Производные 2-арилиндола в качестве ингибиторов npges-1
Номер патента: 15689
Опубликовано: 31.10.2011
Авторы: Фурлотти Гвидо, Поленцани Лоренцо, Колетта Изабелла, Омбрато Розелла, Гарофало Барбара, Каццолла Никола, Гульельмотти Анджело, Мангано Джорджина, Гарроне Беатриче, Мауджери Катерина, Ализи Мария Алессандра













Формула / Реферат
1. Соединение 2-арилиндола, замещенное в положении 5, формулы (I)

где X представляет собой атом галогена или (C1-С3)алкил, трифторметил, нитро, циано, (C1-С3)алкокси или фенильную группу;
Y и Z, которые могут быть одинаковыми или разными, представляют собой Н, или атом галогена, или трифторметил, нитро, (C1-C3)алкокси, СООН, (C1-C3)алкил-СООН или (С2-С3)алкенил-СООН;
W представляет собой атом кислорода или СН2 или NH группу;
R представляет собой атом водорода или (C1-С6)алкильную или (С3-С7)циклоалкильную группу, необязательно замещенную 1-3 гидроксигруппами;
R' представляет собой атом водорода или (C1-С6)алкильную или (С3-С7)циклоалкильную группу;
А представляет собой фенильную, нафтильную или пиридиновую группу, необязательно замещенную 1-3 заместителями, которые могут быть одинаковыми или разными, выбранными из галогена, (C1-С6)алкила, трифторметила, амино, (C1-С3)алкокси, бензилокси, COOR, NHSO2CH3, (C1-C6)алкил-СООН и (С2-С6)алкенил-СООН,
и его физиологически приемлемые аддитивные соли, стереоизомеры, энантиомеры, гидраты, сольваты и полиморфные формы.
2. Соединение по п.1, отличающееся тем, что X представляет собой галоген, (С1-С3)алкил, трифторметил, нитро или (C1-С3)алкокси.
3. Соединение по п.2, отличающееся тем, что X представляет собой Cl, Br, F, трифторметил или нитро.
4. Соединение по п.1, отличающееся тем, что Y и Z независимо друг от друга представляют собой Н, галоген, нитро, СООН, трифторметил или (C1-С3)алкокси.
5. Соединение по п.4, отличающееся тем, что Y и Z независимо друг от друга представляют собой Cl, Br, F, трифторметил, нитро, СООН, метокси или этокси.
6. Соединение по п.1, отличающееся тем, что R представляет собой метил, этил, пропил, изопропил или циклогексил.
7. Соединение по п.1, отличающееся тем, что R' представляет собой Н, метил, этил, пропил, изопропил или циклогексил.
8. Соединение по п.1, отличающееся тем, что А представляет собой фенил, нафтил или пиридин, необязательно замещенные 1 или 2 заместителями, которые могут быть одинаковыми или разными, выбранными из галогена, (C1-С3)алкила, (С1-С3)алкокси и бензилокси.
9. Соединение по п.8, отличающееся тем, что А представляет собой фенил, необязательно замещенный 1 или 2 заместителями, которые могут быть одинаковыми или разными, выбранными из Br, Cl, F, метила, этила, метокси, этокси и бензилокси.
10. Соединение по п.8, отличающееся тем, что А представляет собой нафтил, необязательно замещенный 1 или 2 заместителями, которые могут быть одинаковыми или разными, выбранными из Br, Cl, F, метила, этила, метокси, этокси и бензилокси.
11. Соединение по п.8, отличающееся тем, что А представляет собой пиридин, необязательно замещенный 1 или 2 заместителями, которые могут быть одинаковыми или разными, выбранными из Br, Cl, F, метила, этила, метокси, этокси и бензилокси.
12. Способ получения соединения 2-арилиндола, замещенного в положении 5, формулы (I)

где X представляет собой атом галогена или (C1-С3)алкил, трифторметил, нитро, циано, (C1-С3)алкокси или фенильную группу;
Y и Z, которые могут быть одинаковыми или разными, представляют собой Н или атом галогена, или трифторметил, нитро, (C1-С3)алкокси, СООН, (C1-C3)алкил-СООН или (С2-С3)алкенил-СООН;
W представляет собой атом О или СН2 или NH группу;
R представляет собой атом водорода или (C1-С6)алкильную или (С3-С7)циклоалкильную группу, необязательно замещенную 1-3 гидроксигруппами;
R' представляет собой атом Н или (C1-С6)алкильную или (С3-С7)циклоалкильную группу;
А представляет собой фенильную, нафтильную или пиридиновую группу, необязательно замещенную 1-3 заместителями, которые могут быть одинаковыми или разными, выбранными из галогена, (C1-С6)алкила, трифторметила, амино, (C1-С3)алкокси, бензилокси, COOR, NHSO2CH3, (C1-C6)алкил-СООН и (С2-С6)алкенил-СООН,
отличающийся тем, что:
а) соединение формулы (II)

где X, Y и Z имеют вышеуказанные значения и
Q представляет собой атом галогена или гидроксигруппу,
подвергают взаимодействию с соединением формулы (III)

где R и R' имеют вышеуказанные значения и
G имеет такие же значения, как А, или представляет собой атом водорода,
с образованием соединения формулы (IV)

где X, Y, Z, W, G, R и R' имеют вышеуказанные значения, и
b) когда G представляет собой Н, соединение формулы (IV) подвергают взаимодействию с соединением формулы (V)
![]()
где I представляет собой атом йода и
А имеет вышеуказанные значения,
с образованием соединения формулы (I), и
с) при необходимости получают физиологически приемлемую аддитивную соль соединения формулы (IV) со стадии (а), где G отличен от Н, или соединения формулы (I) со стадии (b).
13. Способ по п.12, отличающийся тем, что стадию (а) осуществляют взаимодействием соединения формулы (II), где Q представляет собой Cl, с амином формулы (III) в присутствии подходящего акцептора кислоты в соответствии со стандартными методиками.
14. Способ по п.12, отличающийся тем, что стадию (а) осуществляют взаимодействием соединения формулы (II), где Q представляет собой ОН, с амином формулы (III) в присутствии подходящего агента сочетания в соответствии со стандартными методиками.
15. Промежуточное соединение формулы (III)

где R представляет собой (C1-С6)алкильную или (С3-С7)циклоалкильную группу, необязательно замещенную 1-3 гидроксигруппами;
R' представляет собой атом Н или (C1-С6)алкильную или (С3-С7)циклоалкильную группу,
G представляет собой фенильную, нафтильную или пиридиновую группу, необязательно замещенную 1-3 заместителями, которые могут быть одинаковыми или разными, выбранными из галогена, (C1-С6)алкила, трифторметила, амино, (C1-С3)алкокси, бензилокси, COOR, NHSO2CH3, (C1-C6)алкил-СООН и (С2-С6)алкенил-СООН,
при условии, однако, что G не является незамещенной фенильной группой, когда R представляет собой метил и R' представляет собой Н.
16. Промежуточное соединение по п.15, отличающееся тем, что R представляет собой метил, этил, пропил, изопропил или циклогексил.
17. Промежуточное соединение по п.15, отличающееся тем, что R' представляет собой Н, метил, этил, пропил, изопропил или циклогексил.
18. Фармацевтическая композиция, содержащая эффективное количество соединения формулы (I)

где X представляет собой атом галогена или (C1-С3)алкил, трифторметил, нитро, циано, (C1-С3)алкокси или фенил;
Y и Z, которые могут быть одинаковыми или разными, представляют собой Н или атом галогена, или трифторметил, нитро, (C1-С3)алкокси, СООН, (C1-C3)алкил-СООН или (С2-С3) алкенил-СООН;
W представляет собой атом О или СН2 или NH группу;
R представляет собой атом водорода или (С1-С6)алкильную или (С3-С7)циклоалкильную группу, необязательно замещенную 1-3 гидроксигруппами;
R' представляет собой атом Н или (C1-С6) алкильную или (С3-С7)циклоалкильную группу;
А представляет собой фенильную, нафтильную или пиридиновую группу, необязательно замещенную 1-3 заместителями, которые могут быть одинаковыми или разными, выбранными из галогена, (C1-С6)алкила, трифторметила, амино, (C1-С3)алкокси, бензилокси, COOR, NHSO2CH3, (C1-C6)алкил-СООН и (С2-С6)алкенил-СООН,
или его физиологически приемлемой аддитивной соли, стереоизомера, энантиомера, гидрата, сольвата или полиморфной формы, и по меньшей мере один фармацевтически приемлемый инертный ингредиент.
Текст
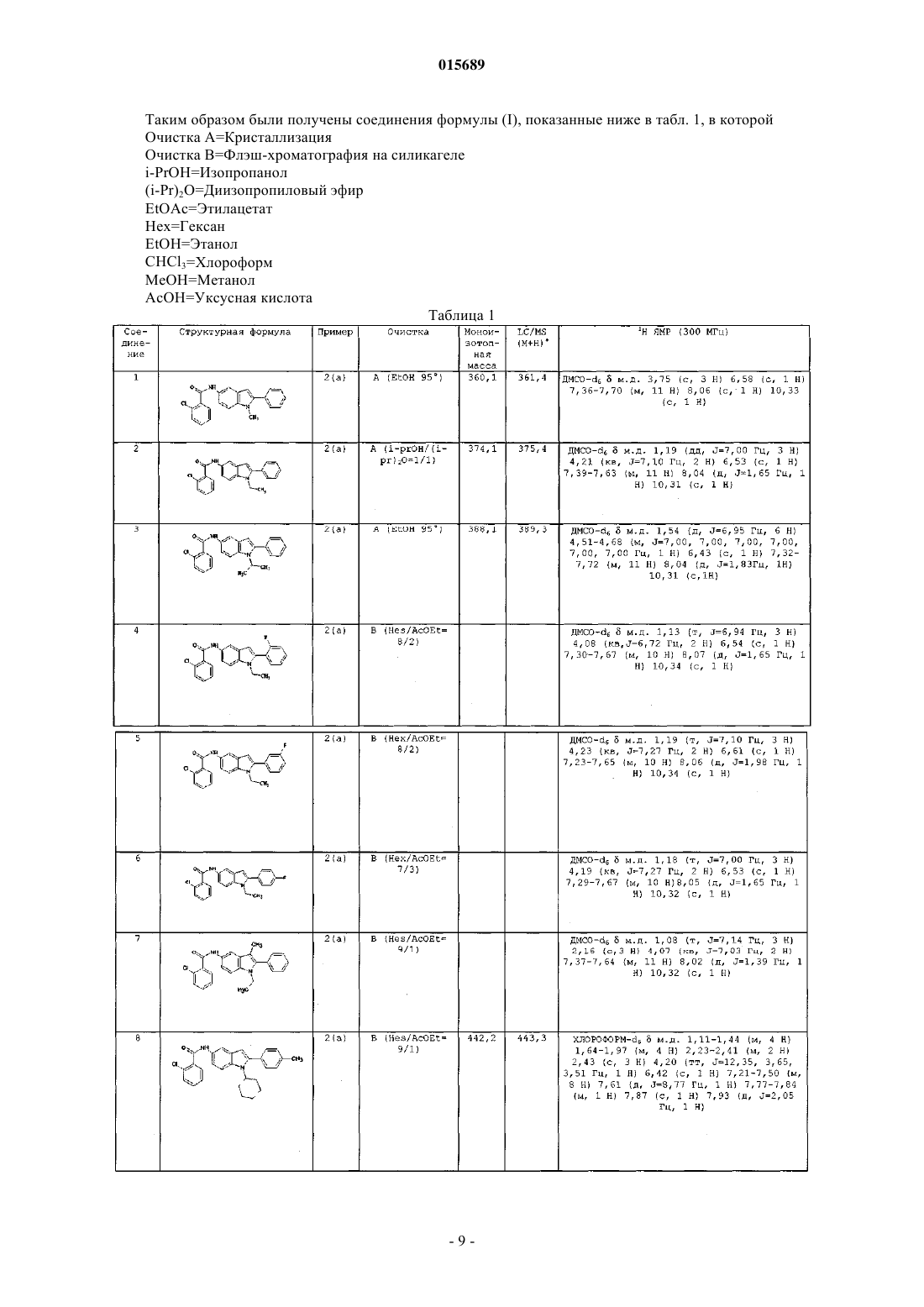
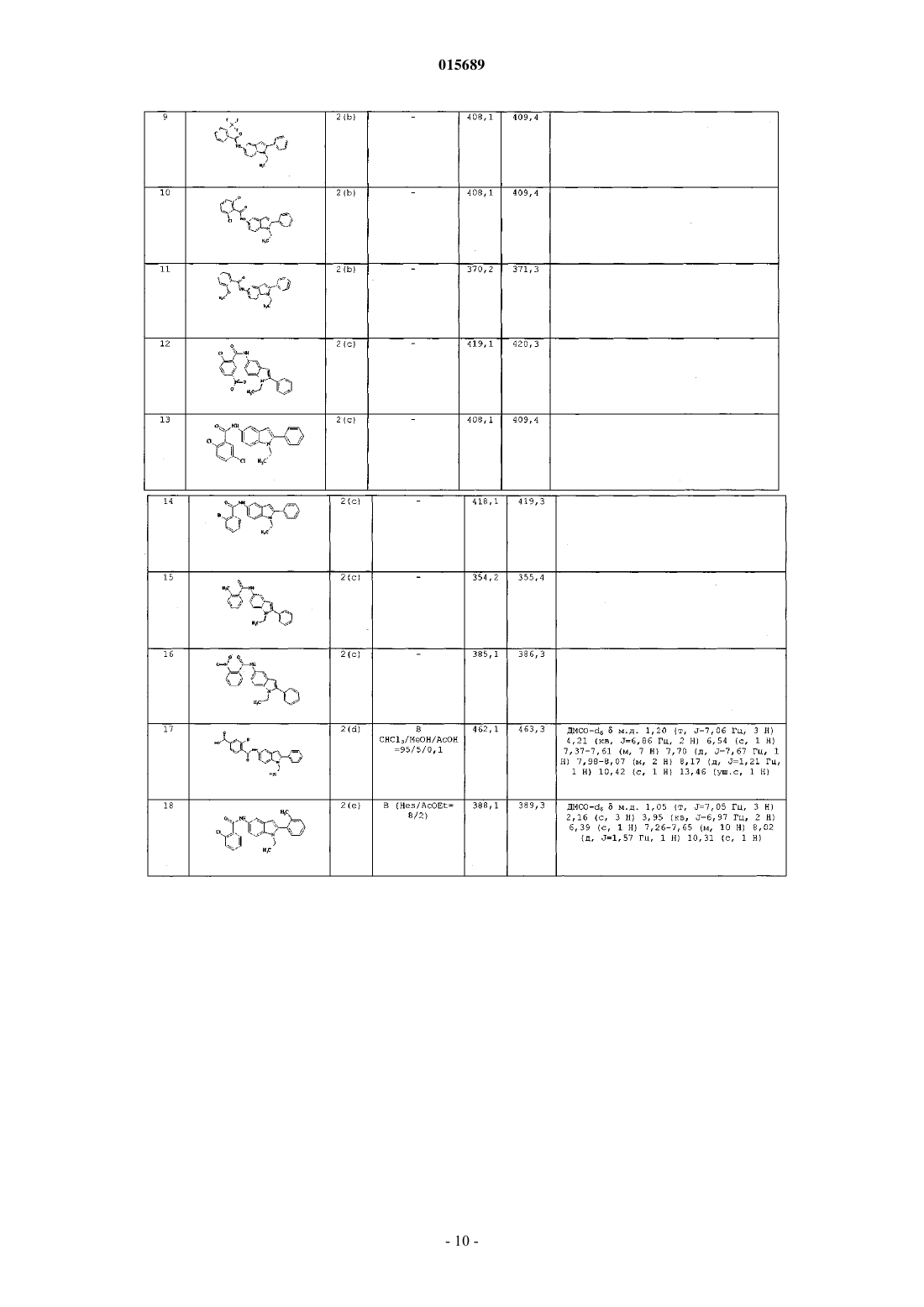
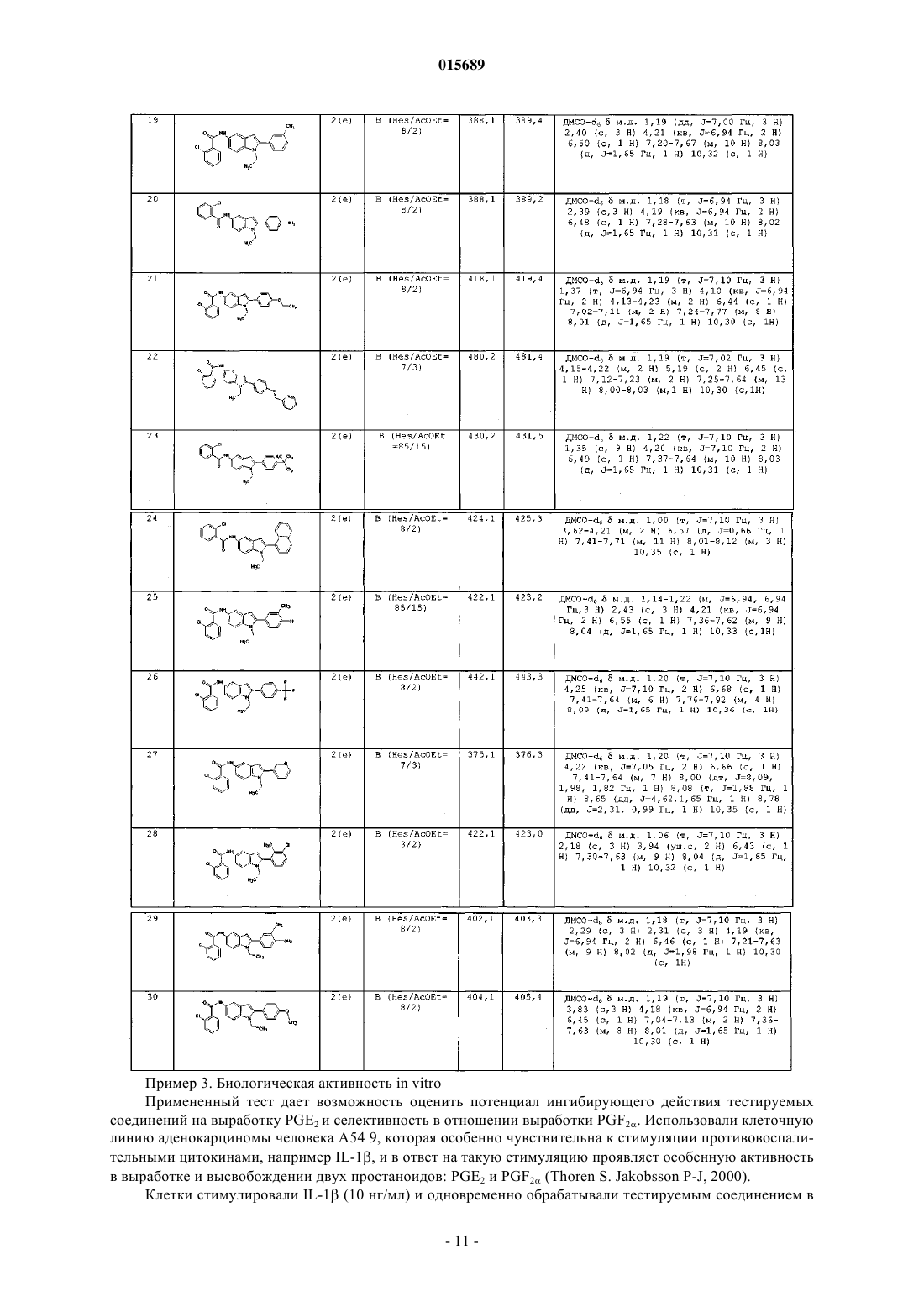
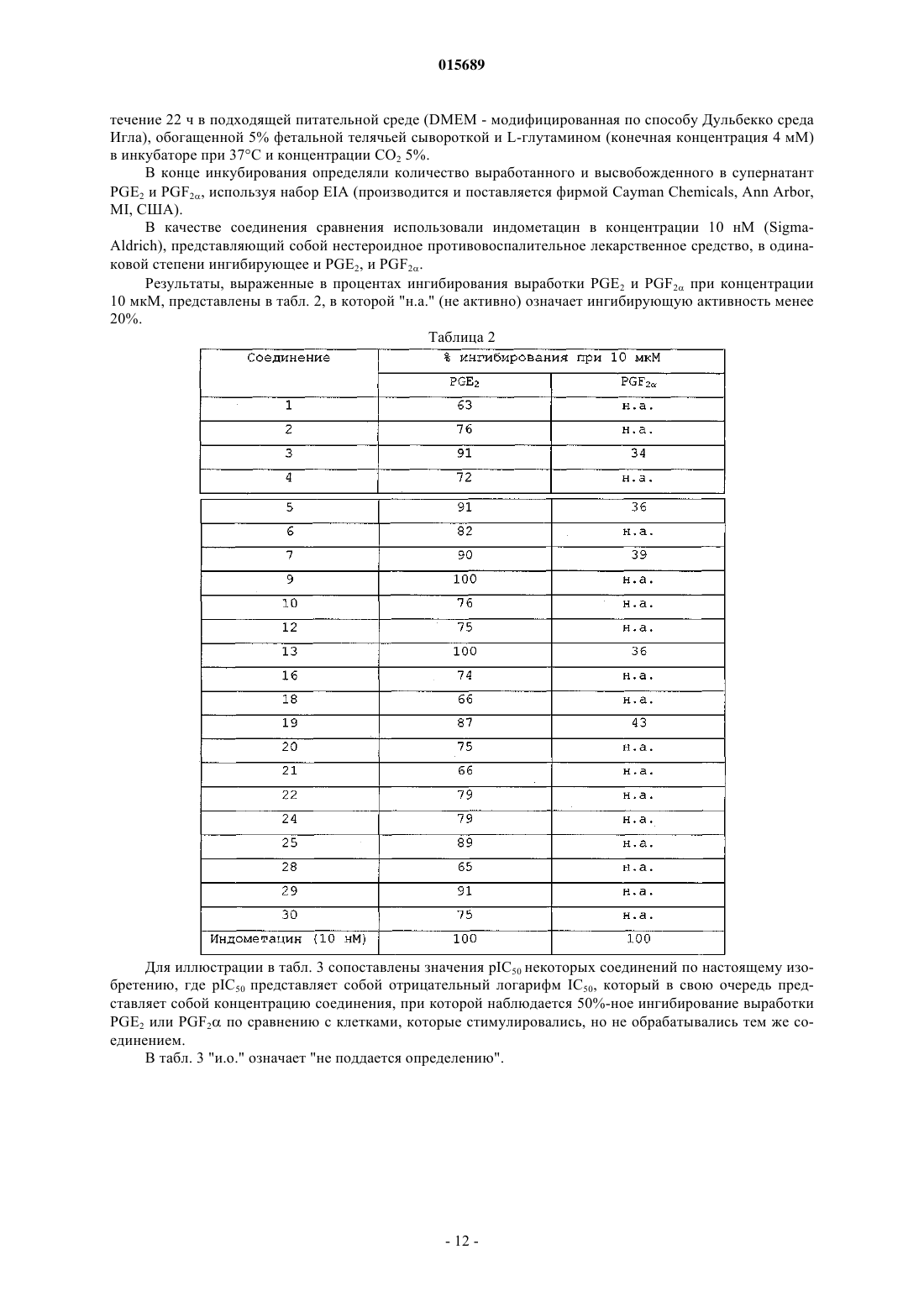
Дата публикации и выдачи патента Номер заявки в котором X, Y, Z, W, A, R и R' имеют значения, указанные в описании, содержащая его фармацевтическая композиция, а также способ их получения и промежуточные соединения.(71)(73) Заявитель и патентовладелец: АЦЬЕНДЕ КИМИКЕ РЬЮНИТЕ АНДЖЕЛИНИ ФРАНЧЕСКО 015689 Настоящее изобретение относится к соединению 2-арилиндола, замещенному в положении 5, к содержащей его фармацевтической композиции, к промежуточным соединениям и к способу его получения. Более конкретно, настоящее изобретение относится к соединению 2-арилиндола, замещенному в положении 5, обладающему ингибирующей активностью в отношении mPGEs-1. Известно, что простагландины (PG) представляют собой окисленные жирные кислоты, высвобождающиеся после синтеза во внеклеточное пространство и затем попадающие в плазму, мочу и другие биологические жидкости. Они являются важными биорегуляторами, но также представляют собой медиаторы воспаления,модулирующие внутриклеточные реакции и межклеточные связи. Простагландины Е 2 (PGE2) выполняют важную физиологическую роль регуляции почечной функции, гомеостаза сосудов, реконструкции костей, возникновения лихорадки, желудочно-кишечной функции и беременности. Помимо перечисленных физиологических функций, PGE2 простагландины выступают в качестве активных медиаторов острого воспаления (включая гипералгезию, расширение сосудов и выброс жидкостей из сосудов: Vane J. R. and Botting R. M. 1997 "Antieinflammatory drugs and theirmechanism of action" Inflamm. Res. 47 (2): p. 78) и хронического воспаления. В частности, особенно высок уровень PGE2 простагландинов при суставных воспалительных патологиях. PGE2 простагландины также играют роль в болевых процессах и являются мощным пиретическим средством (Ayoub S. S. et al., 2004fever: differential transcriptional regulation", Am. J. Physiol. Regul. Integr. Comp. Physiol. 283: R1104-R1117). Ферментом, ответственным за синтез PGE2 простагландинов, является простагландин Е синтаза(PGES), которая превращает эндопероксид PGH2, полученный из арахидоновой кислоты под действием циклооксигеназы, в PGE2. В разных типах клеток была обнаружена активность PGES как в цитозольной фракции, так и мембраносвязанная. Были выявлены три формы фермента (Kudo I. et al. 2005 "Prostaglandin E synthase, a terminal enzymefor prostaglandin E2 biosynthesis", Journal of Biochemistry and Molecular Biology 38, 633-638); среди них микросомный PGES-1 (mPGES-1) представляет собой мембраносвязанный фермент, для активности которого необходим глютатион в качестве необходимого кофактора. Экспрессия mPGES-1 вызывается противовоспалительными стимулами, такими как IL-1 или LPS(Proc. Natl. Acad. SC1. 96: 7220, 1999). Он расположен совместно с СОХ-2 на эндоплазматическом ретикулуме и на оболочке ядра (Lazarus M. et al. 2002 "Biochemical characterization of mouse microsomal prostaglandin E synthase-1 and its colocalization with cyclooxygenase-2 in peritoneal macrophages", Arch. Biochem. Biophys. 397: 336; Murakami M. et al. 2000 "Regulation of prostaglandin E2 biosynthesis by induciblemembrane-associated prostaglandin E2 synthase that acts in concert with cyclooxygenase-2", J. Biol. Chem. 275: 32783; Yamagata K. et al. 2001 "Coexpression of microsomaltype prostaglandin E synthase with cyclooxygenase-2 in brain endothelial cells of rats during endotoxininduced fever", J. NeurosCl. 15; 21 (8): 2669-77). Хотя два указанных фермента (СОХ-2 и mPGES-1) имеют функциональную зависимость и экспрессируются совместно, их скорости возбуждения различаются в нескольких клеточных системах, указывая на различные регуляторные механизмы возбуждения (J. Immunol. 167: 469, 2001). Лекарства, ингибирующие фермент СОХ-2, продемонстрировали эффективность в смягчении воспаления и боли при хронических воспалительных патологиях, таких как артрит, но их длительное использование может вызвать повреждение тканей, обусловленное чрезмерным синтезом цитокинов, например, TNF и IL-1 (Stichtenoth D.O. 2001 "Microsomal prostaglandin E synthase is regulated by proinflammatory cytokines and glucocorticoids in primary rheumatoid synovial cells" J. Immunol. 167: 469). Кроме того, длительное использование данных лекарств связано с побочными эффектами со стороны сердечнососудистой системы. Это привело к изъятию из продажи некоторых селективных ингибиторов СОХ-2 и к пересмотру показаний для всего класса данных лекарств. Недавние исследования направлены на преодоление побочных эффектов ингибиторов СОХ-2 с помощью изучения ингибиторов mPGES-1 в целях разработки лекарств, эффективных при лечении воспаления и боли. Кроме того, многочисленные исследования показали, что PGE2 простагландины являются факторами, провоцирующими развитие опухолей (Castellone M. D. et al. 2005 "Prostaglandin Е 2 promotes coloncancer growth through a novel Gs-Axin-B-catenin", Science 310, 1504-1510; Mehrotra S., et al. 2006 "Microsomal prostaglandin E2 in breast cancer: a potential target for therapy", J. Pathol. 208(3): 356-63; Nakano et al. 2006 "Induction of macrophagic prostaglandin E2 synthesis by glioma cells", J. Neurosurgery 104(4), 574-582),вовлеченными в процессы ангиогенеза, клеточной пролиферации и клеточной миграции. Обнаружено,что селективные ингибиторы FANS и СОХ-2 ингибируют также различные типы опухолей, включая колоректальные опухоли, опухоли пищевода, груди, легкого и мочевого пузыря, путем ингибированияPGE2. PGE2 простагландины, образующиеся из СОХ-2, вызывают рост опухоли за счет связывания с существующими рецепторами и активации сигналов контроля клеточной пролиферации, миграции, апоп-1 015689 тоза и ангиогенеза (Wang D. et al. 2006 "Prostaglandin and cancer" Gut. 55 (l):115-22; Han C. et al. 2006in human hepatocellular carcinoma cells", Journal of Cellular Physiology 207: 261-270). Авторы обнаружили соединение 2-арилиндола, замещенное в положении 5, обладающее селективной ингибирующей активностью в отношении mPGES-1. В первом аспекте настоящее изобретение относится к соединению 2-арилиндола, замещенному в положении 5, формулы (I)X представляет собой атом галогена или (C1-С 3)алкил, трифторметил, нитро, амино, циано, ди(C1 С 3)алкиламино, гидрокси, (C1-С 3)алкокси, фенильную или (C1-С 3)алкилфенильную группу;Y и Z, которые могут быть одинаковыми или разными, представляют собой Н или атом галогена или (C1-С 3)алкил, трифторметил, нитро, амино, ди(C1-С 3)алкиламино, гидрокси, (C1-С 3)алкокси, фенил,СООН, (C1-С 3)алкил-СООН, (С 2-С 3)алкенил-СООН, COOR, CONH2, SO2CH3, SO2NHCH3 или NHSO2CH3 группу;W представляет собой атом кислорода или СН 2 или NH группу;R представляет собой атом водорода или (C1-C6)алкильную или (С 3-С 7)циклоалкильную группу, необязательно замещенную 1-3 гидроксигруппами;R' представляет собой атом водорода или (C1-C6)алкильную или (С 3-С 7)циклоалкильную группу,необязательно замещенную 1-3 гидроксигруппами; А представляет собой фенильную, нафтильную или пиридиновую группу, необязательно замещенную 1-3 заместителями, которые могут быть одинаковыми или разными, выбранными из галогена, (C1C6)алкила, необязательно замещенного 1-3 гидроксигруппами, трифторметила, нитро, амино, ди(C1 С 3)алкиламино, гидрокси, (C1-С 3)алкокси, бензилокси, СООН, COOR, SO2CH3, SO2NHCH3, NHSO2CH3,POR1R2, OPOR1R2, (C1-C6)алкил-СООН, (С 2-С 6)алкенил-СООН, фенила и (C1-С 3)алкилфенила,где, в свою очередь, R1 и R2, которые могут быть одинаковыми или разными, представляют собой(C1-C3)алкил; и его физиологически приемлемым аддитивным солям, стереоизомерам, энантиомерам, гидратам,сольватам и полиморфным формам. Цепь различных алкильных групп, которые могут присутствовать в соединении формулы (I), может быть линейной или разветвленной. В случае некоторых заместителей, соединение формулы (I) по настоящему изобретению может содержать асимметрический атом углерода и, следовательно, может находиться в виде стереоизомеров и энантиомеров. Типичными примерами таких заместителей являются 2-бутанол, 2-метилбутил, 2 бутеновая кислота, 2-метилпропановая кислота и 1,2-пентандиол. Предпочтительно галоген представляет собой бром, хлор или фтор. Предпочтительными значениями X являются галоген, (C1-С 3)алкил, трифторметил, нитро, циано и(C1-С 3)алкокси. Особенно предпочтительными значениями X являются Cl, Br, F, трифторметил и нитро. Предпочтительными значениями Y и Z являются Н, галоген, нитро, СООН, (C1-С 3)алкил, трифторметил и (C1-C3)алкокси. Особенно предпочтительными значениями Y и Z являются Cl, Br, F, трифторметил, нитро, СООН, метил, этил, метокси и этокси. Предпочтительными значениями R являются метил, этил, пропил, изопропил и циклогексил. Предпочтительными значениями R' являются Н, метил, этил, пропил, изопропил и циклогексил. Предпочтительными значениями А являются фенил, нафтил и пиридин, необязательно замещенные 1 или 2 заместителями, которые могут быть одинаковыми или разными, выбранными из галогена, (C1 С 3)алкила, (C1-С 3)алкокси и бензилокси. Первым особенно предпочтительным значением А является фенил, необязательно замещенный 1 или 2 заместителями, которые могут быть одинаковыми или разными, выбранными из Br, Cl, F, метила,этила, метокси, этокси и бензилокси. Вторым особенно предпочтительным значением А является нафтил, необязательно замещенный 1 или 2 заместителями, которые могут быть одинаковыми или разными, выбранными из Br, Cl, F, метила,этила, метокси, этокси и бензилокси. Третьим особенно предпочтительным значением А является пиридин, необязательно замещенный 1 или 2 заместителями, которые могут быть одинаковыми или разными, выбранными из Br, Cl, F, метила,этила, метокси, этокси и бензилокси. В зависимости от природы заместителей, соединение формулы (I) может образовывать аддитивные соли с физиологически приемлемыми органическими или минеральными кислотами или основаниями.-2 015689 Типичными примерами физиологически приемлемых минеральных кислот являются хлористоводородная кислота, бромисто-водородная кислота, серная кислота, фосфорная кислота и азотная кислота. Типичными примерами подходящих физиологически приемлемых органических кислот являются уксусная кислота, аскорбиновая кислота, бензойная кислота, лимонная кислота, фумаровая кислота, молочная кислота, малеиновая кислота, метансульфоновая кислота, щавелевая кислота, паратолуолсульфоновая кислота, янтарная кислота, дубильная кислота и винная кислота. Типичными примерами подходящих физиологически приемлемых минеральных оснований являются аммиак, кальций, магний, натрий и калий. Типичными примерами подходящих физиологически приемлемых органических оснований являются аргинин, бетаин, кофеин, холин, N,N-дибензилэтилендиамин, диэтиламин, 2-диэтиламиноэтанол, 2 диметиламиноэтанол,этаноламин,этилендиамин,N-этилморфолин,N-этилпиперидин,Nметилглюкамин,глюкамин,глюкозамин,гистидин,N-(2-гидроксиэтил)пиперидин,N-(2 гидроксиэтил)пирролидин, изопропиламин, лизин, метилглюкамин, морфолин, пиперазин, пиперидин,теобромин, триэтиламин, триметиламин, трипропиламин и трометамин. Во втором аспекте настоящее изобретение относится к способу получения соединения 2 арилиндола, замещенного в положении 5, формулы (I) где А, X, Y, Z, W, R и R' имеют вышеуказанные значения, и его физиологически приемлемых аддитивных солей, стереоизомеров, энантиомеров, гидратов, сульфатов и полиморфных форм,а) взаимодействием соединения формулы (II)Q представляет собой атом галогена или гидроксигруппу,с соединением формулы (III)R и R' имеют вышеуказанные значения иG имеет такое значение, как А, или представляет собой атом водорода,с образованием соединения формулы (IV)b) когда G представляет собой Н, взаимодействием соединения формулы (IV) с соединением формулы (V) где I представляет собой атом йода, и А имеет вышеуказанные значения,с образованием соединения формулы (I), и с) образованием, при необходимости, физиологически приемлемой аддитивной соли соединения формулы (IV) со стадии (а), где G отличен от Н, или соединения формулы (I) со стадии (b). Очевидно, что соединение формулы (IV), где G отличен от Н, является ничем иным, как соединением формулы (I). Таким образом, на вышеуказанной стадии (с) всегда получают физиологически приемлемую аддитивную соль соединения формулы (I) по настоящему изобретению. Согласно первому варианту осуществления вышеуказанную стадию (а) осуществляют взаимодейст-3 015689 вием соединения формулы (II), где Q представляет собой Cl, с амином формулы (III) в присутствии подходящего акцептора кислоты в соответствии со стандартными методиками. Согласно второму варианту осуществления вышеуказанную стадию (а) осуществляют взаимодействием соединения формулы (II), в котором Q представляет собой ОН, с амином формулы (III) в присутствии подходящего агента сочетания в соответствии со стандартными методиками. Реакцию на вышеуказанной стадии (b) между соединением формулы (IV), где G представляет собой Н, и арилиодидом формулы (V) также осуществляют в соответствии со стандартными методиками. Промежуточные соединения формулы (III) являются новыми. В третьем аспекте настоящее изобретение также относится к соединению формулы (III)R' представляет собой атом водорода или (C1-С 6)алкильную или (С 3-С 7)циклоалкильную группу,необязательно замещенную 1-3 гидроксигруппами,G представляет собой фенильную, нафтильную или пиридиновую группу, необязательно замещенную 1-3 заместителями, которые могут быть одинаковыми или разными, выбранными из галогена, (C1 С 6)алкила, необязательно замещенного 1-3 гидроксигруппами, трифторметила, нитро, амино, ди(C1-С 3) алкиламино, гидрокси, (С 1-С 3)алкокси, бензилокси, СООН, COOR, SO2CH3, SO2NHCH3, NHSO2CH3,POR1R2, OPOR1R2, (C1-C6)алкил-СООН, (С 2-С 6)алкенил-СООН, фенила и (C1-С 3)алкилфенила,где, в свою очередь, R1 и R2, которые могут быть одинаковыми или разными, представляют собой(C1-C3)алкил; при условии, однако, что G не является незамещенной фенильной группой, когда R представляет собой метил и R' представляет собой Н. Предпочтительными значениями R являются метил, этил, пропил, изопропил и циклогексил. Предпочтительными значениями R' являются Н, метил, этил, пропил, изопропил и циклогексил. Первым особенно предпочтительным значением А является фенил, замещенный 1 или 2 заместителями, которые могут быть одинаковыми или разными, выбранными из Br, Cl, F, метила, этила, метокси,этокси и бензилокси. Вторым особенно предпочтительным значением А является нафтил, необязательно замещенный 1 или 2 заместителями, которые могут быть одинаковыми или разными, выбранными из Br, Cl, F, метила,этила, метокси, этокси и бензилокси. Третьим особенно предпочтительным значением А является пиридин, необязательно замещенный 1 или 2 заместителями, которые могут быть одинаковыми или разными, выбранными из Br, Cl, F, метила,этила, метокси, этокси и бензилокси. Исследования биологических свойств соединения формулы (I) по настоящему изобретению показали, что оно обладает неожиданным селективным свойством ингибирования mPGES-1 и ярко выраженным болеутоляющим действием при воспалительной боли. В четвертом аспекте настоящее изобретение относится к фармацевтической композиции, содержащей эффективное количество соединения формулы (I) или его физиологически приемлемой аддитивной соли, стереоизомера, энантиомера, гидрата, сольвата или полиморфной формы и по меньшей мере один фармацевтически приемлемый инертный ингредиент. В настоящем описании и формуле изобретения термин "эффективное количество" относится к количеству, которое обеспечивает поддающееся оценке улучшение в отношении по меньшей мере одного симптома или параметра определенного нарушения. Фармацевтическая композиция по настоящему изобретению пригодна для использования в лечении или профилактике нарушений, связанных с выработкой простагландина Е 2(PGE2), например воспалительных процессов, боли, опухолей, нейродегенеративных заболеваний и атеросклероза. Преимущественно фармацевтическая композиция по настоящему изобретению пригодна для использования в лечении боли при хронических воспалительных патологиях, таких как артрит, или опухолей, особенно колоректальных опухолей, опухолей пищевода, груди, легкого и мочевого пузыря. Предпочтительно фармацевтические композиции по настоящему изобретению получают в подходящих дозированных формах, содержащих эффективную дозу по меньшей мере одного соединения формулы (I) или его физиологически приемлемой аддитивной соли, стереоизомера, энантиомера, гидрата,сольвата или полиморфной формы, и по меньшей мере один фармацевтически приемлемый инертный ингредиент. Примерами подходящих дозированных форм являются таблетки, капсулы, покрытые оболочкой-4 015689 таблетки, гранулы, растворы и сиропы для перорального введения; кремы, мази и антисептические пластыри для местного применения; суппозитории для ректального введения и стерильные растворы для введения инъекцией, или аэрозоли, или препараты для введения в глаза. Дозированные формы могут также содержать другие традиционные ингредиенты, например: консерванты, стабилизаторы, поверхностно-активные вещества, буферные растворы, соли для регулирования осмотического давления, эмульгаторы, подсластители, красители, отдушки и тому подобные. Если это необходимо для определенных видов терапии, фармацевтическая композиция по настоящему изобретению может содержать другие фармакологически активные ингредиенты, одновременное введение которых оказывает благоприятный эффект. Количество соединения формулы (I) или его физиологически приемлемой аддитивной соли, стереоизомера, энантиомера, гидрата, сольвата или полиморфной формы и по меньшей мере одного фармацевтически приемлемого инертного ингредиента в фармацевтической композиции по настоящему изобретению может варьироваться в широких пределах, в зависимости от известных факторов, например типа заболевания, которое необходимо вылечить, тяжести заболевания, массы тела пациента, дозированной формы, выбранного способа введения, количества ежедневных введений и эффективности выбранного соединения формулы (I). Однако специалист в данной области техники может легко определить оптимальное количество в обычном порядке. Обычно, количество соединения формулы (I) или его физиологически приемлемой аддитивной соли, стереоизомера, энантиомера, гидрата, сольвата или полиморфной формы и по меньшей мере одного фармацевтически приемлемого инертного ингредиента в фармацевтической композиции по настоящему изобретению должно быть таким, чтобы обеспечивать уровень введения между 0,0001 и 100 мг/кг/день и более предпочтительно между 0,01 и 10 мг/кг/день. Очевидно, что нет необходимости содержания в фармацевтических препаратах по настоящему изобретению всего количества соединения формулы (I), так как введение указанного эффективного количества можно обеспечить посредством введения нескольких доз фармацевтической композиции по настоящему изобретению. Дозированные формы фармацевтической композиции по настоящему изобретению можно получить согласно методикам, хорошо известным фармацевтическим химикам, включая смешивание, гранулирование, прессование, растворение, стерилизацию и тому подобные. Последующие примеры служат для дополнительной иллюстрации настоящего изобретения, но не ограничивают его. Пример 1. Получение промежуточных соединений а) 1-Метил-2-фенил-1 Н-индол-5-амин К раствору 2-фенил-5-нитроиндола (получен, как описано в J. Org. Chem. (1966), 31 (1), 65-9) (1 г; 4,2 ммоль) в ДМФА (10 мл) добавляли гидрид натрия (50%-ная суспензия) (0,20 г; 4,2 ммоль); затем смесь оставляли перемешиваться на 30 мин. К полученной таким образом смеси затем добавляли по каплям йодметан (0,60 г; 4,2 ммоль), растворенный в ДМФА (10 мл), и полученную смесь оставляли перемешиваться при комнатной температуре на 18 ч. Затем смесь выливали в воду (50 мл) и экстрагировали этилацетатом (250 мл). Органические фазы объединяли, высушивали над Na2SO4 и раствор упаривали при пониженном давлении. Полученный таким образом остаток очищали флэш-хроматографией (элюент: 7/3 гексан/этилацетат), получая 1 г 1-метил-5-нитро-2-фенил-1 Н-индола, который использовали в следующей реакции без какой-либо дальнейшей очистки. 1H ЯМР (300 МГц, ДМСО-d6)м.д. 3,83 (с, 3 Н), 6,88 (д, J = 0,70 Гц, 1 Н), 7,47-7,68 (м, 5 Н), 7,73 (д, J = 9,06 Гц, 1 Н), 8,08 (дд, J = 9,06, 2,34 Гц, 1 Н), 8,59 (д, J = 2,05 Гц, 1 Н). К суспензии 1-метил-5-нитро-2-фенил-1 Н-индола (1 г; 4 ммоль) в 95 этаноле (100 мл) добавляли 10% Pd/C (0,1 г; 0,1 ммоль) и смесь гидрировали в аппарате Парра (30 фунт/кв.дюйм) в течение 4 ч. Реакционную смесь фильтровали и раствор упаривали при пониженном давлении, получая 1-метил 2-фенил-1 Н-индол-5-амин (0,8 г), который использовали без какой-либо дальнейшей очистки. 1b) 1-Этил-2-фенил-1 Н-индол-5-амин Использовали способ, описанный выше в примере 1 а, за исключением того, что вместо иодметана использовали иодэтан. 1-Этил-5-нитро-2-фенил-1 Н-индол: 1 Н ЯМР (300 МГц, ДМСО-d6) 5 м.д. 1,21 (т, J = 7,16 Гц, 3 Н), 4,30 (кв, J = 7,16 Гц, 2 Н), 6,83 (д, J = 0,58 Гц, 1 Н), 7,48-7,63 (м, 5 Н), 7,77 (д, J=9,21 Гц, 1 Н), 8,07 (дд, J = 9,21, 2,34 Гц, 1 Н), 8,58 (д, J = 2,34 Гц, 1 Н). 1-Этил-2-фенил-1 Н-индол-5-амин: 1 Н ЯМР (300 МГц, хлороформ-d)м.д. 1,28 (т, J=6, 94 Гц, 3 Н), 3,59 (ушир, с, 2 Н), 4,13 (кв, J = 7,16 Гц, 2 Н), 6,37 (д, J=0,73 Гц, 1 Н), 6,82 (дд, J = 8,48, 2,19 Гц, 1 Н), 7,09 (д, J = 1,90 Гц, 1 Н), 7,23 (д, J = 8,62c) 1-Изопропил-2-фенил-1 Н-индол-5-амин Использовали способ, описанный выше для стадии а), за исключением того, что вместо йодметана использовали изопропилбромид. 1-Изопропил-5-нитро-2-фенил-1 Н-индол: Моноизотопная масса = 280,1; LC/MS (М+Н)+ = 281,2d) 1-Этил-2-(2-фторфенил)-1 Н-индол-5-амин К суспензии, содержащей ацетат цезия, высушенный в вакууме в течение ночи при 140 С (3,6 г; 19 ммоль), в N,N-диметилацетамиде (ДМА, 5 мл), в инертной атмосфере добавляли ацетат палладия (12 мг; 0,05 ммоль), трифенилфосфин (55 мг; 0,21 ммоль), 1-этил-5-нитро-1 Н-индол (2 г; 10 ммоль) (полученный по методике, описанной в Bioorg. Med. Chem. 13 (2005), 3531-3541) и 1-иод-2-фторбензол (2,53 г; 11 ммоль). Реакционную смесь оставляли перемешиваться при 140 С в инертной атмосфере в течение 18 ч. Затем смесь охлаждали до комнатной температуры, добавляли дихлорметан (50 мл) и полученную таким образом смесь фильтровали под вакуумом через целит. Органический раствор переносили в делительную воронку, промывали Н 2 О (250 мл) и высушивали над Na2SO4. Органический растворитель удаляли упариванием при пониженном давлении, и остаток очищали флэш-хроматографией на силикагеле, получая 1-этил-2-(2-фторфенил)-5-нитро-1 Н-индол (0,7 г), который использовали без какой-либо дальнейшей очистки. 1H ЯМР (300 МГц, ДМСО-d6)м.д. 1,15 (т, J = 7,16 Гц, 3 Н), 4,18 (кв, J = 7,02 Гц, 2 Н), 6,87 (с, 1 Н),7,35-7,50 (м, 2 Н), 7,51-7,70 (м, 2 Н), 7,79 (д, J = 9,06 Гц, 1 Н), 8,10 (дд, J = 9,06, 2,34 Гц, 1 Н), 8,62 (д, J = 2,34 Гц, 1 Н). К суспензии, содержащей 1-этил-2-(2-фторфенил)-5-нитро-1 Н-индол (0,78 г; 2,75 ммоль) в 95 этаноле (100 мл), добавляли 10% Pd/C (0,1 г; 0,1 ммоль), и смесь гидрировали в аппарате Парра (30 фунт/кв.дюйм) в течение 4 ч. Смесь фильтровали и раствор упаривали при пониженном давлении, получая 1-этил-2-(2-фторфенил)-1 Н-индол-5-амин (0,8 г), который использовали без какой-либо дальнейшей очистки.h) 5-Амино-2-фенил-1-циклогексилиндол К суспензии ацетата цезия, высушенного в вакууме в течение ночи при 140 С (1,8 г; 9,5 ммоль), вN,N-диметилацетамиде (5 мл), в инертной атмосфере добавляли ацетат палладия (6 мг; 0,05 ммоль), трифенилфосфин (28 мг; 0,1 ммоль), 1-циклогексилиндол (полученный по методике, описанной в Synthesis 1977, 5, 335-336) (1 г; 5 ммоль) и 1-иод-4-метилбензол (1,26 г; 6 ммоль).-6 015689 Реакционную смесь оставляли перемешиваться при 140 С в инертной атмосфере в течение 18 ч. Затем смесь охлаждали до комнатной температуры и добавляли дихлорметан (50 мл). Реакционную смесь фильтровали под вакуумом через целит. Фильтрат переносили в делительную воронку, и органическую фазу промывали Н 2 О (250 мл) и высушивали над Na2SO4. Органический растворитель удаляли упариванием при пониженном давлении, и остаток очищали флэш-хроматографией на силикагеле (97/3 гексан/этилацетат), получая 1-циклогексил-2-(4-метилфенил)1H-индол (200 мг), который использовали без какой-либо дальнейшей очистки. 1H ЯМР (300 МГц, хлороформ-d)м.д. 1,13-1,98 (м, 8 Н), 2,25-2,41 (м, 2 Н), 2,43 (с, 3 Н), 4,21 (тт, J = 12,42, 3,80 Гц, 1 Н), 6,42 (ушир. с, 1 Н), 7,05-7,11 (м, 1 Н), 7,15 (ддд, J = 7,90, 7,20, 1,30 Гц, 1 Н), 7,22-7,35 (м, 4 Н), 7,57-7,67 (м, 2 Н). К раствору 1-циклогексил-2-(4-метилфенил)-1 Н-индола (100 мг, 0,3 ммоль) в 2 мл концентрированной H2SO4 при 5 С добавляли по каплям раствор NaNO3 (34 мг; 0,4 ммоль) в H2SO4 (1 мл) . По окончании добавления смесь оставляли перемешиваться при 5 С в течение 10 мин. Затем ее выливали в смесь воды и льда (10 мл), и полученное таким образом твердое вещество отфильтровывали и очищали флэш-хроматографией на силикагеле (99/1 гексан/этилацетат), получая 1-циклогексил-2-(4 метилфенил)-5-нитро-1 Н-индол (45 мг), который использовали без какой-либо дальнейшей очистки. 1H ЯМР (300 МГц, хлороформ-d)м.д. 1,15-1,42 (м, 4 Н), 1,64-2,01 (м, 4 Н), 2,16-2,41 (м, 2 Н), 2,45 (с, 3 Н), 4,24 (тт, J = 12,42, 3,80 Гц, 1 Н), 6,59 (с, 1 Н), 7,28-7,35 (м, 4 Н), 7,64 (д, J=9,35 Гц, 1 Н), 8,06 (дд, J = 9,35,2,34 Гц, 1 Н) 8,54 (д, J = 2,34 Гц, 1 Н). К суспензии 1-циклогексил-2-(4-метилфенил)-5-нитро-1 Н-индола (45 мг; 0,13 ммоль) в абсолютном этаноле (5 мл) добавляли дигидрат хлорида олова (152 мг; 0,67 ммоль) и смесь оставляли перемешиваться при 70 С в течение 18 ч. Реакционную смесь охлаждали до комнатной температуры и затем выливали в смесь воды и льда (20 мл), добавляли NaHCO3 (насыщенный раствор) до рН 8 и смесь оставляли перемешиваться на 20 мин. Затем смесь выливали в делительную воронку и экстрагировали этилацетатом (230 мл). Органические фазы объединяли, высушивали над Na2SO4 и удаляли растворитель упариванием при пониженном давлении, получая 5-амино-2-(4-метилфенил)-1-циклогексилиндол (30 мг), который использовали без какой-либо дальнейшей очистки. 1(26,1 г; 202 ммоль), с последующим добавлением по каплям 2-хлорбензоилхлорида (35,4 г; 202 ммоль),растворенного в дихлорметане (50 мл). По завершении добавлений, смесь оставляли перемешиваться при комнатной температуре в течение 2 ч. Затем добавляли воду (400 мл), и органическую фазу отделяли и высушивали над Na2SO4. Органический раствор упаривали при пониженном давлении. Полученный сырой продукт очищали кристаллизацией из этилацетата с образованием целевого продукта (37 г). 1H ЯМР (300 МГц, ДМСО-d6)м.д. 1,35 (т, J = 7,27 Гц, 3 Н), 4,19 (кв, J = 7,05 Гц, 2 Н), 6,41 (дд, J = 2,97, 0,66 Гц, 1 Н), 7,34-7,60 (м, 7 Н), 8,00 (д, J = 1,32 Гц, 1 Н), 10,26 (с, 1 Н) Пример 2. Получение соединений по настоящему изобретению а) Пример первого варианта способа получения К раствору 5-аминоиндола (III) (2 ммоль) в дихлорметане (10 мл) добавляли триэтиламин (2,2 ммоль), с последующим добавлением по каплям ацилхлорида (II) (2,2 ммоль), растворенного в дихлорметане (10 мл). По завершении добавлений смесь оставляли перемешиваться при комнатной температуре в течение 20 ч. Затем добавляли воду (50 мл), и органическую фазу отделяли и высушивали над Na2SO4. Раствор упаривали при пониженном давлении. Полученный сырой продукт очищали, получая соединение (I), в котором X, Y, Z, R, R' и А имеют вышеуказанные значения.b) Пример второго варианта способа получения(3 мл) и ацилхлорид (II) (0,28 ммоль) в дихлорметане (3 мл). Смесь оставляли перемешиваться в течение 20 ч. Смолу Amberlyst A21 затем удаляли фильтрованием и промывали дихлорметаном (5 мл). Органические фазы объединяли, разбавляли диметилформамидом (1 мл) и перемешивали со смолой Amberlyst 15(0,9 г) в течение 5 ч. Данную обработку повторяли дважды. Смолу Amberlyst A21 удаляли фильтрованием и раствор упаривали в центрифуге, получая соединение (I), в котором X, Y, Z, R, R' и А имеют вышеуказанные значения. с) Пример третьего варианта способа получения:(8 мл) и диметилформамиде (0,8 мл) в инертной атмосфере. После перемешивания смеси при комнатной температуре в течение 10 мин добавляли смолу PS-карбодиимид (0,73 г). После перемешивания реакционной смеси в течение 20 ч смолу удаляли фильтрованием и промывали дихлорметаном (25 мл). Раствор упаривали в центрифуге, получая соединение (I), в котором X, Y,Z, R, R' и А имеют вышеуказанные значения,d) Пример четвертого варианта способа получения К раствору бензойной кислоты (II) (10 ммоль) в диметилформамиде (40 мл) при 0 С добавляли при перемешивании 1-гидроксибензотриазол (HOBt) (10 ммоль) и дициклогексилкарбодиимид (ДЦК) (10 ммоль). Смесь оставляли перемешиваться при 0 С в течение 30 мин и добавляли 5-аминоиндол (III) (9 ммоль), растворенный в диметилформамиде (20 мл). Смесь оставляли перемешиваться при 0 С в течение еще 30 мин, и затем при комнатной температуре в течение 18 ч. Смесь фильтровали, добавляли 2 н соляную кислоту до рН 2, и полученный таким образом осадок отфильтровывали и очищали, получая соединение (I), в котором X, Y, Z, R, R' и А имеют вышеуказанные значения. е) Пример пятого варианта способа получения К суспензии ацетата цезия, высушенного в вакууме в течение ночи при 140 С (6,02 ммоль) в N,Nдиметилацетамиде (ДМА) (3 мл), в инертной атмосфере добавляли ацетат палладия (0,017 ммоль), трифенилфосфин (0,067 ммоль), индол (V) (3,35 ммоль) и арилиодид (V) (3,68 ммоль). Реакционную смесь оставляли перемешиваться при 140 С в инертной атмосфере в течение 18 ч. Реакционную смесь охлаждали до комнатной температуры, добавляли дихлорметан (50 мл) и полученную смесь фильтровали под вакуумом через целит. Отфильтрованный органический раствор переносили в делительную воронку. Органическую фазу промывали Н 2 О (250 мл), высушивали над Na2SO4 и упаривали при пониженном давлении. Остаток очищали, получая соединение (I), в котором X, Y, Z, R, R' и А имеют вышеуказанные значения.-8 015689 Таким образом были получены соединения формулы (I), показанные ниже в табл. 1, в которой Очистка А=Кристаллизация Очистка В=Флэш-хроматография на силикагеле Пример 3. Биологическая активность in vitro Примененный тест дает возможность оценить потенциал ингибирующего действия тестируемых соединений на выработку PGE2 и селективность в отношении выработки PGF2. Использовали клеточную линию аденокарциномы человека А 54 9, которая особенно чувствительна к стимуляции противовоспалительными цитокинами, например IL-1, и в ответ на такую стимуляцию проявляет особенную активность в выработке и высвобождении двух простаноидов: PGE2 и PGF2 (Thoren S. Jakobsson P-J, 2000). Клетки стимулировали IL-1 (10 нг/мл) и одновременно обрабатывали тестируемым соединением в- 11015689 течение 22 ч в подходящей питательной среде (DMEM - модифицированная по способу Дульбекко среда Игла), обогащенной 5% фетальной телячьей сывороткой и L-глутамином (конечная концентрация 4 мМ) в инкубаторе при 37 С и концентрации СО 2 5%. В конце инкубирования определяли количество выработанного и высвобожденного в супернатантPGE2 и PGF2, используя набор EIA (производится и поставляется фирмой Cayman Chemicals, Ann Arbor,MI, США). В качестве соединения сравнения использовали индометацин в концентрации 10 нМ (SigmaAldrich), представляющий собой нестероидное противовоспалительное лекарственное средство, в одинаковой степени ингибирующее и PGE2, и PGF2. Результаты, выраженные в процентах ингибирования выработки PGE2 и PGF2 при концентрации 10 мкМ, представлены в табл. 2, в которой "н.а." (не активно) означает ингибирующую активность менее 20%. Таблица 2 Для иллюстрации в табл. 3 сопоставлены значения pIC50 некоторых соединений по настоящему изобретению, где pIC50 представляет собой отрицательный логарифм IC50, который в свою очередь представляет собой концентрацию соединения, при которой наблюдается 50%-ное ингибирование выработкиPGE2 или PGF2 по сравнению с клетками, которые стимулировались, но не обрабатывались тем же соединением. В табл. 3 "и.о." означает "не поддается определению". Пример 4. Биологическая активность in vivo Тестируемое соединение оценивали в мышиной модели судорог, вызываемых уксусной кислотой(Stock J. L. et al., J Clin Inv 2001, 107: 325-331). Данный тест позволяет оценивать антиноцицептивную активность соединений по настоящему изобретению в модели воспалительной боли. В тесте использовали самок мышей CD-I весом 25-30 г. Животным внутрибрюшинно вводили тестовое соединение (0,1-10 мг/кг), суспендированное в метилцеллюлозе (МТС). Контрольным животным тем же образом вводили чистый растворитель (МТС). Через 30 мин после введения животным делали внутрибрюшинную инъекцию уксусной кислоты(0,7 об./об. в физиологическом растворе, 16 мкл/г массы тела) для индуцирования воспалительной боли и определения влияния тестового соединения на ноцицептивный ответ. Сразу после введения уксусной кислоты и в течение следующих 20 мин подсчитывали количество напряжений, которое представляет собой критерий оценки ноцицептивной реакции. Как показано в табл. 4, соединение по настоящему изобретению дозозависимым образом вызывало уменьшение количества напряжений в течение 20 мин после введения уксусной кислоты по сравнению с животными, которым вводили только МТС. Таблица 4 Пример 5. Селективность между изоформами PGES Примененный тест позволяет оценивать способность соединений по настоящему изобретению ингибировать выработку PGE2 в клеточной линии лимфомы человека U-937, которая предпочтительно экспрессирует изоформу фермента (cPGES), отвечающую за выработку PGE2 в базальных условиях, в отсутствии противовоспалительных стимулов. Данная форма фермента отличается от формы, преимущественно экспрессируемой в клетках А 549 (mPGES-1) после провоспалительной стимуляции. Отсутствие ингибирующей активности в отношении PGE2 в данной клеточной модели с уверенностью свидетельствует о селективности соединения по сравнению с формой фермента, отвечающей за выработку PGE2 в присутствии воспалительных стимулов. Результаты, выраженные в процентах ингибирования выработки PGE2, представлены в табл. 5, в которой "н.а." (неактивно) означает ингибирующую активность менее 20%. В качестве соединения сравнения использовался индометацин в концентрации 10 нМ. Обнаружено, что соединения по настоящему изобретению незначительно ингибировали образование PGE2, в основном, благодаря действию cPGES.X представляет собой атом галогена или (C1-С 3)алкил, трифторметил, нитро, циано, (C1-С 3)алкокси или фенильную группу;Y и Z, которые могут быть одинаковыми или разными, представляют собой Н, или атом галогена,или трифторметил, нитро, (C1-C3)алкокси, СООН, (C1-C3)алкил-СООН или (С 2-С 3)алкенил-СООН;W представляет собой атом кислорода или СН 2 или NH группу;R представляет собой атом водорода или (C1-С 6)алкильную или (С 3-С 7)циклоалкильную группу, необязательно замещенную 1-3 гидроксигруппами;R' представляет собой атом водорода или (C1-С 6)алкильную или (С 3-С 7)циклоалкильную группу; А представляет собой фенильную, нафтильную или пиридиновую группу, необязательно замещенную 1-3 заместителями, которые могут быть одинаковыми или разными, выбранными из галогена, (C1 С 6)алкила, трифторметила, амино, (C1-С 3)алкокси, бензилокси, COOR, NHSO2CH3, (C1-C6)алкил-СООН и(С 2-С 6)алкенил-СООН,и его физиологически приемлемые аддитивные соли, стереоизомеры, энантиомеры, гидраты, сольваты и полиморфные формы. 2. Соединение по п.1, отличающееся тем, что X представляет собой галоген, (С 1-С 3)алкил, трифторметил, нитро или (C1-С 3)алкокси. 3. Соединение по п.2, отличающееся тем, что X представляет собой Cl, Br, F, трифторметил или нитро. 4. Соединение по п.1, отличающееся тем, что Y и Z независимо друг от друга представляют собой Н, галоген, нитро, СООН, трифторметил или (C1-С 3)алкокси. 5. Соединение по п.4, отличающееся тем, что Y и Z независимо друг от друга представляют собойCl, Br, F, трифторметил, нитро, СООН, метокси или этокси. 6. Соединение по п.1, отличающееся тем, что R представляет собой метил, этил, пропил, изопропил или циклогексил. 7. Соединение по п.1, отличающееся тем, что R' представляет собой Н, метил, этил, пропил, изопропил или циклогексил. 8. Соединение по п.1, отличающееся тем, что А представляет собой фенил, нафтил или пиридин,необязательно замещенные 1 или 2 заместителями, которые могут быть одинаковыми или разными, выбранными из галогена, (C1-С 3)алкила, (С 1-С 3)алкокси и бензилокси. 9. Соединение по п.8, отличающееся тем, что А представляет собой фенил, необязательно замещенный 1 или 2 заместителями, которые могут быть одинаковыми или разными, выбранными из Br, Cl, F,метила, этила, метокси, этокси и бензилокси. 10. Соединение по п.8, отличающееся тем, что А представляет собой нафтил, необязательно замещенный 1 или 2 заместителями, которые могут быть одинаковыми или разными, выбранными из Br, Cl,F, метила, этила, метокси, этокси и бензилокси.- 14015689 11. Соединение по п.8, отличающееся тем, что А представляет собой пиридин, необязательно замещенный 1 или 2 заместителями, которые могут быть одинаковыми или разными, выбранными из Br, Cl,F, метила, этила, метокси, этокси и бензилокси. 12. Способ получения соединения 2-арилиндола, замещенного в положении 5, формулы (I)X представляет собой атом галогена или (C1-С 3)алкил, трифторметил, нитро, циано, (C1-С 3)алкокси или фенильную группу;Y и Z, которые могут быть одинаковыми или разными, представляют собой Н или атом галогена,или трифторметил, нитро, (C1-С 3)алкокси, СООН, (C1-C3)алкил-СООН или (С 2-С 3)алкенил-СООН;W представляет собой атом О или СН 2 или NH группу;R представляет собой атом водорода или (C1-С 6)алкильную или (С 3-С 7)циклоалкильную группу, необязательно замещенную 1-3 гидроксигруппами;R' представляет собой атом Н или (C1-С 6)алкильную или (С 3-С 7)циклоалкильную группу; А представляет собой фенильную, нафтильную или пиридиновую группу, необязательно замещенную 1-3 заместителями, которые могут быть одинаковыми или разными, выбранными из галогена, (C1 С 6)алкила, трифторметила, амино, (C1-С 3)алкокси, бензилокси, COOR, NHSO2CH3, (C1-C6)алкил-СООН иX, Y и Z имеют вышеуказанные значения иQ представляет собой атом галогена или гидроксигруппу,подвергают взаимодействию с соединением формулы (III)R и R' имеют вышеуказанные значения иG имеет такие же значения, как А, или представляет собой атом водорода,с образованием соединения формулы (IV)I представляет собой атом йода и А имеет вышеуказанные значения,с образованием соединения формулы (I), и с) при необходимости получают физиологически приемлемую аддитивную соль соединения формулы (IV) со стадии (а), где G отличен от Н, или соединения формулы (I) со стадии (b). 13. Способ по п.12, отличающийся тем, что стадию (а) осуществляют взаимодействием соединения формулы (II), где Q представляет собой Cl, с амином формулы (III) в присутствии подходящего акцептора кислоты в соответствии со стандартными методиками. 14. Способ по п.12, отличающийся тем, что стадию (а) осуществляют взаимодействием соединения- 15015689 формулы (II), где Q представляет собой ОН, с амином формулы (III) в присутствии подходящего агента сочетания в соответствии со стандартными методиками. 15. Промежуточное соединение формулы (III)R' представляет собой атом Н или (C1-С 6)алкильную или (С 3-С 7)циклоалкильную группу,G представляет собой фенильную, нафтильную или пиридиновую группу, необязательно замещенную 1-3 заместителями, которые могут быть одинаковыми или разными, выбранными из галогена, (C1 С 6)алкила, трифторметила, амино, (C1-С 3)алкокси, бензилокси, COOR, NHSO2CH3, (C1-C6)алкил-СООН и(С 2-С 6)алкенил-СООН,при условии, однако, что G не является незамещенной фенильной группой, когда R представляет собой метил и R' представляет собой Н. 16. Промежуточное соединение по п.15, отличающееся тем, что R представляет собой метил, этил,пропил, изопропил или циклогексил. 17. Промежуточное соединение по п.15, отличающееся тем, что R' представляет собой Н, метил,этил, пропил, изопропил или циклогексил. 18. Фармацевтическая композиция, содержащая эффективное количество соединения формулы (I)X представляет собой атом галогена или (C1-С 3)алкил, трифторметил, нитро, циано, (C1-С 3)алкокси или фенил;Y и Z, которые могут быть одинаковыми или разными, представляют собой Н или атом галогена,или трифторметил, нитро, (C1-С 3)алкокси, СООН, (C1-C3)алкил-СООН или (С 2-С 3) алкенил-СООН;W представляет собой атом О или СН 2 или NH группу;R представляет собой атом водорода или (С 1-С 6)алкильную или (С 3-С 7)циклоалкильную группу, необязательно замещенную 1-3 гидроксигруппами;R' представляет собой атом Н или (C1-С 6) алкильную или (С 3-С 7)циклоалкильную группу; А представляет собой фенильную, нафтильную или пиридиновую группу, необязательно замещенную 1-3 заместителями, которые могут быть одинаковыми или разными, выбранными из галогена, (C1 С 6)алкила, трифторметила, амино, (C1-С 3)алкокси, бензилокси, COOR, NHSO2CH3, (C1-C6)алкил-СООН и(С 2-С 6)алкенил-СООН,или его физиологически приемлемой аддитивной соли, стереоизомера, энантиомера, гидрата, сольвата или полиморфной формы, и по меньшей мере один фармацевтически приемлемый инертный ингредиент.
МПК / Метки
МПК: C07D 209/10, C07D 401/04, A61K 31/40, C07D 209/12, C07D 209/08, A61K 31/435, A61P 29/00, A61P 35/00
Метки: 2-арилиндола, npges-1, ингибиторов, качестве, производные
Код ссылки
<a href="https://eas.patents.su/17-15689-proizvodnye-2-arilindola-v-kachestve-ingibitorov-npges-1.html" rel="bookmark" title="База патентов Евразийского Союза">Производные 2-арилиндола в качестве ингибиторов npges-1</a>
Сульфонамидные производные в качестве ингибиторов рассасывания костной ткани и ингибиторов адгезии клеток,способ их получения, применение и фармацевтическая композиция

Номер патента: 3102
Опубликовано: 26.12.2002
Авторы: Катбертсон Роберт Эндрю, Карниато Дени, Шойнеманн Карлхайнц, Макдауэлл Роберт, Гадек Томас, Вилл Дэвид Вильям, Кнолле Йохен, Пейман Ануширван, Бодари Сара Кэтрин, Гурвест Жан-Франсуа
МПК: C07D 239/42, A61P 19/10, A61K 31/505...
Метки: фармацевтическая, адгезии, сульфонамидные, костной, клеток,способ, производные, применение, композиция, ингибиторов, ткани, получения, качестве, рассасывания
Формула / Реферат:
1. Сульфонамидные производные общей формулы I где R1 и R2 вместе образуют двухвалентный (С2-С3)алкиленовый радикал; R4 является Н или (С1-С6)алкилом; R5 является (С1-С10)алкилом, (С6-С14)арилом, (C5-C14)гетероарилом или (С6-С14)арил(С1-С10)алкильной группой, где арил, гетероарил или алкил возможно замещены R3, или 2-оксобицикло[2.2.1]гепт-1-илметильной группой; R3 является (C1-C4)алкилом, (C1-C4)алкилокси, галогеном, трифторметилом, циано,...
Гетероариламины-производные пиримидина и пиридазина в качестве ингибиторов гликогенсинтаза-киназы 3-бета (ингибиторов gskз)

Номер патента: 10859
Опубликовано: 30.12.2008
Авторы: Леви Паулус Йоаннес, Фрейн Эдди Жан Эдгар, Лав Кристофер Джон, Де Жонж Марк Рене, Койманс Люсьен Мария Хенрикус, Хэрес Ян, Эмбрехтс Вернер Констант Йохан, Бейнстерс Петер Якобус Йоханнес Антониус, Винкерс Хендрик Мартен, Дильс Гастон Станислас Марселла, Ван Акен Кун Жанн Альфонс, Жанссен Поль Адриан Ян, Виллемс Марк
МПК: A61K 31/505, A61K 31/50, A61K 31/435...
Метки: пиримидина, 3-бета, качестве, gskз, ингибиторов, пиридазина, гликогенсинтаза-киназы, гетероариламины-производные
Формула / Реферат:
1. Соединение формулы его N-оксид, фармацевтически приемлемая аддитивная соль, четвертичный амин и стереохимически изомерная форма, где кольцо А является R1 обозначает водород; X обозначает -O-; -O-C1-6алкил- или прямую связь; Z обозначает прямую связь, -С(=O)- или -NR1-C1-6алкил-; R обозначает водород или R20; R3 обозначает водород; галоген; циано; полигалогенС1-6алкил; R4 обозначает моноциклический, насыщенный или частично насыщенный или...
Производные фталазина в качестве ингибиторов фосфодиэстеразы 4

Номер патента: 3702
Опубликовано: 28.08.2003
Авторы: Гранчини Джанкарло, Норчини Габриеле, Мораццони Габриеле, Наполетано Мауро, Пеллачини Франко
МПК: C07D 237/30, A61K 31/502
Метки: ингибиторов, производные, фталазина, фосфодиэстеразы, качестве
Формула / Реферат:
1. Соединение формулы I где ---- представляет одинарную или двойную связь; B представляет метилен; A представляет пиридин, замещенный одним заместителем или большим количеством заместителей; R представляет два атома водорода или C = O группу, когда ---- представляет одинарную связь, или, когда ---- представляет двойную связь, R представляет водород, необязательно замещенный арил или гетероцикл, (C1-8)алкил, (C2-8)алкенил или (C2-8)алкинил,...
Производные пиридо[2, 1 - а]изохинолина в качестве ингибиторов dpp – iv

Номер патента: 9591
Опубликовано: 28.02.2008
Авторы: Наркизян Робер, Маттей Патрицио, Кун Бернд, Бёрингер Маркус
МПК: A61P 3/10, C07D 471/06
Метки: пиридо[2, производные, ингибиторов, качестве, а]изохинолина
Формула / Реферат:
1. Соединения формулы (I) где R1 представляет собой -C(O)-N(R5)R6 или -N(R5)R6; R2, R3 и R4 каждый независимо представляет собой водород или C1-С6алкокси; R5 представляет собой водород; R6 представляет собой C1-С6алкилкарбонил или С3-С6циклоалкилкарбонил; или R5 и R6 вместе с атомом азота, к которому они присоединены, образуют 4-, 5-, 6- или 7-членное насыщенное или ненасыщенное гетероциклическое кольцо, необязательно содержащее другой...
Производные 6-пиридинил-4-пиримидона в качестве ингибиторов тау-протеинкиназы 1

Номер патента: 13004
Опубликовано: 26.02.2010
Авторы: Фукунага Кендзи, Йокосима Сатоси, Кохара Тосиюки, Уехара Фумиаки, Хики Синсуке, Ватанабе Казутоси
МПК: A61K 31/506, A61P 25/28, A61K 31/505...
Метки: 6-пиридинил-4-пиримидона, ингибиторов, тау-протеинкиназы, производные, качестве
Формула / Реферат:
1. Соединение, представленное формулой (I), его оптически активный изомер или его фармацевтически приемлемая сольгде в указанной формуле каждый символ имеет значения, указанные ниже:R1 представляет метильную группу;R2 представляет атом водорода;R3 представляет атом фтора, замещающий положение 3 пиридинового кольца;q равен 1;R5 представляет группу, представленную следующей формулой (II):где А представляет (С6-С10)-арил или гетероцикл;R6 могут...
Предыдущий патент: Фунгицидные смеси из анилидов 1-метилпиразол-4-илкарбоновой кислоты и азолопиримидиниламинов
Следующий патент: Способы лечения расстройств, выражающихся в разрушительном поведении
Случайный патент: Способ гидрокрекинга парафиновой фракции (варианты)