Гетероциклические соединения в качестве лигандов гистаминовых h3 рецепторов
Номер патента: 22746
Опубликовано: 29.02.2016
Авторы: Кандикере Вишвоттам Нагарадж, Намала Рамбабу, Камбхампати Рамасастри, Ахмад Иштияки, Джасти Венкатесварлу, Муддана Нагешвара Рао, Саралая Раманатха Шрикантха, Схинде Анил Карбхари, Дварампуди Ади Редди, Джаяраджан Прадееп, Кота Лаксман, Нироджи Рамакришна, Гампа Мурлимохан, Тиривеедхи Тарака Нага Винаикумар, Кодру Падмаватхи, Шанмуганатхан Дханалакшми














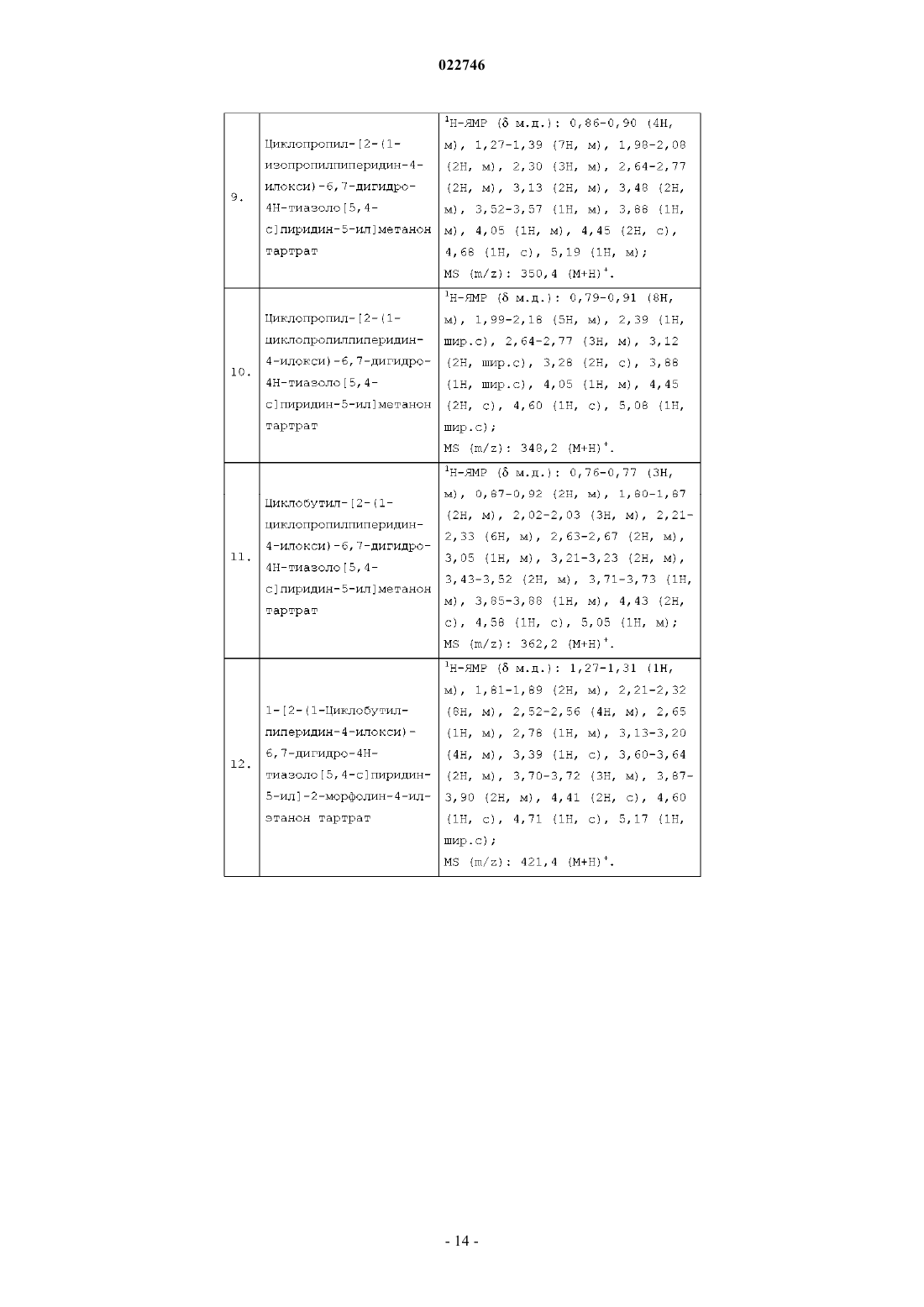






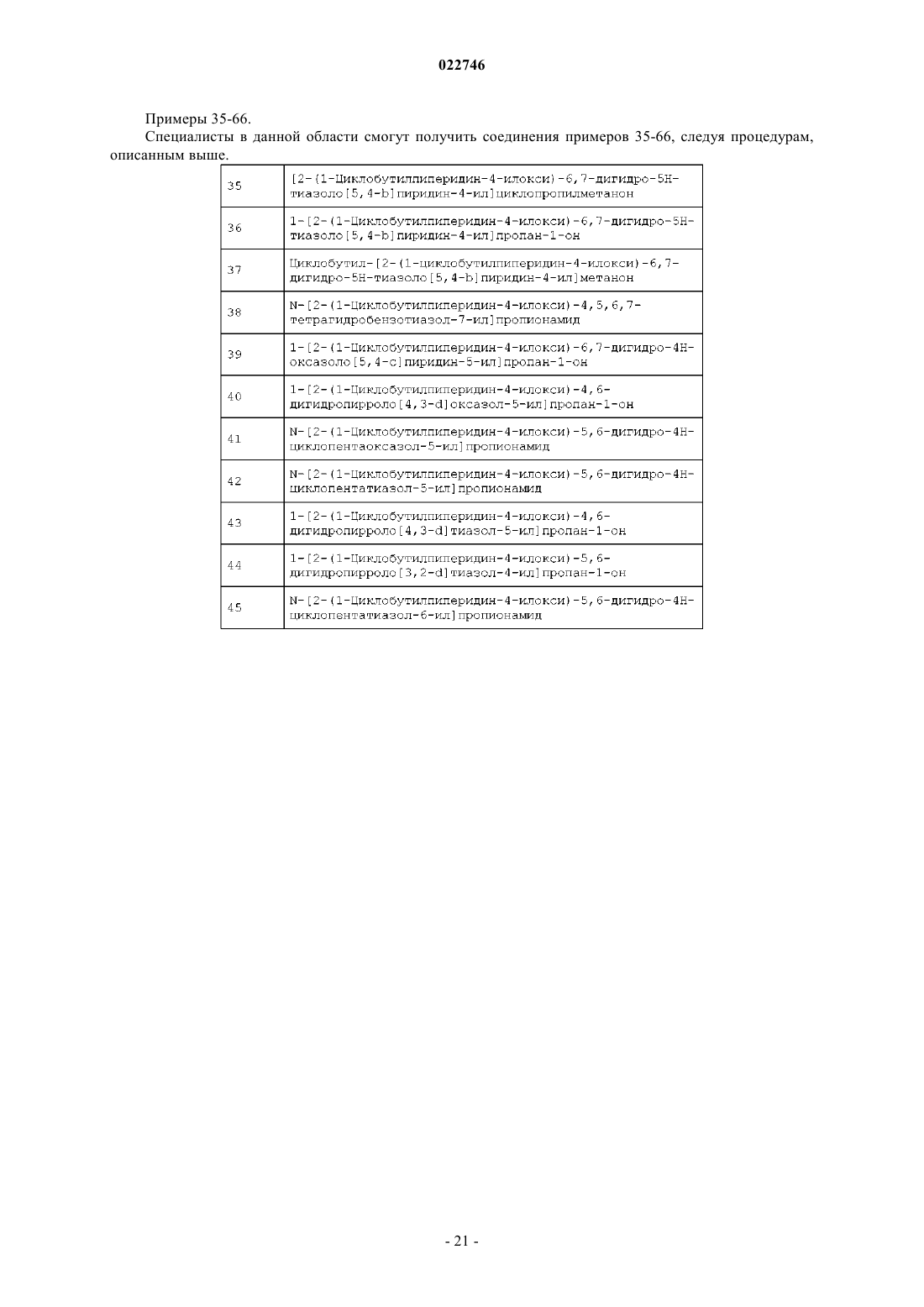







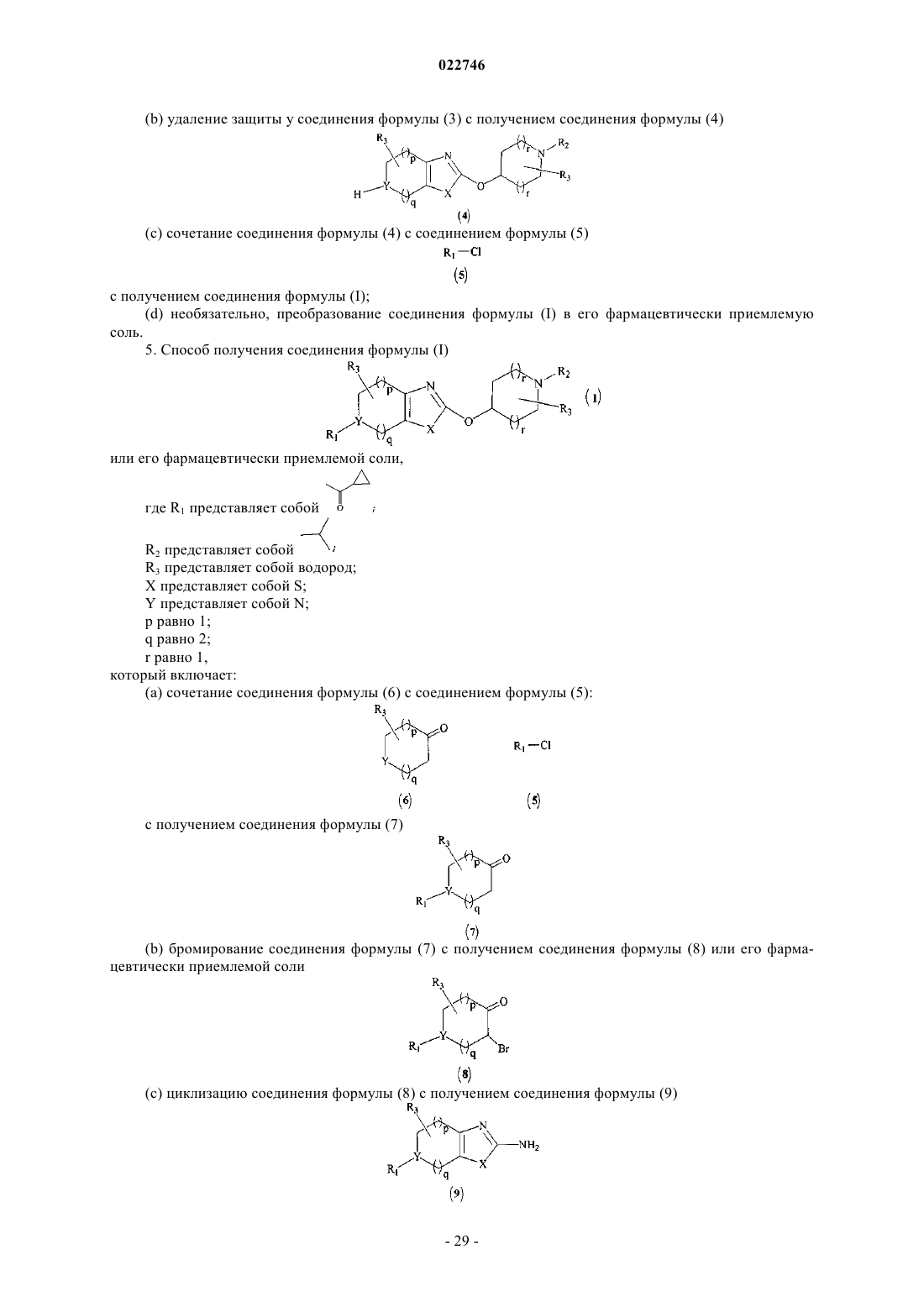
Формула / Реферат
1. Соединение общей формулы (I)

или его фармацевтически приемлемая соль,
где R1 представляет собой:

R2 представляет собой

или

R3 представляет собой водород;
X представляет собой S;
Y представляет собой С или N;
p равно 1;
q равно 1 или 2;
r равно 1.
2. Соединение по п.1, где R2 представляет собой


3. Соединение по п.1, представляющее собой:
1-[2-(1-циклобутилпиперидин-4-илокси)-6,7-дигидро-4H-тиазоло[5,4-с]пиридин-5-ил]пропан-1-он тартрат;
N-[2-(1-циклобутилпиперидин-4-илокси)-4,5,6,7-тетрагидробензотиазол-6-ил]пропионамид;
[2-(1-циклобутилпиперидин-4-илокси)-6,7-дигидро-4H-тиазоло[5,4-с]пиридин-5-ил]циклопропилметанон тартрат;
циклобутил[2-(1-циклобутилпиперидин-4-илокси)-6,7-дигидро-4H-тиазоло[5,4-с]пиридин-5-ил]метанон тартрат;
[2-(1-циклобутилпиперидин-4-илокси)-6,7-дигидро-4H-тиазоло[5,4-с]пиридин-5-ил]-(2-фторфенил)метанон тартрат;
1-[2-(1-циклобутилпиперидин-4-илокси)-6,7-дигидро-4H-тиазоло[5,4-с]пиридин-5-ил]-3-метилбутан-1-он тартрат;
циклобутил[2-(1-изопропилпиперидин-4-илокси)-6,7-дигидро-4H-тиазоло[5,4-с]пиридин-5-ил]метанон;
циклопропил[2-(1-изопропилпиперидин-4-илокси)-6,7-дигидро-4H-тиазоло[5,4-с]пиридин-5-ил]метанон тартрат;
циклопропил[2-(1-циклопропилпиперидин-4-илокси)-6,7-дигидро-4H-тиазоло[5,4-с]пиридин-5-ил]метанон тартрат;
циклобутил[2-(1-циклопропилпиперидин-4-илокси)-6,7-дигидро-4H-тиазоло[5,4-с]пиридин-5-ил]метанон тартрат;
1-[2-(1-циклобутилпиперидин-4-илокси)-6,7-дигидро-4H-тиазоло[5,4-с]пиридин-5-ил]-2-морфолин-4-илэтанон тартрат;
[4-(5-циклобутил-6,7-дигидро-4H-тиазоло[5,4-с]пиридин-2-илокси)пиперидин-1-ил]циклопропилметанон тартрат;
[2-(1-циклобутилпиперидин-4-илокси)-6,7-дигидро-4H-тиазоло[5,4-с]пиридин-5-ил]пиридин-4-илметанон тартрат;
[2-(1-циклобутилпиперидин-4-илокси)-6,7-дигидро-4H-тиазоло[5,4-с]пиридин-5-ил]-(4-метоксифенил)метанон тартрат;
1-[2-(1-циклобутилпиперидин-4-илокси)-6,7-дигидро-4H-тиазоло[5,4-с]пиридин-5-ил]-2-пиперидин-1-илэтанон тартрат;
1-[2-(1-циклобутилпиперидин-4-илокси)-6,7-дигидро-4H-тиазоло[5,4-с]пиридин-5-ил]-2-циклопропилэтанон;
2-(1-циклобутилпиперидин-4-илокси)-5-(2-фторбензолсульфонил)-6,7-дигидро-4H-тиазоло[5,4-с]пиридин тартрат;
2-(1-циклобутилпиперидин-4-илокси)-5-метансульфонил-6,7-дигидро-4H-тиазоло[5,4-с]пиридин;
1-[2-(1-циклобутилпиперидин-4-илокси)-6,7-дигидро-4H-тиазоло[5,4-с]пиридин-5-ил]-2-метилпропан-1-он тартрат;
2-(1-циклобутилпиперидин-4-илокси)-5-(2-трифторметилпиридин-5-ил)-6,7-дигидро-4H-тиазоло[5,4-с]пиридин;
циклопропил[2-(1-изобутилпиперидин-4-илокси)-6,7-дигидро-4H-тиазоло[5,4-с]пиридин-5-ил]метанон тартрат;
[2-(1-циклобутилпиперидин-4-илокси)-6,7-дигидро-4H-тиазоло[5,4-с]пиридин-5-ил]-(2-трифторметилпиридин-5-ил)метанон;
[2-(1-циклобутилпиперидин-4-илокси)-6,7-дигидро-4H-тиазоло[5,4-с]пиридин-5-ил]пиридин-3-илметанон тартрат;
2-(1-циклобутилпиперидин-4-илокси)-5-пиридин-3-ил-6,7-дигидро-4H-тиазоло[5,4-с]пиридин тартрат;
[2-(1-циклобутилпиперидин-4-илокси)-6,7-дигидро-4H-тиазоло[5,4-с]пиридин-5-ил]-(тетрагидропиран-4-ил)метанон;
[2-(1-циклобутилпиперидин-4-илокси)-6,7-дигидро-4H-тиазоло[5,4-с]пиридин-5-ил]морфолин-4-илметанон тартрат;
[2-(1-циклобутилпиперидин-4-илокси)-6,7-дигидро-4H-тиазоло[5,4-с]пиридин-5-ил]пиперидин-1-илметанон гидрохлорид;
6-[2-(1-циклобутилпиперидин-4-илокси)-6,7-дигидро-4H-тиазоло[5,4-с]пиридин-5-ил]никотинамид;
[2-(1-циклобутилпиперидин-4-илокси)-6,7-дигидро-4H-тиазоло[5,4-с]пиридин-5-ил]циклопентилметанон тартрат;
[2-(1-циклобутилпиперидин-4-илокси)-6,7-дигидро-5Н-тиазоло[5,4-b]пиридин-4-ил]циклопропилметанон тартрат и
циклопропил[2-(1-изопропилпиперидин-4-илокси)-4,5,7,8-тетрагидротиазоло[5,4-d]азепин-6-ил]метанон тартрат
или их фармацевтически приемлемую соль.
4. Способ получения соединения формулы (I)

или его фармацевтически приемлемой соли,
где R1 представляет собой:


R2 представляет собой
 или
или
R3 представляет собой водород;
X представляет собой S;
Y представляет собой N;
p равно 1;
q равно 1;
r равно 1,
который включает:
(а) сочетание соединения формулы (1) с соединением формулы (2):

с получением соединения формулы (3)

(b) удаление защиты у соединения формулы (3) с получением соединения формулы (4)

(с) сочетание соединения формулы (4) с соединением формулы (5)
R1 - Cl
с получением соединения формулы (I);
(d) необязательно, преобразование соединения формулы (I) в его фармацевтически приемлемую соль.
5. Способ получения соединения формулы (I)

или его фармацевтически приемлемой соли,
где R1 представляет собой R2 представляет собой
R2 представляет собой
R3 представляет собой водород;
X представляет собой S;
Y представляет собой N;
p равно 1;
q равно 2;
r равно 1,
который включает:
(а) сочетание соединения формулы (6) с соединением формулы (5):

с получением соединения формулы (7)

(b) бромирование соединения формулы (7) с получением соединения формулы (8) или его фармацевтически приемлемой соли

(с) циклизацию соединения формулы (8) с получением соединения формулы (9)

(d) диазотирование соединения формулы (9) с получением соединения формулы (10)

(е) сочетание соединения формулы (10) с соединением формулы (2)

с получением соединения формулы (I),
(f) необязательно, преобразование соединения формулы (I) в его фармацевтически приемлемую соль.
6. Способ получения соединения (I)

или его фармацевтически приемлемой соли,
где R1 представляет собой R2 представляет собой
R2 представляет собой
R3 представляет собой водород;
X представляет собой S;
Y представляет собой С;
p равно 1;
q равно 1;
r равно 1,
который включает:
(а) сочетание соединение формулы (11) с соединением формулы (5):

с получением соединения формулы (12)

(b) сочетание соединения формулы (12) с соединением формулы (2)

с получением соединения формулы (I);
(с) необязательно преобразование соединения формулы (I) в его фармацевтически приемлемую соль.
7. Применение соединения по любому из пп.1-3 для получения лекарственного средства для лечения заболеваний, связанных с гистаминовыми Н3 рецепторами.
8. Применение по п.7, где заболевание, связанное с гистаминовыми H3 рецепторами, выбрано из когнитивных расстройств, расстройств сна, ожирения и боли.
Текст
ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ В КАЧЕСТВЕ ЛИГАНДОВ ГИСТАМИНОВЫХ H3 РЕЦЕПТОРОВ Изобретение относится к гетероциклическим соединениям формулы (I) и их фармацевтически приемлемым солям и к композициям, содержащим такие соединения: Нироджи Рамакришна, Схинде Анил Карбхари, Камбхампати Рамасастри, Намала Рамбабу,Дварампуди Ади Редди, Кота Лаксман, Гампа Мурлимохан, Кодру Падмаватхи, Тиривеедхи Тарака Нага Винаикумар, Кандикере Вишвоттам Нагарадж, Муддана Нагешвара Рао, Саралая Раманатха Шрикантха, Джаяраджан Прадееп,Шанмуганатхан Дханалакшми, Ахмад Иштияки, Джасти Венкатесварлу (IN) Медведев В.Н. (RU) Изобретение также относится к способу получения указанных выше соединений и их фармацевтически приемлемых солей. Соединения формулы (I) являются полезными в лечении различных расстройств, которые связаны с гистаминовыми H3 рецепторами.(71)(73) Заявитель и патентовладелец: СУВЕН ЛАЙФ САЙЕНСИЗ ЛИМИТЕД (IN) Область изобретения Настоящее изобретение относится к гетероциклическим соединениям формулы (I) и их фармацевтически приемлемым солям, способу их получения и композициям, содержащим такие соединения, для лечения различных расстройств, которые связаны с гистаминовыми Н 3 рецепторами: Предпосылки изобретения Гистаминовый H3 рецептор представляет собой связанный с G-белком рецептор (GPCR) и является одним из четырех рецепторов семейства гистамина. Гистаминовый H3 рецептор был идентифицирован в 1983 году, и его клонирование и описание было осуществлено в 1999 году. Гистаминовый Н 3 рецептор экспрессируется в большей степени в центральной нервной системе и в меньшей степени в периферической нервной системе. Известные из литературы данные говорят о том, что гистаминовые H3 рецепторы можно использовать в лечении когнитивных расстройств (British Journal of Pharmacology, 2008, 154(6), 1166-1181), расстройств сна (Trends in Pharmacology Science, 2004, 25(12), 618-625), ожирения (Molecular Interventions,2006, 6 (2), 77-88) и боли (Pain, 2008, 138(1), 61-69). Патенты/патентные публикации US 4695575, EP 0151826, WO 2010/026113, WO 2004/022060 иWO 2003/004480 раскрывают ряд соединений в качестве лигандов гистаминовых H3 рецепторов. Несмотря на то что были раскрыты некоторые лиганды гистаминовых H3 рецепторов, до настоящего времени на рынке не было ни одного соединения в этой области исследований, и все еще существует потребность и область исследований для открытия новых лекарственных средств с новыми химическими структурами для лечения расстройств, связанных с гистаминовыми H3 рецепторами. Краткое описание изобретения Настоящее изобретение относится к новым лигандам гистаминовых H3 рецепторов формулы (I) или их фармацевтически приемлемой соли. Настоящее изобретение относится к применению терапевтически эффективного количества соединения формулы (I) для получения лекарственного средства для лечения различных расстройств, которые связаны с гистаминовыми H3 рецепторами. В частности, соединения по настоящему изобретению являются полезными для лечения различных расстройств, таких как когнитивные расстройства, расстройства сна, ожирение и боль. Еще в одном аспекте изобретение относится к способам применения соединений формулы (I). Еще в одном аспекте изобретение дополнительно относится к способу получения соединений формулы (I) и их фармацевтически приемлемых солей. Репрезентативные соединения по настоящему изобретению включают соединения, указанные ниже,и их фармацевтически приемлемые соли. Настоящее изобретение не должно рассматриваться как ограничивающееся ими: и их фармацевтически приемлемую соль. Подробное описание изобретения Если не указано иное, следующие термины, используемые в описании и в формуле изобретения,имеют значения, указанные ниже. Термин "галоген" означает фтор, хлор, бром или йод. Термин "алкил" означает линейный или разветвленный углеводородный радикал, состоящий исключительно из атомов углерода и водорода, не содержащий никакой ненасыщенности, содержащий от 1 до 8 атомов углерода, и который присоединен к остальной части молекулы посредством простой связи. Примеры "алкильных" групп включают метил, этил, н-пропил, изопропил и т.п. Термин "алкокси" означает алкильную группу, присоединенную через кислородную связь к остальной части молекулы. Примеры "алкокси" групп включают метокси, этокси, пропилокси, изопропилокси и т.п. Термин "галогеналкил" означает линейные или разветвленные алкильные радикалы, содержащие от 1 до 3 атомов углерода. Примеры "галогеналкильных" групп включают фторметил, дифторметил, трифторметил, трифторэтил, фторэтил, дифторэтил и т.п. Термин "галогеналкокси" означает линейные или разветвленные алкоксирадикалы, содержащие от 1 до 3 атомов углерода. Примеры "галогеналкокси" групп включают фторметокси, дифторметокси, трифторметокси, трифторэтокси, фторэтокси, дифторэтокси и т.п. Термин "циклоалкил" означает неароматическое моноциклическое кольцо, включающее от 3 до 8 атомов углерода. Примеры "циклоалкильных" групп включают циклопропил, циклобутил, циклопентил и т.п. Термин "циклоалкилалкил" означает циклоалкильный кольцевой радикал, непосредственно связанный с алкильной группой. Термин "арил" означает любую функциональную группу или заместитель, образованный из простого ароматического кольца. Примеры "арильных" групп включают фенил, нафтил и т.п. Термин "гетероарил" означает органические соединения, которые содержат кольцевую структуру,содержащую в дополнение к углероду такие атомы, как сера, кислород или азот, в качестве части кольца,эти дополнительные атомы могут неоднократно встречаться в кольце. Эти кольца могут представлять собой любые простые ароматические кольца. Примеры "гетероарильных" групп включают пиридин, пиримидин, бензофуранил, бензотиофен, фурил, диоксаланил, пирролил, оксазолил, пиридил, пиридазинил,пиримидинил, пиразинил, хинолинил, индолил и т.п. Термин "гетероциклил" означает неароматическое моноциклическое кольцо, включающее от 2 до 7 атомов углерода, кольцевые структуры таких колец включают от 1 до 3 гетероатомов, эти дополнительные атомы могут неоднократно встречаться в кольце. Примеры "гетероциклильных" групп включают пирролидинил, пиперидинил, пиперазинил, морфолинил и т.п. Термин "гетероциклилалкил" означает гетероциклильный кольцевой радикал, непосредственно связанный с алкильной группой. Термины "лечащий", "лечить" или "лечение" охватывают значения, такие как превентивное, профилактическое и паллиативное лечение. Фраза "фармацевтически приемлемые соли" указывает, что вещество или композиция должны быть совместимы химически и/или токсикологически с другими ингредиентами, составляющими композицию,-4 022746 для лечения которого используют такую композицию. Фраза "терапевтически эффективное количество" означает количество соединения по настоящему изобретению, которое (i) лечит или предотвращает конкретное заболевание, состояние или расстройство;(ii) ослабляет, облегчает или устраняет один или несколько симптомов конкретного заболевания, состояния или расстройств;, (iii) предотвращает или замедляет возникновение одного или нескольких симптомов конкретного заболевания, состояния или расстройства, описанного в настоящем изобретении. Коммерческие реагенты использовали без дополнительной очистки. Комнатная температура относится к 25-40C. Если не указано иное, все масс-спектры были получены с использованием ESI условий. 1 Н-ЯМР спектры записывали при 400 МГц на устройстве Bruker. Дейтерированный хлороформ (99,8%D), метанол или диметилсульфоксид использовали в качестве растворителя. TMS использовали в качестве внутреннего стандарта. Значения химического сдвигавыражали в миллионных долях. Следующие аббревиатуры использовали для мультиплетности для ЯМР сигналов: с=синглет, шир.с=широкий синглет, д=дублет, т=триплет, кв.=квартет, квинт.=квинтет, г=гептет, дд=двойной дублет, дт=двойной триплет, тт=триплет триплетов, м=мультиплет. Хроматография относится к колоночной хроматографии,которую осуществляли с использованием силикагеля 100-200 меш и под давлением азота (флэшхроматография). Для номенклатуры соединений использовали программу Chem Draw Ultra 7.0. Способ получения Соединения формулы (I) можно получить в соответствии со схемой I. Схема I На представленной схеме 1 p имеет значение 1; q имеет значение 1; r имеет значение 1; Y представляет собой N; X представляет собой S; R1 представляет собой -C(O)-R4, -S(O)2-R4, замещенный или незамещенный циклоалкил, арил или гетероарил, и все другие символы имеют значения, определенные выше. Соединение формулы (1) подвергают сочетанию с соединением формулы (2) с получением соединения формулы (3). Соединение формулы (3) подвергают процедуре удаления защиты с получением соединения формулы (4). Соединение формулы (4) подвергают сочетанию с соединением формулы (5) с получением соединения формулы (I). На первой стадии представленного выше способа получения соединение формулы (1) подвергают сочетанию с соединением формулы (2) с получением соединения формул (3). Эту реакцию предпочтительно осуществляют в растворителе, таком как тетрагидрофуран, толуол, этилацетат, дихлорметан, триэтиламин, диметилформамид и т.п. или их смесь, и предпочтительно используют тетрагидрофуран. Реакцию можно осуществить в присутствии основания, такого как гидрид натрия, карбонат натрия, карбонат калия, бикарбонат натрия, гидроксид натрия или их смеси, предпочтительно используют гидрид натрия. Реакцию осуществляют при комнатной температуре. Продолжительность реакции может составлять от 4 до 8 ч, предпочтительно в пределах периода времени от 5 до 7 ч. На второй стадии представленного выше способа получения соединение формулы (3) подвергают процедуре удаления защиты с получением соединения формулы (4). Эту реакцию предпочтительно осуществляют в растворителе, таком как тетрагидрофуран, толуол, этилацетат, дихлорметан, ацетонитрил,1,4-диоксан, диметилформамид и т.п. или их смесь, и предпочтительно используют дихлорметан. Реакцию можно осуществить в присутствии кислоты, такой как трифторуксусная кислота, серная кислота,уксусная кислота, перхлорная кислота, хлористо-водородная кислота и т.п. или их смесь, и предпочтительно используют трифторуксусную кислоту. Реакцию осуществляют при температуре от 60 до 85C,предпочтительно при температуре от 65 до 75C. Продолжительность реакции может составлять от 2 до 6 ч, предпочтительно в пределах периода времени от 3 до 5 ч. На третьей стадии представленного выше способа получения соединение формулы (4) подвергают сочетанию с соединением формулы (5) с получением соединения формулы (I). Эту реакцию предпочтительно осуществляют в растворителе, таком как дихлорметан, тетрагидрофуран, толуол, этилацетат, диметилформамид и т.п. или их смесь, и предпочтительно используют дихлэрметан. Реакцию можно осуществить в присутствии основания, такого как триэтиламин, карбонат калия, диизопропилэтиламин и пиридин, и предпочтительно используют триэтиламин. Реакцию осуществляют при комнатной температуре. Продолжительность реакции может составлять от 15 до 45 мин, предпочтительно в пределах периода времени от 25 до 35 мин. Соединения формулы (1), формулы (2) и формулы (5) могут быть коммерчески доступными или их можно получить традиционными способами или путем модификации с использованием известного способа. Соединения формулы (I) также можно получить с использованием схемы II. Схема II На представленной схеме II p имеет значение 1; q имеет значение 2; r имеет значение 1; Y представляет собой N; X представляет собой S; и все другие символы имеют значения, определенные выше. Соединение формулы (6) подвергают сочетанию с соединением формулы (5) с получением соединения формулы (7). Соединение формулы (7) подвергают бромированию с получением соединения формулы (8). Соединение формулы (8) подвергают циклизации с получением соединения формулы (9). Соединение формулы (9) подвергают диазотированию с получением соединения формулы (10). Соединение формулы (10) подвергают сочетанию с соединением формулы (2) с получением соединения формулы (I). На первой стадии представленного выше способа получения соединение формулы (6) подвергают сочетанию с соединением формулы (5) с получением соединения формулы (7). Эту реакцию предпочтительно осуществляют в растворителе, таком как тетрагидрофуран, толуол, этилацетат, дихлорметан, диметилформамид и т.п. или их смесь, и предпочтительно используют дихлорметан. Реакцию можно осуществить в присутствии основания, такого как триэтиламин, карбонат калия, диизопропилэтиламин и пиридин, и предпочтительно используют триэтиламин. Реакцию осуществляют при комнатной температуре. Продолжительность реакции может составлять от 15 до 45 мин, предпочтительно в пределах периода времени от 25 до 35 мин. На второй стадии представленного выше способа получения соединение формулы (7) подвергают бромированию с получением соединения формулы (8). Реакцию можно осуществить в присутствии кислоты, такой как серная кислота, уксусная кислота, перхлорная кислота, хлористо-водородная кислота и т.п. или их смесь, и предпочтительно используют уксусную кислоту. Реакцию можно осуществить в присутствии агента бромирования, такого как бром, бромид меди(II), бромисто-водородная кислота,N-бромсукцинимид, пиридингидробромид пербромид, тетрабромметан и т.п. или их смесь, и предпочтительно используют бром. Реакцию осуществляют при комнатной температуре. Продолжительность реакции может составлять от 16 до 20 ч, предпочтительно в пределах периода времени от 17 до 19 ч. На третьей стадии представленного выше способа получения соединение формулы (8) подвергают циклизации с получением соединения формулы (9). Эту реакцию предпочтительно осуществляют в растворителе, таком как изопропиловый спирт, тетрагидрофуран, толуол, этилацетат, дихлорметан,триэтиламин, диметилформамид и т.п. или их смесь, и предпочтительно используют изопропиловый спирт. Реакцию можно осуществить в присутствии мочевины или тиомочевины. Реакцию осуществляют при температуре от 60 до 85C, предпочтительно при температуре от 65 до 75C. Продолжительность реакции может составлять от 15 до 45 мин, предпочтительно в пределах периода времени от 25 до 35 мин. На четвертой стадии представленного выше способа получения соединение формулы (9) подвергают диазотированию с получением соединения формулы (10). Эту реакцию предпочтительно осуществляют в растворителе, таком как ацетонитрил, тетрагидрофуран, изопропиловый спирт, толуол, этилацетат, дихлорметан, триэтиламин, диметилформамид и т.п. или их смесь, и предпочтительно используют ацетонитрил. Реакцию можно осуществить в присутствии агента бромирования, такого как бромид меди(II), бромисто-водородная кислота, N-бромсукцинимид, пиридингидробромид пербромид,тетрабромметан и т.п. или их смесь, и предпочтительно используют бромид меди(II). Реакцию можно осуществить в присутствии алкилнитритов, предпочтительно используют трет-бутилнитрит. Реакцию осуществляют при комнатной температуре. Продолжительность реакции может составлять от 15 до 45 мин, предпочтительно в пределах периода времени от 25 до 35 мин. На пятой стадии представленного выше способа получения соединение формулы (10) подвергают сочетанию с соединением формулы (2) с получением соединения формулы (1). Эту реакцию предпочтительно осуществляют в растворителе, таком как тетрагидрофуран, толуол, этилацетат, дихлорметан, триэтиламин, диметилформамид и т.п. или их смесь, и предпочтительно используют тетрагидрофуран. Реакцию можно осуществить в присутствии основания, такого как гидрид натрия, карбонат натрия, карбонат калия, бикарбонат натрия, гидроксид натрия или их смеси, и предпочтительно используют гидрид натрия. Реакцию можно осуществить в присутствии мочевины или тиомочевины. Реакцию осуществляют при температуре от 60 до 85C, предпочтительно при температуре от 65 до 75C. Продолжительность реакции может составлять от 12 до 18 ч, предпочтительно в пределах периода времени от 14 до 16 ч. Соединения формулы (2), формулы (5) и формулы (6) могут быть коммерчески доступными или их можно получить традиционными способами или путем модификации с использованием известного способа. Соединения формулы (I) также можно получить с использованием схемы III. Схема III На представленной схеме III p имеет значение 1; q имеет значение 1; r имеет значение 1; Y представляет собой С; X представляет собой S и все другие символы имеют значения, определенные выше. Соединение формулы (11) подвергают сочетанию с соединением формулы (5) с получением соединения формулы (12). Соединение формулы (12) подвергают сочетанию с соединением формулы (2) с получением соединения формулы (I). На первой стадии представленного выше способа получения соединение формулы (11) подвергают сочетанию с соединением формулы (5) с получением соединения формулы (12). Эту реакцию предпочтительно осуществляют в растворителе, таком как тетрагидрофуран, толуол, этилацетат, дихлорметан, диметилформамид и т.п. или их смесь, и предпочтительно используют дихлорметан. Реакцию можно осуществить в присутствии основания, такого как триэтиламин, карбонат калия, диизопропилэтиламин и пиридин, и предпочтительно используют триэтиламин. Реакцию осуществляют при комнатной температуре. Продолжительность реакции может составлять от 15 до 45 мин, предпочтительно в пределах периода времени от 25 до 35 мин. На второй стадии представленного выше способа получения соединение формулы (12) подвергают сочетанию с соединением формулы (2) с получением соединения формулы (I). Эту реакцию предпочтительно осуществляют в растворителе, таком как тетрагидрофуран, толуол, этилацетат, дихлорметан, триэтиламин, диметилформамид и т.п. или их смесь, и предпочтительно используют диметилформамид. Реакцию можно осуществить в присутствии основания, такого как гидрид натрия, карбонат натрия, карбонат калия, бикарбонат натрия, гидроксид натрия или их смеси, и предпочтительно используют гидрид натрия. Реакцию осуществляют при комнатной температуре. Продолжительность реакции может составлять от 45 до 51 ч, предпочтительно в пределах периода времени от 47 до 49 ч. Соединения формулы (2), формулы (5) и формулы (11) могут быть коммерчески доступными или их можно получить традиционными способами или путем модификации с использованием известного способа. При необходимости, можно осуществить любую одну или несколько из следующих стадий: i) преобразование соединения формулы (I) в другое соединение формулы (I) или ii) образование фармацевтически приемлемой соли. Способ (i) можно осуществить при помощи дальнейших химических модификаций с использованием хорошо известных реакций, таких как окисление, восстановление, введение защиты, удаление защиты, реакция перегруппировки, галогенирование, гидроксилирование, алкилирование, алкилтиолирование, деметилирование, О-алкилирование, О-ацилирование, N-алкилирование, N-алкенилирование,N-ацилирование, N-цианирование, N-сульфонилирование, реакция сочетания с использованием переходных металлов и т.п. В способе (ii) фармацевтически приемлемые соли могут быть получены обычным способом путем взаимодействия с соответствующей кислотой или кислотным производным. Подходящие фармацевтически приемлемые соли будут очевидны для специалистов в данной области, и включают соли, описанные в J. Pharm. Sci., 1977, 66, 1-19, такие как кислотно-аддитивные соли,образованные с неорганическими кислотами, такими как хлористо-водородная, бромисто-водородная,серная, азотная или фосфорная кислота, и органическими кислотами, такими как янтарная, малеиновая,уксусная, фумаровая, лимонная, яблочная, винная, бензойная, п-толуиловая, п-толуолсульфоновая, метансульфоновая или нафталинсульфоновая кислота. Фармацевтически приемлемые соли, составляющие часть настоящего изобретения, можно получить путем обработки соединения формулы (I) 1-6 экв. основания, такого как гидрид натрия, метоксид натрия,этоксид натрия, гидроксид натрия, трет-бутоксид калия, гидроксид кальция, ацетат кальция, хлорид кальция, гидроксид магния, хлорид магния и т.п. Можно использовать растворители, такие как вода, ацетон, простой эфир, ТГФ, метанол, этанол, трет-бутанол, диоксан, изопропанол, изопропиловый эфир или их смеси. Примеры Новые соединения по настоящему изобретению получали в соответствии со следующими экспериментальными процедурами, используя подходящие вещества и подходящие условия. Получение 1. Получение трет-бутилового эфира 2-бром-6,7-дигидро-4H-тиазоло[5,4-с]пиридин-5 карбоновой кислоты. Стадия (i). Получение трет-бутилового эфира 3-бром-4-оксопиперидин-1-карбоновой кислоты. Раствор трет-бутилового эфира 4-оксопиперидин-1-карбоновой кислоты (10 г, 50 ммоль) и алюминийхлорида (0,67 г, 5 ммоль) в тетрагидрофуране (30 мл) и диэтиловом эфире (30 мл) охлаждали до 0C и затем обрабатывали бромом (2,6 мл, 50 ммоль) в течение 30 мин. Перемешивали реакционную массу в течение 24 ч при 0-5C. После завершения реакции полученные твердые вещества фильтровали и маточный раствор концентрировали в вакууме. Полученное неочищенное вещество растирали с диэтиловым эфиром и твердые вещества фильтровали и сушили в вакууме с получением указанного в заголовке соединения (10 г). 1 при кипячении с обратным холодильником в течение 1 ч. После завершения реакции реакционную массу концентрировали и полученное неочищенное вещество растирали с диэтиловым эфиром (50 мл), твердые вещества фильтровали и сушили в вакууме с получением указанного в заголовке соединения (10 г). 1MS (m/z): 256 (М+Н)+. Стадия (iii). Получение трет-бутилового эфира 2-бром-6,7-дигидро-4H-тиазоло[5,4-с]пиридин-5 карбоновой кислоты. Раствор трет-бутилового эфира 2-амино-6,7-дигидро-4H-тиазоло[5,4-с]пиридин-5-карбоновой кислоты (10 г, 40 ммоль, получен на предыдущей стадии) и бромида меди(II) (9,6 г, 43 ммоль) в ацетонитриле (50 мл) охлаждали до 0C. трет-Бутилнитрит (5,1 мл, 43 ммоль) добавляли по каплям в течение 30 мин при 0C. Реакционную массу перемешивали в течение 30 мин и реакционную массу гасили 6 н. раствором хлористо-водородной кислоты. Продукт экстрагировали этилацетатом (3100 мл), объединенные органические слои промывали водой, затем насыщенным солевым раствором и сушили над безводным сульфатом натрия. Органические летучие вещества выпаривали в вакууме. Полученный остаток очищали флэш-хроматографией (этилацетат/н-гексан, 0,5/9,5) с получением указанного в заголовке соединенияMS (m/z): 319,3 (М+Н)+, 321,3 (М+Н)+. Получение 2. Получение трет-бутилового эфира (2-бром-4,5,6,7-тетрагидробензотиазол-6 ил)карбаминовой кислоты. Стадия (i). Получение трет-бутилового эфира (3-бром-4-оксоциклогексил)карбаминовой кислоты. Раствор трет-бутилового эфира (4-оксоциклогексил)карбаминовой кислоты (10 г, 46 ммоль) и алюминийхлорида (0,25 г, 2 ммоль) в тетрагидрофуране (30 мл) и диэтиловом эфире (30 мл) охлаждали до 0C, затем обрабатывали бромом (2,4 мл, 46 ммоль) в течение 30 мин. Реакционную массу перемешивали в течение 24 ч при 0-5C. После завершения реакции полученные твердые вещества фильтровали и нижний фильтрат концентрировали в вакууме. Полученное неочищенное вещество растирали с диэтиловым эфиром и полученные твердые вещества фильтровали и сушили в вакууме с получением указанного в заголовке соединения (9,0 г).MS (m/z): 292,3 (М+Н)+, 294,3 (М+3H)+. Стадия (ii). Получение трет-бутилового эфира 2-амино-4,5,6,7-тетрагидробензотиазол-6 ил)карбаминовой кислоты. Суспензию трет-бутилового эфира 3-бром-4-оксопиперидин-1-карбоновой кислоты (9 г, 31 ммоль,получен на предыдущей стадии) и тиомочевины (2,4 г, 31 ммоль) в изопропаноле (100 мл) нагревали при кипячении с обратным холодильником в течение 1 ч. После завершения реакции реакционную массу концентрировали и полученное неочищенное вещество растирали с диэтиловым эфиром (50 мл), твердые вещества фильтровали и сушили в вакууме с получением указанного в заголовке соединения (9 г). 1(9 г, 33 ммоль, получен на предыдущей стадии) и бромида меди(II) (8,3 г, 37 ммоль) в ацетонитриле(70 мл) охлаждали до 0C. Полученную массу обрабатывали трет-бутилнитритом (4,5 мл, 37 ммоль) в течение 30 мин при 0C. Реакционную массу перемешивали в течение 30 мин и гасили 6 н. раствором хлористо-водородной кислоты. Продукт экстрагировали этилацетатом (3100 мл), объединенные органические слои промывали водой, затем насыщенным солевым раствором и сушили над безводным сульфатом натрия. Органические летучие вещества выпаривали в вакууме. Остаток очищали флэшхроматографией (этилацетат/н-гексан, 0,5/9,5) с получением указанного в заголовке соединения (2,3 г). 1MS (m/z): 333,1 (М+Н)+, 335,3 (М+3H)+. Пример 1. Получение 1-[2-(1-циклобутилпиперидин-4-илокси)-6,7-дигидро-4H-тиазоло[5,4 с]пиридин-5-ил)пропан-1-он тартрата. Стадия (i). Получение трет-бутилового эфира 2-(1-циклобутилпиперидин-4-илокси)-6,7-дигидро 4H-тиазоло[5,4-с]пиридин-5-карбоновой кислоты. 1-Циклобутилпиперидин-4-ол (1,6 г, 10 ммоль) в тетрагидрофуране (20 мл) обрабатывали охлажденной и перемешиваемой суспензией гидрида натрия (0,9 г, 18 ммоль) в тетрагидрофуране (20 мл) медленно в течение 30 мин; реакционную смесь перемешивали в течение 1 ч. Добавляли по каплям раствор трет-бутилового эфира 2-бром-6,7-дигидро-4H-тиазоло[5,4-с]пиридин-5-карбоновой кислоты (3 г,9 ммоль, получен в получении 1) в тетрагидрофуране (30 мл) в течение 15 мин и реакционную смесь на-9 022746 гревали при кипячении с обратным холодильником в течение 6 ч. Реакционную массу гасили ледяной водой и продукт экстрагировали этилацетатом (350 мл). Объединенные органические слои промывали водой, затем насыщенным солевым раствором и сушили над безводным сульфатом натрия. Органические летучие вещества выпаривали в вакууме. Остаток очищали флэш-хроматографией (этилацетат/н-гексан,1/1) с получением указанного в заголовке соединения (2,0 г). 1MS (m/z): 394,2 (М+Н)+. Стадия (ii). Получение 2-(1-циклобутилпиперидин-4-илокси)-4,5,6,7-тетрагидротиазоло[5,4 с]пиридина. Раствор трет-бутилового эфира 2-(1-циклобутилпиперидин-4-илокси)-6,7-дигидро-4H-тиазоло[5,4 с]пиридин-5-карбоновой кислоты (2,0 г, 5 ммоль, получен на предыдущей стадии) в дихлорметане(30 мл) обрабатывали трифторуксусной кислотой (5,0 мл, 50 ммоль) при 0C. Реакционную массу перемешивали в течение 4 ч. После завершения реакции реакционную массу гасили в ледяной воде и доводили pH до 10 с использованием 40% водного раствора гидроксида натрия. Продукт экстрагировали дихлорметаном (350 мл), объединенные органические слои промывали водой, затем насыщенным солевым раствором и сушили над безводным сульфатом натрия. Органические летучие вещества выпаривали в вакууме с получением указанного в заголовке соединения (1,3 г). 1MS (m/z): 294,2 (М+Н)+. Стадия (iii). Получение 1-[2-(1-циклобутилпиперидин-4-илокси)-6,7-дигидро-4H-тиазоло[5,4 с]пиридин-5-ил]пропан-1-она. Раствор 2-(1-циклобутилпиперидин-4-илокси)-4,5,6,7-тетрагидротиазоло[5,4-с]пиридина (1,3 г,4 ммоль, получен на предыдущей стадии) и триэтиламина (1,9 мл, 13 ммоль) в дихлорметане (30 мл) охлаждали до 0C. Добавляли пропионилхлорид (0,4 мл, 5 ммоль) в дихлорметане (5 мл) по каплям в течение 15 мин и реакционную смесь перемешивали в течение 30 мин. Реакционную массу выливали на ледяную воду и продукт экстрагировали этилацетатом (350 мл). Объединенные органические слои промывали водой, затем насыщенным солевым раствором и сушили над безводным сульфатом натрия. Органические летучие вещества выпаривали в вакууме. Остаток очищали флэш-хроматографией (метанол/хлороформ, 2/98) с получением указанного в заголовке соединения (1,0 г). 1MS (m/z): 350,4 (М+Н)+. Стадия (iv). Получение 1-[2-(1-циклобутилпиперидин-4-илокси)-6,7-дигидро-4H-тиазоло[5,4 с]пиридин-5-ил]пропан-1-он тартрата. Раствор 1-[2-(1-циклобутилпиперидин-4-илокси)-6,7-дигидро-4H-тиазоло[5,4-с]пиридин-5 ил]пропан-1-она (0,8 г, 2,3 ммоль, получен на предыдущей стадии) в метаноле (10 мл) обрабатывалиL(+)-винной кислотой (0,34 г, 2,3 ммоль) при 0C. Реакционную массу перемешивали примерно в течение 1 ч, и растворитель выпаривали в вакууме досуха. Твердые вещества промывали диэтиловым эфиром и сушили в вакууме с получением указанного в заголовке соединения (1,1 г). 1(2 мл) и реакционную смесь перемешивали в течение 30 мин. Реакционную массу выливали на ледяную воду и продукт экстрагировали дихлорметаном (315 мл). Объединенные органические слои промывали водой, затем насыщенным солевым раствором и сушили над безводным сульфатом натрия. Органические летучие вещества выпаривали в вакууме с получением указанного в заголовке соединения (2,7 г). 1MS (m/z): 182 (М+Н)+. Стадия (ii). Получение 5-бром-1-циклопропанкарбонилазепан-4-она. Раствор 1-циклопропанкарбонил-азепан-4-она (2,7 г, 14,9 ммоль, получен на предыдущей стадии) в уксусной кислоте (30 мл) охлаждали до 10C и обрабатывали бромом (0,71 мл, 14,9 ммоль) в течение 15 мин. Полученную суспензию перемешивали в течение 18 ч в атмосфере азота. После завершения ре- 10022746 акции смесь концентрировали досуха с получением указанного в заголовке соединения (3,87 г).(2-амино-4,5,7,8-тетрагидротиазоло[5,4-d]азепин-6 ил)циклопропилметанона. Суспензию 5-бром-1-циклопропанкарбонил-азепан-4-она (3,87 г, 14,8 ммоль, получен на предыдущей стадии) и тиомочевины (1,13 г, 14,8 ммоль) в изопропаноле (40 мл) нагревали при кипячении с обратным холодильником в течение 6 ч. После завершения реакции реакционную массу концентрировали,и полученный остаток очищали флэш-хроматографией (метанол/хлороформ, 3/97) с получением указанного в заголовке соединения (0,4 г). 1MS (m/z): 238 (М+Н)+. Стадия (iv). Получение (2-бром-4,5,7,8-тетрагидротиазоло[5,4-d]азепин-6-ил)циклопропилметанона. Раствор (2-амино-4,5,7,8-тетрагидротиазоло[5,4-d]азепин-6-ил)циклопропилметанона (0,4 г,1,68 ммоль, получен на предыдущей стадии) и бромида меди(II) (0,37 г, 1,68 ммоль) в ацетонитриле(40 мл) охлаждали до 0C. трет-Бутилнитрит (0,2 мл, 1,68 ммоль) добавляли по каплям в течение 10 мин при 0C. Реакционную массу перемешивали в течение 30 мин и реакционную массу гасили 3 н. раствором хлористо-водородной кислоты. Продукт экстрагировали этилацетатом (315 мл) и объединенные органические слои промывали водой, затем насыщенным солевым раствором и сушили над сульфатом натрия. Органические летучие вещества выпаривали в вакууме. Полученный таким образом остаток очищали флэш-хроматографией (этилацетат/н-гексан, 7/3) с получением указанного в заголовке соединенияMS (m/z): 301 (М+Н)+. Стадия (v). Получение [2-(4-циклобутилпиперидин-4-илокси)-4,5,7,8-тетрагидротиазоло[5,4d]азепин-6-ил]циклопропилметанона. 1-Циклобутилпиперидин-4-ол (0,04 г, 0,26 ммоль) в тетрагидрофуране (3 мл) обрабатывали охлажденной и перемешиваемой суспензией гидрида натрия (0,021 г, 0,51 ммоль) в тетрагидрофуране (8 мл) медленно в течение 5 мин и реакционную смесь перемешивали в течение 2 ч при комнатной температуре. Добавляли раствор (2-бром-4,5,7,8-тетрагидротиазоло[5,4-d]азепин-6-ил)циклопропилметанона(0,053 г, 0,17 ммоль, получен на предыдущей стадии) в тетрагидрофуране (3 мл) по каплям в течение 5 мин и нагревали при кипячении с обратным холодильником в течение 15 ч. Реакционную массу гасили в ледяной воде и продукт экстрагировали этилацетатом (310 мл). Объединенные органические слои промывали водой, затем насыщенным солевым раствором и сушили над безводным сульфатом натрия. Органические летучие вещества выпаривали в вакууме. Полученный остаток очищали флэшхроматографией (метанол/хлороформ 3/97) с получением указанного в заголовке соединения (0,05 г). 1[2-(4-циклобутилциклогексилокси)-4,5,7,8-тетрагидротиазоло[5,4-d]азепин-6 ил]циклопропилметанона (0,078 г, 0,208 ммоль, получен на предыдущей стадии) в метаноле (5 мл) обрабатывали L(+)-винной кислотой (0,031 г, 0,208 ммоль) при 0C. Реакционную массу перемешивали примерно в течение 1 ч и растворитель выпаривали в вакууме досуха. Твердые вещества промывали диэтиловым эфиром и сушили в вакууме с получением указанного в заголовке соединения (0,1 г). 1(0,50 г, 1,5 ммоль, получен в получении 2) в дихлорметане (30 мл) обрабатывали трифторуксусной кислотой (1,1 мл, 15 ммоль) при 0C. Реакционную массу перемешивали в течение 4 ч. После завершения реакции массу гасили ледяной водой, и доводили рН до 10 с использованием 40% водного раствора гидроксида натрия. Продукт экстрагировали дихлорметаном (350 мл) и объединенные органические слои промывали водой, затем насыщенным солевым раствором и сушили над безводным сульфатом натрия. Органические летучие вещества выпаривали в вакууме с получением указанного в заголовке соединенияMS (m/z): 233,0 (М+Н)+, 235,0 (М+3H)+. Стадия (ii). Получение N-(2-бром-4,5,6,7-тетрагидробензотиазол-6-ил)пропионамида. Раствор 2-бром-4,5,6,7-тетрагидробензотиазол-6-иламина (0,36 г, 1,5 ммоль, получен на предыдущей стадии) и триэтиламина (0,43 мл, ммоль) в дихлорметане (15 мл) охлаждали до 0C. Добавляли пропионилхлорид (0,17 мл, 1,8 ммоль) в дихлорметане (2 мл) и реакционную массу перемешивали в течение 30 мин. После завершения реакции массу выливали на ледяную воду, и продукт экстрагировали этилацетатом (315 мл). Объединенные органические слои промывали водой, затем насыщенным солевым раствором, сушили над безводным сульфатом натрия, и органические летучие вещества выпаривали в вакууме. Остаток очищали флэш-хроматографией (метанол/хлороформ, 2/98) с получением указанного в заголовке соединения (0,4 г). 1MS (m/z): 289,2 (М+Н)+, 291,2 (М+3H)+. Стадия (iii). Получение N-[2-(1-циклобутилпиперидин-4-илокси)-4,5,6,7-тетрагидробензотиазол-6 ил]пропионамида. 1-Циклобутилпиперидин-4-ол (0,25 г, 1,6 ммоль) в N,N-диметилформамиде (5 мл) обрабатывали охлажденной и перемешиваемой суспензией гидрида натрия (0,1 г, 2,08 ммоль) вN,N-диметилформамиде (10 мл) медленно в течение 30 мин и реакционную смесь продолжали перемешивать в течение 1 ч. Раствор N-2-бром-4,5,6,7-тетрагидробензотиазол-6-ил) пропионамида (0,4 г,1,3 ммоль, получен на предыдущей стадии) в N,N-диметилформамиде (5 мл) добавляли по каплям в течение 10 мин и полученную массу перемешивали в течение 48 ч. После завершения реакции смесь гасили в ледяной воде и продукт экстрагировали этилацетатом (315 мл). Объединенные органические слои промывали водой, затем насыщенным солевым раствором, сушили над безводным сульфатом натрия и органические летучие вещества выпаривали в вакууме. Остаток очищали флэш-хроматографией (этилацетат/н-гексан, 1/1) с получением указанного в заголовке соединения (0,068 г). Примеры 4-34. Соединения примеров 4-34 получали, следуя процедурам, описанным в примерах 1-3, с некоторыми некритическими изменениями. Примеры 35-66. Специалисты в данной области смогут получить соединения примеров 35-66, следуя процедурам,описанным выше. Биологические анализы. Пример 67. Анализы связывания и функциональные анализы для гистаминового H3 рецептора человека или крысы. Соединения могут быть оценены в соответствии со следующими процедурами. Материалы и методы. Источник рецептора: фронтальный кортикальный слой головного мозга крысы или рекомбинантная кДНК человека, экспрессируемая в СНО клетках. Радиолиганд: [3 Н] Rметилгистамин. Конечная концентрация лиганда: [3,0 нМ]. Неспецифическая детерминанта: Rметилгистамин (100 мкМ). Ссылочное соединение: Rметилгистамин. Положительный контроль: Rметилгистам.н Условия инкубации. Увеличивающиеся концентрации испытываемых соединений или стандарта инкубировали с мембранными рецепторами и радиолигандом в 5 мМ MgCl2 и 50 мМ TRIS-HCl (рН 7,4) в течение 60 мин при комнатной температуре. Реакцию останавливали путем быстрой вакуум-фильтрации на стекловолоконных фильтрах. Радиоактивность, улавливаемую на фильтрах, определяли и сравнивали с контрольными значениями для установления каких-либо взаимодействий испытываемого соединения(ий) с любым сайтом связывания клонированного рецептора человека либо крысы. Литературная ссылка: Millipore data sheet. Пример 68. Фармакокинетическое испытание грызунов. Самцов крыс Wistar (230-280 г), полученных от NIN (National Institute of Nutrition, Hyderabad, India),использовали в качестве экспериментальных животных. В каждую клетку помещали по три животных. Животных содержали без пищи в течение ночи и поддерживали 12-часовой цикл свет/темнота. Трем крысам вводили New chemical entity (NCE) перорально (3 или 10 мг/кг) и внутривенно (1 или 5 мг/кг) в день 0 и день 2. В каждой временной точке брали кровь из яремной вены. Кровь хранили при 2-8C до использования в анализе. Концентрации NCE соединения в крови определяли с использованием метода LC-MS/MS. Временные точки исследования: перед введение дозы, 0,08, 0,25, 0,5, 1, 2, 4,6, 8 и 24 ч после введения дозы (n=3). Осуществляли определение количеств NCE соединений в крови частично подтвержденным методом LC-MS/MS с использованием процедуры осаждения ацетонитрилом. Определение количествNCE соединений осуществляли в диапазоне значений 1-2000 нг/мл в крови. Исследуемые образцы анализировали с использованием калибровочных образцов в партии и образцов контроля качества параллельно с этой партией. Фармакокинетическое параметры рассчитывали при помощи некомпартментной модели с использованием программы WinNonlin version 5.0.1. Пример 69. Испытание пенетрации в головной мозг грызунов. Самцов крыс Wistar (230-280 г), полученных от NIN (National Institute of Nutrition, Hyderabad, India),использовали в качестве экспериментальных животных. В каждую клетку помещали по три животных. Животным давали воду и пищу ad libitum на протяжении всего эксперимента и поддерживали 12-часовой цикл свет/темнота.New chemical entity (NCE) растворяли в подходящем носителе и вводили перорально (3 или 10 мг/кг). Примерно в точке времени Tmax (т.е. 0,5, 1 и 2 ч) животных умерщвляли. Собирали кровь и ткань головного мозга, и головной мозг гомогенизировали с получением 20% мас./об. Кровь хранили при 2-8C и гомогенат головного мозга замораживали при -20C до использования в анализе. КонцентрацииNCE соединения в крови определяли с использованием метода LC-MS/MS. Определение количеств NCE соединений в крови и гомогенате головного мозга осуществляли частично подтвержденным методом LC-MS/MS с использованием процедуры осаждения ацетонитрилом. Определение количеств NCE соединений осуществляли в диапазоне значений 1-500 нг/мл в крови и гомогенате головного мозга. Исследуемые образцы анализировали с использованием калибровочных образцов в партии и образцов контроля качества параллельно с этой партией. Рассчитывали отношения головной мозг-кровь (Cb/Cp). Пример 70. Модель с задачей распознавания объекта. Свойства соединений по настоящему изобретению по усилению когнитивной функции оценивали с использованием этой модели. Самцов крыс Wister (230-280 г), полученных от N. I. N. (National Institute of Nutrition, Hyderabad, India), использовали в качестве экспериментальных животных. В каждую клетку помещали по 4 животных. Животных поддерживали при 20% пищевой депривации за день до эксперимента и давали воду adlibitum на протяжении всего эксперимента и поддерживали 12-часовой цикл свет/темнота. Также у крыс вырабатывали привыкание к индивидуальным площадкам в течение 1 ч в отсутствие каких-либо объектов. Одна группа из 12 крыс получала носитель (1 мл/кг) перорально, а другая группа животных получала соединение формулы (I) либо перорально, либо интраперитонеально за 1 ч до испытания привычного узнаваемого объекта (Т 1) и выбора (Т 2). Эксперимент осуществляли в 505050 см открытом поле из акрила. В фазе ознакомления (Т 1) крыс помещали индивидуально в открытое поле в течение 3 мин, в котором были размещены два идентичных объекта (пластиковые бутыли, 12,5 (в высоту)5,5 см (в диаметре, покрытых желтой клейкой лентой, по отдельности (a1 и а 2), в двух соседних углах, на расстоянии 10 см от стенок. Через 24 ч испытания (Т 1) на длительную память, тех же крыс помещали на ту же площадку, на которой они находились в Т 1 испытании. В фазе выбора (Т 2) крысам давали исследовать открытое поле в течение 3 мин в присутствии одного знакомого объекта (а 3) и одного нового объекта (b) (стеклянная бутыль янтарного цвета,12 (в высоту)5 см (в диаметре. Знакомые объекты имели одинаковые текстуры, цвета и размеры. В процессе Т 1 и Т 2 испытания, изучения каждого объекта (определенные как фырканье, облизывание, жевание или движение вибрисс при направлении носа в сторону объекта на расстоянии менее 1 см) регистрировали отдельно при помощи секундомера. Сидение на объекте не считалось изучающей активностью,однако это редко наблюдалось. Т 1 представляет собой общее время, затраченное на изучение знакомых объектов (a1+a2). Т 2 представляет собой общее время, затраченное на изучение знакомого объекта и нового объекта(а 3+b). Испытание распознавания объектов осуществляли, как описано в Ennaceur, A., Delacour, J., 1988, A Пример 71. Водный лабиринт Морриса. Свойства соединений по настоящему изобретению по усилению когнитивной функции оценивали с использованием этой модели. Устройство водного лабиринта состояло из круглого бассейна (1,8 м в диаметре, 0,6 м в высоту),сконструированного из черного материала Perspex (TSE systems, Germany), заполненного водой (242C) и расположенного под широкоугольной видеокамерой для наблюдения за животными. 10 см 2 платформу из perspex, находящуюся на 1 см ниже водной поверхности, помещали в центре одной из четырех воображаемых четвертей круга, что оставалось постоянным для всех крыс. Черный Perspex, используемый в конструкции лабиринта и платформы, предполагал отсутствие каких-либо ориентиров внутри лабиринта,направляющих животное на выход из него. В отличие от этого, помещение для обучения предлагало несколько сильных визуальных ориентиров вне лабиринта, способствующих формированию пространственной карты, необходимой для научения избеганию. Использовали автоматическую систему слежения[Videomot 2 (5.51), TSE systems, Germany]. Эта программа анализирует видеоизображения, полученные при помощи цифровой камеры и плат для сбора данных, которые определяли длину пути, скорость плавания и количество заходов и продолжительность времени плавания в каждой четверти круга водного лабиринта. Пример 72. Ингибирование потребления пищи. Свойства соединений по настоящему изобретению против ожирения оценивали с использованием этой модели. Эксперимент продолжался 6 дней. Крыс адаптировали к режиму 18 ч голодания и 6 ч потребления пищи. Животных размещали группами по три животных в каждой клетке, которые были снабжены решетками для режима голодания, и животные голодали в течение 18 ч. Через 18 ч голодающих крыс разделяли, и каждую помещали в отдельную клетку. Крысам давали взвешенное количество пищи в течение 6 ч и измеряли потребление пищи в момент времени 1, 2, 4 и 6 ч. Крыс снова делили на группы и держали без пищи в течение 18 ч. Описанную выше процедуру повторяли в течение 5 дней. Подсчитывали среднее кумулятивное потребление пищи крысами в последние 3 дня. Животных рандомизировали на основании их предыдущего трехдневного потребления пищи. В день эксперимента крысам перорально вводили испытываемые соединения или носитель. Через 60 мин крысам давали пищу и измеряли потребление пищи в момент времени 1, 2, 4 и 6 ч. Потребление пищи крысами, обработанными испытываемым соединением, сравнивали с группой обработки носителем с использованием непарного t-критерия Стьюдента. ФОРМУЛА ИЗОБРЕТЕНИЯ 1. Соединение общей формулы (I) или его фармацевтически приемлемая соль,где R1 представляет собой:[2-(1-циклобутилпиперидин-4-илокси)-6,7-дигидро-5 Н-тиазоло[5,4-b]пиридин-4 ил]циклопропилметанон тартрат и циклопропил[2-(1-изопропилпиперидин-4-илокси)-4,5,7,8-тетрагидротиазоло[5,4-d]азепин-6 ил]метанон тартрат или их фармацевтически приемлемую соль. 4. Способ получения соединения формулы (I) или его фармацевтически приемлемой соли,где R1 представляет собой:(а) сочетание соединения формулы (1) с соединением формулы (2): с получением соединения формулы (3)(b) удаление защиты у соединения формулы (3) с получением соединения формулы (4)(с) сочетание соединения формулы (4) с соединением формулы (5) с получением соединения формулы (I);(d) необязательно, преобразование соединения формулы (I) в его фармацевтически приемлемую соль. 5. Способ получения соединения формулы (I) или его фармацевтически приемлемой соли,где R1 представляет собой(а) сочетание соединения формулы (6) с соединением формулы (5): с получением соединения формулы (7)(b) бромирование соединения формулы (7) с получением соединения формулы (8) или его фармацевтически приемлемой соли(с) циклизацию соединения формулы (8) с получением соединения формулы (9)
МПК / Метки
МПК: C07D 417/12, A61K 31/5517, A61K 31/55, A61P 25/00, A61K 31/437, C07D 498/04, C07D 471/04, C07D 413/12, A61P 23/00, A61K 31/496
Метки: соединения, рецепторов, качестве, гетероциклические, гистаминовых, лигандов
Код ссылки
<a href="https://eas.patents.su/30-22746-geterociklicheskie-soedineniya-v-kachestve-ligandov-gistaminovyh-h3-receptorov.html" rel="bookmark" title="База патентов Евразийского Союза">Гетероциклические соединения в качестве лигандов гистаминовых h3 рецепторов</a>
Пуриновые соединения и их применение в качестве лигандов каннабиноидных рецепторов

Номер патента: 8176
Опубликовано: 27.04.2007
Автор: Гриффит Дейвид Эндрю
МПК: C07D 239/48, A61K 31/52, A61P 3/04...
Метки: соединения, качестве, лигандов, применение, каннабиноидных, пуриновые, рецепторов
Формула / Реферат:
1. Соединение формулы (I) где А представляет собой возможно замещенный фенил или возможно замещенный гетероарил; В представляет собой возможно замещенный фенил или возможно замещенный гетероарил; R1 представляет собой водород, (С1-С4)алкил, галогенозамещенный (С1-С4)алкил или (С1-С4)алкокси; R4 представляет собой (1) группу, имеющую формулу (IA) или формулу (IB) где R4a представляет собой водород или (С1-С3)алкил; R4b и R4b', каждый...
Новые гетероциклические соединения в качестве положительных аллостерических модуляторов метаботропных глутаматных рецепторов

Номер патента: 14904
Опубликовано: 28.02.2011
Авторы: Паломби Джиованни, Гаглиарди Стефания, Фарина Марко, Ле Поул Эммануэль, Рошер Джен-Филиппе
МПК: A61K 31/4439, C07D 401/04, A61P 25/18...
Метки: рецепторов, соединения, модуляторов, глутаматных, метаботропных, гетероциклические, аллостерических, новые, положительных, качестве
Формула / Реферат:
1. Соединение формулы I-Bгде V1и V2 представляют собой независимо атом кислорода или азота;V3 представляет собой атом углерода или азота;R1 и R2независимо друг от друга представляют собой водород;Р представляет собой С6арил, возможно содержащий в качестве заместителя галоген;Q представляет собой арильную или гетероарильную группу формулыгде R3, R4, R5, R6 и R7независимо друг от друга представляют собой водород, галоген, -(С1-С6)алкил или...
Новые гетероциклические соединения в качестве антагонистов метаботропных глутаматных рецепторов 5-го подтипа (мглу5)

Номер патента: 18328
Опубликовано: 30.07.2013
Авторы: Грацьяни Давид, Леонарди Амедео, Мотта Гьянни, Рива Карло, Поггеси Елена, Лонги Маттео Марко
МПК: A61K 31/4545, C07D 211/70, A61P 13/02...
Метки: подтипа, гетероциклические, новые, 5-го, антагонистов, рецепторов, метаботропных, качестве, mglu5, глутаматных, соединения
Формула / Реферат:
1. Соединение, имеющее общую формулу Iв которой Z обозначает группу формулы -CºC-R2 или -CH=CH-R2;R1 обозначает атом водорода или галогена или C1-С6алкильную группу;R2 обозначает выборочно замещенную моно- или бициклическую С2-С9гетероциклическую группу, содержащую от 1 до 3 гетероатомов, выбранных из азота, кислорода и серы,выборочно замещенную фенильную группу,выборочно замещенную С1-С6алкильную группу,выборочно замещенную...
Гетероциклические соединения, содержащие тетрагидропиридиновые или пиперидиновые группы, в качестве антагонистов рецепторов кортикотропин-рилизинг-фактора

Номер патента: 5289
Опубликовано: 30.12.2004
Авторы: Наказато Ацуро, Кумагаи Тосихито, Камео Казуя, Окубо Такетоси
МПК: A61K 31/4365, A61P 43/00, C07D 401/04...
Метки: тетрагидропиридиновые, качестве, антагонистов, содержащие, пиперидиновые, группы, рецепторов, соединения, кортикотропин-рилизинг-фактора, гетероциклические
Формула / Реферат:
1. Тетрагидропиридино- или пиперидиногетероциклическое производное, представленное формулой [I] A-Het [I], где A обозначает группу, представленную следующими формулами [II] или [III]: где группа Y-(CH2)n- формулы [II] находится в положении 4 или 5 и группа Y-C(R0)= формулы [III] находится в положении 3 или 4, R0 обозначает атом водорода, C1-5алкильную группу, C3-8циклоалкильную группу или C3-8циклоалкил-C1-5алкильную группу, n равно целому...
Лечение болезни паркинсона, обструктивного синдрома апноэ во сне, слабоумия с тельцами льюи, сосудистой деменции с помощью не содержащих имидазол алкиламиновых лигандов гистаминовых н3-рецепторов

Номер патента: 16007
Опубликовано: 30.01.2012
Авторы: Шварц Жан -Шарль, Лёкомт Жанн -Мари
МПК: A61K 31/138, A61K 31/15, A61K 31/145...
Метки: паркинсона, содержащих, обструктивного, синдрома, гистаминовых, помощью, лечение, деменции, болезни, льюи, слабоумия, н3-рецепторов, тельцами, имидазол, сосудистой, апноэ, лигандов, сне, алкиламиновых
Формула / Реферат:
1. Применение соединения, выбранного из группы, состоящей из3-фенилпропил-3-пиперидинопропилового эфира;3-(4-хлорфенил)пропил-3-пиперидинопропилового эфира;3-фенилпропил-3-(4-метилпиперидино)пропилового эфира;3-фенилпропил-3-(3,5-цис-диметилпиперидино)пропилового эфира;3-фенилпропил-3-(3,5-транс-диметилпиперидино)пропилового эфира;3-фенилпропил-3-(3-метилпиперидино)пропилового эфира;3-фенилпропил-3-пирролидинопропилового...
Предыдущий патент: Окно
Следующий патент: Способ восстановления носителей данных
Случайный патент: Короткоцепочечный разветвленный полипропилен и способ его получения