1h-индазол-3-карбоксамидные соединения в качестве ингибиторов гликогенсинтазы киназы 3-бета
Номер патента: 24939
Опубликовано: 30.11.2016
Авторы: Драгоне Патриция, Каццолла Никола, Манчини Франческа, Мауджери Катерина, Фурлотти Гвидо, Ализи Мария Алессандра, Омбрато Розелла












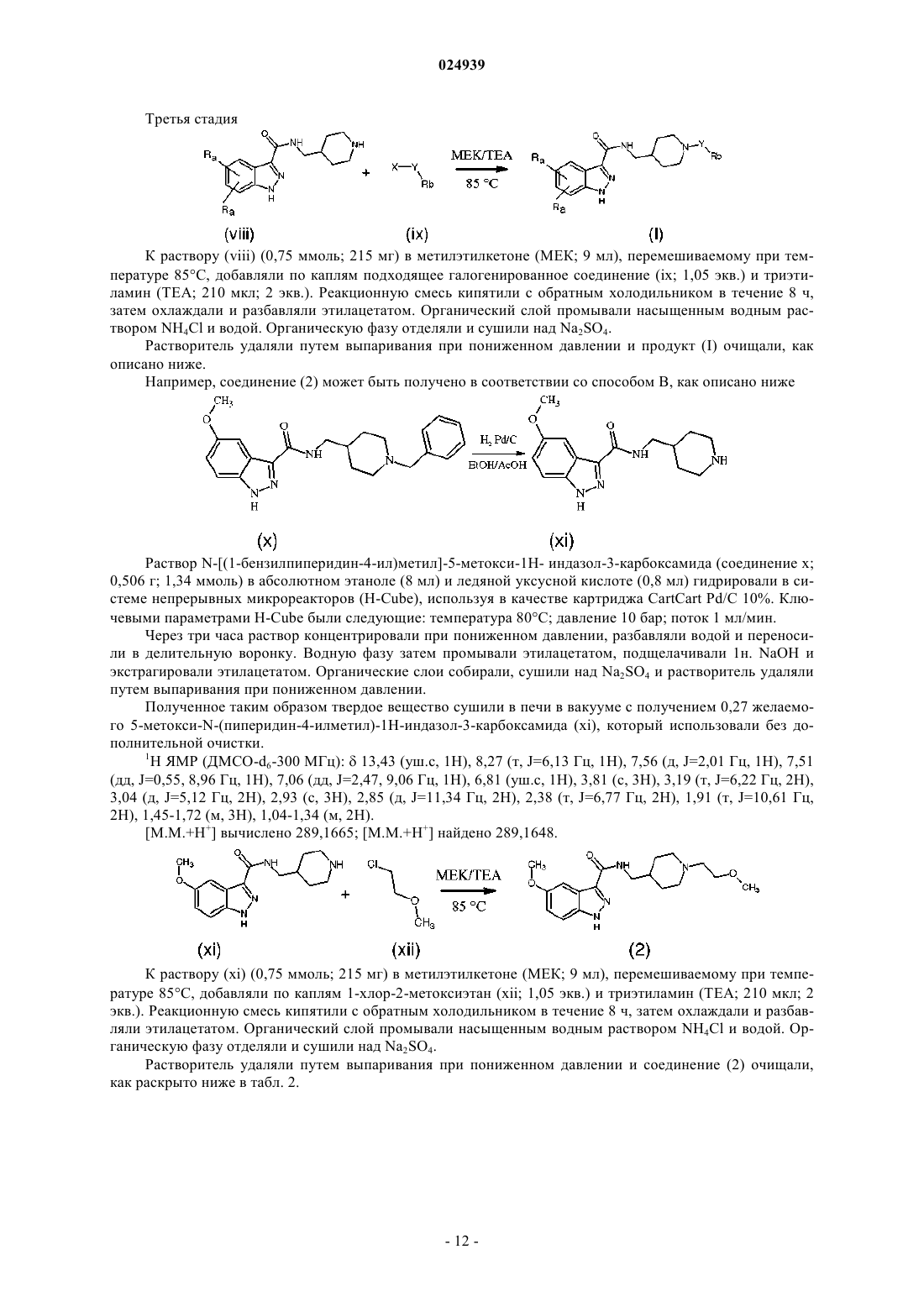

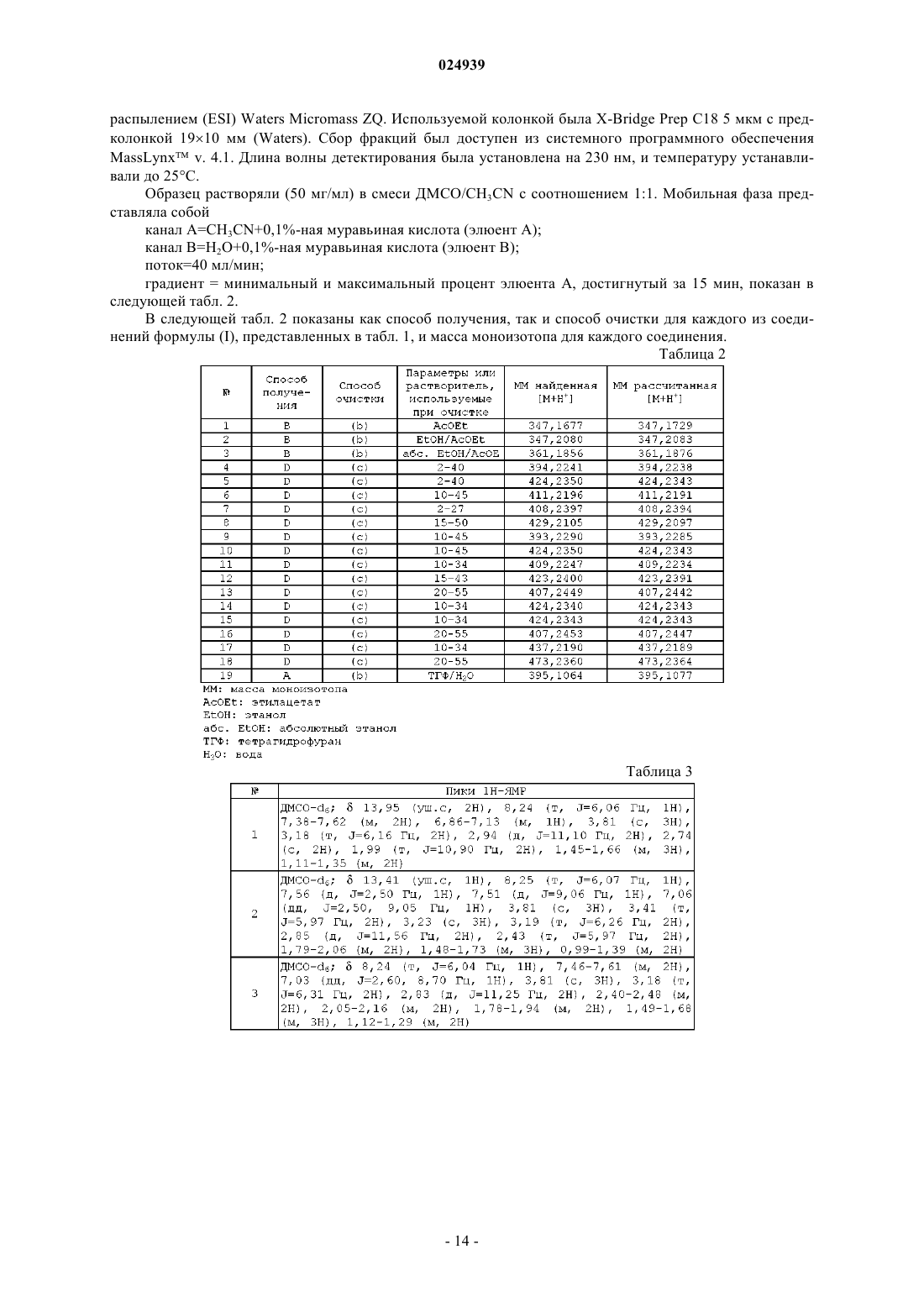
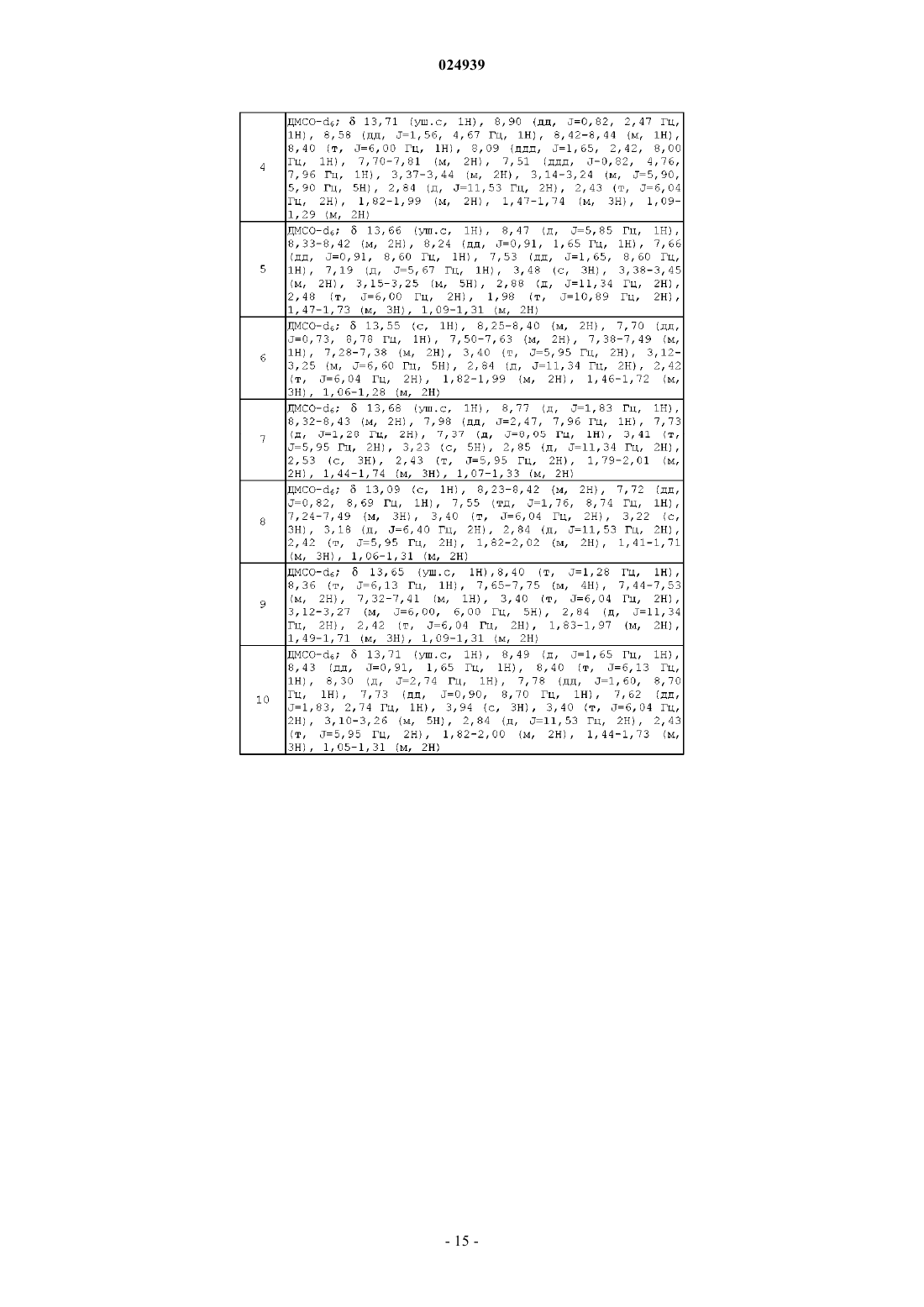
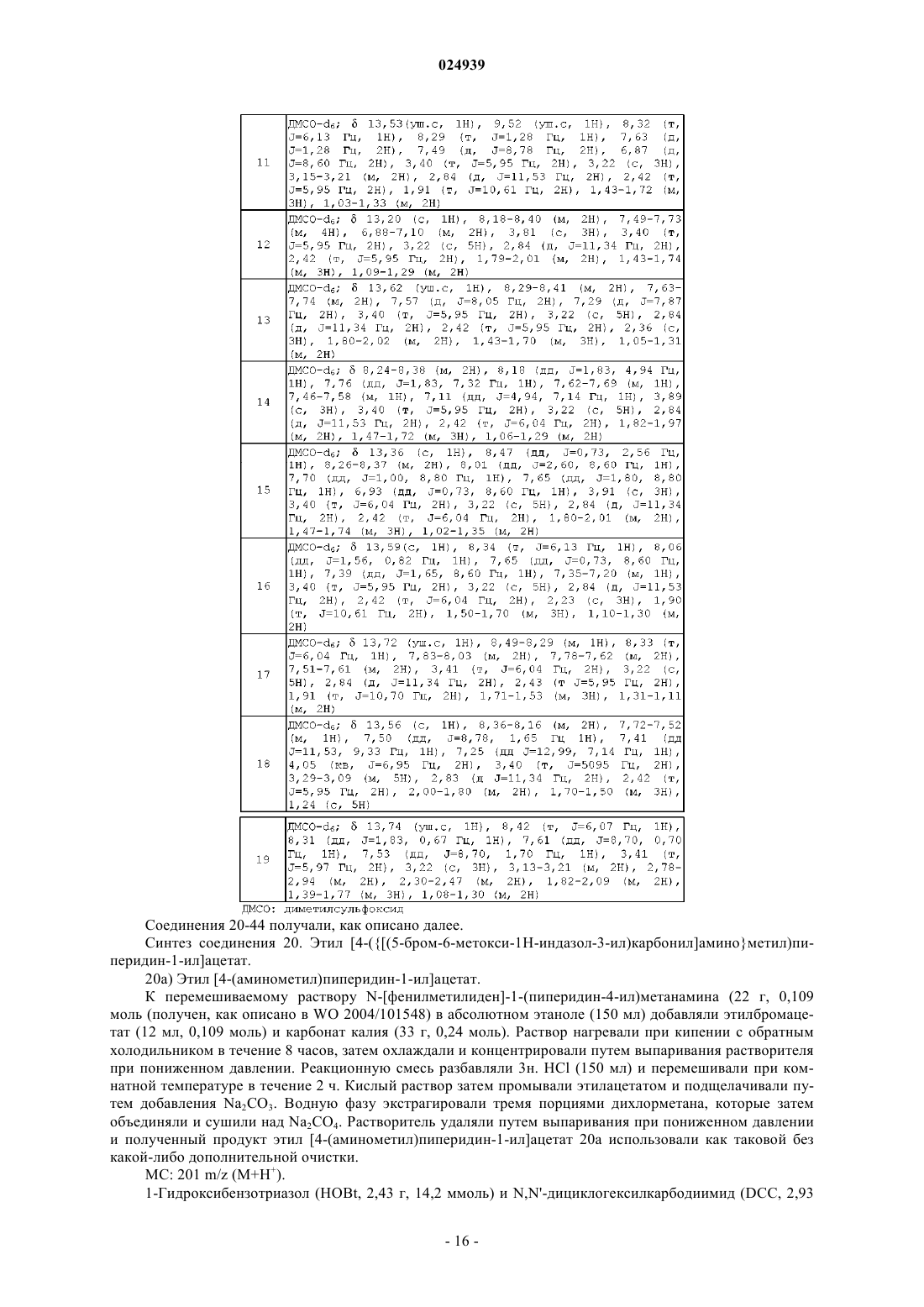







Формула / Реферат
1. 1Н-индазол-3-карбоксамидные соединения, имеющие следующую общую формулу (I):

где Ra и Ra', одинаковые или отличающиеся друг от друга, представляют собой атом водорода; атом галогена; гидроксигруппу; С1-С6 алкильную, С2-С6 алкенильную, С2-С6 алкинильную и С1-С6 алкоксигруппу, необязательно замещенную одним или несколькими заместителями, выбранными из группы, состоящей из галогена, гидрокси, -NH2 и C1-C3 алкокси; карбоциклическое или гетероциклическое кольцо, алифатическое или ароматическое, содержащее от 3 до 12 членов, необязательно замещенное одним или несколькими заместителями, выбранными из группы, состоящей из галогена, гидрокси, C1-C6 алкила, C1-C6 алкокси, -NR1R2, -С(О)ОН, -C(O)OR1 и -C(C)NR1R2;
Y представляет собой связь, С1-С6 алкильную, С2-С6 алкенильную или С2-С6 алкинильную группу, необязательно замещенную одним или несколькими заместителями, выбранными из группы, состоящей из галогена, гидрокси, -NH2 и С1-С3 алкокси;
Rb представляет собой C1-C6 алкоксигруппу; -С(О)ОН; -C(O)OR1; -NO2; -NHC(O)R1;
R1 и R2 представляют собой, независимо, атом водорода, C1-С4 алкильную группу, С2-С4 алкенильную группу, С2-С4 алкинильную группу и фенильную группу;
и их аддитивные соли с фармацевтически приемлемыми органическими и неорганическими кислотами и основаниями.
2. 1Н-индазол-3-карбоксамидные соединения по п.1, где Ra и Ra', одинаковые или отличающиеся друг от друга, представляют собой атом водорода; атом галогена, выбранный из хлора, брома и йода; гидроксигруппу; C1-C6 алкильную и C1-C6 алкоксигруппу, необязательно замещенную одним или несколькими заместителями, выбранными из группы, состоящей из галогена, гидрокси, -NH2 или С1-С3 алкокси; карбоциклическое или гетероциклическое кольцо, насыщенное или ненасыщенное, содержащее от 4 до 10 членов, необязательно замещенное одним или несколькими заместителями, выбранными из группы, состоящей из галогена, гидрокси, С1-С6 алкила, C1-C6 алкокси, -NR1R2, -С(О)ОН, -C(O)OR1 и -С(О)NR1R2; и R1 и R2 представляют собой, независимо, атом водорода, С1-С4 алкильную группу, С2-С4 алкенильную группу, С2-С4 алкинильную группу и фенильную группу.
3. 1Н-индазол-3-карбоксамидные соединения по п.2, где Ra и Ra', одинаковые или отличающиеся друг от друга, представляют собой атом галогена, выбранный из хлора или брома; гидроксигруппу; C1-C6 алкильную группу; C1-C6 алкоксигруппу; или карбоциклическое или гетероциклическое кольцо, насыщенное или ненасыщенное, содержащее от 5 до 6 членов, необязательно замещенное одним или несколькими заместителями, выбранными из группы, состоящей из галогена, гидрокси, C1-C6 алкила, C1-С6 алкокси, -NR1R2 и -СООН; и R1 и R2 представляют собой, независимо, атом водорода, C1-C4 алкильную группу, С2-С4 алкенильную группу, С2-С4 алкинильную группу и фенильную группу.
4. 1Н-индазол-3-карбоксамидные соединения по п.3, где Ra и Ra', одинаковые или отличающиеся друг от друга, представляют собой атом брома, гидроксигруппу; С1-С3 алкоксигруппу; или ненасыщенное карбоциклическое или гетероциклическое кольцо, содержащее 6 членов, необязательно замещенное одним или двумя заместителями, выбранными из группы, состоящей из галогена, гидрокси, C1-С3 алкила, C1-C3 алкокси, -NR1R2 и -СООН; и R1 и R2 представляют собой, независимо, атом водорода, C1-C4 алкильную группу, С2-С4 алкенильную группу, С2-С4 алкинильную группу и фенильную группу.
5. 1Н-индазол-3-карбоксамидные соединения по любому из предшествующих пунктов, где Y представляет собой связь, C1-C6 алкильную группу, необязательно замещенную одним или несколькими заместителями, выбранными из группы, состоящей из галогена, гидрокси, -NH2 и C1-C3 алкокси.
6. 1Н-индазол-3-карбоксамидные соединения по п.5, где Y представляет собой C1-C6 алкильную группу.
7. 1Н-индазол-3-карбоксамидные соединения по п.6, где Y представляет собой С1-С3 алкильную группу.
8. 1Н-индазол-3-карбоксамидные соединения по любому из предшествующих пунктов, где Rb представляет собой С1-С6 алкоксигруппу; -С(О)ОН; -C(O)OR1; -NHCOR1.
9. 1Н-Индазол-3-карбоксамидные соединения по п.8, где Rb представляет собой C1-C6 алкоксигруппу; -С(О)ОН.
10. 1Н-индазол-3-карбоксамидные соединения по п.9, где Rb представляет собой С1-С3 алкоксигруппу, -С(О)ОН.
11. 1Н-индазол-3-карбоксамидные соединения по любому из предшествующих пунктов, где R1 и R2 представляют собой, независимо, С1-С3 алкильную группу.
12. Применение 1Н-индазол-3-карбоксамидных соединений, имеющих следующую общую формулу (I):

где Ra и Ra', одинаковые или отличающиеся друг от друга, представляют собой атом водорода; атом галогена; гидроксигруппу; С1-С6 алкильную, С2-С6 алкенильную, С2-С6 алкинильную и C1-С6 алкоксигруппу, необязательно замещенную одним или несколькими заместителями, выбранными из группы, состоящей из галогена, гидрокси, -NH2 и С1-С3 алкокси; карбоциклическое или гетероциклическое кольцо, алифатическое или ароматическое, содержащее от 3 до 12 членов, необязательно замещенное одним или несколькими заместителями, выбранными из группы, состоящей из галогена, гидрокси, C1-C6 алкила, C1-C6 алкокси, -NR1R2, -С(О)ОН, -C(O)OR1 и -C(O)NR1R2;
Y представляет собой связь, С1-С6 алкильную, С2-С6 алкенильную или С2-С6 алкинильную группу, необязательно замещенную одним или несколькими заместителями, выбранными из группы, состоящей из галогена, гидрокси, -NH2 и С1-С3 алкокси;
Rb представляет собой С1-С6 алкоксигруппу; -С(О)ОН; -C(O)OR1; -NO2; -NHC(O)R1;
R1 и R2 представляют собой, независимо, атом водорода, C1-С4 алкильную группу, С2-С4 алкенильную группу, С2-С4 алкинильную группу и фенильную группу;
и их аддитивных солей с фармацевтически приемлемыми органическими и неорганическими кислотами и основаниями;
при лечении заболевания, вызванного неконтролируемой активацией и/или избыточной экспрессией GSK-3β, выбранного из группы, включающей (i) инсулинрезистентные нарушения; (ii) нейродегенеративные заболевания; (iii) расстройства настроения; (iv) шизофренические расстройства; (v) злокачественные расстройства; (vi) воспаление, (vii) расстройства вследствие токсикомании; (viii) эпилепсии; (ix) невропатическую боль.
13. Применение 1Н-индазол-3-карбоксамида по п.12, где указанные инсулинрезистентные нарушения выбраны из группы, включающей сахарный диабет 2 типа, синдром X, ожирение и синдром поликистозных яичников.
14. Применение 1Н-индазол-3-карбоксамида по п.12, где указанные нейродегенеративные заболевания выбраны из группы, включающей болезнь Паркинсона, болезнь Альцгеймера, болезнь Хантингтона и спинальные нейродегенеративные расстройства.
15. Применение 1Н-индазол-3-карбоксамида по п.14, где указанные спинальные нейродегенеративные расстройства выбраны из группы, включающей амиотрофический латеральный склероз, рассеянный склероз, спинальную мышечную атрофию и нейродегенерацию в связи с повреждением спинного мозга.
16. Применение 1Н-индазол-3-карбоксамида по п.12, где указанные расстройства настроения выбраны из группы, включающей биполярные расстройства и депрессивные расстройства.
17. Применение 1Н-индазол-3-карбоксамида по п.16, где указанные биполярные расстройства выбраны из группы, включающей биполярное расстройство I типа, биполярное расстройство II типа, циклотимию и биполярное расстройство, иным образом не обозначенное (BD-NOS).
18. Применение 1Н-индазол-3-карбоксамида по п.16, где указанные депрессивные расстройства выбраны из группы, включающей большое депрессивное расстройство (MDD), атипичную депрессию (AD), меланхолическую депрессию, психотическую большую депрессию (PMD), кататоническую депрессию, постродовую депрессию (PPD), сезонное аффективное расстройство (SAD), дистимию и депрессивное расстройство, иным образом не обозначенное (DD-NOS).
19. Применение 1Н-индазол-3-карбоксамида по п.12, где указанные расстройства, вызванные токсикоманией, выбраны из группы агрессивных нарушений, вызванных психостимуляторами.
20. Применение 1Н-индазол-3-карбоксамида по п.12, где указанные шизофренические расстройства выбраны из группы, включающей параноидальную шизофрению, шизофрению дезорганизованного типа, кататоническую шизофрению, простую шизофрению, остаточную шизофрению и недифференцированную шизофрению.
21. Применение 1Н-индазол-3-карбоксамида по п.12, где указанные злокачественные расстройства выбраны из группы, включающей рак простаты, поджелудочной железы, яичников и колоректальный рак, а также MLL-связанную лейкемию.
22. Способ лечения патологического состояния, вызванного неконтролируемой активацией и/или избыточной экспрессией GSK-3β, выбранного из группы, включающей (i) инсулинрезистентные нарушения; (ii) нейродегенеративные заболевания; (iii) расстройства настроения; (iv) шизофренические расстройства; (v) злокачественные расстройства; (vi) воспаление, (vii) расстройства вследствие токсикомании; (viii) эпилепсии; (ix) невропатическую боль, путем введения нуждающемуся в этом человеку эффективного количества 1Н-индазол-3-карбоксамида, имеющего следующую общую формулу (I):

где Ra и Ra', одинаковые или отличающиеся друг от друга, представляют собой атом водорода; атом галогена; гидроксигруппу; C1-C6 алкильную, С2-С6 алкенильную, С2-С6 алкинильную и C1-C6 алкоксигруппу, необязательно замещенную одним или несколькими заместителями, выбранными из группы, состоящей из галогена, гидрокси, -NH2 и C1-C3 алкокси; карбоциклическое или гетероциклическое кольцо, алифатическое или ароматическое, содержащее от 3 до 12 членов, необязательно замещенное одним или несколькими заместителями, выбранными из группы, состоящей из галогена, гидрокси, С1-С6 алкила, С1-С6 алкокси, -NR1R2, -C(O)OH, -C(O)OR1 и -C(O)NR1R2;
Y представляет собой связь, C1-C6 алкильную, C2-C6 алкенильную или С2-С5 алкинильную группу, необязательно замещенную одним или несколькими заместителями, выбранными из группы, состоящей из галогена, гидрокси, -NH2 и C1-C3 алкокси;
Rb представляет собой С1-С6 алкоксигруппу; -С(О)ОН; -C(O)OR1; -NO2; -NHC(O)R1;
R1 и R2 представляют собой, независимо, атом водорода, C1-С4 алкильную группу, С2-С4 алкенильную группу, С2-С4 алкинильную группу и фенильную группу; и его аддитивных солей с фармацевтически приемлемыми органическими и неорганическими кислотами и основаниями.
23. Фармацевтическая композиция, содержащая эффективное количество по меньшей мере одного соединения формулы (I), определенного в любом из пп.1-11, выше, его соли с фармацевтически приемлемыми органическими или неорганическими кислотой или основанием и по меньшей мере один инертный фармацевтически приемлемый эксципиент.
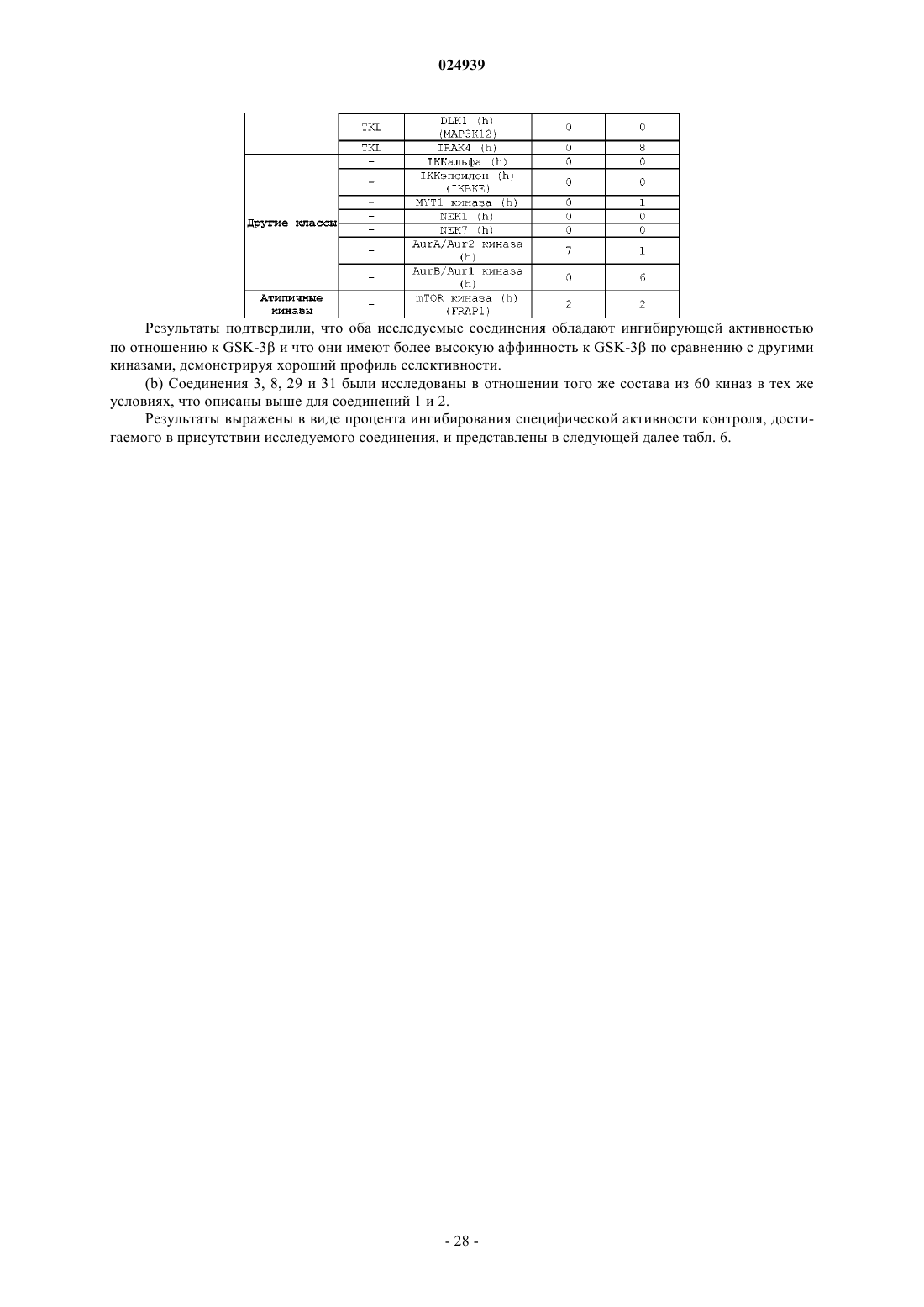
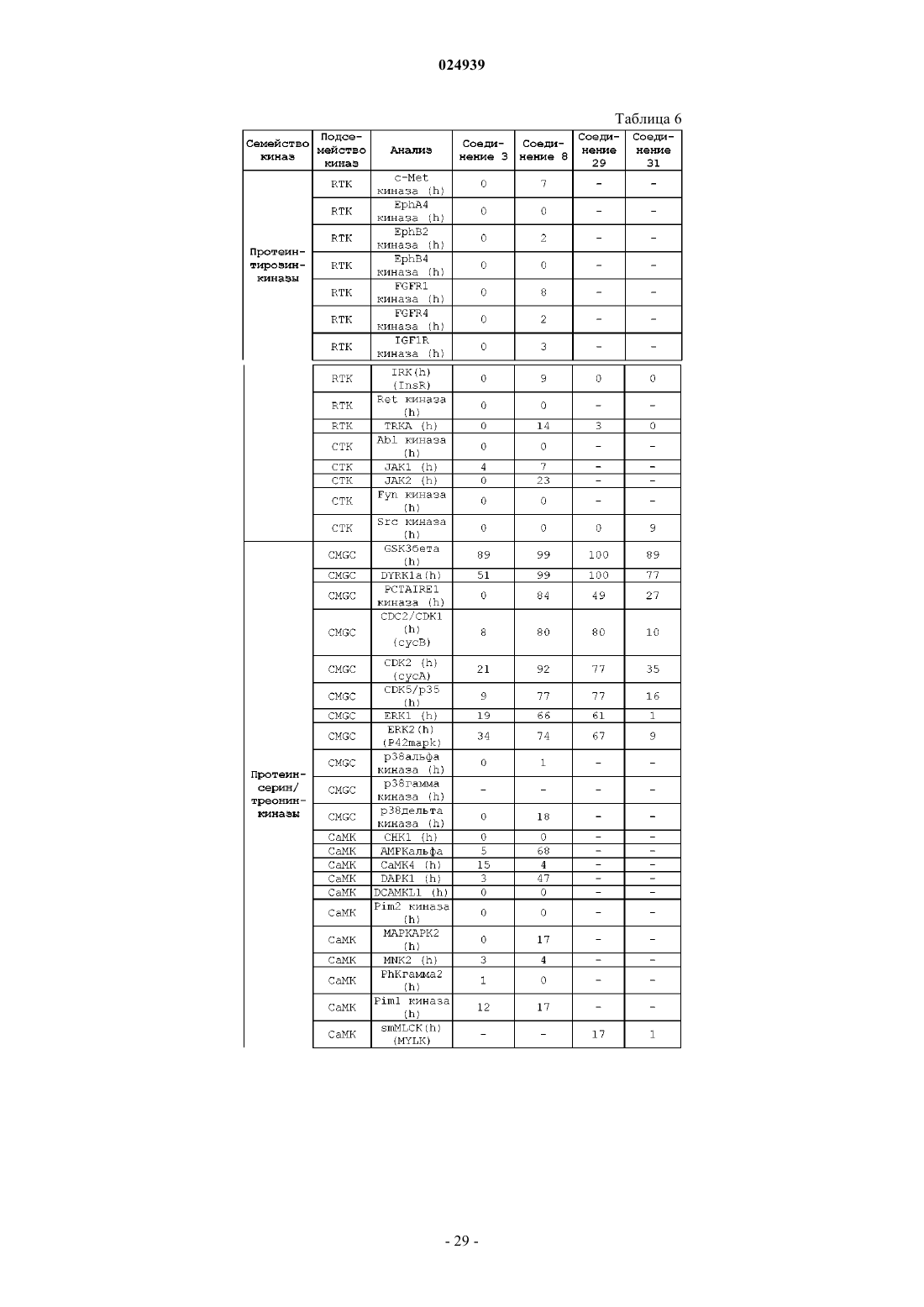
Текст
1H-ИНДАЗОЛ-3-КАРБОКСАМИДНЫЕ СОЕДИНЕНИЯ В КАЧЕСТВЕ ИНГИБИТОРОВ ГЛИКОГЕНСИНТАЗЫ КИНАЗЫ 3-БЕТА Изобретение относится к 1 Н-индазол-3-карбоксамидным соединениям, имеющим следующую общую формулу (I), в качестве ингибиторов гликогенсинтазы киназы 3-бета (GSK-3) и к их применению при лечении связанных с GSK-3 заболеваний, таких как,например, (i) инсулинрезистентные нарушения; (ii) нейродегенеративные заболевания;(71)(73) Заявитель и патентовладелец: АЦЬЕНДЕ КИМИКЕ РЬЮНИТЕ АНДЖЕЛИНИ ФРАНЧЕСКО А.К.Р.А.Ф. С.П.А. (IT) Область изобретения Изобретение относится к 1 Н-индазол-3-карбоксамидным соединениям, действующим в качестве ингибиторов гликогенсинтазы киназы 3-бета (GSK-3), и к их применению при лечении расстройств,связанных с GSK-3, таких как (i) инсулинрезистентные нарушения; (ii) нейродегенеративные заболевания; (iii) расстройства настроения; (iv) шизофренические расстройства; (v) злокачественные расстройства; (vi) воспаление, (vii) расстройства вследствие токсикомании; (viii) эпилепсии; и (ix) невропатическая боль. Уровень техники Протеинкиназы составляют большое семейство структурно родственных ферментов, которые передают фосфатные группы от донорных молекул с высокой энергией (таких как аденозинтрифосфат, АТФ) специфическим субстратам, обычно белкам. После фосфорилирования субстрат подвергается функциональному изменению, с помощью которого киназы могут модулировать различные биологические функции. Обычно протеинкиназы могут быть разделены на несколько групп в соответствии с субстратом, который фосфорилирован. Например, серин/треонин киназы фосфорилируют гидроксильную группу на боковой цепи аминокислот серина или треонина. Гликогенсинтазы киназы 3 (GSK-3) являются постоянно активными многофункциональными ферментами, обнаруженными совсем недавно, принадлежащими к группе серии/треонин киназ. Человеческие GSK-3 кодируются двумя различными и независимыми генами, что приводит к белкам GSK-3- и GSK-3 с молекулярной массой около 51 и 47 кДа соответственно. Две изоформы разделяют почти идентичные последовательности в их киназных доменах, в то время как за пределами киназного домена их последовательности существенно отличаются (Benedetti et al., Neuroscience Letters, 2004,368, 123-126). GSK-3 представляет собой многофункциональный белок серинкиназу, и GSK-3 представляет собой серин-треонин киназу. Было установлено, что GSK-33 широко экспрессируется во всех тканях, с широким распространением экспрессии во взрослом мозге, предполагая фундаментальную роль в нервных сигнальных путях(Grimes and Jope, Progress in Neurobiology, 2001, 65, 391-426). Интерес к киназам 3 гликогенсинтазы возник в связи с их ролью в различных физиологических путях, таких как, например, обмен веществ, клеточный цикл, экспрессия генов, онкогенез стадии эмбрионального развития и нейропротекция (Geetha etal., British Journal Pharmacology, 2009, 156, 885-898). Сначала GSK-3 был идентифицирован из-за его роли в регуляции гликогенсинтазы для превращения глюкозы в гликоген (Embi et al., Eur J. Biochem, 1980, 107, 519-527). GSK-3 показал высокую степень специфичности для гликогенсинтазы. Диабет 2 типа был первым болезненным состоянием, в которое вовлечены GSK-3 ввиду их негативной регуляции некоторых аспектов сигнального пути инсулина. В этом пути 3-фосфоинозитидзависимая протеинкиназа 1 (PDK-1) активирует РКВ, которая, в свою очередь, инактивирует GSK-3. Эта инактивация GSK-3 приводит к дефосфорилированию и активации гликогенсинтазы, которая помогает синтезу гликогена (Cohen et al., FEBS Lett, 1997, 410, 3-10). Более того, селективные ингибиторыGSK-3, как ожидается, усиливают передачу сигнала инсулина в преддиабетических инсулинрезистентных скелетных мышцах крыс, таким образом делая GSK-3 привлекательной мишенью при лечении инсулиновой резистентности скелетной мышцы в преддиабетическом состоянии (Dokken et al., Am J. Physiol. Endocrinol. Metab., 2005, 288, E1188-E1194). Также было установлено, что GSK-3 является возможной лекарственной мишенью при других патологических состояниях, вызванных инсулинрезистентными нарушениями, такими как синдром X,ожирение и синдром поликистозных яичников (Ring D.B. et al., Diabetes, 2003, 52: 588-595). Было обнаружено, что GSK-3 участвует в аномальном фосфорилировании патологического тау при болезни Альцгеймера (Hanger et al., Neurosci. Lett, 1992, 147, 58-62; Mazanetz and Fischer, Nat RevDrug Discov., 2007, 6, 464-479; Hong and Lee, J. Biol. Chem., 1997, 272, 19547-19553). Кроме того, было доказано, что ранняя активация GSK-3, индуцированная аполипопротеином АроЕ 4 и -амилоидом, может привести к апоптозу и гиперфосфорилированию тау (Cedazo-Minguez et al., Journal of Neurochemistry,2003, 87, 1152-1164). Среди других аспектов болезни Альцгеймера сообщалось также об актуальности активации GSK-3 на молекулярном уровне (Hernandez and Avila, FEBS Letters, 2008, 582, 3848-3854). Кроме того, было показано, что GSK-3 участвует в генезисе и поддержании нейродегенеративных изменений, связанных с болезнью Паркинсона (Duka Т. et al., The FASEB Journal, 2009; 23, 2820-2830). В соответствии с этими экспериментальными наблюдениями ингибиторы GSK-3 могут найти применение при лечении нейропатологических последствий и когнитивного дефицита и дефицита внимания, связанных с таутопатией; болезни Альцгеймера; болезни Паркинсона; болезни Хантингтона (участие GSK-3 в таких дефицитах и заболеваниях описано в работе Meijer L. et al., TRENDS Pharm Sci,2004; 25, 471-480); слабоумия, такого как, но не ограничиваясь этим, сосудистая деменция, посттравматическое слабоумие, слабоумие, вызванное менингитом и т.п.; острого инсульта; травматических повре-1 024939 ждений; нарушений мозгового кровообращения; травмы мозга и спинного мозга; периферической нервной системы; ретинопатии и глаукомы (участие GSK-3 в таких состояниях описано в WO 2010/109005). Лечение спинальных нейродегенеративных расстройств, подобных амиотрофическому латеральному склерозу, рассеянному склерозу, спинальной мышечной атрофии и нейродегенерации в связи с повреждением спинного мозга, также было предложено в нескольких исследованиях, связанных с ингибированием GSK-3, как, например, в работах Caldero J. et al., "Lithium prevents excitotoxic cell death of motoneurons in organotypic slice cultures of spinal cord", Neuroscience. 2010 Feb 17;165 (4) :1353-69, Leger B. etmuscle of chronic spinal cord-injured patients", Muscle Nerve, 2009 Jul; 40(1):69-78, и Galimberti D. et al.,"GSK3 genetic variability in patients with Multiple Sclerosis", Neurosci Lett. 2011 Jun 15;497(1):46-8. Кроме того, GSK-3 был связан с расстройствами настроения, такими как биполярные расстройства, депрессия и шизофрения. Ингибирование GSK-3 может быть важной терапевтической мишенью стабилизаторов настроения,и регулирование GSK-3 может быть вовлечено в терапевтические эффекты других препаратов, используемых в психиатрии. Дисрегуляция GSK-3 в расстройствах настроения, биполярное расстройство, депрессия и шизофрения могут иметь несколько эффектов, которые могут ухудшить нейронную пластичность, например модуляцию нейронной архитектуры, нейрогенез, экспрессию генов и способность нейронов реагировать на стрессовые, потенциально летальные состояния (Jope and Roh, Curr. Drug Targets,2006, 7, 1421-1434). Была подчеркнута роль GSK-3 в расстройствах настроения путем изучения лития и вальпроата(Chen et al., J. Neurochem., 1999, 72, 1327-1330; Klein and Melton, Proc. Natl. Acad. Sci. USA, 1996, 93,8455-8459), которые оба являются ингибиторами GSK-3 и используются для лечения расстройства настроения. Имеются также отчеты с генетической точки зрения, подтверждающие роль GSK-3 в физиологии заболеваний биполярного расстройства (Gould, Expert. Opin. Ther. Targets, 2006, 10, 377-392). Сообщалось о снижении уровней белка АКТ 1 и его фосфорилирования GSK-3 в Серин-9 в периферических лимфоцитах и в мозге у людей с шизофренией. Соответственно, этот вывод подтверждает предположение о том, что изменения в сигнальном пути AKT1-GSK-3 способствуют патогенезу шизофрении (Emamian et al., Nat Genet, 2004, 36, 131-137). Кроме того, роль GSK-3 в злокачественном новообразовании является широко признанным явлением. Эффективность малых молекул, которые ингибируют GSK-3, была подтверждена для некоторых специфических методов лечения рака (Jia Luo, Cancer Letters, 2009, 273, 194-200). Экспрессия и активация GSK-3 связаны с прогрессированием рака простаты (Rinnab et al., Neoplasia, 2008, 10, 624-633), и ингибирование GSK3b также было предложено в качестве специфической мишени при раке поджелудочной железы (Garcea et al., Current Cancer Drug Targets, 2007, 7, 209-215) и раке яичников (Qi Cao et al.,Cell Research, 2006, 16 671-677). Раннее ингибирование GSK-3 в раковых клетках толстой кишки активирует р 53-зависимый апоптоз и противодействует росту опухоли (Ghosh et al., Clin Cancer Res 2005, 11,4580-4588). Установление функционального значения GSK-3 в MLL-связанной лейкемии предполагает, что ингибирование GSK-3 может быть перспективной терапией, которая является селективной для трансформированных клеток, которые зависят от избыточной экспрессии НОХ (Birch et al., Cancer Cell, 2010,17, 529-531).GSK-3 участвует в многочисленных воспалительных сигнальных путях, например, среди прочих,ингибирование GSK-3, как было показано, вызывает секрецию противовоспалительного цитокина IL10. В соответствии с этим выводом ингибиторы GSK-3 могут быть полезными для регулирования подавления воспаления (G. Klamer et al., Current Medicinal Chemistry, 2010, 17(26), 2873-2281, Wang et al.,Cytokine, 2010, 53, 130-140). Ингибирование GSK-3 было также показано для ослабления вызванного кокаином поведения у мышей. Введение кокаина мышам, предварительно обработанным ингибитором GSK-3, показало, что фармакологическое ингибирование GSK3 снижает как острые поведенческие реакции на кокаин, так и долгосрочные нейроадаптации к многократному воздействию кокаина (Cocaine-induced hyperactivity andsensitization are dependent on GSK3, Miller J.S. et al. Neuropharmacology. 2009 Jun; 56 (8):1116-23, Epub 2 009 Mar 27). Роль GSK-3 в развитии некоторых форм эпилепсии была продемонстрирована в нескольких исследованиях, в которых предполагалось, что ингибирование GSK-3 может быть путем лечения эпилепсии(Novel glycogen synthase kinase 3 and ubiquitination pathways in progressive myoclonus epilepsy, Lohi H. et al.,Hum Mol. Genet. 2005 Sep 15;14(18):2727-36 and Hyperphosphorylation and aggregation of Tau in laforindeficient mice, an animal model for Lafora disease, Puri R. et al., J. Biol. Chem. 2009 Aug 21;284(34) 22657-63). Связь между ингибированием GSK-3 и лечением невропатической боли была продемонстрирована в работах Mazzardo-Martins L. et al., "Glycogen synthase kinase 3-specific inhibitor AR-A014418 decreasesGu et al., "The Role of Akt/GSK3P Signaling Pathway in Neuropathic Pain in Mice", Poster A525, Anesthesiology 2012 October 13-17, 2012 Washington. Анализ GSK-3, его функции, его терапевтического потенциала и его возможных ингибиторов представлен в обзоре "GSK-3: role in therapeutic landscape and development of modulators" (S. Phukan etal., British Journal of Pharmacology (2010), 160, 1-19). В WO 2004/014864 описаны 1 Н-индазол-3-карбоксамидные соединения в качестве селективных ингибиторов циклинзависимых киназ (CDK). Такие соединения могут быть использованы при лечении злокачественного новообразования с помощью механизма, опосредованного CDK2, и нейродегенеративных заболеваний, в частности болезни Альцгеймера, посредством механизма, опосредованного CDK5, и в качестве противовирусного и противогрибкового средства посредством механизма, опосредованногоCDK7, CDK8 и CDK9. Циклинзависимые киназы (CDK) представляют собой серин/треонин киназы, впервые обнаруженные из-за их роли в регуляции клеточного цикла. CDK также участвуют в регуляции транскрипции, процессинге мРНК и дифференциации нервных клеток. Такие киназы активируют только после их взаимодействия и связывания с регуляторными субъединицами, а именно циклинами. Более того, 1 Н-индазол-3-карбоксамидные соединения были также описаны в качестве анальгетиков при лечении хронической и невропатической боли (см., например, WO 2004/074275 и WO 2004/101548) и в качестве антагонистов рецептора 5-НТ 4, которые могут быть использованы при лечении желудочно-кишечных расстройств, расстройств центральной нервной системы и сердечно-сосудистых заболеваний (см., например, WO 1994/10174). Сущность изобретения Поскольку только недавно GSK-3 был обнаружен в качестве фармакологической мишени, существует большая потребность в поиске соединений, которые селективно ингибируют GSK-3. Заявитель неожиданно обнаружил новые 1 Н-индазол-3-карбоксамидные соединения в соответствии с приведенной ниже формулой (I). Заявитель также неожиданно обнаружил, что указанные новые соединения способны ингибироватьGSK-3 и имеют очень высокое сродство к GSK-3 по сравнению с другими киназами. Таким образом,указанные соединения способны селективно ингибировать GSK-3. Соответственно, соединения в соответствии с настоящим изобретением могут быть использованы при лечении патологических состояний, вызванных неконтролируемой активацией и/или избыточной экспрессией GSK-3, выбранных из группы, включающей (I) инсулинрезистентные нарушения, такие диабет как 2-го типа, синдром X, ожирение и синдром поликистозных яичников; (II) нейродегенеративные заболевания, такие как болезнь Паркинсона, болезнь Альцгеймера, болезнь Хантингтона и спинальные нейродегенеративные расстройства; (III) расстройства настроения, такие как биполярные расстройства и депрессивные расстройства; (IV) шизофренические расстройства; (V) злокачественные расстройства, такие как рак простаты, поджелудочной железы, яичников и колоректальный рак, а также MLLсвязанный лейкоз; (VI) воспаление; (VII) расстройства вследствие токсикомании; (VIII) эпилепсии; и (IX) невропатическую боль. Итак, в первом аспекте, настоящее изобретение относится к 1 Н-индазол-3-карбоксамидным соединениям, имеющим следующую общую формулу (I): где Ra и Ra', одинаковые или отличающиеся друг от друга, представляют собой атом водорода; атом галогена; гидроксигруппу; C1-С 6 алкильную, С 2-С 6 алкенильную, С 2-С 6 алкинильную и C1-С 6 алкоксигруппу,необязательно замещенную одним или несколькими заместителями, выбранными из группы, состоящей из галогена, гидрокси, -NH2 и C1-С 3 алкокси; карбоциклическое или гетероциклическое кольцо, алифатическое или ароматическое, содержащее от 3 до 12 членов, необязательно замещенное одним или несколькими заместителями, выбранными из группы, состоящей из галогена, гидрокси, C1-C6 алкила, C1-C6 алкокси, -NR1R2, -С(О)ОН, -C(O)OR1 и -C(O)NR1R2;Y представляет собой связь, C1-C6 алкильную, С 2-С 6 алкенильную или С 2-С 6 алкинильную группу,необязательно замещенную одним или несколькими заместителями, выбранными из группы, состоящей из галогена, гидрокси, -NH2 и C1-C3 алкокси; ную группу, С 2-С 4 алкинильную группу и фенильную группу; и их аддитивным солям с фармацевтически приемлемыми органическими и неорганическими кислотами и основаниями. Во втором аспекте настоящее изобретение относится к применению 1 Н-индазол-3-карбоксамидных соединений, имеющих следующую общую формулу (I): где Ra и Ra', одинаковые или отличающиеся друг от друга, представляют собой атом водорода; атом галогена; гидроксигруппу; C1-С 6 алкильную, С 2-С 6 алкенильную, С 2-С 6 алкинильную и C1-С 6 алкоксигруппу, необязательно замещенную одним или несколькими заместителями, выбранными из группы,состоящей из галогена, гидрокси, -NH2 и C1-С 3 алкокси; карбоциклическое или гетероциклическое кольцо, алифатическое или ароматическое, содержащее от 3 до 12 членов, необязательно замещенное одним или несколькими заместителями, выбранными из группы, состоящей из галогена, гидрокси, C1-С 6 алкила,C1-С 6 алкокси, -NR1R2, -С(О)ОН, -C(O)OR1 и -C(O)NR1R2;Y представляет собой связь, C1-С 6 алкильную, С 2-С 6 алкенильную или С 2-С 6 алкинильную группу,необязательно замещенную одним или несколькими заместителями, выбранными из группы, состоящей из галогена, гидрокси, -NH2 и C1-С 3 алкокси;R1 и R2 представляют собой, независимо, атом водорода, C1-С 4 алкильную группу, С 2-С 4 алкенильную группу, С 2-С 4 алкинильную группу и фенильную группу; и их аддитивных солей с фармацевтически приемлемыми органическими и неорганическими кислотами и основаниями; при лечении заболевания, вызванного неконтролируемой активацией и/или избыточной экспрессией GSK-3, выбранного из группы, включающей (i) инсулинрезистентные нарушения, такие как сахарный диабет 2 типа, синдром X, ожирение и синдром поликистозных яичников; (ii) нейродегенеративные заболевания, такие как болезнь Паркинсона, болезнь Альцгеймера, болезнь Хантингтона и спинальные нейродегенеративные расстройства;(iii) расстройства настроения, такие как биполярные расстройства и депрессивные расстройства; (iv) шизофренические расстройства; (v) злокачественные расстройства, такие как рак простаты, поджелудочной железы, яичников и колоректальный рак, а также MLL-связанная лейкемия; (vi) воспаление; (vii) расстройства вследствие токсикомании; (viii) эпилепсии; и (ix) невропатическую боль. В другом аспекте настоящее изобретение относится к способу лечения патологического состояния,вызванного неконтролируемой активацией и/или избыточной экспрессией GSK-3, выбранного из группы,включающей (i) инсулинрезистентные нарушения, такие как сахарный диабет 2 типа, синдром X, ожирение и синдром поликистозных яичников; (ii) нейродегенеративные заболевания, такие как болезнь Паркинсона,болезнь Альцгеймера, болезнь Хантингтона и спинальные нейродегенеративные расстройства;(iii) расстройства настроения, такие как биполярные расстройства и депрессивные расстройства; (iv) шизофренические расстройства; (v) злокачественные расстройства, такие как рак простаты, поджелудочной железы, яичников и колоректальный рак, а также MLL-связанная лейкемия; (vi) воспаление; (vii) расстройства вследствие токсикомании; (viii) эпилепсии; и (ix) невропатическую боль, путем введения нуждающемуся в этом человеку эффективного количества 1 Н-индазол-3-карбоксамида, имеющего следующую общую формулу (I): где Ra и Ra', одинаковые или отличающиеся друг от друга, представляют собой атом водорода; атом галогена; гидроксигруппу; C1-C6 алкильную, С 2-С 6 алкенильную, С 2-С 6 алкинильную и C1-С 6 алкоксигруппу, необязательно замещенную одним или несколькими заместителями, выбранными из группы,состоящей из галогена, гидрокси, -NH2 и C1-С 3 алкокси; карбоциклическое или гетероциклическое кольцо, алифатическое или ароматическое, содержащее от 3 до 12 членов, необязательно замещенное одним или несколькими заместителями, выбранными из группы, состоящей из галогена, гидрокси, C1-С 6 алкила,C1-С 6 алкокси, -NR1R2, -С(О)ОН, -C(O)OR1 и -C(O)NR1R2;Y представляет собой связь, C1-С 6 алкильную, С 2-С 6 алкенильную или С 2-С 6 алкинильную группу,необязательно замещенную одним или несколькими заместителями, выбранными из группы, состоящей из галогена, гидрокси, -NH2 и C1-С 3 алкокси;R1 и R2 представляют собой, независимо, атом водорода, C1-С 4 алкильную группу, С 2-С 4 алкенильную группу, С 2-С 4 алкинильную группу и фенильную группу; и его аддитивных солей с фармацевтически приемлемыми органическими и неорганическими кислотами и основаниями. Настоящее изобретение включает также пролекарства, стереоизомеры и энантиомеры соединений формулы (I), описанной выше. Подробное описание изобретения В настоящем описании и в последующей формуле изобретения "C1-6 алкил" предназначен для обозначения линейных или разветвленных алкильных групп, содержащих от 1 до 6 атомов углерода, таких как метил, этил, пропил, изопропил, н-бутил, изобутил, втор-бутил, трет-бутил, н-пентил, втор-пентил, 3 пентил, изо-пентил, нео-пентил, н-гексил, втор-гексил и нео-гексил. В настоящем описании и в последующей формуле изобретения "C1-4 алкил" предназначен для обозначения линейных или разветвленных алкильных групп, содержащих от 1 до 4 атомов углерода, таких как метил, этил, пропил, изопропил, н-бутил, изобутил, втор-бутил и трет-бутил. В настоящем описании и в последующей формуле изобретения "C1-3 алкил" предназначен для обозначения линейных или разветвленных алкильных групп, содержащих от 1 до 3 атомов углерода, таких как метил, этил, пропил и изопропил. В настоящем описании и в последующей формуле изобретения, "С 2-6 алкенил" предназначен для обозначения линейных или разветвленных алкильных групп, содержащих от 2 до 6 атомов углерода и по меньшей мере одну двойную связь, таких как этенил (винил), 1-пропенил, 2-пропенил (аллил), изопропенил, бутенил, пентенил и гексенил. В настоящем описании и в последующей формуле изобретения "С 2-4 алкенил" предназначен для обозначения линейных или разветвленных алкильных групп, содержащих от 2 до 4 атомов углерода и по меньшей мере одну двойную связь, таких как этенил (винил), 1-пропенил, 2-пропенил (аллил), изопропенил и бутенил. В настоящем описании и в последующей формуле изобретения "С 2-6 алкинил" предназначен для обозначения линейных или разветвленных алкильных групп, содержащих от 2 до 6 атомов углерода и по меньшей мере одну тройную связь, таких как этинил, 1-пропинил, 2-пропинил (пропаргил), бутинил,пентинил и гексинил. В настоящем описании и в последующей формуле изобретения "С 2-4 алкинил" предназначен для обозначения линейных или разветвленных алкильных групп, содержащих от 2 до 4 атомов углерода и по меньшей мере одну тройную связь, таких как этинил, 1-пропинил, 2-пропинил (пропаргил) и бутинил. В настоящем описании и в последующей формуле изобретения "C1-6 алкокси" предназначен для обозначения линейных или разветвленных алкоксигрупп, содержащих от 1 до 6 атомов углерода, таких как метокси, этокси, н-пропокси, изо-пропокси, н-бутокси, трет-бутокси, н-пентокси, втор-пентокси,изопентокси и н-гексилокси. В настоящем описании и в последующей формуле изобретения "C1-3 алкокси" предназначен для обозначения линейных или разветвленных алкоксигрупп, содержащих от 1 до 3 атомов углерода, таких как метокси, этокси, н-пропокси и изо-пропокси. В соответствии с предпочтительным вариантом осуществления изобретения значения Ra, Ra' , Rb иY в формуле (I), представленной выше, описаны в данном документе ниже. Предпочтительно, Ra и Ra', одинаковые или отличающиеся друг от друга, представляют собой атом водорода; атом галогена, выбранный из хлора, брома и йода; гидроксигруппу; C1-С 6 алкильную и C1-С 6 алкоксигруппу, необязательно замещенную одним или несколькими заместителями, выбранными из группы, состоящей из галогена, гидрокси, -NH2 или C1-С 3 алкокси; карбоциклическое или гетероциклическое кольцо, алифатическое или ароматическое, содержащее от 4 до 10 членов, необязательно замещенное одним или несколькими заместителями, выбранными из группы, состоящей из галогена, гидрокси, C1-С 6 алкила, C1-С 6 алкокси, -NR1R2, -С(О)ОН, -C(O)OR1 и -C(O)NR1R2. Более предпочтительно, Ra и Ra', одинаковые или отличающиеся друг от друга, представляют собой атом галогена, выбранный из хлора и брома; гидроксигруппу; C1-С 6 алкильную группу; C1-С 6 алкоксигруппу; или карбоциклическое или гетероциклическое кольцо, алифатическое или ароматическое, содержащее от 5 до 6 членов, необязательно замещенное одним или несколькими заместителями, выбранными из группы, состоящей из галогена, гидрокси, C1-С 6 алкила, C1-С 6 алкокси, -NR1R2 и С(О)ОН. Преимущественно, указанное карбоциклическое или гетероциклическое кольцо, алифатическое или ароматическое, содержащее от 5 до 6 членов, выбрано из фенила, пиридина, пиримидина, пиразина, пиридазина, пиррола, фурана, тиофена, оксазола, изоксазола, тиазола, изотиазола, 2 Н-пирана, циклогексила, циклопентила, пиперидина, пиперазина. Еще более предпочтительно, Ra и Ra', одинаковые или отличающиеся друг от друга, представляют со-5 024939 бой атом брома, гидроксигруппу; C1-С 3 алкоксигруппу; или ароматическое карбоциклическое или гетероциклическое кольцо, содержащее 6 членов, необязательно замещенное одним или двумя заместителями,выбранными из группы, состоящей из галогена, гидрокси, C1-С 3 алкила, C1-C3 алкокси, -NR1R2 и -С(О)ОН. В предпочтительном варианте осуществления указанное карбоциклическое или гетероциклическое кольцо, алифатическое или ароматическое, содержащее 6 членов, выбрано из фенила, пиридина, пиримидина, пиразина, пиридазина, 2 Н-пирана, циклогексила, пиперидина, пиперазина. В еще более предпочтительном варианте осуществления изобретения указанное карбоциклическое или гетероциклическое кольцо, алифатическое или ароматическое, содержащее 6 членов, выбрано из фенила, пиридина, пиримидина, 2 Н-пирана, циклогексила. В еще более предпочтительном варианте осуществления изобретения указанное карбоциклическое или гетероциклическое кольцо, алифатическое или ароматическое, содержащее 5 членов, выбрано из оксазола и изоксазола. Предпочтительно, Y представляет собой связь, C1-C6 алкильную группу, необязательно замещенную одним или несколькими заместителями, выбранными из группы, состоящей из галогена, гидрокси,-NH2 и C1-C3 алкокси. Более предпочтительно, Y представляет собой C1-C6 алкильную группу. Еще более предпочтительно, Y представляет собой C1-C3 алкильную группу. Предпочтительно, Rb представляет собой C1-C6 алкоксигруппу; -С(О)ОН; -C(O)OR1 или -NHCOR1. Более предпочтительно, Rb представляет собой C1-C6 алкоксигруппу или -С(О)ОН. Еще более предпочтительно, Rb представляет собой C1-С 3 алкоксигруппу или -С(О)ОН. Предпочтительно, R1 и R2 представляют собой, независимо, атом водорода, С 1-С 4 алкильную группу или фенильную группу. Более предпочтительно, R1 и R2 представляют собой, независимо, C1-С 3 алкильную группу. Еще более предпочтительно, R1 и R2 оба представляют собой метильную группу. Соединения в соответствии с настоящим изобретением предпочтительно используют в виде солей с фармацевтически приемлемыми органическими и неорганическими кислотами или основаниями. Предпочтительно, фармацевтически приемлемые органические кислоты выбраны из группы, состоящей из щавелевой, малеиновой, метансульфоновой, паратолуолсульфоновой, янтарной, лимонной,яблочной, винной и молочной кислоты. Предпочтительно, фармацевтически приемлемые органические основания выбраны из группы, состоящей из трометамина, лизина, аргинина, глицина, аланина и этаноламина. Предпочтительно, фармацевтически приемлемые неорганические кислоты выбраны из группы, состоящей из хлористоводородной, бромистоводородной, фосфорной и серной кислоты. Предпочтительно, фармацевтически приемлемые неорганические основания выбраны из группы,состоящей из гидроксидов или карбонатов щелочных или щелочноземельных металлов, таких как натрий, калий и кальций. Настоящее изобретение включает также пролекарства, стереоизомеры и энантиомеры соединений формулы (I), описанной выше. Как используется в настоящем документе, термин "пролекарство" относится к агенту, который преобразуется в исходное лекарственное средство in vivo путем некоторого физиологического химического процесса (например, пролекарство при установлении физиологического рН преобразуется в желаемую лекарственную форму). Пролекарства часто используют потому, что в некоторых ситуациях они могут быть введены легче, чем исходное лекарственное средство. Они, например, могут быть биодоступны при пероральном введении, тогда как исходное лекарственное средство нет. Пролекарство также может иметь улучшенную растворимость в фармакологических композициях по сравнению с исходным лекарственным средством. Например, без ограничения, пролекарством может быть соединение по настоящему изобретению, когда его вводят в виде сложного эфира ("пролекарство"), чтобы облегчить проникновение через клеточную мембрану, при котором растворимость в воде не удобна, но затем оно подвергается метаболическому гидролизу до карбоновой кислоты, как только попадает внутрь клетки, где растворимость в воде удобна. Пролекарства могут обладать многими полезными свойствами. Например, пролекарство может быть более растворимым в воде, чем конечное лекарственное средство, тем самым облегчая внутривенное введение препарата. Пролекарство может также иметь более высокий уровень пероральной биодоступности по сравнению с конечным препаратом. После введения пролекарство ферментативно или химически расщепляется с высвобождением конечного лекарственного средства в крови или в ткани. Сложноэфирные пролекарства соединений, описанных в данном документе, оговорены особо. Сложный эфир может быть образован из функциональной группы карбоновой кислоты, присоединенной к соединению формулы (I), представленной выше, путем взаимодействия со спиртом или фенолом. Альтернативно, сложный эфир может быть образован из гидроксильной функциональной группы, присоединенной к соединению формулы (I), представленной выше, путем взаимодействия с карбоновой кислотой или с аминокислотой. Не предполагая ограничений, сложный эфир может представлять собой алкиловый сложный эфир, ариловый сложный эфир или гетероариловый сложный эфир. Термин алкил имеет значе-6 024939 ние, обычно понимаемое специалистом в данной области, и относится к линейным, разветвленным или циклическим алкильным группам. С 1-6 алкиловые сложные эфиры являются особенно используемыми,когда алкильная часть эфира имеет от 1 до 6 атомов углерода и включает, но не ограничивается ими, метил, этил, пропил, изопропил, н-бутил, втор-бутил, изо-бутил, трет-бутил, изомеры пентила, изомеры гексила, циклопропил, циклобутил, циклопентил, циклогексил и их комбинации, содержащие от 1-6 атомов углерода. Соединения по настоящему изобретению в соответствии с формулой (I), представленной выше, могут быть использованы при лечении патологического состояния, вызванного неконтролируемой активацией и/или избыточной экспрессией GSK-3, выбранного из группы, включающей (i) инсулинрезистентные нарушения; (ii) нейродегенеративные заболевания; (iii) расстройства настроения; (iv) шизофренические расстройства; (v) злокачественные расстройства; (vi) воспаление; (vii) расстройства вследствие токсикомании; (viii) эпилепсии; и (ix) невропатическую боль. Преимущественно, инсулинрезистентными нарушениями являются сахарный диабет 2 типа, синдром X, ожирение и синдром поликистозных яичников. Преимущественно, острыми и хроническими нейродегенеративными заболеваниями являются болезнь Паркинсона, болезнь Альцгеймера, болезнь Хантингтона и спинальные нейродегенеративные расстройства. Предпочтительно, спинальными нейродегенеративными расстройствами являются амиотрофический латеральный склероз, рассеянный склероз, спинальная мышечная атрофия и нейродегенерация в связи с повреждением спинного мозга. Преимущественно, расстройствами настроения являются биполярные расстройства и депрессивные расстройства. Предпочтительно, биполярными расстройствами являются биполярное расстройство I, биполярное расстройство II, циклотимия и биполярное расстройство, иным образом не обозначенное (BD-NOS). Предпочтительно, депрессивными расстройствами являются большое депрессивное расстройство(SAD), дистимия и депрессивное расстройство, иным образом не обозначенное (DD-NOS). Преимущественно, шизофреническими расстройствами являются параноидальная шизофрения, шизофрения дезорганизованного типа, кататоническая шизофрения, простая шизофрения, остаточная шизофрения и недифференцированная шизофрения. Преимущественно, злокачественными расстройствами являются рак простаты, поджелудочной железы, яичников и колоректальный рак и MLL-связанная лейкемия. Преимущественно, расстройствами вследствие токсикомании являются острые расстройства вследствие злоупотребления психостимуляторами. Обычно 1 Н-индазол-3-карбоксамидные соединения в соответствии с формулой (I), используемые в данном изобретении, вводят в виде фармацевтической композиции. Соответственно, дополнительный аспект настоящего изобретения относится к фармацевтической композиции, содержащей по меньшей мере одно соединение формулы (I), описанной выше, и по меньшей мере один инертный фармацевтически приемлемый эксципиент. Предпочтительно, фармацевтическую композицию по настоящему изобретению получают в виде подходящих лекарственных форм, содержащих эффективное количество по меньшей мере одного соединения формулы (I), описанной выше, его соли с фармацевтически приемлемыми органическими или неорганическими кислотой или основанием, или его пролекарства, и по меньшей мере один инертный фармацевтически приемлемый эксципиент. Примерами подходящих лекарственных форм являются таблетки, капсулы, таблетки с покрытием,гранулы, сиропы и растворы для перорального введения; растворы, помада и мазь для местного применения; лечебные пластыри для трансдермального введения; суппозитории для ректального введения и инъекционные стерильные растворы. Другими подходящими лекарственными формами являются формы с замедленным высвобождением и формы на основе липосом для перорального, инъекционного или трансдермального введения. Стандартные лекарственные формы могут также содержать другие обычно используемые ингредиенты, такие как: консерванты, стабилизаторы, поверхностно-активные вещества, буферы, соли для регулирования осмотического давления, эмульгаторы, подсластители, красители, ароматизаторы и тому подобное. Количество 1 Н-индазол-3-карбоксамида в соответствии с формулой (I) или его фармацевтически приемлемой кислотно-аддитивной соли в фармацевтической композиции по настоящему изобретению может изменяться в широком диапазоне в зависимости от известных факторов, например от типа патологии, тяжести заболевания, массы тела пациента, лекарственной формы, выбранного пути введения, количества введений в день и эффективности выбранного 1 Н-индазол-3-карбоксамидного соединения в соответствии с формулой (I). Тем не менее специалист в данной области может легко определить оптимальное количество по стандартной методике. Как правило, количество соединения формулы (I) или его фармацевтически приемлемой кислотно-7 024939 аддитивной соли в фармацевтической композиции по настоящему изобретению будет таким, чтобы обеспечить уровень введения от 0,0001 до 100 мг/кг/день. Предпочтительно, уровень введения составляет от 0,001 до 50 мг/кг/день и еще более предпочтительно от 0,01 до 10 мг/кг/день. Дозированные формы фармацевтической композиции по настоящему изобретению могут быть получены с помощью методик, которые известны специалистам в фармацевтической химии и включают смешивание, гранулирование, прессование, растворение, стерилизацию и тому подобное. Неограничивающие примеры соединений формулы (I) в соответствии с настоящим изобретением представлены в следующей далее табл. 1. Таблица 1- двойной триплет; (ддд) - двойной двойной дублет; (дтд) - двойной тройной дублет; (м) - мультиплет; J константа связывания; 5 - химический сдвиг (в м.д.). Получение соединений формулы (I). Соединения формулы (I) могут быть получены способами, известными специалистам в данной области, например, следующими способами A-D. Способ А. 1 1-Гидроксибензотриазол (HOBt, 7,4 0 г, 54,8 ммоль) и N,N'-дициклогексилкарбодиимид (DCC, 11 г,53,3 ммоль) добавляли к раствору соответствующим образом замещенной 1 Н-индазол-3-карбоновой кислоты (соединение i, 12 г, 49,8 ммоль) в ДМФА (200 мл) при температуре 0 С. Спустя 1 ч при той же температуре добавляли раствор соответствующим образом 1-замещенного [пиперидин-4-ил]метанамина(соединение ii, 10 г, 58,1 ммоль) в ДМФА (100 мл). Смесь перемешивали при температуре 0 С в течение 2 ч, затем оставляли до достижения комнатной температуры в течение ночи. Смесь разбавляли AcOEt,затем твердый продукт удаляли путем фильтрования. Раствор экстрагировали три раза 2 н. соляной кислотой (HCl). рН кислотной фазы повышали (примерно до 13) с помощью 5 н. NaOH и раствор экстрагировали три раза дихлорметаном (DCM). Органическую фазу сушили над безводным Na2SO4. Растворитель фильтровали, выпаривали при пониженном давлении и остаток очищали подходящим образом. Например, соединение (19) может быть получено в соответствии со способом А, как описано ниже. 1-Гидроксибензотриазол (HOBt, 7,4 0 г, 54,8 ммоль) и N,N'-дициклогексилкарбодиимид (DCC, 11 г,53,3 ммоль) добавляли к раствору 5-бром-1 Н-индазол-3-карбоновой кислоты (соединение iii, 12 г, 49,8 ммоль) в ДМФА (200 мл) при температуре 0 С. Спустя 1 ч при той же температуре добавляли раствор 1[1-(2-метоксиэтил)пиперидин-4-ил]метанамина (соединение iv, 10 г, 58,1 ммоль) в ДМФА (100 мл). Смесь перемешивали при температуре 0 С в течение 2 ч, затем оставляли до достижения комнатной температуры в течение ночи. Смесь разбавляли AcOEt, затем твердый продукт удаляли путем фильтрования. Раствор экстрагировали три раза 2 н. HCl. рН кислотной фазы повышали (примерно до 13) с помощью 5 н.NaOH и раствор экстрагировали три раза DCM. Органическую фазу сушили над безводным Na2SO4. Растворитель фильтровали, выпаривали при пониженном давлении и остаток очищали путем флэшхроматографии (SiO2, CHCl3/МеОН=85/15). Полученное таким образом соединение (19) очищали, как раскрыто в табл. 2, получая 9,5 г твердого продукта. Способ В. Первая стадия К суспензии соответствующего соединения (v) (2,13 г; 0,0061 моль) в толуоле (50 мл) добавляли по каплям раствор 1-(1-бензилпиперидин-4-ил)метанамина (соединение vi; 2,52 г; 0,012 моль), полученного,как описано в WO 94/10174, и триэтиламин (TEA; 3,2 мл; 0,023 моль) в толуоле (10 мл). Реакционную смесь кипятили с обратным холодильником в течение 12 ч и затем фильтровали. Растворитель удаляли путем выпаривания при пониженном давлении и остаток обрабатывали этилацетатом. Органическую фазу переносили в делительную колонку, промывали насыщенным раствором NaHCO3 и водой, разделяли и сушили над Na2SO4. Полученный продукт (vii) подходящим образом кристаллизовали. Вторая стадия Раствор соответствующего N-[(1-бензилпиперидин-4-ил)метил]-1 Н-индазол-3-карбоксамида (соединение vii; 0,506 г; 1,34 ммоль) в абсолютном этаноле (8 мл) и ледяной уксусной кислоте (0,8 мл) гидрировали в системе непрерывных микрореакторов (H-Cube), используя в качестве картриджа CartCartPd/C 10%. Ключевыми параметрами H-Cube были следующие: температура 80 С; давление 10 бар; поток 1 мл/минута. Через три часа раствор концентрировали при пониженном давлении, разбавляли водой и переносили в делительную воронку. Водную фазу затем промывали этилацетатом, подщелачивали 1 н. NaOH и экстрагировали этилацетатом. Органические слои собирали, сушили над Na2SO4 и растворитель удаляли путем выпаривания при пониженном давлении. Полученное таким образом твердое вещество сушили в печи в вакууме с получением 0,27 г желаемого замещенного N-(пиперидин-4-илметил)-1 Н-индазол-3-карбоксамида (viii), который использовали без дополнительной очистки. К раствору (viii) (0,75 ммоль; 215 мг) в метилэтилкетоне (МЕК; 9 мл), перемешиваемому при температуре 85 С, добавляли по каплям подходящее галогенированное соединение (ix; 1,05 экв.) и триэтиламин (TEA; 210 мкл; 2 экв.). Реакционную смесь кипятили с обратным холодильником в течение 8 ч,затем охлаждали и разбавляли этилацетатом. Органический слой промывали насыщенным водным раствором NH4Cl и водой. Органическую фазу отделяли и сушили над Na2SO4. Растворитель удаляли путем выпаривания при пониженном давлении и продукт (I) очищали, как описано ниже. Например, соединение (2) может быть получено в соответствии со способом В, как описано ниже Раствор N-[(1-бензилпиперидин-4-ил)метил]-5-метокси-1 Н- индазол-3-карбоксамида (соединение х; 0,506 г; 1,34 ммоль) в абсолютном этаноле (8 мл) и ледяной уксусной кислоте (0,8 мл) гидрировали в системе непрерывных микрореакторов (H-Cube), используя в качестве картриджа CartCart Pd/C 10%. Ключевыми параметрами H-Cube были следующие: температура 80 С; давление 10 бар; поток 1 мл/мин. Через три часа раствор концентрировали при пониженном давлении, разбавляли водой и переносили в делительную воронку. Водную фазу затем промывали этилацетатом, подщелачивали 1 н. NaOH и экстрагировали этилацетатом. Органические слои собирали, сушили над Na2SO4 и растворитель удаляли путем выпаривания при пониженном давлении. Полученное таким образом твердое вещество сушили в печи в вакууме с получением 0,27 желаемого 5-метокси-N-(пиперидин-4-илметил)-1 Н-индазол-3-карбоксамида (xi), который использовали без дополнительной очистки. 1 К раствору (xi) (0,75 ммоль; 215 мг) в метилэтилкетоне (МЕК; 9 мл), перемешиваемому при температуре 85 С, добавляли по каплям 1-хлор-2-метоксиэтан (xii; 1,05 экв.) и триэтиламин (TEA; 210 мкл; 2 экв.). Реакционную смесь кипятили с обратным холодильником в течение 8 ч, затем охлаждали и разбавляли этилацетатом. Органический слой промывали насыщенным водным раствором NH4Cl и водой. Органическую фазу отделяли и сушили над Na2SO4. Растворитель удаляли путем выпаривания при пониженном давлении и соединение (2) очищали,как раскрыто ниже в табл. 2. Тионилхлорид (SOCl2; 9,3 мл; 0,12 8 моль) добавляли к суспензии соответствующим образом замещенной 1 Н-индазол-3-карбоновой кислоты (соединение i; 2,36 г; 0,0123 моль) в толуоле (77 мл) и реакционную смесь кипятили с обратным холодильником в течение 4 ч. Растворитель удаляли путем выпаривания при пониженном давлении и остаток обрабатывали два раза толуолом с получением 2,13 г желаемого продукта (xiii) 2,10-замещенного 7 Н,14 Н-пиразино[1,2-b:4,5-b']ди-индазол-7,14-диона. Вторая стадия К суспензии (xiii) (5,2 ммоль) в толуоле (40 мл) добавляли по каплям раствор подходящего амина(ii; 2,1 экв.) и триэтиламина (TEA; 3,6 экв.; 2,6 мл). Реакционную смесь кипятили с обратным холодильником в течение 8 ч, затем охлаждали и перемешивали в 2 н. HCl (20 мл) в течение 8 ч. Суспензию переносили в делительную воронку и водную фазу отделяли и подщелачивали 1 н. NaOH. Растворитель удаляли путем выпаривания при пониженном давлении и продукт (I) очищали, как описано ниже. Способ D. Раствор продукта (xiv), подходящим образом замещенную арилбороновую кислоту (соединение xv),[1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладий(II) [Pd(dppf)Cl2], карбонат цезия в 1,4-диоксане и воде (соотношение 3:1) подвергали воздействию микроволнового излучения (MW). Программа была следующей: 3'; T1=160C, T2=130C; максимальная мощность 300 Вт; 45'; T1=160C, T2=130C; максимальная мощность 300 Вт; 5' ; T1=20C, T2=15C. После одного цикла микроволнового облучения растворители удаляли путем выпаривания при пониженном давлении и реакционную смесь разбавляли смесью хлороформа и метанола в соотношении 2:1 и фильтровали. Полученные таким образом продукты (I) очищали, как описано ниже. Способы очистки. Соединения формулы (I), полученные в соответствии с одним из предшествующих описанных способов A-D, могут быть очищены одним из следующих методов (а)-(с).(a) Флэш-хроматография на силикагеле. Флэш-хроматографию проводили на силикагелевом картридже 20-45 мкМ системы Biotage FlashMaster Personal или на силикагелевом картридже 40 мкМ системы флэш-хроматографии Grace Reveleris. Поток=60 мл/мин. Растворителями, используемыми в качестве элюентов, являются метанол и хлороформ.(b) Кристаллизация. Использовали различные кристаллизационные растворители в зависимости от соединения, которое подвергали очистке. Растворители представлены в следующей ниже табл. 2.(c) Система препаративной ЖХ/МС. Система ЖХ/МС, включающая программное обеспечение Waters 2767 Sample manager, детектор абсорбции Waters 2478 dualи одноквадрупольный масс-спектрометр с источником ионизации электро- 13024939 распылением (ESI) Waters Micromass ZQ. Используемой колонкой была X-Bridge Prep C18 5 мкм с предколонкой 1910 мм (Waters). Сбор фракций был доступен из системного программного обеспеченияMassLynx v. 4.1. Длина волны детектирования была установлена на 230 нм, и температуру устанавливали до 25 С. Образец растворяли (50 мг/мл) в смеси ДМСО/CH3CN с соотношением 1:1. Мобильная фаза представляла собой канал A=CH3CN+0,1%-ная муравьиная кислота (элюент А); канал В=Н 2 О+0,1%-ная муравьиная кислота (элюент В); поток=40 мл/мин; градиент = минимальный и максимальный процент элюента А, достигнутый за 15 мин, показан в следующей табл. 2. В следующей табл. 2 показаны как способ получения, так и способ очистки для каждого из соединений формулы (I), представленных в табл. 1, и масса моноизотопа для каждого соединения. Таблица 2 Соединения 20-44 получали, как описано далее. Синтез соединения 20. Этил [4-([(5-бром-6-метокси-1 Н-индазол-3-ил)карбонил]аминометил)пиперидин-1-ил]ацетат. 20 а) Этил [4-(аминометил)пиперидин-1-ил]ацетат. К перемешиваемому раствору N-[фенилметилиден]-1-(пиперидин-4-ил)метанамина (22 г, 0,109 моль (получен, как описано в WO 2004/101548) в абсолютном этаноле (150 мл) добавляли этилбромацетат (12 мл, 0,109 моль) и карбонат калия (33 г, 0,24 моль). Раствор нагревали при кипении с обратным холодильником в течение 8 часов, затем охлаждали и концентрировали путем выпаривания растворителя при пониженном давлении. Реакционную смесь разбавляли 3 н. HCl (150 мл) и перемешивали при комнатной температуре в течение 2 ч. Кислый раствор затем промывали этилацетатом и подщелачивали путем добавления Na2CO3. Водную фазу экстрагировали тремя порциями дихлорметана, которые затем объединяли и сушили над Na2CO4. Растворитель удаляли путем выпаривания при пониженном давлении и полученный продукт этил [4-(аминометил)пиперидин-1-ил]ацетат 20 а использовали как таковой без какой-либо дополнительной очистки. МС: 201 m/z (M+H+). 1-Гидроксибензотриазол (HOBt, 2,43 г, 14,2 ммоль) и N,N'-дициклогексилкарбодиимид (DCC, 2,93 г, 14,2 ммоль) добавляли к раствору 5-бром-6-метокси-1 Н-индазол-3-карбоновой кислоты (3,5 г, 12,9 ммоль) в ДМФА (40 мл) при температуре 0 С. Спустя 1 ч при той же температуре добавляли раствор соединения 20 а (2,6 г, 12,9 ммоль) в ДМФА (25 мл). Смесь перемешивали при температуре 0 С в течение 2 ч, затем оставляли до достижения комнатной температуры в течение ночи. Смесь разбавляли EtOAc и твердый продукт удаляли путем фильтрования. Раствор экстрагировали три раза 2 н. соляной кислотой(HCl). рН кислотной фазы повышали (примерно до 13) с помощью 5 н. NaOH и раствор экстрагировали три раза дихлорметаном (DCM). Органическую фазу сушили над безводным Na2CO4 и растворитель фильтровали и выпаривали при пониженном давлении с получением 1,6 г (3,5 ммоль, 27%-ный выход) этил [4-([(5-бром-6-метокси-1 Н-индазол-3-ил)карбонил]аминометил)пиперидин-1-ил]ацетата (соединение 20). 1H ЯМР (300 МГц, ДМСО-d6)= 13,46 (уш.с, 1 Н), 8,35 (т, J=6,2 Гц, 1 Н), 8,30 (с, 1 Н), 7,12 (с, 1 Н),4,07 (кв, J=7,3 Гц, 2 Н), 3,93 (с, 3 Н), 3,16 (с, 4 Н), 2,81 (д, J=11,0 Гц, 2H), 2,19-2,03 (м, 2 Н), 1,70-1,44 (м,3 Н), 1,31-1,04 (м, 5 Н). МС: 453 m/z (M+H)+. Синтез соединения 21. Гидрат формиата 4-[([6-метокси-5-(пиридин-3-ил)-1 Н-индазол-3 ил]карбониламино)метил]пиперидин-1-илуксусной кислоты. Раствор соединения 20 (200 мг, 0,44 ммоль), пиридин-3-илбороновой кислоты (217 мг, 1,7 7 ммоль),[1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладия(II) [Pd(dppf)Cl2] (81 мг, 0,11 ммоль) и карбоната цезия (57 5 мг, 1,76 ммоль) в 1,4-диоксане и воде (соотношение 3:1, 8 мл) подвергали воздействию микроволнового облучения следующим образом: период времени = 3'; T1=160C, T2=130C; максимальная мощность 300 Вт; период времени = 45'; T1=160C, T2=130C; максимальная мощность 300 Вт; период времени = 5'; T1=20C, T2=15C. После одного цикла микроволнового облучения растворители удаляли путем выпаривания при пониженном давлении и реакционную смесь разбавляли раствором метанола (20 мл), фильтровали через целит и сушили в вакууме. Сырой продукт фильтровали на силикагелевом картридже и промывали хлороформом и метанолом при соотношении 1:1. Полученный твердый продукт растворяли в ДМСО и очищали посредством препаративной ВЭЖХ (канал А=CH3CN+0,1%-ная муравьиная кислота; канал В=Н 2 О+0,1%-ная муравьиная кислота: поток=40 мл/мин; градиент=2-40% элюента А за 15 мин), получая гидрат формиата 4-[([6-метокси-5-(пиридин-3-ил)-1 Н-индазол-3-ил]карбониламино)метил]пиперидин-1-илуксусной кислоты 21 (67 мг, 36%-ный выход). 1H ЯМР (300 МГц, ДМСО-d6)= 13,44 (уш.с, 1 Н), 8,66 (дд, J=0,9, 2,4 Гц, 1 Н), 8,54 (дд, J=1,8, 4,8 Гц,1 Н), 8,42 (т, J=6,2 Гц, 1 Н), 8,01 (с, 1 Н), 7,91-7,85 (м, 1 Н), 7,45 (ддд, J=0,9, 4,8, 7,8 Гц, 1 Н), 7,13 (с, 1 Н),3,86 (с, 3 Н), 3,41 (уш.с, 1 Н), 3,30-3,00 (м, 6 Н), 2,54 (с, 2 Н), 1,73 (д, J=11,0 Гц, 3 Н), 1,52-1,28 (м, 2 Н). МС: 424 m/z (M+H)+. Синтез соединения 22. Гидрат 4-[([6-метокси-5-(5-метоксипиридин-3-ил)-1 Н-индазол-3 ил]карбониламино)метил]пиперидин-1-илуксусной кислоты. Гидрат 4-[([6-метокси-5-(5-метоксипиридин-3-ил)-1 Н-индазол-3-ил]карбониламино)метил]пиперидин-1-илуксусной кислоты 22 получали в соответствии со способом, описанным для соединения 21, исходя из (5-метоксипиридин-3-ил)бороновой кислоты и используя следующие параметры препаративной ВЭЖХ для очистки: канал A=CH3CN+0,1%-ная муравьиная кислота; канал В=Н 2 О+0,1%-ная муравьиная кислота: поток=40 мл/мин; градиент=10%-45% элюента А за 15 мин. Выход: 33 мг, 17%. 1H ЯМР (300 МГц, ДМСО-d6)= 13,46 (уш.с, 1 Н), 8,42 (т, J=6,0 Гц, 1 Н), 8,26 (дд, J=2,0, 6,8 Гц, 2 Н),8,02 (с, 1 Н), 7,43 (дд, J=1,6, 2,7 Гц, 1 Н), 7,13 (с, 1 Н), 3,88 (с, 3 Н), 3,86 (с, 3 Н), 3,98 (уш.с, 1 Н), 3,30-3,01 (м,6 Н), 2,66-2,53 (м, 2 Н), 1,73 (д, J=10,6 Гц, 3 Н), 1,40 (кв, J=11,6 Гц, 2 Н). МС: 454 m/z (M+H)+. Синтез соединения 23. Гидрат 4-[([5-(2,3-дифторфенил)-6-метокси-1 Н-индазол-3-ил]карбониламино)метил]пиперидин-1-илуксусной кислоты. Гидрат 4-[([5-(2,3-дифторфенил)-6-метокси-1 Н-индазол-3-ил]карбониламино)метил]пиперидин 1-илуксусной кислоты 23 получали в соответствии со способом, описанным для соединения 21, исходя из (2,3-дифторфенил)бороновой кислоты и используя следующие параметры препаративной ВЭЖХ для очистки: канал A=CH3CN+0,1%-ная муравьиная кислота; канал В=Н 2 О+0,1%-ная муравьиная кислота: поток=40 мл/мин; градиент=15-50% элюента А за 15 мин. Выход: 48 мг, 24%. 1 Трет-бутил 4-([(5-метокси-1 Н-индазол-3-ил)карбонил]аминометил)пиперидин-1-карбоксилат 24 а получали в соответствии со способом, описанным для соединения 20, исходя из 5-метокси-1 Н-индазол-3 карбоновой кислоты и трет-бутил 4-(аминометил)пиперидин-1-карбоксилата. Выход: 35,2 г, 96%. МС: 389 m/z (M+H)+. 24b) Гидрохлорид 5-метокси-N-(пиперидин-4-илметил)-1 Н-индазол-3-карбоксамида 2 М HCl в Et2O(1,8 л) добавляли к раствору соединения 24 а (92,8 г, 0,24 моль) в МеОН (500 мл). Смесь перемешивали в течение 3 ч при комнатной температуре, затем полученный твердый продукт отфильтровывали и сушили с получением гидрохлорида 5-метокси-N-(пиперидин-4-илметил)-1 Н-индазол-3-карбоксамида 24b (61,1 г, 89%-ный выход). МС: 289 m/z (M+H)+. 24 с) Этил 4-[4-([(5-метокси-1 Н-индазол-3-ил)карбонил]аминометил)пиперидин-1-ил]бутаноат. Смесь соединения 24b (8 г, 2 4,6 ммоль) и карбоната калия (17 г, 123 ммоль) в ацетоне (250 мл) кипятили с обратным холодильником в течение 1 ч, затем добавляли по каплям этил 4-хлорбутаноат (3,62 мл, 25,9 ммоль). Смесь кипятили с обратным холодильником в течение ночи, затем охлаждали и фильтровали. Полученный твердый продукт сушили и очищали посредством препаративной ВЭЖХ (каналA=CH3CN+0,1%-ная муравьиная кислота; канал В=Н 2 О+0,1%-ная муравьиная кислота: поток=40 мл/мин; градиент=10%-45% элюента А за 15 минут) с получением 0,9 г (9%-ный выход) этил 4-[4-([ (5-метокси 1 Н-индазол-3-ил)карбонил]аминометил)пиперидин-1-ил]бутаноата 24 с. МС: 403 m/z (M+H)+. К раствору соединения 24 с (744 мг, 1,85 ммоль) в МеОН (10 мл) добавляли водный раствор NaOH(1M, 3,7 мл). Раствор кипятили с обратным холодильником в течение ночи, затем органический растворитель удаляли в вакууме, остаток разбавляли Н 2 О и рН доводили до значения 5 путем добавления 1 МHCl. Смесь выдерживали при температуре 4 С в течение ночи, затем полученный твердый продукт отфильтровывали, промывали свежей водой и сушили в вакууме с получением 4-[4-([(5-метокси-1 Ниндазол-3-ил)карбонил]аминометил)пиперидин-1-ил]бутановой кислоты 24 (72 мг, 10%-ный выход). 1(т, J=6,1 Гц, 2 Н), 3,00-2,89 (м, 2 Н), 2,81 (т, J=11,4 Гц, 2 Н), 2,32 (т, J=7,1 Гц, 2 Н), 2,01-1,70 (м, 5 Н), 1,641,41 (м, 2 Н). МС: 375 m/z (M+H)+. Синтез соединения 25. Гидрат 4-[([5-(пиримидин-5-ил)-1 Н-индазол-3-ил]карбониламино)метил]пиперидин-1-илуксусной кислоты 25 а) Трет-бутил 4-([(5-бром-1 Н-индазол-3-ил)карбонил]аминометил)пиперидин-1-карбоксилат. Трет-бутил 4- ([(5-бром-1 Н-индазол-3-ил)карбонил]аминометил)пиперидин-1-карбоксилат 25 а получали в соответствии со способом, описанным для соединения 20, исходя из 5-бром-1 Н-индазол-3 карбоновой кислоты и трет-бутил 4-(аминометил)пиперидин-1-карбоксилата. Выход: 40,6 г, 87%. МС: 437 m/z (M+H)+. 25b) Гидрохлорид 5-бром-N-(пиперидин-4-илметил)-1 Н-индазол-3-карбоксамида. Гидрохлорид 5-бром-N-(пиперидин-4-илметил)-1 Н-индазол-3-карбоксамида 25b получали в соответствии со способом, описанным для соединения 24b, исходя из соединения 25 а. Выход: 2 3,8 г, 76%. МС: 337 m/z (M+H)+. 25 с) Этил [4-([(5-бром-1 Н-индазол-3-ил)карбонил]аминометил)пиперидин-1-ил]ацетат. Смесь соединения 25b (2 г, 5,4 ммоль) и карбоната калия (2,3 г, 16, 6 ммоль) в ДМФА (45 мл) перемешивали в течение 1 ч при температуре 70 С. Добавляли по каплям раствор этилбромацетата (0,89 мл, 8 ммоль) в ДМФА (5 мл). Спустя 3 ч выдерживания при температуре 70 С реакционную смесь охлаждали,разбавляли водой и экстрагировали три раза EtOAc. Объединенные органические фазы сушили надNa2SO4 и концентрировали в вакууме. Сырой продукт очищали посредством флэш-хроматографии (силикагель, CHCl3:МеОН 95:5) с получением 710 мг (31%-ный выход) этил [4-([ (5-бром-1 Н-индазол-3 ил)карбонил]аминометил)пиперидин-1-ил]ацетата 25 с. 1(с, 2 Н), 2,81 (д, J=11,2 Гц, 2 Н), 2,22-2,03 (м, 2 Н), 1,72-1,46 (м, 3 Н), 1,31-1,08 (м, 5 Н). МС: 423 m/z (M+H)+. Гидрат 4-[([5-(пиримидин-5-ил)-1 Н-индазол-3-ил]карбониламино)метил]пиперидин-1-илуксусной кислоты 25 получали в соответствии со способом, описанным для соединения 21, исходя из соединения 25 с и пиримидин-5-илбороновой кислоты. Продукт очищали путем кристаллизации в МеОН. Выход: 43 мг, 18%. 1- 18024939 МС: 395 m/z (M+H)+. Синтез соединения 26. Гидрат 4-[([5-(3,5-диметилизоксазол-4-ил)-1 Н-индазол-3-ил]карбониламино)метил]пиперидин-1-илуксусной кислоты. Гидрат 4-[([5-(3,5-диметилизоксазол-4-ил)-1 Н-индазол-3-ил]карбониламино)метил]пиперидин 1-илуксусной кислоты 26 получали в соответствии со способом, описанным для соединения 21, исходя из соединения 25 с и (3,5-диметилизоксазол-4-ил)бороновой кислоты. Продукт очищали путем кристаллизации в МеОН. Выход: 55 мг, 23%. 1H ЯМР (300 МГц, ДМСО-d6)= 13,84 (уш.с, 1 Н), 8,50 (т, J=5,9 Гц, 1 Н), 8,10 (с, 1 Н), 7,72 (д, J=8,6 Гц, 1 Н), 7,40 (дд, J=1,5, 8,8 Гц, 1 Н), 4,10 (уш.с, 1 Н), 3,36-3,03 (м, 6 Н), 2,60 (т, J=11,2 Гц, 2 Н), 2,41 (с, 3 Н),2,22 (с, 3 Н), 1,75 (д, J=11,2 Гц, 3 Н), 1,42 (кв, J=11,4 Гц, 2 Н). МС: 412 m/z (M+H)+. Синтез соединения 27. Гидрат 4-[([5-(2,3-дихлорфенил)-1 Н-индазол-3-ил]карбониламино)метил]пиперидин-1-илуксусной кислоты. Гидрат 4-[([5-(2,3-дихлорфенил)-1 Н-индазол-3-ил]карбониламино)метил]пиперидин-1 илуксусной кислоты 27 получали в соответствии со способом, описанным для соединения 21, исходя из соединения 25 с и (2,3-дихлорфенил)бороновой кислоты и используя следующие параметры препаративной ВЭЖХ для очистки: канал А=CH3CN+0,1%-ная муравьиная кислота; канал В=Н 2 О+0,1%-ная муравьиная кислота: поток=40 мл/мин; градиент=20-55% элюента А за 15 мин. Выход: 42 мг, 15%. 1H ЯМР (300 МГц, ДМСО-d6)= 13,81 (уш.с, 1 Н), 8,50 (т, J=6,0 Гц, 1 Н), 8,17 (дд, J=0,9, 1,6 Гц, 1 Н),7,78-7,60 (м, 2 Н), 7,54-7,34 (м, 3 Н), 4,08 (уш.с, 1 Н), 3,38-2,96 (м, 6 Н), 2,58 (т, J=11,0 Гц, 2H), 1,74 (д,J=11,0 Гц, 3 Н), 1,42 (кв, J=11,6 Гц, 2 Н). МС: 461 m/z (M+H)+. Синтез соединения 28. Гидрат 4-[([5-(3-фторфенил)-1 Н-индазол-3 ил]карбониламино)метил]пиперидин-1-илуксусной кислоты. Гидрат 4-[([5-(3-фторфенил)-1 Н-индазол-3-ил]карбониламино)метил]пиперидин-1-илуксусной кислоты 28 получали в соответствии со способом, описанным для соединения 21, исходя из соединения 25 с и (3-фторфенил)бороновой кислоты и используя следующие параметры препаративной ВЭЖХ для очистки: канал A=CH3CN+0,1%-ная муравьиная кислота; канал В=Н 2 О+0,1%-ная муравьиная кислота: поток=40 мл/мин; градиент=15%-50% элюента А за 15 минут. Выход: 87 мг, 36%. 1(м, 2 Н), 1,75 (д, J=11,0 Гц, 3 Н), 1,42 (кв, J=11,5 Гц, 2 Н). МС: 411 m/z (M+H)+. Синтез соединения 29. 4-[([5-(2,3-Дифторфенил)-1 Н-индазол-3-ил]карбониламино)метил]пиперидин-1-илуксусная кислота. 4-[([5-(2,3-Дифторфенил)-1 Н-индазол-3-ил]карбониламино)метил]пиперидин-1-илуксусную кислоту 29 получали в соответствии со способом, описанным для соединения 21, исходя из соединения 25 с и (2,3-дифторфенил)бороновой кислоты и используя следующие параметры препаративной ВЭЖХ для очистки: канал A=CH3CN+0,1%-ная муравьиная кислота; канал В=Н 2 О+0,1%-ная муравьиная кислота: поток=40 мл/мин; градиент = от 15 до 50% элюента А за 15 мин. Выход: 20 мг, (11,7%). 1(дд, J=0,7, 8,8 Гц, 1 Н), 7,61 (тд, J=1,7, 8,7 Гц, 1 Н), 7,51-7,25 (м, 3 Н), 3,33-3,10 (м, 6 Н), 2,64-2,53 (м, 2 Н),1,74 (д, J=10,5 Гц, 3 Н), 1,54-1,29 (м, 2 Н). Синтез соединения 30. 4-[([5-(5-Метоксипиридин-3-ил)-1 Н-индазол-3-ил]карбониламино)метил]пиперидин-1-илуксусная кислота. 4-[([5-(5-Метоксипиридин-3-ил)-1 Н-индазол-3-ил]карбониламино)метил]пиперидин-1-илуксусную кислоту 30 получали в соответствии со способом, описанным для соединения 21, исходя из соединения 25 с и (5-метоксипиридин-3-ил)бороновой кислоты и используя следующие параметры препаративной ВЭЖХ для очистки: канал A=CH3CN+0,1%-ная муравьиная кислота; канал В=Н 2 О+0,1%-ная муравьиная кислота: поток=40 мл/мин; градиент = от 2 до 40% элюента А за 15 мин. Выход: 47 мг (27,8%). 1(м, 1 Н), 8,30 (д, J=2,7 Гц, 1 Н), 7,81-7,76 (м, 1 Н), 7,76-7,69 (м, J=0,7 Гц, 1 Н), 7,61 (дд, J=1,8, 2,7 Гц, 1 Н),3,93 (с, 3 Н), 3,29-3,12 (м, 6 Н), 2,69-2,55 (м, 2 Н), 1,75 (д, J=11,0 Гц, 3 Н), 1,58-1,27 (м, 2H). Синтез соединения 31. [4-([(5-Этил-1 Н-индазол-3-ил)карбонил]аминометил)пиперидин-1 ил]уксусная кислота. Смесь продукта 25 с (170 мг, 0,4 ммоль), пинаколового эфира винил-бороновой кислоты (0,53 ммоль), [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладия(II) (50 мг, 0,06 ммоль), насыщенного раствора карбоната натрия (1,7 мл) в смеси толуол/этанол (соотношение 1:1, 10 мл) нагревали в микроволновой печи при температуре 150 С, 500 Вт, в течение 2 ч. После фильтрования через целит растворители удаляли при пониженном давлении и сырой продукт элюировали через силикагелевый картридж смесью хлороформ/метанол при соотношении 1:1. Растворители удаляли при пониженном давлении и полученный сырой промежуточный продукт растворяли в этаноле (20 мг/мл) и гидрировали над картриджем 10%Pd/C при температуре 30 С, 1 мл/мин в реакторе для гидрирования Thales Nano H-CUBE с получением[4-([(5-этил-1 Н-индазол-3-ил)карбонил]аминометил)пиперидин-1-ил]уксусной кислоты 31, очищали с использованием следующих параметров препаративной ВЭЖХ: канал A=CH3CN+0, 1%-ная муравьиная кислота; канал В=Н 2 О+0,1%-ная муравьиная кислота: поток=40 мл/мин; градиент = от 10 до 45% элюента А за 15 мин. Выход 170 мг, (41,0%). 1H ЯМР (300 МГц, ДМСО-d6)= 13,48 (уш.с, 1 Н), 8,38 (т, J=6,1 Гц, 1 Н), 7,97 (с, 1 Н), 7,52 (д, J=8,6 Гц, 1 Н), 7,28 (дд, J=1,6, 8,6 Гц, 1 Н), 4,38 (уш.с, 1 Н), 3,33-3,09 (м, 6 Н), 2,73 (кв, J=7,5 Гц, 2 Н), 2,65-2,53 (м,2 Н), 1,83-1,60 (м, 3 Н), 1,54-1.31 (м, 2 Н), 1,23 (т, J=7,5 Гц, 3 Н). Синтез соединения 32. 4-[([5-(Пропан-2-ил)-1 Н-индазол-3-ил]карбониламино)метил]пиперидин 1-илуксусная кислота. 4-[([5-(Пропан-2-ил)-1 Н-индазол-3-ил]карбониламино)метил]пиперидин-1-илуксусную кислоту 32 получали в соответствии со способом, описанным для соединения 31, исходя из пинаколового эфира 1-метилэтилен-бороновой кислоты. Выход=33 мг (7,7%). 1H ЯМР (300 МГц, ДМСО-d6)= 13,45 (уш.с, 1 Н), 8,38 (т, J=6,1 Гц, 1 Н), 8,10-7,86 (м, 1 Н), 7,52 (дд,J=0,5, 8,6 Гц, 1 Н), 7,32 (дд, J=1,6, 8,8 Гц, 1 Н), 4,65 (уш.с, 1 Н), 3,29-3,13 (м, 6 Н), 3,02 (квинт, J=6,8, 13,7 Гц, 1 Н), 2,57 (т, J=11,3 Гц, 2 Н), 1,73 (д, J=10,8 Гц, 3 Н), 1,55-1,32 (м, 2 Н), 1,26 (д, J=7,0 Гц, 6 Н). Синтез соединения 33. 4-[([5-(3,6-Дигидро-2 Н-пиран-4-ил)-1 Н-индазол-3 ил]карбониламино)метил]пиперидин-1-илуксусная кислота. 4-[([5-(3,6-Дигидро-2 Н-пиран-4-ил)-1 Н-индазол-3-ил]карбониламино)метил]пиперидин-1 илуксусную кислоту 33 получали в соответствии со способом, описанным для соединения 31 (без стадии гидрирования), исходя из пинаколового эфира 4-метил-3,6-дигидро-2 Н-пиранил-бороновой кислоты. Выход=150 мг (37,7%). 1[4-([(5-Циклогексил-1 Н-индазол-3-ил)карбонил]аминометил)пиперидин-1-ил]уксусную кислоту 34 получали в соответствии со способом, описанным для соединения 31, исходя из пинаколового эфира циклогексенил-бороновой кислоты. Выход=158 мг (39,6%). 1[4-([(5-Пентил-1 Н-индазол-3-ил)карбонил]аминометил)пиперидин-1-ил]уксусную кислоту 35 получали в соответствии со способом, описанным для соединения 31, исходя из пинаколового эфира 5 пентил-бороновой кислоты. Выход=176 мг (45,6%). 1(м, 11 Н), 0,93-0,77 (м, 3 Н). Синтез соединения 36. 5-Метокси-N-[(1-3-[(фенилкарбонил)амино]пропилпиперидин-4 ил)метил]-1 Н-индазол-3-карбоксамид. 36 а) трет-Бутил 3-[4-([(5-метокси-1 Н-индазол-3-ил)карбонил]аминометил)пиперидин-1 ил]пропилкарбамат. Раствор соединения 24b (1,37 г, 4,36 ммоль) в ДМФА (45 мл) и триэтиламине (1,3 мл, 9,5 ммоль) перемешивали при температуре 80 С в течение 1 ч и затем обрабатывали трет-бутил (3 бромпропил)карбаматом (1,7 г, 7,1 ммоль). Смесь перемешивали в течение ночи при той же температуре. Реакционную смесь затем охлаждали до комнатной температуры и растворитель удаляли путем выпаривания при пониженном давлении. Сырой трет-бутил 3-[4-([(5-метокси-1 Н-индазол-3 ил)карбонил]аминометил)пиперидин-1-ил]пропилкарбамат 36 а использовали на следующей стадии без дополнительной очистки. ЖХ-МС: 446,3 (М+Н)+. 36b) N-[1-(3-аминопропил)пиперидин-4-ил]метил-5-метокси-1 Н-индазол-3-карбоксамид. Раствор сырого трет-бутил 3-[4-([(5-метокси-1 Н-индазол-3-ил)карбонил]аминометил)пиперидин-1 ил]пропилкарбамата 36 а (приблизительно 1,8 г) в CH2Cl2 (15 мл) обрабатывали трифторуксусной кислотой (7 мл) при комнатной температуре в течение ночи. Раствор затем выливали в воду (50 мл) и промывалиCH2Cl2 (320 мл). Кислотную фазу подщелачивали и концентрировали при пониженном давлении. Твердый остаток экстрагировали смесью CH3Cl/СН 3 ОН при соотношении 8/2 (320 мл) и растворитель выпари- 20024939 вали при пониженном давлении. Сырой N-[1-(3-аминопропил)пиперидин-4-ил]метил-5-метокси-1 Ниндазол-3-карбоксамид 36b использовали на следующих стадиях без дополнительной очистки. ЖХ-МС: 34 6,2 (М+Н)+. К раствору сырого N-[1-(3-аминопропил)пиперидин-4-ил]метил-5-метокси-1 Н-индазол-3 карбоксамида 36b (приблизительно 350 мг, 1 ммоль) в ДМСО (1,5 мл) и CH2Cl2 (10 мл) добавляли бензоилхлорид (71 мкл, 0,61 ммоль). Раствор затем перемешивали при комнатной температуре в течение 2 ч. Смесь затем добавляли в воду (20 мл) и экстрагировали CH2Cl2 (310 мл). Объединенные органические фазы концентрировали при пониженном давлении и сырой продукт очищали с помощью флэшхроматографии на силикагеле, используя смесь CHCl3/СН 3 ОН=9: 1 в качестве элюента. Получали 5 метокси-N-[(1-3-[(фенилкарбонил)амино]пропилпиперидин-4-ил)метил]-1 Н-индазол-3-карбоксамид 36N-(1-[3-(бутаноиламино)пропил]пиперидин-4-илметил)-5-метокси-1 Н-индазол-3-карбоксамид 37 получали в соответствии со способом, описанным для соединения 36, исходя из бутаноилхлорида. Выход=7 5 мг (36,8%). 1N-[(1-3-[(2 Е)-бут-2-еноиламино]пропилпиперидин-4-ил)метил]-5-метокси-1 Н-индазол-3 карбоксамид 38 получали в соответствии со способом, описанным для соединения 36, исходя из (2 Е)бут-2-еноилхлорида. Выход=45 мг (51,4%). 1(м, 1 Н), 3,80 (с, 3 Н), 3,50-1,00 (м, 20 Н). ЖХ-МС: 414,25 (М-Н+). Синтез соединения 39. 5-Метокси-N-(1-[3-(пропаноиламино)пропил]пиперидин-4-илметил)-1 Ниндазол-3-карбоксамид. 5-Метокси-N-(1-[3-(пропаноиламино)пропил]пиперидин-4-илметил)-1 Н-индазол-3-карбоксамид 39 получали в соответствии со способом, описанным для соединения 36, исходя из пропаноилхлорида. Выход=90 мг (68,8%). 1N-(1-[3-(бут-2-иноиламино)пропил]пиперидин-4-илметил)-5-метокси-1 Н-индазол-3-карбоксамид 40 получали в соответствии со способом, описанным для соединения 36, исходя из 2-бутиноилхлорида. Выход=17 мг (17,1%). 1-78 С, медленно добавляли раствор 1 М BBr3 в CH2Cl2 (2,2 мл, 2,2 ммоль) (примерно 1 ч). Смеси давали достичь комнатной температуры и перемешивали при данной температуре в течение 2 дней. Смесь затем выливали в насыщенный раствор NaHCO3 и экстрагировали CH2Cl2 (3100 мл). Щелочную фазу подкисляли 1 н. HCl и концентрировали при пониженном давлении. Остаток затем обрабатывали ДМСО (6 мл) и[4-([(5-бром-6-гидрокси-1 Н-индазол-3-ил)карбонил]аминометил)пиперидин-1-ил]уксусную кислоту 41 очищали, используя следующие параметры препаративной ВЭЖХ: канал А=CH3CN+0,1%-ная муравьи- 21024939[4-([(5-Бром-6-метокси-1 Н-индазол-3-ил)карбонил]аминометил)пиперидин-1-ил]уксусную кислоту 42 получали с помощью стадии очистки, описанной для получения соединения 41. Выход 35 мг. 1H ЯМР (300 МГц, ДМСО-d6)= 13,66 (уш.с, 1 Н), 8,45 (т, J=6,1 Гц, 1 Н), 8,30 (с, 1 Н), 7,15 (с, 1 Н),6,80-4,69 (м, 1 Н), 3,93 (с, 3 Н), 3,33-3,10 (м, 6 Н), 2,64 (т, J=11,2 Гц, 2 Н), 1,75 (д, J=10,9 Гц, 3 Н), 1,43 (кв,J=11,6 Гц, 2 Н). Синтез соединения 44. Этил [4-([(5-метокси-1 Н-индазол-3-ил)карбонил]аминометил)пиперидин 1-ил]ацетат. Этил[4-([(5-метокси-1 Н-индазол-3-ил)карбонил]аминометил)пиперидин-1-ил]ацетат получали в соответствии со способом, описанным для соединения 25 с, исходя из гидрохлорида 5-метокси-N(пиперидин-4-илметил)-1 Н-индазол-3-карбоксамида 24b (65%). 1H ЯМР (300 МГц, ДМСО-d6)= 13,54 (с, 1 Н), 8,30 (т, J=6,06 Гц, 1 Н), 7,56 (с, 1 Н), 7,51 (д, J=8,91 Гц, 1 Н), 7,05 (д, J=8,91 Гц, 1 Н), 4,00 (кв, J=7,13 Гц, 2 Н), 3,81 (с, 3 Н), 3,34 (с, 2 Н), 3,17-3,24 (м, 2 Н), 2,812,95 (м, 4 Н), 1,50-1,75 (м, 3 Н), 1,16-1,36 (м, 2 Н), 1,11 (т, J=7,13 Гц, 3 Н). ЖХ-МС: 375,2 (М-Н+). В следующей далее табл. 1 А представлены химические названия и структуры описанных выше соединений 20-44. Таблица 1 А Фармакологические свойства. Фармакологические свойства соединений формулы (I), которые могут быть использованы по настоящему изобретению, оценивались с помощью способов, описанных в следующих разделах. Исследование I. Активность на человеческой GSK-3 (исследование in vitro). Активность на человеческой GSK-3 оценивали с использованием следующих способов (в соответствии с Meijer et al., Chem. Biol., 2003-10:1255-1266). В первом скрининговом анализе соединения были испытаны двукратно при концентрации 10 мкМ. Человеческий рекомбинантный фермент GSK-3 инкубировали в течение 90 мин при 22 С в присутствии соединений или инертного наполнителя в реакционном буфере, содержащем АТФ плюс 100 нМ нефосфорилированного специфического субстрата пептида (Ulight-CFFKNIVTPRTPPPSQGK-амид). Фосфорилирование субстрата определяли с помощью технологии LANCE (PerkinElmer, CT, USA). Результаты, представленные в табл. 4, выражены в виде процента ингибирования специфической активности контроля, достигаемого в присутствии исследуемого соединения (как % ингибирования при 10 мкМ). Во втором анализе те же самые соединения анализировали при пяти концентрациях в диапазоне от 100 мкМ до 10 нм при десятикратных разведениях в двух повторениях. Соединения с 1 по 15 были испытаны с помощью того же первого анализа, соединения 16 до 41 были испытаны в другом анализе, основанном на связывании и перемещении меченого AlexaFluor 647 с дизайном ингибитора киназы, конкурентного по отношению к АТФ, с использованием пакета анализа Eu Kinase технологии LanthaScreenTR-FRET Eu в соответствии с инструкцией завода-изготовителя (Life Technologies, Italy). Результаты этих двух анализов сопоставимы. Значения IC50 (концентрация, вызывающая половину максимального ингибирования специфической активности контроля), представленные в табл. 4, были определены с помощью нелинейного регрессионного анализа кривых ингибирования, построенных из средних дублированных значений, используя построение кривой по уравнению Хилла. Результаты показали, что соединения в соответствии с настоящим изобретением обладают хорошей ингибирующей активностью в этом анализе: при 10 мкМ % ингибирования был больше 70%, и значениеIC50 получалось менее 4,00 мкМ для каждого соединения. Большинство соединений показали значениеIC50 ниже, чем 1,50 мкМ. Исследование II. Селективность по отношению к GSK-3 (исследование in vitro).(А) Соединения 1 и 2 были испытаны в отношении состава из 60 киназ, чтобы оценить их селективность. Анализы были выбраны с учетом рассмотрения разнообразных исследуемых семейств. Исследуемые киназы были представлены следующими подсемействами киназ: протеинсерин/треонинкиназы; протеинтирозинкиназы; другие киназы; и атипичные киназы. Человеческие рекомбинантные киназы инкубировали в присутствии специфических пептидных субстратов плюс АТФ в течение различного времени (10, 15, 30, 60 или 90 мин) при 22 С. Фосфорилированный субстрат отслеживали с помощью технологии LANCE или HTRF (CISBIO, MA, USA). Соединения были испытаны при 10 мкМ с повторением. Результаты выражены в виде процента ингибирования специфической активности контроля, достигаемого в присутствии исследуемого соединения, и представлены в следующей далее табл. 5. Результаты подтвердили, что оба исследуемые соединения обладают ингибирующей активностью по отношению к GSK-3 и что они имеют более высокую аффинность к GSK-3 по сравнению с другими киназами, демонстрируя хороший профиль селективности.(b) Соединения 3, 8, 29 и 31 были исследованы в отношении того же состава из 60 киназ в тех же условиях, что описаны выше для соединений 1 и 2. Результаты выражены в виде процента ингибирования специфической активности контроля, достигаемого в присутствии исследуемого соединения, и представлены в следующей далее табл. 6.
МПК / Метки
МПК: A61P 3/00, A61K 31/454, C07D 401/12, C07D 405/14, A61P 25/00, A61P 35/00, C07D 401/14, A61P 29/00, A61K 31/4545
Метки: соединения, ингибиторов, гликогенсинтазы, 1h-индазол-3-карбоксамидные, качестве, 3-бета, киназы
Код ссылки
<a href="https://eas.patents.su/30-24939-1h-indazol-3-karboksamidnye-soedineniya-v-kachestve-ingibitorov-glikogensintazy-kinazy-3-beta.html" rel="bookmark" title="База патентов Евразийского Союза">1h-индазол-3-карбоксамидные соединения в качестве ингибиторов гликогенсинтазы киназы 3-бета</a>
Триазолопиримидиновые производные в качестве ингибиторов киназы-3 гликогенсинтазы

Номер патента: 10109
Опубликовано: 30.06.2008
Авторы: Фрейн Эдди Жан Эдгар, Вандермасен Неле, Лав Кристофер Джон, Эмбрехтс Вернер Констант Йохан, Коиманс Людвиг Поль, Виллемс Марк, Бейнстерс Петер Якобус Йоханнес Антониус
МПК: C07D 487/04
Метки: киназы-3, ингибиторов, триазолопиримидиновые, качестве, производные, гликогенсинтазы
Формула / Реферат:
1. Соединение формулы (I) его N-оксид, его фармацевтически приемлемая аддитивная соль, его четвертичный амин и его стереохимически изомерная форма, где кольцо А обозначает фенильную, пиридильную, пиримидинильную, пиридазинильную или пиразинильную группу; R1 обозначает водород; арильную группу; формильную группу; C1-6алкилкарбонильную группу; C1-6 алкильную группу; C1-6алкилоксикарбонильную группу; C1-6алкильную группу, замещенную формильной,...
Производные триазолопиримидина в качестве ингибиторов киназы гликогенсинтазы-3

Номер патента: 11300
Опубликовано: 27.02.2009
Авторы: Вандермасен Неле, Виллемс Марк, Эмбрехтс Вернер Констант Йохан, Бейнстерс Петер Якобус Йоханнес Антониус, Коиманс Людвиг Поль, Лав Кристофер Джон, Фрейн Эдди Жан Эдгар
МПК: C07D 487/00
Метки: гликогенсинтазы-3, качестве, триазолопиримидина, киназы, производные, ингибиторов
Формула / Реферат:
1. Производные триазолопиримидина формулы и его фармацевтически приемлемая аддитивная соль, где кольцо A представляет собой фенил; R1 представляет собой водород или C1-6алкил; X1 представляет собой прямую связь или -(CH2)n3-, причем n3 означает целое число, равное 1, 2 или 3; R2 представляет собой C3-7циклоалкил; фенил; 6-членный моноциклический гетероцикл, содержащий N, который может быть замещен C1-6-алкоксикарбонилом; или радикал формулы ...
Гетероариламины в качестве ингибиторов гликогенсинтаза-киназы 3-бета (ингибиторов gsk3)

Номер патента: 7298
Опубликовано: 25.08.2006
Авторы: Хэрес Ян, Лав Кристофер Джон, Дильс Гастон Станислас Марселла, Койманс Люсьен Мария Хенрикус, Де Жонж Марк Рене, Ван Акен Кун Жанн Альфонс, Жанссен Поль Адриан Ян, Бейнстерс Петер Якобус Йоханнес Антониус, Винкерс Хендрик Мартен, Виллемс Марк, Леви Паулус Йоаннес, Эмбрехтс Вернер Констант Йохан, Фрейн Эдди Жан Эдгар
МПК: A61K 31/505, A61K 31/50, A61K 31/435...
Метки: 3-бета, ингибиторов, качестве, гликогенсинтаза-киназы, гетероариламины, gsk3
Формула / Реферат:
1. Соединение формулы его N-оксид, фармацевтически приемлемая аддитивная соль, четвертичный амин и стереохимически изомерная форма, где R1 обозначает водород; X обозначает -O-; -О-С1-4алкил- или прямую связь; Z обозначает прямую связь, -NH-, -С1-4алкил-NH- или -С(=O)-; R2 обозначает водород или фенил; R3 обозначает водород, С1-4алкил, полигалогенC1-6алкил или циано; R4 обозначает моноциклический или бициклический насыщенный гетероцикл;...
Гетероариламины-производные пиримидина и пиридазина в качестве ингибиторов гликогенсинтаза-киназы 3-бета (ингибиторов gskз)

Номер патента: 10859
Опубликовано: 30.12.2008
Авторы: Ван Акен Кун Жанн Альфонс, Де Жонж Марк Рене, Бейнстерс Петер Якобус Йоханнес Антониус, Леви Паулус Йоаннес, Лав Кристофер Джон, Виллемс Марк, Винкерс Хендрик Мартен, Хэрес Ян, Дильс Гастон Станислас Марселла, Фрейн Эдди Жан Эдгар, Эмбрехтс Вернер Констант Йохан, Койманс Люсьен Мария Хенрикус, Жанссен Поль Адриан Ян
МПК: A61K 31/50, A61K 31/435, A61K 31/505...
Метки: 3-бета, ингибиторов, качестве, gskз, пиридазина, пиримидина, гликогенсинтаза-киназы, гетероариламины-производные
Формула / Реферат:
1. Соединение формулы его N-оксид, фармацевтически приемлемая аддитивная соль, четвертичный амин и стереохимически изомерная форма, где кольцо А является R1 обозначает водород; X обозначает -O-; -O-C1-6алкил- или прямую связь; Z обозначает прямую связь, -С(=O)- или -NR1-C1-6алкил-; R обозначает водород или R20; R3 обозначает водород; галоген; циано; полигалогенС1-6алкил; R4 обозначает моноциклический, насыщенный или частично насыщенный или...
Производные амидов в качестве ингибиторов гликогенсинтаза-киназы 3-бета

Номер патента: 7578
Опубликовано: 29.12.2006
Авторы: Де Жонж Марк Рене, Эмбрехтс Вернер Констант Йохан, Фрейн Эдди Жан Эдгар, Жанссен Поль Адриан Ян, Гертс Юго Альфонс Габриель, Лудовики Дональд Вилльям, Хэрес Ян, Леви Паулус Йоаннес, Бейнстерс Петер Якобус Йоханнес Антониус, Виллемс Марк, Дайер Фредерик Франс Дезире, Койманс Люсьен Мария Хенрикус, Лакрамп Жан Фернан Арман, Нюйденс Рони Мария, Кукла Майкл Джозеф, Меркен Марк Юбер
МПК: A61P 25/28, A61P 3/10, A61K 31/505...
Метки: амидов, гликогенсинтаза-киназы, ингибиторов, 3-бета, производные, качестве
Формула / Реферат:
1. Соединение формулы его N-оксид, фармацевтически приемлемая аддитивная соль, четвертичный амин и стереохимически изомерная форма, где R1 означает водород; X означает -O-; -O-С(=O)-NR1-; -O-C1-6алкил-; -О-С2-6алкенил-; -O-C1-6алкил-О- или прямую связь; R2 означает водород, C1-10алкил, R20, причем каждая из указанных групп, представляющих R2, может быть необязательно замещена, где возможно, одним или несколькими заместителями, каждый из...