N-арилпиперидинзамещенные бифенилкарбоксамиды в качестве ингибиторов секреции аполипопротеина в
Номер патента: 8061
Опубликовано: 27.02.2007
Авторы: Рувенс Петер Вальтер Мария, Мерпул Ливен, Бакс Лео Якобус Йозеф






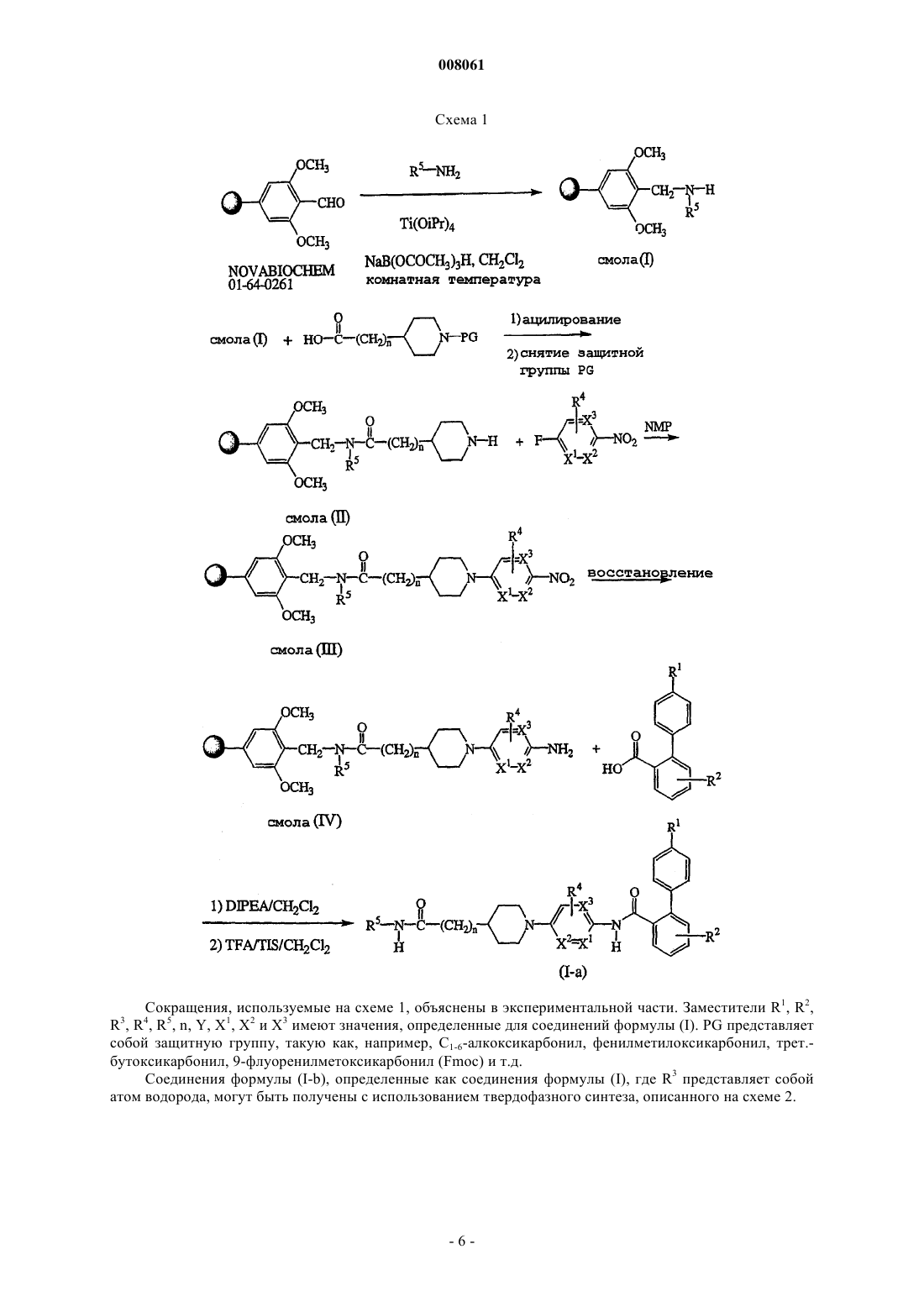
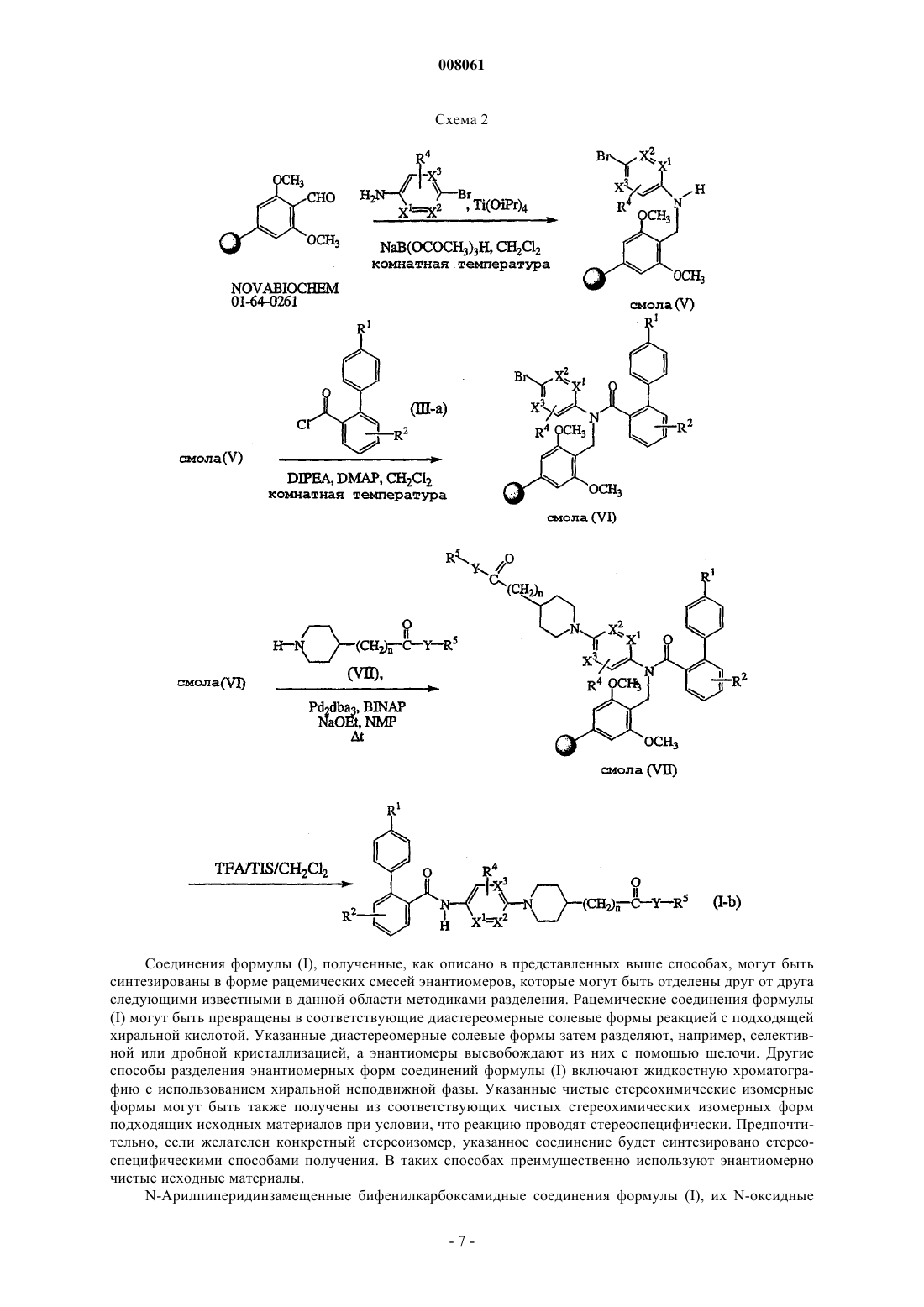
Формула / Реферат
1. Соединение формулы (I)

его N-оксиды, фармацевтически приемлемые кислотно-аддитивные соли и его стереохимические изомерные формы, где
R1 представляет собой атом водорода, C1-4-алкил, атом галогена или полигалоген-С1-4-алкил;
R2 представляет собой атом водорода, C1-4-алкил, атом галогена или полигалоген-С1-4-алкил;
R3 представляет собой атом водорода или C1-4-алкил;
R4 представляет собой атом водорода, C1-4-алкил или атом галогена;
n принимает целые значения 0 или 1;
X1 и X2 или оба представляют собой атом углерода, или, когда один из X1 или X2 представляет собой атом азота, то другой из X1 или X2 представляет собой атом углерода;
X3 представляет собой атом углерода или азота, при условии, что только один из X1 или X2 представляет собой атом азота;
Y представляет собой О или NR6, где R6 представляет собой атом водорода или C1-4-алкил; и
R5 представляет собой атом водорода; C1-6-алкил, необязательно замещенный C1-4-алкокси-, цианогруппой, полигалоген-С1-4-аклилом или арилом; С2-6-алкенил, необязательно замещенный арилом; С3-6-алкинил, необязательно замещенный арилом; арил или гетероарил;
арил представляет собой фенил; фенил, замещенный одним, двумя или тремя заместителями, каждый из которых независимо друг от друга выбран из нитро-, азидо-, цианогруппы, атома галогена, гидроксигруппы, C1-6-алкила, С3-б-циклоалкила, C1-4-алкоксигруппы, полигалоген-С1-6-алкила, амино-, моно- или ди(C1-6-алкил)аминогруппы;
гетероарил представляет собой пиридинил, пиразинил, пиримидинил, пиридазинил, триазинил, триазолил, имидазолил, пиразолил, тиазолил, изотиазолил, оксазолил, пирролил, фуранил или тиенил; и необязательно замещены одним, двумя или тремя заместителями, каждый из которых независимо выбран из нитро-, азидо-, цианогруппы, атома галогена, гидроксигруппы, C1-6-алкила, С3-6-циклоалкила, C1-4-алкилоксигруппы, полигалоген-С1-4-алкила, амино-, моно- или ди(C1-6-алкил)аминогруппы.
2. Соединение по п.1, где X1, X2 и X3 представляют собой атом углерода.
3. Соединение по п.1, где X1 представляет собой атом углерода, X2 представляет собой атом азота и X3 представляет собой атом углерода.
4. Соединение по п.1, где X1 представляет собой атом азота, X2 представляет собой атом углерода и X3 представляет собой атом углерода.
5. Соединение по любому из пп.1-4, где n принимает целое значение 0.
6. Соединение по любому из пп.1-4, где n принимает целое значение 1.
7. Фармацевтическая композиция, содержащая фармацевтически приемлемый носитель и терапевтически эффективное количество соединения по любому из пп.1-6.
8. Способ получения фармацевтической композиции по п.7, включающий тщательное смешивание терапевтически эффективного количества соединения по любому из пп.1-6 с фармацевтически приемлемым носителем.
9. Применение соединения по любому из пп.1-6 в качестве медикамента.
10. Способ получения соединения формулы (I), где промежуточное соединение формулы (II), где R3, R4, R5, n, Y, X1, X2 и X3 имеют значения, определенные в п.1,

вводят в реакцию с бифенилкарбоновой кислотой или галогенидом, имеющими формулу (III), где R1 и R2 имеют значения, определенные в формуле (I) , и Q1 выбран из гидроксигруппы и атома галогена по меньшей мере в одном реакционно-инертном растворителе и необязательно в присутствии подходящего основания

или, если желательно, соединение формулы (I) превращают в кислотно-аддитивную соль, или, наоборот, кислотно-аддитивную соль соединения формулы (I) превращают в свободное основание с помощью щелочи; и, если желательно, получают их стереохимические изомерные формы.
Текст
008061 Настоящее изобретение относится к новым N-арилпиперидинзамещенным бифенилкарбоксамидным соединениям, обладающим ингибирующей секрецию аполипопротеина В активностью и сопутствующей активностью по снижению уровня липидов. Изобретение также относится к способам получения таких соединений, фармацевтическим композициям, содержащим указанные соединения, а также к применению указанных соединений в качестве лекарственного средства для лечения гиперлипидемии, ожирения и диабета II типа. Ожирение является причиной огромного числа серьезных проблем со здоровьем, таких как развитие диабета у взрослых и болезни сердца. Кроме того, потеря веса становится навязчивой идеей у растущего числа людей. В настоящее время широко известна причинная связь между гиперхолестеринемией, которая, в частности, связана с повышенной концентрацией в плазме липопротеинов низкой плотности (далее называются ЛНП, LDL) и липопротеинов очень низкой плотности (далее называются ЛОНП, VLDL), и преждевременным атеросклерозом и/или сердечно-сосудистым заболеванием. Однако в настоящее время для лечения гиперхолестеринемии доступно ограниченное число лекарственных средств. Лекарственные средства, преимущественно используемые для лечения гиперлипидемии, включают смолы, усиливающие секрецию желчных кислот, такие как холестирамин и колестипол, производные фибриновой кислоты, такие как безафибрат, клофибрат, фенофибрат, ципрофибрат и гемфиброзил, никотиновая кислота и ингибиторы синтеза холестерина, такие как ингибиторы редуктазы HMG Софермента-А. Таким образом, все еще остается необходимость в новых снижающих уровень липидов агентах с улучшенной эффективностью и/или действующих посредством других механизмов, чем названные выше лекарства. Плазменные липопротеины представляют собой растворимые в воде комплексы с высокой молекулярной массой, образованные из липидов (холестерин, триглицерид, фосфолипиды) и аполипопротеинов. В зависимости от их плотности (измеренной с помощью ультрацентрифугирования) определено пять основных классов липопротеинов, образованных в печени и/или кишечнике, которые отличаются относительным содержанием липидов и типом аполипопротеинов. К ним относятся ЛНП, ЛОНП, липопротеины промежуточной плотности (далее называются ЛПП, IDL), липопротеины высокой плотности (далее называются ЛВП, LHD) и хиломикроны. Идентифицировано десять основных плазменных аполипопротеинов человека. ЛОНП, которые выделяются печенью и содержат аполипопротеин В (далее называется Аро-В), подвергаются разрушению до ЛНП, которые переносят от 60 до 70% всего сывороточного холестерина. Аро-В также является основным белковым компонентом ЛНП. Повышенные уровни ЛНПхолестерина в сыворотке, обусловленные избыточным синтезом или пониженным метаболизмом, имеют причинную связь с атеросклерозом. Напротив, липопротеины высокой плотности (называемые далее ЛВП), которые содержат аполипопротеин А 1, имеют защитное действие и в обратной зависимости связаны с риском заболевания сердца. Таким образом, отношение ЛВП/ЛНП является удобным методом оценки атерогенного потенциала профиля липидов в плазме человека. Две изоформы аполипопротеинов (аро) В, аро В-48 и аро В-100, являются важными белками в метаболизме липопротеинов человека. Аро В-48, называемый так, поскольку, как оказывается, составляет приблизительно 48% объема аро В-100 на гелях додецилсульфат натрия-полиакриламид, синтезируется в кишечнике человека. Аро В-48 необходим для микросообщества хиломикронов и, следовательно, играет обязательную роль в кишечной абсорбции пищевых жиров. Аро В-100, который продуцируется в печени человека, необходим для синтеза и секреции ЛОНП. ЛНП, которые составляют приблизительно 2/3 холестерина в плазме человека, представляют собой метаболические продукты ЛОНП. Аро В-100 фактически представляет собой единственный белковый компонент ЛНП. Повышенные концентрации аро В-100 и ЛНП холестерина в плазме считаются факторами риска для развития атеросклеротического заболевания коронарной артерии. Большое число генетических и приобретенных заболеваний может быть результатом гиперлипидемии. Они могут быть классифицированы на первичные и вторичные гиперлипидемические состояния. Наиболее традиционными причинами вторичных гиперлипидемий являются сахарный диабет, алкогольная зависимость, лекарства, гипотериоз, хроническая почечная недостаточность, нефротический синдром,холестаз и булимия. Первичные гиперлипидемии также могут быть классифицированы на обычную гиперхолестеринемию, семейную комбинированную гиперлипидемию, семейную гиперхолестеринемию,остаточную гиперлипидемию, хиломикронемический синдром и семейную гипертриглицеридемию. Белок переноса микросомальных триглицеридов (МТР), как известно, катализирует транспорт триглицерида и сложного эфира холестерина за счет предпочтения фосфолипидов, таких как фосфатидилхолин. Показано (D. Sharp et al., Nature (1993) 365:65), что дефект, вызывающий абеталипопротеинемию, находится в гене МТР. Это указывает на то, что МТР необходим для синтеза Аро В-содержащих липопротеинов, таких как ЛОНП, предшественников ЛНП. Из этого следует, что ингибитор МТР мог бы ингибировать синтез ЛОНП и ЛНП, понижая в результате уровни ЛОНП, ЛНП, холестерина и триглицерида у человека. Одной из задач настоящего изобретения является разработка усовершенствованного способа лечения пациента, страдающего ожирением или атеросклерозом, в особенности коронарным атеросклерозом,-1 008061 и в общем случае заболеваниями, которые связаны с атеросклерозом, такими как ишемическая болезнь сердца, заболевание периферических сосудов и церебральное сосудистое заболевание. Еще одной задачей настоящего изобретения является индуцирование регрессии атеросклероза и ингибирование его клинических последствий, в особенности заболеваемости и смертности. Ингибиторы МТР описаны в публикациях WO-00/32582, WO-01/96327 и WO-02/20501. Настоящее изобретение основано на неожиданном открытии, что класс новых N-арилпиперидинзамещенных бифенилкарбоксамидных соединений действует как селективные ингибиторы МТР, то есть эти соединения обладают способностью селективно блокировать МТР на уровне кишечной стенки у млекопитающих и, следовательно, являются потенциальными медицинскими средствами, а именно медицинским средством для лечения гиперлипидемии. Настоящее изобретение дополнительно относится к способам получения указанных N-арилпиперидинзамещенных бифенилкарбоксамидных соединений, а также фармацевтическим композициям, содержащим их. Кроме того, изобретение относится к новым соединениям, которые являются полезными промежуточными соединениями для получения терапевтически активных N-арилпиперидинзамещенных бифенилкарбоксамидных соединений, а также способам получения таких промежуточных соединений. И, наконец, изобретение относится к способу лечения состояния, выбираемого из атеросклероза, панкреатита, ожирения, гиперхолестеринемии, гипертриглицеридемии, гиперлипидемии, диабета и диабета II типа, включающему введение терапевтически активного бифенилкарбоксамидного соединения млекопитающему. Настоящее изобретение относится к семейству новых соединений формулы (I) их N-оксидам, фармацевтически приемлемым кислотно-аддитивным солям и их стереохимическим изомерным формам, гдеR1 представляет собой атом водорода, C1-4-алкил, атом галогена или полигалоген-C1-4-алкил;R2 представляет собой атом водорода, С 1-4-алкил, атом галогена или полигалоген-С 1-4-алкил;R3 представляет собой атом водорода или С 1-4-алкил;R4 представляет собой атом водорода, С 1-4-алкил или атом галогена;n принимает целые значения ноль или 1;X1 и X2 или оба представляют собой атом углерода, или когда один из X1 или X2 представляет собой атом азота, то другой из X1 или X2 представляет собой атом углерода;X3 представляет собой атом углерода или азота при условии, что только один из X1 или X2 представляет собой атом азота;Y представляет собой О или NR6, где R6 представляет собой атом водорода или C1-4-алкил; иR5 представляет собой атом водорода; С 1-6-алкил, необязательно замещенный C1-4-алкокси-, цианогруппой, полигалоген-С 1-4-алкилом или арилом; С 2-6-алкенил, необязательно замещенный арилом; С 3-6 алкинил, необязательно замещенный арилом; арил или гетероарил; арил представляет собой фенил; фенил, замещенный одним, двумя или тремя заместителями, каждый из которых независимо друг от друга выбран из нитро-, азидо-, цианогруппы, атома галогена, гидроксигруппы, С 1-6-алкила, С 3-6-циклоалкила, C1-4-алкоксигруппы, полигалоген-C1-6-алкила, амино-, моноили ди(C1-6-алкил)аминогруппы; гетероарил представляет собой пиридинил, пиразинил, пиримидинил, пиридазинил, триазинил,триазолил, имидазолил, пиразолил, тиазолил, изотиазолил, оксазолил, пирролил, фуранил или тиенил; и необязательно замещен одним, двумя или тремя заместителями, каждый из которых независимо выбран из нитро-, азидо-, цианогруппы, атома галогена, гидроксигруппы, С 1-6-алкила, С 3-6-циклоалкила, C1-4 алкилоксигруппы, полигалоген-С 1-4-алкила, амино-, моно- или ди(C1-6-алкил)аминогруппы. Если не оговорено особо, используются следующие определения: атом галогена является общим определением атомов фтора, хлора, брома и йода;C1-4-алкил определяет линейные и разветвленные насыщенные углеводородные радикалы, содержащие от 1 до 4 атомов углерода, такие как, например, метил, этил, пропил, н-бутил, 1-метилэтил, 2 метилпропил, 1,1-диметилэтил и т.д.;C1-6-алкил включает C1-4-алкил (как определено выше) и его более высокие гомологи, содержащие 5 или 6 атомов углерода, такие как, например, 2-метилбутил, н-пентил, диметилпропил, н-гексил, 2 метилпентил, 3-метилпентил и др.; полигалоген-С 1-4-алкил определяют как полигалогензамещенный С 1-4-алкил, в особенности C1-4 алкил (как определено выше), замещенный 2-6 атомами галогена, такой как дифторметил, трифторметил,трифторэтил и др.;-2 008061 С 2-6-алкенил определяет линейные и разветвленные ненасыщенные углеводородные радикалы, содержащие от 2 до 6 атомов углерода, такие как этенил, пропенил, бутенил, пентенил или гексенил; С 3-6-алкинил определяет линейные и разветвленные углеводородные радикалы, содержащие одну тройную связь и от 3 до 6 атомов углерода, такие как, например, 2-пропинил, 3-бутинил, 2-бутинил, 2 пентинил, 3-пентинил, 3-метил-2-бутинил, 3-гексинил, 2-гексинил и др.;C1-4-алкиламиногруппа определяет первичные аминорадикалы, содержащие от 1 до 6 атомов углерода, такие как, например, метиламино-, этиламино-, пропиламино-, изопропиламино-, бутиламино-,изобутиламиногруппы и т.д.; ди(C1-6-алкил)аминогруппа определяет аминорадикалы, содержащие от 1 до 6 атомов углерода, такие как, например, диметиламино-, диэтиламино-, дипропиламино-, диизопропиламино-, N-метил-N'этиламино-, N-этил-N'-пропиламиногруппы и т.д. Фармацевтически приемлемые кислотно-аддитивные соли, упомянутые выше, как подразумевается,включают терапевтически активные нетоксичные кислотно-аддитивные солевые формы, которые соединения формулы (I) способны образовывать. Фармацевтически приемлемые кислотно-аддитивные соли могут быть удобно получены путем обработки основной формы подходящей кислотой. Подходящими кислотами являются, например, неорганические кислоты, такие как галогенводородные кислоты, например, соляная или бромисто-водородная кислота, серная, азотная, фосфорная и т.д. кислоты; или органические кислоты, такие как, например, уксусная кислота, пропионовая кислота, гидроксиуксусная кислота, молочная кислота, пировиноградная кислота, щавелевая кислота (например, этандионовая), малоновая, янтарная (то есть бутандионовая кислота), малеиновая, фумаровая, яблочная, винная, лимонная, метансульфоновая, этансульфоновая, бензолсульфоновая, п-толуолсульфоновая, цикламиновая, салициловая, п-аминосалициловая, памовая и другие подобные кислоты. И, наоборот, указанные солевые формы могут быть превращены обработкой подходящим основанием в форму свободного основания. Определение кислотно-аддитивная соль, которое использовано выше, включает сольваты, которые соединения формулы (I), а также их соли способны образовывать. Такие сольваты представляют собой, например, гидраты, алкоголяты и др.N-оксидные формы соединений формулы (I), которые могут быть получены известными в данной области способами, означают соединения формулы (I), где атом азота окислен до N-оксида. Определение стереохимические изомерные формы, которое использовалось выше, определяет все возможные изомерные формы, которыми соединения формулы (I) могут обладать. Если не указано или не упомянуто иначе, то химическое название соединений означает смесь всех возможных стереохимических изомерных форм, причем указанные смеси содержат все диастереоизомеры и энантиомеры основной молекулярной структуры. Более конкретно, стереогенные центры могут иметь R- или Sконфигурацию; заместители на бивалентных циклических (частично) насыщенных радикалах могут иметь или цис-, или транс-конфигурацию. Если не указано иначе или не упомянуто иначе, то химическое название соединений означает смесь всех возможных стереоизомерных форм, причем указанные смеси содержат все диастереомеры и энантиомеры основной молекулярной структуры. То же самое применимо к промежуточным соединениям, описанным в работе, которые используются для получения конечных продуктов формулы (I). Определения цис- и транс- используются в данном случае в соответствии с номенклатурой ChemicalAbstracts и относятся к положению заместителей на циклическом фрагменте. Абсолютная стереохимическая конфигурация соединений формулы (I) и промежуточных соединений, используемых для их получения, может быть легко определена специалистом при использовании известных способов, таких как, например, дифракция рентгеновских лучей. Кроме того, некоторые соединения формулы (I) и некоторые промежуточные соединения, используемые при их получении, могут проявлять полиморфизм. Следует понимать, что настоящее изобретение охватывает любую из полиморфных форм, проявляющую свойства, полезные при лечении упомянутых выше состояний. Группа представляющих интерес соединений состоит из соединений формулы (I), для которых применимо одно или несколько из следующих ограничений:b) R2 представляет собой атом водорода или C1-4-алкил;c) R3 представляет собой атом водорода;d) R4 представляет собой атом водорода;e) R5 представляет собой C1-4-алкил или C1-4-алкил, замещенный фенилом. Первую группу конкретных соединений составляют соединения формулы (I), где X1, X2 и X3 представляют собой атом углерода. Вторую группу конкретных соединений составляют соединения формулы (I), где X1 представляет собой атом углерода, X2 представляет собой атом азота, и X3 представляет собой атом углерода. Третью группу конкретных соединений составляют соединения формулы (I), где X1 представляет собой атом азота, X2 представляет собой атом углерода и X3 представляет собой атом углерода.-3 008061 Четвертую группу конкретных соединений составляют соединения формулы (I), где X1 представляет собой атом углерода, X2 представляет собой атом азота и X3 представляет собой атом азота. Пятую группу конкретных соединений составляют соединения формулы (I), где n имеет целое значение 0. Шестую группу конкретных соединений составляют соединения формулы (I), где n имеет целое значение 1. Первую предпочтительную группу соединений составляют соединения формулы (I), где R1 представляет собой C1-4-алкил или трифторметил; R2 представляет собой атом водорода или C1-4-алкил; R3 представляет собой атом водорода; R4 представляет собой атом водорода; R5 представляет собой C1-4 алкил или C1-4-алкил, замещенный фенилом; n имеет целое значение ноль; и X1, X2 и X3 представляют собой атом углерода. Вторую предпочтительную группу соединений составляют соединения формулы (I), где R1 представляет собой C1-4-алкил или трифторметил; R2 представляет собой атом водорода или C1-4-алкил; R3 представляет собой атом водорода; R4 представляет собой атом водорода; R5 представляет собой C1-4 алкил или C1-4-алкил, замещенный фенилом; n имеет целое значение 1; и X1, X2 и X3 представляют собой атом углерода. Третью предпочтительную группу соединений составляют соединения формулы (I), где R1 представляет собой С 1-4-алкил или трифторметил; R2 представляет собой атом водорода или C1-4-алкил; R3 представляет собой атом водорода; R4 представляет собой атом водорода; R5 представляет собой C1-4 алкил или C1-4-алкил, замещенный фенилом; n имеет целое значение 0; X3 представляет собой атом углерода и X1 или X2 представляет собой атом азота и другой из X1 или X2 представляет собой атом углерода. Четвертую предпочтительную группу соединений составляют соединения формулы (I), где R1 представляет собой C1-4-алкил или трифторметил; R2 представляет собой атом водорода или C1-4-алкил; R3 представляет собой атом водорода; R4 представляет собой атом водорода; R5 представляет собой C1-4 алкил или C1-4-алкил, замещенный фенилом; n имеет целое значение 1; X3 представляет собой атом углерода и X1 или X2 представляет собой атом азота и другой X1 или X2 представляет собой атом углерода. Первую более предпочтительную группу соединений составляют соединения предпочтительных групп, где Y представляет собой О. Вторую более предпочтительную группу соединений составляют соединения предпочтительных групп, где Y представляет собой NH. Первый способ получения соединений формулы (I) представляет собой способ, в котором промежуточное соединение формулы (II) где R3, R4, R5, n, Y, X1, X2 и X3 имеют значения, определенные в формуле (I), вводят в реакцию с бифенилкарбоновой кислотой или галогенидом, имеющими формулу (III) где R1 и R2 определены в формуле (I) и Q1 выбран из гидроксигруппы и атома галогена, по меньшей мере, в одном реакционно-инертном растворителе и необязательно в присутствии подходящего основания, причем указанный способ дополнительно необязательно включает превращение соединения формулы (I) в кислотно-аддитивную соль, и/или получение его стереохимических изомерных форм. Когда Q1 представляет собой гидроксигруппу, может быть удобно активировать бифенилкарбоновую кислоту формулы (III) путем добавления эффективного количества активатора реакции. Неограничивающими примерами таких активаторов реакции являются карбонилдиимидазол, диимиды, такие как N,N'дициклогексилкарбодиимид (DCC) или 1-этил-3-(3'-диметиламинопропил)карбодиимид (ЕСС), и их функциональные производные. Во всех названных типах методики ацилирования предпочтительно использовать полярный апротонный растворитель, такой как, например, дихлорметан. Подходящими основаниями для проведения таких процессов являются третичные амины, такие как триэтиламин, триизопропиламин и др. Подходящие температуры для проведения первого способа изобретения обычно находятся в интервале приблизительно от 20 до 140 С в зависимости от конкретного используемого растворителя, и наиболее часто соответствуют температуре кипения растворителя. Второй способ получения бифенилкарбоксамидного соединения изобретения представляет собой способ, в котором промежуточное соединение имеет формулу (IV) где R1, R2, R3, R4, n, X1, X2 и X3 определены в формуле (I) и Q2 выбран из гидроксигруппы и атома галогена, вводят в реакцию с промежуточным соединением (V) формулы R5-Y-H, где R5 и Y определены в формуле (I), по меньшей мере, в одном реакционно-инертном растворителе и необязательно в присутствии, по меньшей мере, одного реагента сочетания и/или подходящего основания, причем указанный способ дополнительно необязательно включает превращение соединения формулы (I) в кислотноаддитивную соль, и/или получение его стереохимических изомерных форм. Когда Q2 представляет собой гидроксигруппу, может быть удобно активировать карбоновую кислоту формулы (IV) путем добавления эффективного количества активатора реакции. Неограничивающими примерами таких активаторов реакции являются карбонилдиимидазол, диимиды, такие как DCC, ЕСС, гидроксибензотриазол, бензотриазол-1-ил-N-окситрис(диметиламино)фосфонийгексафторфосфат (ВОР), тетрапирролидинофосфонийгексафторфосфат, бромтрипирролидинофосфонийгексафторфосфат или их функциональные производные,такие как описанные в публикации Solid-Phase Synthesis: A Practical Guide, edited by Steven A. Katesand Fernando Albericio, Marcel Dekker, Inc., 2000 (ISBN: 0-8247-0359-6), pp. 306-319. Третий способ получения бифенилкарбоксамидного соединения в соответствии с настоящим изобретением представляет собой способ, в котором промежуточное соединение, имеющее формулу (VI) где R1, R2, R3, R4, X1, X2 и X3 определены в формуле (I) и Q3 выбран из атома галогена, В(ОН)2, алкилборонатов и их циклических аналогов, вводят в реакцию с реагентом, имеющим формулу (VII) где n, Y и R5 имеют значения, определенные в формуле (I), по меньшей мере, в одном реакционноинертном растворителе в присутствии, по меньшей мере, одного реагента сочетания на основе переходного металла и/или, по меньшей мере, одного подходящего лиганда, причем указанный способ дополнительно необязательно включает превращение соединения формулы (I) в кислотно-аддитивную соль и/или получение его стереохимических изомерных форм. Такой тип реакции, известный в данной области как реакция Бухвальда, относится к реагентам сочетания на основе подходящего металла и/или подходящих лигандов, например, к соединениям палладия, таким как тетра(трифенилфосфин)палладий, трис(дибензилиденацетон)дипалладий, 2,2'-бис(дифенилфосфино)-1,1'-бинафтил (BINAP) и др., может быть найден,например, в публикациях Tetrahedron Letters (1996), 37(40), 7181-7184 и J.Am.Chem.Soc. (1996), 118:7216. Если Q3 представляет собой В(ОН)2, алкилборонат или его циклический аналог, то в качестве реагента сочетания необходимо использовать ацетат меди в соответствии с публикацией Tetrahedron Letters(1998), 39:2933-6. Соединения формулы (I-а), определенные как соединения формулы (I), где Y представляет собойNH и R3 представляет собой атом водорода, могут быть легко получены с использованием методик твердофазного синтеза, представленных ниже на схеме 1. В общем случае твердофазный синтез включает взаимодействие промежуточного соединения в синтезе с полимерной подложкой. Такое промежуточное соединение, нанесенное на полимер, затем может быть проведено через ряд синтетических стадий. После каждой стадии примеси удаляют фильтрованием смолы и промывками несколько раз различными растворителями. На каждой стадии смола может быть распределена для введения в реакцию с различными промежуточными соединениями на следующей стадии, что обеспечивает синтез большого числа соединений. После последней стадии методики смолу обрабатывают реагентом или способом для отщепления смолы от образца. Более подробное объяснение методики, используемой в твердофазной химии, дано,например, в публикации Handbook of Combinatorial Chemistry: Drugs, Catalysts, Material, edited by K.C. Сокращения, используемые на схеме 1, объяснены в экспериментальной части. Заместители R1, R2,R , R , R5, n, Y, X1, X2 и X3 имеют значения, определенные для соединений формулы (I). PG представляет собой защитную группу, такую как, например, C1-6-алкоксикарбонил, фенилметилоксикарбонил, трет.бутоксикарбонил, 9-флуоренилметоксикарбонил (Fmoc) и т.д. Соединения формулы (I-b), определенные как соединения формулы (I), где R3 представляет собой атом водорода, могут быть получены с использованием твердофазного синтеза, описанного на схеме 2. 3 Соединения формулы (I), полученные, как описано в представленных выше способах, могут быть синтезированы в форме рацемических смесей энантиомеров, которые могут быть отделены друг от друга следующими известными в данной области методиками разделения. Рацемические соединения формулы(I) могут быть превращены в соответствующие диастереомерные солевые формы реакцией с подходящей хиральной кислотой. Указанные диастереомерные солевые формы затем разделяют, например, селективной или дробной кристаллизацией, а энантиомеры высвобождают из них с помощью щелочи. Другие способы разделения энантиомерных форм соединений формулы (I) включают жидкостную хроматографию с использованием хиральной неподвижной фазы. Указанные чистые стереохимические изомерные формы могут быть также получены из соответствующих чистых стереохимических изомерных форм подходящих исходных материалов при условии, что реакцию проводят стереоспецифически. Предпочтительно, если желателен конкретный стереоизомер, указанное соединение будет синтезировано стереоспецифическими способами получения. В таких способах преимущественно используют энантиомерно чистые исходные материалы.N-Арилпиперидинзамещенные бифенилкарбоксамидные соединения формулы (I), их N-оксидные-7 008061 формы, фармацевтически приемлемые соли и стереоизомерные формы обладают подходящей активностью, ингибирующей аполипопротеин В и сопутствующей активностью понижения уровня липидов. Таким образом, настоящие соединения могут быть использованы в качестве медицинского средства, особенно в способе лечения пациентов, страдающих от гиперлипидемии, ожирения, атеросклероза или диабета II типа. В частности, настоящие соединения могут быть использованы для производства медицинского средства для лечения заболеваний, вызванных избытком липопротеинов очень низкой плотности(ЛОНП) или липопротеинов низкой плотности (ЛНП), и особенно заболеваний, вызванных холестерином, связанным с указанными ЛОНП и ЛНП. Основной механизм действия соединений формулы (I), как оказывается, включает ингибирующую МТР (транспортный белок микросомальных триглицеридов) активность в гепатоцитах и эпителиальных клетках кишечника, приводя к повышению продуцирования ЛОНП и хиломикронов, соответственно. Это является новой и инновационной методикой для гиперлипидемии и, как ожидается, понижает ЛНПхолестерин и триглицериды за счет понижения печеночного продуцирования ЛОНП и продуцирования кишечных хиломикронов. Большое число генетических и приобретенных заболеваний может привести к гиперлипидемии. Эти заболевания могут быть разделены на первичное и вторичное гиперлипидемические состояния. Наиболее обычными причинами вторичных гиперлипидемий являются сахарный диабет, алкогольная зависимость, лекарства, гипотериоз, хроническая почечная недостаточность, нефротический синдром, холестаз и булимия. Первичные гиперлипидемии также могут быть классифицированы на обычную гиперхолестеринемию, семейную комбинированную гиперлипидемию, семейную гиперхолестеринемию, остаточную гиперлипидемию, хиломикронемический синдром и семейную гипертриглицеридемию. Настоящие соединения также могут быть использованы для профилактики или для лечения пациентов, страдающих атеросклерозом, в особенности, атеросклерозом сердца и более общих заболеваний, которые относятся к атеросклерозу, таких как ишемическая болезнь сердца, заболевание периферических сосудов, церебральное сосудистое заболевание. Соединения настоящего изобретения могут вызвать обратное развитие атеросклероза и ингибировать клинические последствия атеросклероза, в особенности заболеваемость и смертность. Принимая во внимание применимость соединений формулы (I), следует, что настоящее изобретение также относится к способу лечения теплокровных животных, в том числе людей (в общем называемых пациентами), страдающих от заболеваний, вызванных избытком липопротенов очень низкой плотности (ЛОНП) или липопротеинов низкой плотности (ЛНП), и в особенности заболеваний, вызываемых холестерином, связанным с указанными ЛОНП и ЛНП. Таким образом, предлагается способ лечения пациентов, страдающих от состояний, таких как, например, гиперлипидемия, ожирение, атеросклероз или диабет II типа. Аро В-48, синтезированный кишечником, является необходимым для микросообщества хиломикронов и, следовательно, играет обязательную роль при кишечной абсорбции пищевых жиров. Настоящее изобретение относится к бифенилкарбоксамидным соединениям, которые действуют как селективные ингибиторы МТР на уровне кишечной стенки. Кроме того, настоящее изобретение относится к фармацевтическим композициям, содержащим, по меньшей мере, один фармацевтически приемлемый носитель и фармацевтически эффективное количество N-арилпиперидинзамещенного бифенилкарбоксамидного соединения, имеющего формулу (I). Для получения фармацевтических композиций настоящего изобретения эффективное количество конкретного соединения в форме основания или аддитивной соли в качестве активного ингредиента тщательно смешивают до однородной смеси, по меньшей мере, с одним фармацевтически приемлемым носителем, и этот носитель может иметь большое количество форм в зависимости от формы препарата,необходимой для введения. Такие фармацевтические композиции желательны в стандартной дозированной лекарственной форме предпочтительно для перорального введения, ректального введения, подкожного введения или парентеральной инъекции. Например, при получении композиций в пероральной дозированной форме могут быть использованы любые полезные жидкие фармацевтические носители, такие как, например, вода, гликоли, масла,спирты и т.д., в случае пероральных жидких препаратов, таких как суспензии, сиропы, эликсиры и растворы; или твердые фармацевтические носители, такие как крахмалы, сахара, каолин, смазывающие вещества, связующие вещества, диспергирующие агенты и т.д., в случае порошков, пилюль, капсул и таблеток. Благодаря их простому введению, таблетки и капсулы представляют собой наиболее предпочтительные пероральные дозированные лекарственные формы, в случае которых очевидно используются твердые фармацевтические носители. В случае композиций парентеральных инъекций фармацевтические носители будут преимущественно содержать стерильную воду, хотя другие ингредиенты могут быть включены с целью улучшения растворимости активного ингредиента. Инъецируемые растворы могут быть получены, например, путем использования фармацевтического носителя, содержащего солевой раствор, раствор глюкозы или смесь обоих. Инъецируемые суспензии также могут быть получены путем использования подходящих жидких носителей, суспендирующих агентов и т.д. В композициях, подходящих для подкожного введения, фармацевтический носитель может необязательно содержать повы-8 008061 шающий проницаемость агент и/или подходящий смачивающий агент, необязательно смешанный с небольшими количествами подходящих вспомогательных веществ, которые не оказывают значительного вредного действия на кожу. Указанные вспомогательные вещества могут быть выбраны так, чтобы облегчить использование активного ингредиента на коже и/или чтобы они содействовали получению желаемых композиций. Такие местные композиции могут быть введены различными способами, например,в виде трансдермального пластыря, в виде пятна на коже или мази. Аддитивные соли соединения формулы (I) вследствие их повышенной растворимости в воде в сравнении с соответствующей основной формой, очевидно являются более подходящими при получении водных композиций. Особенно предпочтительно готовить фармацевтические композиции настоящего изобретения в дозированных стандартных лекарственных формах для простоты введения и единообразия дозировок. Определение дозированная стандартная лекарственная форма, используемое в данном случае, относится к физически дискретным единицам, подходящим в качестве единичных дозировок, причем каждая единица содержит заранее определенное количество активного ингредиента, рассчитанное так, чтобы получить желаемый терапевтический эффект, в сочетании с требуемым фармацевтическим носителем. Примерами таких дозированных лекарственных форм являются таблетки (в том числе таблетки с зарубками или таблетки с покрытием), капсулы, пилюли, упаковки порошков, облатки, инъецируемые растворы или суспензии, полные чайные ложки, полные столовые ложки и т.д., и их поделенные кратные части. В случае перорального введения фармацевтические композиции настоящего изобретения могут иметь форму твердых дозированных форм, например, таблеток (как формы для глотания, так и формы для жевания), капсулы или гелевые капы, полученные обычными средствами с фармацевтически приемлемыми наполнителями и носителями, такими как связующие агенты (например, предварительно желатинизированный кукурузный крахмал, поливинилпирролидон, гидроксипропилметилцеллюлоза и др.),наполнители (например, лактоза, микрокристаллическая целлюлоза, фосфат кальция и т.д.), смазывающие вещества (например, стеарат магния, тальк, диоксид кремния и т.д.), диспергирующие агенты (например, картофельный крахмал, натриевая соль гликолята крахмала и т.д.), смачивающие агенты (например, лаурилсульфат натрия) и т.д. На такие таблетки также может быть нанесено покрытие хорошо известными в данной области способами. Жидкие препараты для перорального введения могут иметь форму, например, растворов, сиропов или суспензий, или они могут быть получены в виде сухого продукта для смешивания с водой и/или другим подходящим жидким носителем до применения. Такие жидкие препараты могут быть получены с помощью обычных средств, необязательно с другими фармацевтически приемлемыми добавками, такими как суспендирующие агенты (например, сироп сорбита, метилцеллюлоза, гидроксипропилметилцеллюлоза или гидрированные пищевые жиры), эмульгирующие агенты (например, лецитин или аравийская камедь), неводные носители (например, миндальное масло, масляные сложные эфиры или этиловый спирт), подслащивающие вещества, корригенты, маскирующие вкус или запах агенты и консерванты(например, метил- или пропил-п-гидроксибензоаты или сорбиновая кислота). Фармацевтически приемлемые подслащивающие вещества, которые могут быть использованы в фармацевтических композициях настоящего изобретения, включают предпочтительно, по меньшей мере,одно сильное подслащивающее вещество, такое как аспартам, ацесульфамкалий, цикламат натрия, алитам, дигидрохальконовое подслащивающее вещество, монеллин, стевиозид сахаралозы (4,1',6'-трихлор 4,1',6'-тридеоксигалактосахароза), или, предпочтительно, сахарин, натриевая или кальциевая соль сахарина, и необязательно, по меньшей мере, одно объемное подслащивающее вещество, такое как сорбит,маннит, фруктоза, сахароза, мальтоза, изомальт, глюкоза, сироп гидрированной глюкозы, ксилит, жженый сахар или мед. Сильные подслащивающие вещества обычно используют в низких концентрациях. Например, в случае натриевой соли сахарина указанная концентрация может находиться в интервале приблизительно от 0,04 до 0,1% (мас./об.) из расчета на конечную рецептуру. Объемное подслащивающее вещество может быть эффективно использовано в больших концентрациях, лежащих в интервале приблизительно от 10 до 35%, предпочтительно приблизительно от 10 до 15% (мас./об.). Фармацевтически приемлемые корригенты, которые могут маскировать горький вкус ингредиентов в рецептурах с низкими дозами, представляют собой предпочтительно фруктовые корригенты, такие как вишневый, малиновый, черносмородиновый и клубничный корригент. Комбинация двух корригентов может давать очень хороший результат. В рецептурах с высокой дозой могут требоваться более сильные фармацевтически приемлемые корригенты, такие как Caramel Chocolate, Mint Cool, Fantasy и т.д. Такой корригент может присутствовать в конечной композиции в концентрации, лежащей в интервале приблизительно от 0,05 до 1% (мас./об.). Предпочтительно используют комбинации указанных сильных корригентов. Предпочтительно используют корригент, который не подвергается какому-либо изменению или не теряет вкус и/или цвет в условиях рецептуры.N-Арилпиперидинзамещенные бифенилкарбоксамидные соединения настоящего изобретения могут быть получены для парентерального введения путем инъекции, обычно внутривенной, внутримышечной или подкожной инъекцией, например, путем инъекции ударной дозы или путем внутривенного непрерывного вливания. Рецептуры для инъекции могут быть представлены в виде стандартной дозированной формы, например, в ампулах или контейнерах с множеством доз, включая добавленный консервант. Они-9 008061 могут иметь такие формы, как суспензии, растворы или эмульсии в масляном или водном носителе, и могут содержать рецептурные агенты, такие как изотонирующие, стабилизирующие и/или диспергирующие агенты. С другой стороны, активный ингредиент может присутствовать в порошкообразной форме для смешения перед применением с подходящим носителем, например, со стерильной апирогенной водой. Бифенилкарбоксамидные соединения настоящего изобретения также могут быть приготовлены в ректальных композициях, таких как свечи или удерживающие клизмы, например, содержащие обычные основы для свечей, такие как кокосовое масло и/или другие глицериды.N-Арилпиперидинзамещенные бифенилкарбоксамидные соединения настоящего изобретения могут быть использованы в комбинации с другими фармацевтическими агентами, в частности, фармацевтические композиции настоящего изобретения могут также содержать, по меньшей мере, один дополнительный понижающий уровень липидов агент, приводя таким образом к так называемой комбинированной понижающей уровень липидов терапии. Указанный дополнительный понижающий уровень липидов агент, например, может представлять собой известное лекарство, обычно используемое для лечения гиперлипидемии, такое как, например, усиливающая секрецию желчных кислот смола, производные фибриновой кислоты или никотиновая кислота, которые упоминались ранее при описании уровня техники. Подходящими дополнительными понижающими уровень липидов агентами являются другие ингибиторы биосинтеза холестерина и ингибиторы поглощения холестерина, особенно ингибиторы HMG-CoA редуктазы и ингибиторы HMG-CoA синтазы, ингибиторы гена экспрессии HMG-CoA редуктазы, ингибиторы СЕТР, ингибиторы АСАТ, ингибиторы скваленсинтетазы и др. Любой ингибитор HMG-CoA редуктазы может быть использован в качестве второго соединения при комбинированном лечении настоящего изобретения. Определение ингибитор HMG-CoA редуктазы, используемое в данном описании, если не оговорено особо, относится к соединению, которое ингибирует биотрансформацию гидроксиметилглутарил-кофермента А в мевалоновую кислоту, которая катализируется ферментом HMG-CoA редуктаза. Такие ингибиторы могут быть легко определены специалистом в данной области в соответствии со стандартными методами оценки, то есть Methods of Enzymology(1981), 71:455-509. Примеры соединений описаны, например, в патенте США 4231938 (включая ловастатин), в патенте США 4444784 (включая симвастатин), патенте США 4739073 (включая флувастатин), патенте США 4346227 (включая правастатин), ЕР-А-491226 (включая ривастатин) и в патенте США 4647576 (включая аторвастатин). Любой ингибитор HMG-CoA синтазы может быть использован в качестве второго соединения при комбинированном лечении настоящего изобретения. Определение ингибитор HMG-CoA синтазы, используемое в описании, если не оговорено особо, относится к соединению, которое ингибирует биосинтез гидроксиметилглутарил-кофермента А из ацетил-кофермента А и ацетоацетил-кофермента А, катализируемый ферментом HMG-CoA синтаза. Такие ингибиторы могут быть легко определены специалистом в данной области в соответствии со стандартными способами, то есть Methods of Enzymology (1985),110:19-26. Примеры соединений описаны, например, в патенте США 5120729, который относится к производным бета-лактама, патенте США 5064856, который относится к производным спиролактона,и в патенте США 4847271, который относится к соединениям оксетана. Любой ингибитор гена экспрессии HMG-CoA редуктазы может быть использован в качестве второго соединения при комбинированном лечении в соответствии с настоящим изобретением. Такие агенты могут представлять собой ингибиторы транскрипции HMG-CoA редуктазы, которые блокируют транскрипцию ДНК, или ингибиторы трансляции, которые препятствуют трансляции кодирующей для HMGCoA редуктазы мРНК в белок. Такие ингибиторы могут или непосредственно оказывать влияние на транскрипцию или трансляцию, или могут быть биотрансформированы в соединения, имеющие названные выше характеристики с помощью одного или нескольких ферментов в каскаде синтеза холестерина,или могут привести к накоплению метаболитов, обладающих названной выше активностью. Такая регуляция может быть легко определена специалистом в данной области в соответствии со стандартными способами оценки, то есть Methods of Enzymology (1985), 110:9-19. Примеры соединений описаны, например, в патенте США 5041432 и в публикации E.I.Mercer, Prog.Lip.Res. (1993), 32:357-416. Любой ингибитор СЕТР может быть использован в качестве второго соединения при комбинированном лечении в соответствии с настоящим изобретением. Определение ингибитор СЕТР, используемое в данном случае, если не оговорено особо, относится к соединению, которое ингибирует транспортный белок холестериловых эфиров (СЕТР), опосредованный транспорт различных холестериловых эфиров и триглицеридов от ЛНП до ЛВП и ЛОНП. Примеры таких соединений описаны в патенте США 5512548, J.Antibiot. (1996), 49(8): 815-816, Bioorg.Med.Chem.Lett. (1996), 6:1951-1954. Любой ингибитор АСАТ может быть использован в качестве второго соединения при комбинированном лечении в соответствии с настоящим изобретением. Определение ингибитор АСАТ, используемое в данном случае, если не оговорено особо, относится к соединению, которое ингибирует внутриклеточную этерификацию пищевого холестерина с помощью фермента ацил-СоА:холестеринацилтрансфераза. Такое ингибирование может быть легко определено специалистом в данной области с помощью стандартных способов, то есть способом Heider et al., Journal of Lipid Research (1983), 24:1127. Примеры соединений описаны, например, в патенте США 5510379, в публикациях WO 96/26948 и WO 96/10559.- 10008061 Любой ингибитор скваленсинтетазы может быть использован при комбинированном лечении в соответствии с настоящим изобретением. Определение ингибитор скваленсинтетазы, которое используется в данном случае, если не оговорено особо, относится к соединению, которое ингибирует конденсацию двух молекул фарнесилпирофосфата с образованием сквалена, катализируемую ферментом скваленсинтетаза. Такое ингибирование может быть легко определено специалистом в данной области в соответствии со стандартными способами оценки, то есть Methods of Enzymology (1985), 110:359-373. Примеры соединений описаны, например, в публикациях ЕР-0567026, ЕР-0645378 и ЕР-0645377. Специалист в области лечения гиперлипидемии легко определит терапевтически эффективное количество бифенилкарбоксамидного соединения по результатам представленных далее испытаний. В общем случае предполагается, что терапевтически эффективная доза будет составлять приблизительно от 0,001 до 5 мг/кг массы тела, предпочтительно приблизительно от 0,01 до 0,5 мг/кг массы тела пациента,который принимает лечение. Может быть предпочтительно вводить терапевтически эффективную дозу в форме двух или нескольких субдоз при соответствующих интервалах в течение всего дня. Указанные субдозы могут быть получены в виде отдельных дозированных форм, например, форм, каждая из которых содержит приблизительно от 0,1 до 350 мг, более предпочтительно приблизительно от 1 до 200 мг активного ингредиента на одну стандартную дозированную лекарственную форму. Точная доза и частота введения зависит от конкретного используемого бифенилкарбоксамидного соединения формулы (I), конкретного состояния, которое подвергается лечению, серьезности состояния,подвергающегося лечению, возраста, массы и физического состояния пациента, а также от других медикаментов (включая названные выше дополнительные принижающие уровень липидов агенты), которые пациент может принимать, что хорошо известно специалисту в данной области. Таким образом, указанное ежедневное эффективное количество может быть понижено или повышено в зависимости от реакции подвергающегося лечению пациента и/или в зависимости от оценки врача, прописывающего бифенилкарбоксамидные соединения настоящего изобретения. Интервалы эффективных ежедневных количеств,упомянутые выше, приведены только в качестве руководства. Экспериментальная часть В методиках, описанных ниже, используют следующие сокращения: ДМСО означает диметилсульфоксид; ТГФ означает тетрагидрофуран; ДХМ означает дихлорметан; ДИПЭ означает диизопропиловый эфир; ДМФА означает N,N-диметилформамид; ТФФГ (TFFH) означает гексафторфосфат тетраметилфторформамидиния; N-МП означает N-метил 2-пирролидин; ДИПЭА (DIPEA) означает диизопропилэтиламин; ТФУК означает трифторуксусную кислоту; и ТИС (TIS) означает триизопропилсилан. А. Синтез промежуточных соединений. Пример А.1. а) Смесь 4-(этоксикарбонилметил)пиперидина (0,0222 ммоль) и 2-хлор-5-нитропиридина (0,0222 моль) в ДМСО (40 мл) перемешивают в присутствии Na2CO3 в течение 2 ч. Реакционную смесь охлаждают до комнатной температуры и выливают на смесь лед/вода. Полученный осадок отфильтровывают и промывают водой. Продукт реакции очищают кристаллизацией из смеси этилацетата и гексана, получают этиловый эфир (5'-нитро-3,4,5,6-тетрагидро-2 Н-[1,2']бипиридин-4-ил)уксусной кислоты (промежуточное соединение 1, т.пл. 99-101 С).b) Смесь промежуточного соединения (1) (0,0102 моль) в ТГФ (50 мл) гидрируют в присутствии палладия-на-угле (10%, 0,3 г) в качестве катализатора в течение 30 мин при температуре 50 С. После поглощения водорода (1 экв.) катализатор отфильтровывают и фильтрат упаривают, получают этиловый эфир (5'-амино-3,4,5,6-тетрагидро-2 Н-[1,2']бипиридин-4-ил)уксусной кислоты (промежуточное соединение 2). Пример А.2.a) Смесь 4-(этоксикарбонилметил)пиперидина (0,011 моль) и 1-фтор-4-нитробензола (0,011 моль) в ДМСО (20 мл) перемешивают в присутствии Na2CO3 (0,044 моль) в течение 2 ч при температуре 60 С. Реакционную смесь охлаждают до комнатной температуры и выливают в смесь лед/вода. Полученный осадок отфильтровывают и промывают водой. Реакционный продукт очищают перекристаллизацией из смеси этилацетата и гексана, получают этиловый эфир [1-(4-нитрофенил)пиперидин-4-ил]уксусной кислоты (промежуточное соединение 3, т.пл. 83-85 С).b) Смесь промежуточного соединения (3) (0,0055 моль) в ТГФ (50 мл) гидрируют в присутствии палладия-на-угле (10%, 0,16 г) в качестве катализатора в течение 30 мин при температуре 50 С. После поглощения водорода (1 экв.) катализатор отфильтровывают и фильтрат упаривают, получают этиловый эфир [1-(4-аминофенил)пиперидин-4-ил]уксусной кислоты (промежуточное соединение 4). Пример А.3. Добавляют тионилхлорид (3,6 мл) к прозрачному раствору 4'-(трифторметил)-[1,1'-бифенил]-2 карбоновой кислоты (0,025 моль) в ДМФА (1 мл) и ДХМ (100 мл). Смесь перемешивают и кипятят с обратным холодильником 1 ч. Растворитель упаривают. К остатку добавляют ДХМ (50 мл), затем упаривают, получают 4'-(трифторметил)-[1,1'-бифенил]-2-карбонилхлорид (промежуточное соединение 5). 6-Метил-4'-(трифторметил)-[1,1'-бифенил]-2-карбонилхлорид (промежуточное соединение 6) полу- 11008061 чают аналогично из 6-метил-4'-трифторметилбифенил-2-карбоновой кислоты с использованием описанного выше способа. Пример А. 4. а) Смесь коммерческой смолы Novabiochem 01-64-0261 (5 г), бензиламина (1,765 г) и изопропоксида титана (IV) (4,686 г) в ДХМ (150 мл) осторожно перемешивают один час при комнатной температуре. Добавляют триацетоксиборгидрид натрия (4,5 г) и реакционную смесь перемешивают при комнатной температуре 18 ч. Добавляют метанол (10 мл) и смесь перемешивают 1 ч, затем фильтруют, промывают 1 раз ДХМ, 1 раз метанолом, затем 1 раз ДХМ (50 мл)+ДИПЭА (5 мл), промывают 3 раза вначале ДХМ, затем метанолом, затем сушат, получают 5,23 г смолы (I-а).b) Растворяют моно(9 Н-флуорен-9-илметиловый)эфир пиперидин-1,4-дикарбоновой кислоты (0,3 ммоль) в смеси ДХМ (2 мл) и ДМФА (0,5 мл) и к смеси добавляют смолу (I-а) (150 мг) в ДХМ (1 мл), после чего добавляют ТФФГ (0,3 ммоль) в ДХМ (0,5 мл) и ДИПЭА (0,6 мл) в ДХМ (0,5 мл). Реакционную смесь встряхивают 20 ч при комнатной температуре. Смесь фильтруют, промывают ДХМ (3 раза), СН 3OН (3 раза),ДХМ (3 раза), СН 3 ОН (3 раза), ДХМ (3 раза), СН 3 ОН (3 раза). Добавляют смесь пиперидина в ДМФА(20%, 3 мл) и реакционную смесь встряхивают 3 ч при комнатной температуре. Смесь фильтруют, промывают ДХМ (3 раза), СН 3 ОН (3 раза), ДХМ (3 раза), СН 3 ОН (3 раза), ДХМ (3 раза), СН 3 ОН (3 раза),получают смолу (I-b). с) Добавляют смесь 1-фтор-4-нитробензола (0,5 ммоль) в N-МП (0,5 мл) к смоле (I-b) в N-МП (3 мл). Добавляют ДИПЭА (1 ммоль), растворенный в N-МП (0,5 мл), и реакционную смесь встряхивают 18 ч при температуре 50 С. Реакционную смесь охлаждают, фильтруют, промывают ДХМ (3 раза), СН 3 ОН (3 раза),ДХМ (3 раза), СН 3 ОН (3 раза), ДХМ (3 раза), СН 3 ОН (3 раза), получают смолу (I-с). Пример А.5. а) Гидрат натриевой соли нитромалондиальдегида (0,0143 моль) и полусульфата S-метилизотиурония (0,0254 моль) растворяют в воде (40 мл) и добавляют этиловый эфир пиперидин-4-илуксусной кислоты (0,0214 моль) (полученный путем превращения гидрохлорида этилового эфира пиперидин-4 илуксусной кислоты в свободное основание). Реакционную смесь нагревают на водяной бане в течение 10 мин и оставляют на ночь. Полученный осадок отфильтровывают и промывают водой. Маточные слои обрабатывают NaHCO3 (2 г) и нагревают до 60 С в течение 10 мин, затем смесь охлаждают и оставляют на ночь. И, наконец, полученный осадок отфильтровывают, выделяют этиловый эфир [1-(5-нитропиримидин-2-ил)пиперидин-4-ил]уксусной кислоты (промежуточное соединение 7).b) Раствор промежуточного соединения (7) (0,011 моль) в этилацетате (100 мл) гидрируют при ком- 12008061 натной температуре 16 ч при атмосферном давлении с помощью палладия-на-угле (10%, 0,3 г) в качестве катализатора и водорода (3 экв.). Реакционную смесь отфильтровывают через целит и промывают этилацетатом. Фильтрат упаривают, получают 1,9 г этилового эфира [1-(5-аминопиримидин-2-ил)пиперидин 4-ил]уксусной кислоты (промежуточные соединения 8). В. Синтез конечных соединений. Пример В.1. Раствор промежуточного соединения (6) (0,005 моль) в диоксане (5 мл) добавляют к раствору промежуточного соединения (2) (0,005 моль) в диоксане (15 мл) и триэтиламине (0,005 моль). Реакционную смесь перемешивают при комнатной температуре 1 ч, и затем разбавляют водой. Продукт реакции экстрагируют этилацетатом (100 мл), органический слой промывают рассолом, сушат, упаривают и полученное масло затем очищают колоночной хроматографией на силикагеле с использованием смеси этилацетат/гексан (1:4) в качестве элюента, получают соединение 14 (т.пл. 134-137 С). Пример В.2. Добавляют 4'-(трифторметил)-[1,1'-бифенил]-2-карбоновую кислоту (0,3 ммоль), растворенную в смеси ДХМ и ДМФА (80:20) (1 мл), к смоле (I-d) в ДХМ (1 мл). Добавляют раствор ТФФГ (0,3 ммоль) в ДХМ (1 мл), после чего добавляют раствор ДИПЭА (0,6 ммоль) в ДХМ (1 мл). Реакционную смесь встряхивают в течение 48 ч. Реакционную смесь фильтруют, промывают ДХМ (3 раза), СН 3 ОН (3 раза), ДХМ(3 раза), СН 3OН (3 раза), ДХМ (3 раза) и СН 3 ОН (3 раза). Добавляют ТФУК/ТИС/ДХМ (5:2:93) (4 мл) и смесь встряхивают 1 ч, затем фильтруют. Еще добавляют смесь ТФУК/ТИС/ДХМ (5:2:93) (2 мл), реакционную смесь встряхивают 15 мин и затем фильтруют. Фильтраты сушат продувкой в атмосфере азота при 50 С. Остаток забирают в ДХМ (3 мл) и обрабатывают водным раствором Na2CO3. Органическую фазу очищают с помощью ВЭЖХ на 5 мкм колонке Chromasil (20 мм вн.д. х 150 мм), элюент: от 100% ДХМ до смеси ДХМ/метанол (90/10 в течение 15 мин). Желаемые фракции собирают и органический растворитель упаривают, получают соединение (1). Пример В.3. Растворяют 6-метил-4'-трифторметилбифенил-2-карбоновую кислоту (0,0025 моль) в сухом ДХМ(140 мл) вместе с оксалилхлоридом (2,4 мл) и с несколькими каплями ДМФА при 0 С. Затем порциями добавляют еще 6-метил-4'-трифторметилбифенил-2-карбоновую кислоту (0,0225 моль) в токе азота. Реакционную смесь мягко нагревают до 40 С до получения конечного гомогенного раствора и до остановки выделения газа. Смеси дают охладиться до комнатной температуры, затем фильтруют через воронку Бюхнера. Отфильтрованный остаток растворяют в ДХМ, затем добавляют по каплям при 0 С к раствору промежуточного соединения (4) (0,025 моль) и триэтиламина (3 г) в ДХМ (140 мл). Реакционной смеси позволяют нагреваться до комнатной температуры в течение 90 мин. Осадок отфильтровывают, сушат и очищают с помощью ВЭЖХ на Нуреrрrер С-18, получают соединение (10). Соединение (10) (0,00042 моль) растворяют в 2-пропаноле (5 мл) путем нагревания. Добавляют раствор НСl (6 М) в 2-пропаноле (0,00042 моль) и смесь охлаждают до комнатной температуры, после чего упаривают растворитель. Остаток кристаллизуют из смеси этанола и ДИПЭ, получают аддитивную соль соляной кислоты и соединения (10). Соединение (10) (0,00042 моль) растворяют в 2-пропаноле (5 мл) путем нагревания. Добавляют метансульфоновую кислоту (0,00042 моль) и раствор охлаждают до комнатной температуры. Осадок отфильтровывают и сушат, получают аддитивную соль, метансульфонат соединения (10). Соединение (10) (0,00042 моль) растворяют в 2-пропаноле (5 мл) путем нагревания. Добавляют малеиновую кислоту (0,00042 моль) и раствор охлаждают до комнатной температуры. Осадок отфильтровывают и сушат, получают аддитивную соль, малеат соединения (10). Пример В.4. Соединение (16) (0,0014 моль) суспендируют в этаноле (5 мл), добавляют NH3 (5 мл) и реакционную смесь перемешивают и кипятят в течение ночи. Смесь охлаждают до комнатной температуры и осадок отфильтровывают. Фильтрат упаривают и очищают быстрой колоночной хроматографией, получают соединение (17). Пример В.5. 4'-Трифторметилбифенил-2-карбоновую кислоту (0,0072 моль) в тионилхлориде (2,1 мл) перемешивают и кипятят с обратным холодильником в течение 3 ч в токе азота. Избыток тионилхлорида отгоняют. К остатку добавляют толуол (10 мл) и смесь упаривают досуха на роторном испарителе. Остаток растворяют в ДХМ (10 мл) и охлаждают до 0 С в токе азота. По каплям добавляют раствор промежуточного соединения (8) и триэтиламина (1,1 мл) в ДХМ (10 мл). Реакционной смеси дают медленно нагреться до 20 С, затем перемешивают еще 16 ч. Растворитель упаривают и остаток очищают колоночной хроматографией на силикагеле (элюент: этилацетат/гексан, 1:1), получают 2,76 г соединения (16). В табл. F-1 перечислены соединения, которые получены в приведенных выше примерах. С. Фармакологические примеры. С.1. Количественное определение секреции АроВ. Клетки HepG2 культивируют в 24-луночных планшетах в MEM Rega 3, содержащей 10% фетальной телячьей сыворотки. При слиянии 70% среду заменяют и добавляют испытуемое соединение или носитель (ДМСО, конечная концентрация 0,4%). Через 24 ч выдерживания среду переносят в пробирки Eppendorf и очищают путем центрифугирования. Антитело овцы против любого АроВ добавляют в надосадочную жидкость и смесь выдерживают при 8 С в течение 24 ч. Затем добавляют антитело кролика против антитела овцы и иммунному комплексу позволяют выпадать в осадок в течение 24 ч при 8 С. Иммунный осадок пеллетируют центрифугированием в течение 25 мин при 1320 g и промывают дважды буфером, содержащим 40 мМ Mops, 40 мМ NaH2PO4, 100 мМ NaF, 0,2 мМ DTT, 5 мМ ЭДТУ, 5 мМPMSF. Радиоактивность пеллет оценивают количественно с помощью жидкостного сцинтилляционного счетчика. Полученные значения IС 50 приведены в табл. С.1. Таблица С.1 Значения pIC50 (=-log значений IС 50) С.2. Оценка МТР. Активность МТР измеряют с использованием метода оценки, аналогичного описанному в публикации J.R. Wetterau, D.B. Zilversmit, Chemistry and Physics of Lipids, 38, 205-222 (1985). Чтобы получить донорные и акцепторные везикулы, подходящие липиды в хлороформе помещают в стеклянную пробирку для испытаний и сушат в токе N2. Буфер, содержащий 15 мМ Tris-НСl с рН 7,5, 1 мМ ЭДТУ, 40 мМNaCl, 0,02% NaN3 (оценочный буфер), добавляют к высушенному липиду. Смесь недолго интенсивно перемешивают и липидам затем дают возможность гидратироваться в течение 20 мин на льду. Везикулы затем готовят путем обработки загрузки ультразвуком (Branson 2200) при комнатной температуре в течение максимально 15 мин. Бутилированный гидрокситолуол вводят во все препараты везикул в концентрации 0,1%. Смесь для оценки липидного переноса содержит донорные везикулы (40 нмоль фосфатидилхолина, 7,5% мол. кардиолипина и 0,25% моль три[1-14 С]олеата глицерина), акцепторные везикулы(240 нмоль фосфатидилхолина) и 5 мг БСА в суммарном объеме 675 мкл в 1,5 мл пробирках для микроцентрифугирования. Испытуемые соединения добавляют растворенными в ДМСО (конечная концентрация 0,13%). Через 5 мин предварительного выдерживания при 37 С реакцию начинают путем добавления МТР в 100 мкл буфера для диализа. Реакцию останавливают путем добавления 400 мкл целлюлозыDEAE-52, предварительно уравновешенной в 15 мМ Tris-HCl, рН 7,5, 1 мМ ЭДТУ, 0,02% NaN3 (1:1,об./об.). Смесь перемешивают 4 мин и центрифугируют 2 мин при максимальной скорости в центрифугеEppendorf (4C), чтобы пеллетировать связанные с DEAE-52 донорные везикулы. Подсчитывают радиоактивность аликвот надосадочной жидкости, содержащей акцепторные липосомы, и [14 С]-импульсы используют для расчета процента переноса триглицерида от донорных везикул к акцепторным везикулам. ФОРМУЛА ИЗОБРЕТЕНИЯ 1. Соединение формулы (I) его N-оксиды, фармацевтически приемлемые кислотно-аддитивные соли и его стереохимические изомерные формы, гдеR1 представляет собой атом водорода, C1-4-алкил, атом галогена или полигалоген-С 1-4-алкил;R2 представляет собой атом водорода, C1-4-алкил, атом галогена или полигалоген-С 1-4-алкил;R3 представляет собой атом водорода или C1-4-алкил;R4 представляет собой атом водорода, C1-4-алкил или атом галогена;n принимает целые значения 0 или 1;X1 и X2 или оба представляют собой атом углерода, или, когда один из X1 или X2 представляет собой атом азота, то другой из X1 или X2 представляет собой атом углерода;X3 представляет собой атом углерода или азота, при условии, что только один из X1 или X2 представляет собой атом азота;Y представляет собой О или NR6, где R6 представляет собой атом водорода или C1-4-алкил; иR5 представляет собой атом водорода; C1-6-алкил, необязательно замещенный C1-4-алкокси-, цианогруппой, полигалоген-С 1-4-аклилом или арилом; С 2-6-алкенил, необязательно замещенный арилом; С 3-6 алкинил, необязательно замещенный арилом; арил или гетероарил; арил представляет собой фенил; фенил, замещенный одним, двумя или тремя заместителями, каждый из которых независимо друг от друга выбран из нитро-, азидо-, цианогруппы, атома галогена, гидроксигруппы, C1-6-алкила, С 3-б-циклоалкила, C1-4-алкоксигруппы, полигалоген-С 1-6-алкила, амино-, моноили ди(C1-6-алкил)аминогруппы; гетероарил представляет собой пиридинил, пиразинил, пиримидинил, пиридазинил, триазинил,триазолил, имидазолил, пиразолил, тиазолил, изотиазолил, оксазолил, пирролил, фуранил или тиенил; и необязательно замещены одним, двумя или тремя заместителями, каждый из которых независимо выбран из нитро-, азидо-, цианогруппы, атома галогена, гидроксигруппы, C1-6-алкила, С 3-6-циклоалкила, C1-4 алкилоксигруппы, полигалоген-С 1-4-алкила, амино-, моно- или ди(C1-6-алкил)аминогруппы. 2. Соединение по п.1, где X1, X2 и X3 представляют собой атом углерода.- 16008061 3. Соединение по п.1, где X1 представляет собой атом углерода, X2 представляет собой атом азота иX представляет собой атом углерода. 4. Соединение по п.1, где X1 представляет собой атом азота, X2 представляет собой атом углерода и 3X представляет собой атом углерода. 5. Соединение по любому из пп.1-4, где n принимает целое значение 0. 6. Соединение по любому из пп.1-4, где n принимает целое значение 1. 7. Фармацевтическая композиция, содержащая фармацевтически приемлемый носитель и терапевтически эффективное количество соединения по любому из пп.1-6. 8. Способ получения фармацевтической композиции по п.7, включающий тщательное смешивание терапевтически эффективного количества соединения по любому из пп.1-6 с фармацевтически приемлемым носителем. 9. Применение соединения по любому из пп.1-6 в качестве медикамента. 10. Способ получения соединения формулы (I), где промежуточное соединение формулы (II), где вводят в реакцию с бифенилкарбоновой кислотой или галогенидом, имеющими формулу (III), где R1 и R2 имеют значения, определенные в формуле (I) , и Q1 выбран из гидроксигруппы и атома галогена по меньшей мере в одном реакционно-инертном растворителе и необязательно в присутствии подходящего основания или, если желательно, соединение формулы (I) превращают в кислотно-аддитивную соль, или, наоборот,кислотно-аддитивную соль соединения формулы (I) превращают в свободное основание с помощью щелочи; и, если желательно, получают их стереохимические изомерные формы.
МПК / Метки
МПК: C07D 211/34, A61K 31/445, C07D 211/62, A61P 9/10, A61P 3/06
Метки: качестве, аполипопротеина, секреции, бифенилкарбоксамиды, ингибиторов, n-арилпиперидинзамещенные
Код ссылки
<a href="https://eas.patents.su/18-8061-n-arilpiperidinzameshhennye-bifenilkarboksamidy-v-kachestve-ingibitorov-sekrecii-apolipoproteina-v.html" rel="bookmark" title="База патентов Евразийского Союза">N-арилпиперидинзамещенные бифенилкарбоксамиды в качестве ингибиторов секреции аполипопротеина в</a>
Триамид-замещённые индолы, бензофураны и бензотиофены в качестве ингибиторов микросомального белка, переносящего триглицериды, (мтр) и/или ингибиторов секреции аполипопротеина в (аро в)

Номер патента: 7008
Опубликовано: 30.06.2006
Авторы: Ченг Хенгмиао, Блайз Алан Элвуд, Хуатан Хип, Ли Дзин, Бронк Брайан Скотт, Бертинато Питер, Мейсон Клайв Филип
МПК: A61K 31/404, A61K 31/4439, A61K 31/343...
Метки: триамид-замещённые, бензотиофены, индолы, бензофураны, триглицериды, качестве, микросомального, секреции, ингибиторов, аполипопротеина, аро, белка, переносящего, мтр
Формула / Реферат:
1. Соединение формулы 1b или его фармацевтически приемлемая соль, где R1 представляет собой заместитель в 5 или 6 положении формулы 1b и имеет структуру или, когда R7 представляет собой фенил, пиридил, фенил-Z1-или пиридил-Z1-, необязательно замещенные от одного до пяти, независимо, выбранными R12, то R1 представляет собой (C1-С6)алкил, (С3-С8)циклоалкил, (С5-С10)бициклоалкил, -(CRaRb)tO(C1-C6 алкил), -(СRaRb)tS(C1-C6 алкил), -(CRaRb)rC(O)R15,...
Бифенилкарбоксамиды, полезные в качестве снижающих содержание липидов агентов

Номер патента: 6101
Опубликовано: 25.08.2005
Авторы: Бакс Лео Якобус Йозеф, Мерпул Ливен
МПК: C07C 233/80, A61P 3/06, A61K 31/40...
Метки: агентов, бифенилкарбоксамиды, содержание, липидов, снижающих, полезные, качестве
Формула / Реферат:
1. Соединение формулы (I) N-оксиды, фармацевтически приемлемые кислотно-аддитивные соли и их стереохимически изомерные формы, где p1, p2 и p3 означают целые числа, каждое из которых независимо равно 1-3; каждый из R1 независимо выбирают из группы, включающей водород, C1-4-алкил, C1-4-алкилокси, галоген, гидрокси, меркапто, циано, нитро, C1-4-алкилтио или полигалоген-C1-6-алкил, амино, C1-4-алкиламино и ди(C1-4-алкил)амино; каждый из R2...
Применение 2,6-диизопропилфенилового эфира [(2,4,6-триизопропилфенил)ацетил]сульфаминовой кислоты в качестве ингибитора секреции сальных желез, в частности, для лечения акне и композиции и способы наоснове данного соединения

Номер патента: 4255
Опубликовано: 26.02.2004
Автор: Хоуман Рейнолд
МПК: A61P 17/10, A61K 31/255
Метки: эфира, ингибитора, акне, лечения, кислоты, соединения, секреции, наоснове, частности, применение, сальных, качестве, 2,4,6-триизопропилфенил)ацетил]сульфаминовой, композиции, способы, желез, 2,6-диизопропилфенилового, данного
Формула / Реферат:
1. Фармацевтическая композиция, содержащая ингибирующее акне количество 2,6-диизопропилфенилового эфира [(2,4,6-триизопропилфенил)ацетил]сульфаминовой кислоты и фармацевтически приемлемый носитель. 2. Применение 2,6-диизопропилфенилового эфира [(2,4,6-триизопропилфенил)ацетил]сульфаминовой кислоты для изготовления лекарства для лечения акне. 3. Фармацевтическая композиция, содержащая ингибирующее секрецию сальных желез количество...
Гетероариламины в качестве ингибиторов гликогенсинтаза-киназы 3-бета (ингибиторов gsk3)

Номер патента: 7298
Опубликовано: 25.08.2006
Авторы: Леви Паулус Йоаннес, Койманс Люсьен Мария Хенрикус, Ван Акен Кун Жанн Альфонс, Эмбрехтс Вернер Констант Йохан, Винкерс Хендрик Мартен, Лав Кристофер Джон, Жанссен Поль Адриан Ян, Дильс Гастон Станислас Марселла, Де Жонж Марк Рене, Виллемс Марк, Бейнстерс Петер Якобус Йоханнес Антониус, Хэрес Ян, Фрейн Эдди Жан Эдгар
МПК: A61K 31/505, A61K 31/435, A61K 31/50...
Метки: гетероариламины, 3-бета, гликогенсинтаза-киназы, ингибиторов, gsk3, качестве
Формула / Реферат:
1. Соединение формулы его N-оксид, фармацевтически приемлемая аддитивная соль, четвертичный амин и стереохимически изомерная форма, где R1 обозначает водород; X обозначает -O-; -О-С1-4алкил- или прямую связь; Z обозначает прямую связь, -NH-, -С1-4алкил-NH- или -С(=O)-; R2 обозначает водород или фенил; R3 обозначает водород, С1-4алкил, полигалогенC1-6алкил или циано; R4 обозначает моноциклический или бициклический насыщенный гетероцикл;...
Сульфонамидные производные в качестве ингибиторов рассасывания костной ткани и ингибиторов адгезии клеток,способ их получения, применение и фармацевтическая композиция

Номер патента: 3102
Опубликовано: 26.12.2002
Авторы: Гадек Томас, Бодари Сара Кэтрин, Гурвест Жан-Франсуа, Пейман Ануширван, Карниато Дени, Шойнеманн Карлхайнц, Вилл Дэвид Вильям, Кнолле Йохен, Макдауэлл Роберт, Катбертсон Роберт Эндрю
МПК: A61K 31/505, A61P 19/10, C07D 239/42...
Метки: клеток,способ, получения, костной, ткани, рассасывания, композиция, фармацевтическая, качестве, применение, производные, адгезии, ингибиторов, сульфонамидные
Формула / Реферат:
1. Сульфонамидные производные общей формулы I где R1 и R2 вместе образуют двухвалентный (С2-С3)алкиленовый радикал; R4 является Н или (С1-С6)алкилом; R5 является (С1-С10)алкилом, (С6-С14)арилом, (C5-C14)гетероарилом или (С6-С14)арил(С1-С10)алкильной группой, где арил, гетероарил или алкил возможно замещены R3, или 2-оксобицикло[2.2.1]гепт-1-илметильной группой; R3 является (C1-C4)алкилом, (C1-C4)алкилокси, галогеном, трифторметилом, циано,...
Предыдущий патент: Антагонисты хемокинного рецептора и способы их применения
Следующий патент: Способ получения синтетических промежуточных соединений для пестицидов
Случайный патент: Стелька для обуви