 3 при помощи 1 М HClaq. Эту смесь перемешивали в течение 10 мин и затем выпаривали досуха. Остаток очищали методом препаративной HPLC с получением конечного продукта в виде твердого вещества белого цвета (1 мг, 2%). Степень чистоты по данным LCMS составила 98%; m/z=392 [М+Н]+. 1(1H, m), 8,55 (2 Н, s). Аналоги, показанные на фиг. 4, получали методом, описанным для синтеза соединений (18) и (19). Для каждого аналога приведены данные, его характеризующие.(16) Степень чистоты по данным LCMS составила 98%; m/z=434 [М+Н]+. 1 Н NMR (300 МГц, CD3OD) : 1,03 (6 Н, m), 1,32 (4 Н, m), 1,71-1,95 (15 Н, m), 2,90 (1H, m), 3,01 (2 Н,m), 4,01 (1H, m), 5,37 (1H, m), 8,68 (2 Н, m).(17) Степень чистоты по данным LCMS составила 97%; m/z=366 [М+Н]+. 1 Н NMR (300 МГц, CD3OD) : 0,92 (6 Н, m), 1,19 (3H, m), 1,58 (2 Н, m), 1,75 (4 Н, m), 1,98 (1H, m),2,89 (4 Н, m), 3,70 (1H, m), 8,55 (2 Н, s).(20) Степень чистоты по данным LCMS составила 98%; m/z=454 [М+Н]+. 1 Н NMR (300 МГц, CD3OD) : 1,19-1,62 (10H, m), 1,79-1,92 (6 Н, m), 2,09 (1 Н, m), 2,84 (1H, m), 2,97(21) Степень чистоты по данным LCMS составила 98%; m/z=386 [М+Н]+. 1 Н NMR (300 МГц, CD3OD) : 1,08-1,28 (4 Н, m), 1,77 (2 Н, m), 1,98 (1H, m), 2,68-2,87 (4 Н, m), 4,90(22) Степень чистоты по данным LCMS составила 98%; m/z=464 [М+Н]+. 1 Н NMR (300 МГц, CD3OD) : 1,13 (9 Н, s), 1,56-1,71 (12 Н, m), 1,82 (3H, m), 2,04 (1H, m), 2,91 (3H,m), 3,81 (2 Н, m), 4,14 (1H, m), 5,39 (1 Н, m), 8,55 (2H, m).(23) Степень чистоты по данным LCMS составила 98%; m/z=396 [М+Н]+. 1 Н NMR (300 МГц, CD3OD) : 1,26 (9 Н, s), 1,31 (4 Н, m), 1,94 (2 Н, m), 2,14 (1H, br s), 3,03 (4 Н, m),3,95 (2 Н, m), 4,16 (1H, m), 8,67 (2H, s).(24) Степень чистоты по данным LCMS составила 98%; m/z=408 [М+Н]+. 1 Н NMR (300 МГц, CD3OD) : 1,22-1,33 (3H, m), 1,53-1,73 (11H, m), 2,02 (1H, m), 2,93 (4 Н, m), 3,91(25) Степень чистоты по данным LCMS составила 95%; m/z=340 [М+Н]+. 1 Н NMR (300 МГц, CD3OD) : 1,28 (2 Н, m), 1,97 (2 Н, m), 2,15 (1H, m), 3,07 (4 Н, m), 4,09 (4 Н, m),4,93 (1H, m), 8,67 (2 Н, m).(26) Степень чистоты по данным LCMS составила 87%; m/z=504 [М+Н]+. 1 Н NMR (300 МГц, CD3OD) : 1,02-1,48 (7 Н, br m), 1,68-1,82 (5 Н, m), 2,01 (1H, m), 2,97 (2 Н, m), 3,13 Фиг. 5 Синтез соединений, показанных на фиг. 5, методом, использованным для получения соединений(27) и (28). Стадия 1. Защита при помощи Boc. 4-(Аминометил)бензойную кислоту (10,00 г, 65,36 ммоль) перемешивали с Boc2O (28 г, 130,72 ммоль) в среде H2O (100 мл) и THF (100 мл) при RT. Добавляли насыщенный NaHCO3(aq) до достижения рН, равного 6,0, реакционную смесь перемешивали в течение 16 ч. Затем реакционную смесь подкисляли до pH 3 при помощи 1 М HCl(aq), происходило осаждение твердого продукта. Этот продукт отфильтровывали и сушили с получением твердого вещества белого цвета (16,1 г, 97%); m/z=274 [М+Na]+. Стадия 2. Восстановление кислоты. Продукт со стадии 1 (16,1 г, 64,14 ммоль) перемешивали в THF (300 мл) и диоксане (200 мл) при температуре 0C в атмосфере азота. Затем добавляли LiAlH4, давали реакционной смеси нагреться до RT и перемешивали в течение 16 ч. Затем смесь охлаждали до 0C и обрывали реакцию насыщеннымNH4Claq. Добавляли Na2SO4 и перемешивали смесь в течение 30 мин. Затем смесь фильтровали через целит и концентрировали фильтрат под вакуумом с получением продукта в виде твердого вещества светложелтого цвета (13,1 г, 94%); m/z=260 [М+Na]+. Стадия 3. Окисление спирта. Продукт со стадии 2 (5,87 г, 24,73 ммоль) перемешивали с MnO2 (16,71 г, 192,20 ммоль) в средеDCM в течение 16 ч при RT. Затем реакционную смесь фильтровали через целит и удаляли растворитель под вакуумом с получением продукта в виде желтого масла, которое применяли на следующей стадии без дальнейшей очистки (4,63 г, 80%); m/z=258 [М+Na]+. Продукт со стадии 3 (650 мг, 2,70 ммоль) перемешивали в среде DCE (20 мл) в присутствии промежуточного соединения А (634 мг, 2,70 ммоль) и NaBH(OAc)3 (918 мг, 4,33 ммоль) при RT в атмосфере азота в течение 3 ч. Затем реакционную смесь разбавляли водой (50 мл) и экстрагировали Et2O(2100 мл). Объединенные органические слои высушивали (MgSO4) и удаляли растворитель под вакуумом с получением желаемого продукта в виде коричневого масла, который применяли на следующей стадии без дальнейшей очистки (1,1 г, 98%); m/z=419 [М+Н]+. Стадия 5. Снятие защиты Boc. Продукт со стадии 4 (1,1 г, 2,63 ммоль) перемешивали в среде DCM (5 мл) с 4 М HCl в диоксане(2 мл) при RT в атмосфере азота в течение 3 ч. Растворитель удаляли под вакуумом, остаток высушивали с получением целевого продукта в виде твердого вещества коричневого цвета, представляющего собой соль HCl (670 мг, 100%); m/z=319 [М+Н]+. Стадия 6. Восстановительное аминирование. 2-(4-Оксопиперидин-1-ил)пиримидин-5-карбоновую кислоту (полученную, как описано в примере 1 100 мг, 0,45 ммоль) перемешивали в среде DCE (10 мл) с продуктом со стадии 5 (159 мг, 0,45 ммоль) иNaBH(OAc)3 (191 мг, 0,9 ммоль) при RT в атмосфере азота в течение 64 ч. Затем реакционную смесь разбавляли водой (100 мл) и экстрагировали DCM (2100 мл). Объединенные органические слои высушивали (Na2SO4) и удаляли растворитель под вакуумом. Остаток применяли на следующей стадии без дальнейшей очистки; m/z=524 [М+Н]+. Стадия 7. Образование защищенного гидроксамата. Продукт со стадии 6 (0,45 ммоль) перемешивали в среде DMF (10 мл) с промежуточным соединением Е (621 мкл, 4,50 ммоль), EDCl (103 мг, 0,54 ммоль), HOBt (73 мг, 0,54 ммоль) и Et3N (313 мкл, 2,25 ммоль) при RT в атмосфере азота в течение 40 ч. Реакционную смесь затем разбавляли водой (50 мл) и экстрагировали DCM (2100 мл). Объединенные органические слои высушивали (MgSO4) и удаляли под вакуумом растворитель с получением желаемого продукта в виде желтого масла, который применяли на следующей стадии без дальнейшей очистки; m/z=639 [М+Н]+. Продукт со стадии 7 (0,45 ммоль) перемешивали в DCM (10 мл) с TFA (1 мкл) при RT в течение 30 мин. Затем удаляли растворитель под вакуумом и очищали остаток методом препаративной HPLC с получением продукта в виде твердого вещества розового цвета (15 мг, 6% после проведения двух стадий). Степень чистоты по данным LCMS составила 96%; m/z=639 [М+Н]+. 1 Н NMR (300 МГц, CD3OD) : 1,00 (6 Н, m), 1,61-1,95 (12 Н, m), 2,28 (2 Н, m), 3,04 (3H, m), 4,01 (1H,m), 4,29 (4 Н, m), 5,04 (2 Н, m), 5,34 (1H, m), 7,60 (4 Н, m), 8,70 (2 Н, s). Стадия 9. Гидролиз эфира. Продукт со стадии 7 (0,45 ммоль) перемешивали в среде THF (10 мл) с KOTMS (115 мг, 0,9 ммоль) в течение 96 ч при RT в атмосфере азота. Затем удаляли под вакуумом растворитель и применяли остаток на следующей стадии без дальнейшей очистки; m/z=571 [М+Н]+. Стадия 10. Снятие защиты с гидроксамата с получением Продукт со стадии 9 перемешивали в среде DCM (10 мл) с TFA (1 мл) при RT в течение 30 мин. Затем удаляли растворитель под вакуумом и остаток очищали методом препаративной HPLC с получением желаемого продукта в виде твердого вещества белого цвета (5 мг, 3%); m/z=471 [М+Н]+. 1 Н NMR (300 МГц, d6-DMSO) : 0,9 (6 Н, d, J=4,8 Гц), 1,51 (2 Н, m), 1,71 (2 Н, m), 2,19 (2 Н, m), 3,00(4 Н, m), 4,42 (2 Н, br s), 3,75 (1H, m), 4,22 (4 Н, m), 4,80 (2 Н, m), 7,52 (4 Н, m), 8,71 (2 Н, s), 8,98 (2 Н, br s),11,01 (1H, s). Аналоги, показанные на фиг. 5, были получены методом, описанным для синтеза соединений (27) и(28). Для каждого аналога приведены данные, характеризующие его.(29) Степень чистоты по данным LCMS составила 99%; m/z=565 [М+Н]+. 1 Н NMR (300 МГц, d6-DMSO) : 0,88 (2 Н, m), 1,05-1,30 (5 Н, m), 1,50-1,85 (18 Н, m), 2,18 (2 Н, m),3,00 (2 Н, m), 2,25 (2 Н, m), 4,80 (2 Н, m), 5,17 (2 Н, m), 7,54 (4 Н, m), 8,71 (2 Н, s), 8,93 (2 Н, m), 9,45 (1H, br(30) Степень чистоты по данным LCMS составила 95%; m/z=497 [М+Н]+. 1 Н NMR (300 МГц, CD3OD) : 0,80-1,34 (6 Н, m), 1,49-1,68 (5 Н, m), 1,85 (1H, m), 2,17 (2 Н, m), 2,93(31) Степень чистоты по данным LCMS составила 98%; m/z=569 [М+Н]+. 1 Н NMR (300 МГц, CD3OD) : 1,22 (9 Н, s), 1,51-1,84 (11H, m), 2,18 (2 Н, m), 2,93 (3H, m), 3,44 (1H,m), 3,79 (2 Н, qd, J=7,8, 3,3 Гц), 4,24 (4 Н, m), 4,93 (2 Н, m), 5,22 (1H, m), 7,52 (4H, m), 8,59 (2H, s).(32) Степень чистоты по данным LCMS составила 96%; m/z=501 [М+Н]+. 1 Н NMR (300 МГц, CD3OD) : 1,25 (9 Н, s), 1,62 (2 Н, m), 2,29 (2 Н, m), 3,07 (4 Н, m), 3,85 (2 Н, m), 4,35(33) Степень чистоты по данным LCMS составила 98%; m/z=559 [М+Н]+. 1 Н NMR (300 МГц, CD3OD) : 1,31-1,93 (12 Н, m), 2,29 (2 Н, m), 3,04 (3H, m), 3,56 (1 Н, m), 4,22 (4H,m), 5,17 (2H, m), 5,30 (1H, m), 7,47-7,43 (9H, m), 8,69 (2H, s).(34) Степень чистоты по данным LCMS составила 95%; m/z=491 [М+Н]+. 1 Н NMR (300 МГц, CD3OD) : 1,63 (2 Н, m), 2,29 (2 Н, m), 3,06 (2 Н, m), 3,29 (2 Н, m), 3,56 (1H, m),4,20 (4 Н, m), 5,07 (1H, m), 7,54 (9 Н, m), 8,70 (2 Н, s). Пример 5. Схема 9NaBH3CN (36 мг, 0,58 ммоль) в DCE (10 мл) при RT в атмосфере азота в течение 16 ч. Затем реакционную смесь разбавляли водой (50 мл) и экстрагировали DCM (2100 мл). Объединенные органические слои высушивали (MgSO4) и удаляли под вакуумом растворитель. Остаток очищали методом хроматографии на колонке (0-10% MeOH в DCM) с получением желаемого продукта в виде желтого масла Продукт со стадии 1 (56 мг, 0,08 ммоль) перемешивали в среде DCM (10 мл) с TFA (1 мл) при RT в течение 15 мин. Затем под вакуумом удаляли растворитель и очищали остаток методом препаративнойHPLC с получением желаемого продукта в виде твердого вещества розового цвета (12 мг, 25%); m/z=571 Циклопентил-(2S)-[4-(аминометил)бензил]амино(4-метилциклогексил)ацетат (полученный, как описано в примере 4 - 100 мг, 0,29 ммоль) перемешивали с 2-(4-формилпиперидин-1-ил)-N-(1 изобутоксиэтокси)пиримидин-5-карбоксамидом (полученным, как описано в примере 3 - 110 мг,0,29 ммоль) в среде DCE (10 мл) с NaCNBH3 (36 мг, 0,58 ммоль) при RT в атмосфере азота в течение 16 ч. Затем реакционную смесь разбавляли водой (50 мл) и экстрагировали DCM (2100 мл). Объединенные органические экстракты высушивали (MgSO4) и удаляли под вакуумом растворитель с получением желаемого продукта в виде желтого масла, который применяли на следующей стадии без дальнейшей очистки; m/z=701 [M+Na]+. Стадия 2. Снятие защиты с гидроксамата с получением циклопентил-(2S)-циклогексил-[(4-[(1-[5(гидроксикарбамоил)пиримидин-2-ил]пиперидин-4-илметил)амино]метилбензил)амино]ацетата (38). Продукт со стадии 1 перемешивали в среде DCM (10 мл) с TFA (1 мл) при RT в течение 30 мин. Затем растворитель удаляли под вакуумом и очищали остаток методом препаративной HPLC с получением желаемого продукта в виде твердого вещества пурпурного цвета (14 мг, 8% после проведения двух стадий). Степень чистоты по данным LCMS составила 95%; m/z=579 [М+Н]+. 1
МПК / Метки
МПК: A61P 29/00, A61P 19/02, C07D 239/42, A61P 9/00, A61P 25/28, A61P 7/12, A61K 31/506
Метки: гидроксаматы, ингибиторов, гистон-деацетилазы, качестве
Код ссылки
<a href="https://eas.patents.su/30-17198-gidroksamaty-v-kachestve-ingibitorov-giston-deacetilazy.html" rel="bookmark" title="База патентов Евразийского Союза">Гидроксаматы в качестве ингибиторов гистон-деацетилазы</a>
Сульфонилпроизводные в качестве ингибиторов гистон-деацетилазы

Номер патента: 7099
Опубликовано: 30.06.2006
Авторы: Ван Брандт Свен Францискус Анна, Бакс Лео Якобус Йозеф, Де Винтер Ханс Луи Йос, Анжибо Патрик Рене, Ван Хьюсден Джимми Арнольд Вивиан, Пилатт Изабелль Ноэлль Констанс, Вердонк Марк Густаф Селин, Ван Эмелен Кристоф
МПК: C07D 207/14, A61P 35/00, A61K 31/4545...
Метки: гистон-деацетилазы, ингибиторов, качестве, сульфонилпроизводные
Формула / Реферат:
1. Соединение формулы (I) его N-оксидные формы, его фармацевтически приемлемые соли добавления кислот и его стереохимически изомерные формы, где n равно 0, 1, 2 или 3 и, если n равно 0, то имеется в виду прямая связь; t равно 0, 1, 2, 3 или 4 и, если t равно 0, то имеется в виду прямая связь; каждый Q представляет собой азот или ; каждый Х представляет собой азот или ; каждый Y представляет собой азот или ; каждый Z представляет собой азот...
Сульфонилпроизводные в качестве новых ингибиторов гистон-деацетилазы

Номер патента: 6707
Опубликовано: 24.02.2006
Авторы: Понселе Виржини Софи, Пилатт Изабелль Ноэлль Констанс, Мерпул Ливен, Ван Брандт Свен Францискус Анна, Бакс Лео Якобус Йозеф, Дяткин Алексей Борисович, Ван Эмелен Кристоф, Ван Хьюсден Джимми Арнольд Вивиан, Артс Янине, Де Винтер Ханс Луи Йос, Вердонк Марк Густаф Селин
МПК: C07D 213/82, C07D 207/28, C07D 215/36...
Метки: гистон-деацетилазы, новых, сульфонилпроизводные, качестве, ингибиторов
Формула / Реферат:
1. Соединение формулы (I) его N-оксидные формы, его фармацевтически приемлемые соли добавления кислот и его стереохимически изомерные формы, где n равно 0, 1, 2 или 3 и, если n равно 0, то имеется в виду прямая связь; t равно 0, 1, 2, 3 или 4, и если t равно 0, то имеется в виду прямая связь; каждый Q представляет собой азот или каждый X представляет собой азот или каждый Y представляет собой азот или каждый Z представляет собой азот или ...
Гетероариламины в качестве ингибиторов гликогенсинтаза-киназы 3-бета (ингибиторов gsk3)

Номер патента: 7298
Опубликовано: 25.08.2006
Авторы: Ван Акен Кун Жанн Альфонс, Бейнстерс Петер Якобус Йоханнес Антониус, Жанссен Поль Адриан Ян, Винкерс Хендрик Мартен, Эмбрехтс Вернер Констант Йохан, Де Жонж Марк Рене, Леви Паулус Йоаннес, Койманс Люсьен Мария Хенрикус, Виллемс Марк, Фрейн Эдди Жан Эдгар, Хэрес Ян, Дильс Гастон Станислас Марселла, Лав Кристофер Джон
МПК: A61K 31/50, A61K 31/435, A61K 31/505...
Метки: ингибиторов, гликогенсинтаза-киназы, gsk3, 3-бета, гетероариламины, качестве
Формула / Реферат:
1. Соединение формулы его N-оксид, фармацевтически приемлемая аддитивная соль, четвертичный амин и стереохимически изомерная форма, где R1 обозначает водород; X обозначает -O-; -О-С1-4алкил- или прямую связь; Z обозначает прямую связь, -NH-, -С1-4алкил-NH- или -С(=O)-; R2 обозначает водород или фенил; R3 обозначает водород, С1-4алкил, полигалогенC1-6алкил или циано; R4 обозначает моноциклический или бициклический насыщенный гетероцикл;...
Гидроксаматы как ингибиторы гистондеацетилазы и содержащие их фармацевтические композиции

Номер патента: 12909
Опубликовано: 26.02.2010
Авторы: Фаттори Даниэла, Росси Кристина, Д'андреа Пьеро, Порчеллони Марина, Марастони Елена
МПК: A61K 31/445, A61K 31/4535, A61K 31/4525...
Метки: композиции, ингибиторы, гидроксаматы, содержащие, гистондеацетилазы, фармацевтические
Формула / Реферат:
1. Соединение общей формулы II:где v=1;В означает химическую связь или выбран из группы -CO-, -NR5-CO-, -O-СO-, -SO2-, -NR5-SO2-, либо означает одну из следующих структур:в которой n = 0, 1, 2, a R5 означает H или C1-3-алкил;R1 означает H или выбран из группы: C1-3-алкил, C1-3-ацил либо ацил от одной из следующих кислот: бензойной, фенилуксусной, бензотиофенкарбоновой, индолкарбоновой;R2 означает H или C1-3-алкил;либо R1 и R2 вместе с атомом...
Сульфонамидные производные в качестве ингибиторов рассасывания костной ткани и ингибиторов адгезии клеток,способ их получения, применение и фармацевтическая композиция

Номер патента: 3102
Опубликовано: 26.12.2002
Авторы: Бодари Сара Кэтрин, Катбертсон Роберт Эндрю, Гурвест Жан-Франсуа, Гадек Томас, Кнолле Йохен, Шойнеманн Карлхайнц, Карниато Дени, Пейман Ануширван, Макдауэлл Роберт, Вилл Дэвид Вильям
МПК: A61P 19/10, A61K 31/505, C07D 239/42...
Метки: костной, применение, качестве, рассасывания, ткани, получения, фармацевтическая, композиция, производные, адгезии, ингибиторов, клеток,способ, сульфонамидные
Формула / Реферат:
1. Сульфонамидные производные общей формулы I где R1 и R2 вместе образуют двухвалентный (С2-С3)алкиленовый радикал; R4 является Н или (С1-С6)алкилом; R5 является (С1-С10)алкилом, (С6-С14)арилом, (C5-C14)гетероарилом или (С6-С14)арил(С1-С10)алкильной группой, где арил, гетероарил или алкил возможно замещены R3, или 2-оксобицикло[2.2.1]гепт-1-илметильной группой; R3 является (C1-C4)алкилом, (C1-C4)алкилокси, галогеном, трифторметилом, циано,...