Имидазопиридинзамещённые тропановые производные, обладающие активностью антагониста ccr5 рецептора, для лечения вируса иммунодефицита человека (вич) и воспаления




























Формула / Реферат
1. Соединение формулы (I)

или его фармацевтически приемлемые соль, сольват, где
R1 представляет собой C1-С6алкил и
R2 представляет собой C1-С6алкил или С3-С7циклоалкил, где указанный алкил возможно замещен CF3.
2. Соединение по п.1, где R1 представляет собой С1-С4алкил.
3. Соединение по п.1 или 2, где R1 представляет собой метил.
4. Соединение по любому из пп.1-3, где R2 представляет собой С1-С4алкил, возможно замещенный CF3.
5. Соединение по любому из пп.1-4, где R2 представляет собой метил, этил или изопропил.
6. Соединение по любому из пп.1-3, где R2 представляет собой циклопропил или циклобутил.
7. Соединение по п.1, выбранное из
N-{(1S)-3-[3-эндо-(5-ацетил-2-метил-4,5,6,7-тетрагидро-1H-имидазо[4,5-с]пиридин-1-ил)-8-азабицикло[3.2.1]окт-8-ил]-1-(3-фторфенил)пропил}ацетамида;
N-{(1S)-3-[3-эндо-(5-циклобутанкарбонил-2-метил-4,5,6,7-тетрагидро-1H-имидазо[4,5-с]пиридин-1-ил)-8-азабицикло[3.2.1]окт-8-ил]-1-(3-фторфенил)пропил}ацетамида;
N-{(1S)-3-[3-эндо-(5-циклопропанкарбонил-2-метил-4,5,6,7-тетрагидро-1H-имидазо[4,5-с]пиридин-1-ил)-8-азабицикло[3.2.1]окт-8-ил]-1-(3-фторфенил)пропил}ацетамида;
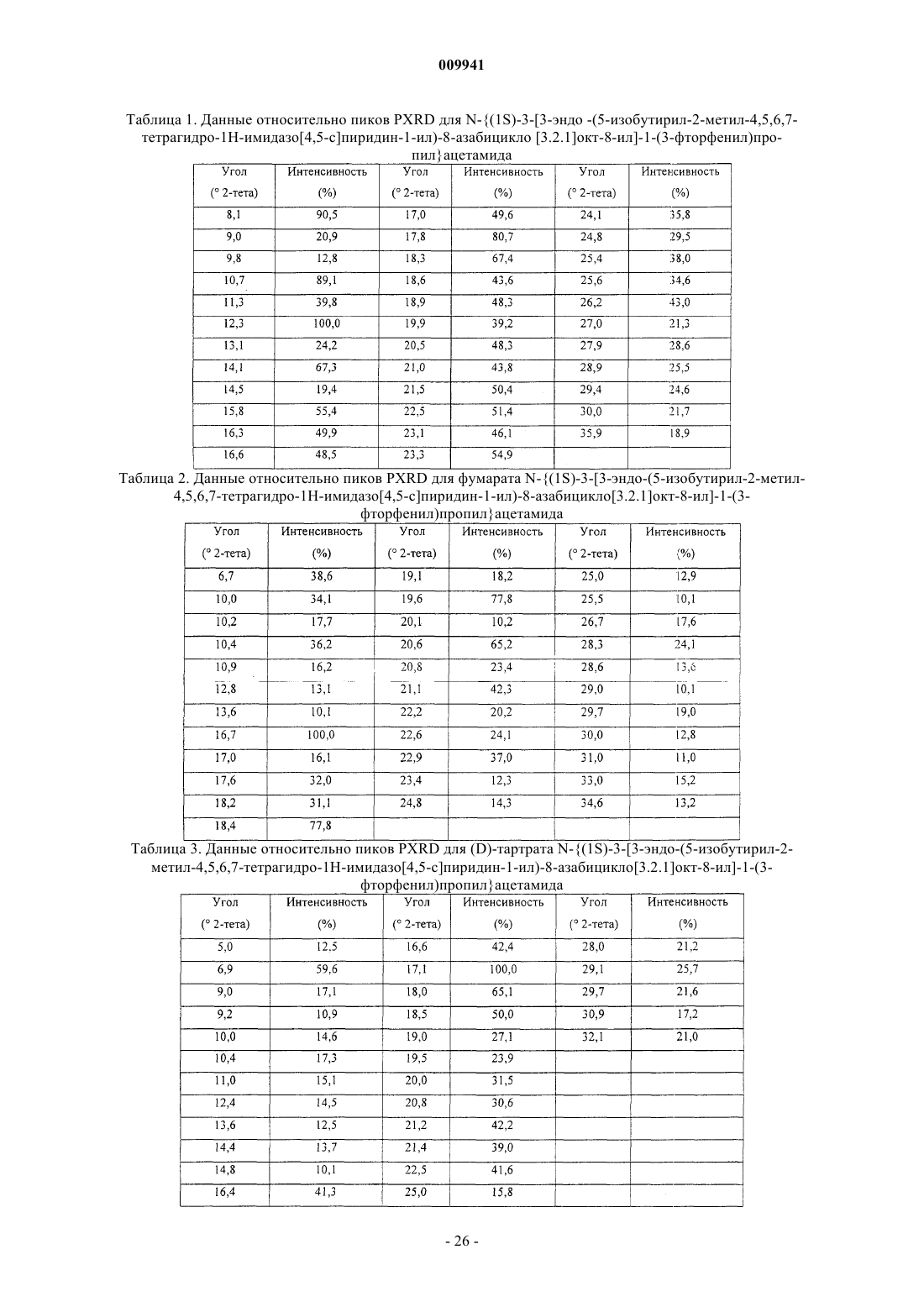
N-{(1S)-3-[3-эндо-(5-изобутирил-2-метил-4,5,6,7-тетрагидро-1H-имидазо[4,5-с]пиридин-1-ил)-8-азабицикло[3.2.1]окт-8-ил]-1-(3-фторфенил)пропил}ацетамида;
N-{(1S)-3-[3-эндо-(2-метил-5-пропионил-4,5,6,7-тетрагидро-1H-имидазо[4,5-с]пиридин-1-ил)-8-азабицикло[3.2.1]окт-8-ил]-1-(3-фторфенил)пропил}ацетамида;
N-{(1S)-3-[3-эндо-(5-бутирил-2-метил-4,5,6,7-тетрагидро-1H-имидазо[4,5-с]пиридин-1-ил)-8-азабицикло[3.2.1]окт-8-ил]-1-(3-фторфенил)пропил}ацетамида;
N-{(1S)-3-[3-эндо-(2-метил-5-(2,2-диметилпропионил)-4,5,6,7-тетрагидро-1H-имидазо[4.5-с]пиридин-1-ил)-8-азабицикло[3.2.1]окт-8-ил]-1-(3-фторфенил)пропил}ацетамида;
N-{(1S)-3-[3-эндо-(2-метил-5-(3,3,3-трифторпропионил)-4,5,6,7-тетрагидро-1H-имидазо[4,5-с]пиридин-1-ил)-8-азабицикло[3.2.1]окт-8-ил]-1-(3-фторфенил)пропил}ацетамида
и их фармацевтически приемлемых солей, сольватов.
8. Фармацевтическая композиция, включающая соединение формулы (I) или его фармацевтически приемлемые соль, сольват по любому из пп.1-7 вместе с одним или более чем одним фармацевтически приемлемым эксципиентом, разбавителем или носителем.
9. Фармацевтическая композиция по п.8, включающая один или более чем один дополнительный терапевтический агент.
10. Применение соединения формулы (I) или его фармацевтически приемлемых соли, сольвата по любому из пп.1-7 в качестве лекарства.
11. Применение соединения формулы (I) или его фармацевтически приемлемых соли, сольвата по любому из пп.1-7 для лечения расстройства, в которое вовлечена модуляция CCR5 рецепторов.
12. Применение по п.11, где расстройство представляет собой вирус иммунодефицита человека (ВИЧ), ретровирусную инфекцию, генетически родственную ВИЧ, синдром приобретенного иммунодефицита (СПИД) или воспалительное заболевание.
13. Применение по п.11, где расстройство представляет собой рассеянный склероз, ревматоидный артрит или отторжение трансплантата.
14. Применение по п.11, где расстройство представляет собой воспалительное заболевание кишечника, эндометриоз, диабет I типа, почечные заболевания, фиброз, хронический панкреатит, воспалительные легочные состояния, энцефалит, хроническую сердечную недостаточность, ишемическую болезнь сердца, псориаз, удар, ожирение, заболевания центральной нервной системы (ЦНС), анемию, рестеноз, атеросклеротические бляшки, атопический дерматит, хронический панкреатит, рак, боль или реакцию на стресс в результате хирургического вмешательства, инфекции, повреждения или другого травматического поражения.
15. Применение по п.11, где расстройство представляет собой вирус гепатита В (HBV), вирус гепатита С (HCV), чуму, поксвирус, токсоплазмоз, микобактериальную, трипаносомную инфекцию, пневмонию или цитоспоридиоз (cytosporidiosis).
16. Применение соединения формулы (I) или его фармацевтически приемлемых соли, сольвата по любому из пп.1-7 для изготовления лекарства для лечения расстройства, в которое вовлечена модуляция CCR5 рецепторов.
17. Способ лечения млекопитающего, страдающего расстройством, в которое вовлечена модуляция CCR5 рецепторов, включающий лечение указанного млекопитающего эффективным количеством соединения формулы (I) или его фармацевтически приемлемых соли, сольвата по любому из пп.1-7.
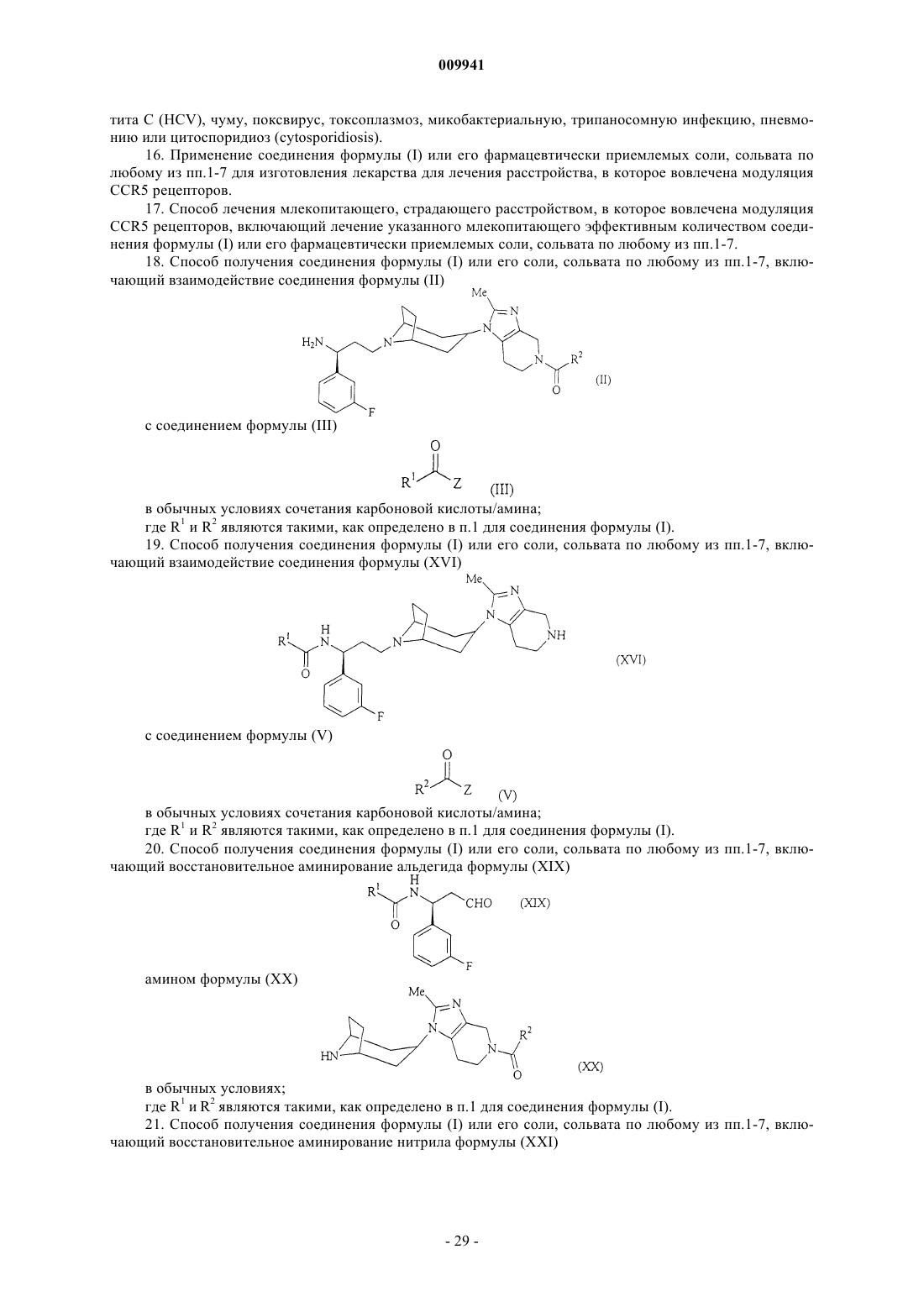
18. Способ получения соединения формулы (I) или его соли, сольвата по любому из пп.1-7, включающий взаимодействие соединения формулы (II)

с соединением формулы (III)

в обычных условиях сочетания карбоновой кислоты/амина;
где R1 и R2 являются такими, как определено в п.1 для соединения формулы (I).
19. Способ получения соединения формулы (I) или его соли, сольвата по любому из пп.1-7, включающий взаимодействие соединения формулы (XVI)

с соединением формулы (V)

в обычных условиях сочетания карбоновой кислоты/амина;
где R1 и R2 являются такими, как определено в п.1 для соединения формулы (I).
20. Способ получения соединения формулы (I) или его соли, сольвата по любому из пп.1-7, включающий восстановительное аминирование альдегида формулы (XIX)

амином формулы (XX)

в обычных условиях;
где R1 и R2 являются такими, как определено в п.1 для соединения формулы (I).
21. Способ получения соединения формулы (I) или его соли, сольвата по любому из пп.1-7, включающий восстановительное аминирование нитрила формулы (XXI)

амином формулы (XX)

в обычных условиях;
где R1 и R2 являются такими, как определено в п.1 для соединения формулы (I).
22. Способ получения соединения формулы (I) или его соли, сольвата по любому из пп.1-7, включающий алкилирование амина формулы (XX)

или его соли соединением формулы (XXII)

в обычных условиях алкилирования;
где R1 и R2 являются такими, как определено в п.1 для соединения формулы (I), и Lg представляет собой уходящую группу, подходящую для алифатического нуклеофильного замещения.
23. Способ получения соединения формулы (I) или его соли, сольвата по любому из пп.1-7, включающий асимметрическое восстановление енамида формулы (XXIII)

в обычных восстанавливающих условиях;
где R1 и R2 являются такими, как определено в п.1 для соединения формулы (I).
24. Способ получения соединения формулы (I) или его соли, сольвата по любому из пп.1-7, включающий взаимодействие амина формулы (II)

или его соли с металлом (то есть депротонированной формы) со сложным эфиром формулы (XXIV)

в обычных условиях;
где R1 и R2 являются такими, как определено в п.1 для соединеэшя формулы (I), и EsGp представляет собой группу, образующую сложный эфир.
25. Комбинация соединения по любому из пп.1-7 или его фармацевтически приемлемых соли или сольвата с одним или более чем одним дополнительным терапевтическим агентом.
26. Комбинация по п.25, где дополнительный терапевтический агент или терапевтические агенты выбраны из агентов, полезных в лечении заболеваний, опосредованных модуляцией CCR5 рецептора или ассоциированных с модуляцией CCR5 рецептора.
27. Комбинация по п.26, где дополнительный терапевтический агент или терапевтические агенты выбраны из агентов, полезных в лечении ВИЧ.
28. Комбинация по любому из пп.25-27, где дополнительный терапевтический агент или терапевтические агенты выбраны из ингибиторов ВИЧ-протеазы, ненуклеозидных ингибиторов обратной транскриптазы, нуклеозидных/нуклеотидных ингибиторов обратной транскриптазы, других CCR5 антагонистов, агентов, которые ингибируют взаимодействие gp120 с CD4, других агентов, которые ингибируют проникновение ВИЧ в клетку-мишень, ингибиторов интегразы и ингибиторов РНКазы H.
Текст
009941 Данное изобретение относится к тропановым производным, к способам их получения, к содержащим их композициям и к их применению. Более конкретно, настоящее изобретение относится к применению 8-азабицикло[3.2.1]октановых производных в лечении ряда расстройств, включающих расстройства, в которые вовлечена модуляция, в частности антагонизм, в отношении хемокиновых CCR5 рецепторов. Соответственно, соединения по данному изобретению полезны в лечении вируса иммунодефицита человека (ВИЧ), такого как ВИЧ-1, и генетически родственных ретровирусных инфекций (и возникающего в результате синдрома приобретенного иммунодефицита, СПИД) и воспалительных заболеваний. Название "хемокин" представляет собой сокращение от "хемотаксических цитокинов". Хемокины включают крупное семейство белков, которые имеют в общем важные структурные признаки и которые обладают способностью привлекать лейкоциты. В качестве хемотаксических факторов для лейкоцитов хемокины играют незаменимую роль в привлечении лейкоцитов в различные ткани организма, что представляет собой процесс, присущий как воспалению, так и реакции организма на инфекцию. Поскольку хемокины и их рецепторы находятся в центре патофизиологии воспаления и инфекционных заболеваний,агенты, которые обладают активностью модулирования, предпочтительно антагонизации, в отношении активности хемокинов и их рецепторов, являются полезными в терапевтическом лечении таких воспалительных и инфекционных заболеваний. Хемокиновый рецептор CCR5 является особенно важным в контексте лечения воспалительных и инфекционных заболеваний. CCR5 представляет собой рецептор для хемокинов, в частности для макрофагальных воспалительных белков (MIP), обозначаемых MIP-1 и MIP-1, и для белка, который регулируется активацией и экспрессируется и секретируется нормальными Т-клетками (regulated upon activationand is normal T-cell expressed and secreted (RANTES. Авторы изобретения обнаружили группу соединений, которые представляют собой сильные и селективные модуляторы, в частности антагонисты CCR5 рецептора. В соответствии с первым аспектом настоящего изобретения предложено соединение формулы (I) или его фармацевтически приемлемые соль, сольват или производное, гдеR2 представляет собой С 1-С 6 алкил или С 3-С 7 циклоалкил, где указанный алкил возможно замещен Термин "алкил" в качестве группы или части группы включает неразветвленные и разветвленные группы. Примеры алкила включают метил, этил, н-пропил, изопропил, н-бутил, изобутил, втор-бутил и трет-бутил. Термин "С 3-С 7 циклоалкил" означает циклопропил, циклобутил, циклопентил, циклогексил или циклогептил. В первом воплощении R1 представляет собой С 1-С 4 алкил. В еще одном воплощении R1 представляет собой метил. В еще одном воплощении R2 представляет собой С 1-С 4 алкил, возможно замещенный CF3. В еще одном воплощении R2 представляет собой циклопропил или циклобутил. В еще одном воплощении R2 представляет собой метил, этил или изопропил. Следует понимать, что данное изобретение охватывает все комбинации воплощений изобретения,как описано выше, соответствующие определению соединений формулы (I). Соединения по данному изобретению включают соединения формулы (I) и их фармацевтически приемлемые соли, сольваты или производные (где производные включают комплексы, пролекарства и изотопно-меченые соединения, а также их соли и сольваты). В еще одном воплощении соединения по данному изобретению представляют собой соединения формулы (I) и их фармацевтически приемлемые соли и сольваты, в частности соединения формулы (I). Следует понимать, что вышеупомянутые соединения по данному изобретению включают их полиморфные формы и изомеры. Фармацевтически приемлемые соли соединений формулы (I) включают их соли присоединения кислоты и основные соли. Подходящие соли присоединения кислоты получают из кислот, которые образуют нетоксичные соли. Примеры включают соли ацетат, аспартат, бензоат, безилат, бикарбонат, бисульфат, борат, бромид,камзилат, карбонат, хлорид, цитрат, эдизилат, эзилат, формиат, фумарат глюцептат, глюконат, глюкуронат, гексафторфосфат, гибензат, гидробромид, гидрохлорид, гидройодид, йодид, изэтионат, лактат, малат, малеат, малонат, мезилат, метилсульфат, нафтилат, 2-напсилат, никотинат, нитрат, оротат, оксалат,пальмитат, памоат, фосфат/гидрофосфат/дигидрофосфат, сахарат, стеарат, сукцинат, сульфат, тартрат,-1 009941 тозилат и трифторацетат. Подходящие основные соли получают из оснований, которые образуют нетоксичные соли. Примеры включают соли алюминия, аргинина, бензатина, кальция, холина, диэтиламина, диоламина, глицина,лизина, магния, меглумина, оламина, калия, натрия, трометамина и цинка. Обзор подходящих солей смотри в "Handbook of Pharmaceutical Salts: Properties, Selection, and Use"by Stahl and Wermuth (Wiley-VCH, Weinheim, Germany, 2002). Фармацевтически приемлемые соли соединений формулы (I) могут быть получены одним или более чем одним из трех способов:(1) путем взаимодействия соединения формулы (I) с желаемой кислотой;(2) путем удаления неустойчивой в кислоте или щелочи защитной группы с подходящего предшественника соединения формулы (I) или путем раскрытия кольца подходящего циклического предшественника, например лактона или лактама, с использованием желаемой кислоты; или(3) путем превращения одной соли соединения формулы (I) в другую посредством взаимодействия с подходящей кислотой или при помощи подходящей ионообменной колонки. Все три взаимодействия, как правило, осуществляют в растворе. Соль можно осадить из раствора и собрать путем фильтрования, или ее можно извлечь путем выпаривания растворителя. Степень ионизации соли может варьировать от полностью ионизированной до почти неионизированной. Соединения по данному изобретению могут существовать как в несольватированной, так и сольватированной формах. Термин "сольват" использован здесь для описания молекулярного комплекса, включающего соединение по данному изобретению и одну или более чем одну молекулу фармацевтически приемлемого растворителя, например этанола. Термин "гидрат" использован в том случае, когда указанный растворитель представляет собой воду. Комплексы включают клатраты, то есть комплексы включения лекарство-носитель, где, в противоположность вышеупомянутым сольватам, лекарство и носитель представлены в стехиометрических или нестехиометрических количествах. Также включены комплексы фармацевтического лекарства, которые содержат два или более чем два органических и/или неорганических компонента, которые могут быть представлены в стехиометрических или нестехиометрических количествах. Получающиеся в результате комплексы могут быть ионизированы, частично ионизированы или неионизированы. Обзор таких комплексов смотри в J. Pharm Sci, 64 (8), 1269-1288 by Haleblian (August 1975). Соединения по настоящему изобретению могут обладать способностью кристаллизоваться в виде более чем одной формы, представляя свойство, известное как полиморфизм, и все такие полиморфные формы ("полиморфы") включены в объем данного изобретения. Полиморфизм, как правило, может возникать как реакция на изменения температуры или давления или обоих факторов, и также может являться следствием вариаций процесса кристаллизации. Полиморфы можно различить по различным физическим характеристикам, и, как правило, для различения полиморфов используют картины дифракции рентгеновских лучей на порошке, свойства растворимости и температуру плавления данного соединения. Некоторые производные соединений формулы (I), которые сами могут обладать небольшой фармакологической активностью или не обладать ей, при введении в организм или на поверхность организма могут превращаться в соединения формулы (I), обладающие желаемой активностью, например, путем гидролитического расщепления. Такие производные называют "пролекарствами". Дополнительную информацию по использованию пролекарств можно найти в "Pro-drugs as Novel Delivery Systems", Vol. 14,ACS Symposium Series (T. Higuchi and W. Stella) и "Bioreversible Carriers in Drug Design", Pergamon Press,1987 (ed. E. B. Roche, American Pharmaceutical Association). Пролекарства в соответствии с изобретением могут быть получены, например, путем замещения соответствующих функциональных групп, представленных в соединениях формулы (I), определенными группировками, известными специалистам в данной области техники как "прогруппировки" в соответствии с тем, как описано, например, в "Design of Prodrugs" by H Bundgaard (Elsevier, 1985). Поскольку соединения формулы (I) содержат функциональную группу вторичного амина (-NHR,где R не представляет собой H), примеры пролекарств в соответствии с данным изобретением включают их амиды, получаемые, например, путем замещения водорода (С 1-С 10)алканоилом. Дополнительные примеры замещающих групп в соответствии с приведенными выше примерами и примеры других типов пролекарств в соответствии с данным изобретением могут быть найдены в вышеупомянутых источниках информации. Кроме того, некоторые соединения формулы (I) сами могут действовать в качестве пролекарств других соединений формулы (I). Кроме того, в объем изобретения также включены метаболиты соединений формулы (I), то есть соединения, образующиеся in vivo после введения лекарства. Некоторые примеры метаболитов в соответствии с данным изобретением включают:(1) когда соединение формулы (I) содержит метильную группу, его гидроксиметильное производное (-СН 3 -СН 2 ОН);(2) когда соединение формулы (I) содержит третичную аминогруппу, его вторичное аминопроиз-2 009941 водное (-NR1 R2-NHR1 или -NHR2);(3) когда соединение формулы (I) содержит вторичную аминогруппу, его первичное производное(4) когда соединение формулы (I) содержит фенильную группировку, его фенольное производное(5) когда соединение формулы (I) содержит амидогруппу, его производное карбоновой кислоты(-CONH2COOH). Соединения формулы (I) могут содержать один или более чем один дополнительный асимметрический атом углерода и поэтому существовать в виде двух или более чем двух стереоизомеров. Имидазольное замещение тропанового кольца в соединениях формулы (I) может иметь либо эндо-, либо экзоконфигурацию и, следовательно, возможны геометрические цис/транс (или Z/E) изомеры. Когда соединение содержит, например, кетогруппу, может возникать таутомерная изомерия ("таутомерия"). Это приводит к тому, что одно соединение может демонстрировать более чем один тип изомерии. В объем настоящего изобретения включены все стереоизомеры соединений формулы (I), включающие все оптические изомеры, геометрические изомеры и таутомерные формы, а также соединения, демонстрирующие более чем один тип изомерии, и смеси одного или более чем одного из них. Также включены соли присоединения кислоты или основания, где противоион является оптически активным,например D-лактат или L-лизин, или рацемическим, например DL-тартрат или DL-аргинин. Имидазольное замещение тропанового кольца в эндо-конфигурации является предпочтительным. Эндо/экзо изомеры могут быть разделены обычными способами, хорошо известными специалистам в данной области техники, например при помощи хроматографии и фракционной кристаллизации. Обычные способы получения/выделения индивидуальных энантиомеров включают хиральный синтез из подходящего оптически чистого предшественника или разделение рацемата (или рацемата соли или производного) с использованием, например, хиральной жидкостной хроматографии высокого давления (ЖХВД). Альтернативно, рацемат (или рацемический предшественник) может быть подвергнут взаимодействию с подходящим оптически активным соединением, например спиртом, или, в случае когда соединение формулы (I) содержит кислотную или основную группировку, кислотой или основанием, таким как винная кислота или 1-фенилэтиламин. Получающаяся в результате диастереомерная смесь может быть разделена путем хроматографии и/или фракционной кристаллизации, и один или оба диастереоизомера преобразованы в соответствующий(е) чистый(е) энантиомер(ы) способами, хорошо известными специалистам в данной области техники. Хиральные соединения по данному изобретению (и их хиральные предшественники) могут быть получены в энантиомерно-обогащенной форме с использованием хроматографии, как правило ЖХВД, на асимметрической смоле с использованием подвижной фазы, состоящей из углеводорода, как правило гептана или гексана, содержащего от 0 до 50% изопропанола, как правило от 2 до 20%, и от 0 до 5% алкиламина, как правило 0,1% диэтиламина. Концентрирование элюата дает обогащенную смесь. Стереоизомерные конгломераты могут быть разделены обычными способами, известными специалистам в данной области техники, смотри, например, "Stereochemistry of Organic Compounds" by E. L.Eliel (Wiley, New York, 1994). Настоящее изобретение также включает все фармацевтически приемлемые меченные изотопами соединения формулы (I), где один или более чем один атом замещен атомами, имеющими тот же атомный номер, но атомную массу или массовое число, отличающиеся от атомной массы или массового числа,обычно обнаруживаемых в природе. Примеры изотопов, подходящих для включения в соединения по данному изобретению, включают изотопы водорода, такие как 2H и 3H, углерода, такие как 11 С, 13C и 14C, хлора, такие как 36Cl, фтора, такие как 18F, йода, такие как 123I и 125I, азота, такие как 13N и 15N, кислорода, такие как 15O, 17O и 18O, фосфора, такие как 32P, и серы, такие как 35S. Некоторые меченные изотопами соединения формулы (I), например соединения, включающие радиоактивный изотоп, полезны в исследованиях распределения лекарства и/или субстрата в тканях. Для этой цели особенно подходят радиоактивные изотопы трития, то есть 3H, и углерода-14, то сеть 14C, ввиду легкости их включения и простых способов их обнаружения. Замещение более тяжелыми изотопами, такими как дейтерий, то есть H, может обеспечить некоторые терапевтические преимущества, являющиеся результатом большей метаболической стабильности,например увеличения периода полувыведения in vivo или уменьшенной потребности в дозах, и, следовательно, в некоторых случаях может быть предпочтительным. Замещение позитрон-излучающими изотопами, такими как 11 С, 18F, 15O и 13N, может быть полезно в исследованиях при помощи позитронной эмиссионной томографии (PET) для определения оккупации рецептора субстратом. Меченные изотопами соединения формулы (I), как правило, могут быть получены обычными способами, известными специалистам в данной области техники, или способами, аналогичными способам,-3 009941 описанным в сопутствующих примерах и подготовительных примерах, с использованием подходящего изотопно-меченого реагента вместо используемого ранее немеченого реагента. Фармацевтически приемлемые сольваты в соответствии с данным изобретением включают сольваты, где кристаллизационный растворитель может быть замещен изотопом, например D2O, d6-ацетон, d6 диметилсульфоксид (ДМСО). Предпочтительные соединения формулы (I) включают соединения примеров 1-8 и их фармацевтически приемлемые соли, сольваты или производные. В следующих общих способах и схемах: если не указано иного, R1 и R2 являются такими, как определено ранее; Z представляет собой ОН или группу, активирующую группу карбоновой кислоты, такую как хлоро- или 1H-имидазол-1-ил; EsGp представляет собой группу, образующую сложный эфир, такую как С 1-6 алкил; Pg представляет собой защитную группу амино, такую как boc; ArLg представляет собой уходящую группу, подходящую для ароматического нуклеофильного замещения, такую как группы,описанные в Jerry March, Advanced Organic Chemistry (4th edition), Wiley Interscience, 1992, стр. 652(включенной в данное описание путем ссылки), например F, Cl, Br, OMe или OEt; boc представляет собой трет-бутоксикарбонил; ДМФ представляет собой N,N-диметилформамид; ДХМ представляет собой дихлорметан; ТГФ представляет собой тетрагидрофуран; Lg представляет собой уходящую группу, подходящую для алифатического нуклеофильного замещения, такую как группы, описанные в Jerry March,там же, стр. 352 (включенной в данное описание путем ссылки), в том числе Cl, Br, I и сульфоновые сложные эфиры (например, тозилат, мезилат и трифлат); WSCDI представляет собой гидрохлорид 1-(3 диметиламинопропил)-3-этилкарбодиимида; DCC представляет собой N,N'-дициклогексилкарбодиимид;HOAT представляет собой 1-гидрокси-7-азабензотриазол; HOBt представляет собой гидрат 1-гидроксибензотриазола; HBTU представляет собой гексафторфосфат O-бензотриазол-1-ил-N,N,N',N'-тетраметилурония. В соответствии с первым способом (А) соединения формулы (I) могут быть получены путем взаимодействия соединения формулы (II) в обычных условиях сочетания карбоновой кислоты/амина. Для удобства данное сочетание осуществляют в условиях, описанных ранее в отношении схемы 1, стадия (1). В соответствии со вторым способом (Б) соединения формулы (I) могут быть получены путем взаимодействия соединения формулы (XVI) в обычных условиях сочетания карбоновой кислоты/амина. Для удобства данное сочетание осуществляют в условиях, описанных ранее в отношении схемы 1a, стадия (1). В соответствии с третьим способом (В) соединения формулы (I) могут быть получены путем восстановительного аминирования альдегида формулы (XIX) в обычных условиях. Для удобства восстановительное аминирование осуществляют в условиях,описанных ранее в отношении схемы 1, стадия (ж). В соответствии с четвертым способом (Г) соединения формулы (I) могут быть получены путем восстановительного аминирования нитрила формулы (XXI) амином формулы (XX) в обычных условиях. Для удобства восстановительное аминирование осуществляют в условиях, описанных ранее в отношении схемы 1, стадия (ж). В соответствии с пятым способом (Д) соединения формулы (I) могут быть получены путем алкилирования амина формулы (XX) соединением формулы (XXII) в обычных условиях алкилирования. Для удобства алкилирование осуществляют в подходящем растворителе, таком как галогеноалкан (например, ДХМ), спирт (например, этанол) или простой эфир(например ТГФ), возможно в присутствии основания, такого как амин (например, триэтиламин или Nэтил-N,N-диизопропиламин); и при температуре от температуры окружающей среды до повышенной температуры (например, температура дефлегмации). В соответствии с еще одним способом (E) соединения формулы (I) могут быть получены путем асимметрического восстановления енамида формулы (XXIII) в обычных условиях восстановления. Для удобства асимметрическое восстановление осуществляют в присутствии переходного металла, такого как Rh, Ru, Pd, Pt, Ir или Ti (например, представленного в количестве 0,001-0,1 моль экв.), хирального лиганда, такого как BINAP (2,2-бис(дифенилфосфино)-1,1'бинафтил), tol-BINAP (2,2-бис(дипаратолилфосфино)-1,1'-бинафтил), Du-PHOS (1,2-бис(2,5-диметилфосфолано)бензол) или Penn-Phos (Р,Р'-1,2-фениленбис(эндо-2,5-диметил-7-фосфабицикло[2.2.1]гептан)(например, представленного в количестве 0,001-0,2 моль экв.), донора водорода, такого как молекулярный водород, фенилсилан, 2-пропанол или формиат аммония, в растворителе, таком как спирт (например, метанол, этанол или 2-пропанол), нитрил (например, ацетонитрил), сложный эфир (например этилацетат), простой эфир (например ТГФ) или толуол, при температуре от 0C до температуры дефлегмации, и возможно при повышенном давлении. В соответствии с еще одним способом (Ж) соединения формулы (I) могут быть получены из амина формулы (II) или его соли с металлом (то есть депротонированная форма) путем взаимодействия со сложным эфиром формулы (XXIV) в обычных условиях. Для удобства реакцию осуществляют в присутствии избытка основания, такого как амин (например, триэтиламин или N-этил-N,N-диизопропиламин), возможного катализатора, в-5 009941 подходящем растворителе, таком как галогеноалкан (например, ДХМ), простой эфир (например, ТГФ),сложный эфир (например, этилацетат) или толуол, в присутствии воды в качестве сорастворителя или без нее. Альтернативно, взаимодействие осуществляют в присутствии ферментативного катализатора в подходящем растворителе, таком как галогеноалкан (например ДХМ), простой эфир (например, ТГФ),сложный эфир (например, этилацетат) или толуол, в присутствии воды в качестве сорастворителя или без нее. Схемы, которые дополнительно иллюстрируют общие способы получения соединений формулы (I) и их промежуточных соединений, следуют далее. Специалисту в данной области техники понятно, что некоторые из методик, описанных в схемах получения соединений формулы (I) или их промежуточных соединений, могут быть неприменимы в отношении некоторых из возможных заместителей. Специалисту в данной области техники также понятно, что может быть необходимо или желательно осуществить описанные в схемах превращения в последовательности, отличающейся от описанной,или модифицировать одно или более чем одно превращение для получения желаемого соединения формулы (I). Специалисту в данной области техники также понятно, что в соответствии с проиллюстрированным на следующих ниже схемах, может быть необходимо или желательно на какой-либо стадии синтеза соединений формулы (I) защищать одну или более чем одну чувствительную группу в молекуле для того,чтобы предотвратить нежелательные побочные реакции. В частности, может быть необходимо или желательно защищать аминогруппы. Защитные группы, применяемые при получении соединений формулы(I), могут быть использованы обычным образом. Смотри, например, группы, описанные в "ProtectiveGroups in Organic Synthesis" by Theodora W. Green and Peter G M Wuts, third edition, (John Wiley and Sons,1999), в частности главу 7, стр. 494-653 ("Protection for the Amino Group"), включенной в данное описание путем ссылки, где также описаны способы удаления таких групп. При получении соединений формулы (I) и их промежуточных соединений используют, в частности,защитные группы амино boc, бензилоксикарбонил, метоксикарбонил, бензил и ацетил. Схема 1-6 009941 С конкретной ссылкой на схему 1 изображенные на ней превращения могут быть осуществлены следующим образом:(а) Замещение уходящей группы на нитропиридине формулы (XV) амином формулы (XIV) для удобства осуществляют в присутствии основания, такого как амин (например, триэтиламин или N-этилN,N-диизопропиламин) или карбонат щелочного металла (например, карбонат натрия или карбонат калия), в растворителе, таком как спирт (например, метанол или этанол), нитрил (например, ацетонитрил) или амид (например ДМФ), и при температуре от температуры окружающей среды до повышенной температуры (например вплоть до приблизительно 120C).(б) Имидазопиридин формулы (XII) может быть получен путем восстановления и циклизации in situ аминонитропиридина формулы (XIII). Восстановление для удобства осуществляют в присутствии восстановителя, такого как железный порошок, в растворителе, таком как карбоновая кислота (например,уксусная кислота), и при температуре от температуры окружающей среды вплоть до приблизительно 120C. Циклизацию промежуточного амино-аминопиридина для удобства осуществляют путем добавления уксусного ангидрида и при повышенной температуре (например, приблизительно 140C).(в) Восстановление имидазопиридина формулы (XII) до имидазопиперидина формулы (XI) для удобства осуществляют путем каталитического гидрирования в присутствии подходящего катализатора,такого как катализатор переходного металла, например платиновый (например, оксид платины) или палладиевый (например, гидроксид палладия или палладий на углероде) катализатор, в растворителе, таком как спирт (например, метанол или этанол) или карбоновая кислота (например, уксусная кислота), при температуре от температуры окружающей среды до повышенной температуры (например, вплоть до 80C) и при повышенном давлении, таком как 150-500 кПа водорода (например, 400 кПа водорода).(г) Имидазопиперидин формулы (XI) может быть защищен путем осуществления взаимодействия с алкилхлорформиатом (например метилхлорформиатом). Это взаимодействие для удобства осуществляют в растворителе, таком как галогеноалкан (например ДХМ) или простой эфир (например ТГФ), с использованием основания, такого как амин (например триэтиламин или N-этил-N,N-диизопропиламин), при комнатной температуре.(д) В качестве альтернативы стадиям (в) и (г) имидазопиридин формулы (XII) обрабатывают алкилхлорформиатом (например, метилхлорформиатом) и аминооснованием (например, триэтиламином илиN-этил-N,N-диизопропиламином) с получением четвертичного промежуточного соединения, которое восстанавливают в обычных условиях. Для удобства метилхлорформиат добавляют к имидазопиридину формулы (XII) в присутствии растворителя, такого как простой эфир (например, ТГФ) или галогеноалкан(например, ДХМ), и при температуре окружающей среды с получением четвертичного промежуточного соединения, которое затем восстанавливают путем добавления галогенида щелочного металла, такого как боргидрид лития, в условиях пониженной температуры (например, приблизительно -70C) Получающееся в результате промежуточное соединение затем восстанавливают путем каталитического гидрирования в присутствии подходящего катализатора, такого как катализатор переходного металла, например палладиевый (например, гидроксид палладия или палладий на углероде) катализатор, в растворителе, таком как спирт (например, метанол или этанол) или карбоновая кислота (например, уксусная кислота), при температуре от температуры окружающей среды до повышенной температуры (например,вплоть до 80C).(е) Когда защитная группа представляет собой защитную группу ацетила или подобную группу, ее удаление для удобства осуществляют путем обработки основанием, таким как гидроксид щелочного металла (например гидроксид натрия или калия), или кислотой, такой как неорганическая кислота (например HCl), и при повышенной температуре, такой как 60-100C.(ж) Соединения формулы (VII) получают путем восстановительного аминирования альдегида формулы (IX) амином формулы (VIII). Для удобства реакцию осуществляют в присутствии кислоты, такой как органическая кислота (например, уксусная кислота), в растворителе, таком как простой эфир (например, ТГФ) или галогеноалкан (например, ДХМ), с использованием гидрида щелочного металла в качестве восстановителя, такого как триацетоксиборгидрид натрия, цианоборгидрид натрия или боргидрид натрия, и при температуре окружающей среды.(з) Когда защиту на стадии (г) осуществляют при помощи метоксикарбонильной группы, ее удаление для удобства осуществляют путем обработки основанием, таким как гидроксид щелочного металла(например, гидроксид натрия или калия), в растворителе, таком как водный спирт (например,Н 2 О/пропан-2-ол), при повышенной температуре (например, 100C).(и) Сочетание кислоты/амина для удобства осуществляют с использованием амина формулы (VI) и хлорангидрида формулы (V), избытка акцептора кислоты, такого как триэтиламин или N-этил-N,Nдиизопропиламин, растворителя, такого как галогеноалкан (например, ДХМ) или простой эфир (например, ТГФ), и при температуре окружающей среды. Альтернативно, сочетание кислоты/амина осуществляют с использованием кислоты формулы (V),активированной реагентами, такими как WSCDI или DCC и HOBt или HOAt, избытка акцептора кислоты, такого как триэтиламин или N-этил-N,N-диизопропиламин, растворителя, такого как галогеноалкан(например, ДХМ) или простой эфир (например, ТГФ), и при температуре окружающей среды.(к) Когда защитная группа представляет собой защитную группу boc, ее удаление для удобства осуществляют в присутствии кислоты, такой как неорганическая кислота (например, безводная HCl) или трифторуксусная кислота, в подходящем растворителе, таком как сложный эфир (например, этилацетат),галогеноалкан (например, ДХМ) или простой эфир (например ТГФ), и при температуре от 0C до температуры окружающей среды.(л) Сочетание кислоты/амина для удобства осуществляют с использованием амина формулы (II) и кислоты или хлорангидрида формулы (III) в условиях, описанных на стадии (и) выше. Специалисту в данной области техники понятно, что одно или более превращений, описанных в схеме 1, могут быть осуществлены в последовательности, отличающейся от описанной последовательности, или могут быть модифицированы для получения желаемого соединения формулы (I). В одном из вариантов схемы 1 соединения формулы (I) могут быть получены путем осуществления стадий с (з) по (л) в другом порядке, как проиллюстрировано на следующей схеме 1 а. Схема 1a Соединения формулы (XX) представляют собой структурные аналоги соединений формулы (I) или их промежуточных соединений и могут быть получены с использованием аналогичных способов. Соединения формул (III), (V), (IX), (XV), (XIX), (XXI), (XXII) и (XXIII) либо представляют собой известные соединения, либо могут быть получены обычными химическими способами, смотри, например, WO 01/90106 (в особенности стр. 5-19), включенную в данное описание путем ссылки. Соединения формулы (I) и их фармацевтически приемлемые соли, сольваты и производные являются полезными, поскольку они обладают фармакологической активностью у животных, включая людей. Более конкретно, они полезны в лечении расстройства, в которое вовлечена модуляция CCR5 рецепторов. Болезненные состояния, представляющие особый интерес, включают ВИЧ, ретровирусные инфекции, генетически родственные ВИЧ, СПИД и воспалительные заболевания. Соединения по данному изобретению могут быть использованы для лечения респираторных расстройств, включая респираторный дистресс-синдром у взрослых (ARDS), бронхит, хронический бронхит,хроническую обструктивную болезнь легких, кистозный фиброз, астму, эмфизему, ринит и хронический синусит. Другие состояния, которые можно лечить, представляют собой состояния, запускаемые, зависящие или каким-либо иным образом связанные с направленной миграцией Т-клеток в различные органы. Полагают, что соединения по данному изобретению могут быть полезны для лечения таких состояний, и в частности состояний, для которых установлена взаимосвязь с CCR5 или хемокинами CCR5, но не ограниченных ими, и более конкретно следующих состояний: рассеянный склероз, артрит, такой как ревматоидный артрит, спондилоартропатии, подагрический артрит, остеоартрит, системная красная волчанка и ювенильный артрит и отторжение трансплантата, в частности трансплантатов целых органов, таких как-8 009941 трансплантаты сердца, легкого, печени, почки и поджелудочной железы (например, аллотрансплантаты почки и легкого), но не ограниченных ими. Другие состояния, для которых установлена подобная взаимосвязь с CCR5 или хемокинами CCR5, включают: воспалительное заболевание кишечника, включая болезнь Крона и неспецифический язвенный колит, эндометриоз, диабет I типа, почечные заболевания,такие как гломерулярное заболевание (например гломерулонефрит), фиброз, такой как печеночный, легочный и почечный фиброз, хронический панкреатит, воспалительные легочные состояния, энцефалит,такой как ВИЧ-энцефалит, хроническую сердечную недостаточность, ишемическую болезнь сердца,псориаз, удар, ожирение, болезни центральной нервной системы (ЦНС), такие как деменции, связанные со СПИДом, и болезнь Альцгеймера, анемию, рестеноз, атеросклеротические бляшки, атопический дерматит, хронический панкреатит, рак, такой как неходжкинская лимфома, саркома Капоши, меланома и рак молочной железы, боль, такую как ноцицептивная боль и невропатическая боль (например, периферическая невропатическая боль), и реакцию на стресс, являющуюся результатом хирургической операции, инфекции, повреждения или другого травматического поражения, но не ограничиваются ими. Инфекционные заболевания, в которые вовлечена модуляция CCR5 рецептора, включают острую и хроническую инфекцию вирусом гепатита В (HBV) и вирусом гепатита С (HCV), бубонную, первичносептическую и легочную чуму, поксвирусную инфекцию, такую как оспа, токсоплазмозную инфекцию,микобактериальную инфекцию, трипаносомную инфекцию, такую как болезнь Шагаса, пневмонию и цитоспоридиоз (cytosporidiosis). Современный обзор возможных применений хемокинов и блокаторов хемокиновых рецепторов смотри в Cascieri, M.A., and Springer, M.S., "The chemokine/chemokine receptor family: potential and progress for therapeutic intervention", Curr. Opin. Chem. Biol., 4(4), 420-7 (August 2000). Соответственно, в еще одном аспекте данного изобретения предложено соединение формулы (I) или его фармацевтически приемлемые соль, сольват или производное для применения в качестве лекарства. В еще одном аспекте данного изобретения предложено соединение формулы (I) или его фармацевтически приемлемые соль, сольват или производное для лечения расстройства, в которое вовлечена модуляция CCR5 рецепторов. В еще одном аспекте данного изобретения предложено соединение формулы (I) или его фармацевтически приемлемые соль, сольват или производное для лечения ВИЧ, ретровирусной инфекции, генетически родственной ВИЧ, СПИД или воспалительного заболевания. В еще одном аспекте данного изобретения предложено соединение формулы (I) или его фармацевтически приемлемые соль, сольват или производное для лечения респираторного расстройства, включая респираторный дистресс-синдром у взрослых (ARDS), бронхит, хронический бронхит, хроническую обструктивную болезнь легких, кистозный фиброз, астму, эмфизему, ринит или хронический синусит. В еще одном аспекте данного изобретения предложено соединение формулы (I) или его фармацевтически приемлемые соль, сольват или производное для лечения рассеянного склероза, ревматоидного артрита или отторжения трансплантата. В еще одном аспекте данного изобретения предложено соединение формулы (I) или его фармацевтически приемлемые соль, сольват или производное для лечения воспалительного заболевания кишечника, эндометриоза, диабета I типа, почечных заболеваний, фиброза, хронического панкреатита, воспалительных легочных состояний, энцефалита, хронической сердечной недостаточности, ишемической болезни сердца, псориаза, удара, ожирения, заболеваний центральной нервной системы (ЦНС), анемии,рестеноза, атеросклеротических бляшек, атопического дерматита, хронического панкреатита, рака, боли или реакции на стресс вследствие хирургического вмешательства, инфекции, повреждения или другого травматического поражения. В еще одном аспекте данного изобретения предложено соединение формулы (I) или его фармацевтически приемлемые соль, сольват или производное для лечения HBV, HCV, чумы, поксвируса, токсоплазмоза, микобактериальной, трипаносомной инфекции, пневмонии или цитоспоридиоза. В еще одном аспекте данного изобретения предложено применение соединения формулы (I) или его фармацевтически приемлемых соли, сольвата или производного для изготовления лекарства для лечения расстройства, в которое вовлечена модуляция CCR5 рецепторов. В еще одном аспекте данного изобретения предложен способ лечения расстройства у млекопитающего, в которое вовлечена модуляция CCR5 рецепторов, включающий лечение указанного млекопитающего эффективным количеством соединения формулы (I) или его фармацевтически приемлемых соли,сольвата или производного. Соединения по данному изобретению могут быть введены в виде кристаллических или аморфных продуктов. Они могут быть получены, например, в виде твердых кусочков, порошков или пленок с использованием способов, таких как осаждение, кристаллизация, сублимационная сушка, распылительная сушка или сушка выпариванием. Для этой цели может быть использована сушка микроволнами или сушка в диапазоне радиочастот. Соединения могут быть введены в отдельности или в комбинации с одним или более чем одним другим соединением по данному изобретению или в комбинации с одним или более чем одним другим-9 009941 лекарством (или в любой их комбинации). Как правило, их вводят в виде препарата в сочетании с одним или более чем одним фармацевтически приемлемым эксципиентом. Термин "эксципиент" использован здесь для описания любого ингредиента, отличного от соединения(ий) по данному изобретению. Выбор эксципиента будет в значительной степени зависеть от факторов, таких как конкретный способ введения,влияние эксципиента на растворимость и стабильность и природа лекарственной формы. Фармацевтические композиции, подходящие для доставки соединений по данному изобретению, и способы их изготовления легко понятны специалистам в данной области техники. Такие композиции и способы их изготовления могут быть найдены, например, в "Remington's Pharmaceutical Sciences", 19thEdition (Mack Publishing Company, 1995). Соединения по данному изобретению могут быть введены перорально. Пероральное введение может включать проглатывание, так что соединение попадает в желудочно-кишечный тракт, или может быть использовано трансбуккальное или подъязычное введение, посредством которого соединение попадает в кровоток непосредственно из полости рта. Препараты, подходящие для перорального введения, включают твердые препараты, такие как таблетки, капсулы, содержащие множество частиц, жидкости или порошки, лепешки (включая лепешки с жидким наполнителем), жевательные резинки, мульти- и наночастицы, гели, твердый раствор, липосому,пленки (включая пленки, адгезивные к слизистой оболочке), овули, спреи и жидкие препараты. Жидкие препараты включают суспензии, растворы, сиропы и эликсиры. Такие препараты могут быть использованы в качестве наполнителей в мягких или твердых капсулах и, как правило, включают носитель, например воду, этанол, полиэтиленгликоль, пропиленгликоль, метилцеллюлозу или подходящее масло, и один или более чем один эмульгатор и/или суспендирующий агент. Жидкие препараты также могут быть изготовлены путем разведения твердого вещества, например, из саше. Соединения по данному изобретению также могут быть использованы в быстрорастворимых, быстрораспадающихся лекарственных формах, таких как формы, описанные в Expert Opinion in TherapeuticPatents, 11 (6), 981-986 by Liang and Chen (2001). Для таблетированных лекарственных форм в зависимости от дозы лекарство может составлять от 0,1 до 80 мас.%, более типично от 1 до 60 мас.%, например от 5 до 50 мас.% относительно лекарственной формы. В дополнение к лекарству таблетки, как правило, содержат разрыхлитель. Примеры разрыхлителей включают крахмалгликолят натрия, натриевую карбоксиметилцеллюлозу, кальциевую карбоксиметилцеллюлозу, натриевую кроскармеллозу, кросповидон, поливинилпирролидон, метилцеллюлозу, микрокристаллическую целлюлозу, гидроксипропилцеллюлозу, замещенную низшим алкилом, крахмал,прежелатинизированный крахмал и альгинат натрия. Как правило, разрыхлитель составляет от 0,1 до 25 мас.%, более типично от 0,5 до 20 мас.%, например от 1 до 15 мас.% относительно лекарственной формы. Связывающие вещества, как правило, используют для придания таблетированному препарату когезионных качеств. Подходящие связывающие вещества включают микрокристаллическую целлюлозу,желатин, сахара, полиэтиленгликоль, природные и синтетические смолы, поливинилпирролидон, прежелатинизированный крахмал, гидроксипропилцеллюлозу и гидроксипропилметилцеллюлезу. Таблетки также могут содержать разбавители, такие как лактозу (моногидрат, высушенный распылением моногидрат, безводный и тому подобное), маннит, ксилит, декстрозу, сахарозу, сорбит, микрокристаллическую целлюлозу, крахмал, карбонат кальция и дигидрат двухосновного фосфата кальция. Таблетки также могут возможно включать поверхностно-активные агенты, такие как лаурилсульфат натрия и полисорбат 80, и скользящие вещества, такие как диоксид кремния и тальк. Если присутствуют поверхностно-активные агенты, они могут составлять от 0,2 до 5 мас.% от массы таблетки, и скользящие вещества могут составлять от 0,2 до 1 мас.% от массы таблетки. Таблетки также, как правило, содержат смазывающие вещества, такие как стеарат магния, стеарат кальция, стеарат цинка, стеарилфумарат натрия и смеси стеарата магния с лаурилсульфатом натрия. Смазывающие вещества, как правило, составляют от 0,25 до 10 мас.%, предпочтительно от 0,5 до 3 мас.% от массы таблетки. Другие возможные ингредиенты включают антиоксиданты, красители, корригенты, консерванты и агенты, маскирующие вкус. Примеры таблеток содержат приблизительно до 80% лекарства, приблизительно от 10 до приблизительно 90 мас.% связывающего вещества, приблизительно от 0 мас.% до приблизительно 85 мас.% разбавителя, приблизительно от 1 мас.% до приблизительно 10 мас.% разрыхлителя и приблизительно от 0,25 мас.% до приблизительно 10 мас.% смазывающего вещества. Таблетируемые смеси могут быть спрессованы непосредственно или при помощи роллера с получением таблеток. Таблетируемые смеси или части смесей альтернативно могут быть гранулированы путем влажной или сухой грануляции или грануляции из расплава, могут быть подвергнуты застыванию после плавления или подвергнуты экструдированию перед таблетированием. Конечный препарат может включать один или более чем один слой и может быть покрыт или не покрыт оболочкой, он даже может быть инкапсулирован. Приготовление таблеток обсуждается в Pharmaceutical Dosage Forms: Tablets, Vol. 1", by H. Lieber- 10009941man and L. Lachman, Marcel Dekker, N.Y., N.Y., 1980 (ISBN 0-8247-6918-X). Твердые препараты для перорального введения могут быть приготовлены для немедленного и/или модифицированного высвобождения. Препараты с модифицированным высвобождением включают замедленное, длительное, прерывистое, контролируемое, направленное и программируемое высвобождение. Подходящие препараты с модифицированным высвобождением для задач в соответствии с данным изобретением описаны в патенте США 6106864. Подробная информация о других подходящих способах высвобождения, таких как высокоэнергетические дисперсии и осмотические и покрытые оболочкой частицы, может быть найдена в Verma et al, Pharmaceutical Technology On-line, 25(2), 1-14 (2001). Применение жевательной резинки для достижения контролируемого высвобождения описано в WO 00/35298. Соединения по данному изобретению также могут быть введены непосредственно в кровоток, в мышцу или во внутренний орган. Подходящие способы парентерального введения включают внутривенное, внутриартериальное, внутрибрюшинное, интратекальное, внутрисуставное, внутриуретральное,внутригрудинное, внутричерепное, внутримышечное и подкожное введение. Подходящие устройства для парентерального введения включают игольные (включая микроигольные) инъекторы, безыгольные инъекторы и средства для инфузии. Препараты для парентерального введения, как правило, представляют собой водные растворы, которые могут содержать эксципиенты, такие как соли, углеводороды и буферы (предпочтительно до рН от 3 до 9), но для некоторых применений они могут быть приготовлены более подходящим образом в виде стерильного неводного раствора или в виде высушенной формы, используемой в сочетании с подходящим разбавителем, таким как стерильная апирогенная вода. Приготовление препаратов для парентерального введения в стерильных условиях, например путем лиофилизации, может быть легко осуществлено с использованием стандартных фармацевтических способов, хорошо известных специалистам в данной области техники. Растворимость соединений по данному изобретению, используемых в приготовлении растворов для парентерального введения, может быть увеличена путем использования подходящих способов приготовления, таких как включение агентов, увеличивающих растворимость. Препараты для парентерального введения могут быть приготовлены для немедленного и/или модифицированного высвобождения. Препараты с модифицированным высвобождением включают замедленное, длительное, прерывистое, контролируемое, направленное и программируемое высвобождение. Таким образом, соединения по данному изобретению могут быть приготовлены в виде твердой, полутвердой формы или тиксотропной жидкости для введения в виде имплантируемого депо-препарата,обеспечивающего модифицированное высвобождение соединения. Примеры таких препаратов включают покрытые лекарством стенты и микросферы поли(DL-молочной/гликолевой кислоты) (PGLA). Соединения по данному изобретению также могут быть введены местно на кожу или слизистые оболочки, то есть дермально или транедермально. Типичные препараты для этой цели включают гели,гидрогели, лосьоны, растворы, кремы, мази, присыпки, повязки, мыла, пленки, кожные пластыри, облатки, имплантаты, губки, волокна, бинты и микроэмульсии. Также могут быть использованы липосомы. Типичные носители включают спирт, воду, минеральное масло, вазелиновое масло, медицинский вазелин, глицерин, полиэтиленгликоль и пропиленгликоль. Могут быть включены агенты, усиливающие проникновение, смотри, например, J Pharm Sci, 88 (10), 955-958 by Finnin and Morgan (October 1999). Другие способы местного введения включают доставку путем электропорации, ионофорез, фонофорез, сонофорез и микроигольную или безыгольную (например, Powderject, Bioject и другие) инъекцию. Препараты для местного введения могут быть приготовлены для немедленного и/или модифицированного высвобождения. Препараты с модифицированным высвобождением включают замедленное,длительное, прерывистое, контролируемое, направленное и программируемое высвобождение. Соединения по данному изобретению также могут быть введены интраназально или путем ингаляции, как правило, в форме сухого порошка (самого по себе, в виде смеси, например в сухой смеси с лактозой, или в виде смешанного компонента в форме частиц, например смешанного с фосфолипидами, такими как фосфатидилхолин) из сухого порошкового ингалятора или в виде аэрозольного спрея из контейнера под давлением, насоса, спрея, пульверизатора (предпочтительно пульверизатора, использующего принципы электродинамики для получения мелкодисперсного тумана), или распылителя, с использованием подходящего пропеллента, такого как 1,1,1,2-тетрафторэтан или 1,1,1,2,3,3,3-гептафторпропан,или без него. Для интраназального применения порошок может включать биоадгезивный агент, например хитозан или циклодекстрин. Контейнер под давлением, насос, спрей, пульверизатор или распылитель содержит раствор или суспензию соединения, включающую, например, этанол (возможно водный этанол) или подходящий альтернативный агент для диспергирования, солюбилизации или пролонгирования высвобождения соединения, пропеллент(ы) в качестве растворителя и возможное поверхностно-активное вещество, такое как сорбитантриолеат, олеиновая кислота или олигомолочная кислота. Перед применением в препарате в виде сухого порошка или суспензии лекарство микронизируют- 11009941 до размера, подходящего для доставки путем ингаляции (как правило, менее чем 5 микрон). Это может быть достигнуто при помощи любого подходящего способа дробления, такого как размол на спиральной струйной мельнице, размол на струйной мельнице с псевдоожиженным слоем, сверхкритическая жидкостная обработка с получением наночастиц, гомогенизация под высоким давлением или сушка распылением. Капсулы (приготовленные, например, из желатина или гидроксипропилметилцеллюлозы (HPMC,блистеры и картриджи для применения в ингаляторе или инсуффляторе могут быть приготовлены таким образом, чтобы содержать порошкообразную смесь соединения по данному изобретению, подходящей порошкообразной основы, такой как лактоза или крахмал, и модификатора характеристик, такого как Lлейцин, маннит или стеарат магния. Лактоза может быть безводной или находиться в форме моногидрата, причем последняя является предпочтительной. Другие подходящие эксципиенты включают декстран,глюкозу, мальтозу, сорбит, ксилит, фруктозу, сахарозу и трегалозу. Подходящий препарат в виде раствора для применения в пульверизаторе с использованием принципов электродинамики для получения мелкодисперсного тумана может содержать от 1 мкг до 20 мг соединения по данному изобретению на одно срабатывание, и объем срабатывания может варьировать от 1 до 100 мкл. Типичный препарат может включать соединение по данному изобретению, пропиленгликоль, стерильную воду, этанол и хлорид натрия. Альтернативные растворители, которые могут быть использованы вместо пропиленгликоля, включают глицерин и полиэтиленгликоль. Подходящие корригенты, такие как ментол и левоментол, или подсластители, такие как сахарин или сахарин натрия, могут быть добавлены в препараты по данному изобретению, предназначенные для ингаляционного/интраназального введения. Препараты для ингаляционного/интраназального введения могут быть приготовлены для немедленного и/или модифицированного высвобождения с использованием, например, поли(DL-молочной/гликолевой кислоты) (PGLA). Препараты с модифицированным высвобождением включают замедленное,длительное, прерывистое, контролируемое, направленное и программируемое высвобождение. В случае сухих порошковых ингаляторов и аэрозолей стандартную дозу определяют посредством клапана, который доставляет отмеренное количество. Дозы в соответствии с изобретением, как правило,устанавливают такими, чтобы вводить отмеренную дозу или "впрыск", содержащий от 1 мкг до 10 мг соединения по данному изобретению. Общая суточная доза, как правило, находится в диапазоне от 1 мкг до 200 мг, которая может быть введена в виде разовой дозы или, более обычно, в виде разделенных доз в течение суток. Соединения по данному изобретению могут быть введены ректально или вагинально, например, в форме суппозитория, пессария или клизмы. Масло какао представляет собой традиционную основу суппозитория, но могут быть использованы различные подходящие альтернативы. Препараты для ректального/вагинального введения могут быть приготовлены для немедленного и/или модифицированного высвобождения. Препараты с модифицированным высвобождением включают замедленное, длительное, прерывистое, контролируемое, направленное и программируемое высвобождение. Соединения по данному изобретению также могут быть введены непосредственно в глаз или ухо,как правило, в форме капель микронизированной суспензии или раствора в изотоничном, стерильном физиологическом растворе с установленным значением рН. Другие препараты, подходящие для глазного и ушного введения, включают мази, биоразрушаемые (например, губки из абсорбируемого геля, коллаген) и биологически неразрушаемые (например, силикон) имплантаты, облатки, линзы и системы в виде множества частиц или везикул, такие как ниосомы или липосомы. Полимер, такой как поперечно-сшитая полиакриловая кислота, поливиниловый спирт, гиалуроновая кислота, целлюлозный полимер, например,гидроксипропилметилцеллюлоза, гидроксиэтилцеллюлоза или метилцеллюлоза, или гетерополисахаридный полимер, например гелановая камедь, могут быть включены вместе с консервантом, таким как хлорид бензалкония. Такие препараты также могут быть доставлены путем ионофореза. Препараты для глазного/ушного введения могут быть приготовлены для немедленного и/или модифицированного высвобождения. Препараты с модифицированным высвобождением включают замедленное, длительное, прерывистое, контролируемое, направленное и программируемое высвобождение. Соединения по данному изобретению можно объединять с растворимыми макромолекулярными компонентами, такими как циклодекстрин и его подходящие производные или полимеры, содержащие полиэтиленгликоль, для улучшения их растворимости, скорости растворения, маскировки вкуса, биодоступности и/или стабильности для применения в любом из вышеупомянутых способов введения. Например, было обнаружено, что комплексы лекарство-циклодекстрин, как правило, полезны для большинства лекарственных форм и путей введения. Могут быть использованы как комплексы включения, так и комплексы без включения. Альтернативно прямому комплексообразованию с лекарством,циклодекстрин может быть использован в качестве вспомогательного вещества, то есть как носитель,разбавитель или солюбилизатор. Для этих целей наиболее часто используют альфа-, бета- и гаммациклодекстрины, примеры которых можно найти в международных заявках на патентыWO 91/11172, WO 94/02518 и WO 98/55148.- 12009941 Ввиду того, что может быть желательно вводить соединение по данному изобретению в комбинации с другим терапевтическим агентом, например, с целью лечения конкретного заболевания или состояния, в объем настоящего изобретения входит то, чтобы две или более чем две фармацевтические композиции, по меньшей мере одна из которых содержит соединение по данному изобретению, могли быть для удобства объединены в форме набора, подходящего для совместного введения этих композиций. Таким образом, набор по данному изобретению включает две или более чем две отдельные фармацевтические композиции, по меньшей мере одна из которых содержит соединение формулы (I) или его фармацевтически приемлемые соль, сольват или производное, и средства для раздельного хранения указанных композиций, такие как контейнер, разделенный флакон или разделенный пакет из фольги. Пример такого набора представляет собой хорошо известная блистерная упаковка, используемая для упаковки таблеток, капсул, и тому подобное. Набор по данному изобретению особенно подходит для введения различных лекарственных форм,например, для перорального и парентерального введения, для введения раздельных композиций с различными интервалами введения доз, или для титрования отдельных композиций одна относительно другой. Для того чтобы способствовать соблюдению больным режима и схемы лечения, набор, как правило включает указания по введению и может быть снабжен так называемой памяткой. Для введения пациентам, людям, имеющим массу приблизительно 65-70 кг, общая суточная доза соединения по данному изобретению, как правило, находится в диапазоне от 1 до 10000 мг, таком как 101000 мг, например 25-500 мг, в зависимости, конечно, от способа введения, возраста, состояния и массы пациента, и в любом случае остается на усмотрение лечащего врача. Общая суточная доза может быть введена в виде разовой или разделенных доз. Соответственно, в еще одном аспекте данного изобретения предложена фармацевтическая композиция, включающая соединение формулы (I) или его фармацевтически приемлемые соль, сольват или производное вместе с одним или более чем одним фармацевтически приемлемым эксципиентом, разбавителем или носителем. Соединения формулы (I) и их фармацевтически приемлемые соли, сольваты и производные обладают преимуществом, которое заключается в том, что они более селективны, обладают более быстрым началом действия и являются более сильнодействующими, лучше всасываются, более стабильны, более устойчивы к метаболизму, обладают уменьшенным "пищевым эффектом", обладают улучшенным профилем безопасности или другими более желательными свойствами (например, относящимися к растворимости или гигроскопичности) по сравнению с соединениями предшествующего уровня техники. В частности, соединения по данному изобретению обладают уменьшенной ингибирующей активностью в отношении HERG калиевого канала. Было идентифицировано, что пролонгирование длительности сердечного потенциала действия (пролонгирование QT) является следствием действия на HERG калиевый канал (Expert Opinion of Pharmacotherapy, 2, стр. 947-973, 2000). Известно, что пролонгированиеQT обладает потенциальной склонностью вызывать фатальные сердечные аритмии трепетания-мерцания(Torsades de Pointes, TdP). Предлагая соединения, которые дополнительно демонстрируют уменьшенную ингибирующую активность в отношении HERG калиевого канала при сравнимой или улучшенной фармакокинетике, в данном изобретении предложены соединения, которые представляют собой терапевтически эффективные антагонисты CCR5, дополнительно обладающие улучшенной безопасностью в отношении сердца. Соединения формулы (I) и их фармацевтически приемлемые соли, сольваты и производные могут быть введены сами по себе или как часть комбинированного лечения. Таким образом, в объем настоящего изобретения включены воплощения, включающие совместное введение, в дополнение к соединению по данному изобретению, одного или более чем одного дополнительного терапевтического агента, и композиции, которые содержат, в дополнение к соединению по данному изобретению, один или более чем один дополнительный терапевтический агент. Такие множественные схемы приема лекарства, часто называемые комбинированным лечением, могут быть использованы в лечении и профилактике любого из заболеваний или состояний, опосредованных или ассоциированных с модуляцией хемокинового CCR5 рецептора, в частности инфицирования вирусом иммунодефицита человека, ВИЧ. Применение такого комбинированного лечения особенно подходит для лечения и профилактики инфекции и размножения вируса иммунодефицита человека, ВИЧ, и родственных патогенных ретровирусов у пациента, нуждающегося в лечении или имеющего риск их возникновения. Способность таких ретровирусных патогенов развиваться в течение относительно короткого периода времени в штаммы, резистентные к какой-либо монотерапии, вводимой указанному пациенту, хорошо известна в литературе. Рекомендованное лечение ВИЧ представляет собой комбинацию лечения лекарствами, называемую Высокоактивной АнтиРетровирусной Терапией, или BAAPT. При BAAPT объединяют три или более чем три лекарства против ВИЧ. Таким образом, способы лечения и фармацевтические композиции по настоящему изобретению могут использовать соединение по данному изобретению в форме монотерапии, но указанные способы и композиции также могут быть использованы в форме комбинированного лечения, в котором одно или более чем одно соединение по данному изобретению вводят совместно в комбинации с одним или более- 13009941 чем одним дополнительным терапевтическим агентом, таким как агенты, более подробно описанные ниже. В еще одном воплощении данного изобретения комбинации по настоящему изобретению включают лечение соединением формулы (I) или его фармацевтически приемлемыми солью, сольватом или производным и одним или более чем одним дополнительным терапевтическим агентом, выбранным из следующих: ингибиторы ВИЧ-протеаз (PI), включая индинавир, ритонавир, саквинавир, нелфинавир, лопинавир, ампренавир, атазанавир, типранавир, AG1859 и TMC 114, но не ограничиваясь ими; ненуклеозидные ингибиторы обратной транскриптазы (NNRTI), включая: невирапин; делавирдин; каправирин; эфавиренц; 5-[3,5-диэтил-1-(2-гидроксиэтил)-1H-пиразол-4-ил]оксиизофталонитрил или его фармацевтически приемлемые соли, сольваты или производные; 5-[3-циклопропил-1-(2-гидроксиэтил)-5-метил-1Hпиразол-4-ил]оксиизофталонитрил или его фармацевтически приемлемые соли, сольваты или производные; GW-8248; GW-5634 и TMC 125, но не ограничиваясь ими; нуклеозидные/нуклеотидные ингибиторы обратной транскриптазы (NRTI), включая зидовудин, диданозин, зальцитабин, ставудин, ламивудин, абакавир, адефовир дипивоксил, тенофовир, эмтрицитабин и аловудин, но не ограничиваясь ими; другие антагонисты CCR5, включаяN-(1S)-3-[3-(3-изопропил-5-метил-4H-1,2,4-триазол-4-ил)экзо-8-азабицикло[3.2.1]окт-8-ил]-1 фенилпропил-4,4-дифторциклогексанкарбоксамид или его фармацевтически приемлемые соли, сольваты или производные,метил-1-эндо-8-[(3S)-3-(ацетиламино)-3-(3-фторфенил)пропил]-8-азабицикло[3.2.1]окт-3-ил-2 метил-4,5,6,7-тетрагидро-1H-имидазо[4,5-с]пиридин-5-карбоксилат или его фармацевтически приемлемые соли, сольваты или производные,метил-3-эндо-8-[(3S)-3-(ацетамидо)-3-(3-фторфенил)пропил]-8-азабицикло[3.2.1]окт-3-ил-2 метил-4,5,6,7-тетрагидро-3H-имидазо[4,5-с]пиридин-5-карбоксилат или его фармацевтически приемлемые соли, сольваты или производные,этил-1-эндо-8-[(3S)-3-(ацетиламино)-3-(3-фторфенил)пропил]-8-азабицикло[3.2.1]окт-3-ил-2 метил-4,5,6,7-тетрагидро-1H-имидазо[4,5-с]пиридин-5-карбоксилат или его фармацевтически приемлемые соли, сольваты или производные,Sch D, ONO4128, GW873140, AMD-887 и CMPD-167; агенты, которые ингибируют взаимодействиеgp120 с CD4, включая BMS806, BMS-488043, метиламид 5-(1S)-2-[(2R)-4-бензоил-2-метилпиперазин-1 ил]-1-метил-2-оксоэтокси-4-метоксипиридин-2-карбоновой кислоты и 4-(1S)-2-[(2R)-4-бензоил-2 метилпиперазин-1-ил]-1-метил-2-оксоэтокси-3-метокси-N-метилбензамид; другие агенты, которые ингибируют проникновение ВИЧ в клетку-мишень, включая энфувиритид, T1249, PRO 542 и PRO 140, но не ограничиваясь ими; ингибиторы интегразы, включая L-870810, но не ограничиваясь им; и ингибиторы РНКазы H. Также в объем настоящего изобретения включены комбинации соединения формулы (I) или его фармацевтически приемлемых солей, сольватов или производных вместе с одним или более чем одним дополнительным терапевтическим агентом, независимо выбранным из группы, состоящей из ингибиторов пролиферации, например гидроксимочевины; иммуномодуляторов, таких как гранулоцитарномакрофагальный колониестимулирующий фактор роста (например, сарграмостим), модуляторов тахикининового рецептора (например, антагонистов нейрокинина-1 (NK1 и различных форм интерферона или производных интерферона; других агонистов/ангагонистов хемокинового рецептора, таких как антагонисты CXCR4 (например, AMD-070); агентов, которые по существу ингибируют, прекращают или уменьшают вирусную транскрипцию или репликацию РНК, таких как ингибиторы tat (транскрипционного транс-активатора) или nef (негативного регуляторного фактора); агентов, которые по существу ингибируют, прекращают или уменьшают трансляцию одного или более чем одного белка, экспрессируемого вирусом (включая негативную регуляцию экспрессии белка или антагонизм одного или более чем одного белка, но не ограничиваясь ими), отличного от обратной транскриптазы, такого как Tat или Nef; агентов,которые оказывают влияние на экспрессию, в частности осуществляют негативную регуляцию экспрессии, CCR5 рецептора; хемокинов, которые индуцируют интернализацию CCR5 рецептора, таких какMIP-1, MIP-1, RANTES и их производные; и других агентов, которые ингибируют вирусную инфекцию или улучшают состояние или исход лечения ВИЧ-инфицированных индивидуумов посредством различных механизмов. Агенты, которые влияют (в частности оказывают негативную регуляцию) на экспрессию CCR5 рецептора, включают иммуносупрессоры, такие как ингибиторы кальцинейрина (например, такролимус и циклоспорин А); стероиды; агенты, которые препятствуют продуцированию цитокинов или передаче сигналов от цитокинов, такие как ингибиторы Janus киназы (JAK) (например, ингибиторы JAK-3, включая 3-(3R,4R)-4-метил-3-[метил-(7H-пирроло [2,3-d]пиримидин-4-ил)амино]пиперидин-1-ил-3-оксопропионитрил) и их фармацевтически приемлемые соли, сольваты или производные; антитела к цитокинам (например антитела, которые ингибируют рецептор интерлейкина-2 (ИЛ-2), включая басиликсимаб и даклизумаб); и агенты, которые препятствуют клеточной активации или клеточному циклу, такие как рапамицин. Также в объем настоящего изобретения включены комбинации соединения формулы (I) или его- 14009941 фармацевтически приемлемых солей, сольватов или производных вместе с одним или более чем одним дополнительным терапевтическим агентом, который замедляет скорость метаболизма соединения по данному изобретению, тем самым приводя к увеличенному воздействию в организме пациента. Увеличение воздействия таким образом известно как бустинг. Оно обладает преимуществом увеличения эффективности соединения по данному изобретению или уменьшения дозы, требующейся для достижения той же самой эффективности, как и эффективность небустерной дозы. Метаболизм соединений по данному изобретению включает окислительные процессы, осуществляемые ферментами Р 450 (CYP450), в частности CYP 3 А 4 и конъюгации уридилдифосфатглюкуронозилтрансферазы и сульфатирующих ферментов. Таким образом, среди агентов, которые могут быть использованы для увеличения воздействия соединения по настоящему изобретению на пациента, находятся агенты, которые действуют как ингибиторы по меньшей мере одной изоформы ферментов цитохромов Р 450 (CYP450). Изоформы CYP450, которые могут быть благоприятным образом ингибированы, включают CYP1 А 2, CYP2D6, CYP2C9, CYP2C19 иCYP3A4, но не ограничиваются ими. Подходящие агенты, которые могут быть использованы для ингибирования CYP 3 А 4, включают ритонавир, саквинавир или кетоконазол, но не ограничиваются ими. Специалисту в данной области техники понятно, что комбинированное лекарственное лечение, как описано выше, может включать два или более чем два соединения, имеющие один и тот же или отличный механизм действия. Таким образом, только для иллюстрации, комбинация может включать соединение по данному изобретению и: один или более чем один нуклеозидный ингибитор обратной транскриптазы (NRTI); один или более чем один NRTI и PI; один или более чем один NRTI и другой антагонистCCR5; PI; PI и NNRTI; NNRTI; и так далее. В дополнение к требованию терапевтической эффективности, для которой может быть необходимо применение терапевтических агентов в дополнение к соединениям по данному изобретению, могут существовать дополнительные разумные обоснования, которые заставляют или очень рекомендуют применение комбинации соединения по данному изобретению и другого терапевтического агента, например, в лечении заболеваний или состояний, которые непосредственно вытекают или косвенно сопутствуют основному или лежащему в основе заболеванию или состоянию, модулируемому хемокиновым CCR5 рецептором. Например, когда основное заболевание или состояние, модулируемое хемокиновым CCR5 рецептором, представляет собой ВИЧ-инфекцию и размножение ВИЧ, может быть необходимо или по меньшей мере желательно лечить вирус гепатита С (HCV), вирус гепатита В (HBV), вирус папилломы человека (HPV), оппортунистические инфекции (включая бактериальные и грибковые инфекции), новообразования и другие состояния, которые возникают как результат состояния нарушенного иммунитета у пациента, которого лечат. Другие терапевтические агенты могут быть использованы с соединениями по данному изобретению, например, для того чтобы обеспечить иммуностимуляцию или для лечения боли и воспаления, которые сопутствуют исходной и основной ВИЧ-инфекции. Соответственно, терапевтические агенты для применения в комбинации с соединениями формулы(I) и их фармацевтически приемлемыми солями, сольватами и производными, также включают: интерфероны, пэгилированные интерфероны (например, пэгинтерферон альфа-2 а и пэгинтерферон альфа-2b),ламивудин, рибавирин и эмтрицитабин для лечения гепатита; противогрибковые средства, такие как флюконазол, фосфлюконазол, итраконазол и вориконазол; антибактериальные средства, такие как азитромицин и кларитромицин; интерфероны, даунорубицин, доксорубицин и паклитаксель для лечения саркомы Капоши, связанной со СПИДом; и цидофовир, фомивирзен, фоскарнет, ганцикловир и вальцит для: лечения цитомегаловирусного (ЦМВ) ретинита. Дополнительные комбинации для применения в соответствии с данным изобретением включают комбинацию соединения формулы (I) или его фармацевтически приемлемых соли, сольвата или производного с антагонистом CCRl, таким как ВХ-471; агонистом бета-адренорецептора, таким как сальметерол; кортикостероидным агонистом, таким как пропионат флютиказона; антагонистом лейкотриена D4(LTD4), таким как монтелукаст; мускариновым антагонистом, таким как тиотропия бромид; ингибитором фосфодиэстеразы 4 (PDE4), таким как циломиласт или рофлюмиласт; ингибитором циклооксигеназы-2 (СОХ-2), таким как целекоксиб, вальдекоксиб или рофекоксиб; альфа-2-дельта лигандом, таким как габапентин или прегабалин; бета-интерфероном, таким как REBIF; модулятором рецептора фактора некроза опухолей (TNF), таким как ингибитор TNF-альфа (например, адалимумаб); ингибитором 3 гидрокси-3-метилглутарилкоэнзим A (HMG CoA) редуктазы, таким как статин (например, аторвастатин); или иммуносупрессором, таким как циклоспорин, или макролидом, таким как такролимус. В вышеописанных комбинациях соединение формулы (I) или его фармацевтически приемлемые соль, сольват или производное и другой(ие) терапевтический(е) агент(ы) могут быть введены, в отношении лекарственных форм, раздельно или в сочетании друг с другом и, в отношении времени введения,либо одновременно, либо последовательно. Таким образом, введение одного составляющего агента может быть осуществлено перед, одновременно или после введения другого(их) составляющего(их) агента(ов). Соответственно, в еще одном аспекте данного изобретения предложена фармацевтическая композиция, включающая соединение формулы (I) или его фармацевтически приемлемые соль, сольват или производное и один или более чем один дополнительный терапевтический агент.- 15009941 Следует понимать, что все ссылки на лечение, используемые в данном описании, включают целебное, паллиативное и профилактическое лечение. Данное изобретение проиллюстрировано следующими примерами и подготовительными примерами, в которых могут быть использованы следующие сокращения: ч означает час,мин означает минуту,LRMS означает масс-спектр низкого разрешения,HRMS означает масс-спектр высокого разрешения,APCI+ означает химическую ионизацию при атмосферном давлении,ESI+ означает ионизацию электрораспылением,ЯМР означает ядерный магнитный резонанс,тcx означает тонкослойную хроматографию,Me означает метил. Пример 1. N-[1S)-3-[3-Эндо-(5-Ацетил-2-метил-4,5,6,7-тетрагидро-1H-имидазо-[4,5-с]пиридин-1 ил)-8-азабицикло[3.2.1]окт-8-ил]-1-(3-фторфенил)пропилацетамид К перемешиваемому раствору амина N-(1S)-3-[3-эндо-(2-метил-4,5,6,7-тетрагидро-1H-имидазо[4,5-с]пиридин-1-ил)-8-азабицикло[3.2.1]окт-8-ил]-1-(3-фторфенил)пропилацетамида (94 мг, 0,21 ммоль) в дихлорметане (5 мл) при комнатной температуре добавляли ацетилхлорид (18 мкл, 0,26 ммоль),а затем N,N-диизопропилэтиламин (45 мкл, 0,26 ммоль). Через 15 ч реакционную смесь разбавляли дихлорметаном (5 мл) и водой (5 мл) и затем пропускали через картридж для разделения фаз. Органический компонент концентрировали путем пропускания потока азота над раствором и получающуюся в результате смесь очищали с использованием картриджа Mega Bond ElutFlash Si (10 г, Varian), элюируя смесью дихлорметан:метанол:концентрированный водный аммиак (95:5:0,5, по объему) с получением указанного в заголовке соединения в виде белой пены (80 мг, 79%).[]D -21,7 (2,12 мг/мл в MeOH). Примеры 2-3. Соединения этих примеров были получены в соответствии со способом, описанным выше для примера 1, с использованием N-(1S)-3-[3-эндо-(2-метил-4,5,6,7-тетрагидро-1H-имидазо[4,5-с] пиридин-1-ил)-8-азабицикло[3.2.1]окт-8-ил]-1-(3-фторфенил)пропилацетамида и соответствующего соединения формулы (V). Все LRMS представляли собой ионизацию электрораспылением. Пример 2. К перемешиваемому раствору амина N-(1S)-3-[3-эндо-(2-метил-4,5,6,7-тетрагидро-1H-имидазо[4,5-с]пиридин-1-ил)-8-азабицикло[3.2.1]окт-8-ил]-1-(3-фторфенил)пропилацетамида (19,9 г, 45,3 ммоль) в тетрагидрофуране (500 мл) добавляли триэтиламин (7,0 мл, 50,0 ммоль) с последующим добавлением по каплям изобутирилхлорида (5,3 мл, 50 ммоль). Через 1 ч добавляли по каплям вторую порцию изобутирилхлорида (0,5 мл, 5,0 ммоль). Через 0,5 ч реакционную смесь концентрировали до приблизительно 300 мл и добавляли этилацетат (200 мл). Реакционную смесь промывали 10% водным растворомK2CO3 (200 мл; мас./об.). Водную фазу отделяли и экстрагировали этилацетатом (100 мл). Органические компоненты объединяли, промывали рассолом (100 мл), сушили (MgSO4) и концентрировали до получения маловязкого масла, которое только начинало образовывать пену. Остаток растворяли в этилацетате(100 мл) и нагревали до 90C. К горячему раствору добавляли воду (0,5 мл) и смесь оставляли медленно охлаждаться до комнатной температуры. Осадок собирали путем фильтрования, промывали этилацетатом (50 мл) и сушили при пониженном давлении с получением указанного в заголовке соединения в виде белого твердого вещества (20,9 г, 90%).[]D -23,4 (1,64 мг/мл в MeOH). Примеры 5-7. Соединения этих примеров были получены в соответствии со способом, описанным выше для примера 1, с использованием N-(1S)-3-[3-эндо-(2-метил-4,5,6,7-тетрагидро-1H-имидазо[4,5-с]пиридин-1 ил)-8-азабицикло[3.2.1]окт-8-ил]-1-(3-фторфенил)пропилацетамида и соответствующего соединения формулы (V). Все LRMS представляли собой электрораспыление, за исключением Примера 6, который представлял собой химическую ионизацию при атмосферном давлении. Пример 5. К перемешиваемому раствору 3,3,3-трифторпропионовой кислоты (15 мкл, 1,71 ммоль) в дихлорметане (2 мл) добавляли гексафторфосфат О-бензотриазол-1-ил-N,N,N',N'-тетраметилурония (65 мг, 1,71 ммоль), триэтиламин (47 мкл, 3,41 ммоль), а затем амин N-(1S)-3-[3-эндо-(2-метил-4,5,6,7-тетрагидро 1H-имидазо[4,5-с]пиридин-1-ил)-8-азабицикло[3.2.1]окт-8-ил]-1-(3-фторфенил)пропилацетамид (50 мг,1,14 ммоль). Реакционную смесь нагревали при 30C в течение 15 ч, концентрировали путем пропускания потока азота над раствором и очищали посредством препаративной ЖХВД (колонка Phenomenex C18 15x10 см 10 мкм, скорость потока 20 мл/мин, детектирование при 225 нм, градиент подвижной фазы от 95:5 до 5:95 А:Б, (А: 0,1% диэтиламин в Н 2 О, Б: MeCN с получением указанного в заголовке соединения в виде смолы (20 мг). К раствору N-(1S)-3-[3-эндо-(5-изобутирил-2-метил-4,5,6,7-тетрагидро-1H-имидазо[4,5-с]пиридин 1-ил)-8-азабицикло[3.2.1]окт-8-ил]-1-(3-фторфенил)пропилацетамида (300 мг, 0,59 ммоль) в тетрагидрофуране (2 мл) при кипячении с обратным холодильником по каплям добавляли раствор фумаровой кислоты (68 мг, 0,59 ммоль) в горячем этаноле (2 мл). Через 48 ч добавляли тетрагидрофуран (2 мл) и через 48 ч кристаллы собирали путем фильтрования, промывали тетрагидрофураном (2 мл) и сушили на воздухе с получением указанного в заголовке соединения в виде белого порошка (110 мг, 30%). Обнаружено С 62,04; H 7,26; N, 10,70. C33H44FN5O6. 0,75 H2O требует С 62,00; H 7,17; N 10,96. Пример 10. N-(1S)-3-[3-эндо-(5-Изобутирил-2-метил-4,5,6,7-тетрагидро-1H-имидазо[4,5-с]пиридин-1-ил)-8-азабицикло[3.2.11 окт-8-ил]-1-(3-фторфенил)пропилацетамидN-(1S)-3-[3-эндо-(2-Метил-4,5,6,7-тетрагидро-1H-имидазо[4,5-с]пиридин-1-ил)-8-азабицикло[3.2.1] окт-8-ил]-1-(3-фторфенил)пропилацетамид (264,4 г, 0,60 моль) добавляли к пропан-2-олу (2,67 л) с последующим добавлением карбоната калия (92,7 г, 0,67 моль) и получающуюся в результате смесь охлаждали до 15 С. Затем в течение 10 мин добавляли изобутирилхлорид (97,8 г, 0,91 моль), поддерживая температуру ниже 25 С, и после перемешивания в течение 10 мин реакцию завершали. Затем добавляли карбонат калия (267,2 г) в воде (2,40 л) и получающиеся в результате 2 фазы разделяли. Водный слой затем экстрагировали этилацетатом (2,67 л) и объединенные органические фазы промывали насыщенным водным хлоридом натрия (1,32 л) и водой (800 мл), затем концентрировали в вакууме. Остаток разбавляли этилацетатом (1,07 л) и вновь концентрировали в вакууме. Повторяли эту стадию повторного растворения и концентрирования и получающийся в результате остаток обрабатывали этилацетатом до достижения общего объема 1,07 л. Его охлаждали до 0-5C, перемешивали в течение 2 ч, фильтровали и промывали охлажденным этилацетатом (2130 мл). Твердый продукт затем сушили в вакуумной печи при 50C с получением указанного в заголовке соединения (269,8 г, 0,53 моль, 88,0%). Пример 11. Фумарат N-(1S)-3-[3-эндо-(5-Изобутирил-2-метил-4,5,6,7-тетрагидро-1H-имидазо[4,5 с]пиридин-1-ил)-8-азабицикло[3.2.1]окт-8-ил]-1-(3-фторфенил)пропилацетамидаN-(1S)-3-[3-эндо-(5-Изобутирил-2-метил-4,5,6,7-тетрагидро-1H-имидазо[4,5-с]пиридин-1-ил)-8 азабицикло[3.2.1]окт-8-ил]-1-(3-фторфенил)пропилацетамид (554,0 г, 1,08 моль) добавляли к пропан-2 олу (13,85 л) с последующим добавлением фумаровой кислоты (126,2 г, 1,09 моль) и получающуюся в результате смесь нагревали до температуры дефлегмации и перемешивали в течение 20 мин. Этот раствор затем осветляли путем фильтрования при этой температуре перед концентрированием в вакууме до объема растворителя 4 мл/г, в расчете на исходный N-(1S)-3-[3-эндо-(5-изобутирил-2-метил-4,5,6,7 тетрагидро-1H-имидазо[4,5-с]пиридин-1-ил)-8-азабицикло[3.2.1]окт-8-ил]-1-(3-фторфенил)пропилацетамид. Его охлаждали до 0-5C, перемешивали в течение 2 ч, фильтровали и промывали охлажденным пропан-2-олом (2550 мл). Твердый продукт затем сушили в вакуумной печи при 50C с получением указанного в заголовке соединения (495,9 г, 0,79 моль, 72,9%). Пример 12. Соль (D)-тартрат N-(1S)-3-[3-эндо-(5-изобутирил-2-метил-4,5,6,7-тетрагидро-1Hимидазо[4,5-с]пиридин-1-ил)-8-азабицикло[3.2.1]окт-8-ил]-1-(3-фторфенил)пропилацетамида получали в соответствии со способом, описанным выше для соли фумарата из примера 9, с использованием N(1S)-3-[3-эндо-(5-изобутирил-2-метил-4,5,6,7-тетрагидро-1H-имидазо[4,5-с]пиридин-1-ил)-8-азабицикло[3.2.1]окт-8-ил]-1-(3-фторфенил)пропилацетамида и D-винной кислоты. Данные по пикам дифракции рентгеновских лучей на порошке (PXRD) представлены ниже. Подготовительный пример 1. 8-Бензил-8-азабицикло[3.2.1]октан-3-он Раствор 2,5-диметокситетрагидрофурана (50 г, 378 ммоль) в соляной кислоте (0,025 н., 160 мл) охлаждали до 0C в течение 16 ч. Добавляли гидрохлорид бензиламина (65 г, 453 ммоль), кетомалоновую кислоту (55 г, 377 ммоль) и водный раствор ацетата натрия (300 мл, 0,69 M) и реакционную смесь перемешивали при комнатной температуре в течение одного часа. Смесь нагревали до 50C в течение еще 90 мин, затем охлаждали в ледяной бане и подщелачивали 2 н. раствором гидроксида натрия до рН 12. Слои разделяли и водную фазу экстрагировали этилацетатом (3300 мл). Объединенные органические экс- 19009941 тракты промывали водой, сушили (MgSO4), фильтровали и упаривали при пониженном давлении. Получающееся в результате коричневое масло перегоняли при пониженном давлении (126 С/399,97 Па (3 мм рт.ст. с получением указанного в заголовке соединения в виде беловатого твердого вещества (37,81 г).(18,2 г, 83,4 ммоль) и 20% мас./мас. гидроксида палладия на углероде (3,0 г) в этилацетате (165 мл) перемешивали в течение 4 часов при комнатной температуре в атмосфере водорода при 269 кПа. Смесь фильтровали через Arbocel и растворитель удаляли при пониженном давлении. Остаток очищали путем колоночной хроматографии на силикагеле, используя градиент элюции гексана:простого эфира (от 100:0 до 50:50) с получением указанного в заголовке соединения в виде бесцветного масла, которое кристаллизовалось при стоянии (16,2 г). 1 Раствор трет-бутил-3-оксо-8-азабицикло[3.2.1]октан-8-карбоксилата (10,0 г, 44,4 ммоль), бензиламина (4,85 мл, 49,7 ммоль) и триацетоксиборгидрида натрия (14,11 г, 66,6 ммоль) перемешивали в течение 16 часов при комнатной температуре в смеси ледяная уксусная кислота:дихлорметан (1:9 об./об., 290 мл). Растворители выпаривали при пониженном давлении и остаток растворяли в этилацетате (200 мл),затем промывали насыщенным водным раствором карбоната натрия (50 мл) и водой (50 мл). Органический раствор сушили (MgSO4, фильтровали и упаривали при пониженном давлении. Остаток очищали путем колоночной хроматографии на силикагеле, используя в качестве элюента смесь дихлорметан:метанол:концентрированный водный аммиак (98:2:0,25) с получением указанного в заголовке соединения в виде белого твердого вещества (7,00 г). 1 Н ЯМР (400 МГц, CDCl3): : 1.42-1.48 (11H, m), 1.52-1.61 (2 Н, m), 1.85-2.19 (5 Н, m), 2.95-3.03 (1H,m), 3.74 (2 Н, s), 4.03-4.23 (2 Н, m), 7.20-7.26 (1H, m), 7.26-7.32 (4 Н, m) млн-1. Подготовительный пример 4. трет-Бутил-3-эндоамино-8-азабицикло[3.2.1]октан-8-карбоксилат Смесь трет-бутил-3-(бензиламино)-эндо-8-азабицикло[3.2.1]октан-8-карбоксилата (7,00 г, 22,1 ммоль), формиата аммония (7,00 г, 111 ммоль) и 20% мас./мас. гидроксида палладия на углероде (0,70 г) в этаноле (200 мл) нагревали до 50C до прекращения образования газа. Охлажденную смесь фильтровали через Arbocel и фильтрат упаривали при пониженном давлении. Остаток очищали путем колоночной хроматографии на силикагеле с использованием градиента элюции дихлорметана:метанола:концентрированного водного аммиака (от 98:2:0,25 до 95:5:0,5) с получением указанного в заголовке соединения в виде бесцветного масла (4,70 г). трет-Бутил-3-аминоэндо-8-азабицикло[3.2.1]октан-8-карбоксилат (3,0 г, 13,2 ммоль), 4-этокси-3- 20009941 нитропиридингидрохлорид (2,7 г, 13,2 ммоль) и N-этил-N,N-диизопропиламин (1,89 г, 14,6 ммоль) растворяли в 1-метил-2-пирролидиноне (5 мл) и нагревали при 120C в течение 18 часов. Охлажденную реакционную смесь разбавляли этилацетатом (150 мл) и промывали водой (350 мл), насыщенным водным раствором гидрокарбоната натрия (50 мл) и рассолом (30 мл). Органический слой сушили (MgSO4) и растворитель удаляли путем выпаривания при пониженном давлении. Остаток растирали с диэтиловым эфиром и фильтровали с получением указанного в заголовке соединения в виде желтого твердого вещества (1,5 г). трет-Бутил-3-эндо-[(3-нитро-4-пиридинил)амино]-8-азабицикло[3.2.1]октан-8-карбоксилат (4,40 г,12,6 ммоль) и железный порошок (2,11 г, 37,8 ммоль) растворяли в ледяной уксусной кислоте (50 мл) и смесь нагревали до 60C в течение 2 ч. Затем добавляли уксусный ангидрид (8 мл) и смесь нагревали до 140C в течение 18 ч. Охлажденную реакционную смесь фильтровали через слой Arbocel и растворитель удаляли при пониженном давлении. Остаток распределяли между дихлорметаном (200 мл) и водой(200 мл) и рН смеси доводили до значения 9 с использованием 2 н. водного раствора гидроксида натрия. Смесь снова фильтровали через слой Arbocel и органическую фазу отделяли. Водный слой экстрагировали дихлорметаном (100 мл) и объединенные органические экстракты сушили (MgSО 4). Растворитель выпаривали при пониженном давлении и остаток растирали с этилацетатом, фильтровали и сушили(MgSO4) с получением указанного в заголовке соединения в виде белого твердого вещества (3,27 г).(2,47 г, 8,7 ммоль) в этаноле (20 мл) добавляли бензилбромид (1,78 г, 10,4 ммоль) и смесь перемешивали при комнатной температуре в течение 48 ч. Реакционную смесь затем охлаждали до -70C и порциями в течение 10 мин добавляли боргидрид натрия (0,33 г, 8,7 ммоль). Через 1 ч при -70C реакционную смесь оставляли нагреваться до -40C, затем снова охлаждали до -70C и добавляли дополнительное количество боргидрида натрия (0,33 г, 8,7 ммоль). Через еще 1 ч при -70C добавляли воду (10 мл) и реакционную смесь оставляли нагреваться до комнатной температуры. Этанол выпаривали при пониженном давлении и водный остаток экстрагировали дихлорметаном (325 мл). Объединенные органические экстракты сушили (MgSO4) и растворитель выпаривали при пониженном давлении. Остаток очищали путем колоночной флэш-хроматографии на силикагеле, элюируя градиентом растворителей этилацетат:метанол:диэтиламин (от 100:0:2, по объему, до 98:2:2, затем 95:5:2). Фракции, содержащие продукт,упаривали с получением указанного в заголовке соединения в виде белой пены (2,23 г).[4,5-с]пиридин (2,23 г, 5,89 ммоль) растворяли в 6 н. водной соляной кислоте (30 мл) и кипятили с обратным холодильником в течение 18 ч. рН охлажденной реакционной смеси доводили до значения 10 путем добавления 2 н. водного раствора гидроксида натрия и экстрагировали дихлорметаном (250 мл). Объединенные органические экстракты сушили (MgSO4) и растворитель выпаривали при пониженном давле- 21009941 нии. Остаток очищали путем колоночной флэш-хроматографии на силикагеле, элюируя градиентом растворителей дихлорметан : метанол:диэтиламин (от 100:0:0,5, по объему, до 93:7:1). Фракции, содержащие продукт, упаривали с получением сказанного в заголовке соединения в виде белой пены (1,47 г). Гидрид диизобутилалюминия (1M в дихлорметане, 39 мл, 39 ммоль) охлаждали до -78C и по каплям добавляли к раствору метил-(3S)-3-[(трет-бутоксикарбонил)амино]-3-(3-фторфенил)пропаноата (WO 0039125, стр.60, получение 12) (5,4 г, 18,2 ммоль) в дихлорметане (100 мл) при -78 С. Реакционную смесь перемешивали в течение 30 мин при -78C, затем добавляли метанол (50 мл, предварительно охлажденный до -78C). Реакционную смесь перемешивали в течение 30 мин, затем добавляли 2 н. соляную кислоту (250 мл). Эту бифазную смесь оставляли нагреваться до комнатной температуры, слои разделяли и органический слой сушили (MgSO4), фильтровали и упаривали при пониженном давлении с получением указанного в заголовке соединения в виде прозрачного бесцветного масла, 4,8 г. К перемешиваемому раствору 1-эндо-(8-азабидикло[3.2.1]окт-3-ил)-5-бензил-2-метил-4,5,6,7-тетрагидро-1H-имидазо[4,5-с]пиридина (2,16 г, 6,4 ммоль) и трет-бутил-(1S)-1-(3-фторфенил)-3-оксопропилкарбамата (2,06 г, 7,7 ммоль), растворенного в дихлорметане (25 мл), в атмосфере азота при комнатной температуре добавляли уксусную кислоту (0,39 г, 6,4 ммоль). Затем добавляли триацетоксиборгидрид натрия (1,63 г, 7,7 ммоль) и реакционную смесь выдерживали при комнатной температуре в течение 2 ч. Реакционную смесь распределяли между насыщенным водным раствором гидрокарбоната натрия (50 мл) и дихлорметаном (50 мл). Органическую фазу удаляли и водную фазу промывали дихлорметаном (50 мл). Объединенные органические фазы сушили (MgSO4 и растворитель выпаривали при пониженном давлении. Остаток очищали путем колоночной флэш-хроматографии на силикагеле, элюируя градиентом растворителей дихлорметан:метанол:концентрированный водный аммиак (от 99:1:0,1, по объему, до 96:4:0,4). Фракции, содержащие продукт, упаривали с получением указанного в заголовке соединения в виде белой пены (2,56 г). Смесь трет-бутил-(1S)-3-[3-эндо-(5-бензил-2-метил-4,5,6,7-тетрагидро-1H-имидазо[4,5-с]пиридин 1-ил)-8-азабицикло[3.2.1]окт-8-ил]-1-(3-фторфенил)пропилкарбамата (2,55 г, 4,34 ммоль), формиата аммония (2,73 г, 43,4 ммоль) и 20% мас./мас. гидроксида палладия на углероде (0,25 г) в этаноле (35 мл) нагревали до 60C. Через 1 ч добавляли дополнительное количество формиата аммония (0,63 г, 10,1 ммоль) и нагревание продолжали при 60C в течение еще 2 ч. Охлажденную реакционную смесь затем фильтровали через Arbocel и фильтрат упаривали при пониженном давлении. Остаток распределяли- 22009941 между дихлорметаном (100 мл) и насыщенным водным раствором гидрокарбоната натрия (50 мл), органическую фазу отделяли и промывали водой (30 мл). Органический слой сушили (MgSO4) и растворитель упаривали при пониженном давлении. Остаток очищали путем колоночной флэш-хроматографии на силикагеле, элюируя градиентом растворителей дихлорметан:метанол:концентрированный водный аммиак (от 99:1:0,1 до 93:7:1) с получением указанного в заголовке соединения в виде белой пены (1,50 г). Метилхлорформиат (0,167 г, 1,76 ммоль) добавляли к раствору трет-бутил-(1S)-3-[3-эндо-(2-метил 4,5,6,7-тетрагидро-1H-имидазо[4,5-с]пиридин-1-ил)-8-азабицикло[3.2.1]окт-8-ил]-1-(3-фторфенил) пропилкарбамата (0,80 г, 1,60 ммоль) в дихлорметане (10 мл) в атмосфере азота при комнатной температуре. Реакционную смесь перемешивали при комнатной температуре в течение 1,5 ч и затем промывали насыщенным водным раствором гидрокарбоната натрия (10 мл). Органическую фазу удаляли и водный слой экстрагировали дополнительным количеством дихлорметана (210 мл). Объединенные дихлорметановые экстракты сушили (MgSO4) и растворитель удаляли при пониженном давлении. Остаток очищали путем колоночной флэш-хроматографии на силикагеле,элюируя смесью растворителей дихлорметан:метанол:концентрированный водный аммиак (от 99:1:0,1 до 93:7:1) с получением указанного в заголовке соединения в виде белой пены (0,84 г). Газообразный хлорид водорода барботировали через раствор метил-1-эндо-(8-(3S)-3-[(трет-бутоксикарбонил)амино]-3-(3-фторфенил)пропил-8-азабицикло[3.2.1]окт-3-ил)-2-метил-4,5,6,7-тетрагидро 1H-имидазо[4,5-с]пиридин-5-карбоксилата (0,83 г, 1,50 ммоль) в дихлорметане (15 мл) при 0C до насыщения раствора. Реакционную смесь затем оставляли нагреваться до комнатной температуры и перемешивали в течение одного часа. Растворитель выпаривали при пониженном давлении и остаток суспендировали в дихлорметане (10 мл). Этот процесс повторяли трижды с получением указанного в заголовке соединения в виде белого твердого вещества (0,82 г).[4,5-с]пиридин-5-карбоксилата (0,409 г, 0,72 ммоль) и триэтиламина (0,33 г, 3,25 ммоль), растворенного в дихлорметане (10 мл), в атмосфере азота при комнатной температуре и реакционную смесь перемешивали в течение 2 часов. Затем раствор промывали водой (10 мл), 1 н. раствором гидроксида натрия (10 мл) и рассолом (10 мл). Органическую фазу отделяли, сушили (MgSO4) и растворитель удаляли при пониженном давлении. Остаток очищали путем колоночной флэш-хроматографии на силикагеле, элюируя градиентом растворителей дихлорметан:метанол:концентрированный водный аммиак (от 99:1:0,1, по объему, до 97:3:0,3). Фракции, содержащие продукт, упаривали с получением указанного в заголовке соединения в виде белой пены (0,24 г). К перемешиваемому раствору метил-1-эндо- 8-[(3S)-3-(ацетиламино)-3-(3-фторфенил)пропил]-8 азабицикло[3.2.1]окт-3-ил-2-метил-4,5,6,7-тетрагидро-1H-имидазо[4,5-с]пиридин-5-карбоксилата (13,27 г, 26,7 ммоль) в пропан-2-оле (80 мл) добавляли 2 М водный раствор гидроксида натрия (120 мл) и смесь кипятили с обратным холодильником в течение 48 ч. После охлаждения до комнатной температуры смесь экстрагировали этилацетатом (2200 мл). Объединенные органические компоненты промывали рассолом (150 мл), сушили (MgSO4) и концентрировали при пониженном давлении. Неочищенную смесь продуктов очищали путем колоночной флэш-хроматографии, элюируя смесью дихлорметан:метанол: концентрированный водный аммиак (90:10:1, затем 80:20:1, по объему) с получением указанного в заголовке соединения в виде белой пены (8,54 г, 73%).LRMS (химическая ионизация при атмосферном давлении): m/z [MH+] 440. Подготовительный пример 16. N-(1S)-3-[3-Эндо-(2-метил-4,5,6,7-тетрагидро-1H-имидазо[4,5-с] пиридин-1-ил)-8-азабицикло[3.2.11 окт-8-ил]-1-(3-фторфенил)пропилацетамид(L)-Тартрат метил-1-эндо-8-[(3S)-3-(ацетиламино)-3-(3-фторфенил)пропил]-8-азабицикло[3.2.1] окт-3-ил-2-метил-4,5,6,7-тетрагидро-1H-имидазо[4,5-с]пиридин-5-карбоксилата (WO 03/084954, пример 46) (898 г, 1,39 моль) добавляли к дихлорметану (4,5 л) и воде (4,5 л). Затем добавляли водный раствор гидроксида натрия (10 М, 450 мл) и получающуюся в результате смесь перемешивали в течение 15 мин. Две фазы разделяли и водный слой экстрагировали дополнительным количеством дихлорметана (2,25 л). Объединенные органические вещества затем концентрировали и получающееся в результате масло растворяли в пропан-2-оле (4,5 л). Затем добавляли водный гидроксид натрия (2 М, 6,93 л, 13,9 моль) и эту бифазную смесь кипятили с обратным холодильником в течение 65 ч. После охлаждения до комнатной температуры фазы разделяли и водный слой экстрагировали этилацетатом (4,5 л). Объединенные органические слои затем промывали насыщенным водным раствором хлорида натрия (4,5 л) и концентрировали в вакууме. Остаток разбавляли этилацетатом (9 л) и вновь концентрировали в вакууме. Наконец добавляли дополнительное количество этилацетата (4,5 л) и получающуюся в результате суспензию перемешивали при 0-5C в течение 1 ч, фильтровали и промывали охлажденным этилацетатом (2450 мл). Твердый продукт затем сушили в вакуумной печи при 40C с получением указанного в заголовке соединения (547,2 г, 1,24 моль, 89,8%). Данные LRMS указанного в заголовке соединения были идентичны данным для соединения, указанного в заголовке подготовительного примера 15. Биологические данные Способность соединений формулы (I) и их фармацевтически приемлемых солей, сольватов и производных модулировать активность хемокинового рецептора продемонстрирована с использованием методики, известной из уровня техники, такой как методика с использованием анализа связывания CCR5 с последующими процедурами, описанными Combadiere et al. в J. Leukoc. Biol., 60, 147-52 (1996); и/или- 24009941 путем использования анализов мобилизации внутриклеточного кальция, как описано теми же авторами. Клеточные линии, экспрессирующие интересуемый рецептор, включают линии, которые в природе экспрессируют рецептор, такие как PM-1, или стимулированные интерлейкином-2 (IL-2) лимфоциты периферической крови (PBL), или клетки, сконструированные для экспрессии рекомбинантного рецептора,такие как клетки яичника китайского хомячка (CHO) 300.19, L1.2 или НЕК-293. Соединение примера 4 при тестировании с использованием анализа связывания CCR5 в соответствии с Combadiere et al (выше) продемонстрировало значения IC50 7,5 нМ (MIP-1), 7,3 нМ (MP-1) и 6,7 нМ (RANTES). Соединение примера 5 при тестировании с использованием анализа связывания CCR5 в соответствии с Combadiere et al (выше) продемонстрировало значения IC50 2,7 нМ (MIP-1), 2,4 нМ (MIP-1) и 1,9 нМ (RANTES). Соединения всех примеров при тестировании с использованием анализа мобилизации внутриклеточного кальция в соответствии с Combadiere et al (выше) представляли собой сильные антагонисты со значениями IC50 менее чем 100 нМ (MIP-1). Фармакологическая активность соединений формулы (I) и их фармацевтически приемлемых солей,сольватов и производных дополнительно продемонстрирована с использованием анализа индуцированного gpl60 межклеточного слияния с определением значений IC50 соединений против слияния с ВИЧ-1. В анализе индуцированного gpl60 межклеточного слияния используют клеточную линию HeLa Р 4 и клеточную линию CHO-Tat10. Клеточная линия HeLa Р 4 экспрессирует CCR5 и CD4 и была трансфицирована LTR-галактозидазой ВИЧ-1. Среды для этой клеточкой линии представляют собой модифицированную по Дульбекко среду Игла (D-MEM) (без L-глутамина), содержащую 10% фетальную телячью сыворотку(FCS), 2 мМ L-глутамин пенициллин/стрептомицин (Pen/Strep; 100 Е/мл пенициллина плюс 10 мг/мл стрептомицина) и 1 мкг/мл пуромицина. Клеточная линия CHO представляет собой Tat (транскрипционный транс-активатор)экспрессирующий клон, полученный из клеточной линии CHO JRR17.1, которая была трансфицирована плазмидойpTat puro. Среда для этой клеточной линии представляет собой обогащенную среду для культуры клеток млекопитающих, исходно разработанной в Roswell Park Memorial Institute RPMI1640 (без L-глутамина),содержащей 10% FCS, 2 мМ L-глутамин, 0,5 мг/мл гигромицина В и 12 мкг/мл пуромицина. Линия CHOJRR17.1 экспрессирует gp160 (JRFL) и представляет собой клон, который был выбран благодаря его способности к слиянию с клеточной линией, экспрессирующей CCR5/CD4. При слиянии клеток Tat, присутствующий в клетках CHO, способен трансактивировать длинный концевой повтор (LTR) ВИЧ-1, присутствующий в клетках HeLa, приводя к экспрессии фермента галактозидазы. Затем эту экспрессию измеряют с использованием набора для анализа -галактозидазы в качестве репортера Fluor Асе (Bio-Rad кат.170-3150). Этот набор представляет собой количественный флуоресцентный анализ, который определяет уровень экспрессии -галактозидазы с использованием в качестве субстрата 4-метилумбеллиферулгалактопиранозида (MUG). -Галактозидаза гидролизует флуорогенный субстрат, приводя в результате к высвобождению флуоресцирующей молекулы 4 метилумбеллиферона (4MU). Флуоресценцию 4-метилумбеллиферона затем измеряют на фотометре с использованием длины волны возбуждения 360 нм и длины волны испускания 460 нм. Соединения, которые ингибируют слияние, вызывают уменьшение сигнала, и после растворения в подходящем растворителе и разбавления культуральной средой кривая зависимости доза-ответ для каждого соединения может быть использована для расчета значений IC50. Все соединения примеров по данному изобретению имеют значения IC50 в соответствии с вышеописанным способом менее чем 10 нМ. Соединения примеров 1 и 5 имеют значения IC50, соответственно 130 и 120 пм. Данные дифракции рентгеновских лучей на порошке (PXRD) Все картины PXRD были получены на порошковом дифрактометре Bruker D5000 в пределах диапазона угла 2-тета 2-55 с шагом 0,02. Образцы вращали, в то же время облучая их рентгеновскими лучами CuK1 (длина волны равна 1,5046 ангстрем), отфильтрованными через графитовый монохроматор ( равен 0,15405 нм), и с использованием рентгеновской трубки, функционирующей при 40 кВ/40 мА. Дифрактометр калибровали с использованием стандартного кварцевого образца перед и после сбора данных для каждого образца. Основные пики (в градусах 2-тета) картин PXRD для примеров 10, 11 и 12 проиллюстрированы в следующих таблицах.- 25009941 Таблица 1. Данные относительно пиков PXRD для N-(1S)-3-[3-эндо -(5-изобутирил-2-метил-4,5,6,7 тетрагидро-1H-имидазо[4,5-с]пиридин-1-ил)-8-азабицикло [3.2.1]окт-8-ил]-1-(3-фторфенил)пропилацетамида Таблица 2. Данные относительно пиков PXRD для фумарата N-(1S)-3-[3-эндо-(5-изобутирил-2-метил 4,5,6,7-тетрагидро-1H-имидазо[4,5-с]пиридин-1-ил)-8-азабицикло[3.2.1]окт-8-ил]-1-(3 фторфенил)пропилацетамида Таблица 3. Данные относительно пиков PXRD для (D)-тартрата N-(1S)-3-[3-эндо-(5-изобутирил-2 метил-4,5,6,7-тетрагидро-1H-имидазо[4,5-с]пиридин-1-ил)-8-азабицикло[3.2.1]окт-8-ил]-1-(3 фторфенил)пропилацетамида- 26009941 Моделирование картины PXRD для фумарата N-(1S)-3-[3-эндо-(5-изобутирил-2-метил-4,5,6,7 тетрагидро-1H-имидазо[4,5-с]пиридин-1-ил)-8-азабицикло[3.2.1]окт-8-ил]-1-(3-фторфенил)пропилацетамида включало углы 2-тета, межплоскостное расстояние и относительные интенсивности, которые рассчитывали из структуры его монокристалла с использованием "Reflex Powder Diffraction" модуль Accelrys Materials Studio [версия 2.2]. Подходящие параметры модели представляли собой: Длина волны равна 1,540562(Cu K) Поляризационный фактор равен 0,5 Профиль псевдо-Фойгта (U равен 0,01, V равен -0,001, W равен 0,002) Основные пики (в градусах 2-тета) моделированной картины PXRD для фумарата N-(1S)-3-[3 эндо-(5-изобутирил-2-метил-4,5,6,7-тетрагидро-1H-имидазо[4,5-с]пиридин-1-ил)-8-азабицикло[3.2.1]окт 8-ил]-1-(3-фторфенил)пропилацетамида представлены в таблице 4. Таблица 4. Моделированные данные относительно пиков для фумарата N-(1S)-3-[3-эндо-(5 изобутирил-2-метил-4,5,6,7-тетрагидро-1H-имидазо[4,5-с]пиридин-1-ил)-8-азабицикло[3.2.1]окт-8-ил]-1(3-фторфенил)пропилацетамида Данные дифференциальной сканирующей калориметрии Все данные дифференциальной сканирующей калориметрии (ДСК) были получены на приборе для ДСК Perkin Elmer PYRIS Diamond с автоматическим пробоотборником, с использованием газового потока азота. Образцы помещали в алюминиевые чашечки объемом 50 мкл с отверстиями и крышками и нагревали от 10 до 300C со скоростью 20C мин-1.N-1S)-3-[3-эндо-(5-Изобутирил-2-метил-4,5,6,7-тетрагидро-1H-имидазо[4,5-с]пиридин-1-ил)-8 азабицикло[3.2.1]окт-8-ил]-(3-фторфенил)пропилацетамид Размер образца 3,016 мг Эндотермический пик при 118C - плавление Фумарат N-1S)-3-[3-эндо-(5-изобутирил-2-метил-4,5,6,7-тетрагидро-1H-имидазо[4,5-с]пиридин-1 ил)-8-азабицикло[3.2.1]окт-8-ил]-1-(3-фторфенил)пропил)ацетамида Размер образца 2,905 мг Эндотермический пик при 219 С - плавление Эндотермическое явление при 228 С Экзотермическое явление при 246C(D)-Тартрат N-1S)-3-[3-эндо-(5-изобутирил-2-метил-4,5,6,7-тетрагидро-1H-имидазо[4,5-с]пиридин 1-ил)-8-азабицикло[3.2.1]окт-8-ил]-1-(3-фторфенил)пропилацетамида Размер образца 2,979 мг Эндотермический пик при 217Cплавление или его фармацевтически приемлемые соль, сольват, гдеR2 представляет собой C1-С 6 алкил или С 3-С 7 циклоалкил, где указанный алкил возможно замещен 2. Соединение по п.1, где R1 представляет собой С 1-С 4 алкил. 3. Соединение по п.1 или 2, где R1 представляет собой метил. 4. Соединение по любому из пп.1-3, где R2 представляет собой С 1-С 4 алкил, возможно замещенный 5. Соединение по любому из пп.1-4, где R2 представляет собой метил, этил или изопропил. 6. Соединение по любому из пп.1-3, где R2 представляет собой циклопропил или циклобутил. 7. Соединение по п.1, выбранное изN-(1S)-3-[3-эндо-(2-метил-5-(3,3,3-трифторпропионил)-4,5,6,7-тетрагидро-1H-имидазо[4,5-с]пиридин-1-ил)-8-азабицикло[3.2.1]окт-8-ил]-1-(3-фторфенил)пропилацетамида и их фармацевтически приемлемых солей, сольватов. 8. Фармацевтическая композиция, включающая соединение формулы (I) или его фармацевтически приемлемые соль, сольват по любому из пп.1-7 вместе с одним или более чем одним фармацевтически приемлемым эксципиентом, разбавителем или носителем. 9. Фармацевтическая композиция по п.8, включающая один или более чем один дополнительный терапевтический агент. 10. Применение соединения формулы (I) или его фармацевтически приемлемых соли, сольвата по любому из пп.1-7 в качестве лекарства. 11. Применение соединения формулы (I) или его фармацевтически приемлемых соли, сольвата по любому из пп.1-7 для лечения расстройства, в которое вовлечена модуляция CCR5 рецепторов. 12. Применение по п.11, где расстройство представляет собой вирус иммунодефицита человека(ВИЧ), ретровирусную инфекцию, генетически родственную ВИЧ, синдром приобретенного иммунодефицита (СПИД) или воспалительное заболевание. 13. Применение по п.11, где расстройство представляет собой рассеянный склероз, ревматоидный артрит или отторжение трансплантата. 14. Применение по п.11, где расстройство представляет собой воспалительное заболевание кишечника, эндометриоз, диабет I типа, почечные заболевания, фиброз, хронический панкреатит, воспалительные легочные состояния, энцефалит, хроническую сердечную недостаточность, ишемическую болезнь сердца, псориаз, удар, ожирение, заболевания центральной нервной системы (ЦНС), анемию, рестеноз,атеросклеротические бляшки, атопический дерматит, хронический панкреатит, рак, боль или реакцию на стресс в результате хирургического вмешательства, инфекции, повреждения или другого травматического поражения. 15. Применение по п.11, где расстройство представляет собой вирус гепатита В (HBV), вирус гепа- 28009941 тита С (HCV), чуму, поксвирус, токсоплазмоз, микобактериальную, трипаносомную инфекцию, пневмонию или цитоспоридиоз (cytosporidiosis). 16. Применение соединения формулы (I) или его фармацевтически приемлемых соли, сольвата по любому из пп.1-7 для изготовления лекарства для лечения расстройства, в которое вовлечена модуляцияCCR5 рецепторов, включающий лечение указанного млекопитающего эффективным количеством соединения формулы (I) или его фармацевтически приемлемых соли, сольвата по любому из пп.1-7. 18. Способ получения соединения формулы (I) или его соли, сольвата по любому из пп.1-7, включающий взаимодействие соединения формулы (II) в обычных условиях сочетания карбоновой кислоты/амина; где R1 и R2 являются такими, как определено в п.1 для соединения формулы (I). 19. Способ получения соединения формулы (I) или его соли, сольвата по любому из пп.1-7, включающий взаимодействие соединения формулы (XVI) в обычных условиях сочетания карбоновой кислоты/амина; где R1 и R2 являются такими, как определено в п.1 для соединения формулы (I). 20. Способ получения соединения формулы (I) или его соли, сольвата по любому из пп.1-7, включающий восстановительное аминирование альдегида формулы (XIX) в обычных условиях; где R1 и R2 являются такими, как определено в п.1 для соединения формулы (I). 21. Способ получения соединения формулы (I) или его соли, сольвата по любому из пп.1-7, включающий восстановительное аминирование нитрила формулы (XXI) в обычных условиях; где R1 и R2 являются такими, как определено в п.1 для соединения формулы (I). 22. Способ получения соединения формулы (I) или его соли, сольвата по любому из пп.1-7, включающий алкилирование амина формулы (XX) или его соли соединением формулы (XXII) в обычных условиях алкилирования; где R1 и R2 являются такими, как определено в п.1 для соединения формулы (I), и Lg представляет собой уходящую группу, подходящую для алифатического нуклеофильного замещения. 23. Способ получения соединения формулы (I) или его соли, сольвата по любому из пп.1-7, включающий асимметрическое восстановление енамида формулы (XXIII) в обычных восстанавливающих условиях; где R1 и R2 являются такими, как определено в п.1 для соединения формулы (I). 24. Способ получения соединения формулы (I) или его соли, сольвата по любому из пп.1-7, включающий взаимодействие амина формулы (II) или его соли с металлом (то есть депротонированной формы) со сложным эфиром формулы (XXIV) в обычных условиях; где R1 и R2 являются такими, как определено в п.1 для соединения формулы (I), и EsGp представляет собой группу, образующую сложный эфир. 25. Комбинация соединения по любому из пп.1-7 или его фармацевтически приемлемых соли или
МПК / Метки
МПК: C07D 471/04, A61K 31/4188, A61P 31/18
Метки: иммунодефицита, обладающие, вируса, активностью, вич, рецептора, антагониста, имидазопиридинзамещённые, человека, воспаления, производные, тропановые, лечения
Код ссылки
<a href="https://eas.patents.su/30-9941-imidazopiridinzameshhyonnye-tropanovye-proizvodnye-obladayushhie-aktivnostyu-antagonista-ccr5-receptora-dlya-lecheniya-virusa-immunodeficita-cheloveka-vich-i-vospaleniya.html" rel="bookmark" title="База патентов Евразийского Союза">Имидазопиридинзамещённые тропановые производные, обладающие активностью антагониста ccr5 рецептора, для лечения вируса иммунодефицита человека (вич) и воспаления</a>
Производные пиразола для лечения инфекции вируса иммунодефицита человека (вич)

Номер патента: 7184
Опубликовано: 25.08.2006
Авторы: Маубрей Чарлз Эрик, Прайс Дейвис Энтони, Стаппл Пол Энтони, Селби Мэттью Данкан, Джоунз Лин Хауард
МПК: A61K 31/415, A61K 31/4162, A61K 31/4155...
Метки: инфекции, вируса, производные, пиразола, иммунодефицита, человека, лечения, вич
Формула / Реферат:
1. Соединение формулы (I) или его фармацевтически приемлемые соль, сольват или производное, где либо R1 представляет собой Н, С1-С6алкил, С3-С7циклоалкил, фенил, бензил, галогено, -CN, -OR7, -CO2R10, -CONR5R10, R8 или R9, причем указанные С1-С6алкил, С3-С7циклоалкил, фенил и бензил возможно замещены галогено, -CN, -OR10, S(O)xR10, -CO2R10, -CONR5R10, -OCONR5R10, -NR5CO2R10, -NR10R11, -NR5COR10, -SO2NR5R10,-NR5CONR5R10, -NR5SO2R10 или R10; и R2...
Синтез 1,3 – оксаселеноланнуклеозидов, обладающих активностью против вируса иммунодефицита человека и против вируса гепатита-b

Номер патента: 5097
Опубликовано: 28.10.2004
Авторы: Шинази Раймонд Ф., Ду Джинфа, Чу Чунг К.
МПК: A61P 31/18, C07D 329/00, A61K 31/513...
Метки: иммунодефицита, обладающих, активностью, человека, вируса, против, гепатита-b, оксаселеноланнуклеозидов, синтез
Формула / Реферат:
1. 1,3-Оксаселеноланлактон формулы где R обозначает водород, ацил, моно-, ди- или триэфир фосфорной кислоты, стабилизированный фосфат или остаток липида, или его фармацевтически приемлемая соль. 2. Способ получения 1,3-оксаселеноланлактона формулы где R обозначает водород, ацил, моно-, ди- или триэфир фосфорной кислоты, стабилизированный фосфат или остаток липида, или его фармацевтически приемлемой соли, включающий a) получение димера селенола...
Производные хиноксалин-2,3-диона и их фармацевтически приемлемые соли, фармацевтические композиции, обладающие антагонистической активностью в отношении рецептора глютамата и противосудорожной активностью, способы лечения пациентов, страдающих от удара или заболеваний, при помощи этих соединений.

Номер патента: 762
Опубликовано: 24.04.2000
Авторы: Никам Шам, Корнберг Брайэн Эдвард, Рафферти Майкл Фрэнсис
МПК: C07D 241/44
Метки: заболеваний, активностью, глютамата, фармацевтически, пациентов, композиции, этих, лечения, соли, способы, антагонистической, производные, соединений, фармацевтические, обладающие, удара, рецептора, приемлемые, помощи, страдающих, противосудорожной, хиноксалин-2,3-диона, отношении
Формула / Реферат:
1. Производные хиноксалин-2,3-диона общей формулы I где R - группа - NR4R5, где R4 и R5 независимо друг от друга означают водород, низший алкил, незамещенный или замещенный низшим алкилом, низший циклоалкил, который может содержать 1 или 2 атома кислорода в качестве гетероатома, R1 и R2 независимо друг от друга означают водород или нитрогруппу, R3 - низший алкил, при этом радикалы R3 и R-CH2- могут каждый находиться в положениях 5 или...
Производные пиримидина в качестве ингибиторов репликации вируса иммунодефицита человека

Номер патента: 2973
Опубликовано: 26.12.2002
Авторы: Хо Чих Юнг, Андрис Кунрад Йозеф Лодевейк Марсель, Янссен Марсель Огюст Констант, Де Корт Барт, Хэрес Ян, Лудовики Дональд Уилльям, Ван Акен Кун Жанн Альфонс, Де Жонж Марк Рене, Янссен Поль Адриан Жан, Койманс Люсьен Мария Хенрикус, Кукла Майкл Джозеф
МПК: C07D 239/50, A61P 31/18, A61K 31/505...
Метки: иммунодефицита, человека, качестве, пиримидина, ингибиторов, репликации, вируса, производные
Формула / Реферат:
1. Применение соединения формулы его N-оксида, фармацевтически приемлемой соли присоединения или стереохимически изомерной формы, где А является СН или N; n равен 0, 1, 2, 3 или 4; Q является водородом или -NR1R2; R1 и R2, каждый независимо, выбран из водорода, гидрокси, C1-12aлкила, С1-12алкилокси, С1-12алкилкарбонила, С1-12алкилоксикарбонила, где каждая из указанных С1-12алкильных групп необязательно и каждая независимо может быть замещена...
Производные триазола, обладающие противогрибковой активностью, предназначенные для человека и животных

Номер патента: 3189
Опубликовано: 27.02.2003
Авторы: Фраире Кристина, Скиоппакасси Джованна, Наполетано Маоро, Альбини Энрико
МПК: A61K 31/4196, C07D 249/08, A61P 31/10...
Метки: предназначенные, животных, активностью, человека, производные, триазола, противогрибковой, обладающие
Формула / Реферат:
1. Соединение, выбранное из группы, содержащей (2R,3S) 1-(1H-1,2,4-триазолил)-2-(2,4-дифторфенил)-3-этил-4-(1,1,2,2-тетрафторэтокси)-2-бутанол, (2R,3S) 1-(1H-1,2,4-триазолил)-2-(2,4-дихлорфенил)-3-метил-4-(1,1,2,2-тетрафторэтокси)-2-бутанол, (2R,3R) 1-(1H-1,2,4-триазолил)-2-(2,4-дихлорфенил)-3-метил-4-(1,1,2,2-тетрафторэтилтио)-2-бутанол, (2R,3S) 1-(1H-1,2,4-триазолил)-2-(2,4-дифторфенил)-3-метил-4-(1,1,2,2-тетрафторэтокси)-2-бутанол, (2R)...
Предыдущий патент: Производные малонамида, блокирующие активность γ- секретазы
Следующий патент: Азабициклические производные в качестве антагонистов мускаринового рецептора
Случайный патент: Устройство для электрогидравлической системы управления паровой турбиной