Производные хиназолинона в качестве ингибиторов parp
Номер патента: 12837
Опубликовано: 30.12.2009
Авторы: Мертенс Йозефус Каролус, Кенни Людо Эдмон Жозефин, Ван Дюн Якобус Альфонсус Йозефус, Гийемон Жером Эмиль Жорж, Ваутерс Вальтер Баудевейн Леопольд, Сомерс Мария Викторина Франциска
















Формула / Реферат
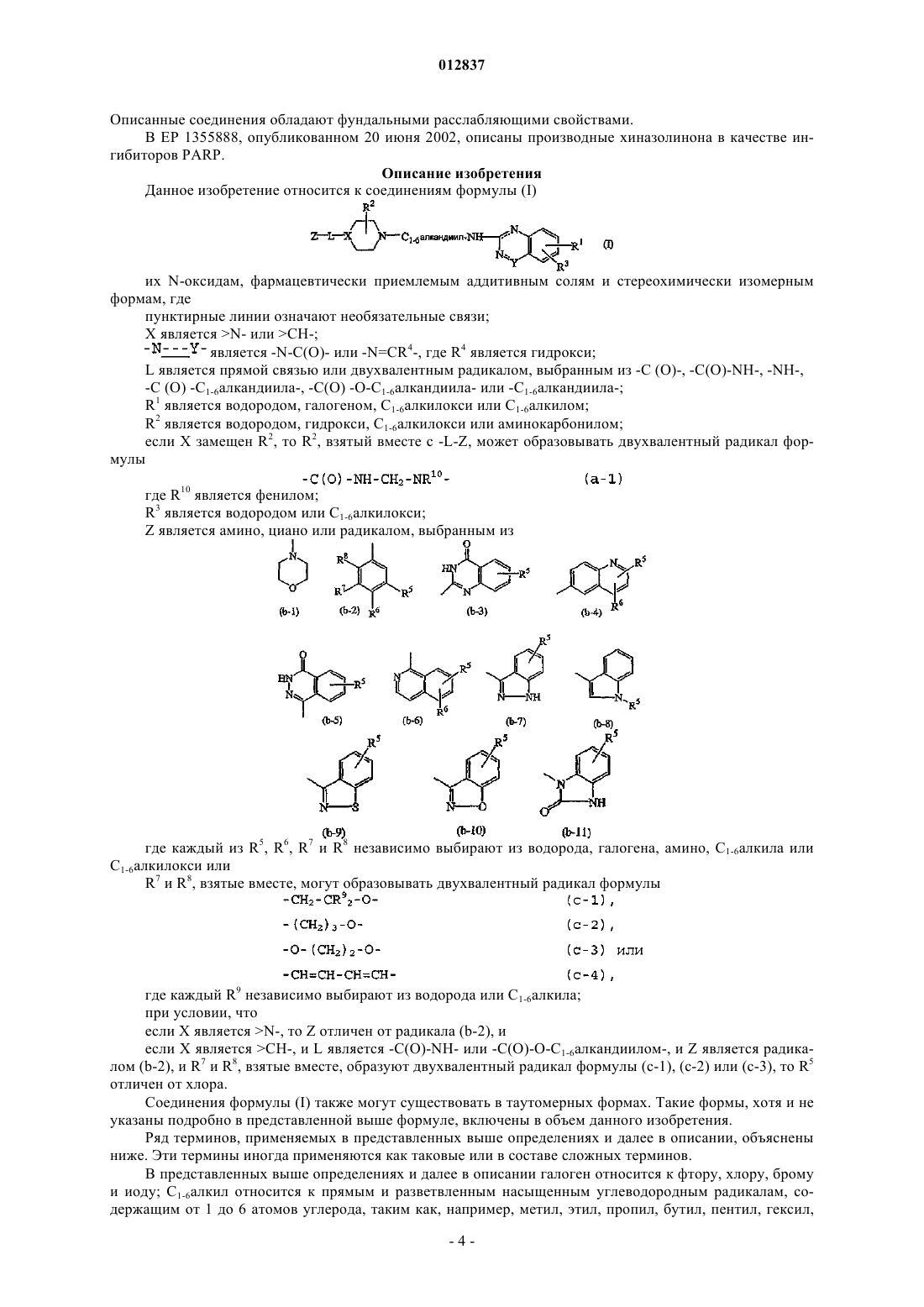
1. Соединение формулы (I)

его N-оксиды, фармацевтически приемлемые аддитивные соли и стереохимически изомерные формы, где
пунктирные линии означают необязательные связи;
X является >N- или >СН-;
![]() является -N-C(O)- или -N=CR4-, где R4 является гидрокси;
является -N-C(O)- или -N=CR4-, где R4 является гидрокси;
L является прямой связью или двухвалентным радикалом, выбранным из -С(О)-, -C(O)-NH-, -NH-,
-С(О)-O-С1-6алкандиила- или -C1-6алкандиила-;
R1 является водородом, галогеном, C1-6алкилокси или C1-6алкилом;
R2 является водородом, гидрокси или C1-6алкилокси;
R3 является водородом или C1-6алкилокси;
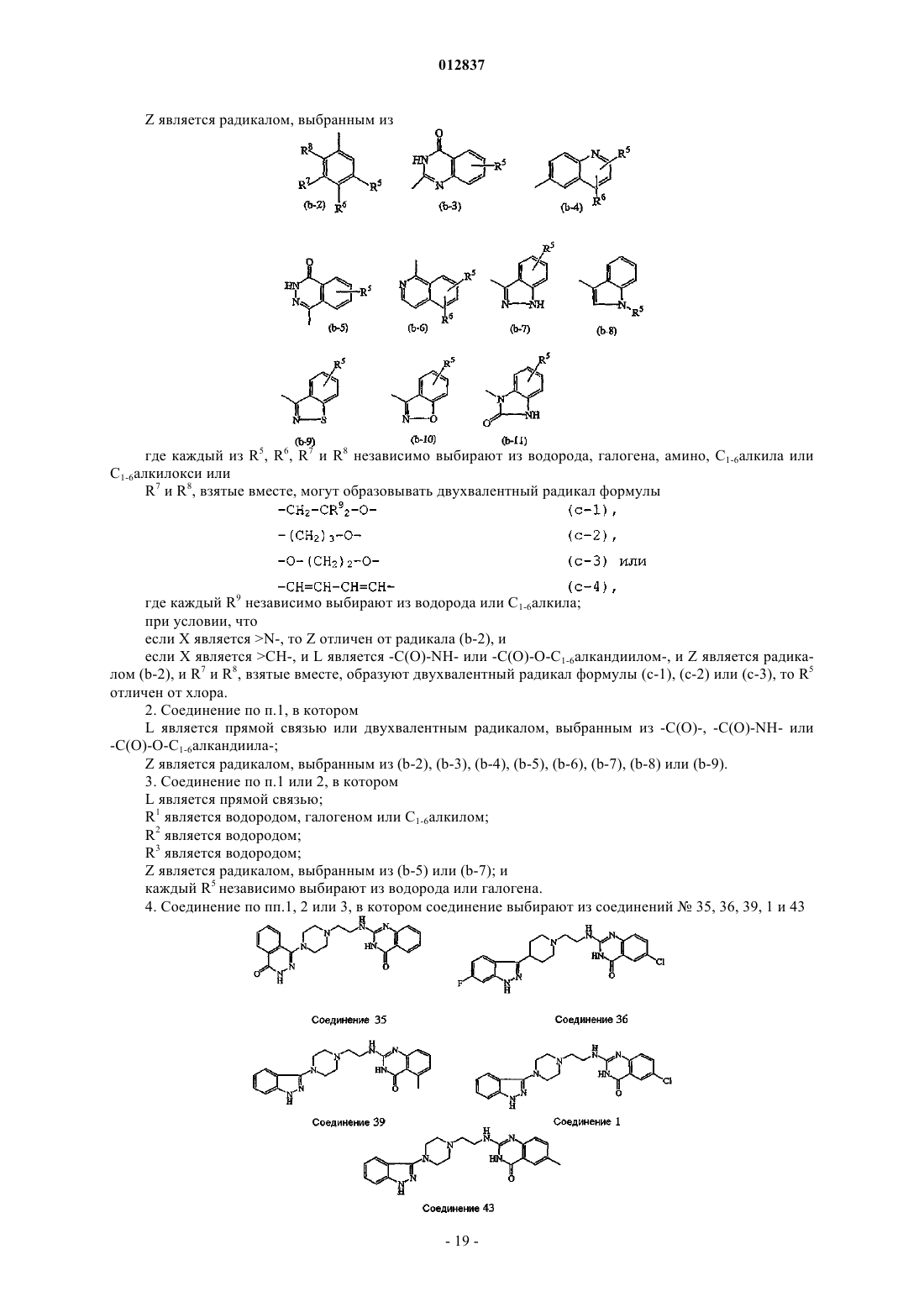
Z является радикалом, выбранным из

где каждый из R5, R6, R7 и R8 независимо выбирают из водорода, галогена, амино, C1-6алкила или
C1-6алкилокси или
R7 и R8, взятые вместе, могут образовывать двухвалентный радикал формулы

где каждый R9 независимо выбирают из водорода или C1-6алкила;
при условии, что
если X является >N-, то Z отличен от радикала (b-2), и
если X является >СН-, и L является -C(O)-NH- или -С(O)-O-C1-6алкандиилом-, и Z является радикалом (b-2), и R7 и R8, взятые вместе, образуют двухвалентный радикал формулы (с-1), (с-2) или (с-3), то R5 отличен от хлора.
2. Соединение по п.1, в котором
L является прямой связью или двухвалентным радикалом, выбранным из -С(О)-, -С(О)-NH- или
-С(О)-O-C1-6алкандиила-;
Z является радикалом, выбранным из (b-2), (b-3), (b-4), (b-5), (b-6), (b-7), (b-8) или (b-9).
3. Соединение по п.1 или 2, в котором
L является прямой связью;
R1 является водородом, галогеном или C1-6алкилом;
R2 является водородом;
R3 является водородом;
Z является радикалом, выбранным из (b-5) или (b-7); и
каждый R5 независимо выбирают из водорода или галогена.
4. Соединение по пп.1, 2 или 3, в котором соединение выбирают из соединений ь 35, 36, 39, 1 и 43


5. Соединение по п.1, в котором Z является радикалом, выбранным из (b-3), (b-4), (b-5), (b-6), (b-7), (b-8), (b-9), (b-10) или (b-11).
6. Применение соединения по любому из пп.1-5 в качестве лекарственного средства.
7. Фармацевтическая композиция, содержащая фармацевтически приемлемые носители и в качестве активного ингредиента терапевтически эффективное количество соединения по любому из пп.1-5.
8. Способ получения фармацевтической композиции по п.7, где фармацевтически приемлемые носители и соединение по любому из пп.1-5 тщательно смешивают.
9. Применение соединения для производства лекарственного средства для лечения заболевания, медиированного TANK и PARP, в котором указанным соединением является соединение формулы (I)

его N-оксиды, фармацевтически приемлемые аддитивные соли и стереохимически изомерные формы, где
пунктирные линии означают необязательные связи;
X является >N- или >СН-;
![]() является -N-C(O)- или -N=CR4-, где R4 является гидрокси;
является -N-C(O)- или -N=CR4-, где R4 является гидрокси;
L является прямой связью или двухвалентным радикалом, выбранным из -С(О)-, -C(O)-NH-, -NH-,
-С(О)-O-C1-6алкандиила- или -C1-6алкандиила-;
R1 является водородом, галогеном, C1-6алкилокси или C1-6алкилом;
R2 является водородом, гидрокси или C1-6алкилокси;
R3 является водородом или C1-6алкилокси;
Z является радикалом, выбранным из

где каждый из R5, R6, R7 и R8 независимо выбирают из водорода, галогена, амино, C1-6алкила или
C1-6алкилокси или
R7 и R8, взятые вместе, могут образовывать двухвалентный радикал формулы

где каждый R9 независимо выбирают из водорода или C1-6алкила.
10. Применение соединения по любому из пп.1-5 для производства лекарственного средства для ингибирования роста раковых клеток.
11. Применение по п.9 или 10, где лечение включает химиосенсибилизацию.
12. Применение по п.9 или 10, где лечение включает радиосенсибилизацию.
13. Комбинация соединения формулы (I) по любому из пп.1-5 с химиотерапевтическим агентом.
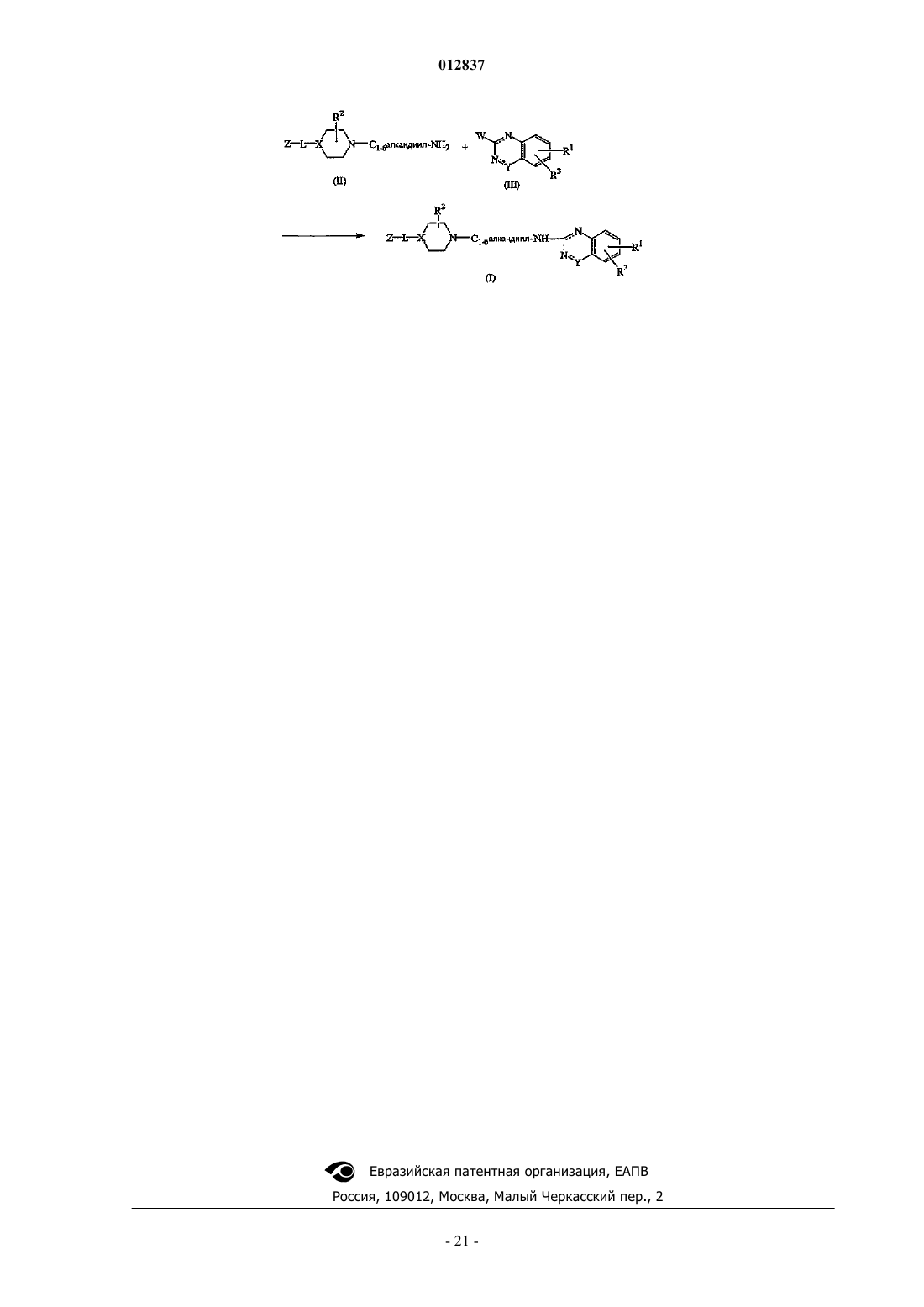
14. Способ получения соединения по п.1, отличающийся взаимодействием промежуточного соединения формулы (II) с промежуточным соединением формулы (III), в котором W является подходящей уходящей группой, с получением соединения формулы (I-а), где L1 является -С1-6алкандиил-NH- и обе пунктирные линии могут быть связями, в инертном к реакции растворителе с добавлением соответствующего основания

Текст
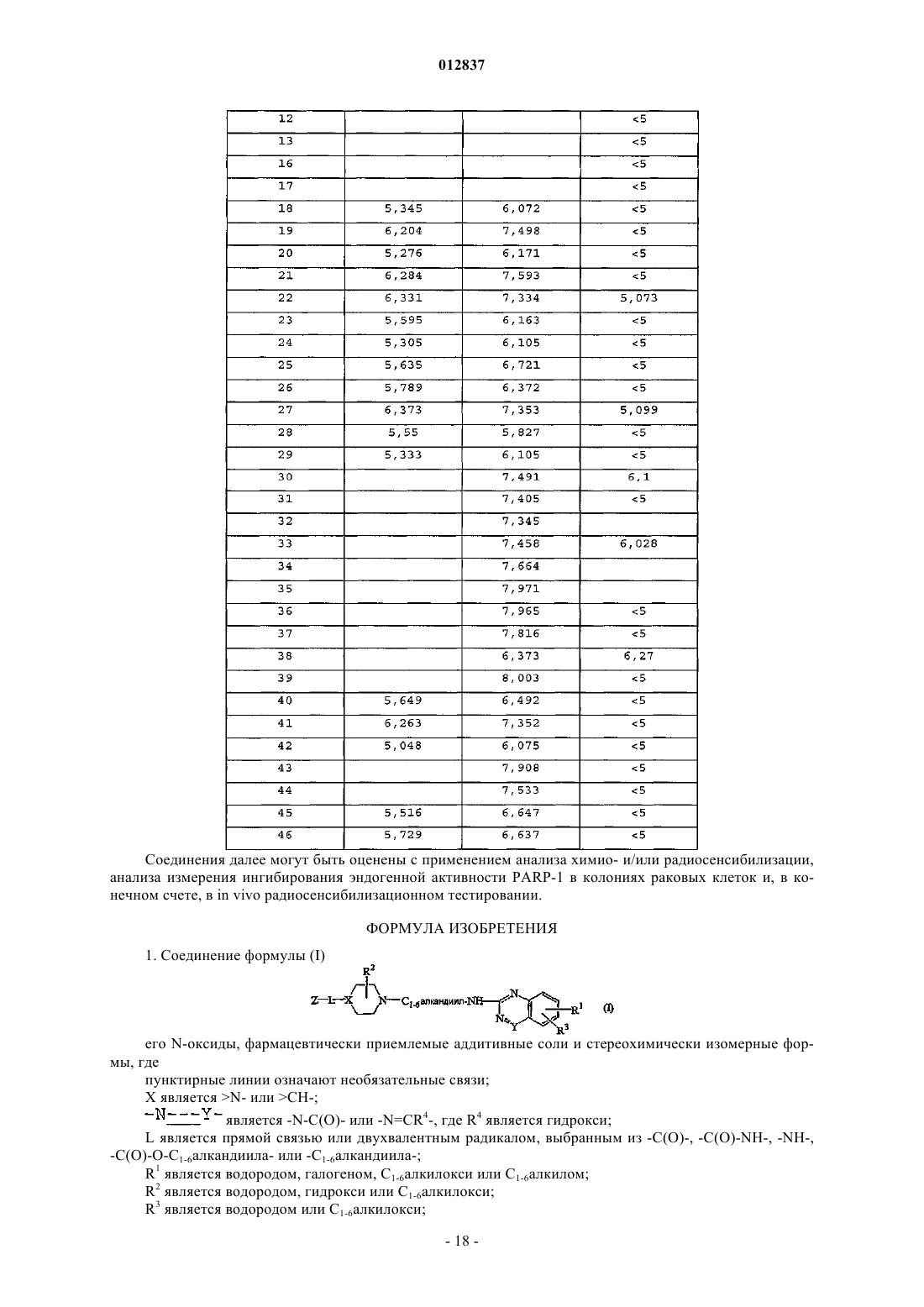
012837 Область техники Данное изобретение относится к ингибиторам PARP и представляет соединения и композиции, содержащие раскрытые соединения. Далее данное изобретение представляет способы применения раскрытых ингибиторов PARP, например, в качестве лекарственного средства. Предшествующий уровень техники Ядерный фермент поли(ADP-рибоза)полимераза-1 (PARP-1) представляет собой член семейства ферментов PARP. Это увеличивающееся семейство ферментов состоит из таких PARP, как, например,PARP-1, PARP-2, PARP-3 и Vault-PARP, и танкираз (TANK), таких как, например, TANK-1, TANK-2 иTANK-3. PARP также обозначают как поли(аденозин 5'-дифосфорибоза)полимераза или PARSPARP-1 является основным ядерным белком 116 кДа, состоящим из трех доменов: N-концевого ДНК-связывающего домена, содержащего два цинковых пальца, аутомодификационного домена и С-концевого каталитического домена. Он присутствует практически во всех эукариотах. Фермент синтезирует поли(ADP-рибозу), разветвленный полимер, который может состоять из около 200 единицADP-рибозы. Белковые акцепторы поли(ADP-рибозы) непосредственно или опосредованно вовлечены в сохранение целостности ДНК. Они включают гистоны, топоизомеразы, ДНК и РНК полимеразы, ДНК лигазы и Са 2+- и Mg2+-зависимые эндонуклеазы. PARP белок экспрессируется в значительных количествах во многих тканях, наиболее заметно в иммунной системе, сердце, мозге и линиях зародышевых клеток. В нормальных физиологических условиях активность PARP минимальна. Однако повреждение ДНК вызывает немедленную активацию PARP практически в 500-кратном размере. Танкиразы (TANK) идентифицированы как компоненты человеческого теломерного комплекса. Было предположено, что они также играют роль в транспорте везикул и могут служить в качестве каркаса для белков, вовлеченных в различные другие клеточные процессы. Теломеры, которые являются существенными для сохранения и стабильности хромосом, сохраняются теломеразой, специализированной обратной транскриптазой. TANK представляют собой (ADP-рибоза)трансферазы, которые имеют некоторые характеристики как сигнальных, так и цитоскелетных белков. Они содержат PARP домен, который катализирует поли-ADP-рибозилирование субстратных белков, стерильного альфа-мотива, общего с некоторыми сигнальными молекулами, и ANK-домен, который содержит 24 повтора анкирина, которые являются гомологами цитоскелетного белка анкирина. ANK-домен взаимодействует с теломерным белком, связывающим теломерный повтор фактором-1 (TRF-1). Поэтому эти белки названы TRF1 взаимодействующими, анкирин-родственными ADP-рибоза-полимеразами (TANK). Одной из наиболее специфичных функций TANK является ADP-рибозилирование TRF-1. Функция человеческого теломера требует два теломер-специфических ДНК-связывающих белка, TRF-1 и TRF-2.TRF-2 защищает концы хромосом и TRF-1 регулирует длину теломера. ADP-рибозилирование ингибирует способность TRF-1 связываться с теломерной ДНК. Такое поли-ADP-рибозилирование TRF-1 высвобождает TRF-1 из теломеров, открывая теломерный комплекс и обеспечивая доступ к теломеразе. Поэтому TANK действует в качестве положительного регулятора длины теломера, позволяя удлинение теломеров посредством теломеразы. Среди многих функций, приписываемых PARP, и особенно PARP-1, имеется его основная роль в способствовании репариции ДНК путем ADP-рибозилирования и, следовательно, координирования ряда белков репарации ДНК. В результате активации PARP значительно уменьшаются уровни NAD+. Продолжительная активация PARP ведет к тяжелому истощению NAD+ в клетках, страдающих от значительного повреждения ДНК. Короткий период полураспада поли(ADP-рибозы) дает быструю интенсивность кругооборота. Как только поли(ADP-рибоза) сформируется, она быстро деградирует с помощью конститутивно активной поли(ADP-рибозы)гликогидролазы (PARG), вместе с фосфодиэстеразой иPARP и PARG образуют цикл, который превращает большое количество NAD+ в ADP-рибозу. Менее чем за час чрезмерное стимулирование PARP может привести к падению NAD+ и АТФ менее чем до 20% от нормального уровня. Такой сценарий особенно пагубен при ишемии, когда потеря кислорода уже чрезмерно подрывает выход клеточной энергии. Дальнейшее образование свободных радикалов во время реперфузии считается основной причиной повреждения тканей. Некоторое падение АТФ, которое типично во многих органах во время ишемии и реперфузии, может быть связано с уменьшением NAD+ вследствие круговорота поли(ADP-рибозы). Таким образом, ожидается, что ингибирование PARP иPARG сохраняет уровень клеточной энергии, тем самым усиливая выживание ишемических тканей после инсульта. Синтез поли(ADP-рибозы) также вовлечен в индуцированную экспрессию ряда генов, существенных для воспалительной реакции. Ингибиторы PARP подавляют вырабатывание индуцибельной синтазы окиси азота (iNOS) в макрофагах, селектина Р-типа и молекулы-1 межклеточной адгезии (ICAM-1) в эндотелиальных клетках. Такая активность лежит в основе сильного противовоспалительного действия,вызываемого ингибиторами PARP. Ингибирование PARP способно снижать некроз путем предотвращения транслокации и инфильтрации нейтрофилов в поврежденных тканях.PARP активируется поврежденными фрагментами ДНК и, при активации, катализирует присоеди-1 012837 нение вплоть до 100 единиц ADP-рибозы к множеству ядерных белков, включая гистоны и сами PARP. Во время основных клеточных стрессов значительная активация PARP может быстро привести к повреждению или смерти клеток из-за истощения запасов энергии. Так как четыре молекулы АТФ используются для каждой регенерированной молекулы NAD+, NAD+ истощается при значительной активацииPARP, при попытках повторно синтезировать NAD+ АТФ также может истощиться. Было описано, что активация PARP играет ключевую роль как в NMDA-, так и NO-вызванной нейротоксичности. Это было продемонстрировано в корковых культурах и в гиппокампальных срезах, где предупреждение токсичности прямо соотносится с эффективностью ингибирования PARP. Так была определена потенциальная роль ингибиторов PARP в лечении нейродегенеративных заболеваний и травм головы, даже если точный механизм действия все еще не был выяснен. Так же было продемонстрировано, что единичные инъекции ингибиторов PARP снижают размер инфаркта, вызванного ишемией и реперфузией сердца или скелетных мышц у кроликов. В этих исследованиях единичная инъекция 3-аминобензамида (10 мг/кг), либо за минуту до окклюзии, либо за минуту до реперфузии, вызывает подобное снижение размера инфаркта в сердце (32-42%), в то время как 1,5-дигидроксиизохинолин (1 мг/кг), другой ингибитор PARP, снижает размер инфаркта в сравнимой степени (38-48%). Эти результаты позволяют сделать разумное предположение, что ингибиторы PARP могут спасти ранее ишемическое сердце или поврежденную реперфузией ткань скелетных мышц. Активация PARP также может применяться в качестве меры повреждения после нейротоксичного инсульта в результате воздействия любого из следующих индукторов, таких как глутамат (через стимулирование рецептора NMDA), реакционноспособные промежуточные соединения кислорода, амилоидного -белка, N-метил-4-фенил-1,2,3,6-тетрагидропиридин (МРТР) или его активный метаболитN-метил-4-фенилпиридин (МРР+), которые участвуют в патологических состояниях, таких как удар, болезнь Альцгеймера и болезнь Паркинсона. Другие исследования были продолжены для анализа роли активации PARP в мозжечковых нервных клетках-зернах in vitro и в МРТР нейротоксичности. Избыточное воздействие на нервы глутамата, который служит преобладающим нейротрансмиттером центральной нервной системы и действует на рецепторы N-метил-D-аспартата (NMDA) и рецепторов других подтипов, наиболее часто возникает в результате удара или других нейродегенеративных процессов. Лишенные кислорода нейроны выделяют глутамат в больших количествах во время ишемического инсульта мозга, например во время удара или сердечного приступа. Такое избыточное выделение глутамата, в свою очередь, вызывает чрезмерное стимулирование (эксцитотоксичность) N-метил-D-аспартата(NMDA), АМРА, Каината и MGR рецепторов, которые открывают ионные каналы и вызывают неконтролируемый поток ионов (например, Са 2+ и Na+ в клетки и K+ из клеток), что приводит к чрезмерному стимулированию нейронов. Чрезмерно стимулированные нейроны выделяют больше глутамата, создавая контур обратной связи или эффект домино, который в конце концов вызывает повреждение или смерть клеток из-за образования протеаз, липаз и свободных радикалов. Избыточная активация рецепторов глутамата вовлечена в различные неврологические заболевания и состояния, включая эпилепсию, удар, болезнь Альцгеймера, болезнь Паркинсона, боковой амиотрофический склероз (ALS), болезнь Хантингтона, шизофрению, хроническую боль, ишемию и потерю нейронов вследствие гипоксии, гипогликемии,ишемии, травмы и повреждения нервов. Воздействие глутамата и стимулирование также рассматривается как основа для навязчивых расстройств, особенно зависимости от лекарственных средств. Доказательства включают открытия, сделанные для многих видов животных, а также для культур коры головного мозга, подвергнутых воздействию глутамата или NMDA, о том, что антагонисты рецептора глутамата(т.е. соединения, которые блокируют связывание глутамата или активацию его рецептора) блокируют повреждение нервов вследствие инсульта сосудов. Попытки предотвратить токсичность ex cito блокированием NMDA, АМРА, каината и MGR рецепторов оказались трудными, так как каждый рецептор имеет множество мест, с которыми может связываться глутамат, и, следовательно, поиск эффективной смеси антагонистов или универсального антагониста, предотвращающего связывание глутамата со всеми рецепторами и позволяющего проверить эту теорию, затруднен. Более того, многие композиции, которые эффективны при блокировании рецепторов, также токсичны для животных. В настоящее время не существует известного эффективного лечения глутаматных аномальностей. Стимулирование рецепторов NMDA глутаматом, например, активирует фермент синтазу нейронной окиси азота (nNOS), что приводит к образованию окиси азота (NO), которая также медиирует нейротоксичность. NMDA нейротоксичность может быть предотвращена обработкой ингибиторами синтазы окиси азота (NOS) или через целевое генетическое разрушение nNOS in vitro. Другим применением ингибиторов PARP является лечение повреждений периферийных нервов и результирующего патологического болевого синдрома, известного как невропатическая боль, такая как боль, вызываемая хроническим повреждением сокращений (CCI) общего седалищного нерва, и при котором происходит транссинаптическое изменение спинного роговидного отростка спинного мозга, характеризуемое возникновением гиперхроматоза цитоплазмы и нуклеоплазмы (так называемые темные нейроны). Также существует доказательство того, что ингибиторы PARP полезны для лечения воспалительного заболевания кишечника, такого как колит. Более конкретно, колит вызывают у крыс внутрипросвет-2 012837 ным введением гаптена тринитробензола сульфоновой кислоты в 50% этаноле. Обработанные крысы получают 3-аминобензамид, специфичный ингибитор активности PARP. Ингибирование активности PARP снижает воспалительную реакцию и восстанавливает морфологию и энергетический статус дистальной части толстой кишки. Другие доказательства позволяют предположить, что ингибиторы PARP пригодны для лечения артрита. Далее похоже, что ингибиторы PARP пригодны для лечения диабетов. Было показано, что ингибиторы PARP пригодны для лечения эндотоксического шока или септического шока. Ингибиторы PARP также применяют для продления ресурса и пролиферативной способности клеток, включая лечение заболеваний, таких как старение кожи, болезнь Альцгеймера, атеросклероз, остеоартрит, остеопороз, мышечная дистрофия, дегенеративные заболевания скелетных мышц, включая репликативное старение, возрастную дегенерацию мышц, иммунное старение, СПИД и другие иммунные возрастные заболевания; и для изменения экспрессии генов в стареющих клетках. Также известно, что ингибиторы PARP, такие как 3-аминобензамид, действуют на общую репарацию ДНК в ответ, например, на перекись водорода или ионизирующее излучение. Основная роль PARP в репарации оборванных цепей ДНК хорошо известна, особенно если вызывается непосредственно ионизирующим излучением или опосредованно после ферментной репарации повреждений ДНК, вызванных метилирующими агентами, ингибиторами топоизомеразы I и другими химиотерапевтическими агентами, такими как цисплатин и блеомицин. Множество исследований с применением нокаутированных мышей, моделей трансдоминантного ингибирования (сверхэкспрессии ДНКсвязывающего домена), антисмысловых ингибиторов и ингибиторов с низким молекулярным весом продемонстрировали роль PARP в репарации и выживании клеток после повреждения ДНК. Ингибирование ферментативной активности, опосредованной PARP, должно привести к усилению чувствительности опухолевых клеток к повреждающему ДНК лечению. Было показано, что ингибиторы PARP являются эффективными при радиосенсибилизации (гипоксических) опухолевых клеток и являются эффективными в предотвращении восстановления опухолевых клеток после потенциально летального и сублетального повреждения ДНК после радиационной терапии,предположительно благодаря их способности предотвращать воссоединение разрывов цепей ДНК и влиять на некоторые сигнальные пути повреждения ДНК. Ингибиторы PARP применялись для лечения рака. Кроме того, в патенте США 5177075 описано несколько изохинолинов, применяемых для усиления летального воздействия ионизирующего излучения или химиотерапевтических агентов на опухолевые клетки. В "Effect of 6-(5-Phenanthridinone), an Inhibitorof Poly(ADP-ribose)Polymerase, on Cultured Tumor Cells", Oncol. Res., 6:9, 399-403 (1994) обсуждается ингибирование активности PARP, снижение пролиферации опухолевых клеток и значительный синергетический эффект при совместной обработке опухолевых клеток с алкилирующим лекарственным средством. Обзоры известного уровня техники представлены у Li and Zhang in IDrugs, 2001, 4 (7): 804-812, byAme et al. in Bioassays, 2004, 26: 882-883 and by Nguewa et al., in Progress in BiophysicMolecular Biology, 2005, 88: 143-172. До сих пор существует необходимость в эффективных и мощных ингибиторах PARP, более конкретно в ингибиторах PARP-1, которые дают минимальные побочные эффекты. В данном изобретении представлены соединения, композиции и способы ингибирования активности PARP для лечения рака и/или профилактики повреждения клеток, тканей и/или органов вследствие повреждения или смерти клеток, вызванных, например, некрозом или апоптозом. Соединения и композиции в соответствии с данным изобретением особенно полезны для усиления эффективности химиотерапии и радиотерапии, где первичным эффектом лечения является повреждение ДНК в целевых клетках. Обзор известного уровня техники В GB 1062357, опубликованном 22 марта 1967, описаны производные хиназолона, обладающие антигипертензивным действием. В DE 2258561, опубликованном 20 июня 1973, описаны замещенные производные пиридинона с антигипертензивным действием. В ЕР 13612, опубликованном 11 ноября 1983, описаны замещенные производные пиперидинилалкилхиназолина. Описанные соединения являются антагонистами серотонина. В ЕР 669919, опубликованном 9 июня 1994, описаны диметилбензофураны и диметилбензопираны в качестве антагонистов 5-НТ 3. Более конкретно, описаны соединения 8, 4, 5, 10, 11, 12, 13, 15, 16,17 и 14 настоящей заявки. В US 5374637, опубликованном 20 декабря 1994, описаны производные бензамида. Описанные соединения обладают свойствами, стимулирующими желудочно-кишечную моторику. В частности, описаны соединения 8, 6 и 9 настоящей заявки. В ЕР 885190, опубликованном 23 декабря 1998, описаны 1,4-дизамещенные производные пиперидина, обладающие гастрокинетическими свойствами. В частности, описано соединение 7 настоящей заявки. В ЕР 1036073, опубликованном 17 июня 1999, описаны замещенные производные хиназолиндиона.-3 012837 Описанные соединения обладают фундальными расслабляющими свойствами. В ЕР 1355888, опубликованном 20 июня 2002, описаны производные хиназолинона в качестве ингибиторов PARP. Описание изобретения Данное изобретение относится к соединениям формулы (I) их N-оксидам, фармацевтически приемлемым аддитивным солям и стереохимически изомерным формам, где пунктирные линии означают необязательные связи;L является прямой связью или двухвалентным радикалом, выбранным из -С (О)-, -C(O)-NH-, -NH-,-С (О) -С 1-6 алкандиила-, -С(О) -O-C1-6 алкандиила- или -С 1-6 алкандиила-;R2 является водородом, гидрокси, С 1-6 алкилокси или аминокарбонилом; если X замещен R2, то R2, взятый вместе с -L-Z, может образовывать двухвалентный радикал формулы где R10 является фенилом;R3 является водородом или С 1-6 алкилокси; где каждый из R5, R6, R7 и R8 независимо выбирают из водорода, галогена, амино, С 1-6 алкила или С 1-6 алкилокси илиR7 и R8, взятые вместе, могут образовывать двухвалентный радикал формулы где каждый R9 независимо выбирают из водорода или С 1-6 алкила; при условии, что если X является N-, то Z отличен от радикала (b-2), и если X является СН-, и L является -C(O)-NH- или -С(O)-O-С 1-6 алкандиилом-, и Z является радикалом (b-2), и R7 и R8, взятые вместе, образуют двухвалентный радикал формулы (с-1), (с-2) или (с-3), то R5 отличен от хлора. Соединения формулы (I) также могут существовать в таутомерных формах. Такие формы, хотя и не указаны подробно в представленной выше формуле, включены в объем данного изобретения. Ряд терминов, применяемых в представленных выше определениях и далее в описании, объяснены ниже. Эти термины иногда применяются как таковые или в составе сложных терминов. В представленных выше определениях и далее в описании галоген относится к фтору, хлору, брому и иоду; С 1-6 алкил относится к прямым и разветвленным насыщенным углеводородным радикалам, содержащим от 1 до 6 атомов углерода, таким как, например, метил, этил, пропил, бутил, пентил, гексил,-4 012837 1-метилэтил, 2-метилпропил, 2-метилбутил, 2-метилпентил и подобные; С 1-6 алкандиил относится к двухвалентным прямым и разветвленным насыщенным углеводородным радикалам, содержащим от 1 до 6 атомов углерода, таким как, например, метилен, 1,2-этандиил, 1,3-пропандиил, 1,4-бутандиил, 1,5 пентандиил, 1,6-гександиил, и к их разветвленным изомерам, таким как 2-метилпентандиил, 3 метилпентандиил, 2,2-диметилбутандиил, 2,3-диметилбутандиил и подобные. Термин фармацевтически приемлемые соли означает фармацевтически приемлемые кислотноили основно-аддитивные соли. Фармацевтически приемлемые кислотно- или основно-аддитивные соли в данном описании включают терапевтически активные нетоксичные кислотно- и нетоксичные основноаддитивные соли, которые могут образовывать соединения формулы (I). Соединения формулы (I), которые имеют основные свойства, могут быть превращены в их фармацевтически приемлемые кислотноаддитивные соли обработкой указанной основной формы соответствующей кислотой. Подходящие кислоты включают, например, неорганические кислоты, такие как галогенводородные кислоты, например хлористо-водородная или бромисто-водородная кислота; серная; азотная; фосфорная и подобные кислоты; или органические кислоты, такие как, например, уксусная, пропановая, гидроксиуксусная, молочная,пировиноградная, щавелевая, малоновая, янтарная (т.е. бутандиовая кислота), малеиновая, фумаровая,яблочная, винная, лимонная, метансульфоновая, этансульфоновая, бензолсульфоновая, п-толуолсульфоновая, цикламиновая, салициловая, п-аминосалициловая, памовая и подобные кислоты. Соединения формулы (I), которые имеют кислотные свойства, могут быть превращены в их фармацевтически приемлемые основно-аддитивные соли обработкой кислотной формы подходящим органическим или неорганическим основанием. Подходящие основные соли могут включать, например, аммониевые соли, соли щелочных и щелочно-земельных металлов, например соли лития, натрия, калия, магния,кальция и подобных, соли с органическими основаниями, например соли бензатина, N-метил-Dглюкамина, гидрабамина, и соли с аминокислотами, такие как, например, аргинин, лизин и подобные. Термины кислотно- или основно-аддитивная соль включают гидратные и сольватные аддитивные формы, которые могут образовывать соединения формулы (I). Примеры таких солей включают, например, гидраты, алкоголяты и подобные. Термин стереохимически изомерные формы соединений формулы (I) в данном описании определяет все возможные соединения, образованные теми же атомами, связанные теми же последовательностями связей, но имеющие различные трехмерные структуры, которые не являются взаимозаменяемыми,которые могут образовывать соединения формулы (I). Если не указано иначе, химические обозначения соединений охватывают смесь всех возможных стереохимически изомерных форм, которые указанное соединение может иметь. Указанная смесь может содержать все диастереомеры и/или энантиомеры основной молекулярной структуры указанного соединения. Все стереохимически изомерные формы соединений формулы (I), в чистой форме и в смеси друг с другом, включены в объем данного изобретения.N-оксиды соединений формулы (I) включают соединения формулы (I), в которых один или несколько атомов азота окислены до так называемого N-оксида, особенно те N-оксиды, в которых один или более атомов азота пиперидина или пиперазина N-окислены. Применяемый в данном описании термин соединения формулы (I) включает также N-оксиды,фармацевтически приемлемые кислотно- или основно-аддитивные соли и все стереоизомерные формы. В GB 1062357 описаны производные хиназолона, обладающие антигипертензивными свойствами. ВDE 2258561 описаны замещенные производные пиридинона с антигипертензивным действием. В ЕР 13612 описаны замещенные производные пиперидинилалкилхиназолина, которые являются антагонистами серотонина. В ЕР 669919 описаны диметилбензофураны и диметилбензопираны в качестве антагонистов 5-НТ 3. В US 5374637 описаны производные бензамида, которые обладают свойствами, стимулирующими моторику желудочно-кишечного тракта. В ЕР 885190 описаны 1,4-дизамещенные производные пиперидина, обладающие гастрокинетическими свойствами. В ЕР 1036073 описаны замещенные производные хиназолиндиона, которые обладают фундальными расслабляющими свойствами. Неожиданно было обнаружено, что соединения в соответствии с данным изобретением демонстрируют ингибирующее действие в отношении PARP. Первая группа представляющих интерес соединений включает те соединения формулы (I), к которым применяется одно или более из следующих ограничений:f) каждый R5 и R6 независимо выбирают из водорода или амино. Вторая группа представляющих интерес соединений включает те соединения формулы (I), к которым применяется одно или более из следующих ограничений:b) L является прямой связью или двухвалентным радикалом, выбранным из -С(О)-, -NH-,-С(О)-С 1-6 алкандиила- или С 1-6 алкандиила-;d) каждый R5 независимо выбирают из водорода, фтора, иода, брома, амино, С 1-6 алкила или С 1-6 алкилокси;e) каждый R6 независимо выбирают из водорода, хлора, иода, брома, амино, С 1-6 алкила или С 1-6 алкилокси. Третья группа представляющих интерес соединений включает те соединения формулы (I), к которым применяется одно или более из следующих ограничений:a) L является прямой связью или двухвалентным радикалом, выбранным из -С(О)- или -C(O)-NH-;d) каждый R5, R6, R7 и R8 независимо выбирают из водорода, галогена, С 1-6 алкила или С 1-6 алкилокси илиe) R7 и R8, взятые вместе, могут образовывать двухвалентный радикал формулы (с-1) или (с-4). Четвертая группа представляющих интерес соединений включает те соединения формулы (I), к которым применяется одно или более из следующих ограничений:a) L является прямой связью или двухвалентным радикалом, выбранным из -С(O)-, -C(O)-NH- или С(О)-O-С 1-6 алкандиила-;d) каждый R5, R6, R7 и R8 независимо выбирают из водорода, галогена, амино, C1-6 алкила или С 1-6 алкилокси илиe) R7 и R8, взятые вместе, могут образовывать двухвалентный радикал формулы (с-1), (с-2), (с-3) или (с-4). Пятая группа представляющих интерес соединений включает те соединения формулы (I), к которым применяется одно или более из следующих ограничений:f) каждый R5 независимо выбирают из водорода или галогена. Группа предпочтительных соединений включает те соединения формулы (I), в которых L является прямой связью или двухвалентным радикалом, выбранным из -С(O)-, -C(O)-NH- или -С(О)-O-C1-6 алкандиила-; R2 является водородом, гидрокси или С 1-6 алкилокси; Z является радикалом, выбранным из (b-2),(b-3), (b-4), (b-5), (b-6), (b-7), (b-8) или (b-9); каждый R5, R6, R7 и R8 независимо выбирают из водорода,галогена, амино, С 1-6 алкила или C1-6 алкилокси или R7 и R8, взятые вместе, могут образовывать двухвалентный радикал формулы (с-1), (с-2), (с-3) или (с-4). Группа более предпочтительных соединений включает те соединения формулы (I), в которых L является прямой связью; R1 является водородом, галогеном или С 1-6 алкилом; R2 является водородом; R3 является водородом; Z является радикалом, выбранным из (b-5) или (b-7); и каждый R5 независимо выбирают из водорода или галогена. Наиболее предпочтительными соединениями являются соединения 35, 36, 39, 1 и 43.-6 012837 Соединения формулы (I) могут быть получены общими методами, описанными в ЕР 1036073,ЕР 885190, US 5374637, ЕР 669919 и ЕР 13612. Исходные материалы и некоторые промежуточные соединения являются известными соединениями и коммерчески доступны или могут быть получены обычными реакционными методиками, известными в данной области техники. Некоторые способы получения более подробно представлены ниже. Другие способы получения конечных соединений формулы (I) описаны в примерах. Соединения формулы (I) могут быть получены взаимодействием промежуточного соединения формулы (II) с промежуточным соединением формулы (III), в котором W является подходящей уходящей группой, такой как, например, галоген, например фтор, хлор, бром или иод, или сульфонилокси радикалом, таким как метилсульфонилокси, 4-метилфенилсульфонилокси и подобные. Реакция может проводиться в инертном к реакции растворителе, таком как, например, спирт, например метанол, этанол,2-метоксиэтанол, пропанол, бутанол и подобные; простой эфир, например 4,4-диоксан, 1,1'-оксибиспропан и подобные; или кетон, например 4-метил-2-пентанон, N,N-диметилформамид, нитробензол и подобные. Добавление подходящего основания, такого как, например, карбонат или гидрокарбонат щелочного или щелочно-земельного металла, например триэтиламин или карбонат натрия, может применяться для связывания кислоты, которая высвобождается во время реакции. Небольшое количество подходящего иодида металла, например иодида натрия или калия, может быть добавлено для ускорения реакции. Перемешивание может улучшить скорость реакции. Реакция подходящим образом может проводиться при температуре в интервале от комнатной температуры до температуры кипения с обратным холодильником реакционной смеси и, при желании, реакция может проводиться при повышенном давлении. Соединения формулы (I) также могут быть превращены друг в друга известными в данной области техники реакциями или трансформациями функциональных групп. Некоторые из подобных трансформаций уже описаны выше. Другие примеры включают гидролиз сложных эфиров карбоновой кислоты до соответствующей карбоновой кислоты или спирта; гидролиз амидов до соответствующих карбоновых кислот или аминов; гидролиз нитрилов до соответствующих амидов; аминогруппы на имидазоле или фениле могут быть заменены на водород известными в данной области техники реакциями диазотирования и последующей заменой диазогруппы на водород; спирты могут быть превращены в сложные эфиры и простые эфиры; первичные амины могут быть превращены во вторичные или третичные амины; двойные связи могут быть гидрированы до соответствующих одинарных связей; иодный радикал на фенильной группе может быть превращен в группу сложного эфира введением моноокиси углерода в присутствии подходящего палладиевого катализатора. Данное изобретение также относится к соединению формулы (I), такому как описано выше, для применения в медицине. Соединения в соответствии с данным изобретением обладают ингибирующими свойствами в отношении PARP, как можно видеть из экспериментальной части, представленной ниже. Термин PARP в данном описании относится к белку, обладающему поли-ADP-рибозилирующим действием. В пределах значения этого термина PARP включают все белки, кодированные parp геном, их мутанты и их альтернативные фрагменты белков. Кроме того, в данном описании термин PARP включает аналоги PARP, гомологи PARP и аналоги PARP других животных. Термин PARP включает, но не ограничен этим, PARP-1. В значение данного термина также могут быть включены PARP-2, PARP-3, Vault-PARP (PARP-4), PARP-7 (TiPARP), PARP-8, PARP-9 (Bal),PARP-10, PARP-11, PARP-12, PARP-13, PARP-14, PARP-15, PARP-16, TANK-1, TANK-2 и TANK-3. Соединения, которые ингибируют как PARP-1, так и танкиразу 2, могут обладать преимущественными свойствами в том, что они имеют улучшенное действие, ингибирующее рост в раковых клетках. Данное изобретение также включает применение соединений при получении лекарственных средств для лечения любых описанных здесь заболеваний и расстройств у животных, где указанные соединения являются соединениями формулы (I)-7 012837 их N-оксидами, фармацевтически приемлемыми аддитивными солями и стереохимически изомерными формами, где пунктирные линии означают необязательные связи;L является прямой связью или двухвалентным радикалом, выбранным из -С(О)-, -C(O)-NH-, -NH-,-С(О)-С 1-6 алкандиила-, -С(О)-О-С 1-6 алкандиила- или -С 1-6 алкандиила-;R2 является водородом, гидрокси, C1-6 алкилокси или аминокарбонилом; если X замещен R2, то R2, взятый вместе с -L-Z, может образовывать двухвалентный радикал формулы где R10 является фенилом;R3 является водородом или С 1-6 алкилокси; где каждый из R5, R6, R7 и R8 независимо выбирают из водорода, галогена, амино, С 1-6 алкила или С 1-6 алкилокси илиR7 и R8, взятые вместе, могут образовывать двухвалентный радикал формулы где каждый R9 независимо выбирают из водорода или С 1-6 алкила. Далее данное изобретение также относится к применению соединения, описанного выше, для производства лекарственного средства для лечения расстройства, медиированного PARP. В частности, данное изобретение относится к применению соединения, описанного выше, для производства лекарственного средства для лечения расстройства, медиированного PARP. Соединения, которые ингибируют как PARP-1, так и TANK-2, могут обладать преимущественными свойствами в том, что они имеют повышенное рост-ингибирующее действие в раковых клетках. В свете их PARP-связывающих свойств соединения в соответствии с данным изобретением могут применяться в качестве ссылочных (контрольных) соединений или изотопных индикаторов, когда один из атомов молекулы может быть заменен, например, радиоактивным изотопом. Для получения фармацевтических композиций в соответствии с данным изобретением эффективное количество определенного соединения в виде основно- или кислотно-аддитивной соли в качестве активного ингредиента объединяют в однородной смеси с фармацевтически приемлемым носителем, где носитель может иметь множество форм в зависимости от формы композиции, желаемой для введения. Такие фармацевтические композиции имеют, желательно, единичную дозированную форму, подходящую предпочтительно для перорального, ректального, чрескожного или парентерального введения. Например,при получении композиций в пероральной дозированной форме может применяться любая обычная фармацевтическая среда, такая как, например, вода, гликоли, масла, спирты и подобные для оральных жидких композиций, таких как суспензии, сиропы, эликсиры и растворы; или твердые носители, такие как крахмалы, сахара, каолин, лубриканты, связующие агенты, дезинтегрирующие агенты и подобные-8 012837 для порошков, пилюль, капсул и таблеток. Благодаря простоте их введения, таблетки и капсулы являются наиболее преимущественной пероральной дозированной формой, и для них, очевидно, применяются твердые фармацевтические носители. Для парентеральных композиций носитель обычно включает стерильную воду, по крайней мере, в большей части, хотя могут быть включены и другие ингредиенты, например, для придания растворимости. Могут быть получены, например, растворы для инъекций, в которых носитель представляет собой физиологический раствор, раствор глюкозы или смесь физиологического раствора и раствора глюкозы. Также могут быть получены суспензии для инъекций, в которых применяются подходящие жидкие носители, суспендирующие агенты и подобные. В композициях, подходящих для чрескожного введения, носитель необязательно содержит агент, улучшающий проникновение и/или подходящий увлажняющий агент, необязательно объединенный с подходящими добавками любой природы в небольших пропорциях, где добавки не должны оказывать значительное вредное влияние на кожу. Такие добавки могут способствовать нанесению на кожу и/или могут быть полезны для получения желаемых композиций. Такие композиции могут вводиться различными путями, например в виде чрескожного пластыря, точечно или в виде мази. Особенно преимущественно получать указанные фармацевтические композиции в виде единичных дозированных форм для облегчения введения и однородности дозирования. Единичные дозированные формы, указанные в описании и формуле изобретения,относятся к физически дискретным единицам, подходящим для введения в виде единичной дозы, где каждая единица содержит предопределенное количество активного ингредиента, рассчитанное для оказания желаемого действия в сочетании с требуемым фармацевтическим носителем. Примеры таких единичных дозированных форм включают таблетки (включая рифленые и таблетки с оболочкой), капсулы,пилюли, пакетики с порошком, брикеты, растворы для инъекций или суспензий, полная чайная ложка,полная столовая ложка и подобные, и их делимые множества. Соединения в соответствии с данным изобретением могут лечить или предотвращать повреждение тканей в результате повреждения или смерти клеток вследствие некроза или апоптоза; могут улучшать повреждения нервных или сердечно-сосудистых тканей, включая повреждения вследствие очаговой ишемии, инфаркта миокарда и реперфузии; могут лечить различные заболевания и состояния, вызываемые или обостряемые активностью PARP; могут продлевать или увеличивать ресурс или пролиферативную способность клеток; могут изменять экспрессию генов стареющих клеток; могут радиосенсибилизировать и/или химиосенсибилизировать клетки. В общем, ингибирование активности PARP бережет клетки от потери энергии, предотвращая, в случае нервных клеток, необратимую деполяризацию нейронов и таким образом обеспечивая нейропротективное действие. По указанным выше причинам данное изобретение также относится к способу введения терапевтически эффективного количества указанных выше соединений в количестве, эффективном для ингибирования активности PARP, для лечения или предупреждения повреждения тканей в результате повреждения или смерти клеток вследствие некроза или апоптоза, для воздействия на активность нейронов, не медиированную NMDA токсичностью, для воздействия на активность нейронов, медиированную NMDA токсичностью, для лечения повреждения нервной ткани вследствие ишемии и реперфузии, неврологических расстройств и нейродегенеративных заболеваний; для профилактики или лечения инсульта сосудов; для лечения или профилактики сердечно-сосудистых заболеваний; для лечения других состояний и/или расстройств, таких как возрастная мышечная дегенерация, СПИД и другие иммунные возрастные заболевания, воспаление, подагра, артрит, атеросклероз, общая атрофия, рак, дегенеративные заболевания скелетных мышц, включая репликативное старение, диабет, травма головы, воспалительное заболевание кишечника (такое как колит и болезнь Крона), мышечная дистрофия, остеоартрит, остеопороз, хроническая и/или острая боль (такая как невропатическая боль), почечная недостаточность, ишемия сетчатки,септический шок (такой как эндотоксический шок) и старение кожи, для продления ресурса и пролиферативной способности клеток; для изменения экспрессии генов в стареющих клетках; для химиосенсибилизации и/или радиосенсибилизации (гипоксических) опухолевых клеток. Данное изобретение также относится к лечению заболеваний и состояний у животных, которое включает введение указанному животному терапевтически эффективного количества указанных выше соединений. В частности, данное изобретение относится к способу лечения, профилактики или ингибирования неврологических расстройств у животных, который включает введение указанному животному терапевтически эффективного количества указанных выше соединений. Неврологическое расстройство выбирают из группы, включающей периферийную невропатию, вызванную физическим повреждением или болезненным состоянием, травматическое повреждение мозга, физическое повреждение спинного мозга,удар, связанный с повреждением мозга, очаговую ишемию, общую ишемию, реперфузию, демиелинизирующее заболевание и неврологическое расстройство, связанное с нейродегенерацией. Данное изобретение также относится к применению соединений формулы (I) для ингибирования активности PARP, для лечения, предупреждения или ингибирования повреждения тканей в результате повреждения клеток вследствие некроза или апопотоза, для лечения, профилактики или ингибирования неврологического расстройства у животного. Термин профилактика нейродегенерации включает способность предотвращать нейродегенерацию у пациентов, у которых нейродегенеративное заболевание только что диагностировано, или при-9 012837 риске развития нового дегенеративного заболевания, и для профилактики дальнейшей нейродегенерации у пациентов, которые уже страдают от или имеют симптомы нейродегенеративного заболевания. Термин лечение в данном описании относится к любому лечению заболевания и/или состояния у животного, особенно человека, и включает: (i) профилактику заболевания и/или состояния от возникновения у пациента, который может быть предрасположен к заболеванию и/или состоянию, но у которого оно еще не диагностировано; (ii) ингибирование заболевания и/или состояния, например остановка его развития; (iii) облегчение заболевания и/или состояния, например вызов регрессии заболевания и/или состояния. Термин радиосенсибилизатор в данном описании определяет молекулу, предпочтительно молекулу с низким молекулярным весом, вводимую животным в терапевтически эффективных количествах для повышения чувствительности клеток к ионизирующему излучению и/или для способствования лечению заболеваний, которые лечат ионизирующим излучением. Заболевания, которые лечат ионизирующим излучением, включают неопластические заболевания, доброкачественные и злокачественные опухоли и раковые клетки. Лечение ионизирующим излучением других заболеваний, не перечисленных выше, также включено в объем данного изобретения. Термин химиосенсибилизатор в данном описании определяет молекулу, предпочтительно молекулу с низким молекулярным весом, вводимую животным в терапевтически эффективных количествах для повышения чувствительности клеток к химиотерапии и/или для способствования лечению заболеваний, которые лечат химиотерапевтическими агентами. Заболевания, которые лечат химиотерапией,включают неопластические заболевания, доброкачественные и злокачественные опухоли и раковые клетки. Лечение химиотерапией других заболеваний, не перечисленных выше, также включено в объем данного изобретения. Соединения, композиции и способы в соответствии с данным изобретением особенно полезны для лечения или профилактики повреждения тканей в результате смерти или повреждения клеток вследствие некроза или апоптоза. Соединения в соответствии с данным изобретением могут быть противораковыми агентами, где данный термин также включает агенты против роста опухолевых клеток и противоопухолевые агенты. Например, способы в соответствии с данным изобретением применяются для лечения рака и химиосенсибилизации и/или радиосенсибилизации опухолевых клеток при раковых заболеваниях, таких как АСТН-вырабатывающие опухоли, острая лимфоцитарная лейкемия, острая нелимфатическая лейкемия,рак коры надпочечника, рак мочевого пузыря, рак мозга, рак груди, рак шейки матки, хроническая лимфоцитарная лейкемия, хроническая миелоцитарная лейкемия, рак ободочной и прямой кишки, лимфома Т-клеток кожи, эндометриальный рак, рак пищевода, саркома Эвинга, рак желчного пузыря, лейкоз ворсистых клеток, рак головы и шеи, лимфома Ходжкина, саркома Капоши, рак почек, рак печени, рак легких (мелкоклеточный и немелкоклеточный), злокачественный брюшинный выпот, злокачественный плевральный выпот, меланома, мезотелиома, множественная миелома, нейробластома, неходжкинская лимфома, остеосаркома, рак яичников, рак яичника (эмбриона), рак простаты, рак поджелудочной железы, рак полового члена, ретинобластома, рак кожи, саркома мягких тканей, карциномы плоских клеток,рак желудка, рак яичек, рак щитовидной железы, трофобластические неоплазмы, рак матки, рак влагалища, рак вульвы и опухоль Вилма. Следовательно, соединения в соответствии с данным изобретением могут применяться как радиосенсибилизатор и/или химиосенсибилизатор. Известно, что радиосенсибилизаторы повышают чувствительность раковых клеток к токсическому действию ионизирующего излучения. Несколько механизмов способа действия радиосенсибилизаторов предложены в литературе, включая радиосенсибилизаторы гипоксических клеток (например, соединения 2-нитроимидазола и соединения диоксида бензотриазина), имитирующие кислород или, альтернативно,действующие как биовосстановительные агенты при гипоксии; радиосенсибилизаторы негипоксических клеток (например, галогенированные пиримидины), которые могут быть аналогами ДНК оснований и предпочтительно внедряться в ДНК раковых клеток и тем самым способствовать вызванному радиацией распаду молекул ДНК и/или оберегать нормальные механизмы репарации ДНК; и различные другие вероятные механизмы действия были предположены для радиосенсибилизаторов при лечении заболеваний. Многие протоколы лечения рака в настоящее время включают радиосенсибилизаторы в сочетании с облучением рентгеновскими лучами. Примеры активизированных рентгеновскими лучами радиосенсибилизаторов включают, но не ограничены ими, следующие: метронидазол, мизонидазол, дезметилмизонидазол, пимонидазол, этанидазол, ниморазол, митомицин С, RSU 1069, SR 4233, ЕО 9, RB 6145, никотинамид, 5-бромдезоксиуридин (BUdR), 5-иоддезоксиуридин (IUdR), бромдезоксицитидин, фтордезоксиуридин (FudR), гидроксимочевину, цисплатин и их терапевтически эффективные аналоги и производные. Фотодинамическая терапия (ФДТ) раковых заболеваний включает видимый свет в качестве излучающего активатора сенсибилизирующего агента. Примеры фотодинамических агентов радиосенсибилизаторов включают, но не ограничены ими, следующие: производные гематопорфирина, фотофрин, производные бензопорфирина, этиопорфирин олова, феоборбид-а, бактериохлорофилл-а, нафталоцианины,фталоцианины, фталоцианин цинка и их терапевтически эффективные аналоги и производные.- 10012837 Радиосенсибилизаторы могут вводиться в комбинации с терапевтически эффективным количеством одного или более других соединений, включая, но не ограничиваясь ими, соединения, которые способствуют внедрению радиосенсибилизаторов в целевые клетки; соединения, которые контролируют поток терапевтических агентов, питательных веществ и/или кислорода в целевые клетки; химиотерапевтические агенты, которые действуют на опухоль с или без дополнительного облучения; или другие терапевтически эффективные соединения для лечения рака или других заболеваний. Примеры дополнительных терапевтических агентов, которые могут применяться в комбинации с радиосенсибилизаторами, включают, но не ограничены ими, 5-фторурацил, лейковорин, 5'-амино-5'-дезокситимидин, кислород, карбоген, агенты трансфузии эритроцитов, перфторированные углероды (например, Fluosol 10 DA), 2,3-DPG,BW12C, блокаторы кальциевого канала, пентоксифиллин, антиангиогенезные соединения, гидралазин иLBSO. Примеры химиотерапевтических агентов, которые могут применяться в комбинации с радиосенсибилизаторами, включают, но не ограничены ими, адриамицин, камптотецин, карбоплатин, цисплатин,даунорубицин, доцетаксел, доксорубицин, интерферон (альфа, бета, гамма), интерлейкин 2, иринотекан,паклитаксел, топотекан и их терапевтически эффективные аналоги и производные. Химиосенсибилизаторы могут вводиться в комбинации с терапевтически эффективным количеством одного или более других соединений, включая, но не ограничиваясь ими, соединения, которые способствуют внедрению химиосенсибилизаторов в целевые клетки; соединения, которые контролируют поток терапевтических агентов, питательных веществ и/или кислорода в целевые клетки; химиотерапевтические агенты, которые действуют на опухоль; или другие терапевтически эффективные соединения для лечения рака или других заболеваний. Примеры дополнительных терапевтических агентов, которые могут применяться в комбинации с химиосенсибилизаторами, включают, но не ограничены ими, метилирующие агенты, ингибиторы топоизомеразы I и другие химиотерапевтические агенты, такие как цисплатин и блеомицин. Соединения формулы (I) также могут применяться для определения или идентификации PARP, более конкретно рецептора PARP-1. Для этой цели соединения формулы (I) могут быть мечеными. Такие индикаторы могут быть выбраны из группы, включающей радиоизотопы, спин-индикаторы, индикаторы антигена, ферментные индикаторы флуоресцентной группы или хемилюминесцентной группы. Специалист в данной области техники легко определит эффективное количество из результатов тестирования, представленных ниже. В общем, считается, что эффективное количество составляет от 0,001 до 100 мг/кг массы тела, в частности от 0,005 до 10 мг/кг массы тела. Может быть удобным вводить требуемую дозу два, три, четыре или более раз с соответствующими интервалами в течение суток. Такие субдозы могут быть в виде единичных дозированных форм, например, содержащих от 0,05 до 500 мг, в частности от 0,1 мг до 200 мг активного ингредиента на единичную дозированную форму. Экспериментальная часть Далее ДХМ означает дихлорметан, ДМФ означает N,N-диметилформамид, МеОН означает метанол, МИК означает метилизобутилкетон, МЭК означает метилэтилкетон, ТЭА означает триэтиламин и ТГФ означает тетрагидрофуран. А. Получение промежуточных соединений. Пример А 1. а) Получение промежуточного соединения 1 Смесь 3-(1-пиперазинил)-1 Н-индазола (0,11 моль), хлорацетонитрила (0,16 моль) и ТЭА (13 г) в толуоле (200 мл) и ацетонитриле (200 мл) перемешивают и кипятят с обратным холодильником в течение 3 ч. Охлажденную реакционную смесь промывают водой (250 мл). Органический слой отделяют, сушат(MgSO4), фильтруют и растворитель выпаривают. Остаток растворяют в трихлорметане и очищают, пропуская через двуокись кремния на стеклянном фильтре (элюент: трихлорметан/МеОН 90/10). Наиболее чистую фракцию собирают и растворитель выпаривают. Остаток кристаллизуют из ацетонитрила. Кристаллы отфильтровывают и сушат с получением 26 г (99%) промежуточного соединения 1, температура плавления 136 С.b) Получение промежуточного соединения 2 Смесь промежуточного соединения 1 (0,11 моль) в NH3/MeOH (600 мл) гидрируют при температуре 50 С с применением никеля Ренея (4 г) в качестве катализатора. После поглощения Н 2 (2 экв.) катализатор отфильтровывают, и фильтрат выпаривают. Остаток кристаллизуют из ацетонитрила. Кристаллы отфильтровывают и сушат с получением 21 г (77,5%) промежуточного соединения 2, температура плавления 121 С.- 11012837 Пример А 2. а) Получение промежуточного соединения 3 Фосфорилхлорид (110,9 мл) по каплям добавляют при температуре 5 С к ДМФ (81,5 мл). Смесь перемешивают до полного растворения. Добавляют этиловый эфир 4-[(1-оксобутил)амино]бензойной кислоты (0,34 моль). Смесь перемешивают при температуре 100 С в течение 15 ч, затем охлаждают до комнатной температуры и выливают в ледяную воду. Осадок отфильтровывают, промывают водой и сушат с получением 42,35 г (47%) промежуточного соединения 3.b) Получение промежуточного соединения 4 Смесь промежуточного соединения 3 (0,1606 моль) в метилате натрия, 30% раствора в МеОН (152,8 мл) и МеОН (400 мл) перемешивают и кипятят с обратным холодильником в течение 15 ч, затем охлаждают и выливают в ледяную воду. Осадок отфильтровывают, промывают водой и помещают в ДХМ. Органический слой отделяют, сушат (MgSO4), фильтруют и растворитель выпаривают досуха с получением 31,64 г (85%) промежуточного соединения 4. с) Получение промежуточного соединения 5 Тетрагидроалюминат лития (0,1288 моль) порциями добавляют при температуре 0 С в потоке N2 к раствору промежуточного соединения 4 (0,1288 моль) в ТГФ (263 мл). Смесь перемешивают в течение 30 мин, выливают в ледяную воду и экстрагируют ДХМ. Органический слой отделяют, сушат (MgSO4),фильтруют и растворитель выпаривают досуха с получением 27,4 г (98%) промежуточного соединения 5.d) Получение промежуточного соединения 6 Метансульфонилхлорид (0,104 моль) по каплям добавляют при температуре 0 С в потоке N2 к смеси промежуточного соединения 5 (0,069 моль) и ТЭА (0,207 моль) в ДХМ (120 мл). Смесь перемешивают при температуре 0 С в течение 4 ч. Растворитель выпаривают досуха (без нагревания). Продукт применяют без дальнейшей очистки с получением 20,4 г промежуточного соединения 6. Пример A3. а) Получение промежуточного соединения 7 Этиловый эфир 4-(2-аминоэтил)-1-пиперазинкарбоновой кислоты (0,0586 моль) и 2-(метилтио)4(1 Н)-хиназолинон (0,0588 моль) нагревают при температуре 180 С в течение 2 ч при перемешивании,обрабатывая жавелевой водой, и затем помещают в ДХМ и МеОН. Растворитель выпаривают досуха. Остаток очищают хроматографией на колонке с силикагелем (15-35 мкм) (элюент: ДХМ/МеОН/NH4OH 94/6/0,5). Чистые фракции собирают и растворитель выпаривают. Маслянистый остаток кристаллизуют из диэтилового эфира. Осадок отфильтровывают и сушат с получением промежуточного соединения 7,температура плавления 138 С.b) Получение промежуточного соединения 8 Смесь промежуточного соединения 7 (0,0223 моль) и гидроксида калия (0,223 моль) в 2-пропаноле(100 мл) перемешивают и кипятят с обратным холодильником в течение 4 дней. Растворитель выпаривают досуха. Остаток помещают в МеОН при перемешивании при температуре 60 С. Соль отфильтровывают. Растворитель выпаривают с получением 6,5 г промежуточного соединения 8. В. Получение конечных соединений. Пример В 1. Получение соединения 1(0,00097 моль) нагревают при температуре 160 С в течение 1 ч, затем помещают в воду и карбонат калия 10% и экстрагируют ДХМ/МеОН 90/10. Органический слой отделяют, сушат (MgSO4), фильтруют и рас- 12012837 творитель выпаривают. Остаток (0,3 г) очищают хроматографией на колонке с силикагелем (15-40 мкм)(элюент: ДХМ/МеОН/NH4OH 92/8/0,5). Чистые фракции собирают и растворитель выпаривают. Остаток кристаллизуют из МЭК и DIPE. Осадок отфильтровывают и сушат с получением 0,2 г (58%) соединения 1, температура плавления 186 С. Пример В 2. Получение соединения 2 Смесь 1-(3-аминопропил)-4-(4-хлорфенил)-4-пиперидинола (0,015 моль) и 2-хлор-4(1 Н)-хиназолинона (0,018 моль) в диметилацетамиде (5 мл) перемешивают при температуре 120 С в течение 1 ч. Реакционную смесь охлаждают, растворяют в ДХМ и промывают водным аммиаком. Органический слой отделяют, сушат (MgSO4), фильтруют и растворитель выпаривают. Остаток очищают хроматографией на колонке с силикагелем (элюент: ДХМ/(MeOH/NH3) 92/8). Чистые фракции собирают и растворитель выпаривают. Остаток суспендируют в DIPE. Осадок отфильтровывают и сушат (вакуум; 70 С), с получением 3,72 г (60%) соединения 2, температура плавления 178,4 С. Пример В 3. Получение соединения 3 Смесь промежуточного соединения 6 (0,0124 моль), промежуточного соединения 8 (0,0137 моль) и карбоната калия (0,0373 моль) в ДМФ (80 мл) перемешивают при температуре 60 С в течение 1 ч, выливают в ледяную воду и перемешивают при комнатной температуре в течение 30 мин. Осадок отфильтровывают, промывают водой и помещают в 2-пропанон. Осадок отфильтровывают и сушат с получением 1,5 г (26%) соединения 3, температура плавления 118 С. В табл. F-1 перечислены соединения, полученные по методике одного из указанных выше примеров.In vitro анализ сцинтилляционной близости (SPA) на ингибирующее действие PARP-1. Соединения в соответствии с данным изобретением тестируют в in vitro анализе, основанном на технологии SPA (собственность Amersham Pharmacia Biotech). В принципе исследование основано на признанной технологии SPA для определения поли(ADP-рибозил)ирования биотинилированных белковмишеней, т.е. гистонов. Такое рибозилирование вызывается применением активированного никированной ДНК фермента PARP-1 и [3 Н]-никотинамида аденина динуклеотида ([3 Н]-NAD+) в качестве донораADP-рибозила. В качестве индуктора активности фермента PARP-1 получают никированную ДНК. Для этого 25 мг ДНК (поставщик: Sigma) растворяют в 25 мл буфера для ДНКазы (10 мМ Tris-HCl, pH 7,4; 0,5 мг/мл альбумина бычьей сыворотки (АБС); 5 мМ MgCl26 Н 2 О и 1 мМ KCl) к которому добавляют 50 мкл раствора ДНКазы (1 мг/мл в 0,15 М NaCl). После инкубирования в течение 90 мин при температуре 37 С реакцию останавливают добавлением 1,45 г NaCl, с последующим дополнительным инкубированием при температуре 58 С в течение 15 мин. Реакционную смесь охлаждают на льду и диализуют при температуре 4 С в течение, соответственно, 1,5 и 2 ч с 1,5 л 0,2 М KCl и дважды с 1,5 л 0,01 М KCl в течение 1,5 и 2 ч соответственно. Смесь аликвотируют и хранят при температуре -20 С. Гистоны (1 мг/мл, тип II-A, поставщик: Sigma) биотинилируют с применением набора для биотинилирования от Amersham, и хранят алик- 15012837 воты при температуре -20 С. Маточный раствор 100 мг/мл SPA шариков поли(винилтолуола) (PVT) (поставщик: Amersham) получают в ФРФБ. Маточный раствор [3 Н]-NAD+ получают добавлением 120 мкл[3 Н]-NAD+ (0,1 мКи/мл, поставщик: NEN) к 6 мл инкубационного буфера (50 мМ Tris/HCl, рН 8,0; 0,2 мМ DTT; 4 мМ MgCl2). Раствор 4 мМ NAD+ (поставщик: Roche) получают в инкубационном буфере (из 100 мМ маточного раствора в воде, хранящегося при температуре -20 С). Фермент PARP-1 получают по методикам известного уровня техники, например клонированием и экспрессией белка, начиная с кДНК печени человека. Информацию, относящуюся к применению белковой последовательности ферментаPARP-1, включая литературные ссылки, можно найти в базе данных Swiss-Prot под номером первичного доступа Р 09874. Биотинилированные гистоны и шарики PVT-SPA смешивают и предварительно инкубируют в течение 30 мин при комнатной температуре. Фермент PARP-1 (концентрация зависит от партии) смешивают с никированной ДНК, и смесь предварительно инкубируют в течение 30 мин при температуре 4 С. Равные части полученного раствора гистонов/PVT-SPA шариков и раствора фермента PARP1/ДНК смешивают, и 75 мкл этой смеси вместе с 1 мкл соединения в ДМСО и 25 мкл [3 Н]-NAD+ добавляют в каждую ячейку в 96-ячеечный титровальный микропланшет. Конечные концентрации в инкубационной смеси составляют 2 мкг/мл для биотинилированных гистонов, 2 мг/мл для PVT-SPA шариков, 2 мкг/мл для никированной ДНК и от 5 до 10 мкг/мл для фермента PARP-1. После инкубирования смеси в течение 15 мин при комнатной температуре реакцию останавливают добавлением 100 мкл 4 мМ NAD+ в инкубационном буфере (конечная концентрация 2 мМ) и планшеты смешивают. Шарики оставляют осаждаться в течение по крайней мере 15 мин, и планшеты переносят в TopCountNXT (Packard) для сцинтилляционного считывания, значения выражают как импульсы в минуту(имп./мин). Для каждого эксперимента параллельно запускают контроль (содержащий фермент PARP-1 и ДМСО без соединения), пустую инкубационную смесь (содержащую ДМСО, но не содержащую фермент PARP-1 или соединение) и образцы (содержащие фермент PARP-1 и соединение, растворенное в ДМСО). Все тестируемые соединения растворяют и в конечном счете далее разводят в ДМСО. В первом случае соединения тестируют в концентрации 10-5 М. Когда соединения показывают активность при 10-5 М, получают кривую доза-реакция, в которой соединения тестируют при концентрациях от 10-5 М и 10-8 М. В каждом тесте пустое значение вычитают из значений контроля и образца. Контрольный образец представляет максимальную активность фермента PARP-1. Для каждого образца количество имп./мин выражают как процент от среднего значения имп./мин для контроля. В подходящий момент значенияIC50 (концентрация лекарственного средства, необходимая для снижения активности фермента PARP-1 до 50% от контрольного) рассчитывают с применением линейной интерполяции между экспериментальными значениями непосредственно выше и ниже 50% уровня. Действие тестируемых соединений выражают как pIC50 (отрицательный логарифм значения IC50). В качестве ссылочного (контрольного) соединения применяют 4-амино-1,8-нафталимид для подтверждения анализа SPA. Тестируемые соединение демонстрируют ингибирующее действие при исходной тестируемой концентрации 10-5 М (см. табл. 2).In vitro фильтрационный анализ на ингибирующее действие PARP-1. Соединения в соответствии с данным изобретением тестируют с применением in vitro фильтрационного анализа для оценки активности PARP-1 (инициированного в присутствии никированной ДНК) через поли(ADP-рибозил)ирующее действие его гистона с применением [32 Р]-NAD в качестве донораADP-рибозила. Радиоактивные рибозилированные гистоны осаждают трихлоруксусной кислотой (ТХК) в 96-ячеечных фильтровальных планшетах, и введенный [32 Р] измеряют на сцинтилляционном счетчике. Получают смесь гистонов (маточный раствор: 5 мг/мл в Н 2 О), NAD+ (маточный раствор: 100 мМ в Н 2 О) и [32 Р]-NAD+ в инкубационном буфере (50 мМ Tris/HCl, pH 8; 0,2 мМ DTT; 4 мМ MgCl2). Также получают смесь фермента PARP-1 (5-10 мкг/мл) и никированной ДНК. Никированную ДНК получают как описано в in vitro SPA для ингибирующего действия PARP-1. Семьдесят пять мкл смеси ферментаPARP-1/ДНК вместе с 1 мкл соединения в ДМСО и 25 мкл смеси гистоны-NAD+/[32P]-NAD+ добавляют в каждую ячейку 96-ячеечного фильтровального планшета (0,45 мкм, поставщик: Millipore). Конечные концентрации в инкубационной смеси составляют 0,2 мкг/мл для гистонов, 0,1 мМ для NAD+, 200 мкМ(0,5 мкС) для [32 Р]-NAD+ и 2 мкг/мл для никированной ДНК. Планшеты инкубируют в течение 15 мин при комнатной температуре, и реакцию останавливают добавлением 10 мкл ледяной 100% ТХК с последующим добавлением 10 мкл ледяного раствора АБС (1% в Н 2 О). Белковую фракцию осаждают в течение 10 мин при температуре 4 С, и планшеты фильтруют в вакууме. Планшеты последовательно промывают, каждую ячейку, 1 мл 10% ледяной ТХК, 1 мл 5% ледяной ТХК и 1 мл 5% ТХК при комнатной температуре. Наконец, в каждую ячейку добавляют 100 мкл сцинтилляционного раствора (Microscint 40,Packard), и планшеты переносят в TopCountNXT (поставщик: Packard) для сцинтилляционного считывания, и значения выражают как импульсы в минуту (имп./мин). Для каждого эксперимента параллельно запускают контроль (содержащий фермент PARP-1 и ДМСО без соединения), пустую инкубационную смесь (содержащую ДМСО, но не содержащую фермент PARP-1 или соединение) и образцы (содержащие фермент PARP-1 и соединение, растворенное в ДМСО). Все тестируемые соединения растворяют и в конечном счете далее разводят в ДМСО. В первом случае соединения тестируют в концентрации 10-5 М. Когда соединения показывают активность при 10-5 М, получают кривую доза-реакция, в которой- 16012837 соединения тестируют при концентрациях от 10-5 М и 10-8 М. В каждом тесте пустое значение вычитают из значений контроля и образца. Контрольный образец представляет максимальную активность фермента PARP-1. Для каждого образца количество имп./мин выражают как процент от среднего значения имп./мин для контроля. В подходящий момент значения IC50 (концентрация лекарственного средства,необходимая для снижения активности фермента PARP-1 до 50% от контрольного) рассчитывают с применением линейной интерполяции между экспериментальными значениями непосредственно выше и ниже 50% уровня. Действие тестируемых соединений выражают как pIC50 (отрицательный логарифм значения IC50). В качестве ссылочного соединения применяют 4-амино-1,8-нафталимид для подтверждения фильтрационного анализа. Тестируемые соединение демонстрируют ингибирующее действие при исходной тестируемой концентрации 10-5 М (см. табл. 2).In vitro анализ сцинтилляционной близости (SPA) на ингибирующее действие TANK-2. Соединения в соответствии с данным изобретением тестируют в in vitro анализе, основанном на технологии SPA с применением планшетов Ni Flash (96 или 384 ячейки). В принципе исследование основано на признанной технологии SPA для определения авто-поли(ADP-рибозил)ирования белкаTANK-2 с применением [3 Н]-никотинамидадениндинуклеотида ([3 Н]-NAD+) в качестве донора ADPрибозила. Маточный раствор [3 Н]-NAD+/NAD получают добавлением 64,6 мкл [3 Н]-NAD+ (0,1 мКи/мл, поставщик: Perkin Elmer) и 46,7 мл маточного-NAD (10,7 мМ, хранится при температуре -20 С, поставщик:Roche) к 1888,7 мкл буфера для исследований (60 мМ Tris/HCl, рН 7,4; 0,9 мМ DTT; 6 мМ MgCl2). Фермент TANK-2 получают, как описано в ЕР 1238063. 60 мкл буфера для исследований вместе с 1 мкл соединения в ДМСО, 20 мкл [3 Н] -NAD+/NAD и 20 мкл фермента TANK-2 (конечная концентрация 6 мкг/мл) добавляют в каждую ячейку в 96-ячеечный флэш-планшет с покрытием Ni (Perkin Elmer). После инкубирования смеси в течение 120 мин при комнатной температуре реакцию останавливают добавлением 60 мкл останавливающего раствора (42,6 мг NAD в 6 мл Н 2 О). Планшеты накрывают герметиком для планшетов и помещают в TopCountNXT (Packard) для сцинтилляционного считывания. Значения выражают как импульсы в минуту (имп./мин). Для каждого эксперимента параллельно запускают контроль (содержащий фермент TANK-2 и ДМСО без соединения), пустую инкубационную смесь (содержащую ДМСО, но не содержащую фермент TANK-2 или соединение) и образцы (содержащие ферментTANK-2 и соединение, растворенное в ДМСО). Все тестируемые соединения растворяют и в конечном счете далее разводят в ДМСО. В первом случае соединения тестируют в концентрации 10-5 М. Когда соединения показывают активность при 10-5 М, получают кривую доза-реакция, в которой соединения тестируют при концентрациях от 10-5 М и 10-8 М. В каждом тесте пустое значение вычитают из значений контроля и образца. Контрольный образец представляет максимальную активность фермента TANK-2. Для каждого образца количество имп./мин выражают как процент от среднего значения имп./мин для контроля. В подходящий момент значения IC50 (концентрация лекарственного средства, необходимая для снижения активности фермента TANK-2 до 50% от контрольного) рассчитывают с применением линейной интерполяции между экспериментальными значениями непосредственно выше и ниже 50% уровня. Действие тестируемых соединений выражают как pIC50 (отрицательный логарифм значения IC50). В качестве ссылочного соединения применяют 3-аминобензамид и 4-амино-1,8-нафталимид для подтверждения анализа SPA. Данный анализ представлен для 96-ячеечных планшетов. В анализе с применением 384-ячеечных планшетов применяются те же самые конечные концентрации и объемы адаптируют. Результаты с применением 96-ячеечного планшета, при их наличии, представлены в табл. 2, в противном случае показаны результаты анализа в 384-ячеечном планшете. Таблица 2 Соединения далее могут быть оценены с применением анализа химио- и/или радиосенсибилизации,анализа измерения ингибирования эндогенной активности PARP-1 в колониях раковых клеток и, в конечном счете, в in vivo радиосенсибилизационном тестировании. ФОРМУЛА ИЗОБРЕТЕНИЯ 1. Соединение формулы (I) его N-оксиды, фармацевтически приемлемые аддитивные соли и стереохимически изомерные формы, где пунктирные линии означают необязательные связи;L является прямой связью или двухвалентным радикалом, выбранным из -С(О)-, -C(O)-NH-, -NH-,-С(О)-O-С 1-6 алкандиила- или -C1-6 алкандиила-;R3 является водородом или C1-6 алкилокси; где каждый из R5, R6, R7 и R8 независимо выбирают из водорода, галогена, амино, C1-6 алкила илиR7 и R8, взятые вместе, могут образовывать двухвалентный радикал формулы где каждый R9 независимо выбирают из водорода или C1-6 алкила; при условии, что если X является N-, то Z отличен от радикала (b-2), и если X является СН-, и L является -C(O)-NH- или -С(O)-O-C1-6 алкандиилом-, и Z является радикалом (b-2), и R7 и R8, взятые вместе, образуют двухвалентный радикал формулы (с-1), (с-2) или (с-3), то R5 отличен от хлора. 2. Соединение по п.1, в которомL является прямой связью или двухвалентным радикалом, выбранным из -С(О)-, -С(О)-NH- илиZ является радикалом, выбранным из (b-5) или (b-7); и каждый R5 независимо выбирают из водорода или галогена. 4. Соединение по пп.1, 2 или 3, в котором соединение выбирают из соединений 35, 36, 39, 1 и 43- 19012837 5. Соединение по п.1, в котором Z является радикалом, выбранным из (b-3), (b-4), (b-5), (b-6), (b-7),(b-8), (b-9), (b-10) или (b-11). 6. Применение соединения по любому из пп.1-5 в качестве лекарственного средства. 7. Фармацевтическая композиция, содержащая фармацевтически приемлемые носители и в качестве активного ингредиента терапевтически эффективное количество соединения по любому из пп.1-5. 8. Способ получения фармацевтической композиции по п.7, где фармацевтически приемлемые носители и соединение по любому из пп.1-5 тщательно смешивают. 9. Применение соединения для производства лекарственного средства для лечения заболевания, медиированного TANK и PARP, в котором указанным соединением является соединение формулы (I) его N-оксиды, фармацевтически приемлемые аддитивные соли и стереохимически изомерные формы, где пунктирные линии означают необязательные связи;L является прямой связью или двухвалентным радикалом, выбранным из -С(О)-, -C(O)-NH-, -NH-,-С(О)-O-C1-6 алкандиила- или -C1-6 алкандиила-;R3 является водородом или C1-6 алкилокси; где каждый из R5, R6, R7 и R8 независимо выбирают из водорода, галогена, амино, C1-6 алкила илиR7 и R8, взятые вместе, могут образовывать двухвалентный радикал формулы где каждый R9 независимо выбирают из водорода или C1-6 алкила. 10. Применение соединения по любому из пп.1-5 для производства лекарственного средства для ингибирования роста раковых клеток. 11. Применение по п.9 или 10, где лечение включает химиосенсибилизацию. 12. Применение по п.9 или 10, где лечение включает радиосенсибилизацию. 13. Комбинация соединения формулы (I) по любому из пп.1-5 с химиотерапевтическим агентом. 14. Способ получения соединения по п.1, отличающийся взаимодействием промежуточного соединения формулы (II) с промежуточным соединением формулы (III), в котором W является подходящей уходящей группой, с получением соединения формулы (I-а), где L1 является -С 1-6 алкандиил-NH- и обе пунктирные линии могут быть связями, в инертном к реакции растворителе с добавлением соответствующего основания
МПК / Метки
МПК: C07D 405/12, A61K 31/517, A61P 25/00, C07D 403/12, C07D 239/91, C07D 401/12
Метки: производные, хиназолинона, ингибиторов, качестве
Код ссылки
<a href="https://eas.patents.su/22-12837-proizvodnye-hinazolinona-v-kachestve-ingibitorov-parp.html" rel="bookmark" title="База патентов Евразийского Союза">Производные хиназолинона в качестве ингибиторов parp</a>
Производные хиназолиндиона в качестве ингибиторов parp

Номер патента: 11552
Опубликовано: 28.04.2009
Авторы: Кенни Людо Эдмон Жозефин, Сомерс Мария Викторина Франциска, Ваутерс Вальтер Баудевейн Леопольд, Мертенс Йозефус Каролус, Ван Дюн Якобус Альфонсус Йозефус
МПК: C07D 401/04, A61K 31/517, A61P 35/00...
Метки: производные, ингибиторов, хиназолиндиона, качестве
Формула / Реферат:
1. Соединение формулы (I) его N-оксидные формы, его фармацевтически приемлемые аддитивные соли и его стереохимически изомерные формы, где каждый X независимо обозначает или и когда X означает то Y означает каждый Y независимо обозначает или за исключением случая, когда X обозначает и тогда Y обозначает L1 означает прямую связь или двухвалентный радикал, выбранный из -C1-6алкандиила-, L2 означает прямую связь или двухвалентный радикал,...
Сульфонамидные производные в качестве ингибиторов рассасывания костной ткани и ингибиторов адгезии клеток,способ их получения, применение и фармацевтическая композиция

Номер патента: 3102
Опубликовано: 26.12.2002
Авторы: Пейман Ануширван, Бодари Сара Кэтрин, Вилл Дэвид Вильям, Шойнеманн Карлхайнц, Гурвест Жан-Франсуа, Кнолле Йохен, Гадек Томас, Карниато Дени, Макдауэлл Роберт, Катбертсон Роберт Эндрю
МПК: A61P 19/10, A61K 31/505, C07D 239/42...
Метки: производные, клеток,способ, костной, ингибиторов, качестве, композиция, фармацевтическая, получения, рассасывания, адгезии, применение, ткани, сульфонамидные
Формула / Реферат:
1. Сульфонамидные производные общей формулы I где R1 и R2 вместе образуют двухвалентный (С2-С3)алкиленовый радикал; R4 является Н или (С1-С6)алкилом; R5 является (С1-С10)алкилом, (С6-С14)арилом, (C5-C14)гетероарилом или (С6-С14)арил(С1-С10)алкильной группой, где арил, гетероарил или алкил возможно замещены R3, или 2-оксобицикло[2.2.1]гепт-1-илметильной группой; R3 является (C1-C4)алкилом, (C1-C4)алкилокси, галогеном, трифторметилом, циано,...
Гетероариламины-производные пиримидина и пиридазина в качестве ингибиторов гликогенсинтаза-киназы 3-бета (ингибиторов gskз)

Номер патента: 10859
Опубликовано: 30.12.2008
Авторы: Бейнстерс Петер Якобус Йоханнес Антониус, Жанссен Поль Адриан Ян, Виллемс Марк, Эмбрехтс Вернер Констант Йохан, Ван Акен Кун Жанн Альфонс, Хэрес Ян, Винкерс Хендрик Мартен, Лав Кристофер Джон, Фрейн Эдди Жан Эдгар, Койманс Люсьен Мария Хенрикус, Де Жонж Марк Рене, Дильс Гастон Станислас Марселла, Леви Паулус Йоаннес
МПК: A61K 31/505, A61K 31/50, A61K 31/435...
Метки: gskз, гликогенсинтаза-киназы, пиридазина, ингибиторов, качестве, пиримидина, 3-бета, гетероариламины-производные
Формула / Реферат:
1. Соединение формулы его N-оксид, фармацевтически приемлемая аддитивная соль, четвертичный амин и стереохимически изомерная форма, где кольцо А является R1 обозначает водород; X обозначает -O-; -O-C1-6алкил- или прямую связь; Z обозначает прямую связь, -С(=O)- или -NR1-C1-6алкил-; R обозначает водород или R20; R3 обозначает водород; галоген; циано; полигалогенС1-6алкил; R4 обозначает моноциклический, насыщенный или частично насыщенный или...
Производные фталазина в качестве ингибиторов фосфодиэстеразы 4

Номер патента: 3702
Опубликовано: 28.08.2003
Авторы: Мораццони Габриеле, Пеллачини Франко, Норчини Габриеле, Наполетано Мауро, Гранчини Джанкарло
МПК: C07D 237/30, A61K 31/502
Метки: фталазина, качестве, фосфодиэстеразы, ингибиторов, производные
Формула / Реферат:
1. Соединение формулы I где ---- представляет одинарную или двойную связь; B представляет метилен; A представляет пиридин, замещенный одним заместителем или большим количеством заместителей; R представляет два атома водорода или C = O группу, когда ---- представляет одинарную связь, или, когда ---- представляет двойную связь, R представляет водород, необязательно замещенный арил или гетероцикл, (C1-8)алкил, (C2-8)алкенил или (C2-8)алкинил,...
Производные хиназолина в качестве ингибиторов киназы

Номер патента: 5809
Опубликовано: 30.06.2005
Авторы: Панди Анджали, Цукуда Еидзи, Иде Синити, Ирие Дзунко, Ода Содзи, Скарборо Роберт М., Номото Юдзи, Мацуно Кендзи, Итимура Митио
МПК: C07D 403/12
Метки: ингибиторов, качестве, хиназолина, киназы, производные
Формула / Реферат:
1. Азотсодержащее гетероциклическое соединение формулы в которой R1 обозначает радикал, выбранный из группы, в которую входят -CN, -O-метил, -O-этил, -O-пропил, -O-изопропил, -O-бутил, -O-трет-бутил, -O-изоамил, 1-нафтилокси, 2-нафтилокси, 4-индолилокси, 5-индолилокси, 5-изохинолилокси и их пространственные изомеры и гомологи; R2 и R4 каждый представляют собой -O-CH3 или -O(-CH2)n-R3; причем, когда R2 представляет собой -O-CH3, R4 представляет...
Предыдущий патент: Способ получения водорастворимых дитерпенов и их применение
Следующий патент: Синергетически термостабильная композиция масла для жарки и способ ее производства
Случайный патент: Заградительное устройство