Полициклические соединения как эффективные антагонисты альфа2-адренорецептора
Номер патента: 8537
Опубликовано: 29.06.2007
Авторы: Дин Белл Дэвид, Хаапалинна Антти, Толванен Арто, Йокела Рея, Карьялайнен Арто, Ратилайнен Яри, Саллинен Юкка























Формула / Реферат
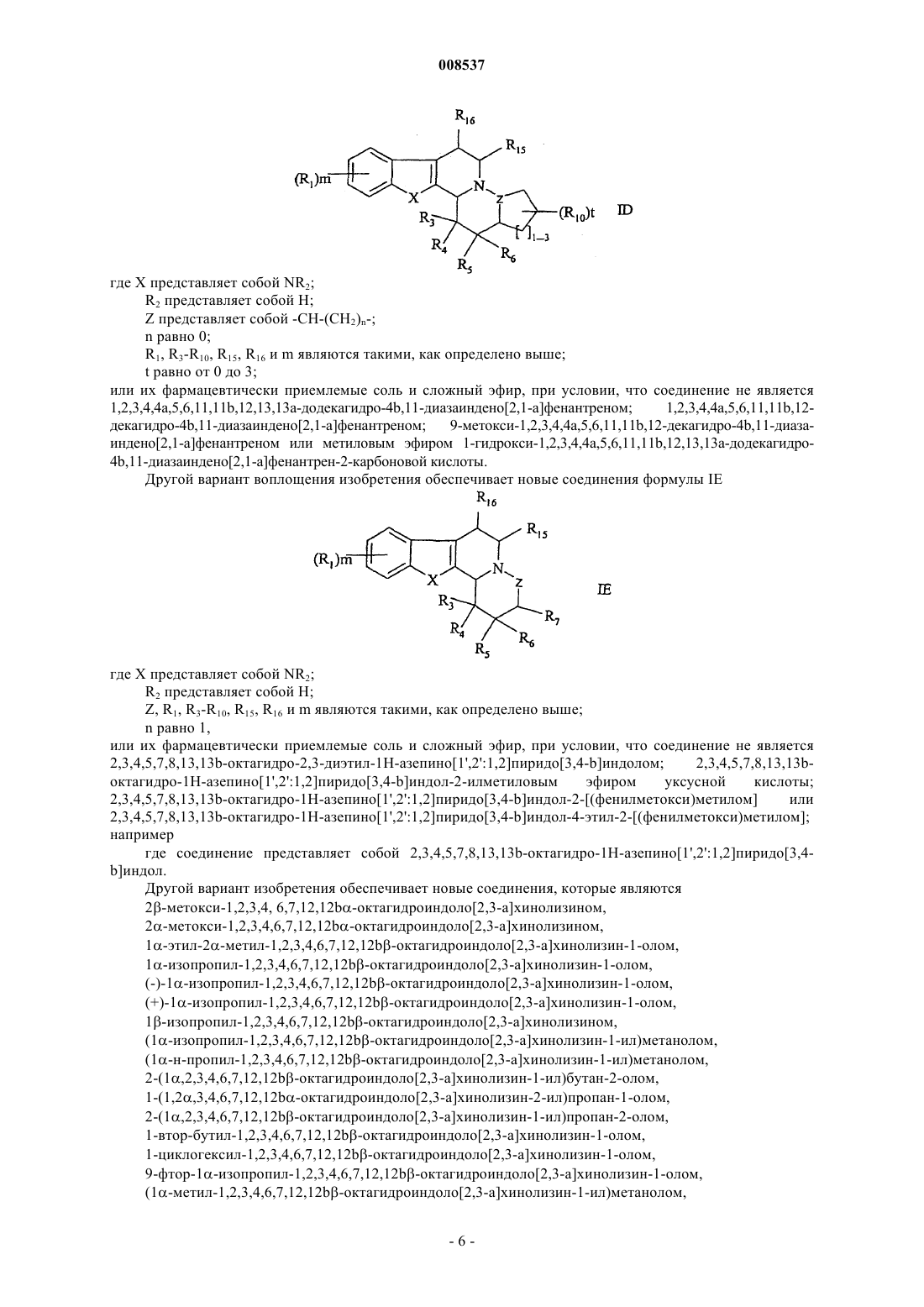
1. Соединение формулы IA

где X представляет собой CR2R2', О или S;
Z представляет собой -CHR8-(CH2)n- или простую связь;
R1 представляет собой гидрокси, (C1-C6)алкил, (C1-C6)алкокси, галоген, галоген(C1-C6)алкил, (C1-C6)алкокси-СО-, CN, NO2, NH2, моно- или ди(C1-C6)алкиламино или карбоксильную группу;
R2 и R2' представляют собой независимо Н, гидрокси или (C1-C6)алкил или R2 и R2' образуют вместе с атомами углерода, образующими кольцо, к которым они присоединены, карбонильную группу;
R3 представляет собой гидрокси, (C1-C6)алкил, (C2-C6)алкенил, гидрокси(C1-C6)алкил, (C1-C6)алкокси, (C1-C6)алкокси(C1-C6)алкил, гидрокси(C1-C6)алкокси(C1-C6)алкил, (C3-C7)циклоалкил, (C3-C7)циклоалкил(C1-C6)алкил, арил, арил(C1-C6)алкил, арилокси, арил(C1-C6)алкокси, арилокси(C1-C6)алкил, арил(C1-C6)алкокси(C1-C6)алкил, галоген(C1-C6)алкил, NH2, амино(C1-C6)алкил, моно- или ди(C1-C6)алкиламино, моно- или ди(C1-C6)алкиламино(C1-C6)алкил, (C1-C6)алкил-СО-, (C1-C6)алкил-СО-O-, (C1-C6)алкил-СО-O-(C1-C6)алкил, (C1-C6)алкокси-СО-, (C1-C6)алкокси-СО-(C1-C6)алкил, (C1-C6)алкокси-СО-(C1-C6)алкокси(C1-C6)алкил, карбамоил, моно- или ди(C1-C6)алкилкарбамоил, карбоксил или (C1-C6)алкил-S-(C1-C6)алкил, где указанный (C3-C7)циклоалкил или арил являются незамещенными или замещенными 1 или 2 заместителями, каждый из которых независимо представляет собой гидрокси, (C1-C6)алкил, галоген, (C1-C6)алкокси, NH2, CN или NO2, или один из R3 или R4 и R6 вместе образуют связь между атомами кольца, к которым они присоединены;
R4 представляет собой гидрокси, (C1-C6)алкил, гидрокси(C1-C6)алкил, (C1-C6)алкокси или (C1-C6)алкокси(C1-C6)алкил;
R5 представляет собой Н;
R6 представляет собой Н или R6 образует связь между атомом кольца, к которому он присоединен, и атомом кольца, к которому присоединен R7;
R7 представляет собой Н, гидрокси, (C1-C6)алкил, гидрокси(C1-C6)алкил, (C1-C6)алкокси или (C1-C6)алкокси(C1-C6)алкил;
R8 представляет собой Н;
R15 представляет собой Н, (C1-C6)алкил, (С2-C6)алкенил, гидрокси(C1-C6)алкил, (C1-C6)алкокси(C1-C6)алкил, гидрокси(C1-C6)алкокси(C1-C6)алкил, галоген(C1-C6)алкил, амино(C1-C6)алкил, моно- или ди(C1-C6)алкиламино(C1-C6)алкил, (C1-C6)алкил-СО-, (C1-C6)алкил-СО-O-(C1-C6)алкил, (C1-C6)алкокси-СO-, (C1-C6)алкокси-СО-(C1-C6)алкил, (C1-C6)алкокси-СО-(C1-C6)алкокси(C1-C6)алкил, карбамоил, моно- или ди(C1-C6)алкилкарбамоил или карбоксильную группу;
R16 представляет собой Н или (C1-C6)алкил;
R7 и R8 присоединены к атомам углеродного кольца, которые являются соседними;
m равно 0-2; и
n равно 0 или 1,
или его фармацевтически приемлемая соль или сложный эфир.
2. Соединение по п.1, где X представляет собой CR2R2'.
3. Соединение по п.1, где X представляет собой О.
4. Соединение по п.1, где X представляет собой S.
5. Соединение по любому из пп.1-4, где R3 представляет собой гидрокси, (C1-C6)алкил, гидрокси(C1-C6)алкил, (C1-C6)алкокси(C1-C6)алкил, (C1-C6)алкокси-СО- или (C1-C6)алкил-СО-О-(C1-C6)алкил и R4 представляет собой (C1-C6)алкил или гидрокси(C1-C6)алкил.
6. Соединение по любому из пп.1-5, где R3 представляет собой гидрокси, гидрокси(C1-C6)алкил или (C1-C6)алкокси(C1-C6)алкил и R4 представляет собой (C1-C6)алкил.
7. Соединение по п.1, где соединение представляет собой
1a-метил-1,3,4,5,6,11b-гексагидро-2Н-11-окса-4а-азабензо[а]флуорен-1-ол,
(1a-метил-1,3,4,5,6,11bb-гексагидро-2Н-11-окса-4а-азабензо[а]флуорен-1-ил)метанол,
(-)-(1a-метил-1,3,4,5,6,11bb-гексагидро-2Н-11-окса-4а-азабензо[а]флуорен-1-ил)метанол,
(+)-(1a-метил-1,3,4,5,6,11bb-гексагидро-2Н-11-окса-4а-азабензо[а]флуорен-1-ил)метанол,
1a-изопропил-1,3,4,5,6,11b-гексагидро-2Н-11-окса-4а-азабензо[а]флуорен-1-ол,
1a-этил-1,3,4,5,6,11ba-гексагидро-2Н-11-окса-4а-азабензо[а]флуорен-1-ол,
(1a-этил-1,3,4,5,6,11bb-гексагидро-2Н-11-окса-4а-азабензо[а]флуорен-1-ил)метанол,
(1-гидроксиметил-1,3,4,5,6,11b-гексагидро-2Н-11-окса-4а-азабензо[а]флуорен-1-ил)метанол,
1-метоксиметил-1a-метил-1,3,4,5,6,11bb-гексагидро-2Н-11-окса-4а-азабензо[а]флуорен,
(-)-1-метоксиметил-1a-метил-1,3,4,5,6,11bb-гексагидро-2Н-11-окса-4а-азабензо[а]флуорен,
(+)-1-метоксиметил-1a-метил-1,3,4,5,6,11bb-гексагидро-2Н-11-окса-4а-азабензо[а]флуорен,
этиловый эфир 1a-метил-1,3,4,5,6,11ba-гексагидро-2Н-11-окса-4а-азабензо[а]флуорен-1-карбоновой кислоты,
1-этоксиметил-1a-метил-1,3,4,5,6,11bb-гексагидро-2Н-11-окса-4а-азабензо[а]флуорен,
(1a-метил-1,3,4,5,6,11ba-гексагидро-2Н-11-окса-4а-азабензо[а]флуорен-1-ил)метанол,
(-)-(1a-метил-1,3,4,5,6,11ba-гексагидро-2Н-11-окса-4а-азабензо[а]флуорен-1-ил)метанол,
(+)-(1a-метил-1,3,4,5,6,11ba-гексагидро-2Н-11-окса-4а-азабензо[а]флуорен-1-ил)метанол,
метиловый эфир 1a-этил-1,3,4,5,6,11ba-гексагидро-2Н-11-окса-4а-азабензо[а]флуорен-1-карбоновой кислоты,
1-метоксиметил-1a-метил-1,3,4,5,6,11ba-гексагидро-2Н-11-окса-4а-азабензо[а]флуорен,
(-)-1-метоксиметил-1a-метил-1,3,4,5,6,11ba-гексагидро-2Н-11-окса-4а-азабензо[а]флуорен,
(+)-1-метоксиметил-1a-метил-1,3,4,5,6,11ba-гексагидро-2Н-11-окса-4а-азабензо[а]флуорен,
(1a-этил-1,3,4,5,6,11ba-гексагидро-2Н-11-окса-4а-азабензо[а]флуорен-1-ил)метанол,
1a-метил-1,3,4,5,6,11bb-гексагидро-2Н-11-окса-4а-азабензо[а]флуорен-1-илметиловый эфир уксусной кислоты или
(1a-метил-1,2,3,4,6,7,12,12ba-октагидроиндено[2,1-а]хинолизин-1-ил)метанол.
8. Соединение по любому из пп.1, 3, 5, 6 или 7, где соединение является [(-)-(1a-метил-1,3,4,5,6,11bb-гексагидро-2Н-11-окса-4а-азабензо[а]флуорен-1-ил)метанолом].
9. Соединение по любому из пп.1, 3, 5, 6 или 8, где соединение представляет собой [(-)-1-метоксиметил-1a-метил-1,3,4,5,6,11bb-гексагидро-2Н-11-окса-4а-азабензо[а]флуорен].
10. Фармацевтическая композиция, содержащая по меньшей мере одно соединение по любому из пп.1-9 и фармацевтически приемлемый разбавитель, носитель и/или эксципиент.
11 Применение соединения по любому из пп.1-9 для изготовления лекарственного средства, пригодного в качестве селективного антагониста альфа2С.
12. Применение по п.11, где лекарственное средство используется для лечения психического растройства, вызванного стрессом, болезни Паркинсона, дипрессии, шизофрении, синдрома гиперактивности с дифицитом внимания, посттравматического стресса, тревожного расстройства, синдрома Тауретта, блефароспазма и других фокальных нарушений тонуса, височной эпилепсии с психозом, психоза, обусловленного действием наркотиков, заболевания Хантингтона, заболеваний, вызванных колебаниями половых гормонов, или панического расстройства.
Текст
008537 Настоящее изобретение относится к фармакологически активным производным арилхинолизина и родственным соединениям, к их фармацевтически приемлемым солям и сложным эфирам, а также к фармацевтическим композициям, содержащим их, и к их использованию в качестве альфа 2 антагонистов. Некоторые соединения, проявляющие альфа адренергическую активность, хорошо известны в данной области. Как также общеизвестно и принято в данной области, такие соединения могут быть использованы для лечения большого множества заболеваний и состояний периферической системы и центральной нервной системы (ЦНС). Альфа адренергические рецепторы могут быть разделены на фармакологической основе на альфа 1 и альфа 2-адренорецепторы, которые оба могут быть разделены на подтипы. Три генетически кодируемых подтипа, а именно альфа 2 А-, альфа 2 В- и альфа 2 С-адренорецепторы, обнаружены у человека. Соответственно, альфа 2-адренорецепторы человека были подразделены на три фармакологических подтипа,известных как альфа 2 А-, альфа 2 В- и альфа 2 С-адренорецепторы. Четвертый, фармакологически определенный подтип, альфа 2D известен у грызунов и некоторых других млекопитающих и он соответствует генетически определенным альфа 2 А-адренорецепторам. Подтипы альфа 2-адренорецепторов имеют разное распределение в тканях и функциональные роли. Например, тогда как альфа 2 А-адренорецепторы широко экспрессируются в различных тканях, альфа 2 Садренорецепторы сконцентрированы в ЦНС и, видимо, они играют роль в модулировании специфических, опосредованных ЦНС поведенческих и физиологических реакций. Соединения, которые являются неспецифичными к любому из приведенных выше подтипов альфа 2 и соединения, которые являются специфичными к определенным подтипам альфа 2, уже известны. Например, атипамезол представляет собой неспецифический альфа 2-антагонист. Атипамезол описан, например, в ЕР-А-183492 (сравните р.13, соединение XV) и Haapalinna, A. et al., Naunyn-Schmiedeberg'sArch. Pharmacol. 356 (1997), 570-582. Патент США 5902807 описывает соединения, которые являются селективными антагонистами подтипа альфа 2 С и могут быть использованы в лечении психических заболеваний, например психических расстройств, индуцированных стрессом. Такие соединения включают,например, МК-912 и ВАМ-1303. Кроме того, WO-A-9928300 описывает замещенные производные имидазола, имеющие агонист-подобную активность в отношении альфа 2 В- или 2 В/2 С-адренорецепторов. Кроме того, WO 01/64645 относится к производным хинолина, полезных в качестве альфа 2 антагонистов, а также к селективным агентам альфа 2 С-антагонистам. Описания всех документов, цитированных выше в данном разделе, включены в данное описание в виде ссылки. Некоторые производные арилхинолизина и связанные соединения описаны в литературе, некоторые из которых обладают полезным фармацевтическим действием. Например, патенты США 4806545 и 4044012 описывают 1,1-дизамещенные индоло[2,3-а]хинолизидины как полезные в качестве сосудорасширяющих и противогипоксических агентов. Далее замещенные производные арилхинолизина, описанные, например, в патенте США 4686226, обладающие альфа 2-адренорецептор антагонистической активностью, являются полезными, например, в качестве антидепрессантов, гипотензивных или противодиабетических агентов или ингибиторов агрегации тромбоцитов. Кроме того, патент США 3492303 относится к индоло[2,3-а]хинолизидинам, полезным в качестве супрессоров центральной нервной системы. Молекулярное моделирование мишеней для синтеза альфа 1 А и альфа 2 селективных лигандов обсуждается уGriffith, R. et al., J. Comput.-Aided Mol. Design 13 (1999), 69-78. Целью настоящего изобретения является обеспечение новых антагонистов альфа 2 адренорецепторов, которые могут быть использованы для лечения заболеваний или состояний периферической или центральной нервной системы, при которых показано, что альфа 2-антагонисты являются полезными. Соответственно, целью настоящего изобретения является обеспечение новых соединений для использования в качестве агентов - альфа 2-антагонистов в лечении млекопитающих, включая людей и животных. Также изобретение обеспечивает соединения, полезные в качестве агентов - альфа 2 С-антагонистов для лечения различных расстройств или состояний центральной нервной системы, для которых показано,что альфа 2 С-антагонисты являются полезными. Фиг. 1 а и b показывают результаты двух отдельных тестов двигательной активности, где двигательную активность мышей исследовали после инъекций растворителя (носителя) или амфетамина (амф)(4 мкмоль/кг). Мышей заранее обрабатывали (за 20 мин до амфетамина) растворителем, подтипом неселективного альфа 2-антагониста атипамезолом (1 мкмоль/кг) или альфа 2 С-антагонистами, соединением K(3 мкмоль/кг) (фиг. 1 а) или соединением L (3 мкмоль/кг) (фиг. 1b). р 0,05, р 0,01 и р 0,001 по сравнению с группой растворитель+амф - (односторонний ANOVA+LSD-тест). Фиг. 2 показывает индуцированный альфа 2-агонистом седативный эффект (измеряемый как двигательное угнетение) у мышей. Неселективный альфа 2-антагонист атипамезол (Ати, Ati) противодействовал седативным эффектам альфа 2-подтипа неселективного агониста дексмедетомидина (Декс; 50 нмоль/кг п/к), тогда как селективные альфа 2-антагонисты не имели достоверных эффектов(раств.=растворитель). (р 0,001, по сравнению с Декс+растворитель). Фиг. 3 показывает действие альфа 2 С-селективных антагонистов соединения K (3 мкмоль/кг) и со-1 008537 единения L (3 мкмоль/кг), неселективного антагониста атипамезола (10 мкмоль/кг) и стандартных антидепрессантов дезипрамина (10 мкмоль/кг) и флуоксетина (10 мкмоль/кг) в тесте принудительного плавания у крыс. Все соединения, кроме атипамезола, увеличивали активность (р 0,001, по сравнению с растворителем). Фиг. 4 а и 4b показывают действие соединений K и L на рефлекс испуга и его опережающее ингибирование у крыс.(Раств.=растворитель). Звездочки как на фиг. 1; сравнения проводили между ФЦК (РСР, фенилциклидин)+растворитель и ФЦК+соединения K и L. Фиг. 5 а и 5b показывают эффект неселективного антагониста атипамезола (Ати) на рефлекс испуга и его опережающее ингибирование у крыс в присутствии фенилциклидина (ФЦК);(раств.=растворитель). Звездочки как на фиг. 1, сравнения с группой растворитель+ФЦК. Один вариант воплощения настоящего изобретения охватывает применение соединений формулы IR2 и R2' представляют собой независимо Н, гидрокси или (C1-C6)алкил или R2 и R2' образуют, вместе с атомами углерода, образующими кольцо, к которым они присоединены, карбонильную группу;(C1-C6)алкил-S-(C1-C6)алкил, где указанный (C3-C7)циклоалкил или арил является незамещенным или замещен 1 или 2 заместителями, каждый из которых независимо представляет собой гидрокси, (C1C6)алкил, галоген, (C1-C6)алкокси, NH2, CN или NO2, или один из R3 или R4 и R6 вместе образуют связь между атомами кольца, к которым они присоединены;R5 представляет собой Н, гидрокси, (C1-C6)алкил, (С 2-C6)алкенил, (C1-C6)алкокси, (C1C6)алкокси(C1-C6)алкил, (C3-C7)циклоалкил, (C3-C7)циклоалкил (C1-C6)алкил, арил, арил(C1-C6)алкил,арилокси, арил(C1-C6)алкокси, арилокси(C1-C6)алкил, арил(C1-C6)алкокси (C1-C6)алкил, галоген(C1C6)алкил, (C1-C6)алкил-СО-O-, (C1-C6)алкил-СО-O-(C1-C6)алкил, (C1-C6)алкокси-СО-(C1-C6)алкокси(C1C6)алкил, карбамоил, моно- или ди(C1-C6)алкилкарбамоил, карбоксил или (C1-C6)алкил-S-(C1-C6)алкил,где указанный (C3-C7)циклоалкил или арил является замещенным или незамещен 1 или 2 заместителями,каждый из которых независимо представляет собой гидрокси, (C1-C6)алкил, галоген, (C1-C6)алкокси,NH2, CN или NO2, или R4 и R5 образуют, вместе с атомами углерода, образующими кольцо, к которым они присоединены, конденсированное пяти-семичленное насыщенное карбоциклическое кольцо, замещенное 1-3 заместителями R9, каждый из которых независимо представляет собой гидрокси, (C1C6)алкил, галоген, NH2, NO2, (C3-C7)циклоалкил, гидрокси(C1-C6)алкил, галоген(C1-C6)алкил, амино(C1C6)алкил, моно- или ди(C1-C6)алкиламино, моно- или ди(C1-C6)алкиламино (C1-C6)алкил, (C1-C6)алкокси,(C1-C6)алкокси(C1-C6)алкил, карбоксил, (C1-C6)алкил-СО-, (C1-C6)алкил-СО-О-, (C1-C6)алкокси-СО-, (C1C6)алкокси-СО-(C1-C6)алкил, карбамоил моно- или ди(C1-C6)алкилкарбамоил или оксо;R6 представляет собой Н, гидрокси, (C1-C6)алкил, (C1-C6)алкокси или (C1-C6)алкокси(C1-C6)алкил или R6 образует связь между атомом кольца, к которому он присоединен, и атомом кольца, к котором присоединен R7;-2 008537 карбоциклическое кольцо, незамещенное или замещенное 1-3 заместителем(ями) R10, каждый из которых независимо представляет собой гидрокси, (C1-C6)алкил, галоген, NH2, NO2, (C3-C7)циклоалкил, гидрокси(C1-C6)алкил, галоген(C1-C6)алкил, амино(C1-C6)алкил, моно- или ди(C1-C6)алкиламино, моно- или ди(C1-C6)алкиламино (C1-C6)алкил, (C1-C6)алкокси, (C1-C6)алкокси (C1-C6)алкил, карбоксил, (C1C6)алкил-СО-, (C1-C6)алкил-СО-O-, (C1-C6)алкокси-СО-, (C1-C6)алкокси-СО-(C1-C6)алкил, карбамоил,моно- или ди(C1-C6)алкилкарбамоил или оксо;n равно 0 или 1,или его фармацевтически приемлемых солей или сложных эфиров, при условии, что соединение не является 1,2,3,4,5,10b-гексагидро-10-тиа-3 а-азациклопента[а]фтором, для получения лекарственного средства для лечения заболеваний или состояний, при которых показано, что альфа 2-антагонисты являются эффективными. В возможной подгруппе соединений формулы I X представляет собой NR2. В другой возможной подгруппе соединений формулы I m равно 0, n равно 0, R2 представляет собой Н, R3 представляет собой Н, гидрокси, (C1-C6)алкил, гидрокси(C1-C6)алкил, (C1-C6)алкокси (C1-C6)алкил,(C3-C7)циклоалкил, галоген(C1-C6)алкил, (C1-C6)алкил-СО-, (C1-C6)алкил-СО-О-(C1-C6)алкил, (C1C6)алкокси-СО- или (C1-C6)алкокси-СО-(C1-C6)алкил, R4 представляет собой Н, гидрокси, (C1-C6)алкил или гидрокси(C1-C6)алкил, R5 представляет собой Н, гидрокси, (C1-C6)алкил или (C1-C6)алкокси, R6 представляет собой Н или (C1-C6)алкил и R7 представляет собой Н, (C1-C6)алкил или гидрокси(C1-C6)алкил. В другой возможной подгруппе соединений формулы I R3 представляет собой Н или (C1-C6)алкил иR4 представляет собой гидрокси или гидрокси(C1-C6)алкил. В другой возможной подгруппе соединений формулы I R4 и R5 образуют, вместе с атомами углерода, образующими кольцо, к которым они присоединены, конденсированное шестичленное насыщенное карбоциклическое кольцо. В другой возможной подгруппе соединений формулы I R4 и R6 вместе образуют связь между атомами кольца, к которым они присоединены, или R6 образует связь между атомом кольца, к которому он присоединен, и атомом кольца, к которому присоединен R7. В еще одной возможной подгруппе соединений формулы I соединение представляет собой 1-этил-1,2,3,4,6,7,12,12b-октагидроиндоло[2,3-а]хинолизин-1-ол,(1-этил-1,2,3,4,6,7,12,12b-октагидроиндоло[2,3-а]хинолизин-1-ил)метанол,1-метил-1,2,3,4,6,7,12,12b-октагидроиндоло[2,3-а]хинолизин-1-ол,(1-метил-1,2,3,4,6,7,12,12b-октагидроиндоло[2,3-а]хинолизин-1-ил)метанол или 3,4,4 а,5,6,7,8,13,13b,13 с-декагидро-2 Н-6 а,13-диазаиндено[1,2-c]фенантрен-1-он. В другой возможной подгруппе соединений формулы I X представляет собой CR2R2'. В следующей возможной подгруппе соединений формулы I X представляет собой S. В еще одной возможной подгруппе соединений формулы I X представляет собой О. Когда X представляет собой О, одна возможная подгруппа соединений формулы I включает R5 и R6,как определено в описании применения соединений формулы I выше. Другой возможной подгруппой соединений формулы I, когда X представляет собой О, является таковая, где R5 представляет собой Н, гидрокси, (C1-C6)алкил, (С 2-C6)алкенил, (C1-C6)алкокси, (C1C6)алкокси (C1-C6)алкил, (C3-C7)циклоалкил, (C3-C7)циклоалкил (C1-C6)алкил, арил, арил(C1-C6)алкил,арилокси, арил(C1-C6)алкокси, арилокси(C1-C6)алкил, арил(С 1-C6)алкокси (C1-C6)алкил, галоген(C1C6)алкил, (C1-C6)алкил-СO-O-, (C1-C6)алкил-СО-O-(C1-C6)алкил, (C1-C6)алкокси-СО-(C1-C6)алкокси(C1C6)алкил, карбамоил, моно- или ди(C1-C6)алкилкарбамоил, карбоксил или (C1-C6)алкил-S-(C1-C6)алкил иR6 представляет собой Н, гидрокси, (C1-C6)алкил, (C1-C6)алкокси или (C1-C6)алкокси(C1-C6)алкил. Другой вариант воплощения изобретения обеспечивает соединения формулы IAZ, R1, R2, R2', R3-R10, R15 и R16, m и n являются такими, как определено выше,или их фармацевтически приемлемые соль или сложный эфир, при условии, чтоa) когда X представляет собой О, m равно 0 и n равно 0, тогда R3-R8 все не являются одновременно водородом;b) соединение не является 1,2,3,4,5,10b-гексагидро-10-тиа-3 а-азациклопента[а]флуореном; 1,3,4,5,6,11b-гексагидро-2 Н-11-тиа-4 а-азабензо[а]флуореном; 1-(1,3,4,5,6,11b-гексагидро-2 Н-11-тиа-4 аазабензо[а]флуорен-1-ил)этаноном или метиловым эфиром 1,3,4,5,6,11b-гексагидро-2 Н-11-тиа-4 аазабензо[а]флуорен-1-карбоновой кислоты; например где X представляет собой CR2R2'; или где X представляет собой О; или где X представляет собой S; или где R3 представляет собой гидрокси, (C1-C6)алкил, гидрокси(C1-C6)алкил, (C1-C6)алкокси (C1C6)алкил, (C1-C6)алкокси-СO- или (C1-C6)алкил-СО-O-(C1-C6)алкил и R4 представляет собой Н, (C1C6)алкил или гидрокси(C1-C6)алкил; или где R3 представляет собой гидрокси, гидрокси(C1-C6)алкил или (C1-C6)алкокси(C1-C6)алкил и R4 представляет собой (C1-C6)алкил; или где R4 и R5 образуют, вместе с атомами углерода кольца, к которым они присоединены, конденсированное шестичленное насыщенное карбоциклическое кольцо; или где соединение представляет собой 1-метил-1,3,4,5,6,11b-гексагидро-2 Н-11-окса-4 а-азабензо[а]флуорен-1-ол,(1-метил-1,3,4,5,6,11b-гексагидро-2 Н-11-окса-4 а-азабензо[а]флуорен-1-ил)метанол,(-)-(1-метил-1,3,4,5,6,11b-гексагидро-2 Н-11-окса-4 а-азабензо[а]флуорен-1-ил)метанол,(+)-(1-метил-1,3,4,5,6,11b-гексагидро-2 Н-11-окса-4 а-азабензо[а]флуорен-1-ил)метанол,1-изопропил-1,3,4,5,6,11b-гексагидро-2 Н-11-окса-4 а-азабензо[а]флуорен-1-ол,1-этил-1,3,4,5,6,11b-гексагидро-2 Н-11-окса-4 а-азабензо[а]флуорен-1-ол,(1-этил-1,3,4,5,6,11b-гексагидро-2 Н-11-окса-4 а-азабензо[а]флуорен-1-ил)метанол,1-метил-1,3,4,6,11b-гексагидро-2 Н-11-окса-4 а-азабензо[а]флуорен,(1-гидроксиметил-1,3,4,5,6,11b-гексагидро-2 Н-11-окса-4 а-азабензо[а]флуорен-1-ил)-метанол,1-метоксиметил-1-метил-1,3,4,5,6,11b-гексагидро-2 Н-11-окса-4 а-азабензо[а]флуорен,(-)-1-метоксиметил-1-метил-1,3,4,5,6,11b-гексагидро-2 Н-11-окса-4 а-азабензо[а]флуорен,(+)-1-метоксиметил-1-метил-1,3,4,5,6,11b-гексагидро-2 Н-11-окса-4 а-азабензо[а]флуорен,этиловый эфир 1-метил-1,3,4,5,6,11b-гексагидро-2 Н-11-окса-4 а-азабензо[а]флуорен-1-карбоновой кислоты,1-этоксиметил-1-метил-1,3,4,5,6,11b-гексагидро-2 Н-11-окса-4 а-азабензо[а]флуорен,(1-метил-1,3,4,5,6,11b-гексагидро-2 Н-11-окса-4 а-азабензо[а]флуорен-1-ил)метанол,(-)-(1 а-метил-1,3,4,5,6,11b-гексагидро-2 Н-11-окса-4 а-азабензо[а]флуорен-1-ил)метанол,(+)-(1-метил-1,3,4,5,6,11b-гексагидро-2 Н-11-окса-4 а-азабензо[а]флуорен-1-ил)метанол,метиловый эфир 1-этил-1,3,4,5,6,11b-гексагидро-2 Н-11-окса-4 а-азабензо[а]флуорен-1-карбоновой кислоты,1-метоксиметил-1-метил-1,3,4,5,6,11b-гексагидро-2 Н-11-окса-4 а-азабензо[а]флуорен,(-)-1-метоксиметил-1-метил-1,3,4,5,6,11b-гексагидро-2 Н-11-окса-4 а-азабензо[а]флуорен,(+)-1-метоксиметил-1-метил-1,3,4,5,6,11b-гексагидро-2 Н-11-окса-4 а-азабензо[а]флуорен,(1-этил-1,3,4,5,6,11b-гексагидро-2 Н-11-окса-4 а-азабензо[а]флуорен-1-ил)метанол,1-метил-1,3,4,5,6,11b-гексагидро-2 Н-11-окса-4 а-азабензо[а]флуорен-1-илметиловый эфир уксусной кислоты или(1-метил-1,2,3,4,6,7,12,12b-октагидроиндено[2,1-а]хинолизин-1-ил)метанол. Другой вариант воплощения изобретения обеспечивает новые соединения формулы IBZ, R1, R3-R10, R15, R16, m и n являются такими, как определено выше,или их фармацевтически приемлемые соль и сложный эфир, при условии, чтоa) когда m равно 0 или R1 представляет собой метокси и R4 представляет собой Н или этил, тогда R3 не является метокси-СО;b) соединение не является 12-метил-1,2,3,4,6,7,12,12b-октагидроиндоло[2,3-а]хинолизином; 1-этил 12-метил-1,2,3,4,6,7,12,12b-октагидроиндоло[2,3-а]хинолизином; 2,3-диэтил-12-метил-1,2,3,4,6,7,12,12bоктагидроиндоло[2,3-а]хинолизином; 12-метил-1,2,3,4,6,7,12,12b-октагидроиндоло[2,3-а]хинолизин-1 олом; 2-(1-этил-12-метил-1,2,3,4,6,7,12,12b-октагидроиндоло[2,3-а]хинолизин-1-ил)этанолом; 11-метил 2,3,5,6,11,11b-гексагидро-1 Н-индолизино[8,7-b]индолом; (11-метил-2,3,5,6,11,11b-гексагидро-1 Н-индолизино[8,7-b]индол-1-ил)метанолом; (1,11-диэтил-2,3,5,6,11,11b-гексагидро-1 Н-индолизино[8,7-b]индол 1-ил)метанолом или метиловым эфиром 3-(1-этил-12-метил-1,2,3,4,6,7,12,12b-октагидроиндоло[2,3 а]хинолизин-1-ил)пропионовой кислоты; например где R3 представляет собой гидрокси, (C1-C6)алкил, гидрокси(C1-C6)алкил или (C1-C6)алкокси(C1C6)алкил и R4 представляет собой Н, (C1-C6)алкил или гидрокси(C1-C6)алкил; или где соединение представляет собой 1-этил-12-метил-1,2,3,4,6,7,12b-октагидроиндоло[2,3 а]хинолизин-1-ол или 1-этил-12-этил-1,2,3,4,6,7,12b-октагидроиндоло[2,3-а]хинолизин-1-ол. Другой вариант воплощения изобретения обеспечивает новые соединения формулы IСr равно от 1 до 3; или их фармацевтически приемлемые соль и сложный эфир, при условии, что соединение не является 10 метил-5,7,7 а,8,9,10,11,11 а,11b,12-декагидро-6 Н-6 а,12-диазаиндено[1,2-а]флуореном; метиловым эфиром 3-гидрокси-1,2,3,4,4 а,5,6,7,8,13,13b,13 с-додекагидро-6 а,13-диазаиндено[1,2-c]фенантрен-4-карбоновой кислоты; метил-3-этил-1,2,3 а,4,6,7,12b,12 с-октагидро-3H,12 Н-индоло[2,3-g]циклопент[а]индолизин-2 карбоксилатом; метил-1,2,3 а,4,6,7,12b,12 с-октагидро-3H,12 Н-индоло[2,3-g]циклопент[а]индолизин-2 карбоксилатом или 12 с-этил-1,3 а,4,6,7,12b,12 с-октагидроциклопент[1,2]индолизино[8,7-b]индол-3(2 Н)оном; например где r равно 1 и R3 представляет собой Н, гидрокси, (С 1-C6)алкил или гидрокси(C1-C6)алкил; или где соединение представляет собой 3,4,4 а,5,6,7,8,13,13b,13 с-декагидро-2 Н-6 а,13 диазаиндено[1,2-c]фенантрен-1-он, 1,2,3,4,5,6,7,8,13,13b-декагидро-6 а,13-диазаиндено[1,2-с]фенантрен,1,2,3,4,4 а,5,6,7,8,13,13b,13 с-додекагидро-6 а,13-диазаиндено[1,2-c]фенантрен-1-иловый эфир уксусной кислоты или 1,2,3,4,4 а,5,6,7,8,13,13b,13 с-додекагидро-6 а,13-диазаиндено[1,2-c]фенантрен-1 иловый эфир уксусной кислоты. Другой вариант воплощения изобретения обеспечивает новые соединения формулы IDt равно от 0 до 3; или их фармацевтически приемлемые соль и сложный эфир, при условии, что соединение не является 1,2,3,4,4 а,5,6,11,11b,12,13,13 а-додекагидро-4b,11-диазаиндено[2,1-а]фенантреном; 1,2,3,4,4 а,5,6,11,11b,12 декагидро-4b,11-диазаиндено[2,1-а]фенантреном; 9-метокси-1,2,3,4,4 а,5,6,11,11b,12-декагидро-4b,11-диазаиндено[2,1-а]фенантреном или метиловым эфиром 1-гидрокси-1,2,3,4,4 а,5,6,11,11b,12,13,13 а-додекагидро 4b,11-диазаиндено[2,1-а]фенантрен-2-карбоновой кислоты. Другой вариант воплощения изобретения обеспечивает новые соединения формулы IEn равно 1,или их фармацевтически приемлемые соль и сложный эфир, при условии, что соединение не является 2,3,4,5,7,8,13,13b-октагидро-2,3-диэтил-1 Н-азепино[1',2':1,2]пиридо[3,4-b]индолом; 2,3,4,5,7,8,13,13bоктагидро-1 Н-азепино[1',2':1,2]пиридо[3,4-b]индол-2-илметиловым эфиром уксусной кислоты; 2,3,4,5,7,8,13,13b-октагидро-1 Н-азепино[1',2':1,2]пиридо[3,4-b]индол-2-[(фенилметокси)метилом] или 2,3,4,5,7,8,13,13b-октагидро-1 Н-азепино[1',2':1,2]пиридо[3,4-b]индол-4-этил-2-[(фенилметокси)метилом]; например где соединение представляет собой 2,3,4,5,7,8,13,13b-октагидро-1 Н-азепино[1',2':1,2]пиридо[3,4b]индол. Другой вариант изобретения обеспечивает новые соединения, которые являются 2-метокси-1,2,3,4, 6,7,12,12b-октагидроиндоло[2,3-а]хинолизином,2-метокси-1,2,3,4,6,7,12,12b-октагидроиндоло[2,3-а]хинолизином,1-этил-2-метил-1,2,3,4,6,7,12,12b-октагидроиндоло[2,3-а]хинолизин-1-олом,1-изопропил-1,2,3,4,6,7,12,12b-октагидроиндоло[2,3-а]хинолизин-1-олом,(-)-1-изопропил-1,2,3,4,6,7,12,12b-октагидроиндоло[2,3-а]хинолизин-1-олом,(+)-1-изопропил-1,2,3,4,6,7,12,12b-октагидроиндоло[2,3-а]хинолизин-1-олом,1-изопропил-1,2,3,4,6,7,12,12b-октагидроиндоло[2,3-а]хинолизином,(1-изопропил-1,2,3,4,6,7,12,12b-октагидроиндоло[2,3-а]хинолизин-1-ил)метанолом,(1-н-пропил-1,2,3,4,6,7,12,12b-октагидроиндоло[2,3-а]хинолизин-1-ил)метанолом,2-(1,2,3,4,6,7,12,12b-октагидроиндоло[2,3-а]хинолизин-1-ил)бутан-2-олом,1-(1,2,3,4,6,7,12,12b-октагидроиндоло[2,3-а]хинолизин-2-ил)пропан-1-олом,2-(1,2,3,4,6,7,12,12b-октагидроиндоло[2,3-а]хинолизин-1-ил)пропан-2-олом,1-втор-бутил-1,2,3,4,6,7,12,12b-октагидроиндоло[2,3-а]хинолизин-1-олом,1-циклогексил-1,2,3,4,6,7,12,12b-октагидроиндоло[2,3-а]хинолизин-1-олом,9-фтор-1-изопропил-1,2,3,4,6,7,12,12b-октагидроиндоло[2,3-а]хинолизин-1-олом,(1-метил-1,2,3,4,6,7,12,12b-октагидроиндоло[2,3-а]хинолизин-1-ил)метанолом,-6 008537(-)-(1-метил-1,2,3,4,6,7,12,12b-октагидроиндоло[2,3-а]хинолизин-1-ил)метанолом,(+)-(1-метил-1,2,3,4,6,7,12, 2b-октагидроиндоло[2,3-а]хинолизин-1-ил)метанолом,(1-этил-1,4,6,7,12,12b-гексагидроиндоло[2,3-а]хинолизин-1-ил)метанолом,3,4-диметил-1,2,3,4,6,7,12,12b-октагидроиндоло[2,3-а]хинолизином,(1,2,3,4,6,7,12,12b-октагидроиндоло[2,3-а]хинолизин-2-ил)пропан-2-олом,(1,2,3,4,6,7,12,12b-октагидроиндоло[2,3-а]хинолизин-2-ил)пропан-2-олом,(2-этил-1,2,3,4,6,7,12,12b-октагидроиндоло[2,3-а]хинолизин-2-ил)метанолом,(2-этил-1,2,3,4,6,7,12,12b-октагидроиндоло[2,3-а]хинолизин-2-ил)метанолом,этиловым эфиром (1 этил-1,2,3,4,6,7,12,12b-октагидроиндоло[2,3-а]хинолизин-1-илметокси) уксусной кислоты,1-(2-этил-1,2,3,4,6,7,12,12b-октагидроиндоло[2,3-а]хинолизин-2-ил)этаноном,1-(2-этил-1,2,3,4,6,7,12,12b-октагидроиндоло[2,3-а]хинолизин-2-ил)этанолом,2-(2-этил-1,2,3,4,6,7,12,12b-октагидроиндоло[2,3-а]хинолизин-2-ил)пропан-2-олом,2-(3-этил-1,2,3,4,6,7,12,12b-октагидроиндоло[2,3-а]хинолизин-2-ил)пропан-2-олом,(3-этил-2-метил-1,2,3,4,6,7,12,12b-октагидроиндоло[2,3-а]хинолизин-1-ил)метанолом,3-этил-1,2-диметил-1,2,3,4,6,7,12,12b-октагидроиндоло[2,3-а]хинолизином,1,2-диметил-1,2,3,4,6,7,12,12b-октагидроиндоло[2,3-а]хинолизин-1-олом,(1-этил-2-метил-1,2,4,4,6,7,12,12b-октагидроиндоло[2,3-а]хинолизин-3-ил)метанолом,метиловым эфиром 1 гидроксиметил-1-метил-1,2,3,4,6,7,12,12b-октагидроиндоло[2,3-а]хинолизин-6-карбоновой кислоты,5,6,7,7 а,8,9,10,11,11 а,11b-декагидро-12-окса-6 а-азаиндено[1,2-а]флуореном,2,3,4,4 а,5,6,7,8,13b,13 с-декагидро-1 Н-13-окса-6 а-азаиндено[1,2-c]фенантреном,2,3,4,4 а,5,6,7,8,13b,13 с-декагидро-1 Н-13-окса-6 а-азаиндено[1,2-c]фенантреном,2,3,4,4 а,5,6,7,8,13,13b-декагидро-1 Н-6 а,13-диазаиндено[1,2-c]фенантрен-13 с-олом,(-)-2,3,4,4 а,5,6,7,8,13,13b-декагидро-1 Н-6 а,13-диазаиндено[1,2-c]фенантрен-13 с-олом,(+)-2,3,4,4 а,5,6,7,8,13,13b-декагидро-1 Н-6 а,13-диазаиндено[1,2-c]фенантрен-13 с-олом,(2,3,4,4 а,5,6,7,8,13,13b-декагидро-1 Н-6 а,13-диазаиндено[1,2-c]фенантренил)-13 с-метанолом или 5,6,7,7 а,11,11b,12-декагидро-6 а,12-диазаиндено[1,2-а]флуорен-11 а-олом. Термины, используемые в данном описании, имеют следующие значения. Термин "галоген", как используется в данном описании, как таковой или как часть другой группы,относится к хлору, брому, фтору или йоду. Термин "карбоксил", как используется в данном описании, относится к группе -СООН. Термин "оксо", как используется в данном описании, относится к группе =O. Термин "(C1-C6)алкил", как используется в данном описании, как таковой или как часть другой группы, относится к линейной или разветвленной углеродной цепи, имеющей 1-6 атомов углерода. Характерные примеры (C1-C6)алкила включают, но не ограничиваются ими, метил, этил, н-пропил, изопропил, н-бутил, изобутил, втор-бутил, трет-бутил, н-пентил, изопентил, неопентил, н-гексил и подобные. Термин "(C2-C6)алкенил", как используется в данном описании, как таковой или как часть другой группы, относится к радикалу с линейной или разветвленной углеродной цепью, имеющему 2-6 атомов углерода и содержащему двойную связь(и). Термин "(C3-C7)циклоалкил", как используется в данном описании, как таковой или как часть другой группы, относится к насыщенной циклической углеводородной группе, содержащей 3-7 атомов углерода. Характерные примеры циклоалкила включают, но не ограничиваются ими, циклопропил, циклобутил, циклопентил, циклогексил и подобные. Термин "(C3-C7)циклоалкил (C1-C6)алкил", как используется в данном описании, относится к (C3C7)циклоалкильной группе, как определено в данном описании, присоединенной к фрагменту основной молекулы посредством (C1-C6)алкильной группы, как определено в данном описании. Термин "арил", как используется в данном описании, как таковой или как часть другой группы, относится к моноциклической или бициклической ароматической группе, содержащей 6-12 атомов углерода. Характерные примеры арилов включают, но не ограничиваются ими, фенил, нафтил и подобные. Термин "арил(C1-C6)алкил", как используется в данном описании, как таковой или как часть другой группы, относится к арильной группе, как определено в данном описании, присоединенной к фрагменту основной молекулы посредством (C1-C6)алкильной группы, как определено в данном описании. Термин "арилокси", как используется в данном описании, как таковой или как часть другой группы,относится к арильной группе, как определено в данном описании, присоединенной к фрагменту основной молекулы посредством группы -O-. Термин "арил(C1-C6)алкокси", как используется в данном описании, как таковой или как часть другой группы, относится к арильной группе, как определено в данном описании, присоединенной к фрагменту основной молекулы посредством (C1-C6)алкоксигруппы, как определено в данном описании. Термин "арилокси(C1-C6)алкил", как используется в данном описании, относится к арилоксигруппе,-7 008537 как определено в данном описании, присоединенной к фрагменту основной молекулы посредством (C1C6)алкильной группы, как определено в данном описании. Термин "арил(C1-C6)алкокси (C1-C6)алкил, как используется в данном описании, относится к арил(C1-C6)алкоксигруппе, как определено в данном описании, присоединенной к фрагменту основной молекулы посредством (1-C6)алкильной группы, как определено в данном описании. Термин "гидрокси", как используется в данном описании, как таковой или как часть другой группы,относится к группе -ОН. Термин "гидрокси(C1-C6)алкил", как используется в данном описании, как таковой или как часть другой группы, относится по меньшей мере к одной гидроксигруппе, как определено в данном описании,присоединенной к фрагменту основной молекулы посредством (C1-C6)алкильной группы, как определено в данном описании. Характерные примеры гидрокси(C1-C6)алкила включают, но не ограничиваются ими,гидроксиметил, 2-гидроксиэтил, 1-гидроксиэтил, 3-гидроксипропил, 1-гидроксипропил, 1-метил-1 гидроксиэтил, 1-метил-1-гидроксипропил и подобные. Термин "галоген(C1-C6)алкил", как используется в данном описании, относится к одному или более галогену, как определено в данном описании, присоединенному к фрагменту основной молекулы посредством (C1-C6)алкильной группы, как определено в данном описании. Характерные примеры галоген(C1-C6)алкила включают, но не ограничиваются ими, фторметил, дифторметил, трифтортил, 2 хлорэтил, 3-бромпропил и подобные. Термин "амино", как используется в данном описании, как таковой или как часть другой группы,относится к -NH2 группе. Термин "амино(C1-C6)алкил", как используется в данном описании, относится к аминогруппе, как определено в данном описании, присоединенной к фрагменту основной молекулы посредством (C1C6)алкильной группы, как определено в данном описании. Характерные примеры амино(C1-C6)алкила включают, но не ограничиваются ими, аминометил, 2-аминоэтил, 1-аминоэтил, 3-аминопропил, 2 аминопропил, 4-аминобутил, 1-метил-1-аминоэтил и подобные. Термин "моно- или ди(C1-C6)алкиламино", как используется в данном описании, как таковой или как часть другой группы, относится к одной или двум (C1-C6)алкильным группам(е), как определено в данном описании, присоединенным к фрагменту основной молекулы посредством аминогруппы, как определено в данном описании. Характерные примеры моно- или ди(C1-C6)алкиламино включают, но не ограничиваются ими, метиламино, этиламино, пропиламино, бутиламино, диметиламино, диэтиламино,N-этил-N-дитиламино и подобные. Термин "моно- или ди(C1-C6)алкиламино(C1-C6)алкил", как используется в данном описании, относится к моно- или ди(C1-C6)алкиламиногруппе, как определено в данном описании, присоединенной к фрагменту основной молекулы посредством (C1-C6)алкильной группы, как определено в данном описании. Характерные примеры моно- или ди(C1-C6)алкиламино(C1-C6)алкила включают, но не ограничиваются ими, N,N-диметиламинометил, N,N-диэтиламинометил, N-метиламиноэтил, N-метиламинопропил,N-этил-N-метиламинометил и подобные. Термин "(C1-C6)алкокси", как используется в данном описании, как таковой или как часть другой группы, относится к (C1-C6)алкилу, как определено в данном описании, присоединенному к фрагменту основной молекулы посредством -О- группы. Характерные примеры (C1-C6)алкокси включают, но не ограничиваются ими, метокси, этокси, пропокси, бутокси, изобутокси, втор-бутокси, трет-бутокси и подобные. Термин "(C1-C6)алкокси (C1-C6)алкил", как используется в данном описании, как таковой или как часть другой группы, относится по меньшей мере к одной (C1-C6)алкоксигруппе, как определено в данном описании, присоединенной к фрагменту основной молекулы посредством (C1-C6)алкильной группы,как определено в данном описании. Характерные примеры (C1-C6)алкокси(C1-C6)алкила включают, но не ограничиваются ими, метоксиметил, этоксиметил, 2-метоксиэтил, 2-этоксиэтил, 3,3-диметоксипропил,2,4-диметоксибутил и подобные. Термин "гидрокси(C1-C6)алкокси", как используется в данном описании, как таковой или как часть другой группы, относится к гидроксигруппе, как определено в данном описании, присоединенной к фрагменту основной молекулы посредством (C1-C6)алкоксигруппы, как определено в данном описании. Термин "гидрокси(C1-C6)алкокси(C1-C6)алкил", как используется в данном описании, относится к гидрокси(C1-C6)алкоксигруппе, как определено в данном описании, присоединенной к фрагменту основной молекулы посредством (C1-C6)алкильной группы, как определено в данном описании. Термин "карбамоил", как используется в данном описании, как таковой или как часть другой группы, относится к -CONH2 группе. Термин "моно- или ди(C1-C6)алкилкарбамоил", как используется в данном описании, относится к одной или двум (C1-C6)алкильным группам(е), как определено в данном описании, присоединенным к фрагменту основной молекулы посредством групп -HNCO- или -NCO-. Характерные примеры моно- или ди(C1-C6)алкилкарбамоила включают, но не ограничиваются ими, N-метилкарбамоил, N-этилкарбамоил,N-пропилкарбамоил, N,N-диметилкарбамоил, N,N-диэтилкарбамоил и подобные. На соединения формулы I и IA, IB, IС, ID и IE, а также их фармацевтически приемлемые соли и-8 008537 сложные эфиры, ссылаются ниже как на соединения по изобретению, если не указано иначе. В объем изобретения входят все возможные стереоизомеры соединений, включая геометрические изомеры, например Z и Е изомеры (цис- и транс-изомеры), и оптические изомеры, например диастереоизомеры и энантиомеры. Кроме того, изобретение включает как отдельные изомеры, так и их любые смеси, например рацемические смеси. Отдельные изомеры могут быть получены с использованием соответствующих изомерных форм исходного вещества или они могут быть разделены после получения окончательного соединения в соответствии с обычными способами разделения. Для разделения оптических изомеров, например энантиомеров, из их смеси, могут быть использованы обычные способы разделения,например фракционная кристаллизация. Фармацевтически приемлемые соли, например аддитивные соли кислоты с органическими и неорганическими кислотами, хорошо известны в области фармацевтики. Неограничивающие примеры таких солей включают хлориды, бромиды, сульфаты, нитраты, фосфаты, сульфонаты, форматы, тартраты, малеаты, цитраты, бензоаты, салицилаты и аскорбаты. Фармацевтически приемлемые сложные эфиры, когда применяются, могут быть получены известными методами с использованием фармацевтически приемлемых кислот, которые являются обычными в области фармацевтики и которые сохраняют фармакологические свойства свободной формы. Неограничивающие примеры таких сложных эфиров включают сложные эфиры алифатических или ароматических спиртов, например метиловый, этиловый, пропиловый, изопропиловый, бутиловый, изобутиловый, втор-бутиловый и трет-бутиловый сложные эфиры. Соединения по изобретению могут быть получены аналогично или в соответствии с методами, известными в литературе с использованием подходящих исходных веществ. Исходные вещества формул II,III и IV являются коммерчески доступными или могут быть получены посредством множества известных путей синтеза, известных в литературе. Например, используемыми исходными веществами являются арилалкиламины формулы (II) где R1 является таким, как определено выше, и X представляет собой NH, О, СН 2 или S. Когда X представляет собой О, амины формулы (II) могут быть получены, например, в соответствии со способом, описанным в описании патента США 4710504. Когда X представляет собой CH2, соединения формулы (II) могут быть получены, как описано в J. Med. Chem. 10 (1967), 856-859. Когда X представляет собой S, соединения формулы (II) могут быть получены декарбоксилированием соответствующего 3-(тианафтен-3-ил)-L-аланина. Другими используемыми исходными веществами являются соединения формулы (III) где R3 является таким, как определено выше, и R11 представляет собой ОН или галоген. Кроме того, используемыми исходными веществами являются соединения формулы (IV) где R3-R7 и Z являются такими, как определено выше, и Y представляет собой О или NH. Соединения формулы (IV) могут быть получены в соответствии со способами, описанными вTetrahedron 33 (1977), 1803-1808. Аналогично, соответствующие хлорангидриды могут быть использованы вместо лактонов (Y=O). Когда R3 и R5 образуют кольцо, соединения формулы (IV) получают частичным восстановлением их соответствующих ангидридов. В общем, соединения формулы (I), где X представляет собой NH, О или S, могут быть получены,например, аналогично или в соответствии со следующей схемой реакции 1. где R1, R3-R7 и Z являются такими, как определено выше. В соответствии со схемой I амиды (V), полученные в результате алкилирования аминов (II) соединениями формулы (III), преобразуют в енамины (VII) через бета-карболины (V) реакцией БишлераНапиеральски для образования кольца D, воздействием соединений формулы (VI) с 1,3-дигалоалканами в основных условиях, как описано в Gazz. Chim. Ital. 111 (1981), 257-267. На последней стадии соединения формулы (I) получают 1) окислением енаминов (VII) с использованием йодида калия, йода и воздуха или 2) взаимодействием енаминов (VII) с формальдегидом в присутствии основания Хенига при 60 С. Другой путь получения соединений формулы (I), где X представляет собой NR2, О, СН 2 или S, проиллюстрирован на схеме 2. Схема 2 где X представляет собой NR2, О, СН 2 или S, R3-R7 и Z являются такими, как определено выше. На схеме 2 арилалкиламины формулы (II), где X представляет собой NH, О, CH2 или S, подвергают взаимодействию с соединениями формулы (IV) или соответствующим хлорангидридом с получением- 10008537 амидов (VIII), как описано в Tetrahedron 33 (1977), 1803-1808. Реакция циклизации промежуточных продуктов (VIII) Бишлера-Напиеральски приводит к енаминам (IX), которые преобразуют в соединения формулы (I). Соединения формулы (I), где X представляет собой NH, могут быть алкилированы алкилгалидами в присутствии подходящего основания при комнатной температуре (Heterocycles 27 (1988) 1179-1190) в соответствии со следующей схемой 3. Схема 3 где R1-R7 и Z являются такими, как определено выше. Следующий способ получения соединений формулы (I) проиллюстрирован на схеме 4. Схема 4 где R2 представляет собой ВОС и R1, R5 и R6 являются такими, как определено выше. На схеме 4 пиридин алкилируют триптофилбромидами (X) с получением солей пиридина (XI), который затем подвергают частичному восстановлению с получением соединения формулы (XII). Защита соединений формулы (XII) с использованием ди-т-бутилбикарбоната в основных условиях дает соединения формулы (XIII). Реакция Полоновски-Пуатье полученных промежуточных продуктов и их циклизация с использованием МеОН/HCl дает соединения формулы (I). Следующий способ получения соединений формулы (I), где X представляет собой О, S или NH, R1 и R3-R8 являются такими, как определено выше, показан на следующей схеме 5. Схема 5- 11008537 На схеме 5 окислительная циклизация производного (XIV) с ацетатом ртути в соответствии со способом, описанным в Heterocycles 32 (1991) 489-497, дает енамин (XV). Данный промежуточный продукт может быть окислен или обработан формальдегидом, как на схеме 1, или восстановлен борогидридом натрия с получением соединений формулы (I). Следующий способ получения соединений формулы (I), где R6 и R7 образуют связь, проиллюстрирован на схеме 6. Схема 6 где X представляет собой NH и R3 представляет собой низший алкил. Применение способа, описанного в J. Org. Chem. 52 (1987), 353-356, гетеро-реакция ДиельсаАльдера 3,4-дигидрокарболина (XVI) с диеновым сложным эфиром (XVII), полученным реакцией Виттига, как описано в Can. J. Chem. 65 (1987), 670-682, дает соединения формулы (XVIII), которые затем восстанавливают до спиртов формулы (I). Следующий способ получения соединений формулы (I) проиллюстрирован на схеме 7. Схема 7 где X, R1, R3, R7 и Z являются такими, как определено выше. R12 может быть Н или OCH3 и R13 может быть алкильной или арильной группой. На схеме 7 соединения формулы (XIX), когда R12 представляет собой Н, получают, как описано в J.Chem. Soc, Chem. Commun. (1995), 2317-2318, и соединения формулы (XIX), когда R12 представляет собой OCH3, получают, как описано в J. Chem. Soc. (С) (1971), 736-743. Соединения формулы (XIX) подвергают взаимодействию с реактивами Гриньяра с получением соединений формулы (I). Когда R12 в формуле (XIX) представляет собой Н, другие группы R13 в формуле (I) также являются Н. Новый способ получения определенных соединений формулы (I) показан на схеме 8. Схема 8 где X, R1, R3 и z являются такими, как определено выше. R14 представляет собой низшую алкильную группу.Chem. Soc. (С) (1971), 736-743, депротонируется сильным основанием с получением аниона (XXI). Этот анион алкилируют и затем циклизуют с кислотой с получением соединений формулы (XXII). Восстановление (XXII) LiAlH4 затем дает соединения формулы (I). Разделение рацемических соединений формулы (I) можно проводить, например, путем преобразования соединений формулы (I) в смесь солей их диастереоизомеров путем реакции с оптически активной кислотой, такой как D-винная кислота, дибензоил-О-винная кислота и др., и путем разделения диастереоизомеров кристаллизацией. Для специалиста является очевидным, что в вышеуказанных реакциях любые исходные вещества или промежуточные продукты могут быть защищены, если необходимо, способом, хорошо известным в области химии. Любую защищенную функциональную группу впоследствии подвергают снятию защиты обычным образом. Необходимо отметить, что вышеуказанные синтетические пути предназначены для иллюстрации получения соединений по изобретению и такое получение не означает его ограничение, т.е. другие синтетические способы, которые находятся в рамках общих знаний специалиста, также являются возможными. Соединения по изобретению могут быть преобразованы, при необходимости, в форму их фармацевтически приемлемых соли или сложного эфира с использованием способов, хорошо известных в данной области. Настоящее изобретение будет объяснено более детально следующими примерами. Примеры предназначены только с целью иллюстрации и не ограничивают объем изобретения, определенный в формуле изобретения. Пример 1. 1-Пропил-4,9-дигидро-3Hкарболин 8,00 г (50,0 ммоль) триптамина растворяли в 150 мл этилацетата и медленно добавляли 4,80 мл(52,0 ммоль) н-масляной кислоты. После стояния в течение 4 ч при 0 С реакционную смесь фильтровали с получением 12,30 г (49,5 ммоль) бутирата триптамина, который был расплавленным. Расплав нагревали при 200 С и выдерживали 30 мин при такой температуре. Образовавшуюся воду удаляли с использованием прибора Дина-Старка. После охлаждения расплав смешивали с 120 мл толуола, добавляли 23,5 мл (257,7 ммоль) свежеперегнанного оксихлорида фосфора и реакционную смесь кипятили с обратным холодильником в течение 4 ч. Раствор выпаривали в вакууме и темное масло смешивали с 20% раствором уксусной кислоты (350 мл). Твердое вещество отфильтровывали, водный раствор делали щелочным 25% гидроксидом аммония при охлаждении и экстрагировали дихлорметаном (350 мл). Комбинированные органические фазы сушили над сульфатом натрия, осушающее вещество отфильтровывали и фильтрат выпаривали с получением указанного в заголовке соединения, которое очищали колоночной хроматографией (силикагель, дихлорметан/метанол, 95:5). ЯМР: 1,00 (т, 3H), 1,75 (м, 2 Н), 2,66 (т, 2 Н), 2,87 (т, 2 Н), 3,90 (т, 2 Н), 7,00-7,62 (м, 4 Н), 8,94 (ушир.с,1 Н). МС: 212 (28%), 211 (12%), 197 (25%), 184 (100%), 169 (13%). Пример 2. 1-Изобутил-4,9-дигидро-3Hкарболин Повторяли методику примера 1, используя изовалериановую кислоту вместо н-масляной кислоты. ЯМР: 0,98 (д, 6 Н), 2,16 (м, 1 Н), 2,54 (д, 2 Н), 2,86 (т, 2 Н), 3,89 (т, 2 Н), 7,00-7,62 (м, 4 Н), 8,60 (ушир.с,1 Н). МС: 226 (16%), 211 (18%), 184 (100%), 169 (13%). Пример 3. 1-Бутил-4,9-дигидро-3Hкарболин Повторяли методику примера 1, используя н-валериановую кислоту вместо н-масляной кислоты. ЯМР: 1,00 (т, 3H), 7,00-7,62 (м, 4 Н), 8,64 (ушир.с, 1 Н). МС: 226 (18%), 211 (18%), 184 (100%), 169 (14%). Пример 4. 1-(2-Метилбутил)-4,9-дигидро-3Hкарболин Повторяли методику примера 1, используя 3-метилвалериановую кислоту вместо н-масляной кислоты. ЯМР: 0,84 (т, 3H), 0,87 (д, 3H), 7,05-7,60 (м, 4 Н), 12,2 (ушир. с, 1 Н). МС: 240 (9%), 225 (10%), 211 (10%), 185 (13%), 184 (100%), 183 (14%), 155 (24%). Пример 5. 1-Циклогексилметил-4,9-дигидро-3Hкарболин Повторяли методику примера 1, используя 3-циклогексилуксусную кислоту вместо н-масляной кислоты. ЯМР: 1,0-1,9 (м, 11 Н), 2,56 (д, 2 Н), 2,85 (м, 2 Н), 3,88 (м, 2 Н), 7,14-7,63 (м, 4 Н), 8,55 (ушир.с, 1 Н),МС: 266 (8%), 185 (15%), 184 (100%), 183 (12%), 155 (17%). Пример 6. 1-Изопропил-1,2,3,4,6,7,12,12b-октагидроиндоло[2,3-а]хинолизин 2,56 г (11,5 ммоль) 4,9-дигидро-1-изобутил-3-H-пиридо[3,4-b]индола (пример 2), 2 мл N-этилдиизопропиламина и 1,35 мл (13,8 ммоль) 1-бром-3-хлорпропана растворяли в 50 мл ацетонитрила. Смесь кипятили с обратным холодильником в атмосфере аргона в течение 8 ч. После выпаривания рас- 13008537 творителя добавляли 20 мл метанола и 1,3 г (34,5 ммоль) борогидрида натрия. Реакционную смесь перемешивали в течение 1 ч при комнатной температуре и затем добавляли 20 мл воды. Реакционную смесь экстрагировали дихлорметаном (350 мл). Комбинированные органические фазы высушивали над сульфатом натрия, осушающее вещество отфильтровывали и фильтрат выпаривали с получением указанного в заголовке соединения, которое очищали колоночной хроматографией (силикагель, дихлорметан/метанол, 95:5). ЯМР: 1,02 (ушир.с, 6 Н), 7,11 (т, 1 Н), 7,18 (т, 1 Н), 7,35 (д, 1 Н), 7,48 (д, 1 Н), 7,85 (ушир.с, 1 Н). МС: 267 (100%), 253 (20%), 197 (35%), 170 (30%), 169 (30%). Пример 7. 2-(1,2,3,4,6,7,12,12b-Октагидроиндоло[2,3-а]хинолизин-1-ил)бутан-2-ол К раствору 190 мг (0,7 ммоль) 1-(1,2,3,4,6,7,12,12b-октагидроиндоло[2,3-а]хинолизин-1-ил)этанона(Tetrahedron Lett. 30 (1989), 719-722) в 5 мл дихлорметана при -60 С добавляли 0,11 мл (0,8 ммоль) бромида этилмагния (1,0 М). Реакционную смесь перемешивали 30 мин при этой температуре и 2 ч при комнатной температуре. Затем добавляли воду (10 мл) и реакционную смесь экстрагировали дихлорметаном(350 мл). Комбинированные органические фазы сушили над сульфатом натрия, осушающее вещество отфильтровывали и фильтрат выпаривали с получением указанного в заголовке соединения, которое очищали колоночной хроматографией (силикагель, дихлорметан/метанол, 95:5). ЯМР: 0,97 (т, 3H), 1,30 (с, 3H), 4,69 (ушир.с, 1 Н), 7,00-7,50 (м, 4 Н), 8,36 (ушир.с, 1 Н). МС: 297 (100%), 281 (30%), 269 (35%), 225 (28%), 197 (45%), 170 (35%), 169 (34%). Пример 8. 2-(1,2,3,4,6,7,12,12b-Октагидроиндоло[2,3-а]хинолизин-1-ил)пропан-2-ол Методику примера 7 повторяли, используя бромид метилмагния (избыток) вместо бромида этилмагния. ЯМР: 1,37 (с, 3H), 1,42 (с, 3H), 4,73 (ушир.с, 1 Н), 7,00-7,50 (м, 4 Н), 8,18 (ушир.с, 1 Н). МС: 283 (100%), 267 (42%), 225 (33%), 197 (60%), 170 (50%), 169 (50%). Пример 9. 1-Изопропил-1,2,3,4,6,7,12,12b-октагидроиндоло[2,3-а]хинолизин-1-ол (соединение А) 5,13 г (23,0 ммоль) 4,9-дигидро-1-изобутил-3-H-пиридо[3,4-b]индола, 4 мл N-этилдиизопропиламина и 2,7 мл (27,6 ммоль) 1-бром-3-хлорпропана растворяли в 100 мл ацетонитрила. Смесь кипятили с обратным холодильником в атмосфере аргона в течение 8 ч. Темный раствор концентрировали до масла, которое обрабатывали 20% гидроксида натрия. Через 10 мин перемешивания раствор экстрагировали дихлорметаном (350 мл). Комбинированные органические фазы высушивали над сульфатом натрия, осушающее вещество отфильтровывали и фильтрат выпаривали с получением соответствующего енамина, который растворяли в 100 мл ацетонитрила. Добавляли 7,0 г (27,6 ммоль) йода и 4,6 г (27,6 ммоль) йодида калия. Реакционную смесь перемешивали в темноте на воздухе в течение 3 ч. После выпаривания растворителя добавляли 50 мл метанола и, при охлаждении, 2,6 г (69 ммоль) борогидрида натрия. Реакционную смесь перемешивали в течение 1 ч при комнатной температуре и затем добавляли 20 мл воды. Реакционную смесь экстрагировали дихлорметаном (350 мл). Комбинированные органические фазы сушили над сульфатом натрия, осушающее вещество отфильтровывали и фильтрат выпаривали с получением указанного в заголовке соединения, которое очищали колоночной хроматографией (силикагель, дихлорметан/метанол, 95:5). ЯМР: 0,47 (д, 3H), 0,90 (д, 3H), 3,48 (ушир.с, 1 Н), 7,00-7,50 (м, 4 Н), 8,92 (ушир.с, 1 Н). МС: 284 (14%), 239 (13%), 171 (100%), 170 (16%), 169 (33%). Пример 10. 1-Этил-2-метил-1,2,3,4,6,7,12,12b-октагидроиндоло[2,3-а]хинолизин-1-ол (соединение В) Повторяли методику примера 9, используя 4,9-дигидро-1-пропил-3H-пиридо[3,4-b]индол вместо 4,9-дигидро-1-изобутил-3-H-пиридо[3,4-b]индола и 1,3-дибромбутан вместо 1-бром-3-хлорпропана. ЯМР: 0,69 (т, 3H), 1,00 (д, 3H), 3,20 (ушир.с, 1 Н), 7,00-7,60 (м, 4 Н), 9,04 (ушир.с, 1 Н). МС: 284 (5%), 267 (15%), 225 (100%), 210 (15%), 195 (15%), 182 (72%), 171 (41%), 170 (22%), 169(32%). Пример 11. 9-Фтор-1-изопропил-1,2,3,4,6,7,12,12b-октагидроиндоло[2,3-а]хинолизин-1-ол Повторяли методику примера 9, используя 6-фтор-1-изобутил-4,9-дигидро-3H-пиридо[3,4-b]индол(полученный из 5-фтортриптамина, как описано в примере 2), вместо 4,9-дигидро-1-изобутил-3Hпиридо[3,4-b]индола. ЯМР: 0,45 (д, 3H), 0,89 (д, 3H), 3,32 (с, 1 Н), 6,8-7,25 (м, 3H), 8, 94 (ушир.с, 1 Н). МС: 302 (26%), 203 (13%), 189 (100%), 161 (26%). Пример 12. 1-втор-Бутил-1,2,3,4,6,7,12,12b-октагидроиндоло[2,3-а]хинолизин-1-ол (смесь изомеров) (соединение С) Повторяли методику примера 9, используя 1-(2-метилбутил)-4,9-дигидро-3H-пиридо[3,4-b]индол вместо 4,9-дигидро-1-изобутил-3 Р-пиридо[3,4-b]индола. ЯМР: 0,48 (д, 3H, основной изомер), 0,69 (т, 3H, второстепенный изомер), 0,82 (т, 3H, основной изомер), 0,92 (д, 3H, второстепенный изомер), 3,30 (с, 1 Н), 7,0-7,5 (м, 4 Н), 8,88 (ушир.с, 1 Н, второстепенный изомер), 8,93 (ушир.с, 1 Н, основной изомер). МС: 298 (23%), 172 (24%), 171 (100%), 170 (15%), 169 (23%), 143 (29%).- 14008537 Пример 13. 1-Циклогексил-1,2,3,4,6,7,12,12b-октагидроиндоло[2,3-а]хинолизин-1-ол Повторяли методику примера 9, используя 1-циклогексилметил-4,9-дигидро-3H-пиридо[3,4b]индол вместо 4,9-дигидро-1-изобутил-3H-пиридо[3,4-b]индола. ЯМР: 3,35 (ушир.с, 1 Н), 7,02-7,55 (м, 4 Н), 8,98 (ушир.с, 1 Н). МС: 324 (21%), 172 (12%), 171 (100%), 170 (10%), 169 (15%), 143 (22%). Пример 14. (1-Изопропил-1,2,3,4,6,7,12,12b-октагидроиндоло[2,3-а]хинолизин-1-ил)метанол Методику примера 9 повторяли, используя вместо окисления с использованием йода и йодида калия, получаемый енамин обрабатывали 40% водным формальдегидом, реакционную смесь кипятили с обратным холодильником в течение 3 ч и растворитель выпаривали. Осадок разбавляли этилацетатом и промывали насыщенным раствором соли. Органическую фазу сушили над сульфатом натрия, осушитель отфильтровывали и фильтрат выпаривали с получением указанного в заголовке соединения, которое очищали колоночной хроматографией (силикагель, дихлорметан/метанол, 98:2). ЯМР: 0,58 (ушир.с, 3H), 0,82 (д, 3H), 3,07 (ушир.с, 1 Н), 3,62 (д, 1 Н), 4,13 (д, 1 Н), 7,00-7,50 (м, 4 Н),9,41 (ушир.с, 1 Н). МС: 298 (100%), 297 (55%), 281 (60%), 170 (75%), 169 (52%). Пример 15. (1-н-Пропил-1,2,3,4,6,7,12,12b-октагидроиндоло[2,3-а]хинолизин-1-ил)метанол Повторяли методику примера 14, используя 4,9-дигидро-1-бутил-3-H-пиридо[3,4-b]индол вместо 4,9-дигидро-1-изобутил-3-H-пиридо[3,4-b]индола. ЯМР: 0,81 (т, 3H), 3,34 (ушир.с, 1 Н), 3,65 (д, 1 Н), 3,82 (д, 1 Н), 7,00-7,50 (м, 4 Н), 10,07 (ушир.с, 1 Н). МС: 298 (100%), 297 (65%), 281 (67%), 170 (75%), 169 (52%). Пример 16. (1-Метил-1,2,3,4,6,7,12,12b-октагидроиндоло[2,3-а]хинолизин-1-ил)метанол Повторяли методику примера 14, используя 1-этил-4,9-дигидро-3H-пиридо[3,4-b]индол вместо 4,9 дигидро-1-изобутил-3H-пиридо[3,4-b]индола. ЯМР: 0,91 (с, 3H), 3,37 (ушир.с, 1 Н), 3,70 (д, 1 Н), 3,76 (д, 1 Н), 7,0-7,6 (м, 4 Н), 9,78 (ушир.с, 1 Н). МС: 270 (97%), 269 (100%), 253 (53%), 197 (48%), 170 (68%), 169 (62%). Пример 17. (1-Этил-1,4,6,7,12,12b-гексагидроиндоло[2,3-а] хинолизин-1-ил)метанол Смесь 0,34 г (2,0 ммоль) 3,4-дигидрокарболина и 0,39 г (2,5 ммоль) этил 2-этилпента-2,4 диеноата в 5 мл хлорбензола кипятили с обратным холодильником в течение 16 ч. Растворитель выпаривали и осадок подвергали колоночной хроматографии (силикагель, дихлорметан/метанол, 99:1) с получением промежуточного продукта сложного эфира. Данный продукт восстанавливали обычным методом с алюмогидридом лития в сухом тетрагидрофуране с получением указанного в заголовке соединения. ЯМР: 0,82 (т, 3H), 3,69 (д, 1 Н), 3,70 (ушир.с, 1 Н), 3,90 (д, 1 Н), 5,42 (ддд, 1 Н), 5,97 (ддд, 1 Н), 7,0-7,5(м, 4 Н), 10,02 (ушир.с, 1 Н). МС: 282 (31%), 171 (14%), 170 (100%), 169 (52%). Пример 18. 2-Метокси-1,2,3,4,6,7,12,12b-октагидроиндоло[2,3-а]хинолизин и 2-метокси 1,2,3,4,6,7,12,12b-октагидроиндоло[2,3-а]хинолизин (соединение D) 1,16 г (14,7 ммоль) пиридина и 3,0 г (13,4 ммоль) триптофилбромида растворяли в 15 мл сухого диэтилового эфира. Реакционную смесь нагревали при перемешивании при 60 С до завершения выпаривания растворителя. Затем смесь нагревали до 100 С в течение 2 ч с получением соответствующей соли пиридинбромида. Его растворяли в 100 мл метанола и добавляли 1,52 г (40,1 ммоль) борогидрида натрия частями при охлаждении. Реакционную смесь перемешивали при комнатной температуре в течение 4 ч, с последующим добавлением 20 мл воды. Реакционную смесь экстрагировали дихлорметаном (330 мл). Комбинированные органические фазы высушивали над сульфатом натрия, осушающее вещество отфильтровывали и фильтрат выпаривали. Осадок растворяли в 100 мл сухого дихлорметана и добавляли 2,91 г (13,3 ммоль) ди-т-бутилбикарбоната и 0,149 г (1,2 ммоль) 4-(диметиламино)пиридина. Реакционную смесь перемешивали в течение 2 ч при комнатной температуре в атмосфере аргона. Растворитель выпаривали и осадок очищали колоночной хроматографией (силикагель, дихлорметан/метанол, 98:2). Полученное вязкое масло растворяли в 40 мл дихлорметана и добавляли 2,54 г (13,3 ммоль) mCPBA. Раствор перемешивали в течение 2 ч при 0 С, затем растворитель выпаривали и неочищенный продукт очищали колоночной хроматографией (силикагель, дихлорметан/метанол, 98:2) с получением Воc Nbоксида. К перемешиваемому раствору 0,59 г (1,7 ммоль) Воc Nb-оксида в 15 мл дихлорметана при 0 С медленно добавляли 3,0 мл трифторуксусного ангидрида. Охлаждающую баню удаляли и перемешивание продолжали в течение 2 ч при кт, затем растворитель выпаривали. Добавляли метанол, насыщенный газообразным хлористым водородом (20 мл), и смесь кипятили с обратным холодильником в течение 2 ч. Обработка щелочью и очистка колоночной хроматографией (силикагель, дихлорметан/метанол, 98:2) давала два простых эфира. 2-Метокси-1,2,3,4,6,7,12,12b-октагидроиндоло[2,3-а]хинолизин: ЯМР: 1,54 (ддд, 1 Н), 3,24 (дд, 1 Н), 3,38 (дддд, 1 Н), 3,43 (с, 3H), 7,00-7,50 (м, 4 Н), 7,77 (ушир.с, 1 Н). МС: 256 (100%), 255 (86%), 255 (59%), 197 (35%), 169 (30%). 2-Метокси-1,2,3,4,6,7,12,12b-октагидроиндоло[2,3-а]хинолизин:(J. Chem. Soc. Chem. Commun. 22 (1995), 2317-2318) в 2 мл дихлорметана при -60 С добавляли 0,22 мл(1,7 ммоль) 1 М бромида этилмагния. Реакционную смесь перемешивали в течение 4 ч в атмосфере аргона. Обработка водным гидроксидом натрия с последующим экстрагированием дихлорметаном и очисткой колоночной хроматографией (силикагель, дихлорметан/метанол, 98:2) давала указанное в заголовке соединение. ЯМР: 1,02 (т, 3H), 1,93 (ушир.д, 1 Н), 2,30 (ушир.д, 1 Н), 6,80-7,40 (м, 4 Н). МС: 284 (95%), 283 (100%), 225 (80%), 169 (36%). Пример 20. (1,2,3,4,6,7,12,12b-Октагидроиндоло[2,3-а]хинолизин-2-ил)пропан-2-ол К раствору 88 мг (0,31 ммоль) метилового эфира 1,2,3,4,6,7,12,12b-октагидроиндоло[2,3 а]хинолизин-2-карбоновой кислоты в 3 мл сухого тетрагидрофурана по каплям добавляли 1 мл (3,0 ммоль) раствора хлорида метилмагния (3 М в тетрагидрофуране). Затем полученный раствор кипятили с обратным холодильником в течение 90 мин. Затем смесь обрабатывали как в примере 7 с получением неочищенного спирта, который очищали колоночной хроматографией (силикагель, дихлорметан/метанол, 95:5) с получением указанного в заголовке соединение. ЯМР: 1,20 (с, 3H), 1,25 (с, 3H), 3,28 (ушир.д, 1 Н), 7,0-7,5 (м, 4 Н). МС: 284 (86%), 283 (65%), 225 (100%). Пример 21. (1,2,3,4,6,7,12,12b-Октагидроиндоло[2,3-а]хинолизин-2-ил)пропан-2-ол Как в примере 20, 64 мг (0,23 ммоль) метилового эфира 1,2,3,4,6,7,12,12b-октагидроиндоло[2,3-а]хинолизин-2-карбоновой кислоты в 3 мл сухого тетрагидрофурана и 0,7 мл (2,1 ммоль) раствора хлорида метилмагния (3 М в тетрагидрофуране) кипятили с обратным холодильником в течение 90 мин. Обработка, как указано выше, давала, после колоночной хроматографии (силикагель, дихлорметан/метанол, 90:10), указанное в заголовке соединение. ЯМР: 1,17 (с, 3H), 1,18 (с, 3H), 4,57 (ушир.с, 1 Н), 7,0-7,5 (м, 4 Н), 8,65 (ушир.с, 1 Н). МС: 284 (58%), 283 (53%), 225 (100%). Пример 22. (2-Этил-1,2,3,4,6,7,12,12b-октагидроиндоло[2,3-а]хинолизин-2-ил)метанол К перемешиваемому раствору 0,36 г (3,6 ммоль) диизопропиламина в 4 мл сухого тетрагидрофурана при -50 С добавляли 2,0 мл (3,6 ммоль) н-бутиллития (1,8 М в гексанах). Смеси давали нагреться до-30 С (15 мин), затем охлаждали до -70 С. При такой температуре добавляли 0,64 г (3,6 ммоль) гексаметилфосфорамида. Перемешивание продолжали в течение 30 мин при такой температуре, затем добавляли 0,42 г (1,48 ммоль) метил 1-[2-(3-индолил)этил]-1,2,5,6-тетрагидропиридин-4-карбоксилата в 7 мл тетрагидрофурана. После перемешивания в течение 20 мин при -70 С смеси давали нагреться до -40 С (15 мин). При такой температуре добавляли 0,3 г (3,6 ммоль) этилйодида и перемешивание продолжали в течение 1 ч. Затем охлаждающую баню удаляли и, после дополнительных 15 мин, смесь гасили 5% аммиаком. Водный слой экстрагировали дихлорметаном (320 мл) и комбинированные органические слои промывали водой. Сушка над сульфатом натрия, фильтрование и выпаривание растворителя давали неочищенный енамин, который растворяли в 50 мл метанола, насыщенного хлористым водородом, и полученный раствор перемешивали в течение 16 ч при комнатной температуре. Растворитель выпаривали и осадок обрабатывали водным гидрокарбонатом натрия. После обычной экстракции (дихлорметан) растворитель выпаривали с получением неочищенного продукта, который подвергали колоночной хроматографии (силикагель, дихлорметан/метанол, 98:2) с получением промежуточного продукта сложного эфира, метилового эфира 2-этил-1,2,3,4,6,7,12,12b-октагидроиндоло[2,3-а]хинолизин-2-карбоновой кислоты. Такое соединение затем обрабатывали алюмогидридом лития в сухом тетрагидрофуране обычным образом с получением, после колоночной хроматографии (силикагель, дихлорметан/метанол, 95:5), указанного в заголовке спирта. ЯМР: 0,90 (т, 3H), 3,29 (д, 1 Н), 3,43 (д, 1 Н), 3,52 (ушир.д, 1 Н), 7,0-7,5 (м, 4 Н). МС: 284 (100%), 283 (98%), 253 (33%), 197 (37%), 170 (33%), 169 (40%), 156 (34%). Пример 23. (2-Этил-1,2,3,4,6,7,12,12b-октагидроиндоло[2,3-а]хинолизин-2-ил)метанол Раствор 51 мг (0,16 ммоль) промежуточного продукта сложного эфира, полученного в примере 22,(метиловый эфир 2 а-этил-1,2,3,4,6,7,12,12b-октагидроиндоло[2,3-а]хинолизин-2-карбоновой кислоты) в 4 мл трифторуксусной кислоты кипятили с обратным холодильником в атмосфере аргона в течение 16 ч. Кислоту выпаривали и осадок обрабатывали водным гидрокарбонатом натрия. После обычного экстрагирования (дихлорметан) получали неочищенную смесь (20:80) двух диастереомеров, метилового эфира 2-этил-1,2,3,4,6,7,12,12b-октагидроиндоло[2,3-а]хинолизин-2-карбоновой кислоты и метилового эфира 2-этил-1,2,3,4,6,7,12,12b-октагидроиндоло[2,3-а]хинолизин-2-карбоновой кислоты. Последний изомер отделяли колоночной хроматографией (силикагель, дихлорметан/метанол, 99:1) и затем восстанавливали обычным образом алюмогидридом лития в сухом тетрагидрофуране. Очистка, как указано выше, затем давала указанный в заголовке спирт.- 16008537 ЯМР: 0,87 (т, 3H), 3,51 (д, 1 Н), 3,78 (д, 1 Н), 7,0-7,5 (м, 4 Н). МС: 284 (95%), 283 (100%), 253 (30%), 197 (30%), 170 (17%), 169 (23%), 156 (19%). Пример 24. 1-(2-Этил-1,2,3,4,6,7,12,12b-октагидроиндоло[2,3-а]хинолизин-2-ил)этанон и 2-(2 этил-1,2,3,4,6,7,12,12b-октагидроиндоло[2,3-а]хинолизин-2-ил)пропан-2-ол Как в примере 20, 230 мг (0,74 ммоль) промежуточного продукта сложного эфира, полученного в примере 22 (метиловый эфир 2-этил-1,2,3,4,6,7,12,12b-октагидроиндоло[2,3-а]хинолизин-2-карбоновой кислоты) в сухом тетрагидрофуране (9 мл) и 3,7 мл (11,1 ммоль) хлорида метилмагния (3 М в тетрагидрофуране) кипятили с обратным холодильником в течение ночи. Обычная обработка давала, после колоночной хроматографии (силикагель, дихлорметан/метанол, 98:2-95:5), смесь двух соединений 5:1. 1-(2-Этил-1,2,3,4,6,7,12,12b-октагидроиндоло[2,3-а]хинолизин-2-ил)этанон: ЯМР: 0,82 (т, 3H), 2,14 (с, 3H), 3,44 (ушир.д, 1 Н), 7,05-7,50 (м, 4 Н), 8,05 (ушир.с, 1 Н). МС: 296 (83%), 295 (62%), 253 (100%), 184 (95%). 2-(2-Этил-1,2,3,4,6,7,12,12b-октагидроиндоло[2,3-а]хинолизин-2-ил)пропан-2-ол: ЯМР: 1,05 (т, 3H), 1,24 (с, 6 Н), 3,42 (ушир.д, 1 Н), 7,05-7,50 (м, 4 Н), 7,88 (ушир.с, 1 Н). МС: 312 (48%), 311 (37%), 253 (100%). Пример 25. 1-(2-Этил-1,2,3,4,6,7,12,12b-октагидроиндоло[2,3-а]хинолизин-2-ил)этанол Кетон, полученный в вышеуказанной реакции, восстанавливали борогидридом натрия в метаноле обычным образом с получением указанного в заголовке спирта, как неразделимой смеси диастереомеров. ЯМР: 0,95 (т, 3H, второстепенный), 1,18 (т, 3H, основной), 3,61 (кв., 1 Н, второстепенный), 3,67 (кв.,1 Н, основной). МС: 298 (100%), 297 (64%), 253 (87%). Пример 26. 2,3,4,5,7,8,13,13b-Октагидро-1 Н-азепино[1',2':1,2]пиридо[3,4-b]индол (соединение Е) К раствору 0,20 г (1,2 ммоль) триптамина в 5,0 мл ксилола добавляли 0,14 г (1,2 ммоль) капролактама. Смесь кипятили с обратным холодильником в течение 7 ч. После выпаривания растворителя осадок растворяли в 5,0 мл толуола, добавляли 0,65 мл свежеперегнанного оксихлорида фосфора и реакционную смесь кипятили с обратным холодильником в течение 9 ч. Раствор выпаривали в вакууме и осадок смешивали с 20% раствором уксусной кислоты (310 мл). Твердое вещество отфильтровывали, и водный раствор делали щелочным (рН 11) 25% гидроксидом аммония при охлаждении и экстрагировали дихлорметаном (320 мл). К комбинированным органическим слоям добавляли 6,0 мл 4M гидроксида натрия и эту смесь кипятили с обратным холодильником в течение 1 ч. Органическую фазу сушили над сульфатом натрия, осушающее вещество отфильтровывали и фильтрат концентрировали с получением масла, которое растворяли в 30 мл метанола. К холодному раствору добавляли 0,2 г (5,6 ммоль) борогидрида натрия. Смесь перемешивали при комнатной температуре в течение 1 ч. Медленно добавляли воду и реакционную смесь экстрагировали дихлорметаном (320 мл). Комбинированные органические фазы сушили над сульфатом натрия, осушающее вещество отфильтровывали и растворитель выпаривали с получением указанного в заголовке соединения, которое очищали колоночной хроматографией (силикагель, дихлорметан/метанол, 95:5). ЯМР: 4,03 (ушир.д, 1 Н), 7,11-7,46 (м, 4 Н), 8,05 (ушир.с, 1 Н). МС: 240 (52%), 239 (100%), 198 (10%), 170 (24%). Пример 27. 1-Этил-12-метил-1,2,3,4,6,7,12b-октагидроиндоло[2,3-а]хинолизин-1-ол К раствору 0,05 г (0,1 ммоль) 1-этил-1-гидрокси-1,2,3,4,6,7,12,12b-октагидроиндоло[2,3-а] хинолизина и 0,05 г (0,9 ммоль) KOH в 1,0 мл ацетона добавляли 0,02 мл (0,3 ммоль) йодметана. Реакционную смесь перемешивали при кт в течение 1 ч. Медленно добавляли воду и реакционную смесь экстрагировали дихлорметаном (320 мл). Комбинированные органические фазы сушили над сульфатом натрия, осушающее вещество отфильтровывали и фильтрат выпаривали с получением указанного в заголовке соединения, которое очищали колоночной хроматографией (силикагель, дихлорметан/метанол,95:5). ЯМР: 0,71 (т, 3H), 1,01 (м, 2 Н), 3,59 (ушир.с, 1 Н), 3,72 (с, 3H), 7,00-7,50 (м, 4 Н). МС: 284 (21%), 283 (100%), 185 (60%), 170 (10%). Пример 28. 1-Этил-12-этил-1,2,3,4,6,7,12b-октагидроиндоло[2,3-а]хинолизин-1-ол Методику примера 27 повторяли, используя йодэтан вместо йодметана. ЯМР: 0,71 (т, 3H), 1,00 (м, 2 Н), 1,07 (т, 3H), 3,60 (с, 1 Н), 4,20 (м, 1 Н), 4,64 (м, 1 Н), 7,00-7,50 (м, 4 Н). МС: 298 (29%), 297 (19%), 199 (100%), 171 (33%). Пример 29. 1-Метил-1,3,4,5,6,11b-гексагидро-2 Н-11-окса-4 а-азабензо[а]флуорен-1-ол К раствору 0,48 г (3,0 ммоль) 2-(3-бензо[b]фуранил)этиламина в 5,0 мл ксилола добавляли 0,34 г(3,0 ммоль) -метилвалеролактона. Смесь кипятили с обратным холодильником в течение 7,5 ч. После выпаривания растворителя осадок растворяли в 6,0 мл толуола, добавляли 0,72 мл свежеперегнанного оксихлорида фосфора и реакционную смесь кипятили с обратным холодильником в течение 11 ч. Раствор выпаривали в вакууме и полученное масло смешивали с 20% раствором уксусной кислоты (320 мл). Твердое вещество отфильтровывали, водный раствор делали щелочным (рН 11) с помощью 25%- 17008537 гидроксида аммония при охлаждении и экстрагировали дихлорметаном (320 мл). К комбинированным органическим фазам добавляли 12,5 мл 4 М гидроксида натрия и эту смесь кипятили с обратным холодильником в течение 1 ч. Органическую фазу сушили над сульфатом натрия, осушающее вещество отфильтровывали и фильтрат концентрировали с получением соответствующего енамина, который окисляли, как описано в примере 9. ЯМР: 1,18 (с, 3H), 3,25 (ушир.д, 1 Н), 7,10-7,50 (м, 4 Н). МС: 257 (25%), 242 (10%), 172 (100%). Пример 30. (1-Метил-1,3,4,5,6,11b-гексагидро-2 Н-11-окса-4 а-азабензо[а]флуорен-1-ил)метанол Методику примера 29 повторяли, за исключением того, что к образовавшемуся енамину медленно добавляли 40% водный формальдегид. Реакционную смесь кипятили с обратным холодильником в течение 3,5 ч и растворитель выпаривали. Осадок разбавляли этилацетатом и промывали насыщенным раствором соли. Органическую фазу сушили над сульфатом натрия, осушающее вещество отфильтровывали и фильтрат выпаривали с получением указанного в заголовке соединения, которое очищали колоночной хроматографией (силикагель, дихлорметан/метанол, 98:2). ЯМР: 0,89 (с, 3H), 3,40 (ушир.с, 1 Н), 3,62 (д, 1 Н), 4,29 (д, 1 Н), 7,10-7,50 (м, 4 Н). МС: 271 (69%), 270 (100%), 198 (45%), 171 (52%), 170 (60%) . Пример 31. 1-Изопропил-1,3,4,5,6,11b-гексагидро-2 Н-11-окса-4 а-азабензо[а]флуорен-1-ол Повторяли методику примера 9, используя 2-(3-бензо[b]фуранил)этиламин вместо триптамина. ЯМР: 1,00 (м, 6 Н), 7,25 (м, 2 Н), 7,44 (м, 2 Н). МС: 285 (23%), 242 (10%), 198 (10%), 186 (23%), 172 (100%). Пример 32. 1-Этил-1,3,4,5,6,11b-гексагидро-2 Н-11-окса-4 а-азабензо[а]флуорен-1-ол Повторяли методику примера 29, используя -этилвалеролактон вместо -метилвалеролактона. ЯМР: 0,73 (т, 3H), 3,22 (ушир.с, 1 Н), 7,00-7,30 (м, 2 Н), 7,40-7,55 (м, 2 Н). МС: 271 (15%), 186 (18%), 173 (11%), 172 (100%), 170 (28%). Пример 33. (1-Этил-1,3,4,5,6,11b-гексагидро-2 Н-11-окса-4 а-азабензо[а]флуорен-1-ил)метанол Повторяли методику примера 30, используя -этилвалеролактон вместо -метилвалеролактона. ЯМР: 0,62 (т, 3H), 3,48 (ушир.с, 1 Н), 3,52 (д, 1 Н), 4,06 (д, 1 Н), 7,00-7,30 (м, 2 Н), 7,40-7,55 (м, 2 Н). МС: 285 (56%), 284 (100%), 268 (19%), 198 (36%), 172 (20%), 171 (44%), 170 (54%). Пример 34. 5,6,7,7 а,11,11b,12-Декагидро-6 а,12-диазаиндено[1,2-а]флуорен-11 а-ол Повторяли методику примера 29, используя вместо 2-(3-бензо[b]фуранил)этиламина и -метил-валеролактона триптамин и гексагидроизобензофуран-1-он. ЯМР: 4,45 (ушир.д, 1 Н), 7,00-7,60 (м, 4 Н), 9,11 (ушир.с, 1 Н). МС: 296 (8%), 143 (100%), 130 (81%). Пример 35. 1,2,3,4,4 а,5,6,7,8,13-Декагидро-6 а,13-диaзaиндeнo[1,2-c]фенантрен К раствору 0,356 г (1,26 ммоль) N-[2-(3-индолил)этил]декагидроизохинолина в 20 мл этанола добавляли раствор 1,6 г ацетата ртути и 1,88 г дигидрата динатриевой этилендиаминтетрауксусной кислоты в 40 мл воды и полученную смесь кипятили с обратным холодильником в течение 3 ч. Охлажденную смесь делали основной с помощью разбавленного гидроксида аммония (рН 11) и затем экстрагировали дихлорметаном (330 мл). Комбинированные органические слои сушили над сульфатом натрия, фильтровали и растворитель выпаривали с получением неочищенного енамина (смеси региоизомеров), который непосредственно использовали на следующей стадии (см. пример 36). Чистый енамин мог быть получен колоночной хроматографией (силикагель, дихлорметан/метанол/ триэтиламин, 98:1:1). Пример 36. 2,3,4,4,5,6,7,8,13,13b-Декагидро-1 Н-6 а,13-диазаиндено[1,2-с]фенантрен-13 с-ол (соединение F) Как в примере 9, 0,42 г (1,51 ммоль) неочищенного енамина примера 35 обрабатывали 0,21 г йодида калия и 0,32 г йода в 30 мл ацетонитрила. После восстановления 0,29 г борогидрида натрия в 30 мл метанола неочищенный продукт очищали колоночной хроматографией (диоксид кремния, дихлорметан/метанол, 99:1) с получением чистого спирта. ЯМР: 3,18 (ушир.с, 1 Н), 7,0-7,55 (м, 4 Н), 9,18 (ушир.с, 1 Н). МС: 296 (25%), 295 (10%), 185 (15%), 171 (100%). Пример 37. (2,3,4,4,5,6,7,8,13,13b-Декагидро-1 Н-6 а,13-диазаиндено[1,2-с]фенантренил)-13 сметанол Раствор 150 мг (1,51 ммоль) вышеуказанного чистого енамина (из примера 35), 2 мл 36% водного формальдегида и 0,2 мл N-этилдиизопропиламина в 10 мл ацетонитрила кипятили с обратным холодильником в течение 3 ч. После обработки неочищенный продукт очищали колоночной хроматографией (силикагель, дихлорметан/метанол, 98:2) с получением чистого спирта. ЯМР: 3,29 (ушир.с, 1 Н), 3,98 (д, 1 Н), 4,17 (д, 1 Н), 7,0-7,5 (м, 4 Н), 10,05 (ушир.с, 1 Н). МС: 310 (88%), 309 (100%), 293 (34%), 197 (67%), 184 (35%), 170 (90%), 169 (77%). Пример 38. 3,4-Диметил-1,2,3,4,6,7,12,12b-октагидроиндоло[2,3-а]хинолизин- 18008537 К раствору 0,422 г (1,65 ммоль) N-[2'-(3'-индолил)этил]-2,3-диметилпиперидина в 25 мл этанола добавляли 2,1 г ацетата ртути и 2,46 г дигидрата динатриевой соли этилендиаминтетрауксусной кислоты в 50 мл воды и полученную смесь кипятили с обратным холодильником в течение 3 ч. Охлажденную смесь делали основной с помощью разбавленного гидроксида аммония и затем экстрагировали дихлорметаном. Сушка над сульфатом натрия, фильтрование и выпаривание растворителя давали неочищенный енамин,который растворяли в 30 мл метанола и охлаждали на ледяной бане. Добавляли несколько капель уксусной кислоты с последующим добавлением 0,322 г борогидрида натрия по частям. После перемешивания в течение 1,5 ч смесь обрабатывали обычным образом с получением неочищенного продукта, который очищали колоночной хроматографией (силикагель, дихлорметан/метанол 98,5:1,5). ЯМР: 0,89 (д, 3H), 0,96 (д, 3H), 3,76 (ушир.д, 1 Н), 7,0-7,5 (м, 4 Н), 7,71 (ушир.с, 1 Н). МС: 254 (95%), 253 (100%), 239 (30%), 170 (31%), 169 (36%). Пример 39. Этиловый эфир (1-этил-1,2,3,4,6,7,12,12b-октагидроиндоло[2,3-а]хинолизин-1 илметокси)уксусной кислоты Раствор 0,02 г (0,07 ммоль) (1-этил-1,2,3,4,6,7,12,12b-октагидроиндоло[2,3-а]хинолизин-1 ил)метанола (Gazz. Chim. Ital. 111 (1981), 257-267) в смеси N,N-диметилформамид-толуол (1 мл, 1:1) добавляли к 6,8 мг (0,28 ммоль) гидрида натрия, предварительно промытому гептаном. Реакционную смесь перемешивали при комнатной температуре в течение 1 ч и затем по каплям добавляли этилбромацетат(0,009 мл, 0,084 ммоль) в толуоле (1 мл). Перемешивание продолжали в течение 3 ч при комнатной температуре. Медленно добавляли воду и реакционную смесь экстрагировали дихлорметаном (320 мл). Комбинированные органические фазы сушили над сульфатом натрия, осушающее вещество отфильтровывали и фильтрат выпаривали с получением указанного в заголовке соединения, которое очищали колоночной хроматографией (силикагель, дихлорметан/метанол, 5:5). ЯМР: 0,61 (т, 3H), 1,28 (т,3H), 3,49 (с, 1 Н), 4,25 (д, 1 Н), 4,30 (кв., 2 Н), 4,55 (д, 1 Н), 6,93 (т, 1 Н), 7,03(т, 1 Н), 7,35 (д, 1 Н), 7,38 (д, 1 Н), 10,64 (с, 1 Н). МС: 370 (40%), 369 (30%), 283 (12%), 267 (100%), 197 (12%), 170 (12%), 169 (16%). Пример 40. 5,6,7, 7 а,8,9,10,11,11a, 11b-Декагидро-12-окса-6 а-азаиндено[1,2-а]флуорен К раствору 0,70 г (0,43 ммоль) 2-(3-бензо[b]фуранил)этиламина в 30 мл хлорбензола добавляли 0,13 г (0,87 ммоль) цис-1,2-циклогександикарбонового ангидрида. Смесь облучали в микроволновой печи(1000 Вт, Т=130 С) в течение 30 мин. Хлорбензол заменяли этанолом (5 мл) и добавляли 82,7 мг (2,18 ммоль) борогидрида натрия. Смесь перемешивали при кт в течение 18 ч, затем добавляли воду и продукт выделяли обычным образом. Добавляли трифторуксусную кислоту (0,12 мл, 1,53 ммоль) в дихлорметане(10 мл) и реакционную смесь перемешивали в течение 2 ч при кт. Обработка щелочью (4 М гидроксида натрия) давала амидный промежуточный продукт, который растворяли в диэтиловом эфире (15 мл), добавляли 0,1 г (2,63 ммоль) алюмогидрида лития и реакционную смесь кипятили с обратным холодильником в течение 1,5 ч. Медленно добавляли воду при охлаждении. После обычного экстрагирования неочищенный продукт очищали колоночной хроматографией (силикагель, дихлорметан/метанол, 90:10). ЯМР: 1,77 (м, 2 Н), 2,07 (м, 1 Н), 2,29 (м, 1 Н), 2,48 (м, 1 Н), 3,04 (м, 1 Н), 3,20 (м, 1 Н), 4,13 (с, 1 Н),7,18-7,50 (м, 4 Н). МС: 267 (46%), 266 (100%), 185 (52%), 170 (12%). Пример 41. 1-Метил-1,3,4,6,11b-гексагидро-2 Н-11-окса-4 а-азабензо[а]флуорен (соединение G) Повторяли методику примера 26, используя -метилвалеролактон и 2-(3-бензо[b] фуранил)этиламин вместо -капролактама и триптамина соответственно. ЯМР: 0,88 (д, 3H), 3,34 (ушир.с, 1 Н), 7,19-7,43 (м, 4 Н). МС: 241 (40%), 240 (50%), 226 (100%), 198 (10%), 170 (68%), 170 (24%). Пример 42. (1-Гидроксиметил-1,3,4,5,6,11b-гексагидро-2 Н-11-окса-4 а-азабензо[а]флуорен-1-ил)метанол Повторяли методику примера 41, используя -валеролактон вместо -метилвалеролактона, и полученный енамин обрабатывали формальдегидом, как в примере 14. ЯМР: 3,30 (д, 1 Н), 3,76 (д, 1 Н), 3,79 (д, 1 Н), 3,82 (с, 1 Н), 4,31 (д, 1 Н), 7,18-7,50 (м, 4 Н). МС: 287 (56%), 286 (60%), 270 (40%), 256 (100%), 198 (34%), 172 (26%), 170 (54%). Пример 43. 1-Метоксиметил-1-метил-1,3,4,5,6,11b-гексагидро-2 Н-11-окса-4 а-азабензо[а]флуорен Раствор 173,1 мг (0,64 ммоль) спирта, описанного в примере 30, в 5 мл тетрагидрофурана, добавляли к 153,0 мг (6,38 ммоль) гидрида натрия, предварительно промытого гептаном. Реакционную смесь перемешивали при 35 С в течение 1 ч с последующим добавлением по каплям раствора 0,04 мл (0,64 ммоль) йодметана в тетрагидрофуране (5 мл). Перемешивание продолжали в течение 1 ч. Медленно добавляли воду и реакционную смесь экстрагировали дихлорметаном (320 мл). Комбинированные органические фазы сушили над сульфатом натрия, осушающее вещество отфильтровывали и фильтрат выпаривали с получением указанного в заголовке соединения, которое очищали колоночной хроматографией- 19008537 2,3,4,4 а,5,6,7,8,13b,13 с-декагидро-1 Н-13-окса-6 а-азаиндено[1,2-с]фенантрен Методику примера 41 повторяли, используя цис-октагидроизохромен-1-он вместо -метил-валеролактона. Два изомера разделяли колоночной хроматографией (силикагель, этилацетат/гептан,70:30). 2,3,4,4 а,5,6,7,8,13b,13 с-Декагидро-1 Н-13-окса-6 а-азаиндено[1,2-c]фенантрен: ЯМР: 3,28 (с, 1 Н), 7,17-7,53 (м, 4 Н). МС: 281 (40%), 280 (100%), 238 (15%), 198 (12%), 170 (24%). 2,3,4,4 а,5,6,7,8,13b,13 с-Декагидро-1 Н-13-окса-6 а-азаиндено[1,2-c]фенантрен: ЯМР: 2,75 (д, 1 Н), 7,15-7,43 (м, 4 Н). МС: 281 (38%), 280 (100%), 198 (16%), 170 (30%). Пример 45. Этиловый эфир 1-метил-1,3,4,5,6,11b-гексагидро-2 Н-11-окса-4 а-азабензо[а]флуорен 1-карбоновой кислоты К смеси 0,375 г (2,33 ммоль) 2-(3-бензо[b]фуранил)этиламина и триэтиламина (0,97 мл, 7,0 ммоль) в дихлорметане (3 мл) добавляли 0,56 г (2,33 ммоль) 5-хлор-2-этоксикарбонил-2-метилвалероилхлорида(полученного в соответствии со способом, описанным для соответствующего 2-этил производного в J.Org. Chem. 45 (1980), 32-34) в дихлорметане (4 мл). После перемешивания при комнатной температуре в течение 45 мин добавляли воду и смесь экстрагировали дихлорметаном. Сушка над сульфатом натрия,отфильтровывание осушающего вещества и выпаривание растворителя давало неочищенный амид, который очищали колоночной хроматографией (этилацетат/гептан, 1:1). Чистый амид (0,3 г, 0,82 ммоль) растворяли в толуоле (3 мл) и добавляли 0,38 мл (4,1 ммоль) оксихлорида фосфора. Смесь кипятили с обратным холодильником в течение 2 ч, затем выпаривали досуха. Осадок растворяли метанолом (3 мл) и добавляли 57 мг (1,5 ммоль) борогидрида натрия по частям. После перемешивания в течение 1 ч добавляли воду и смесь экстрагировали этилацетатом. Сушка над сульфатом натрия с последующим фильтрованием и выпариванием давала неочищенный сложный эфир, который очищали колоночной хроматографией (силикагель, этилацетат/гептан, 1:1). ЯМР: 0,65 (т, 3H), 1,55 (с, 3H), 3,30 (ушир.с, 1 Н), 7,16-7,50 (м, 4 Н). МС: 313 (70%), 312 (100%), 284 (22%), 240 (32%), 198 (80%), 171 (35%), 170 (95%). Пример 46. 1-Этоксиметил-1-метил-1,3,4,5,6,11b-гексагидро-2 Н-11-окса-4 а-азабензо[а]флуорен Повторяли методику примера 43, используя йодэтан вместо йодметана. ЯМР: 0,74 (с, 3H), 1,17 (т, 3H), 3,37 (с, 1 Н), 3,38 (д, 1 Н), 3,54 (кв., 2 Н), 3,96 (д, 1 Н), 7,10-7,60 (м, 4 Н). МС: 299 (70%), 298 (92%), 270 (40%), 254 (100%), 198 (34%), 171 (86%), 170 (72%). Пример 47. (1-Метил-1,3,4,5,6,11b-гексагидро-2 Н-11-окса-4 а-азабензо[а]флуорен-1-ил)метанол К суспензии 0,31 г (8,23 ммоль) алюмогидрида лития в сухом тетрагидрофуране (10 мл) добавляли 0,86 г (2,74 ммоль) сложного эфира, описанного в примере 45, в сухом тетрагидрофуране (10 мл). Реакционную смесь кипятили с обратным холодильником в течение 1 ч. Медленно добавляли воду и реакционную смесь экстрагировали этилацетатом (320 мл). Комбинированные органические фазы сушили над сульфатом натрия, фильтровали и фильтрат выпаривали с получением желаемого продукта, который очищали колоночной хроматографией (силикагель, этилацетат/гептан 50:50). ЯМР: 1,30 (с, 3H), 2,98 (ушир.с, 1 Н), 3,21 (д, 1 Н), 3,69 (д, 1 Н), 4,33 (с, 1 Н), 7,15-7,55 (м, 4 Н). МС: 271 (52%), 270 (100%), 198 (34%), 172 (20%), 171 (44%), 170 (66%). Пример 48. (1-Метил-1,2,3,4,6,7,12,12b-октагидроиндено[2,1-а]хинолизин-1-ил)метанол Повторяли методики, описанные в примерах 4 5 и 47, используя 2-(3H-инден-1-ил)этиламин вместо 2-(3-бензо[b]фуранил)этиламина. ЯМР: 0,82 (с, 3H), 3,07 (ушир.с, 1 Н), 3,23 (д, 1 Н), 3,39 (д, 1 Н), 3,52 (д, 1 Н), 3,70 (д, 1 Н), 7,05-7,35 (м,4 Н). МС: 269 (43%), 268 (100%), 252 (29%), 196 (36%), 168 (40%). Пример 49. Метиловый эфир 1-этил-1,3,4,5,6,11b-гексагидро-2 Н-11-окса-4 а-азабензо[а]флуорен 1-карбоновой кислоты. Повторяли методику примера 45, используя хлорид 5-хлор-2-этоксикарбонил-2-этилвалероила вместо хлорида 5-хлор-2-этоксикарбонил-2-метилвадероила. ЯМР: 0,90 (т, 3H), 6,90-7,58 (м, 4 Н). МС: 327 (72%), 326 (100%), 312 (20%), 298 (20%), 254 (30%), 198 (54%), 172 (60%), 170 (90%). Пример 50. 1-Метоксиметил-1-метил-1,3,4,5,6,11b-гексагидро-2 Н-11-окса-4 а-азабензо[а]флуорен Повторяли методику примера 43, используя спирт, описанный в примере 47, в качестве исходного соединения. ЯМР: 1,44 (с, 3H), 2,99 (д, 1 Н), 3,15 (ушир.с, 1 Н), 3,22 (с, 3H), 3,70 (д, 1 Н), 7,18-7,50 (м, 4 Н). МС: 285 (84%), 284 (100%), 270 (14%), 254 (92%), 198 (34%), 171 (74%), 170 (50%). Пример 51. (1-этил-1,3,4,5,6,11b-гексагидро-2 Н-11-окса-4 а-азабензо[а]флуорен-1-ил)метанол Повторяли методику примера 47, используя сложный эфир, описанный в примере 49, в качестве исходного соединения. ЯМР: 1,00 (т, 3H), 2,93 (м, 2 Н), 3,29 (с, 1 Н), 7,15-7,60 (м, 4 Н).- 20008537 МС: 286 (90%), 285 (68%), 284 (100%), 268 (16%), 198 (22%), 171 (22%), 170 (36%). Пример 52. Метиловый эфир 1 гидроксиметил-1-метил-1,2,3,4,6,7,12,12b-октагидроиндоло[2,3 а]хинолизин-6-карбоновой кислоты Повторяли методику получения (1-этил-1,2,3,4,6,7,12,12b-октагидроиндоло[2,3-а]хинолизин-1 ил)метанола (Gazz. Chim. Ital. 111 (1981), 257-267), используя метиловый эфир L-триптофана вместо триптамина. ЯМР: 0,74 (с, 3H), 3,39 (с, 3H), 3,46 (д, 1 Н), 3,97 (д, 1 Н), 4,38 (ушир.с, 1 Н), 7,00-7,50 (м, 4 Н), 8,90(ушир.с, 1 Н). МС: 328 (26%), 327 (100%), 299 (38%), 268 (32%), 170 (10%), 169 (24%). Пример 53. Разделение 1-изопропил-1,2,3,4,6,7,12,12b-октагидроиндоло[2,3-а]хинолизин-1-ола Раствор 0,3 г (1,1 ммоль) -1-изопропил-1,2,3,4,6,7,12,12b-октагидроиндоло[2,3-а]хинолизин-1 ола и 0,16 г (1,1 ммоль) L-винной кислоты в 15 мл ацетона кипятили с обратным холодильником в течение 30 мин. При стоянии при комнатной температуре в течение ночи выпадало в осадок 200 мг твердого вещества. После двойной перекристаллизации из метанола собранную соль L-тартрата разделяли между дихлорметаном и 10% раствором гидроксида натрия, сушили над сульфатом натрия и выпаривали с получением 116,6 мг (-)-1-изопропил-1,2,3,4,6,7,12,12b-октагидроиндоло[2,3-а]хинолизин-1-ола с[]D=-64,5 (с, 0,011 в CHCl3). Другой энантиомер (+)-1-изопропил-1,2,3,4,6,7,12,12b-октагидроиндоло[2,3-а] хинолизин-1-ол []D=+64,5 (с, 0,011 в CHCl3) выделяли из маточной жидкости таким же образом. Пример 54. Разделение (1-метил-1,3,4,5,6,11b-гексагидро-2 Н-11-окса-4 а-азабензо[а]флуорен-1 ил)метанола Методику примера 53 повторяли, используя ангидрид (+)-диацетил-L-винной кислоты и изопропанол вместо L-винной кислоты и ацетона. Оптическую чистоту разделенных энантиомеров подтверждали хиральной ВЭЖХ (колонка: DAICEL CHEMICAL INDUSTRIES, LTD CHIRACEL OJ, размеры 0,4625 см, поток: 0,5 мл/мин, подвижная фаза: н-гексан (Merck Uvasol для спектроскопии)/изопропанол(Rathburn, для градиентной ВЭЖХ) (100:20), определение УФ при 272 нм, время удерживания: 8,8 мин[(-)-(1-метил-1,3,4,5,6,11b-гексагидро-2 Н-11-окса-4 а-азабензо[а]флуорен-1-ил)метанол]). Пример 55. Разделение (1-метил-1,3,4,5,6,11b-гексагидро-2 Н-11-окса-4 а-азабензо[а]флуорен-1 ил)метанола Методику примера 53 повторяли, используя моногидрат (-)ди-п-толуоил-L-винной кислоты и этилацетат вместо L-винной кислоты и ацетона. Оптическую чистоту разделенных энантиомеров подтверждали хиральной ВЭЖХ (колонка: DAICEL CHEMICAL INDUSTRIES, LTD CHIRACEL OJ, размерами 0,4625 см, поток: 0,8 мл/мин, подвижная фаза: н-гексан (Merck Uvasol для спектроскопии)/изопропанол(Rathburn, для градиентной ВЭЖХ) (180:20), определение УФ при 254 нм, время удерживания: 7,8 мин[(-)-(1-метил-1,3,4,5,6,11b-гексагидро-2 Н-11-окса-4 а-азабензо[а]флуорен-1-ил)метанол]). Пример 56. Энантиомеры 1-метоксиметил-1-метил-1,3,4,5,6,11b-гексагидро-2 Н-11-окса-4 аазабензо[а]флуорена Методику примера 43 повторяли, используя чистые энантиомеры, (+)-(1-метил-1,3,4,5,6,11bгексагидро-2 Н-11-окса-4 а-азабензо[а]флуорен-1-ил)метанол и (-)-(1-метил-1,3,4,5,6,11b-гексагидро 2 Н-11-окса-4 а-азабензо[а]флуорен-1-ил)метанол, соответственно, из примера 54 вместо спирта, описанного в примере 30. Оптическую чистоту продуктов подтверждали хиральной ВЭЖХ (колонка: ROCKLAND TECHNOLOGIES, INS ULTRON ES-OVM, размеры 4,615 см, поток: 0,8 мл/мин, подвижная фаза: 0,04 М KH2PO4 (рН 4,6)/ацетонитрил (Merck Lichrosolv Isocratic класс для жидкостной хроматографии)(90:10), время удерживания 3,8 мин [(-)-1-метоксиметил-1-метил-1,3,4,5,6,11b-гексагидро-2 Н-11-окса 4 а-азабензо[а]флуорен] и 5,8 мин [(+)-1-метоксиметил-1 а-метил-1,3,4,5,6,11b-гексагидро-2 Н-11-окса-4 аазабензо[а]флуорен]). Пример 57. Энантиомеры 1-метоксиметил-1-метил-1,3,4,5,6,11b-гексагидро-2 Н-11-окса-4 аазабензо[а]флуорена Методику примера 43 повторяли, используя чистые знантиомеры, (+)-(1-метил-1,3,4,5,6,11 гексагидро-2 Н-11-окса-4 а-азабензо[а]флуорен-1-ил)метанол и (-)-(1-метил-1,3,4,5,6,11b-гексагидро 2 Н-11-окса-4 а-азабензо[а]флуорен-1-ил)метанол, соответственно, из примера 55 вместо спирта, описанного в примере 30. Оптическую чистоту продуктов подтверждали хиральной ВЭЖХ (колонка: DAICELCHEMICAL INDUSTRIES, LTD CHIRACEL OJ, размеры 0,4625 см, поток: 0,8 мл/мин, подвижная фаза: н-гексан (Merck Uvasol для спектроскопии), время удерживания 5,6 мин [(+)-1-метоксиметил-1-метил 1,3,4,5,6,11b-гексагидро-2 Н-11-окса-4 а-азабензо[а]флуорен] и 6,3 мин [(-)-1-метоксиметил-1-метил 1,3,4,5,6,11b-гексагидро-2 Н-11-окса-4 а-азабензо[а]флуорен]). Следующие известные соединения могут быть получены аналогично или в соответствии со способами, известными в литературе.(1,2,3,4,6,7,12,12b-Октагидроиндоло[2,3-а]хинолизин-1-ил)метанол (соединение О) получали восстановлением соответствующего сложного эфира, синтез которого описан в Tetrahedron 52 (1996),9925. 1-(1,2,3,4,6,7,12,12b-Октагидроиндоло[2,3-а]хинолизин-1-ил)этанол (соединение Р) получали восстановлением его соответствующего кетона, синтез которого описан в Tetrahedron Lett. 30 (1989), 719. 1-Пропил-1,2,3,4,6,7,12,12b-октагидроиндоло[2,3-а]хинолизин получали в соответствии со способом, описанным в J. Org. Chem. 34 (1969), 330. 1-Этил-1-метил-1,2,3,4,6,7,12,12b-октагидроиндоло[2,3-а]хинолизин получали в соответствии со способом, описанным в J. Chem. Res. (S) (1995), 382. 2-трет-Бутил-1,2,3,4,6,7,12,12b-октагидроиндоло[2,3-а]хинолизин и 2-трет-бутил-1,2,3,4,6,7,12,12bоктагидроиндоло[2,3-а]хинолизин (соединение Q) получали в соответствии со способом, описанным в Tetrahedron 45 (1989), 3975. 2-трет-Бутил-1,4,6,7,12,12b-гексагидроиндоло[2,3-а]хинолизин и 2-трет-бутил-3,4,6,7,12,12bгексагидроиндоло[2,3-а]хинолизин получали в соответствии со способом, описанным в Tetrahedron 47(-)-1-Этил-1,2,3,4,6,7,12,12b-октагидроиндоло[2,3-а]хинолизин-1-ол и (+)-1-этил-1,2,3,4,6,7,12,12bоктагидроиндоло[2,3-а]хинолизин-1-ол получали разделением их рацемической смеси (соединение K). Как уже было указано выше, соединения по настоящему изобретению проявляют интересные фармакологические свойства, а именно, они проявляют афинность в отношении альфа 2-адренорецепторов. Указанная фармакологическая активность соединений по изобретению продемонстрирована с помощью фармакологических тестов, представленных ниже. Эксперимент I. Связывание радиоактивного лиганда с альфа 2-адренорецепторами Примеры связывающей способности альфа 2-адренорецепторов соединений, включенных в настоящее изобретение, показаны в табл. 1. Многие из этих соединений являются высокоафинными лигандами в отношении всех альфа 2-рецепторов, но некоторые из них проявляют селективность в отношении альфа 2 С-подтипу.- 22008537 Таблица 1 Рассчитанные значения Ki исследований связывания радиоактивных лигандов с использованием клеток,экспрессирующих подтипы человеческих альфа 2-адренорецепторов Эксперимент II. Антагонизм in vitro альфа 2-адренорецепторов Функциональная активность двух соединений (K и L), проявляющих альфа 2-селективность в экспериментах связывания, определяли как способность соединений ингибировать эпинефринстимулированное связывание 35S-GTPS с G белками (Jasper, J.R. et al., Biochem. Pharmacol. 55(7) (1998),1035-44) в мембранах клеток СНО, стойко трансфицированных подтипами человеческих альфа 2 адренорецепторов. Антагонистическая активность соединения K и соединения L представлена в табл. 2. Результаты показывают, что эти соединения являются селективными антагонистами альфа 2 С-подтипов. Таблица 2 Средняя антагонистическая активность (Kв) соединения K и соединения L на подтипах человеческих альфа 2-адренорецепторовIn vivo действие альфа 2 С-селективных соединений В настоящее время в данной области не известно, какие эффекты in vivo могут быть обусловлены селективным альфа 2 С-антагонизмом. На основании доступных знаний и предшествующего опыта авторы выбрали две различные модели поведения, а именно, модель d-амфетамин-стимулированной двигательной активности и тест принудительного плавания, с целью продемонстрировать специфическое альфа 2 С-антагонистическое действие в ЦНС мышей и крыс in vivo. Выбор этих методов главным образом основан на опубликованных гипотезах о теоретических эффектах альфа 2 С-антагонистов; в отсутствие подходящих лигандов, эти гипотезы были основаны на исследованиях, включающих мышей с генетически измененной экспрессией альфа 2 С-адренорецептора (Scheinin, M. et al., Life Sci 68(19-20) (2001),2277-85). Эксперимент III. Тест D-амфетамин-стимулированной двигательной активности Генетически модифицированные мыши, имеющие нефункционирующие альфа 2 С-адренорецепторы(альфа 2 С-"нокаут"; альфа 2 С-KO), являются более чувствительными к действию, усиливающему двигательную активность психостимулятора d-амфетамина и, с другой стороны, повышенная экспрессия альфа 2 С-адренорецептора у мышей (alpha2C-OE) приводит к противоположному эффекту, т.е. уменьшению действия стимулятора (Scheinin, M. et al., Life Sci 68(19-20) (2001), 2277-85). Следовательно, может быть предложена гипотеза, что альфа 2 С-антагонисты будут усиливать двигательное действие d-амфетамина. Вышеуказанное предположение было протестировано введением группам мышей (n=10-12/доза группа)амфетамина (4 мкмоль/кг п/к) отдельно или вместе с альфа 2 С-антагонистами (3 мкмоль/кг п/к) по настоящему изобретению или с альфа 2-подтипом неселективного сильного альфа 2-антагониста (1 мкмоль/кг п/к) (Haapalinna, A. et al., Naunyn-Schmiedeberg's Arch. Pharmacol. 356 (1997) 570-582), и путем последующего измерения двигательной активности у мышей с помощью автоматизированной системы с инфракрасным фотолучем, созданной для изучения активности (PAS CageRack, SanDiego Instruments, SanDiego, CA., USA). Как ожидали, оба тестируемых альфа 2 С селективных антагониста увеличивали активность мышей (фиг. 1 а, b), как ожидали для альфа 2 С-антагонистов. Подтип неселективного альфа 2 антагониста также усиливал действие d-амфетамина. Тестируемые соединения не влияли на исходную двигательную активность мыши (в дозах между 0,1-10 мг/кг п/к). Эксперимент IV. Антагонизм седативного эффекта, индуцированного альфа 2-агонистом Одним из явных эффектов неселективных альфа 2-агонистов у грызунов является их способность вызывать сильный седативный эффект. Этот эффект, измеряемый как двигательное ингибирование альфа 2-агонистом дексмедетомидином не изменялось у мышей с генетически измененной экспрессией альфа 2 С (Scheinin, M. et al., Life Sci 68(19-20) (2001), 2277-85). С другой стороны, альфа 2-агонист не имел седативного эффекта у мышей с генетически нарушенным альфа 2 А-адренорецептором (Hunter, J.С. et al.,British Journal of Pharmacology 122(7) (1997), 1339-44). Следовательно, так как седативный эффект альфа 2-агонистов обычно связан с альфа 2-адренорецептором, ожидали, что альфа 2 С-антагонисты значительно не будут модулировать седативный эффект, индуцированный альфа 2-агонистами. Это предположение было протестировано в эксперименте, где дексмедетомидин вводили мышам, ранее получившим альфа 2 С-антагонисты соединение K или соединение L, или подтип неселективного антагониста атипамезол (Haapalinna, A. et al., Naunyn-Schmiedeberg's Arch. Pharmacol. 356 (1997) 570-582). Как ожидали, альфа 2 С-антагонисты не имели четкого действия, тогда как атипамезол эффективно противодействовал эффекту дексмедетомидина. Этот результат демонстрирует отсутствие альфа 2 А-антагонизма у альфа 2 С селективных соединений по настоящему изобретению (фиг. 2). Эксперимент V. Тест принудительного плавания Тест принудительного плавания (ТПП (FST), т.е. тест Порсольта) обычно используют в фармакологическом скрининге новых антидепрессантов. В данном тесте антидепрессанты повышают активность животных по сравнению с необработанными контрольными. Оказалось, что мыши альфа 2 С-KO являются более активными, а мыши альфа 2 С-ОЕ являются менее активными при ТПП (патент США 5902807 иScheinin, M. et al., Life Sci 68(19-20) (2001), 2277-85). Следовательно, исследовали, обладает ли селективный альфа 2 С-антагонист антидепрессантподобной активностью (например, свойством повышения активности) в ТПП. На фиг. 3 показано как оба альфа 2 С-соединения повышали активность в данном тесте,как ожидалось на основании исследований на трансгенных мышах (Scheinin, M. et al., Life Sci. 68(19-20)(2001), 2277-85) и как сообщалось о недавно разработанном альфа 2 С-антагонисте (WO 01/64645). Также положительные контрольные вещества дезипрамин и флуоксетин (клинически эффективные антидепрессанты) были активными. Подтип неселективного альфа 2-антагониста атипамезол не обладал антидепрессант-подобным эффектом, как ожидалось (WO 01/64645). Эксперимент VI. Опережающее ингибирование рефлекса испуга Опережающее ингибирование (ОИ (PPI рефлекса испуга относится к уменьшению реакции испуга, вызываемого непугающим стимулом низкой интенсивности (опережающий импульс), который представляют коротко перед раздражителем, вызывающим испуг. ОИ может быть использован как рабочая мера сенсомоторного проведения и, видимо, присутствует у всех млекопитающих, включая крыс и людей (Swerdlow, N.R. et al. The archives of general psychiatry 51 (1994), 139-154). Нормально функционирующий ОИ может быть нарушен психостимуляторами, такими как d-амфетамин или фенилциклидин(ФЦК), и меняться на противоположный клинически эффективными антипсихотическими агентами. В предыдущем исследовании, мутация альфа 2 С-KO была ассоциирована с ослабленным ОИ, тогда как альфа 2 С-OЕ демонстрировала повышенный ОИ. Другими словами, генетически измененная экспрессия альфа 2 С у мышей была ассоциирована с изменениями в ОИ в отношении, предполагающем, что альфа 2 С снижает ОИ (Scheinin, M. et al. Life Sci. 68(19-20) (2001), 2277-85). Такая гипотеза была протестирована с соединениями K и L отдельно и против нарушения ОИ ФЦК. Группам крыс (n=10/группа) вводили альфа 2 С-антагонисты за 20 мин, и ФЦК или растворитель за 10 мин до измерения звуковой реактивности испуга и ОИ в тест-системе, созданной для исследования испуга (SR-LAB, San Diego Instruments, CA, USA). Было обнаружено, что альфа 2 С-антагонисты оказались способны ослаблять нарушение ОИ, вызываемое ФЦК (фиг. 3). Это было неожиданным и противоречило гипотезе, основанной на трансгенных исследованиях. Неселективный альфа 2-антагонист атипамезол способствовал отличным эффектам от тех, которые наблюдались при селективных альфа 2 С- 24008537 антагонистах: атипамезол не усиливал ОИ, но он увеличивал рефлекс испуга, как таковой (т.е. испуг без опережающих импульсов) (фиг. 4). В заключении, результаты, представленные в данном разделе, показывают, что такие антагонисты,которые классифицированы как альфа 2 С-селективные в соответствии с in vitro экспериментами, видимо,функционируют как альфа 2 С-селективные антагонисты также in vivo таким же образом, как прогнозировали на основании доступных знаний альфа 2 С-антагонизма. Однако обнаружение того, что альфа 2 Сантагонисты не снижают ОИ, как предсказывали, а наоборот, повышают ОИ, могло рассматриваться как неожиданное и это добавляет новое значение в настоящее время предложенную применимость соединений по настоящему изобретению. Соединения по настоящему изобретению могут быть использованы для лечения любого заболевания или состояния, при которых показано, что альфа 2-антагонисты являются эффективными. Соединения также могут быть использованы для обращения эффектов, индуцируемых альфа 2-агонистами. Соответственно, соединения по изобретению могут быть полезны в лечении различных расстройств центральной нервной системы (ЦНС), т.е. различных неврологических, психиатрических и когнитивных расстройств (таких как депрессия, тревожные расстройства, посттравматические стрессорные расстройства, шизофрения, болезнь Паркинсона и другие расстройства движения). Более того, они могут быть использованы в лечении различных периферических расстройств, например, диабета, ортостатической гипотензии, липолитических расстройств (таких как ожирение), болезни Рейно и мужских и женских сексуальных дисфункций. Селективные альфа 2 С-антагонисты по настоящему изобретению могут быть использованы для лечения различных расстройств или состояний ЦНС-системы, где показано, что альфа 2 С-антагонисты являются полезными, например, для облегчения симптомов различных психических расстройств, вызываемых стрессом, болезнью Паркинсона, депрессией, негативными симптомами шизофрении, расстройствами дефицита внимания и гиперактивности, посттравматическими стрессовыми расстройствами и тревожными расстройствами. Кроме того, из-за новых и ранее неопубликованных обнаруженных эффектов настоящих альфа 2 Сантагонистов на ФЦК-нарушенную ОИ, альфа 2 С-селективные соединения также могут использоваться для лечения расстройств и состояний, ассоциированных с дефицитом сенсорно-двигательной селекции,особенно расстройствами и состояниями, где дефицит сенсорно-двигательной селекции приводит к сенсорному погружению и когнитивному разложению, вызывая дисфункцию внимания и ощущения. Такие расстройства и состояния включают, но не ограничиваются ими, шизофрению, обсессивнокомпульсивные расстройства, синдром Туретта, блефароспазм и другие очаговые дистонии, эпилепсию височной доли с психозом, психозы, индуцированные лекарственными препаратами (например, психоз,вызываемый хроническим применением допаминергических агентов) (Braff, D.L. et al. Psychopharmacology (Berl) 156(2-3) (2001), 234-258), болезнь Геттингтона, болезнь Паркинсона, расстройства, вызываемые колебаниями уровней половых гормонов (такими как пременструальный синдром) и панические расстройства. Далее, симптомы, которые обычно связаны с приведенными выше расстройствами или состояниями, включают, но не ограничиваются ими, галлюцинации, иллюзии, паратимию, тревожное возбуждение,психотические когнитивные нарушения (включая дефициты в мыслях и речи), социальноевыпадение и симптомы отмены (включая делирий), связанные с прекращением курения сигарет или злоупотребления алкоголем или лекарственными препаратами. Такие симптомы также могут наблюдаться у животных в необычных обстоятельствах, например, во время расставания с хозяевами или во время транспортировки. Из-за селективности их действия альфа 2 С-антагонисты по изобретению имеют меньше или не имеют нежелательных побочных эффектов, присущих неселективному альфа 2-антагонизму, таких как повышение артериального давления, частоты сердечных сокращений, секреции слюны, секреции желудочно-кишечного тракта, тревоги и стартовой реактивности как таковой (Ruffolo, R.R.J., et al. Annu RevPharmacol Toxicol 32 (1993) 243-279). Соединение по изобретению можно вводить, например, энтерально, местно или парентерально посредством любой фармацевтической композиции, применимой для указанного введения, и содержащей по меньшей мере одно активное соединение формулы I в фармацевтически приемлемых и эффективных количествах вместе с фармацевтически приемлемыми разбавителями, носителями и/или эксципиентами,известными в данной области. Получение таких фармацевтических композиций хорошо известно в данной области. Терапевтическая доза для введения пациенту, нуждающемуся в лечении, будет варьироваться в зависимости от вводимого соединения, вида, возраста и пола субъекта, подвергаемого лечению, особенного лечимого состояния, а также пути и способа введения и легко определяется специалистом в данной области. Соответственно, типичная дозировка для перорального введения составляет от 5 мкг/кг до 100 мг/кг в день и для парентерального введения от 0,5 мкг/кг до 10 мг/кг для взрослого млекопитающего. Настоящее изобретение, кроме того, обеспечивает соединение по изобретению или его сложный эфир или соль для применения в качестве альфа 2-антагониста. Кроме того, обеспечивают способ лечения- 25008537 заболеваний или состояний, при которых показано, что альфа 2-антагонисты являются полезными, например способ лечения заболеваний или состояний центральной нервной системы. В таком способе терапевтически эффективное количество соединения по изобретению вводят субъекту, нуждающемуся в таком лечении. Также обеспечивают применение соединений по изобретению с получением лекарственного средства для использования при вышеуказанных показаниях. Специалист в данной области должен принять во внимание, что варианты воплощения изобретения,описанные в данном изобретении, могут быть модифицированы без отклонений от общей идеи изобретения. Специалисту в данной области также будет очевидно, что изобретение не ограничивается определенными описанными вариантами воплощения, но имеет целью также включать модификации вариантов воплощения изобретения, которые не меняют существа и входят в объем изобретения. ФОРМУЛА ИЗОБРЕТЕНИЯ 1. Соединение формулы IAR2 и R2' представляют собой независимо Н, гидрокси или (C1-C6)алкил или R2 и R2' образуют вместе с атомами углерода, образующими кольцо, к которым они присоединены, карбонильную группу;R3 представляет собой гидрокси, (C1-C6)алкил, (C2-C6)алкенил, гидрокси(C1-C6)алкил, (C1C6)алкокси, (C1-C6)алкокси(C1-C6)алкил, гидрокси(C1-C6)алкокси(C1-C6)алкил, (C3-C7)циклоалкил, (C3C7)циклоалкил(C1-C6)алкил, арил, арил(C1-C6)алкил, арилокси, арил(C1-C6)алкокси, арилокси(C1C6)алкил, арил(C1-C6)алкокси(C1-C6)алкил, галоген(C1-C6)алкил, NH2, амино(C1-C6)алкил, моно- или ди(C1-C6)алкиламино, моно- или ди(C1-C6)алкиламино(C1-C6)алкил, (C1-C6)алкил-СО-, (C1-C6)алкил-СОO-, (C1-C6)алкил-СО-O-(C1-C6)алкил, (C1-C6)алкокси-СО-, (C1-C6)алкокси-СО-(C1-C6)алкил, (C1C6)алкокси-СО-(C1-C6)алкокси(C1-C6)алкил, карбамоил, моно- или ди(C1-C6)алкилкарбамоил, карбоксил или (C1-C6)алкил-S-(C1-C6)алкил, где указанный (C3-C7)циклоалкил или арил являются незамещенными или замещенными 1 или 2 заместителями, каждый из которых независимо представляет собой гидрокси,(C1-C6)алкил, галоген, (C1-C6)алкокси, NH2, CN или NO2, или один из R3 или R4 и R6 вместе образуют связь между атомами кольца, к которым они присоединены;R6 представляет собой Н или R6 образует связь между атомом кольца, к которому он присоединен, и атомом кольца, к которому присоединен R7;n равно 0 или 1,или его фармацевтически приемлемая соль или сложный эфир. 2. Соединение по п.1, где X представляет собой CR2R2'. 3. Соединение по п.1, где X представляет собой О. 4. Соединение по п.1, где X представляет собой S. 5. Соединение по любому из пп.1-4, где R3 представляет собой гидрокси, (C1-C6)алкил, гидрокси(C1- 26008537C6)алкил, (C1-C6)алкокси(C1-C6)алкил, (C1-C6)алкокси-СО- или (C1-C6)алкил-СО-О-(C1-C6)алкил и R4 представляет собой (C1-C6)алкил или гидрокси(C1-C6)алкил. 6. Соединение по любому из пп.1-5, где R3 представляет собой гидрокси, гидрокси(C1-C6)алкил или(C1-C6)алкокси(C1-C6)алкил и R4 представляет собой (C1-C6)алкил. 7. Соединение по п.1, где соединение представляет собой 1-метил-1,3,4,5,6,11b-гексагидро-2 Н-11-окса-4 а-азабензо[а]флуорен-1-ол,(1-метил-1,3,4,5,6,11b-гексагидро-2 Н-11-окса-4 а-азабензо[а]флуорен-1-ил)метанол,(-)-(1-метил-1,3,4,5,6,11b-гексагидро-2 Н-11-окса-4 а-азабензо[а]флуорен-1-ил)метанол,(+)-(1-метил-1,3,4,5,6,11b-гексагидро-2 Н-11-окса-4 а-азабензо[а]флуорен-1-ил)метанол,1-изопропил-1,3,4,5,6,11b-гексагидро-2 Н-11-окса-4 а-азабензо[а]флуорен-1-ол,1-этил-1,3,4,5,6,11b-гексагидро-2 Н-11-окса-4 а-азабензо[а]флуорен-1-ол,(1-этил-1,3,4,5,6,11b-гексагидро-2 Н-11-окса-4 а-азабензо[а]флуорен-1-ил)метанол,(1-гидроксиметил-1,3,4,5,6,11b-гексагидро-2 Н-11-окса-4 а-азабензо[а]флуорен-1-ил)метанол,1-метоксиметил-1-метил-1,3,4,5,6,11b-гексагидро-2 Н-11-окса-4 а-азабензо[а]флуорен,(-)-1-метоксиметил-1-метил-1,3,4,5,6,11b-гексагидро-2 Н-11-окса-4 а-азабензо[а]флуорен,(+)-1-метоксиметил-1-метил-1,3,4,5,6,11b-гексагидро-2 Н-11-окса-4 а-азабензо[а]флуорен,этиловый эфир 1-метил-1,3,4,5,6,11b-гексагидро-2 Н-11-окса-4 а-азабензо[а]флуорен-1 карбоновой кислоты,1-этоксиметил-1-метил-1,3,4,5,6,11b-гексагидро-2 Н-11-окса-4 а-азабензо[а]флуорен,(1-метил-1,3,4,5,6,11b-гексагидро-2 Н-11-окса-4 а-азабензо[а]флуорен-1-ил)метанол,(-)-(1-метил-1,3,4,5,6,11b-гексагидро-2 Н-11-окса-4 а-азабензо[а]флуорен-1-ил)метанол,(+)-(1-метил-1,3,4,5,6,11b-гексагидро-2 Н-11-окса-4 а-азабензо[а]флуорен-1-ил)метанол,метиловый эфир 1-этил-1,3,4,5,6,11b-гексагидро-2 Н-11-окса-4 а-азабензо[а]флуорен-1-карбоновой кислоты,1-метоксиметил-1-метил-1,3,4,5,6,11b-гексагидро-2 Н-11-окса-4 а-азабензо[а]флуорен,(-)-1-метоксиметил-1-метил-1,3,4,5,6,11b-гексагидро-2 Н-11-окса-4 а-азабензо[а]флуорен,(+)-1-метоксиметил-1-метил-1,3,4,5,6,11b-гексагидро-2 Н-11-окса-4 а-азабензо[а]флуорен,(1-этил-1,3,4,5,6,11b-гексагидро-2 Н-11-окса-4 а-азабензо[а]флуорен-1-ил)метанол,1-метил-1,3,4,5,6,11b-гексагидро-2 Н-11-окса-4 а-азабензо[а]флуорен-1-илметиловый эфир уксусной кислоты или(1-метил-1,2,3,4,6,7,12,12b-октагидроиндено[2,1-а]хинолизин-1-ил)метанол. 8. Соединение по любому из пп.1, 3, 5, 6 или 7, где соединение является [(-)-(1-метил 1,3,4,5,6,11b-гексагидро-2 Н-11-окса-4 а-азабензо[а]флуорен-1-ил)метанолом]. 9. Соединение по любому из пп.1, 3, 5, 6 или 8, где соединение представляет собой [(-)-1 метоксиметил-1-метил-1,3,4,5,6,11b-гексагидро-2 Н-11-окса-4 а-азабензо[а]флуорен]. 10. Фармацевтическая композиция, содержащая по меньшей мере одно соединение по любому из пп.1-9 и фармацевтически приемлемый разбавитель, носитель и/или эксципиент. 11 Применение соединения по любому из пп.1-9 для изготовления лекарственного средства, пригодного в качестве селективного антагониста альфа 2 С. 12. Применение по п.11, где лекарственное средство используется для лечения психического растройства, вызванного стрессом, болезни Паркинсона, дипрессии, шизофрении, синдрома гиперактивности с дифицитом внимания, посттравматического стресса, тревожного расстройства, синдрома Тауретта,блефароспазма и других фокальных нарушений тонуса, височной эпилепсии с психозом, психоза, обусловленного действием наркотиков, заболевания Хантингтона, заболеваний, вызванных колебаниями половых гормонов, или панического расстройства.
МПК / Метки
МПК: C07D 471/04, C07D 491/147, A61K 31/55, A61P 3/06, A61P 15/10, A61P 25/00, A61P 9/02, C07D 495/14, A61K 31/4375, C07D 455/00, A61P 3/10
Метки: антагонисты, альфа2-адренорецептора, соединения, эффективные, полициклические
Код ссылки
<a href="https://eas.patents.su/30-8537-policiklicheskie-soedineniya-kak-effektivnye-antagonisty-alfa2-adrenoreceptora.html" rel="bookmark" title="База патентов Евразийского Союза">Полициклические соединения как эффективные антагонисты альфа2-адренорецептора</a>
Соединения, эффективные в качестве агонистов &beta- 2 – адренорецептора и ингибиторов фосфодиэстеразы pde4

Номер патента: 5824
Опубликовано: 30.06.2005
Авторы: Стерк Герт, Ван-Дер-Лан Ивонне, Тиммерманн Хендрик, Эльтце Манфрид, Хатцельманн Армин, Кристианс Йоханнес, Брундель Паулус, Бундшу Даниела
МПК: C07D 237/14, A61P 11/06, A61K 31/50...
Метки: соединения, beta, фосфодиэстеразы, агонистов, адренорецептора, ингибиторов, эффективные, качестве
Формула / Реферат:
1. Соединения формулы I где Ar1означает фенильный радикал формулы (Ia), замещенный R1 и R2, или радикал дигидробензофуранил формулы (Ib), замещенный R3, R4 и R5 где R1 означает C1-C4алкокси или C1-C4алкокси, который полностью или преимущественно замещен фтором, R2 означает C1-C8алкокси или C1-C4алкокси, который полностью или преимущественно замещен фтором, R3 означает C1-C4алкокси, R4 и R5 вместе, включая два атома углерода, к которым...
Новые полициклические соединения и их применение

Номер патента: 7868
Опубликовано: 27.02.2007
Авторы: Чаттерджи Санкар, Биховски Рон, Данн Дерек, Атор Марк А., Хадкинс Роберт Л.
МПК: A61K 31/407, A61P 25/00, A61K 31/4035...
Метки: соединения, применение, полициклические, новые
Формула / Реферат:
1. Соединение формулы IIIa где каждый А и В обозначает, независимо, С(=O), СН(ОН) или CH2; Е и F вместе с атомами углерода, к которым они присоединены, образуют незамещенную С4-С7-циклоалкильную группу; R1 обозначает водород, низший алкил, низший алкил, имеющий по меньшей мере один заместитель J; формил; ацетил, низший алканоил, низший алканоил, имеющий по меньшей мере один заместитель J, низший алкилсульфонил, низший арилсульфонил, остаток...
Соединения четвертичного аммония как антагонисты тахикинина

Номер патента: 2424
Опубликовано: 25.04.2002
Авторы: Монаган Сандра Марина, Алкер Дэвид, Бернс Кристофер Джон
МПК: C07D 453/02, A61P 1/06, A61K 31/439...
Метки: аммония, соединения, антагонисты, тахикинина, четвертичного
Формула / Реферат:
1. Соединение формулы где R представляет собой фенил, С3-С7 циклоалкил или гетероарил, каждый из которых является необязательно сопряженным с бензольным циклом или с С3-С7 циклоалкилом и необязательно замещенным, включая фрагмент, сопряженный с бензольным циклом или с С3-С7 циклоалкилом, 1-3 заместителями, каждый из которых независимо выбран из C1-C4 алкила, фтор(C1-C4) алкила, C1-C4 алкоксигруппы, фтор (C1-C4)алкоксигруппы, феноксигруппы,...
Производные имидазола, обладающие аффиностью по отношению к альфа2 рецепторам

Номер патента: 8251
Опубликовано: 27.04.2007
Авторы: Ниеми Рику, Хуусконен Юхани, Ярвинен Томи
МПК: A61K 31/4164, A61K 31/415, A61P 3/06...
Метки: аффиностью, производные, имидазола, рецепторам, отношению, обладающие, альфа2
Формула / Реферат:
1. Соединение общей формулы I или его фармацевтически приемлемые соли или гидраты, где R представляет собой незамещенный или замещенный низший алкил, незамещенный или замещенный арил, незамещенный или замещенный циклоалкил, незамещенный или замещенный гетероарил, незамещенную или замещенную низшую алкиламиногруппу или насыщенную пяти- или шестичленную гетероциклическую группу, содержащую 1 или 2 атома азота. 2. Соединение по п.1, где R...
Эффективные противоопухолевые лекарственные средства

Номер патента: 5564
Опубликовано: 28.04.2005
Авторы: Такахаси Наото, Вейтмен Стив, Д'инкальчи Маурицио, Гешер Андреас, Джавацци Рафаэлла, Фэрклот Глинн Томас
МПК: A61P 35/00, A61K 45/06
Метки: противоопухолевые, лекарственные, средства, эффективные
Формула / Реферат:
1. Применение ET-743 для изготовления лекарственного препарата для эффективного лечения опухоли посредством комбинированной терапии, включающей применение ET-743 в синергической комбинации с другим противоопухолевым лекарственным средством. 2. Применение противоопухолевого лекарственного средства для изготовления лекарственного препарата для эффективного лечения опухоли посредством комбинированной терапии, включающей применение...