Пиперидинилкарбонилпирролидины и их применение в качестве агонистов меланокортина
Номер патента: 11159
Опубликовано: 27.02.2009
Авторы: Лансделл Марк, Хепворт Дэвид, Фрэйдет Дэвид Себастьен, Калабрезе Эндрю Энтони




























Формула / Реферат
1. Соединение общей формулы

или его фармацевтически приемлемая соль, гидрат, сольват или изомер,
где R1 представляет собой -(C1-C6)алкил, -(C3-C8)циклоалкил, фенил или пиридил, где каждый из фенила и пиридила необязательно замещен одной или несколькими группами, выбранными из -(C1-C4)алкила, галогена, -OR6, -CN и CF3;
R2 представляет собой OH;
R3 представляет собой Н, -(C1-C6)алкил, -(C3-C8)циклоалкил, (C1-C2)алкил (C3-C8)циклоалкил или гетероциклическую группу, выбранную из пиридила, пиримидинила, пиразинила, пиридазинила и тетрагидропиранила, где алкильная группа необязательно замещена -OR6;
R4 и R5, каждый независимо, представляют собой Н или галоген;
R6 представляет собой Н, CH3 или CH2CH3.
2. Соединение по п.1, имеющее общую формулу

где R1, R2, R3, R4 и R5 являются такими, как они определены в п.1, и где стереохимия групп в положениях 3 и 4 пирролидинового кольца является транс относительно друг друга.
3. Соединение по п.1, имеющее общую формулу

где R1, R2, R3, R4 и R5 являются такими, как они определены в п.1, и где стереохимия метильных групп в положениях 3 и 5 пиперидинового кольца является цис относительно друг друга.
4. Соединение по п.3, где стереохимия групп в положениях 3 и 4 пирролидинового кольца такова, что они находятся в транс-положении относительно друг друга.
5. Соединение по п.1, имеющее общую формулу

где R1, R2, R3, R4 и R5 являются такими, как они определены в п.1, и где стереохимия групп в положениях 3 и 4 пирролидинового кольца является транс-конфигурацией относительно друг друга и стереохимия метильных групп в положениях 3 и 5 пиперидинового кольца является цис-конфигурацией относительно друг друга.
6. Соединение по п.1, имеющее общую формулу

где R1, R2, R3, R4 и R5 являются такими, как они определены в п.1, и где стереохимия метильных групп в положениях 3 и 5 пиперидинового кольца является цис-конфигурацией относительно друг друга и где R1 группа в положении 4 находится в транс-положении относительно метильных групп в положениях 3 и 5 пиперидинового кольца и R2 группа находится в цис-положении относительно метильных групп.
7. Соединение по п.6, где стереохимия групп в положениях 3 и 4 пирролидинового кольца является транс-конфигурацией относительно друг друга.
8. Соединение по любому из предшествующих пунктов, имеющее общую формулу

где R1, R2, R3, R4 и R5 являются такими, как они определены в п.1, и где стереохимия групп в положениях 3 и 4 пирролидинового кольца является транс-конфигурацией относительно друг друга, стереохимия метильных групп в положениях 3 и 5 пиперидинового кольца является цис-конфигурацией относительно друг друга и где R1 группа в положении 4 находится в транс-положении относительно метильных групп в положениях 3 и 5 пиперидинового кольца и R2 группа находится в цис-положении относительно метильных групп.
9. Соединение по п.1, имеющее общую формулу

где R1, R2, R3, R4 и R5 являются такими, как они определены в п.1, и где стереохимия групп в положениях 3 и 4 пирролидинового кольца является транс-конфигурацией относительно друг друга, стереохимия метильных групп в положениях 3 и 5 пиперидинового кольца является цис-конфигурацией относительно друг друга и где R1 группа в положении 4 находится в транс-положении относительно метильных групп в положениях 3 и 5 пиперидинового кольца, R2 группа находится в цис-положении относительно метильных групп и R4 и R5 находятся в положениях 2 и 4 фенильного кольца.
10. Соединение по любому из пп.1-9, где R1 представляет собой н-пропил, изопропил, н-бутил, метоксиметил, циклопропил, циклогексил, фенил, 3-фторфенил, 4-фторфенил, 4-хлорфенил, 4-метилфенил, 4-метоксифенил, 2,6-дифторфенил, 2,4-дифторфенил, 3,4-дифторфенил, пиридин-2-ил, 4-хлорпиридин-2-ил или пиридин-3-ил.
11. Соединение по любому из пп.1-10, где R1 представляет собой пиридин-2-ил, 4-хлорпиридин-2-ил, фенил, 3-фторфенил, 4-фторфенил, 4-хлорфенил, 4-метилфенил, 4-метоксифенил, 2,6-дифторфенил, 2,4-дифторфенил или 3,4-дифторфенил.
12. Соединение по любому пп.1-11, где R3 представляет собой водород, этил, изопропил, н-пропил, н-бутил, трет-бутил, изобутил, 2-метоксиэтил, циклопентил, циклобутил, циклопентилметил, пиридин-2-ил, пиридин-3-ил, пиридазин-3-ил, 6-гидрокси-1-метилпиридазин-3-ил, пиразинил, пиримидин-5-ил, пиримидин-2-ил, пиримидин-4-ил или тетрагидропиран-4-ил.
13. Соединение по любому из пп.1-12, где R4 представляет собой Н, F или Cl и R5 представляет собой F или Cl.
14. Соединение по любому из пп.1-12, где фенильная группа, имеющая заместители R4 и R5, представляет собой 2,4-замещенную фенильную группу, в которой каждая из R4 и R5 групп независимо выбрана из F или Cl; или 4-монозамещенную фенильную группу, в которой R4 представляет собой Н и R5 представляет собой F или Cl.
15. Соединение по п.14, где фенильная группа, имеющая заместители R4 и R5, представляет собой 4-хлорфенил или 2,4-дифторфенил.
16. Соединение по п.5, где
R1 представляет собой фенильную, 3-фторфенильную, 4-фторфенильную, 2,6-дифторфенильную, 2,4-дифторфенильную, 3,4-дифторфенильную или пиридин-2-ильную группу;
R2 представляет собой OH;
R3 представляет собой Н;
R4 представляет собой Н или F и
R5 представляет собой F или Cl.
17. Соединение по п.16, выбранное из
гидрохлорида (3R,4R,5S)-1-{[(3S,4R)-4-(2,4-дифторфенил)пирролидин-3-ил]карбонил}-4-(4-фторфенил)-3,5-диметилпиперидин-4-ола;
гидрохлорида (3R,4R,5S)-4-(2,4-дифторфенил)-1-{[(3S,4R)-4-(2,4-дифторфенил)пирролидин-3-ил]карбонил}-3,5-диметилпиперидин-4-ола;
гидрохлорида (3R,4R,5S)-1-{[(3S,4R)-4-(2,4-дифторфенил)пирролидин-3-ил]карбонил}-3,5-диметил-4-фенилпиперидин-4-ола;
гидрохлорида (3R,4R,5S)-1-{[(3S,4R)-4-(4-хлорфенил)пирролидин-3-ил]карбонил}-3,5-диметил-4-фенилпиперидин-4-ола
и их фармацевтически приемлемых солей, гидратов или сольватов.
18. Соединение по п.5, где
R1 представляет собой фенильную или пиридин-2-ильную группу;
R2 представляет собой OH;
R3 представляет собой гетероциклическую группу, выбранную из пиридин-2-ильной, пиридин-3-ильной, пиридазин-3-ильной, пиразинильной, пиримидин-5-ильной, пиримидин-4-ильной, пиримидин-2-ильной и тетрагидропиран-4-ильной;
обе из R4 и R5 групп представляют собой F.
19. Соединение по п.18, выбранное из
гидрохлорида (3R,4R,5S)-1-{[(3S,4R)-4-(2,4-дифторфенил)-1-пиридин-2-илпирролидин-3-ил]карбонил}-3,5-диметил-4-фенилпиперидин-4-ола;
гидрохлорида (3R,4R,5S)-1-{[(3S,4R)-4-(2,4-дифторфенил)-1-пиридин-3-илпирролидин-3-ил]карбонил}-3,5-диметил-4-фенилпиперидин-4-ола;
гидрохлорида (3R,4R,5S)-1-{[(3S,4R)-4-(2,4-дифторфенил)-1-пиридазин-3-илпирролидин-3-ил]карбонил}-3,5-диметил-4-фенилпиперидин-4-ола;
гидрохлорида (3R,4R,5S)-1-{[(3S,4R)-4-(2,4-дифторфенил)-1-пиримидин-4-илпирролидин-3-ил]карбонил}-3,5-диметил-4-фенилпиперидин-4-ола;
гидрохлорида (3R,4R,5S)-1-{[(3S,4R)-4-(2,4-дифторфенил)-1-пиридазин-3-илпирролидин-3-ил]карбонил}-3,5-диметил-4-пиридин-2-илпиперидин-4-ола;
и их фармацевтически приемлемых солей, гидратов или сольватов.
20. Соединение по п.5, где
R1 представляет собой фенил, 4-фторфенил, 4-хлорфенил, 3-фторфенил, 2,4-дифторфенил, 3,4-дифторфенил, пиридин-2-ил;
R2 представляет собой OH;
R3 представляет собой трет-бутил, изопропил, этил;
обе из R4 и R5 групп представляют собой F.
21. Соединение по п.20, выбранное из
(3R,4R,5S)-1-{[(3S,4R)-1-трет-бутил-4-(2,4-дифторфенил)пирролидин-3-ил]карбонил}-3,5-диметил-4-фенилпиперидин-4-ола;
гидрохлорида (3R,4R,5S)-1-{[(3S,4R)-1-трет-бутил-4-(2,4-дифторфенил)пирролидин-3-ил]карбонил}-3,5-диметил-4-фенилпиперидин-4-ола;
гидрохлорида (3R,4R,5S)-1-{[(3S,4R)-4-(2,4-дифторфенил)-1-изопропилпирролидин-3-ил]карбонил}-3,5-диметил-4-фенилпиперидин-4-ола;
гидрохлорида (3R,4R,5S)-1-{[(3S,4R)-1-трет-бутил-4-(2,4-дифторфенил)пирролидин-3-ил]карбонил}-4-(3,4-дифторфенил)-3,5-диметилпиперидин-4-ола;
гидрохлорида (3R,4R,5S)-1-{[(3S,4R)-1-трет-бутил-4-(2,4-дифторфенил)пирролидин-3-ил]карбонил}-4-(4-фторфенил)-3,5-диметилпиперидин-4-ола;
гидрохлорида (3R,4R,5S)-1-{[(3S,4R)-4-(2,4-дифторфенил)-1-изопропилпирролидин-3-ил]карбонил}-4-(4-фторфенил)-3,5-диметилпиперидин-4-ола;
гидрохлорида (3R,4R,5S)-1-{[(3S,4R)-1-трет-бутил-4-(2,4-дифторфенил)пирролидин-3-ил]карбонил}-4-(4-хлорфенил)-3,5-диметилпиперидин-4-ола;
гидрохлорида (3R,4R,5S)-1-{[(3S,4R)-1-трет-бутил-4-(2,4-дифторфенил)пирролидин-3-ил]карбонил}-4-(2,4-дифторфенил)-3,5-диметилпиперидин-4-ола;
гидрохлорида (3R,4R,5S)-1-{[(3S,4R)-1-трет-бутил-4-(2,4-дифторфенил)пирролидин-3-ил]карбонил}-3,5-диметил-4-пиридин-2-илпиперидин-4-ола;
гидрохлорида (3R,4R,5S)-1-{[(3S,4R)-1-трет-бутил-4-(2,4-дифторфенил)пирролидин-3-ил]карбонил}-3,5-диметил-4-пропилпиперидин-4-ола;
гидрохлорида (3R,4R,5S)-4-(4-хлорфенил)-1-{[(3S,4R)-4-(2,4-дифторфенил)-1-изопропилпирролидин-3-ил]карбонил}-3,5-диметилпиперидин-4-ола;
гидрохлорида (3R,4R,5S)-4-(3,4-дифторфенил)-1-{[(3S,4R)-4-(2,4-дифторфенил)-1-изопропилпирролидин-3-ил]карбонил}-3,5-диметилпиперидин-4-ола;
гидрохлорида (3R,4R,5S)-4-(2,4-дифторфенил)-1-{[(3S,4R)-4-(2,4-дифторфенил)-1-изопропилпирролидин-3-ил]карбонил}-3,5-диметилпиперидин-4-ола;
гидрохлорида (3R,4R,5S)-1-{[(3S,4R)-4-(2,4-дифторфенил)-1-этилпирролидин-3-ил]карбонил}-4-(3-фторфенил)-3,5-диметилпиперидин-4-ола.
22. Соединение по п.1, выбранное из (3R,4R,5S)-1-{[(3S,4R)-1-трет-бутил-4-(2,4-дифторфенил)пирролидин-3-ил]карбонил}-3,5-диметил-4-фенилпиперидин-4-ола и его фармацевтически приемлемых солей кислот.
23. Соединение по п.1, представляющее собой гидрохлорид (3R,4R,5S)-1-{[(3S,4R)-1-трет-бутил-4-(2,4-дифторфенил)пирролидин-3-ил]карбонил}-3,5-диметил-4-фенилпиперидин-4-ола.
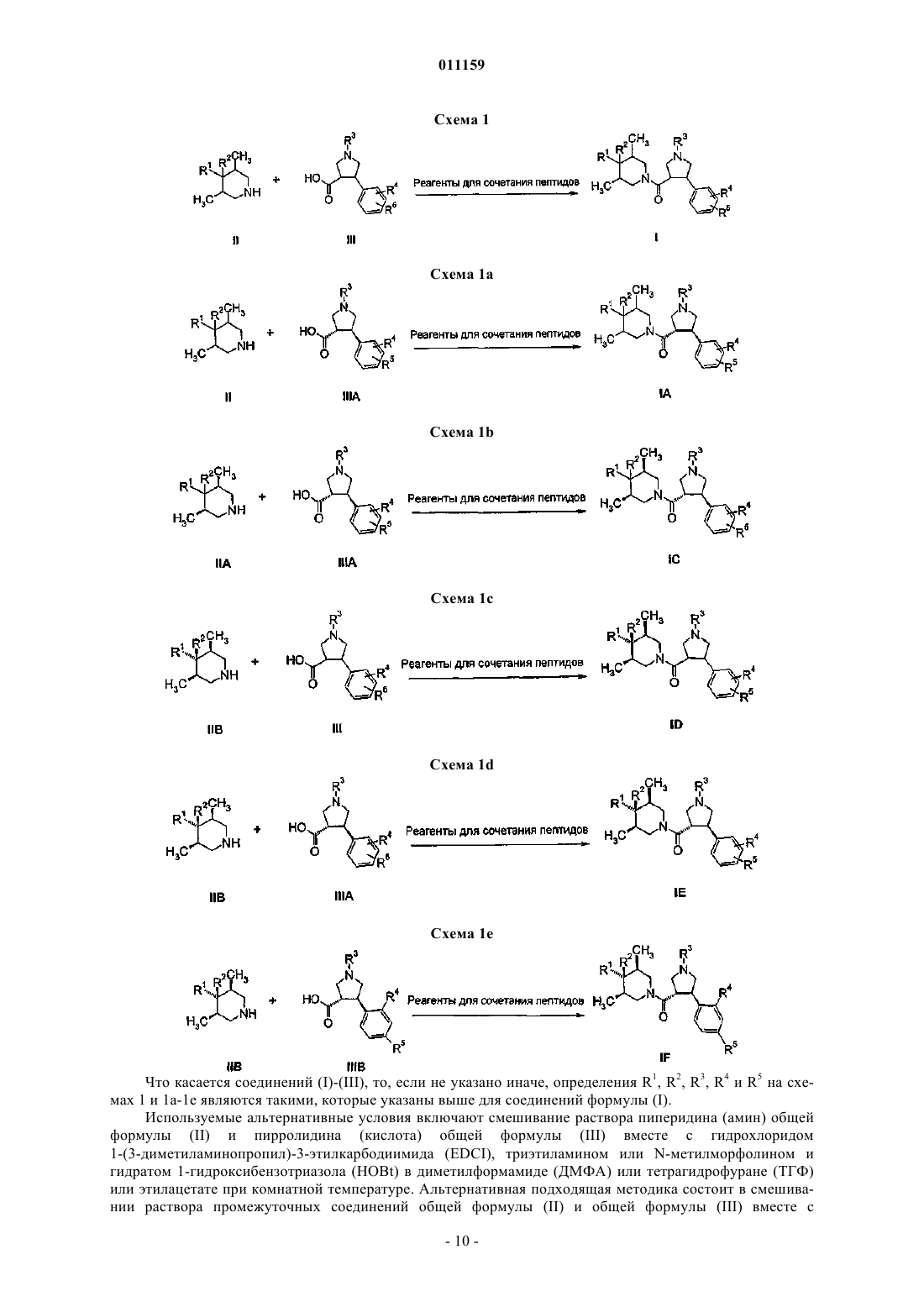
24. Способ получения соединения формулы (I), определенного в п.1, включающий реакцию сочетания соединений (II) и (III)

где R1, R2, R3, R4 и R5 являются такими, как они определены в п.1.
25. Фармацевтическая композиция, содержащая соединение формулы (I), определенное в любом из предшествующих пунктов, или его фармацевтически приемлемую соль, гидрат, сольват или изомер, вместе с одним или несколькими фармацевтически приемлемыми эксципиентами, разбавителями или носителями.
26. Фармацевтическая композиция по п.25, содержащая одно или несколько дополнительных лекарственных веществ.
27. Фармацевтическая композиция по п.26, где дополнительное лекарственное вещество представляет собой одно или несколько средств, выбранных из ингибиторов PDE5; ингибиторов NEP; селективных агонистов или модуляторов D3 или D4; модуляторов рецептора эстрогена и/или агонистов эстрогена и/или антагонистов эстрогена; заменителей тестостерона, тестостерона или тестостеронового имплантата; эстрогена; эстрогена и медроксипрогестерона или медроксипрогестеронацетата (МПА) или эстрогена и метилированного тестостеронового средства гормонзаместительной терапии.
28. Фармацевтическая композиция по п.26, где дополнительное лекарственное вещество представляет собой ингибитор PDE5 или ингибитор NEP.
29. Применение соединения формулы (I), определенного в любом из предшествующих пунктов, или его фармацевтически приемлемой соли, гидрата, сольвата или изомера в качестве лекарственного средства.
30. Применение по п.29 при лечении женской половой дисфункции, мужской эректильной дисфункции, ожирения или диабета.
Текст
011159 Настоящее изобретение относится к новому классу соединений-агонистов меланокортина MCR4 и,в особенности, к соединениям-агонистам, селективным к MCR4, к их применению в медицине, к содержащим их композициям, к способам их получения и к промежуточным соединениям, используемым в таких способах. Настоящее изобретение в особенности относится к классу соединений-агонистов MCR4,применимых для лечения половых дисфункций и/или ожирения. Соединения настоящего изобретения применимы при лечении мужских и женских половых дисфункций, включающих гипоактивное половое влечение, нарушение полового возбуждения, оргазменное расстройство и/или боль у женщин в половых органах, мужскую эректильную дисфункцию, а также ожирения (путем снижения аппетита, повышения интенсивности обмена веществ, снижения всасывания жира или уменьшения потребности в углеводах) и сахарного диабета (путем повышения толерантности к глюкозе, и/или снижения резистентности к инсулину). Соединения изобретения применимы при лечении других заболеваний, расстройств или состояний, включающих, но без ограничения, гипертензию, гиперлипидемию, остеоартрит, рак, заболевание желчного пузыря, приступы апноэ во сне, депрессию, тревогу,компульсивное побуждение, неврозы, бессонницу/расстройство сна, токсикоманию, боль, жар, воспаление, иммунную модуляцию, ревматоидный артрит, задубление кожи, акне (угри) и другие кожные заболевания, а также для нейрозащитной активации и улучшения познавательной способности и памяти,включая лечение болезни Альцгеймера. Соединения настоящего изобретения являются в особенности подходящими для лечения женской половой дисфункции, мужской эректильной дисфункции, ожирения и диабета. Желательные свойства соединений-агонистов MCR4 настоящего изобретения включают желательную эффективность в отношении MCR4, которая подробно обсуждена в последующем описании; селективность к MCR4 в сравнении с селективностью к MCR1 и/или MCR5 и/или MCR3, которая подробно обсуждена в последующем описании; желательную эффективность в отношении MCR4 и селективность к MCR4 в сравнении с MCR1 и/или MCR5 и/или MCR3; хорошие биофармацевтические свойства, такие как физическая стабильность; растворимость; подходящая метаболическая стабильность. Сущность изобретения Настоящее изобретение относится к соединениям формулы или их фармацевтически приемлемым солям, гидратам, сольватам или изомерам,где R1 представляет собой -(C1-C6)алкил, -(C3-C8)циклоалкил, фенил или пиридил, где каждый из фенила и пиридила необязательно замещен одной или несколькими группами, выбранными из -(C1-C4)алкила,галогена, -OR6, -CN и CF3;R3 представляет собой Н, -(C1-C6)алкил, -(C3-C8)циклоалкил, (C1-C2)алкил (C3-C8)циклоалкил или гетероциклическую группу, выбранную из пиридила, пиримидинила, пиразинила, пиридазинила и тетрагидропиранила, где алкильная группа необязательно замещена -OR6;R6 представляет собой Н, CH3 или CH2CH3. В соответствии с предпочтительным вариантом настоящее изобретение относится к соединению,имеющему общую формулу где R1, R2, R3, R4 и R5 являются такими, как они определены выше, и стереохимия групп в положениях 3 и 4 пирролидинового кольца является транс относительно друг друга. В соответствии с еще одним предпочтительным вариантом настоящее изобретение относится к соединению, имеющему общую формулу где R1, R2, R3, R4 и R5 являются такими, как они определены выше, и где стереохимия метильных групп в положениях 3 и 5 пиперидинового кольца является цис относительно друг друга. Предпочтительно в соединение формулы (IB) стереохимия групп в положениях 3 и 4 пирролидинового кольца такова, что они находятся в транс-положении относительно друг друга. Следующую предпочтительную группу соединений настоящего изобретения представляют соединения, имеющие общую формулу где R1, R2, R3, R4 и R5 являются такими, как они определены выше, стереохимия групп в положениях 3 и 4 пирролидинового кольца является транс-конфигурацией относительно друг друга и стереохимия метильных групп в положениях 3 и 5 пиперидинового кольца является цис-конфигурацией относительно друг друга. В соответствии со следующим предпочтительным вариантом настоящее изобретение относится к соединению, имеющему общую формулу где R1, R2, R3, R4 и R5 являются такими, как они определены выше, стереохимия метильных групп в положениях 3 и 5 пиперидинового кольца является цис-конфигурацией относительно друг друга, и R1 группа в положении 4 находится в транс-положении относительно метильных групп в положениях 3 и 5 пиперидинового кольца, и R2 группа находится в цис-положении относительно метильных групп. Предпочтительно в соединении формулы (ID) стереохимия групп в положениях 3 и 4 пирролидинового кольца является транс-конфигурацией относительно друг друга. В соответствии с еще одним предпочтительным вариантом осуществления настоящее изобретение относится к соединению, имеющему общую формулу где R1, R2, R3, R4 и R5 являются такими, как они определены выше, и где стереохимия групп в положениях 3 и 4 пирролидинового кольца является транс-конфигурацией относительно друг друга, стереохимия метильных групп в положениях 3 и 5 пиперидинового кольца является цис-конфигурацией относительно друг друга, и где R1 группа в положении 4 находится в транс-положении относительно метильных групп в положениях 3 и 5 пиперидинового кольца, и R2 группа находится в цис-положении относительно метильных групп. В следующем предпочтительном аспекте настоящее изобретение относится к соединению, имеющему общую формулу где R1, R2, R3, R4 и R5 являются такими, как они определены выше, и где стереохимия групп в положениях 3 и 4 пирролидинового кольца является транс-конфигурацией относительно друг друга, стереохимия метильных групп в положениях 3 и 5 пиперидинового кольца является цис-конфигурацией относительно друг друга, и где R1 группа в положении 4 находится в транс-положении относительно метильных групп в положениях 3 и 5 пиперидинового кольца, R2 группа находится в цис-положении относительно метильных групп и R4 и R5 находятся в положениях 2 и 4 фенильного кольца. В соединениях настоящего изобретения R1 предпочтительно представляет собой н-пропил,изопропил, н-бутил, метоксиметил, циклопропил, циклогексил, фенил, 3-фторфенил, 4-фторфенил,4-хлорфенил, 4-метилфенил, 4-метоксифенил, 2,6-дифторфенил, 2,4-дифторфенил, 3,4-дифторфенил,пиридин-2-ил, 4-хлорпиридин-2-ил или пиридин-3-ил. Более предпочтительно R1 представляет собой пиридин-2-ил, 4-хлорпиридин-2-ил, фенил,3-фторфенил, 4-фторфенил, 4-хлорфенил, 4-метилфенил, 4-метоксифенил, 2,6-дифторфенил,2,4-дифторфенил или 3,4-дифторфенил. Предпочтительно в соединениях настоящего изобретения R3 представляет собой водород, этил,изопропил, н-пропил, н-бутил, трет-бутил, изобутил, 2-метоксиэтил, циклопентил, циклобутил, циклопентилметил, пиридин-2-ил, пиридин-3-ил, пиридазин-3-ил, 6-гидрокси-1-метилпиридазин-3-ил, пиразинил, пиримидин-5-ил, пиримидин-2-ил, пиримидин-4-ил или тетрагидропиран-4-ил, a R4 представляет собой Н, F или Cl и R5 представляет собой F или Cl. Предпочтительную группу соединений настоящего изобретения составляют соединения, где фенильная группа, имеющая заместители R4 и R5, представляет собой 2,4-замещенную фенильную группу,в которой каждая из R4 и R5 групп независимо выбрана из F или Cl; или 4-монозамещенную фенильную группу, в которой R4 представляет собой Н и R5 представляет собой F или Cl. Предпочтительной является фенильная группа, имеющая заместители R4 и R5, т.е. представляющая собой 4-хлорфенил или 2,4-дифторфенил. Следующую предпочтительную группу соединений настоящего изобретения представляют соединения, гдеR5 представляет собой F или Cl. Конкретными предпочтительными соединениями настоящего изобретения являются соединения,выбранные из гидрохлорида (3R,4R,5S)-1-[(3S,4R)-4-(2,4-дифторфенил)пирролидин-3-ил]карбонил-4-(4-фторфенил)-3,5-диметилпиперидин-4-ола; гидрохлорида(3R,4R,5S)-4-(2,4-дифторфенил)-1-[(3S,4R)-4-(2,4-дифторфенил)пирролидин-3 ил]карбонил-3,5-диметилпиперидин-4-ола; гидрохлорида (3R,4R,5S)-1-[(3S,4R)-4-(2,4-дифторфенил)пирролидин-3-ил]карбонил-3,5-диметил-4 фенилпиперидин-4-ола; гидрохлорида (3R,4R,5S)-1-[(3S,4R)-4-(4-хлорфенил)пирролидин-3-ил]карбонил-3,5-диметил-4 фенилпиперидин-4-ола и их фармацевтически приемлемых солей, гидратов или сольватов. Предпочтительную группу соединений настоящего изобретения составляют также соединения, гдеR1 представляет собой фенильную или пиридин-2-ильную группу;R3 представляет собой гетероциклическую группу, выбранную из пиридин-2-ильной, пиридин-3 ильной, пиридазин-3-ильной, пиразинильной, пиримидин-5-ильной, пиримидин-4-ильной, пиримидин-2 ильной и тетрагидропиран-4-ильной; обе из R4 и R5 групп представляют собой F. Следующую группу конкретных предпочтительных соединений настоящего изобретения составляют соединения, выбранные из гидрохлорида (3R,4R,5S)-1-[(3S,4R)-4-(2,4-дифторфенил)-1-пиридин-2-илпирролидин-3-ил]карбонил 3,5-диметил-4-фенилпиперидин-4-ола;(3R,4R,5S)-1-[(3S,4R)-4-(2,4-дифторфенил)-1-пиридазин-3-илпирролидин-3-ил]карбонил-3,5-диметил-4-пиридин-2-илпиперидин-4-ола и их фармацевтически приемлемых солей, гидратов или сольватов. В соединениях настоящего изобретенияR1 предпочтительно представляет собой фенил, 4-фторфенил, 4-хлорфенил, 3-фторфенил,2,4-дифторфенил, 3,4-дифторфенил, пиридин-2-ил;R3 представляет собой трет-бутил, изопропил, этил; обе из R4 и R5 групп представляют собой F. Конкретными предпочтительными соединениями настоящего изобретения являются соединения,выбранные из(3R,4R,5S)-1-[(3S,4R)-1-трет-бутил-4-(2,4-дифторфенил)пирролидин-3-ил]карбонил-3,5-диметил 4-фенилпиперидин-4-ола; гидрохлорида (3R,4R,5S)-1-[(3S,4R)-1-трет-бутил-4-(2,4-дифторфенил)пирролидин-3-ил]карбонил-3,5 диметил-4-фенилпиперидин-4-ола; гидрохлорида (3R,4R,5S)-1-[(3S,4R)-4-(2,4-дифторфенил)-1-изопропилпирролидин-3-ил]карбонил-3,5 диметил-4-фенилпиперидин-4-ола; гидрохлорида (3R,4R,5S)-1-[(3S,4R)-1-трет-бутил-4-(2,4-дифторфенил)пирролидин-3-ил]карбонил-4(3,4-дифторфенил)-3,5-диметилпиперидин-4-ола; гидрохлорида (3R,4R,5S)-1-[(3S,4R)-1-трет-бутил-4-(2,4-дифторфенил)пирролидин-3-ил]карбонил-4(4-фторфенил)-3,5-диметилпиперидин-4-ола; гидрохлорида (3R,4R,5S)-1-[(3S,4R)-4-(2,4-дифторфенил)-1-изопропилпирролидин-3-ил]карбонил-4(4-фторфенил)-3,5-диметилпиперидин-4-ола; гидрохлорида (3R,4R,5S)-1-[(3S,4R)-1-трет-бутил-4-(2,4-дифторфенил)пирролидин-3-ил]карбонил-4(4-хлорфенил)-3,5-диметилпиперидин-4-ола; гидрохлорида (3R,4R,5S)-1-[(3S,4R)-1-трет-бутил-4-(2,4-дифторфенил)пирролидин-3-ил]карбонил-4(2,4-дифторфенил)-3,5-диметилпиперидин-4-ола; гидрохлорида (3R,4R,5S)-1-[(3S,4R)-1-трет-бутил-4-(2,4-дифторфенил)пирролидин-3-ил]карбонил-3,5 диметил-4-пиридин-2-илпиперидин-4-ола; гидрохлорида (3R,4R,5S)-1-[(3S,4R)-1-трет-бутил-4-(2,4-дифторфенил)пирролидин-3-ил]карбонил-3,5 диметил-4-пропилпиперидин-4-ола; гидрохлорида (3R,4R,5S)-4-(4-хлорфенил)-1-[(3S,4R)-4-(2,4-дифторфенил)-1-изопропилпирролидин-3 ил]карбонил-3,5-диметилпиперидин-4-ола; гидрохлорида (3R,4R,5S)-4-(3,4-дифторфенил)-1-[(3S,4R)-4-(2,4-дифторфенил)-1-изопропилпирролидин-3-ил]карбонил-3,5-диметилпиперидин-4-ола; гидрохлорида (3R,4R,5S)-4-(2,4-дифторфенил)-1-[(3S,4R)-4-(2,4-дифторфенил)-1-изопропилпирролидин-3-ил]карбонил-3,5-диметилпиперидин-4-ола; гидрохлорида (3R,4R,5S)-1-[(3S,4R)-4-(2,4-дифторфенил)-1-этилпирролидин-3-ил]карбонил-4-(3 фторфенил)-3,5-диметилпиперидин-4-ола. Особенно предпочтительным соединением настоящего изобретения является (3R,4R,5S)-1[(3S,4R)-1-трет-бутил-4-(2,4-дифторфенил)пирролидин-3-ил]карбонил-3,5-диметил-4-фенилпиперидин-4-ол и его фармацевтически приемлемые соли кислот. Другим особо предпочтительным соединением настоящего изобретения является гидрохлорид(3R,4R,5S)-1-[(3S,4R)-1-трет-бутил-4-(2,4-дифторфенил)пирролидин-3-ил]карбонил-3,5-диметил-4 фенилпиперидин-4-ола. Настоящее изобретение далее относится к способу получения соединения формулы (I), включающему реакцию сочетания соединений (II) и (III) где R1, R2, R3, R4 и R5 являются такими, как они определены выше.-4 011159 В следующем аспекте настоящее изобретение относится к фармацевтической композиции, содержащей соединение формулы (I), определенное выше, или его фармацевтически приемлемую соль, гидрат, сольват или изомер, вместе с одним или несколькими фармацевтически приемлемыми эксципиентами, разбавителями или носителями. Фармацевтическая композиция настоящего изобретения может содержать одно или несколько дополнительных лекарственных веществ. Предпочтительно фармацевтическая композиция содержит дополнительное лекарственное вещество, представляющее собой одно или несколько средств, выбранных из ингибиторов PDE5; ингибиторовNEP; селективных агонистов или модуляторов D3 или D4; модуляторов рецептора эстрогена, и/или агонистов эстрогена, и/или антагонистов эстрогена; заменителей тестостерона, тестостерона или тестостеронового имплантата; эстрогена; эстрогена и медроксипрогестерона или медроксипрогестеронацетата(МПА) или эстрогена и метилированного тестостеронового средства гормонзаместительной терапии. В одном из вариантов осуществления фармацевтическая композиция содержит дополнительное лекарственное вещество, представляющее собой ингибитор PDE5 или ингибитор NEP. Кроме того, настоящее изобретение касается применения соединения формулы (I), определенного выше, или его фармацевтически приемлемой соли, гидрата, сольвата или его изомера в качестве лекарственного средства. Предпочтительно соединение формулы (I) применяется при лечении женской половой дисфункции,мужской эректильной дисфункции, ожирения или диабета. Определения. Подходящие для использования в данном изобретении гетероциклические группы представляют собой 4-10-членные моно- или бициклические гетероарильные кольца, содержащие от одного до трех гетероатомов, выбранных из N, S и O и их комбинаций, и где бициклические гетероарильные кольца являются 9-10-членными кольцевыми системами, которые могут представлять собой два гетероарильных кольца, конденсированных вместе, или гетероарильное кольцо, конденсированное к арильному кольцу. Подходящие для использования в данном изобретении бициклические гетероарильные группы включают бензимидазолильную, бензотриазолильную, бензотиазолильную, индазолильную, индолильную, имидазопиридинильную, имидазопиримидинильную, пирролопиридинильную, хинолинильную,изохинолинильную, хиназолинильную, нафтиридинильную и пиридопиримидинильную группы. Для использования в данном изобретении предпочтительны моноциклические 5-6-членные гетероарильные кольца, содержащие один или три гетероатома, выбранные из N и O и их комбинаций. Подходящие для использования в данном изобретении 5-членные моноциклические гетероарильные группы включают триазинильную, оксадиазинильную, оксазолильную, тиазолильную, тиадиазолильную, фурильную, тиенильную и пирролильную и имидазолильную группы. Подходящие для использования в данном изобретении 6-членные моноциклические гетероарильные группы включают пиридинильную, пиримидинильную, пиридазинильную и пиразинильную группы. Предпочтительные гетероциклические кольца R1 представляют собой моноциклические 5-6 членные гетероарильные кольца, содержащие один или два гетероатома, выбранные из N и O и их комбинаций. Более предпочтительные гетероциклические кольца R1 представляют собой моноциклические 5-6-членные гетероарильные кольца, содержащие один или 2 гетероатома N. В особенности предпочтительные в данном изобретении гетероциклические кольца R1 представляют собой моноциклические 6 членные гетероарильные кольца, содержащие один или два гетероатома N, такие как пиридинил и пиримидинил. В особенности предпочтительная гетероарильная группа R1 в данном изобретении является пиридинильной группой. Предпочтительные гетероциклические кольца R3 представляют собой моноциклические 5-6 членные гетероарильные кольца, содержащие один или два гетероатома, выбранные из N и O и их комбинаций, такие как тетрагидропиранильная, пиридинильная, пиридазинильная, пиразинильная и пиримидинильная группы. Более предпочтительные гетероциклические кольца R3 представляют собой моноциклические 5-6-членные гетероарильные кольца, содержащие один или два гетероатома N. Наиболее предпочтительны в качестве гетероциклических колец R3 моноциклические 6-членные гетероарильные кольца, содержащие один или два гетероатома N, такие как пиридинильная, пиридазинильная, пиразинильная и пиримидинильная группы. В особенности предпочтительные для использования в настоящем изобретении 6-членные моноциклические гетероарильные группы R3 представляют собой пиридин-2-ильную, пиридин-3-ильную,пиридазин-3-ильную, пиразинильную, пиримидин-5-ильную и пиримидин-2-ильную группы. В особенности предпочтительные для использования в данном изобретении 6-членные моноциклические гетероарильные группы R3 включают пиридин-2-ильную, пиридин-3-ильную и пиридазин-3-ильную группы. Из данных групп наиболее предпочтительна пиридазин-3-ильная группа. Подходящие конденсированные кольцевые системы, образованные взятыми вместе R4 и R5, могут представлять собой карбоциклические кольцевые системы или гетероциклические кольцевые системы,содержащие до двух гетероатомов, выбранных из O, N или S. Включая фенильное кольцо, к которому-5 011159 они присоединены, предпочтительные кольцевые системы, которые R4 и R5 могут образовывать, представляют собой индан, 1,2,3,4-тетрагидронафталин, индолил, индазолил, нафтил, хинолил, бензотиазолил, бензимидазолил, бензо[1,3]диоксолан, 2,3-дигидробензо[1,4]диоксин, 2,3-дигидробензофуран,2,3-дигидробензотиофен и 1,3-дигидроизобензофуран. В вышеуказанных определениях алкильная, алкенильная и алкинильная группы, имеющие три или более атомов углерода, и алканоильные группы, имеющие четыре или более атомов углерода, могут представлять собой прямую цепь или разветвленную цепь, если не указано иначе. Так например, С 4 алкильный заместитель может быть в форме нормального бутила (н-бутил), изобутила (i-бутил), вторичного бутила (втор-бутил) или третичного бутила (трет-бутил). Для избежания сомнений, когда R1 и/или R3 является необязательно замещенной алкильной группой, такая(ие) алкильная(ые) группа(ы) может(гут) быть дополнительно не замещена(ы) затем дополнительной (незамещенной) алкильной группой. Кроме того, когда R3 замещена алкенильной или алкинильной группой, атом углерода (указанной ненасыщенной группы), который непосредственно связан с атомом N, сам может быть ненасыщенным. Термин галоген включает Cl, Br, F и I. Когда в данном описании используется термин арил, он включает шести-десятичленные карбоциклические ароматические группы, такие как фенил и нафтил. Настоящее изобретение дополнительно включает соединения общих формул (IA)-(IF), а также их смеси, которые будут детально обсуждены в последующем описании. Для избежания сомнений все ссылки на соединения общей формулы (I), такие как, например, соли, полиморфы, пролекарства или их оптические, геометрические и таутомерные изомеры, предназначены для включения соединений, имеющих общие формулы (IA)-(IF), если специально не указано иначе. Фармацевтически приемлемые соли соединений формулы (I) включают их кислотно-аддитивные соли и основно-аддитивные соли. Фармацевтически приемлемая соль соединения формулы (I) может быть легко получена смешиванием вместе растворов соединения формулы (I) и желательной кислоты или основания. Соль может быть осаждена из раствора и собрана фильтрацией или может быть извлечена выпариванием растворителя. Подходящие кислотно-аддитивные соли образованы из кислот, образующих нетоксичные соли. Примеры включают следующие соли: ацетат, адипат, аспартат, бензоат, безилат, бикарбонат/карбонат,бисульфат/сульфат, борат, камзилат, цитрат, цикламат, эдизилат, эзилат, формиат, фумарат, глюцептат,глюконат, глюкуронат, гексафторфосфат, гибензат, гидрохлорид/хлорид, гидробромид/бромид, гидроиодид/иодид, изетионат, лактат, малат, малеат, малонат, мезилат, метилсульфат, нафтилат, 2-напсилат, никотинат, нитрат, оротат, оксалат, пальмитат, памоат, фосфат/кислый фосфат/дигидрофосфат, пироглутамат, сахарат, стеарат, сукцинат, таннат, тартрат, тозилат, трифторацетат и ксинофоат. Подходящие основно-аддитивные соли образованы из оснований, образующих нетоксичные соли. Примеры включают соли алюминия, аргинина, бензатина, кальция, холина, диэтиламина, диоламина,глицина, лизина, магния, меглумина, оламина, калия, натрия, трометамина и цинка. Могут быть также образованы полусоли кислот и оснований, например гемисульфат и гемикальциевая соль. Для обзора подходящих солей см. Handbook of Pharmaceutical Salts: Properties, Selection and Use byStahl and Wermuth (Wiley-VCH, 2002). Фармацевтически приемлемые соли соединений формулы (I) могут быть получены одним или несколькими из трех способов:(i) взаимодействием соединения формулы (I) с требуемой кислотой или основанием;(ii) удалением кислотно- или основно-лабильной защитной группы из подходящего предшественника соединения формулы (I) или размыканием кольца подходящего циклического предшественника,например лактона или лактама, с использованием требуемой кислоты или основания или(iii) превращением одной соли соединения формулы (I) в другую взаимодействием с соответствующей кислотой или основанием или посредством подходящей ионообменной колонки. Все три реакции обычно осуществляют в растворе. Образовавшаяся соль может быть осаждена и собрана фильтрацией или может быть извлечена выпариванием растворителя. Степень ионизации в полученной соли может изменяться от полностью ионизованной до почти неионизованной. Соединения изобретения могут существовать в континууме твердых состояний: от полностью аморфного до полностью кристаллического. Термин аморфный относится к состоянию, в котором вещество утрачивает дальний порядок на молекулярном уровне и в зависимости от температуры может проявлять физические свойства твердого вещества или жидкости. Обычно такие вещества не дают характерные рентгенограммы и хотя проявляют свойства твердого вещества, более формально описаны как жидкость. При нагревании происходят изменения свойств: от свойств, характерных для твердого вещества, до свойств, характерных для жидкости, которые характеризуют изменение состояния, обычно переход второго порядка (стеклование). Термин кристаллический относится к твердой фазе, в которой вещество имеет регулярно-упорядоченную внутреннюю структуру на молекулярном уровне и дает характерную рентгенограмму с определенными пиками. Такие вещества при достаточном нагреве будут также проявлять свойства жидкости, но изменение от твердого состояния до жидкого характеризуется-6 011159 фазовым превращением, обычно переход первого порядка (точка плавления). Соединения изобретения могут также существовать в несольватированных и сольватированных формах. Использованный в данном описании термин сольват описывает молекулярный комплекс, содержащий соединение изобретения и одну или несколько молекул фармацевтически приемлемого растворителя, например этанола. Термин гидрат используется в том случае, когда растворителем является вода. Приемлемая на практике система классификации органических гидратов является такой, которая определяет гидраты с изолированными участками, каналами или гидраты, координированные с ионом металла - см. Polymorphism in Pharmaceutical Solids by K.R. Morris (Ed. H.G. Brittain, Marcel Dekker, 1995). Гидраты с изолированными участками являются такими, в которых молекулы воды изолированы от прямого контакта друг с другом за счет вмешивания органических молекул. В гидратах с каналами молекулы воды лежат в каналах решетки, где они находятся по соседству с другими молекулами воды. В гидратах, координированных с ионом металла, молекулы воды связаны с ионом металла. Когда растворитель или вода прочно связаны, комплекс будет иметь четко определенную стехиометрию, не зависящую от влажности. Однако, когда растворитель или вода слабо связаны, как это имеет место в сольватах с каналами и гигроскопических соединениях, содержание воды/растворителя будет зависеть от влажности и условий сушки. В таких случаях будет нормой нестехиометрия. В область изобретения включены также многокомпонентные комплексы (иные, чем соли и сольваты), в которых лекарственное вещество и по меньшей мере один другой компонент присутствуют в стехиометрических или нестехиометрических количествах. Комплексы такого типа включают клатраты(комплексы включения лекарственное вещество-хозяин) и сокристаллы. Последние обычно определяют как кристаллические комплексы нейтральных молекулярных компонентов, которые связаны вместе посредством нековалентных взаимодействий, но могут также представлять собой комплекс нейтральной молекулы с солью. Сокристаллы могут быть получены кристаллизацией из расплава, перекристаллизацией из растворителей или физическим совместным измельчением компонентов - см. Chem. Commun, 17,1889-1896, by О. Almarsson and M.J. Zaworotko (2004). Для общего обзора многокомпонентных комплексов см. J. Pharm. Sci., 64 (8), 1269-1288 by Haleblian (August 1975). Соединения изобретения при подвержении их соответствующим условиям могут также существовать в мезоморфном состоянии (мезофаза или жидкий кристалл). Мезоморфное состояние является промежуточным между действительным кристаллическим состоянием и действительным жидким состоянием (или расплав или раствор). Мезоморфизм, возникающий в результате изменения температуры, описывается как термотропный и мезоморфизм, являющийся результатом присоединения второго компонента, такого как вода или другой растворитель, описывается как лиотропный. Соединения, которые имеют возможность образовывать лиотропные мезофазы, описаны как амфифильные и состоят из молекул, которые обладают ионной (такой как -COO-Na+, -COO-K+ или -SO3-Na+) или неионной (такой как-N-N+(CH3)3) полярной головной группой. Для получения подробной информации см. Crystals and thePolarizing Microscope by N.H. Hartshorne and A. Stuart, 4th Edition (Edward Arnold, 1970). Далее все ссылки на соединения формулы (I) включают ссылки на их соли, сольваты, многокомпонентные комплексы и жидкие кристаллы и на сольваты, многокомпонентные комплексы и жидкие кристаллы их солей. Соединения изобретения включают соединения формулы (I), определенные выше, их полиморфы и кристаллические формы и изомеры (включая оптические, геометрические и таутомерные изомеры), которые будут определены в последующем описании, и изотопно-меченные соединения формулы (I). Так называемые пролекарства соединений формулы (I) представляют собой некоторые производные соединений формулы (I), которые могут сами иметь низкую фармакологическую активность или не иметь таковую, и при введении в организм или на тело могут быть превращены, например, гидролитическим расщеплением в соединения формулы (I), имеющие желательную активность. Такие производные относятся к пролекарствам. Дополнительная информация об использовании пролекарств может быть найдена в Pro-drugs as Novel Delivery Systems, vol. 14, ACS Symposium Series (T. Higuchi and W. Stella)Association). Пролекарства могут быть получены, например, заменой присутствующих в соединениях формулы(I) соответствующих функциональных групп некоторыми группами, известными специалистам в данной области как прогруппы, которые раскрыты, например, в Design of Prodrugs by H. Bundgaard (Elsevier,1985). Некоторые примеры пролекарств соединений формулы (I) включают:(i) когда соединение формулы (I) содержит функциональную группу карбоновой кислоты (-COOH) или ее сложный эфир, например соединение, в котором водород функциональной группы карбоновой кислоты в соединении формулы (I) заменен -(C1-C8)алкилом;(ii) когда соединение формулы (I) содержит функциональную группу спирта (-OH), его простой эфир, например соединение, в котором водород функциональной группы спирта в соединении формулы-7 011159 группой, предпочтительное пролекарство в данном случае представляет собой простой эфир и(iii) когда соединение формулы (I) содержит функциональную первичную или вторичную аминогруппу (-NH2 или -NHR, где RН), например, где R3=Н, его предпочтительное пролекарство представляет собой его амид, например соединение, в котором один или оба атома водорода функциональной аминогруппы соединения формулы (I) заменен(ы) -(C1-C10)алканоилом, предпочтительно-(C1-C6)алканоилом, более предпочтительно метил-, этил- или пропилалканоилом. В особенности предпочтительные пролекарства представляют собой простые эфиры, простые-(C1-C4)алкиловые эфиры и сложные -(C1-C4)алкиловые эфиры соединений общей формулы (I), при этом в особенности предпочтительны сложные эфиры. Сложноэфирные пролекарства подробно раскрыты вDesign of ester prodrugs to enhance oral absorption of poorly permeable compounds: Challenges to the discovery scientist, Current Drug Metabolism, (2003), 4(6), 461-485. Дополнительные примеры заменяющих групп в соответствии с упомянутыми выше примерами и примерами других типов пролекарств могут быть найдены в вышеприведенных ссылках. И, наконец, некоторые соединения формулы (I) могут сами действовать в качестве пролекарств других соединений формулы (I). В настоящем описании раскрываются также метаболиты соединений формулы (I), т.е. соединения,образованные in vivo при введении лекарственного вещества. Некоторые примеры метаболитов в соответствии с изобретением включают:(i) когда соединение формулы (I) содержит метильную группу, его гидроксиметильное производное(ii) когда соединение формулы (I) содержит алкоксильную группу, его гидроксильное производное(iii) когда соединение формулы (I) содержит третичную аминогруппу, его производное с вторичной аминогруппой (-NR7R8-NHR7 или -NHR8, где R7 и R8 являются различными группами);(iv) когда соединение формулы (I) содержит вторичную аминогруппу, его производное с первичной аминогруппой (-NHR7-NH2);(v) когда соединение формулы (I) содержит фенильную группу, его фенольное производное(vi) когда соединение формулы (I) содержит амидную группу, его производное карбоновой кислоты(-CONH2COOH). Соединения формулы (I), содержащие один или несколько асимметрических атомов углерода, могут существовать в виде двух или более стереоизомеров. Когда соединение формулы (I) содержит алкенильную или алкениленовую группу, возможны геометрические цис/транс- (или Z/E) изомеры. Когда структурные изомеры являются взаимопревращаемыми из-за низкоэнергетического барьера, может иметь место таутомерная изомерия (таутомерия). Она может принять форму протонной таутомерии в соединениях формулы (I), содержащих, например, имино-, кето- или оксимную группу, или так называемой валентной таутомерии в соединениях, содержащих ароматическую группу. Логически следует, что одно соединение может проявлять более одного вида изомерии. В объем настоящего изобретения включены все стереоизомеры, геометрические изомеры и таутомерные формы соединений формулы (I), в том числе соединения, проявляющие более одного вида изомерии, и смеси одного или нескольких из указанных изомеров и таутомеров. Включены также кислотноаддитивные соли или основно-аддитивные соли, в которых противоион является оптически активным,например d-лактат или l-лизин, или рацемическая смесь, например dl-тартрат или dl-аргинин. В объем настоящего изобретения специально включены стереоизомерные смеси соединений,имеющих формулу (I), или диастереомерно обогащенный или диастереомерно чистый изомер соединения формулы (I), или энантиомерно обогащенный или энантиомерно чистый изомер соединения формулы (I). Цис/транс-изомеры могут быть разделены традиционными методами, хорошо известными специалистам в данной области, например хроматографией и фракционированной кристаллизацией. Традиционные методы получения/выделения отдельных энантиомеров включают хиральный синтез из подходящего оптически чистого предшественника или разделение рацемата (рацемата соли или производного) с использованием, например, высокоэффективной жидкостной хроматографии (ВЭЖХ). Альтернативно, рацемат (или предшественник рацемической смеси) может взаимодействовать с подходящим оптически активным соединением, например спиртом или, в случае, когда соединение формулы (I) содержит кислотную или основную группу, - с кислотой или основанием, такими как винная кислота или 1-фенилэтиламин. Полученная диастереоизомерная смесь может быть разделена хроматографией и/или фракционированной кристаллизацией и один или оба из диастереоизомеров превращены в соответствующий(ие) чистый(ые) энантиомер(ы) средствами, известными специалистам в данной области. Хиральные соединения изобретения (и их хиральные предшественники) могут быть получены в энантиомерно обогащенной форме с использованием хроматографии, обычно ВЭЖХ, или асимметрической смолы с подвижной фазой, состоящей из углеводорода, обычно гептана или гексана, содержащей от-8 011159 0 до 50 об.% изопропанола, обычно от 2 до 20 об.% и от 0 до 5 об.% алкиламина. Концентрирование элюата дает обогащенную смесь. Абсолютный состав подвижной фазы будет зависеть от выбранной хиральной стационарной фазы (асимметрическая смола). Приготовление 2, которое подробно раскрыто в последующем описании, является примером такого метода разделения. Когда рацемат кристаллизуется, возможны кристаллы двух различных типов. Первый тип представляет собой рацемическое соединение (истинный рацемат), упомянутое выше, при этом получают гомогенную форму кристалла, содержащего оба энантиомера в эквимолярных количествах. Второй тип является рацемической смесью или конгломератом, в котором получены две формы кристаллов в эквимолярных количествах, каждая из которых содержит один энантиомер. Хотя обе из кристаллических форм, присутствующих в рацемической смеси, имеют идентичные физические свойства, они могут иметь другие физические свойства по сравнению с истинным рацематом. Рацемические смеси могут быть разделены традиционными методами, известными специалистам в данной области, см., например, Stereochemistry of Organic Compounds by E.L. Eliel and S.H. Wilen (Wiley,1994). Таким образом, настоящее изобретение дополнительно относится к соединениям общих формул(IA)-(IF), указанным выше. Настоящее изобретение включает все фармацевтически приемлемые изотопно-меченные соединения формулы (I), в которых один или несколько атомов заменены атомами, имеющими такой же атомный номер, но атомную массу или массовое число, отличающееся от атомной массы или массового числа, обычно встречаемого в природе. Примеры изотопов, подходящих для включения в соединения изобретения, включают изотопы водорода, такие как 2 Н и 3 Н, углерода, такие как 11C, 13C и 14C, хлора, такие как 36Cl, фтора, такие как 18F,иода, такие как 123I и 125I, азота, такие как 13N и 15N, кислорода, такие как 15O, 17O и 18O, фосфора, такие как 32 Р и серы, такие как 35S. Некоторые изотопно-меченные соединения формулы (I), например, такие, которые включают радиоактивный изотоп, применимы в исследованиях распределения лекарственного вещества и/или субстрата в тканях. Радиоактивные изотопы трития, т.е. 3 Н, и углерода-14, т.е. 14 С, являются в особенности применимыми для данной цели с точки зрения легкости их включения и готовых средств обнаружения. Замещение более тяжелыми изотопами, такими как дейтерий, т.е. 2 Н, может дать некоторые терапевтические выгоды в результате более высокой метаболической стабильности, например, увеличенный период полураспада in vivo или сниженные требования к дозировке лекарственного средства и, следовательно, может быть предпочтительным в некоторых обстоятельствах. Замещение изотопами, излучающими позитрон, такими как 11 С, 18F, 15O и 13N, может быть применено в исследованиях топографией позитронного излучения (ТПИ) (PET) для проверки занятости субстрата рецептором. Изотопно-меченные соединения формулы (I) могут быть обычно получены традиционными методиками, известными специалистам в данной области, или способами, аналогичными тем, которые описаны в последующих примерах и приготовлениях, с использованием подходящего изотопно-меченного реагента вместо прежде использованного немеченного реагента. Фармацевтически приемлемые сольваты в соответствии с изобретением включают такие, в которых растворитель кристаллизации может быть изотопно-замещенным, например, D2O, d6-ацетон, d6-ДМСО. В настоящем описании раскрываются также соединения общей формулы (IG) и общей формулы в которой R1, R2, R3, R4 и R5 являются такими, как они определены выше. Представленные ниже схемы иллюстрируют способы синтеза соединений формулы (I). Схема 1 иллюстрирует получение соединений формулы (I) реакцией пептидного сочетания промежуточных соединений (II) и (III) и, в случае необходимости, добавлением подходящего основания и/или добавки (такой как гидрат 1-гидроксибензотриазола или 4-диметиламинопиридин). Схема 1 а иллюстрирует получение соединений формулы (IA) пептидным сочетанием промежуточных соединений (II) и (IIIA). Подобно, схемы, 1b-1 е иллюстрируют получение соединений формул (IC),(ID), (IE) и (IF) пептидным сочетанием промежуточных соединений (IIA) и (IIIA); (IIB) и (III); (IIB) и(IIIA) и (IIB) и (IIIB) соответственно. Соединения формул (IB), (IG) и (IH) могут быть получены подобным путем из релевантных промежуточных соединений Что касается соединений (I)-(III), то, если не указано иначе, определения R1, R2, R3, R4 и R5 на схемах 1 и 1 а-1 е являются такими, которые указаны выше для соединений формулы (I). Используемые альтернативные условия включают смешивание раствора пиперидина (амин) общей формулы (II) и пирролидина (кислота) общей формулы (III) вместе с гидрохлоридом 1-(3-диметиламинопропил)-3-этилкарбодиимида (EDCI), триэтиламином или N-метилморфолином и гидратом 1-гидроксибензотриазола (HOBt) в диметилформамиде (ДМФА) или тетрагидрофуране (ТГФ) или этилацетате при комнатной температуре. Альтернативная подходящая методика состоит в смешивании раствора промежуточных соединений общей формулы (II) и общей формулы (III) вместе с- 10011159 гексафторфосфатом O-бензотриазол-1-ил-N,N,N',N'-тетраметилурония (HBTU) или циклическим ангидридом 1-пропилфосфоновой кислоты в CH2Cl2 или EtOAc. Вместо вышеуказанных растворителей может быть использован любой подходящий инертный растворитель, при этом термин инертный растворитель означает растворитель, не содержащий карбоновую кислоту или первичный или вторичный амин. Следует использовать по меньшей мере 1 экв. каждого из реагентов сочетания и, в случае необходимости, может быть использован избыток любого одного реагента или оба вышеуказанных приема. Таким образом, в соответствии с дополнительным вариантом в настоящем изобретении предлагается способ получения соединений общей формулы (I), включающий пептидное сочетание пиперидина(амин) общей формулы (II) с пирролидином (кислота) общей формулы (III). В соответствии с предпочтительным вариантом в настоящем изобретении предлагается способ получения соединений общей формулы (IA), включающий пептидное сочетание пиперидина (амин) общей формулы (II) с пирролидином (кислота) общей формулы (IIIA). В соответствии с более предпочтительным вариантом в настоящем изобретении предлагается способ получения соединений общей формулы (ID), включающий пептидное сочетание пиперидина (амин) общей формулы (IIB) с пирролидином (кислота) общей формулы (III). В соответствии с еще более предпочтительным вариантом в настоящем изобретении предлагается способ получения соединений общей формулы (IC), включающий пептидное сочетание пиперидина (амин) общей формулы (IIA) с пирролидином (кислота) общей формулы (IIIA). В соответствии с еще одним предпочтительным вариантом в настоящем изобретении предлагается способ получения соединений общей формулы (IE), включающий пептидное сочетание пиперидина (амин) общей формулы (IIB) с пирролидином(кислота) общей формулы (IIIA). В соответствии с особенно предпочтительным вариантом в настоящем изобретении предлагается способ получения соединений общей формулы (IF), включающий пептидное сочетание пиперидина (амин) общей формулы (IIB) с пирролидином (кислота) общей формулы (IIIB). В соответствии с еще одним другим вариантом в настоящем изобретении предлагаются промежуточные соединения общей формулы (II), (IIA) и (IIB) в которых R1 и R2 являются такими, как они определены выше. В данном изобретении предпочтительны промежуточные соединения формулы (II), более предпочтительно формулы (IIA) и в особенности предпочтительно формулы (IIB), где R2=OH и R1 - монозамещенный фенил или 2,6- или 3,4- или 2,4-дизамещенный фенил или пиридинильная группа, где фенилзамещающие группы выбраны из F, Cl и OCH3. В особенности предпочтительную группу промежуточных соединений в данном изобретении представляют соединения формулы (II), более предпочтительно формулы (IIA) и в особенности предпочтительно формулы (IIB), в которой R2=-OH и R1 представляет собой фенил, 4-фторфенил, 3,4-дифторфенил,4-хлорфенил, 3-фторфенил, 4-метилфенил, 4-метоксифенил, 2,6-дифторфенил, 2,4-дифторфенил,3,4-дифторфенил или пиридин-2-ил. В соответствии с предпочтительным вариантом в настоящем изобретении предлагаются промежуточные соединения общей формулы (IIB), в которой R2=-OH и R1 представляет собой фенил, 4-фторфенил, 3,4-дифторфенил, 3-фторфенил, 4-метилфенил, 4-метоксифенил,2,6-дифторфенил, 2,4-дифторфенил, 3,4-дифторфенил или пиридин-2-ил. В соответствии с еще одним дополнительным вариантом в настоящем изобретении предлагаются промежуточные соединения общих формул (III), (IIIA) и (IIIB) в которых R3, R4 и R5 являются такими, как они определены выше. В данном изобретении предпочтительны промежуточные соединения формулы (II), более предпочтительно формулы (IIA), наиболее предпочтительно формулы (IIB), где R4 является Н или F или Cl; R5 является F или Cl и R3 представляет собой Н или -(C2-C4)алкил, -(C3-C8)циклоалкил,(C1-C2)алкил(C3-C8)циклоалкил или гетероциклическую группу. Предпочтительную группу промежуточных соединений в данном изобретении представляют соединения формулы (III), более предпочтительно формулы (IIIA) и в особенности формулы (IIIB), где R3 представляет собой -Н, i-Pr, Et или гетероциклическую группу, выбранную из пиридин-2-ильной, пиридин-3-ильной, пиридазин-3-ильной, пиразинильной, пиримидин-5-ильной, пиримидин-4-ильной, пиримидин-2-ильной или тетрагидропиран-4-ильной- 11011159 групп. В соответствии с другим вариантом в настоящем изобретении предложен способ получения соединений общей формулы (I), более предпочтительно общей формулы (IC), еще более предпочтительно общей формулы (IE) и в особенности предпочтительно общей формулы (IF) пептидным сочетанием промежуточных соединений (II) и (III), предпочтительно (IIA) и (IIIA), более предпочтительно (IIB) и (IIIA),в особенности (IIB) и (IIIB), где R2 является -OH; R1 представляет собой фенил, 4-фторфенил,3,4-дифторфенил, 3-фторфенил, 4-метилфенил, 4-метоксифенил, 2,6-дифторфенил, 2,4-дифторфенил,3,4-дифторфенил или пиридин-2-ил; R3 представляет собой -Н, t-Bu, i-Pr, Et или гетероциклическую группу, выбранную из пиридин-2-ильной, пиридин-3-ильной, пиридазин-3-ильной, пиразинильной, пиримидин-5-ильной, пиримидин-4-ильной, пиримидин-2-ильной или тетрагидропиран-4-ильной групп;R4 является Н, Cl или F и R5 является Cl или F. В соответствии с предпочтительным способом данного изобретения соединения общей формулы(IF) получают пептидным сочетанием промежуточных соединений (IIA) и (IIIA), в которых R2 является-OH; R1 представляет собой фенил, 4-фторфенил, 3,4-дифторфенил, 3-фторфенил, 4-метилфенил,4-метоксифенил, 2,6-дифторфенил, 2,4-дифторфенил, 3,4-дифторфенил или пиридин-2-ил; R3 представляет собой -Н, t-Bu, i-Pr, Et или гетероциклическую группу, выбранную из пиридин-2-ильной, пиридин 3-ильной, пиридазин-3-ильной, пиразинильной, пиримидин-5-ильной, пиримидин-4-ильной, пиримидин 2-ильной или тетрагидропиран-4-ильной групп; R4 является H, Cl или F и R5 является Cl или F. В соответствии с более предпочтительным способом данного изобретения соединения общей формулы (IF) получают пептидным сочетанием промежуточных соединений (IIA) и (IIIA), где R2 является-OH; R1 представляет собой фенил, 4-фторфенил, 3,4-дифторфенил, 3-фторфенил, 2,4-дифторфенил или 3,4-дифторфенил или пиридин-2-ил; R3 является t-Bu, i-Pr или Et и оба из R4 и R5 являются F. Схема 2 иллюстрирует альтернативный путь получения соединений общей формулы (I), имеющих диапазон R3 групп за счет использования стратегии защитных групп. Соединения общих формул (IA)(IF) могут быть также получены, когда необходимо, в соответствии с путем, показанным на схеме 2, в котором использованы соответствующие промежуточные соединения (II), (IIA) или (IIB) с соответствующим защищенным амином формулы (IV), (IVA) или (IVB) соответственно Схема 2 Что касается соединений (I), (II), (IV) и (V) на схеме 2 или (IVA) или (IVB), показанных ниже, в том случае, если не указано иначе, определения R1, R2, R3, R4 и R5 являются такими, как они указаны для соединений формулы (I). PG является азотзащитной группой На схеме 2 сочетают аминные промежуточные соединения общей формулы (II) и защищенные пирролидиновокислотные промежуточные соединения общей формулы (IV) с использованием стандартных методов сочетания пептидов, которые были прежде описаны для схемы 1, при этом получают связанное и защищенное промежуточное соединение общей формулы (V), из которого с использованием стандарт- 12011159 ных методов снятия защиты может быть удалена азотзащитная группа с получением соединения общей формулы (I), в котором R3=Н. Могут быть использованы любые подходящие азотзащитные группы (которые раскрыты в Protecting Groups in Organic Synthesis 3rd Edition T.W. Greene and P.G. Wuts, WileyInterscience, 1999). Общеизвестная азотзащитная группа (PG), подходящая для использования в данном изобретении, представляет собой трет-бутоксикарбонил, который легко удаляется обработкой кислотой,такой как трифторуксусная или хлористо-водородная кислота, в органическом растворителе, таком как дихлорметан или 1,4-диоксан. Альтернативные (группе Н) группы могут быть введены в R3 с использованием традиционных методов алкилирования. Подходящие методы алкилирования вторичных аминов включают в себя:(i) взаимодействие с альдегидом и гидридным восстановителем, таким как триацетоксиборгидрид натрия, необязательно в присутствии уксусной кислоты в инертном растворителе, таком как дихлорметан или ацетонитрил;(ii) взаимодействие с алкилгалогенидом или соответствующим образом активированным производным спирта (например, в виде сложного эфира сульфокислоты) в присутствии основания (такого как триэтиламин) в инертном растворителе. Арильные и гетероарильные группы могут быть введены в R3 замещением подходящей удаляемой группы из гетероароматического предшественника. Подходящие удаляемые группы включают галогены. В некоторых случаях для получения необходимых продуктов для проведения реакции сочетания могут потребоваться катализаторы на основе переходного металла (например, палладий, медь) или необязательно в комбинации с фосфиновым лигандом, таким как 1,1'-бинафталин-2,2'-диил-бисдифенилфосфин. Схема 3 а иллюстрирует путь получения пирролидиновокислотных промежуточных соединений общей формулы (III) из ненасыщенных сложноэфирных промежуточных соединений общей формулы Что касается соединений (III), (VI), (VII)-(XIII) на схеме 3 а, в том случае, если не указано иначе, определения R1, R2, R3, R4 и R5 являются такими, как они указаны выше для соединений формулы (I). PG2 является подходящей защитной группой для карбоновой кислоты. Соединения общей формулы (VI) могут быть получены реакцией Виттига (Wittig) или подобным олефинированием альдегидного промежуточного соединения общей формулы (X) подходящим илидом,например метил(трифенилфосфоранилиден)ацетатом или анионом фосфоната, например, таким, который получен в результате депротонирования триметилфосфоноацетата, предпочтительно в виде трансизомера. В литературе описано множество альтернативных способов получения ненасыщенных сложноэфирных промежуточных соединений общей формулы (VI), включающих этерификацию предшественника производного коричной кислоты (VII) с использованием стандартных методов этерификации или реакцию Хека (Heck), включающую взаимодействие ароматического галогенида (VIII) с подходящим акрилатным эфиром, таким как трибутилакрилат (IX), в присутствии палладиевого катализатора и подходящего основания, такого как триэтиламин.- 13011159 Образовавшееся Е-олефиновое промежуточное соединение общей формулы (VI) будет подвергаться [3+2]-азометинилидному циклоприсоединению за счет взаимодействия с соединением общей формулы (XI) с получением пирролидина, имеющего почти исключительно транс-конфигурацию. Для данной реакции требуется инертный растворитель, такой как дихлорметан или толуол или тетрагидрофуран, и активация одним или несколькими средствами:(1) кислотным катализатором, таким как ТФУ (TFA);(2) десилилирующим агентом, таким как фторид серебра;(3) нагревом. Альтернативно, получают пирролидин с почти исключительно цис-конфигурацией взаимодействием соединения общей формулы (XI) с ненасыщенным сложным эфиром или кислотой Z-олефиновой конфигурации. Такие Z-олефины могут быть получены посредством восстановления алкина по Линдлару(Lindlar) или олефинированием в соответствии с реакцией Стилла-Дженнари (Still-Gennari). Соединение общей формулы (XII), полученное в результате реакции циклоприсоединения, представляет собой рацемат, в связи с чем может потребоваться разделение на его составляющие энантиомеры, которое может быть осуществлено препаративной ВЭЖХ с использованием хиральной стационарной фазы. Альтернативно, кислотное промежуточное соединение общей формулы (III) может быть разделено стандартными методами (например, образование диастереоизомерных производных взаимодействием с энантиомерно чистым реагентом, разделение образовавшихся диастереоизомеров физическими методами и расщепление с получением кислоты (III. Промежуточные соединения общей формулы (XII) могут быть превращены в соединения общей формулы (III) гидролизом сложного эфира. Для осуществления такого превращения доступно множество методов (см. Advanced Organic Chemistry: Reactions, Mechanisms, and Structure, Fourth Edition. March,Jerry, 1992, p. 378-383, published by Wiley, New York, N.Y. USA). При особой обработке соединения общей формулы (XII) водным раствором гидроксида щелочного металла, такого как гидроксид лития, гидроксид натрия или гидроксид калия, в подходящем органическом растворителе будут получены соответствующие соединения общей формулы (III). В таких реакциях также предпочтительно используются смешивающиеся с водой органические сорастворители (такие как 1,4-диоксан или тетрагидрофуран). Предпочтительный метод гидролиза такого сложного эфира включает обработку сложного эфира триметилсиланолатом калия в инертном растворителе, таком как диэтиловый эфир, при комнатной температуре. При необходимости для способствования гидролизу реакционная смесь может быть нагрета. Гидролиз сложного эфира может быть также осуществлен с использованием кислой среды, например нагревом сложного эфира в водном растворе кислоты,такой как хлористо-водородная кислота. Некоторые сложные эфиры более выгодно гидролизуются в кислой среде, например трет-бутиловые или бензгидриловые сложные эфиры. Такие сложные эфиры могут быть расщеплены обработкой безводными кислотами, такими как трифторуксусная кислота или хлористо-водородная кислота, в инертном органическом растворителе, таком как дихлорметан. Схема 3b иллюстрирует альтернативный путь получения одиночного энантиомера пирролидиновокислотного промежуточного соединения общей формулы (III) из ненасыщенных сложноэфирных промежуточных соединений общей формулы (VI) с использованием оксазолидинона в качестве хирального вспомогательного вещества. Кислота формулы (XVIII) может быть получена гидролизом ненасыщенного сложного эфира (VI) и для получения промежуточного соединения формулы (XVIII) в качестве хирального вспомогательного вещества (в котором R предпочтительно представляет собой фенил, третичный бутил или изопропил) может быть использован оксазолидинон. Альтернативно, взаимодействие соединения формулы (VI) (когда R=СО-трет-Bu) с литиевой солью оксазолидинона в подходящем растворителе (например, ТГФ) может также обеспечить получение соединения формулы (XVIII). Соединение формулы (XVIII) будет подвергаться [3+2]-азометинилидному циклоприсоединению за счет взаимодействия с соединением общей формулы (XI) с получением диастереоизомеров (XX) и(XVIX), которые могут быть разделены хроматографией или кристаллизацией и гидролизованы с получением пирролидина формулы (III). Схема 4 показывает, что синтез защищенных пирролидиновокислотных промежуточных соединений общей формулы (IV) может быть осуществлен с использованием метода, подобного способу, раскрытому выше для промежуточного соединения общей формулы (III), за исключением того, что промежуточное соединение общей формулы (XIIA) содержит азотзащитную группу, которая может быть впоследствии удалена в ходе синтеза. После удаления защитной группы с использованием подходящих традиционных методов могут быть введены альтернативные R3 группы методами, показанными на схеме 2. Пирролидины общей формулы (IV), имеющие азотзащитную группу, могут быть также получены энантиоселективно с использованием оксазолидинона в качестве вспомогательного вещества способом,подобным тому, который показан на схеме 3b. Схема 4 Что касается соединений (VI), (XIA), (XIIA), (XII) и (IV) на схеме 4, в том случае, если не указано иначе, определения R1, R2, R3, R4 и R5 являются такими, как они указаны выше для соединений формулы(I). В формулах (XIA), (XIIA) и (IV) PG выбрана из подходящих азотзащитных групп. В формулах (VI),(XIIA), (VII) PG2 выбрана из подходящих защитных групп для карбоновой кислоты. Синтез азометинилидных предшественников общей формулы (XI) может быть осуществлен как показано на схеме 5. Первичный амин общей формулы (XIII) может быть алкилирован обработкой хлорметилтриметилсиланом, необязательно в чистом виде или в инертном растворителе, и, при необходимости,- 15011159 реакционная смесь может быть нагрета. Полученное промежуточное соединение (XIV) может быть затем подвергнуто взаимодействию с формальдегидом в метаноле и в присутствии подходящего основания,такого как карбонат калия или трет-бутиламин, с получением промежуточного соединения (XI). Для получения подобных промежуточных соединений (XIA), содержащих азотзащитную группу, может быть затем использована подобная последовательность реакций. Схема 5 Что касается соединений (XIII), (XIIIA), (XIV), (XIVA), (XIA) и (XI) на схеме 5, в том случае, если не указано иначе, определения R3 являются такими, как они указаны выше для соединений формулы (I). В формулах (XIIIA), (XIVA), (XIA) PG выбрана из подходящих азотзащитных групп. Как показано на схеме 6, пиперидиновые промежуточные соединения общей формулы (II), в которой R2=OH, могут быть получены присоединением металлоорганических нуклеофилов к кетонам общей формулы (XV), содержащим подходящую азотзащитную группу, при этом образуются промежуточные соединения общей формулы (XVI). Такое нуклеофильное присоединение обычно осуществляют при низкой температуре в безводном простом эфире или неполярном растворителе с использованием реактива Гриньяра, литийорганического соединения или другого подходящего металлоорганического реагента. Указанные металлоорганические реагенты могут быть получены обменом металл-галоген с использованием подходящего галогенидного предшественника Y-Br или Y-I и н-бутиллития или трет-бутиллития. Подходящие защитные группы включают Bn, который может быть удален гидрированием, или Boc, который может быть удален обработкой кислотой, такой как ТФУ, или РМВ, который может быть удален обработкой DDQ, CAN или хлорэтилхлорформиатом, с получением желательного пиперидинового промежуточного соединения общей формулы (II). При использовании некоторых защитных групп и при определенных условиях защитная группа может быть лабильной для обработки металлоорганическим реагентом и поэтому оба превращения могут быть осуществлены в одну стадию, например, когда PG=Boc,защитная группа может быть иногда отщеплена, если промежуточные соединения формулы (VII) обрабатывают металлоорганическим реагентом. Схема 6 Что касается соединений (XV), (XVI) и (II) на схеме 6, в том случае, если не указано иначе, определения R1 являются такими, как они указаны выше для соединений формулы (I). В формулах (XV), (XVI)PG выбрана из подходящих азотзащитных групп. Как показано на схеме 7, когда используется (3R,5S)-1-бензил-3,5-диметилпиперидин-4-он, стереохимии присоединения благоприятствует размещение гидроксильной группы в продукте в цисконфигурации относительно двух метильных групп. Регулируемое присоединение к карбонильным системам, таким как указанная, описано в литературе (Journal of Medicinal Chemistry (1964), 7(6), p. 726-8). Схема 7 Что касается соединений (XV), (XXI) и (II) на схеме 7, в том случае, если не указано иначе, определения R1 являются такими, как они указаны выше для соединений формулы (I). В формулах (XV), (XXI)PG выбрана из подходящих азотзащитных групп. Кроме этого, схема 8 иллюстрирует, что при форсированных восстановительных условиях, таких как гидрирование при высоком давлении или температуре или присутствие сильной кислоты плюс триэтилсилана, промежуточные соединения общей формулы (II), в которой R2=OH, могут быть превращены в другие промежуточные соединения общей формулы (II), в которой R2=Н. В некоторых случаях для способствования указанному превращению может потребоваться защита атома азота пиперидина. Таким образом, промежуточные соединения общей формулы (XVI) могут быть превращены в другие промежуточные соединения общей формулы (XXII), в которой R2=Н, и затем для получения соединений общей формулы (II), в которой R2=Н, удаляют защитную группу. Схема 8 Что касается соединений (XVI) и (II) на схеме 8, в том случае, если не указано иначе, определенияR1 являются такими, как они указаны выше для соединений формулы (I). В формулах (II) и (XXIII) PG выбрана из подходящих азотзащитных групп. Кроме этого, схема 9 иллюстрирует, что промежуточные соединения общей формулы (II), в которойR2=OH, могут быть превращены в дополнительные промежуточные соединения общей формулы (II), в которой R2=OMe. Такое превращение может быть осуществлено стандартным синтезом простого эфира по Вильямсону (Williamson). Т.е. спиртовая группа в соединениях общей формулы (II), в которой R2=OH,может быть депротонирована сильным основанием, таким как гидрид натрия, в безводном растворителе,таком как тетрагидрофуран или диметилформамид, и образовавшийся анион подвергают взаимодействию с йодметаном и, при необходимости, реакционную смесь нагревают. Для способствования указанному превращению может потребоваться защита атома азота пиперидина, и таким образом промежуточные соединения общей формулы (XVI), где R2=OH, могут быть превращены в дополнительные промежуточные соединения общей формулы (XXV), в которой R2=ОМе, и затем, как показано на схеме 9, для получения соединений общей формулы (II), в которой R2=ОМе, удаляют защитную группу. Схема 9 Что касается соединений (XVI) и (II) на схеме 9, в том случае, если не указано иначе, определенияR1 являются такими, как они указаны выше для соединений формулы (I). В формулах (II) и (XVI) PG выбрана из подходящих азотзащитных групп. Специалистам в данной области понятно, что, кроме защиты азотных групп, которая обсуждалась выше, в различные периоды времени синтеза соединений формулы (I) может быть необходимой защита других групп, таких как, например, гидроксильные группы, подходящей защитной группой, которую- 17011159 впоследствии удаляют. Методы удаления любой конкретной защитной группы зависят от защитной группы. Примеры методики защиты/снятия защиты см. в Protective groups in Organic synthesis TWGreene and PGM Wutz. Так, например, когда гидроксильная группа защищена в виде простого метилового эфира, условия снятия защиты включают кипячение с обратным холодильником в 48% водном растворе HBr или перемешивание с борантрибромидом в дихлорметане. Альтернативно, когда гидроксильная группа защищена в виде простого бензилового эфира, условия снятия защиты включают гидрирование в атмосфере водорода в присутствии палладиевого катализатора. В соответствии с предпочтительным вариантом в настоящем изобретении предлагаются способы получения соединений общей формулы (I) с использованием методов, аналогичных тем, которые раскрыты для получения соединений примера 1 с помощью приготовлений 1-5 и 12-16, и более предпочтительно примера 5 с помощью приготовлений 1, 21, 22b, 4, 5 и 12-16, имеющих определенную в данном изобретении стереохимию. В соответствии с дополнительным вариантом в настоящем изобретении независимо предлагается промежуточное соединение приготовления 1; и/или промежуточное соединение приготовления 2; и/или промежуточное соединение приготовления 3; и/или промежуточное соединение приготовления 4; и/или промежуточное соединение приготовления 5; и/или промежуточное соединение приготовления 21; и/или промежуточное соединение приготовления 22b; и/или промежуточное соединение приготовления 12; и/или промежуточное соединение приготовления 13; и/или промежуточное соединение приготовления 14; и/или промежуточное соединение приготовления 15; и/или промежуточное соединение приготовления 16. Представленные выше общие механизмы реакций получения новых исходных продуктов, использованных в предшествующих методах, являются традиционными, и подходящие реагенты и условия реакций для их выполнения или приготовления, а также методики выделения требуемых продуктов будут хорошо понятны специалистам в данной области из ссылок на литературные источники и представленных в описании примеров и приготовлений. Активность к MCR4. Соединения настоящего изобретения полезны в качестве агонистов MCR4 при лечении различных болезненных состояний. Вышеуказанные агонисты MCR4 предпочтительно показывают фармакологическую эффективность на рецепторе МС 4, выраженную в виде EC50, менее примерно 1000 нМ, более предпочтительно менее 150 нМ, еще более предпочтительно менее примерно 100 нМ, все же более предпочтительно менее примерно 50 нМ и в особенности менее примерно 10 нМ, где измерение фармакологической эффективности в виде ЕС 50 на MCR4 может быть осуществлено с использованием раскрытых в последующем описании протоколов С или Е. Соединения в соответствии с настоящим изобретением, включающие соединения примеров 12, 20, 16, 48, 1, 5, 6, 22, 13, 9, 10, 50, 14, 17, 19, 53, 40, 15, 52, 51, 8, 33, 31, 34, 35, 36, 42, 44 и 47, испытывали на активность и было найдено, что они показывают фармакологическую эффективность на рецепторе МС 4 менее примерно 150 нМ. Таким образом, в соответствии с дополнительным вариантом в настоящем изобретении предлагаются соединения формулы (I), имеющие функциональную эффективность в отношении рецептора МС 4 менее примерно 150 нМ. Испытывали на активность предпочтительную группу соединений в соответствии с настоящим изобретением, включающую соединения примеров 1, 5, 6, 22, 13, 9, 17, 19, 53, 15, 52, 51, 8, 31, 34, 35, 42, 44 и 47, и было найдено, что они показывают функциональную эффективность в отношении рецептора МС 4 менее примерно 50 нМ. Испытывали на активность дополнительную предпочтительную группу соединений в соответствии с настоящим изобретением, включающую соединения примеров 1, 5, 9, 19, 8, 31, 34, 35, 42 и 47, и было найдено, что они показывают функциональную эффективность в отношении рецептора МС 4 менее примерно 10 нМ. Предпочтительные соединения данного изобретения проявляют функциональную эффективность в отношении рецептора MCR4, определенную выше в данном описании, и являются селективными кMCR4 по сравнению к MCR1. Вышеуказанные агонисты MCR4 предпочтительно имеют селективность кMCR4 по сравнению к MCR1, при этом вышеуказанные агонисты рецептора MCR4 по меньшей мере примерно в 10 раз, предпочтительно по меньшей мере примерно в 20 раз, более предпочтительно по меньшей мере примерно в 30 раз, еще более предпочтительно по меньшей мере примерно в 100 раз, все же более предпочтительно по меньшей мере примерно в 300 раз, все же даже более предпочтительно по меньшей мере примерно в 500 раз и в особенности предпочтительно по меньшей мере примерно в 1000 раз более функционально селективны к рецептору MCR4 по сравнению к рецептору MCR1, где оценки относительной селективности основаны на измерении функциональной эффективности в отношенииMCR1 и MCR4, которое может быть осуществлено с использованием раскрытых в последующем описании протоколов А и С или Е. Соединения в соответствии с настоящим изобретением, включающие соединения примеров 1, 5, 6, 13, 10, 50, 14, 17, 33, 31 и 35, показывают функциональную эффективность в отношении рецептора MCR4 и в результате испытаний было найдено, что они показывают селективность к MCR4 примерно в 10 раз большую, чем к MCR1. Таким образом, в соответствии с дополнительным вариантом в настоящем изобретении предлагаются соединения формулы (I), которые показывают функциональную эффективность в отношении рецептора MCR4 и показывают селективность к MCR4 при- 18011159 мерно в 10 раз большую, чем к MCR1. Предпочтительная группа соединений в соответствии с настоящим изобретением, включающая соединения примеров 1, 5, 13, 14, 17, 31 и 35, показывает функциональную эффективность в отношении рецептора MCR4 и в результате испытаний было найдено, что они показывают селективность к MCR4 примерно в 30 раз большую, чем к MCR1. Дополнительная предпочтительная группа соединений в соответствии с настоящим изобретением, включающая соединения примеров 13, 14, 31 и 35, показывает функциональную эффективность в отношении рецептора MCR4 и в результате испытаний было найдено, что они показывают селективность к MCR4 примерно в 100 раз большую, чем к MCR1. Агонисты MCR4 предпочтительно имеют селективность к MCR4 по сравнению к MCR3, при этом вышеуказанные агонисты рецептора MCR4 по меньшей мере примерно в 10 раз, предпочтительно по меньшей мере примерно в 30 раз, более предпочтительно по меньшей мере примерно в 100 раз, все же более предпочтительно по меньшей мере примерно в 300 раз, все же даже более предпочтительно по меньшей мере примерно в 500 раз и в особенности предпочтительно по меньшей мере примерно в 1000 раз более функционально селективны к рецептору MCR4 по сравнению к рецептору MCR3, где оценки относительной селективности основаны на измерении функциональной эффективности в отношенииMCR3 и MCR4, которое может быть осуществлено с использованием раскрытых в последующем описании протоколов А и В или Е. Соединения примеров 1, 2 и 3 показывают функциональную эффективность в отношении рецептора MCR4 и в результате испытаний было найдено, что они показывают селективность к MCR4 примерно в 30 раз большую, чем к MCR3. Предпочтительные соединения данного изобретения показывают функциональную эффективность в отношении рецептора MCR4, определенную выше в данном описании, и являются селективными кMCR4 по сравнению к MCR5. Вышеуказанные агонисты MCR4 предпочтительно имеют селективность кMCR4 по сравнению к MCR5, при этом вышеуказанные агонисты рецептора MCR4 по меньшей мере примерно в 10 раз, предпочтительно по меньшей мере примерно в 30 раз, более предпочтительно по меньшей мере примерно в 100 раз, еще более предпочтительно по меньшей мере примерно в 300 раз, все же даже более предпочтительно по меньшей мере примерно в 500 раз и в особенности предпочтительно по меньшей мере примерно в 1000 раз более функционально селективны к рецептору MCR4 по сравнению к рецептору MCR5, где оценки относительной селективности основаны на измерении функциональной эффективности, которое может быть осуществлено с использованием раскрытых в последующем описании протоколов D и Е. Соединения в соответствии с настоящим изобретением, включающие соединения примеров 1, 5, 6, 22, 13, 9, 10, 50, 14, 17, 19, 53, 15, 52, 51, 33, 31, 35, 42 и 44, показывают функциональную эффективность в отношении рецептора MCR4 и в результате испытаний было найдено, что они показывают селективность к MCR4 примерно в 10 раз большую, чем к MCR5. Таким образом, в соответствии с дополнительным вариантом в настоящем изобретении предлагаются соединения формулы(I), которые показывают функциональную эффективность в отношении рецептора MCR4 и показывают селективность к MCR4 примерно в 10 раз большую, чем к MCR5. Предпочтительная группа соединений в соответствии с настоящим изобретением, включающая соединения примеров 1, 5, 22, 13, 9, 50, 17, 19,53, 15, 52, 31, 33, 35, 42 и 44, показывает функциональную эффективность в отношении рецептора MCR4 и в результате испытаний было найдено, что они показывают селективность к MCR4 примерно в 100 раз большую, чем к MCR5. Дополнительная предпочтительная группа соединений в соответствии с настоящим изобретением, включающая соединения примеров 22, 13, 19, 15, 35, 42 и 44, показывает функциональную эффективность в отношении рецептора MCR4 и в результате испытаний было найдено, что они показывают селективность к MCR4 примерно в 300 раз большую, чем к MCR5. Агонисты MCR4 предпочтительно имеют селективность к MCR4 по сравнению к MCR1 и MCR3,при этом вышеуказанные агонисты рецептора MCR4 по меньшей мере примерно в 10 раз, предпочтительно по меньшей мере примерно в 30 раз, более предпочтительно по меньшей мере примерно в 100 раз, еще более предпочтительно по меньшей мере примерно в 300 раз, все же даже более предпочтительно по меньшей мере примерно в 1000 раз более функционально селективны к рецептору MCR4 по сравнению к рецепторам MCR1 и MCR3. Предпочтительные соединения данного изобретения показывают функциональную эффективность в отношении рецептора MCR4, определенную выше в данном описании, и являются селективными кMCR4 по сравнению к MCR1 и MCR5. Вышеуказанные агонисты MCR4 предпочтительно имеют селективность к MCR4 по сравнению к MCR1 и MCR5, при этом вышеуказанные агонисты рецептора MCR4 по меньшей мере примерно в 10 раз, предпочтительно по меньшей мере примерно в 30 раз, более предпочтительно по меньшей мере в 100 раз, еще более предпочтительно по меньшей мере примерно в 300 раз, все же даже более предпочтительно по меньшей мере примерно в 500 раз и в особенности предпочтительно по меньшей мере примерно в 1000 раз более функционально селективны к рецептору MCR4 по сравнению с рецепторами MCR1 и MCR5. Соединения в соответствии с настоящим изобретением, включающие соединения примеров 1, 5, 6,13, 10, 50, 14, 17, 33, 31 и 35, показывают функциональную эффективность в отношении рецептораMCR4 и в результате испытаний было найдено, что они показывают селективность к рецептору MCR4 примерно в 10 раз большую по сравнению с селективностью к рецепторам MCR1 и MCR5. Таким обра- 19011159 зом, в соответствии с дополнительным вариантом в настоящем изобретении предлагаются соединения формулы (I), которые показывают функциональную эффективность в отношении рецептора MCR4 и показывают селективность к рецептору MCR4 примерно в 10 раз большую по сравнению с селективностями к рецепторам MCR1 и MCR5. Предпочтительная группа соединений в соответствии с настоящим изобретением, включающая соединения примеров 1, 5, 13, 31 и 35, показывает функциональную эффективность в отношении рецептора MCR4, и в результате испытаний было найдено, что они показывают селективность к рецептору MCR4 примерно в 100 раз большую по сравнению с селективностями к рецепторам MCR1 и MCR5. Агонисты MCR4 предпочтительно имеют селективность к MCR4 по сравнению к MCR3 и MCR5,при этом вышеуказанные агонисты рецептора MCR4 по меньшей мере примерно в 10 раз, предпочтительно по меньшей мере примерно в 30 раз, более предпочтительно по меньшей мере примерно в 100 раз, все же более предпочтительно по меньшей мере примерно в 300 раз, наиболее предпочтительно по меньшей мере примерно в 1000 раз более функционально селективны к рецептору MCR4 по сравнению с селективностями к рецепторам MCR3 и MCR5. Кроме их роли при лечении половой дисфункции, соединения настоящего изобретения являются,вероятно, эффективными для ряда дополнительных показаний, раскрытых в данном описании. Использованные в данном описании термины лечение, лечить или терапия предназначены для включения в себя как предупреждения заболевания, так и лечения, т.е. они включают превентивное и паллиативное лечение указанных состояний. Соединения изобретения применимы при лечении заболеваний, расстройств или состояний, включающих, но без ограничения, лечение мужских и женских половых дисфункций, включающих гипоактивное сексуальное влечение, нарушение полового возбуждения, оргазменное расстройство и/или боль у женщин в половых органах, мужскую эректильную дисфункцию, ожирение (путем снижения аппетита,повышения интенсивности обмена веществ, снижения всасывания жира или уменьшения потребности в углеводах), сахарный диабет (путем повышения толерантности к глюкозе, снижения резистентности к инсулину), гипертензию, гиперлипидемию, остеоартрит, рак, заболевание желчного пузыря, приступы апноэ во сне, депрессию, тревогу, компульсивное побуждение, неврозы, бессонницу/расстройство сна,токсикоманию, боль, жар, воспаление, иммунную модуляцию, ревматоидный артрит, задубление кожи, акне (угри) и другие кожные заболевания, а также для нейрозащитной активации и улучшения познавательной способности и памяти, включая лечение болезни Альцгеймера. Некоторые соединения формулы (I) показывают высокоспецифическую активность к рецептору меланокортина-4, что делает их в особенности полезными при лечении мужской и женской половых дисфункций, а также ожирения. Соединения настоящего изобретения применимы при лечении мужской и женской половой дисфункции, в особенности мужской эректильной дисфункции. Женская половая дисфункция (ЖПД) (FSD) включает нарушение полового возбуждения у женщин(FSAD), нарушения полового влечения, такие как гипоактивное сексуальное влечение (потеря интереса к сексу) и оргазменные расстройства, такие как аноргазмия (отсутствие способности достигать оргазма). Мужская половая дисфункция включает мужскую эректильную дисфункцию (МЭД) (MED) и эякуляторные расстройства, такие как аноргазмия (отсутствие способности достигать оргазма) или нарушения полового влечения, такие как гипоактивное сексуальное влечение (потеря интереса к сексу). Соединения настоящего изобретения являются в особенности применимыми при лечении женских половых дисфункций, включающих гипоактивное сексуальное влечение, нарушение полового возбуждения, оргазменное расстройство, боль у женщин в половых органах и мужскую эректильную дисфункцию. Соединения настоящего изобретения являются в особенности подходящими для лечения женских половых дисфункций, мужской эректильной дисфункции, ожирения и диабета. Мужская эректильная дисфункция (МЭД) (MED). Соединения настоящего изобретения применимы при лечении мужской половой дисфункции, в особенности мужской эректильной дисфункции. Мужскую эректильную дисфункцию (МЭД), иначе известную как мужское эректильное расстройство, определяют как неспособность достигать и/или поддерживать эрекцию полового члена, необходимую для удовлетворительного полового сношения (NIHConsensus Development Panel on Impotence, 1993). Установлено, что распространенность эректильной дисфункции (ЭД) (ED) всех степеней (минимальная, умеренная и полная импотенция) составляет 52% у мужчин в возрасте от 40 до 70 лет, при этом ее более высокие степени наблюдаются у мужчин старше 70 лет (Melman et al., 1999, J. Urology, 161, p. 511). Данное состояние оказывает значительное отрицательное действие на качество жизни человека и его партнера и часто вызывают повышенную тревогу и напряжение, которые могут привести к депрессии и низкой самооценке. Два десятилетия назад МЭД рассматривали в основном как физиологическое расстройство (Benet et al., 1994, Comp. Ther., 20: 669-673), тогда как в настоящее время известно, что у большинства индивидуумов имеется лежащая в ее основе органическая причина. В результате были сделаны большие успехи в установлении механизма нормальной эрекции полового члена и патофизиологии МЭД.- 20011159 Эрекция полового члена представляет собой гемодинамическое событие, зависящее от баланса сокращения и расслабления гладкой мышцы пещеристых тел и сосудистой сети полового члена (Lerner etal., 1993, J. Urology, 149, 1256-1255). Гладкая мышца пещеристых тел отнесена также в данном описании к корпоральной гладкой мышце или во множественном смысле к пещеристым телам. Расслабление гладкой мышцы пещеристых тел приводит к усиленному кровотоку в трабекулярные промежутки пещеристых тел, что вызывает их наполнение кровью и сдавливание дренирующих вен. В результате происходит значительное повышение кровяного давления, которое приводит к эрекции (Naylor, 1998, J. Urology,81, 424-431). Изменения, происходящие во время процесса эрекции, являются комплексными и требуют высококоординированной регуляции периферической и центральной нервной систем и эндокринной системы(Naylor, 1998, J. Urology, 81, 424-431). Сокращение корпоральной гладкой мышцы модулируется симпатической норадренергической иннервацией через активацию постсинаптических 1 адренорецепторов. МЭД может быть связана с повышением эндогенного тонуса гладкой мышцы пещеристого тела. Однако процесс расслабления корпоральной гладкой мышцы частично опосредован неадренергической, нехолинергической (НАНХ) (NANC) нейтротрансмиссией. В половом члене найден ряд других НАНХ нейротрансмиттеров, иных, чем NO,такие как пептид, связанный с геном кальцитонина (CGRP), и вазоактивный интестинальный пептид(ВИП) (VIP). Основным расслабляющим фактором, ответственным за опосредование указанного расслабления, является оксид азота (NO), который синтезируется из L-аргинина с помощью синтазы оксида азота (COA) (NOS) (Taub et al., 1993, Urology, 42, 698-704). Предполагается, что снижение тонуса корпоральной гладкой мышцы может помогать NO вызывать расслабление пещеристого тела. Во время полового возбуждения у мужчин NO высвобождается из нейронов и эндотелия и связывается с растворимой гуанилатциклазой (рГЦ) (sGC), расположенной в клетках гладкой мышцы и эндотелии, и активирует ее,что приводит к повышению уровней внутриклеточного циклического гуанозин 3',5'-монофосфата(цГМФ) (cGMP). Повышение уровней цГМФ приводит к расслаблению пещеристого тела вследствие снижения внутриклеточной концентрации кальция ([Ca2+]i) через неизвестные механизмы, которые, как предполагается, включают активацию протеинкиназы G (возможно, благодаря активации насосов Ca2+ и активированным Ca2+ каналам K+). В центральной нервной системе выявлены многочисленные потенциальные участки для модуляции сексуального поведения. Предполагается, что основные нейротрансмиттеры включают серотонин,норэпинефрин, окситоцин, оксид азота, допамин и меланокортины, например альфа-меланоцитстимулирующий гормон. Имитированием действий одного из данных основных нейротрансмиттеров можно регулировать сексуальную функцию. Меланокортины представляют собой пептиды, происходящие от проопиомеланокортинов (ПОМК)(РОМС), которые связываются со связанными G-протеином рецепторами (GPCR) меланокортинового семейства рецепторов и активируют их. Меланокортины регулируют разное число физиологических процессов, включающих половую функцию и сексуальное поведение, потребность в пище и обмен веществ. Имеется пять рецепторов меланокортина MCR1, MCR2, MCR3, MCR4 и MCR5, которые клонированы и экспрессированы в различных тканях. MCR1 специфически экспрессирован в меланоцитах и клетках меланомы, MCR2 является рецептором АСТН и экспрессирован в ткани надпочечника, MCR3 предпочтительно экспрессирован в головном мозге и лимбической системе, MCR4 обширно экспрессирован в головном мозге и спинном мозге и MCR5 экспрессирован в головном мозге и во многих периферических тканях, включающих кожу, жировую ткань, скелетные мышцы и лимфоидную ткань. MCR3 может быть вовлечен в регуляцию половой функции, потребление пищи и термогенез. Было показано,что активация MCR4 вызывает эрекцию полового члена у грызунов и инактивация MCR4 вызывает ожирение (обзор в Hadley, 1999, Ann NY Acad. Sci., 885: 1-21, Wikberg et al. 2000, Pharmacol Res., 42(5), 393420). Было найдено, что синтетические агонисты рецептора меланокортина инициируют эрекцию у мужчин с психогенной эректильной дисфункцией (Wessells et al., Int. J. Impot. Res. 2000 Oct.; 12 Suppl. 4: S749). Wessels et al. описывают действие меланотана II (MT II), неселективного агониста рецептора меланокортина, на мужчин с эректильной дисфункцией (ЭД). MT II вводили 20 мужчинам с психогенной и органической ЭД, при этом проводили двойной слепой перекрестный эксперимент, в котором в качестве контроля использовали плацебо. Ригидность полового члена регистрировали в течение 6 ч с использованием сканера RigiScan. Степень сексуального влечения и побочные действия регистрировали в соответствии с опросным листом. В отсутствие сексуальной стимуляции меланотан II вызывал эрекцию полового члена у 17 из 20 мужчин. Субъекты, подвергнутые эксперименту в среднем в течение 41 мин, имели ригидность 80%. O повышенном сексуальном влечении сообщалось после ведения 13/19 (68%) доз МТ II по сравнению с 4/21 (19%) плацебо (Р 0,01). Часто сообщалось о побочных действиях МТ II, выражающихся в тошноте и зевоте; при дозе 0,025 мг/кг массы тела у 12,9% субъектов была сильная тошнота. Побочные неблагоприятные реакции, наблюдаемые после введения MT II, могут быть результатом активации MC-1R, MC-2R, MC-3R и/или MC-5R.- 21011159 В данном изобретении предполагается, что селективный агонист MCR4 может быть введен перорально (включая буккальное или подъязычное введение) и будет эффективным при лечении женской половой дисфункции или мужской эректильной дисфункции, но при этом не оказывает значительных побочных действий, таких как те, которые наблюдались Wessels et al., т.е. селективное средство будет более толерантным. Другой синтетический пептидный аналог альфа-MSH представляет собой палатин РТ-141. Он является агонистом рецепторов меланокортина, включающих MCR3 и MCR4. Molinoff et al. (Ann N.Y. Acad.Sci. (2003), 994, 96-102) описывают, каким образом введение РТ-141 крысам и приматам, не являющимися человеком, приводит к эрекциям полового члена. Системное введение РТ-141 крысам активирует нейроны в гипоталамусе за счет повышения c-Fos иммунореактивности. Нейроны в этом же самом участке центральной нервной системы поглощают вирус псевдобешенства, впрыскиваемый в пещеристое тело полового члена крыс. Введение РТ-141 (внутриназально или подкожно) здоровому человеку и больным с эректильной дисфункцией приводило к быстрому, зависящему от дозы, повышению эректильной активности. Применение РТ-141 для половой дисфункции раскрыто в патентах США : 5576290, 6579968 и заявке США 2002/0107182 А 1. Пептиды, такие как МТ II или РТ-141, кроме того, интенсивно метаболизируются в кишечнике и, как таковые, их наиболее эффективно вводят парентеральным путем, например подкожно, внутривенно, внутриназально или внутримышечно, поскольку они не всасываются в большой круг кровообращения, когда их вводят перорально. Следовательно, желательно разработать соединения-агонисты MCR4 для лечения мужской и женской половой дисфункции, подходящие для пероральной доставки (включая буккальное или подъязычное введение), и/или уменьшить или устранить нежелательные побочные действия, такие как тошнота. В данном изобретении предполагается, что селективные агонисты MCR4 в соответствии с настоящим изобретением будут показывать пероральную биодоступность и, как таковые, их можно будет дополнительно вводить перорально (включая буккальное или подъязычное введение). Имеется ряд сообщений, показывающих, что селективные агонисты MCR4 повышают эректильную активность у крыс (Martin et al., 2002, Eur. J. Pharmacol., 454 (1), 71-79; Van Der Ploeg et al., 2002, Proc.N-[(3R)-1,2,3,4-тетрагидроизохинолиний-3-илкарбонил]-(1R)-1-(4 хлорбензил)-2-[4-циклогексил-4-(1 Н-1,2,4-триазол-1-илметил)пиперидин-1-ил]-2-оксоэтиламин (1), который является сильнодействующим селективным агонистом рецептора меланокортина 4 подтипаCragnolini et al. (Neuropeptides, 34 (3-4), 211-5) показали, что альфа-MSH значительно улучшает сексуальное поведение у крыс-самок при лордозе после инъекции в вентромедиальное ядро головного мозга. Кроме того, они показали, что HS014 (предполагаемый антагонист MCR4, Vergoni 1998, Eur. J. Pharmacol. 362(2-3), 95-101) в зависимости от дозы блокирует просексуальное действие альфа-MSH при лордозе у крыс-самок. Способы стимуляции сексуальной ответной реакции у женщин с использованием различных меланотропных пептидов (подобных МТ II) раскрыты в патенте США 6051555.MCR4 является, по существу, инициатором мужского и женского сексуального поведения. Соответственно, настоящее изобретение относится к применению соединения формулы (I) при получении лекарственного средства для лечения мужской и женской половой дисфункции и в особенности мужской эректильной дисфункции. Больные с МЭД от слабой до сильновыраженной будут получать пользу от лечения соединениями настоящего изобретения. Однако в ранних исследованиях предполагается, что степень реагирования больных со слабой, умеренной и сильновыраженной МЭД может быть больше при использовании комбинации селективный агонист MCR4/ингибитор PDE5. Термины слабая, умеренная и сильновыраженная МЭД известны специалистам в данной области, а терапевтические рекомендации могут быть найдены в The Journal of Urology, vol. 151, 54-61) (Jan. 1994). В ранних исследованиях предполагается, что нижеуказанные группы больных с МЭД будут получать пользу от лечения селективным агонистом MCR4 и/или PDE5i (или другой комбинацией, указанной в последующем описании). Такие группы больных, которые подробно раскрыты в Clinical Andrology,vol. 23,4, p. 773-782 и главе 3 книги I Eardley and K. Sethia "Erectile Dysfunction-Current Investigationand Management", published by Mosby-Wolfe, включают больных, имеющих следующие расстройства: психогенную, органическую, сосудистую, эндокринологическую, нейрогенную, артериогенную, вызванную лекарственным веществом половую дисфункцию (лактогенную) и половую дисфункцию, связанную с кавернозными факторами, в особенности веногенными факторами. Соответственно, настоящее изобретение относится к применению соединения формулы (I) при изготовлении лекарственного средства в комбинации с ингибитором PDE5 для лечения мужской эректильной дисфункции. Подходящие ингибиторы PDE5 раскрыты в последующем описании.- 22011159 Женская половая дисфункция (ЖПД) (FSD). Соединения настоящего изобретения применимы при лечении женской половой дисфункции(ЖПД) (FSD), в особенности FSAD. В соответствии с изобретением ЖПД может быть определена как трудность или неспособность женщины находить удовлетворение в сексуальном выражении. Термин ЖПД является собирательным термином для нескольких разных женских сексуальных расстройств (Leiblum, S.R. (1998) - Definition andUrology, 54, 385-391). Женщина может утратить половое влечение, иметь трудности с половым возбуждением или оргазмом, боль при половом акте или комбинацию из указанных проблем. ЖПД могут вызвать некоторые типы заболеваний, медикаментозное лечение, травмы или психологические проблемы. Лечение при разработке нацелено на лечение специфических подтипов ЖПД, предпочтительно на нарушение полового влечения и полового возбуждения. Категории ЖПД лучше всего определяют сопоставлением их с фазами сексуальной реакции здоровой женщины: влечение, возбуждение и оргазм (Leiblum, S.R. (1998) - Definition and classification of female sexual disorders. Int. J. Impotence Res., 10, S104-S106). Влечение или либидо является побуждением к сексуальному выражению. Его проявления часто включают сексуальные мысли в компании интересующего партнера или при подвержении другому эротическому раздражителю. Возбуждение является сосудистой реакцией на сексуальную стимуляцию, важным компонентом которой является прилив крови к половым органам, и включает увеличенное вагинальное смазывание, растяжение влагалища и повышенное восприятие/чувствительность половых органов. Оргазм представляет собой освобождение от сексуального напряжения, которое достигает высшей точки во время возбуждения. Следовательно, ЖПД возникает, когда женщина имеет неадекватную или неудовлетворительную реакцию на любой из указанных фаз, обычно это влечение, возбуждение или оргазм. Категории ЖПД включают гипоактивное сексуальное влечение, нарушение полового возбуждения, оргазменные расстройства и боль в половых органах. Хотя соединения изобретения будут улучшать реакцию половых органов на сексуальную стимуляцию (как это имеет место при нарушении полового возбуждения у женщин), поступая так можно также снять сопутствующую боль, дистресс и дискомфорт, связанные с половым актом и таким образом лечить другие женские сексуальные расстройства. Гипоактивное сексуальное влечение присутствует в том случае, если женщина не имеет влечения или имеет незначительное сексуальное влечение и не имеет или имеет небольшое число сексуальных мыслей или фантазий. Такой тип ЖПД может быть вызван низкими уровнями тестостерона, обусловленными или естественной менопаузой, или менопаузой, связанной с хирургическим вмешательством. Другие причины включают болезнь, медикаментозное лечение, усталость, депрессию и тревогу. Нарушение женского полового возбуждения (FSAD) характеризуется неадекватной реакцией половых органов на сексуальную стимуляцию. Половые органы не подвергаются приливу крови, который характеризует нормальное сексуальное возбуждение. Влагалищные стенки плохо смазываются и поэтому половой акт является болезненным. Оргазм может быть задержан. Нарушение полового возбуждения может быть вызвано пониженным содержанием эстрогена в менопаузе или после родов и во время лактации, а также заболеванием с сосудистыми компонентами, таким как диабет и атеросклероз. Другие причины являются результатом лечения мочегонными средствами, антигистаминными средствами, антидепрессантами, например селективными ингибиторами обратного захвата серотонина (SSRI), или антигипертензивными средствами. Боль в половых органах (включает диспареунию и вагинизм) характеризуется болью, возникающей в результате проникновения, и может быть вызвана медикаментозным лечением, которое снижает смазывание половых органов, эндометриозом, воспалением тазовых органов, воспалительным заболеванием кишечника или заболеванием мочевых путей. Как указывалось выше, предполагается, что MCR4 является инициатором сексуального поведения. Клитор рассматривается как гомолог полового члена (Levin, R.J. (1991), Ехр. Clin. Endocrinol., 98, 61-69); такой же механизм, который обеспечивает эректильную реакцию у мужчин, продуцирует при ЖПД прилив крови к женским половым органам с сопутствующим эффектом. Кроме того, имеются изменения процептивности и восприимчивости (лордоз). Таким образом, в соответствии с предпочтительным аспектом изобретения предлагается применение соединения формулы (I) при получении лекарственного средства для лечения или профилактики женской половой дисфункции, в особенности гипоактивного сексуального влечения, нарушения полового возбуждения, оргазменного расстройства и боли в половых органах. Соединения формулы (I) предпочтительно применимы при лечении или профилактике нарушения полового возбуждения, оргазменного расстройства и гипоактивного сексуального влечения, и наиболее предпочтительно при лечении или профилактике нарушения полового возбуждения. В предпочтительном варианте соединения формулы (I) применимы при лечении субъекта с нарушением женского полового возбуждения и сопутствующего гипоактивного сексуального влечения.- 23011159 Диагностическое и статистическое руководство (DSM) IV Американской психиатрической ассоциации определяет нарушение женского полового возбуждения (FSAD), какстойкую или повторяющуюся неспособность достигать или поддерживать до завершения половой активности адекватный ответ смазка-набухание при половом возбуждении. Нарушение должно вызывать заметный дистресс или межличностные трудности. Половое возбуждение состоит из вазогиперемии в тазе, влагалищного смазывания и расширения и набухания наружных половых органов. Нарушение вызывает заметный дистресс и/или межличностные трудности.FSAD представляет собой широко распространенное половое расстройство, поражающее женщин предменопаузального, перименопаузального и постменопаузального периода жизни ( гормонзаместительная терапия (ГЗТ) (HRT. Оно связано с сопутствующими расстройствами, такими как депрессия,сердечно-сосудистые заболевания, диабет и мочеполовые расстройства (UG). Первичные последствия FSAD включают отсутствие прилива крови/набухания, отсутствие смазывания и отсутствие доставляющего удовольствие полового ощущения. Вторичные последствия FSAD включают пониженное сексуальное влечение, боль во время полового акта и трудности в достижении оргазма. Недавно было сделано предположение, что по меньшей мере у части больных с симптомами FSAD имеются сосудистые заболевания (Goldstein et al., Int. J. Impot. Res., 10, S84-S90, 1998), при этом данные,полученные при испытаниях на животных, поддерживают такую точку зрения (Park et al., Int. J. Impot.R.J. Levin указывает, что посколькумужские и женские половые органы эмбриологически развиваются из общей зачаточной ткани, это доказывает, что [такие] мужские и женские половые структуры являются гомологами друг друга. Следовательно, клитор является гомологом полового члена и половые губы являются гомологами мошоночного мешочка (Levin, R.J. (1991), Exp. Clin. Endocrinol., 98, 6169). Лекарства-кандидаты для лечения FSAD, которые исследуют на их эффективность, представляют собой лекарственные средства для терапии первичной эректильной дисфункции, которые ускоряют кровообращение в мужских половых органах. Соединения настоящего изобретения являются выгодными за счет обеспечения средств для восстановления нормального полового возбуждения, а именно повышенного прилива крови к половым органам, приводящего к наполнению кровью влагалища, клитора и половых губ. В результате улучшается смазывание влагалища вследствие транссудации плазмы, повышается вагинальная эластичность и половая чувствительность. Следовательно, настоящее изобретение предлагает средства для восстановления или потенцирования нормального полового возбуждения. Таким образом, согласно предпочтительному аспекту изобретения предлагается применение соединения формулы (I) при изготовлении лекарственного средства для лечения или профилактики нарушения женского полового возбуждения. Термин женские половые органы в соответствии с мнением авторов данного изобретения означает следующее. Половые органы состоят из внутренних и наружных органов. Внутренние органы расположены в тазе и состоят из яичников, маточных труб, матки и влагалища. Наружные органы находятся на поверхности мочеполовой диафрагмы и ниже тазовой дуги. Они включают лобок, большие половые губы и малые половые губы, клитор, преддверие, луковицы преддверия и большие (бартолиновые) железы преддверия влагалища (Gray's Anatomy, CD. Clemente, 13th American Edition). Соединения изобретения находят применение в следующих группах населения, больных ЖПД: молодые, пожилые женщины, женщины предменопаузального, перименопаузального и постменопаузального периода жизни с гормонзаместительной терапией или без таковой. Соединения изобретения находят применение для больных ЖПД, возникающей в результате:(i) васкулогенных этиологий, например сердечно-сосудистых или атеросклеротических заболеваний, гиперхолестеринемии, курения сигарет, диабета, гипертензии, облучения и промежностной травмы,травматического повреждения сосудистой системы подвздошно-подчревной половой области;(ii) нейрогенных этиологий, таких как травмы спинного мозга или заболевания центральной нервной системы, включающие рассеянный склероз, диабет, паркинсонизм, инсульты, периферические невропатии, травму или радикальную операцию в области таза;(iii) гормональных/эндокринных этиологий, таких как дисфункция системы гипоталамус/гипофиз/половая железа или дисфункция яичников, дисфункция поджелудочной железы, хирургическая или лекарственная кастрация, андрогенная недостаточность, высокие уровни циркулирующего пролактина, например гиперпролактинемия, естественная менопауза, преждевременное угасание функции яичников, гипер- и гипотиреоз;(iv) психогенных этиологий, таких как депрессия, обсессивно-компульсивное расстройство, тревога, послеродовая депрессия/Baby Blues, эмоциональные стрессы, супружеская дискордантность, дисфункциональные позы, сексуальные фобии, религиозное угнетение или посттравматические переживания; и/или(v) вызванной лекарственным веществом половой дисфункции в результате: терапии селективными ингибиторами обратного захвата серотонина (SSRI) и другими антидепрессантами (трициклическими и специальными транквилизаторами), антигипертензивных терапий, приема симпатолитических средств,затяжной терапии пероральными противозачаточными средствами. Соединения настоящего изобретения могут быть доставлены в комбинации со вспомогательным активным веществом для лечения половой дисфункции, ожирения или диабета. Подходящие вспомогательные активные вещества, предназначенные для использования в комбинациях настоящего изобретения, включают: 1) соединения, которые модулируют действие натрийуретических факторов, в особенности предсердного натрийуретического фактора (известного также как предсердный натрийуретический пептид),натрийуретических факторов В типа и С типа, такие как ингибиторы нейтральной эндопептидазы, и в особенности соединения, раскрытые в WO 02/02513, WO 02/03995, WO 02/079143 и ЕР-А-1258474, и главным образом соединение примера 22, представленного в WO 02/079143, которым является(2S)-2-[(1-3-[4-(хлорфенил)пропил]аминокарбонил)циклопентил]метил-4-метоксибутановая кислота; 2) соединения, которые ингибируют ангиотензинпревращающий фермент, такие как энаприл, и комбинированные ингибиторы ангиотензинпревращающего фермента и нейтральной эндопептидазы,такие как омапатрилат; 3) субстраты для NO-синтазы, такие как L-аргинин; 4) средства, снижающие содержание холестерина, такие как статины (например, аторвастатин/торговая марка Liptor) и фибраты; 5) модуляторы эстрогенного рецептора, и/или агонисты эстрогена, и/или антагонисты эстрогена,предпочтительно ралоксифен или лазофоксифен, (-)-цис-6-фенил-5-[4-(2-пирролидин-1-илэтокси)фенил]5,6,7,8-тетрагидронафталин-2-ол и его фармацевтически приемлемые соли, получение которых подробно раскрыто в WO 96/21656; 6) ингибитор ФДЭ (PDE), в особенности ингибитор PDE 2, 3, 4, 5, 7 или 8, предпочтительно ингибитор PDE2 или PDE5 и наиболее предпочтительно ингибитор PDE5 (см. в последующем описании),указанные ингибиторы предпочтительно имеют ИК 50 против соответственного фермента менее 100 нМ(при условии, что ингибиторы PDE3 и -4 вводят только местно или инъекцией в половой член); 7) вазоактивный кишечный белок (VIP), миметический VIP, аналог VIP, в особенности опосредованный одним или более рецепторами VIP подтипов VPAC1, VPAC или РАСАР (пептид, активирующий гипофизарную аденилатциклазу), один или несколько агонистов рецептора VIP или аналог VIP (например, Ro-125-1553) или фрагмент VIP, один или несколько антагонистов -адренорецептора в комбинации с VIP (например, инвикорп, авиптадил); 8) агонист, антагонист или модулятор серотонинового рецептора, в особенности агонисты, антагонисты или модуляторы 5 НТ 1 А (включая VML 670 [WO 02/074288] и фибансерина [заявка США 2003/0104980]), 5 НТ 2 А, 5 НТ 2 С, 5 НТ 3 и/или 5 НТ 6 рецепторов, включая такие, которые раскрыты вWO 09902159, WO 00002550 и/или WO 00028993; 9) заменители тестостерона (включающие дегидроандростендион), тестостерон (например, Tostrelle, LibiGel), дигидротестостерон или тестостероновый имплантат; 10) селективные модуляторы андрогенового рецептора, например LGD-2226; 11) эстроген, эстроген и медроксипрогестерон или медроксипрогестеронацетат (МПА) (МРА) (т.е. в виде комбинации) или эстроген и метилированное тестостероновое средство гормонзаместительной терапии (например, ГЗТ (HRT), в особенности премарин, ценестин, эстрофеминал, эквин, эстрак, эстрофен,элесте соло, эстрин, эстрадерм TTS, эстрадерм матрикс, дерместрил, премпфаз, примпро, премпак, премик, эстратест, эстратест HS, тиболон); 12) модулятор переносчиков норадреналина, допамина и/или серотонина, такой как бупропион,GW-320659; 13) агонист или модулятор рецепторов окситоцина/вазопрессина, предпочтительно селективный агонист или модулятор окситоцина; и 14) агонист или модулятор рецепторов допамина, предпочтительно селективный агонист или модулятор D3 или D4, например апоморфин. В данном изобретении предпочтительны комбинации соединений настоящего изобретения и одного или более дополнительных лекарственных веществ, выбранных из ингибиторов PDE5; ингибиторов NEP; селективных агонистов или модуляторов D3 или D4; модуляторов эстрогенного рецептора, и/или агонистов эстрогена, и/или антагонистов эстрогена; заменителей тестостерона, тестостерона или тестостеронового имплантата; эстрогена, эстрогена и медроксипрогестерона или медроксипрогестеронацетата(МПА) или эстрогена и метилированного тестостеронового средства гормонзаместительной терапии. Предпочтительные комбинации для лечения МЭД представляют собой комбинации соединений настоящего изобретения и одного или более ингибиторов PDE5 и/или ингибиторов NEP. Предпочтительные комбинации для лечения ЖПД представляют собой комбинации соединений настоящего изобретения и ингибиторов PDE5, и/или ингибиторов NEP, и/или селективных агонистов либо модуляторов D3 или D4 и/или модуляторов эстрогенного рецептора, агонистов эстрогена, антагонистов- 25011159 эстрогена, и/или заменителей тестостерона, тестостерона, тестостеронового имплантата, и/или эстрогена,эстрогена и медроксипрогестерона или медроксипрогестеронацетата (МПА), эстрогена и метилированного тестостеронового средства гормонзаместительной терапии. В особенности предпочтительные ингибиторы PDE5 в указанных комбинированных продуктах,предназначенных для терапии МЭД или ЖПД, представляют собой силденафил, тадалафил, варденафил и 5-[2-этокси-5-(4-этилпиперазин-1-илсульфонил)пиридин-3-ил]-3-этил-2-[2-метоксиэтил]-2,6-дигидро 7 Н-пиразол[4,3-d]пиримидин-7-он. В особенности предпочтительные ингибиторы NEP в таких комбинированных продуктах, предназначенных для лечения МЭД или ЖПД, представляют собой соединения, приведенные в примерахWO 02/079143. Предпочтительные комбинированные продукты данного изобретения, предназначенные для лечения МЭД или ЖПД, представляют собой комбинацию силденафила, тадалафила, варденафила или 5-[2-этокси-5-(4-этилпиперазин-1-илсульфонил)пиридин-3-ил]-3-этил-2-[2-метоксиэтил]-2,6-дигидро-7 Нпиразол[4,3-d]пиримидин-7-она с соединением примера 1 данного изобретения и/или комбинацию любого из соединений, приведенных в примерах WO 02/079143, с соединением примера 1 данного изобретения. Приведенные в данном изобретении перекрестные ссылки на соединения, содержащиеся в патентах и заявках на патент, которые могут быть использованы в соответствии с изобретением, представлены для обозначения терапевтически активных соединений, определенных в формуле изобретения (в особенности в п.1 формулы) и конкретных примерах (все из которых включены в данное описание посредством ссылок). Если вводят комбинацию активных веществ, тогда они могут быть введены одновременно, по отдельности или последовательно. Вспомогательные вещества - ингибиторы PDE5. В особенности предпочтительные в данном изобретении вспомогательные активные вещества представляют собой ингибиторы PDE5. Пригодность любого отдельного ингибитора цГМФ PDE5 может быть легко определена оценкой его эффективности и селективности с использованием методов, раскрытых в литературе, и последующей оценкой его токсичности, абсорбции, метаболизма, фармакокинетики и т.д. в соответствии со стандартной фармацевтической практикой. Значения ИК 50 (IC50) для ингибиторов цГМФ PDE5 могут быть определены с использованием анализа PDE5 (см. ниже). Ингибиторы цГМФ PDE5, использованные в фармацевтических комбинациях в соответствии с настоящим изобретением, являются предпочтительно селективными к ферменту PDE5. Они предпочтительно являются селективными (при пероральном введении) по сравнению с селективностью к PDE3,более предпочтительно по сравнению с селективностью к PDE3 и PDE4. Ингибиторы цГМФ PDE5 изобретения (при пероральном введении) предпочтительно имеют степень селективности в 100 раз больше,более предпочтительно в 300 раз больше к PDE5 по сравнению с селективностью к PDE3 и более предпочтительно по сравнению с селективностью к PDE3 и PDE4. Степени селективности могут быть легко определены специалистом в данной области. Значения ИК 50 для фермента PDE3 и PDE4 могут быть определены с использованием известной из литературы установленной методики, см. S.A. Ballard et al., Journal of Urology, 1998, vol. 159, p. 2164-2171, которая подробно рассмотрена в последующем описании. Подходящие для использования в соответствии с настоящим изобретением ингибиторы цГМФ(i) 5-[2-этокси-5-(4-метил-1-пиперазинилсульфонил)фенил]-1-метил-3-н-пропил-1,6-дигидро-7 Нпиразол[4,3-d]пиримидин-7-он (силденафил), известный также как 1-3-(6,7-дигидро-1-метил-7-оксо-3 пропил-1 Н-пиразол[4,3-d]пиримидин-5-ил)-4-этоксифенил]сульфонил]-4-метилпиперазин(+)-3-этил-5-[5-(4-этилпиперазин-1-илсульфонил)-2-(2-метокси-1(R)-метилэтокси)пиридин-3 ил]-2-метил-2,6-дигидро-7 Н-пиразол[4,3-d]пиримидин-7-он, известный также как 3-этил-5-5-[4-этилпиперазин-1-илсульфонил]-2-([(1R)-2-метокси-1-метилэтил]окси)пиридин-3-ил-2-метил-2,6-дигидро 7 Н-пиразол[4,3-d]пиримидин-7-он (см. WO 99/54333);(vi) 5-[2-этокси-5-(4-этилпиперазин-1-илсульфонил)пиридин-3-ил]-3-этил-2-[2-метоксиэтил]-2,6 дигидро-7 Н-пиразол[4,3-d]пиримидин-7-он, известный также как 1-6-этокси-5-[3-этил-6,7-дигидро-2-(2 метоксиэтил)-7-оксо-2 Н-пиразол[4,3-d]пиримидин-5-ил]-3-пиридилсульфонил-4-этилпиперазин(6R,12aR)-2,3,6,7,12,12 а-гексагидро-2-метил-6-(3,4-метилендиоксифенил)пиразин[2',1':6,1]пиридо[3,4-b]индол-1,4-дион (тадалафил, IC-351, Cialis), т.е. соединение примеров 78 и 95 опубликованной международной заявки WO 95/19978, а также соединение примеров 1, 3, 7 и 8;(xii) 2-[2-этокси-5-(4-этилпиперазин-1-ил-1-сульфонил)фенил]-5-метил-7-пропил-3 Н-имидазо[5,1f][1,2,4]триазин-4-он (варденафил) известный также как 1-3-(3,4-дигидро-5-метил-4-оксо-7 пропилимидазо-[5,1-f]-as-триазин-2-ил)-4-этоксифенил]сульфонил]-4-этилпиперазин, т.е. соединение примеров 20, 19, 337 и 336 опубликованной международной заявки WO 99/24433;(xiv) соединение примера 11 опубликованной международной заявки WO 93/07124;(xix) 1-6-этокси-5-[3-этил-6,7-дигидро-2-(2-метоксиэтил)-7-оксо-2 Н-пиразол[4,3-d]пиримидин-5 ил]-3-пиридилсульфонил-4-этилпиперазин и их фармацевтически приемлемые соли и сольваты. Пригодность любого отдельного ингибитора PDE5 может быть легко определена оценкой его эффективности и селективности с использованием методов, раскрытых в литературе, и последующей оценкой его токсичности, абсорбции, метаболизма, фармакокинетики и т.д. в соответствии со стандартной фармацевтической практикой. Ингибиторы PDE5 предпочтительно имеют значение ИК 50 менее 100 нМ, более предпочтительно менее 50 нМ, наиболее предпочтительно менее 10 нМ. Ингибиторы PDE5, использованные в фармацевтических комбинациях в соответствии с настоящим изобретением, являются предпочтительно селективными к ферменту PDE5. Они предпочтительно имеют селективность к PDE5 в 100 раз большую, более предпочтительно в 300 раз большую, чем к PDE3. Более предпочтительно ингибитор PDE5 имеет селективность к PDE5 в 100 раз большую, более предпочтительно в 300 раз большую, чем к PDE3 и PDE4. Степени селективности могут быть легко определены специалистом в данной области из релевантных значений ИК 50. Значения ИК 50 для фермента PDE3 иPDE4 могут быть определены с использованием известной из литературы установленной методики, такой как методика, раскрытая в S.A. Ballard et al., Journal of Urology, 1998, vol. 159, p. 2164-2171. Значения ИК 50 для PDE5 фермента могут быть определены с использованием известной из литературы установленной методики и такой, которая раскрыта в WO 01/27113. Данные по половой активности in vivo. Данные эффективности соединения примера 1 в отношении MCR4 in vivo получали селективной активацией рецептора меланокортина MCR4 с использованием методики, оценивающей самопроизвольную эрекцию полового члена у находящихся в сознании крыс. Эректильные реакции регистрировали измерением внутрикавернозного давления с использованием хирургически имплантированного телеметрического устройства (ТА 11 РА-С 40, 8 мм катетер с модифицированным 3 мм кончиком, доступный от фирмы Data Sciences International Inc.). Повышение внутрикавернозного давления является показателем эрекции полового члена, так как повышение кавернозного давления представляет собой существенное гемодинамическое событие во время инициирования и поддержания эрекции полового члена. Отдельные детали хирургических процедур, приобретение данных и анализ, использованный для измерения повышения внутрикавернозного давления могут быть найдены во всех подробностях в Bernabe J., Rampin О., Sachs В.D., Giuliano F. Intracavernous pressure during erectionin rats: an integrative approach based on telemetric recording, Am. J. Physiol. 1999 Feb.; 276 (2 Pt 2): R441-9. Тестируемые животные (крысы) обитали в естественной среде (во время темного цикла суток) в течение 18 ч перед базисной оценкой эректильной функции. Перед введением тестируемого вещества оценивали базовую эректильную активность (B) в течение 10 мин после введения носителя с использованием телеметрического регистрирования внутрикавернозного давления. После подкожного введения носи- 27011159 теля примера 1 (в таком же носитителе) оценивали эрекции пениса (с использованием телеметрического регистрирования внутрикавернозного давления) в течение 10-минутных периодов с интервалами 30, 60 и 90 мин после дозирования соединения. Соединение примера 1 давало зависимое от дозы увеличение числа эрекций полового члена, когда его дозировали подкожно (s.c.) дозой 1-100 мкг/кг массы тела (см. фиг. 1 и 2). Животные, которым вводили носитель (базис), показывали минимальную эректильную активность (см. фиг. 1). Фиг. 1 иллюстрирует результаты предварительных исследований и на ней представлено сравнение числа эрекций, наблюдаемых в течение 10-минутного периода, начиная через 60 мин после s.c. введения животным соединения примера 1 дозой 1, 10 и 100 мкг/кг, имеющим базовую эректильную активность(В). Данные на фиг. 1 показывают, что при всех тестируемых дозах соединение примера 1 повышает эректильную активность в сравнении с базовой эректильной активностью (В). Фиг. 1 дополнительно иллюстрирует, что соединение примера 1 увеличивает число самопроизвольных эрекций у находящихся в сознании крыс в зависимости от дозы. Максимальная эффективная доза, наблюдаемая в данном предварительном исследовании, составляла 1 мкг/кг при s.c. Фиг. 2 иллюстрирует результаты дополнительного, более детального исследования и на ней показано сравнение числа эрекций, наблюдаемых в течение 10-минутного периода, начиная через 30 мин послеs.c. дозирования животным соединения примера 1 дозой 1, 10 и 100 мкг/кг, имеющим базовую эректильную активность (введен носитель). Данные на фиг. 2 показывают, что при всех тестируемых дозах соединение примера 1 повышает эректильную активность в сравнении с базовой эректильной активностью(введен носитель). Фиг. 2 дополнительно иллюстрирует, что соединение примера 1 увеличивает число самопроизвольных эрекций у находящихся в сознании крыс в зависимости от дозы. Максимальная эффективная доза, наблюдаемая в данном дополнительном, более детальном исследовании, составляла 10 мкг/кг при s.c. В предварительном исследовании наблюдаемая максимальная эффективная доза соединения примера 1 составляла 1 мкг/кг при s.c. и в дополнительном, более детальном исследовании наблюдаемая максимальная эффективная доза составляла 10 мкг/кг при s.c. Число наблюдаемых эрекций в двух данных исследованиях не отличалось значительно и при указанных уровнях доз. В результате обоих представленных исследований может быть независимо сделано одинаковое заключение, состоящее в том, что соединение примера 1 увеличивает число самопроизвольных эрекций у находящихся в сознании крыс в зависимости от дозы. В данных экспериментах предполагается, что разница, наблюдаемая в дозе, при которой достигается максимальный эффект в предварительном и более детальном исследовании, обусловлена отражением ожидаемой биологической изменчивости, связанной с моделью животного такого типа. Данные, показанные на фиг. 1 и 2, подтверждают, что рецепторы MCR4 вовлечены в вызывание и поддержание эрекции полового члена и поэтому предполагается, что селективные агонисты MCR4 в соответствии с настоящим изобретением могут предоставить благоприятную возможность для лечения мужской эректильной дисфункции. Анализ. Стимуляция аденилатциклазы с последующей активацией рецептора является широко используемой мерой функциональной активности ряда рецепторных систем. В функциональном анализе для измерения циклического АМФ (цАМФ) (сАМР) используются клетки почки человеческого эмбриона (HEK),которые стабильно экспрессируют рецептор человеческого меланокортина MCR1, MCR3 или MCR4. Активация рецепторов MCR1, MCR3 или MCR4 стимулирует аденилатциклазу, генерируя цАМФ, уровень которого измеряют с использованием наборов для анализа AlphaScreen (PerkinElraer). Набор для анализа цАМФ AlphaScreen состоит из донорных гранул, акцепторных гранул и биотинилированного цАМФ, который связывает различные гранулы вместе. Возбуждение данного связанного комплекса при 680 нм на микропластине анализатора Fusion- приводит к излучению света в интервале 520-620 нм. Генерированный в анализе цАМФ конкурирует с биотинилированным цАМФ за участки связывания на акцепторных гранулах, предотвращая связывание донорных и акцепторных гранул и, следовательно, уменьшение излучения света.(i) Методика стандартного функционального анализа MCR1, MCR3 и MCR4 [протокол А, В и С анализа соответственно]. Концепция анализа. Определение активности соединений в соответствии с настоящим изобретением против подтипов рецептора человеческого MCR1, MCR3 и MCR4 осуществляли с использованием трех иммортализованных клеточных линий почки человеческого эмбриона, которые биологически разработаны для экспрессии подтипов рецептора человеческого меланокортина MCR1, MCR3 или MCR4. Указанные клеточные линии разработаны с использованием протоколов, похожих на те, которые описаны в общих чертах- 28011159 Такая вызванная соединением активация этих рецепторов MCR1, MCR3 или MCR4 приводит к стимуляции клеточного фермента аденилатциклазы, которая, в свою очередь, приводит к клеточной генерации и внутриклеточной аккумуляции циклического аденозинмонофосфата (цАМФ). Найдено, что величина приростов внутриклеточного цАМФ пропорциональна степени, с которой тестируемое соединение активировало присутствующие в вышеуказанных клеточных линиях рецепторы MCR1, MCR3 илиMCR4. Внутриклеточные уровни цАМФ вычисляли количественно с использованием коммерчески доступных наборов для анализа AlphaScree от PerkinElmer. Подробный протокол анализа и объяснение концепции, лежащей в основе данного набора, доступны через веб-сайт PerkinElmer(www.perkinelmer.com). Приведенный ниже протокол предоставляет сущность такой информации. Количество внутриклеточного цАМФ, продуцированного вызванной соединением активацией рецепторов MCR1, MCR3 и MCR4 в указанных трех клеточных линиях измеряли с использованием микропластинчатого анализатора Fusion-, установленного для стимуляции при длине волны 680 нм и для измерения энергии, испускаемой при длинах волн в диапазоне 520-620 нм. Вызванные соединением приросты активации рецептора MCR1, MCR3 или MCR4 затем вычисляли количественно в виде уменьшения количества света, излученного при длинах волн в диапазоне 520-620 нм. Затем осуществляли анализ полученных данных с использованием программы для подбора по кривым и кажущейся эффективности тестируемого соединения (выраженной в виде ЕС 50 и определенной в виде эффективной концентрации соединения, которая вызывает 50% максимального эффекта, индуцированного соединением), экстраполированной из подобранной кривой. Материалы. От фирмы Perkin Elmer: набор для анализа цАМФ AlphaScreen,кат. 6760600 М, микропластинчатый анализатор Fusion- (установленный для стимуляции при длине волны 680 нм и для регистрации света, излученного при длинах волн в диапазоне 520-620 нм). От фирмы Invitrogen: фосфатный буферный солевой раствор (ФБСР) (PBS) (w/o Ca2+ и Mg2+),кат. 14190-094; среда Дульбекко, модифицированная Иглом (DMEM), с высоким содержанием глюкозы, кат. 21969-035; сбалансированный солевой раствор Хэнка (HBSS),кат. 14065-049; генетицин,кат. 10131-027. От фирмы Sigma: бычий сывороточный альбумин (БСА) (BSA),кат А 7030;(N-(2-гидроксиэтил)пиперазин-N'-(2-этансульфоновая кислота) (HEPES),кат. Н 0887; жидкость для клеточной диссоциации,кат. С 5914; диметилсульфоксид (ДМСО),кат. D8418; циклический аденозинмонофосфат (цАМФ), кат. А 9501; 3-изобутил-1-метилксантин (IBMX),кат. I5879; 1 М раствор хлорида магния (MgCl2),кат. М 1028; трипан голубой,кат. Т-8154; камера для подсчета клеток (Bright-line 35962-9). От РРА лаборатории GmbH: сыворотка плода теленка (СПТ) (FCS),кат. А 15-043. От фирмы Gilson: пипетки емкостью от 10 до 1000 мкл. От фирмы Hereaus: инкубатор клеток Hera Cell CO2. От фирмы Medical Air Technology: шкаф для микробиологического хранения II класса Bio-Mat2. От фирмы Bachem: -меланоцитстимулирующий гормон -MSH,кат. Н 1075, использованный в качестве положительного контроля. Буферы. Стимулирующий буфер (согласно протоколу AlphaScreen): HBSS, дополненный 0,5 мМ IBMX,5 мМ HEPES, 0,1% (мас. /об.) БСА и 10 мМ MgCl2. Буфер для лизиса (согласно протоколу AlphaScreen): 5 мМ раствор HEPES, дополненный 0,1% (мас./об.) БСА и 0,3% (об./об.) Tween-20. Детекторная смесь (согласно протоколу AlphaScreen): буфер для лизиса, дополненный биотинилированным цАМФ (10 нМ) и донорными гранулами (10 мкг/мл), поставляемый в комплекте для анализа цАМФ AlphaScreen. Потребительские товары. От фирмы Fisher: 384-луночные планшеты для анализа с несвязующей поверхностью, кат. DPS-172-020Q. От фирмы Costar: стерильные пипетки объемом от 2 до 50 мл со стерильными кончиками от Р 10 до Р 1000; стерильные резервуары,кат. 4878; колбы, имеющие колпачок с отверстием, Т 225, кат.3001. Приготовление соединений. Для проведения стандартного функционального анализа MCR1, MCR3 и MCR4 соединения сначала растворяли в ДМСО с получением концентрации соединения 4 мМ и затем дополнительно разводили для- 29011159 анализа в стимулирующем буфере с получением действительных концентраций, которые в 2 раза больше, чем те, которые желательны в качестве конечной концентрации для анализа. Изменения культуры клеток с течением времени. Подробно раскрытые выше три клеточные линии HEK, экспрессирующие подтипы человеческих рецепторов MCR1, MCR3 или MCR4, выращивали в колбах Т 225, имеющих колпачок с отверстием, содержащих 50 мл питательной среды (DMEM, дополненная 10% (об./об.) СПТ, 2 мМ L-глутамина, 25 мМHEPES и 1,0 мг/мл генетицина), сохраняли в инкубаторе для клеток при температуре 37 С и в среде, содержащей 5% CO2. Клетки собирали, когда они достигали 80-90% слияния, сначала удалением имеющейся питательной среды и затем промывкой ФБСР, который предварительно нагревали до температуры 37 С. Затем ФБСР удаляли и в колбу прибавляли 5 мл жидкости для клеточной диссоциации. Колбы инкубировали в течение 5 мин в инкубаторе для клеток, установленном на температуру 37 С, и в атмосфере, содержащей 5% CO2, для отделения клеток. Клетки удаляли со дна колбы введением в колбу острого надрезывающего приспособления. После удаления клеток добавляли предварительно нагретую до температуры 37 С питательную среду, клетки повторно суспендировали и слегка перемешивали для отбора пипеткой одноклеточной взвеси. Затем определяли количество клеток в полученной взвеси клеток с использованием камеры для подсчета клеток и взвесь клеток, или использовали для экспериментирования,или переносили в новую колбу Т 225 для сохранения культуры клеток. Методика анализа. Использованная методика анализа была, по существу, такой, которая раскрыта в методике с использованием набора AlphaScreen (www.perkinelmer.com), однако для облегчения манипулирования жидкостью все анализируемые объемы удваивали. Во-первых, 10 мкл растворов тестируемого соединения переносили в 384-луночные планшеты с несвязывающей поверхностью для проведения анализа. Во-вторых, анализируемые клетки собирали как указано выше.(i) Для проведения стандартного функционального анализа MCR1 приготавливали взвесь клеток с концентрацией 3105 клеток/мл в стимулирующем буфере (дополненном 10 мкл/мл анти-цАМФ раствора акцепторных гранул, поставляемого в наборе для анализа цАМФ AlphaScreen).(ii) Для проведения стандартного функционального анализа MCR3 приготавливали взвесь клеток с концентрацией 5104 клеток/мл в стимулирующем буфере (дополненном 10 мкл/мл анти-цАМФ раствора акцепторных гранул, поставляемого в наборе для анализа цАМФ AlphaScreen).(iii) Для проведения стандартного функционального анализа MCR4 приготавливали взвесь клеток с концентрацией 1105 клеток/мл в стимулирующем буфере (дополненном 10 мкл/мл анти-цАМФ раствора акцепторных гранул, поставляемого в наборе для анализа цАМФ AlphaScreen). Затем 10 мкл взвеси клеток переносили в каждую лунку 384-луночного планшета с несвязывающей поверхностью. После чего анализируемые планшеты инкубировали в темноте при комнатной температуре в течение 30 мин. В-третьих, реакцию завершали добавлением 30 мкл детекторной смеси в каждую лунку. Планшеты инкубировали всю ночь в темноте при комнатной температуре перед переносом в микропластинчатый анализатор Fusion- для количественного определения.(ii) Методика стандартного функционального анализа MCR5 и усовершенствованная методика функционального анализа MCR4 [протоколы D и Е анализа соответственно]. Концепция анализа. Определение активности соединения против подтипа человеческого рецептора MCR5 осуществляли с использованием иммортализованной клеточной линии яичников хомячка (CHO-K1), которую биологически разработали для стабильной стойкой экспрессии рекомбинантного человеческого рецептора MCR5 и репортера гена -лактамазы (CHO-K1-MCR5-CREлактамаза). Подобно, с использованием усовершенствованной методики анализа определяли также активность соединений против подтипа человеческого рецептора MCR4 с использованием иммортализованной клеточной линии CHO-K1, которую разработали для стабильной экспрессии рекомбинантного человеческого рецептора MCR4 и репортера гена-лактамазы (CHO-K1-MCR4-CREлактамаза). Указанные клеточные линии разработаны с использованием протоколов, похожих на те, которые описаны в общих чертах Zaccolo et al. (Zaccolo, M., (2000),Nature, 2(1); 25-29). Вызванная соединением активация рецепторов MCR5 или MCR4 в указанных двух клеточных линиях стимулировала продуцирование и внутриклеточную аккумуляцию фермента -лактамазы. Количество продуцированного фермента -лактамазы было прямо пропорционально степени, с которой тестируемое соединение активировало рецепторы MCR5 или MCR4, присутствующие в данных клетках, и количественное определение осуществляли с использованием набора для анализа репортера гена-лактамазы, который коммерчески доступен от фирмы Invitrogen Life Technologies. Подробное описание технологии и технических средств и протоколов анализа доступно из веб-сайта Invitrogen(www.invitrogen.com). Представленный ниже протокол раскрывает сущность такой методики анализа.
МПК / Метки
МПК: A61K 31/454, C07D 401/14, C07D 401/06, A61P 15/00
Метки: пиперидинилкарбонилпирролидины, агонистов, применение, качестве, меланокортина
Код ссылки
<a href="https://eas.patents.su/30-11159-piperidinilkarbonilpirrolidiny-i-ih-primenenie-v-kachestve-agonistov-melanokortina.html" rel="bookmark" title="База патентов Евразийского Союза">Пиперидинилкарбонилпирролидины и их применение в качестве агонистов меланокортина</a>
Производные 3-ароилиндола и их применение в качестве агонистов рецепторов cb2

Номер патента: 5854
Опубликовано: 30.06.2005
Авторы: Конжи Кристиан, Ринальди Мирей, Барт Франсис, Верне Клод, Васс Фабьенн, Гийомон Кароль
МПК: A61K 31/404, A61P 43/00, C07D 209/12...
Метки: 3-ароилиндола, рецепторов, качестве, производные, применение, агонистов
Формула / Реферат:
1. Соединение формулы где Ar представляет собой a) фенил, моно-, ди- или тризамещенный одной или более чем одной группой, выбранной из атома галогена, (C1-C4)алкила, трифторметила, амино, нитро, гидроксила, (C1-C4)алкокси, (C1-C4)алкилсульфанила или (C1-C4)алкилсульфонила; b) нафтил, который не замещен или замещен единожды или дважды атомом галогена, (C1-C4)алкилом или трифторметилом; A представляет собой C2-C6алкиленовый радикал; Y...
Новые замещенные 4-фенил-4-[1н-имидазол-2-ил] пиперидиновые производные и их применение в качестве селективных непептидных агонистов дельта-опиоидов

Номер патента: 6507
Опубликовано: 29.12.2005
Авторы: Мерт Тео Франс, Ленартс Йозеф Элизабет, Янссенс Франс Эдуард, Гомес-Санчес Антонио, Фернандес-Гадеа Франсиско Хавьер
МПК: A61K 31/445, C07D 401/04, A61P 25/04...
Метки: 4-фенил-4-[1н-имидазол-2-ил, применение, дельта-опиоидов, качестве, агонистов, непептидных, новые, производные, замещенные, пиперидиновые, селективных
Формула / Реферат:
1. Соединение формулы (I) где A=B представляет собой двухвалентный радикал с p-связью; X представляет собой ковалентную связь, -CH2- или CH2CH2-; R1 представляет собой водород, гидрокси, алкилокси, алкилкарбонилокси, Ar-окси, Het-окси, Ar-карбонилокси, Het-карбонилокси, Ar-алкилокси, Het-алкилокси, алкил, полигалогеналкил, алкилоксиалкил, Ar-алкил, Het-алкил, Ar, Het, тио, алкилтио, Ar-тио, Het-тио или NR9R10, где R9 и R10, каждый независимо,...
Производные бензил (иден)-лактамов, их получение и применение в качестве селективных (ант)агонистов рецепторов 5-нт1а и/или 5-нт1d

Номер патента: 1485
Опубликовано: 23.04.2001
Автор: Хауард Гарри Р.
МПК: A61K 31/395, A61P 25/00, C07D 209/34...
Метки: применение, производные, иден)-лактамов, качестве, рецепторов, 5-нт1d, антагонистов, 5-нт1а, селективных, получение, бензил
Формула / Реферат:
1. Соединение формулы I, изображенной ниже, где R1 представляет собой 2-диметиламиноэтоксигруппу или группу формулы G1, G2, G3, G4 или G5, которые изображены ниже, где Е представляет собой кислород, серу, SO или SO2; R6 и R7 независимо выбраны из водорода, (С1-С6)алкила, [(С2-С4)алкил]арила, где арильная группировка представляет собой фенил или нафтил, и гетероарила-(СН2)q, где гетероарильная группировка выбрана из пиридила, пиримидила,...
Карбоксамиды в качестве агонистов 5-ht1f

Номер патента: 1718
Опубликовано: 27.08.2001
Автор: Флаф Майкл Эдвард
МПК: A61K 31/40, C07D 209/88, A61P 25/06...
Метки: карбоксамиды, качестве, агонистов, 5-ht1f
Формула / Реферат:
1. Соединение формулы I где R1 и R2 независимо представляют водород, C1-C4алкил или -СН2СН2-арил, где арил представляет фенил, фенил, монозамещенный галогеном, или 1-(C1-C6алкил)пиразол-4-ил; R3 представляет C3-C6циклоалкил или гетероцикл; n = 1 или 2; и их фармацевтически приемлемые соли и гидраты. 2. Соединение по п.1, где n = 1. 3. Соединение по п.2, где R1 и R2 независимо представляют C1-C4алкил. 4. Фармацевтическая композиция, которая...
Тетрагидробензазепиновые производные в качестве агонистов дофаминовых рецепторов d1

Номер патента: 4745
Опубликовано: 26.08.2004
Автор: Тилбрук Гари
МПК: C07D 223/16, A61P 25/16, A61K 31/55...
Метки: качестве, рецепторов, тетрагидробензазепиновые, агонистов, дофаминовых, производные
Формула / Реферат:
1. Соединение-агонист дофаминовых рецепторов D1, общей формулы I где R1 представляет собой галоген; R2 представляет собой водород, метил или низший алкенил с 3-5 атомами углерода; R3 и R4 вместе образуют фурановое, дигидрофурановое, тиофеновое, дигидротиофеновое, циклопентановое или циклогексановое кольцо и R5 представляет собой водород; или R4 и R5 вместе образуют фурановое, дигидрофурановое, тиофеновое, дигидротиофеновое, циклопентановое или...
Предыдущий патент: Замещённые глюкопиранозилом бензольные производные, содержащие эти соединения лекарственные средства, их применение и способ их получения
Следующий патент: Композиция для трансдермальной доставки гормонов без необходимости использования агентов, усиливающих проникновение, и ее применения
Случайный патент: Способ разработки мощного пологого калийного пласта с прослоем каменной соли