Производные бензоазепиноксиуксусной кислоты и их применение в качестве агонистов дельта-ppar для повышения hdl-c и снижения ldl-c и холестерина
Номер патента: 16583
Опубликовано: 30.06.2012
Авторы: Демарест Кит Т., Шэнь Лань, Чжан Янь, Гээ-Хун Ко, Лу Сунфэн, Пелтон Патрисия



















Формула / Реферат
1. Соединение формулы (I)

где X означает ковалентную связь, О или S;
R1 и R2 независимо выбирают из группы, состоящей из Н, С1-8алкила и C1-8алкила, замещенного гидроксильной группой, или R1, R2 и атом углерода, к которому они присоединены, могут вместе образовывать С3-7циклоалкил;
R3 означает Н;
R4 и R5 независимо выбирают из Н и C1-8алкила;
R6 и R7 независимо выбирают из группы, состоящей из Н, галогена, C1-3алкила, C1-3алкила, замещенного галогеном, C1-3алкокси и C1-3алкокси, замещенного галогеном;
n равно 1 и
Q выбирают из группы, состоящей из

и его энантиомеры, диастереомеры, сольваты или фармацевтически приемлемые соли.
2. Соединение по п.1, в котором R1 и R2 независимо выбирают из группы, состоящей из Н и C1-8алкила, или R1, R2 и атом углерода, к которому они присоединены, могут вместе образовывать С3-5циклоалкил.
3. Соединение по п.1, в котором R1 и R2 независимо выбирают из группы, состоящей из Н и СН3, или R1, R2 и атом углерода, к которому они присоединены, могут вместе образовывать .
.
4. Соединение по п.1, в котором R6 и R7 независимо выбирают из группы, состоящей из Н, галогена, C1-3алкила, замещенного галогеном, C1-3алкокси и C1-3алкокси, замещенного галогеном.
5. Соединение по п.1, в котором R6 означает Н и R7 выбирают из группы, состоящей из галогена, C1-3алкила, замещенного галогеном, и C1-3алкокси, замещенного галогеном.
6. Соединение по п.5, в котором R7 выбирают из группы, состоящей из F, CF3 и -O-CF3.
7. Соединение по п.1, в котором R5 означает Н, СН3 или -СН2СН3.
8. Соединение по п.1, в котором Q выбирают из группы, состоящей из

9. Соединение по п.1, в котором Q выбирают из группы, состоящей из

10. Соединение по п.1, в котором X означает О.
11. Соединение по п.1, в котором
X означает О;
R1, R2, R4 и R5 независимо выбирают из группы, состоящей из Н и C1-3алкила, или R1, R2 и атом углерода, к которому они присоединены, могут вместе образовывать
и
R6 и R7 независимо выбирают из группы, состоящей из Н, галогена, C1-3алкокси, C1-3алкила, замещенного галогеном и C1-3алкокси, замещенного галогеном;
и его энантиомеры, диастереомеры, сольваты или фармацевтически приемлемые соли.
12. Соединение формулы (Ia)

где R1 и R2 независимо выбирают из группы, состоящей из Н и C1-8алкила, или R1, R2 и атом углерода, к которому они присоединены, могут вместе образовывать C3-5циклоалкил;
R6 и R7 независимо выбирают из группы, состоящей из Н, C1-3алкила, галогена и C1-3алкила, замещенного галогеном;
R4 и R5 независимо выбирают из группы, состоящей из Н и C1-8алкила; и
Q выбирают из группы, состоящей из

и его энантиомеры, диастереомеры, сольваты или фармацевтически приемлемые соли.
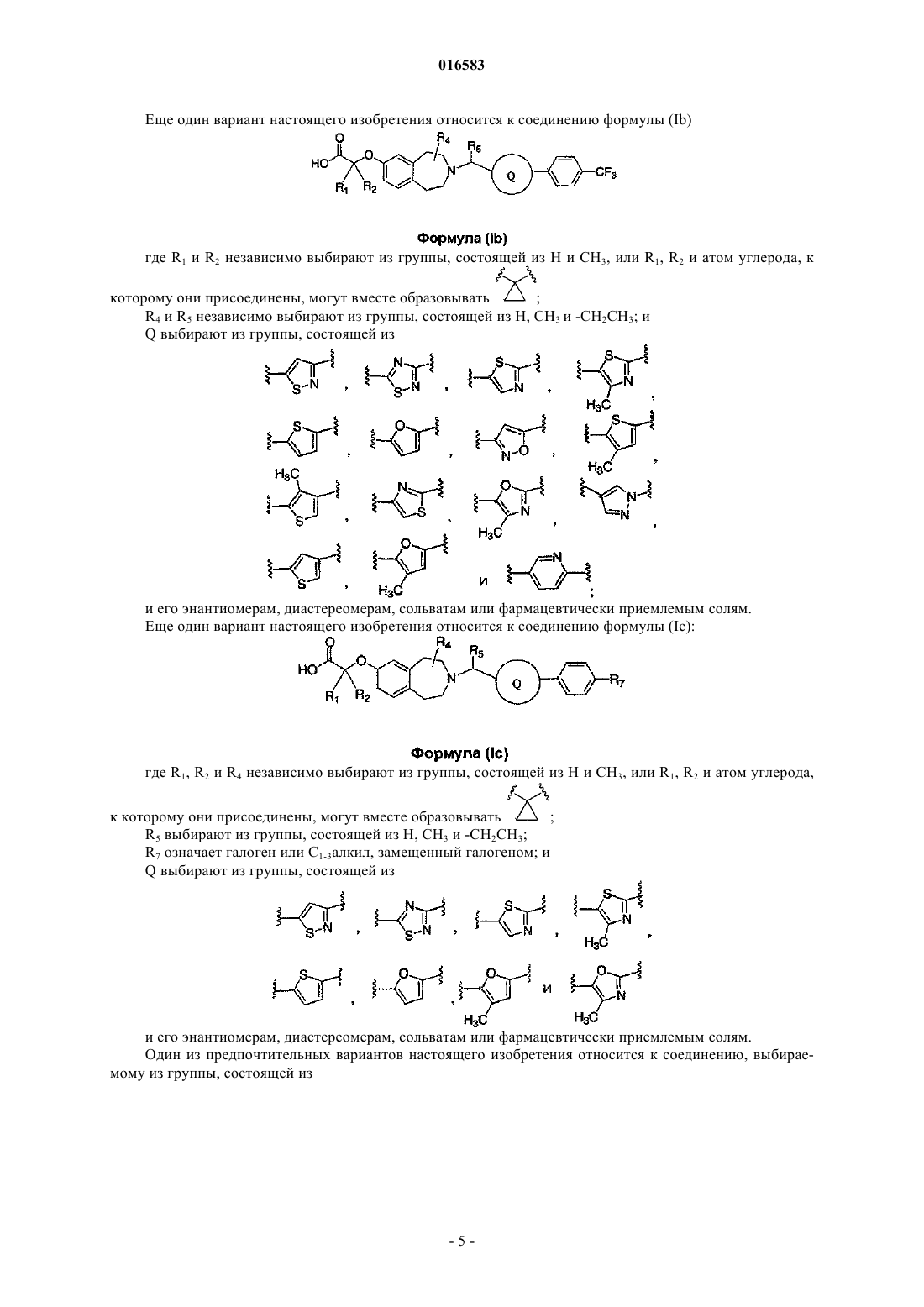
13. Соединение формулы (Ib)
 где R1 и R2 независимо выбирают из группы, состоящей из Н и СН3, или R1, R2 и атом углерода, к которому они присоединены, могут вместе образовывать
где R1 и R2 независимо выбирают из группы, состоящей из Н и СН3, или R1, R2 и атом углерода, к которому они присоединены, могут вместе образовывать
R4 и R5 независимо выбирают из группы, состоящей из Н, СН3 и -СН2СН3; и
Q выбирают из группы, состоящей из

и его энантиомеры, диастереомеры, сольваты или фармацевтически приемлемые соли.
14. Соединение формулы (Ic)

где R1, R2 и R4 независимо выбирают из группы, состоящей из Н и СН3, или R1, R2 и атом углерода, к которому они присоединены, могут вместе образовывать ,
,
R5 выбирают из группы, состоящей из Н, СН3 и -СН2СН3;
R7 означает галоген или C1-3алкил, замещенный галогеном; и
Q выбирают из группы, состоящей из

и его энантиомеры, диастереомеры, сольваты или фармацевтически приемлемые соли.
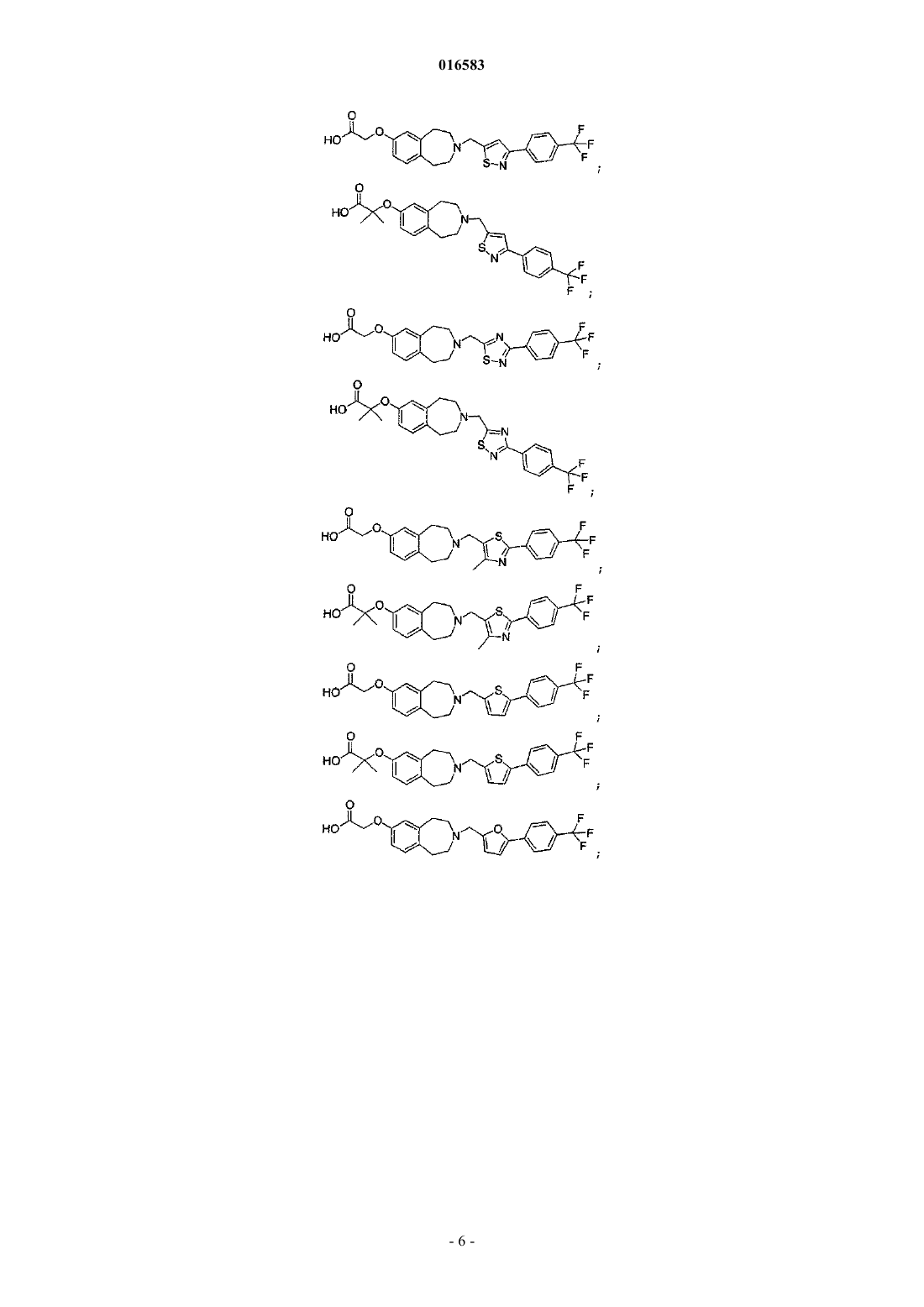
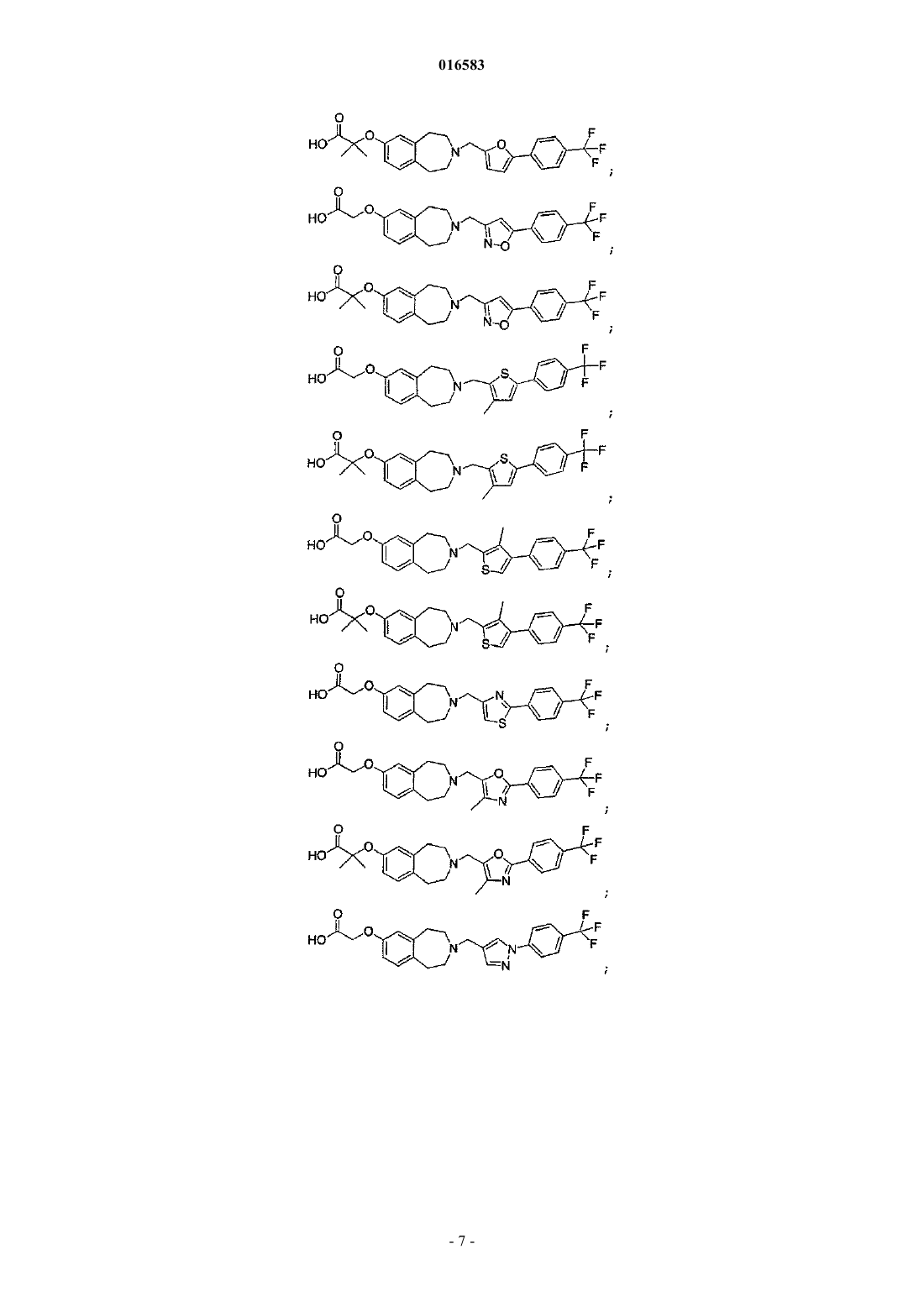
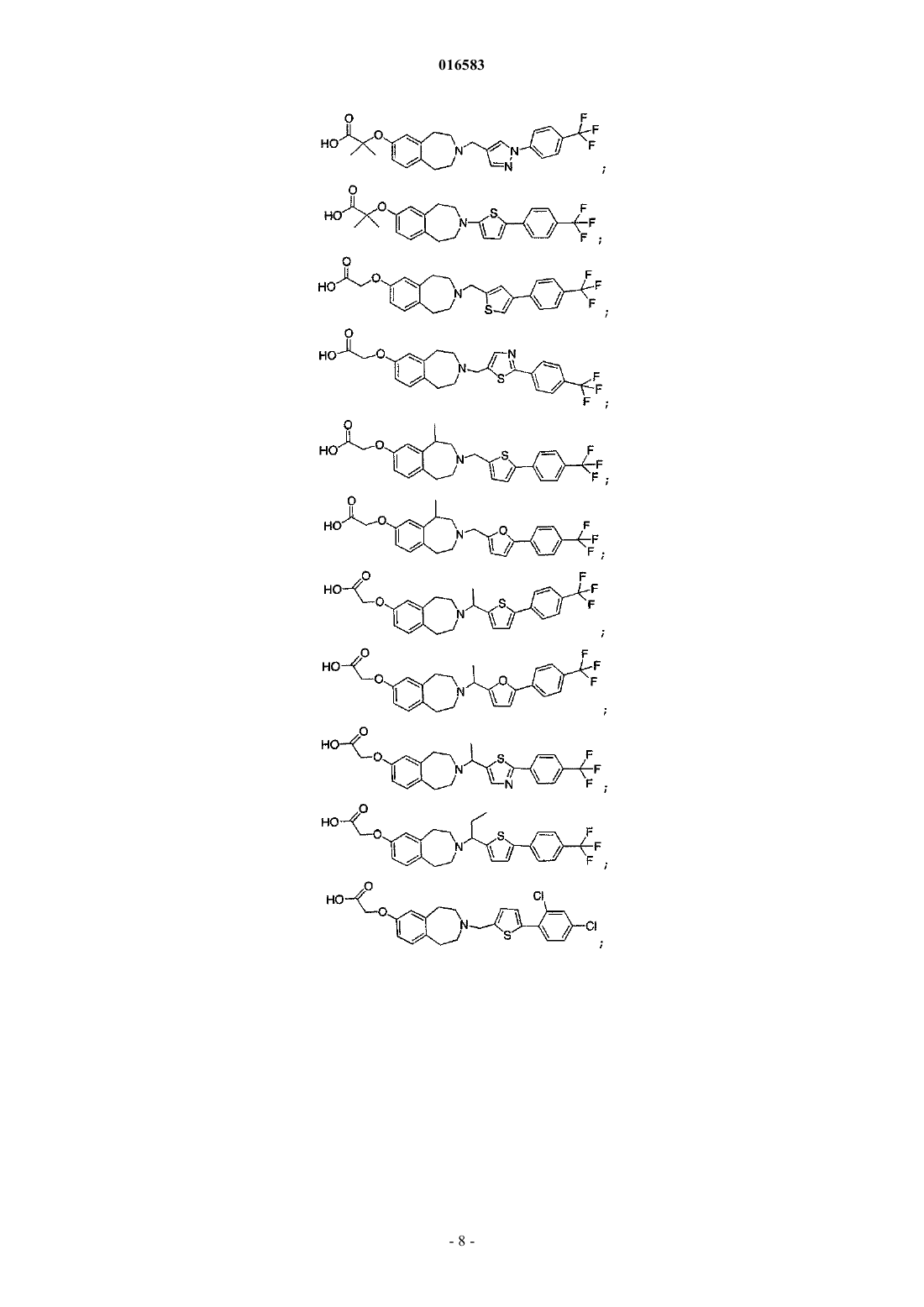
15. Соединение, выбираемое из группы, состоящей из




и его энантиомеры, диастереомеры, сольваты или фармацевтически приемлемые соли.
16. Соединение, выбираемое из группы, состоящей из


которое, по существу, не содержит соответствующий другой энантиомер.
17. Соединение формулы

18. Фармацевтическая композиция, содержащая соединение, соль или сольват по любому из пп.1-17 в смеси с фармацевтически приемлемым носителем, наполнителем или разбавителем.
19. Способ лечения или предотвращения заболевания или состояния, которое подвержено воздействию модуляции рецепторов PPAR, у млекопитающего, который включает введение млекопитающему, нуждающемуся в таком лечении или предотвращении, терапевтически эффективного количества соединения, соли или сольвата по п.1.
20. Способ лечения или предотвращения заболевания или состояния, которое подвержено воздействию модуляции дельта-PPAR, у млекопитающего, который включает введение млекопитающему, нуждающемуся в таком лечении или предотвращении, терапевтически эффективного количества соединения, соли или сольвата по п.1.
21. Способ по п.19 или 20, в котором указанное терапевтически эффективное количество включает дозу в пределах от около 0,1 до около 15000 мг.
22. Способ по п.19 или 20, в котором указанное терапевтически эффективное количество включает дозу в пределах от около 50 до около 1000 мг.
23. Способ по п.19 или 20, в котором указанное терапевтически эффективное количество включает дозу в пределах от около 100 до около 1000 мг.
24. Способ лечения или предотвращения заболевания или состояния, выбираемого из группы, включающей диабет, нефропатию, невропатию, ретинопатию, синдром поликистоза яичников, гипертензию, ишемию, удар, спастический колит, воспаление, катаракту, сердечно-сосудистые заболевания, метаболический X синдром, гипер-LDL-холестеринемию, дислипидемию (включая гипертриглицеридемию, гиперхолестеринемию, смешанную гиперлипидемию и гипо-HDL-холестеринемию), атеросклероз и ожирение, который включает стадию введения млекопитающему, нуждающемуся в таком лечении, терапевтически эффективного количества соединения, соли или сольвата по п.1.
25. Способ по п.24, в котором указанное терапевтически эффективное количество включает дозу в пределах от около 0,1 до около 15000 мг.
26. Способ по п.24, в котором указанное терапевтически эффективное количество включает дозу в пределах от около 50 до около 1000 мг.
27. Способ по п.24, в котором указанное терапевтически эффективное количество включает дозу в пределах от около 100 до около 1000 мг.
28. Соединение формулы (I)

где X означает ковалентную связь, О или S;
R1 и R2 независимо выбирают из группы, состоящей из Н, C1-8алкила и C1-8алкила, замещенного гидроксильной группой, или R1, R2 и атом углерода, к которому они присоединены, могут вместе образовывать C3-7циклоалкил;
R3 означает Н;
R4 и R5 независимо выбирают из Н и C1-8алкила;
R6 и R7 независимо выбирают из группы, состоящей из Н, галогена, C1-3алкила, C1-3алкила, замещенного галогеном, C1-3алкокси и C1-3алкокси, замещенного галогеном;
n равно 1 и
Q означает С6-10арил;
и его энантиомеры, диастереомеры, сольваты или фармацевтически приемлемые соли.
29. Соединение формулы (I)

где X означает ковалентную связь, О или S;
R1 и R2 независимо выбирают из группы, состоящей из Н, C1-8алкила и C1-8алкила, замещенного гидроксильной группой, или R1, R2 и атом углерода, к которому они присоединены, могут вместе образовывать С3-7циклоалкил;
R3 означает Н;
R4 и R5 независимо выбирают из Н и C1-8алкила;
R6 и R7 независимо выбирают из группы, состоящей из Н, галогена, C1-3алкила, C1-3алкила, замещенного галогеном, C1-3алкокси и C1-3алкокси, замещенного галогеном;
n равно 1 и
Q выбирают из группы, состоящей из

и его энантиомеры, диастереомеры, сольваты или фармацевтически приемлемые соли.
30. Соединение формулы (I)

где X означает ковалентную связь, О или S;
R1 и R2 независимо выбирают из группы, состоящей из Н, C1-8алкила и C1-8алкила, замещенного гидроксильной группой, или R1, R2 и атом углерода, к которому они присоединены, могут вместе образовывать C3-7циклоалкил;
R3 означает Н;
R4 и R5 представляют собой Н;
R6 и R7 независимо выбирают из группы, состоящей из Н, галогена, C1-3алкила, C1-3алкила, замещенного галогеном, C1-3алкокси и C1-3алкокси, замещенного галогеном;
n равно 2 и
Q выбирают из группы, состоящей из

и его энантиомеры, диастереомеры, сольваты или фармацевтически приемлемые соли.
31. Соединение по пп.28, 29 или 30, выбираемое из группы, состоящей из


Текст
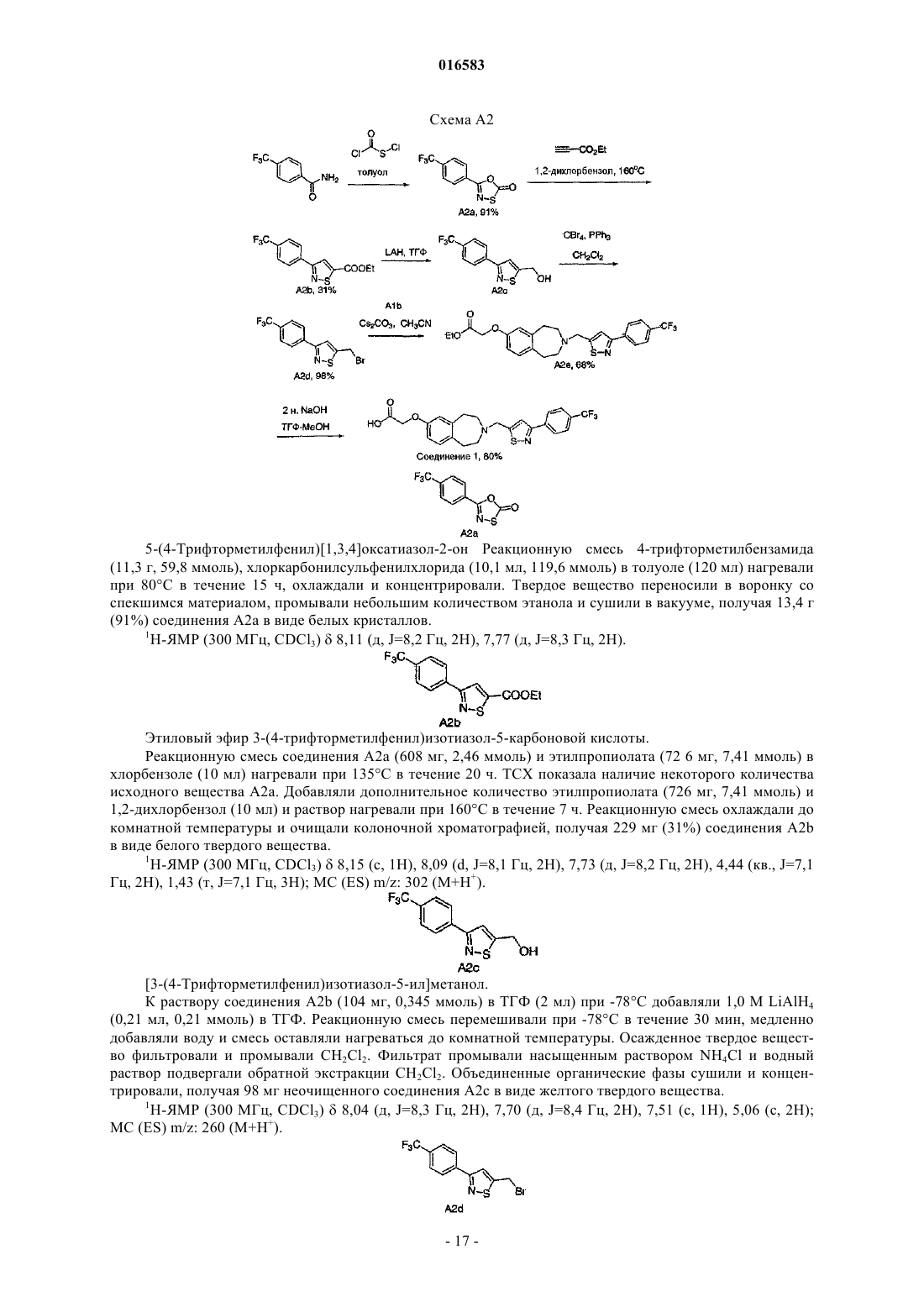
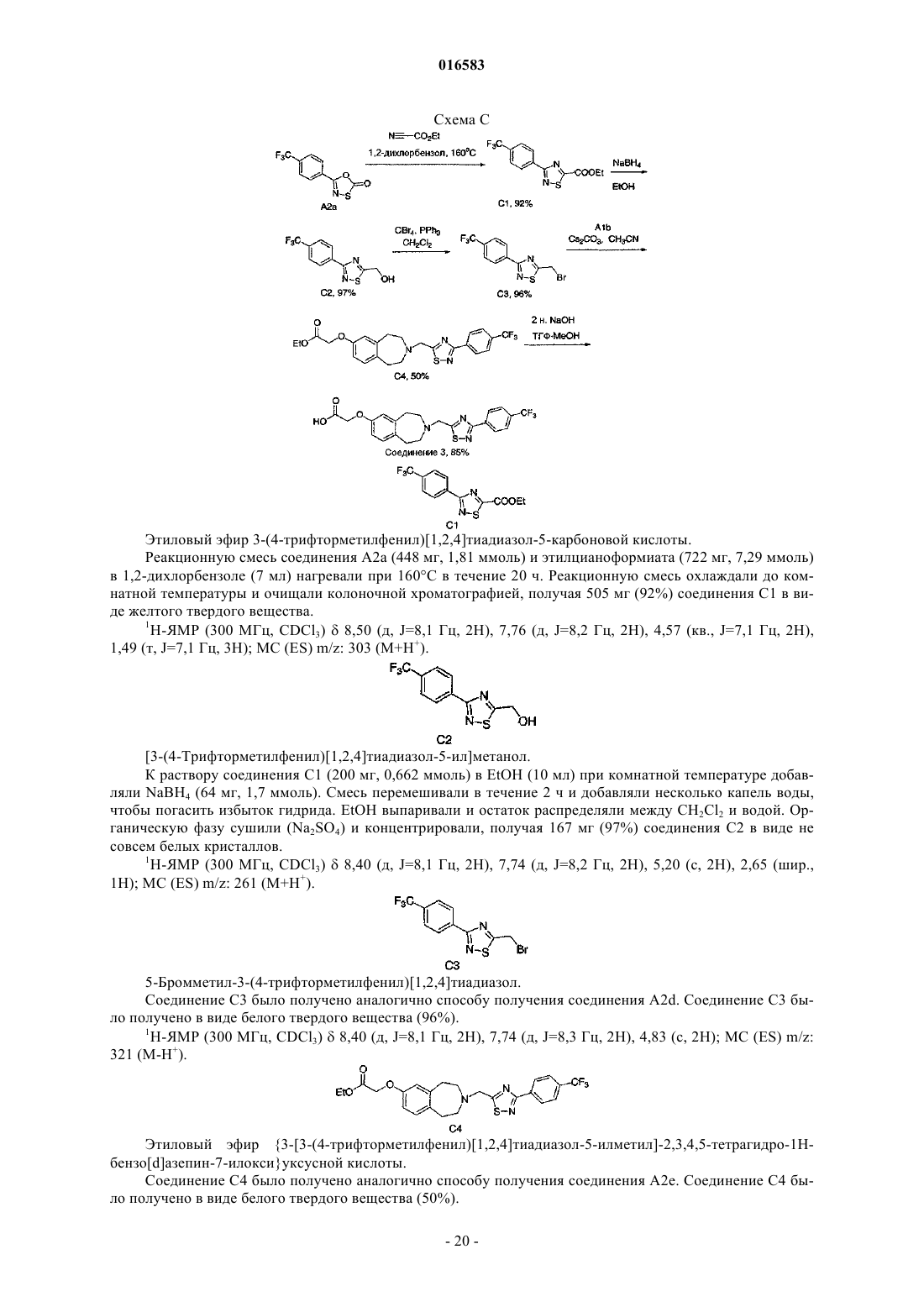
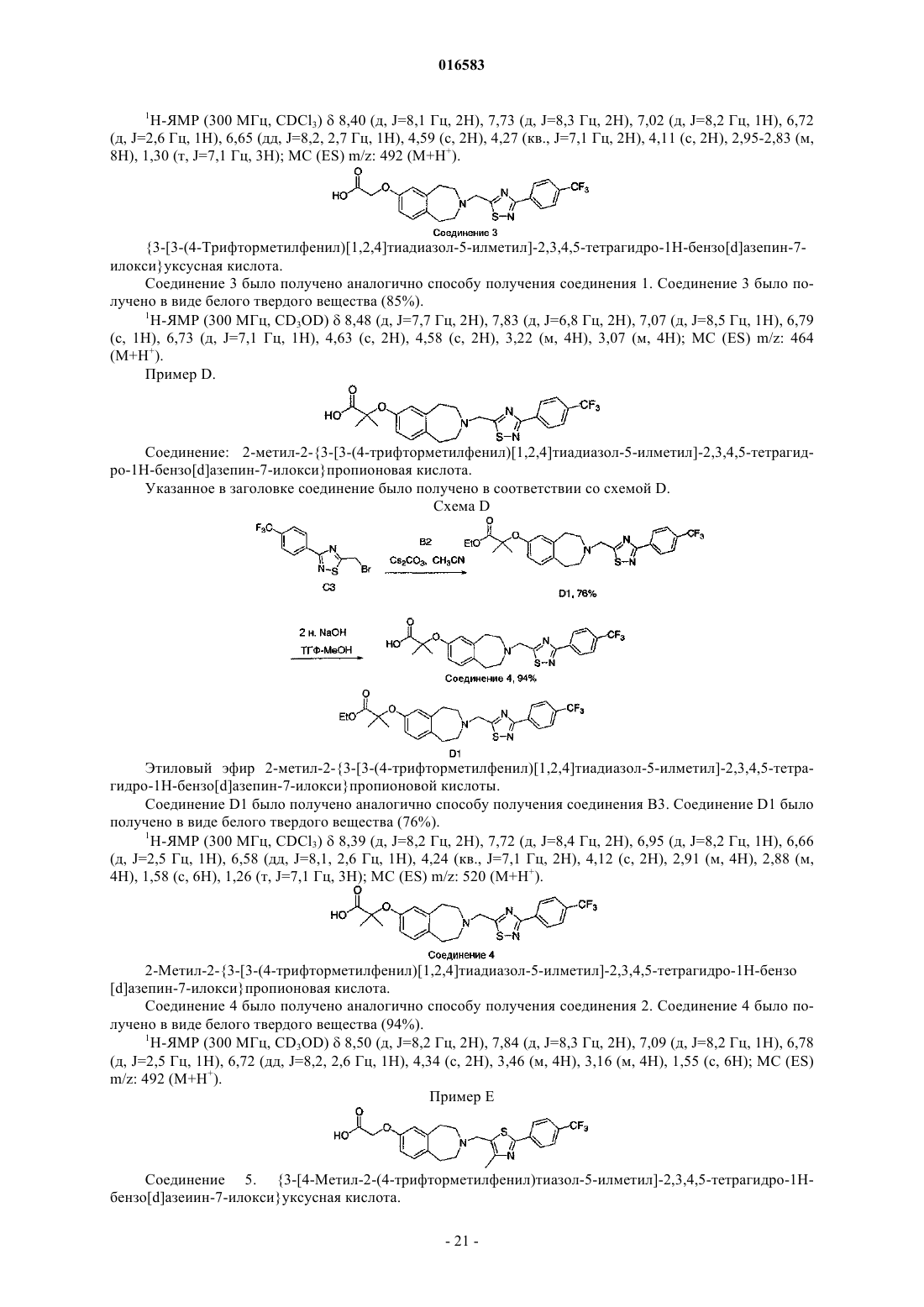
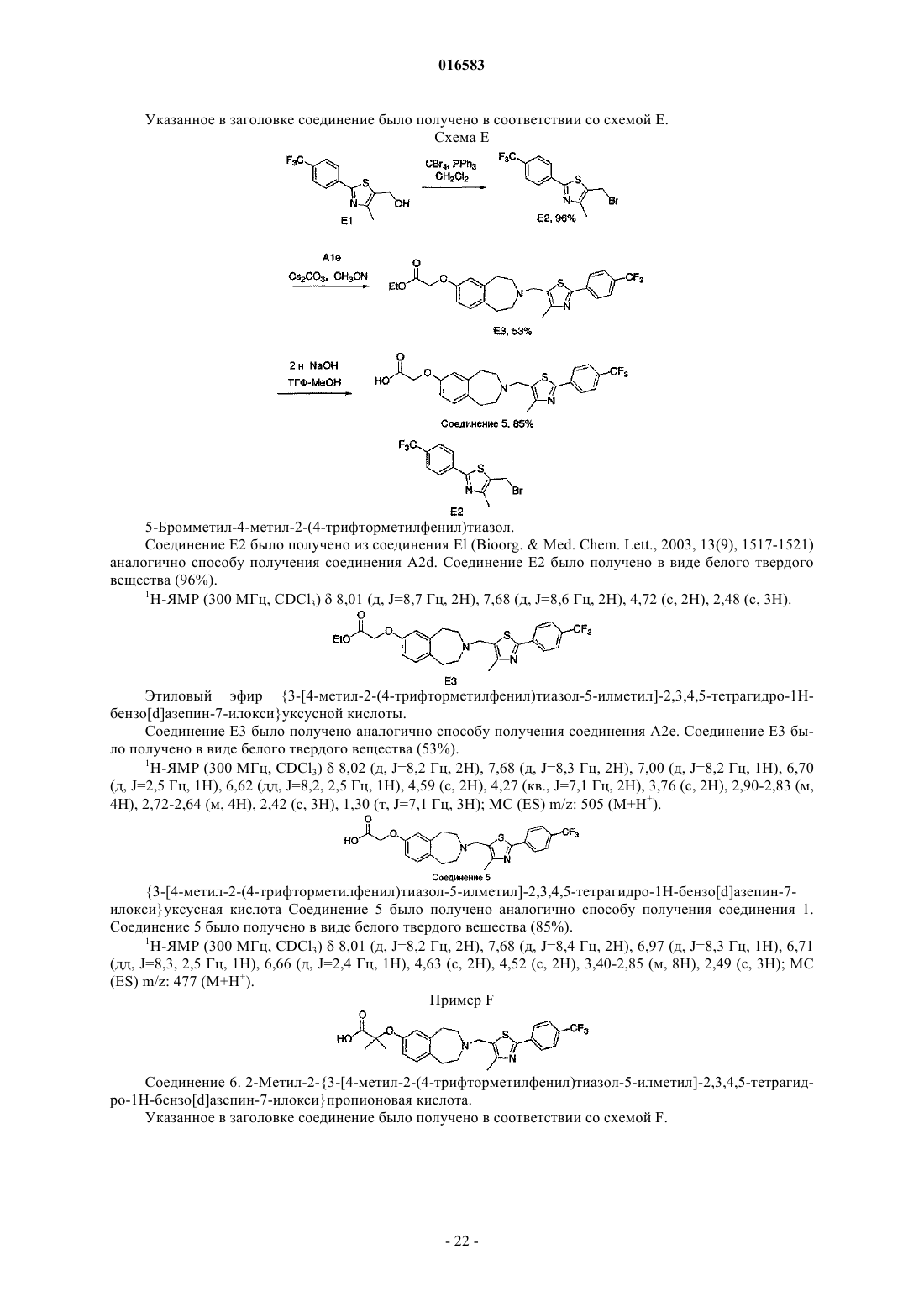
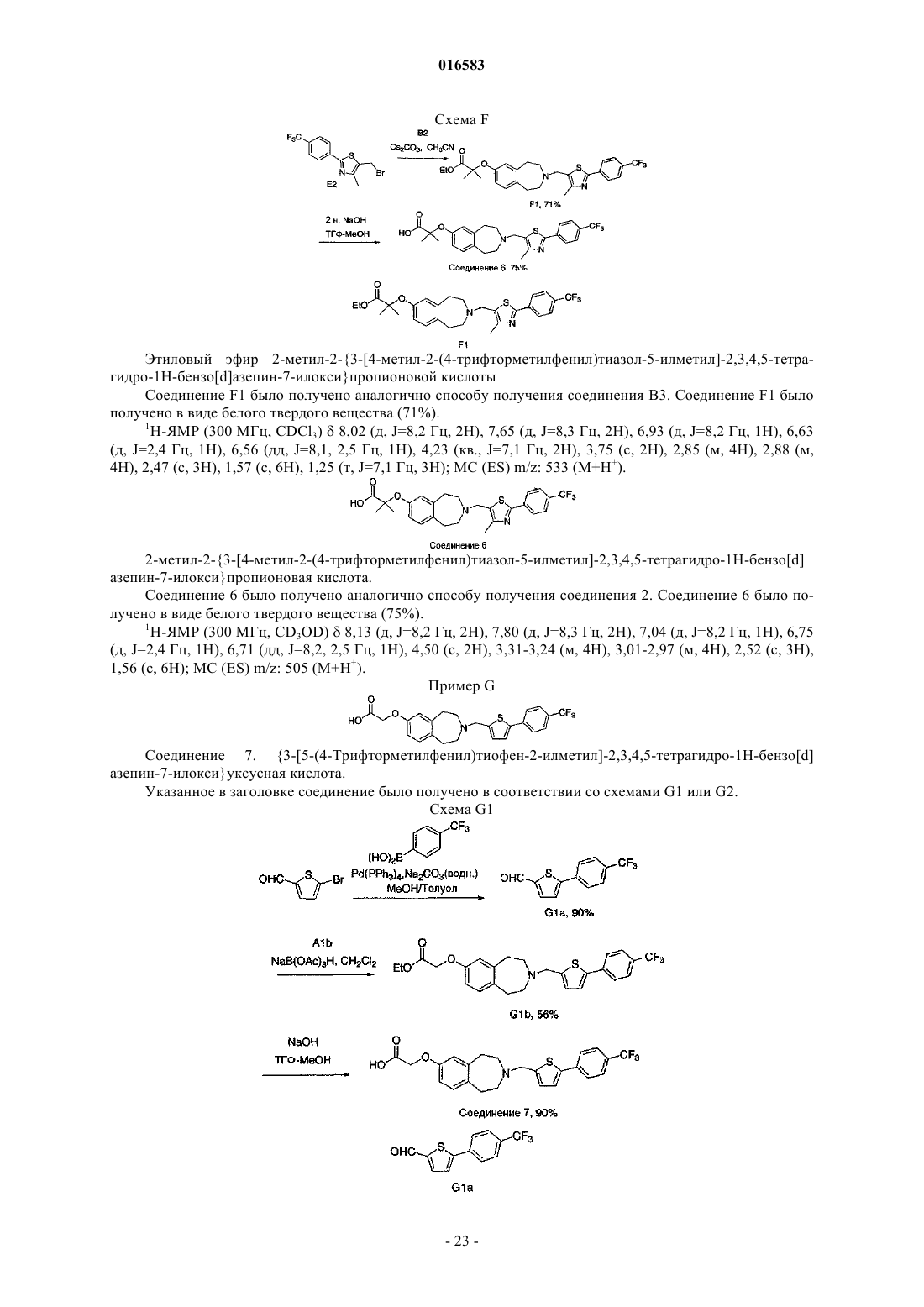
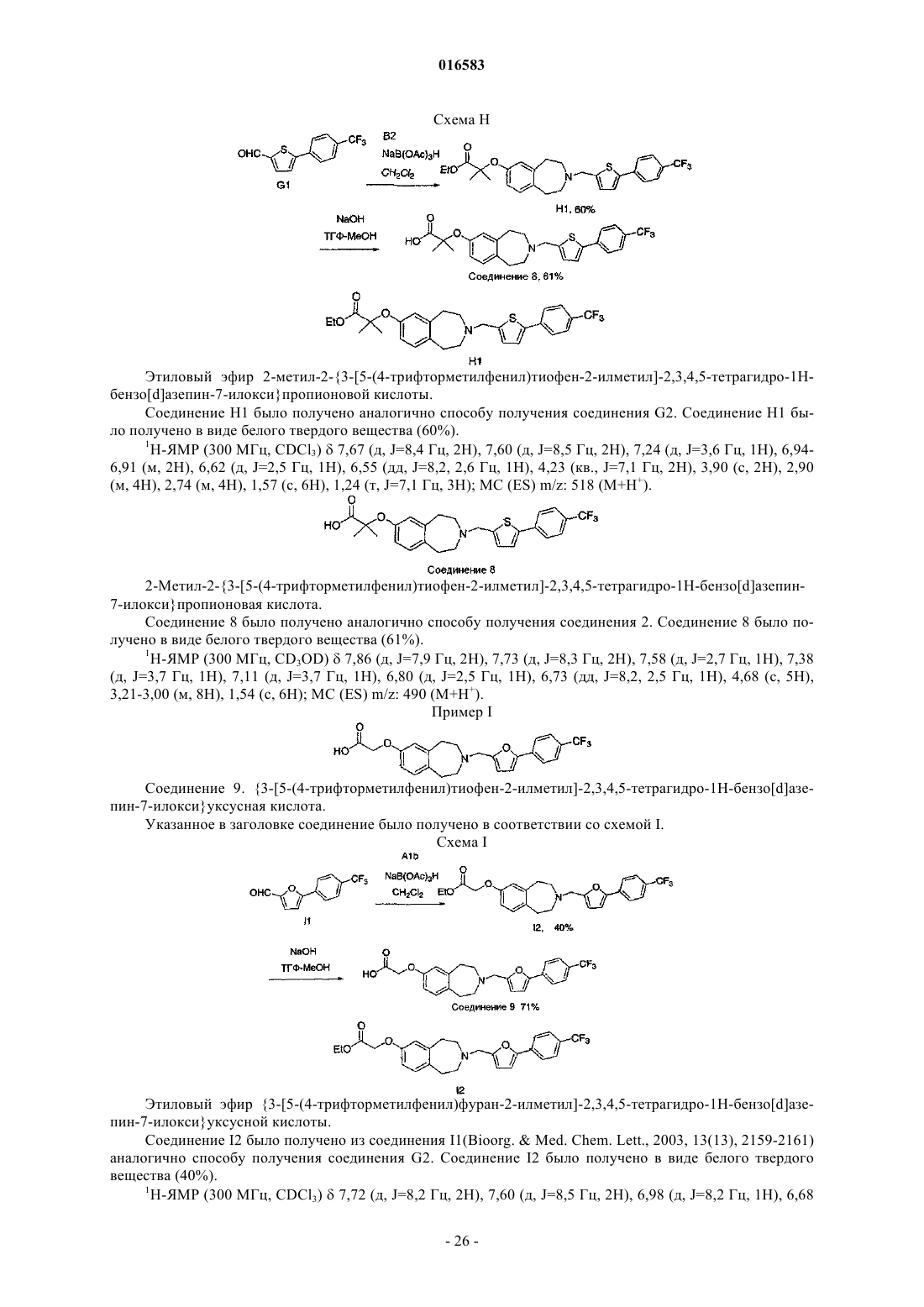
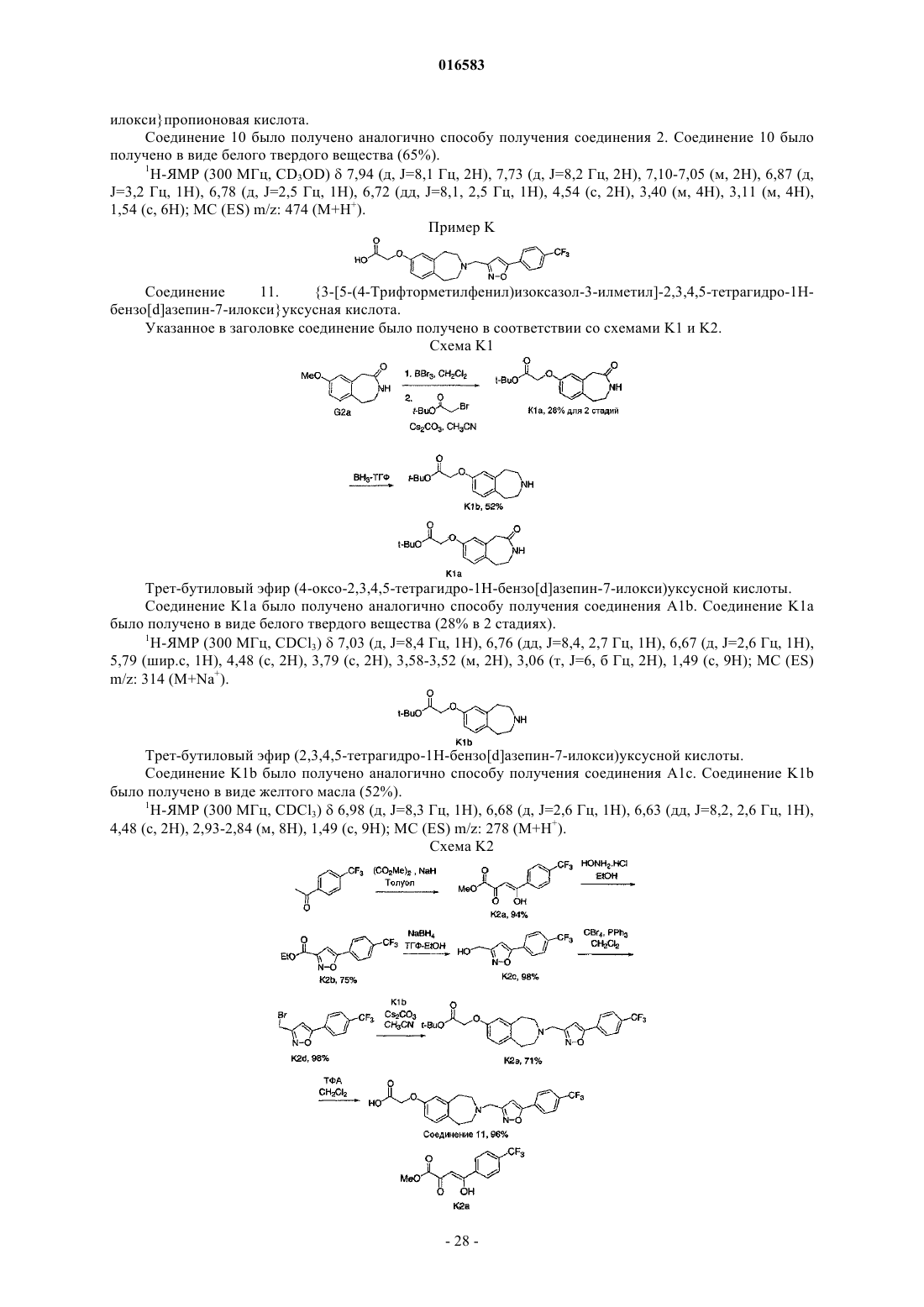
ПРОИЗВОДНЫЕ БЕНЗОАЗЕПИНОКСИУКСУСНОЙ КИСЛОТЫ И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ АГОНИСТОВ ДЕЛЬТА-PPAR ДЛЯ ПОВЫШЕНИЯ HDL-C И СНИЖЕНИЯ Изобретение относится к соединениям формулы (I), пригодным для использования в качестве агонистов PPAR. Данное изобретение относится также к фармацевтическим композициям,содержащим соединения по настоящему изобретению, и способам их применения для лечения одного или нескольких состояний, которые включают, не ограничиваясь ими, диабет, нефропатию,невропатию, ретинопатию, синдром поликистоза яичников, гипертензию, ишемию, удар,спастический колит, воспаление, катаракту, сердечно-сосудистые заболевания, метаболический 016583 Настоящая заявка притязает на приоритет предварительной заявки на патент США 60/793001, поданной 18 апреля 2006 г., которая полностью включена в настоящее описание изобретения в качестве ссылки. Исследование и разработка описанного ниже изобретения не финансировались государством. Уровень техники Рецепторы, активируемые пролифератором пероксисом (PPAR), считаются сенсорами обмена веществ, регулирующими экспрессию генов, участвующих в глюкозном и липидном гомеостазе. Указанные рецепторы являются членами надсемейства ядерных рецепторов гетеродимеров RXR и факторами транскрипции, активируемыми лигандами. Агонисты подтипов PPAR (например, гемфиброзил) иPPAR (например, авандиа) применяют для лечения соответственно дислипидемий и диабета. Данные рецепторы имеют различную степень распределения в тканях, при этом PPAR в большей степени экспрессируется в печени, PPAR экспрессируется в жировой ткани и PPAR, который распространен особенно широко, экспрессируется у взрослых крыс (Braissant et al., 1996) и у человека во многих различных тканях, имеющих отношение к липидному обмену и включающих печень, почки, жировую ткань брюшной полости и скелетные мышцы (Auboeuf et al., 1997). В научной литературе недавно были опубликованы данные о сильнодействующих лигандах дляPPAR, позволяющие лучше понять функцию данного рецептора в липидном обмене (Barak et al., 2002;Oliver et al., 2001; Tanaka et al., 2003; Wang et al., 2003). Указанные соединения повышали холестерин липопротеина высокой плотности (HDL-C) и снижали содержание триглицеридов, оказывая незначительное воздействие на глюкозу, у мышей db/db (Leibowitz et al., 2000) и макак-резусов (Oliver et al.,2001) (хотя у обезьян наблюдалось снижение уровней инсулина). HDL-C удаляет холестерин из периферических клеток в соответствии с процессом, именуемым обратным транспортом холестерина. Первая и ограничивающая скорость стадия, которая представляет собой перенос клеточного холестерина и фосфолипидов на аполипопротеин A-I, являющийся компонентом HDL, опосредована ATP-связывающим кластерным транспортером A1 (ABCA1) (Lawn et al., 1999). Активация PPAR, по-видимому, повышает уровни HDL-C благодаря регуляции транскрипции ABCA1 (Oliver et al., 2001). Поэтому индуцируя мРНК АВСА 1 в макрофагах, агонисты PPAR5 могут повышать уровни HDL-C у субъектов и удалять излишек холестерина из макрофагов с высоким содержанием липидов, что является одним из основных факторов развития атеросклеротического поражения. Подобный подход должен стать альтернативой терапии лекарственными средствами на основе статинов, которые оказывают незначительное воздействие на уровни HDL-C и главным образом снижают уровни LDL-C, или фибратами, которые являются единственными коммерчески доступными агонистами PPAR, и характеризуются низкой эффективностью,вызывая лишь умеренное повышение уровней HDL-C. Кроме того, подобно фибратам, агонисты PPAR также уменьшают содержание триглицеридов, являющихся дополнительным фактором риска возникновения сердечно-сосудистых заболеваний. Примеры известных агонистов дельта-PPAR, которые в разной степени пригодны для лечения гиперлипидемии, диабета или атеросклероза, включают L-165041 (Leibowitz et al., 2000) и GW501516(Oliver et al., 2001). Существует постоянная потребность в новых агонистах дельта-PPAR. Кроме того,существует потребность в новых агонистах дельта-PPAR, которые повышают уровень HDL-C и снижают уровень LDL-C и/или холестерина. Существует также потребность в новых агонистах дельта-PPAR для лечения диабета, нефропатии, невропатии, ретинопатии, синдрома поликистоза яичников, гипертензии,ишемии, удара, спастического колита, воспаления, катаракты, сердечно-сосудистых заболеваний, метаболического X синдрома, гипер-LDL-холестеринемии, дислипидемии (включая гипертриглицеридемию,гиперхолестеринемию, смешанную гиперлипидемию и гипо-HDL-холестеринемию), атеросклероза, ожирения и других нарушений, связанных с липидным обменом и осложнениями энергетического гомеостаза. Сущность изобретения Настоящее изобретение относится к соединению формулы (I)R1 и R2 независимо выбирают из группы, состоящей из Н, C1-8 алкила и C1-8 алкила, замещенного гидроксильной группой, или R1, R2 и атом углерода, к которому они присоединены, могут вместе образовывать С 3-7 циклоалкил;R4 и R5 независимо выбирают из Н и C1-8 алкила;R6 и R7 независимо выбирают из группы, состоящей из Н, галогена, C1-3 алкила, C1-3 алкила, заме-1 016583 щенного галогеном, C1-3 алкокси и C1-3 алкокси, замещенного галогеном; и к его энантиомерам, диастереомерам, сольватам или фармацевтически приемлемым солям. Настоящее изобретение также относится к фармацевтическим композициям, содержащим указанное выше соединение, соль или сольват в смеси с фармацевтически приемлемым носителем, наполнителем или разбавителем. Кроме того, изобретение относится к способу лечения или предотвращения заболевания или состояния, которое подвержено воздействию модуляции рецепторов PPAR, у млекопитающего, который включает введение млекопитающему, нуждающемуся в таком лечении или предотвращении, терапевтически эффективного количества указанного выше соединения, соли или сольвата. Подробное описание изобретения Приведенные ниже подчеркнутые термины имеют следующие значения в настоящем описании изобретения:"Алкил" означает насыщенный или ненасыщенный, одновалентный углеводородный радикал с разветвленной цепью, прямой цепью или циклической структурой, полученный в результате удаления одного атома водорода у одного атома углерода исходного алкана, алкена или алкина. Типичные алкильные группы включают, не ограничиваясь ими, метил; этилы, такие как этанил, этенил, этинил; пропилы, такие как пропан-1-ил, пропан-2-ил, циклопропан-1-ил, проп-1-ен-1-ил, проп-1-ен-2-ил, проп-2-ен-1-ил,циклопроп-1-ен-1-ил; циклопроп-2-ен-1-ил, проп-1-ин-1-ил, проп-2-ин-1-ил и т.д.; бутилы, такие как бутан-1-ил, бутан-2-ил, 2-метилпропан-1-ил, 2-метилпропан-2-ил, циклобутан-1-ил, бут-1-ен-1-ил, бут-1 ен-2-ил, 2-метилпроп-1-ен-1-ил, бут-2-ен-1-ил, бут-2-ен-2-ил, бута-1,3-диен-1-ил, бута-1,3-диен-2-ил,циклобут-1-ен-1-ил, циклобут-1-ен-3-ил, циклобута-1,3-диен-1-ил, бут-1-ин-1-ил, бут-1-ин-3-ил, бут-3 ин-1-ил и т.д.; и тому подобные. Для определения конкретных уровней насыщенности используется номенклатура "алканил", "алкенил" и/или "алкинил". В предпочтительных вариантах осуществления изобретения алкильными группами является (C1-C6) алкил, причем (C1-C3) является особенно предпочтительным. Циклическим алкилом может быть, например, С 3-10 алкил; циклоалкилом предпочтительно является С 3-7 циклоалкил."Арил" означает одновалентный ароматический углеводородный радикал, полученный в результате удаления одного атома водорода у одного атома углерода исходной ароматической кольцевой системы. Типичные арильные группы включают, не ограничиваясь ими, радикалы, полученные из ацеантрилена,аценафтилена, ацефенантрилена, антрацена, азулена, бензола, хризена, коронена, флуорантена, флуорена, гексацена, гексафена, гексалена, as-индацена, s-индацена, индана, индена, нафталина, октацена, октафена, окталена, овалена, пента-2,4-диена, пентацена, пенталена, пентафена, перилена, феналена, фенантрена, пицена, плейадена, пирена, пирантрена, рубицена, трифенилена, тринафталина и тому подобных. В предпочтительных вариантах осуществления изобретения арильной группой является (C5-20) арил, причем (С 5-10) является особенно предпочтительным. Особенно предпочтительными арильными группами являются фенильные и нафтильные группы."Алкилокси" означает насыщенный одновалентный углеводородный радикал спирта с разветвленной цепью, прямой цепью или циклической структурой, полученный в результате удаления атома водород у атома кислорода гидроксида спирта. Типичные алкилоксигруппы включают, не ограничиваясь ими, метилокси-; этилокси-; пропилоксигруппы, такие как пропан-1-илокси (СН 3 СН 2 СН 2 О-), пропан-2 илокси СН 3)2 СНО-), циклопропан-1-илокси и т.д.; бутилоксигруппы, такие как бутан-1-илокси, бутан 2-илокси, 2-метилпропан-1-илокси, 2-метилпропан-2-илокси, циклобутан-1-илокси и т.д.; и тому подобные. В предпочтительных вариантах осуществления изобретения алкилоксигруппами являются (C18)алкилоксигруппы, причем (C1-3) являются особенно предпочтительными. Термин "замещенный" служит для определения радикала, в котором один или несколько атомов водорода независимо заменены одинаковыми или различными заместителями, например гидроксилом или-2 016583 галогеном. Применительно к заместителям термин "независимо" означает, что при возможности использования нескольких заместителей такие заместители могут быть одинаковыми или различными. В любой структуре, включающей символ , такой символ означает расположение одной или нескольких открытых валентностей, где часть структуры присоединяется к остальной части молекулы. В настоящем описании изобретения сначала указывается концевая часть обозначенной боковой цепи и затем функциональная группа, примыкающая к точке присоединения. Таким образом, например,заметитель, являющийся "фенилС 1-6 алканиламинокарбонилС 1-6 алкилом", означает группу формулы: Настоящее изобретение относится к соединению формулы (I)R1 и R2 независимо выбирают из группы, состоящей из Н, C1-8 алкила и C1-8 алкила, замещенного гидроксильной группой, или R1, R2 и атом углерода, к которому они присоединены, могут вместе образовывать С 3-7 циклоалкил;R4 и R5 независимо выбирают из Н и C1-8 алкила;R6 и R7 независимо выбирают из группы, состоящей из Н, галогена, C1-3 алкила, C1-3 алкила, замещенного галогеном, C1-3 алкокси и C1-3 алкокси, замещенного галогеном; и его энантиомерам, диастереомерам, сольватам или фармацевтически приемлемым солям. Настоящее изобретение, в частности, относится к соединению формулы (I), в котором R1 и R2 независимо выбирают из группы, состоящей из Н и C1-8 алкила, или R1, R2 и атом углерода, к которому они присоединены, могут вместе образовывать С 3-5 циклоалкил. Другой вариант настоящего изобретения относится к соединению формулы (I), в котором R1 и R2 независимо выбирают из группы, состоящей из Н и СН 3, или R1, R2 и атом углерода, к которому они присоединены, могут вместе образовывать Другой вариант настоящего изобретения относится к соединению формулы (I), в котором R6 и R7 независимо выбирают из группы, состоящей из Н, галогена, C1-3 алкила, замещенного галогеном, C1-3 алкокси и C1-3 алкокси, замещенного галогеном. Другой вариант настоящего изобретения относится к соединению формулы (I), в котором R6 означает Н и R7 выбирают из группы, состоящей из галогена, C1-3 алкила, замещенного галогеном, и C1-3 алкокси, замещенного галогеном. Другой вариант настоящего изобретения относится к соединению формулы (I), в котором R7 выбирают из группы, состоящей из F, CF3 и -O-CF3. Другой вариант настоящего изобретения относится к соединению формулы (I), в котором R5 означает Н, СН 3 или -СН 2 СН 3. Другой вариант настоящего изобретения относится к соединению формулы (I), в котором Q выбирают из группы, состоящей из Другой вариант настоящего изобретения относится к соединению формулы (I), в котором Q выбирают из группы, состоящей из Предпочтительный вариант настоящего изобретения относится к соединению формулы (I), в котором X означает О. Другой вариант настоящего изобретения относится к соединению формулы (I), в котором X означает О;R1, R2, R4 и R5 независимо выбирают из группы, состоящей из Н и C1-3 алкила, или R1, R2 и атом углерода, к которому они присоединены, могут вместе образовывать и R6 и R7 независимо выбирают из группы, состоящей из Н, галогена, C1-3 алкокси, C1-3 алкила, замещенного галогеном и C1-3 алкокси, замещенного галогеном; и его энантиомерам, диастереомерам, сольватам или фармацевтически приемлемым солям. Еще один вариант настоящего изобретения относится к соединению формулы (Ia)R1 и R2 независимо выбирают из группы, состоящей из Н и C1-8 алкила, или R1, R2 и атом углерода, к которому они присоединены, могут вместе образовывать С 3-5 циклоалкил;R6 и R7 независимо выбирают из группы, состоящей из Н, C1-3 алкила, галогена и C1-3 алкила, замещенного галогеном;R4 и R5 независимо выбирают из группы, состоящей из Н и C1-8 алкила; и и его энантиомерам, диастереомерам, сольватам или фармацевтически приемлемым солям.-4 016583 Еще один вариант настоящего изобретения относится к соединению формулы (Ib) где R1 и R2 независимо выбирают из группы, состоящей из Н и СН 3, или R1, R2 и атом углерода, кR4 и R5 независимо выбирают из группы, состоящей из Н, СН 3 и -СН 2 СН 3; и и его энантиомерам, диастереомерам, сольватам или фармацевтически приемлемым солям. Еще один вариант настоящего изобретения относится к соединению формулы (Ic): где R1, R2 и R4 независимо выбирают из группы, состоящей из Н и СН 3, или R1, R2 и атом углерода,; к которому они присоединены, могут вместе образовыватьR7 означает галоген или C1-3 алкил, замещенный галогеном; и и его энантиомерам, диастереомерам, сольватам или фармацевтически приемлемым солям. Один из предпочтительных вариантов настоящего изобретения относится к соединению, выбираемому из группы, состоящей из и его энантиомерам, диастереомерам, сольватам или фармацевтически приемлемым солям. Другой вариант настоящего изобретения относится к соединению, выбираемому из группы, состоящей из которое, по существу, не содержит соответствующий другой энантиомер. Одним из предпочтительных вариантов настоящего изобретения является соединение формулы Другой вариант настоящего изобретения относится к фармацевтической композиции, содержащей одно из описанных выше соединений, или его соль или сольват в смеси с фармацевтически приемлемым носителем, наполнителем или разбавителем. Другой вариант настоящего изобретения относится к способу лечения или предотвращения заболевания или состояния, которое подвержено воздействию модуляции рецепторов PPAR, у млекопитающего, который включает введение млекопитающему, нуждающемуся в таком лечении или предотвращении,терапевтически эффективного количества одного из описанных выше соединений, или его соли или сольвата. Более предпочтительно, указанное заболевание или состояние подвержено воздействию модуляции дельта-PPAR. Указанное терапевтически эффективное количество включает дозу в пределах от около 0,1 до около 15000 мг, предпочтительно от около 50 до около 1000 мг, более предпочтительно от около 100 до около 1000 мг. Другой вариант настоящего изобретения относится к способу лечения или предотвращения заболе-9 016583 вания или состояния, выбираемого из группы, включающей диабет, нефропатию, невропатию, ретинопатию, синдром поликистоза яичников, гипертензию, ишемию, удар, спастический колит, воспаление, катаракту, сердечно-сосудистые заболевания, метаболический X синдром, гипер-LDL-холестеринемию,дислипидемию (включая гипертриглицеридемию, гиперхолестеринемию, смешанную гиперлипидемию и гипо-HDL-холестеринемию), атеросклероз и ожирение, который включает стадию введения млекопитающему, нуждающемуся в таком лечении, терапевтически эффективного количества одного из описанных выше соединений, его соли или сольвата. В способе по изобретению указанное терапевтически эффективное количество включает дозу в пределах от около 0,1 до около 15000 мг, предпочтительно от около 50 до около 1000 мг, более предпочтительно от около 100 до около 1000 мг. Другой вариант настоящего изобретения относится к соединению формулыR1 и R2 независимо выбирают из группы, состоящей из Н, С 1-8 алкила и C1-8 алкила, замещенного гидроксильной группой, или R1, R2 и атом углерода, к которому они присоединены, могут вместе образовывать С 3-7 циклоалкил;R4 и R5 независимо выбирают из Н и C1-8 алкила;R6 и R7 независимо выбирают из группы, состоящей из Н, галогена, C1-3 алкила, C1-3 алкила, замещенного галогеном, C1-3 алкокси и C1-3 алкокси, замещенного галогеном;Q означает С 6-10 арил; и его энантиомерам, диастереомерам, сольватам или фармацевтически приемлемым солям. Еще один вариант настоящего изобретения относится к соединению формулы (I)R1 и R2 независимо выбирают из группы, состоящей из Н, C1-8 алкила и C1-8 алкила, замещенного гидроксильной группой, или R1, R2 и атом углерода, к которому они присоединены, могут вместе образовывать С 3-7 циклоалкил;R4 и R5 независимо выбирают из Н и C1-8 алкила;R6 и R7 независимо выбирают из группы, состоящей из Н, галогена, C1-3 алкила, C1-3 алкила, замещенного галогеном, C1-3 алкокси и C1-3 алкокси, замещенного галогеном; и его энантиомерам, диастереомерам, сольватам или фармацевтически приемлемым солям. Другой вариант настоящего изобретения относится к соединению формулы (I)R1 и R2 независимо выбирают из группы, состоящей из Н, C1-8 алкила и C1-8 алкила, замещенного гидроксильной группой, или R1, R2 и атом углерода, к которому они присоединены, могут вместе образовывать С 3-7 циклоалкил;R6 и R7 независимо выбирают из группы, состоящей из Н, галогена, C1-3 алкила, C1-3 алкила, замещенного галогеном, C1-3 алкокси и C1-3 алкокси, замещенного галогеном; и его энантиомерам, диастереомерам, сольватам или фармацевтически приемлемым солям. Предпочтительный вариант настоящего изобретения относится к соединению, выбираемому из группы, состоящей из Фармацевтические композиции согласно изобретению могут быть использованы для лечения состояния, прямо или косвенно опосредованного дельта-PPAR. Способы согласно настоящему изобретению предназначены для лечения или предотвращения указанных выше заболеваний или состояний у млекопитающего, более конкретно - у человека. Соединения по настоящему изобретению могут присутствовать в форме фармацевтически приемлемых солей. Соли соединений по настоящему изобретению, предназначенные для применения в медицине, являются нетоксичными "фармацевтически приемлемыми солями" (Ref. International J. Pharm.,1986, 33, 201-217; J. Pharm. Sci., 1997 (Jan), 66, 1, 1). Однако при получении соединений по настоящему изобретению или их фармацевтически приемлемых солей могут быть использованы другие соли, хорошо известные специалистам в данной области. Типичные органические или неорганические кислоты включают, не ограничиваясь ими, хлористо-водородную, бромисто-водородную, иодисто-водородную, перхлорную, серную, азотную, фосфорную, уксусную, пропионовую, гликолевую, молочную, янтарную,малеиновую, фумаровую, яблочную, винную, лимонную, бензойную, миндальную, метансульфоновую,гидроксиэтансульфоновую, бензолсульфоновую, щавелевую, памовую, 2-нафталинсульфоновую, паратолуолсульфоновую, циклогексансульфамовую, салициловую, сахариновую или трифторуксусную кислоту. Типичные органические или неорганические основания включают, не ограничиваясь ими, основные или катионные соли, такие как бензатин, хлорпрокаин, холин, диэтаноламин, этилендиамин, меглумин, прокаин, алюминий, кальций, литий, магний, калий, натрий и цинк. Соединения по настоящему изобретению, имеющие по меньшей мере один хиральный центр, могут существовать в виде энантиомеров. Соединения, имеющие два или более хиральных центров, могут дополнительно существовать в виде диастереомеров. Очевидно, что все такие изомеры и их смеси входят в объем настоящего изобретения. Кроме того, некоторые кристаллические формы указанных соединений могут существовать в виде полиморфов, которые также входят в объем настоящего изобретения. Некоторые соединения могут образовывать сольваты с водой (то есть гидраты) или с обычными органическими растворителями. Если в процессе получения соединений по настоящему изобретению образуется смесь стереоизомеров, такие изомеры могут быть разделены стандартными методами, такими как препаративная хроматография. Соединения могут быть получены в рацемической форме, либо отдельные энантиомеры могут- 11016583 быть получены путем энантиоспецифического синтеза или разделения. Соединения, например, могут быть разделены на составляющие их энантиомеры стандартными методами, такими как образование диастереомерных пар в результате образования соли с оптически активной кислотой, такой как (-)-дипаратолуоил-d-винная кислота и/или (+)-ди-паратолуоил-1-винная кислота с последующим выполнением фракционированной кристаллизации и восстановлением свободного основания. Соединения могут быть также разделены путем образования диастереомерных сложных эфиров или амидов с последующим хроматографическим разделением и удалением дополнительного хирального центра. Альтернативно соединения по настоящему изобретению могут быть разделены в колонке для хиральной ВЭЖХ. В контексте настоящего описания изобретения выражение "по существу, не содержит" означает наличие менее 25%, предпочтительно менее 10%, более предпочтительно менее 5%, еще предпочтительнее менее 2% и наиболее предпочтительно менее 1% изомера, высчитанное по приведенным ниже формулам. В любом процессе получения соединений по настоящему изобретению может быть необходимо и/или желательно защитить восприимчивые или реакционноспособные группы в любых рассмотренных молекулах. Такая задача может быть решена при помощи стандартных защитных групп, описанных в публикациях Protective Groups in Organic Chemistry, ed. J.F.W. McOmie, Plenum Press, 1973, и T.W. GreeneP.G.M. Wuts, Protective Groups in Organic Synthesis, John WileySons, 1991. Защитные группы могут быть удалены на последующей стадии методами, известными в данной области. Хотя соединения по настоящему изобретению (в том числе их фармацевтически приемлемые соли и фармацевтически приемлемые сольваты) могут быть введены отдельно, обычно их вводят в смеси с фармацевтическим носителем, наполнителем или разбавителем, выбираемым с учетом предполагаемого способа введения и стандартной фармацевтической или ветеринарной практики. Таким образом, настоящее изобретение относится к фармацевтическим и ветеринарным композициям, содержащим соединения формулы (I) и один или несколько фармацевтически приемлемых носителей, наполнителей или разбавителей. В качестве примера можно отметить, что в фармацевтических композициях по настоящему изобретению соединения по настоящему изобретению могут быть смешаны с любыми приемлемыми связывающими веществами, смазывающими веществами, суспендирующими агентами, покрывающими агентами и/или солюбилизирующими агентами. Таблетки и капсулы, содержащие соединения по настоящему изобретению, можно вводить по одной, двум или более за один прием. Кроме того, указанные соединения можно вводить в препаратах пролонгированного действия. Альтернативно соединения общей формулы (I) можно вводить путем ингаляции или в форме суппозитория или вагинального суппозитория, либо их можно применять местно в форме лосьона, раствора,крема, мази или присыпки. Альтернативным способом чрескожного введения является использование кожного пластыря. Например, указанные соединения могут быть введены в крем, состоящий из водной эмульсии полиэтиленгликолей или жидкого парафина. Соединения по настоящему изобретению можно также вводить в концентрации от 1 до 10 мас.% в мазь на основе белого воска или бесцветного мягкого парафина вместе с требуемыми стабилизаторами и консервантами. В некоторых применениях указанные композиции предпочтительно вводят перорально в форме таблеток, содержащих наполнители, такие как крахмал или лактоза, в капсулах или оболочках отдельно или в смеси с наполнителями либо в форме эликсиров, растворов или суспензий, содержащих ароматизаторы или красители. Композиции (а также отдельные соединения) можно также вводить парентерально, например внутриполостно, внутривенно, внутримышечно или подкожно. В данном случае композиции содержат приемлемый носитель или разбавитель. Композиции, предназначенные для парентерального введения, лучше всего использовать в форме стерильного водного раствора, который может содержать другие вещества, например достаточное количество солей или моносахаридов для получения изотонического раствора по отношению к крови. Композиции, предназначенные для трансбуккального или подъязычного введения, могут иметь форму таблеток или лепешек, получаемых стандартным методом. В качестве другого примера можно привести фармацевтические композиции, содержащие одно или несколько соединений по настоящему изобретению, в которых активный ингредиент может быть получен путем однородного смешивания одного или нескольких соединений с фармацевтическим носителем стандартными методами приготовления фармацевтических препаратов. Носитель может находиться в- 12016583 различных формах в зависимости от требуемого способа введения (например, перорального, парентерального). Таким образом, для жидких препаратов для перорального введения, таких как суспензии,эликсиры и растворы, приемлемыми носителями и добавками являются вода, гликоли, масла, спирты,ароматизаторы, консерванты, стабилизаторы, красители и тому подобные; для твердых препаратов для перорального введения, таких как порошки, капсулы и таблетки, приемлемыми носителями и добавками являются крахмалы, сахара, разбавители, гранулирующие агенты, смазывающие вещества, связывающие вещества, дезинтеграторы и тому подобные. На твердые препараты для перорального введения могут быть также нанесены покрытия из таких веществ, как сахара или энтеросолюбильные покрытия, позволяющие изменить основное место абсорбции. В композиции для парентерального введения для улучшения растворимости или сохранности может быть добавлен носитель, обычно состоящий из стерильной воды и других ингредиентов. Инъецируемые суспензии или растворы могут быть также получены при использовании водных носителей наряду с соответствующими добавками. Соединения по настоящему изобретению можно вводить в виде однократной суточной дозы, либо общая суточная доза может быть разделена на несколько доз, вводимых два, три или четыре раза в сутки. Кроме того, соединения по настоящему изобретению можно вводить в интраназальной форме путем местного применения приемлемых интраназальных наполнителей или в виде чрескожных пластырей, хорошо известных специалистам в данной области. Специалисту в данной области должно быть также известно, что терапевтически эффективная доза активных соединений по настоящему изобретению или фармацевтической композиции, содержащей такие соединения, может изменяться в соответствии с требуемым эффектом. Поэтому предназначенные для введения оптимальные дозы могут быть легко определены и изменены в зависимости от конкретного вводимого соединения, способа введения, эффективности препарата и тяжести заболевания. Кроме того,доза может быть доведена до соответствующего терапевтического уровня с учетом факторов, характерных для конкретного субъекта, подвергаемого лечению, которые включают возраст, массу тела, режим питания субъекта и продолжительность введения. Таким образом, приведенные выше дозы являются иллюстративными для среднестатистического случая. Конечно, могут быть индивидуальные ситуации,требующие введения более высокой или более низкой дозы, которые также входят в объем настоящего изобретения. Соединения по настоящему изобретению можно вводить, используя любые вышеуказанные композиции и схемы приема или принятые в данной области композиции и схемы приема, когда применение соединений по настоящему изобретению необходимо для лечения нуждающегося в таком лечении субъекта. Настоящее изобретение относится также к фармацевтической упаковке или набору, состоящему из одного или нескольких контейнеров, заполненных одним или несколькими ингредиентами фармацевтических и ветеринарных композиций по настоящему изобретению. К таким контейнерам может необязательно прилагаться уведомление в форме, установленной государственным агентством, регулирующим изготовление, применение или продажу фармацевтических средств или биологических продуктов, которое должно содержать информацию о разрешении, предоставленном данным агентством на изготовление, применение или продажу данного средства для введения человеку. Фармацевтическую композицию, предназначенную для перорального введения, предпочтительно получают в форме таблеток, содержащих 0,01, 10,0, 50,0, 100, 150, 200, 250 и 500 мг активного ингредиента для симптоматического выбора дозы для нуждающегося в лечении субъекта. Примеры заболеваний, входящих в объем настоящего изобретения, включают, не ограничиваясь ими, диабет, нефропатию, невропатию, ретинопатию, синдром поликистоза яичников, гипертензию,ишемию, удар, спастический колит, воспаление, катаракту, сердечно-сосудистые заболевания, метаболический X синдром, гипер-LDL-холестеринемию, дислипидемию (включая гипертриглицеридемию, гиперхолестеринемию, смешанную гиперлипидемию и гипо-HDL-холестеринемию), атеросклероз, ожирение и другие заболевания, обусловленные липидным обменом и осложнениями энергетического гомеостаза. Соединения по настоящему изобретению могут быть использованы также в качестве агонистов дельта-PPAR для лечения, предотвращения или подавления развития заболевания, прямо или косвенно опосредованного дельта-PPAR. Соединения по настоящему изобретению особенно пригодны для лечения диабета, нефропатии,невропатии, ретинопатии, синдрома поликистоза яичников, гипертензии, ишемии, удара, спастического колита, воспаления, катаракты, сердечно-сосудистых заболеваний, метаболический X синдром, гиперLDL-холестеринемии, дислипидемии (включая гипертриглицеридемию, гиперхолестеринемию, смешанную гиперлипидемию и гипо-HDL-холестеринемию), атеросклероза, ожирения и других заболеваний,обусловленных липидным обменом и осложнениями энергетического гомеостаза. Терапевтически эффективная доза соединений по настоящему изобретению, применяемых для лечения вышеуказанных заболеваний и состояний, может быть определена специалистами в данной области при помощи общепринятых животных моделей. Такая доза, по-видимому, должна находиться для среднестатистического человека (70 кг) в пределах от около 0,01 до около 15000 мг активного ингреди- 13016583 ента, вводимого 1-4 раза в сутки. Общие методы синтеза Типичные соединения по настоящему изобретению могут быть синтезированы в соответствии с описанными ниже общими методами синтеза, а также приведенными далее иллюстративными примерами. Так как схемы носят иллюстративный характер, настоящее изобретение не ограничено представленными химическими реакциями и условиями их выполнения. Получение различных исходных веществ,показанных на схемах, находится в компетенции специалистов в данной области. Схема 1. Общие методы синтеза соединений формулы (Id) На схеме 1, где R1, R2, R3, R4, R5, R6, R7, X и Q имеют указанные выше значения, показано несколько общих методов синтеза соединений формулы Id. Например, в методе 1 соответствующее соединение формулы Id может быть получено путем алкилирования замещенного бензоазепина 1-В соединением формулы 1-С, где Y может быть удаляемой группой, такой как Br, Cl, I, мезилат и т.д., в основных условиях, таких как Cs2CO3 в CH3CN. В методе 2 соединение формулы Id получают путем восстановительного аминирования соединения 1-В арилальдегидом 1-D (например, R5 означает Н) при использованииNaBH(OAc)3; или осуществляя взаимодействие соединения 1-В с арилкетоном (например, R5 означаетC1-C3 алкил) с образованием основания Шиффа, которое затем восстанавливают NaCNBH3. В методе 3 соединение формулы Id получают, осуществляя взаимодействие соединения 1-В с арилальдегидом 1-Е с образованием основания Шиффа, которое затем подвергают взаимодействию с алкилорганическими реагентами, такими как реагенты Гриньяра, CH3Li или медьорганический реагент. Схема 2. Общие методы синтеза соединений формулы (Ie)- 14016583 На схеме 2, где R1, R2, R3, R4, R5, R6, R7, X и Q имеют указанные выше значения, показано несколько общих методов синтеза соединений формулы Ie. Например, в методе 1 соединение формулы Ie получают путем восстановительного аминирования соединения 1-В арилальдегидом 2-А (например, R5 означает Н) при использовании NaBH(OAc)3; или осуществляя взаимодействие соединения 1-В с арилкетоном (например, R5 означает C1-C3 алкил) с образованием основания Шиффа, которое затем восстанавливаютNaCNBH3. В методе 2 соединение формулы Ie получают, осуществляя взаимодействие соединения 1-В с арилальдегидом 2-В с образованием основания Шиффа, которое затем подвергают взаимодействию с алкилорганическими реагентами, такими как реагенты Гриньяра, CH3Li или медьорганический реагент. Хиральные соединения формулы (I) могут быть разделены на энантиомеры при помощи хроматографии в хиральной стационарной фазе. Альтернативно основные соединения по настоящему изобретению могут быть превращены в диастереомерные соли путем смешивания с хиральной кислотой и разделения на энантиомеры при помощи фракционированной кристаллизации. Соответствующий продукт, полученный на каждой стадии способа, обычно желательно отделить от других компонентов реакционной смеси и подвергнуть очистке, прежде чем использовать его в качестве исходного вещества на следующей стадии. Методы разделения обычно включают упаривание, экстракцию, осаждение и фильтрацию. Методы очистки обычно включают хроматографию на колонках (Still,W.C. et al., J. Org. Chem. 1978, 43, 2921), тонкослойную хроматографию, кристаллизацию и перегонку. Структуры конечных продуктов, промежуточных соединений и исходных веществ подтверждены спектроскопическими, спектрометрическими и аналитическими методами, которые включают спектроскопию ядерного магнитного резонанса (ЯМР), масс-спектрометрию (МС) и жидкостную хроматографию(ВЭЖХ). В описании способов получения соединений по настоящему изобретению этиловый эфир, тетрагидрофуран и диоксан являются общими примерами эфирных растворителей; бензол, толуол, гексаны и гептаны являются типичными углеводородными растворителями; и дихлорметан и дихлорэтан являются типичными галогенированными углеводородными растворителями. В тех случаях, когда продукт выделяют в виде кислотно-аддитивной соли, свободное основание может быть получено методами, известными специалистам в данной области. В тех случаях, когда продукт выделяют в виде кислотноаддитивной соли, соль может содержать один или несколько эквивалентов кислоты. Энантиомеры соединений по настоящему изобретению могут быть разделены при помощи хиральной ВЭЖХ. Аббревиатуры: Ас - СН 3 С(О)-; водн. - водный; Соединение 1. 3-[3-(4-Трифторметилфенил)изотиазол-5-илметил]-2,3,4,5-тетрагидро-1 Н-бензо[d] азепин-7-илоксиуксусная кислота. Указанное в заголовке соединение было получено в соответствии со схемами А 1 и А 2. Схема А 1 Соединение A1a может быть получено в соответствии с опубликованными способами (патент США 4659705 и заявка на европейский патент 204349).CH2Cl2, 11,4 мл, 11,4 ммоль). Реакционную смесь оставляли нагреваться до комнатной температуры и перемешивали при комнатной температуре в течение 5 ч. Реакционную смесь гасили, медленно добавляя МеОН (5 мл). Затем реакционную смесь концентрировали, получая желтое твердое вещество. Смесь вышеуказанного неочищенного фенола, этилбромацетата (950 мг, 5,69 ммоль) и Cs2CO3 (2,47 г, 7,58 ммоль) в CH3CN (15 мл) перемешивали при 80 С в течение 20 ч. Смесь охлаждали до комнатной температуры, распределяли между EtOAc и H2O и водный слой экстрагировали EtOAc. Объединенные органические фазы сушили (Na2SO4), концентрировали и очищали колоночной хроматографией, получая 350 мг (35%) соединения A1b в виде белого твердого вещества. 1 Н-ЯМР (300 МГц, CDCl3)7,04 (д, J=8,4 Гц, 1 Н), 7,77 (дд, J=8,4, 2,7 Гц, 1 Н), 6,69 (д, J=2,5 Гц, 1 Н),5,83 (шир.с, 1 Н), 4,59 (с, 2 Н), 4,27 (кв., J=7,1 Гц, 2 Н), 3,80 (с, 2 Н), 3,58-3,52 (м, 2 Н), 3,06 (т, J=6,0 Гц, 2 Н),1,30 (т, J=7,1 Гц, 3H); МС (ES) m/z: 286 (M+Na+). Этиловый эфир (2,3,4,5-тетрагидро-1 Н-бензо[d]азепин-7-илокси)уксусной кислоты. 1 М борана в ТГФ (1 мл, 1,02 ммоль) по каплям добавляли к охлаждаемому льдом и перемешиваемому раствору соединения A1b (90 мг, 0,342 ммоль) в ТГФ (5 мл). Смесь продолжали перемешивать при 0 С в течение 1 ч и затем при комнатной температуре в течение 20 мин. Раствор снова охлаждали до 0 С и медленно добавляли 1 н. HCl (2 мл), чтобы удалить избыток борана. Раствор перемешивали при комнатной температуре в течение 15 мин и концентрировали для удаления ТГФ. Водный раствор промывалиEtOAc, подщелачивали Na2CO3 до рН больше 10 и экстрагировали EtOAc. Объединенные органические фазы сушили (Na2SO4), концентрировали и сушили в вакууме, получая 43 мг (48%) соединения A1c в виде желтого гелеобразного масла. 1 Н-ЯМР (300 МГц, CDCl3)6,99 (д, J=8,4 Гц, 1 Н), 6,70 (с, 1 Н), 6,60 (д, J=8,4 Гц, 1 Н), 4,59 (с, 2 Н),4,28 (кв., J=7,1 Гц, 2 Н), 2,97 (шир.м, 4 Н), 2,84 (шир.м, 4 Н), 2,67 (с, 1 Н), 1,30 (т, J-7,1 Гц, 3H); МС (ES)(11,3 г, 59,8 ммоль), хлоркарбонилсульфенилхлорида (10,1 мл, 119,6 ммоль) в толуоле (120 мл) нагревали при 80 С в течение 15 ч, охлаждали и концентрировали. Твердое вещество переносили в воронку со спекшимся материалом, промывали небольшим количеством этанола и сушили в вакууме, получая 13,4 г Этиловый эфир 3-(4-трифторметилфенил)изотиазол-5-карбоновой кислоты. Реакционную смесь соединения А 2 а (608 мг, 2,46 ммоль) и этилпропиолата (72 6 мг, 7,41 ммоль) в хлорбензоле (10 мл) нагревали при 135 С в течение 20 ч. ТСХ показала наличие некоторого количества исходного вещества А 2 а. Добавляли дополнительное количество этилпропиолата (726 мг, 7,41 ммоль) и 1,2-дихлорбензол (10 мл) и раствор нагревали при 160 С в течение 7 ч. Реакционную смесь охлаждали до комнатной температуры и очищали колоночной хроматографией, получая 229 мг (31%) соединения А 2b в виде белого твердого вещества. 1 Н-ЯМР (300 МГц, CDCl3)8,15 (с, 1 Н), 8,09 (d, J=8,1 Гц, 2 Н), 7,73 (д, J=8,2 Гц, 2 Н), 4,44 (кв., J=7,1 Гц, 2 Н), 1,43 (т, J=7,1 Гц, 3H); МС (ES) m/z: 302 (М+Н+).(0,21 мл, 0,21 ммоль) в ТГФ. Реакционную смесь перемешивали при -78 С в течение 30 мин, медленно добавляли воду и смесь оставляли нагреваться до комнатной температуры. Осажденное твердое вещество фильтровали и промывали CH2Cl2. Фильтрат промывали насыщенным раствором NH4Cl и водный раствор подвергали обратной экстракции CH2Cl2. Объединенные органические фазы сушили и концентрировали, получая 98 мг неочищенного соединения А 2 с в виде желтого твердого вещества. 1 Н-ЯМР (300 МГц, CDCl3)8,04 (д, J=8,3 Гц, 2 Н), 7,70 (д, J=8,4 Гц, 2 Н), 7,51 (с, 1 Н), 5,06 (с, 2 Н); МС (ES) m/z: 260 (М+Н+).CH2Cl2 (25 мл) при 0 С добавляли PPh3 (1,3 г, 5,01 ммоль) и CBr4 (1,7 г, 5,01 ммоль). Реакционную смесь оставляли нагреваться до комнатной температуры и перемешивали в течение 4 ч при комнатной температуре. Реакционную смесь концентрировали и очищали колоночной хроматографией, получая 1,36 г(98%) соединения A2d в виде белого твердого вещества. Этиловый эфир 3-[3-(4-трифторметилфенил)изотиазол-5-илметил]-2,3,4,5-тетрагидро-1 Н-бензо[d] азепин-7-илоксиуксусной кислоты. Смесь соединения A1e (15 мг, 0,060 ммоль), соединения A2d (23 мг, 0,072 ммоль) и Et3N (20 мг,0,18 ммоль) в CH3CN (1 мл) перемешивали в течение 5 ч. Добавляли EtOAc и H2O и водный слой экстрагировали EtOAc. Объединенные органические фазы сушили (Na2SO4), концентрировали и очищали колоночной хроматографией, получая 20 мг (68%) соединения А 2 е в виде белого твердого вещества. 1 3-[3-(4-Трифторметилфенил)изотиазол-5-илметил]-2,3,4,5-тетрагидро-1 Н-бензо[d]азепин-7 илоксиуксусная кислота. Смесь соединения А 2 е (20 мг, 0,041 ммоль) и 2 М NaOH (41 мкл, 0,082 ммоль) в ТТФ-МеОН (0,6 мл-0,2 мл) перемешивали в атмосфере N2 в течение 2 ч и концентрировали. Добавляли CH2Cl2 и воду и смесь подкисляли концентрированной HCl. Органическую фазу отделяли и водную фазу экстрагировалиCH2Cl2. Объединенные органические слои сушили (Na2SO4), концентрировали и очищали колоночной хроматографией, получая 15 мг (80%) соединения 1 в виде белого твердого вещества. 1 Соединение 2. 2-Метил-2-3-[3-(4-трифторметилфенил)изотиазол-5-илметил]-2,3,4,5-тетрагидро 1 Н-бензо[d]азепин-7-илоксипропионовая кислота. Указанное в заголовке соединение было получено в соответствии со схемой В. Схема ВCH2Cl2, 62,8 мл, 62,8 ммоль). Смесь оставляли нагреваться до комнатной температуры и перемешивали при комнатной температуре в течение 5 ч. Реакционную смесь гасили, медленно добавляя МеОН (5 мл). Затем реакционную смесь концентрировали, получая желтое твердое вещество. Смесь вышеуказанного неочищенного фенола, этилбромизобутирата (6,1 г, 31,4 ммоль) и Cs2CO3(20,8 г, 63,8 ммоль) в CH3CN (200 мл) перемешивали при 80 С в течение 20 ч. Смесь охлаждали до комнатной температуры, распределяли между EtOAc и H2O и водный слой экстрагировали EtOAc. Объединенные органические фазы сушили (Na2SO4), концентрировали и очищали колоночной хроматографией,получая 4,2 г (70%) соединения В 1 в виде белого твердого вещества. 1 Н-ЯМР (300 МГц, CD3OD)7,03 (д, J=8,4 Гц, 1 Н), 6,68 (дд, J=8,3, 2,6 Гц, 1 Н), 6,63 (д, J=2,6 Гц,1 Н), 4,21 (кв., J=7,1 Гц, 2 Н), 3,76 (с, 2 Н), 3,57-3,52 (м, 2 Н), 3,04 (т, J=6,0 Гц, 2 Н), 1,53 (с, 6 Н), 1,24 (т,J=7,1 Гц, 3H); МС (ES) m/z: 314 (M+Na+). Этиловый эфир 2-метил-2-(2,3,4,5-тетрагидро-1 Н-бензо[d]азепин-7-илокси)пропионовой кислоты. 1 М борана в ТГФ (20,5 мл, 20,5 ммоль) по каплям добавляли к охлаждаемому льдом и перемешиваемому раствору соединения В 1 (2,0 г, 6,85 ммоль) в ТГФ (20 мл). Смесь продолжали перемешивать при комнатной температуре в течение 1 ч. Раствор снова охлаждали до 0 С и медленно добавляли 1 н.HCl (25 мл), чтобы удалить избыток борана. Раствор перемешивали при комнатной температуре в течение 15 мин и концентрировали для удаления ТГФ. Водный раствор промывали EtOAc, подщелачивалиNa2CO3 до рН больше 10 и экстрагировали EtOAc. Объединенные органические фазы сушили (Na2SO4) и концентрировали, получая 1,5 г (79%) соединения В 2 в виде бесцветного гелеобразного масла. 1 Н-ЯМР (300 МГц, CDCl3)6,95 (д, J=8,4 Гц, 1 Н), 6,64 (д, J=2,5 Гц, 1 Н), 6,55 (дд, J=8,4, 2,6 Гц, 1 Н),4,22 (кв., J=7,1 Гц, 2 Н), 2,99 (м, 2 Н), 2,88 (м, 2 Н), 1,59 (с, 6 Н), 1,25 (т, J=7,1 Гц, 3H); МС (ES) m/z: 278 Этиловый эфир 2-метил-2-3-[3-(4-трифторметилфенил)изотиазол-5-илметил]-2,3,4,5-тетрагидро 1 Н-бензо[d]азепин-7-илоксипропионовой кислоты. Соединение В 3 было получено аналогично способу получения соединения А 2 е при замене соединения A1b соединением В 2 (белое твердое вещество, 72%). 1 Н-ЯМР (300 МГц, CDCl3)8,05 (д, J=8,4 Гц, 2 Н), 7,69 (д, J=8,4 Гц, 2 Н), 7,55 (с, 1 Н), 6,93 (д, J=8,2 Гц, 1 Н), 6,64 (д, J=2,5 Гц, 1 Н), 6,57 (дд, J=8,2, 2,5 Гц, 1 Н), 4,23 (кв., J=7,1 Гц, 2 Н), 4,05 (с, 2 Н), 2,92 (м,4 Н), 2,80 (м, 4 Н), 1,57 (с, 6 Н), 1,25 (т, J=7,1 Гц, 3H); МС (ES) m/z: 519 (М+Н+). 2-Метил-2-3-[3-(4-трифторметилфенил)изотиазол-5-илметил]-2,3,4,5-тетрагидро-1 Н-бензо[d]азепин-7-илоксипропионовая кислота. Соединение 2 было получено аналогично способу получения соединения 1. Соединение 2 было получено в виде белого твердого вещества (100%). 1 Н-ЯМР (300 МГц, CD3OD)8,21 (д, J=8,1 Гц, 2 Н), 8,06 (с, 1 Н), 7,80 (д, J=8,2 Гц, 2 Н), 7,09 (д, J=8,2 Гц, 1 Н), 6,78 (д, J=2,5 Гц, 1 Н), 6,72 (дд, J=8,2, 2,5 Гц, 1 Н), 4,74 (с, 2 Н), 3,33 (м, 4 Н), 3,11 (м, 4 Н), 1,54 (с,6 Н); МС (ES) m/z: 491 (М+Н+). Пример С Соединение 3. 3-[3-(4-Трифторметилфенил)[1,2,4]тиадиазол-5-илметил]-2,3,4,5-тетрагидро-1 Нбензо[d]азепин-7-илоксиуксусная кислота. Указанное в заголовке соединение было получено в соответствии со схемой С. Этиловый эфир 3-(4-трифторметилфенил)[1,2,4]тиадиазол-5-карбоновой кислоты. Реакционную смесь соединения А 2 а (448 мг, 1,81 ммоль) и этилцианоформиата (722 мг, 7,29 ммоль) в 1,2-дихлорбензоле (7 мл) нагревали при 160 С в течение 20 ч. Реакционную смесь охлаждали до комнатной температуры и очищали колоночной хроматографией, получая 505 мг (92%) соединения С 1 в виде желтого твердого вещества. 1[3-(4-Трифторметилфенил)[1,2,4]тиадиазол-5-ил]метанол. К раствору соединения С 1 (200 мг, 0,662 ммоль) в EtOH (10 мл) при комнатной температуре добавляли NaBH4 (64 мг, 1,7 ммоль). Смесь перемешивали в течение 2 ч и добавляли несколько капель воды,чтобы погасить избыток гидрида. EtOH выпаривали и остаток распределяли между CH2Cl2 и водой. Органическую фазу сушили (Na2SO4) и концентрировали, получая 167 мг (97%) соединения С 2 в виде не совсем белых кристаллов. 1 Н-ЯМР (300 МГц, CDCl3)8,40 (д, J=8,1 Гц, 2 Н), 7,74 (д, J=8,2 Гц, 2 Н), 5,20 (с, 2 Н), 2,65 (шир.,1 Н); МС (ES) m/z: 261 (М+Н+). 5-Бромметил-3-(4-трифторметилфенил)[1,2,4]тиадиазол. Соединение С 3 было получено аналогично способу получения соединения A2d. Соединение С 3 было получено в виде белого твердого вещества (96%). 1 Н-ЯМР (300 МГц, CDCl3)8,40 (д, J=8,1 Гц, 2 Н), 7,74 (д, J=8,3 Гц, 2 Н), 4,83 (с, 2 Н); МС (ES) m/z: 321 (М-Н+). Этиловый эфир 3-[3-(4-трифторметилфенил)[1,2,4]тиадиазол-5-илметил]-2,3,4,5-тетрагидро-1 Нбензо[d]азепин-7-илоксиуксусной кислоты. Соединение С 4 было получено аналогично способу получения соединения А 2 е. Соединение С 4 было получено в виде белого твердого вещества (50%). 3-[3-(4-Трифторметилфенил)[1,2,4]тиадиазол-5-илметил]-2,3,4,5-тетрагидро-1 Н-бензо[d]азепин-7 илоксиуксусная кислота. Соединение 3 было получено аналогично способу получения соединения 1. Соединение 3 было получено в виде белого твердого вещества (85%). 1 Н-ЯМР (300 МГц, CD3OD)8,48 (д, J=7,7 Гц, 2 Н), 7,83 (д, J=6,8 Гц, 2 Н), 7,07 (д, J=8,5 Гц, 1 Н), 6,79 Соединение: 2-метил-2-3-[3-(4-трифторметилфенил)[1,2,4]тиадиазол-5-илметил]-2,3,4,5-тетрагидро-1 Н-бензо[d]азепин-7-илоксипропионовая кислота. Указанное в заголовке соединение было получено в соответствии со схемой D. Схема D Этиловый эфир 2-метил-2-3-[3-(4-трифторметилфенил)[1,2,4]тиадиазол-5-илметил]-2,3,4,5-тетрагидро-1 Н-бензо[d]азепин-7-илоксипропионовой кислоты. Соединение D1 было получено аналогично способу получения соединения В 3. Соединение D1 было получено в виде белого твердого вещества (76%). 1[d]азепин-7-илоксипропионовая кислота. Соединение 4 было получено аналогично способу получения соединения 2. Соединение 4 было получено в виде белого твердого вещества (94%). 1- 21016583 Указанное в заголовке соединение было получено в соответствии со схемой Е. Схема Е 5-Бромметил-4-метил-2-(4-трифторметилфенил)тиазол. Соединение Е 2 было получено из соединения El (Bioorg.Med. Chem. Lett., 2003, 13(9), 1517-1521) аналогично способу получения соединения A2d. Соединение Е 2 было получено в виде белого твердого вещества (96%). 1 Этиловый эфир 3-[4-метил-2-(4-трифторметилфенил)тиазол-5-илметил]-2,3,4,5-тетрагидро-1 Нбензо[d]азепин-7-илоксиуксусной кислоты. Соединение Е 3 было получено аналогично способу получения соединения А 2 е. Соединение Е 3 было получено в виде белого твердого вещества (53%). 1 Н-ЯМР (300 МГц, CDCl3)8,02 (д, J=8,2 Гц, 2 Н), 7,68 (д, J=8,3 Гц, 2 Н), 7,00 (д, J=8,2 Гц, 1 Н), 6,70 3-[4-метил-2-(4-трифторметилфенил)тиазол-5-илметил]-2,3,4,5-тетрагидро-1 Н-бензо[d]азепин-7 илоксиуксусная кислота Соединение 5 было получено аналогично способу получения соединения 1. Соединение 5 было получено в виде белого твердого вещества (85%). 1 Соединение 6. 2-Метил-2-3-[4-метил-2-(4-трифторметилфенил)тиазол-5-илметил]-2,3,4,5-тетрагидро-1 Н-бензо[d]азепин-7-илоксипропионовая кислота. Указанное в заголовке соединение было получено в соответствии со схемой F. Этиловый эфир 2-метил-2-3-[4-метил-2-(4-трифторметилфенил)тиазол-5-илметил]-2,3,4,5-тетрагидро-1 Н-бензо[d]азепин-7-илоксипропионовой кислоты Соединение F1 было получено аналогично способу получения соединения В 3. Соединение F1 было получено в виде белого твердого вещества (71%). 1 Н-ЯМР (300 МГц, CDCl3)8,02 (д, J=8,2 Гц, 2 Н), 7,65 (д, J=8,3 Гц, 2 Н), 6,93 (д, J=8,2 Гц, 1 Н), 6,63 2-метил-2-3-[4-метил-2-(4-трифторметилфенил)тиазол-5-илметил]-2,3,4,5-тетрагидро-1 Н-бензо[d] азепин-7-илоксипропионовая кислота. Соединение 6 было получено аналогично способу получения соединения 2. Соединение 6 было получено в виде белого твердого вещества (75%). 1 Н-ЯМР (300 МГц, CD3OD)8,13 (д, J=8,2 Гц, 2 Н), 7,80 (д, J=8,3 Гц, 2H), 7,04 (д, J=8,2 Гц, 1 Н), 6,75 Соединение 7. 3-[5-(4-Трифторметилфенил)тиофен-2-илметил]-2,3,4,5-тетрагидро-1 Н-бензо[d] азепин-7-илоксиуксусная кислота. Указанное в заголовке соединение было получено в соответствии со схемами G1 или G2. Схема G1- 23016583 5-(4-Трифторметилфенил)тиофен-2-карбальдегид. Смесь 5-бромтиофен-2-карбоксиальдегида (2 г, 10,5 ммоль), 4-трифторметилбензолбороновой кислоты (2,19 г, 11,5 ммоль), Pd(PPh4)3 (605 мг, 0,52 ммоль) и 2 н. Na2CO3 (21 мл, 42 ммоль) в толуоле/МеОН (30 мл/15 мл) дегазировали N2 и затем перемешивали при 80 С в течение 18 ч. Реакционную смесь охлаждали до комнатной температуры, распределяли между EtOAc и H2O и водный слой экстрагировали EtOAc. Объединенные органические фазы сушили (Na2SO4), концентрировали и очищали колоночной хроматографией, получая соединение G1a (2,4 г, 90%) в виде белого твердого вещества. 1 Этиловый эфир 3-[5-(4-трифторметилфенил)тиофен-2-илметил]-2,3,4,5-тетрагидро-1 Н-бензо[d] азепин-7-илоксиуксусной кислоты. Смесь соединения A1b (100 мг, 0,401 ммоль) и соединения Gla (113 мг, 0,442 ммоль) в дихлорметане (4 мл) перемешивали при комнатной температуре в течение 1,5 ч. Добавляли Na(OAc)3BH (170 мг,0,803 ммоль) и полученную смесь перемешивали в течение 17 ч. Добавляли насыщенный растворNaHCO3 и полученный раствор экстрагировали CH2Cl2. Объединенные органические фазы сушили(Na2SO4), концентрировали и очищали колоночной хроматографией, получая соединение G1b (110 мг,56%) в виде белого твердого вещества. 1 3-[5-(4-трифторметилфенил)тиофен-2-илметил]-2,3,4,5-тетрагидро-1 Н-бензо[d]азепин-7-илокси уксусная кислота. Соединение 7 было получено аналогично способу получения соединения 1. Соединение 7 было получено в виде белого твердого вещества (90%). 1 Н-ЯМР (300 МГц, CD3OD)7,85 (д, J=8,2 Гц, 2 Н), 7,72 (д, J=8,4 Гц, 2 Н), 7,56 (д, J=3,7 Гц, 1 Н), 7,36- 24016583 7-Метокси-2,3,4,5-тетрагидро-1 Н-бензо[d]азепин. 1 М борана в ТГФ (1 мл, 1,02 ммоль) по каплям добавляли к охлаждаемому льдом и перемешиваемому раствору A1a (1,91 г, 10 ммоль) в ТГФ (50 мл). Соединение A1a было получено в соответствии с опубликованными способами (заявка на европейский патент 204349). Баню со льдом удаляли и раствор нагревали с обратным холодильником в течение 3 ч. Реакционную смесь снова охлаждали до 0 С, добавляли МеОН (2 мл), перемешивали при комнатной температуре в течение 35 мин и концентрировали. Белый твердый остаток обрабатывали 6 н. HCl (50 мл) и смесь нагревали с обратным холодильником в течение 1 ч и затем при комнатной температуре в течение ночи. Водный раствор промывали Et2O, подщелачивали 5 н. NaOH до рН больше 10 и экстрагировали EtOAc. Объединенные органические фазы промывали насыщенным раствором соли, сушили (Na2SO4), концентрировали и сушили в вакууме, получая соединение G2b (1,17 г, 66%) в виде прозрачного желтого масла. 1 7-Метокси-3-[5-(4-трифторметилфенил)тиофен-2-илметил]-2,3,4,5-тетрагидро-1 Н-бензо[d]азепин. К раствору соединения G2b (2,02 г, 11,4 ммоль) и соединения G1a (2,92 г, 11,4 ммоль) в дихлорметане (50 мл) добавляли АсОН (0,65 мл, 11,4 ммоль). Реакционную смесь перемешивали в течение 1,5 ч. Добавляли Na(OAc)3BH (3,62 г, 17,1 ммоль) и реакционную смесь перемешивали еще 20 ч. Добавляли 2 н.NaOH (рН приблизительно 11) и раствор экстрагировали EtOAc. Объединенные органические фазы сушили (Na2SO4), концентрировали и очищали колоночной хроматографией, получая соединение G2c (2,23 г, 47%) в виде желтого твердого вещества. 1 Н-ЯМР (300 МГц, CDCl3)7,68 (д, J=8,2 Гц, 2 Н), 7,61 (д, J=8,5 Гц, 2 Н), 7,24 (д, J=3,8 Гц, 1 Н), 7,01 3-[5-(4-Трифторметилфенил)тиофен-2-илметил]-2,3,4,5-тетрагидро-1 Н-бензо[d]азепин-7-ол. Смесь соединения G2c (2,21 г, 5,30 ммоль), HBr (48%, 6,0 мл, 53,0 ммоль) и Bu4NBr (171 мг, 0,53 ммоль) в АсОН (6 мл) перемешивали при 100 С в течение 16,5 ч. Добавляли насыщенный раствор K2CO3 до рН приблизительно 10 и раствор экстрагировали EtOAc. Объединенные органические фазы сушили(Na2SO4) и концентрировали, получая соединение G2d (1,77 г, 83%) в виде бежевого твердого вещества. 1 Н-ЯМР (400 МГц, CDCl3)7,66 (д, J=8,4 Гц, 2 Н), 7,61 (д, J=8,4 Гц, 2 Н), 7,25 (д, J=3,6 Гц, 1 Н), 6,97 Этиловый эфир 3-[5-(4-трифторметилфенил)тиофен-2-илметил]-2,3,4,5-тетрагидро-1 Н-бензо[d] азепин-7-илоксиуксусной кислоты. К раствору NaH (60% в минеральном масле, 317 мг, 7,93 ммоль) в ТГФ (12 мл) добавляли соединение G2d (1,07 г, 2,64 ммоль) в ТГФ (5 мл) и этилбромацетат (0,35 мл, 3,17 ммоль). Реакционную смесь перемешивали при температуре кипения с обратным холодильником в течение 1 ч, охлаждали, гасили насыщенным раствором NH4Cl и распределяли между простым эфиром и водой. Органическую фазу сушили (Na2SO4), концентрировали и очищали колоночной хроматографией, получая соединение G1b (1,02 г, 79%), которое далее превращали в соединение 7 в соответствии с приведенным выше описанием. Пример Н Соединение 8. 2-Метил-2-3-[5-(4-трифторметилфенил)тиофен-2-илметил]-2,3,4,5-тетрагидро-1 Нбензо[d]азепин-7-илоксипропионовая кислота. Указанное в заголовке соединение было получено в соответствии со схемой Н. Этиловый эфир 2-метил-2-3-[5-(4-трифторметилфенил)тиофен-2-илметил]-2,3,4,5-тетрагидро-1 Нбензо[d]азепин-7-илоксипропионовой кислоты. Соединение H1 было получено аналогично способу получения соединения G2. Соединение H1 было получено в виде белого твердого вещества (60%). 1 Н-ЯМР (300 МГц, CDCl3)7,67 (д, J=8,4 Гц, 2 Н), 7,60 (д, J=8,5 Гц, 2 Н), 7,24 (д, J=3,6 Гц, 1 Н), 6,946,91 (м, 2 Н), 6,62 (д, J=2,5 Гц, 1 Н), 6,55 (дд, J=8,2, 2,6 Гц, 1 Н), 4,23 (кв., J=7,1 Гц, 2 Н), 3,90 (с, 2 Н), 2,90 2-Метил-2-3-[5-(4-трифторметилфенил)тиофен-2-илметил]-2,3,4,5-тетрагидро-1 Н-бензо[d]азепин 7-илоксипропионовая кислота. Соединение 8 было получено аналогично способу получения соединения 2. Соединение 8 было получено в виде белого твердого вещества (61%). 1 Н-ЯМР (300 МГц, CD3OD)7,86 (д, J=7,9 Гц, 2 Н), 7,73 (д, J=8,3 Гц, 2 Н), 7,58 (д, J=2,7 Гц, 1 Н), 7,38 Соединение 9. 3-[5-(4-трифторметилфенил)тиофен-2-илметил]-2,3,4,5-тетрагидро-1 Н-бензо[d]азепин-7-илоксиуксусная кислота. Указанное в заголовке соединение было получено в соответствии со схемой I. Схема I Этиловый эфир 3-[5-(4-трифторметилфенил)фуран-2-илметил]-2,3,4,5-тетрагидро-1 Н-бензо[d]азепин-7-илоксиуксусной кислоты. Соединение I2 было получено из соединения I1(Bioorg.Med. Chem. Lett., 2003, 13(13), 2159-2161) аналогично способу получения соединения G2. Соединение I2 было получено в виде белого твердого вещества (40%). 1 Н-ЯМР (300 МГц, CDCl3)7,72 (д, J=8,2 Гц, 2 Н), 7,60 (д, J=8,5 Гц, 2 Н), 6,98 (д, J=8,2 Гц, 1 Н), 6,68 3-[5-(4-Трифторметилфенил)тиофен-2-илметил]-2,3,4,5-тетрагидро-1 Н-бензо[d]азепин-7 илоксиуксусная кислота. Соединение 9 было получено аналогично способу получения соединения 1. Соединение 9 было получено в виде белого твердого вещества (71%). 1 Н-ЯМР (300 МГц, CD3OD)7,89 (д, J=8,2 Гц, 2 Н), 7,69 (д, J=8,3 Гц, 2 Н), 6,98-6,94 (м, 2 Н), 6,756,69 (м, 3H), 4,44 (с, 2 Н), 4,33 (с, 2 Н), 3,14 (м, 4 Н), 2,82 (м, 4 Н); МС (ES) m/z: 446 (М+Н+). Пример J Соединение 10. 2-Метил-2-3-[5-(4-трифторметилфенил)фуран-2-илметил]-2,3,4,5-тетрагидро-1 Нбензо[d]азепин-7-илоксипропионовая кислота. Указанное в заголовке соединение было получено в соответствии со схемой J. Схема J[5-(4-Трифторметилфенил)фуран-2-ил]метанол. Соединение J1 было получено аналогично способу получения соединения Е 1. Соединение J1 было получено в виде белого твердого вещества (75%). 1 Этиловый эфир 2-метил-2-3-[5-(4-трифторметилфенил)фуран-2-илметил]-2,3,4,5-тетрагидро-1 Нбензо[d]азепин-7-илоксипропионовой кислоты. Смесь соединения J1 (80 мг, 0,33 ммоль), метансульфонилхлорида (38 мг, 0,33 ммоль) и триэтиламина (230 мкл, 1,65 ммоль) в CH2Cl2 (2 мл) перемешивали при комнатной температуре в течение 1,5 ч. Добавляли соединение В 2 (50 мг, 0,165 ммоль) в CH3CN (1 мл) и раствор перемешивали в течение ночи в атмосфере N2. Полученную смесь концентрировали и очищали колоночной хроматографией(EtOAc/гексан), получая 10 мг (12%, 2 стадии) соединения J2 в виде белого твердого вещества. 1 Н-ЯМР (300 МГц, CDCl3)7,72 (д, J=8,2 Гц, 2 Н), 7,60 (д, J=8,4 Гц, 2 Н), 6,91 (д, J=8,2 Гц, 1 Н), 6,69 2-метил-2-3-[5-(4-трифторметилфенил)фуран-2-илметил]-2,3,4,5-тетрагидро-1 Н-бензо[d]азепин-7- 27016583 илоксипропионовая кислота. Соединение 10 было получено аналогично способу получения соединения 2. Соединение 10 было получено в виде белого твердого вещества (65%). 1 Соединение 11. 3-[5-(4-Трифторметилфенил)изоксазол-3-илметил]-2,3,4,5-тетрагидро-1 Нбензо[d]азепин-7-илоксиуксусная кислота. Указанное в заголовке соединение было получено в соответствии со схемами K1 и K2. Схема K1 Трет-бутиловый эфир (4-оксо-2,3,4,5-тетрагидро-1 Н-бензо[d]азепин-7-илокси)уксусной кислоты. Соединение K1 а было получено аналогично способу получения соединения A1b. Соединение K1a было получено в виде белого твердого вещества (28% в 2 стадиях). 1 Н-ЯМР (300 МГц, CDCl3)7,03 (д, J=8,4 Гц, 1 Н), 6,76 (дд, J=8,4, 2,7 Гц, 1 Н), 6,67 (д, J=2,6 Гц, 1 Н),5,79 (шир.с, 1 Н), 4,48 (с, 2 Н), 3,79 (с, 2 Н), 3,58-3,52 (м, 2 Н), 3,06 (т, J=6, б Гц, 2 Н), 1,49 (с, 9 Н); МС (ES) Трет-бутиловый эфир (2,3,4,5-тетрагидро-1 Н-бензо[d]азепин-7-илокси)уксусной кислоты. Соединение K1b было получено аналогично способу получения соединения A1c. Соединение K1b было получено в виде желтого масла (52%). 1- 28016583 Метиловый эфир 4-гидрокси-2-оксо-4-(4-трифторметилфенил)бут-3-еновой кислоты. К раствору 1-(4-трифторметилфенил)этанона (2,0 г, 10,6 ммоль) и диметилоксилата (1,63 г, 13,8 ммоль) в толуоле (50 мл) при 0 С порциями добавляли NaH (60%, 636 мг, 15,9 ммоль). Смесь перемешивали при комнатной температуре в течение 1 ч и затем при 60 С в течение 2 ч. Реакционную смесь охлаждали до комнатной температуры и гасили, медленно добавляя H2O. Смесь подкисляли 1 н. HCl и экстрагировали EtOAc. Объединенные органические фазы сушили (Na2SO4), концентрировали и очищали колоночной хроматографией, получая 2,74 г (94%) соединения К 2 а в виде белого твердого вещества. 1 Н-ЯМР (300 МГц, CDCl3)8,11 (д, J=8,3 Гц, 2 Н), 7,77 (д, J=8,3 Гц, 2 Н), 7,10 (с, 2 Н), 3,97 (с, 3H); МС (ES) m/z: 297 (M+Na+). Этиловый эфир 5-(4-трифторметилфенил)изоксазол-3-карбоновой кислоты. К раствору соединения K2 а (2,7 г, 9,85 ммоль) в EtOH (40 мл) добавляли гидрохлорид гидроксиламина (2,05 г, 29,5 ммоль). Смесь перемешивали при комнатной температуре в течение 1 ч и затем при 80 С в течение 2 ч. Реакционную смесь охлаждали до комнатной температуры, осадок фильтровали и промывали EtOH. Белое твердое вещество сушили в вакууме, получая 2,0 г (75%) соединения K2b. 1[5-(4-Трифторметилфенил)изоксазол-3-ил]метанол. Соединение K2 с было получено аналогично способу получения соединения С 2. Соединение K2 с было получено в виде белого твердого вещества (98%). 1 Н-ЯМР (300 МГц, CDCl3)7,90 (д, J=8,2 Гц, 2 Н), 7,73 (д, J=8,5 Гц, 2 Н), 6,70 (с, 1 Н), 4,84 (с, 2 Н); МС (ES) m/z: 244 (М+Н+). 3-Бромметил-5-(4-трифторметилфенил)изоксазол. Соединение K2d было получено аналогично способу получения соединения A2d. Соединение K2d было получено в виде белого твердого вещества (98%). 1 Н-ЯМР (300 МГц, CDCl3)7,91 (д, J=8,3 Гц, 2 Н), 7,75 (д, J=8,3 Гц, 2 Н), 6,73 (с, 2 Н), 4,49 (с, 2 Н); МС (ES) m/z: 306 (М+Н+). трет-Бутиловый эфир 3-[5-(4-трифторметилфенил)изоксазол-3-илметил]-2,3,4,5-тетрагидро-1 Нбензо[d]азепин-7-илоксиуксусной кислоты. Соединение K2 е было получено аналогично способу получения соединения А 2 е. Соединение K2 е было получено в виде белого твердого вещества (71%). 1 3-[5-(4-Трифторметилфенил)изоксазол-3-илметил]-2,3,4,5-тетрагидро-1 Н-бензо[d]азепин-7 илоксиуксусная кислота. К раствору соединения K2 е (38,5 мг, 0,07 6 ммоль) в CH2Cl2 (1 мл) добавляли трифторуксусную кислоту (0,1 мл). Смесь перемешивали при комнатной температуре в течение 15 ч,концентрировали и очищали колоночной хроматографией, получая 30 мг (96%) соединения 11 в виде белого твердого вещества. 1 Н-ЯМР (300 МГц, CD3OD)8,08 (д, J=8,2 Гц, 2 Н), 7,86 (д, J=8,3 Гц, 2 Н), 7,17-7,13 (м, 2 Н), 6,85 (д,J=23 Гц, 1 Н), 6,78 (дд, J=8,2, 2,6 Гц, 1 Н), 4,64 (с, 4 Н), 3,5 (м, 4 Н), 3,18 (м, 4 Н); МС (ES) m/z: 447 (М+Н+). Пример L.
МПК / Метки
МПК: A61P 3/10, A61K 31/55, A61P 1/18, C07D 223/16, A61P 3/06
Метки: применение, ldl-c, качестве, повышения, hdl-c, холестерина, агонистов, производные, бензоазепиноксиуксусной, кислоты, снижения, дельта-ppar
Код ссылки
<a href="https://eas.patents.su/30-16583-proizvodnye-benzoazepinoksiuksusnojj-kisloty-i-ih-primenenie-v-kachestve-agonistov-delta-ppar-dlya-povysheniya-hdl-c-i-snizheniya-ldl-c-i-holesterina.html" rel="bookmark" title="База патентов Евразийского Союза">Производные бензоазепиноксиуксусной кислоты и их применение в качестве агонистов дельта-ppar для повышения hdl-c и снижения ldl-c и холестерина</a>
Новые замещенные 4-фенил-4-[1н-имидазол-2-ил] пиперидиновые производные и их применение в качестве селективных непептидных агонистов дельта-опиоидов

Номер патента: 6507
Опубликовано: 29.12.2005
Авторы: Гомес-Санчес Антонио, Фернандес-Гадеа Франсиско Хавьер, Ленартс Йозеф Элизабет, Мерт Тео Франс, Янссенс Франс Эдуард
МПК: C07D 401/04, A61K 31/445, A61P 25/04...
Метки: агонистов, применение, производные, пиперидиновые, качестве, замещенные, непептидных, 4-фенил-4-[1н-имидазол-2-ил, дельта-опиоидов, селективных, новые
Формула / Реферат:
1. Соединение формулы (I) где A=B представляет собой двухвалентный радикал с p-связью; X представляет собой ковалентную связь, -CH2- или CH2CH2-; R1 представляет собой водород, гидрокси, алкилокси, алкилкарбонилокси, Ar-окси, Het-окси, Ar-карбонилокси, Het-карбонилокси, Ar-алкилокси, Het-алкилокси, алкил, полигалогеналкил, алкилоксиалкил, Ar-алкил, Het-алкил, Ar, Het, тио, алкилтио, Ar-тио, Het-тио или NR9R10, где R9 и R10, каждый независимо,...
Феноксиуксусные кислоты в качестве активаторов ppar дельта

Номер патента: 15717
Опубликовано: 31.10.2011
Авторы: Поливка Зденек, Могенсен Йохн Патрик, Гавранек Мирослав, Пигера Павел, Петтерссон Ингрид, Сауерберг Пер
МПК: C07C 323/16, A61P 3/10, A61K 31/44...
Метки: кислоты, качестве, дельта, феноксиуксусные, активаторов
Формула / Реферат:
1. Соединение общей формулы (I)где представляет собой двойную связь либо с Е-, либо с Z-замещением;X1 представляет собой C1-6-алкил, необязательно замещенный одним или несколькими заместителями, выбранными из морфолино, пиперазино, пиперидино или пирролидино;Х2 представляет собой фенилен, необязательно замещенный одним или несколькими заместителями, выбранными из галогена или C1-6-алкила, необязательно замещенного одним или несколькими атомами...
Производные феноксиуксусных кислот, применимые в качестве двойных агонистов активируемого пероксисомным пролифератором рецептора ( ppar )

Номер патента: 9804
Опубликовано: 28.04.2008
Авторы: Шэнь Лань, Ко Джи-Хун, Ван Айхуа, Чжан Янь
МПК: C07D 285/08, A61K 31/433, A61P 31/10...
Метки: качестве, применимые, производные, агонистов, пероксисомным, пролифератором, кислот, активируемого, двойных, рецептора, феноксиуксусных
Формула / Реферат:
1. Соединение формулы (I) где m равно 1, 2 или 3; n равно 0 или 1; X означает S или О; Y означает S, СН2 или О; R1 и R2 независимо выбраны из Н, C1-4алкила, C1-3алкокси, галогена и -NRaRb, где каждый из Ra и Rb независимо выбран из Н и C1-4 алкила; каждый из R3 и R4 независимо выбран из Н, галогена, циано, C1-4алкила, C1-3алкокси и NRcRd, где каждый из Rc и Rd независимо выбран из Н и C1-4алкила и где по меньшей мере один из R3 и R4 не Н; и...
Производные 3-ароилиндола и их применение в качестве агонистов рецепторов cb2

Номер патента: 5854
Опубликовано: 30.06.2005
Авторы: Ринальди Мирей, Васс Фабьенн, Конжи Кристиан, Барт Франсис, Гийомон Кароль, Верне Клод
МПК: C07D 209/12, A61P 43/00, A61K 31/404...
Метки: агонистов, рецепторов, применение, производные, качестве, 3-ароилиндола
Формула / Реферат:
1. Соединение формулы где Ar представляет собой a) фенил, моно-, ди- или тризамещенный одной или более чем одной группой, выбранной из атома галогена, (C1-C4)алкила, трифторметила, амино, нитро, гидроксила, (C1-C4)алкокси, (C1-C4)алкилсульфанила или (C1-C4)алкилсульфонила; b) нафтил, который не замещен или замещен единожды или дважды атомом галогена, (C1-C4)алкилом или трифторметилом; A представляет собой C2-C6алкиленовый радикал; Y...
Производные пирролопиридина и их применение в качестве модуляторов ppar-рецепторов

Номер патента: 14185
Опубликовано: 29.10.2010
Авторы: Барт Мартин, Доде Пьер, Лежендр Кристиан, Бине Жан, Пупардин-Оливье Оливия, Бубиа Бенаисса
МПК: A61K 31/437, A61P 3/06, C07D 471/04...
Метки: ppar-рецепторов, качестве, производные, модуляторов, пирролопиридина, применение
Формула / Реферат:
1. Производное пирролопиридина, характеризующееся тем, что оно выбрано из:i) соединений формулыгде R1и R2, каждый независимо, представляют собой атом водорода, атом галогена, группу C1-C3-алкил или C1-C4-алкокси либо группу CF3;R3 и R4, каждый независимо, представляют собой атом водорода или группу C1-C4-алкил;R представляет собой атом водорода или группу C1-C3-алкил;n=1, 2 или 3;X представляет собой одинарную связь или атом кислорода;Ar...
Предыдущий патент: Производные лактамида, способ их получения и применение
Следующий патент: Новые гены, родственные гену глутаминилциклазы
Случайный патент: Способы лечения инфекций, вызываемых вирусом гепатита дельта, с использованием β-l-2'-дезоксинуклеозидов