Соединения и композиции в качестве ингибиторов протеинкиназы
Номер патента: 19966
Опубликовано: 30.07.2014
Авторы: Лу Вэньшо, Мишелли Пьер-Ив, Марсилдж Томас Х., Уно Тецуо, Цзян Тао, Чэнь Бэй, Джин Юнхо, Пэй Вэй







Формула / Реферат
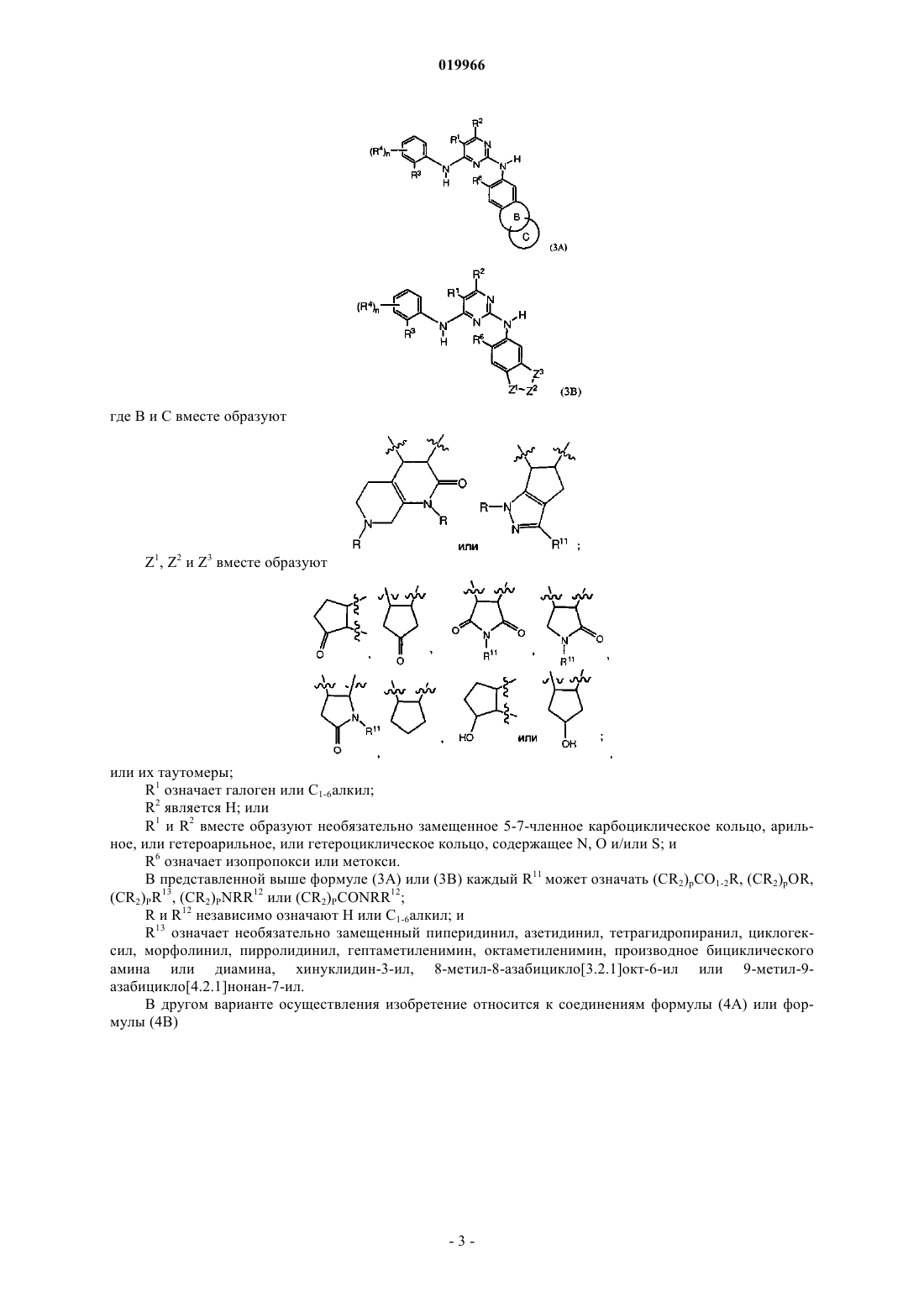
1. Соединение формулы (1)

или его фармацевтически приемлемые соли, где W является

А1 и А4 означают С;
каждый А2 и А3 означает С;
R1 означает галоген или C1-6алкил;
R2 является Н;
R3 является SO2(C1-6алкил);
R4, R7 и R10 независимо означают Н;
R6 означает OR12;
R, R5 и R5¢ независимо означают Н или C1-6алкил;
один из R8 и R9 означает (CR2)qY, а другой означает C1-6алкил;
R12 означает C1-6алкил или 5-7-членное гетероциклическое кольцо, содержащее N, О и/или S;
Y означает 3-12-членное карбоциклическое кольцо, 5-12-членное арильное или 5-12-членное гетероарильное или гетероциклическое кольцо, содержащее N, О и/или S и присоединенное к А2 или А3 или к обоим через атом углерода указанного выше гетероарильного кольца, когда q в (CR2)qY равно 0; каждое из которых является незамещенным или замещено C1-6алкилом, необязательно содержащим атомы N или О или их комбинацию; гидрокси C1-8алкилом; амино; гетероциклическим кольцом, которое может быть замещено C1-6алкилом; арилом, который может быть замещен C1-6алкилом или галогеном; или гетероарилом;
n равно 1и
q равно 0.
2. Соединение по п.1, где R5 и R5' независимо означают Н.
3. Соединение по п.1, где R6 является OR12 и R12 означает C1-6алкил.
4. Соединение по п.1, где указанное выше соединение выбирают из группы, состоящей из следующих:

5-метил-N2-[4-метил-5-(1-метил-1H-пиразол-4-ил)-2-(пиперидин-4-илокси)фенил]-N4-[2-(пропан-2-сульфонил)фенил]пиримидин-2,4-диамин,

N2-(5-(6-(3-диметиламино)пропокси)пиридин-3-ил)-2-изопропокси-4-метилфенил)-N4-(2-(изопропилсульфонил)фенил-5-метилпиримидин-2,4-диамин,

N2-(2-изопропокси)-(4-метил-5-(6-(4-метилпиперазин-1-ил)пиридин-3-ил)фенил)-N4-(2-(изопропилсульфонил)фенил)-5-метилпиримидин-2,4-диамин,

N2-(2-изопропокси)-(4-метил-5-(6-морфолинопиридин-3-ил)фенил)-N4-(2-(изопропилсульфонил)фенил-5)-метилпиримидин-2,4-диамин,

N2-(2-изопропокси-4-метил-5-(6-(пиперазин-1-ил)пиридин-3-ил)фенил)-N4-(2-(изопропилсульфонил)фенил)-5-метилпиримидин-2,4-диамин,

N2-(2-изопропокси-4-метил-5-(2-(4-метилпиперазин-1-ил)пиридин-4-ил)фенил)-N4-(2-(изопропилсульфонил)фенил)-5-метилпиримидин-2,4-диамин,

5-хлор-N2-(2-изопропокси-4-метил-5-(3-метилизоксазол-5-ил)фенил)-N4-(2-(изопропилсульфонил)фенил)пиримидин-2,4-диамин,

5-хлор-N2-(2-изопропокси-4-метил-5-(5-метил-1H-пиразол-3-ил)фенил)-N4-(2-(изопропилсульфонил)фенил)пиримидин-2,4-диамин,

5-хлор-N2-(2-изопропокси-4-метил-5-(2-метил-1H-имидазол-4-ил)фенил)-N4-(2-(изопропилсульфонил)фенил) пиримидин-2,4-диамин,

(5-(5-(4-(2-(изопропилсульфонил)фениламино)-5-метилпиримидин-2-иламино)-4-изопропокси-2-метилфенил)изоксазол-3-ил)метанол,

N2-(5-(3-((диметиламино)метил)изоксазол-5-ил)-2-изопропокси-4-метилфенил)-N4-(2-(изопропилсульфонил)фенил)-5-метилпиримидин-2,4-диамин,

5-хлор-N2-(2-изопропокси-5-метил-4-(6-(4-метилпиперазин-1-ил)пиридин-3-ил)фенил)-N4-(2-(изопропилсульфонил)фенил) пиримидин-2,4-диамин,
или его фармацевтически приемлемая соль.
5. Соединение по п.1, где указанным выше соединением является соединение формулы (2)

где R1 означает галоген или C1-6алкил;
R2 является Н;
R6 является изопропокси или метокси;
Y означает необязательно замещенный C3-7циклоалкил, C3-7циклоалкенил или фенил; или Y означает пиридил, пиразолил, изоксазолил, имидазолил, тиазолил, бензимидазолил; каждый из которых является незамещенным или замещен C1-6алкилом, необязательно содержащим атомы N или С или их комбинацию; гидрокси C1-8алкилом; амино; гетероциклическим кольцом, которое может быть замещено C1-6алкилом; арилом, который может быть замещен C1-6алкилом или галогеном; или гетероарилом; и
R, R3, R4 р и q имеют значения, определенные в п.1.
6. Соединение по п.5, где указанное выше соединение выбирают из группы, состоящей из следующих:

N2-(4-((1R,2S,4S)-2,4-бис(диметиламино)циклогексил)-2-изопропокси-5-метилфенил)-5-хлор-N4-(2-(изопропилсульфонил)фенил)пиримидин-2,4-диамин,

N2-(4-((1R,4R)-4-(диметиламино)циклогексил)-2-изопропокси-5-метилфенил)-N4-(2-(изопропилсульфонил)фенил)-5-метилпиримидин-2,4-диамин,

N2-(4-((1S,4S)-4-(диметиламино)циклогексил)-2-изопропокси-5-метилфенил)-N4-(2-(изопропилсульфонил)фенил)-5-метилпиримидин-2,4-диамин,

(цис,транс) 5-хлор-N2-{2-изопропокси-5-метил-4-[4-(4-метилпиперазин-1-ил)циклогексил]фенил}-N4-[2-(пропан-2-сульфонил)фенил]пиримидин-2,4-диамин,

(цис,транс) 5-хлор-N2-{4-[4-(2-диметиламиноэтиламино)циклогексил]-2-изопропокси-5-метилфенил}-N4-[2-(пропан-2-сульфонил)фенил]пиримидин-2,4-диамин,

4-[4-(4-{5-хлор-4-[2-(пропан-2-сульфонил)фениламино]пиримидин-2-иламино}-5-изопропокси-2-метилфенил)циклогексил]пиперазин-2-он,

5-хлор-N2-[4-(4-диметиламиноциклогексил)-2-изопропокси-5-метилфенил]-N4-[2-(пропан-2-сульфонил)фенил]пиримидин-2,4-диамин,

5-хлор-N2-[2-изопропокси-5-метил-4-(4-метиламиноциклогексил)фенил]-N4-[2-(пропан-2-сульфонил)фенил]пиримидин-2,4-диамин,

5-хлор-N2-[2-изопропокси-5-метил-4-(4-пирролидин-1-илциклогексил)фенил]-N4-[2-(пропан-2-сульфонил)фенил]пиримидин-2,4-диамин,

5-хлор-N2-[2-изопропокси-4-{4-[(2-метоксиэтил)метиламино]циклогексил}-5-метилфенил]-N4-[2-(пропан-2-сульфонил)фенил]пиримидин-2,4-диамин,

5-хлор-N2-(4-((1S,4S)-4-(диметиламино)циклогексил)-2-изопропокси-5-метилфенил)-N4-(2-(изопропилсульфонил)фенил)пиримидин-2,4-диамин,

5-хлор-N2-(4-((1R,4R)-4-(диметиламино)циклогексил)-2-изопропокси-5-метилфенил)-N4-(2-(изопропилсульфонил)фенил)пиримидин-2,4-диамин,

5-хлор-N2-(2-изопропокси-5-метил-4-((1S,4S)-4-(4-метилпиперазин-1-ил)циклогексил)фенил)-N4-(2-(изопропилсульфонил)фенил) пиримидин-2,4-диамин,

5-хлор-N2-(2-изопропокси-5-метил-4-((1R,4R)-4-(4-метилпиперазин-1-ил)циклогексил)фенил)-N4-(2-(изопропилсульфонил)фенил)пиримидин-2,4-диамин,

5-хлор-N2-(2-изопропокси-5-метил-4-((1S,4S)-4-(метиламино)циклогексил)фенил)-N4-(2-(изопропилсульфонил)фенил)пиримидин-2,4-диамин,

5-хлор-N2-(2-изопропокси-5-метил-4-((1R,4R)-4-(метиламино)циклогексил)фенил)-N4-(2-(изопропилсульфонил)фенил)пиримидин-2,4-диамин,

5-хлор-N2-(2-изопропокси-4-метил-5-(1Н-пиразол-3-ил)фенил)-N4-(2-(изопропилсульфонил)фенил)пиримидин-2,4-диамин,

N2-(4-(1Н-бензо[d]имидазол-2-ил)-2-изопропокси-5-метилфенил)-5-хлор-N4-(2-(изопропилсульфонил)фенил)пиримидин-2,4-диамин,

5-хлор-N4-(2-(изопропилсульфонил)фенил)-N2-(2-метокси-(5-метил-4-фенил)фенил)пиримидин-2,4-диамин,

5-хлор-N4-(2-(изопропилсульфонил)фенил)-N2-(2-метокси-5-метил-4-(4-метилфенил)фенил)пиримидин-2,4-диамин,

5-хлор-N4-(2-(изопропилсульфонил)фенил)-N2-(2-метокси-5-метил-4-(4-фторфенил)фенил)пиримидин-2,4-диамин,

5-хлор-N4-(2-(изопропилсульфонил)фенил)-N2-(2-метокси-5-метил-4-(2,4-дифторфенил)фенил)пиримидин-2,4-диамин,

5-хлор-N2-(2-изопропокси-5-метил-4-((1S,4S)-4-(пирролидин-1-ил)циклогексил)фенил)-N4-(2-(изопропилсульфонил)фенил)пиримидин-2,4-диамин,

5-хлор-N2-(2-изопропокси-5-метил-4-((1R,4R)-4-(пирролидин-1-ил)циклогексил)фенил)-N4-(2-(изопропилсульфонил)фенил)пиримидин-2,4-диамин,

2-(N-((1R,4R)-4-(4-(4-(2-(изопропилсульфонил)фениламино)-5-хлорпиримидин-2-иламино)-5-изопропокси-2-метилфенил)циклогексил)-N-метиламино)этанол,

N2-(4-((1S,4S)-4-(N-(2-метоксиэтил)-N-метиламино)циклогексил)-2-изопропокси-5-метилфенил)-5-хлор-N4-(2-(изопропилсульфонил)фенил)пиримидин-2,4-диамин,

N2-(4-((1R,4R)-4-(N-(2-метоксиэтил)-N-метиламино)циклогексил)-2-изопропокси-5-метилфенил)-5-хлор-N4-(2-(изопропилсульфонил)фенил)пиримидин-2,4-диамин,

5-хлор-N2-(4-((1R,4R)-4-(диметиламино)циклогексил)-2-метокси-5-метилфенил)-N4-(2-(изопропилсульфонил)фенил)пиримидин-2,4-диамин,

5-хлор-N2-(4-((1S,4S)-4-(диметиламино)циклогексил)-2-метокси-5-метилфенил)-N4-(2-(изопропилсульфонил)фенил)пиримидин-2,4-диамин,
или его фармацевтицески приемлемая соль.
7. Соединение, выбранное из группы, включающей:

N2-{2-изопропокси-4-метил-5-[1-(2-морфолин-4-илэтил)-1H-пиразол-4-ил]фенил}-5-метил-N4-2-(пропан-2-сульфонил)фенилпиримидин-2,4-диамин,

N2-(5-(6-(2-морфолиноэтиламино)пиридин-3-ил)-2-изопропокси-4-метилфенил)-N4-(2-(изопропилсульфонил)фенил)-5-метилпиримидин-2,4-диамин,

N2-(2-изопропокси-4-метил-5-(3-((пиперазин-1-ил)метил)изоксазол-5-ил)фенил)-N4-(2-(изопропилсульфонил)фенил)-5-метилпиримидин-2,4-диамин,

N2-(2-изопропокси-4-метил-5-(3-((4-(2-морфолиноэтил)пиперазин-1-ил)метил)изоксазол-5-ил)фенил)-N4-(2-(изопропилсульфонил)фенил-5-метилпиримидин-2,4-диамин,

(5-(5-(4-(2-изопропилсульфонил)фениламино)-5-хлорпиримидин-2-иламино)-4-изопропокси-2-метилфенил)-N-(2-морфолиноэтил)изоксазол-3-карбоксамид,

(5-(5-(4-(2-(изопропилсульфонил)фениламино)-5-хлорпиримидин-2-иламино)-4-изопропокси-2-метилфенил)изоксазол-3-ил)(4-этилпиперазин-1-ил)метанон,

(5-(5-(4-(2-(изопропилсульфонил)фениламино)-5-хлорпиримидин-2-иламино)-4-изопропокси-2-метилфенил)-N-метоксиизоксазол-3-карбоксамид.
8. Фармацевтическая композиция, содержащая терапевтически эффективное количество соединения по любому из пп.1-7 и фармацевтически приемлемый носитель.
9. Способ ингибирования киназы анапластической лимфомы, включающий введение в клеточную или тканевую систему или млекопитающему субъекту терапевтически эффективного количества соединения по любому из пп.1-7 или его фармацевтически приемлемых солей, оказывающих ингибирующее действие на указанную выше киназу.
10. Применение соединения по любому из пп.1-7 или его фармацевтической композиции для производства лекарственного средства, предназначенного для лечения состояния, опосредованного киназой анапластической лимфомы, где указанным выше состоянием является аутоиммунное заболевание, вторичная болезнь, инфекционное заболевание или нарушение клеточной пролиферации.
11. Применение соединения по любому из пп.1-7 или его фармацевтической композиции для производства лекарственного средства, предназначенного для лечения нарушения клеточной пролиферации, где указанным выше нарушением клеточной пролиферации является лимфома, остеосаркома, меланома или опухоли молочной железы, почки, простаты, толстой кишки, щитовидной железы, яичника, поджелудочной железы, неврональная опухоль, опухоли легкого, матки или опухоль желудочно-кишечного тракта, немелкоклеточная карцинома легких или нейробластома.
12. Применение по п.11, где указанным нарушением клеточной пролиферации является немелкоклеточная карцинома легких.
13. Применение по п.11, где указанным нарушением клеточной пролиферации является нейробластома.
14. Применение по п.11, где указанным вторым терапевтическим средством является химиотерапевтическое средство.
Текст
Изобретение относится к новым производным пиримидина и пиридина, к их фармацевтическим композициям и к способам применения таких соединений. Например, производные пиримидина и пиридина согласно изобретению могут быть использованы для лечения, улучшения или предотвращения состояния, которое отвечает на ингибирование активности киназы анапластической лимфомы (ALK), очагово-адгезивной киназы (FAK), дзета-цепь-ассоциированной протеинкиназы 70 (ZAP-70), инсулинподобного фактора роста (IGF-1R) или их комбинаций. По данной заявке испрашивается приоритет согласно предварительной заявке США 60/869299,поданной 8 декабря 2006 г., которая во всей своей полноте включена в настоящее описание посредством ссылки. Изобретение относится к ингибиторам протеинкиназы, более конкретно к новым пиримидиновым и пиридиновым производным и к их фармацевтическим композициям, а также к применению их в качестве фармацевтических средств. Киназа анапластической лимфомы (ALK), член суперсемейства инсулиновых рецепторных тирозинкиназ, непосредственно связана с онкогенезом в гемопоэтических и негемопоэтических опухолях. Аномальная экспрессия белков-рецепторов ALK полной длины была отмечена в нейробластомах и глиобластомах; и слитые белки ALK обнаружены в анапластической крупноклеточной лимфоме. Изучение слитых белков ALK повышает возможность новых методов терапевтического воздействия на пациентов с ALK-позитивными злокачественными опухолями. (Pulford et al., Cell. Mol. Life Sci. 61:2939-2953(2004. Очагово-адгезивная киназа (FAK) является ключевым ферментом в интегрин-опосредованном каскаде реакций передачи сигнала в клетку (D. Schlaepfer et al., Prog. Biophys. Mol. Biol. 1999, 71, 43578). Пусковым механизмом в каскаде реакций передачи сигнала является аутофосфорилирование тирозинаY397. Фосфорилированный тирозин Y397 является SH2-участком присоединения для Src-семейства тирозинкиназ; связанная c-Src киназа фосфорилирует другие остатки тирозина в FAK. Среди них фосфорилированный тирозин Y925 становится связывающим участком для SH2-участка малого адапторного белка Grb2. Данное прямое связывание Grb2 с FAK является одной из ключевых стадий активации нисходящих [в пути передачи сигнала] мишеней, таких как каскад киназ Ras-ERK2/MAP. Дзета-цепь-ассоциированная протеинкиназа 70 (ZAP-70), член семейства тирозиновых протеинкиназ, имеет потенциальное прогностическое значение при хроническом лимфолейкозе (CLL). Известно,что протеинкиназа ZAP-70, которая является важной в Т-клеточной и NK-клеточной передаче сигнала,но отсутствует в нормальных периферических В-клетках, экспрессируется в большинстве немутировавших клеток при CLL с более неблагоприятным прогнозом и отсутствует в большинстве случаев клеток с мутировавшими генами IgVH. Протеинкиназа ZAP-70 также экспрессируется в меньшинстве случаев других В-клеточных опухолей. (Orchard et al., Leuk. Lymphoma 46:1689-98 (2005. Процесс передачи сигнала инсулиноподобного фактора роста (IGF-1) в значительной степени вовлекается в развитие опухолей, где рецептор IGF-1 (IGF-1R) выступает в качестве доминирующего фактора. Рецептор IGF-1R играет важную роль в опухолевой трансформации и выживаемости злокачественных клеток, но только отчасти участвует в нормальном клеточном росте. Было предположено, что таргетирование рецептора IGF-1R является перспективным средством терапии рака (Larsson et al., Br. J. Cancer 92:2097-2101 (2005. В связи с тем что выясняется роль ALK, FAK, ZAP-70 и IGF-1R, связанная с заболеваниями, существует потребность в соединениях, которые могут быть полезными при лечении или предотвращении заболевания, которое отвечает на ингибирование ALK, FAK, ZAP-70 и/или IGF-1R. Изобретение относится к новым производным пиримидина и пиридина, к их фармацевтическим композициям и к их применению в качестве фармацевтических средств. В одном аспекте изобретение относится к соединению формулы (1) или к его фармацевтически приемлемым солям, где W является А 1 и А 2 независимо означают С или N; каждый А 2 и А 3 означает С или один из А 2 и А 3 является N, когда R6 и R7 образуют кольцо; В и С означают независимо необязательно замещенное 5-7-членное карбоциклическое кольцо,арильное, гетероарильное или гетероциклическое кольцо, содержащее N, О или S;R1 и R2 независимо означают галоген, OR12, NR(R12), SR12 или необязательно замещенный C1-6 ал-1 019966 кил, C2-6 алкенил или C2-6 алкинил; или один из R1 и R2 является Н;R4, R6, R7 и R10 независимо означают необязательно замещенный C1-6 алкил, С 2-6 алкенил или C2-6 алкинил; OR12, NR(R12), галоген, нитро, SO2R12, (CR2)pR13 или X; или R4, R7 и R10 означают независимо Н;R8 и R9 независимо означают С 1-6 алкил, C2-6 алкенил или С 2-6 алкинил, галоген или X, или один из R8 9 и R является Н, когда R1 и R2 образуют кольцо; и при условии, что один из R8 и R9 является X; альтернативно, R1 и R2 или R6 и R7, R7 и R8 или R8 и R9, когда присоединены к атому углерода, могут образовывать необязательно замещенное 5-7-членное моноциклическое или конденсированное карбоциклическое кольцо, арильное или гетероарильное или гетероциклическое кольцо, содержащее N, О и/или S; или R7, R8, R9 и R10 отсутствуют, когда присоединены к атому N;R12 и R13 независимо означают необязательно замещенное 3-7-членное насыщенное или частично ненасыщенное карбоциклическое кольцо или 5-7-членное гетероциклическое кольцо, содержащее N, О и/или S; арил или гетероарил; или R12 означает Н, С 1-6 алкил;Y означает необязательно замещенное 3-12-членное карбоциклическое кольцо, 5-12-членное арильное или 5-12-членное гетероарильное или гетероциклическое кольцо, содержащее N, О и/или S и присоединенное к А 2 или А 3 или к обоим через атом углерода указанного выше гетероарильного или гетероциклического кольца, когда q в (CR2)qY равно 0; иn, p и q независимо равны 0-4. В представленной выше формуле (1) R1 может означать галоген или С 1-6 алкил; R2 является Н илиNH2; или R1 и R2 вместе образуют необязательно замещенное 5-6-членное арильное или гетероарильное или гетероциклическое кольцо, содержащее 1-3 атома азота. В других примерах R3 в формуле (1) может означать SO2R12, SO2NH2, SO2NRR12, CO2NH2, CONRR12, CO1-2R12 или циано; и R12 означает C1-6 алкил,необязательно замещенный С 3-7 циклоалкил, С 3-7 циклоалкенил, пирролидинил, пиперазинил, пиперидинил, морфолинил или азетидинил. В других примерах R5, R5', R7 и R10 в формуле (1) независимо означают Н, и n равно 0. В других примерах R6 в формуле (1) может означать галоген или OR12, и R12 означаетC1-6 алкил. В одном варианте осуществления изобретение относится к соединениям формулы (2) где R1 означает галоген или С 1-6 алкил;R1 и R2 вместе образуют необязательно замещенное 5-6-членное гетероарильное или гетероциклическое кольцо, содержащее один или два атома азота;R6 является изопропокси или метокси; один из R8 и R9 является (CR2)qY, и другой является C1-6 алкилом, циано, CO1-2R12, CONR(R12) илиY означает необязательно замещенный С 3-7 циклоалкил, С 3-7 циклоалкенил или фенил; или Y означает пиридил, пиразолил, изоксазолил, имидазолил, тиазолил, бензимидазолил, пирролидинил, пиперазинил, пиперидинил, морфолинил, азетидинил, гептаметиленимин или октаметиленимин, каждый из которых присоединен к фенильному кольцу через атом углерода, когда q в (CR2)qY равно 0; n равно 0-1; иq равно 0-4. В указанной выше формуле (2) один из R8 и R9 может означать (CR2)qY, и другой является С 1-6 алкилом; n и q независимо равны 0. В других примерах Y означает пирролидинил, пиперидинил, азетидинил. В других примерах R1 означает галоген или C1-6 алкил; и R2 является Н. В другом варианте осуществления изобретение относится к соединениям формулы (3 А) или (3 В)R1 означает галоген или С 1-6 алкил;R1 и R2 вместе образуют необязательно замещенное 5-7-членное карбоциклическое кольцо, арильное, или гетероарильное, или гетероциклическое кольцо, содержащее N, О и/или S; иR6 означает изопропокси или метокси. В представленной выше формуле (3 А) или (3 В) каждый R11 может означать (CR2)pCO1-2R, (CR2)pOR,(CR2)PR13, (CR2)PNRR12 или (CR2)PCONRR12;R13 означает необязательно замещенный пиперидинил, азетидинил, тетрагидропиранил, циклогексил, морфолинил, пирролидинил, гептаметиленимин, октаметиленимин, производное бициклического амина или диамина, хинуклидин-3-ил, 8-метил-8-азабицикло[3.2.1]окт-6-ил или 9-метил-9 азабицикло[4.2.1]нонан-7-ил. В другом варианте осуществления изобретение относится к соединениям формулы (4 А) или формулы (4 В) где R1 означает галоген или C1-6 алкил;R1 и R2 вместе образуют необязательно замещенное 5-7-членное карбоциклическое кольцо, арильное или гетероарильное или гетероциклическое кольцо, содержащее N, О и/или S;R6 является изопропокси или метокси; и В 2 и В 3 независимо означают необязательно замещенный 5-6-членный арил или гетероарил, содержащий N, О или S. В другом аспекте изобретение относится к соединениям формулы (5) или к их фармацевтически приемлемым солям,где W является А 1 и А 4 независимо означают С или N; каждый А 2 и А 3 означает С, или один из А 2 и А 3 представляет собой N, когда R6 и R7 образуют кольцо; В и С независимо означают, необязательно замещенное 5-7-членное карбоциклическое кольцо,арильное, гетероарильное или гетероциклическое кольцо, содержащее N, О или S;Z1, Z2 и Z3 независимо представляют собой NR11, C=O, CR-OR, (CR2)1-2 или =C-R12;R1 и R2 независимо означают галоген, OR12, NR(R12), SR12 или необязательно замещенный C11 2 6 алкил, C2-6 алкенил или С 2-6 алкинил; или один из R и R является Н; 3 12 12X; при условии, что R6 и R7 оба не являются Н;R8 и R9 означают независимо С 1-6 алкил, C2-6 алкенил или С 2-6 алкинил, галоген или X, или один из R8 9 и R является Н; и при условии, что один из R8 и R9 является X; альтернативно, R1 и R2 или R6 и R7, R7 и R8 или R9 и R10, когда присоединены к атому углерода, могут образовывать необязательно замещенное 5-7-членное моноциклическое или конденсированное карбоциклическое кольцо, арильное или гетероарильное или гетероциклическое кольцо, содержащее N, О и/или S; или R7, R8, R9 и R10 отсутствуют, когда присоединены к атому N;R12 и R13 независимо означают необязательно замещенное 3-7-членное насыщенное или частично ненасыщенное карбоциклическое кольцо или 5-7-членное гетероциклическое кольцо, содержащее N, О и/или S; арил или гетероарил; или R12 означает Н, C1-6 алкил;Y означает необязательно замещенное 3-12-членное карбоциклическое кольцо, 5-12-членное арильное или 5-12-членное гетероарильное или гетероциклическое кольцо, содержащее N, О и/или S, и присоединенное к А 2 или А 3 или к обоим через атом углерода указанного выше гетероарильного или гетероциклического кольца, когда q в (CR2)qY равно 0; и n, p и q независимо равны 0-4. В другом аспекте настоящее изобретение относится к фармацевтическим композициям, содержащим соединение формулы (1), (2), (3 А), (3 В), (4 А), (4 В) или (5) и фармацевтически приемлемый эксципиент. В другом аспекте настоящее изобретение относится к способам модуляции тирозинкиназ ALK,FAK, ZAP-70 и/или IGF-1R, включающим введение в систему или субъекту, нуждающемуся в этом, терапевтически эффективного количества соединения формулы (1), (2), (3 А), (3 В), (4 А), (4 В) или (5) или его фармацевтически приемлемых солей или фармацевтических композиций, посредством которых происходит модуляция указанных выше тирозинкиназ ALK, FAK, ZAP-70 и/или IGF-1R. Изобретение также относится к способам лечения, улучшения или предотвращения состояния, которое отвечает на ингибирование тирозинкиназ ALK, FAK, ZAP-70 и/или IGF-1R, включающим введение в систему или субъекту,нуждающемуся в этом, эффективного количества соединения формулы (1), (2), (3 А), (3 В), (4 А), (4 В) или(5) или его фармацевтически приемлемых солей или фармацевтических композиций, и необязательно в комбинации со вторым терапевтическим средством, оказывающих лечебное действие на указанное выше состояние. Альтернативно, настоящее изобретение относится к применению соединения формулы (1),(2),(3 А), (3 В), (4 А), (4 В) или (5) в производстве лекарственного средства для лечения состояния, опосредованного ALK, FAK, ZAP-70 и/или IGF-1R. В отдельных вариантах осуществления изобретения соединения согласно изобретению могут быть использованы сами по себе или в комбинации с другим терапевтическим средством для лечения состояния, опосредованного ALK, где указанным выше состоянием является аутоиммунное заболевание, вторичная болезнь, инфекционное заболевание или нарушение пролиферации клеток. Кроме того, изобретение относится к способам лечения нарушения клеточной пролиферации,включающим введение в систему или субъекту, нуждающемуся в этом, эффективного количества соединения формулы (1), (2), (3 А), (3 В), (4 А), (4 В) или (5) или его фармацевтически приемлемых солей или фармацевтических композиций, и необязательно в комбинации со вторым терапевтическим средством,оказывающих лечебное действие на указанное выше состояние. Альтернативно, настоящее изобретение относится к применению соединения формулы (1), (2), (3 А), (3 В), (4 А), (4 В) или (5) в производстве лекарственного средства, предназначенного для лечения нарушения клеточной пролиферации. В отдельных примерах соединения согласно изобретению могут быть использованы сами по себе или в комбинации с химиотерапевтическим средством для лечения нарушения клеточной пролиферации, которое включает, но не ограничивается ими, лимфому, остеосаркому, меланому или опухоль молочной железы,опухоли почки, простаты, толстой кишки, щитовидной железы, яичника, поджелудочной железы, легкого, матки или опухоль желудочно-кишечного тракта. В указанных выше способах применения соединений согласно изобретению соединение формулы(1), (2), (3 А), (3 В), (4 А), (4 В) или (5) может быть введено в систему, включающую клетки или ткани, или млекопитающему субъекту, такому как человек или животное."Алкил" относится к фрагменту и служит структурным элементом других групп, например галоидзамещенный алкил и алкокси, и может быть с прямой или разветвленной цепью. Необязательно замещенный алкил, алкенил или алкинил, как использовано в данном описании, может быть необязательно галогенированным (например, CF3) или может иметь один или более углеродов, который замещен или заменен гетероатомом, таким как NR, О или S (например, -ОСН 2 СН 2 О-, алкилтиолы, тиоалкокси, алкиламины и т.д.)."Арил" относится к моноциклическому или конденсированному бициклическому ароматическому кольцу, содержащему атомы углерода. "Арилен" означает двухвалентный радикал, производный арильной группы. Например, арильной группой может быть фенил, инденил, инданил, нафтил или 1,2,3,4 тетрагидронафталенил, которая может быть необязательно замещена в орто-, мета- или пара-положении."Гетероарил", как использовано в данном описании, относится к термину, который определен выше для арила, где один или несколько членов кольца являются гетероатомом. Примеры гетероарилов включают, но не ограничиваются ими, пиридил, пиразинил, индолил, индазолил, хиноксалинил, хинолинил,бензофуранил, бензопиранил, бензотиопиранил, бензо[1.3]диоксол, имидазолил, бензоимидазолил, пиримидинил, фуранил, оксазолил, изоксазолил, триазолил, бензотриазолил, тетразолил, пиразолил, тиенил, пирролил, изохинолинил, пуринил, тиазолил, тетразинил, бензотиазолил, оксадиазолил, бензоксадиазолил и т.д."Карбоциклическое кольцо", как использовано в данном описании, относится к насыщенному или частично ненасыщенному, моноциклическому, конденсированному бициклическому или соединенному мостиковой связью полициклическому кольцу, содержащему атомы углерода, которые могут быть необязательно замещены, например, =O. Примеры карбоциклических колец включают, но не ограничиваются ими, циклопропил, циклобутил, циклопентил, циклогексил, циклопропилен, циклогексанон и т.д."Гетероциклическое кольцо", как использовано в данном описании, относится к термину, который определен для карбоциклического кольца выше, где один или более углеродов кольца является гетероатомом. Например, гетероциклическое кольцо может содержать N, О, S, -N=, -S-, -S(O), -S(O)2- или -NR-,где R может быть водородом, C1-4 алкилом или защитной группой. Примеры гетероциклических колец включают, но не ограничиваются ими, морфолино, пирролидинил, пирролидинил-2-он, пиперазинил,пиперидинил, пиперидинилон, 1,4-диокса-8-азаспиро[4.5]дека-8-ил, 1,2,3,4-тетрагидрохинолинил и т.д. Гетероциклические кольца, как использовано в данном описании, могут охватывать бициклические амины и бициклические диамины. Термины "совместное введение", или "комбинированное введение", или тому подобное, как использовано в данном описании, подразумевают как охватывающие введение подобранных терапевтических средств одному больному и предназначены для включения схем лечения, в которых средства необязательно вводят одним и тем же способом введения или в одно и то же время. Термин "фармацевтическая комбинация", как использовано в данном описании, относится к продукту, полученному при смешивании или комбинировании активных ингредиентов, и включает как связанные, так и несвязанные комбинации активных ингредиентов. Термин "связанная комбинация" означает, что активные ингредиенты, например соединение формулы (1) и соагент, оба введены больному одновременно в виде одной формы или дозировки. Термин "несвязанная комбинация" означает, что активные ингредиенты, например соединение формулы (1) и соагент, оба введены больному в виде отдельных форм, либо одновременно, конкурентно, либо последовательно с неопределенными интервалами времени, где такое введение способствует доставке терапевтически эффективных уровней активных ингредиентов в организм пациента. Последнее также относится к терапии смесями, например, введение трех или более активных ингредиентов. Термин "терапевтически эффективное количество" означает количество заданного соединения, которое будет вызывать биологический или медицинский ответ в клетке, ткани, органе, системе, животном или человеке, которое будет определять исследователь, ветеринар, врач или другой клиницист. Термин "введение" или "применение" заданного соединения означает доставку соединения согласно изобретению или его пролекарств субъекту, нуждающемуся в таком лечении. Изобретение предоставляет новые пиримидиновые и пиридиновые производные и их фармацевтические композиции, и способы применения таких соединений. В одном аспекте изобретение относится к соединению формулы (1) или к его фармацевтически приемлемым солям; где А 1 и А 4 независимо означают С или N; каждый А 2 и А 3 является С, или один из А 2 и А 3 является N, когда R6 и R7 образуют кольцо; В и С независимо означают необязательно замещенное 5-7-членное карбоциклическое кольцо,арильное, гетероарильное или гетероциклическое кольцо, содержащее N, О или S;R1 и R2 независимо означают галоген, OR12, NR(R12), SR12 или необязательно замещенный C1-6 алкил, C2-6 алкенил или C2-6 алкинил; или один из R1 и R2 является Н;R4, R6, R7 и R10 независимо означают необязательно замещенный C1-6 алкил, C2-6 алкенил или С 2-6 алкинил; OR12, NR(R12), галоген, нитро, SO2R12, (CR2)PR13 или X; или R4, R7 и R10 независимо означают Н;R8 и R9 независимо означают C1-6 алкил, C2-6 алкенил, С 2-6 алкинил, галоген или X, или один из R8 иR является Н, когда R1 и R2 образуют кольцо; и при условии, что один из R8 и R9 является X; альтернативно, R1 и R2 или R6 и R7, R7 и R8 или R9 и R10, когда присоединены к атому углерода, могут образовывать необязательно замещенное 5-7-членное моноциклическое или конденсированное карбоциклическое кольцо, арильное или гетероарильное или гетероциклическое кольцо, содержащее N, О и/или S; или R7, R8, R9 и R10 отсутствуют, когда присоединены к атому N;R12 и R13 независимо означают необязательно замещенное 3-7-членное насыщенное или частично ненасыщенное карбоциклическое кольцо или 5-7-членное гетероциклическое кольцо, содержащее N, О и/или S; арил или гетероарил; или R12 означает Н, С 1-6 алкил;Y означает необязательно замещенное 3-12-членное карбоциклическое кольцо, 5-12-членное арильное или 5-12-членное гетероарильное или гетероциклическое кольцо, содержащее N, О и/или S, и присоединенное к А 2 или А 3 или к обоим через атом углерода указанного выше гетероарильного или гетероциклического кольца, когда q в (CR2)qY равно 0; иn, p и q независимо равны 0-4. В одном варианте осуществления изобретение относится к соединениям формулы (2) 9 где R1 означает галоген или С 1-6 алкил;R1 и R2 вместе образуют необязательно замещенное 5-6-членное гетероарильное или гетероциклическое кольцо, содержащее один или два атома азота;R6 является изопропокси или метокси; один из R8 и R9 является (CR2)qY, и другой является C1-6 алкилом, циано, CO1-2R12, CONR(R12) илиY означает необязательно замещенный С 3-7 циклоалкил, С 3-7 циклоалкенил или фенил; или Y означает пиридил, пиразолил, изоксазолил, имидазолил, тиазолил, бензимидазолил, пирролидинил, пиперазинил, пиперидинил, морфолинил, азетидинил, гептаметиленимин или октаметиленимин, каждый из которых присоединен к фенильному кольцу через атом углерода, когда q в (CR2)qY равно 0;q равно 0-4. В другом варианте осуществления изобретение относится к соединениям формулы (3 А) или (3 В)R1 означает галоген или C1-6 алкил;R1 и R2 вместе образуют необязательно замещенное 5-7-членное карбоциклическое кольцо, арильное или гетероарильное или гетероциклическое кольцо, содержащее N, О и/или S; иR6 означает изопропокси или метокси. В другом варианте осуществления изобретение относится к соединениям формулы (4 А) или формулы (4 В) где R1 означает галоген или C1-6 алкил;R1 и R2 вместе образуют необязательно замещенное 5-7-членное карбоциклическое кольцо, арильное или гетероарильное или гетероциклическое кольцо, содержащее N, О и/или S; иR6 является изопропокси или метокси; и В 2 и В 3 независимо означают необязательно замещенный 5-6-членный арил или гетероарил, содержащий N, О или S. В другом аспекте изобретение относится к соединениям формулы (5) или к их фармацевтически приемлемым солям; А 1 и А 4 независимо означают С или N; каждый А 2 и А 3 означает С, или один из А 2 и А 3 является N, когда R6 и R7 образуют кольцо; В и С независимо означают необязательно замещенное 5-7-членное карбоциклическое кольцо,арильное, гетероарильное или гетероциклическое кольцо, содержащее N, О или S;R1 и R2 независимо означают галоген, OR12, NR(R12), SR12 или необязательно замещенный С 1-6 алкил, C2-6 алкенил или C2-6 алкинил; или один из R1 и R2 является Н;X; при условии, что R6 и R7 оба не являются Н;R8 и R9 независимо означают С 1-6 алкил, C2-6 алкенил, С 2-6 алкинил, галоген или X, или один из R8 и 9R является Н; и при условии, что один из R8 и R9 является X; альтернативно, R1 и R2 или R6 и R7, R7 и R8 или R9 и R10, когда присоединены к атому углерода, могут образовывать необязательно замещенное 5-7-членное моноциклическое или конденсированное карбоциклическое кольцо, арильное, или гетероарильное, или гетероциклическое кольцо, содержащее N, О и/или S; или R7, R8, R9 и R10 отсутствуют, когда присоединены к атому N;R12 и R13 независимо означают необязательно замещенное 3-7-членное насыщенное или частично ненасыщенное карбоциклическое кольцо или 5-7-членное гетероциклическое кольцо, содержащее N, О и/или S; арил или гетероарил; или R12 означает Н, С 1-6 алкил;Y означает необязательно замещенное 3-12-членное карбоциклическое кольцо, 5-12-членное арильное или 5-12-членное гетероарильное или гетероциклическое кольцо, содержащее N, О и/или S, и присоединенное к А 2, или А 3, или к обоим через атом углерода указанного выше гетероарильного или гетероциклического кольца, когда q в (CR2)qY равно 0; иn, р и q независимо равны 0-4. В любой указанной выше формуле Y или R13 может независимо означать гетероциклические кольца, которые могут быть представлены бициклическим амином или бициклическим диамином. Примеры бициклического амина и бициклических диаминов включают, но не ограничиваются ими, необязательно замещенный гексаметиленимин; гептаметиленимин; хинуклидин; 3-азабицикло[3.3.0]октан; 3,8-диазабицикло[3.2.1]октан; октагидро-1H-пиридо[3,4-C]азепин; октагидропирролизин; 6-азабицикло[3.2.1]октан; 3-азабицикло[3.2.1]октан; 2,5-диазабицикло[2.2.1]гептан; 1-азабицикло[2.2.1]гептан; 2-азабицикло[2.2.1] гептан; 1,4-диазабицикло[4.4.0]декан; 1,4-диазабицикло[4.3.0]нонан; 1-азабицикло[3.2.1]октан; 3-азабицикло[3.3.0]октан; 8-азабицикло[3.2.1]октан; 3,9-диазабицикло[4.2.1]нонан; октагидропирроло[3,4C]пиррол; октагидропирроло[3,4-B]пиррол; гексагидропирроло[3,2-B]пиррол; гексагидропирроло[3,2C]пиррол; 1,4-диазациклооктан; 1,5-диазациклооктан; 3,7-диазабицикло[4.2.0]октан; 3,7-диазабицикло[3.3.1]нонан; октагидропирроло[3,4-C]пиридин; октагидропирроло[3,4-B]пиридин; октагидроциклопента[C]пирролидин; гексагидроциклопента[C]пирролидин; 8-азабицикло[3.2.1]октан; декагидрохинолин; декагидроизохинолин; декагидропиридо[3,4-B]азепин; декагидропиридо[4,3-B]азепин; 9 азабицикло[3.3.1]нонан; биспидин; 3-азабицикло[3.1.0]гексан; 8-азабицикло[3.2.1]октан; 2-азабицикло[3.3.1]нонан; тетрагидрохинолин; тетрагидроизохинолин; 2,5-диазабицикло[2.2.2]октан; декагидро 2,7-нафтиридин; 1,4-диазепан; азонан; октагидро-1H-индол; октагидро-1H-изоиндол; 2-азабицикло[3.3.0]октан; 6-азабицикло[3.2.1]октан; 7-азабицикло[2.2.1]гептан; декагидропиразино[1,2 а]азепин; 3,8-диазабицикло[3.2.1]октан; 3-азабицикло[3.2.1]октан; 3-азатрицикло[4.2.1.0(2,5)]нонан; 2,6 диазаспиро[3.5]нонан; 6-азабицикло[2.1.0]гексан и т.д. В каждой из указанных выше формул любые асимметричные атомы углерода могут иметь (R)-, (S)или (R,S)-конфигурацию. Таким образом, соединения могут существовать в виде смесей изомеров или в виде чистых изомеров, например в виде чистых энантиомеров или диастереомеров. Изобретение также охватывает возможные таутомеры соединений согласно изобретению. В каждой из указанных выше формул любой необязательно замещенный фрагмент может быть замещен С 1-6 алкилом, C2-6 алкенилом или C3-6 алкинилом, причем любой из них может быть необязательно галогенирован или необязательно иметь углерод, который может быть заменен или замещен N, S, О или их комбинацией (например, гидроксиС 1-8 алкил, С 1-8 алкоксиС 1-8 алкил); галогеном, амино, амидино, С 1-6 алкокси; гидроксилом, метилендиокси, карбокси; C1-8 алкилкарбонилом, C1-8 алкоксикарбонилом, карбамоилом, C1-8 алкилкарбамоилом, сульфамоилом, циано, оксо, нитро или необязательно замещенным карбоциклическим кольцом, гетероциклическим кольцом, арилом или гетероарилом, как описано ранее. Соединения согласно изобретению и их фармацевтически приемлемые соли проявляют ценные фармакологические свойства при исследовании in vitro с помощью бесклеточного анализа киназной активности и анализа на клетках и, следовательно, полезны в качестве фармацевтических средств. В одном аспекте соединения формулы (1), (2), (3 А), (3 В), (4 А), (4 В) или (5) могут ингибировать тирозинкиназную активность киназы анапластической лимфомы (ALK) и слитого белка NPM-ALK. Такая тирозинпротеинкиназа является продуктом слияния генов нуклеофосмина (NPM) и ALK, и проявляет при этом тирозинпротеинкиназную активность киназы ALK независимо от лиганда. ПротеинкиназаNPM-ALK играет ключевую роль в передаче сигнала в ряде гематопоэтических клеток и других клетках человека, приводящую к гематологическим и неопластическим заболеваниям, например в клетках анапластической крупноклеточной лимфомы (ALCL) и неходжкинской лимфомы (NHL), особенно вALK+NHL или Alkomas, в воспалительных миофибробластных опухолях (IMT) и нейробластомах.(Duyster et al. 2001 Oncogene 20, 5623-5637). Кроме NPM-ALK другие гибридные гены были идентифицированы при гематологических и неопластических заболеваниях, например TPM3-ALK (слияние немышечного тропомиозина с ALK). Ингибирование тирозинкиназной активности ALK можно продемонстрировать известными методами, например, с использованием рекомбинантного киназного домена ALK по аналогии с исследованием киназы VEGF-R, описанным Wood et al., Cancer Res. 60, 2178-2189 (2000). В общем, исследование фермента in vitro с использованием тирозинпротеинкиназы GST-ALK осуществляют в 96-луночных планшетах методом связывания на фильтрах в 20 мМ трис-HCl буфере, рН 7,5, 3 мМ MgCl2, 10 мМMnCl2, 1 мМ DTT, 0,1 мкКи/образец (=30 мкл) [-33 Р]-ATP, 2 мкМ ATP, 3 мкг/мл поли(Glu, Tyr 4:1)полиEY (Sigma P-0275), 1% ДМСО, 25 нг фермента ALK. Образцы инкубируют в течение 10 мин при температуре окружающей среды. Реакции останавливают путем добавления 50 мкл 125 мМ EDTA и реакционную смесь переносят в многолуночный планшет для проведения высокоэффективного скрининга MAIPMultiscreen (Millipore, Bedford, MA, USA), предварительно смоченный метанолом, и регидратируют в течение 5 мин водой. После промывания (0,5% Н 3 РО 4) планшеты просчитывают на жидкостном сцинтилляционном счетчике. Величины IC50 рассчитывают методом линейной регрессии, позволяющим оценить процент ингибирования. Соединения формулы (1), (2), (3 А), (3 В), (4 А), (4 В) или (5) могут эффективно ингибировать рост мышиных клеток BaF3, отличающихся повышенной экспрессией NPM-ALK человека (DSMZ DeutscheSamming von Mikroorganismen und Zelikulturen GmbH, Germany). Экспрессия протеинкиназы NPM-ALK может быть достигнута путем трансфекции линии клеток BaF3 экспрессирующим вектором pClneo(Promega Corp., Madison WI, USA), кодирующим NPM-ALK и последующего отбора устойчивых клетокG418. Выживаемость нетрансфицированных клеток BaF3 зависят от IL-3. В отличие от этого, клеткиBaF3, экспрессирующие NPM-ALK (названные в описании в дальнейшем BaF3-NPM-ALK), могут пролиферировать в отсутствие IL-3, поскольку они получают сигнал к пролиферации через киназу NPMALK. Следовательно, предполагаемые ингибиторы киназы NPM-ALK устраняют сигнал к росту и могут приводить к антипролиферативной активности. Однако противопролиферативная активность предполагаемых ингибиторов NPM-ALK-киназы может быть подавлена при добавлении IL-3, который обеспечивает сигналы к росту посредством независимого от NPM-ALK механизма. Аналогичная клеточная система с использованием киназы FLT3 также была описана (см., Е. Weisberg et al. Cancer Cell; 1, 433-443(2002. Ингибирующая активность соединений согласно изобретению может быть определена следующим образом. В общем, BaF3-NPM-ALK-клетки (15000/лунка титрационного микропланшета) переносят в 96 луночные титрационные микропланшеты. Испытуемые соединения, растворенные в диметилсульфоксиде (ДМСО), добавляют в ряде концентраций (серия разведений) таким образом, чтобы конечная концентрация ДМСО составляла не более 1% (об./об.). После добавления планшеты инкубируют в течение двух дней, в течение которых контрольные культуры без испытуемого соединения претерпевают два цикла клеточного деления. Рост BaF3-NPM-ALK-клеток измеряют путем окрашивания красителем YOPRO[T. Idziorek et al. J. Immunol. Methods; 185:249-258 (1995)]: 25 мкл буфера для лизиса, содержащего 20 мМ цитрата натрия, рН 4,0, 26,8 мМ хлорида натрия, 0,4% NP40, 20 мМ EDTA и 20 мМ добавляют к каждой лунке. Лизис клеток завершается в течение 60 мин при комнатной температуре, и общее количество красителя YOPRO, связанного с ДНК, определяют путем измерения с использованием 96-луночного планшет-ридера Cytofluor II (PerSeptive Biosystems) со следующими установочными параметрами: возбуждение (нм) 485/20 и испускание (нм) 530/25. Величины IC50 могут быть определены с помощью компьютерной системы по формуле(ABS означает абсорбцию) Величина IC50 в указанных экспериментах представлена в виде такой концентрации исследуемого тестируемого соединения, которая приводит к количеству клеток, которое на 50% ниже, чем количество клеток, существующее при использовании контроля без ингибитора. Соединения согласно изобретению в свободном виде или в виде фармацевтически приемлемой соли могут проявлять значительные фармакологические свойства, например, как показано испытаниями in vitro, описанными в данном описании. В общем, соединения согласно изобретению имеют величины IC50 от 1 нМ до 10 мкМ. В некоторых примерах соединения согласно изобретению имеют величины IC50 от 0,01 до 5 мкМ. В других примерах соединения согласно изобретению имеют величины IC50 от 0,01 до 1 мкМ, или более конкретно от 1 нМ до 1 мкМ. В других примерах соединения согласно изобретению имеют величины IC50 менее чем 1 нМ или более чем 10 мкМ. Соединения согласно изобретению могут оказывать ингибирование, выраженное в процентах, составляющее выше 50%, или в других вариантах осуществления изобретения могут оказывать ингибирование в процентах, составляющее выше чем приблизительно 70%, в отношении ALK при 10 мкМ. Противопролиферативное действие соединений согласно изобретению также можно определить для линии клеток лимфомы человека KARPAS-299 (DSMZ Deutsche Sammiung von Mikroorganismen und Zelikulturen GmbH, Braunschweig, Germany, данные описаны WG Dirks et al. в Int. J. Cancer 100, 49-56(2002, используя аналогичную методологию, как описано выше для линии клеток BaF3-NPM-ALK. В некоторых вариантах осуществления изобретения соединения согласно изобретению могут оказывать ингибирующее действие с IC50 в области приблизительно от 0,01 до 1 мкМ. Действие соединений согласно изобретению на автофосфорилирование ALK можно определить на линии клеток лимфомы человека KARPAS-299 посредством иммуноблоттинга, как описано WG Dirks et al. в Int. J. Cancer 100, 49-56(2002). В другом аспекте соединения согласно изобретению могут ингибировать очагово-адгезивную киназу (FAK) и могут быть использованы в качестве фармацевтических средств, предназначенных для лечения состояний, вызванных нарушением каскадов реакций передачи сигнала, связанным с FAK, таких как лечение отдельных опухолей. Ингибирование эндогенной киназы FAK, участвующей в передаче сигнала,приводит к снижению подвижности клеток и в некоторых случаях вызывает гибель клеток. С другой стороны, усиление FAK-зависимой передачи сигнала посредством экзогенной экспрессии повышает подвижность клеток. Кроме того, повышенная экспрессия FAK наблюдается в инвазивных и метастатических эпителиальных, мезенхимальных опухолях, опухолях щитовидной железы и простаты. Поэтому ингибитор FAK, вероятно, может быть лекарственным средством против роста опухолей и метастазирования. Таким образом, соединения согласно изобретению могут оказаться полезными для предотвращения и/или лечения позвоночных и более конкретно млекопитающих, пораженных неопластическим заболеванием, в частности, которым является опухоль молочной железы, рак кишки (толстая и прямая), рак желудка и рак яичника и простаты, немелкоклеточный рак легкого, мелкоклеточный рак легкого, рак печени, меланома, бластома мочевого пузыря и рак головы и шеи. Связь между ингибированием FAK и иммунной системой описана, например, G.A. van Seventer etal., в Eur. J. Immunol. 2001, 31, 1417-1427. Поэтому соединения согласно изобретению являются полезными, например, для предотвращения и/или лечения позвоночных и более конкретно млекопитающих,имеющих нарушения иммунной системы, заболевания и расстройства, опосредованные Т-лимфоцитами,B-лимфоцитами, тучными клетками и/или эозинофилами, например острое или хроническое отторжение алло- или ксенотрансплантатов органа или ткани, атеросклероз, окклюзия сосуда вследствие сосудистого повреждения, такого как ангиопластика, рестеноз, гипертензия, сердечная недостаточность, хроническое обструктивное легочное заболевание, заболевание ЦНС, такое как болезнь Альцгеймера или амиотрофический боковой склероз; рак; инфекционное заболевание, такое как СПИД; септический шок или респираторный дистресс-синдром взрослых, ишемия/реперфузионное повреждение, например инфаркт миокарда, удар, ишемия кишки, почечная недостаточность или геморрагический шок или травматический шок. В другом аспекте соединения согласно изобретению могут ингибировать дзета-цепьассоциированный белок 70 (ZAP-70). Взаимодействие агентов согласно изобретению с тирозинпротеинкиназой ZAP-70 может быть продемонстрировано, например, их способностью предотвращать фосфорилирование LAT-11 (линкер для активации Т-клетки) посредством тирозинпротеинкиназы ZAP-70 в водном растворе. Поэтому соединения согласно изобретению могут быть пригодными для предотвращения или лечения нарушений или заболеваний, где ингибирование ZAP-70 играет некоторую роль. Соединения согласно изобретению также могут ингибировать рецептор 1 инсулиноподобного фактора роста (IGF-1R) и могут быть пригодными для лечения IGF-1R-опосредованных заболеваний. Примеры IGF-1R-опосредованных заболеваний включают, но не ограничиваются ими, пролиферативные заболевания, такие как опухоли, например, опухоли молочной железы, почки, простаты, колоректальные опухоли, опухоли щитовидной железы, яичника, поджелудочной железы, нейрональные опухоли, опухоли легкого, матки и желудочно-кишечные опухоли, а также остеосаркомы и меланомы. Эффективность соединений согласно изобретению как ингибиторов тирозинкиназной активности IGF-1R может быть продемонстрирована с помощью иммуноанализа с захватом ELISA. В указанном анализе определяют активность соединений согласно изобретению против аутофосфорилирования IGF-1R, индуцированного(IGF-1). Соединения согласно изобретению также могут быть использованы при лечении и/или предотвращении острых и хронических воспалительных заболеваний и расстройств или аутоиммунных заболеваний, таких как ревматоидный артрит, остеоартрит, системная красная волчанка, зоб Хашимото, рассеянный склероз, миастения гравис, диабет (тип I и II) и связанные с ним нарушения, респираторных заболеваний, таких как астма, или воспалительных поражений печени, воспалительного поражения почечных клубочков, кожных проявлений иммунологически опосредованных нарушений или заболеваний, воспалительных и гиперпролиферативных кожных заболеваний (таких как псориаз, атонический дерматит,аллергический контактный дерматит, контактный дерматит, обусловленный раздражителем, и также экзематозный дерматит, себорейный дерматит), воспалительных заболеваний глаз, например синдрома Шегрена, кератоконъюктивита или увеита, воспалительного заболевания кишечника, болезни Крона или язвенного колита. В соответствии с изложенным выше настоящее изобретение предоставляет:(1) соединение согласно изобретению для применения в качестве фармацевтического средства;(2) соединение согласно изобретению для применения в качестве ингибитора ALK, ингибитораFAK, ингибитора ZAP-70 и/или ингибитора IGF-1R, например для применения при любом из отдельных показаний, описанных выше;(3) фармацевтическую композицию, например, для применения при любом из показаний, описанных выше, содержащую соединение согласно изобретению в качестве активного ингредиента вместе с одним или несколькими фармацевтически приемлемыми разбавителями или носителями;(4) способ лечения по любому отдельному показанию, описанному выше, субъекта, нуждающегося в этом, включающий введение эффективного количества соединения согласно изобретению или фармацевтической композиции, содержащей данное соединение;(5) применение соединения согласно изобретению для производства лекарственного средства,предназначенного для лечения или предотвращения заболевания или состояния, при котором активацияALK, FAK, ZAP-70 и/или IGF-1R играет некоторую роль или непосредственно связана с заболеванием или состоянием;(6) способ, как определено выше в п. (4), включающий совместное введение, например, одновременно или в последовательности, терапевтически эффективного количества соединения согласно изобретению и одного или более других дополнительных лекарственных средств, где указанные дополнительные лекарственные средства являются пригодными при любом из отдельных показаний, описанных выше;(7) комбинацию, содержащую терапевтически эффективное количество соединения согласно изобретению и одно или более других дополнительных лекарственных средств, где указанные дополнительные лекарственные средства являются пригодными при любом из отдельных показаний, описанных выше;(8) применение соединения согласно изобретению для производства лекарственного средства,предназначенного для лечения или предотвращения заболевания, которое отвечает на ингибирование киназы анапластической лимфомы;(9) применение согласно п. (8), где заболевание, подвергаемое лечению, выбирают из анапластической крупноклеточной лимфомы, неходжкинской лимфомы, воспалительных миофибробластных опухолей, нейробластом и неопластических заболеваний;(10) применение по п. (8) или (9), где соединением является фармацевтически приемлемая соль любого одного из примеров;(11) способ лечения заболевания, которое отвечает на ингибирование киназы анапластической лимфомы, особенно заболевания, выбранного из анапластической крупноклеточной лимфомы, неходжкинской лимфомы, воспалительных миофибробластных опухолей, нейробластом и неопластических заболеваний, включающий введение эффективного количества соединения согласно изобретению или его фармацевтически приемлемой соли. Введение и фармацевтические композиции Как правило, соединения согласно изобретению будут вводиться в терапевтически эффективном количестве любым из обычных и подходящих способов, известных в данной области, либо отдельно,либо в комбинации с одним или более терапевтическими средствами. Терапевтически эффективное количество может широко варьировать в зависимости от тяжести заболевания, возраста и состояния здоровья субъекта, эффективности используемого соединения и других факторов, известных специалистам в данной области. Например, для лечения неопластических заболеваний и нарушений иммунной системы требуемая дозировка также будет изменяться в зависимости от способа введения, отдельного состояния,- 12019966 подвергаемого лечению, и желаемого эффекта. Обычно удовлетворительные результаты, относящиеся ко всему организму, как показано, получают при суточных дозировках приблизительно от 0,01 до 100 мг/кг массы тела или в особенности приблизительно от 0,03 до 2,5 мг/кг массы тела. Указанная суточная дозировка у высшего млекопитающего, например человека, может составлять диапазон приблизительно от 0,5 до 2000 мг или более конкретно приблизительно от 0,5 до 100 мг, без труда введенная, например, в разделенных дозах вплоть до четырех раз в день или в форме замедленного действия. Подходящие единичные дозированные формы для перорального введения содержат приблизительно от 1 до 50 мг активного ингредиента. Соединения согласно изобретению могут быть введены в виде фармацевтических композиций любым подходящим способом, например внутрь тонкой кишки, например перорально, например в виде таблеток или капсул; парентерально, например в виде инъецируемых растворов или суспензий; или местно, например в виде лосьонов, гелей, мазей или кремов, или в виде назальных препаратов или суппозиториев. Фармацевтические композиции, содержащие соединение согласно настоящему изобретению в свободном виде или в виде фармацевтически приемлемой соли в комбинации по меньшей мере с одним фармацевтически приемлемом носителем или разбавителем, могут быть получены обычными способами смешивания, грануляции, нанесения покрытия, растворения или лиофилизации. Например, фармацевтические композиции, содержащие соединение согласно изобретению в комбинации по меньшей мере с одним фармацевтически приемлемым носителем или разбавителем, могут быть получены обычным способом смешивания с фармацевтически приемлемым носителем или разбавителем. Единичные дозированные формы для перорального введения содержат, например, приблизительно от 0,1 до 500 мг активного вещества. В одном варианте осуществления изобретения фармацевтические композиции представляют собой растворы активного ингредиента, включающие суспензии или дисперсии, такие как изотоничные водные растворы. В случае лиофилизированных композиций, содержащих только активный ингредиент или вместе с носителем, таким как маннит, дисперсии или суспензии, могут быть получены непосредственно перед применением. Фармацевтические композиции могут быть стерилизованными и/или содержать адъюванты, такие как консервирующие, стабилизирующие, смачивающие или эмульгирующие средства,растворы активаторов, соли для регуляции осмотического давления и/или буферы. Подходящие консерванты включают, но не ограничиваются ими, антиоксиданты, такие как аскорбиновая кислота, или бактерицидные средства, такие как сорбиновая кислота или бензойная кислота. Растворы или суспензии также могут содержать повышающие вязкость средства, включающие, но не ограниченные ими, натрийкарбоксиметилцеллюлозу, карбоксиметилцеллюлозу, декстран, поливинилпирролидон, желатины или солюбилизаторы, например твин 80 (полиоксиэтилен(20)сорбитанмоноолеат). Суспензии в масле могут содержать в качестве масляного компонента растительные, синтетические или полусинтетические масла, стандартные для инъекций. Примеры включают жидкие сложные эфиры жирных кислот, которые содержат в качестве кислотного компонента длинноцепочечную жирную кислоту, имеющую от 8 до 22 атомов углерода, или в некоторых вариантах осуществления изобретения от 12 до 22 атомов углерода. Подходящие жидкие сложные эфиры жирных кислот содержат, но не ограничиваются ими, лауриновую кислоту, тридекановую кислоту, миристиновую кислоту, пентадекановую кислоту, пальмитиновую кислоту, маргариновую кислоту, стеариновую кислоту, арахидоновую кислоту,бегеновую кислоту или соответствующие ненасыщенные кислоты, например олеиновую кислоту, элаидиновую кислоту, эруковую кислоту, брассидиновую кислоту и линоленовую кислоту, и если желательно, могут включать антиоксиданты, например витамин Е, 3-каротен или 3,5-ди-ретбутилгидрокситолуол. Спиртовой компонент указанных выше сложных эфиров жирных кислот может иметь шесть атомов углерода и может быть моновалентным или поливалентным, например, моно-, диили тривалентным спиртом. Подходящие спиртовые компоненты включают, но не ограничиваются ими,метанол, этанол, пропанол, бутанол или пентанол или их изомеры; этиленгликоль или глицерин. Другие подходящие сложные эфиры жирных кислот включают, но не ограничиваются ими, этилолеат, изопропилмиристат, изопропилпальмитат, LABRAFIL М 2375 (полиоксиэтиленглицерин),LABRAFIL М 1944 CS (ненасыщенные полигликолизированные глицериды, полученные путем алкоголиза масла из косточек абрикоса и содержащие глицериды и сложный эфир полиэтиленгликоля),LABRASOL (насыщенные полигликолизированные глицериды, полученные путем алкоголиза ТСМ и содержащие глицериды и сложный эфир полиэтиленгликоля; все доступны от GaKefosse, France) и/илиMIGLYOL 812 (триглицерид насыщенных жирных кислот с длиной цепи от С 8 до C12 от HlS AG,Germany) и растительные масла, такие как хлопковое масло, миндальное масло, оливковое масло, касторовое масло, кунжутное масло, соевое масло или арахисовое масло. Фармацевтические композиции для перорального введения могут быть получены, например, путем комбинирования активного ингредиента с одним или несколькими твердыми носителями, и если желательно, путем грануляции образовавшейся смеси и обработки смеси или гранул посредством включения дополнительных эксципиентов с образованием таблеток или сердцевин таблеток. Подходящие носители включают, но не ограничиваются ими, наполнители, такие как сахара, например лактоза, сахароза, маннит или сорбит, препараты целлюлозы и/или фосфаты кальция, например трикальцийфосфат или вторичный кислый фосфат кальция, и также связующие средства, такие как крахмалы, например кукурузный, пшеничный, рисовый или картофельный крахмал, метилцеллюлоза,гидроксипропилметилцеллюлоза, натрийкарбоксиметилцеллюлоза и/или поливинилпирролидон, и/или если желательно, дезинтегрирующие средства, такие как приведенные выше крахмалы, карбоксиметилкрахмал, поперечно-сшитый поливинилпирролидон, альгиновая кислота и ее соль, такая как альгинат натрия. Дополнительные эксципиенты включают стабилизаторы потока и лубриканты, например, кремниевую кислоту, тальк, стеариновую кислоту или ее соли, такие как стеарат магния или кальция, и/или полиэтиленгликоль или его производные. Сердцевины таблеток могут быть покрыты подходящими оболочками, необязательно энтеросолюбильными, при использовании для этого, в частности, концентрированных растворов сахара, которые могут содержать аравийскую камедь, тальк, поливинилпирролидон, полиэтиленгликоль и/или диоксид титана, или при использовании растворов для покрытий в подходящих органических растворителях или смесях растворителей, или для получения сердцевин таблеток с энтеросолюбильными покрытиями, могут быть использованы растворы подходящих препаратов целлюлозы, таких как фталат ацетилцеллюлозы или фталат гидроксипропилметилцеллюлозы. Красители или пигменты могут быть добавлены к таблеткам или покрытиям таблеток, например, с целью распознавания или для обозначения различных доз активного ингредиента. Фармацевтические композиции для перорального введения также могут включать твердые капсулы,содержащие желатин, и капсулы в мягкой упаковке, содержащие желатин и пластификатор, такой как глицерин или сорбит. Твердые капсулы могут содержать активный ингредиент в виде гранул, например,в смеси с наполнителями, такими как кукурузный крахмал, связующими веществами и/или веществами,способствующими скольжению, такими как тальк или стеарат магния, и необязательно стабилизаторами. В мягких капсулах активный ингредиент может быть растворенным или суспендированным в подходящих жидких эксципиентах, таких как жидкие масла, парафиновое масло или жидкие полиэтиленгликоли или сложные эфиры жирных кислот и этиленгликоля или пропиленгликоля, к которым также могут быть добавлены стабилизаторы и детергенты, например полиоксиэтиленовые эфиры сорбита и жирной кислоты. Фармацевтическими композициями, подходящими для ректального введения, являются, например,суппозитории, содержащие комбинацию активного ингредиента и основы суппозитория. Подходящими основами суппозиториев являются, например, природные или синтетические триглицериды, углеводороды парафинового ряда, полиэтиленгликоли или высшие алканолы. Фармацевтические композиции, подходящие для парентерального введения, могут содержать водные растворы активного ингредиента в водорастворимом виде, например растворимой в воде соли, или водные инъецируемые суспензии, которые содержат повышающие вязкость вещества, например натрийкарбоксиметилцеллюлозу, сорбит и/или декстран и, если желательно, стабилизаторы. Активный ингредиент, необязательно вместе с эксципиентами, также может быть в лиофилизированном виде и может быть переведен в раствор непосредственно перед парентеральным введением при добавлении подходящих растворителей. Растворы, которые используют, например, для парентерального введения, также могут быть использованы в качестве инфузионных растворов. Получение инъецируемых препаратов обычно осуществляют в стерильных условиях, как, например, заполнение ампул или пузырьков, и запаивание контейнеров. Соединения согласно изобретению могут быть введены в виде одного активного ингредиента или вместе с другими лекарственными средствами, применяемыми от неопластических заболеваний или используемых в схемах лечения с иммуномодулирующим эффектом. Например, соединения согласно изобретению могут быть использованы в соответствии с настоящим изобретением в комбинации с фармацевтическими композициями, эффективными при различных заболеваниях, как описано выше, например,с циклофосфамидом, 5-фторурацилом, флударабином, гемцитабином, цисплатином, карбоплатином, винкристином, винбластином, этопозидом, иринотеканом, паклитакселом, доцетакселом, ритуксаном, доксорубицином, гефитинибом или иматинибом; или также с циклоспоринами, рапамицинами, аскомицинами или их иммуносупрессорными аналогами, например циклоспорином А, циклоспорином G, FK-506,сиролимусом или эверолимусом, кортикостероидами, например преднизоном, циклофосфамидом, азатиопреном, метотрексатом, солями золота, сульфасалазином, противомалярийными средствами, бреквинаром, лефлуномидом, мизорибином, микофеноловой кислотой, микофенолятом, мофетилом, 15 деоксиспергуалином, иммуносупрессорными моноклональными антителами, например моноклональными антителами к рецепторам лейкоцитов, например МНС, CD2, CD3, CD4, CD7, CD25, CD28, CD40,CD45, CD58, CD80, CD86, CD152, ICOS, LFA-1, VLA-4 или их лигандам, или с другими иммуномодулирующими соединениями, например CTLA41g. Изобретение также предоставляет для фармацевтических комбинаций, например, набор, содержащий а) первое средство, являющееся соединением согласно изобретению, как изложено в данном описании, в свободном виде или в виде его фармацевтически приемлемой соли, и b) по меньшей мере одно дополнительное средство. Набор может включать инструкции для его применения. Способы получения соединений согласно изобретению соединения формулы (1) могут быть получены согласно следующей реакционной схеме I, на которой каждый заместитель определен в разделе Соединение формулы (1) может быть синтезировано путем взаимодействия соединения формулы(6) с соединением формулы (7) в присутствии палладиевого катализатора (например, ацетата палладия и тому подобное), лиганда (например, ксантфоса [фосфорорганическое соединение] и тому подобное) и основания (например, карбоната цезия и тому подобное) в подходящем растворителе (например, ТГФ и тому подобное). Реакция протекает при температуре в области приблизительно от 70 до 180C, и для завершения реакции может потребоваться от 10 мин до 8 ч. Альтернативно, соединение формулы (1) может быть синтезировано путем взаимодействия соединения формулы (6) с соединением формулы (7) в присутствии кислоты (например, HCl, TsOH и тому подобное) в подходящем растворителе (например, 2-пропаноле и тому подобное). Реакция протекает при температуре в области приблизительно от 70 до 150C, и для завершения реакции может потребоваться вплоть до 12 ч. Дополнительные способы получения соединений согласно изобретению Соединения согласно изобретению, включая их соли, также получают в виде гидрата, или их кристаллы могут включать, например, растворитель, используемый для кристаллизации (представлены в виде сольватов). Обычно соли могут быть преобразованы в соединения в свободном виде, например, при обработке подходящими основными средствами, например карбонатами щелочных металлов, гидрокарбонатами щелочных металлов или гидроксидами щелочных металлов, такими как карбонат калия или гидроксид натрия. Основываясь на тесной взаимосвязи между новыми соединениями в свободном виде и их солями, включая соли, которые могут быть использованы в качестве промежуточных соединений,например, при очистке и идентификации новых соединений, любую ссылку на свободные соединения,приведенную в описании выше и ниже, следует относить также к их соответствующим солям, по необходимости. Соли соединений согласно изобретению с солеобразующей группой могут быть получены способом, известным как таковым. Таким образом, аддитивные соли кислоты соединений формулы (1), (2),(3 А), (3 В), (4 А), (4 В) или (5) могут быть получены обработкой кислотой или подходящим анионообменником. Фармацевтически приемлемые соли соединений согласно изобретению могут быть получены,например, в виде аддитивных солей кислоты, с органическими или неорганическими кислотами, из соединений формулы (1), (2), (3 А), (3 В), (4 А), (4 В) или (5) с основным атомом азота. Подходящие неорганические кислоты включают, но не ограничиваются ими, галогенсодержащие кислоты, такие как хлористо-водородная кислота, серную кислоту или фосфорную кислоту. Подходящие органические кислоты включают, но не ограничиваются ими, карбоновую, сульфоновую или сульфаминовую кислоты, например, уксусную кислоту, пропионовую кислоту, октановую кислоту, декановую кислоту, додекановую кислоту, гликолевую кислоту, молочную кислоту, фумаровую кислоту, янтарную кислоту, адипиновую кислоту, пимелиновую кислоту, пробковую кислоту, азелаиновую кислоту, яблочную кислоту, винную кислоту, лимонную кислоту, аминокислоты, такие как глутаминовая кислота или аспарагиновая кислота, малеиновую кислоту, гидроксималеиновую кислоту, метилмалеиновую кислоту,циклогексанкарбоновую кислоту, амадантанкарбоновую кислоту, бензойную кислоту, салициловую кислоту, 4-аминосалициловую кислоту, фталевую кислоту, фенилуксусную кислоту, миндальную кислоту,коричную кислоту, метан- или этансульфокислоту, 2-гидроксиэтансульфокислоту, этан-1,2 дисульфокислоту, бензолсульфокислоту, 2-гафталинсульфокислоту, 1,5-нафталиндисульфокислоту, 2-,3- или 4-метилбензолсульфокислоту, метилсерную кислоту, этилсерную кислоту, додецилсерную кислоту, N-циклогексилсульфамовую кислоту, N-метил, N-этил или N-пропилсульфамовую кислоту, или другие органические протонные кислоты, такие как аскорбиновая кислота. С целью выделения и очистки также можно использовать фармацевтически неприемлемые соли,например пикраты или перхлораты. Для терапевтических целей используют только фармацевтически приемлемые соли или свободные соединения (где применяют в виде фармацевтических препаратов). Соединения согласно изобретению в неокисленном виде могут быть получены из N-оксидов соединений согласно изобретению путем обработки восстановителем (например, серой, диоксидом серы, три- 15019966 фенилфосфином, литийборгидридом, натрийборгидридом, трихлоридом фосфора, трибромидом или тому подобное) при температуре от 0 до 80C. Пролекарственные производные соединений согласно изобретению могут быть получены способами, известными специалисту в данной области (например, для дальнейших подробностей см. Saulnier etal., (1994), Bioorganic and Medicinal Chemistry Letters, Vol. 4, p. 1985). Например, соответствующие пролекарства могут быть получены путем взаимодействия соединения, не являющегося производным соединения согласно данному изобретению, с подходящим карбамилирующим средством (например, 1,1 ацилоксиалкилкарбанохлоридатом, паранитрофенилкарбонатом или тому подобное). Защищенные производные соединений согласно изобретению могут быть получены способами, известными специалисту в данной области. Подробное описание способов, используемых для присоединения защитных групп и для их удаления, можно найти в публикации T.W. Greene, "Protecting Groups inOrganic Chemistry", 3rd edition, John Wiley and Sons, Inc., 1999. Соединения согласно изобретению могут быть получены в виде их индивидуальных стереоизомеров путем взаимодействия рацемической смеси соединения с оптически активным разделяющим реагентом с образованием пары диастереоизомерных соединений, разделения диастереомеров и выделения оптически чистых энантиомеров. Разделение энантиомеров можно проводить при использовании ковалентных диастереомерных производных соединений согласно изобретению или при использовании диссоциируемых комплексов (например, кристаллических диастереомерных солей). Диастереомеры обладают различными физическими свойствами (например, точки плавления, точки кипения, растворимости, реакционная способность и т.д.) и могут быть легко разделены исходя из преимущества указанных выше различий. Диастереомеры могут быть разделены фракционной кристаллизацией, хроматографией или методами разделения/разрешения, основанными на различиях в растворимости. Затем выделяют оптически чистый энантиомер, наряду с разделяющим реагентом, любыми практическими способами, которые не должны приводить к рацемизации. Подробное описание методов, используемых для разделения стереоизомеров соединений, присутствующих в их рацемической смеси, можно найти в публикации JeanJacques, Andre Collet, Samuel H. Wilen, "Enantiomers, Racemates and Resolutions", John Wiley and Sons, Inc.,1981. Таким образом, соединения согласно изобретению могут быть получены способом, который включает:(a) получение соединения, следуя реакционной схеме I, и(b) необязательно преобразование соединения согласно изобретению в фармацевтически приемлемую соль;(c) необязательно преобразование солевой формы соединения согласно изобретению в форму, отличную от солевой;(d) необязательное преобразование неокисленной формы соединения согласно изобретению в фармацевтически приемлемый N-оксид;(e) необязательное преобразование N-оксида соединения согласно изобретению в его неокисленную форму;(f) необязательно отделение индивидуального изомера соединения согласно изобретению от смеси изомеров;(g) необязательно преобразование недериватизированного соединения согласно изобретению в его фармацевтически приемлемое пролекарство; и(h) необязательно преобразование пролекарства соединения согласно изобретению в его недериватизированную форму. Поскольку получение исходных веществ подробно не описано, соединения являются известными или могут быть получены аналогично способам, известным в данной области, или как описано в примерах ниже. Специалисту в данной области будет понятно, что представленные выше преобразования только представляют собой конкретные примеры способов получения соединений согласно настоящему изобретению, и что другие хорошо известные способы могут быть использованы аналогичным образом. Настоящее изобретение подтверждено, но не ограничено, следующими примерами, которые иллюстрируют получение соединений согласно изобретению. Получение промежуточных соединений Промежуточное соединение 1. 2-Хлор-N-(2-(изопропилсульфонил)фенил)-5-метилпиримидин-4 амин К суспензии 730 мг NaH в смеси ДМФА/ДМСО (25/2,5 мл) добавляли по каплям при 0C 2,53 г го в 10 мл ДМФА/ДМСО (отношение 9/1), добавляли по каплям. Раствор нагревали до комнатной температуры и перемешивали в течение ночи. После обработки неочищенный продукт непосредственно кристаллизовали из холодного CH3CN несколькими порциями с получением 2-хлор-N-(2-(изопропилсульфонил)фенил)-5-метилпиримидин-4-амина в виде окрашенных в светло-кремовый цвет кристаллов: Используя методику, аналогично описанной для синтеза 2-хлор-N-(2-(изопропилсульфонил)фенил)5-метилпиримидин-4-амина, выделяли 2,5-дихлор-N-(2-(изопропилсульфонил)фенил)пиримидин-4-амин в виде твердого вещества, окрашенного в кремовый цвет: ES-MC m/z 346, О (М+Н+). Промежуточное соединение 3. 2-Хлор-4-фтор-5-нитротолуол К раствору 100 г (0,7 моль) 2-хлор-4-фтортолуола в 250 мл концентрированной H2SO4 добавляли порциями 85 г (0,875 моль) KNO3 при 0C (добавление всего количества KNO3 оканчивали в течение 1 ч). Смесь красноватого цвета нагревали при комнатной температуре в течение ночи, реакцию гасили добавлением измельченного льда и реакционную смесь экстрагировали EtOAc. Органические слои объединяли, сушили над MgSO4 и концентрировали. Затем неочищенное масло очищали через рыхлый толстый слой диоксида кремния (элюент: смесь 97/3 гексаны/EtOAc), с получением 2-хлор-4-фтор-5 нитротолуола в виде бледно-желтого масла, которое затвердевало при стоянии. 1(0,659 моль, 5 экв.) Cs2CO3. Смесь перемешивали при 60C в течение ночи и большую часть 2-пропанола упаривали при пониженном давлении. Добавляли воду и раствор экстрагировали EtOAc. Органические слои объединяли, сушили над MgSO4, концентрировали и неочищенный продукт фильтровали через слой диоксида кремния (элюент: смесь 95/5 гексаны/EtOAc), с получением 2-хлор-4-изопропокси-5 нитротолуола в виде бледно-желтого рыхлого твердого вещества. Промежуточное соединение 5. Пинаконовый сложный эфир 2-метил-4-нитро-5-изопропоксифенилбороновой кислоты Смесь 5,09 г 2-хлор-4-изопропокси-5-нитротолуола (0,02216 моль), 6,20 г (0,02437 моль) пинакондиборана, 595 мг (0,00212 моль) PCy3, 1,014 г (0,00108 моль) Pd2dba3 и 3,16 г (0,0322 моль) KOAc в 100 мл сухого диоксана нагревали до 100C в течение ночи. После охлаждения до комнатной температуры темный раствор фильтровали через целит и растворитель упаривали при пониженном давлении. Неочищенное масло очищали хроматографией на колонке с силикагелем (элюент: смесь 95/5 гексаны/EtOAc) с получением пинаконового сложного эфира 2-метил-4-нитро-5-изопропоксифенилбороновой кислоты в виде масла, которое затвердевало при стоянии. 1 Раствор N-трет-бутоксикарбонил-4-пиперидона (10,17 г, 0,05 моль) в ТГФ (100 мл) добавляли по каплям в охлажденный (-78C) энергично перемешиваемый раствор LDA (40 мл 1,5 М раствора в циклогексанах, 0,06 моль) в ТГФ (100 мл) в атмосфере N2. Реакционную смесь выдерживали при -78C в течение 30 мин до добавления раствора фенилтрифторсульфонимида (19,85 г, 0,055 моль) в ТГФ (50 мл). Затем реакционную смесь нагревали до комнатной температуры и перемешивали в течение 3 ч. Реакцию гасили при 0C добавлением 100 мл насыщенного водного раствора NH4Cl и фильтровали через целит. Фильтрат добавляли к 100 мл EtOAc и слои разделяли. Органический слой промывали Н 2 О, сушили надMgSO4 и концентрировали. Неочищенный продукт очищали колоночной флэш-хроматографией (в качестве элюента 0-30% смесь EtOAc в гексанах и проверяли ТСХ с окрашиванием 2% KMnO4 в EtOH) с получением трет-бутилового эфира 4-трифторметансульфонилокси-3,6-дигидро-2 Н-пиридин-1-карбоновой кислоты в виде желтого масла. Промежуточное соединение 7. трет-Бутиловый эфир 4-(5-изопропокси-2-метил-4-нитрофенил)-3,6 дигидро-2 Н-пиридин-1-карбоновой кислоты К раствору пинаконового эфира 2-метил-4-нитро-5-изопропоксифенилбороновой кислоты (2,04 г,6,4 ммоль) и трет-бутилового эфира 4-трифторметансульфонилокси-3,6-дигидро-2 Н-пиридин-1 карбоновой кислоты (3,2 г, 9,6 ммоль) в 110 мл DME/H20 (10:1 об./об.) добавляли Pd(PPh3)4 (365 мг, 0,32 ммоль) и Cs2CO3 (4,2 г, 12,8 ммоль). Реакционную смесь нагревали в атмосфере N2 при 80C в течение ночи. После охлаждения до комнатной температуры реакционную смесь фильтровали через целит, фильтрат разбавляли 100 мл EtOAc, последовательно промывали Н 2 О, насыщенным раствором соли и в конце концентрировали в вакууме. Неочищенный продукт очищали флэш-хроматографией на силикагеле (в качестве элюента 5-15% смесь EtOAc в гексанах) с получением трет-бутилового эфира 4-(5-изопропокси 2-метил-4-нитрофенил)-3,6-дигидро-2 Н-пиридин-1-карбоновой кислоты в виде желтого масла. 1 Смесь 2-хлор-4-фтор-5-нитробензойной кислоты (5,0 г, 22,8 ммоль) и карбоната цезия (29,7 г, 91,1 ммоль) в 2-пропаноле (100 мл) нагревали при 50C в течение ночи. Растворитель удаляли в вакууме и добавляли 100 мл воды. Концентрированный водный раствор HCl добавляли по каплям к указанному выше раствору при 0C вплоть до рН 2. Продукт выпадал в виде осадка, который выделяли фильтрованием, промывали водой и сушили в вакууме с получением 2-хлор-4-изопропокси-5-нитробензойной кислоты. Пример 1. 6-5-Хлор-4-[2-(пропан-2-сульфонил)фениламино]пиримидин-2-иламино-5-изопропокси-2-пиперидин-4-ил-2,3-дигидроизоиндол-1-он (178) К раствору 2-хлор-4-изопропокси-5-нитробензойной кислоты (промежуточное соединение 8, 10 г,38,5 ммоль) в DCM (200 мл) и ДМФА (1 мл) медленно добавляли тионилхлорид (9,17 г, 77 ммоль) посредством шприца. Смесь перемешивали в течение 3 ч и затем концентрировали досуха. Полученное белое твердое вещество, 2-хлор-4-изопропокси-5-нитробензоилхлорид, сушили в вакууме. К смеси третбутилового эфира 4-аминопиперидин-1-карбоновой кислоты (1,44 г, 7,2 ммоль) и триэтиламина (3 мл,21,6 ммоль) в DCM (100 мл) медленно добавляли 2-хлор-4-изопропокси-5-нитробензоилхлорид (2 г, 7,2 ммоль), растворенный в DCM (10 мл), посредством шприца. Смесь перемешивали при комнатной температуре в течение 3 ч и затем концентрировали. Полученное твердое вещество растворяли в этилацетате и промывали водой и насыщенным раствором соли в указанном порядке. После упаривания растворителя указанное в заголовке соединение получали в виде светло-желтого твердого вещества, которое непосредственно использовали на следующей стадии без очистки. Стадия 3: трет-бутиловый эфир 4-(4-изопропокси-5-нитро-2-винилбензоиламино)пиперидин-1 карбоновой кислоты К смеси трет-бутилового эфира 4-(2-хлор-4-изопропокси-5-нитробензоиламино)пиперидин-1 карбоновой кислоты (7,2 ммоль), полученного на предыдущей стадии, дибутилового эфира винилбороновой кислоты (1,72 г, 9,4 ммоль) и карбоната натрия (5,34 г, 50,4 ммоль) в смеси ТГФ/Н 2 О (100/25 мл) добавляли дихлорбис(трифенилфосфин)палладий(II) (442 мг, 5 ммол.%). Смесь продували N2 в течение 3 мин и нагревали при 90C в атмосфере N2 в течение ночи в круглодонной колбе, снабженной конденсатором. Смесь охлаждали до комнатной температуры и выливали в насыщенный раствор хлорида аммония. Смесь экстрагировали этилацетатом (3100 мл). Органические экстракты объединяли, промывали насыщенным раствором соли и концентрировали. Неочищенный продукт очищали колоночной хроматографией на силикагеле (40% этилацетат в гексанах), с получением трет-бутилового эфира 4-(4 изопропокси-5-нитро-2-винилбензоиламино)пиперидин-1-карбоновой кислоты в виде белого твердого вещества. Стадии 4, 5 и 6: трет-бутиловый эфир 4-(5-изопропокси-6-нитро-1-оксо-1,3-дигидроизоиндол-2 ил)пиперидин-1-карбоновой кислоты трет-Бутиловый эфир 4-(4-изопропокси-5-нитро-2-винилбензоиламино)пиперидин-1-карбоновой кислоты, полученный на предыдущей стадии (1,9 г, 4,38 ммоль), растворяли в DCM (100 мл) и охлаждали до -78C. O3 (газ) барботировали через раствор до тех пор, пока раствор не становился серо-голубым. Затем раствор продували N2 (газ) до тех пор, пока голубое окрашивание не исчезало. Раствор нагревали до комнатной температуры и обрабатывали трифенилфосфин-смолой (5 г), предварительно набухавшей вDCM (100 мл). Через 30 мин смесь фильтровали, фильтрат концентрировали и образовавшийся остаток растворяли в смеси DCM/ТФУК (100 мл/25 мл). К данной смеси добавляли триэтилсилан (4,6 мл, 17,5 ммоль). Образовавшуюся смесь перемешивали при комнатной температуре в течение ночи. Реакционную смесь концентрировали и вновь растворяли в DCM. DCM-раствор промывали 1 н. водным раствором HCl(320 мл). Объединенный водный слой обрабатывали конц. водным раствором NaOH до рН 12. Водный слой экстрагировали этилацетатом (330 мл). Объединенные органические слои промывали насыщенным раствором соли и сушили над сульфатом натрия. Твердое вещество светло-желтого цвета получали после упаривания органического растворителя. Твердое вещество растворяли в смеси метанола и триэтиламина (100 мл, 9:1 об./об.). К данной смеси добавляли ди-трет-бутилбикарбонат (680 мг, 3,1 ммоль). После перемешивания при 50 С в течение 30 мин смесь концентрировали и очищали колоночной флэш-хроматографией на силикагеле (элюент: смесь 40-50% этилацетата в гексанах) с получением трет-бутилового эфира 4-(5-изопропокси-6-нитро-1-оксо 1,3-дигидроизоиндол-2-ил)пиперидин-1-карбоновой кислоты в виде белого твердого вещества. 1H ЯМР (400 МГц, CDCl3)8,19 (с, 1 Н), 7,11 (с, 1 Н), 4,74 (кв., 1 Н), 4,45-4,38 (м, 1 Н), 4,35 (с, 2 Н),2,90-2,80 (м, 2 Н), 1,85-1,81 (м, 2 Н), 1,66-1,63 (м, 2 Н), 1,48 (с, 9 Н), 1,42 (д, 6 Н). Стадии 7, 8 и 9. К раствору трет-бутилового эфира 4-(5-изопропокси-6-нитро-1-оксо-1,3 дигидроизоиндол-2-ил)пиперидин-1-карбоновой кислоты, полученного на предыдущей стадии (850 мг, 2 ммоль), в метаноле добавляли Pd/C (10% на углероде, 100 мг). Смесь подвергали гидрированию при 1 атм газа водорода. Через 4 ч смесь фильтровали и концентрировали. Полученный анилин в виде желтого твердого вещества использовали на следующей стадии без дополнительной очистки. К смеси неочищенного продукта (2 ммоль) с предыдущей стадии (2,5-дихлорпиримидин-4-ил)-[2-(пропан-2 сульфонил)фенил]амина (промежуточное соединение 2, 770 мг, 2,2 ммоль), карбоната цезия (1,3 г, 4 ммоль) и ксантфоса (115 мг, 0,2 ммоль) в ТГФ (20 мл) добавляли ацетат палладия (22 мг, 5 ммол.%) в трубке для СВЧ. Смесь продували N2 в течение 3 мин. Плотно закрытую трубку нагревали при 150C в течение 20 мин при микроволновом облучении. Смесь охлаждали, фильтровали и концентрировали. Остаток очищали колоночной флэш-хроматоргафией на силикагеле (элюент: смесь 65% этилацетат в гексанах) с получением желтого твердого вещества. Твердое вещество обрабатывали DCM/ТФУК (1/1, 10 мл) в течение 1 ч с последующим концентрированием в вакууме. Конечная очистка препаративной ОФ ЖХМС приводила к 6-5-хлор-4-[2-(пропан-2-сульфонил)фениламино]пиримидин-2-иламино-5 изопропокси-2-пиперидин-4-ил-2,3-дигидроизоиндол-1-ону (178) в виде белого твердого вещества. 1 К раствору 5-изопропокси-6-нитро-2-пиперидин-4-илиндан-1-она (пример 1, стадия 5) в ТГФ (5 мл) и метанола (5 мл) добавляли последовательно формальдегид (104,2 мкл, 1,39 ммоль) и 10 капель АсОН. Реакционную смесь перемешивали при комнатной температуре в течение 1 ч, затем добавляли цианоборгидрид натрия (175,1 мг, 2,78 ммоль) одной порцией и реакционную смесь перемешивали в течение дополнительных 30 мин. Реакцию гасили насыщенным водным раствором NH4Cl и реакционную смесь концентрировали в вакууме с получением маслянистого остатка. Полученное масло распределяли междуEtOAc и насыщенным раствором соли, органический экстракт сушили над Na2SO4, фильтровали и концентрировали в вакууме. Хроматографией на силикагеле (смесь 5% МеОН в DCM) получали 5 изопропокси-2-(1-метилпиперидин-4-ил)-6-нитроиндан-1-он; МС m/z 333,2 (М+1). Стадии 2 и 3. Следуя методикам, описанным выше (пример 1, стадии 7 и 8), с использованием продукта со стадии 1 получали указанное в заголовке соединение 6-5-хлор-4-[2-(пропан-2 сульфонил)фениламино]пиримидин-2-иламино-5-изопропокси-2-(1-метилпиперидин-4-ил)-2,3-дигидроизоиндол-1-он (181) в виде белого твердого вещества. 1 К смеси 4-хлор-5-метил-2-нитрофенола (3,752 г, 20,0 ммоль), трет-бутилового эфира 4 гидроксипиперидин-1-карбоновой кислоты (4,83 г, 24 ммоль) и трифенилфосфина (6,23 г, 24 ммоль) в 75 мл ТГФ добавляли в несколько порций диизопропилазодикарбоксилат (4,73 мл, 245 ммоль) при 22C в течение 1 ч. Реакционную смесь перемешивали при той же температуре в течение дополнительных 2 ч. Реакционную смесь концентрировали в вакууме. Остаток брали в 50 мл эфира и оставляли при 22C в течение 14 ч. Образовавшиеся кристаллы удаляли фильтрованием. Фильтрат концентрировали в вакууме и остаток очищали на колонке с 330 г SiO2 (ISCO), используя в качестве элюента градиент от 20 до 40% смеси этилацетат в гексанах с получением трет-бутилового эфира 4-(4-хлор-5-метил-2 нитрофенокси)пиперидин-1-карбоновой кислоты в виде вязкого темно-желтого масла. МС (ES+): 315,1 Смесь трет-бутилового эфира 4-(4-хлор-5-метил-2-нитрофенокси)пиперидин-1-карбоновой кислоты с предыдущей стадии (375,8 мг, 1,01 ммоль), 1-метил-4-(4,4,5,5-тетраметил[1.3.2]диоксаборолан-2-ил)1H-пиразола (Boron Molecular, 224,4 мг 1,08 ммоль), моногидрата триосновного фосфата калия (392 мг),Pd2(dba)3 (45 мг) и дициклофосфинбифенила (43 мг) в 4 мл смеси 1,4-диоксан/H2O (3/1) нагревали в плотно закрытой трубке при 150C в течение 20 мин при микроволновом облучении. Реакционную смесь фильтровали через небольшой слой целита, сушили над Na2SO4 и концентрировали. Остаток очищали,используя колонку с SiO2 (ISCO), с получением трет-бутилового эфира 4-[5-метил-4-(1-метил-1Hпиразол-4-ил)-2-нитрофенокси]пиперидин-1-карбоновой кислоты. МС (ES+): 417,3 (МН+), 439,2 (MNa+). Стадии 3, 4 и 5. Используя методики, аналогично описанным в примере 1 синтеза (стадии 7, 8 и 9),и конечную очистку препаративной ОФ ЖХ-МСЧ, получали 5-метил-N2-[4-метил-5-(1-метил-1Hпиразол-4-ил)-2-(пиперидин-4-илокси)фенил]-N4-[2-(пропан-2-сульфонил)фенил]пиримидин-2,4-диамин Смесь 1-(4-изопропокси-2-метил-5-нитрофенил)этанона (0,788 г, 3,32 ммоль), этиленгликоля (1,8 мл) и моногидрата п-толуолсульфокислоты (6,3 мг) в 60 мл бензола нагревали с обратным холодильником с ловушкой Дина-Старка в течение 18 ч. Реакционную смесь разбавляли 100 мл этилацетата и последовательно промывали каждый раз по 100 мл водного насыщенного раствора NaHCO3, Н 2 О и насыщенного раствора соли. Органическую фазу сушили над безводным Na2SO4, фильтровали и концентрировали в вакууме с получением 2-(4-изопропокси-2-метил-5-нитрофенил)-2-метил[1.3]диоксолана в виде желтых кристаллов. МС (ES+): 282,2 (МН+). Стадии 2 и 3: 5-хлор-N2-[2-изопропокси-4-метил-5-(2-метил [1.3]диоксолан-2-ил)фенил]-N4-[2(пропан-2-сульфонил)фенил]пиримидин-2,4-диамин Используя методики, аналогично описанным в примере 1 (стадии 7 и 8) синтеза, с использованием 2-(4-изопропокси-2-метил-5-нитрофенил)-2-метил[1.3]диоксолана с предыдущей стадии в качестве исходного вещества и очистку хроматографией на силикагеле (градиент от 2 до 20% EtOAc в гексанах),получали 5-хлор-N2-[2-изопропокси-4-метил-5-(2-метил[1.3]диоксолан-2-ил)фенил]-N4-[2-(пропан-2 сульфонил)фенил]пиримидин-2,4-диамин в виде белого твердого вещества. МС (ES+): 561,2 (МН+). Стадия 4. Раствор 5-хлор-N2-[2-изопропокси-4-метил-5-(2-метил[1.3]диоксолан-2-ил)фенил]-N4-[2(пропан-2-сульфонил)фенил]пиримидин-2,4-диамина с предыдущей стадии (84 мг, 0,15 ммоль) в 5 мл 1,4-диоксана обрабатывали 1 мл водного 1 н. раствора HCl при 22C в течение 2 ч. Реакционную смесь обрабатывали и получали 1-(5-5-хлор-4-[2-(пропан-2-сульфонил)фениламино]пиримидин-2-иламино 4-изопропокси-2-метилфенил)этанон (36). МС (ES+): 517,2 (МН+). Пример 5. N2-2-изопропокси-4-метил-5-[1-(2-морфолин-4-илэтил)-1H-пиразол-4-ил]фенил-5 метил-N4-[2-(пропан-2-сульфонил)фенил]пиримидин-2,4-диамин (37) 4-Бром-3-метилфенол (1,122 г, 6,00 ммоль) и Yb(CF3SO3)3 (372 мг) в 30 мл дихлорметана обрабатывали 0,38 мл конц. раствора HNO3 при 22C. После перемешивания при той же температуре в течение 1 ч дополнительно добавляли 0,1 мл конц. раствора HNO3 и реакционную смесь перемешивали в течение дополнительного часа. Реакционную смесь промывали Н 2 О, сушили над безводным сульфатом магния,фильтровали и концентрировали. Остаток очищали на колонке с SiO2 (ISCO), с получением смеси 4 бром-5-метил-2-нитрофенола в виде желтых кристаллов и его региоизомера в качестве побочного продукта в виде оранжевых кристаллов. Стадия 2: 1-бром-4-изопропокси-2-метил-5-нитробензол(0,262 мл) и трифенилфосфина (894 мг) в 10 мл ТГФ добавляли диизопропилазодикарбоксилат (0,671 мл) при 22C. Реакционную смесь концентрировали в вакууме. Остаток очищали на колонке с SiO2 (ISCO) с получением 1-бром-4-изопропокси-2-метил-5-нитробензола в виде ярко-желтого твердого вещества. Стадия 3: 5-бром-2-изопропокси-4-метилфениламин К 1-бром-4-изопропокси-2-метил-5-нитробензолу с предыдущей стадии (0,734 г, 2,68 ммоль) и железу (порошок, 325 меш, 1,05 г) в 20 мл этанола добавляли 1 мл 1 н. водного раствора HCl при охлаждении в бане со льдом. После указанного добавления реакционную смесь нагревали с обратным холодильником в течение 2 ч. Затем дополнительные 0,5 г железа добавляли и реакционную смесь нагревали с обратным холодильником в течение дополнительных 2 ч. Реакционную смесь охлаждали и фильтровали через слой целита. Фильтрат концентрировали в вакууме с получением 5-бром-2-изопропокси-4 метилфениламина в виде оранжевого масла. Продукт использовали на следующей стадии без очистки. Стадия 4: N2-(5-бром-2-изопропокси-4-метилфенил)-5-метил-N4-[2-(пропан-2-сульфонил)фенил] пиримидин-2,4-диамин 5-Бром-2-изопропокси-4-метилфениламин с предыдущей стадии (537 мг, 2,20 ммоль) и 2-хлор-N-(2(изопропилсульфонил)фенил)-5-метилпиримидин-4-амин (промежуточное соединение 1, 652 мг, 2,00 ммоль) в присутствии метансульфокислоты (0,143 мл) в 4 мл 2-пропанола конденсировали при 140C в течение 30 мин в плотно закрытой трубке при микроволновом облучении. После обработки получалиN2-(5-бром-2-изопропокси-4-метилфенил)-5-метил-N4-[2-(пропан-2-сульфонил)фенил]пиримидин-2,4 диамин. МС (ES+): 535,1 (МН+). Стадия 5. Смесь N2-(5-бром-2-изопропокси-4-метилфенил)-5-метил-N4-[2-(пропан-2-сульфонил) фенил]пиримидин-2,4-диамина с предыдущей стадии (53 мг, 0,099 ммоль), 4-2-[4-(4,4,5,5 тетраметил[1.3.2]диоксаборолан-2-ил)пиразол-1-ил]этилморфолина (Boron Molecular, 61 мг 0,20 ммоль),K3PO4 (58 мг), Pd2(dba)3 (10 мг) и трициклогексилфосфина (8 мг) в 1 мл смеси 1,4-диоксан/Н 2 О (3/1 об./об.) нагревали в плотно закрытой трубке при 150C в течение 20 мин при микроволновом облучении. Реакционную смесь фильтровали через небольшой слой целита и концентрировали. Конечной очисткой препаративной ОФ ЖХ-МС получали N2-2-изопропокси-4-метил-5-[1-(2-морфолин-4-илэтил)-1 Нпиразол-4-ил]фенил-5-метил-N4-[2-(пропан-2-сульфонил)фенил]пиримидин-2,4-диамин (37). МС (ES+): 634,3 (МН+). Пример 6. 5-Хлор-N2-(2-изопропокси-5-метил-4-морфолин-4-илметилфенил)-N4-[2-(пропан-2 сульфонил)фенил]пиримидин-2,4-диамин (60) К смеси 1-хлор-2-метил-4-нитро-5-изопропоксибензола (промежуточное соединение 4,870 мг, 3,77 ммоль), дибутилового эфира винилбороновой кислоты (1,24 мл, 5,6 ммоль) и карбоната натрия (2,8 г,26,4 ммоль) в смеси ТГФ/Н 2 О (20/5 мл) добавляли дихлорбис(трифенилфосфин)палладий(II) (132 мг, 5 ммол.%). Реакционную трубку плотно закрывали, смесь продували N2 в течение 3 мин и затем реакционную смесь нагревали при 90C в атмосфере N2 в течение ночи. Реакционную смесь охлаждали до комнатной температуры и выливали в насыщенный водный раствор хлорида аммония. Неочищенную реакционную смесь экстрагировали этилацетатом (3100 мл). Органические экстракты объединяли, промывали насыщенным раствором соли и концентрировали. Неочищенный продукт очищали хроматографией на силикагеле (смесь 10% этилацетат в гексанах) с получением 1-метил-5-нитро-4-пропокси-2 винилбензола в виде желтого твердого вещества. 1-Метил-5-нитро-4-пропокси-2-винилбензол, полученный на предыдущей стадии (360 мг, 1,63 ммоль), растворяли в DCM (20 мл) и охлаждали до -78C. O3(газ) барботировали в раствор до тех пор, пока цвет раствора не становился серо-голубым. Затем раствор продували N2 (газ) до исчезновения голубого окрашивания. Раствор нагревали до комнатной температуры и обрабатывали трифенилфосфин-смолой (2 г), предварительно набухавшей в DCM (30 мл). Через 30 мин смесь фильтровали и фильтрат концентрировали с получением 2-метил-4-нитро-5 пропоксибензальдегида в виде желтого твердого вещества. Стадии 3 и 4: 2-изопропокси-5-метил-4-морфолин-4-илметилфениламин К раствору 2-метил-4-нитро-5-пропоксибензальдегида, полученного на предыдущей стадии (34 мг,0,152 ммоль), в смеси МеОН/ТГФ (0,5/0,5 мл) добавляли уксусную кислоту (5 капель). Смесь перемешивали при комнатной температуре в течение 1 ч. Затем добавляли цианоборгидрид натрия (20 мг, 0,30 ммоль). После перемешивания в течение 30 мин реакцию гасили добавлением насыщенного водного раствора хлорида натрия. Реакционную смесь экстрагировали этилацетатом (35 мл). Органические фазы объединяли и концентрировали с получением продукта амина в виде масла желтого цвета, которое непосредственно использовали на следующей стадии без очистки. К раствору продукта, полученного на предыдущей стадии, в метаноле (5 мл) добавляли Pd/C (10% на углероде, 2 мг). Смесь подвергали гидрированию при 1 атм водорода. Через 4 ч смесь фильтровали и концентрировали. Полученный анилиновый продукт (желтое твердое вещество) использовали на следующей стадии без дополнительной очистки. Стадия 5. К смеси анилинового продукта, полученного на предыдущей стадии (0,152 ммоль), (2,5 дихлорпиримидин-4-ил)-[2-(пропан-2-сульфонил)фенил]амина (промежуточное соединение 2, 52 мг,0,152 ммоль), карбоната цезия (99 мг, 0,30 ммоль) и ксантфоса (8 мг, 0,02 ммоль) в ТГФ (2 мл) добавляли ацетат палладия (2 мг, 5 ммол.%) в трубке для СВЧ. Смесь продували N2 в течение 3 мин и затем плотно закрытую трубку нагревали при 150C в течение 20 мин при микроволновом облучении. Реакционную смесь фильтровали, концентрировали и очищали препаративной хроматографией ОФ ЖХ-МС с массспектрометрией в "триггерном режиме" с получением указанного в заголовке соединения 5-хлор-N2-(2 изопропокси-5-метил-4-морфолин-4-илметилфенил)-N4-[2-(пропан-2-сульфонил)фенил]пиримидин-2,4 диамина (60) в виде желтого твердого вещества. 1 4-Пиридинборную кислоту (147 мг, 1,20 ммоль, 1,1 экв.) растворяли в смеси 2:1 об./об. диоксан и Н 2 О (15 мл) и N2 пропускали через раствор в течение 5 мин. Трис(дибензилиденацетон)дипалладий(0) (100 мг, 0,109 ммоль, 0,1 экв.), 2-дициклогексинфосфин 2'-6'-диметоксибифенил (112 мг, 0,272 ммоль, 0,25 экв.), 1-хлор-5-изопропокси-2-метил-4-нитробензол(промежуточное соединение 4, 250 мг, 1,09 ммоль, 1,0 экв.) и K3PO4 (462 мг, 2,18 ммоль, 2,0 экв.) добавляли в атмосфере N2. Реакционный сосуд плотно закрывали и нагревали посредством микроволнового облучения до 150C в течение 20 мин. После охлаждения до комнатной температуры реакционную смесь разбавляли этилацетатом и промывали 1 н. водным раствором NaOH (2), затем органический слой сушили над Na2SO4 и фильтровали. После концентрирования неочищенный продукт очищали хроматографией на силикагеле (градиент от гексанов до смеси 30% этилацетат в гексанах), с получением 4-(5 изопропокси-2-метил-4-нитрофенил)пиридина в виде твердого вещества коричневого цвета: ES-MC m/z 273,1 (М+Н+). Стадии 2 и 3: трет-бутиловый эфир 4-(4-амино-5-изопропокси-2-метилфенил)пиперидин-1 карбоновой кислоты 4-(5-Изопропокси-2-метил-4-нитрофенил)пиридин с предыдущей стадии (438 мг, 1,61 ммоль), растворенный в уксусной кислоте (30 мл), обрабатывали ТФУК (0,24 мл, 3,22 ммоль) и PtO2 (176 мг, 40% мас./мас.). Реакционную смесь энергично перемешивали при 1 атм Н 2 в течение 36 ч. Реакционную смесь фильтровали и фильтрат концентрировали в вакууме. Образовавшийся остаток разбавляли этилацетатом и промывали 1 н. водным раствором NaOH (2), затем органический слой сушили над Na2SO4 и фильтровали. После концентрирования неочищенный продукт (391 мг) растворяли в безводном CH2Cl2 (30 мл). ТЭА добавляли (0,44 мл, 3,15 ммоль, 2 экв.) с последующим добавлением Boc2O (344 мг, 1,57 ммоль, 1 экв.). Реакционную смесь перемешивали при комнатной температуре в течение 30 мин. Реакционную смесь концентрировали в вакууме. Образовавшийся остаток очищали хроматографией на силикагеле(градиент от гексанов до смеси 30% этилацетат в гексанах) с получением трет-бутилового эфира 4-(4 амино-5-изопропокси-2-метилфенил)пиперидин-1-карбоновой кислоты в виде липкой пены: ES-MC m/z 293,1 (M-tBu+H)+. Стадии 4 и 5. трет-Бутиловый эфир 4-(4-амино-5-изопропокси-2-метилфенил)пиперидин-1 карбоновой кислоты (170 мг, 0,488 ммоль) с предыдущей стадии, (2,5-дихлорпиримидин-4-ил)-[2(пропан-2-сульфонил)фенил]амин (промежуточное соединение 2, 169 мг, 0,488 ммоль, 1 экв.), ксантфос(28 мг, 0,049 ммоль, 0,1 экв.), ацетат палладия (5,5 мг, 0,024 ммоль, 0,05 экв.) и Cs2CO3 (477 мг, 1,46 ммоль, 3 экв.) растворяли в безводном ТГФ (6 мл). Газ N2 барботировали через реакционную смесь в течение 5 мин, затем реакционный сосуд плотно закрывали и нагревали посредством микроволнового облучения до 150C в течение 20 мин. Реакционную смесь фильтровали и фильтрат концентрировали в вакууме. После концентрирования неочищенный продукт очищали хроматографией на силикагеле (градиент от гексанов до смеси 30% этилацетат в гексанах) с получением трет-бутилового эфира 4-(4-5-хлор 4-[2-(пропан-2-сульфонил)фениламино]пиримидин-2-иламино-5-изопропокси-2-метилфенил)пиперидин-1-карбоновой кислоты в виде желтой пены: ES-MC m/z 658,3 (М+Н+). Полученный продукт (105 мг,0,160 ммоль) растворяли в CH2Cl2 (3 мл) и обрабатывали ТФУК (3 мл). Через 45 мин реакционную смесь концентрировали в вакууме. Добавляли раствор 1 н. HCl в Et2O (5 мл 2), что приводило к осаждению продукта в виде HCl-соли. Растворитель удаляли декантированием. Полученный 5-хлор-N2-(2 изопропокси-5-метил-4-пиперидин-4-илфенил)-N4-[2-(пропан-2-сульфонил)фенил]пиримидин-2,4 диамин (66) сушили в высоком вакууме с получением не совсем белого порошка. 1 4-(5-Изопропокси-2-метил-4-нитрофенил)пиридин (пример 7, стадия 1, 217 мг, 0,797 ммоль) растворяли в безводном ТГФ (9 мл). Иодметан (0,10 мл, 1,61 ммоль, 2 экв.) добавляли и реакционную смесь перемешивали при 40C в плотно закрытой трубке в течение 2 дней. Летучие компоненты удаляли в вакууме с получением 4-(5-изопропокси-2-метил-4-нитрофенил)-1-метилпиридинийиодида в виде твердого вещества коричневого цвета: ES-MC m/z 287,1 (М+). Стадии 2 и 3: 2-изопропокси-5-метил-4-(1-метилпиперидин-4-ил)фениламин 4-(5-Изопропокси-2-метил-4-нитрофенил)-1-метилпиридинийиодид с предыдущей стадии (0,697 ммоль) растворяли в СН 3 ОН (20 мл) и охлаждали до 0C. NaBH4 (264 мг, 6,97 ммоль, 10 экв.) медленно добавляли. По окончании добавления охлаждающую баню отставляли и реакционную смесь перемешивали при комнатной температуре в течение 1 ч. Реакцию гасили путем медленного добавления 1 н. водного раствора HCl (14 мл). Растворитель СН 3 ОН частично удаляли в вакууме. Образовавшийся остаток распределяли между EtOAc и 1 н. водным раствором NaOH. Дополнительно добавляли 50%-ный водный раствор NaOH до тех пор, пока рН водного слоя не составил рН 12. Слой EtOAc промывали 1 н. водным раствором NaOH (2), затем органический слой сушили над Na2SO4, фильтровали и концентрировали в вакууме. После концентрирования неочищенный продукт (175 мг) растворяли в уксусной кислоте (10 мл). Добавляли ТФУК (0,15 мл, 3 экв.) и PtO2 (53 мг, 30% мас./мас.) и реакционную смесь помещали во встряхиватель Пара при давлении газа Н 2 50 фунт/кв.дюйм в течение 14 ч. Реакционную смесь фильтровали и фильтрат концентрировали в вакууме. Образовавшийся остаток распределяли между EtOAc и 1 н. водным раствором NaOH. Дополнительно добавляли 50%-ный водный раствор NaOH до тех пор, пока рН водного слоя не составил рН 12. Слой EtOAc промывали 1 н. водным раствором NaOH (2), затем органический слой сушили над Na2SO4, фильтровали и концентрировали в вакууме с получением 2 изопропокси-5-метил-4-(1-метилпиперидин-4-ил)фениламина, который использовали на стадии 4 без дальнейшей очистки: ES-MC m/z 263,2 (М+Н+). Стадия 4. Используя методики, аналогично описанным в примере 7 (стадия 4) синтеза, и конечную очистку препаративной ОФ ЖХ-МС получали 5-хлор-N2-[2-изопропокси-5-метил-4-(1-метилпиперидин 4-ил)фенил]-N4-[2-(пропан-2-сульфонил)фенил]пиримидин-2,4-диамин (67) в виде бледно-желтого порошка: (HCl соль, ДМСО-d6+следы D2O)8,28 (с, 1 Н), 8,19 (д, 1 Н), 7,86 (д, 1 Н), 7,66 (дд, 1 Н), 7,45 (дд,1 Н), 7,37 (с, 1 Н), 6,77 (с, 1 Н), 4,56-4,49 (м, 1 Н), 3,51-3,37 (м, 3 Н), 3,16-3,08 (м, 2 Н), 2,98-2,88 (м, 1 Н), 2,77(0,04 мл, 0,262 ммоль, 3 экв.) добавляли с последующим добавлением 2-бромэтанола (0,019 мл, 0,262 ммоль, 3 экв.), растворенного в безводном ДМФА (0,7 мл). Реакционный сосуд плотно закрывали и нагревали при 70C в течение 12 ч. После охлаждения до комнатной температуры реакционную смесь раз- 26019966 бавляли этилацетатом и промывали 1 н. водным раствором NaOH (5), затем органический слой сушили над Na2SO4 и фильтровали. После концентрирования неочищенный продукт очищали препаративной ОФ ЖХ-МС, с получением 2-[4-(4-5-хлор-4-[2-(пропан-2-сульфонил)фениламино]пиримидин-2-иламино 5-изопропокси-2-метилфенил)пиперидин-1-ил]этанола (72) в виде порошка желтого цвета: ES-MC m/z 602,2 (М+Н+). Пример 10. 2-[4-(6-5-Хлор-4-[2-(пропан-2-сульфонил)фениламино]пиримидин-2-иламино-5 изопропокси-1-оксо-1,3-дигидроизоиндол-2-ил)пиперидин-1-ил]ацетамид (149) К смеси 6-5-хлор-4-[2-(пропан-2-сульфонил)фениламино]пиримидин-2-иламино-5-изопропокси 2-пиперидин-4-ил-2,3-дигидроизоиндол-1-она (пример 1, 20 мг, 0,033 ммоль) и триэтиламина (23 мкл,0,165 ммоль) в ДМФА (1,5 мл) добавляли 2-бромацетамид (10 мг, 0,066 ммоль). Смесь перемешивали при 60C в течение 4 ч. Реакционную смесь фильтровали и фильтрат очищали препаративной хроматографией ОФ ЖХ-МС с масс-спектрометрией в "триггерном режиме" (mass-trigger) с получением указанного в заголовке соединения 2-[4-(6-5-хлор-4-[2-(пропан-2-сульфонил)фениламино]пиримидин-2 иламино-5-изопропокси-1-оксо-1,3-дигидроизоиндол-2-ил)пиперидин-1-ил]ацетамида (136) в виде белого твердого вещества. 1 К смеси 6-5-хлор-4-[2-(пропан-2-сульфонил)фениламино]пиримидин-2-иламино-5-изопропокси 2-пиперидин-4-ил-2,3-дигидроизоиндол-1-она (пример 1, 20 мг, 0,033 ммоль) и триэтиламина (23 мкл,0,165 ммоль) в ДМФА (1,5 мл) добавляли диметилкарбамилхлорид (11 мг, 0,1 ммоль). Смесь перемешивали при комнатной температуре в течение 1 ч. Реакционную смесь фильтровали и фильтрат очищали препаративной хроматографией ОФ ЖХ-МС с масс-спектрометрией в "триггерном режиме" с получением указанного в заголовке соединения диметиламида 4-(6-5-хлор-4-[2-(пропан-2-сульфонил)фениламино]пиримидин-2-иламино-5-изопропокси-1-оксо-1,3-дигидроизоиндол-2-ил)пиперидин-1-карбоновой кислоты (155) в виде белого твердого вещества. 1 Смесь N2-(5-бром-2-изопропокси-4-метилфенил)-5-метил-N4-[2-(пропан-2-сульфонил)фенил]пиримидин-2,4-диамина (пример 5, стадия 4, 110 мг, 0,21 ммоль), этинилтриметилсилана (0,14 мл), N,Nдиизопропилэтиламина (0,10 мл), PdCl2(PhCN)2 (12 мг), tBu3PHBF4 (17 мг) и Cul (4 мг) в 1 мл 1,4 диоксана перемешивали при 22C в течение 20 ч с последующим нагреванием при 60C в течение дополнительных 2 ч. Реакционную смесь фильтровали через небольшой слой целита и концентрировали. Остаток очищали на колонке с 4 г SiO2 (ISCO), используя в качестве элюента градиент от 0 до 20% этилацетата в гексанах, с получением N2-(2-изопропокси-4-метил-5-триметилсиланилэтинилфенил)-5-метил-N4-[2(пропан-2-сульфонил)фенил]пиримидин-2,4-диамина в виде вязкого масла желтого цвета. Стадия 2. Раствор N2-(2-изопропокси-4-метил-5-триметилсиланилэтинилфенил)-5-метил-N4-[2(пропан-2-сульфонил)фенил]пиримидин-2,4-диамина с предыдущей стадии (0,102 г, 0,18 ммоль) в 2,5 мл ТГФ обрабатывали TBAF (0,5 мл, 1 М в ТГФ) и 30 мкл АсОН при 22C в течение 2 ч. Реакционную смесь концентрировали и остаток очищали, используя колонку с 4 г SiO2 (ISCO), с получением N2-(5-этинил-2 изопропокси-4-метилфенил)-5-метил-N4-[2-(пропан-2-сульфонил)фенил]пиримидин-2,4-диамина (257). МС (ES+): 479,2 (МН+). Пример 13. Этиловый эфир 4-(6-5-хлор-4-[2-(пропан-2-сульфонил)фениламино]пиримидин-2 иламино-5-изопропокси-1-оксо-1,3-дигидроизоиндол-2-ил)пиперидин-1-карбоновой кислоты (175) К смеси 6-5-хлор-4-[2-(пропан-2-сульфонил)фениламино]пиримидин-2-иламино-5-изопропокси 2-пиперидин-4-ил-2,3-дигидроизоиндол-1-она (пример 1, 15 мг, 0,025 ммоль) и триэтиламина (37,5 мкл,0,25 ммоль) в ДМФА (0,5 мл) добавляли этиловый эфир хлоругольной кислоты (5,4 мг, 0,05 ммоль). Смесь перемешивали при комнатной температуре в течение 1 ч. Неочищенную реакционную смесь очищали препаративной хроматографией ОФ ЖХ-МС с масс-спектрометрией в "триггерном режиме" с получением указанного в заголовке соединения этилового эфира 4-(6-5-хлор-4-[2-(пропан-2 сульфонил)фениламино]пиримидин-2-иламино-5-изопропокси-1-оксо-1,3-дигидроизоиндол-2-ил)пиперидин-1-карбоновой кислоты (175) в виде белого твердого вещества. 1 трет-Бутиловый эфир 4-(4-изопропокси-5-нитро-2-винилбензоиламино)пиперидин-1-карбоновой кислоты (пример 1, стадия 3, 1,2 г, 2,77 ммоль), растворенный в 50 мл DCM, охлаждали до -78C. Газ озон барботировали через раствор до тех пор, пока исходное вещество не израсходовалось, и затем газ азот барботировали через раствор в течение 5 мин. Реакционную смесь нагревали до комнатной температуры. Трифенилфосфин-смолу (2,77 г) в 10 мл DCM добавляли и образовавшуюся смесь перемешивали в течение 1,5 ч. Смолу удаляли фильтрованием и фильтрат концентрировали. Хроматографией на силикагеле (5% МеОН в DCM) получали трет-бутиловый эфир 4-(1-гидрокси-6-изопропокси-5-нитро-3-оксо 1,3-дигидроизоиндол-2-ил)пиперидин-1-карбоновой кислоты; МС m/z 336,2 (М-Вос+Н+). Стадии 2, 3 и 4: 5-изопропокси-2-(1-метилпиперидин-4-ил)-6-нитроиндан-1,3-дион К раствору трет-бутилового эфира 4-(1-гидрокси-6-изопропокси-5-нитро-3-оксо-1,3 дигидроизоиндол-2-ил)пиперидин-1-карбоновой кислоты с предыдущей стадии (173,9 мг, 0,4 ммоль) в ДМФА (4 мл) добавляли дихромат пиридиния (286,5 мг, 0,8 ммоль) одной порцией. После перемешивания при комнатной температуре в течение 2 ч реакционную смесь выливали в 25 мл воды и продукт экстрагировали EtOAc. Органические экстракты сушили над Na2SO4, фильтровали и концентрировали в вакууме, с получением неочищенного трет-бутилового эфира 4-(5-изопропокси-6-нитро-1,3 диоксоизоиндол-2-ил)пиперидин-1-карбоновой кислоты, который непосредственно использовали на следующей стадии без дальнейшей очистки. К раствору трет-бутилового эфира 4-(5-изопропокси-6-нитро-1,3-диоксоизоиндол-2-ил)пиперидин 1-карбоновой кислоты, полученного на предыдущей стадии, в 3 мл DCM добавляли ТФУК (3 мл). Реакционную смесь перемешивали при комнатной температуре в течение 1 ч. После концентрирования добавляли воду (5 мл) к неочищенный реакционной смеси, образовавшуюся смесь нейтрализовали до рН 8 путем добавления NaHCO3 и продукт экстрагировали DCM. Органические экстракты сушили надNa2SO4, фильтровали и концентрировали в вакууме, с получением неочищенного 5-изопропокси-6 нитро-2-пиперидин-4-илизоиндол-1,3-диона, который непосредственно использовали на следующей стадии без дальнейшей очистки. К раствору 5-изопропокси-6-нитро-2-пиперидин-4-илизоиндол-1,3-диона, полученного на предыдущей стадии, в ТГФ (5 мл) и метаноле (5 мл) добавляли последовательно формальдегид (30 мкл, 0,4 ммоль) и 2 капли АсОН. Реакционную смесь перемешивали при комнатной температуре в течение 1 ч,затем раствор цианоборгидрида натрия (50,4 мг, 0,8 ммоль) добавляли одной порцией и образовавшуюся смесь перемешивали в течение дополнительных 30 мин. Реакцию гасили путем добавления насыщенного водного раствора NH4Cl с последующим концентрированием в вакууме, с получением маслянистого остатка. Полученное масло распределяли между EtOAc и насыщенным раствором соли. Органический экстракт сушили над Na2SO4, фильтровали и концентрировали. Хроматография на силикагеле (5% МеОН в(М+1). Стадии 5 и 6. Следуя описанным выше методикам (пример 1, стадии 7 и 8), с использованием продукта со стадии 4, получали указанное в заголовке соединение 5-5-хлор-4-[2-(пропан-2 сульфонил)фениламино]пиримидин-2-иламино-6-изопропокси-2-(1-метилпиперидин-4-ил)изоиндол 1,3-дион (176) в виде желтого твердого вещества. 1
МПК / Метки
МПК: C07D 471/04, C07D 473/16, C07D 487/08, C07D 451/02, C07D 413/12, C07D 403/12, C07D 213/74, C07D 487/04, C07D 239/95, C07D 239/48, C07D 401/12, C07D 417/12, C07D 401/14, C07D 403/14, C07D 453/02
Метки: соединения, композиции, качестве, протеинкиназы, ингибиторов
Код ссылки
<a href="https://eas.patents.su/30-19966-soedineniya-i-kompozicii-v-kachestve-ingibitorov-proteinkinazy.html" rel="bookmark" title="База патентов Евразийского Союза">Соединения и композиции в качестве ингибиторов протеинкиназы</a>
Соединения и композиции в качестве ингибиторов протеинкиназы

Номер патента: 17405
Опубликовано: 28.12.2012
Авторы: Лу Вэньшо, Чэнь Бэй, Марсилдж Томас Х., Джин Юнхо, Уно Тецуо, Цзян Тао, Мишелли Пьер-Ив, Пэй Вэй
МПК: C07D 239/95, C07D 213/74, C07D 239/48...
Метки: ингибиторов, качестве, соединения, протеинкиназы, композиции
Формула / Реферат:
1. Соединение формулы (2)или его фармацевтически приемлемые соли, гдеR1 означает галоген или С1-6алкил;R2 является Н;R3 является (CR2)0-2SO2R12, (CR2)0-2SO2NRR12, (CR2)0-2C(O)O0-1R12, (CR2)0-2CONRR12, CO2NH2 или циано;R4 является С1-6алкил, С2-6алкенил или С2-6алкинил, OR12, NR(R12), галоген, нитро, SO2R12, (CR2)pR13 или X; или R4 означает Н;R6 является изопропокси или метокси;один из R8 и R9 является (CR2)qY и другой является С1-6алкилом,...
Соединения и композиции в качестве ингибиторов протеинкиназы

Номер патента: 19941
Опубликовано: 30.07.2014
Авторы: Уно Тецуо, Мишелли Пьер-Ив, Лу Вэньшо, Джин Юнхо, Пэй Вэй, Цзян Тао, Марсилдж Томас Х., Чэнь Бэй
МПК: C07D 213/74, C07D 239/95, C07D 239/48...
Метки: ингибиторов, композиции, качестве, соединения, протеинкиназы
Формула / Реферат:
1. Соединение формулы (1)или его фармацевтически приемлемые соли, где W являетсяА1 и А4 независимо означают С;каждый А2 и А3 означает С;R1 и R2 вместе образуют 5-6-членный арил, гетероарил или гетероциклил, содержащий 1-3 атома азота, каждый из которых необязательно может быть замещен C1-6алкокси;R3 является (CR2)0-2SO2R12;R4, R7 и R10 независимо означают Н;R, R5 и R5' независимо означают Н или C1-6алкил;R6 означает галоген или О(C1-6алкил);один...
Соединения и композиции в качестве ингибиторов протеинкиназы

Номер патента: 18282
Опубликовано: 28.06.2013
Авторы: Пэй Вэй, Марсилдже Томас Х., Чжу Сюфэн, Гэ Йонсюй, Хуан Чэнь, Ву Баогэнь, Мишелли Пьерр-Ив, Чэнь Бэй, Цзян Тао, Гао Чжаобо, Нгуйен Трук Нгок, Ли Юньчэн
МПК: A61K 31/505, A61K 31/506, A61K 31/519...
Метки: соединения, протеинкиназы, композиции, качестве, ингибиторов
Формула / Реферат:
1. Соединение формулы (1)или его физиологически приемлемая соль,где R1 и R2 представляют собой H;R3 представляет собой галоген;R4 представляет собой H;альтернативно, R3 и R4 вместе с атомами углерода, к которым они присоединены, могут образовывать 5-6-членное кольцо, содержащее 1-3 гетероатома азота и необязательно замещенное 1-2 группами R10, где R10 представляет собой галоген, C1-6алкил, фенил или NR2;R5 представляет собой C1-6алкил или...
Соединения и композиции в качестве ингибиторов протеинкиназы

Номер патента: 18484
Опубликовано: 30.08.2013
Авторы: Чжу Сюфэн, Мишелли Пьерр-Ив, Цзян Тао, Ву Баогэнь, Нгуйен Трук Нгок, Пэй Вэй
МПК: A61K 31/519, A61P 31/00, A61K 31/53...
Метки: качестве, соединения, композиции, ингибиторов, протеинкиназы
Формула / Реферат:
1. Соединение формул (1) или (2)или его физиологически приемлемая соль;где X представляет собой 5-6-членный гетероарил, замещенный C1-6алкилом; 6-членное гетероциклическое кольцо, содержащее NR6 или О; или (CR2)0-4CR(NRR7)(CO2R7);Y представляет собой S(O)0-2R8;Z1 представляет собой N или CH;Ph представляет собой фенил иВ представляет собой 5-6-членное кольцо, необязательно содержащее NR6, О, =O или S;R1, R2, R3 и R4 представляют собой H;R5...
Пиразолзамещённые аминогетероарильные соединения в качестве ингибиторов протеинкиназы

Номер патента: 11725
Опубликовано: 28.04.2009
Авторы: Пэйриш Мейсон Алан, Трэн -Дюбе Мишелль Бич, Шэнь Хун, Кунг Пэй -Пэй, Мэн Джерри Цзялунь, Цуй Дзингронг Жан, Цзя Лэй, Фанк Ли Эндрю, Намбу Митчелл Дэвид
МПК: C07D 401/04, A61K 31/4439, A61P 35/00...
Метки: качестве, соединения, ингибиторов, пиразолзамещённые, аминогетероарильные, протеинкиназы
Формула / Реферат:
1. Соединение формулы 1 где Y представляет собой N или CR12; А представляет собой С6-12 арил, и А необязательно замещен одной или несколькими группами R3; R1 выбран из необязательно, замещенных одной, двумя или тремя группами R13; R2 представляет собой водород; каждый R3 независимо представляет собой галоген, C1-12 алкил, C2-12 алкенил, C2-12 алкинил, С3-12 циклоалкил, С6-12 арил, 3-12-членную гетероалициклическую группу, содержащую 1 или 2...
Предыдущий патент: Комбинация ингибитора протеазы ns3 hcv с интерфероном и рибавирином
Следующий патент: Ковалентный конъюгат полиэтиленгликоля с гормоном роста человека
Случайный патент: Антифрикционный материал, способ его получения и элемент узла трения