Производные фталазина в качестве ингибиторов parp
Номер патента: 14955
Опубликовано: 29.04.2011
Авторы: Сомерс Мария Викторина Франциска, Мевеллек Лоранс Анн, Кенни Людо Эдмон Жозефин, Ваутерс Вальтер Баудевейн Леопольд, Ван Дюн Якобус Альфонсус Йозефус, Мертенс Йозефус Каролус























Формула / Реферат
1. Соединение формулы (I)

его формы N-оксидов, фармацевтически приемлемые аддитивные соли и стереохимически изомерные формы,
где пунктирная линия обозначает необязательную связь;
n равен 0, 1, 2 или 3; если n равен 0, тогда подразумевается прямая связь;
Q представляет собой -С(=O)- или -CR3-, где R3 представляет собой галоген или C1-6-алкил; если Q представляет собой -CR3-, пунктирная линия обозначает связь;
каждый X независимо представляет собой -N< или -СН<; если X представляет собой -СН<, тогда Y представляет собой -N< или -NH-, и если X представляет собой -N<, тогда Y представляет собой -СН< или
-СН2-;
каждый Y независимо представляет собой -N<, -NH-, -CH< или -СН2-;
L1 представляет собой прямую связь или двухвалентный радикал, выбранный из -С1-6-алкандиил-NH-, -NH- или -NH-С1-6-алкандиил-NH-;
L2 представляет собой прямую связь или двухвалентный радикал, выбранный из -C1-6-алкандиила-, -С2-6-алкендиила-, карбонила или -C1-6-алкандиила-, замещенного одним заместителем, выбранным из группы, состоящей из гидрокси или арила;
R1 представляет собой водород, нитро, галоген или амино;
R2 представляет собой водород, С1-6-алкил или арил-C1-6-алкил;
центральный фрагмент ![]() также может быть связан мостиковой связью (т.е. образовывать бициклическую группу) с этиленовой мостиковой связью;
также может быть связан мостиковой связью (т.е. образовывать бициклическую группу) с этиленовой мостиковой связью;
Z представляет собой водород, гидрокси, С1-6-алкил, C1-6-алкилокси, арилокси, амино, циано, арил-C1-6-алкиламино или бензтиазолил(C1-6-алкил)амино либо кольцевую систему, выбранную из

где R4и R5, каждый независимо, выбраны из водорода, галогена, С1-6-алкила, С1-6-алкилокси или тригалогенметила;
арил представляет собой фенил или фенил, замещенный одним или двумя заместителями, каждый из которых независимо выбран из галогена, С1-6-алкила или С1-6-алкилокси;
при условии, что если Q представляет собой -С(=O)- и X представляет собой -N<, и Y представляет собой
-СН< или -СН2-, и L1представляет собой прямую связь, и L2представляет собой прямую связь или двухвалентный радикал -С1-6-алкандиил- или -С1-6-алкандиил-, замещенный гидрокси, и R1 представляет собой водород, и R2 представляет собой водород или С1-6-алкил, тогда Z отличен от водорода, гидрокси или С1-6-алкила; и
при условии исключения соединений

2. Соединение по п.1, где n равен 1 или 2; Q представляет собой -С(=O)-; Y представляет собой -N<, -NH- или -СН<; L1 представляет собой прямую связь или двухвалентный радикал -NH-; L2 представляет собой прямую связь или двухвалентный радикал, выбранный из карбонила, -С1-6-алкандиила- или -C1-6-алкандиила-, замещенного гидрокси; R1 представляет собой водород; R2 представляет собой водород, арил-C1-6-алкил; Z представляет собой водород, С1-6-алкилокси, арилокси, амино или кольцевую систему, выбранную из (а-2) или (а-3); и R4 и R5, каждый независимо, выбраны из водорода или галогена.
3. Соединение по пп.1 и 2, где соединение представляет собой соединение № 41, 13, 62, 26, 64, 2, 8, 9, 34, 11, 15 и 10.

4. Соединение по п.1, где X представляет собой -СН<.
5. Применение соединения по любому одному из предшествующих пунктов в качестве лекарственного средства.
6. Фармацевтическая композиция, содержащая фармацевтически приемлемые носители и терапевтически эффективное количество соединения по любому из пп.1-4 в качестве активного ингредиента.
7. Способ получения фармацевтической композиции по п.6, где фармацевтически приемлемые носители и соединение по любому из пп.1-4 смешивают в однородную смесь.
8. Применение соединения для изготовления лекарственного средства для лечения нарушений, опосредованных PARP, где указанное соединение представляет собой соединение формулы (I)

его формы N-оксидов, фармацевтически приемлемые аддитивные соли и стереохимически изомерные формы, где
пунктирная линия обозначает необязательную связь;
n равен 0, 1, 2 или 3; если n равен 0, тогда подразумевается прямая связь;
Q представляет собой -С(=O)- или -CR3-, где R3 представляет собой галоген или C1-6-алкил; если Q представляет собой -CR3-, пунктирная линия обозначает связь;
X независимо представляет собой -N< или -СН<; если X представляет собой -СН<, тогда Y представляет собой -N< или -NH-; и когда X представляет собой -N<, тогда Y представляет собой -СН< или -СН2-;
L1 представляет собой прямую связь или двухвалентный радикал, выбранный из -С1-6-алкандиил-NH-, -NH- или -NH-С1-6-алкандиил-NH-;
L2 представляет собой прямую связь или двухвалентный радикал, выбранный из -C1-6-алкандиила-, -С2-6-алкендиила-, карбонила или -С1-6-алкандиила-, замещенного одним заместителем, выбранным из группы, состоящей из гидрокси или арила;
R1 представляет собой водород, нитро, галоген или амино;
R2 представляет собой водород, C1-6-алкил или арил-C1-6-алкил;
центральный фрагмент ![]() также может быть связан мостиковой связью (т.е. образовывать бициклическую группу) с этиленовой мостиковой связью;
также может быть связан мостиковой связью (т.е. образовывать бициклическую группу) с этиленовой мостиковой связью;
Z представляет собой водород, гидрокси, C1-6-алкил, C1-6-алкилокси, арилокси, амино, циано, арил-C1-6-алкиламино или бензтиазолил(C1-6-алкил)амино либо кольцевую систему, выбранную из

где R4и R5, каждый независимо, выбраны из водорода, галогена, C1-6-алкила, C1-6-алкилокси или тригалогенметила;
арил представляет собой фенил или фенил, замещенный одним или двумя заместителями, каждый из которых независимо выбран из галогена, C1-6-алкила или C1-6-алкилокси.
9. Применение по п.8 ингибитора PARP формулы (I) для изготовления лекарственного средства для лечения нарушений, опосредованных PARP-1.
10. Применение по п.8 или 9, где лечение включает химиосенсибилизацию.
11. Применение по п.8 или 9, где лечение включает радиосенсибилизацию.
12. Способ получения соединения по п.1, включающий получение соединения формулы (I-c) или соединений формулы (I), где L2представляет собой прямую связь или двухвалентный радикал -С1-6-алкандиил- и Z представляет собой амино, начиная с соединения формулы (I-e), где Z-L2- представляет собой арил-C1-6-алкил (например) или арил-C1-6-алкиламино

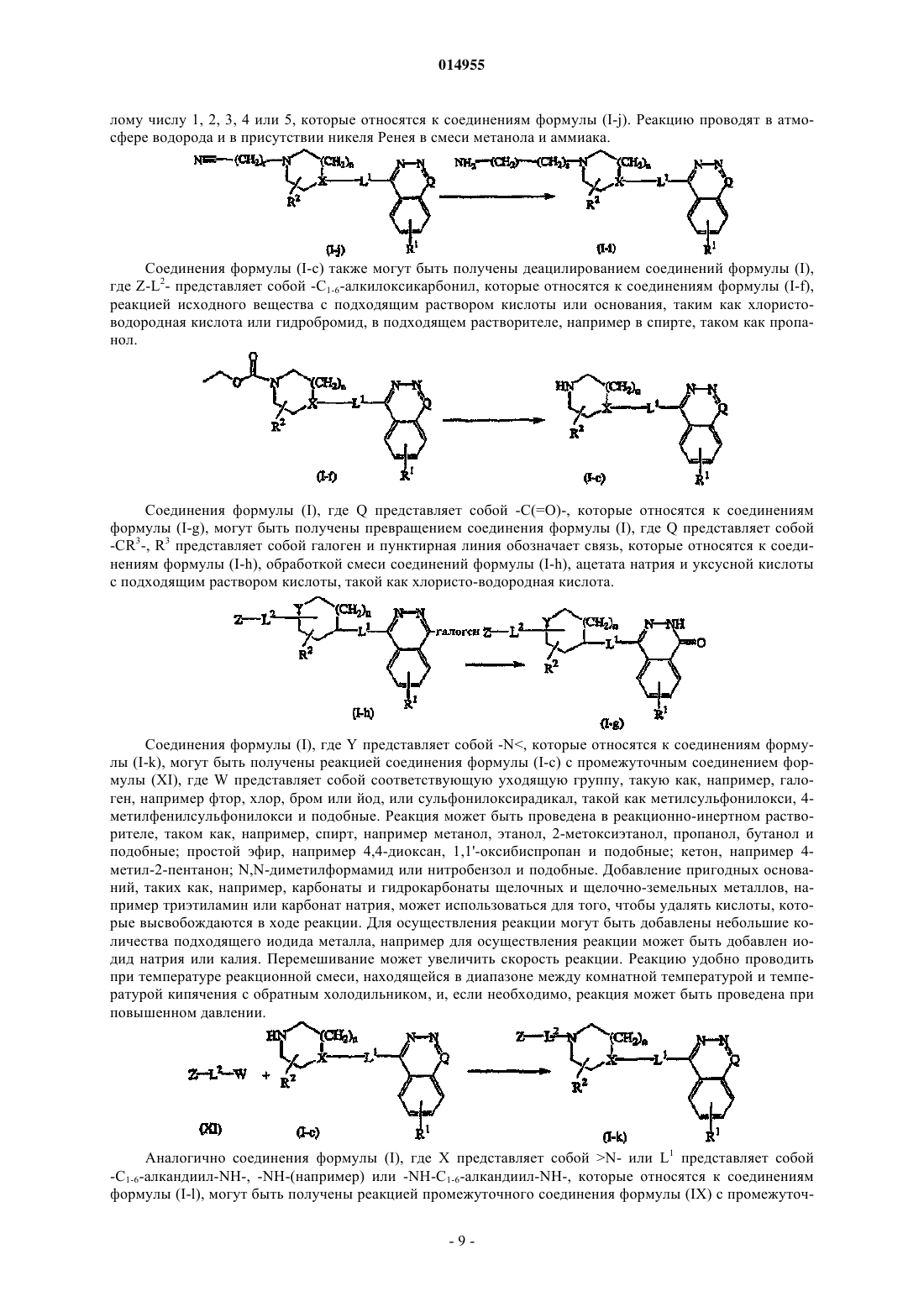
13. Способ получения соединения по п.1, включающий превращение соединений формулы (I-j) в соединения формулы (I-i)

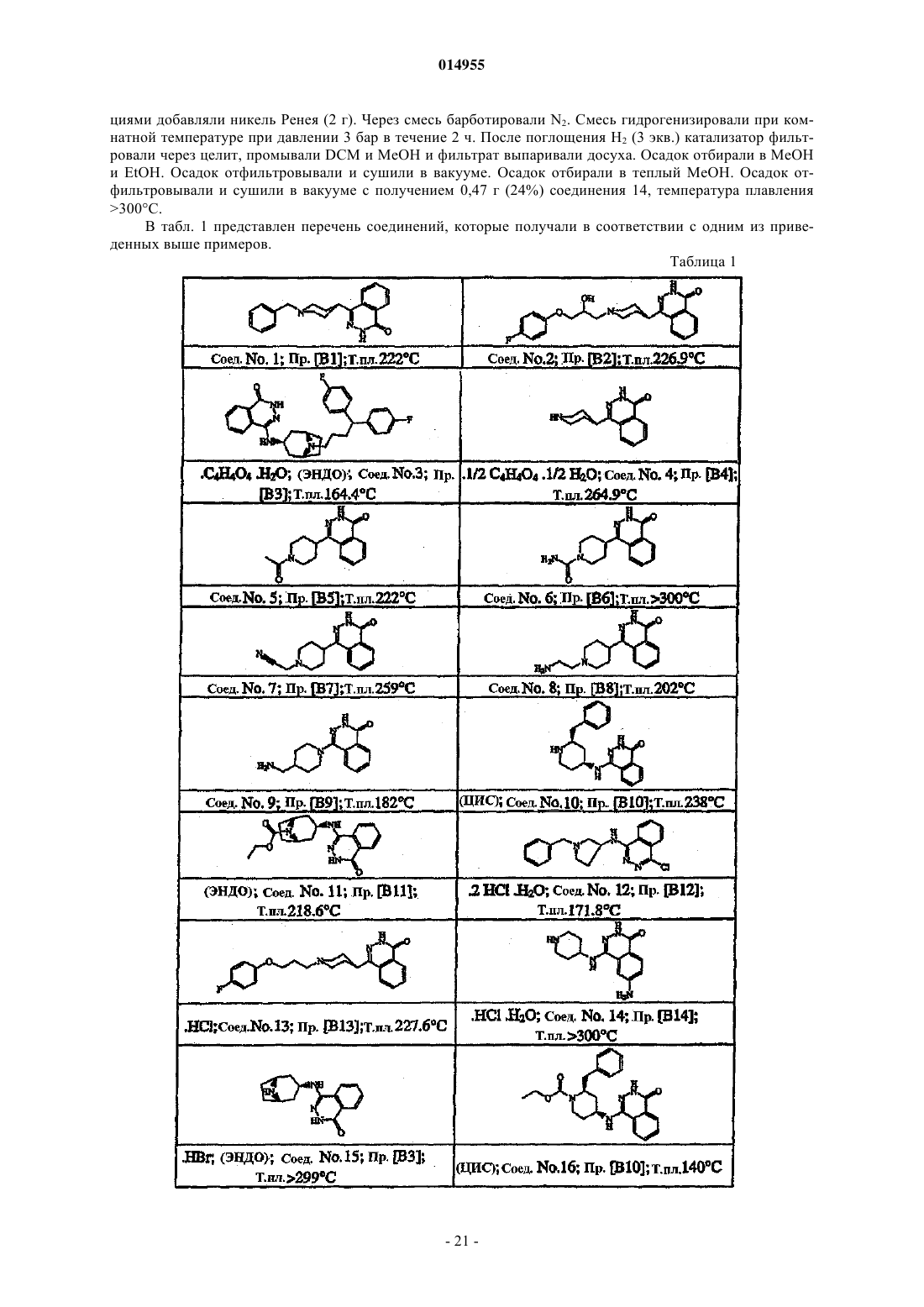
14. Способ получения соединения по п.1, включающий деацилирование соединений формулы (I-f) с образованием соединений формулы (I-c)

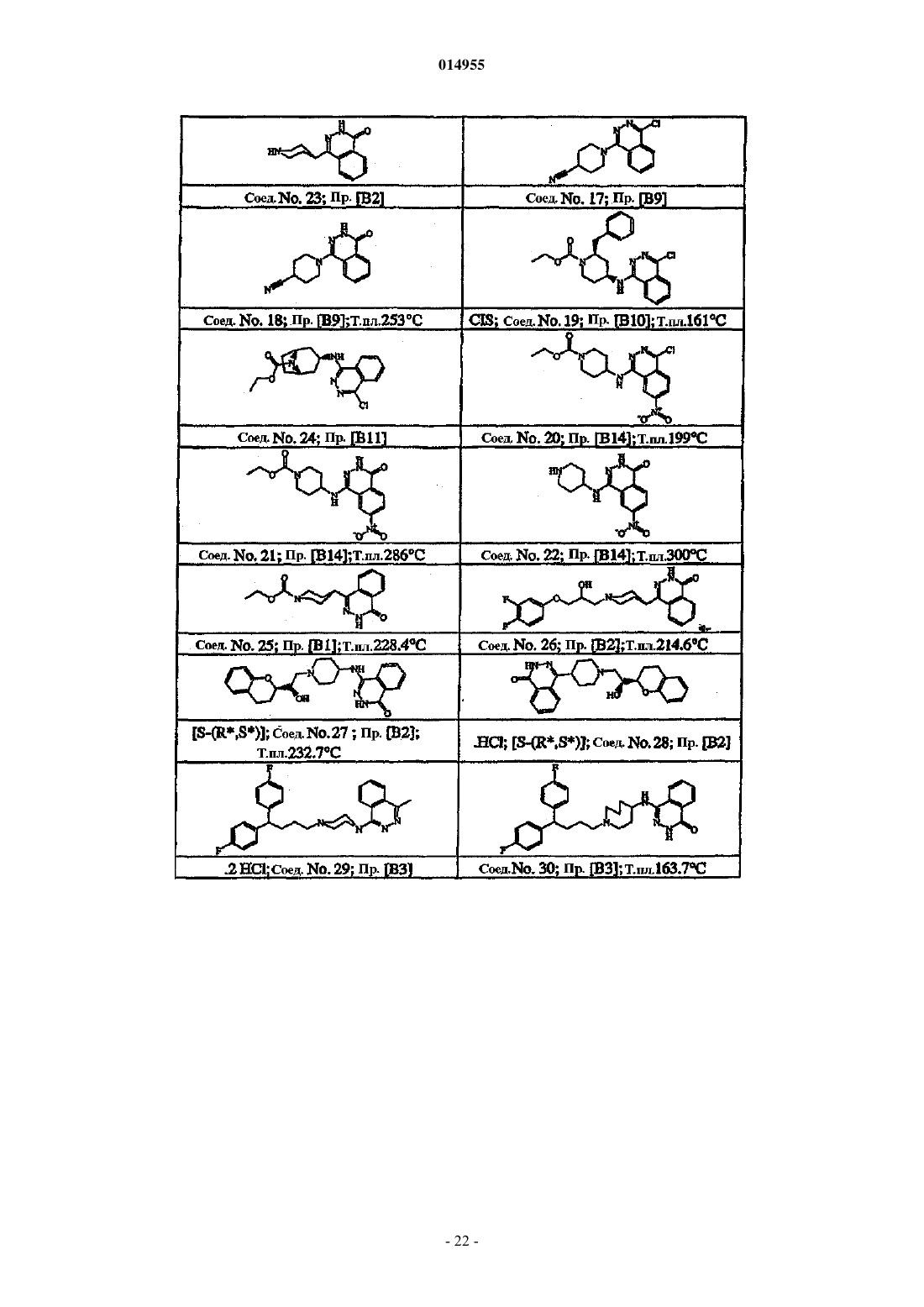
15. Способ получения соединения по п.1, включающий взаимодействие соединения формулы (I-c) с промежуточным соединением формулы (XI), где W представляет собой соответствующую уходящую группу, с образованием соединений формулы (I-k)

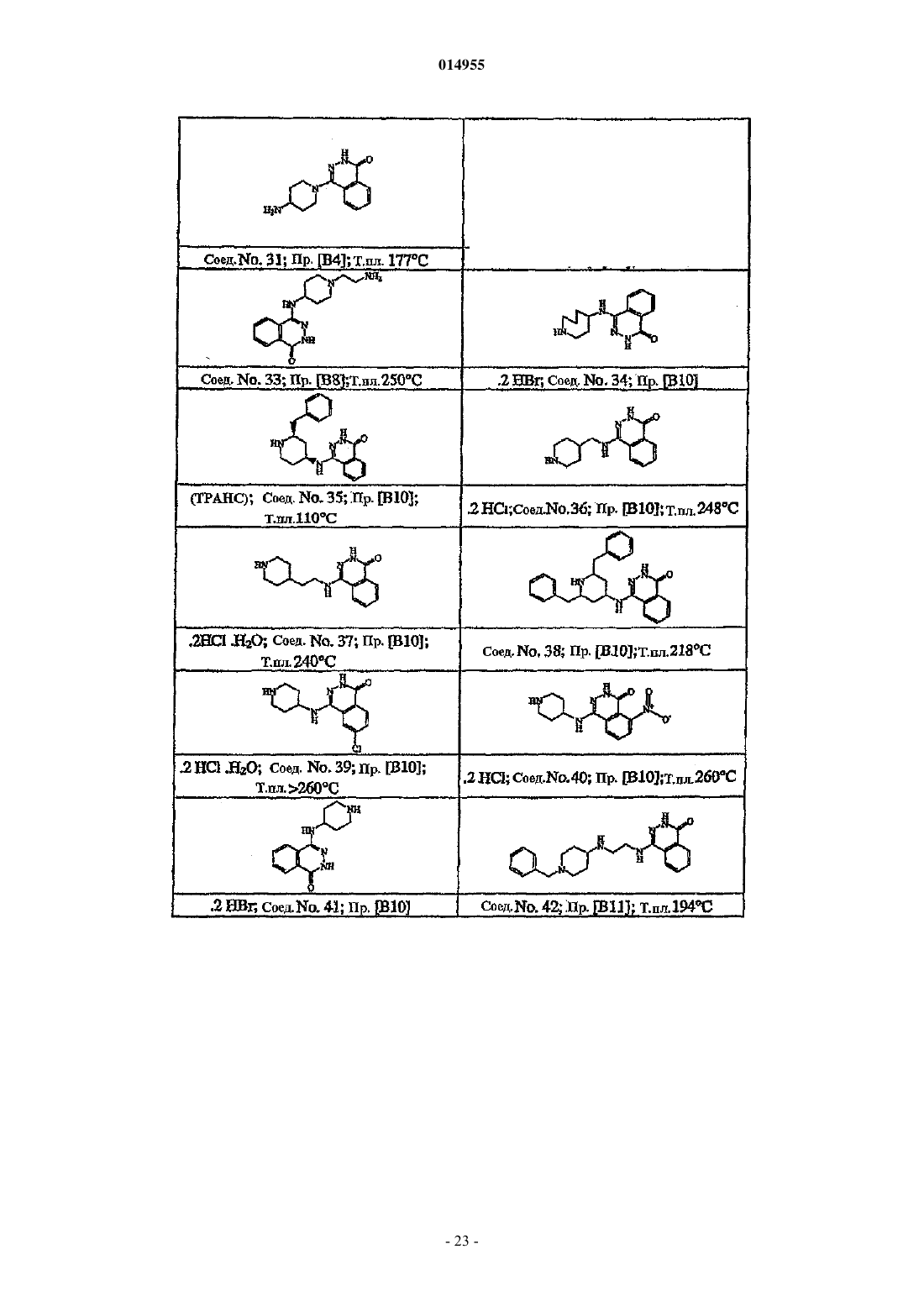
16. Способ получения соединения по п.1, включающий взаимодействие промежуточного соединения формулы (IX) с промежуточным соединением формулы (X) с образованием соединения формулы (I-l)

Текст
Настоящее изобретение относится к соединениям формулы (I), их применению в качестве ингибиторов PARP, а также к фармацевтическим композициям, содержащим указанные соединения формулы (I), где R1, R2, L1, L2, X, Y, Q и Z определены в описании. Мевеллек Лоранс Анн (FR), Кенни Людо Эдмон Жозефин, Мертенс Йозефус Каролус, Ван Дюн Якобус Альфонсус Йозефус,Сомерс Мария Викторина Франциска,Ваутерс Вальтер Баудевейн Леопольд (BE) 014955 Настоящее изобретение относится к ингибиторам PARP и представляет соединения и композиции,содержащие описанные соединения. Более того, настоящее изобретение относится к применению описанных ингибиторов PARP, например, в качестве лекарственного средства. Ядерный фермент поли(АДФ-рибоза)полимераза-1 (PARP-1) является членом семейства ферментовPARP. Это увеличивающееся семейство ферментов состоит из PARP, таких как, например, PARP-1PARP-1 является основным ядерным белком с массой 116 кДа, состоящим из трех доменов: Nконцевого ДНК-связывающего домена, содержащего два цинковых пальца, домена аутомодификации и С-концевого каталитического домена. Он представлен практически у всех эукариотов. Фермент осуществляет синтез поли(АДФ-рибозы), разветвленного полимера, который состоит из более чем 200 остатков АДФ-рибозы. Белковые акцепторы поли(АДФ-рибозы) непосредственно или косвенно вовлечены в поддержание целостности ДНК. Они включают гистоны, топоизомеразы, ДНК и РНК полимеразы, ДНКлигазы и Са 2+- и Mg2+-зависимые эндонуклеазы. Белок PARP экспрессируется на высоком уровне во многих тканях, особенно в клетках иммунной системы, сердца, головного мозга и в клетках зародышевой линии. В нормальных физиологических условиях активность PARP минимальна. Однако повреждение ДНК приводит к незамедлительному повышению активации PARP вплоть до 500 раз. Танкиразы (TANK) были идентифицированы в качестве компонентов теломерного комплекса человека. Также предполагалось, что они участвуют в везикулярном транспорте и могут служить в качестве каркаса для белков, вовлеченных во множество других клеточных процессов. Теломеры, которые необходимы для поддержания целостности и стабильности хромосом, поддерживаются с помощью теломеразы, специализированной обратной транскриптазы. TANK представляют собой (АДФ-рибоза)трансферазу с некоторыми свойствами как сигнальных белков, так и белков цитоскелета. Они содержат домен PARP,который катализирует полиАДФ-рибозилирование субстратных белков, альфа-мотив стерильности, который принадлежит определенным сигнальным молекулам, и домен ANK, который содержит 24 анкириновых повтора, гомологичных белку цитоскелета анкирину. Домен ANK взаимодействует с теломерным белком, связывающим теломерные повторы фактором-1 (TRF-1). Таким образом, указанные белки называют взаимодействующими с TRF1, анкиринсвязанными полимеразами АДФ-рибозы (TANK). Одной из более конкретных функций TANK является АДФ-рибозилирование в TRF-1. Для осуществления функции теломер у человека необходимо два специфичных для теломер ДНК-связывающих белка, TRF-1 и TRF-2. TRF-2 защищает концы хромосом, TRF-1 регулирует длину теломеры. Рибозилирование АДФ приводит к ингибированию способности TRF-1 связываться с теломерной ДНК. Это рибозилирование поли-АДФ в TRF-1 приводит к отсоединению TRF-1 от теломер, открывая теломерный комплекс и делая возможным доступ для теломеразы. Таким образом, TANK функционирует как положительный регулятор длины теломеры, делая возможной элонгацию теломер теломеразой. Среди множества функций, приписываемых PARP и особенно PARP-1, его главная роль состоит в облегчении репарации ДНК посредством рибозилирования АДФ и, таким образом, координирования ряда белков репарации ДНК. В результате активации PARP уровни NAD+ значительно снижаются. Чрезмерная активация PARP приводит к тяжелому истощению запасов NAD+ в клетках, в которых произошло крупное повреждение ДНК. Короткое время полураспада поли(АДФ-рибозы) приводит к большой скорости кругооборота. После образования поли(АДФ-рибозы) она быстро деградируется конститутивно активной поли(АДФ-рибоза)гликогидролазой (PARG) совместно с фосфодиэстеразой и (АДФрибоза)лиазой белка. PARP и PARG образуют цикл, который превращает большое количество NAD+ в АДФ-рибозу. Менее чем за один час избыточная стимуляция PARP может привести к падению уровняNAD+ и АТФ до менее чем 20% от нормального уровня. Такой сценарий является особенно опасным при ишемии, когда недостаток кислорода уже существенно нарушил выработку энергии. Последующая продукция свободных радикалов в процессе реперфузии, как полагают, является основной причиной повреждения в тканях. Частично снижение уровня АТФ, которое является типичным для множества органов в процессе ишемии и реперфузии, может быть связано с истощением NAD+ вследствие кругооборота поли(АДФ-рибозы). Таким образом, ожидается, что ингибирование PARP или PARG будет сохранять уровень энергии клетки, тем самым увеличивая выживаемость ишемизированных тканей после повреждения. Синтез поли(АДФ-рибозы) также вовлечен в индукцию экспрессии ряда генов, необходимых для воспалительного ответа. Ингибиторы PARP подавляют продукцию индуцибельной синтетазы оксида азота (iNOS) в макрофагах, селектина Р-типа и молекулы межклеточной адгезии-1 (ICAM-1) в эндотелиальных клетках. Такая активность лежит в основе сильных противовоспалительных эффектов, проявляемых ингибиторами PARP. Ингибирование PARP может привести к уменьшению некроза посредством предотвращения транспорта и инфильтрации нейтрофилов в поврежденные ткани.PARP активируется поврежденными фрагментами ДНК и после активации катализирует присоединение вплоть до 100 остатков АДФ-рибозы ко множеству ядерных белков, включая гистоны и самуPARP. При значительных стрессовых воздействиях на клетки чрезмерная активация PARP может быстро-1 014955 привести к повреждению и гибели клеток вследствие истощения запасов энергии. Так как на восстановление каждой молекулы NAD+ расходуется четыре молекулы АТФ, и массовая активация PARP приводит к истощению запасов NAD+, то при попытках ресинтезировать NAD+ запасы АТФ также истощаются. Сообщалось, что активация PARP играет ключевую роль в нейротоксичности, индуцируемой какNMDA, так и NO. Это было показано на кортикальной культуре и на срезах гиппокампа, где предотвращение токсичности прямо коррелирует со способностью ингибировать PARP. Возможная роль ингибиторов PARP для лечения нейродегенеративных заболеваний и травмы головы, таким образом, была установлена даже при отсутствии объяснения их механизма действия. Аналогично было показано, что разовые инъекции ингибиторов PARP приводили к снижению размера области инфаркта вследствие ишемии и реперфузии в сердце или в скелетной мышце у кроликов. В этих исследованиях однократная инъекция 3-аминобензамида (10 мг/кг) либо за 1 мин до окклюзии, либо за 1 мин до реперфузии приводила к сходному снижению размера области инфаркта в сердце (32-42%), а 1,5-дигидроксиизохинолин (1 мг/кг), еще один ингибитор PARP, вызывал снижение размера области инфаркта на сопоставимом уровне (38-48%). Эти результаты делают целесообразным предположение о том,что ингибиторы PARP могут восстанавливать предварительно ишемизированное сердце или скелетную мышечную ткань после реперфузионного повреждения. Активацию PARP также можно использовать как показатель повреждения после нейротоксических повреждений вследствие воздействия любого из следующих ниже индукторов, таких как глутамат (через стимуляцию рецепторов NMDA), реакционноспособные промежуточные соединения кислорода, амилоидный белок, N-метил-4-фенил-1,2,3,6-тетрагидропиридин (МРТР) или его активный метаболит Nметил-4-фенилпиридин (МРР+), который участвует в развитии патологических состояний, таких как инсульт, болезнь Альцгеймера и болезнь Паркинсона. В других исследованиях было продолжено исследование роли активации PARP на зернистых клетках мозжечка in vitro и в нейротоксичности с МРТР. Чрезмерное воздействие на нейроны глутамата, который выполняет роль преобладающего нейротрансмиттера центральной нервной системы и действует через рецепторы N-метил-D-аспартата (NMDA) и другие подтипы рецепторов, часто происходит как результат инсульта или другого нейродегенеративного процесса. Лишенные кислорода нейроны в больших количествах высвобождают глутамат в процессе ишемического повреждения головного мозга, такого как инсульт, или после инфаркта миокарда. Это избыточное высвобождение глутамата, в свою очередь, вызывает избыточную стимуляцию (эксайтотоксичность) рецепторов N-метил-D-аспартата (NMDA), АМРА, Kainate и MGR, которая приводит к открытию ионных каналов и обеспечивает неконтролируемый поток ионов (например, Са 2+ и Na+ в клетки и K+ из клеток), что приводит к чрезмерной стимуляции нейронов. Нейроны при чрезмерной стимуляции секретируют большее количество глутамата, создавая цепь с обратной связью или эффект "домино", который, в конечном счете, приводит к повреждению клетки или смерти вследствие продукции протеаз, липаз и свободных радикалов. Чрезмерная активация рецепторов глутамата вовлечена в различные неврологические заболевания и состояния, включая эпилепсию, инсульт, болезнь Альцгеймера, болезнь Паркинсона, боковой амиотрофический склероз (ALS), болезнь Гентингтона, шизофрению, хроническую боль, ишемию и потерю нейронов после гипоксии, гипогликемии, ишемии, травмы и повреждения нерва. Воздействие глутамата и стимуляция глутаматом также принимают участие в качестве основы в компульсивных нарушениях, в частности, в зависимости от лекарственных средств. Доказательства того, что антагонисты рецепторов глутамата (т.е. соединения, которые блокируют связывание глутамата или активацию глутаматом его рецепторов) блокируют повреждение нейронов после сосудистого инсульта,включают результаты исследований на животных многих видов, а также на церебральной кортикальной культуре, обработанной глутаматом или NMDA. При попытках предотвратить эксайтотоксичность блокированием рецепторов NMDA, AMPA, Kainate и MGR столкнулись со сложностями вследствие того,что каждый рецептор имеет множество участков, с которыми может связываться глутамат, и, таким образом, поиск эффективного сочетания антагонистов или универсального антагониста для предотвращения связывания глутамата со всеми рецепторами и для того, чтобы сделать возможным исследование этой теории, является затрудненным. Более того, множество композиций, которые являются эффективными в отношении блокирования рецепторов, также являются токсичными для животных. По существу,в настоящее время не существует известного эффективного способа лечения аномалий, связанных с глутаматом. Стимуляция глутаматом рецепторов NMDA, например, приводит к активации фермента, нейрональной синтетазы оксида азота (nNOS), что приводит к образованию оксида азота (NO), который также участвует в нейротоксичности. Нейротоксичность, связанную с NMDA, можно предотвратить введением ингибиторов синтетазы оксида азота (NOS) или посредством направленного генетического поврежденияnNOS in vitro. Другое использование ингибиторов PARP представляет собой лечение повреждений периферических нервов и являющегося их следствием синдрома патологической боли, известного как невропатическая боль, такая как боль, вызванная повреждением вследствие длительного сдавления (CCI) общего седалищного нерва, при котором происходит транссинаптическое изменение дорзального рога спинного-2 014955 мозга, которое характеризуется гиперхроматозом цитоплазмы и нуклеоплазмы (так называемые "темные" нейроны). Также существуют доказательства того, что ингибиторы PARP полезны для лечения воспалительных нарушений кишечника, таких как колиты. Конкретно, колиты вызывали у крыс посредством введения в просвет гаптена тринитробензолсульфоновой кислоты в 50% этаноле. После введения крысам вводили 3-аминобензамид, специфический ингибитор активности PARP. Ингибирование активности PARP приводило к снижению воспалительного ответа и восстановлению морфологии и активности дистального кишечника. Кроме того, доказательства позволяют предположить, что ингибиторы PARP полезны для лечения артрита. Кроме того, ингибиторы PARP, по-видимому, полезны для лечения диабета. Было показано, что ингибиторы PARP полезны для лечения эндотоксического шока или септического шока. Ингибиторы PARP также используются для увеличения срока жизни и пролиферативной способности клеток, включая лечение заболеваний, таких как старение кожи, болезнь Альцгеймера, атеросклероз,остеоартрит, остеопороз, мышечная дистрофия, дегенеративные заболевания скелетной мускулатуры,включающие репликативные возрастные изменения, связанную с возрастом мышечную дегенерацию,возрастные изменения иммунной системы, СПИД и другие заболевания иммунной системы, связанные со старением; и для изменения экспрессии генов в стареющих клетках. Также известно, что ингибиторы PARP, такие как 3-аминобензамид, действуют на репарацию ДНК в целом, например, в ответ на воздействие пероксида водорода или ионизирующей радиации. Ключевая роль PARP в репарации разрывов цепей ДНК твердо установлена особенно при репарации, вызванной непосредственно ионизирующей радиацией или косвенно после ферментативной репарации повреждений ДНК, вызванных метилирующими агентами, ингибиторами топоизомеразы I и другими химиотерапевтическими средствами, такими как цисплатин и блеомицин. Множество исследований с использованием "нокаута" мышей, моделей с трансдоминантным ингибированием (сверхэкспрессия ДНК-связывающего домена), антисмысловых и низкомолекулярных ингибиторов показало роль PARP в репарации и выживании клеток после индукции повреждения ДНК. Ингибирование ферментативной активности PARP должно привести к усилению чувствительности клеток опухоли к способам лечения с повреждением ДНК. Сообщалось, что ингибиторы PARP эффективны в радиосенсибилизации (гипоксических) клеток опухоли и эффективны в предотвращении восстановления клеток опухоли при потенциально летальном и сублетальном повреждении ДНК после лучевой терапии, по-видимому, вследствие их способности предотвращать воссоединение разрывов ДНК и вследствие их воздействия на ряд сигнальных путей, связанных повреждением ДНК. Ингибиторы PARP используются для лечения злокачественной опухоли. Кроме того, в патенте США 5177075 описаны несколько изохинолинов, используемых для усиления летальных эффектов ионизирующей радиации или химиотерапевтических средств на клетки злокачественной опухоли. WeltinCells", Oncol. Res., 6:9, 399-403 (1994), описывают ингибирование активности PARP, снижение пролиферации опухолевых клеток и выраженный синергический эффект при дополнительном введении в опухолевые клетки алкилирующего лекарственного средства. Обзоры уровня техники опубликованы Li и Zhang в IDrugs 2001, 4(7): 804-812, Ame et al. в Bioassays 2004, 26: 882-883 и Nguewa et al. в Progress in BiophysicMolecular Biology 2005, 88: 143-172. Сохраняется потребность в эффективных и мощных ингибиторах PARP и, более конкретно, ингибиторах PARP-1, которые вызывают минимальные побочные действия. Настоящее изобретение относится к соединениям, композициям для ингибирования активности PARP и способам ингибирования активности PARP для лечения злокачественной опухоли и/или профилактики повреждения клеток, тканей и/или органа или гибели вследствие, например, некроза или апоптоза. Соединения и композиции по настоящему изобретению полезны, в особенности, для усиления эффективности химиотерапии и лучевой терапии, где основной эффект лечения представляет собой эффект вследствие повреждения ДНК в клетках-мишенях. Синтез производных аминофталазинона описан Komendy et al. в Acta Chimica Academiae Scientiarum Hungaricae 1981, 106(2): 155-66 и в ActaChimica Hungarica 1983, 112(1): 65-82. В ЕР 156433, опубликованном 2 октября 1985 г., описаны пиридазинамины. Описанные соединения обладают противовирусными свойствами. Более конкретно, описаны соединения 77, 78, 79, 80, 81, 82,83 и 84 в настоящем изобретении.-3 014955 Данное изобретение относится к соединениям формулы (I) к их формам N-оксидов, фармацевтически приемлемым аддитивным солям и стереохимически изомерным формам,где пунктирная линия обозначает необязательную связь;n равен 0, 1, 2 или 3, если n равен 0, тогда подразумевается прямая связь;Q представляет собой -C(=O)- или -CR3-, где R3 представляет собой галоген или C1-6-алкил; если Q представляет собой -CR3-, пунктирная линия обозначает связь; каждый X независимо представляет собой -N или -СН; если X представляет собой -СН, тогда Y представляет собой -N или -NH-; каждый Y независимо представляет собой -N, -NH-, -СН или -CH2-; за исключением случаев, когда X представляет собой -СН, тогда Y представляет собой -N илиL1 представляет собой прямую связь или двухвалентный радикал, выбранный из -С 1-6-алкандиилNH-, -NH- или -NH-С 1-6-алкандиил-NH-;L2 представляет собой прямую связь или двухвалентный радикал, выбранный из -С 1-6-алкандиила-,-С 2-6-алкендиила-, карбонила или -C1-6-алкандиила-, замещенного одним заместителем, выбранным из группы, состоящей из гидрокси или арила;R2 представляет собой водород, C1-6-алкил или арил-C1-6-алкил; также может быть связан мостиковой связью (т.е. образовывать центральный фрагмент бициклическую группу) с этиленовой мостиковой связью; где R4 и R5, каждый независимо, выбраны из водорода, галогена, С 1-6-алкила, C1-6-алкилокси или тригалогенметила; арил представляет собой фенил или фенил, замещенный одним или двумя заместителями, каждый из которых независимо выбран из галогена, C1-6-алкила или C1-6-алкилокси; при условии, что если Q представляет собой -C(=O)- и X представляет собой -N, и Y представляет собой -СН или -CH2-, и L1 представляет собой прямую связь, и L2 представляет собой прямую связь или двухвалентный радикал -C1-6-алкандиил- или -C1-6-алкандиил-, замещенный гидрокси, и R1 представляет собой водород, и R2 представляет собой водород или С 1-6-алкил, тогда Z отличен от водорода, гидрокси или С 1-6-алкила; при условии исключения соединений Соединения формулы (I) могут также существовать в их таутомерных формах. Подразумевается,что такие формы, хотя и не явно показанные в приведенной выше формуле, входят в объем настоящего изобретения. Ряд терминов, используемых в указанных выше определениях и в дальнейшем, разъяснены ниже. Эти термины иногда используются сами по себе или в составных терминах. Как используется в указанных выше определениях и в дальнейшем, галоген является общим названием для фтора, хлора, брома и йода; тригалогенметил относится к метилу, содержащему три одинаковых или различных заместителя галогена, например трифторметил; C1-6-алкил относится к неразветвленным и разветвленным насыщенным углеводородным радикалам с количеством атомов углерода от 1 до 6, таким как, например, метил, этил, пропил, бутил, пентил, гексил, 1-метилэтил, 2-метилпропил, 2 метилбутил, 2-метилпентил и подобные; -C1-6-алкандиил- относится к двухвалентным неразветвленным и разветвленным насыщенным углеводородным радикалам с количеством атомов углерода от 1 до 6, таким как, например, метилен, 1,2-этандиил, 1,3-пропандиил, 1,4-бутандиил, 1,5-пентандиил, 1,6 гександиил и их разветвленным изомерам, таким как 2-метилпентандиил, 3-метилпентандиил, 2,2 диметилбутандиил, 2,3-диметилбутандиил и подобные; -С 2-6-алкендиил- относится к двухвалентным неразветвленным и разветвленным углеводородным радикалам, содержащим одну двойную связь и имеющим от 2 до 6 атомов углерода, таким как, например, этендиил, 2-пропендиил, 3-бутендиил, 2 пентендиил, 3-пентендиил, 3-метил-2-бутендиил и подобные. Под термином "фармацевтически приемлемые соли" подразумевают фармацевтически приемлемые кислотно- или основно-аддитивные соли. Под фармацевтически приемлемыми кислотно- или основноаддитивными солями, как указано выше, подразумевают, что они включают терапевтически активные нетоксичные формы кислотно-аддитивной соли и нетоксичные формы основно-аддитивной соли, которые соединения формулы (I) способны образовывать. Соединения формулы (I), которые имеют свойства оснований, могут быть превращены в их фармацевтически приемлемые кислотно-аддитивные соли обработкой указанной основной формы соответствующей кислотой. Подходящие кислоты включают, например, неорганические кислоты, такие как галогенводородные кислоты, например хлористо-водородная или бромисто-водородная кислота; серная; азотная; фосфорная и другие кислоты; или органические кислоты, такие как, например, уксусная, пропионовая, гидроксиуксусная, молочная, пировиноградная, щавелевая, малоновая, янтарная (т.е. бутандиовая кислота), малеиновая, фумаровая, яблочная, виннокаменная, лимонная, метансульфоновая, этансульфоновая, бензолсульфоновая, п-толуолсульфоновая,цикламовая, салициловая, п-аминосалициловая, памовая и сходные кислоты. Соединения формулы (I) с кислотными свойствами можно превращать в их фармацевтически приемлемые основно-аддитивные соли обработкой указанной кислотной формы соответствующим органическим или неорганическим основанием. Подходящие формы основных солей включают, например, соли аммония, соли щелочных и щелочно-земельных металлов, например соли лития, натрия, калия, магния,кальция и подобные, соли с органическими основаниями, например соли бензатина, N-метил-Dглюкамина, гидрабамина и соли с аминокислотами, такими как, например, аргинин, лизин и подобные. Термины "кислотная" или "основная аддитивная соль" также включают гидраты и аддитивные сольватные формы, которые соединения формулы (I) способны образовывать. Примерами таких форм являются, например, гидраты, алкоголяты и подобные. Термин "стереохимически изомерные формы соединений формулы (I)", как используется в данном-5 014955 описании, относится ко всем возможным соединениям, состоящим из одних и тех же атомов, связанных одной и той же последовательностью связей, но с отличающимися трехмерными структурами, не являющимися равнозначными, которыми соединения формулы (I) могут обладать. Если не упомянуто или не указано иное, химическое обозначение соединения включает смесь всех возможных стереохимически изомерных форм, которыми указанное соединение может обладать. Указанная смесь может содержать все диастереомеры и/или энантиомеры базовой молекулярной структуры указанного соединения. Подразумевается, что все стереохимически изомерные формы соединений формулы (I) как в чистой форме, так и в смеси друг с другом входят в объем настоящего изобретения. Под формами N-оксидов соединений формулы (I) подразумевают, что они включают соединения формулы (I), где один или несколько атомов азота окислены до так называемых N-оксидов, в частности до N-оксидов, в которых один или несколько атомов азота пиперидина или пиперазина N-оксидированы. Под термином "соединения формулы (I)", используемым в данном описании, подразумевают, что он также включает формы N-оксидов, фармацевтически приемлемые кислотно- или основно-аддитивные соли и все стереоизомерные формы.Komendy et al. описывают только синтез производных аминофталазинона. Соединения, описанные в ЕР 156433, обладают противовирусными свойствами. Более конкретно, описаны соединения 77-84 настоящего описания. Неожиданно было обнаружено, что соединения по настоящему изобретению проявляют ингибирующую активность в отношении PARP. Первая группа из рассматриваемых соединений состоит из таких соединений формулы (I), где налагаются одно или несколько следующих ниже ограничений:b) R2 представляет собой водород или арил-C1-6-алкил;c) Z представляет собой водород, C1-6-алкил, C1-6-алкилокси, арилокси, амино, циано, арил-C1-6 алкиламино или бензтиазолил(C1-6-алкил)амино либо кольцевую систему, выбранную из (а-1), (а-2), (а-3),(а-4), (а-5), (а-6), (а-7), (а-8), (а-9), (а-10). Вторая группа рассматриваемых соединений состоит из таких соединений формулы (I), где налагаются одно или несколько следующих ограничений:a) Q представляет собой -CR3-, где R3 представляет собой галоген или С 1-6-алкил;c) каждый Y независимо представляет собой -N или -NH-;d) L1 представляет собой двухвалентный радикал, выбранный из -С 1-6-алкандиил-NH-, -NH- илиe) L2 представляет собой двухвалентный радикал, выбранный из -С 2-6-алкендиила-, карбонила илиh) Z представляет собой С 1-6-алкилокси, арилокси, амино, циано, арил-C1-6-алкиламино или бензтиазолил(C1-6-алкил)амино либо кольцевую систему, выбранную из (а-1), (а-2), (а-3), (а-4), (а-5), (а-6), (а-7),(а-8), (а-9), (а-10). Третья группа рассматриваемых соединений состоит из таких соединений формулы (I), где налагается одно или несколько следующих ниже ограничений:d) каждый Y независимо представляет собой -СН или -СН 2-;e) L1 представляет собой двухвалентный радикал, выбранный из -С 1-6-алкандиил-NH-, -NH- илиf) L2 представляет собой двухвалентный радикал, выбранный из -C1-6-алкандиила-, -С 2-6 алкендиила-, карбонила или -C1-6-алкандиила-, замещенного одним заместителем, выбранным из гидрокси или арила;i) Z представляет собой водород, гидрокси, С 1-6-алкил, C1-6-алкилокси, арилокси, амино, циано,арил-C1-6-алкиламино или бензтиазолил(C1-6-алкил)амино либо кольцевую систему, выбранную из (а-1),(а-2), (а-4), (а-5), (а-6), (а-7), (а-8), (а-9), (а-10). Группа предпочтительных соединений состоит из таких соединений формулы (I), где налагается одно или несколько следующих ограничений:c) L1 представляет собой прямую связь или двухвалентный радикал -NH-;d) L2 представляет собой прямую связь или двухвалентный радикал, выбранный из карбонила, -C1-6 алкандиила- или -C1-6-алкандиила-, замещенного гидрокси;g) Z представляет собой водород, С 1-6-алкилокси, арилокси, амино или кольцевую систему, выбранную из (а-2) или (а-3);h) R4 и R5, каждый независимо, выбраны из водорода или галогена. Группа наиболее предпочтительных соединений состоит из таких соединений формулы (I), где n равен 1 или 2; Q представляет собой -С(=O)-; Y представляет собой -N, -NH- или -СН; L1 представляет собой прямую связь или двухвалентный радикал -NH-; L2 представляет собой прямую связь или двухвалентный радикал, выбранный из карбонила, -C1-6-алкандиила- или -C1-6-алкандиила-, замещенного гидрокси; R1 представляет собой водород; R2 представляет собой водород, арил-C1-6-алкил; Z представляет собой водород, С 1-6-алкилокси, арилокси, амино или кольцевую систему, выбранную из (а-2) или (а-3); R4 и R5, каждый независимо, выбраны из водорода или галогена. Наиболее предпочтительными соединениями являются соединения 41, 13, 62, 26, 64, 2, 8, 9, 34,11, 15 и 10. Соединения формулы (I) могут быть получены в соответствии с основными способами, описанными в ЕР 156433. Исходные вещества и некоторые промежуточные соединения являются известными соединениями и коммерчески доступны или могут быть получены в соответствии с традиционными методиками, главным образом, известными в данной области. Некоторые способы получения более подробно описаны ниже. Другие способы получения конечных соединений формулы (I) описаны в примерах. Соединения формулы (I), где L1 представляет собой связь, X представляет собой -СН- и Q представляет собой -С(=O)-, которые относятся к соединениям формулы (I-а), могут быть получены взаимодействием промежуточного соединения формулы (II) с гидразином. Реакцию можно проводить в подходящем растворителе, таком как, например, спирт, например метанол, этанол, пропанол и подобных. Соединения формулы (I), где Y представляет собой -N и L2 представляет собой двухвалентный радикал -C1-6-алкандиил-, замещенный гидрокси, которые относятся к соединениям формулы (I-b), могут быть получены реакцией промежуточного соединения формулы (V), где t равен целому числу 0, 1, 2, 3 или 4, с соединением формулы (I), где L2 представляет собой прямую связь и Z представляет собой водород, которые относятся к соединениям формулы (I-c). Реакцию можно проводить в реакционно-инертном растворителе, таком как, например, N,N-диметилформамид и подобных или спирт, например пропанол и подобных. Может использоваться добавление соответствующего основания, такого как, например, карбонат или гидрокарбонат щелочного или щелочно-земельного металла, например триэтиламина или карбоната натрия. Соединения формулы (I), где Y представляет собой -N и L2 представляет собой необязательно замещенный двухвалентный радикал -C1-6-алкандиил-, которые относятся к соединениям формулы (I-d),могут быть получены восстановительным N-алкилированием соединений формулы (I-c), где r равен целому числу 0, 1, 2, 3, 4 или 5, с соответствующим карбонильным промежуточным соединением формулы(VIII). Указанную реакцию восстановительного N-алкилирования можно удобно проводить посредством каталитической гидрогенизации перемешиваемой и нагреваемой смеси реагирующих веществ в подходящем реакционно-инертном органическом растворителе в соответствии с известными в данной области методами каталитической гидрогенизации. Подходящими растворителями являются, например, спирты,например метанол, этанол, 2-пропанол и подобные; циклические простые эфиры, например 1,4-диоксан и подобные; галогенированные углеводороды, например трихлорметан и подобные; N,Nдиметилформамид; диметилсульфоксид и подобные; или смесь 2 или более таких растворителей. Под термином "известные в данной области методы каталитической гидрогенизации" подразумевают, что реакцию проводят в атмосфере водорода и в присутствии подходящего катализатора, такого как, например, палладий на активированном угле, платина на активированном угле и подобные. В целях предотвращения нежелательной дополнительной гидрогенизации определенных функциональных групп в участвующих в реакции веществах и продуктах реакции может быть предпочтительным добавление в реакционную смесь подходящего каталитического яда, например тиофена и подобных. Известные в данной области методы каталитической гидрогенизации, как описано выше, также могут быть использованы для получения соединений формулы (I-c) (например) или соединений формулы(I), где L2 представляет собой прямую связь или двухвалентный радикал -C1-6-алкандиил- и Z представляет собой амино, начиная с соединений формулы (I), где Z-L2- представляет собой арил-C1-6-алкил (например) или арил-C1-6-алкиламино, которые относятся к соединениям формулы (I-е). Методы каталитической гидрогенизации также могут использоваться для получения соединений формулы (I), где Z-L2- представляет собой аминоС 1-6-алкил, которые относятся к соединениям формулы(I-i), превращением соединений формулы (I), где Z-L2- представляет собой циано(СН 2)r, где r равен це-8 014955 лому числу 1, 2, 3, 4 или 5, которые относятся к соединениям формулы (I-j). Реакцию проводят в атмосфере водорода и в присутствии никеля Ренея в смеси метанола и аммиака. Соединения формулы (I-c) также могут быть получены деацилированием соединений формулы (I),где Z-L2- представляет собой -C1-6-алкилоксикарбонил, которые относятся к соединениям формулы (I-f),реакцией исходного вещества с подходящим раствором кислоты или основания, таким как хлористоводородная кислота или гидробромид, в подходящем растворителе, например в спирте, таком как пропанол. Соединения формулы (I), где Q представляет собой -С(=O)-, которые относятся к соединениям формулы (I-g), могут быть получены превращением соединения формулы (I), где Q представляет собой-CR3-, R3 представляет собой галоген и пунктирная линия обозначает связь, которые относятся к соединениям формулы (I-h), обработкой смеси соединений формулы (I-h), ацетата натрия и уксусной кислоты с подходящим раствором кислоты, такой как хлористо-водородная кислота. Соединения формулы (I), где Y представляет собой -N, которые относятся к соединениям формулы (I-k), могут быть получены реакцией соединения формулы (I-c) с промежуточным соединением формулы (XI), где W представляет собой соответствующую уходящую группу, такую как, например, галоген, например фтор, хлор, бром или йод, или сульфонилоксирадикал, такой как метилсульфонилокси, 4 метилфенилсульфонилокси и подобные. Реакция может быть проведена в реакционно-инертном растворителе, таком как, например, спирт, например метанол, этанол, 2-метоксиэтанол, пропанол, бутанол и подобные; простой эфир, например 4,4-диоксан, 1,1'-оксибиспропан и подобные; кетон, например 4 метил-2-пентанон; N,N-диметилформамид или нитробензол и подобные. Добавление пригодных оснований, таких как, например, карбонаты и гидрокарбонаты щелочных и щелочно-земельных металлов, например триэтиламин или карбонат натрия, может использоваться для того, чтобы удалять кислоты, которые высвобождаются в ходе реакции. Для осуществления реакции могут быть добавлены небольшие количества подходящего иодида металла, например для осуществления реакции может быть добавлен иодид натрия или калия. Перемешивание может увеличить скорость реакции. Реакцию удобно проводить при температуре реакционной смеси, находящейся в диапазоне между комнатной температурой и температурой кипячения с обратным холодильником, и, если необходимо, реакция может быть проведена при повышенном давлении. Аналогично соединения формулы (I), где X представляет собой N- или L1 представляет собой-С 1-6-алкандиил-NH-, -NH-(например) или -NH-С 1-6-алкандиил-NH-, которые относятся к соединениям формулы (I-l), могут быть получены реакцией промежуточного соединения формулы (IX) с промежуточ-9 014955 ным соединением формулы (X), где W представляет собой описанную выше группу. Соединения формулы (I) также могут быть превращены друг в друга известными в данной области реакциями или трансформациями функциональных групп. Некоторые из таких трансформаций уже описаны выше. Другие примеры представляют собой гидролиз сложных эфиров карбоновых кислот до соответствующих карбоновых кислот или спиртов; гидролиз амидов до соответствующих карбоновых кислот или аминов; гидролиз нитрилов до соответствующих амидов; аминогруппы имидазола или фенила могут быть заменены водородом известными в данной области реакциями диазотирования и последующей заменой диазогрупп водородом; спирты могут быть превращены в сложные и простые эфиры; первичные амины могут быть превращены во вторичные или третичные амины; двойные связи могут быть гидрогенизированы до соответствующей одинарной связи; радикал йода на фенильной группе может быть превращен в сложноэфирную группу вставкой монооксида углерода в присутствии подходящего палладиевого катализатора. Промежуточные соединения формулы (II) могут быть получены реакцией промежуточного соединения формулы (III) с подходящим 1(3H)-изобензофураноном формулы (IV) в смеси натрия и подходящего растворителя, такого как, например, спирт, например, этанол и подобные. Настоящее изобретение также относится к соединениям для применения в качестве лекарственного средства, где указанные соединения представляют собой соединения формулы (I) и к их формам N-оксидов, фармацевтически приемлемым аддитивным солям и стереохимически изомерным формам, где пунктирная линия обозначает необязательную связь;n равен 0, 1, 2 или 3, если n равен 0, тогда подразумевается прямая связь;Q представляет собой -С(=O)- или -CR3-, где R3 представляет собой галоген или С 1-6-алкил; если Q представляет собой -CR3-, тогда пунктирная линия обозначает связь; каждый X независимо представляет собой -N или -СН; если X представляет собой -СН, тогда Y представляет собой -N или -NH-; каждый Y независимо представляет собой -N, -NH-, -СН или -CH2-; за исключением случаев, когда X представляет собой -СН, тогда Y представляет собой -N илиL1 представляет собой прямую связь или двухвалентный радикал, выбранный из -C1-6-алкандиилNH-, -NH- или -NH-С 1-6-алкандиил-NH-;L2 представляет собой прямую связь или двухвалентный радикал, выбранный из -C1-6-алкандиила-,-С 2-6-алкендиила-, карбонила или -C1-6-алкандиила-, замещенного одним заместителем, выбранным из гидрокси или арила;R2 представляет собой водород, C1-6-алкил или арил-C1-6-алкил; центральный фрагмент также может быть связан мостиковой связью (т.е. образовывать бициклическую группу) с этиленовой мостиковой связью; где R4 и R5, каждый независимо, выбраны из водорода, галогена, C1-6-алкила, C1-6-алкилокси или тригалогенметила; арил представляет собой фенил или фенил, замещенный одним или двумя заместителями, каждый из которых независимо выбран из галогена, C1-6-алкила или С 1-6-алкилокси; при условии, что если n равен 1 и X представляет собой -N, и Y представляет собой -N, и L1 и L2 представляют собой прямую связь, и R1 и R2 представляют собой водород, и Q представляет собой -CR3-,где R3 представляет собой хлор, тогда Z отличен от кольцевой системы (а-3). Как можно видеть из экспериментальной части, приведенной ниже, соединения по настоящему изобретению обладают свойствами ингибирования PARP. Термин "PARP" используется в данном описании для обозначения белка с активностью в отношении полиАДФ-рибозилирования. В объеме значения этого термина PARP включает все белки, кодируемые геном PARP, их мутантные формы и их белки, полученные в результате альтернативного сплайсинга. Кроме того, как используется здесь, термин "PARP" включает аналоги PARP, гомологи и аналоги других животных. Термин "PARP" включает, но не ограничивается им, PARP-1. В объем значения этого термина могут быть включены PARP-2, PARP-3, Vault-PARP (PARP-4), PARP-7 (TiPARP), PARP-8, PARP-9 (Bal),PARP-10, PARP-11, PARP-12, PARP-13, PARP-14, PARP-15, PARP-16, TANK-1, TANK-2 и TANK-3. Соединения, которые ингибируют и PARP-1, и танкиразу-2, могут обладать предпочтительными свойствами в том отношении, что они обладают усиленной активностью в отношении ингибирования роста злокачественных клеток. Настоящее изобретение также включает применение соединений для получения лекарственного средства для лечения любого заболевания и нарушения у описанных здесь животных, где указанные соединения представляют собой соединения формулы (I) их формы N-оксидов, фармацевтически приемлемые аддитивные соли и стереохимически изомерные формы, где пунктирная линия обозначает необязательную связь;n равен 0, 1, 2 или 3, и если n равен 0, тогда подразумевается прямая связь;Q представляет собой -С(=O)- или -CR3-, где R3 представляет собой галоген или С 1-6-алкил; и если Q представляет собой -CR3-, тогда пунктирная линия обозначает связь; каждый X независимо представляет собой -N или -СН; если X представляет собой -СН, тогда Y представляет собой -N или -NH-; каждый Y независимо представляет собой -N, -NH-, -СН или -СН 2-; за исключением случаев, когда X представляет собой -СН, тогда Y представляет собой -N илиL1 представляет собой прямую связь или двухвалентный радикал, выбранный из -С 1-6-алкандиилNH-, -NH- или -NH-С 1-6-алкандиил-NH-;L2 представляет собой прямую связь или двухвалентный радикал, выбранный из -C1-6-алкандиила-,-С 2-6-алкендиила-, карбонила или -C1-6-алкандиила-, замещенного одним заместителем, выбранным из гидрокси или арила;R2 представляет собой водород, C1-6-алкил или арил-C1-6-алкил; также может быть связан мостиковой связью (т.е. образовывать центральный фрагмент бициклическую группу) с этиленовой мостиковой связью; где R4 и R5, каждый независимо, выбраны из водорода, галогена, C1-6-алкила, C1-6-алкилокси или тригалогенметила; арил представляет собой фенил или фенил, замещенный одним или двумя заместителями, каждый из которых независимо выбран из галогена, C1-6-алкила или C1-6-алкилокси. С точки зрения свойств в отношении связывания PARP соединения по настоящему изобретению могут использоваться в качестве контрольных соединений или соединений, содержащих изотопный индикатор, в случае которых один из атомов молекулы может быть заменен, например, радиоактивным изотопом. Для получения фармацевтических композиций по данному изобретению эффективное количество конкретного соединения в форме кислотной или основной аддитивной соли в качестве активного ингредиента комбинируют в однородную смесь с фармацевтически приемлемым носителем, который может принимать широкое разнообразие форм в зависимости от желаемой формы препарата для введения. Желательно, чтобы эти фармацевтические композиции находились в единичных дозированных формах,предпочтительно пригодных для перорального, ректального, подкожного введения или парентеральной инъекции. Например, при получении композиций в виде пероральной дозированной формы могут использоваться любые из обычных фармацевтических сред, таких как, например, вода, гликоли, масла,спирты и подобные, в случае пероральных жидких препаратов, таких как суспензии, сиропы, эликсиры и растворы; или твердые носители, такие как крахмалы, сахара, каолин, смазывающие вещества, связующие вещества, дезинтегрирующие агенты и подобные, в случае порошков, пилюль, капсул и таблеток. Вследствие простоты введения таблетки и капсулы представляют собой наиболее предпочтительные пероральные единичные дозированные формы, в случае которых обычно используют твердые фармацевтические носители. В случае композиций для парентерального введения носитель обычно содержит стерильную воду, по меньшей мере, в большинстве случаев, хотя для обеспечения растворимости в состав могут быть включены другие ингредиенты. Например, могут быть получены растворы для инъекций, в которых носитель содержит физиологический раствор, раствор глюкозы или смесь физиологического раствора и раствора глюкозы. Также могут быть получены суспензии для инъекций, в случае которых могут использоваться подходящие жидкие носители, суспендирующие агенты и подобные. В композициях, пригодных для подкожного введения, носитель необязательно содержит агент, увеличивающий проницаемость, и/или подходящий увлажняющий агент, необязательно комбинированный с подходящими добавками любой природы в небольших количествах, при этом добавки не приводят к значительным вредным воздействиям на кожу. Указанные добавки могут облегчать введение через кожу и/или могут быть полезными для получения желаемых композиций. Эти композиции могут быть введены различными способами, например в виде трансдермального пластыря, в виде капель, в виде мази. Особенно предпочтительным является изготовление вышеуказанных фармацевтических композиций в единичных дозированных формах для простоты введения и единообразия дозирования. Единичные дозированные формы, как используется в описании и формуле изобретения, относятся к физически дискретным единицам,подходящим в качестве единичных доз, где каждая единица содержит предопределенное количество активного ингредиента, вычисленное для получения желаемого терапевтического эффекта, вместе с необходимым фармацевтическим носителем. Примерами таких единичных дозированных форм являются таблетки (включая шероховатые или покрытые таблетки), капсулы, пилюли, пакетики с порошками, пластинки, растворы или суспензии для инъекций, формы для введения с помощью чайных и столовых ложек и подобные и их отдельные кратные единицы. С помощью соединений по настоящему изобретению можно лечить или предотвращать повреждение тканей в результате повреждения или смерти клеток вследствие некроза или апоптоза; можно уменьшать повреждение нервной ткани или тканей сердечно-сосудистой системы, включая повреждение после локальной ишемии, инфаркта миокарда и повреждение при реперфузии; можно лечить различные заболевания и состояния, вызванные или усиленные вследствие активности PARP; можно продлевать или увеличивать длительность жизни или пролиферативную способность клеток; можно изменять экс- 12014955 прессию генов стареющих клеток; можно повышать в клетках радиочувствительность и/или чувствительность к химиотерапевтическим средствам. Как правило, ингибирование активности PARP защищает клетки от потери энергии, предотвращая, в случае нервных клеток, необратимую деполяризацию нейронов и, таким образом, обеспечивая нейропротекцию. По следующим ниже причинам настоящее изобретение, кроме того, относится к способу введения терапевтически эффективного количества указанных выше соединений в количестве, достаточном для ингибирования активности PARP, для лечения или профилактики повреждения тканей вследствие повреждения или смерти клеток вследствие некроза или апоптоза, для воздействия на активность нейронов,на опосредуемую токсичностью NMDA, для воздействия на активность нейронов, опосредуемую токсичностью NMDA, для лечения повреждения нервной ткани вследствие ишемии и повреждения при реперфузии, неврологических нарушений и нейродегенеративных заболеваний; для профилактики или лечения сосудистого инсульта; для лечения или профилактики сердечно-сосудистых нарушений; для лечения других состояний и/или нарушений, таких как мышечная дегенерация, связанная со старением,СПИД и другие заболевания иммунной системы, связанные со старением, воспаление, подагра, артрит,атеросклероз, кахексия, злокачественная опухоль, дегенеративные заболевания скелетной мускулатуры,включающие нарушения репликации, связанные со старением, диабет, травму головы, воспалительные нарушения кишечника (такие как колиты и болезнь Крона), мышечная дистрофия, остеоартрит, остеопороз, хроническая и/или острая боль (такая как невропатическая боль), почечная недостаточность, ишемия сетчатки, септический шок (такой как эндотоксический шок), и старение кожи, для продления жизни и пролиферативной способности клеток; для изменения экспрессии генов стареющих клеток; для химиосенсибилизации и/или радиосенсибилизации (гипоксических) злокачественных клеток. Настоящее изобретение также относится к лечению заболеваний и состояний у животных, которое включает введение указанному животному терапевтически эффективного количества указанных выше соединений. В частности, настоящее изобретение относится к способу лечения, профилактики или замедления развития неврологического нарушения у животного, который включает введение указанному животному терапевтически эффективного количества указанных выше соединений. Неврологическое нарушение выбрано из группы, состоящей из периферической невропатии вследствие физического повреждения или болезненного состояния, травматического повреждения головного мозга, физического повреждения спинного мозга, инсульта, ассоциированного с повреждением головного мозга, местной ишемии, общей ишемии, повреждения при реперфузии, демиелинизирующего заболевания и неврологического нарушения, связанного с нейродегенерацией. Настоящее изобретение также включает применение соединений формулы (I) для ингибирования активности PARP для лечения, профилактики или замедления повреждения тканей в результате повреждения или гибели клеток вследствие некроза или апоптоза, для лечения, профилактики или замедления развития неврологического нарушения у животных. Термин "профилактика нейродегенерации" включает способность предотвращать нейродегенерацию у пациентов, которым недавно поставили диагноз нейродегенеративного заболевания, или при риске развития нового дегенеративного заболевания и для предотвращения дальнейшей нейродегенерации у пациентов, которые уже страдают от нейродегенеративного заболевания или имеют симптомы нейродегенеративного заболевания. Как используется в данном описании, термин "лечение" включает любой способ лечения заболевания и/или состояния у животного, в частности у человека, и включает: (i) профилактику заболевания и/или состояния у субъекта, который может быть предрасположен к заболеванию и/или состоянию, которое еще не диагностируется; (ii) замедление развития заболевания и/или состояния, т.е. прекращение его развития; (iii) смягчение заболевания и/или состояния, т.е. осуществление ослабления симптомов заболевания и/или состояния. Как используется в данном описании, термин "радиосенсибилизатор" определяется как молекула,предпочтительно низкомолекулярная молекула, которую вводят животным в терапевтически эффективных количествах для повышения чувствительности клеток к ионизирующей радиации и/или для обеспечения лечения заболеваний, которые поддаются лечению ионизирующей радиацией. Заболевания, которые поддаются лечению ионизирующей радиацией, включают заболевания, относящиеся к новообразованиям, доброкачественные и злокачественные опухоли и злокачественные клетки. Лечение ионизирующей радиацией других заболеваний, которые не приведены в данном описании, также охватывается настоящим изобретением. Как используется в данном описании, термин "химиосенсибилизатор" определяется как молекула,предпочтительно низкомолекулярная молекула, которую вводят животным в терапевтически эффективных количествах для повышения чувствительности клеток к химиотерапии и/или для обеспечения лечения заболеваний, которые поддаются лечению химиотерапевтическими средствами. Заболевания, которые поддаются лечению химиотерапией, включают заболевания, относящиеся к новообразованиям, доброкачественным и злокачественным опухолям и злокачественным клеткам. Лечение химиотерапией других заболеваний, которые не приведены в данном описании, также охватывается настоящим изобретением.- 13014955 Соединения, композиции и способы по настоящему изобретению являются, в частности, полезными для лечения или профилактики повреждения тканей в результате гибели или повреждения клеток вследствие некроза или апоптоза. Соединения по настоящему изобретению могут представлять собой "средства против злокачественной опухоли", и этот термин также включает "средства против роста опухолевых клеток" и "средства против новообразований". Например, способы по данному изобретению полезны для лечения злокачественных опухолей и химиосенсибилизации и/или радиосенсибилизации клеток опухоли при злокачественных опухолях, таких как опухоли, продуцирующие АСТН, острый лимфоцитарный лейкоз, острый нелимфоцитарный лейкоз, рак коры надпочечников, рак мочевого пузыря, злокачественная опухоль мозга, рак молочной железы, рак шейки матки, хронический лимфоцитарный лейкоз, хронический миелоцитарный лейкоз, рак ободочной и прямой кишки, Т-клеточная лимфома кожи, рак эндометрия, рак пищевода, саркома Юинга, рак желчного пузыря, волосатоклеточный лейкоз, рак головы и шеи, лимфома Ходжкина, саркома Капоши, рак почки, рак печени, рак легкого (мелкоклеточный и/или немелкоклеточный), злокачественный перитонеальный выпот, злокачественный плевральный выпот, меланома, мезотелиома, множественная миелома, нейробластома, неходжскинская лимфома, остеосаркома, рак яичника,рак яичника из зародышевых клеток, рак предстательной железы, рак поджелудочной железы, рак полового члена, ретинобластома, рак кожи, саркома мягких тканей, плоскоклеточный рак, рак желудка, рак яичка, рак щитовидной железы, трофобластическое новообразование, рак тела матки, рак влагалища, рак наружных женских половых органов и опухоль Вильмса. Таким образом, соединения по настоящему изобретению могут быть использованы в качестве "радиосенсибилизаторов" и/или "химиосенсибилизаторов". Известно, что радиосенсибилизаторы повышают чувствительность злокачественных клеток к токсическим эффектам ионизирующей радиации. В литературе предполагается несколько механизмов для способа действия радиосенсибилизаторов, включая радиосенсибилизаторы гипоксических клеток (например, соединения 2-нитроимидазола и соединения диоксида бензотриазина), имитирующие кислород или альтернативно ведущие себя подобно биологическим восстановителям при гипоксии; радиосенсибилизаторы негипоксических клеток (например, галогенированные пиримидины), которые могут быть аналогами оснований ДНК и предпочтительно встраиваются в ДНК злокачественных опухолей и, таким образом, обеспечивают индуцируемый облучением разрыв молекулы ДНК и/или предотвращают развитие нормальных механизмов репарации ДНК; и различные другие возможные механизмы действия могут быть предположены для радиосенсибилизаторов при лечении заболевания. В настоящее время во множестве различных протоколов лечения радиосенсибилизаторы используются совместно с облучением рентгеновскими лучами. Примеры активируемых рентгеновскими лучами радиосенсибилизаторов включают, но не ограничиваются ими, следующие: метронидазол, мизонидазол,десметилмизонидазол, пимонидазол, этанидазол, ниморазол, митомицин С, RSU 1069, SR 4233, EO9, RB 6145, никотинамид, 5-бромдезоксиуридин (BUdR), 5-йоддезоксиуридин (IUdR), ромдезоксицитидин,фтордезоксиуридин (FudR), гидроксимочевину, цисплатин и терапевтически эффективные аналоги и производные указанных выше веществ. При фотодинамической терапии (PDT) злокачественных опухолей в качестве радиационного активатора сенсибилизатора используют видимый свет. Примеры фотодинамических радиосенсибилизаторов включают, но не ограничиваются ими, следующие: производные гематопорфирина, фотофрин, производные бензопорфирина, этиопорфирин олова, феофорбид-а, бактериохлорофилл-а, нафталоцианины,фталоцианины, фталоцианин цинка и терапевтически эффективные аналоги и производные указанных выше веществ. Радиосенсибилизаторы могут быть введены вместе с терапевтически эффективным количеством одного или нескольких других соединений, включая, но не ограничиваясь ими, соединения, которые обеспечивают введение радиосенсибилизаторов в клетки-мишени; соединения, которые контролируют доставку лекарственных средств, пищевых добавок и/или кислорода в клетки-мишени; химиотерапевтические средства, которые воздействуют на опухоль с дополнительным облучением или без него; или другие терапевтически эффективные соединения для лечения злокачественной опухоли или другого заболевания. Примеры дополнительных терапевтических средств, которые могут использоваться совместно с радиосенсибилизаторами, включают, но не ограничиваются ими, 5-фторурацил, лейковорин, 5'амино-5'-дезокситимидин, кислород, карбоген, трансфузию эритроцитов, перфторуглероды (например,флюосол 10 DA), 2,3-DPG, BW12C, блокаторы кальциевых каналов, пентоксифиллин, антиангиогенные соединения, гидралазин и LBSO. Примеры химиотерапевтических средств, которые могут использоваться совместно с радиосенсибилизаторами, включают, но не ограничиваются ими, адриамицин, камптотецин, карбоплатин, цисплатин, даунорубицин, доцетаксел, доксорубицин, интерферон (альфа, бета, гамма), интерлейкин-2, иринотекан, паклитаксел, топотекан и терапевтически эффективные аналоги и производные указанных выше средств. Химиосенсибилизаторы могут быть введены совместно с терапевтически эффективным количеством одного или нескольких других соединений, включая, но не ограничиваясь ими, соединения, которые обеспечивают транспорт химиосенсибилизаторов в клетки-мишени; соединения, которые контролируют- 14014955 доставку лекарственных средств, пищевых добавок и/или кислорода в клетки-мишени; химиотерапевтические средства, которые воздействуют на опухоль, или другие терапевтически эффективные соединения для лечения злокачественной опухоли или другого заболевания. Примеры дополнительных терапевтических средств, которые могут применяться совместно с химиосенсибилизаторами, включают, но не ограничиваются ими, метилирующие агенты, ингибиторы топоизомеразы I и другие химиотерапевтические средства, такие как цисплатин и блеомицин. Соединения формулы (I) также могут использоваться для обнаружения или идентификации PARP и, более конкретно, рецептора PARP-1. Для этой цели к соединениям формулы (I) можно присоединять метку. Указанная метка может быть выбрана из группы, состоящей из радиоизотопной, спиновой метки,антигенной метки, ферментной метки, флуоресцентной группы или хемилюминесцентной группы. Специалисты в данной области легко могут определить эффективное количество из результатов теста, представленного ниже. Как правило, предусматривается, что эффективное количество может составлять от 0,001 до 100 мг/кг массы тела, в частности от 0,005 до 10 мг/кг массы тела. Может быть целесообразным введение требуемой дозы в виде двух, трех, четырех или более субдоз в течение суток с соответствующими интервалами. Указанные субдозы могут быть включены в состав в качестве единичных дозированных форм, например, содержащих от 0,05 до 500 мг, в частности от 0,1 мг до 200 мг активного ингредиента в единичной дозированной форме. Следующие ниже примеры иллюстрируют настоящее изобретение. Экспериментальная часть. В дальнейшем в данном описании "DCM" определяется как дихлорметан, "DIPE" определяется как диизопропиловый эфир, "ДМФА" определяется как N,N-диметилформамид, "EtOH" определяется как этанол, "EtOAc" определяется как этилацетат, "МеОН" определяется как метанол и "TEA" определяется как триэтиламин, "ТГФ" определяется как тетрагидрофуран. А. Получение промежуточных соединений. Пример А 1. Получение промежуточного соединения 1 К растворенной смеси натрия (0,2 моль) в абсолютном EtOH (400 мл) добавляли 1-(фенилметил)-4 пиперидинон (0,2 моль) и 1(3H)-изобензофуранон (0,2 моль). Смесь нагревали до температуры кипения с обратным холодильником и кипятили с обратным холодильником в течение одной ночи. Смесь выпаривали, добавляли воду и экстрагировали толуолом. Водный слой нейтрализовали уксусной кислотой и экстрагировали DCM. Органический слой сушили, фильтровали и выпаривали с получением 30,3 г(51,7%) промежуточного соединения 1. Пример А 2. а) Получение промежуточного соединения 2 К смеси этилового эфира 4-оксо-2-(фенилметил)-1-пиперидинкарбоновой кислоты (0,14 моль) и бензиламина (0,14 моль) в EtOH (300 мл) при комнатной температуре добавляли тетракис(изопропанолят)титан (50 мл) и смесь перемешивали при комнатной температуре в течение 6 ч. Добавляли раствор гидробората натрия (5,3 г) в EtOH (150 мл) и смесь перемешивали при комнатной температуре в течение 18 ч. Смесь гидролизовали водой, фильтровали через целит и выпаривали. Осадок отбирали в DCM и промывали водой. Органический слой отделяли, сушили (MgSO4), фильтровали и растворитель выпаривали. Осадок (38,5 г) очищали колоночной хроматографией с силикагелем (20-45 мкм)(элюент: циклогексан/EtOAc 60/40). Очищенные фракции собирали и растворитель выпаривали с получением 10 г (27%) промежуточного соединения 2.b) Получение промежуточного соединения 3 Промежуточное соединение 2 (0,0571 моль) в EtOH (300 мл) гидрогенизировали при 50 С в присутствии Pd/C (10 г) в качестве катализатора в течение одной ночи при давлении 3 бар в аппарате Парра.- 15014955 После поглощения Н 2 (1 экв.) катализатор отфильтровывали через целит, промывали EtOH и фильтрат выпаривали с получением 13,4 г (93%) промежуточного соединения 3. В. Получение конечных соединений. Пример В 1. Смесь промежуточного соединения 1 (0,098 моль) и моногидрата гидразина (0,22 моль) в EtOH(350 мл) перемешивали и кипятили с обратным холодильником в течение одной ночи. Смесь выпаривали, добавляли воду и экстрагировали DCM. Органический слой сушили, фильтровали и выпаривали. Осадок очищали колоночной хроматографией с силикагелем (элюент: CHCl3/MeOH 98,5/1,5). Очищенные фракции собирали и выпаривали. Часть (2,5 г) осадка (10,5 г, 33,5%) кристаллизовали из 2 пропанола с получением 1,5 г (20,1%) соединения 1, температура плавления 222 С. Пример В 2.a) Получение промежуточного соединения 23 Смесь соединения 1 (0,028 моль) в МеОН (150 мл) гидрогенизировали в присутствии Pd/C 10% (2 г) в качестве катализатора при 50 С. После поглощения Н 2 (1 экв.) катализатор фильтровали через hyflo и фильтрат выпаривали с получением соединения 23.(150 мл) перемешивали и кипятили с обратным холодильником в течение одной ночи. Смесь охлаждали при перемешивании и кристаллизовали. Осадок отфильтровывали и сушили с получением 2,4 г (60,3%) соединения 2, температура плавления 226,9 С. Пример В 3. Смесь 4-фтор(4-фторфенил)бензолбутаналя (0,045 моль), соединения 15 (0,045 моль) и ацетата калия (6 г) в МеОН (250 мл) гидрогенизировали в течение одной ночи при 50 С в присутствии 10% Pd/C(3 г) в качестве катализатора в присутствии 4% раствора тиофена (1 мл). После поглощения H2 (1 экв.) катализатор отфильтровывали и фильтрат выпаривали. Осадок растворяли в DCM. Органический раствор промывали водным раствором аммиака, сушили (MgSO4), фильтровали и растворитель выпаривали. Осадок очищали колоночной хроматографией с силикагелем (элюент: DCM/MeOH 96/4). Необходимые фракции собирали и растворитель выпаривали. Часть (3 г) осадка (18 г, 78%) растворяли в 2-пропаноле и превращали в соль (Е)-2-бутендиовой кислоты (1:1). Осадок отфильтровывали и сушили с получением 2,9 г (60%) соединения 3, температура плавления 164,4 С. Смесь соединения 1 (0,028 моль) в МеОН (150 мл) гидрогенизировали в присутствии 10% Pd/C (2 г) в качестве катализатора при 50 С. После поглощения Н 2 (1 экв.) катализатор отфильтровывали и фильтрат выпаривали. Часть (1,5 г) осадка (6,8 г, 100%) растворяли в 2-пропаноле и превращали в соль (Е)-2 бутендиовой кислоты (2:1) в 2-пропаноле с получением 0,7 г (37,2%) соединения 4, температура плавления 264,9 С. Пример В 5. Получение соединения 5 К смеси соединения 4 (0,00436 моль) и TEA (0,00872 моль) в DCM (10 мл) при комнатной температуре по каплям добавляли ангидрид уксусной кислоты (0,00523 моль). Смесь перемешивали при комнатной температуре в течение 2 ч и выливали в ледяную воду. Добавляли DCM. Смесь подкисляли 1 н. HCl и экстрагировали DCM. Органический слой отделяли, подщелачивали 10% карбонатом калия, сушили К смеси соединения 4 (0,00436 моль) в DCM (20 мл) при комнатной температуре по каплям добавляли изоцианаттриметилсилан (0,00523 моль). Смесь перемешивали при комнатной температуре в течение 2 ч. Растворитель выпаривали досуха. Осадок отбирали в теплый МеОН. Осадок отфильтровывали и сушили в вакууме. Осадок (0,8 г, 67%) отбирали в МеОН и DCM. Смесь перемешивали. Осадок отфильтровывали и сушили в вакууме с получением 0,55 г (46%) соединения 6, температура плавления 300 С. Пример В 7. Получение соединения 7 Смесь соединения 4 (0,013 моль), хлорацетонитрила (0,014 моль) и карбоната натрия (0,065 моль) в ДМФА (150 мл) перемешивали при 70 С в течение 3 ч, охлаждали, выливали в ледяную воду и экстрагировали EtOAc. Органический слой отделяли, сушили (MgSO4), фильтровали и растворитель выпаривали досуха. Осадок (6,1 г) очищали колоночной хроматографией с силикагелем (15-40 мкм) (элюент:DCM/MeOH/NH4OH 98/2/0,1). Очищенные фракции собирали и растворитель выпаривали. Осадок отбирали в МеОН. Осадок отфильтровывали и сушили с получением 0,33 г (10%) соединения 7, температура плавления 259 С. Пример В 8. Получение соединения 8 Смесь соединения 7 (0,0027 моль) в 7 н. МеОН/NH3 (30 мл) гидрогенизировали при давлении 3 бар в течение 24 ч в присутствии никеля Ренея (0,73 г) в качестве катализатора. После поглощения H2 (2 экв.) катализатор фильтровали через целит и фильтрат выпаривали. Осадок (0,48 г) очищали колоночной хроматографией с силикагелем (15-40 мкм) (элюент: DCM/MeOH/NH4OH от 85/14/1 до 83/15/2). Очищенные фракции собирали и растворитель выпаривали. Осадок кристаллизовали из диэтилового эфира. Осадок(MgSO4), фильтровали и растворитель выпаривали досуха. Осадок (16 г) очищали колоночной хроматографией с силикагелем (20-45 мкм) (элюент: DCM/MeOH 98/2). Очищенные фракции собирали и растворитель выпаривали с получением 7,5 г (61,1%) соединения 17. Смесь соединения 17 (0,024 моль) и ацетата натрия (0,036 моль) в уксусной кислоте (77 мл) перемешивали и кипятили с обратным холодильником в течение 2 ч. Растворитель выпаривали досуха. Осадок отбирали в воду. Смесь подщелачивали твердым карбонатом калия. Добавляли DCM. Твердое вещество отфильтровывали и сушили с получением 4,67 г (76%) соединения 18, температура плавления 253 С. Смесь соединения 18 (0,016 моль) в 7 н. MeOH/NH3 (150 мл) гидрогенизировали при комнатной температуре при давлении 3 бар в течение 18 ч в присутствии никеля Ренея (4,2 г) в качестве катализатора. После поглощения Н 2 (2 экв.) катализатор фильтровали через целит и фильтрат выпаривали досуха. Осадок (5,3 г) очищали колоночной хроматографией с силикагелем (15-40 мкм) (элюент:DCM/MeOH/NH4OH 88/12/1). Очищенные фракции собирали и растворитель выпаривали. Осадок кристаллизовали из DIPE. Осадок отфильтровывали и сушили с получением: 2,8 г (67,7%) соединения 9,температура плавления 182 С. Пример В 10. а) Получение соединения 19 Смесь 1,4-дихлорфталазина (0,00502 моль), промежуточного соединения 3 (0,00452 моль) и карбоната натрия (0,01004 моль) в ДМФА (15 мл) перемешивали при 130 С в течение 5 ч, доводили до комнатной температуры, выливали в ледяную воду и экстрагировали посредством DCM. Органический слой отделяли, промывали водой, сушили (MgSO4), фильтровали и растворитель выпаривали. Осадок (1,2 г) очищали колоночной хроматографией с силикагелем (15-40 мкм) (элюент: DCM/MeOH/NH4OH от 99/1/0,1 до 85/15/0,1). Очищенные фракции собирали и растворитель выпаривали. Осадок (0,7 г, 33%) кристаллизовали из ацетонитрила и диэтилового эфира. Осадок отфильтровывали и сушили в вакууме с получением 0,32 г (15%) соединения 19, температура плавления 161 С. Смесь соединения 19 (0,0087 моль) и ацетата натрия (0,01306 моль) в уксусной кислоте (40 мл) перемешивали и кипятили с обратным холодильником в течение 4 ч. Растворитель выпаривали досуха. До- 18014955 бавляли 10% хлористо-водородную кислоту (40 мл). Смесь перемешивали и кипятили с обратным холодильником в течение 1 ч и затем доводили до комнатной температуры. Добавляли DCM. Смесь подщелачивали разбавленным раствором NH4OH и экстрагировали DCM. Органический слой отделяли, промывали водой, сушили (MgSO4), фильтровали и растворитель выпаривали. Осадок очищали колоночной хроматографией с силикагелем (15-40 мкм) (элюент: DCM/MeOH 97,5/2,5). Необходимые фракции собирали и растворитель выпаривали. Осадок (1,4 г, 40%) кристаллизовали из диэтилового эфира. Осадок отфильтровывали и сушили в вакууме с получением 0,95 г (27%) соединения 16, температура плавления 140 С. Смесь соединения 16 (0,00492 моль) в 12 н. HCl (50 мл) перемешивали и кипятили с обратным холодильником в течение одной ночи и затем доводили до комнатной температуры. Растворитель выпаривали досуха. Осадок отбирали в EtOAc. Смесь подщелачивали 10% карбонатом калия и экстрагировали EtOAc и небольшим количеством EtOH. Органический слой отделяли, сушили (MgSO4), фильтровали и растворитель выпаривали. Осадок отбирали в ацетонитрил. Осадок отфильтровывали и сушили в вакууме с получением 1,29 г (79%) соединения 10, температура плавления 238 С. Пример В 11. Смесь этилового эфира 3-амино-8-азабицикло[3,2,1]октан-8-карбоновой кислоты (0,25 моль), 1,4 дихлорфталазина (0,25 моль) и карбоната натрия (0,25 моль) в ДМФА (600 мл) перемешивали в течение 4 ч при 130 С. Реакционную смесь охлаждали и выливали в воду. Осадок отфильтровывали, промывали водой, затем растворяли в DCM. Органический раствор сушили (MgSO4), фильтровали и растворитель выпаривали. Осадок кристаллизовали из смеси 2-пропанол/DIPE. Осадок отфильтровывали и сушили с получением 65 г (72%) соединения 24. Смесь соединения 24 (0,16 моль) и ацетата натрия (0,16 моль) в уксусной кислоте (800 мл) перемешивали и кипятили с обратным холодильником в течение 5 ч. Растворитель выпаривали. К осадку добавляли 10% хлористо-водородную кислоту (800 мл), реакционную смесь перемешивали и кипятили с обратным холодильником в течение 1 ч, затем охлаждали до комнатной температуры и полученный в результате осадок отфильтровывали, промывали водой, затем сушили. Часть (4 г) осадка (30 г) кристаллизовали из 2-пропанола. Осадок отфильтровывали и сушили с получением 2,5 г (34%) соединения 11,температура плавления 218,6 С. Пример В 12. Получение соединения 12 Смесь 1,4-дихлорфталазина (0,13 моль), 1-(фенилметил)-3-пирролидинамина (0,12 моль) и карбоната натрия (0,26 моль) в ДМФА (150 мл) перемешивали в атмосфере N2 при 130 С в течение 4 ч. Смесь охлаждали, выливали в лед и экстрагировали DCM. Органический слой промывали насыщенным раствором, сушили (MgSO4), отфильтровывали и выпаривали. Осадок очищали колоночной хроматографией с силикагелем (элюент: DCM/MeOH от 100/0 до 97/3). Очищенные фракции собирали и выпаривали. Часть осадка (38 г, 93,5%) растворяли в 2-пропаноле и превращали в гидрохлоридную соль (1:2) в 2-пропаноле с получением 2,43 г соединения 12, температура плавления 171,8 С.(1:1) в 2-пропаноле. Осадок отфильтровывали и промывали 2-пропанолом и DIPE. Осадок сушили при комнатной температуре с получением 1,2 г (14,4%) соединения 13, температура плавления 227,6 С. Пример В 14.(0,0557 моль),этилового эфира 4-амино-1 пиперидинкарбоновой кислоты, (0,0501 моль) и карбоната натрия (0,0836 моль) в ДМФА (150 мл) перемешивали при 130 С в течение одной ночи и затем доводили до комнатной температуры. Растворитель выпаривали досуха. Осадок отбирали в DCM. Смесь выливали в ледяную воду и экстрагировали посредством DCM. Органический слой отделяли, промывали водой, сушили (MgSO4), фильтровали и растворитель выпаривали. Осадок очищали колоночной хроматографией с силикагелем (20-45 мкм) (элюент:DCM/MeOH/NH4OH 98/2/0,4). Очищенные фракции собирали и растворитель выпаривали. Осадок (6,5 г) кристаллизовали из диэтилового эфира. Осадок отфильтровывали и сушили в вакууме с получением 6,2 г Смесь соединения 20 (0,0155 моль) и ацетата натрия (0,0233 моль) в уксусной кислоте (50 мл) перемешивали и кипятили с обратным холодильником в течение 3 ч. Растворитель выпаривали досуха. Добавляли 10% хлористо-водородную кислоту (50 мл). Смесь перемешивали и кипятили с обратным холодильником в течение 1 ч, доводили до комнатной температуры, выливали в ледяную воду, подщелачивали концентрированным раствором NH4OH и перемешивали. Осадок отфильтровывали, промывали водой,промывали 2-пропаноном и диэтиловым эфиром и сушили в вакууме с получением 5,3 г (95%) соединения 21, температура плавления 286 С. с) Получение соединения 22 Смесь соединения 21 (0,11 моль) в 10 н. хлористо-водородной кислоте (100 мл) перемешивали и кипятили с обратным холодильником в течение одной ночи и доводили до комнатной температуры. Растворитель выпаривали досуха. Осадок отбирали в МеОН и EtOH. Смесь перемешивали. Осадок отфильтровывали и сушили в вакууме с получением 3,5 г (98%) соединения 22, температура плавления 300 С.N2 барботировали через смесь соединения 22 (0,00614 моль) в МеОН (30 мл) и ТГФ (30 мл). Пор- 20014955 циями добавляли никель Ренея (2 г). Через смесь барботировали N2. Смесь гидрогенизировали при комнатной температуре при давлении 3 бар в течение 2 ч. После поглощения H2 (3 экв.) катализатор фильтровали через целит, промывали DCM и МеОН и фильтрат выпаривали досуха. Осадок отбирали в МеОН и EtOH. Осадок отфильтровывали и сушили в вакууме. Осадок отбирали в теплый МеОН. Осадок отфильтровывали и сушили в вакууме с получением 0,47 г (24%) соединения 14, температура плавления 300 С. В табл. 1 представлен перечень соединений, которые получали в соответствии с одним из приведенных выше примеров. Таблица 1 Фармакологический пример. Сцинтилляционный анализ сближения (SPA) in vitro на ингибиторную активность PARP-1. Соединения по настоящему изобретению тестировали в анализе in vitro, основанном на методе SPA(запатентованной Amersham Pharmacia Biotech). В принципе, анализ основан на общепринятом методе SPA для определения полиАДФрибозилирования биотинилированных белков-мишеней, т.е. гистонов. Это рибозилирование индуцируют, используя фермент PARP-1, активированный ДНК с одноцепочечными разрывами, и [3 Н]никотинамидадениндинуклеотид ([3 Н]-NAD+) в качестве донора АДФ-рибозилов. В качестве индуктора активности фермента PARP-1 получали ДНК с одноцепочечными разрывами. Для этого 25 мг ДНК (поставщик: Sigma) растворяли в 25 мл буфера для ДНКазы (10 мМ Tris-HCl,pH 7,4; 0,5 мг/мл бычьего сывороточного альбумина (BSA); 5 мМ MgCl26 Н 2 О и 1 мМ KCl) к которому добавляли 50 мкл раствора ДНКазы (1 мг/мл в 0,15 М NaCl). После инкубации в течение 90 мин при- 25014955 37 С, реакцию останавливали добавлением 1,45 г NaCl с последующей дополнительной инкубацией при 58 С в течение 15 мин. Реакционную смесь охлаждали во льду и проводили диализ при 4 С в течение соответственно 1,5 и 2 ч против 1,5 л 0,2 М KCl и дважды против 1,5 л 0,01 М KCl в течение 1,5 и 2 ч соответственно. Смесь разделяли на аликвоты и хранили при -20 С. Гистоны (1 мг/мл, тип II-А, поставщик: Sigma) биотинилировали с использованием набора для биотинилирования Amersham и хранили,разделенными на аликвоты, при -20 С. Получали исходный раствор 100 мг/мл сфер SPA из поливинилтолуола (PVT) (поставщик: Amersham) в PBS. Исходный раствор [3H]-NAD+ получали при добавлении 120 мкл [3H]-NAD+ (0,1 мКи/мл, поставщик: NEN) к 6 мл буфера для инкубации (50 мМ Tris/HCl, pH 8; 0,2 мМ DTT; 4 мМ MgCl2). Получали раствор 4 мМ NAD+ (поставщик: Roche) в буфере для инкубации(из 100 мМ исходного раствора в воде, хранящегося при -20 С). Фермент PARP-1 получали с использованием известных в данной области методик, т.е. клонированием и экспрессией белка, с начальным этапом с использованием кДНК печени человека. Информацию, касающуюся используемой белковой последовательности фермента PARP-1, включая ссылки на литературные источники, можно найти в базе данных Swiss-Prot с первичным номером доступа Р 09874. Биотинилированные гистоны и сферы PVTSPA смешивали и предварительно инкубировали в течение 30 мин при комнатной температуре. ФерментPARP-1 (концентрация зависела от партии) смешивали с ДНК с одноцепочечными разрывами и смесь предварительно инкубировали в течение 30 мин при 4 С. Равные части раствора гистоны/сферы PVTSPA и раствора фермент PARP-1/ДНК смешивали и в 96-луночные микротитрационные планшеты добавляли 75 мкл полученной смеси вместе с 1 мкл соединения в ДМСО и 25 мкл [3 Н]-NAD+ на лунку. Конечные концентрации в инкубационной смеси составляли 2 мкг/мл для биотинилированных гистонов,2 мг/мл для сфер PVT-SPA, 2 мкг/мл для ДНК с одноцепочечными разрывами и между 5 и 10 мкг/мл для фермента PARP-1. После инкубации смеси в течение 15 мин при комнатной температуре реакцию останавливали добавлением 100 мкл 4 мМ NAD+ в буфере для инкубации (конечная концентрация 2 мМ) и содержимое планшетов перемешивали. Сферам давали возможность осаждаться в течение по меньшей мере 15 мин и планшеты переносили в TopCountNXT (Packard) для сцинтилляционного измерения, значения выражались как число импульсов в минуту (cpm). Для каждого эксперимента параллельно исследовали контроли (содержащие фермент PARP-1 и ДМСО без соединения), простую инкубацию (включающую ДМСО, но без ферментаPARP-1 или соединения) и образцы (включающие фермент PARP-1 и соединение, растворенное в ДМСО). Все тестируемые соединения растворяли и с течением времени дополнительно растворяли в ДМСО. В первом примере соединения тестировали при концентрации 10-5 М. Если соединения показывали активность при 10-5 М, строили кривую доза-ответ, где соединения тестировали при концентрациях между 10-5 и 10-8 М. В каждом тесте из значений для контроля и образца вычитали значения для простого раствора. Контрольный образец показывал максимальную активность фермента PARP-1. Для каждого образца количество cpm выражали в виде процентной доли от среднего значения cpm для контролей. Когда это целесообразно, вычисляли значения IC50 (концентрация лекарственного средства, необходимая для снижения активности фермента PARP-1 до 50% от активности в контроле) с использованием линейной интерполяции между экспериментальными точками непосредственно выше и ниже уровня 50%. Эффекты тестируемых соединений представлены в виде pIC50 (значение отрицательного логарифма значения IC50). Для проверки анализа SPA в качестве контрольного соединения использовали 4-амино-1,8 нафталимид. Тестируемые соединения показывали ингибиторную активность при начальной исследуемой концентрации 10-5 М (см. табл. 2). Анализ фильтрования in vitro на ингибиторную активность PARP-1. Соединения по настоящему изобретению тестировали в анализе фильтрования in vitro, оценивая активность PARP-1 (запускаемую в присутствии ДНК с одноцепочечными разрывами) посредством его активности в отношении полиАДФ-рибозилирования гистонов с использованием [32P]-NAD в качестве донора АДФ-рибозилов. Радиоактивные рибозилированные гистоны осаждали трихлоруксусной кислотой (ТСА) в 96-планшетах для фильтрации и проводили измерение встроенного [32 Р] с использованием сцинтилляционного счетчика. Получали смесь гистонов (исходный раствор: 5 мг/мл в Н 2 О), NAD+ (исходный раствор: 100 мМ в Н 2 О), и [32 Р]-NAD+ в буфере для инкубации (50 мМ Tris/HCl, рН 8; 0,2 мМ DTT; 4 мМ MgCl2). Также получали смесь фермента PARP-1 (5-10 мкг/мл) и ДНК с одноцепочечными разрывами. ДНК с одноцепочечными разрывами получали, как описано для SPA in vitro для ингибирующей активности PARP-1. 75 мкл смеси фермент PARP-1/ДНК совместно с 1 мкл соединения в ДМСО и 25 мкл смеси гистоныNAD+/[32P]-NAD+ добавляли на лунку 96-луночного планшета для фильтрования (0,45 мкм, поставщикMillipore). Конечные концентрации в инкубационной смеси составляли 2 мкг/мл для гистонов, 0,1 мМ для NAD+, 200 мкМ (0,5 мкКи) для [32P]-NAD+ и 2 мкг/мл для ДНК с одноцепочечными разрывами. Планшеты инкубировали в течение 15 мин при комнатной температуре, реакцию останавливали добавлением 10 мкл ледяного 100% ТСА и затем добавляли 10 мкл ледяного раствора BSA (1% в Н 2 О). Белковой фракции давали возможность выпасть в осадок в течение 10 мин при 4 С и планшеты фильтровали в вакууме. Планшеты последовательно промывали для каждой лунки 1 мл 10% ледяного ТСА, 1 мл 5% ледяного ТСА и 1 мл 5% ТСА при комнатной температуре. Наконец, в каждую лунку добавляли 100 мкл- 26014955 раствора для сцинтилляции (Microscint 40, Packard), планшеты переносили в TopCountNXT (поставщик: Packard) для сцинтилляционных измерений и значения выражали в количестве импульсов в минуту(cpm). Для каждого эксперимента параллельно исследовали контроли (содержащие фермент PARP-1 и ДМСО без соединения), простую инкубацию (включающую ДМСО, но без фермента PARP-1 или соединения) и образцы (включающие фермент PARP-1 и соединение, растворенное в ДМСО). Все тестируемые соединения растворяли и с течением времени дополнительно разбавляли в ДМСО. В первом примере, соединения тестировали при концентрации 10-5 М. Если соединения показывали активность при 10-5 М, строили кривую доза-ответ, где соединения тестировали при концентрациях между 10-5 и 10-8 М. В каждом тесте из значений для контроля и образца вычитали значения для простого раствора. Контрольный образец показывал максимальную активность фермента PARP-1. Для каждого образца количество cpm выражали в виде процентной доли от среднего значения cpm для контролей. Когда это целесообразно, вычисляли значения IC50 (концентрация лекарственного средства, необходимая для снижения активности фермента PARP-1 до 50% от активности в контроле) с использованием линейной интерполяции между экспериментальными точками непосредственно выше и ниже уровня 50%. Эффекты тестируемых соединений представлены в виде pIC50 (значение отрицательного логарифма значения IC50). Для проверки анализа фильтрования в качестве контрольного соединения использовали 4-амино-1,8 нафталимид. Тестируемые соединения показывали ингибиторную активность при начальной исследуемой концентрации 10-5 М (см. табл. 2). Сцинтилляционный анализ сближения (SPA) in vitro на ингибиторную активность TANK-2. Соединения по настоящему изобретению тестировали в анализе in vitro, основанном на методе SPA с использованием планшетов Ni Flash (96- или 384-луночных). В принципе, анализ основан на методе SPA для определения полиАДФ-рибозилирования белкаTANK-2 с использованием и [3 Н]-никотинамидадениндинуклеотида ([3H]-NAD+) в качестве донора АДФрибозилов. Исходный раствор [3 Н]-NAD+/NAD получали добавлением 64,6 мкл [3H]-NAD+ (0,1 мКи/мл, поставщик: Perkin Elmer) и 46,7 мкл исходного раствора NAD (10,7 мМ, хранящегося при -20 С, поставщикRoche) к 1888,7 мкл буфера для анализа (60 Мм Tris/HCl, pH 7,4; 0,9 мМ DTT; 6 мМ MgCl2). ФерментTANK-2 получали, как описано в ЕР 1238063. 60 мкл буфера для анализа совместно с 1 мкл соединения в ДМСО, 20 мкл [3 Н] -NAD+/NAD и 20 мкл фермента TANK-2 (конечная концентрация 6 мкг/мл) добавляли на лунку в 96-луночный планшет с покрытием Ni (Perkin Elmer). После инкубации смеси в течение 120 мин при комнатной температуре реакцию останавливали добавлением 60 мкл стоп-реагента (42,6 мгNAD в 6 мл Н 2 О). Планшеты накрывали приспособлением для накрывания планшетов и помещали вTopCountNXT (Packard) для сцинтилляционного измерения. Значения выражали в количестве импульсов в минуту (cpm). Для каждого эксперимента параллельно исследовали контроли (содержащие фермент TANK-2 и ДМСО без соединения), простую инкубацию (включающую ДМСО, но без ферментаTANK-2 или соединения) и образцы (включающие фермент TANK-2 и соединение, растворенное в ДМСО). Все тестируемые соединения растворяли и с течением времени дополнительно разбавляли в ДМСО. В первом примере соединения тестировали при концентрации 10-5 М. Если соединения показывали активность при 10-5 М, строили кривую доза-ответ, где соединения тестировали при концентрациях между 10-5 и 10-8 М. В каждом тесте из значений для контроля и образца вычитали значения для пустого раствора. Контрольный образец показывал максимальную активность фермента TANK-2. Для каждого образца количество cpm выражали в виде процентной доли от среднего значения cpm для контроля. Когда это целесообразно, вычисляли значения IC50 (концентрация лекарственного средства, необходимая для снижения активности фермента TANK-2 до 50% от активности в контроле) с использованием линейной интерполяции между экспериментальными точками непосредственно выше и ниже уровня 50%. Эффекты тестируемых соединений представлены в виде pIC50 (значение отрицательного логарифма значения IC50). Для проверки анализа SPA в качестве контрольных соединений использовали 3-аминобензамид и 4-амино-1,8-нафталимид. В данном случае описан анализ с использованием 96-луночных планшетов. В анализе с использованием 384-луночных планшетов использовали такие же конечные концентрации и значения были адаптированы. Если результаты исследований с 96-луночными планшетами были доступны, эти результаты включали в табл. 2, в ином случае показаны результаты анализа с 384-луночными планшетами. Соединения, кроме того, можно оценивать в анализе химиосенсибилизации или радиосенсибилизации клеток, в анализе, при котором измеряют ингибирование эндогенной активности PARP-1 в клеточных линиях злокачественной опухоли и, наконец, в тесте радиосенсибилизации in vivo. ФОРМУЛА ИЗОБРЕТЕНИЯ 1. Соединение формулы (I) его формы N-оксидов, фармацевтически приемлемые аддитивные соли и стереохимически изомерные формы,где пунктирная линия обозначает необязательную связь;n равен 0, 1, 2 или 3; если n равен 0, тогда подразумевается прямая связь;Q представляет собой -С(=O)- или -CR3-, где R3 представляет собой галоген или C1-6-алкил; если Q представляет собой -CR3-, пунктирная линия обозначает связь; каждый X независимо представляет собой -N или -СН; если X представляет собой -СН, тогда Y представляет собой -N или -NH-, и если X представляет собой -N, тогда Y представляет собой -СН или -СН 2-; каждый Y независимо представляет собой -N, -NH-, -CH или -СН 2-;L1 представляет собой прямую связь или двухвалентный радикал, выбранный из -С 1-6-алкандиилNH-, -NH- или -NH-С 1-6-алкандиил-NH-;L2 представляет собой прямую связь или двухвалентный радикал, выбранный из -C1-6-алкандиила-, С 2-6-алкендиила-, карбонила или -C1-6-алкандиила-, замещенного одним заместителем, выбранным из группы, состоящей из гидрокси или арила; также может быть связан мостиковой связью (т.е. образовывать
МПК / Метки
МПК: A61K 31/502, C07D 403/14, C07D 403/12, A61P 31/00, C07D 401/04
Метки: производные, качестве, фталазина, ингибиторов
Код ссылки
<a href="https://eas.patents.su/30-14955-proizvodnye-ftalazina-v-kachestve-ingibitorov-parp.html" rel="bookmark" title="База патентов Евразийского Союза">Производные фталазина в качестве ингибиторов parp</a>
Производные фталазина в качестве ингибиторов фосфодиэстеразы 4

Номер патента: 3702
Опубликовано: 28.08.2003
Авторы: Норчини Габриеле, Пеллачини Франко, Мораццони Габриеле, Наполетано Мауро, Гранчини Джанкарло
МПК: C07D 237/30, A61K 31/502
Метки: фосфодиэстеразы, качестве, фталазина, ингибиторов, производные
Формула / Реферат:
1. Соединение формулы I где ---- представляет одинарную или двойную связь; B представляет метилен; A представляет пиридин, замещенный одним заместителем или большим количеством заместителей; R представляет два атома водорода или C = O группу, когда ---- представляет одинарную связь, или, когда ---- представляет двойную связь, R представляет водород, необязательно замещенный арил или гетероцикл, (C1-8)алкил, (C2-8)алкенил или (C2-8)алкинил,...
Производные хиназолиндиона в качестве ингибиторов parp

Номер патента: 11552
Опубликовано: 28.04.2009
Авторы: Ваутерс Вальтер Баудевейн Леопольд, Ван Дюн Якобус Альфонсус Йозефус, Сомерс Мария Викторина Франциска, Мертенс Йозефус Каролус, Кенни Людо Эдмон Жозефин
МПК: A61P 35/00, C07D 401/04, A61K 31/517...
Метки: качестве, хиназолиндиона, ингибиторов, производные
Формула / Реферат:
1. Соединение формулы (I) его N-оксидные формы, его фармацевтически приемлемые аддитивные соли и его стереохимически изомерные формы, где каждый X независимо обозначает или и когда X означает то Y означает каждый Y независимо обозначает или за исключением случая, когда X обозначает и тогда Y обозначает L1 означает прямую связь или двухвалентный радикал, выбранный из -C1-6алкандиила-, L2 означает прямую связь или двухвалентный радикал,...
Производные хиназолинона в качестве ингибиторов parp

Номер патента: 12837
Опубликовано: 30.12.2009
Авторы: Мертенс Йозефус Каролус, Ван Дюн Якобус Альфонсус Йозефус, Кенни Людо Эдмон Жозефин, Ваутерс Вальтер Баудевейн Леопольд, Сомерс Мария Викторина Франциска, Гийемон Жером Эмиль Жорж
МПК: A61K 31/517, A61P 25/00, C07D 239/91...
Метки: качестве, ингибиторов, хиназолинона, производные
Формула / Реферат:
1. Соединение формулы (I) его N-оксиды, фармацевтически приемлемые аддитивные соли и стереохимически изомерные формы, где пунктирные линии означают необязательные связи; X является >N- или >СН-; является -N-C(O)- или -N=CR4-, где R4 является гидрокси; L является прямой связью или двухвалентным радикалом, выбранным из -С(О)-, -C(O)-NH-, -NH-, -С(О)-O-С1-6алкандиила- или -C1-6алкандиила-; R1 является водородом, галогеном,...
Сульфонамидные производные в качестве ингибиторов рассасывания костной ткани и ингибиторов адгезии клеток,способ их получения, применение и фармацевтическая композиция

Номер патента: 3102
Опубликовано: 26.12.2002
Авторы: Гурвест Жан-Франсуа, Карниато Дени, Бодари Сара Кэтрин, Шойнеманн Карлхайнц, Вилл Дэвид Вильям, Макдауэлл Роберт, Кнолле Йохен, Катбертсон Роберт Эндрю, Гадек Томас, Пейман Ануширван
МПК: C07D 239/42, A61K 31/505, A61P 19/10...
Метки: сульфонамидные, рассасывания, клеток,способ, ингибиторов, фармацевтическая, получения, композиция, применение, ткани, качестве, костной, производные, адгезии
Формула / Реферат:
1. Сульфонамидные производные общей формулы I где R1 и R2 вместе образуют двухвалентный (С2-С3)алкиленовый радикал; R4 является Н или (С1-С6)алкилом; R5 является (С1-С10)алкилом, (С6-С14)арилом, (C5-C14)гетероарилом или (С6-С14)арил(С1-С10)алкильной группой, где арил, гетероарил или алкил возможно замещены R3, или 2-оксобицикло[2.2.1]гепт-1-илметильной группой; R3 является (C1-C4)алкилом, (C1-C4)алкилокси, галогеном, трифторметилом, циано,...
Гетероариламины-производные пиримидина и пиридазина в качестве ингибиторов гликогенсинтаза-киназы 3-бета (ингибиторов gskз)

Номер патента: 10859
Опубликовано: 30.12.2008
Авторы: Ван Акен Кун Жанн Альфонс, Жанссен Поль Адриан Ян, Винкерс Хендрик Мартен, Дильс Гастон Станислас Марселла, Фрейн Эдди Жан Эдгар, Виллемс Марк, Бейнстерс Петер Якобус Йоханнес Антониус, Хэрес Ян, Леви Паулус Йоаннес, Лав Кристофер Джон, Де Жонж Марк Рене, Койманс Люсьен Мария Хенрикус, Эмбрехтс Вернер Констант Йохан
МПК: A61K 31/50, A61K 31/505, A61K 31/435...
Метки: gskз, пиридазина, 3-бета, гетероариламины-производные, пиримидина, ингибиторов, качестве, гликогенсинтаза-киназы
Формула / Реферат:
1. Соединение формулы его N-оксид, фармацевтически приемлемая аддитивная соль, четвертичный амин и стереохимически изомерная форма, где кольцо А является R1 обозначает водород; X обозначает -O-; -O-C1-6алкил- или прямую связь; Z обозначает прямую связь, -С(=O)- или -NR1-C1-6алкил-; R обозначает водород или R20; R3 обозначает водород; галоген; циано; полигалогенС1-6алкил; R4 обозначает моноциклический, насыщенный или частично насыщенный или...