Ингибиторы цистеинпротеазы
Номер патента: 19277
Опубликовано: 28.02.2014
Авторы: Пелькман Микаэль, Иванов Владимир Леонардович, Айеса Сусана, Нильссон Магнус, Вехлинг Хорст, Хьюитт Эллен, Линд Петер, Оден Лурдес, Канберг Пиа, Классон Бьерн, Бельфраге Анна Карин, Йенссон Даниель, Грабовска Урсула





















Формула / Реферат
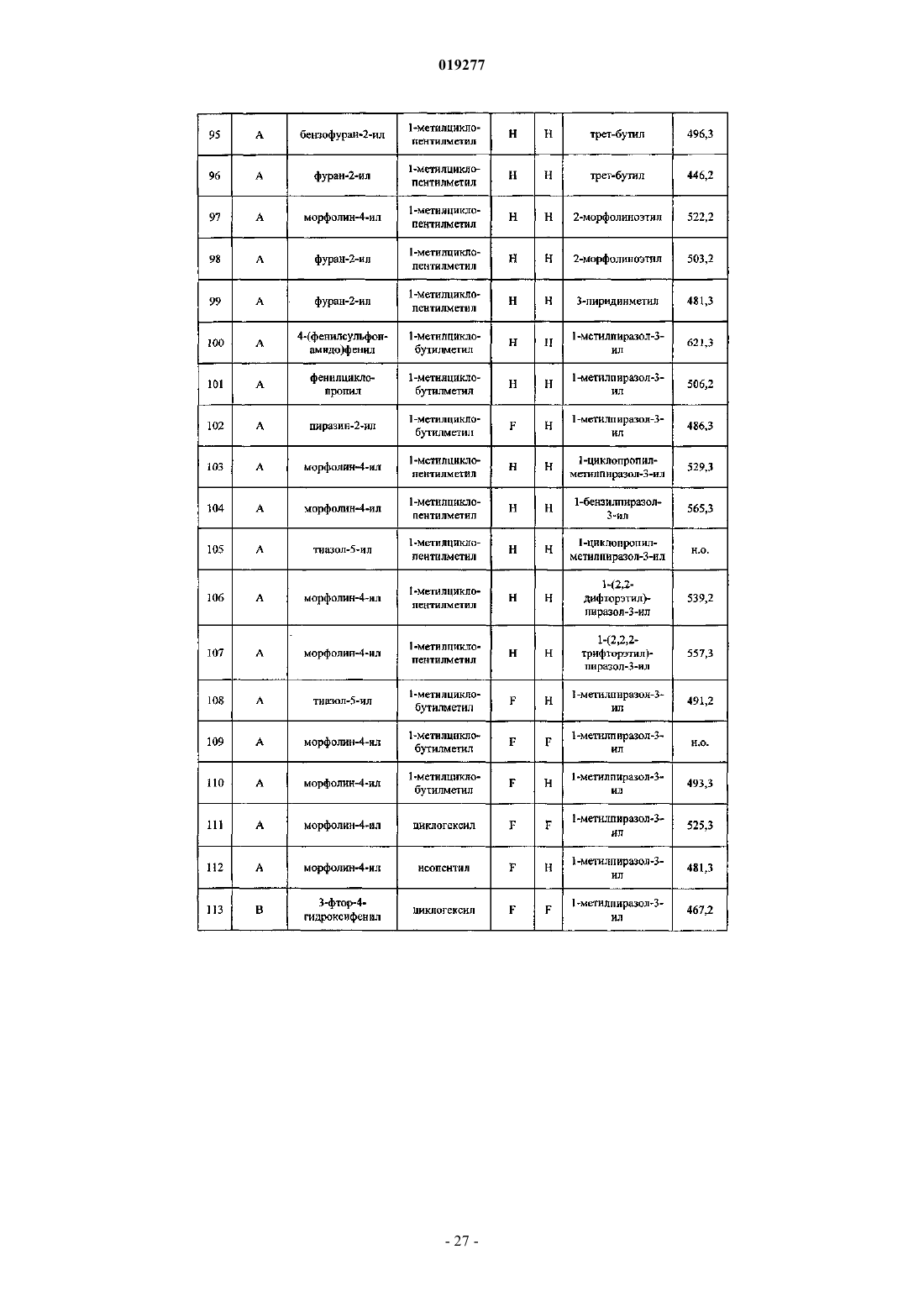
1. Соединение формулы I
в которой
R1a представляет собой Н и
R1b представляет собой метил, циклопропил, 1-фенилэтил или пиразол-1-ил, причем циклопропил или фенил необязательно замещены не более чем тремя заместителями, независимо выбранными из С1-С4алкила, галогена, С1-С4галогеналкила, С1-С4алкокси, С1-С4галогеналкокси, С1-С4алкоксикарбонила, С1-С4алкилкарбонила, амино, С1-С4алкиламина, С1-С4диалкиламина, С1-С4алкилсульфонила, С1-С4алкилсульфониламино, аминокарбонила, аминосульфонила, RxOC (=O)-С0-С2алкилена (где Rx представляет собой Н или C1-С4алкил), или С3-С6циклоалкил-С0-С2алкилена или бензила (циклоалкил или бензил необязательно замещены 1-3 заместителями, выбранными из С1-С4алкила, галогена, C1-С4галогеналкила, С1-С4алкокси, С1-С4галогеналкокси), и пиразол-1-ил необязательно замещен С1-С4алкилом, галогеном, С1-С4галогеналкилом, С1-С4алкокси или циклопропилом;
R2a и R2b независимо выбраны из Н, галогена, С1-С4алкила, С1-С4галогеналкила, С1-С4алкокси; или
R2a и R2b вместе с атомом углерода, к которому они присоединены, образуют С3-С6циклоалкил;
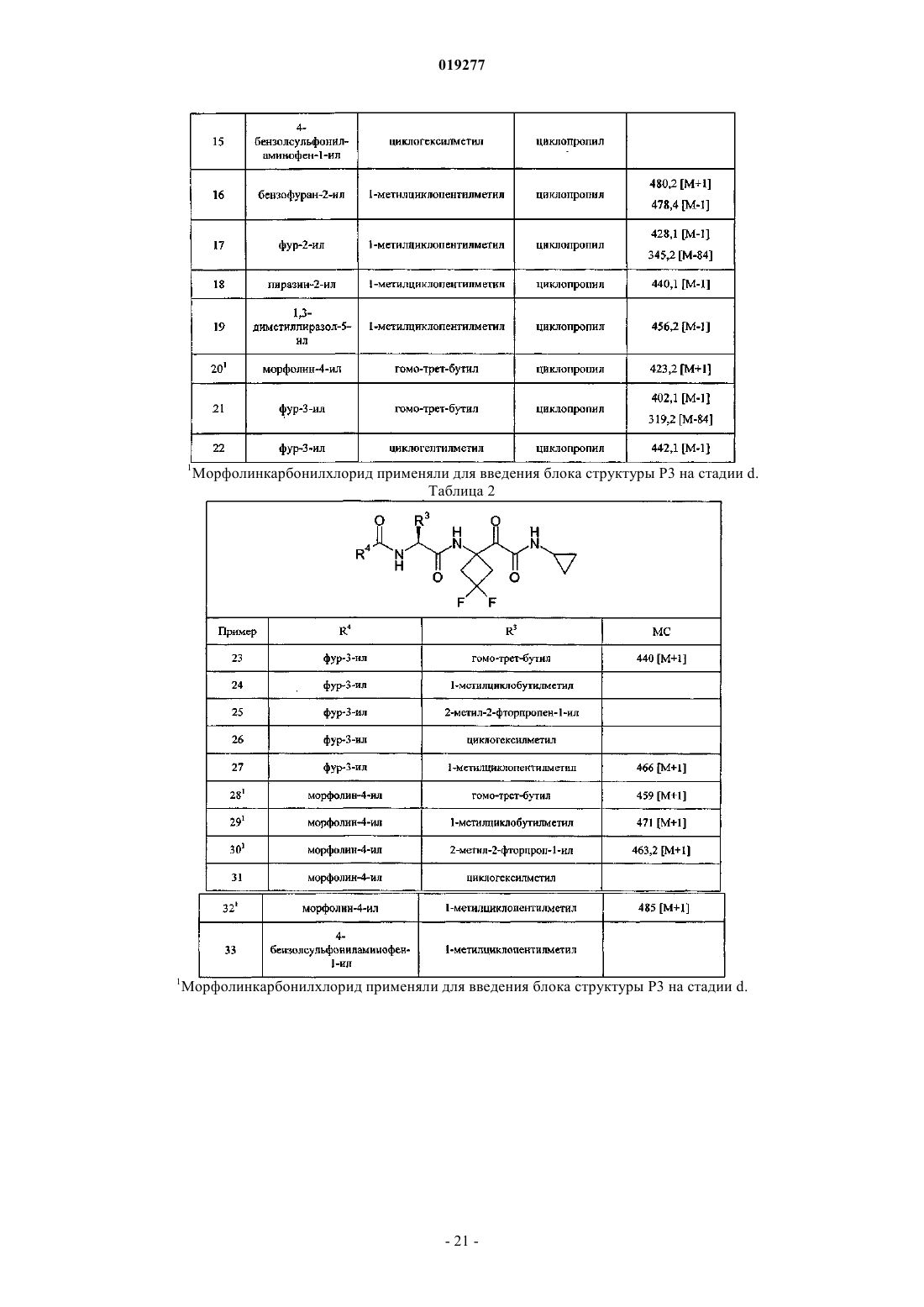
R3 представляет собой неопентил, циклобутилметил, 1-метилциклобутилметил или 1-метилциклопентилметил, каждый из которых необязательно замещен одним или двумя атомами F или ОМе;
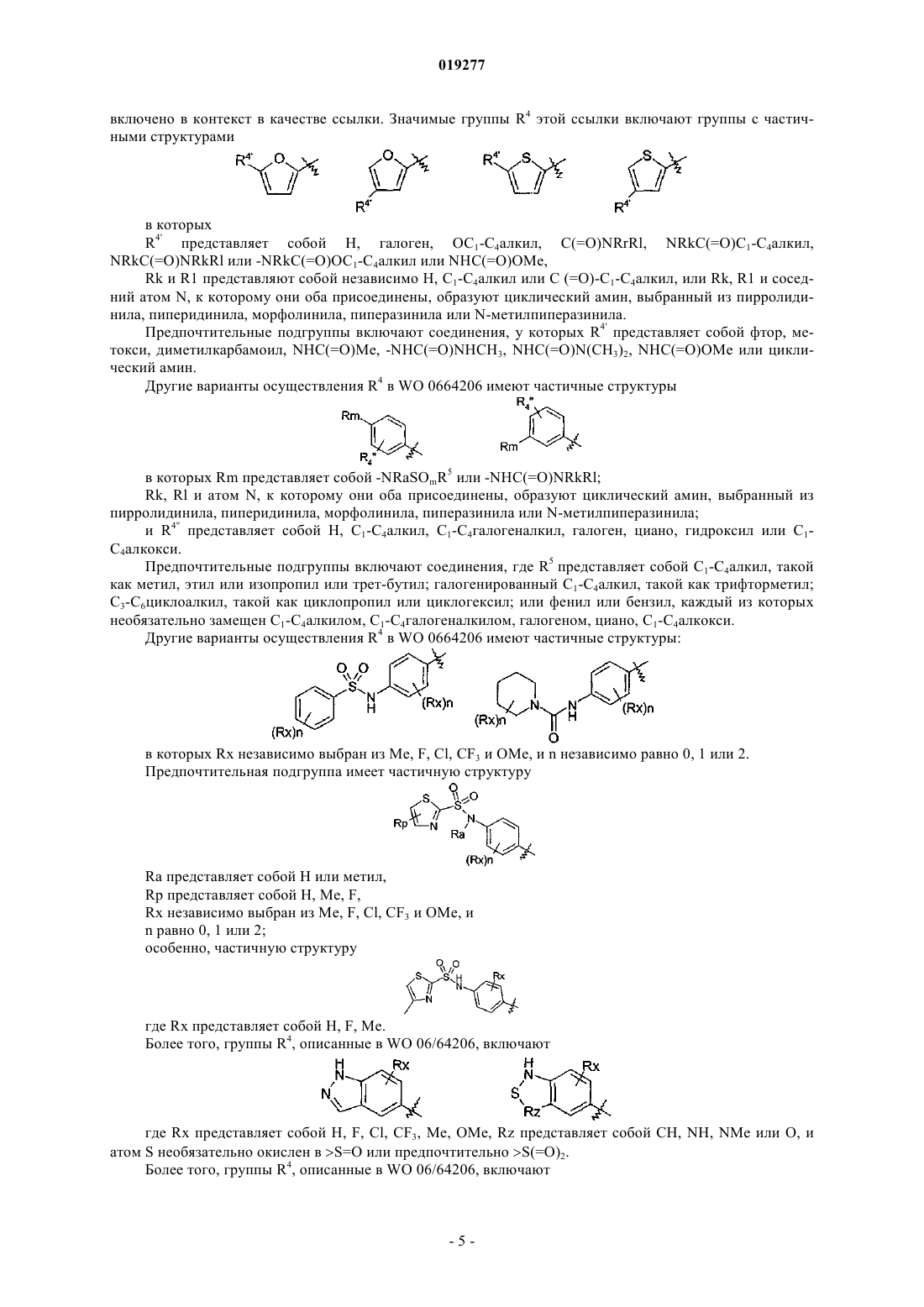
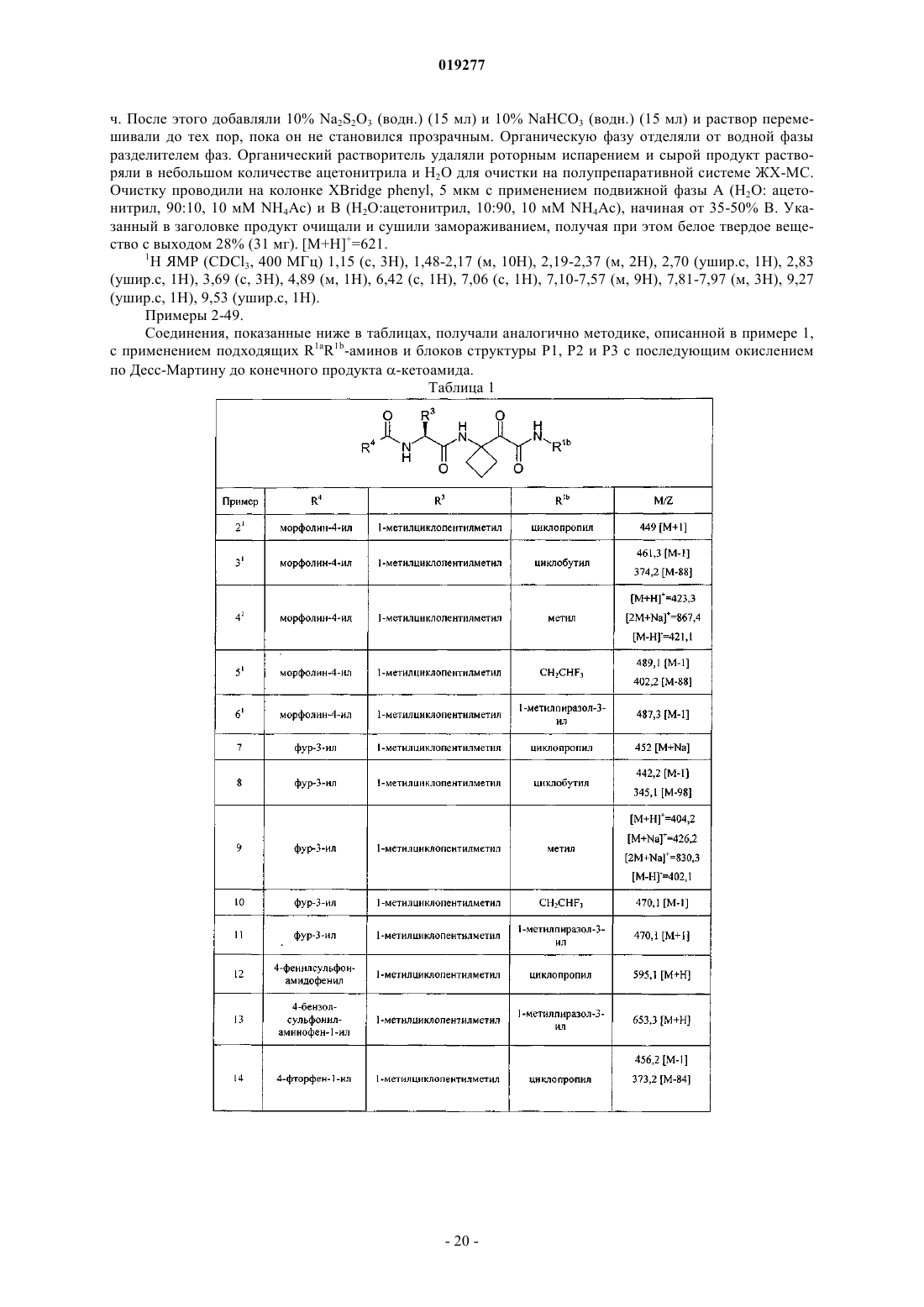
R4 представляет собой Het или карбоциклил, каждый из которых необязательно замещен 1-3 заместителями, независимо выбранными из галогена, азидо, циано, гидрокси, оксо, С1-С4алкила, C1-С4галогеналкила, С1-С4алкокси, С1-С4галогеналкокси, C1-С4алкоксикарбонила, С1-С4алкилкарбонила, С1-С4алкилсульфонила, С1-С4алкилсульфониламино, аминосульфонила, -NRkRl, -C(=O)NRkRl, -NRkC(=O)Rl, NRkC(=O)ORl,
-NRk(C=O)NRkRl, где оксо в качестве заместителя может присутствовать только в том случае, если это позволяет валентность,
где Rk и Rl независимо представляют собой Н, С1-С4алкил, или один из них представляет собой Н, и другой представляет собой -С(=O)С1-С4алкил;
и/или где Het или карбоциклильная группа необязательно замещена группой формулы -X-R5;
где X представляет собой сульфонамидо;
R5 представляет собой Н, С1-С4алкил или моноциклическое кольцо, выбранное из С3-С6циклоалкила, С3-С6циклоалкенила, фенила, пирролидинила, пиперидинила, морфолинила, тиоморфолинила, пиперазинила, индолинила, пиранила, тетрагидропиранила, тетрагидротиопиранила, тиопиранила, фуранила, тетрагидрофуранила, тиенила, пирролила, оксазолила, изоксазолила, тиазолила, имидазолила, пиридинила, пиримидинила, пиразинила, пиридазинила, тетразолила, пиразолила, индолила, причем С1-С4алкил или моноциклическое кольцо необязательно замещены одним-тремя заместителями, выбранными из галогена, азидо, циано, гидрокси, С1-С4алкила, C1-С4галогеналкила, С1-С4алкокси, С1-С4галогеналкокси, C1-С4алкоксикарбонила, С1-С4алкилкарбонила, амино, С1-С4алкиламино, С1-С4диалкиламино, С1-С4алкилсульфонила, С1-С4алкилсульфониламино, аминокарбонила, аминосульфонила;
Het представляет собой стабильную, моноциклическую или бициклическую, насыщенную, частично насыщенную или ароматическую систему колец, содержащую 1-4 гетероатома, независимо выбранные из О, S и N, причем каждое кольцо имеет 5 или 6 атомов кольца;
карбоциклил представляет собой С3-С6циклоалкил, С5-С6циклоалкенил или фенил;
или его фармацевтически приемлемая соль, гидрат или N-оксид.
2. Соединение по п.1, в котором оба R2a и R2b представляют собой F.
3. Соединение по п.1, в котором R2a представляет собой хлор, фтор, трифторметил или метокси и R2b представляет собой Н.
4. Соединение по п.1, в котором R3 представляет собой 1-фторциклобутилметил.
5. Соединение по любому из пп.1-4, в котором R4 представляет собой морфолинил, пиперидинил, пиперазинил, циклопентил, циклогексил или пиридинил, каждый из которых необязательно замещен галогеном, гидрокси, С1-С4алкилом, С1-С4галогеналкилом, С1-С4алкокси, С1-С4галогеналкокси, амино, С1-С4алкиламино, ди(С1-С4алкил)амино или NRkS(=O)mRq;
где Rk представляет собой Н или С1-С4алкил;
Rq представляет собой С1-С4алкил, Het или карбоциклил, каждый из которых необязательно замещен С1-С4алкилом, галогеном, С1-С4галогеналкилом, С1-С4алкокси; и
m равно 0, 1 или 2.
6. Соединение по п.5, в котором R4 представляет собой морфолин-4-ил.
7. Соединение по п.5, в котором R4 представляет собой пиперазин-1-ил или пиперидин-1-ил, каждый из которых замещен в 4-положении, или пиперидин-4-ил, замещенный в 1 положении; где в каждом случае заместитель выбран из -NHS (=O)2карбоциклила или NHS(=O)Het,
где карбоциклил или Het необязательно замещен галогеном, С1-С4алкилом, С1-С4галогеналкилом или С1-С4алкилокси.
8. Соединение по п.5, в котором R4 представляет собой циклогексил или пиперазин-1-ил, замещенный в 4-положении галогеном, амино или гидрокси.
9. Соединение по любому из пп.1-4, в котором R4 представляет собой замещенный фенил.
10. Соединение по п.9, в котором фенил замещен 1-3 заместителями, независимо выбранными из галогена, гидрокси, C1-С4алкила, С1-С4галогеналкила, циано, С1-С4алкилС(=O)NH- или С1-С4алкокси.
11. Соединение по п.10, в котором фенил замещен м-фтором, п-фтором, п-гидрокси, п-гидрокси-м-хлором, п-гидрокси-м-фтором, п-гидрокси-м-метокси, п-гидрокси-м-метилом, бис-п-хлор-п-гидрокси, м-циано, п-ацетамидо или о-фтор-п-гидрокси.
12. Фармацевтическая композиция, содержащая соединение по любому из предшествующих пунктов и фармацевтически приемлемый носитель.
13. Применение соединения по любому из пп.1-11 для профилактики или лечения расстройства, характеризующегося аномальной экспрессией или активацией катепсина S.
14. Применение по п.13, где расстройство выбрано из:
a) псориаза;
b) аутоиммунных заболеваний, включающих идиопатическую тромбоцитопеническую пурпуру (ITP), ревматоидный артрит (RA), рассеянный склероз (MS), тяжелую миастению (MG), синдром Шегрена, болезнь Граве и системную красную волчанку (SLE); или
c) неаутоиммунных заболеваний, включающих аллергический ринит, астму, атеросклероз, хроническое обструктивное заболевание легких (CORD) и хроническую боль.
15. Способ профилактики или лечения расстройства, характеризующегося аномальной экспрессией или активацией катепсина S, включающий введение эффективного количества соединения по любому из пп.1-11 человеку или животному, пораженному расстройством или имеющему риск возникновения расстройства.
16. Способ по п.15, где расстройством является:
a) псориаз;
b) аутоиммунное заболевание, выбранное из группы, состоящей из идиопатической тромбоцитопенической пурпуры (ITP), ревматоидного артрита (RA), рассеянного склероза (MS), тяжелой миастении (MG), синдрома Шегрена, болезни Граве и системной красной волчанки (SLE); или
c) неаутоиммунное заболевание, выбранное из группы, состоящей из аллергического ринита, астмы, атеросклероза, хронического обструктивного заболевания легких (CORD) и хронической боли.
17. Соединение формулы II
где R2a и R2b независимо выбраны из Н, галогена, С1-С4алкила, С1-С4галогеналкила, С1-С4алкокси;
PG представляет собой Н или формил, ацетил, бензоил, пивалоил, трет-бутилацетил, фенилсульфонил, бензил, трет-бутоксикарбонил или бензилоксикарбонил;
PG* представляет собой Н или С1-С4алкиловый или бензиловый эфир.
Текст
Соединения формулы I, в которой R1a представляет собой Н и R1b представляет собой С 1-С 4 алкил,карбоциклил или Het; или R1a и R1b вместе обозначают насыщенный циклический амин с 3-6 атомами кольца; R2a и R2b представляют собой Н, галоген, С 1-С 4 алкил, C1-С 4 галогеналкил, С 1 С 4 алкокси; или R2a и R2b вместе с атомом углерода, к которому они присоединены, образуют С 3 С 6 циклоалкил; R3 представляет собой разветвленную C5-С 10 алкильную цепь, С 2-С 4 галогеналкил или С 3-С 7 циклоалкилметил, R4 представляет собой Het, карбоциклил, необязательно замещенный,как указано в описании, и их фармацевтически приемлемые соли, гидраты и N-оксиды являются ингибиторами катепсина S и являются применимыми при лечении псориаза, аутоиммунных расстройств и других расстройств, таких как астма, атеросклероз, CORD и хроническая боль. Область техники, к которой относится изобретение Данное изобретение относится к ингибиторам катепсина S и их применению в способах лечения расстройств, связанных с катепсином S, таких как аутоиммунные расстройства, аллергия и состояния хронической боли. Уровень техники изобретения Папаиновое суперсемейство цистеинпротеаз широко распространено в разных видах организмов,включающих млекопитающих, позвоночных, простейших, растения и бактерии. В этом суперсемействе описан ряд ферментов катепсинов млекопитающих, включающих катепсины В, F, Н, K, L, О, S и W, причем аномальная регуляция их активности задействована в ряде метаболических расстройств, в том числе артрите, мышечной дистрофии, воспалении, гломерулонефрите и прорастании опухоли. Патогенные,подобные катепсину ферменты включают бактериальные гингипаины, малярийные фалципаины I, II, III и т.д. и цистеинпротеазы из Pneumocystis carinii, Trypanosoma cruzei и brucei, Crithidia fusiculata, Schistosoma spp. В WO 97/40066 описано применение ингибиторов против катепсина S. Предполагается, что ингибирование указанного фермента предупреждает или лечит заболевание, вызванное активностью протеазы. Катепсин S является очень активной цистеинпротеазой, принадлежащей к суперсемейству папаина. Его первичная структура на 57, 41 и 45% гомологична катепсину L и Н человека и цистеинпротеазе папаину растений, соответственно, хотя только на 31% гомологична катепсину В. Его обнаруживают в основном в В-клетках, дендритных клетках и микрофагах, и это ограниченное присутствие предполагает потенциальное участие данного фермента в патогенезе дегенеративного заболевания. Кроме того, обнаружено, что деструкция Ii протеолизом требуется молекулам класса II МНС для связывания антигенных пептидов и для переноса образовавшегося комплекса к поверхности клеток. Кроме того, обнаружено, что катепсин S является основным ферментом в В-клетках для эффективного протеолиза Ii, необходимого для придания молекулам класса II компетентности для связывания пептидов. Следовательно, ингибирование этого фермента может быть применимым при модуляции ограниченной классом II иммунной реакции (WO 97/40066). Другими расстройствами, в которых принимает участие катепсин S, являются астма, хроническое обструктивное легочное заболевание, эндометриоз и хроническая боль. Краткое описание изобретения Согласно первому аспекту изобретения предложено соединение формулы IR1b представляет собой C1-С 6 алкил, необязательно замещенный 1-3 заместителями, независимо выбранными из галогена, гидрокси, циано, азидо, С 1-С 4 галогеналкила, С 1-С 4 алкокси, С 1-С 4 галогеналкокси,С 1-С 4 алкоксикарбонила, С 1-С 4 алкилкарбонила, амина, С 1-С 4 алкиламина, С 1-С 4 диалкиламина, С 1 С 4 алкилсульфонила, С 1-С 4 алкилсульфониламино, аминокарбонила, аминосульфонила, карбоциклила иR1b представляет собой карбоциклил или Het; илиR1a и R1b вместе с атомом N, к которому они присоединены, образуют насыщенный циклический амин с 3-6 атомами кольца; где карбоциклил, Het или циклический амин необязательно замещены 1-3 заместителями, независимо выбранными из галогена, гидрокси, циано, азидо, С 1-С 4 алкила, С 1-С 4 галогеналкила, С 1-С 4 алкокси,С 1-С 4 галогеналкокси, С 1-С 4 алкоксикарбонила, C1-С 4 алкилкарбонила, амино, С 1-С 4 алкиламино, С 1 С 4 диалкиламино, C1-С 4 алкилсульфонила, С 1-С 4 алкилсульфониламино, аминокарбонила, аминосульфонила, RxOC(=O)-С 0-С 2 алкиленила (где Rx представляет собой Н, С 1-С 4 алкил или С 1-С 4 галогеналкил),фенила, бензила или С 3-С 6 циклоалкил-С 0-С 2 алкиленила; где фенильный, бензильный или циклоалкильный остаток необязательно замещен 1-3 заместителями, независимо выбранными из галогена, С 1-С 4 алкила, С 1-С 4 агалогеналкила или С 1-С 4 алкокси;R2a и R2b независимо выбраны из H, галогена, С 1-С 4 алкила, С 1-С 4 галогеналкила, С 1-С 4 алкокси; илиR3 представляет собой C5-С 10 алкил, необязательно замещенный 1-3 заместителями, независимо выбранными из галогена, C1-С 4 галогеналкила, С 1-С 4 алкокси или С 1-С 4 галогеналкокси; илиR3 представляет собой С 2-С 4 алкильную цепь по меньшей мере с 2 атомами хлора или 3 атомами фтора в качестве заместителей; илиR4 представляет собой Het или карбоциклил, каждый из которых необязательно замещен 1-3 заместителями, независимо выбранными из галогена, азидо, циано, гидрокси, оксо, С 1-С 4 алкила, С 1-1 019277 С 4 галогеналкила, С 1-С 4 алкокси, С 1-С 4 галогеналкокси, С 1-С 4 алкоксикарбонила, С 1-С 4 алкилкарбонила, С 1 С 4 алкилсульфонила, С 1-С 4 алкилсульфониламино, аминосульфонила, -NRkRl, -C(=O)NRkRl, -NRkC(=O)Rl,NRkC(=O)ORl, -NRk(C=O)NRkRl, где оксо в качестве заместителя может присутствовать, только если это позволяет валентность,где Rk и Rl независимо представляют собой Н, С 1-С 4 алкил или один из них представляет собой Н и другой представляет собой -С (=O)С 1-С 4 алкил; и/или где Het или карбоциклильная группа необязательно замещена группой формулы -X-R5; где X представляет собой С 1-С 4 алкилен или 1-4-членный линкер, содержащий 0-3 метиленовые группы, расположенные рядом с СН(СН 3), C(CH3)2, CF2, этеном, этином, С 0-С 4 алкиламино, С 0 С 4 алкиламидо, сульфонамидо, аминосульфонилом, сложным эфиром, простым эфиром, мочевиной или карбаматной функциональной группой или с любой стороны таких групп;R5 представляет собой Н, С 1-С 4 алкил или моноциклическое кольцо, выбранное из С 3 С 6 циклоалкила, С 3-С 6 циклоалкенила, фенила, пирролидинила, пиперидинила, морфолинила, тиоморфолинила, пиперазинила, индолинила, пиранила, тетрагидропиранила, тетрагидротиопиранила, тиопиранила, фуранила, тетрагидрофуранила, тиенила, пирролила, оксазолила, изоксазолила, тиазолила, имидазолила, пиридинила, пиримидинила, пиразинила, пиридазинила, тетразолила, пиразолила, индолила, причем С 1-С 4 алкил или моноциклическое кольцо необязательно замещено одним-тремя заместителями, выбранными из галогена, азидо, циано, гидрокси, С 1-С 4 алкила, C1-С 4 галогеналкила, С 1-С 4 алкокси, С 1 С 4 галогеналкокси, C1-С 4 алкоксикарбонила, С 1-С 4 алкилкарбонила, амино, С 1-С 4 алкиламино, С 1 С 4 диалкиламино, С 1-С 4 алкилсульфонила, C1-С 4 алкилсульфониламино, аминокарбонила, аминосульфонила;Het представляет собой стабильную, моноциклическую или бициклическую, насыщенную, частично насыщенную или ароматическую систему колец, содержащую 1-4 гетероатома, независимо выбранные из О, S и N, причем каждое кольцо имеет 5 или 6 атомов кольца; карбоциклил представляет собой С 3-С 6 циклоалкил, С 5-С 6 циклоалкенил или фенил; или его фармацевтически приемлемая соль, гидрат или N-оксид. В некоторых вариантах осуществления R1a представляет собой Н, и R1b представляет собой С 1 С 4 алкил, такой как этил, изопропил, трет-бутил или предпочтительно метил, необязательно замещенный одним или несколькими заместителями, указанными выше, предпочтительно 1-3 атомами галогена (например, F) или С 1-С 4 алкилоксигруппой (например, метоксигруппой). В других вариантах осуществления R1b представляет собой метил, циклопропил, 1-фенилэтил или 5- или 6-членное гетероциклическое кольцо, содержащее 1-3 атома азота и 0 или 1 атом серы, причем циклопропил, фенил или гетероциклическое кольцо необязательно замещено не более чем тремя заместителями, независимо выбранными из С 1-С 4 алкила, галогена, С 1-С 4 галогеналкила, С 1-С 4 алкокси, C1 С 4 галогеналкокси, С 1-С 4 алкоксикарбонила, С 1-С 4 алкилкарбонила, амина, С 1-С 4 алкиламина, С 1 С 4 диалкиламина, С 1-С 4 алкилсульфонила, С 1-С 4 алкилсульфониламино, аминокарбонила, аминосульфонила, RxOOC-C0-С 2 алкилена (где Rx представляет собой Н или С 1-С 4 алкил) или С 3-С 6 циклоалкил-С 0 С 2 алкилена или бензила (циклоалкил или фенильное кольцо бензила необязательно замещено 1-3 заместителями, выбранными из С 1-С 4 алкила, галогена, С 1-С 4 галогеналкила, С 1-С 4 алкокси, С 1 С 4 галогеналкокси). Примеры 5- или 6-членного ароматического гетероциклила для R1b включают пиридил или пиримидил и особенно пирролил, пиразолил, имидазолил, тиазолил, тиадиазолил, триазолил или тетразолил,каждый из которых необязательно замещен С 1-С 4 алкилом (например, Me), галогеном (например, F), С 1 С 4 галогеналкилом (например, CF3), С 1-С 4 алкокси (например, МеО), С 3-С 6 циклоалкил-С 0-С 1 алкиленом(например, циклопропилом или циклопропилметилом), бензилом или С 0-С 2 алкилен-СООН или его С 1 С 4 алкиловыми эфирами. Примером ароматического гетероциклила является 1-метилпиразол-5-ил. Обычно согласно данному варианту осуществления гетероциклическое кольцо представляет собой пирролил, пиразолил, имидазолил, триазолил, тиазолил или тиадиазолил, каждый из которых необязательно замещен С 1-С 4 алкилом, галогеном, С 1-С 4 галогеналкилом, С 1-С 4 алкокси, С 3-С 6 циклоалкилом или С 3-С 6 циклоалкилметилом. Обычно согласно данному варианту осуществления гетероциклическое кольцо представляет собой пиразол-1-ил, необязательно замещенный С 1-С 4 алкилом, галогеном, C1-С 4 галогеналкилом или циклопропилом. Следующим типичным значением для R1b согласно данному варианту осуществления является метил или циклопропил. В других вариантах осуществления R1a представляет собой Н, и R1b представляет собой метил или этил, который замещен в 1 положении циклической группой, такой как фенил, или R1b представляет собой моноциклический гетероциклил, такой как пирролидинил, пиперидинил, морфолинил, тиоморфолинил, пиперазинил, индолинил, пиранил, тетрагидропиранил, тетрагидротиопиранил, тиопиранил, фуранил, тетрагидрофуранил, тиенил, пирролил, оксазолил, изоксазолил, тиазолил, имидазолил, пиридинил,пиримидинил, пиразинил, пиридазинил, тетразолил, пиразолил, индолил и тому подобное. Фенил или гетероциклил необязательно замещены, например, 1-3 заместителями, независимо выбранными из гид-2 019277 рокси, амино, С 1-С 4 алкила, галогена, С 1-С 4 галогеналкила, С 1-С 4 алкокси, амино, С 1-С 4 алкиламина, С 1 С 4 диалкиламина и т.д. Примером заместителя является 1-фенилэтил. В других вариантах осуществления R1a представляет собой Н и R1b представляет собой С 3 С 6 циклоалкил, предпочтительно циклобутил или циклопропил, необязательно замещенный, как описано выше. Циклоалкил предпочтительно является незамещенным или замещенным 1-3 заместителями, выбранными из галогена (например, 1 или 2 атомами фтора), гидрокси, С 1-С 4 алкила (например, 1 или 2 метилами), С 1-С 4 галогеналкила (например, группой CF3), С 1-С 4 алкокси (например, группой MeO), C1 С 4 алкиламином (например, группой -NHMe), С 1-С 4 диалкиламином (например, группой -N(Me)2) и т.д. Примером циклоалкила является циклопропил или моно- или гем-фторциклопропил. В некоторых вариантах осуществления R1a представляет собой Н, и R1b представляет собой 6- или предпочтительно 5-членное ароматическое гетероциклическое кольцо, содержащее 1-3 атома азота и 0 или 1 атом серы и необязательно замещенное, как указано выше. Гетероциклическое кольцо предпочтительно связано с соседним атомом азота альфа-кетоамидной группы через атом углерода гетероциклического кольца. Примеры заместителей включают С 1-С 4 алкил, галоген, С 1-С 4 галогеналкил, С 1-С 4 алкокси,С 1-С 4 галогеналкокси, С 1-С 4 алкоксикарбонил, С 1-С 4 алкилкарбонил, амин, С 1-С 4 алкиламин, С 1 С 4 диалкиламин, С 1-С 4 алкилсульфонил, C1-С 4 алкилсульфониламино, аминокарбонил, аминосульфонил,RxOOC-C0-С 2 алкилен (где Rx представляет собой Н или С 1-С 4 алкил) или С 3-С 6 циклоалкил-С 0-С 2 алкилен или бензил (циклоалкильное или фенильное кольцо бензильной группы необязательно замещено 1-3 заместителями, выбранными из С 1-С 4 алкила, галогена, C1-С 4 галогеналкила, С 1-С 4 алкокси, С 1 С 4 галогеналкокси). В некоторых вариантах осуществления R1a и R1b и атом N, к которому они присоединены, образуют 3-6-членный циклический амин, такой как азиридин, азетидин, пирролидин и предпочтительно морфолин, пиперазин или пиперидин. Эти циклические амины могут быть незамещенными или замещенными,как описано выше, предпочтительно 1-3 заместителями, выбранными из галогена (например, 1 или 2 атомами фтора), гидрокси, С 1-С 4 алкила (например, 1 или 2 метилами), С 1-С 4 галогеналкила (например,группой CF3), С 1-С 4 алкокси (например, группой МеО), С 1-С 4 алкиламина (например, группой -NHMe), С 1 С 4 диалкиламина (например, группой -N(Me)2) и т.д. В некоторых вариантах осуществления оба из R2a и R2b представляют собой водород. Однако в данном изобретении предпочтительно, чтобы по меньшей мере один из R2a и R2b был замещен, как указано выше. Дальнейшие предпочтительные варианты осуществления включают соединения, у которых один изR2a и R2b представляет собой Н, и другой представляет собой Cl, F, CF3 или МеО. В других вариантах осуществления один из R2a и R2b представляет собой Н и другой представляет собой F. Особенно предпочтительными согласно этому варианту осуществления являются соединения,имеющие стереохимию, показанную в формуле В некоторых вариантах осуществления оба из R2a и R2b представляют собой F. Соединения этого аспекта имеют формулу В других вариантах осуществления один из R2a и R2b представляет собой Н и другой представляет собой хлор, фтор, трифторметил или метокси. В других вариантах осуществления R2a и R2b вместе с атомом углерода, к которому они присоединены, образуют С 3-С 6 циклоалкил. Некоторые варианты осуществления имеют R3 в виде циклоалкилалкила, необязательно замещенного, например, галогеном (таким как F) или алкокси (например, МеО). Примеры циклоалкилалкилов включают 1-метилциклопентилметил, 1-метилциклогексилметил, 1-метилциклобутилметил, 1-метил-3,3 дифторциклобутилметил, 1-метил-4,4-дифторциклогексилметил, циклопропилметил или 1-метил-3,3 дифторциклопентилметил. Предпочтительные значения R3 включают трет-бутилметил или циклобутилметил, или 1 метилциклобутилметил или 1-метилциклопентилметил, каждый из которых необязательно замещен одним или двумя атомами F или ОМе. Репрезентативными значениями являются 1-фторциклобутилметил и 1-фторциклопентилметил. Другие варианты осуществления имеют R3 в виде алкила с неразветвленной или разветвленной це-3 019277 пью с 5-10 атомами углерода, необязательно замещенного 1-4 атомами галогена (например, Cl или F) или С 1-С 4 алкокси (например, МеО). Примеры алкилов включают 2,2-диметилпропил, 3,3-диметилпентил,2,2,3,3-тетраметилбутил. Примеры галогенированных алкилов включают 2,2-дихлорэтил, 3,3,3 трифторпропил, 2,2-трифторметилэтил и 2,2,2-трифторэтил. В некоторых вариантах осуществления R4 представляет собой морфолинил, пиперидинил, пиперазинил, циклопентил, циклогексил или пиридинил, каждый из которых необязательно замещен галогеном,гидрокси, С 1-С 4 алкилом, С 1-С 4 галогеналкилом, C1-С 4 алкокси, С 1-С 4 галогеналкокси, амино, С 1 С 4 алкиламино, ди(С 1-С 4 алкил)амино или NRkS(=O)mRq; где Rk представляет собой Н или С 1-С 4 алкил;Rq представляет собой С 1-С 4 алкил, Het или карбоциклил, каждый из которых необязательно замещен С 1-С 4 алкилом, галогеном, С 1-С 4 галогеналкилом, С 1-С 4 алкокси; иm равно 0, 1 или 2. В других вариантах осуществления R4 представляет собой необязательно замещенный тиазолил, такой как тиазолил-5-ил, необязательно замещенный С 1-С 4 алкилом (например, Me), галогеном (например,F) или С 1-С 4 алкокси (например, МеО). В других вариантах осуществления R4 представляет собой морфолин-4-ил. В некоторых вариантах осуществления R4 связан с соседней амидной группой цепи через атом азота кольца, тем самым образуя функциональную группу мочевины. Репрезентативным видом R4 является морфолин-4-ил. Следующие репрезентативные виды имеют частичную структуру в которой X представляет собой С и Rt представляет собой гидрокси, фтор, С 1-С 4 алкил, например,гем-метил, С 1-С 4 алкокси (например, МеО), С 1-С 4 галогеналкил (например, CF3) или функциональную группу NRk-S(=O)2RS, в которой Rs представляет собой С 1-С 4 алкил, Het или карбоциклил, каждый из которых необязательно замещен 1-3 С 1-С 4 алкилами (например, Me), атомами галогена (например, F), С 1 С 4 галогеналкилами (например, CF3) или С 1-С 4 алкокси (например, МеО). Альтернативно, X представляет собой N, и Rt представляет собой С 1-С 4 алкил (например, Me) или функциональную группу -NRkS(=O)2RS, в которой Rs представляет собой С 1-С 4 алкил, Het или карбоциклил, каждый из которых необязательно замещен 1-3 С 1-С 4 алкилами (например, Me), атомами галогена (например, F), С 1 С 4 галогеналкилами (например, CF3) или С 1-С 4 алкокси (например, МеО). В некоторых вариантах осуществления R4 представляет собой пиперазин-1-ил или пиперидин-1-ил,каждый из которых замещен в 4-положении, или пиперидин-4-ил, замещенный в 1 положении; в каждом случае заместитель выбран из NHS (=O)2 карбоциклила или NHS(=O)Het, где карбоциклил или Het необязательно замещен галогеном, С 1-С 4 алкилом, С 1-С 4 алогеналкилом или С 1-С 4 алкилокси. В других вариантах осуществления R4 представляет собой циклогексил или пиперазин-1-ил, замещенный в 4-положении галогеном, амино, С 1-С 4 алкиламино, ди(С 1-С 4 алкил)амино или гидрокси. В некоторых вариантах осуществления R4 представляет собой фенил, который замещен 1-3 заместителями, независимо выбранными из галогена, гидрокси, С 1-С 4 алкила, С 1-С 4 галогеналкила, циано, С 1 С 4 алкилС(=O)NH- и С 1-С 4 алкокси. Репрезентативные виды R4 включают фенил, замещенный м-фтором, п-фтором, п-гидрокси, пгидрокси-м-хлором, п-гидрокси-м-фтором, п-гидрокси-м-метокси, п-гидрокси-м-метилом, бис-п-хлор-пгидрокси, м-циано, п-ацетамидо или о-фтор-п-гидрокси. Как указано выше, R4 может быть замещен группой формулы X-R5, в которой R5 представляет собой Н, необязательно замещенный С 1-С 4 алкил или необязательно замещенное моноциклическое кольцо,которое отделено от кольца R4 двухвалентной линкерной (связывающей) группой X. Линкерная группа X может содержать С 1-С 4 алкилен с неразветвленной цепью, такой как этилен или метилен. Альтернативно, линкерная группа может содержать двухвалентную функциональную группу,выбранную из СН(СН 3), С(СН 3)2, CF2, этена, этина, С 0-С 4 алкиламина, С 0-С 4 алкиламида, сульфонамида,сложного эфира, простого эфира, мочевины или карбамата, которая может быть связана непосредственно с R4 и/или R5 или может иметь одну или несколько метиленовых групп между функциональной группой и R4 и/или между функциональной группой и R5. Общую длину линкерной группы (включая любые метиленовые группы между функциональной группой и R4 и/или R5) образуют 1-4 атома цепи, предпочтительно от одного до трех. Двухвалентные функциональные группы в линкерной группе X, содержащие несколько гетероатомов, такие как амид, сульфонамид, сложный эфир или карбамат, могут быть расположены в любой ориентации, например, ориентации -O(C=O)NH- или -NH(=O)O- в случае карбамата. Выражения С 0 С 4 алкиламин и С 0-С 4 алкиламид в контексте линкерной группы X означают, что алкильная группа (или Н в случае Со) ответвляется от атома азота, например, -NH-, -N(СН 3)-, -NHC(=O)-, -N(CH3)C(=O)-,-С(=O)N(CH3)- и т.д. Более того, группы R4 включают группы, описанные в заявке WO 0664206, содержание которой-4 019277 включено в контекст в качестве ссылки. Значимые группы R4 этой ссылки включают группы с частичными структурамиR4' представляет собой Н, галоген, ОС 1-С 4 алкил, C(=O)NRrRl, NRkC(=О)С 1-С 4 алкил,NRkC(=O)NRkRl или -NRkC(=O)ОС 1-С 4 алкил или NHC(=O)OMe,Rk и R1 представляют собой независимо Н, С 1-С 4 алкил или С (=O)-С 1-С 4 алкил, или Rk, R1 и соседний атом N, к которому они оба присоединены, образуют циклический амин, выбранный из пирролидинила, пиперидинила, морфолинила, пиперазинила или N-метилпиперазинила. Предпочтительные подгруппы включают соединения, у которых R4' представляет собой фтор, метокси, диметилкарбамоил, NHC(=O)Me, -NHC(=O)NHCH3, NHC(=O)N(СН 3)2, NHC(=O)OMe или циклический амин. Другие варианты осуществления R4 в WO 0664206 имеют частичные структурыRk, Rl и атом N, к которому они оба присоединены, образуют циклический амин, выбранный из пирролидинила, пиперидинила, морфолинила, пиперазинила или N-метилпиперазинила; и R4 представляет собой Н, С 1-С 4 алкил, С 1-С 4 галогеналкил, галоген, циано, гидроксил или С 1 С 4 алкокси. Предпочтительные подгруппы включают соединения, где R5 представляет собой С 1-С 4 алкил, такой как метил, этил или изопропил или трет-бутил; галогенированный С 1-С 4 алкил, такой как трифторметил; С 3-С 6 циклоалкил, такой как циклопропил или циклогексил; или фенил или бензил, каждый из которых необязательно замещен С 1-С 4 алкилом, С 1-С 4 галогеналкилом, галогеном, циано, С 1-С 4 алкокси. Другие варианты осуществления R4 в WO 0664206 имеют частичные структуры: в которых Rx независимо выбран из Me, F, Cl, CF3 и ОМе, и n независимо равно 0, 1 или 2. Предпочтительная подгруппа имеет частичную структуруRa представляет собой Н или метил,Rp представляет собой Н, Me, F,Rx независимо выбран из Me, F, Cl, CF3 и ОМе, и где Ry представляет собой Н, С 1-С 4 алкил, амино, NHC1-С 4 алкил (такой как метиламино), N(С 1 С 4 алкил)2 (такой как диметиламино), NHC(=O)С 1-С 4 алкил (такой как ацетамидо); атомы азота кольца необязательно замещены С 1-С 4 алкилом (таким как метил, этил или трет-бутил) или С(=O)С 1-С 4 алкилом (таким как ацетил); и где Rx представляет собой независимо Н, F, Cl, CF3 или OMe; один или оба из атомов азота кольца необязательно замещены С 1-С 4 алкилом (таким как метил, этил или трет-бутил) или C(=O)C1-С 4 алкилом (таким как ацетил); О' отсутствует (т.е. присутствуют 2 атома водорода) или представляет собой О. Далее типичная группа R4 включает где Het представляет собой 5- или 6-членный, насыщенный, частично ненасыщенный или ароматический гетероцикл, содержащий 1-3 гетероатома, независимо выбранные из S, О и N. Соединения формулы I характеризуются различными подходящими фармацевтическими свойствами и проявляют по меньшей мере одно улучшенное свойство по сравнению с соединениями известного уровня техники. В частности, ингибиторы настоящего изобретения превосходят по одному или нескольким следующим, относящимся к фармакологии свойствам, т.е. активности, пониженной цитотоксичности, улучшенным фармакокинетикам, приемлемой дозе и загрузке препаративной формы. Без желания быть связанным каким-либо образом с теорией или описанием экспериментальных способов связывания для конкретных фрагментов ингибиторов, обозначения P1, P2 и Р 3, применяемые в контексте, представлены только для удобства и имеют их общепринятые значения, и считается, что они означают те части ингибитора, которые занимают субсайты S1, S2 и S3, соответственно, фермента, гдеS1 является соседним с сайтом расщепления и S3 удален от сайта расщепления. Следующий аспект изобретения содержит способ применения соединения формулы I для профилактики или лечения заболеваний, вызванных аномальной экспрессией или активацией катепсина, т.е. заболеваний или состояний, облегчаемых или модифицируемых ингибированием катепсина S, предпочтительно без существенного сопутствующего ингибирования других членов суперсемейства папаина. В следующем аспекте изобретения предложено применение соединений формулы I для профилактики или лечения заболеваний, вызванных аномальной экспрессией или активацией катепсина, т.е. заболеваний или состояний, облегчаемых или модифицируемых ингибированием катепсина S, предпочтительно без значительного сопутствующего ингибирования других членов суперсемейства папаина. В другом аспекте изобретения предложено применение соединений формулы I для изготовления лекарственного средства для профилактики или лечения заболеваний, вызванных аномальной экспрессией или активацией катепсина S, т.е. заболеваний или состояний, облегчаемых или модифицируемых ингибированием катепсина S, предпочтительно без значительного сопутствующего ингибирования других членов папаинового суперсемейства. Примеры таких заболеваний или состояний, определяемых только что в предыдущих трех абзацах,включают заболевания и состояния, перечисленные в WO 97/40066, такие как аутоиммунные заболевания, аллергии, такие как астма и сенная лихорадка, рассеянный склероз, ревматоидный артрит и тому подобное. Следующим примером является лечение эндометриоза и особенно хронической боли, как описано в WO 03/20287. В изобретении дополнительно предлагается применение соединений формулы IV в терапии и при изготовлении лекарственного средства для лечения заболеваний или состояний, которые облегчают или развитие которых замедляют ингибированием катепсина S. В одном из вариантов осуществления способы применяют для лечения млекопитающих, особенно людей с риском аутоиммунного заболевания или пораженных аутоиммунным заболеванием. Термин "аутоиммунитет" означает феномен, в котором иммунная реакция хозяина направляется против своих собственных компонентов организма, приводя к патологии. Многие аутоиммунные реакции человека ассоциируются с определенными МНС-комплексами класса II. Эта ассоциация имеет место вследствие того,что структуры, распознаваемые Т-клетками, клетками, которые вызывают аутоиммунитет, являются комплексами, состоящими из молекул МНС класса II и антигенных пептидов. Аутоиммунное заболевание может иметь место, когда Т-клетки реагируют с молекулами МНС класса II хозяина и когда образуют комплекс с пептидами, образованными из продуктов собственных генов хозяина. Если образование этих комплексов МНС класса II/антигенные пептиды ингибируется, аутоиммуная реакция ослабляется или подавляется. Любое аутоиммунное заболевание, в котором играют роль комплексы МНС классаII/антигенные пептиды, можно лечить согласно способам настоящего изобретения. Такие аутоиммунные заболевания включают, например, инсулинзависимый юношеский сахарный диабет, рассеянный склероз, обыкновенную пузырчатку, болезнь Граве, тяжелую миастению, системную красную волчанку, ревматоидный артрит и тироидит Хашимото. В других вариантах осуществления способы применяют для лечения млекопитающих, особенно людей с риском аллергических реакций или людей, пораженных аллергическими реакциями. Термин"аллергическая реакция" означает феномен, при котором иммунная реакция хозяина к конкретному антигену является ненужной или непропорциональной, приводящей к патологии. Аллергии являются хорошо известными в данной области, и термин "аллергическая реакция" применяют в контексте в соответствии со стандартным применением в медицинской сфере. Примеры аллергий включают, но не ограничиваются перечисленным, аллергии на пыльцу, "амброзию" (сорняк), водных животных, имеющих панцирь, домашних животных (например, кошек и собак),жало пчел, аллергены клещей домашней пыли и тому подобное. Другой, особенно рассматриваемой аллергической реакцией является аллергическая реакция, которая вызывает астму. Аллергические реакции могут иметь место у человека вследствие того, что Т-клетки распознают комплексы МНС классаII/антигенные пептиды. Если образование этих комплексов МНС класса II/антигенные пептиды ингибируется, аутоиммунная реакция ослабляется или подавляется. Любую аллергическую реакцию, в которой играют роль комплексы МНС класса II/антигенные пептиды, можно лечить согласно способам настоящего изобретения. Иммуносупрессия способами настоящего изобретения будет обычно профилактическим или терапевтическим лечением тяжелых или опасных для жизни аллергических реакций, которые могут возникать во время астматических приступов или анафилактического шока. В других вариантах осуществления способы применяют для лечения млекопитающих, особенно людей, которых подвергали или собираются подвергнуть трансплантации органа или пересадке ткани. При трансплантации ткани (например, почки, легкого, печени, сердца) или пересадке кожи, когда имеется несовместимость между генотипами МНС класса II (типы HLA) донора и реципиента, может быть тяжелая "аллогенная" иммунная реакция против тканей донора, которая является результатом присутствия несобственных или аллогенных молекул МНА класса II, презентирующих антигенные пептиды на поверхности донорных клеток. До степени, при которой эта реакция зависит от образования комплексов МНС класса II/антигенные пептиды, ингибирование катепсина S может подавлять эту реакцию и ослаблять отторжение ткани. Ингибитор катепсина S можно применять отдельно или в сочетании с другими терапевтическими агентами, например, в качестве вспомогательного средства для циклоспорина А и/или антилимфоцитного гамма-глобулина, для достижения иммуносупрессии и стимуляции жизнеспособности трансплантата. Введение предпочтительно выполняют системным введением его хозяину до и/или после хирургической операции. Альтернативно или дополнительно, может быть эффективной перфузия в орган или ткань донора либо до, либо после трансплантации или пересадки. Указанные выше варианты осуществления иллюстрировали механизмом действия МНС класса II,но изобретение не ограничивается этим механизмом действия. Супрессия катепсина S, как лечениеCOPD или хронической боли, например, может совсем не включать участие МНС класса II. Родственный аспект изобретения относится к способу лечения пациента, подвергаемого терапии,где терапия вызывает у пациента иммунную реакцию, предпочтительно вредную иммунную реакцию,содержащему введение пациенту соединения формулы I или его фармацевтически приемлемой соли, Nоксида или гидрата. Иммунная реакция обычно опосредуется молекулами МНС класса II. Соединение данного изобретения можно вводить до терапии, одновременно с терапией или после терапии. Терапия обычно включает лечение биологическим средством, таким как белок, предпочтительно антитело, более предпочтительно моноклональное антитело. Более предпочтительно биологическим средством является Ремикад, Ребакто, Реферон А, фактор VIII, фактор VII, Бетасерон, Эпоген, Энбрел, интерферон бета, Ботокс, Фабразим, Элспар, Церезим, Миоблок, Алдуразирн, Верлума, интерферон альфа, Гумира, Аранесп, Зевалин или ОКТЗ. Альтернативно, лечение включает применение гепарина, гепарина с низкой молекулярной массой, прокаинамида или гидралазина. Анализы для оценки ингибиторов катепсина S при лечении хронической боли, включающей невропатическую или воспалительную боль, описаны в WO 03/20287. В настоящее время предпочтительные показания, излечимые согласно настоящему изобретению,включают псориаз; аутоиммунные показания, включающие идиопатическую тромбоцитопеническую пурпуру (ITP),ревматоидный артрит (RA), рассеянный склероз (MS), тяжелую миастению (MG), синдром Шегрена, болезнь Граве и системную красную волчанку (SLE); неаутоиммунные показания, включающие аллергический ринит, астму, атеросклероз, хроническое обструктивное заболевание легких (CORD) и хроническую боль. Соединения изобретения могут образовывать соли, которые образуют дополнительный аспект изобретения. Подходящие фармацевтически приемлемые соли соединений изобретения включают соли органических кислот, особенно карбоновых кислот, включающих, но не ограничивающихся перечисленным, ацетат, трифторацетат, лактат, глюконат, цитрат, тартрат, малеат, малат, пантотенат, изетионат,адипат, альгинат, аспартат, бензоат, бутират, диглюконат, циклопентанат, глюкогептанат, глицерофосфат, оксалат, гептаноат, гексаноат, фумарат, никотинат, памоат, пектинат, 3-фенилпропионат, пикрат,пивалат, пропионат, тартрат, лактобионат, пиволат, камфорат, ундеканоат и сукцинат, соли органических сульфоновых кислот, такие как метансульфонат, этансульфонат, 2-гидроксиэтансульфонат, камфорасульфонат, 2-нафталинсульфонат, бензолсульфонат, п-хлорбензолсульфонат и п-толуолсульфонат, и неорганических кислот, такие как гидрохлорид, гидробромид, гидроиодид, сульфат, бисульфат, гемисульфат, тиоцианат, персульфат, соли фосфорной и сульфоновой кислот. Соединения изобретения можно в некоторых случаях выделять в виде гидрата. Гидраты обычно получают перекристаллизацией из смеси водного/органического растворителей с применением такого органического растворителя, как диоксин, тетрагидрофуран или метанол. Гидраты можно также генерировать in situ введением соответствующего кетона пациенту.N-оксиды соединений изобретения можно получать способами, известными среднему специалисту в данной области техники. N-оксиды можно получать, например, обработкой неокисленной формы соединения изобретения окисляющим агентом (например, трифторперуксусной кислотой, пермалеиновой кислотой, пербензойной кислотой, перуксусной кислотой, мета-хлорпероксибензойной кислотой или тому подобное) в подходящем инертном органическом растворителе (например, галогенированном углеводороде, таком как дихлорметан) приблизительно при 0 С. Альтернативно, N-оксиды соединений изобретения можно получить из N-оксида подходящего исходного вещества. Соединения изобретения в неокисленной форме можно получить из N-оксидов соответствующих соединений изобретения обработкой восстанавливающим агентом (например, серой, диоксидом серы,трифенилфосфином, литийборогидридом, натрийборогидридом, бихлоридом, трибромидом фосфора и т.д.) в подходящем инертном органическом растворителе (например, ацетонитриле, этаноле, водном диоксане или т.д.) при 0-80 С. Настоящее изобретение включает также меченные изотопом соединения формулы I или любой подгруппы формулы I, в которых один или несколько атомов заменены изотопом этого атома, т.е. атомом,имеющим такое же атомное число, но атомную массу, отличную от атомной массы, обычно находимой в природе. Примеры изотопов, которые можно включить в соединения формулы I или любую подгруппу формулы I, включают, но не ограничиваются перечисленным, изотопы водорода, такие как 2 Н и 3 Н (обозначенные также D для дейтерия и Т для трития, соответственно), углерода, такие как 11 С, 13 С и 14 С, азота, такие как 13N и 15N, кислорода, такие как 15O, 17O и 18O, фосфора, такие как 31 Р и 32 Р, серы, такие как 35I, I, I и 131I. Выбор изотопа, включенного в меченное изотопом соединение, будет зависеть от конкретного применения такого соединения. Например, для анализов распределения в ткани лекарственного средства или субстрата наиболее применимыми обычно являются соединения, в которые включен радиоактивный изотоп, такой как 3 Н или 14 С. Для применений с целью получения радиоизображения, например позитронноэмиссионной томографии (PET), будет применимым изотоп, испускающий позитроны, такой как 11 С, 18F,13N или О. Включение более тяжелого изотопа, такого как дейтерий, т.е. 2 Н, может обеспечить более высокую метаболическую стабильность соединения формулы I или любой подгруппы формулы I, которая может привести, например, к повышенному полупериоду существования соединения in vivo или пониженным требованиям доз. Меченные изотопами соединения формулы I или любой подгруппы формулы I можно получить способами, аналогичными способам, описанным ниже на схемах и/или в примерах данного описания, с применением подходящего, меченного изотопом реагента или исходного вещества вместо соответствующего немеченного изотопом реагента или исходного вещества или общепринятыми методиками, известными специалисту в данной области техники. Следует отметить, что положения радикалов в любой молекулярной части, применяемой в определениях, могут быть в любом положении такой части, при условии, что она является химически стабильной. Применяемые в данном описании следующие термины имеют значения, указанные ниже.Cm-Cn-алкил, применяемый в отдельности или в составных выражениях, таких как Cm-Cnгалогеналкил,Cm-Cn-алкилкарбонил,Cm-Cn-алкиламин,Cm-Cn-алкилсульфонил,Cm-Cnалкилсульфониламино и т.д., представляет собой неразветвленный или разветвленный алкильный радикал, имеющий указанное число атомов углерода, например С 1-С 4 алкил означает алкильный радикал,имеющий от 1 до 4 атомов углерода. Предпочтительными алкильными радикалами для применения в настоящем изобретении являются С 1-С 4 алкилы, включающие метил, этил, н-пропил, изопропил, трет-8 019277 бутил, н-бутил и изобутил. Метил и трет-бутил обычно являются предпочтительными. С 1-С 6 алкил имеет соответствующее значение, включающее также все изомеры пентила и гексила с неразветвленной или разветвленной цепью. Другие радикалы Cm-Cn-алкила, такого как С 5-С 10 алкил, имеют соответствующее значение. Термин Me означает метил, МеО означает метокси, Et означает этил и Ас означает ацетил. С 0-С 2 алкилен, применяемый в составных выражениях, таких как С 3-С 6 циклоалкил-С 0-С 2 алкилен,относится к двухвалентному радикалу, образованному из метильной или этильной группы, или в случаеC0 С 0-С 2 алкилен означает связь. С 1-С 4 галогеналкил относится к С 1-С 4 алкилу, у которого по меньшей мере один атом С замещен галогеном, предпочтительно хлором или фтором. Обычно предпочтительным является трифторметил. С 1-С 4 алкокси представляет собой радикал С 1-С 4 алкил-O, у которого С 1-С 4 алкил имеет значения,указанные выше, и включает метокси, этокси, н-пропокси, изопропокси, трет-бутокси, н-бутокси и изобутокси. Обычно предпочтительными являются метокси и изопропокси. С 1-С 6 алкокси имеет соответствующее значение, расширяемое для включения всех изомеров с неразветвленной или разветвленной цепью пентокси и гексокси. Другие радикалы Cm-Cn-алкокси, такие как C5-С 10 алкокси, имеют соответствующее значение. Имеется в виду, что термин С 1-С 4 галогеналкокси, применяемый в данном описании, включает С 1 С 4 алкокси, где по меньшей мере один С-атом замещен одним или несколькими атомами галогена, обычно хлора или фтора. Во многих случаях предпочтительным является трифторметил. С 1-С 4 алкоксикарбонил означает радикал С 1-С 4 алкил-O-C(=O). Термин оксо" представляет собой =O, т.е. карбонильная группа образуется при присоединении =O к атому углерода. Карбоциклил включает циклопентил, циклогексил и особенно циклопропил и циклобутил. Карбоциклил дополнительно включает циклопентенил и циклогексенил, в каждом случае с одной двойной связью. Часто предпочтительным карбоциклилом является фенил. Термин "циклический амин" включает азиридин, азетидин, пирролидин, пиперидин, пиперазин и морфолинил.Het представляет собой стабильную, моноциклическую или бициклическую, насыщенную, частично насыщенную или ароматическую систему колец, содержащую 1-4 гетероатома, независимо выбранные из O, S и N, причем каждое кольцо имеет 5 или 6 атомов кольца. Примеры ароматических Het включают фуран, тиофен, пиррол, имидазол, пиразол, триазол, тетразол, тиазол, оксазол, изоксазол, оксадиазол, тиадиазол, изотиазол, пиридин, пиридазин, пиразин, пиримидин, хинолин, изохинолин, бензофуран,бензотиофен, индол, индазол и тому подобное. Примеры ненасыщенных Het включают тетрагидрофуран,пиран, дигидропиран, 1,4-диоксан, 1,3-диоксан, пиперидин, пирролидин, морфолин, тетрагидротиопиран, тетрагидротиофен, 2 Н-пиррол, пирролин, пиразолин, имидазолин, тиазолидин, изоксазолидин и тому подобное. Соединения изобретения включают ряд групп, таких как ОН, NH или СООН, к которым можно присоединить общепринятые пролекарственные фрагменты. Пролекарства обычно гидролизуются in vivo с высвобождением "родительского" соединения в плазме, печени или стенках кишечника. Подходящими пролекарствами являются сложные эфиры гидроксильных групп, таких как фенольная гидроксильная группа у R4, или аминных функциональных групп, таких как аминная функциональная группа сульфонамида. Предпочтительные фармацевтически приемлемые эфиры включают эфиры, полученные из С 1 С 6 карбоновых кислот, такие как ацетил или пивалоил, или необязательно замещенной бензойной кислоты, предпочтительно незамещенные или замещенные заместителями широкого диапазона, описанными для R1a, обычно 1-3 атомами галогена (например, F), С 1-С 4 алкильными (например, Me), C1 С 4 галогеналкильными (например, CF3) или С 1-С 4 алкилоксигруппами (например, МеО). Предпочтительные сульфонамидные пролекарства включают аминоацилы, образованные из С 1-С 6 карбоновых кислот,таких как ацетил или пивалоил, или эфиров необязательно замещенной бензойной кислоты, предпочтительно незамещенных или замещенных заместителями широкого диапазона, как описано для R1a, обычно 1-3 атомами галогена (например, F), С 1-С 4 алкильными (например, Me), С 1-С 4 галогеналкильными (например, CF3) или C1-С 4 алкилоксигруппами (например, МеО). Если не указывается или не оговаривается особо, химическое название соединения включает смесь всех возможных стереохимически изомерных форм, которые может иметь указанное соединение. Указанная смесь может содержать все диастереомеры и/или энантиомеры основной молекулярной структуры указанного соединения. Предполагается, что все стереохимически изомерные формы соединений настоящего изобретения как в чистой форме, так и в виде смеси друг с другом, включены в объем настоящего изобретения. Чистые стереоизомерные формы соединений и промежуточных продуктов, указанные в данном описании, определяются как изомеры, по существу, свободные от других энантиомерных или дистереомерных форм той же самой основной молекулярной структуры указанных соединений или промежуточных продуктов. В частности, термин "стереоизомерно чистые" относится к соединениям или промежуточным продуктам, имеющим стереоизомерный избыток по меньшей мере 80% (т.е. минимум 90% одно-9 019277 го изомера и максимум 10% других возможных изомеров) и до стереоизомерного избытка 100% (т.е. 100% одного изомера и отсутствие другого), более конкретно соединениям или промежуточным продуктам, имеющим стереоизомерный избыток от 90 до 100%, еще более конкретно, имеющим стереоизомерный избыток от 94 до 100% и наиболее конкретно, имеющим стереоизомерный избыток от 97 до 100%. Аналогичным образом следует понимать термины "энантиомерно чистый" и "диастереомерно чистый",но кроме того эти термины имеют отношение к энантиомерному избытку и диастереомерному избытку,соответственно, рассматриваемой смеси. Соединения изобретения можно получать в виде их индивидуальных стереоизомеров реакцией рацемической смеси соединения с оптически активным разделяющим агентом с образованием пары диастереомерных соединений, разделением диастереомеров и выделением оптически чистого энантиомера. Хотя разделение энантиомеров можно проводить с применением ковалентных диастереомерных производных соединений формулы I, предпочтительными являются диссоциируемые комплексы (т.е. кристаллические диастереомерные соли). Диастереомеры имеют различные физические свойства (например,точки плавления, точки кипения, растворимости, реакционные способности и т.д.), и их можно легко разделить с использованием этих различий. Диастереомеры можно разделить хроматографией, например ВЭЖХ или предпочтительно способами разделения/расщепления на составляющие, основанными на различиях в растворимости. Оптически чистый энантиомер затем выделяют вместе с разделяющим агентом любым практическим способом, который не приведет к рацемизации. Более подробное описание методик, приемлемых для разделения стереоизомеров соединений из их рацемической смеси, можно найти в Jean Jacques Andre Collet, Samuel H. Wilen, Enantiomers, Racemates and Resolutions, John WileySons, Inc. (1981). Хотя активный агент можно вводить отдельно, предпочтительным является присутствие его как части фармацевтического препарата. Такой препарат будет содержать указанный выше активный агент вместе с одним или несколькими приемлемыми носителями/эксципиентами и необязательно другими терапевтическими ингредиентами. Носитель(и) должен быть приемлемым в смысле совместимости с другими ингредиентами препарата и не должен быть вредным для реципиента. Препараты включают препараты, подходящие для ректального, назального, местного (в том числе трансбуккального и сублингвального), вагинального или парентерального (в том числе подкожного,внутримышечного, внутривенного и внутрикожного) введения, но предпочтительным препаратом является перорально вводимый препарат. Препараты можно преимущественно представить в виде стандартной лекарственной формы, например таблеток и капсул с пролонгированным высвобождением, их можно получать любыми способами, хорошо известными в области фармации. Такие способы включают стадию объединения указанного выше активного агента с носителем. Обычно препараты получают образованием однородной и тесной смеси активного агента с жидкими носителями или тонкоизмельченными твердыми носителями или обоими и затем, если необходимо, формованием продукта. Изобретение относится к способам получения фармацевтической композиции, содержащим получение сочетания или смеси соединения формулы I или его фармацевтически приемлемой соли с фармацевтически приемлемым носителем или наполнителем. Если изготовление фармацевтических препаратов включает тесное смешивание фармацевтических эксципиентов и активного ингредиента в форме соли, то часто предпочтительным является применение эксципиентов, которые не являются основными по природе, т.е. являются либо кислотными, либо нейтральными. Препараты для перорального введения настоящего изобретения можно представить в виде дискретных единиц (для разового приема), таких как капсулы, саше или таблетки, каждая из которых содержит предварительно определенное количество активного агента; в виде порошка или гранул; в виде раствора или суспензии активного агента в водной жидкости или неводной жидкости; или в виде жидкой эмульсии типа масло-в-воде или жидкой эмульсии типа воды-в-масле и в виде болюса и т.д. Что касается композиций для перорального введения (например, таблеток и капсул), термин подходящий носитель включает наполнители, такие как обычные эксципиенты, например, связывающие агенты, например сироп, аравийская камедь, желатин, сорбит, трагакант, поливинилпирролидон (повидон),метилцеллюлоза, этилцеллюлоза, натрийкарбоксиметилцеллюлоза, гидроксипропилметилцеллюлоза,сахароза и крахмал; наполнители и носители, например, кукурузный крахмал, желатин, лактоза, сахароза, микрокристаллическая целлюлоза, каолин, маннит, дикальцийфосфат, хлорид натрия и альгиновая кислота; и смазывающие вещества, такие как стеарат магния, стеарат натрия и стеараты других металлов,стеарат глицерина, стеариновая кислота, силиконовая жидкость, тальк, воски, масла и коллоидальный диоксид кремния. Можно также применять корригенты, такие как перечная мята, винтергриновое масло,вишневый корригент или тому подобное. Может быть желательным добавлением красящего агента, чтобы изготовить легко идентифицируемую лекарственную форму. Таблетки могут быть покрыты оболочкой способами, хорошо известными в данной области. Таблетки можно изготовить прессованием или формованием, необязательно с одним или несколькими вспомогательными ингредиентами. Прессованные таблетки можно изготовить прессованием в подходящей машине активного агента в сыпучей форме, такой как порошок или гранулы, необязательно смешанных со связывающим веществом, смазывающим веществом, инертным разбавителем, консерван- 10019277 том, поверхностно-активным веществом или диспергирующим агентом. Формованные таблетки можно изготовить формованием в подходящей машине смеси порошкообразного соединения, увлажненного инертным жидким разбавителем. Таблетки можно необязательно покрыть оболочкой или сделать на них риску и можно изготовить их так, чтобы обеспечить медленное или регулируемое высвобождение активного агента. Другие препараты, подходящие для перорального введения, включают лепешки, содержащие активный агент в подходящей основе, обычно сахарозе и аравийской камеди или трагаканте, пастилки, содержащие активный агент в инертной основе, такой как желатин и глицерин, или сахароза и аравийская камедь; и жидкости для полоскания рта, содержащие активный агент в подходящем жидком носителе. Как и в случае всех фармацевтических препаратов, подходящая доза для соединений или препаратов изобретения будет зависеть от показания, тяжести заболевания, размера и метаболической активности и пациента, способа введения и ее легко определяют общепринятыми испытаниями на животных. Обычно требуемыми и достижимыми являются дозы, обеспечивающие внутриклеточные (для ингибирования физиологических протеаз папаинового суперсемейства) концентрации порядка 0,01-100 мкМ, более предпочтительно 0,01-10 мкМ, такие как 0,1-5 мкМ. Соединения изобретения получают с помощью различных химических способов синтеза в растворе и на твердой фазе. Типичной первой стадией является получение блока структуры Р 1 формулы II в которой R2a и R2b имеют значения, указанные выше, PG представляет собой общепринятую Nзащитную группу, такую как Boc, CBz или Fmoc, и PG представляет собой Н или общепринятую карбоксизащитную группу, такую как С 1-С 4 алкиловый или бензиловый эфир. Эти блоки структуры являются новыми и составляют следующий аспект изобретения. Блоки структуры формулы II обычно получают, как описано ниже на схеме 1. Схема 1 Подходящим исходным веществом является N-защищенная циклобутиламинокислота, некоторые из таких кислот являются коммерчески доступными или их можно получить, как показано в нижеследующих примерах или как описано Allan et al. в J. Med. Chem., 1990, 33(10), 2905-2915. Карбоновую кислоту(1a) превращают посредством синтеза Вайнреба вN,Одиметилгидроксамовую кислоту (1b), которая дает соответствующий альдегид (1 с). Альдегид можно также получить восстановлением карбоксильной функциональной группы циклобутиламинокислоты и окислением в условиях Десс-Мартина. Альдегид (1 с) можно затем подвергнуть реакции с подходящим изоцианидом по реакции Пассерини с получением требуемого -гидрокси-R1aR1b-амида, у которого R1a представляет собой Н (Id). Однако в случае, когда подходящий изоцианид не является легко доступным,альтернативно можно применять трет-бутилизоцианид, чтобы таким образом получить трет-бутиламид. Последующий гидролиз амида затем дает требуемый блок структуры Р 1, -гидроксикарбоновую кислоту(1 е). Обычно сильно кислотные условия, требуемые для гидролиза амида, также приводят к потере NBoc-защиты, если ее применяют. Поэтому амин можно применять непосредственно для конденсации с блоком структуры Р 2, или же, если амин необходимо хранить, его можно снова защитить. Блок структуры Р 1, полученный таким образом, затем наращивают по С- или N-концу, как показано ниже на схеме 2. С-конец обычно наращивают сначала реакцией блока структуры формулы II с R1aR1b-амином, у которого R1a и R1b представляют собой Rla и Rlb, соответственно, или их синтоны (выбранные с учетом чувствительности функциональной группы R1b к условиям удлинения Р 3, указанным ниже). Реакция протекает по общеизвестным правилам химического синтеза пептидов, обсуждаемым ниже. Полученный таким образом продукт Р 1-основная боковая цепь после этого освобождают от защиты у N-конца и наращивают блоками структуры Р 2 и затем Р 3 с применением общепринятых способов химического синтеза пептидов. Например, остаток Р 2 можно ввести при помощи Вос-Р 2-ОН с применением стандартных условий конденсации, таких как HATU, DIPEA в ДМФА. Концевую Вос-защиту снова удаляют ацетилхлоридом в метаноле, и остаток Р 3 вводят при помощи Р 3-ОН с применением стандартных условий конденсации, таких как HATU, DIPEA в ДМФА. Широкий диапазон подходящим образом защищенных L-аминокислот, подходящих для блоков структур Р 2, или карбоциклических или гетероциклических карбоновых кислот, подходящих для блоков структур Р 3, являются коммерчески доступными или доступными посредством простых химических способов или как показано в WO 06/064286. Блоки структур Р 3 и Р 2 можно альтернативно сначала конденсировать и затем подвергать реакции с продуктом Р 1-основная боковая цепь. Конечные стадии обычно содержат превращение синтонов R4/R1a/R1b (если они присутствуют) в их конечную форму и, наконец, окисление альфа-гидроксиамидной функциональной группы с применением условий реакции Десс-Мартина с получением требуемого альфа-кетоамидного соединения формулы I. Наращивание обычно проводят в присутствии подходящего агента конденсации, например, гексафторфосфата бензотриазол-1-илокситриспирролидинофосфония (РуВОР), гексафторфосфата Обензотриазол-1-ил-N,N,N',N'-тетраметилурония (HBTU), гексафторфосфата О-(7-азабензотриазол-1-ил)1,1,3,3-тетраметилурония (HATU), гидрохлорида 1-(3-диметиламинопропил)-3-этилкарбодиимида (EDC) или 1,3-дициклогексилкарбодиимида (DCC), необязательно в присутствии 1-гидроксибензотриазола(НОВТ) и основания, такого как N,N-диизопропилэтиламин, триэтиламин, N-метилморфолин и тому подобное. Реакцию обычно проводят при 20-30 С, предпочтительно приблизительно при 25 С, и для завершения ее требуется 2-24 ч. Подходящими растворителями реакции являются инертные органические растворители, такие как галогенированные органические растворители (например, метиленхлорид, хлороформ и тому подобное), ацетонитрил, N,N-диметилформамид, растворители типа простых эфиров, такие как тетрагидрофуран, диоксан и тому подобное. Альтернативно, указанную выше стадию наращивания реакцией конденсации можно проводить сначала превращением блока структуры Р 3/Р 2 в активированное производное кислоты, такое как сукцинимидный эфир, и затем реакцией его с амином Р 1. Обычно требуется 2-3 ч для завершения реакции. Условия, применяемые в этой реакции, зависят от природы активированного производного кислоты. Например, если оно является хлорангидридом кислоты, реакцию проводят в присутствии подходящего основания (например, триэтиламина, диизопропилэтиламина, пиридина и тому подобное). Подходящими растворителями реакции являются полярные органические растворители, такие как ацетонитрил, N,Nдиметилформамид, дихлорметан или их любая подходящая смесь. Термин "N-защитная группа" или "N-защищенная", применяемый в данном описании, относится к таким группам, предназначенным для защиты N-конца аминокислоты или пептида или защиты аминогруппы от нежелательных реакций во время синтетических методик. Обычно применяемые N-защитные группы описаны в публикации Greene, "Protective Groups in Organic Synthesis" (John WileySons, NewYork, 1981), которая тем самым включена в качестве ссылки. N-защитные группы включают ацильные группы, такие как формил, ацетил, пропионил, пивалоил, трет-бутилацетил, 2-хлорацетил, 2-бромацетил,трифторацетил, трихлорацетил, фталил, о-нитрофеноксиацетил, -хлорбутирил, бензоил, 4-хлорбензоил,4-бромбензоил, 4-нитробензоил и тому подобное; сульфонильные группы, такие как бензолсульфонил, птолуолсульфонил и тому подобное; карбаматобразующие группы, такие как бензилоксикарбонил, пхлорбензилоксикарбонил,п-метоксибензилоксикарбонил,п-нитробензилоксикарбонил,2-нитробензилоксикарбонил,п-бромбензилоксикарбонил,3,4-диметоксибензилоксикарбонил,4-метоксибензилоксикарбонил,2-нитро-4,5-диметоксибензилоксикарбонил,3,4,5-триметоксибензил- 12019277 оксикарбонил,1-(п-бифенилил)-1-метилэтоксикарбонил-диметил-3,5-диметоксибензилоксикарбонил, бензгидрилоксикарбонил, трет-бутоксикарбонил, диизопропилметоксикарбонил, изопропилоксикарбонил, этоксикарбонил, метоксикарбонил, аллилоксикарбонил, 2,2,2-трихлорэтоксикарбонил,феноксикарбонил, 4-нитрофеноксикарбонил, флуоренил-9-метоксикарбонил, циклопентилоксикарбонил,адамантилоксикарбонил, циклогексилоксикарбонил, фенилтиокарбонил и тому подобное; алкильные группы, такие как бензил, трифенилметил, бензилоксиметил и тому подобное; и силильные группы, такие как триметилсилил и тому подобное. Предпочтительные N-защитные группы включают формил,ацетил, бензоил, пивалоил, трет-бутилацетил, фенилсульфонил, бензил (bz), трет-бутоксикарбонил(ВОС) и бензилоксикарбонил (Cbz). Гидрокси- и/или карбоксизащитные группы также широко рассматриваются в указанной выше публикации Greene и включают простые эфиры, такие как метиловый, замещенный метиловый эфир, такой как метоксиметиловый, метилтиометиловый, бензилоксиметиловый, трет-бутоксиметиловый, 2 метоксиэтоксиметиловый эфир и тому подобное, силиловые простые эфиры, такие как триметилсилиловый (TMS), трет-бутилдиметилсилиловый (TBDMS), трибензилсилиловый, трифенилсилиловый, третбутилдифенилсилиловый, триизопропилсилиловый эфиры и тому подобное, замещенные этиловые простые эфиры, такие как 1-этоксиметиловый, 1-метил-1-метоксиэтиловый, трет-бутиловый, аллиловый,бензиловый, п-метоксибензиловый, дифенилметиловый, трифенилметиловый эфир и тому подобное,аралкиловые группы, такие как тритил и пиксил (производные 9-гидрокси-9-фенилксантена, особенно хлорид). Сложноэфирные гидроксизащитные группы включают такие сложные эфиры, как формиат, бензилформиат, хлорацетат, метоксиацетат, феноксиацетат, пивалоат, адамантоат, мезитоат, бензоат и тому подобное. Гидроксизащитные группы карбонатов включают метил, винил, аллил, циннамил, бензил и тому подобное. Подробное описание вариантов осуществления Различные варианты осуществления изобретения теперь будут описаны только путем иллюстрации с обращением к следующим примерам. Ниже в примерах обычно применяют следующие системы (инструменты). Спектры ядерного магнитного резонанса (ЯМР) регистрировали на инструменте Varian Gemini 7 Tesla 300 МГц или инструменте Bruker Avance 400 МГц в указанном растворителе. Химические сдвиги указаны в м.д. в сторону более высокого поля или более низкого поля относительно тетраметилсилана (TMS). Множественности резонанса указаны как с, д, т, м, ушир. и каж. для синглета, дублета, триплета, мультиплета, уширенной и кажущейся полосы соответственно. Спектры масс-спектрометрии (МС) регистрировали на инструментеWaters 996 и Micromass ZMD. Анализы высокоэффективной жидкостной хроматографией (ВЭЖХ) проводили с применением системы Hewlett Packard 1100 Series HPLC, снабженной колонкой Zorbax SB-C8,4,6 мм 15 см. Колоночную хроматографию проводили с применением силикагеля 60 (230-400 мешASTM, Merck) и тонкослойную хроматографию (ТСХ) проводили на пластинках для ТСХ, предварительно покрытых силикагелем 60 F254 (Merck). Получение блока структуры 1 (ВВ 1), блока структуры Р 1(ВВ 1-а) К раствору 1-трет-бутоксикарбониламиноциклобутанкарбоновой кислоты (3 г, 13,94 ммоль) в сухом ДМФА (50 мл) добавляли соль N,O-диметилгидроксиламинHCl (1,36 г, 13,94 ммоль) и DIEA (9,21 мл, 55,75 ммоль). Реакционную колбу охлаждали до 0 С и спустя 10 мин к раствору добавляли HATU(5,30 г, 13,94 ммоль) (при добавлении раствор становился желтым). Спустя 2 ч ДМФА удаляли роторным испарением при пониженном давлении. Остаток растворяли в 100 мл EtOAc и дважды промывали 10% лимонной кислотой (водной) и насыщенным раствором NaHCO3 (водным). Органическую фазу сушилиNa2SO4, фильтровали и упаривали на диоксиде кремния. Продукт очищали флэш-хроматографией (гептан:этилацетат (1:1, получая при этом продукт в виде бесцветного масла, которое медленно кристаллизовалось (3,13 г), с выходом 87%. Стадия b) трет-Бутиловый эфир (1-формилциклобутил)карбаминовой кислоты (ВВ 1-b)LiAlH4 (202 мг, 5,33 ммоль) добавляли к раствору амида Вайнреба ВВ 1-а (1,10 г, 4,27 ммоль) в су- 13019277 хом диэтиловом простом эфире (35 мл) при 0 С. Раствор перемешивали в течение 15 мин перед тем, как реакцию гасили медленным добавлением монокалиевой соли винной кислоты (насыщ., водный раствор) и перемешивали в течение 10 мин. Раствор выливали в делительную воронку и водную фазу экстрагировали дважды этилацетатом. Объединенные органические фазы промывали 0,5 M HCl (3 раза), NaHCO3(водным) (2 раза) и насыщенным раствором соли (1 раз). Органическую фазу сушили Na2SO4, фильтровали и упаривали на диоксиде кремния. Продукт очищали флэш-хроматографией (гептан:этилацетат(4:13:1, получая при этом продукт в виде белых кристаллов (0,647 г) с выходом 76%. Стадия с) трет-Бутиловый эфир [1-(трет-бутилкарбамоилгидроксиметил)циклобутил]карбаминовой кислоты (ВВ 1-с) ВВ 1-b (1,75 г, 8,78 ммоль) растворяли в CH2Cl2 (18 мл) и охлаждали на ледяной бане в атмосфере инертного газа. Добавляли пиридин (2,85 мл) с последующим добавлением трет-бутилизоцианида (1,50 мл, 13,3 ммоль). Затем по каплям добавляли трифторуксусную кислоту (1,35 мл, 17,5 ммоль) на протяжении 30 мин. Желтый раствор перемешивали при к.т. на протяжении ночи. Смесь концентрировали,разбавляли EtOAc (100 мл) и промывали последовательно 1 н НС 1 (50 мл), насыщенным NaHCO3 (50 мл) и насыщенным NaCl (250 мл), сушили (Na2SO4) и концентрировали в вакууме. Полученный неочищенный продукт обрабатывали ТГФ (2,5 мл) и 1M LiOH в смеси 3/1 МеОН-вода (2,5 мл) при к.т. Анализ проводили ТСХ (3/1 петролейный эфир-EtOAc). После времени реакции 45 мин добавляли 1 н HCl (2,5 мл), воду (10 мл) и EtOAc (20 мл) и слои разделяли. Органическую фазу промывали насыщенным NaHCO3 (20 мл) и затем насыщенным NaCl (220 мл), сушили (Na2SO4) и концентрировали. Флэшхроматография (75 г диоксида кремния, смесь 5/1-1/1 петролейный эфир:EtOAc) давала белое твердое вещество (2,36 г, 89%). Стадия d) (1-трет-Бутоксикарбониламиноциклобутил)гидроксиуксусная кислота (ВВ 1)BB1-c (1,30 г, 4,33 ммоль) кипятили с обратным холодильником с 6 н HCl (40 мл) до завершения гидролиза амида, проводя мониторинг гидролиза с помощью ЖХ-МС. Смесь упаривали, упаривая несколько раз вместе с водой. К остатку добавляли 1M NaOH (15 мл) и основный раствор перемешивали в вакууме в течение 15 мин. Добавляли Boc2O (1,92 г, 8,80 ммоль) в диоксане (10 мл), поддерживая рН 1011, и смесь перемешивали при к.т. на протяжении ночи. Смесь разбавляли водой (50 мл), подкисляли 1 нHCl до рН 3 на ледяной бане и затем экстрагировали EtOAc (250 мл, затем 30 мл). Органическую фазу промывали насыщенным NaCl (50 мл), сушили (Na2SO4) и упаривали, получая при этом неочищенный блок структуры ВВ 1, Р 1 (0,649 г). 1H ЯМР (400 МГц, d6-ДМСО)6,88 (ушир.с, 1H), 4,15 (с, 1H), 2,40 (ушир.м, 2H), 1,98 (ушир.м, 2 Н),1,80 (ушир.м, 2 Н), 1,35 (с, 9 Н); МС ES+ m/z 146 (100%), 190 (50%). Получение блока структуры 2, альтернативного блоку структуры P1 (BB2) Стадия а) 1-Бром-3-хлорпропан-2-илокси)метил)бензол (ВВ 2-а) К перемешиваемой смеси бензилбромида (185 г, 1,08 моль) и хлорида ртути (1,5 г) добавляли эпихлоргидрин (100 г, 1,08 моль). Реакционную смесь нагревали в течение 12 ч при 100 С. Анализ ТСХ подтвердил образование продукта. Продукт выделяли из темно-коричневой реакционной смеси колоночной хроматографией с применением петролейного эфира в качестве элюента. Система для ТСХ: петролейный эфир:этилацетат (9:1), Rf=0,7. Выход 148 г, 51%. Стадия b) Диэтиловый эфир 3-бензилоксициклобутан-1,1-дикарбоновой кислоты (ВВ 2-b) К перемешиваемой суспензии гидрида натрия (22,5 г, 0,562 моль) в 800 мл сухого диоксана по каплям добавляли диэтилмалонат (90 г, 0,562 моль) на протяжении 20 мин. После того, как это добавление было завершено, по каплям на протяжении 20 мин добавляли ВВ 2-а (148 г, 0,56 моль). Смесь затем кипятили с обратным холодильником в течение 24 ч. После охлаждения до комнатной температуры к смеси добавляли гидрид натрия (22,5 г, 0,562 моль) в небольшом количестве диоксана (20 мл) и кипячение с обратным холодильником продолжали в течение дополнительных 48 ч. Растворитель частично удаляли при пониженном давлении и смесь обрабатывали 800 мл воды. Эту смесь затем экстрагировали этилаце- 14019277 татом (500 мл 3), экстракты сушили (Na2SO4) и концентрировали в вакууме и остаток очищали колоночной хроматографией с применением смеси петролейный эфир:этилацетат (10%), которая давала указанное в заголовке соединение. Система для ТСХ: петролейный эфир:этилацетат (9:1), Rf=0,3. Выход 100 г, 58%. Стадия с) Диэтил-3-гидроксициклобутан-1,1-дикарбоксилат (ВВ 2-С) К раствору соединения ВВ 2-b (40 г) в EtOH (500 мл) добавляли 10% палладий на угле (4 г) и смесь гидрировали в течение 3,5 ч при 50 фунтов/кв. дюйм (344738 Па) и при комнатной температуре. Катализатор удаляли фильтрованием, промывали этилацетатом, EtOH, и растворитель затем удаляли при пониженном давлении. Остаток очищали хроматографией на силикагеле с применением смеси гексан/этилацетат в качестве элюента, получая при этом указанное в заголовке соединение. Система для ТСХ: петролейный эфир:этилацетат (9:1), Rf=0,3. Выход 18 г, 64%. Стадия d) Диэтил-3-оксоциклобутан-1,1-дикарбоксилат (ВВ 2-d) К раствору соединения ВВ 2-с (18 г, 0,0833 моль) в DCM (200 мл) добавляли РСС (37 г, 0,176 моль) и смесь перемешивали в течение 4 ч при комнатной температуре. Раствор фильтровали через колонку силикагеля и остаток промывали смесью DCM/MeOH, 98/2, и затем фильтровали через аналогичную колонку. Объединенные фракции упаривали при пониженном давлении, получая при этом требуемое соединение (11 г, 62%). Стадия е) Диэтил-3,3-дифторциклобутан-1,1-дикарбоксилат (ВВ 2-е) К охлажденному раствору соединения BB2-d (11 г, 0,0513 моль) в сухом DCM (150 мл) добавляли по каплям раствор DAST (18,72 г, 0,116 моль) и смесь перемешивали при комнатной температуре на протяжении ночи. Смесь добавляли к ледяной воде и экстрагировали три раза DCM. Раствор сушили сульфатом натрия и упаривали при пониженном давлении. Остаток очищали хроматографией на силикагеле с применением смеси гексан/этилацетат в качестве элюента, получая при этом указанное в заголовке соединение (7,7 г, 64%). Стадия f) 1-(Этоксикарбонил)-3,3-дифторциклобутанкарбоновая кислота (BB2-f) Соединение ВВ 2-е (7,7 г, 0,0325 моль) растворяли в охлажденном льдом 0,5 M растворе (30 мл) гидроксида калия в этаноле и воде (6 мл). Смесь перемешивали при комнатной температуре на протяжении ночи. Добавляли воду и большую часть этанола удаляли при пониженном давлении. Смесь подкисляли 2 М HCl и экстрагировали три раза этилацетатом. Органическую фазу сушили сульфатом натрия и упаривали при пониженном давлении, получая при этом требуемое соединение (5,8 г, 86%). Стадия g) Этил-1-(трет-бутоксикарбониламино)-3,3-дифторциклобутанкарбоксилат (ВВ 2-g) К раствору соединения BB2-f (5,8 г, 0,0273 моль) в сухом диоксане (100 мл) добавляли трет-бутанол(24,4 мл), DPPA (7,87 г, 0,027 моль) и TEA (2,87 г, 0,0284 моль) и смесь кипятили с обратным холодильником в течение 5 ч. Добавляли этилацетат (приблизительно 200 мл) и органическую фазу промывали дважды 5% лимонной кислотой и насыщенным гидрокарбонатом натрия. Раствор сушили и упаривали при пониженном давлении. Требуемый продукт выделяли хроматографией на силикагеле с применением смеси гексан/этилацетат (4 г, 51,4%). Стадия h) трет-Бутил-3,3-дифтор-1-(гидроксиметил)циклобутилкарбамат (BB2-h) К охлажденному льдом раствору соединения ВВ 2-g (4 г, 0,0143 моль) в сухом ТГФ (100 мл) медленно добавляли 2 M раствор борогидрида лития (30 мл) и смеси давали возможность нагреться до комнатной температуры. Смесь перемешивали в течение 3 ч при комнатной температуре. Добавляли ледяную воду и 5% лимонную кислоту и смесь экстрагировали три раза DCM. Органическую фазу сушили(Na2SO4), фильтровали и упаривали при пониженном давлении, что давало указанное в заголовке соединение (3,1 г, 91%). Стадия i) трет-Бутил-3,3-дифтор-1-формилциклобутилкарбамат (BB2-i) К раствору соединения BB2-h (3,1 г, 0,0130 моль) в сухом DCM (100 мл) добавляли периодинан Десс-Мартина (19,9 г, 0,0470 моль) и смесь перемешивали в течение 3 ч при комнатной температуре. Добавляли этилацетат (200 мл) и органическую фазу промывали дважды 10% раствором тиосульфата натрия, дважды 0,5 М раствором NaOH и насыщенным раствором соли. Органическую фазу сушили и упаривали при пониженном давлении. Остаток очищали хроматографией на силикагеле с применением смеси гексан/этилацетат в качестве элюента, получая при этом указанное в заголовке соединение (2,7 г,87%). Стадияj) трет-Бутил-1-(2-(трет-бутиламино)-1-гидрокси-2-оксоэтил)-3,3-дифторциклобутилкарбамат (BB2-j) К охлажденному льдом раствору соединения BB2-i (1,5 г, 0,0064 моль) в сухом DCM (100 мл) добавляли трет-бутилизоцианат (0,81 г, 0,009 моль) и пиридин (2,04 г, 0,027 моль). На протяжении периода 10 мин добавляли трифторуксусную кислоту (1,58 г, 0,015 моль). Смесь перемешивали в течение 5 ч при комнатной температуре. Добавляли этилацетат, и органическую фазу промывали дважды 5% лимонной кислотой и насыщенным раствором соли. Органическую фазу упаривали и растворяли в диоксане (50 мл). Добавляли 1M раствор LiOH (100 мл) и смесь перемешивали на протяжении ночи при комнатной температуре. Добавляли 5% лимонную кислоту и смесь экстрагировали три раза этилацетатом. Органическую фазу промывали насыщенным раствором соли, сушили (Na2SO4), фильтровали и упаривали при пониженном давлении. Продукт очищали хроматографией на силикагеле с применением смеси гексан/этилацетат в качестве элюента (1,0 г, 46%). Стадия k) 2-(1-трет-Бутоксикарбониламино)-3,3-дифторциклобутил)-2-гидроксиуксусная кислота(ВВ 2) Соединение BB2-j (1 г) растворяли в 6 н HCl (40 мл) и кипятили с обратным холодильником в течение 24 ч, после чего ТСХ показала, что реакция достигла завершения. Реакционную смесь концентрировали в вакууме, и остаток растворяли в смеси ТГФ:Н 2 О (7:3, 50 мл), добавляли TEA (1,8 мл, 0,012 моль) и Вос-ангидрид (2,6 г, 0,012 моль). Смесь перемешивали при к.т. в течение 8 ч, когда ТСХ подтвердила,что реакция достигла завершения. Реакционную смесь концентрировали в вакууме и остаток очищали колоночной хроматографией с применением 5% метанола в хлороформе, которая давала указанное в заголовке соединение (0,6 г, 72%). 1H ЯМР (400 МГц, d6-ДМСО)7,30 (ушир.с, 1 Н), 4,11 (с, 1 Н), 2,90 (ушир.м, 2 Н), 2,61 (ушир.м, 2 Н),1,35 (с, 9 Н); МС ES+ m/z 281 (100%). Получение блока структуры 3 - альтернативного блоку структуры Р 1 (BB3)(0,04 г, 0,085 ммоль) нагревали в течение 12 ч при 150 С. Неочищенный продукт очищали колоночной хроматографией (силикагель 60-120 меш, элюент 1% EtOAc в петролейном эфире), которая давала указанное в заголовке соединение в виде вязкой жидкости (50 г, выход 70%). Стадия b) Диэтил-3-(бензилокси)циклобутан-1,1-дикарбоксилат (ВВ 3-b) В трехгорлую колбу, снабженную мешалкой, капельной воронкой и парциальным конденсатором горячего орошения, помещали NaH (4,6 г, 0,192 моль) в сухом диоксане (150 мл). К этой перемешиваемой реакционной смеси по каплям на протяжении 30 мин добавляли диэтилмалонат (30,75 г, 0,192 моль). После завершения добавления по каплям на протяжении периода 30 мин добавляли соединение ВВ 3-а(50 г, 0,19 моль). Реакционную смесь кипятили с обратным холодильником в течение 24 ч. После охлаждения до комнатной температуры к реакционной смеси добавляли NaH (4,6 г, 0,192 моль) и сухой диоксан (40 мл) и смесь далее кипятили с обратным холодильником в течение еще 48 ч. Растворитель частично удаляли при пониженном давлении и смесь обрабатывали водой (150 мл). Продукт экстрагировали диэтиловым эфиром (3100 мл), органический слой промывали насыщенным раствором соли и сушили над безводным Na2SO4. Раствор концентрировали в вакууме, и сырой продукт очищали колоночной хроматографией (силикагель 60-120 меш, элюент 2% EtOAc в петролейном эфире), которая давала указанное в заголовке соединение в виде вязкой жидкости (33 г, выход 57%). Система для ТСХ: 15% EtOAc в петролейном эфире, Rf=0,5. Стадия с) Диэтил-3-гидроксициклобутан-1,1-дикарбоксилат (ВВ 3-с) К раствору соединения ВВ 3-b (33 г, 0,108 моль) в EtOH (300 мл) добавляли 10% палладий на угле(10 г) и смесь гидрировали в течение 48 ч при давлении 50 фунтов/кв.дюйм (344738 Па) и комнатной температуре. Катализатор удаляли фильтрованием через слой целита и тщательно промывали EtOAc. Растворитель удаляли при пониженном давлении. Продукт очищали хроматографией на силикагеле (силикагель 60-120 меш, элюент 20% EtOAc в петролейном эфире), которая давала продукт 3 в виде вязкой жидкости (12 г, выход 51%). Система для ТСХ: 30% EtOAc в петролейном эфире, Rf=0,3 г. Стадия d) Диэтил-3-фторциклобутан-1,1-дикарбоксилат (ВВ 3-d) Соединение ВВ 3-с (0,8 г, 0,0037 моль) растворяли в сухом DCM (16 мл) и охлаждали до 0 С. К холодному раствору по каплям добавляли DAST (1,8 г, 0,011 моль). Реакционную смесь нагревали до комнатной температуры и перемешивали в течение 12 ч. Реакционную смесь гасили холодным насыщенным раствором NaHCO3. Сырой продукт экстрагировали DCM (100 мл). Органический слой промывали 10% раствором NaHCO3, водой и затем насыщенным раствором соли и сушили над безводным Na2SO4. Рас- 16019277 твор концентрировали в вакууме и сырой продукт очищали колоночной хроматографией (силикагель 60120 меш, элюент 1-2% EtOAc в петролейном эфире), которая давала указанное в заголовке соединение в виде бледно-желтой жидкости (460 мг, выход 57%). Система для ТСХ: 10% EtOAc в петролейном эфире,Rf=0,4. Стадия е) 1-(Этоксикарбонил)-3-фторциклобутанкарбоновая кислота (ВВ 3-е) Соединение BB3-d (0,46 г, 0,0021 моль) растворяли в охлажденном льдом 0,5 М растворе гидроксида калия в EtOH (4,2 мл) и воде (1,4 мл). Смесь перемешивали при комнатной температуре на протяжении ночи. Добавляли воду и большую часть этанола удаляли при пониженном давлении. Смесь подкисляли 2 н HCl и экстрагировали EtOAc (350 мл). Органическую фазу сушили над безводным Na2SO4. Раствор концентрировали в вакууме, получая при этом неочищенное указанное в заголовке соединение(0,35 г, неочищенный продукт), которое применяли как таковое для следующей стадии. Система для ТСХ: 50% EtOAc в петролейном эфире, Rf=0,3. Стадия f) Этил-1-(трет-бутоксикарбониламино)-3-фторциклобутанкарбоксилат (ВВ 3-f) К раствору соединения ВВ 3-е (0,35 г, 0,0018 моль) в сухом диоксане (6 мл) добавляли трет-бутанол(1,8 мл), дифенилфосфорилазид (0,56 г, 0,002 моль) и триэтиламин (0,2 г, 0,002 моль) и смесь кипятили с обратным холодильником в течение 5 ч. После завершения реакции к реакционной смеси добавлялиEtOAc (60 мл) и органический слой промывали 5% лимонной кислотой (220 мл) с последующим промыванием насыщенным NaHCO3 (50 мл). Органический раствор упаривали при пониженном давлении. К остатку добавляли EtOAc (100 мл), и органический слой промывали насыщенным раствором соли и сушили над безводным Na2SO4. Раствор концентрировали в вакууме, и сырой продукт очищали колоночной хроматографией (силикагель 60-120 меш, элюент 5-10% EtOAc в петролейном эфире), которая давала указанное в заголовке соединение в виде белых кристаллов (0,27 г, выход 56%). Система для ТСХ: 20% EtOAc в петролейном эфире, Rf=0,4. Стадия g) трет-Бутил-3-фтор-1-(гидроксиметил)циклобутилкарбамат (ВВ 3-g) К охлажденному льдом раствору соединения BB3-f (0,27 г, 0,001 моль) в сухом ТГФ (10 мл) медленно добавляли 2 M раствор литийборогидрида (2 мл, 0,004 моль) и смеси давали возможность нагреться до комнатной температуры. Смесь перемешивали в течение 3 ч при комнатной температуре. Реакционную смесь гасили ледяной водой (2 мл) и 5% лимонной кислотой (5 мл) и сырой продукт экстрагировали DCM (250 мл). Органический слой промывали насыщенным раствором соли и сушили над безводным Na2SO4. Раствор концентрировали в вакууме, и сырой продукт очищали колоночной хроматографией (силикагель 60-120 меш, элюент 15-18% EtOAc в петролейном эфире), которая давала указанное в заголовке соединение в виде белого твердого вещества (90 мг, выход 39%). Система для ТСХ: 50%EtOAc в петролейном эфире, Rf=0,5. Стадия h) трет-Бутил-3-фтор-1-формилциклобутилкарбамат (BB3-h) К дегазированному раствору соединения ВВ 3-g (90 мг, 0,0004 моль) в сухом DCM (4,5 мл) добавляли периодинан Десс-Мартина (0,21 г, 0,0005 моль) и смесь перемешивали в течение 3 ч при комнатной температуре. Добавляли EtOAc (30 мл) и органический слой промывали 10% раствором тиосульфата натрия (210 мл), 0,5M NaOH (20 мл) и насыщенным раствором соли. Органический слой сушили над безводным Na2SO4. Раствор концентрировали в вакууме, и сырой продукт очищали колоночной хроматографией (силикагель 60-120 меш, элюент 10-15 EtOAc в петролейном эфире), которая давала указанное в заголовке соединение в виде белого кристаллического твердого вещества (75 мг, выход 87%). Система для ТСХ: 20% EtOAc в петролейном эфире, Rf=0,4. Стадия i) трет-Бутил-1-(2-(трет-бутиламино)-1-гидрокси-2-оксоэтил)-3-фторциклобутилкарбамат(ВВ 3-i) К охлажденному льдом раствору BB3-h (1,3 г, 0,0059 моль) в сухом DCM (25 мл) добавляли третбутилизоцианид (0,75 г, 0,0089 моль) и сухой пиридин (2,6 мл). На протяжении периода десяти минут добавляли трифторуксусную кислоту (0,9 мл, 0,0118 моль), поддерживая температуру 0 С. Реакционную смесь медленно нагревали до комнатной температуры и перемешивали в течение 16 ч. Добавляли EtOAc(50 мл), и органическую фазу промывали дважды 5% лимонной кислотой и насыщенным раствором соли. Органическую фазу упаривали, и неочищенный продукт растворяли в ТГФ (25 мл). Добавляли 1M раствор LiOH в смеси MeOH-Н 2 О (3:2, об./об.) (2,6 мл) и смесь перемешивали в течение 2 ч при комнатной температуре. Реакционную смесь гасили 5% лимонной кислотой и смесь экстрагировали этилацетатом (225 мл). Органический слой промывали насыщенным раствором соли и сушили над безводнымNa2SO4. Растворитель выпаривали в вакууме, получая при этом указанное в заголовке соединение, которое было достаточно чистым для применения в следующей стадии (1,6 г, выход 84%). Система для ТСХ: 20% EtOAc в петролейном эфире, Rf=0,3. Стадия j) 2-(1-(трет-Бутоксикарбониламино)-3-фторциклобутил)-2-гидроксиуксусная кислота (ВВ 3) Соединение ВВ 3-i (1,6 г, 0,005 моль) кипятили с обратным холодильником с 6 н HCl (60 мл) в течение 16 ч до завершения гидролиза амида. Растворитель выпаривали при пониженном давлении и выпаривали вместе с водой несколько раз. Продукт растворяли в смеси ТГФ:Н 2 О (7:3, об./об., 50 мл), раствор охлаждали до 0 С и добавляли Et3N (2,1 мл, 0,015 моль) с последующим добавлением ди-трет- 17019277 бутилдикарбоната (2,18 г, 0,01 моль). Смесь перемешивали при комнатной температуре на протяжении ночи (мониторинг рН проводили с регулярными интервалами и рН поддерживали 11 на всем протяжении реакции). Реакционную смесь нейтрализовали 1 н HCl и продукт экстрагировали EtOAc (250 мл). Органический слой промывали насыщенным раствором соли и сушили над безводным Na2SO4. Растворитель выпаривали при пониженном давлении с последующей очисткой колоночной хроматографией (силикагель 60-120 меш, элюент 5% МеОН в CHCl3), которая давала указанный в заголовке блок структуры Р 1 в виде твердого вещества (0,65 г, выход 50%). Система для ТСХ: 30% МеОН в CHCl3, Rf=0,3. 1(ушир.м, 5 Н), 1,36 (с, 9 Н); МС ES+ m/z 262 (100%). Блок структуры 4 - примерный блок структуры Р 1-основная боковая цепь (ВВ 4)(получен восстановлением этил-1-(трет-бутилокси)карбонил]амино]-3 гидроксициклобутан-1-карбоксилата, как описано в J. Med. Chem., 1990, 33(10), 2905-2915) и протоновый пористый материал (N,N,N',N'-тетраметилнафталин-1,8-диамин) (1,63 г, 6,04 ммоль) растворяли в DCM(18 мл), охлаждали до 0 С, сразу при энергичном перемешивании добавляли триметилоксонийбортетрафторид (447 мг, 3,02 ммоль) в виде твердого вещества. Реакционную смесь перемешивали в течение 3 ч и разбавляли DCM (50 мл) и при энергичном перемешивании добавляли 20 мл насыщенного раствора соли. Органическую фазу промывали бикарбонатом натрия, насыщенным раствором соли, сушили над сульфатом натрия, упаривали и очищали на короткой колонке диоксида кремния (DCM в качестве элюента). Образовавшийся продукт растворяли в 5 мл ТГФ, добавляли 4,5 мл 1 М раствор фторида тетрабутиламмония в ТГФ (1 М), смесь перемешивали при комнатной температуре в течение 4,5 ч. При проведении мониторинга ТСХ раствор упаривали на диоксиде кремния, очищали на диоксиде кремния (от смеси 1:1 EtOAc-гексан до неразбавленного EtOAc), получая при этом указанное в заголовке соединение(251 мг, 72%). ЖХ/МС 232 (M+1). Стадия b) трет-Бутил-1-формил-3-метоксициклобутилкарбамат (ВВ 4-b) Спирт ВВ 4-а растворяли в 20 мл DCM, сразу добавляли периодинан Десс-Мартина. Смесь перемешивали в течение 2,5 ч, разбавляли 50 мл DCM и добавляли 20 мл 10% Na2S2O3, перемешивали, промывали бикарбонатом натрия, насыщенным раствором соли, сушили над сульфатом натрия. Очищали на диоксиде кремния (от смеси 1:1 EtOAc-гексан до неразбавленного EtOAc), получая при этом указанное в заголовке соединение (500 мг, 59%). Стадия с) трет-Бутил-1-(2-(циклопропиламино)-1-гидрокси-2-оксоэтил)-3-метоксициклобутилкарбамат (ВВ 4) Альдегид ВВ 4-b (498 мг, 1,56 ммоль) растворяли в сухом DCM (8 мл). В условиях перемешивания добавляли пиридин (0,52 мл) с последующим добавлением циклопропилизонитрила. Реакционную смесь помещали на ледяную баню и по каплям в течение 20 мин добавляли 0,25 мл TFA. Реакционную смесь перемешивали на протяжении ночи. Промывали 1 M HCl, бикарбонатом натрия, насыщенным раствором соли, сушили над сульфатом натрия, упаривали, растворяли в диоксане и перемешивали с гидроксидом лития на протяжении ночи и нейтрализовали лимонной кислотой. Из образовавшегося раствора продукт экстрагировали EtOAc. Продукт очищали на диоксиде кремния (EtOAc-гексан, от 1:3 до 1:1), получая при этом 263 мг указанного в заголовке соединения (54%). ЖХ/МС 314 (M+1). Стадия а) трет-Бутил-1-(1-гидрокси-2-(1-метил-1 Н-пиразол-3-иламино)-2-оксоэтил)циклобутилкарбамат (1-а) 1-Метил-1 Н-пиразол-3-амин (158 мг, 1,63 ммоль) и DIEA (1,08 мл, 6,52 ммоль) добавляли к раствору 2-(1-(трет-бутоксикарбониламино)циклобутил)-2-гидроксиуксусной кислоты (400 мг, 1,63 ммоль) в ДМФА (20 мл). Раствор охлаждали до 0 С и спустя 10 мин добавляли HATU (620 мг, 1,63 ммоль). После выдерживания приблизительно в течение 2 ч при к.т. анализ ЖХ-МС показал присутствие продукта и отсутствие исходного соединения, и растворитель удаляли роторным испарением. Неочищенный продукт растворяли в 40 мл EtOAc и промывали 25 мл насыщ. NaHCO3 (водный). Органическую фазу сушили Na2SO4, фильтровали и упаривали досуха. Сырой продукт очищали на колонке с 25 г диоксида кремния на Biotage Flashmaster с применением элюирования с градиентом смеси гептан:этилацетат, 1:1, что давало указанное в заголовке соединение в виде белого твердого вещества (488 мг, выход 92%).[М+Н]+=325. Стадия b) трет-Бутил-(2S)-1-(1-(1-гидрокси-2-(1-метил-1 Н-пиразол-3-иламино)-2-оксоэтил)циклобутиламино)-3-(1-метилциклобутил)-1-оксопропан-2-илкарбамат (1-b) Предварительно приготовленную смесь 9:1 метанола и ацетилхлорида (10 мл) добавляли к соединению 1-а (244 мг, 0,752 ммоль) и раствор перемешивали в течение 6 ч. Растворитель затем удаляли роторным испарением и сырой продукт выдерживали в высоком вакууме на протяжении ночи. Гидрохлоридную соль образовавшегося продукта незащищенный блок структуры Р 1-основная боковая цепь растворяли в ДМФА (15 мл) и к раствору добавляли (S)-2-(трет-бутоксикарбониламино)-3-(1 метилциклобутил)пропановую кислоту (193 мг, 7,52 ммоль) и DIEA. Раствор охлаждали до 0 С и спустя 10 мин добавляли HATU (620 мг, 1,63 ммоль) и раствору давали возможность достичь к.т. После приблизительно 2 ч растворитель удаляли роторным испарением. Сырой продукт растворяли в 40 мл EtOAc и промывали 25 мл насыщ. NaHCO3 (водный). Органическую фазу сушили Na2SO4, фильтровали и упаривали досуха. Сырой продукт очищали на колонке с 25 г диоксида кремния с применением элюирования с градиентом смесью гептан:этилацетат, что давало указанное в заголовке соединение (235 мг, 67%).[М+Н]+=464. Стадия с) N-2S)-1-(1-(1-Гидрокси-2-(1-метил-1 Н-пиразол-3-иламино)-2-оксоэтил)циклобутиламино)-3-(1-метилциклобутил)-1-оксопропан-2-ил)-4-(фенилсульфонамидо)бензамид (1-c) Предварительно приготовленную смесь 9:1 метанола и ацетилхлорида (8 мл) добавляли к соединению 1-b (100 мг, 0,216 ммоль), и раствор перемешивали в течение 6 ч. Растворитель затем удаляли роторным испарением, и сырой продукт выдерживали в высоком вакууме на протяжении ночи. Гидрохлоридную соль образовавшегося незащищенного продукта Р 2-Р 1-основная боковая цепь растворяли в ДМФА (15 мл), к раствору добавляли 4-(фенилсульфонамидо)бензойную кислоту (193 мг, 7,52 ммоль) иDIEA и раствор охлаждали до 0 С. Спустя 10 мин добавляли HATU (620 мг, 1,63 ммоль), и раствору давали возможность достичь к.т. После приблизительно 2 ч растворитель удаляли роторным испарением,сырой продукт растворяли в EtOAc (40 мл) и промывали насыщ. NaHCO3 (водный) (25 мл). Органическую фазу сушили Na2SO4, фильтровали и упаривали досуха. Сырой продукт очищали на колонке с 25 г диоксида кремния на Biotage Flashmaster с применением элюирования с градиентом смесьюDCM:метанол, что давало указанное в заголовке соединение (112 мг, 83%). [М+Н]+ = 623. Стадия d) (S)-N-(1-(1-(2-(1-Метил-1 Н-пиразол-3-иламино)-2-оксоацетил)циклобутиламино)-3-(1 метилциклобутил)-1-оксопропан-2-ил)-4-(фенилсульфонамидо)бензамид (1) Соединение 1-e (112 мг, 0,180 ммоль) растворяли в дихлорметане (15 мл) и к раствору добавляли периодинан Десс-Мартина (114 мг, 0,270 ммоль). Мутную реакционную смесь перемешивали в течение 4 ч. После этого добавляли 10% Na2S2O3 (водн.) (15 мл) и 10% NaHCO3 (водн.) (15 мл) и раствор перемешивали до тех пор, пока он не становился прозрачным. Органическую фазу отделяли от водной фазы разделителем фаз. Органический растворитель удаляли роторным испарением и сырой продукт растворяли в небольшом количестве ацетонитрила и H2O для очистки на полупрепаративной системе ЖХ-MC. Очистку проводили на колонке XBridge phenyl, 5 мкм с применением подвижной фазы А (H2O: ацетонитрил, 90:10, 10 мМ NH4Ac) и В (H2O:ацетонитрил, 10:90, 10 мМ NH4Ac), начиная от 35-50% В. Указанный в заголовке продукт очищали и сушили замораживанием, получая при этом белое твердое вещество с выходом 28% (31 мг). [M+Н]+=621. 1 Н ЯМР (CDCl3, 400 МГц) 1,15 (с, 3 Н), 1,48-2,17 (м, 10 Н), 2,19-2,37 (м, 2 Н), 2,70 (ушир.с, 1 Н), 2,83(ушир.с, 1 Н), 9,53 (ушир.с, 1 Н). Примеры 2-49. Соединения, показанные ниже в таблицах, получали аналогично методике, описанной в примере 1,с применением подходящих R1aR1b-аминов и блоков структуры P1, P2 и Р 3 с последующим окислением по Десс-Мартину до конечного продукта -кетоамида. Таблица 1 Морфолинкарбонилхлорид применяли для введения блока структуры Р 3 на стадии d. Таблица 2 Морфолинкарбонилхлорид применяли для введения блока структуры Р 3 на стадии d. Морфолинкарбонилхлорид применяли для введения блока структуры Р 3 на стадии d. Таблица 4 Морфолинкарбонилхлорид применяли для введения блока структуры Р 3 на стадии d. Стереохимия при хиральном центре, к которому присоединены R2aR2b, не определена. Стадия а) трет-Бутиловый эфир [1-(циклопропилкарбамоилгидроксиметил)-3,3-дифторциклобутил]карбаминовой кислоты (50-а) Циклопропиламин (1 экв., 1,63 ммоль) и DIEA (4 экв., 6,52 ммоль) добавляли к раствору ВВ 2 (1 экв., 1,63 ммоль) в ДМФА (8 мл/ммоль). Раствор охлаждали до 0 С и спустя 10 мин добавляли HATU (1 экв., 1,63 ммоль). После выдерживания приблизительно 2 ч при к.т. анализ ЖХ-МС показал присутствие продукта и отсутствие исходного соединения, и растворитель удаляли роторным испарением. Сырой продукт растворяли в 40 мл EtOAc и промывали 25 мл насыщ. NaHCO3 (водн.). Органическую фазу сушили Na2SO4, фильтровали и упаривали досуха. Сырой продукт очищали на колонке с 25 г диоксида кремния на Biotage Flashmaster, получая при этом указанный в заголовке продукт в виде белого твердого вещества (92%). Стадия[1-1-[гидрокси-(1-метил-1 Н-пиразол-3-илкарбамоил)метил]циклобутилкарбамоил-2-(1-метилциклопентил)этил]карбаминовой кислоты (50-b) Соединение 50-а (50 мг, 0,156 ммоль) растворяли в растворе метанол:ацетилхлорид, 9:1 (1,5 мл) при 0 С. Раствор перемешивали при к.т. в течение 16 ч, затем концентрировали и дважды упаривали вместе сDCM. Полученный остаток растворяли в безводном ДМФА (1 мл) и затем добавляли при 0 С к холодному раствору (S)-2-(трет-бутоксикарбониламино)-3-(1-фторциклопентил)пропановой кислоты (получена,как описано в примере 8 WO 2006/064286) (45 мг, 0,165 ммоль) и HATU (63 мг, 0,165 ммоль) в сухом ДМФА (2 мл). Добавляли DIEA (130 мкл, 0,75 ммоль) и реакционную смесь перемешивали при 0 С в течение 30 мин, затем при к.т. в течение 2 ч. Раствор концентрировали в вакууме, остаток растворяли вDCM (3 мл) и вводили в колонку с диоксидом кремния (10 г). Соединение очищали флэшхроматографией (гептан:этилацетат, 75:25-25:75), которая давала указанное в заголовке соединение (74 мг, 95%) в виде смеси диастереомеров. МС m/z 478,2 (M+Н)+. Стадия-Гидроксиамид 50-b окисляли согласно способу, описанному в примере 1, стадия d. Очистка флэш-хроматографией давала указанное в заголовке соединение в виде смеси диастереомеров. МС m/z 476,2 [М+Н]+. Стадия d) N-[1-(1-Циклопропиламинооксалил-3,3-дифторциклобутилкарбамоил)-2-(1-фторциклопентил)этил]-3-фтор-4-гидроксибензамид (50) Карбамат 50-с (71 мг, 0,15 ммоль) растворяли в растворе метанол:ацетилхлорид, 9:1, (1,5 мл) при 0 С. Раствор перемешивали при комнатной температуре в течение 16 ч, затем концентрировали и упаривали дважды вместе с DCM. Сырой продукт растворяли в сухом ДМФА (1 мл) при 0 С и затем добавляли к холодному раствору 4-гидрокси-3-фторбензойной кислоты (25 мг, 0,165 ммоль) и HATU (63 мг,0,165 ммоль) в сухом ДМФА (2 мл). Добавляли DIEA (130 мкл, 0,75 ммоль) и реакционную смесь перемешивали при 0 С в течение 30 мин, затем при к.т. в течение 2 ч. Раствор концентрировали в вакууме и остаток растворяли в DCM (3 мл) и очищали флэш-хроматографией на колонке с диоксидом кремния (10 г) с элюированием (DCM:MeOH, 100:0-92:8) и затем препаративной хроматографией (градиент 20-70%,подвижная фаза: смесь ацетонитрил-вода, 1% NH4OH), получая при этом указанное в заголовке соединение (4,2 мг, 6%). МС m/z 514,1494 (M-HF)+. Чистота 91% при определении аналитической ЖХ-МС. Соединения, показанные в таблице ниже, получали аналогично методике, указанной либо в способе А, либо в способе В, с применением подходящих R1aR1b-аминов, блоков структур Р 1 и Р 2 и Р 3-кислот. Морфолинкарбонилхлорид применяли для введения Р 3-части на стадии d способа А. РуВор применяли в качестве агента сочетания на стадии b способа А. 3 Вос-группу удаляли с применением 4 M раствора HCl-диоксан на стадии d способа В. 4R R-амин получали восстановлением соответствующего нитросоединения. 7 РуВор применяли в качестве агента сочетания на стадии а способа А. 8 Вос-группу удаляли с применением смеси TFA/вода/триизопропилсилан, 9,5:0,5:0,5, на стадии с способа А. 9 трет-Бутилдифенилсилилгидроксизащищенное производное Р 3-кислоты применяли на стадии d способа В. Требуемое соединение получали десилилированием, проводимым обработкой TBAF в ТГФ. 10 Стереохимию у хирального центра, к которому присоединены R2aR2b, не определяли. 2 Соль метиловый эфир 2-амино-3-(1-метилциклопентил)пропионовой кислотыHCl (123-а)(S)-2-(трет-Бутоксикарбониламино)-3-(1-метилциклопентил)пропановую кислоту (272 мг, 1 ммоль) растворяли в МеОН (1 мл). По каплям при комнатной температуре добавляли 4 М HCl (3 мл) в диоксане. Спустя приблизительно 3 ч растворитель удаляли роторным испарением и остаток выпаривали вместе с МеОН (2) для удаления избыточного HCl. Полученное соединение применяли на последующих стадиях без дополнительной очистки. Стадия b) Литиевая соль 3-(1-метилциклопентил)-2-[(пирролидин-1-карбонил)амино]пропионовой кислоты (123-b) Соединение 123-а (19 мг, 86 мкмоль) растворяли в ТГФ (1 мл) и добавляли триэтиламин (3 экв.) и пирролидин-1-карбонилхлорид (1 экв.). Реакционную смесь нагревали до 50 С в герметизированной трубке в течение 16 ч. Анализ ЖХ/МС показал 90% превращение. К реакционному раствору добавлялиEtOAc и органическую фазу промывали 0,1 M HCl (водн.) (3). Органический слой сушили (MgSO4),фильтровали и растворитель удаляли в вакууме. Образовавшийся сырой метиловый сложный эфир растворяли в ТГФ и добавляли 1 М LiOH в метаноле (3 экв.). Раствор перемешивали при комнатной температуре в течение 16 ч. Анализ ЖХ/МС показал полный гидролиз эфира, и растворитель удаляли в вакууме, получая при этом литиевую соль, которую применяли в следующей стадии без дополнительной очистки. Стадия мл), который предварительно перемешивали при комнатной температуре в течение 10 мин. Смесь перемешивали при комнатной температуре в течение 16 ч и затем к реакционной смеси добавляли DCM и органическую фазу промывали 0,1 М HCl (водн.) (2) и 10% NaHCO3 (водн.) (2). Органическую фазу сушили и концентрировали в вакууме, и остаток очищали препаративной ЖХ/МС. Полученный спирт снова растворяли в DCM (1,5 мл) и в виде одной порции при комнатной температуре добавляли периодинан Десс-Мартина (1,5 экв., мкмоль). Реакционную смесь перемешивали при комнатной температуре в течение 2 ч, после этого времени анализ ЖХ/МС показал полное окисление. Реакционную смесь разбавляли DCM и раствор промывали смесью 1:1 10% Na2S2O3 (водн.) и 10% NaHCO3 (водн.). Органический слой пропускали с элюированием через сепаратор с гидрофобной фазой и концентрировали в вакууме. Остаток очищали препаративной ЖХ/МС, получая при этом требуемое соединение (выход 5,1 мг,ЖХ/МС: tR=4,97 мин, 491,14 [М+Н]+). Биологические примеры Определение протеолитической каталитической активности катепсина К Общепринятые анализы катепсина К проводят с применением рекомбинантного фермента человека,такого как фермент, описанный в PDB. Стандарт ID BC016058; мРНК; HUM; 1699 ВР. Катепсин К homo sapiens DE (пикнодисостозис), мРНК (клон кДНК MGC: 23107DR SWISS-PROT; P43235. Рекомбинантный катепсин К можно экспрессировать в различных коммерчески доступных системах экспрессии, включающих системы Е. coli, Pichia и Baculovirus. Очищенный фермент активировали удалением пропоследовательности общепринятыми способами. Стандартные условия анализа для определения кинетических констант включали применение флуорогенного пептидного субстрата, обычно H-D-Ala-Leu-Lys-AMC, и такие константы определяли либо в 100 мМ Mes/Трис, рН 7,0, содержащей 1 мМ EDTA и 10 мМ 2-меркаптоэтанол, либо в 100 мМ фосфате Na, мМ EDTA, 0,1% ПЭГ 4000, рН 6,5, или 100 мМ ацетате Na, рН 5,5, содержащем 5 мМ EDTA и 20 мМ цистеин, в каждом случае необязательно с 1 М DTT в качестве стабилизатора. Применяемая концентрация фермента была 5 нМ. Исходный раствор субстрата получали как 10 мМ раствор в ДМСО. Скрининги проводили при фиксированной концентрации субстрата 60 мкМ и подробные кинетические исследования проводили с удвоенными разведениями субстрата от 250 мкМ. Общую концентрацию ДМСО при анализе поддерживали ниже 3%. Все анализы проводили при температуре окружающей среды. Мониторинг флуоресценции продукта (возбуждение при 390 нм, эмиссия при 460 нм) проводили флуоресцентным планшет-ридером Labsystems Fluoroskan Ascent. Кривые прогресса флуоресценции продукта генерировали на протяжении 15 мин после генерации продукта АМС. Определение Ki катепсина S В анализах применяли экспрессированный бакуловирусом катепсин S человека и флуоресцентный субстрат boc-Val-Leu-Lys-АМС, доступный от Bachem, в формате 384-луночного планшета, в котором можно испытывать 7 испытуемых соединений параллельно с позитивным контролем, содержащим в качестве компаратора известный ингибитор катепсина S. Разведения субстрата 280 мкл/лунку 12,5% ДМСО добавляют в ряды В-Н двух столбцов полипропиленового планшета с 96 глубокими лунками. 70 мкл/лунку субстрата добавляют в ряд А. 2250 мкл/лунку буфера для анализа(100 мМ фосфат Na, 100 мМ NaCl, рН 6,5) добавляют в ряд А, смешивают и вдвое разводят вдоль планшета до ряда Н. Разведения ингибиторов 100 мкл/лунку буфера для анализа добавляют к 2-5 и 7-12 столбцам 4 рядов полипропиленового планшета с 96 лунками с V-образным дном. 200 мкл/лунку буфера для анализа добавляют к столбцам 1 и 6. Первое испытуемое соединение, полученное в ДМСО, добавляют к лункам столбца 1 верхнего ряда,обычно при объеме для обеспечения величины, превышающей в 10-30 раз первоначально определенную приблизительную (ориентировочную) величину Ki. Приблизительную Ki вычисляют из предварительного опыта, в котором 10 мкл/лунку 1 мМ boc-VLK-AMC (разведение 1/10 10 мМ исходного раствора в ДМСО, разведенного в буфере для анализа) заливают в лунки рядов В-Н и 20 мкл/лунку в лунки ряда А 96-луночного планшета Microfluor. 2 мкл каждого 10 мМ испытуемого соединения добавляют в отдельную лунку ряда А, столбцы 1-10. В каждую лунку рядов В-Н добавляют 90 мкл буфера для анализа,содержащего 1 мМ DTT и 2 нМ катепсина S, и 180 мкл в лунки ряда А. Смешивают содержимое лунок ряда А с применением многоканальной пипетки и разводят в два раза до ряда G. Смешивают содержимое ряда Н и считывают данные во флуоресцентном спектрофотометре. Показания являются данными Prism,подогнанными для уравнения конкурентного ингибирования, при устанавлении S=100 мкМ и KM=100 мкМ для получения оценки Ki вплоть до максимума 100 мкМ. Второе испытуемое соединение добавляют к лункам столбца 6 верхнего ряда, третье соединение
МПК / Метки
МПК: C07D 231/12, A61K 31/343, A61P 37/00, A61P 11/00, A61P 17/06, A61K 31/5375, C07D 307/80, C07D 239/80, C07C 235/74, A61K 31/415, C07D 233/64, C07D 275/00, C07C 215/20, A61K 31/145, A61K 31/341, C07D 295/20, C07D 307/46, C07D 275/03, C07D 241/12, C07D 213/71, C07C 311/21, C07D 215/20, A61K 31/16, A61K 31/4965
Метки: цистеинпротеазы, ингибиторы
Код ссылки
<a href="https://eas.patents.su/30-19277-ingibitory-cisteinproteazy.html" rel="bookmark" title="База патентов Евразийского Союза">Ингибиторы цистеинпротеазы</a>
Ингибиторы цистеинпротеазы

Номер патента: 1937
Опубликовано: 22.10.2001
Авторы: Ло Кастро Стефен, Вебер Дэниел Ф., Ру Ю., Маркьюс Роберт В.
МПК: C07D 403/14, A61P 19/02, A61K 31/535...
Метки: ингибиторы, цистеинпротеазы
Формула / Реферат:
1. Соединения формулы где А представляет С(О) или СН(ОН); R1 представляет R2 представляет Н, C1-6-алкил, С3-6-циклоалкил-С0-6-алкил, Аr-С0-6-алкил, Het-C0-6-алкил, R5C(O)-, R5C(S)-, R5SO2-, R5OC(O)-, R5R'NC(O)-, R5R'NC(S)-, адамантил-С(О)- или R'' представляет Н, C1-6-алкил, Аr-С0-6-алкил или Het-C0-6-алкил; R''' представляет Н, C1-6-алкил, С3-6-циклоалкил-С0-6-алкил, Аr-С0-6-алкил или Het-C0-6-алкил; каждый R3 независимо представляет Н,...
Ингибиторы акт и p70 s6-киназы

Номер патента: 18947
Опубликовано: 29.11.2013
Авторы: Джозеф Саджан, Дэлли Роберт Дин, Шеферд Тимоти Алан
МПК: A61P 35/00, A61K 31/52, C07D 473/00...
Метки: s6-киназы, ингибиторы, акт
Формула / Реферат:
1. Соединение формулыгде X представляет собой F, Cl, CF3, CN или Н;Y представляет собой F, Н или Cl;R1 и R2 независимо представляют собой Н, С1-С4алкил или СН2СН2ОН; или R1 и R2 вместе с атомом азота, к которому они присоединены, образуют пирролидиновое кольцо, необязательно замещенное гидроксиметилом в положении 2 или гидроксилом в положении 3, или азетидиновое кольцо, замещенное гидроксилом в положении 3;R3 представляет собой Н или ОН;R6...
Ингибиторы тромбина

Номер патента: 2767
Опубликовано: 29.08.2002
Авторы: Хёффкен Ханс Вольфганг, Бём Ханс-Йоахим, Мак Хельмут, Хорнбергер Вильфрид, Пфайффер Томас, Козер Штефан, Зайтц Вернер, Цирке Томас
МПК: A61P 7/02, A61K 38/55, C07K 5/06...
Метки: тромбина, ингибиторы
Формула / Реферат:
1. Ингибиторы тромбина формулы (I) где R1 - алкил с 1-20 атомами углерода, фенил- или нафтилалкил с 1-10 атомами углерода в алкильной части, группа R2OOC-(CH2), где R2 означает водород или алкил с 1-10 атомами углерода, А - остаток a-аминокислоты формулы (II) где R3 - водород, алкил с 1-8 атомами углерода, циклоалкил с 3-7 атомами углерода, фенил и нафтил, незамещенные или замещенные алкилом или алкоксилом, каждый с 1-4 атомами углерода, или...
Бифенилсульфонамидные ингибиторы матричных металлопротеиназ.

Номер патента: 1561
Опубликовано: 23.04.2001
Авторы: Слискович Драго Роберт, О'брайн Петрик Майкель
МПК: A61P 9/10, C07C 311/19, A61K 31/18...
Метки: ингибиторы, матричных, металлопротеиназ, бифенилсульфонамидные
Формула / Реферат:
1. Соединение формулы I где R1 означает алкил с числом углеродных атомов от 1 до 6, галоген, нитрогруппу, (CH2)0-4-NR4R5, цианогруппу, OR4, R2 означает водород или алкил с числом углеродных атомов от 1 до 6, замещенный по выбору следующими группами: фенил, замещенный фенил, фенокси-, замещенная фенокси-, NR4R5, OR6, карбокси-, карбоксиамидо-, тио-, метилтио-, индол, имидазол и фталимидо-; R3 означает ОН, ОС1-С6-алкил или NHOH; R4 означает...
Ингибиторы p70 s6 киназы

Номер патента: 16445
Опубликовано: 30.05.2012
Авторы: Хольст Кристиан Л., Чжуан Цзяньпин, Шеферд Тимоти Алан, Дэлли Роберт Дин, Джозеф Саджан
МПК: A61K 31/437, A61P 35/00, C07D 487/04...
Метки: ингибиторы, киназы
Формула / Реферат:
1. Соединение формулыгде Y представляет собой CH;Z1 и Z2 представляют собой независимо CR3 или N при условии, что Z1 и Z2, оба, не являются N;R1 представляет собой H или CH3;R2 представляет собой фенил, содержащий первый заместитель, выбранный из галогена и трифторметила, и необязательно дополнительно содержащий второй заместитель, при этом указанный второй заместитель представляет собой галоген;R3 представляет собой водород, галоген,...
Предыдущий патент: Программируемый контрастный душ
Следующий патент: Способы и составы для борьбы с эктопаразитами
Случайный патент: Устройство для сохранения равновесия крыльев на многосекционной жатке