Производные хиназолина в качестве ингибиторов киназы
Номер патента: 5809
Опубликовано: 30.06.2005
Авторы: Панди Анджали, Ода Содзи, Цукуда Еидзи, Номото Юдзи, Ирие Дзунко, Мацуно Кендзи, Итимура Митио, Иде Синити, Скарборо Роберт М.




Формула / Реферат
1. Азотсодержащее гетероциклическое соединение формулы

в которой R1 обозначает радикал, выбранный из группы, в которую входят -CN, -O-метил, -O-этил, -O-пропил, -O-изопропил, -O-бутил, -O-трет-бутил, -O-изоамил, 1-нафтилокси, 2-нафтилокси, 4-индолилокси, 5-индолилокси, 5-изохинолилокси и их пространственные изомеры и гомологи;
R2 и R4 каждый представляют собой -O-CH3 или -O(-CH2)n-R3; причем, когда R2 представляет собой -O-CH3, R4 представляет собой -O(-CH2)n-R3, а когда R2 представляет собой -O(-CH2)n-R3, R4 представляет собой -O-CH3;
R3 обозначает радикал, выбранный из группы, в которую входят -OH, -O-CH3, -O-CH2-CH3, -NH2, -N(CH3)2, -NH(-CH2-Ph), -NH(Ph), -CN,

n равно 2 или 3;
и все его фармацевтически приемлемые изомеры, соли, гидраты, сольваты и пролекарственные производные.
2. Соединение по п.1, выбранное из группы, в которую входят
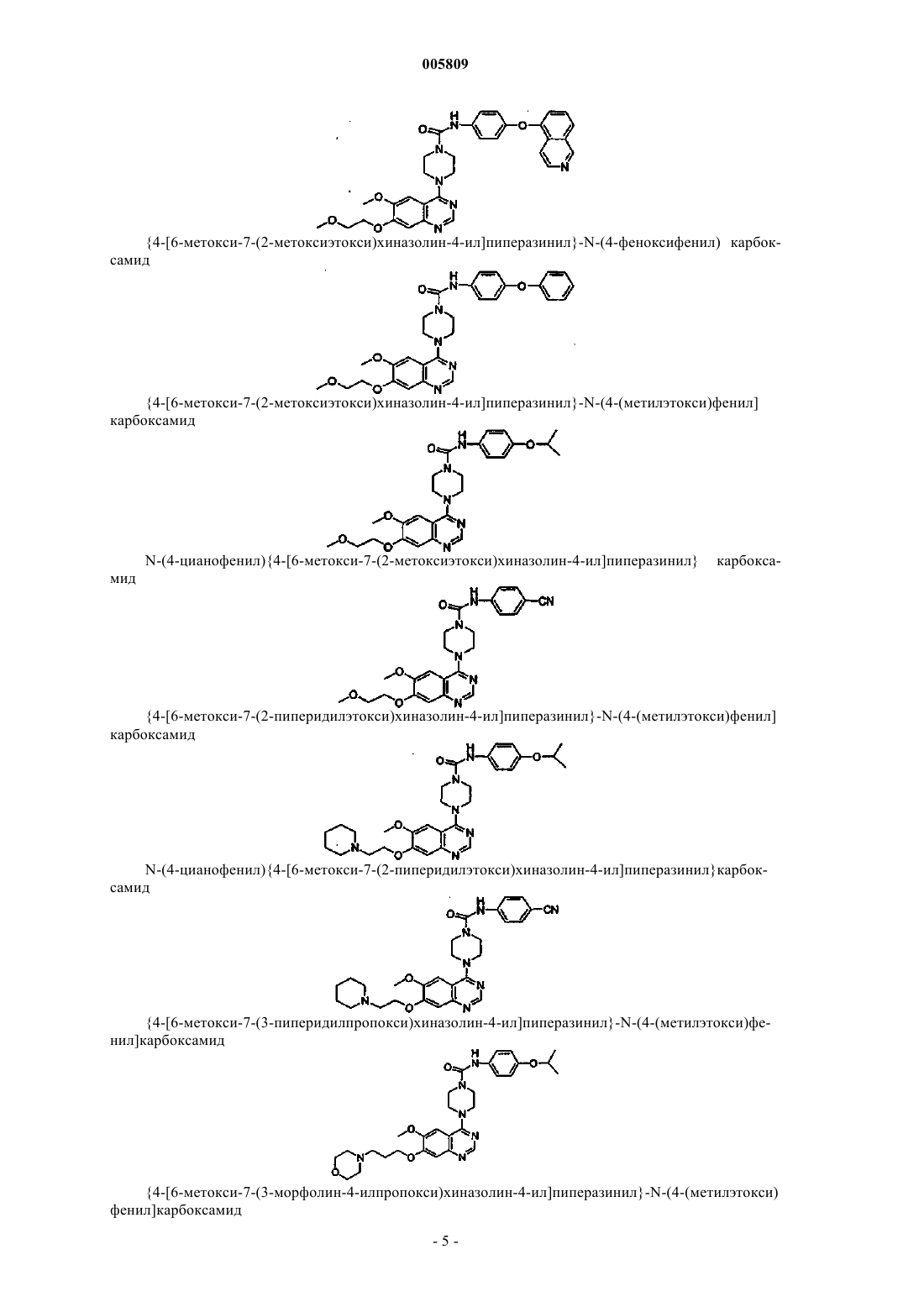
N-(4-индол-5-илоксифенил){4-[6-метокси-7-(2-метоксиэтокси)хиназолин-4-ил]пиперазинил}карбоксамид

N-(4-индол-4-илоксифенил){4-[6-метокси-7-(2-метоксиэтокси)хиназолин-4-ил]пиперазинил}карбоксамид

{4-[6-метокси-7-(2-метоксиэтокси)хиназолин-4-ил]пиперазинил}-N-(4-(2-нафтилокси)фенил)карбоксамид

N-(4-(5-изохинолилокси)фенил){4-[6-метокси-7-(2-метоксиэтокси)хиназолин-4-ил]пиперазинил}карбоксамид

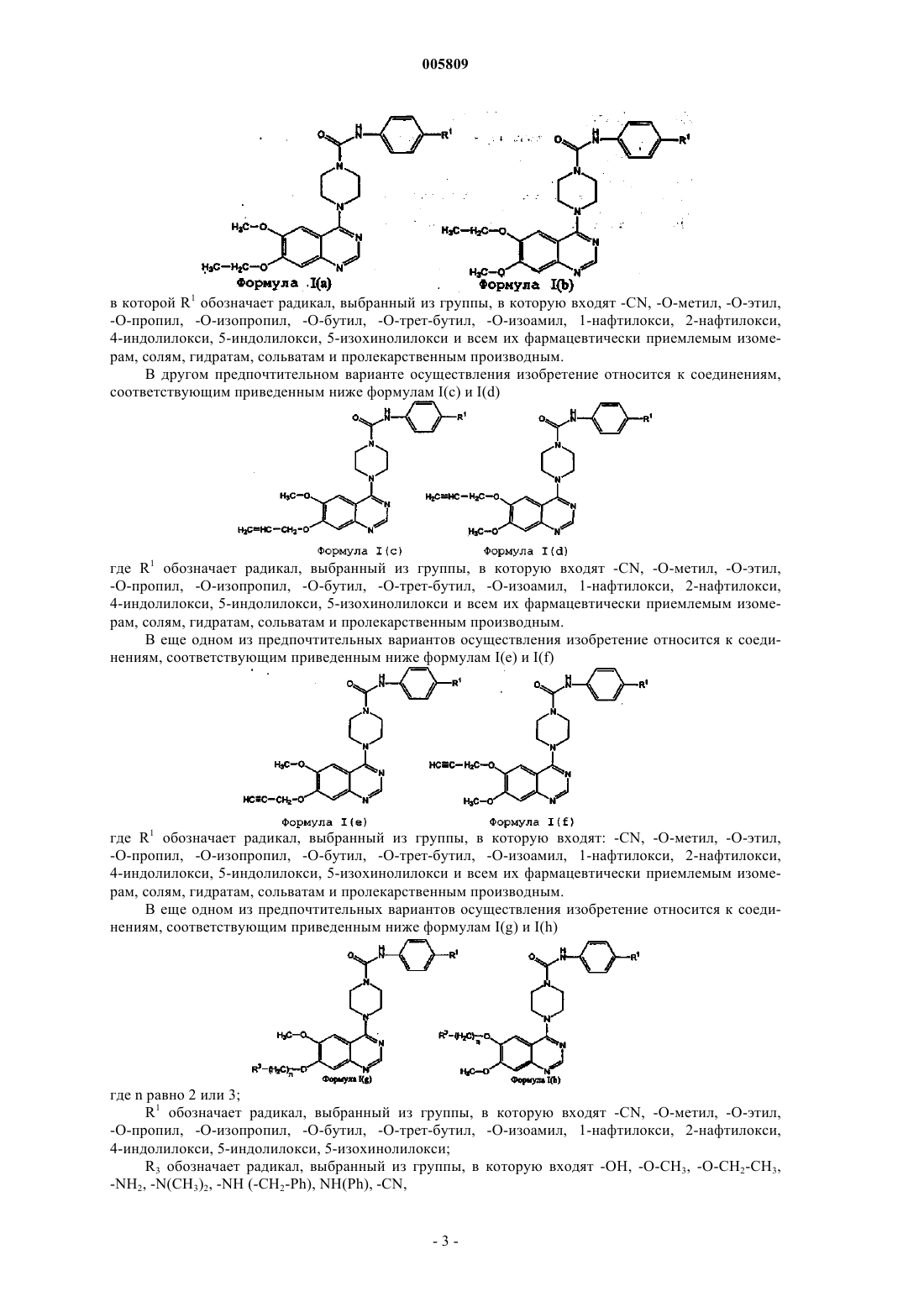
{4-[6-метокси-7-(2-метоксиэтокси)хиназолин-4-ил]пиперазинил}-N-[4-(метилэтокси)фенил]карбоксамид

N-(4-цианофенил){4-[6-метокси-7-(2-метоксиэтокси)хиназолин-4-ил]пиперазинил}карбоксамид

{4-[6-метокси-7-(2-пиперидилэтокси)хиназолин-4-ил]пиперазинил}-N-[4-(метилэтокси)фенил]карбоксамид

N-(4-цианофенил){4-[6-метокси-7-(2-пиперидилэтокси)хиназолин-4-ил]пиперазинил}карбоксамид

{4-[6-метокси-7-(3-пипередилпропокси)хиназолин-4-ил]пиперазинил}-N-[4-(метилэтокси)фенил]карбоксамид

{4-[6-метокси-7-(3-морфолин-4-илпропокси)хиназолин-4-ил]пиперазинил}-N-[4-(метилэтокси)фенил]карбоксамид

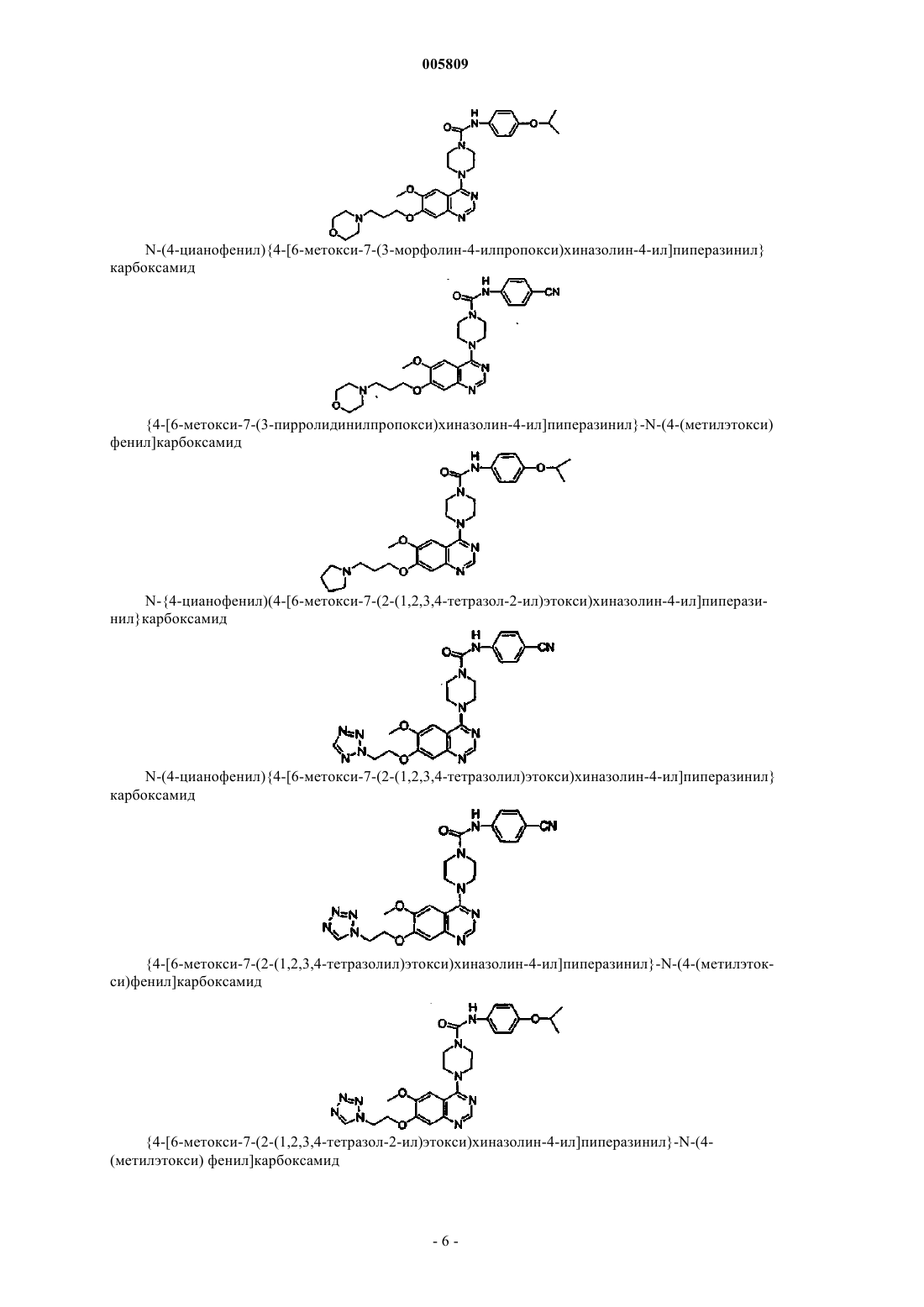
N-(4-цианофенил){4-[6-метокси-7-(3-морфолин-4-илпропокси)хиназолин-4-ил]пиперазинил}карбоксамид

{4-[6-метокси-7-(3-пирролидинилпропокси)хиназолин-4-ил]пиперазинил}-N-[4-(метилэтокси)фенил]карбоксамид

N-(4-цианофенил){4-[6-метокси-7-(2-(1,2,3,4-тетразол-2-ил)этокси)хиназолин-4-ил]пиперазинил}карбоксамид

N-(4-цианофенил) {4-[6-метокси-7-(2-(1,2,3,4-тетразолил)этокси)хиназолин-4-ил]пиперазинил}карбоксамид

{4-[6-метокси-7-(2-(1,2,3,4-тетразолил)этокси)хиназолин-4-ил]пиперазинил}-N-[4-(метилэтокси)фенил]карбоксамид

{4-[6-метокси-7-(2-(1,2,3,4-тетразол-2-ил)этокси)хиназолин-4-ил]пиперазинил}-N-[4-(метилэтокси)фенил]карбоксамид

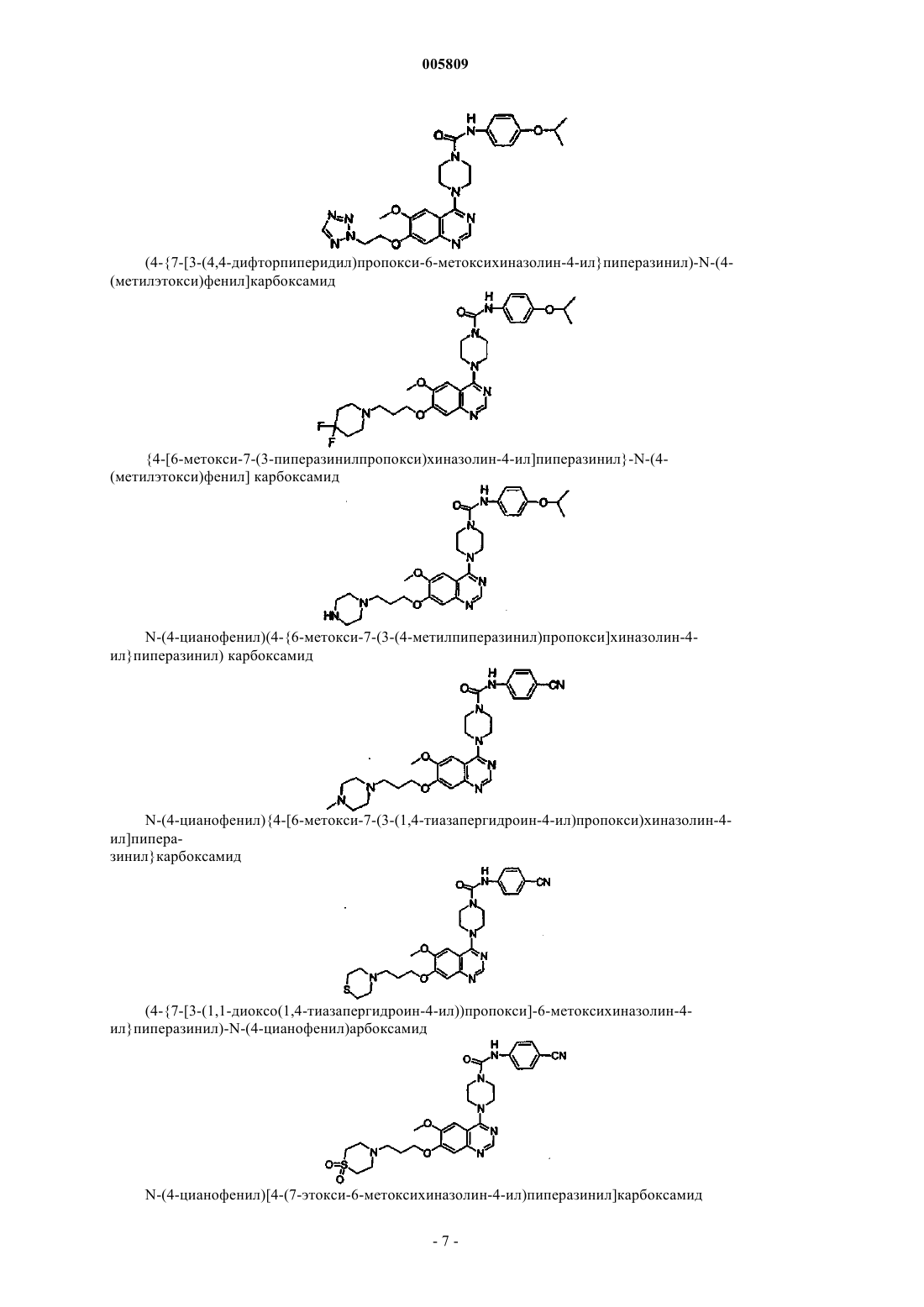
(4-{7-[3-(4,4-дифторпиперидил)пропокси-6-метокси]хиназолин-4-ил}пиперазинил)-N-[4-(метилэтокси)фенил]карбоксамид

{4-[6-метокси-7-(3-пиперазинилпропокси)хиназолин-4-ил]пиперазинил}-N-[4-(метилэтокси)фенил]карбоксамид

N-(4-цианофенил)(4-{6-метокси-7-[3-(4-метилпиперазинил)пропокси]хиназолин-4-ил}пиперазинил)карбоксамид

N-(4-цианофенил){4-[6-метокси-7-(3-(1,4-тиазапергидроин-4-ил)пропокси)хиназолин-4-ил]пиперазинил}карбоксамид
(4-{7-[3-(1,1-диоксо(1,4-тиазапергидроин-4-ил))пропокси]-6-метоксихиназолин-4-ил}пиперазинил)-N-(4-цианофенил)карбоксамид

N-(4-цианофенил)[4-(7-этокси-6-метоксихиназолин-4-ил)пиперазинил]карбоксамид

[4-(7-этокси-6-метоксихиназолин-4-ил)пиперазинил]-N-[4-(метилэтокси)фенил]карбоксамид

[4-(7-этокси-6-метоксихиназолин-4-ил)пиперазинил]-N-[4-(индол-4-илокси)фенил]карбоксамид

(4-{6-метокси-7-[3-(2-метилпиперидил)пропокси]хиназолин-4-ил}пиперазинил)-N-[4-(метилэтокси)фенил]карбоксамид

(4-{6-метокси-7-[3-(4-метилпиперидил)пропокси]хиназолин-4-ил}пиперазинил)-N-[4-(метилэтокси)фенил]карбоксамид

N-(4-цианофенил)[4-{6-метокси-7-[3-(2-метилпиперидил)пропокси]хиназолин-4-ил}пиперазинил]карбоксамид

N-(4-цианофенил)[4-{6-метокси-7-[3-(4-метилпиперидил)пропокси]хиназолин-4-ил}пиперазинил]карбоксамид

и их фармацевтически приемлемые изомеры, соли, гидраты, сольваты и пролекарственные производные.
3. Фармацевтическая композиция, содержащая эффективное количество азотсодержащего гетероциклического соединения по п.1 или 2 или его фармацевтически приемлемой соли и фармацевтически приемлемый разбавитель или носитель.
4. Способ ингибирования фосфорилирования рецептора ФРТ у больного, включающий стадию введения больному композиции по п.3.
5. Способ ингибирования аномального клеточного роста и клеточного блуждания у больного и таким образом предупреждения или лечения клеточно-пролиферативного заболевания, включающий стадию введения больному композиции по п.3.
6. Способ по п.5, в котором упомянутое клеточно-пролиферативное заболевание принадлежит к группе, в которую входят артериосклероз, васкулярная реобструкция, рестеноз, рак и гломерулосклероз.
7. Соединение по п.1, имеющее формулу

и все его фармацевтически приемлемые изомеры, соли, гидраты, сольваты, пролекарственные производные.
8. Соединение по п.1, имеющее следующую формулу

и все его фармацевтически приемлемые изомеры, соли, гидраты, сольваты, пролекарственные производные.
9. Соединение по п.1, имеющее следующую формулу

и все его фармацевтически приемлемые изомеры, соли, гидраты, сольваты, пролекарственные производные.
10. Соединение по п.1, имеющее следующую формулу

и все его фармацевтически приемлемые изомеры, соли, гидраты, сольваты, пролекарственные производные.
Текст
005809 Область техники, к которой относится изобретение Настоящее изобретение относится к азотсодержащим гетероциклическим соединениям и их фармацевтически приемлемым солям, которые обладают ингибирующей активностью в отношении фосфорилирования киназ, ингибирующего активность последних. Изобретение относится также к способу ингибирования киназ и лечению болезненных состояний у млекопитающих путем ингибирования фосфорилирования киназ. Известно, что ФРТ (фактор роста тромбоцитов) действует как отягчающий фактор в отношении клеточно-пролиферативных болезней, таких как артериосклероз, васкулярная реобструкция после подкожной коронарной ангиопластики или операции с шунтированием, рак гломерулонефрит,гломерулосклероз, псориаз и артикулярный ревматизм [Cell, 46, 155-169 (1986); Science, 253, 11291132 (1991); Nippon Rinsho (Japanese J. of Clinical Medicine), 50, 3038-3045 (1992); Nephrol DialTransplant, 10, 787-795 (1995); Kidney International, 43 (Suppl.39), 86-89 (1993); Journal of Rheumatology, 21, 1507-1511 (1994); Scandinavian Journal of Immunology, 27, 285-294 (1988) и т.д.]. Из используемых в качестве лекарственных средств производных хиназолина в патенте ЮАР 67 06512 (1968) описан в качестве бронхо-дилататора амид N,N-диметил-4-(6,7-диметокси-4 хиназолинил)-1-пиперазинкарбоновой кислоты. Производные диметоксихиназолина описаны в качестве ингибиторов фосфорилирования рецептора фактора роста эпидермиса (ФРЭ) в японской опубликованной нерассмотренной патентной заявке 208911/93 и WO 96/09294. Производные хинолина, обладающие активностью агониста рецептора бензодиазепина, описаны в Pharmacology(1966), a используемые в качестве антипаразитарных агентов производные хинолина описаны в Indian Journal of Chemistry, 26B, 550-555 (1987). Известные в настоящее время ингибиторы фосфорилирования рецептора ФРТ включают бисмоно- или бициклические арильные соединения и гетероарильные соединения (WO 92/20642), производные хиноксалина [Cancer Research, 54, 6106 (1994)], производные пиримидина (японская опубликованная нерассмотренная патентная заявка 87834/94) и производные диметоксихинолина [Abstracts of the 16th Annual Meeting of the Pharmaceutical Society of Japan (Kanazawa) (1966), 2, p.275, 29(C2) 15-2]. Сущность изобретения Настоящее изобретение относится к азотсодержащим гетероциклическим соединениям и их фармацевтически приемлемым солям, которые обладают ингибирующей активностью в отношении фосфорилирования киназ, ингибирующего активность последних. Особенно значительным согласно изобретению ингибированием киназ является ингибирование рецепторных тирозинкиназ, включающих рецептор фактора роста тромбоцитов (ФРТ), Flt3, CSF-1R, рецептора фактора роста эпидермиса (ФРЭ), фактора роста фибробластов (ФРФ), рецептора фактора роста сосудистого эндотелия (РФРСЭ) и проч. Другим типом киназного ингибирования согласно изобретению является ингибиторная активность в отношении безрецепторных тирозинкиназ, включающих src и abl и т.п. Третьим типом киназного ингибирования согласно изобретению является ингибиторная активность в отношении серин/треонинкиназ, включающих такие киназы, как МАРК, МЕК, и циклинзависимые киназы (ЦЗК, CDK), способствующие клеточной пролиферации, АКТ и CDK такие, которые способствуют клеточному выживанию, и NIK, регулирующие воспалительные реакции. Ингибирование таких киназ может быть использовано для лечения болезней, включающих клеточное выживание,пролиферацию и миграцию, в том числе сердечно-сосудистое заболевание, такое как артериосклероз и васкулярная реобструкция, рак, гломерулосклероз, фиброзные заболевания и воспаление, а также для общего лечения клеточно-пролиферативных заболеваний. В одном из своих предпочтительных вариантов осуществления изобретение относится к соединениям и их фармацевтически приемлемым солям, которые ингибируют или предотвращают ингибирование фосфорилирования по меньшей мере одного рецептора ФРТ по меньшей мере одной тирозинкиназой. Такого рода киназное ингибирование рецептора ФРТ может препятствовать аномальному клеточному росту и клеточному блужданию, благодаря чему такие соединения обладают ценностью для профилактики или лечения клеточно-пролиферативных заболеваний, таких как артериосклероз, васкулярная реобструкция, рак и гломерулосклероз. Настоящее изобретение относится к азотсодержащим гетероциклическим соединениям, представленным следующей формулой (I):R1 обозначает радикал, выбранный из группы, в которую входят: -CN, -X, -СХ 3, -R5, -CO2R5,-C(O)R5, -SO2R5, линейный или разветвленный -О-(C1-C8)-алкил, -О-фенил, -О-нафтил, -О-индолил и -О-изохинолинил;R5 обозначает водород или линейный или разветвленный C1-C8-алкил;R6 обозначает -ОН, -X или линейный или разветвленный C1-C8-алкил; п равно 2 или 3;R3 обозначает радикал, выбранный из группы, в которую входят: -ОН, -О-СНз, -О-СН 2-СН 3,-NH2, -N(CH3)2, -NH (-CH2-Ph), -NH(Ph), -CN, и всем их фармацевтически приемлемым изомерам, солям, гидратам, сольватам и пролекарственным производным. Наиболее предпочтительными соединениями, соответствующими приведенной выше формуле,являются соединения, у которых R1 является радикалом, выбранным из группы, в которую входятCN, -O-метил, -О-этил, -О-пропил, -O-изопропил, -О-бутил, -О-трет-бутил, -O-изоамил,1-нафтилокси, 2-нафтилокси, 4-индолилокси, 5-индолилокси, 5-изохинолилокси и их изомеры положения и гомологи, а также все фармацевтически приемлемые изомеры, соли, гидраты, сольваты и пролекарственные производные этих соединений. Наиболее предпочтительными являются также соединения, у которых R2 и R4 различны и один 2 из R и R4 означает О-СН 3, а также все фармацевтически приемлемые изомеры, соли, гидраты, сольваты и пролекарственные производные этих соединений. Фармацевтически приемлемые соли соединений формулы I включают фармацевтически приемлемые солевые аддукты с кислотами, соли с металлами, аммониевые соли, солевые аддукты с органическими аминами, солевые аддукты с аминокислотами и т.п. Примерами фармацевтически приемлемых солевых аддуктов с кислотами соединений формулы I являются солевые аддукты с неорганическими кислотами, такие как гидрохлорид, сульфат и фосфат, и солевые аддукты с органическими кислотами, такие как ацетат, малеат, фумарат, тартрат,цитрат и метансульфонат. Примерами фармацевтически приемлемых солей с металлами являются соли с щелочными металлами, такие как натриевая соль и калиевая соль, соли с щелочноземельными металлами, такие как магниевая соль и кальциевая соль, алюминиевая соль и цинковая соль. Примерами фармацевтически приемлемых аммониевых солей являются соль аммония и соль тетраметиламмония. Примеры фармацевтически приемлемых солевых аддуктов с органическими аминами включают соли с гетероциклическими аминами, такие как соли с морфолином и пиперидином. Примерами фармацевтически приемлемых солевых аддуктов с аминокислотами являются соли с лизином, глицином и фенилаланином. В одном из предпочтительных вариантов осуществления изобретение относится к соединениям, соответствующим приведенным ниже формулам II(а) и I(b) в которой R1 обозначает радикал, выбранный из группы, в которую входят -CN, -О-метил, -О-этил,-О-пропил, -О-изопропил, -О-бутил, -O-трет-бутил, -О-изоамил, 1-нафтилокси, 2-нафтилокси,4-индолилокси, 5-индолилокси, 5-изохинолилокси и всем их фармацевтически приемлемым изомерам, солям, гидратам, сольватам и пролекарственным производным. В другом предпочтительном варианте осуществления изобретение относится к соединениям,соответствующим приведенным ниже формулам I(с) и I(d) где R1 обозначает радикал, выбранный из группы, в которую входят -CN, -О-метил, -О-этил,-О-пропил, -О-изопропил, -О-бутил, -O-трет-бутил, -O-изоамил, 1-нафтилокси, 2-нафтилокси,4-индолилокси, 5-индолилокси, 5-изохинолилокси и всем их фармацевтически приемлемым изомерам, солям, гидратам, сольватам и пролекарственным производным. В еще одном из предпочтительных вариантов осуществления изобретение относится к соединениям, соответствующим приведенным ниже формулам I(е) и I(f) где R1 обозначает радикал, выбранный из группы, в которую входят: -CN, -O-метил, -O-этил,-О-пропил, -О-изопропил, -О-бутил, -O-трет-бутил, -O-изоамил, 1-нафтилокси, 2-нафтилокси,4-индолилокси, 5-индолилокси, 5-изохинолилокси и всем их фармацевтически приемлемым изомерам, солям, гидратам, сольватам и пролекарственным производным. В еще одном из предпочтительных вариантов осуществления изобретение относится к соединениям, соответствующим приведенным ниже формулам I(g) и I(h) и всем их фармацевтически приемлемым изомерам, солям, гидратам, сольватам и пролекарственным производным. Фармацевтически приемлемые соли соединений формулы I включают фармацевтически приемлемые солевые аддукты с кислотами, соли с металлами, аммониевые соли, солевые аддукты с органическими аминами, солевые аддукты с аминокислотами и т.п. Настоящее изобретение не ограничивается перечисленными выше соединениями. Предусматриваются аналоги бициклических соединений. Ниже в качестве особо предпочтительных вариантов осуществления настоящего изобретения приведены соединения, выбранные из группы, в которую входят и всем их фармацевтически приемлемым изомерам, солям, гидратам, сольватам и пролекарственным производным.- 12005809 Получение соединений Соединения могут быть получены с использованием методов и последовательностей операций,в общем виде описанных в W0 98/14431, опубликованном 12 сентября 1998 г., который включен в настоящую заявку в качестве справочного материала. Исходные материалы также могут быть синтезированы или приобретены как описано в названном документе. При необходимости могут быть использованы уходящие группы, такие как галоген, низший алкокси, низший алкилтио, низший алкилсульфонилокси, арилсульфонилокси и т.д., за исключением стадии реакции, за которой следует удаление защитных групп. Подходящими защитными группами являются, в частности, защитные группы, описанные Т. W. Greene в Protective Groups in Organic Synthesis, John WileySons Inc.(1981) и т.д., такие как этоксикарбонил, трет-бутоксикарбонил, ацетил и бензил. Защитные группы могут быть введены и удалены с помощью обычных методов, используемых в синтетической органической химии [напр., Т. W. Greene, Protective Groups in Organic Synthesis,John WileySons Inc. (1981)]. Если в таких процессах определенные группы изменяются в условиях применяемого метода или не подходят для осуществления метода, желаемое соединение может быть получено с использованием способов введения или удаления защитных групп, которые традиционно используются в синтетической органической химии [напр., T.W. Greene, Protective Groups in Organic Synthesis, JohnWileySons Inc. (1981)] и т.д. Превращение содержащихся в заместителях функциональных групп может быть выполнено известными способами [напр., R. С. Larock, Comprehensive Organic Transformations (1989)] в дополнение к описанным выше процессам, а некоторые из активных соединений формулы I могут быть использованы в качестве промежуточных соединений для последующего синтеза новых производных, соответствующих формуле I. Промежуточные и целевые соединения в описанных выше процессах могут быть выделены и очищены с помощью способов очистки, традиционно используемых в синтетической органической химии, например нейтрализации, фильтрации, экстракции, промывки, сушки, упаривания, перекристаллизации и различных типов хроматографии. Промежуточные соединения могут быть использованы для последующей реакции и без очистки. Некоторые соединения формулы I могут существовать в виде таутомеров, и настоящее изобретение охватывает все возможные изомеры, включая таутомеры и их смеси. В тех случаях, когда хиральные атомы углерода являются причиной существования двух разных энантиомеров, рассматриваются оба энантиомера, а также способы разделения двух энантиомеров. В том случае, когда желательна соль какого-либо соединения формулы I, и это соединение получено в виде желаемой соли, последняя может быть подвергнута очистке как таковая. В том случае, когда какое-либо соединение формулы I получено в свободной форме и желательна его соль,соединение формулы I растворяют или суспендируют в подходящем органическом растворителе,после чего для образования соли добавляют кислоту или основание. Приведенные ниже не ограничивающие изобретения схемы реакций I и II иллюстрируют предпочтительные варианты осуществления изобретения в том, что касается получения соответствующих изобретению соединений. Схема I Синтез трет-бутилового эфира 4-[6-метокси-7-(фенилметокси)хиназолин-4-ил]пиперазинкарбоновой кислоты дает промежуточный продукт, который может быть использован в синтезе разных соединений (схема может быть применена для получения бициклических изомеров положения), как описано выше для формулы I. Ванилиновую кислоту бензилируют с последующим нитрованием дымящей азотной кислотой при приблизительно 100 С. Нитрогруппу восстанавливают с помощью восстанавливающего агента, такого как хлорид олова и ему подобного, и затем циклизуют с помощью основания, такого как формамид, при повышенной температуре преимущественно в пределах от 100 до 200 С с образованием хиназолинона. Синтез 4-хлорхиназолинона проводят, обрабатывая хиназолинон галогенирующими агентами, такими как тионилхлорид, оксалилхлорид и хлорокись- 13005809 фосфора, в присутствии растворителя, такого как толуол или четыреххлористый углерод. Этот промежуточный продукт получают обработкой 4-хлорхиназолинона Вос-пиперазином в подходящем растворителе, таком как изопропиловый спирт, ацетонитрил или ТГФ, при комнатной температуре или при температуре кипения в течение 1-6 ч в присутствии триэтиламинового или пиридинового основания. Схема II Приведенная в качестве иллюстрации cхема II иллюстрирует синтез различных промежуточных соединений на основе замещенной мочевины из промежуточного соединения, полученного поcхеме I или другими способами. Промежуточное соединение из схемы I (или его бициклический изомер положения) дебензилируют в условиях гидрирования с последующим алкилированием с помощью разных замещенных алкилгалогенидов. Удаление защитной группы Воc осуществляют с помощью трифторуксусной кислоты с последующим действием различных изоцианатов с образованием конечных производных мочевины. В тех случаях, когда изоцианаты не являются коммерческими продуктами, промежуточное пиперазиновое соединение может быть обработано фосгеном с образованием карбамоилхлоридного промежуточного соединения, которое вводят во взаимодействие с различными замещенными анилинами. На пиперазиновое промежуточное соединение можно также подействовать п-нитрофенилхлорформиатом с образованием нитрофенилкарбаматного промежуточного соединения, которое при взаимодействии с различными анилинами может дать желаемые мочевины. Если производное мочевины имеет концевую группу NH2 (или группу NH2, у которой один или более атомов водорода замещены способным к замене заместителем), такое соединение может быть использовано в качестве промежуточного соединения для получения производного мочевины с концевыми группами -NH-Ph-R1. Аналогичным образом, если на фенильной группе желательна другая группа R1, замещаемая уходящая группа, находящаяся в п-положении фенила, может быть заменена в результате реакции сочетания с введением данного заместителя R1 в соответствии с приведенным выше описанием для формулы I. Такого рода последовательности операций для получения заявленных соединений являются лишь иллюстрацией предпочтительного аспекта изобретения. Другие последовательности операций и адаптации станут очевидными рядовому специалисту из схем реакций и структур соединений по изобретению. Предполагается, что такие последовательности операций входят в рамки настоящего изобретения. Соединения формулы I и их фармацевтически приемлемые соли могут существовать в виде аддуктов с водой (гидратов) или различными растворителями, которые также входят в рамки настоящего изобретения. Для лучшего иллюстрирования настоящего изобретения предлагаются следующие не ограничивающие изобретения примеры. Пример 1. Промежуточное соединение трет-бутиловый эфир 4-[6-метокси-7-(2 пиперидилэтокси) хиназолин-4-ил]пиперазинкарбоновой кислоты было получено с использованием приведенных ниже последовательностей операций, описанных в общем виде в схемах реакций I и II. Стадия А. К раствору ванилиновой кислоты (25 г, 149 ммоль) в ДМФ (300 мл) добавляют К 2 СО 3 (102,7 г, 744 ммоль) и BnBr (44,2 г, 372 ммоль) и перемешивают образовавшуюся суспензию при комнатной температуре в течение ночи. Реакционную смесь фильтруют, добавляют этилацетат,промывают раствор солевым раствором, высушивают и упаривают. Очистка хроматографией на силикагеле дает 55 г (96%) промежуточного продукта. MS(ES) 349 (М+Н)+. Стадия В. К раствору защищенного бензилом продукта стадии А (20 г, 57,4 ммоль) в CH2Cl2(100 мл) при -10 С медленно прибавляют уксусную кислоту (100 мл). К полученному холодному раствору медленно прибавляют конц. НNО 3 (25,8 г, 574,4 ммоль), нагревают реакционную смесь до комнатной температуры и затем кипятят в течение ночи при 100 С. После этого выливают реакционную смесь на лед, экстрагируют продукт этилацетатом, промывают солевым раствором и высушивают над MgSO4. Растворитель удаляют в вакууме, получая искомый промежуточный продукт в виде желтого твердого вещества (21,8 г, 96,5%). MS(ES) 416 (M+Na). Стадия С. К раствору нитро-продукта стадии В (10,9 г, 27,7 ммоль) в этилацетате (100 мл) добавляют SnCl2.H2O (18,7 г, 83,1 ммоль) и нагревают реакционную смесь при 50 С в течение ночи. После охлаждения реакционную смесь фильтруют через целит, фильтрат промывают 10%-нымNаНСО 3 и экстрагируют этилацетатом. Органические слои высушивают и упаривают с образованием промежуточного амино-продукта в виде коричневого твердого вещества (9,5 г, 95%). MS(ES) 364(М+Н). Стадия D. Аминопродукт (3 г, 8,3 ммоль) стадии С растворяют в формамиде (20 мл). К раствору добавляют формиат аммония (781 мг, 12,4 ммоль) и нагревают реакционную смесь в течение 4 ч при 150 С. В течение этого времени по данным ЖХВР (жидкостной хроматографии высокого разрешения) расходуется все количество исходного материала. После охлаждения реакционную смесь выливают в воду с образованием осадка кремового цвета. Собранный с помощью фильтрации осадок является искомым промежуточным продуктом: циклизованного 7-бензилокси-6-метокси-4 хиназолинона (1,9 г, 81%). MS (ES)283 (M+H). Стадия Е. Смесь 7-бензилокси-6-метокси-4-хиназолинона (1 г, 3,5 ммоль, со стадии D), тионилхлорида (5 мл) и ДМФ (5 капель) греют при кипении в течение 4 ч. После охлаждения избыток тионилхлорида удаляют упариванием и полученный осадок подвергают перегонке с толуолом, получая в результате промежуточный 4-хлор-6-метокси-7-бензилоксихиназолин в виде желтого твердого вещества (652 мг, 62%). MS (ES) 301 (М+Н). Стадия F. К раствору 4-хлор-6-метокси-7-бензилоксихиназолина (1,8 г, 6 ммоль) в ТГФ (20 мл) добавляют Вос-пиперазин (2,2 г, 12 ммоль) и затем N,N-диизопропилэтиламин (DIEA) (4,2 мл, 24 ммоль), после чего нагревают реакционную смесь в течение ночи при 50 С. Растворитель упаривают, остаток растворяют в воде и экстрагируют продукт этилацетатом. Этилацетатный слой высушивают, фильтруют и упаривают, получая промежуточный третбутиловый эфир 4-[6-метокси-7-фенилметокси)хиназолин-4-ил]пиперазинкарбоновой кислоты в виде белого твердого вещества (2,2 г, 81%). MS(ES) 451 (М+Н). Стадия G. Бензилокси-соединение стадии F (500 мг, 1,1 ммоль) растворяют в этаноле (5 мл),добавляют Рd(OH)2/С (50 мг) и помещают смесь в гидрогенизатор Парра при давлении Н 2 3,5 кг/см 2. Реакционную смесь фильтруют через целит, промывают этанолом и упаривают растворитель, получая промежуточный дебензилированный продукт (400 мг, 98%). MS(ES) 361 (М+Н). Стадия Н. К раствору трет-бутилового эфира 4-[7-гидрокси-6-метоксихиназолин-4-ил]пиперазинкарбоновой кислоты (1,8 г, 5 ммоль) и Cs2CO3 (3,3 г, 10 ммоль) в ДМФ (10 мл) добавляют 1-хлорэтилтозилат (1,8 мл, 10 ммоль). Смесь перемешивают в течение ночи при комнатной температуре, упаривают растворитель и очищают сырой остаток с помощью ЖХВР с обращенной фазой,получая в качестве целевого продукта трет-бутиловый эфир 4-[6-метокси-7-(2-хлорэтокси)хиназолин-4-ил]пиперазинкарбоновой кислоты (850 мг, 40%). MS(ES) 423 (М+Н). Стадия I. К раствору исходного материала из стадии Н (450 мг, 1,2 ммоль) в ДМФ (10 мл) добавляют пиперидин (1,2 мл, 12 ммоль) и нагревают реакционную смесь в течение ночи при 80 С. Раствор упаривают и очищают сырой остаток с помощью ЖХВР с обращенной фазой, получая в качестве целевого продукта трет-бутиловый эфир 4-[6-метокси-7-(2-пиперидилэтокси)хиназолин-4- 15005809 ил]пиперазинкарбоновой кислоты (310 мг, 55%). MS(ES) 472 (М+Н). Пример 2. N-(4-Цианофенил)4-[6-метокси-7-(2-пиперидилэтокси)хиназолин-4-ил]пиперазинилкарбоксамид был получен с использованием промежуточного соединения, полученного в примере 1, и следующей последовательности операций, описанной в общем виде в схеме реакций II. К трет-бутиловому эфиру 4-[6-метокси-7-(2-пиперидилэтокси)хиназолин-4-ил]пиперазинкарбоновой кислоты (из примера 1 (стадия I), 111 мг, 0,3 ммоль) добавляют 4 н. НСl в диоксане (1 мл) и перемешивают реакционную смесь 1 ч при комнатной температуре. Упаривают растворитель и несколько раз азеотропируют с пентаном, получая продукт с удаленной Вос-группой. К этому остатку добавляют ДМФ (2 мл) и затем 4-цианофенилизоцианат (75 мг, 0,45 ммоль), после чего реакционную смесь перемешивают при комнатной температуре в течение ночи. Растворитель упаривают и очищают остаток с помощью ЖХВР с обращенной фазой, получая в качестве целевого продукта N(4-цианофенил)4-[6-метокси-7-(2-пиперидилэтокси)хиназолин-4-ил]пиперазинилкарбоксамид в виде белого твердого вещества (89 мг, 59%). MS(ES) 516 (М+Н). Примеры 3, 4. N-(4-Цианофенил)4-[6-метокси-7-(2-(метокси)этокси)хиназолин-4-ил]пиперазинил карбоксамид. был получен с использованием последовательности операций, описанных в примерах 1 и 2, за исключением того, что в качестве алкилирующего агента вместо 1-хлорэтокситозилата был использован 1-бромэтилметиловый эфир. Целевой продукт получен с выходом соизмеримым с выходом, указанным в примерах 1 и 2. Фармакологическая активность соединений настоящего изобретения была установлена с использованием, в частности, следующих тест-процедур. Биологический тест-анализ Типа 1 Ингибирующий эффект соединений на аутофосфорилирование рецептора фактора роста тромбоцитов -PDGF.(1) Испытание на фосфорилирование на клетках HR5. Клеточная линия HR5 является клеточной линией клеток СНО, приготовленной с помощью клеточной инженерии для суперэкспресии р-PDGFR (рецептор ФРТ) человека. Эта клеточная линия может быть получена в АТСС. Уровень экспрессии -PDGFR в клетках HR5 имеет порядок 5 х 104 рецепторов на клетку. Для испытания на фосфорилирование в соответствии с изобретением клеткиHR5 выращивали до слияния на микропластинах с 96 лунками в стандартных условиях культивирования ткани с последующим сывороточным голоданием в течение 16 ч. Находящиеся в покое клетки инкубировали 30 мин при 37 С без или с увеличивающимися концентрациями тестируемого соединения (0,01-30 мкм) с последующим добавлением в течение 10 мин 8 нМ PDGF ВВ. Клетки были лизированы в системе 100 мМ Трис, рН 7,5, 750 мМ NaCl, 0,5% Тритон Х-100, 10 мМ пирофосфат натрия, 50 мМ NaF, 10 мкм/мл апротинин, 10 мкм/мл лейпептин, 1 мМ фенилметилсульфонилфторид, 1 мМ ванадат натрия, после чего лизат осветляли в течение 5 мин центрифугированием при 15000 г. Осветленные лизаты были перенесены на вторую микропластину, каждая лунка которой была предварительно покрыта 500 нг 1 В 5 В 11 антиPDGF mAb и инкубировали 2 ч при комнатной температуре. После трехкратной промывки связывающим буфером (0,3% желатина, 15 мМ Hepes рН 7,5, 100 мМ NaCl, 0,01% Tween) было добавлено 250 нг/мл поликлонального антифосфотирозинового антитела кролика (Transduction Laboratory) и пластины инкубировали 60 мин при 37 С. После этого каждую лунку трижды промывали связывающим буфером и инкубировали 60 мин при 37 С с 1 мкг/мл лошадиного противокроличьего анти-антитела, коньюгированного с пероксидазой хрена (Boehringer Mannheim). В промытые после этого лунки был добавлен ABTS(Sigma) и за скоростью образования субстрата следили при 650 нм. Результаты испытания приведены в виде IC50 (что означает концентрацию соединения по изобретению, ингибирующую на 50% фосфорилирование рецептора ФРТ) в сравнении с контрольными клетками, не подвергнутыми действию соединения по изобретению. Примеры таких результатов в виде IC50 в проведенном на HR5 испытании соединений по изобретению представлены ниже в табл. 1.(2) Испытание на фосфорилирование на MG63. Клеточная линия MG63 является клеточной линией остеосаркомной опухоли человека, которая может быть получена в АТСС. В испытании измеряется фосфорилирование эндогенного -PDGFR в клетках MG63. Условия испытания такие же, как описаны для клеток HR5, за исключением того,что проводилось PDGF-BB-стимулирование в присутствии или в отсутствие 45%-ной плазмы человека. Результаты испытания на MG63 приведены в виде IС 5 о (означающего концентрацию соединения по изобретению, ингибирующую на 50% фосфорилирование рецептора ФРТ) в сравнении с контрольными клетками, не подвергнутыми действию соединения по изобретению. Примеры таких результатов в виде IС 50 в проведенном на MG63 испытании соединений по изобретению представлены ниже в табл. 1. Результаты испытаний соединений из примеров 2 и 4 представлены в приведенной ниже табл. 1. Таблица 1 Биологический тест-анализ Типа 2 Ингибирование роста клеток гладких мышц Клетки гладких мышц сосудов выделяют из аорты свиньи методом эксплантации и используют для теста. Клетки помещают в лунки 96-луночной пластины (8000 клеток на 1 лунку) и культивируют в течение 4 дней в модифицированной Dulbeccois среде Игла (DMEM; Nissui PharmaceuticalCo., Ltd.), содержащей 10% фетальной сыворотки быка (FBS; Hyclon). После этого клетки культивируют 3 дня в DMEM, содержащей 0,1% FBS и синхронизируют в стационарной фазе клеточного роста. В каждую лунку добавляют DMEM, содержащую 0,1% FBS, и разные концентрации испытуемого образца и вызывают рост клеток с помощью PDGF-BB (SIGMA, конечная концентрация 20 нг/мл). После культивирования в течение 3 дней измеряют клеточный рост с помощью аналитического набора для клеточного роста (Boehringer Mannheim) по методу ХТТ [J. Immunol. Methods,142, 257-265 (1991)] и вычисляют степень клеточного роста по следующему уравнению: Степень клеточного роста = 100 х 1-(М-РО)/(Р 100-РО), где Р 100 обозначает поглощение реагента ХТТ при стимулировании с помощью PDGF-BB; РО обозначает поглощение реагента ХТТ без стимулирования с помощью PDGF-BB; и М обозначает поглощение реагента ХТТ после добавления образца при стимулировании с помощью PDGF-BB. Результаты теста выражены в виде концентрации тестируемого соединения, которая ингибирует клеточный рост на 50% (IC50) . Биологический тест-анализ Типа 3 Ингибирующий эффект в отношении гипертрофии сосудистой интимы Крыс-самцов SD (вес 375-445 г, Charles River, золотой стандарт) анестезируют с помощью пентобарбитала натрия (50 мг/кг внутрибрюшинно), после чего на шее каждого животного делают медиальный надрез и затем производят ретроградное введение баллонного катетера (2F, EdwardsLaboratories) в левую наружную сонную артерию. После семикратного повторения такой операции катетер извлекают, левую наружную сонную артерию перевязывают и зашивают рану. Тестируемое соединение суспендируют в 0,5%-ном растворе Tween 80 в водном растворе хлорида натрия до концентрации 20 мг/мл в случае внутрибрюшинного введения и в 0,5%-ном растворе метилцеллюлозы 400 до концентрации 6 мг/мл в случае перорального введения. Суспензию вводят один раз в сутки в случае внутрибрюшинного применения и один или два раза в сутки в случае перорального применения в течение 15 дней, начиная с дня, предшествующего повреждению баллонным катетером. На 14-й день после повреждения баллонным катетером животное забивают и удаляют его левую наружную сонную артерию. Ткани фиксируют формалином, покрывают парафином и делают срезы,которые окрашивают с помощью Elastica Wangeeson. Площадь поперечного сечения сосудистых тканей (интимы и средней оболочки стенки сосуда) измеряют с помощью анализатора изображений(Luzex F, NIRECO) и принимают отношение площадей интимы и средней оболочки (I/M) в качестве- 17005809 меры гипертрофии сосудистой интимы. Из полученных результатов очевидно, что применение соединений настоящего изобретения значительно ингибирует гипертрофию сосудистой интимы. Биологический тест-анализ Типа 4 Оценка с использованием модели адъювантного артрита крысы Мертвые клетки Mycobacterium bacterium (Difco Laboratories Inc.) разрушают в агатовой ступке и суспендируют в жидком парафине до конечной концентрации 6,6 мг/мл, после чего стерилизуют паром высокого давления. Затем 100 мл суспензии инъецируют подкожно в правую заднюю стопу каждого животного из группы 8-недельных крыс-самок Льюис (Charles River Japan) (по 6 животных в группе) с целью инициирования адъювантного артрита. Тестируемое соединение суспендируют в 0,5%-ном растворе метилцеллюлозы до конечной концентрации 3 мг/мл и, начиная с момента непосредственно перед инициированием артрита, суспензию вводят перорально в количестве от 100 мл/100 г веса тела 1 раз в сутки 5 дней в неделю. Контрольной группе вводят 0,5%-ный раствор метилцеллюлозы. Нормальная группа не получает адъювантную обработку и не подвергается введению тестируемого соединения. Введение тестируемого соединения продолжают до 18-го дня после адъювантной обработки. На 17-й день подсчитывают число лейкоцитов в периферической крови, а на 18-й день забирают всю кровь и производят вскрытие. Измеряют и рассчитывают изменение веса тела со временем, изменение отека в задней стопе со временем, вес селезенки и тимуса, число лейкоцитов в периферической крови, содержание гидроксипролина в моче, содержание глюкозаминогликана в моче, концентрацию SH в сыворотке, содержание монооксида азота в сыворотке и концентрацию мукопротеина в сыворотке. Объем каждой из задних ступней измеряют с использованием прибора для измерения отека задней ступни крыс(ТК-1-1, Unicom). Число лейкоцитов в периферической крови подсчитывают с помощью автоматического многоканального счетчика кровяных клеток (Sysmex К-2000, Toa Iyo Denshi Co., Ltd.). Содержание гидроксипролина в моче измеряют по методу, описанному Ikeda et al. в Annual Report ofTokyo Metropolitan Research Laboratories P.H., 36, 277 (1985), a содержание глюкозаминогликана измеряют по методу, описанному Moriyama et al. в Hinyo Kiyo, 40, 565 (1994) и Klompmakers et al. вal. в Inflammation, 17, 595 (1993), а концентрацию монооксида азота измеряют по методу, описанному Tracey et al. , Journal of PharmacologyExperimental Therapeutics, 272, 1011 (1995). Концентрацию мукопротеина измеряют с помощью Aspro GP Kit (Otsuka Pharmaceutical Co., Ltd.). Степень ингибирования для каждого показания рассчитывают в соответствии со следующим уравнением:% ингибирования = (Контрольная группа - Группа, получившая соединение)/(Контрольная группа Нормальная группа) х 100 Из результатов, полученных в приведенных испытаниях, очевидно, что соединение по изобретению ингибирует возникновение адъювантного артрита. Биологический тест-анализ Типа 5 Активность по модели мезангиального пролиферативного гломерулонефрита Антимоноклональное антитело крысы Thy-1.1 OX-7 (Sedaren) вводят внутривенно через вену хвоста крысам-самцам Wister-Kyoto (Charles River Japan, 160 г, 6 животных в группе) в количестве 1,0 мл/кг. Тестируемое соединение суспендируют в 0,5%-ном растворе метилцеллюлозы и полученную суспензию вводят каждой из крыс дважды в сутки в течение 7 дней, начиная со дня перед введением ОХ-7. На 7-й день после введения ОХ-7, когда рост мезангиальных клеток и гипертрофия внеклеточного матрикса становятся заметными, у каждой крысы удаляют почку, фиксируют ее в течение 6 ч 20%-ным забуференным формалином и делают срезы. Последние подвергают иммунотканевому окрашиванию с помощью антитела РС 10 (DAKO) против внутриядерного антигена пролиферативных клеток. После сравнительного окрашивания окрашивающим раствором метилового зеленого с использованием диаминобензидина в качестве цветного проявителя вносят частицы парафина. В срезе почки наблюдают половину клубочков и рассчитывают число клеток в одном клубочке, которые являются положительными по отношению к внутриядерному антигену пролиферативных клеток. Испытание на значимость разницы проводят с помощью теста Уилкинсона. Из полученных результатов очевидно, что соединения настоящего изобретения проявляют облегчающий эффект в отношении мезангиального пролиферативного гломерулонефрита. Соединения формулы I и их фармацевтически приемлемые соли могут применяться как таковые, но обычно предпочтительно их применение в виде фармацевтически приемлемых композиций,которые используют для животных и для человека. Предпочтительно применять такой способ введения, который наиболее эффективен для лечения. В частности, введение осуществляют перорально или неперорально, используя в последнем случае интраректальное, интраоральное, подкожное, внутримышечное или внутривенное введение. Примерами применяемых форм являются капсулы, таблетки, гранулы, порошки, сиропы,эмульсии, свечи и инъекции. Пригодные для перорального применения жидкие композиции, такие как эмульсии и сиропы,- 18005809 могут быть приготовлены с использованием воды, cахаров, таких как сахароза, сорбит и фруктоза,гликолей таких как полиэтиленгликоль и пропиленгликоль, масел таких как кунжутное масло, оливковое масло и соевое масло, консервантов, таких как бензоаты, вкусовых добавок, таких как земляничный привкус и мятное масло, и т.д. Капсулы, таблетки, порошки и гранулы могут быть приготовлены с использованием наполнителей, таких как лактоза, глюкоза, сахароза и маннит, распадающихся агентов, таких как крахмал и альгинат натрия, смазочных средств, таких как стеарат магния и тальк, связующих, таких как поливиниловый спирт, гидроксипропилцеллюлоза и желатин, поверхностно-активных веществ, таких как эфиры жирных кислот, пластификаторов, таких как глицерин, и т.д. Композиции, пригодные для неперорального применения, преимущественно включают стерилизованный водный препарат, содержащий активное соединение, которое является изотоническим по отношению к крови реципиента. В частности, инъекции приготовляют с использованием носителя, включающего солевой раствор, раствор глюкозы, смесь солевого раствора и раствора глюкозы. Композиции для местного применения приготовляют растворением или суспендированием активного соединения в одном или более видах растворителей, таких как минеральное масло, нефтепродукт и многоатомный спирт или другие основы, используемые для наружных лекарств. Композиции для кишечного применения приготовляют с использованием обычных носителей таких как масло какао, гидрированный жир или гидрированная карбоновая кислота жира, и предлагают в виде свечей. Композиции для неперорального применения могут быть также составлены таким образом,чтобы они содержали один или более видов добавок, выбранных из гликолей, масел, вкусовых добавок, консервантов (включая антиоксиданты) , наполнителей, распадающихся агентов, смазочных средств, связующих, ПАВ и пластификаторов, которые используют для приготовления композиций для перорального применения. Эффективная доза и порядок применения для каждого из соединений формулы I или их фармацевтически приемлемых солей могут варьировать в зависимости от способа применения, возраста и веса больного и от характера и стадии подлежащих лечению заболеваний. Однако обычно целесообразно применять соединения формулы I или их фармацевтически приемлемые соли в суточной дозе от 0,01 до 1000 мг для взрослого, преимущественно от 5 до 500 мг, за один прием или несколькими частями. Все соединения настоящего изобретения могут быть непосредственно применены для лечения киназозависимых заболеваний млекопитающих в роли ингибиторов киназы, в частности ингибиторов тирозинкиназы. Особо предпочтительными являются соединения, имеющие IC50 в пределах от 10 нМ до 10 м. Еще более предпочтительны соединения, имеющие IC50 в пределах от 10 нМ до 1 м. Наиболее предпочтительны соединения, которые имеют значение IC50 ниже 1 м. Могут быть выбраны конкретные соединения настоящего изобретения, обладающие способностью специфически ингибировать один из трех типов протеинкиназы (например киназу, которая фосфорилирует тирозин, киназу, которая фосфорилирует тирозин и треонин, и киназу, которая фосфорилирует треонин). Болезни, зависимые от тирозинкиназы, включают гиперпролиферативную дисфункцию, которую вызывает или поддерживает аномальная активность тирозинкиназы. В число примеров входят псориаз, легочный фиброз, гломерулонефрит, рак, атеросклероз и антиангиопоэз(например, раковый рост и диабетическая ретинопатия). Имеющиеся сведения о зависимости между другими классами киназ и конкретными болезнями недостаточны. Однако соединения, обладающие специфической протеинкиназо-ингибирующей активностью, обладают ценным лечебным эффектом. Таким же образом были определены и другие классы киназ. Кверцетин, генистеин и стауроспорин, являющиеся ингибиторами протеинкиназ, ингибируют наряду с тирозинкиназой многие другие типы протеинкиназ. Однако из-за отсутствия у них специфичности они проявляют высокую цитотоксичность. Таким образом, ингибитор протеинкиназ (или ингибитор других классов киназ), который способен проявлять нежелательные побочные эффекты из-за отсутствия селективности, может быть идентифицирован с помощью обычного теста, в котором измеряется цитотоксичность. Принимая во внимание изложенное выше описание, предполагается, что изобретение может быть реализовано рядовым специалистом. Приведенные выше примеры не являются ограничительными в том смысле, что специалист, обладающий обычными познаниями в изложенном выше предмете, сможет без труда предусмотреть другие модификации и варианты изобретения, не отступая от его основной концепции. Такие модификации и варианты также входят в рамки настоящего изобретения. Хотя настоящее изобретение в целях ясности и понимания было в определенной степени детализировано с помощью приведенных примеров, рядовому специалисту ясно, что, не отступая от сущности изобретения и не выходя за его рамки, могут быть произведены различные модификации и предложены равноценные варианты. Следует принять во внимание, что приведенные выше обсуждение и примеры представляют всего лишь детализированное описание некоторых предпочти- 19005809 тельных вариантов осуществления изобретения. Все патенты, журнальные статьи и другие обсуждаемые или упоминаемые выше документы полностью включены в настоящую заявку в качестве ссылочного материала. ФОРМУЛА ИЗОБРЕТЕНИЯ 1. Азотсодержащее гетероциклическое соединение формулыR1 обозначает радикал, выбранный из группы, в которую входят -CN, -О-метил, -О-этил,-О-пропил, -О-изопропил, -О-бутил, -O-трет-бутил, -O-изоамил, 1-нафтилокси, 2-нафтилокси, 4 индолилокси, 5-индолилокси, 5-изохинолилокси и их пространственные изомеры и гомологи;R2 и R4 каждый представляют собой -О-СН 3 или -О(-СН 2)n-R3; причем, когда R2 представляет собой -О-СН 3, R4 представляет собой -O(-CH2)n-R3, а когда R2 представляет собой -O(-CH2)n-R3, R4 представляет собой -О-СН 3;n равно 2 или 3; и все его фармацевтически приемлемые изомеры, соли, гидраты, сольваты и пролекарственные производные. 2. Соединение по п.1, выбранное из группы, в которую входят и их фармацевтически приемлемые изомеры, соли, гидраты, сольваты и пролекарственные производные. 3. Фармацевтическая композиция, содержащая эффективное количество азотсодержащего гетероциклического соединения по п.1 или 2 или его фармацевтически приемлемой соли и фармацевтически приемлемый разбавитель или носитель. 4. Способ ингибирования фосфорилирования рецептора ФРТ у больного, включающий стадию введения больному композиции по п.3. 5. Способ ингибирования аномального клеточного роста и клеточного блуждания у больного и таким образом предупреждения или лечения клеточно-пролиферативного заболевания, включающий стадию введения больному композиции по п.3. 6. Способ по п.5, в котором упомянутое клеточно-пролиферативное заболевание принадлежит к группе, в которую входят артериосклероз, васкулярная реобструкция, рестеноз, рак и гломерулосклероз. 7. Соединение по п.1, имеющее формулу и все его фармацевтически приемлемые изомеры, соли, гидраты, сольваты, пролекарственные производные. 8. Соединение по п.1, имеющее следующую формулу и все его фармацевтически приемлемые изомеры, соли, гидраты, сольваты, пролекарственные производные. 9. Соединение по п.1, имеющее следующую формулу и все его фармацевтически приемлемые изомеры, соли, гидраты, сольваты, пролекарственные производные. 10. Соединение по п.1, имеющее следующую формулу и все его фармацевтически приемлемые изомеры, соли, гидраты, сольваты, пролекарственные производные.
МПК / Метки
МПК: C07D 403/12
Метки: киназы, ингибиторов, производные, хиназолина, качестве
Код ссылки
<a href="https://eas.patents.su/27-5809-proizvodnye-hinazolina-v-kachestve-ingibitorov-kinazy.html" rel="bookmark" title="База патентов Евразийского Союза">Производные хиназолина в качестве ингибиторов киназы</a>
Сульфонамидные производные в качестве ингибиторов рассасывания костной ткани и ингибиторов адгезии клеток,способ их получения, применение и фармацевтическая композиция

Номер патента: 3102
Опубликовано: 26.12.2002
Авторы: Шойнеманн Карлхайнц, Пейман Ануширван, Катбертсон Роберт Эндрю, Гадек Томас, Макдауэлл Роберт, Бодари Сара Кэтрин, Карниато Дени, Гурвест Жан-Франсуа, Вилл Дэвид Вильям, Кнолле Йохен
МПК: C07D 239/42, A61K 31/505, A61P 19/10...
Метки: применение, фармацевтическая, сульфонамидные, ингибиторов, получения, адгезии, качестве, композиция, ткани, костной, производные, клеток,способ, рассасывания
Формула / Реферат:
1. Сульфонамидные производные общей формулы I где R1 и R2 вместе образуют двухвалентный (С2-С3)алкиленовый радикал; R4 является Н или (С1-С6)алкилом; R5 является (С1-С10)алкилом, (С6-С14)арилом, (C5-C14)гетероарилом или (С6-С14)арил(С1-С10)алкильной группой, где арил, гетероарил или алкил возможно замещены R3, или 2-оксобицикло[2.2.1]гепт-1-илметильной группой; R3 является (C1-C4)алкилом, (C1-C4)алкилокси, галогеном, трифторметилом, циано,...
Производные тетрагидрофуро[2,3-c]пиридина, способ их получения и применение в качестве ингибиторов металлопротеазы

Номер патента: 2527
Опубликовано: 27.06.2002
Авторы: Пьер Ален, Бонне Жаклин, Де Нантей Гийом, Атасси Ганем, Бенуа Ален, Сабатини Массимо
МПК: A61P 19/02, C07D 491/048, A61K 31/4355...
Метки: тетрагидрофуро[2,3-c]пиридина, металлопротеазы, производные, способ, ингибиторов, качестве, получения, применение
Формула / Реферат:
1. Соединения формулы (I) где R1 - атом водорода или галогена, либо линейный или разветвленный (C1-С6)алкил, либо линейная или разветвленная (C1-C6)алкоксигруппа, R2 - гидрокси, линейная или разветвленная (C1-C6)алкокси или -NHОН-группа, Ar1 - фениленовая или бифениленовая группа, Х - атом кислорода или серы, NR-группа, -Сº С-группа или связь, R - атом водорода, либо линейная или разветвленная (C1-С6)алкильная группа, n - целое число от 0...
Производные гидроксамовой кислоты в качестве ингибиторов металлопротеаз матрикса (мпм)

Номер патента: 2882
Опубликовано: 31.10.2002
Авторы: Уитлок Гэвин Алистер, Дак Кевин Нил
МПК: A61P 17/00, C07D 211/16, A61K 31/4425...
Метки: ингибиторов, мпм, качестве, гидроксамовой, кислоты, матрикса, производные, металлопротеаз
Формула / Реферат:
1. Соединение формулы (I) или его фармацевтически или ветеринарно приемлемая соль, или фармацевтически или ветеринарно приемлемый сольват любого из них, где пунктирная линия обозначает возможную связь; А представляет собой С или СН; В представляет собой СН2, О или отсутствует; R1 и R2, каждый независимо, выбраны из водорода, С1-С6алкила, возможно замещенного С1-С4алкокси или фенилом, и С1-С6алкенила; или вместе с атомом углерода, к которому они...
Производные фталазина в качестве ингибиторов фосфодиэстеразы 4

Номер патента: 3702
Опубликовано: 28.08.2003
Авторы: Мораццони Габриеле, Гранчини Джанкарло, Норчини Габриеле, Наполетано Мауро, Пеллачини Франко
МПК: C07D 237/30, A61K 31/502
Метки: фосфодиэстеразы, производные, фталазина, ингибиторов, качестве
Формула / Реферат:
1. Соединение формулы I где ---- представляет одинарную или двойную связь; B представляет метилен; A представляет пиридин, замещенный одним заместителем или большим количеством заместителей; R представляет два атома водорода или C = O группу, когда ---- представляет одинарную связь, или, когда ---- представляет двойную связь, R представляет водород, необязательно замещенный арил или гетероцикл, (C1-8)алкил, (C2-8)алкенил или (C2-8)алкинил,...
Производные пирролидина в качестве ингибиторов фосфодиэстеразы, специфичной к циклическому амф

Номер патента: 5686
Опубликовано: 28.04.2005
Авторы: Бэрджесс Лоренс И., Джоунс Закари С., Мартинс Тимоти Дж., Ньюхаус Брэдли Дж., Шлахтер Стивен, Фаулер Керри У., Одинго Джошуа, Оливер Эйми, Кесицки Эдвард А., Годино Джон Дж.
МПК: C07D 295/20, A61K 31/40
Метки: амф, производные, качестве, специфичной, фосфодиэстеразы, циклическому, ингибиторов, пирролидина
Формула / Реферат:
1. Соединение, имеющее формулу где R1 выбран из группы, включающей водород, C1-6алкил, мостиковый алкил, выбранный из группы, включающей норборнил, адамантил, бицикло[2.2.2]октил, бицикло[3.2.1]гептил, бицикло[3.2.1]октил, бицикло[4.1.0]гептил, бицикло[3.1.0]гексил и декагидронафтил, замещенный или незамещенный арил, выбранный из группы включающей фенил, нафтил, бифенил, тетрагидронафтил, инданил, 2-хлорфенил, 3-хлорфенил, 4-хлорфенил,...
Предыдущий патент: Средства и способ определения геометрии подземной трещины во время или после проведения гидроразрыва
Следующий патент: Бензоилсульфонамиды и сульфонилбензамидины для применения в качестве противоопухолевых агентов
Случайный патент: Способ построения геологических моделей подземных осадочных объёмов