Производные 2-гетероариламинопиримидина в качестве ингибиторов киназ
Номер патента: 17392
Опубликовано: 28.12.2012
Авторы: Кьянелли Донателла, Ли Сяолинь, Лю Сяодон, Набакка Джульет, Мольтени Валентина, Лорен Джон






Формула / Реферат
1. Соединение формулы (1)

или его фармацевтически приемлемая соль,
где один из Х1, Х2, Х3, Х4, Х5 и Х 6 обозначает N, а другие обозначают CR3, и кольцо Е присоединено к NR, R2 и R3 через атом углерода,
Ar обозначает фенил,
R1 и R2 независимо обозначают С1-С6алкил или (CR2)kOR7,
R3 обозначает -L-NR4R5, -X-NR-C(O)R8 или -X-NR-C(O)NR4R5,
где L обозначает -Х-С(О) или (CR2)1-4 и
X обозначает (CR2)j или [C(R)(CR2OR)];
R4, R5 независимо обозначают Н, С1-С6алкил, который необязательно замещен галогеном, гидроксильной группой или (CR2)k-R6,
R6 обозначает незамещенное 5-10-членное гетероарильное кольцо, содержащее 1-2 гетероатома N, или 5-10-членное гетероарильное кольцо, содержащее 1-2 гетероатома N, замещенное метилом, или незамещенное 5-7-членное гетероциклическое кольцо, содержащее 1-2 гетероатома, независимо выбранных из N, О и S, и необязательно содержащее 1 или 2 =О, или 5-7-членное гетероциклическое кольцо, содержащее 1-2 гетероатома, независимо выбранных из N, О и S, которое замещено этилом или циклопропилом, и необязательно содержащее 1 или 2 =О,
R7 и R8 независимо обозначают (CR2)k-R6 или С1-С6алкил, который необязательно замещен галогеном; или R7 обозначает Н,
в другом варианте R4 и R5 вместе с атомом N в каждом NR4R5 образуют незамещенное 4-7-членное гетероциклическое кольцо, содержащее 1-2 гетероатома N, необязательно содержащее =О, или 4-7-членное гетероциклическое кольцо, содержащее 1-2 гетероатома N, замещенное 1-3 группами R11, и необязательно содержащее =О,
R11 обозначает (CR2)kS(O)1-2R8, (CR2)k-OR7 или CO2R7,
каждый R обозначает Н или С1-С6алкил,
каждый k равен 0-6 и
j и m независимо равны 0-4.
2. Соединение по п.1, где R1 обозначает С1-С6алкоксигруппу или галогеналкокси, содержащую 1-6 атомов фтора.
3. Соединение по п.2, где R1 обозначает ОСН3, OCHF2, OCF3, OCH2CF3, OCF2CH3 или OCH2CF3.
4. Соединение по любому из пп.1-3, где R2, если присутствует, обозначает С1-С6алкил.
5. Соединение по любому из пп.1-4, где
R3 обозначает -L-NR4R5, -X-NR-C(O)R8 или -X-NR-C(O)NR4R5,
L обозначает -Х-С(О),
X обозначает (CR2)j и
j равен 0.
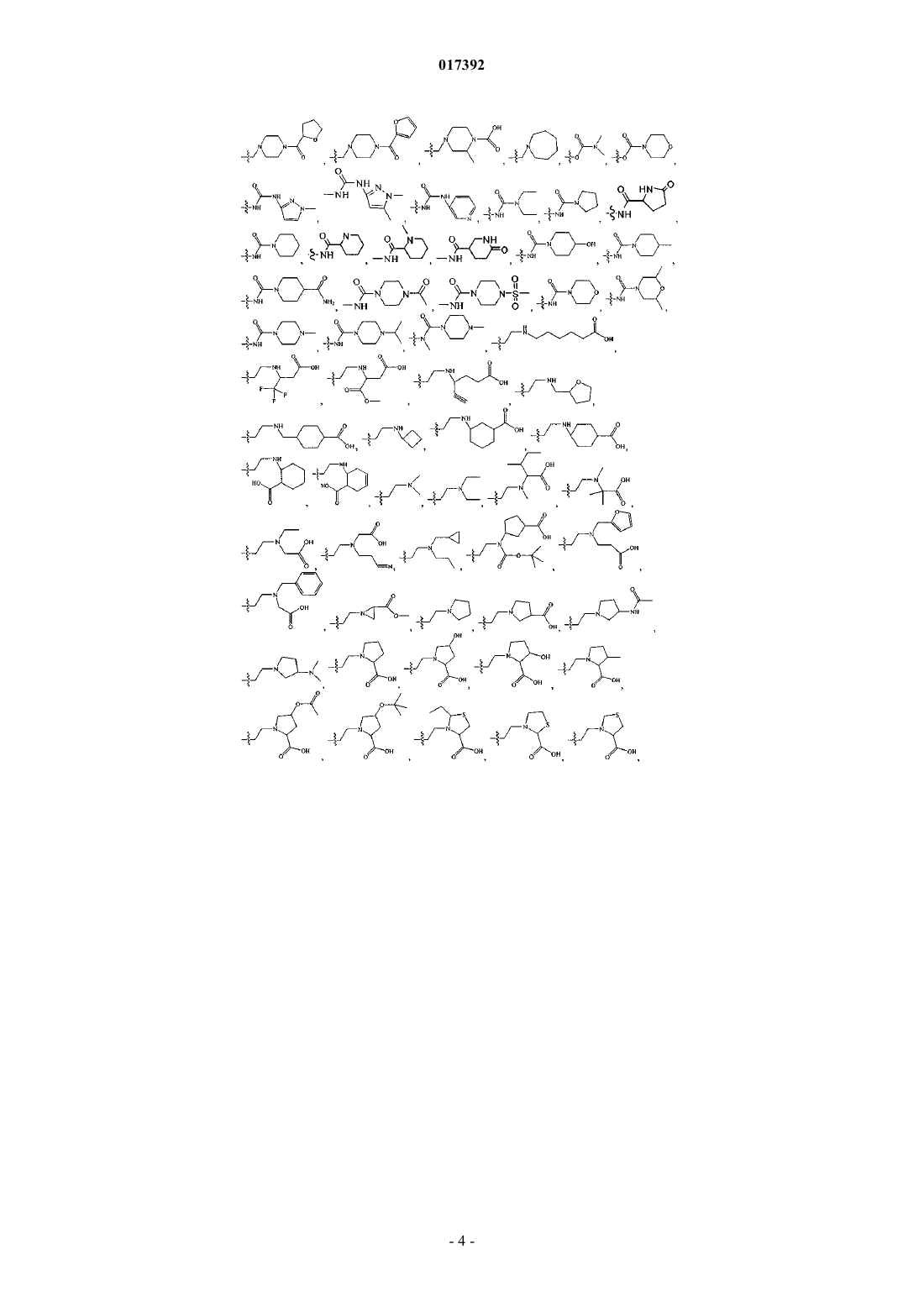
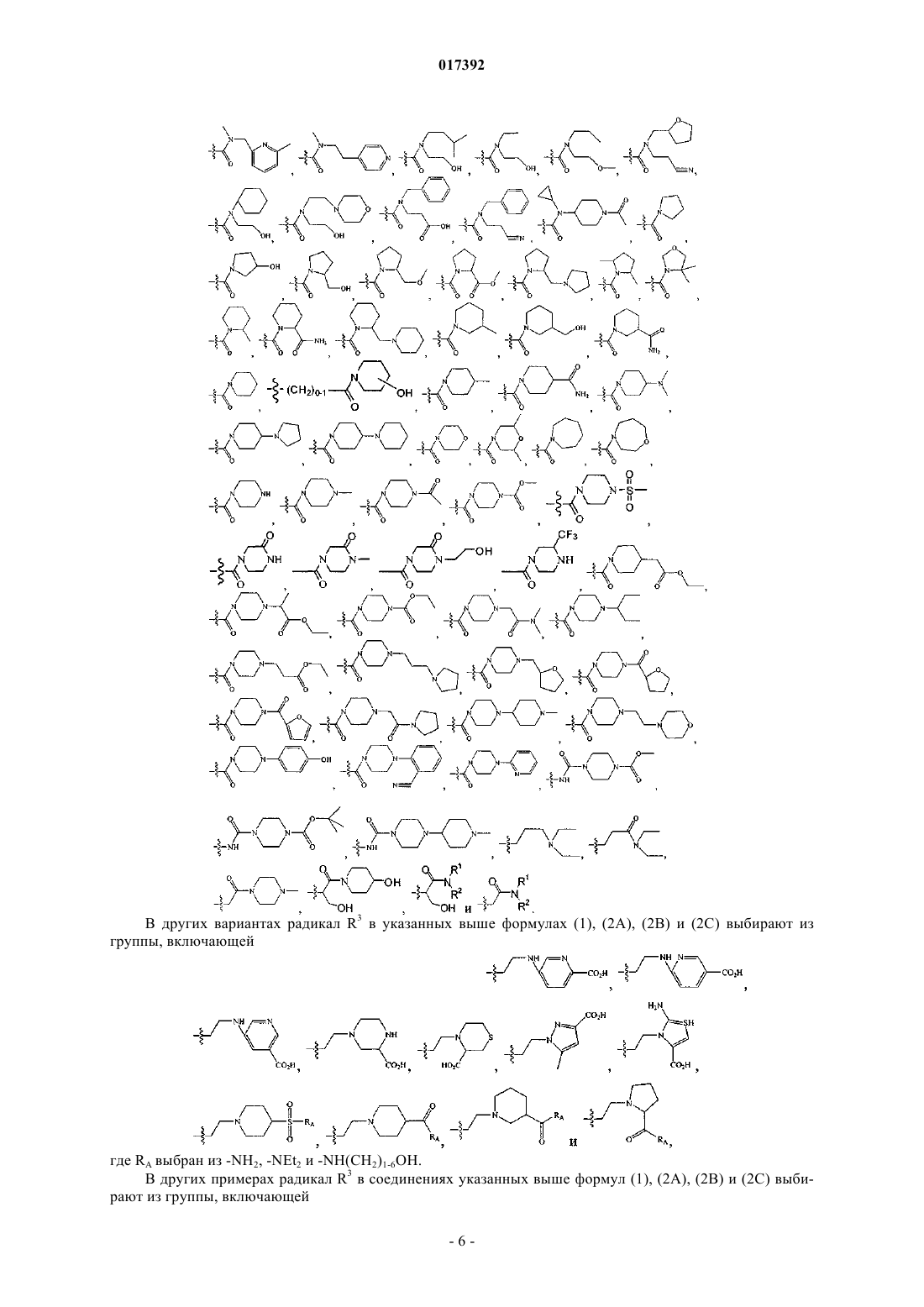
6. Соединение по любому из пп.1-5, где R3 выбирают из группы, включающей



7. Соединение по п.1, где указанное соединение выбирают из соединений формулы (2А), (2В) и (2С)

где R1 обозначает С1-С6алкоксигруппу или галогеналкокси, содержащую 1-6 атомов фтора,
R2, если присутствует, обозначает С1-С6алкил,
R3 обозначает -L-NR4R5, -X-NR-C(O)R8 или -X-NR-C(O)NR4R5,
L обозначает -Х-С(О),
X обозначает (CR2)j,
R4 и R5 независимо обозначают Н, С1-С6алкил, который необязательно замещен галогеном, гидроксильной группой или R6; или R4 и R5 вместе с атомом N образуют пиперазинил, пирролидинил или пиперидинил, каждый из которых необязательно замещен =О или 1-2 группами R11,
R11 обозначает (CR2)kS(O)1-2R8,
R8 обозначает (CR2)k-R6 или С1-С6алкил, который необязательно замещен галогеном,
R6 обозначает незамещенное 5-10-членное гетероарильное кольцо, или 5-10-членное гетероарильное кольцо, замещенное метилом, или незамещенное 5-7-членное гетероциклическое кольцо, необязательно содержащее 1 или 2 =О,
R обозначает Н или С1-С6алкил,
j равен 0,
k равен 0-4 и
m равен 0-1.
8. Соединение по п.1, где указанное соединение выбирают из группы, включающей


9. Фармацевтическая композиция, включающая терапевтически эффективное количество соединения по любому из пп.1-8 и фармацевтически приемлемый носитель.
Текст
В настоящем изобретении предлагаются соединения формулы (I) и их фармацевтические композиции, которые используют в качестве ингибиторов протеинкиназ,а также способы применения указанных соединений для лечения, снижения интенсивности симптомов или профилактики состояния связанного с аномальной или разрегулированной активностью киназы. В некоторых вариантах осуществления настоящего изобретения предлагаются способы применения указанных соединений для лечения, снижения интенсивности симптомов или профилактики заболеваний или нарушений, при которых наблюдается аномальная активность киназ c-kit, PDGFR, PDGFR, CSF1R, Abl, BCR-Abl, CSK, JNK1, JNK2, p38, p70S6K,TGF, SRC, EGFR, trkB, FGFR3, Fes, Lck, Syk, RAF, MKK4, MKK6, SAPK2, BRK, Fms, KDR, craf или b-raf. 017392 Перекрестные ссылки на родственные заявки В настоящей заявке испрашивается приоритет в связи с предварительной заявкой 60/957240, поданной 22 августа 2007 г., которая в полном объеме включена в настоящее описание в качестве ссылки. Область изобретения В настоящем изобретении предлагаются ингибиторы протеинкиназ и способы применения указанных соединений. Более конкретно в настоящем изобретении предлагаются ингибиторы киназ c-kit иPDGFR и их применение для лечения и профилактики заболеваний, опосредованных киназами c-kit иPDGFR. Предпосылки создания настоящего изобретения Протеинкиназы представляют собой большое семейство белков, играющих главную роль в регуляции множества различных клеточных процессов и обеспечении контроля функционирования клетки. Частичный список указанных киназ включает, но не ограничиваясь только ими, рецепторные тирозинкиназы, например рецепторную киназу фактора роста тромбоцитов (PDGFR), рецепторную киназу фактора стволовых клеток, киназу c-kit, рецептор фактора роста нервов, киназу trkB и рецепторную киназу роста фибробластов FGFR3, нерецепторную тирозинкиназу, такую как киназа Abl и гибридная киназа BCRAbl, Fes, Lck и Syk, а также серин/треонинкиназы, такие как киназы b-RAF, MAP (например, MKK6) иSAPK2. Аномальная активность киназы наблюдается при различных заболеваниях, включая доброкачественные и злокачественные пролиферативные заболевания, а также заболевания, которые развиваются из-за аномальной активации иммунной или нервной систем. Описание вариантов осуществления настоящего изобретения В настоящем изобретении предлагаются соединения и их фармацевтические композиции, которые можно использовать в качестве ингибиторов протеинкиназ. В одном объекте настоящего изобретения предлагаются соединения формулы (1) или их фармацевтически приемлемые соли,где 1-4 X1, X2, X3, X4, X5 и Х 6 обозначают N, а остальные радикалы обозначают CR3, кольцо Е присоединено к NR, R2 и R3 через атом углерода,Ar обозначает необязательно замещенный 5-6-членный арил или гетероарил при условии, что Ar не обозначает имидазолил,R1 и R2 независимо обозначают С 1-С 6 алкил, С 2-С 6 алкенил или С 2-С 6 алкинил, каждый из которых необязательно замещен атомом галогена, аминогруппой или гидроксильной группой, галоген, цианогруппу, нитрогруппу, (CR2)kOR7, (CR2)kO(CR2)1-4R7, (CR2)kSR7, (CR2)kNR9R10, (CR2)kC(O)O0-1R7, OC(O)R7,(CR2)kC(S)R7, (CR2)kC(O)NR9R10, (CR2)kC(O)NR(CR2)0-6C(O)O0-1R7, (CR2)kNRC(O)O0-1R7,(CR2)kS(O)1-2NR9R10, (CR2)kS(O)1-2R8, (CR2)kNRS(O)1-2R8 или (CR2)kR6, или любые две соседние группы R2 вместе с атомами, к которым они присоединены, образуют необязательно замещенное 5-8-членное карбоциклическое, гетероциклическое, арильное или гетероарильное кольцо,R3 обозначает -L-NR4R5, -X-NR-C(O)R8 или -X-NR-C(O)NR4R5, где L обозначает -Х-С(О), -Х-ОС(О),а X обозначает (CR2)j или [C(R)(CR2OR)],-SO0-2(CR2)j, (CR2)1-4, -O(CR2)1-4, илиR4, R5, R9 и R10 независимо обозначают Н, С 1-С 6 алкил, С 2-С 6 алкенил или С 2-С 6 алкинил, каждый из которых необязательно замещен галогеном, аминогрупой, гидроксильной группой, алкоксигруппой, цианогруппой, карбоксильной группой или R6, (CR2)kCN, (CR2)1-6NR7R7, (CR2)1-6OR7, (CR2)kC(O)O0-1R7,(CR2)kC(O)NR7R7 или (CR2)k-R6,R6 обозначает необязательно замещенный С 3-С 7 циклоалкил, С 6-С 10 арил или 5-10-членный гетероарил или 5-7-членное гетероциклическое кольцо,R7 и R8 независимо обозначают (CR2)k-R6 или С 1-С 6 алкил, С 2-С 6 алкенил или С 2-С 6 алкинил, каждый из которых необязательно замещен атомом галогена, аминогруппой, амидогруппой, гидроксильной группой, алкоксигруппой, цианогруппой, карбоксильной группой или группой R6, или R7 обозначает Н,в другом варианте R4 и R5 вместе с атомом N в каждой группе NR4R5, R7 и R7 вместе с атомом N в группе NR7R7 или R9 и R10 вместе с атомом N в группе NR9R10 могут образовывать 4-7-членное гетероциклическое кольцо, необязательно замещенное 1-3 группами R11 и необязательно содержащее NR12, О,S, =O или двойную связь,R11 обозначаетAr обозначает фенил, а X обозначает (CR2)0. В некоторых примерах кольцо Е в указанной выше формуле (1) обозначает пиридинил. В других примерах Ar обозначает фенил. В еще одном варианте R2, если он присутствует, обозначает галоген, С 1 С 6 алкил, С 1-С 6 алкоксигруппу, гидроксигруппу или CO2R7, a R7 обозначает Н или С 1-С 6 алкил. В других примерах R3 обозначает -L-NR4R5, -X-NR-C(O)R8 или -X-NR-C(O)NR4R5, L обозначает -Х-С(О), X обозначает (CR2)j, a j равен 0. В одном варианте осуществления настоящего изобретения предлагаются соединения формулы (2 А),(2 В) или (2 С) где R1 обозначает С 1-С 6 алкоксигруппу или галогеналкил, содержащий 1-6 атомов фтора,R2, если он присутствует, обозначает С 1-С 6 алкил,R3 обозначает -L-NR4R5, -X-NR-C(O)R8 или -X-NR-C(O)NR4R5,L обозначает -Х-С(О),X обозначает (CR2)j,R4 и R5 независимо обозначают Н, С 1-С 6 алкил, С 2-С 6 алкенил или С 2-С 6 алкинил, каждый из которых необязательно замещен атомом галогена, аминогруппой, гидроксильной группой, алкоксигруппой, цианогруппой, карбоксильной группой или группой R6, или R4 и R5 вместе с атомом N образуют пиперазинил, пирролидинил или пиперидинил, каждый из которых необязательно замещен группой =0 или 1-2 группами R11,j равен 0,k равен 0-4,m равен 0-1, аR, R6, R8 и R11 имеют значения, как определено для формулы (1). В формулах (1), (2 А), (2 В) и (2 С) R1 может обозначать С 1-С 6 алкоксигруппу или галогеналкил, содержащий 1-6 атомов фтора. Например, R1 может обозначать ОСН 3, OCHF2, OCF3, OCH2CF3, OCF2CH3 или OCH2CF3. В формулах (1), (2 А), (2 В) и (2 С) R3 выбирают из следующих групп: В других вариантах радикал R3 в указанных выше формулах (1), (2 А), (2 В) и (2 С) выбирают из группы, включающей где RA выбран из -NH2, -NEt2 и -NH(CH2)1-6OH. В других примерах радикал R3 в соединениях указанных выше формул (1), (2 А), (2 В) и (2 С) выбирают из группы, включающей В другом варианте настоящего изобретения предлагаются фармацевтические композиции, содержащие терапевтически эффективное количество соединения формул (1), (2 А), (2 В) или (2 С) и фармацевтически приемлемый эксципиент. В еще одном варианте настоящего изобретения предлагаются способы модуляции активности киназы, которые заключаются во введении в систему или субъекту, нуждающемуся в таком лечении, терапевтически эффективного количества соединения формул (1), (2 А), (2 В) или (2 С) или их фармацевтически приемлемых солей или фармацевтически приемлемых композиций и таким образом в модуляции указанной активности киназ. В одном варианте предлагаются способы модуляции киназ c-kit, PDGFR, PDGFR, CSF1R, Abl,BCR-Abl, CSK, JNK1, JNK2, p38, p70S6K, TGF, SRC, EGFR, trkB, FGFR3, Fes, Lck, Syk, RAF, MKK4,MKK6, SAPK2, BRK, Fms, KDR, c-raf или b-raf. В некоторых вариантах предлагаются способы модуляции киназ c-kit, PDGFR или PDGFR, более конкретно в настоящем изобретении предлагаются способы, согласно которым соединения формул (1), (2 А), (2 В) или (2 С), или их фармацевтически приемлемые соли, или фармацевтические композиции напрямую контактируют с киназами PDGFR или PDGFR invitro или in vivo. В настоящем изобретении также предлагаются способы лечения заболевания или состояния, при котором модуляция активности киназы может предотвращать, подавлять или снижать интенсивность патологии и/или симптоматологии заболевания или состояния, и указанные способы заключаются во введении субъекту терапевтически эффективного количества соединения формул (1), (2 А), (2 В) или (2 С),или их фармацевтически приемлемых солей, или их фармацевтических композиций, необязательно в комбинации со вторым терапевтическим агентом. Примеры терапевтических агентов, которые можно использовать в комбинации с соединением по настоящему изобретению, включают, но не ограничиваясь только ими, противофиброзный агент, пирфенидон, такролимус, противовоспалительный агент, кортикостероид, хромолин, антагонист лейкотриена, блокатор IgE, бронхолитическое средство, агонист 2, ксантины, антихолинергический или химиотерапевтический агент. При совместном введении с вторым терапевтическим агентом соединение формул (1), (2 А), (2 В) или (2 С), или их фармацевтически приемлемые соли, или фармацевтические композиции можно вводить до введения второго терапевтического агента,одновременно с ним или после его введения. В одном варианте предлагаются способы лечения заболевания или состояния, модулируемых киназами c-kit, PDGFR, PDGFR, CSF1R, Abl, BCR-Abl, CSK, JNK1, JNK2, p38, p70S6K, TGF, SRC, EGFR,trkB, FGFR3, Fes, Lck, Syk, RAF, MKK4, MKK6, SAPK2, BRK, Fms, KDR, c-raf или b-raf. В отдельных примерах предлагаются способы лечения заболевания или состояния, которые модулируются киназамиPDGFR, PDGFR или c-kit. В некоторых примерах предлагаются способы лечения заболеваний или-7 017392 состояний, которые модулируются киназой c-kit. Примеры заболевания или состояния, опосредованного киназами, которое можно лечить с использованием соединений и композиций по настоящему изобретению, включают, но не ограничиваясь только ими, заболевание, связанное с тучными клетками, аллергическое заболевание, синдром раздраженной кишки, фиброзные заболевания, неопластические нарушения, воспалительное нарушение, аутоиммунное нарушение, заболевание трансплантат против хозяина, метаболический синдром, нарушение, связанное с центральной нервной системой, нейродегенеративное нарушение, боль, алкогольную или наркотическую зависимость, рак, сердечно-сосудистое заболевание и заболевание, связанное с прионной инфекцией. Примеры заболеваний, связанных с тучными клетками, которые можно лечить с использованием соединений и композиций по настоящему изобретению, включают, но не ограничиваясь только ими, аллергические нарушения (включая астму и атопический дерматит), крапивницу, угри и угри, вызванные пропионибактериями, прогрессирующую оссифицирующую фибродисплазию, воспаление и разрушение тканей, вызванное воздействием химического и биологического оружия (например, сибирская язва и сернистый иприт), муковисцидоз, заболевания почек, воспалительные заболевания мышц, ВИЧ, диабет типа II, церебральную ишемию, мастоцитоз, лекарственную зависимость и абстинентный синдром, заболевания центральной нервной системы, предотвращение или снижение выпадения волос, бактериальные инфекции, интерстициальный цистит, воспалительные заболевания кишечника (например, болезнь Крона, неспецифический язвенный колит, неопределенный колит и инфекционный колит), ангиогенез опухоли, аутоиммунные заболевания, воспалительные заболевания, рассеянный склероз и потерю костной массы. Примеры аллергических заболеваний, которые можно лечить с использованием соединений и композиций по настоящему изобретению, включают, но не ограничиваясь только ими, астму, атопический дерматит, аллергический ринит, аллергичекий синусит, анафилактический синдром, крапивницу, отек Квинке, аллергический контактный дерматит, узловатую эритему, полиформную эритерму, кожный некрозирующий венулит, воспаление кожи вследствие укуса насекомых и инвазию кровососущих паразитов. Синдром раздраженной кишки представляет собой функциональное нарушение желудочнокишечного тракта, характеризующееся болями в брюшной полости и изменением функционирования кишечника. Боли обычно ослабевают при дефекации и могут быть связаны с большей или меньшей частотой формирования стула, изменением консистенции стула, запором или недержанием стула, чувством неполного опорожнения, выделением слизи или вздутием живота. Фиброзное заболевание в настоящем контексте включает все состояния, связанные с образованием и отложением компонентов внеклеточного матрикса, прежде всего во внутренних органах, включая почки, сердце, легкие, печень, кожу и суставы. Примеры фиброзных заболеваний, которые можно лечить с использованием соединений и композиций по настоящему изобретению, включают, но не ограничиваясь только ими, склеродермию, легочный фиброз, идиопатический легочный фиброз, первичную легочную гипертензию (например, артериальную легочную гипертензию), фиброз печени, фиброз почек, фиброз сердца, цирроз печени, фиброз костного мозга, гепатит С и стеатогепатит неалкогольного происхождения. Примеры неопластических нарушений, которые можно лечить с использованием соединений и композиций по настоящему изобретению, включают, но не ограничиваясь только ими, мастоцитоз, стромальную опухоль желудочно-кишечного тракта, мелкоклеточный рак легких, немелкоклеточный рак легких, острый миелоидный лейкоз, острый лимфоцитарный лейкоз, миелодипластический синдром,хронический миелогенный лейкоз, колоректальную карциному, желудочную карциному, рак яичек, глиобластому и астроцитому. Примеры воспалительных заболеваний, которые можно лечить с использованием соединений и композиций по настоящему изобретению, включают, но не ограничиваясь только ими, ревматоидный артрит, конъюктивит, ревматоидный спондилит, остеоартрит и подагрический артрит. Примеры аутоиммунных заболеваний, которые можно лечить с использованием соединений и композиций по настоящему изобретению, включают, но не ограничиваясь только ими, рассеянный склероз,псориаз, воспалительное заболевание кишечника, синдром воспаленного кишечника, язвенный колит,болезнь Крона, ревматоидный артрит, полиартрит, очаговую или системную склеродермию, системную красную волчанку, дискоидную красную волчанку, кожную волчанку, дерматомиозит, полимиозит, синдром Шегрена, узелковый панартерит, аутоиммунную энтеропатию и пролиферативный гломерулонефрит. Примеры заболеваний трансплантат против хозяина, которые можно лечить с использованием соединений и композиций по настоящему изобретению, включают, но не ограничиваясь только ими, отторжение трансплантатов органов, таких как почки, поджелудочная железа, печень, сердце, легкие и костной мозг. Примеры метаболических синдромов, которые можно лечить с использованием соединений и композиций по настоящему изобретению, включают, но не ограничиваясь только ими, диабет типа I и II и ожирение.-8 017392 Примеры заболеваний, связанных с центральной нервной системой, которые можно лечить с использованием соединений и композиций по настоящему изобретению, включают, но не ограничиваясь только ими, депрессию, дистимическое расстройство, циклотимическое расстройство, анорексию, булимию, предменструальный синдром, синдром постменопаузы, задержку умственного развития, потерю внимания, пессимизм, возбуждение, самоосуждение и сниженное половое влечение, тревожное состояние, психиатрические заболевания и шизофрению. Примеры депрессивных состояний, которые можно лечить с использованием соединений и композиций по настоящему изобретению, включают, но не ограничиваясь только ими, биполярную депрессию,тяжелую или меланхолическую депрессию, атипичную депрессию, резистентную депрессию и сезонную депрессию. Примеры тревожных состояний, которые можно лечить с использованием соединений и композиций по настоящему изобретению, включают, но не ограничиваясь только ими, тревогу, связанную с гипервентиляцией легких или сердечной аритмией, фобические расстройства, обессивно-компульсивное расстройство, расстройства, связанные с посттравматическим стрессом, острым стрессом и генерализованное тревожное расстройство. Примеры психиатрических заболеваний, которые можно лечить с использованием соединений и композиций по настоящему изобретению, включают, но не ограничиваясь только ими, приступы паники, включая психоз, бредовое расстройство, конверсионные расстройства,фобии, мании, делирий, диссоциативные эпизоды, включая диссоциативную амнезию, диссоциативную реакцию бегства и диссоциативное суицидальное поведение, самоотрицание, жестокое или агрессивное поведение, травму, пограничное расстройство личности и острый психоз, такой как шизофрения, включая параноидальную шизофрению, дезорганизованную шизофрению, кататоническую шизофрению и недифференцированную шизофрению. Примеры нейродегенеративных заболеваний, которые можно лечить с использованием соединений и композиций по настоящему изобретению, включают, но не ограничиваясь только ими, остеоартрит,болезнь Альцгеймера, болезнь Паркинсона, болезнь Гентингтона, прионные заболевания, заболевание двигательных нейронов и боковой амиотрофический склероз. Примеры болевых состояний, которые можно лечить с использованием соединений и композиций по настоящему изобретению, включают, но не ограничиваясь только ими, острую боль, послеоперационную боль, хроническую боль, ноцицептивную боль, боль при раке, невропатическую боль и психогенный болевой синдром. Примеры расстройств, вызванных употреблением психоактивных веществ, которые можно лечить с использованием соединений и композиций по настоящему изобретению, включают, но не ограничиваясь только ими, наркотическую зависимость, привыкание к чрезмерному употреблению лекарственных средств, лекарственную зависимость, абстинентный синдром и передозировку лекарственных средств. Примеры раковых заболеваний, которые можно лечить с использованием соединений и композиций по настоящему изобретению, включают, но не ограничиваясь только ими, глиому, меланому, желудочнокишечную стромальную опухоль, мелкоклеточный рак легкого, колоректальный рак и другие солидные опухоли. Примеры сердечно-сосудистых заболеваний, которые можно лечить с использованием соединений и композиций по настоящему изобретению, включают, но не ограничиваясь только ими, стенокардию,инфаркт миокарда, застойную сердечную недостаточность, кардиомиопатию, гипертензию, артериальный и венозный стеноз. Более конкретно соединения по настоящему изобретению можно использовать для лечения и профилактики астмы, атопического нейродермита, крапивницы, синдрома раздраженной кишки или фиброзных заболеваний, включая, но не ограничиваясь только ими, склеродермию, легочный фиброз, идиопатический легочный фиброз, первичную легочную гипертензию, первичную легочную артериальную гипертензию,идиопатическую артериальную гипертензию, фиброз печени, фиброз почек и фиброз сердца. Кроме того, в настоящем изобретении предлагается применение соединений формул (1), (2 А), (2 В) или (2 С) или их фармацевтических композиций, необязательно в комбинации с вторым терапевтическим агентом, для получения лекарственных средств, предназначенных для лечения заболеваний и состояний,модулируемых киназами c-kit, PDGFR, PDGFR, CSF1R, Abl, BCR-Abl, CSK, JNK1, JNK2, p38, p70S6K,TGF, SRC, EGFR, trkB, FGFR3, Fes, Lck, Syk, RAF, MKK4, MKK6, SAPK2, BRK, Fms, KDR, c-raf илиb-raf, и более конкретно для получения лекарственных средств, предназначенных для лечения заболеваний и состояний, модулируемых киназами PDGFR, PDGFR или c-kit. Согласно вышеупомянутым способам применения соединений по настоящему изобретению соединения формул (1), (2 А), (2 В) или (2 С) можно вводить в систему, содержащую клетки или ткани. В другом варианте соединения формул (1), (2 А), (2 В) или (2 С) можно вводить человеку или животному. Определения Термин алкил, использованный в данном контексте, обозначает остаток или структурный элемент каких-либо других групп, например галогензамещенный алкил и алкоксигруппу с прямой или разветвленной цепью. Необязательно замещенный алкил, алкенил или алкинил, использованные в данном контексте, необязательно содержат атом галогена (например, CF3) или один или более атомов углерода, за-9 017392 мещенных или замененных на гетероатом, такой как NR, О или S (например, -ОСН 2 СН 2 О-, алкилтиол,тиоалкоксигруппа, алкиламины и т.п.). Термин арил, использованный в данном контексте, обозначает моноциклическое или конденсированное бициклическое ароматическое кольцо, содержащее атомы углерода. Например, арил может обозначать фенил или нафтил. Термин арилен, использованный в данном контексте, обозначает двухвалентный радикал, образованный из арильной группы. Термин гетероарил, использованный в данном контексте, обозначает арил, как описано выше, при этом один или более атомов в цикле обозначают гетероатом. Примеры гетероарильных групп включают(но не ограничиваются только ими) пиридил, индолил, индазолил, хиноксалинил, хинолинил, бензофуранил, бензопиранил, бензотиопиранил, бензо[1,3]диоксол, имидазолил, бензимидазолил, пиримидинил,фуранил, оксазолил, изоксазолил, триазолил, тетразолил, пиразолил, тиенил и т.п. Термин карбоциклическое кольцо, использованный в данном контексте, обозначает насыщенное или частично ненасыщенное моноциклическое, конденсированное бициклическое или мостиковое полициклическое кольцо, содержащее атомы углерода, необязательно замещенные, например, группой =О. Примеры карбоциклических колец включают, но не ограничиваясь только ими, циклопропил, циклобутил, циклопентил, циклогексил, циклопропилен, циклогексанон и т.п. Термин гетероциклическое кольцо, использованный в данном контексте, обозначает карбоциклическое кольцо, как описано выше, где один или более атомов углерода обозначают гетероатом. Например, гетероциклическое кольцо может содержать группы N, О, S, -N=, -S-, -S(O), -S(O)2- или -NR-, где R обозначает водород, C1-С 4 алкил или защитную группу. Примеры гетероциклических колец включают, но не ограничиваясь только ими, морфолино, пирролидинил, пирролидинил-2-он, пиперазинил, пиперидинил, пиперидинилон, 1,4-диокса-8-азаспиро[4,5]дец-8-ил и т.п. Использованный в данном контексте термин атом Н обозначает в составе любой группы заместителя (например, в группе CH2) все пригодные изотопы, например Н, 2 Н и 3 Н. Если не указано иное, то термин необязательно замещенный заместитель обозначает, что заместитель обозначает группу, которая необязательно замещена одной или более группами, независимо выбранными, например, из следующих групп: необязательно замещенный атомом галогена алкил, алкенил,алкинил, алкоксигруппа, алкиламин, алкилтиогруппа, алкинил, амид, аминогруппа, включая моно- и дизамещенные аминогруппы, арил, арилоксигруппа, арилтиогруппа, карбонил, карбоциклил, цианогруппа,циклоалкил, галоген, гетероалкил, гетероалкенил, гетероалкинил, гетероарил, гетероциклил, гидроксигруппа, изоцианатогруппа, изотиоцианатогруппа, меркаптогруппа, нитрогруппа, О-карбамоил, Nкарбамил, О-тиокарбамил, N-тиокарбамил, С-амидогруппа, N-амидогруппа, S-сульфонамидогруппа, Nсульфонамидогруппа, С-карбоксигруппа, О-карбоксигруппа, пергалогеналкил, перфторалкил, силил,сульфонил, тиокарбонил, тиоцианатогруппа, тригалогенметансульфонил и их защищенные соединения. Защитные группы, которые можно использовать для защиты указанных выше заместителей, известны и приведены в справочных изданиях, таких как Greene и Wuts, Protective Groups in Organic Synthesis, 3-е изд., John WileySons, New York, NY (1999) и Kocienski, Protective Groups, Thieme Verlag, New York,NY (1994), включенных в данных контекст в полном объеме качестве ссылок. Термин совместное введение, использованный в данном контексте, обозначает введение выбранных терапевтических агентов одному пациенту и обозначает курс лечения, при котором агенты необязательно вводят одним и тем же способом или в одно и то же время. Термин фармацевтическая комбинация, использованный в данном контексте, обозначает продукт,полученный при смешивании активных ингредиентов, и включающий фиксированные и нефиксированные комбинации активных ингредиентов. Термин фиксированная комбинация обозначает, что активные ингредиенты, т.е. соединение формулы (1) и совместный агент, вводят пациенту одновременно в форме единой дозы или лекарственной формы. Термин нефиксированная комбинация обозначает, что активные ингредиенты, например соединение формулы (1) и совместный агент, вводят пациенту в виде отдельных форм одновременно, или совместно, или последовательно, без определенных временных ограничений, причем указанное введение обеспечивает терапевтически эффективные уровни активных ингредиентов в организме пациента. Последнее также относится к комбинированной терапии, например введению трех или более активных ингредиентов. Термин терапевтически эффективное количество обозначает количество соединения, которое вызывает биологическую или лечебную ответную реакцию в клетке, ткани, органе, системе, в организме животного или человека, по мнению исследователя, ветеринара, врача или другого специалиста. Термин введение соединения обозначает доставку соединения по настоящему изобретению и его пролекарства субъекту, нуждающемуся в лечении. Если в данном изобретении не указано иное, киназу выбирают из следующих киназ: c-kit, PDGFR,PDGFR, CSF1R, Abl, BCR-Abl, CSK, JNK1, JNK2, p38, p70S6K, TGF, SRC, EGFR, trkB, FGFR3, Fes,Lck, Syk, RAF, MKK4, MKK6, SAPK2, BRK, Fms, KDR, c-raf и b-raf, включая киназы дикого типа и мутантные формы (т.е. отличающиеся одной или несколькими аминокислотами по сравнению с последовательностью дикого типа).- 10017392 Способы осуществления настоящего изобретения В настоящем изобретении предлагаются соединения и их фармацевтические композиции, которые можно применять в качестве ингибиторов протеинкиназ. В одном объекте настоящего изобретения предлагаются соединения формулы (1) или его фармацевтически приемлемая соль,где 1-4 X1, X2, X3, X4, X5 и Х 6 обозначают N, а остальные группы обозначают CR3, кольцо Е присоединено к группам NR, R2 и R3 через атом углерода,Ar обозначает необязательно замещенный 5-6-членный арил или гетероарил при условии, что Ar не обозначает имидазолил,R1 и R2 независимо обозначают С 1-С 6 алкил, С 2-С 6 алкенил или С 2-С 6 алкинил, каждый из которых необязательно замещен галогеном, аминогруппой или гидроксильной группой, галоген, цианогруппу,нитрогруппу, (CR2)kOR7, (CR2)kO(CR2)1-4R7, (CR2)kSR7, (CR2)kNR9R10, (CR2)kC(O)O0-1R7, OC(O)R7,(CR2)kC(S)R7, (CR2)kC(O)NR9R10, (CR2)kC(O)NR(CR2)0-6C(O)O0-1R7, (CR2)kNRC(O)O0-1R7,(CR2)kS(O)1-2NR9R10, (CR2)kS(O)1-2R8, (CR2)kNRS(O)1-2R8 или (CR2)kR6, или любые две соседние группы R2 вместе с атомами, к которым они присоединены, могут образовывать необязательно замещенное 5-8 членное карбоциклическое, гетероциклическое, арильное или гетероарильное кольцо,R3 обозначает -L-NR4R5, -X-NR-C(O)R8 или -X-NR-C(O)NR4R5, где L обозначает -Х-С(О), -Х-ОС(О),X обозначает (CR2)j или [C(R)(CR2OR)],-SO0-2(CR2)j, (CR2)1-4, -O(CR2)1-4 илиR4, R5, R9 и R10 независимо обозначают Н, С 1-С 6 алкил, С 2-С 6 алкенил или С 2-С 6 алкинил, каждый из которых необязательно замещен галогеном, аминогруппой, гидроксильной группой, алкоксигруппой,цианогруппой, карбоксильной группой или группами R6; (CR2)kCN, (CR2)1-6NR7R7, (CR2)1-6OR7,(CR2)kC(O)O0-1R7, (CR2)kC(O)NR7R7 или (CR2)k-R6,R6 обозначает необязательно замещенный С 3-С 7 циклоалкил, С 6-С 10 арил или 5-10-членный гетероарил или 5-7-членное гетероциклическое кольцо,R7 и R8 независимо означают (CR2)k-R6 или С 1-С 6 алкил, С 2-С 6 алкенил, С 2-С 6 алкинил, каждый из которых необязательно замещен атомом галогена, аминогруппой, амидогруппой, гидроксигруппой, алкоксигруппой, цианогруппой, карбоксильной группой или R6, или R7 обозначает Н,в другом варианте R4 и R5 вместе с атомом N в каждой группе NR4R5, R7 и R7 вместе с атомом N в группе NR7R7 или R9, R10 вместе с атомом N в группе NR9R10 необязательно образуют 4-7-членное гетероциклическое кольцо, необязательно замещенное 1-3 группами R11, необязательно содержащими группы NR12, О, S, -О или двойную связь,R11 обозначает(CR2)kNRS(O)1-2R8,R12 обозначает H, R8, -(CR2)1-4CO2R7, (CR2)k-C(O)-(CR2)k-R8, (CR2)kC(O)NR7R7, (CR2)kC(O)NR(CR2)07 7 7 7 8 8 6C(O)O0-1R , (CR2)1-4NRC(O)O0-1R , (CR2)kS(O)1-2NR R , (CR2)kS(O)1-2R или (CR2)kNRS(O)1-2R ,каждый радикал R обозначает Н или С 1-С 6 алкил,каждый k равен 0-6,j и m независимо равны 0-4,при условии, что R8 в группе -X-NR-C(O)R8 не обозначает фенил, если Ar обозначает фенил, а X обозначает (CR2)0. В одном варианте настоящего изобретения предлагаются соединения формул (2 А), (2 В) или (2 С)R2, если присутствует, обозначает С 1-С 6 алкил,R3 обозначает -L-NR4R5, -X-NR-C(O)R8 или -X-NR-C(O)NR4R5,L обозначает -Х-С(О),X обозначает (CR2)j,R4 и R5 независимо обозначают Н, С 1-С 6 алкил, С 2-С 6 алкенил или С 2-С 6 алкинил, каждый из которых необязательно замещен атомом галогена, аминогруппой, гидроксигруппой, алкоксигруппой, цианогруппой, карбоксильной группой или R6, или R4 и R5 вместе с атомом N образуют пиперазинил, пирролидинил или пиперидинил, каждый из которых необязательно замещен группой =О или 1-2 группами R11,j равен 0,k равен 0-4,m равен 0-1, аR, R6, R8 и R11 определены для формулы (1). В каждой из вышеописанных формул любой асимметричный атом углерода может находиться в(R)-, (S)- или (R,S)-конфигурации. Соединения могут присутствовать в виде смеси изомеров или индивидуальных изомеров, например в виде индивидуальных энантиомеров или диастереомеров. Настоящее изобретение также включает возможные таутомеры соединений по настоящему изобретению. Настоящее изобретение также включает все пригодные изотопные формы соединений по настоящему изобретению или их фармацевтически приемлемых солей. Изотопные формы соединений по настоящему изобретению или их фармацевтически приемлемых солей обозначают соединения, в которых по крайней мере один атом заменен на атом с одинаковым атомным номером, но атомной массой, отличающейся от атомной массы существующего в природе атома. Примеры изотопов, которые могут содержаться в соединениях по настоящему изобретению и их фармацевтически приемлемых солях, включают,но не ограничиваясь только ими, изотопы водорода, углерода, азота и кислорода, такие как 2 Н, 3 Н, 11 С,13 С, 14 С, 15N, 17 О, 18 О, 35S, 18F, 36Cl и 123I. Некоторые изотопные формы соединений по настоящему изобретению и их фармацевтически приемлемых солей, например, включающие радиоактивные изотопы 3 Н или 14 С, используют в исследованиях по распределению лекарственных агентов и/или субстратов в тканях. В конкретных примерах изотопы 2 Н, 3 Н и 14 С используют в связи с простым методом их получения и детектирования. В других примерах использование в качестве заместителей таких изотопов, как 2 Н,обеспечивает определенные терапевтические преимущества в связи с лучшей метаболической стабильностью, например за счет увеличения периода полураспада in vivo или снижения требуемой дозы. Изотопные формы соединения по настоящему изобретению или их фармацевтически приемлемых солей обычно получают и использованием стандартных методик и соответствующих реагентов. Изотопные формы соединения могут изменять метаболизм соединений и/или незначительно изменять физические свойства, такие как гидрофобность и т.п. Изотопные формы соединения могут увеличивать эффективность и безопасность, биодоступность и период полураспада, изменять степень связывания с белками,биораспределение, увеличивать содержание активных метаболитов и/или снижать образование реакционноспособных или токсичных метаболитов. В каждой из описанных выше формул каждый необязательно замещенный остаток может содержать заместители, выбранные из группы, включающей С 1-С 6 алкил, С 2-С 6 алкенил или С 3-С 6 алкинил, каждый из которых необязательно замещен галогеном или необязательно содержит атом углерода, который заменен на атомы N, S, О, или их комбинацию (например, гидрокси(С 1-С 8)алкил, С 1-С 8 алкокси(С 1 С 8)алкил), галоген, аминогруппу, амидиногруппу, С 1-С 6 алкоксигруппу, гидроксил, метилендиоксигруппу, карбоксигруппу, С 1-С 3 алкилкарбонил, С 1-С 8 алкоксикарбонил, карбамоил, С 1-С 8 алкилкарбамоил,сульфамоил, цианогруппу, оксогруппу, нитрогруппу или необязательно замещенное карбоциклическое кольцо, гетероциклическое кольцо, арил или гетероарил, как описано выше. Соединения формул (1), (2 А), (2 В) или (2 С) в свободной форме или в форме фармацевтически приемлемой соли характеризуются различными фармакологическими свойствами, например, как установлено по данным испытаний in vitro, как описано в данном контексте. Значение IC50 в описанных экспериментах обозначает концентрацию исследуемого соединения, при которой число клеток снижается на 50% по сравнению с контролем в отсутствие ингибитора. В общем случае соединения по настоящему изобретению характеризуются значениями IC50 от 1 нМ до 10 мкМ. В некоторых примерах соединения по настоящему изобретению характеризуются значениями IC50 от 0,01 до 5 мкМ. В других примерах соединения по настоящему изобретению характеризуются значением IC50 от 0,01 до 1 мкМ, более предпочтительно от 1 нМ до 1 мкМ. В других примерах соединения по настоящему изобретению характеризуются значениями IC50 менее 1 нМ или более 10 мкМ. Степень ингибирования соединений по настоящему изобретению в % составляет более 50% или в других вариантах более приблизительно 70% в отношении одной или более следующих киназ при концентрации 10 мкМ: c-kit, PDGFR, PDGFR, CSF1R, Abl,BCR-Abl, CSK, JNK1, JNK2, p38, p70S6K, TGF, SRC, EGFR, trkB, FGFR3, Fes, Lck, Syk, RAF, MKK4,MKK6, SAPK2, BRK, Fms, KDR, c-raf или b-raf. Соединения по настоящему изобретению также можно применять для лечения опосредованного киназами состояния или заболевания, такого как заболевание, опосредованное киназами c-kit, PDGFR,- 12017392PDGFR, CSF1R, Abl, BCR-Abl, CSK, JNK1, JNK2, p38, p70S6K, TGF, SRC, EGFR, trkB, FGFR3, Fes,Lck, Syk, RAF, MKK4, MKK6, SAPK2, BRK, Fms, KDR, c-raf или b-raf. Более конкретно соединения по настоящему изобретению можно использовать для лечения и профилактики астмы, атопического дерматита, крапивницы, синдрома раздраженной кишки или фиброзных заболеваний, включая, но не ограничиваясь только ими, склеродермию, легочный фиброз, идиопатический легочный фиброз, первичную легочную гипертензию, первичную легочную артериальную гипертензию, идиопатическую артериальную гипертензию, фиброз печени, почек и сердца. Фармакология и применение Активность соединений по настоящему изобретению оценивали с использованием панели киназ(дикого типа и/или мутантных форм), и указанные соединения модулируют активность по крайней мере одной киназы в панели. Таким образом, соединения по настоящему изобретению можно использовать для лечения заболеваний или нарушений, в развитии которых принимают участие киназы (в развитии патологии или симптомологии указанных заболеваний). Примеры киназ, активность которых ингибируется соединениями и композициями, описанными в данном контексте, и в отношении которых можно использовать описанные в данном контексте способы, включают, не ограничиваясь только ими, киназыc-Kit. Тучные клетки представляют собой элементы ткани, образующиеся из определенной субпопуляции кроветворных стволовых клеток, экспрессирующих антигены CD34, c-kit и CD13. Тучные клетки характеризуются гетерогенностью не только в отношении расположения и структуры ткани, но и на функциональном и гистохимическом уровнях. Незрелые тучные клетки-предшественники циркулируют в кровотоке и распределяются по различным тканям. Такие дифференциация и пролиферация контролируются цитокинами, одним из важнейших является фактор стволовых клеток, также известный как лиганд Kit,фактор Стила или фактор роста тучных клеток. Рецептор фактора стволовых клеток кодируется протоонкогеном, c-kit, который экспрессируется в кроветворных клетках-предшественниках, тучных клетках,половых клетках, интерстициальных клетках Кахаля и некоторых опухолях человека, а также экспрессируется некроветворными клетками. Тирозинкиназы представляют собой белки рецепторного или нерецепторного типа, которые превращают концевой фосфат из АТФ в остатки тирозина в белке, таким образом активируя или инактивируя пути передачи сигнала. Рецептор фактора стволовых клеток, c-kit, представляет собой трансмембранную рецепторную протеинтирозинкиназу типа III, которая инициирует рост клеток и каскады пролиферационной передачи сигнала в ответ на связывание с фактором стволовых клеток. Лигирование рецептора c-kit в присутствии фактора стволовых клеток приводит к его димеризации с последующим трансфосфорилированием, что, в свою очередь, приводит к накоплению и активации различных внутрицитоплазматических субстратов. Указанные активированные субстраты приводят к активации множества внутриклеточных сигнальных путей, ответственных за пролиферацию и активацию клеток. Известно, что такие белки принимают участие в различных клеточных механизмах, которые в случае нарушения приводят к заболеваниям, таким как аномальная клеточная пролиферация и миграция, а также воспаление. Указанные соединения по настоящему изобретению подавляют клеточные процессы, включающие фактор стволовых клеток, например ингибируют автофосфорилирование рецептора фактора стволовых клеток и стимулируемую фактором стволовых клеток активацию киназы MAPK (митоген-активированной протеинкиназы). Активность рецепторной протеинтирозинкиназы c-kit регулируется в нормальных клетках, нормальная функциональная активность генного продукта c-kit имеет значение для поддержания нормального кроветворения, меланогенеза, гаметогенеза и роста и дифференциации тучных клеток. Кроме важной роли в нормальной физиологической активности клетки c-kit играет роль в биологических аспектах развития определенных видов рака у человека и разрегулированная активность киназы c-kit принимает участие в развитии патогенеза рака у человека и определенных видов опухолей. Пролиферация роста опухолевых клеток, опосредованная киназой c-kit, может происходить в результате специфической мутации полипептида c-kit, которая приводит к независимой активации лигандов или аутокринной стимуляции рецептора. В первом случае мутации, вызывающие конститутивную активацию активности киназы c-kit в отсутствие связывания с фактором стволовых клеток, приводят к развитию злокачественных опухолей у человека, включая опухоли половых клеток, тучных клеток, желудочно-кишечные стромальные опухоли, мелкоклеточный рак легких, меланому, рак молочной железы, острый миелогенный лейкоз, нейробластому и мастоцитоз. Тучные клетки, содержащиеся в тканях пациента, принимают участие в развитии или вносят вклад в развитие заболеваний, таких как аутоиммунные заболевания (рассеянный склероз, ревматоидный артрит, воспалительные заболевания кишечника), аллергические заболевания, развитие кровеносных сосудов в опухоли, воспалительные заболевания и интерстициальный цистит. Аллергические заболевания включают, но не ограничиваясь только ими, аллергический ринит, аллергический синусит, анафилакти- 13017392 ческий синдром, крапивницу, отек Квинке, атопический дерматит, аллергический контактный дерматит,узловатую эритему, множественную эритему, кожный некротирующий венулит, воспаления кожи вследствие укуса насекомых и астму. Астма характеризуется обструкцией дыхательных путей, бронхиальной гиперчувствительностью, воспалением дыхательных путей и включает бронхиальную астму и аллергическую астму. В случае указанных заболеваний тучные клетки принимают участие в разрушении тканей в результате высвобождения смеси различных протеаз и медиаторов, таких как гистамин, нейтральные протеазы,липидсодержащие медиаторы (простагландины, тромбоксаны и лейкотриены) и различные цитокины(ИЛ-1, ИЛ-2, ИЛ-3, ИЛ-4, ИЛ-5, ИЛ-6, ИЛ-8, TNF-A, GM-CSF, MIP-LA, MIP-1b, MIP-2 и IFN-). Активация тучных клеток индуцирует различные эффекторные ответные реакции, такие как секреция аллергических медиаторов, протеаз, хемокинов, таких как МСР-1 и RANTES, лейкотриенов, простагландинов,нейротрофинов, индукция транскрипции генов цитокинов (ИЛ-4, ИЛ-5, ИЛ-6, ИЛ-13, TNFA и GM-CSF). Указанные медиаторы способствуют формированию астматического фенотипа при влиянии на эндотелиальные клетки, клетки гладкой мускулатуры и фибробласты, а также на клеточный матрикс и при накоплении других воспалительных клеток. Тучные клетки могут участвовать в развитии астмы, как установлено при лечении моноклональными и гуманизированными антителами anti-IgE. Принцип лечения антителами anti-IgE заключается в специфическом воздействии на IgE, что приводит к инактивации свободного anti-IgE и остановке дальнейшего продуцирования IgE. Кроме того, поскольку уровни IgE являются основным регулятором уровня экспрессии рецептора IgE, FceRI, цель такого лечения заключается в снижении экспрессии FceRI на тучных клетках и базофилах и, следовательно, в снижении способности указанных клеток к активации. Снижение экспрессии FceRI на базофилах было установлено при лечении антителами anti-IgE. Снижение экспрессии FceRI на базофилах связано со снижением способности базофилов секретировать медиаторы при активации. Ингибиторы c-kit также можно использовать для лечения инсулиннезависимого сахарного диабета,также известного как диабет типа II, хронического заболевания, при котором инсулин не ускоряет захват глюкозы клетками, что приводит к увеличению уровня глюкозы в крови. Указанное заболевание поражает приблизительно 100 миллионов человек во всем мире, 75% из которых при установке диагноза страдали от ожирения. В течение многих лет неспособность регулировать захват глюкозы приводит к развитию диабета типа II и к необходимости регулировать уровень глюкозы в крови с использованием лекарственных средств. В конечном итоге нерегулируемый уровень глюкозы в крови приводит к повреждению кровеносных сосудов, почек и глаз, а также к развитию сердечно-сосудистых заболеваний. Указанные повреждения ткани играют роль в повышении смертности у людей, страдающих от диабета. Кроме того, активация тучных клеток различными стимуляторами, такими как стресс, травма, инфекция, а также нейромедиаторы, может приводить к обострению химического дисбаланса и к нарушениям ЦНС. Более конкретно дегрануляция тучных клеток стимулируется обычными нейромедиаторами,такими как нейротензин, соматостатин, вещество Р и ацетилхолин, факторами роста или выживания,прежде всего NGF, TGFL. Тучными клетками, принимающими участие в ответной реакции на такие стимулы, являются тучные клетки мозга, а также другие тучные клетки, из которых их содержимое высвобождается в кровоток, который в конечном итоге доставляет их в сенсорные, моторные или мозговые нейроны. Тучные клетки мозга окрашиваются аналогично тучным клеткам соединительной ткани, но характеризуются секреторными свойствами тучных клеток слизистой оболочки, что свидетельствует об особой субпопуляции специфичных тучных клеток. После активации тучных клеток высвобождающиеся гранулы высвобождают различные факторы,способные модулировать и изменять нейропередачу сигнала и выживаемость нейронов. Среди таких факторов важным является серотонин, так как повышение уровня свободного серотонина наблюдается у пациентов в состоянии депрессии. В другом варианте внезапное повышение уровня серотонина может наблюдаться после периода недостатка серотонина, что приводит к возникновению болей и мигрени. В связи с этим предполагается, что тучные клетки усиливают аутокринный или паракринный способ разрегулирования нейротрансмиссии. Например, вызванное состоянием тревоги или стрессом высвобождение нейромедиаторов, таких как серотонин, активирует тучные клетки, которые, в свою очередь, высвобождают содержимое их гранул, что в дальнейшем вызывает химический дисбаланс в мозге и заболевания ЦНС. Другие медиаторы, высвобождаемые тучными клетками, подразделяются на вазоактивные, ноцицептивные, провоспалительные и другие нейромедиаторы. Вместе указанные факторы вызывают серьезные нарушения активности нейронов: сенсорных, моторных нейронов или нейронов ЦНС. Кроме того, у пациентов, страдающих мастоцитозом, наблюдается склонность к развитию заболеваний ЦНС по сравнению с нормальной популяцией. Указанный факт можно объяснить наличием активирующей мутации рецептора c-kit, при этом происходит индукция дегрануляции тучных клеток и выброс факторов, которые приводят к химическому дисбалансу и изменению нейропередачи. В некоторых случаях активированные тучные клетки также могут принимать участие в разрушении- 14017392 нервных тканей при высвобождении смеси различных протеаз и медиаторов, которые подразделяются на три группы: предварительно ассоциированные с гранулами медиаторы (гистамин, протеогликаны и нейтральные протеазы), содержащие липиды протеазы (простагландины, тромбоксаны и лейкотриены) и различные цитокины (ИЛ-1, ИЛ-2, ИЛ-3, ИЛ-4, ИЛ-5, ИЛ-6, ИЛ-8, TNF-A, GM-CSF, MIP-LA, MIP-1b,MIP-2 и IFN-). Высвобождение медиаторов из активированных тучных клеток (TNF-А, гистамина, лейкотриенов, простагландинов и т.п.), а также протеаз может вызывать: 1) воспаление и расширение кровеносных сосудов и 2) процесс разрушения нервной ткани. Ингибирование активности c-kit снижает пролиферацию клеток, снижая количество тучных клеток, ответственных за развитие заболеваний и/или состояний. Таким образом, можно ожидать, что ингибиторы c-kit играют важную роль при лечении c-kitзависимых заболеваний и/или состояний, таких как заболевания ЦНС. Установлено также, что тучные клетки принимают участие или вносят вклад в развитие симптомов лекарственной зависимости или абстиненции. Лекарственная зависимость развивается в результате так называемой переносимости, которая заключается в необходимости увеличения дозы лекарственного средства для поддержания наиболее полного эффекта, и физической зависимости, которая проявляется в привыкании организма к лекарственному средству. Если прием лекарственного средства прекращают,субъект может ощущать неприятный синдром отмены лекарственного средства (абстинентный синдром). Активация тучных клеток при введении различных лекарственных средств, включая, но не ограничиваясь только ими, производные салициловой кислоты, производные морфина, опиоиды, героин, амфетамины, алкоголь, никотин, анальгетики, анестетики и транквилизаторы, приводит к дегрануляции тучных клеток, при которой усиливается химический дисбаланс, что является причиной лекарственной зависимости и синдрома отмены лекарственного средства. После активации тучных клеток высвобождающиеся гранулы высвобождают различные факторы, модулирующие и изменяющие нейропередачу. К указанным факторам относится морфин, который связывается или хранится в гранулах тучных клеток. Табачный дым также вызывает выделение медиаторов из тучных клеток собаки и модулирует продуцирование простагландина, что приводит к развитию астмы. Кроме того, пациенты, страдающие от мастоцитоза, в большей степени предрасположены к развитию расстройств, вызванных употреблением психоактивных лекарственных средств, по сравнению с нормальной популяцией. Этот факт можно объяснить присутствием активирующих мутаций рецептора с-kit, что вызывает дегрануляцию тучных клеток и выброс факторов, способствующих усилению химического дисбаланса и изменению нейропередачи. В настоящее время отсутствует способ лечения, обеспечивающий снижение интенсивности синдрома лекарственной зависимости и позволяющий устранить у индивидуалов нарушения, связанные со злоупотреблением лекарственными средствами. Ингибиторы c-kit можно использовать для лечения заболеваний, связанных со злоупотреблением лекарственными средствами, прежде всего, наркотической и лекарственной зависимости, привыкания к лекарственным средствам, зависимости от лекарственных средств, синдрома отмены и передозировки лекарственных средств, и способ лечения заключается во введении соединения, которое снижает число тучных клеток у субъекта, нуждающегося в таком лечении. Киназа c-kit характеризуется гомологией с рецептором PDGF (тромбоцитарного фактора роста) и рецептором CSF-1 (c-Fms). Данные исследований различных эритроидных и миелоидных линий клеток свидетельствуют об экспрессии гена c-kit на ранних стадиях дифференциации (см. Andre и др., Oncogene 4, сс. 1047-1049 (1989. Некоторые опухоли, например глиобластомные клетки, также характеризуются выраженной экспрессией c-kit. Тромбоцитарный фактор роста (PDGF). Тромбоцитарный фактор роста (PDGF) играет важную роль как на стадии нормального роста, так и на стадии патологической пролиферации клеток. Соединения по настоящему изобретению могут ингибировать активность рецептора PDGF (PDGFR) и их можно использовать в качестве агентов для лечения незлокачественных пролиферативных заболеваний, таких как склеродермия и другие фиброзные заболевания, атеросклероз, тромбоз или псориаз. Соединения по настоящему изобретению также можно использовать в качестве подавляющих образование опухоли веществ, например, при мелкоклеточном раке легких, глиоме, саркоме, опухоли предстательной железы, и опухоли кишки, молочной железы, и яичников. В одном варианте соединения по настоящему изобретению можно использовать для лечения и профилактики фиброзных нарушений или заболеваний, состояний, связанных с образованием и отложением компонентов межклеточного матрикса во внутренних органах, включая почки, сердце, легкие, печень,кожу и суставы. В различных исследованиях в качестве основного действующего соединения при развитии фиброзной ответной реакции на повреждение ткани используют PDGFR, например: 1) наблюдается положительная регуляция в альвеолярных макрофагах у пациентов, страдающих идиопатическим легочным фиброзом; 2) рецептор PDGF является одним из первых генов, которые положительно регулируются после активации печеночных звездчатых клеток с образованием миофибробластов, что является основной стадией при развитии фиброза печени; 3) при склеродермии наблюдается значительная положительная регуляция PDGF и его рецепторов,- 15017392 а также в процессе почечного фиброгенеза; 4) PDGF индуцируется при повреждении и/или провоспалительными цитокинами или по аутокринному механизму на миобластах, активируя их пролиферацию, дифференциацию и миграцию. Такие миобласты затем секретируют межклеточные матричные белки и коллаген, что приводит к образованию шрамов и прогрессирующему повреждению органов. Секреция TGFP также в значительной степени способствует продуцированию коллагена в процессе фиброгенеза; 5) трансген PDGF-C вызывает развитие фиброза печени у мышей, в то же время растворимый доминантно-негативный вариант PDGFR предотвращает фиброз печени у крыс; 6) введение PDGF-B в почки ускоряет развитие признаков почечного фиброгенеза у крыс. Фиброзные заболевания или состояния, которые можно лечить с использованием соединений по настоящему изобретению, включают фиброзные заболевания легких, такие как легочный фиброз (или интерстициальное заболевание легких или интерстициальный легочный фиброз), идиопатический легочный фиброз, первичную легочную гипертензию, идиопатическую легочную артериальную гипертензию,фиброзный элемент пневмокониоза (связанный с воздействием внешних вредных факторов, таких как курение, асбест, хлопковый очес, каменная пыль, рудничная пыль и другие частицы), легочный саркоидоз, фиброзирующий альвеолит, фиброзный или гипертрофический элемент муковисцидоза, хроническое обструктивное заболевание легких, респираторный дистресс-синдром у взрослых и эмфизему. Соединения по настоящему изобретению также можно применять для лечения и профилактики заболеваний, при которых наблюдается фиброзная гипертрофия почек (почечный фиброз), печени (печеночный фиброз),сердца (сердечный фиброз), предстательной железы (например, доброкачественная гипертрофия предстательной железы), плевры (плеврит, плевральный фиброз), поджелудочной железы и кожи и/или мышечных тканей, например склеродермия, эозинофильный фасциит, дисковые повреждения, связанные с волчанкой или дискоидной волчанкой, или хирургические спайки. Другие фиброзные заболевания или нарушения, которые можно лечить с использованием соединений по настоящему изобретению, включают, но не ограничиваясь только ими, системный склероз, смешанное заболевание соединительной ткани, фибродисплазию, фиброзно-кистозную мастопатию, саркоидоз, миозит(например, полимиозит, первичный идиопатический полимиозит, детский полимиозит, дерматомиозит,детский дерматомиозит, первичный идиопатический дерматомиозит у взрослых, миозит с телами включения, полимиозит или дерматомиозит, связанный со злокачественными опухолями), заболевания, при которых наблюдается фиброзно-васкулярная интимальная гипертрофия, такие как васкулит (включая васкулит коронарных артерий), узелковый полиартриит или височный артериит, заболевания, при которых наблюдается фиброзная гипертрофия нервной ткани, такие как церебросклероз, кольцевидный склероз, диффузный склероз и лобарный склероз, и заболевания, при которых наблюдается фиброзная гипертрофия или фиброз стенок кишечника, такие как воспаление кишечника, включая болезнь Крона. Кроме того, соединения по настоящему изобретению также можно использовать для защиты стволовых клеток, например, для предотвращения гемотоксического воздействия хемиотерапевтических агентов, таких как 5-фторурацил, а также для лечения астмы и гиперэозинофилии. Соединения по настоящему изобретению также можно применять для лечения заболеваний, чувствительных к ингибированию рецепторной киназы PDGF. Соединения по настоящему изобретению также оказывают благоприятное действие при лечении заболеваний, развивающихся в результате трансплантации, например аллогенной трансплантации, прежде всего при отторжении ткани, например облитеративном бронхиолите, т.е. хроническом отторжении аллотрансплантата легкого. В отличие от пациентов, не страдающих облитеративным бронхиолитом, у пациентов, страдающих от указанного заболевания, наблюдается повышенный уровень PDGF в бронхоальвеолярной промывной жидкости. Соединения по настоящему изобретению также можно эффективно использовать для лечения заболеваний, связанных с миграцией и пролиферацией клеток гладкой мускулатуры сосудов (PDGF и PDGFR также часто принимают участие в развитии указанных процессов), таких как рестеноз и атеросклероз. Указанные эффекты и их последствия, действующие на пролиферацию или миграцию клеток гладкой мускулатуры сосудов in vitro и in vivo, можно оценивать при введении соединений по настоящему изобретению и при изучении их влияния на утолщение интимы сосудов при механическом повреждении inCSF1R (FMS). Белок, кодируемый указанным геном, является рецептором колониестимулирующего фактора 1,цитокина, который контролирует продуцирование, дифференциацию и функционирование макрофагов.CSFR1 опосредует большинство, если не все биологические действия данного цитокина. Кодируемый белок является трансмембранной рецепторной тирозинкиназой и принадлежит к семейству рецепторных тирозинпротеинкиназ CSF1/PDGF. Мутации указанного гена связывают с предрасположенностью к злокачественным миелоидным заболеваниям (см., например, Casas и др., Leuk. Lymphoma 44, сс. 1935-1941Abl. Trk, Svk, Ras, Raf, MAPK, TGF, FGFR3, c-Src, SAPK, Lck, Fes, Csk. Тирозинкиназа Абельсона (т.е. Abl, c-Abl) принимает участие в регуляции клеточного цикла, в клеточной ответной реакции на генотоксический стресс и в передаче информации о клеточном окружении через интегриновый сигнал. Белок Abl выполняет сложную функцию клеточного модуля, который интегрирует сигнал из различных вне- и внутриклеточных источников, и управляет клеточным циклом и апоптозом. Тирозинкиназа Абельсона включает различные подтипы, такие как гибридный белок (онкопротеин) BCR-Abl с разрегулированной активностью тирозинкиназы, или v-Abl. Гибридный белок BCR-Abl образуется в результате реципрокной транслокации, при которой происходит гибридизация протоонкогена Abl с геном Bcr. BCR-Abl трансформирует В-клетки за счет увеличения митогенной активности. Такое увеличение приводит к снижению чувствительности к апоптозу, а также к изменению адгезии и хоминга клеток-предшественников хронического миелогенного лейкозаBCR-Abl играет важную роль в патогенезе, в 95% случаев ХМЛ и в 10% случаев острого лимфоцитарного лейкоза (ОЛЛ). Гливек STI-571 является ингибитором онкогенной тирозинкиназы BCR-Abl и используется для лечения ХМЛ. Однако у некоторых пациентов на стадии бластного криза ХМЛ развивается резистентность к STI-571 из-за мутаций в последовательности киназы BCR-Abl. В настоящее время выявлено более 22 мутаций, таких как G250E, E255V, T315I, F317L и М 351 Т. Соединения по настоящему изобретению ингибируют киназу abl, например киназу v-abl. Соединения по настоящему изобретению также ингибируют киназу BCR-Abl дикого типа и мутантные формы киназы BCR-Abl, и, таким образом, их можно использовать для лечения Bcr-abl-положительных форм рака и опухолей, таких как лейкозы (прежде всего ХМЛ и ОЛЛ, в развитии которых принимает участие прежде всего апоптозные механизмы). Соединения по настоящему изобретению также можно эффективно использовать для обработки лейкозных стволовых клеток, указанные соединения можно использовать для очистки указанных клеток in vitro после удаления таких клеток (например, при удалении костного мозга) и реимплантации клеток после их очистки от раковых клеток (например, реимплантация очищенных клеток костного мозга). Семейство trk нейротрофиновых рецепторов (trkA, trkB, trkC) повышает выживаемость, ускоряет рост и дифференциацию нейрональных и ненейрональных тканей. Белок TrkB экспрессируется клетками нейроэндокринного типа в тонком кишечнике и ободочной кишке, в -клетках поджелудочной железы, в моноцитах и макрофагах лимфатических узлов и селезенки и в гранулярных слоях эпидермиса (Shibayama и Koizumi (1996. Экспрессия белка TrkB связана с нежелательным прогрессированием опухоли Вильмса и нейробластом. Кроме того, TrkB экспрессируется в раковых клетках предстательной железы,но не экспрессируется в нормальных клетках. Сигнальный путь рецепторов trk включает каскад активации MAPK через Shc, активированных генов Ras, ERK-1 и ERK-2, и путь передачи сигнала PLC-1 (Sugimoto и др., (2001.Syk является тирозинкиназой, которая играет важную роль в дегрануляции тучных клеток и активации эозинофилов. Соответственно, киназа Syk принимает участие в развитии различных аллергических заболеваний, прежде всего, астмы. Установлено, что киназа Syk связывается с фосфорилированной цепью рецептора FcR1 через N-концевые домены SH2 и играет важную роль в следующих сигнальных путях. Сигнальный путь Ras-Raf-MEK-ERK опосредует ответную реакцию клеток на сигналы роста. В результате мутации Ras образуется его онкогенная форма в 15% случаях рака человека. Семейство Raf принадлежит к серин/треонинпротеинкиназам и включает три белка: A-Raf, B-Raf и c-Raf (или Raf-1). BRaf может играть важную роль при формировании некоторых опухолей и при этом нет необходимости в активации аллеля Ras (Nature 417, сс. 949-954 (2002. Мутации киназы B-Raf выявлены во множестве злокачественных меланом. Существующие способы лечения меланомы ограничены по своей эффективности, особенно на поздних стадиях меланомы. Соединения по настоящему изобретению, кроме того, подавляют клеточные процессы с участием киназы b-Raf и являются новыми перспективными агентами, предназначенными для лечения рака человека, прежде всего, лечения меланомы. Митогенактивированные протеинкиназы (MAPK) являются членами консервативных путей передачи сигнала, которые активируют факторы транскрипции, факторы трансляции и другие молекулымишени в ответ на различные внеклеточные сигналы. MAPK активируются при фосфорилировании двойной фосфорилируемой последовательности Thr-X-Tyr в присутствии митоген-активированной протеинкиназы (MKK). У высших эукариотов физиологическая роль сигнала киназы MAPK коррелирует с такими клеточными процессами, как пролиферация, онкогенез, рост и дифференциация. Соответственно,способность регулировать передачу сигнала через указанные пути (прежде всего через MKK4 и MKK6) можно использовать для разработки новых способов лечения и профилактики заболеваний человека, связанных с сигнальным путем MAPK, таких как воспалительные заболевания, аутоиммунные заболевания и рак. Многочисленные формы р 38 MAPK (, , , ), каждая из которых кодируется отдельным геном,- 17017392 образуют часть каскада киназ, принимающих участие в ответной реакции клеток на различные стимулы,включая осмотический стресс, УФ-излучение и опосредованные цитокинами явления. Предполагается,что четыре указанные изоформы р 38 регулируют различные аспекты внутриклеточной передачи сигнала. Их активация является частью каскада передачи сигналов, которые приводят к синтезу и продуцированию противовоспалительных цитокинов, например TNF. р 38 действует при фосфорилировании субстратов, расположенных в следующих путях, включая другие киназы и факторы транскрипции. Установлено, что агенты, ингибирующие р 38 киназу, блокируют продуцирование цитокинов, включая, но не ограничиваясь только ими, TNF, ИЛ-6, ИЛ-8 и ИЛ-1. Установлено, что моноциты периферической крови (МЦПК) экспрессируют и секретируют противовоспалительные цитокины при стимуляции липополисахаридами in vitro. Ингибиторы р 38 эффективно блокируют этот эффект, если МЦПК перед стимуляцией липополисахаридами предварительно обработать указанными соединениями. Ингибиторы р 38 проявляют эффективность на моделях воспалительных заболеваний у животных. Деструктивное действие многих заболеваний и состояний на организм связано с избыточным продуцированием противовоспалительных цитокинов. Способность ингибитора р 38 регулировать указанное сверхпродуцирование можно использовать при разработке модифицирующих заболевание агентов. Установлено, что молекулы, блокирующие функцию р 38, проявляют эффективность для подавления резорбции костной ткани, воспаления и других иммунных и воспалительных патологий. Таким образом, безопасный и эффективный ингибитор р 38 представляет собой агент для лечения истощающих заболеваний, на которые можно воздействовать за счет модуляции сигнального пути р 38. Следовательно,соединения по настоящему изобретению, ингибирующие активность р 38, можно использовать для лечения воспаления, остеоартрита, ревматоидного артрита, рака, аутоиммунных заболеваний и для лечения других опосредованных цитокинами заболеваний. Трансформирующий фактор роста(TGF) относится к подсемейству белков, включающему, например, TGF1, TGF2 и TGF3, которые являются плеотропными модуляторами роста и дифференциации клеток, эмбрионального развития и развития костной ткани, образования внеклеточного матрикса,кроветворения, иммунных и воспалительных ответных реакций. Члены семейства TGF инициируют внутриклеточные сигнальные пути, приводящие в конечном итоге к экспрессии генов, которые регулируют клеточный цикл, контролируют пролиферативные ответные реакции или связаны с белками внеклеточного матрикса, которые опосредуют сигнальный путь внешняя среда-клетка, адгезию, миграцию клеток и межклеточную коммуникацию. Следовательно, соединения по настоящему изобретению, которые являются ингибиторами внутриклеточных сигнальных путей TGF, можно использовать в качестве терапевтических средств для лечения фибропролиферативных заболеваний, включая заболевания почек, связанные с разрегулированной активностью TGF, и повышенный фиброз, включая гломерулонефрит (ГН), например мезангиальный пролиферативный ГН, иммунный ГН и ГН мениска. Другие почечные состояния включают диабетическую нефропатию, почечный интерстициальный фиброз, почечный фиброз у пациентов после трансплантации, которым вводят циклоспорин, и нефропатию, связанную с ВИЧ. Коллагеновые сосудистые нарушения включают прогрессирующий системный склероз, полимиозит, склеродермию, дерматомиозит, эозинофильный фасциит, очаговую склеродермию или заболевания, связанные с синдромом Рейно. Фиброз легких, развивающийся в результате избыточной активности TGF, включает респираторный дистресс-синдром, хроническое обструктивное заболевание легких у взрослых, идиопатический легочный фиброз и интерстициальный легочный фиброз, часто связанный с аутоиммунными нарушениями,такими как системная красная волчанка и склеродермия, химический контакт или аллергии. Другим аутоиммунным нарушением, связанным с фибропролиферативными процессами, является ревматоидный артрит. Фибропролиферативные состояния могут быть связаны с глазными хирургическими операциями. Такие операции включают реплантацию сетчатки, сопровождающуюся пролиферативной витреоретинопатией, удаление катаракты с имплантацией искусственного хрусталика и дренирование после операции глаукомы. Установлено, что рецептор 3 фактора роста фибропластов оказывает отрицательное регуляторное действие на рост костной ткани и подавляет пролиферацию хондроцитов. Различные мутации в рецепторе 3 фактора роста фибропластов вызывают летальную дисплазию. Одна мутация, TDII FGFR3, характеризуется конститутивной активностью тирозинкиназы, которая активирует фактор транскрипции Stat1,вызывая экспрессию ингибитора клеточного цикла, остановку роста и аномальное развитие костной ткани (Su и др., Nature, 386, сс. 288-292 (1997. Кроме того, FGFR3 экспрессируется в большинстве случаев рака типа множественной миеломы. Киназа c-Src передает онкогенные сигналы многих рецепторов. Например, сверхэкспрессия EGFR или HER2/neu в опухолях приводит к конститутивной активации киназы c-Src, что является характерным для злокачественной клетки, но не наблюдается в нормальной клетке. С другой стороны, мыши с дефицитом экспрессии киназы c-Src представляют собой фенотип остеопетроза, что свидетельствует о важной роли c-Src в функционировании остеокластов и возможном участии в развитии ассоциированных нару- 18017392 шений. Семейство рибосомальных протеинкиназ S6 включает по крайней мере 8 членов (RSK1, RSK2,RSK3, RSK4, MSK1, MSK2, p70S6K и p70S6Kb). Рибосомальные протеинкиназы S6 выполняют важные плеотропные функции, в том числе играют важную роль в регуляции трансляции мРНК при биосинтезе белков (Eur. J. Biochem., 267 (21), сс. 6321-6330 (2000, ноябрь), Exp. Cell. Res., 253 (1), сс. 100-109 (1999,25 ноября), Mol. Cell. Endocrinol., 151 (1-2), сс. 65-77 (1999, 25 мая. Кроме того, фосфорилирование рибосомального белка S6 киназой p70S6 связано с регуляцией клеточной подвижности (Immunol. Cell.Biol., 78 (4), сс. 447-451 (2000, август и клеточного роста (Prog. Nucleic Acid Res. Mol. Biol., 65, cc. 101127 (2000 и, следовательно, является важным фактором при метастазировании опухолей, иммунной ответной реакции и регенерации тканей, а также при других патологических состояниях. Стресс-активируемые протеинкиназы (SAPK, так называемые N-концевые киназы jun или киназы JNK) представляют собой семейство протеинкиназ, которые принимают участие на предпоследней стадии пути передачи сигнала, при этом происходит активация фактора транскрипции c-jun и экспрессия генов, регулируемых c-jun. Прежде всего, c-jun принимает участие в транскрипции генов, которые кодируют белки, связанные с репарацией ДНК, поврежденной вследствие генотоксических факторов. Агенты,которые ингибируют активность SAPK в клетке, предотвращают репарацию ДНК и сенсибилизируют клетку к противораковым терапевтическим агентам, которые действуют за счет индукции повреждения ДНК.Lck принимает участие в передаче сигнала Т-клетками. У мышей с отсутствием гена Lck наблюдается низкий уровень тимоцитов. Функция Lck в качестве положительного активатора передачи сигнала Т-клетками свидетельствует о том, что ингибиторы Lck можно использовать для лечения аутоиммунного заболевания, такого как ревматоидный артрит.Fes интенсивно экспрессируется в миелоидных кроветворных клетках и принимает участие в путях передачи сигнала дифференциации и выживания миелоидных лейкоцитов. CSK принимает участие в развитии рака, прежде всего, колоректального рака и рака молочной железы. В соответствии с указанным выше в настоящем изобретении предлагается также способ профилактики или лечения любого описанного выше заболевания или нарушения у субъекта, нуждающегося в указанном лечении, причем способ заключается в том, что указанному субъекту вводят терапевтически эффективное количество (см. раздел Способы введения и фармацевтические композиции ниже) соединения формулы (1), (2 А), (2 В) или (С) или его фармацевтически приемлемой соли. Для любого из указанных выше применений требуемые дозировки изменяются в зависимости от способа введения, конкретного состояния, подлежащего лечению, и требуемого действия. Способы введения и фармацевтические композиции. Использованный в данном контексте термин фармацевтическая композиция обозначает смесь соединения по настоящему изобретению с другими химическими компонентами, такими как носители, стабилизаторы, разбавители, диспергирующие агенты, суспендирующие агенты, загустители, и/или эксципиентами. Фармацевтическая композиция ускоряет введение соединения в организм. Фармацевтические композиции, содержащие соединение по настоящему изобретению, можно вводить в терапевтически эффективных количествах в виде фармацевтических композиций в любой стандартной форме и любым известным способом, включая, не ограничиваясь только ими: внутривенное, пероральное, ректальное, аэрозольное, парентеральное, офтальмологическое, легочное, чрескожное, вагинальное, ушное, назальное и местное введение. Соединение предпочтительно вводят местным способом, а не системным, например инъекцией соединения напрямую в орган, в большинстве случаев в составе с замедленным всасыванием или с замедленным высвобождением. Кроме того, фармацевтическую композицию, содержащую соединение по настоящему изобретению, можно вводить в виде системы направленной доставки лекарственных средств,например в виде липосомы с покрытием из органоспецифических антител. Липосомы доставляются в мишень и селективно поглощаются органом. Кроме того, фармацевтические композиции, содержащие соединение по настоящему изобретению, можно вводить в форме состава с быстрым высвобождением, в форме состава с замедленным высвобождением или в форме состава с промежуточной скоростью высвобождения. Для перорального введения соединение по настоящему изобретению можно перерабатывать, например, при простом смешивании активных соединений с известными фармацевтически приемлемыми носителями или эксципиентами. Указанные носители позволяют перерабатывать соединения, описанные в данном контексте, в таблетки, порошки, пилюли, драже, капсулы, жидкости, гели, сиропы, эликсиры,суспензии, эмульсии и т.п., которые пациент, нуждающийся в лечении, проглатывает. Фармацевтические препараты для перорального введения получают при смешивании одного или более твердых эксципиентов с одним или более соединений, описанных в данном контексте, необязательно измельчении полученной смеси и при переработке смеси гранул с последующим добавлением при необходимости пригодных вспомогательных веществ, при этом получают таблетки или ядра драже. Пригодными эксципиентами являются, прежде всего, наполнители, такие как сахара, включая лактозу,- 19017392 сахарозу, маннит или сорбит, препараты целлюлозы, такие как, например, кукурузный крахмал, пшеничный крахмал, рисовый крахмал, картофельный крахмал, желатин, трагакантовая камедь, метилцеллюлоза, микрокристаллическая целлюлоза, гидроксипропилметилцеллюлоза, натриевая соль карбоксиметилцеллюлозы, или другие соединения, такие как поливинилпирролидон (ПВП или повидон) или фосфат кальция. При необходимости можно добавлять дезинтегрирующие агенты, такие как сшитая натриевая соль кроскармеллозы, поливинилпирролидон, агар, или альгиновая кислота или ее соль, такая как альгинат натрия. На ядра драже наносят также пригодные покрытия. Для этой цели используют концентрированные растворы сахара, которые необязательно содержат аравийскую камедь, тальк, поливинилпирролидон, гель карбопол, полиэтиленгликоль, и/или диоксид титана, растворы глазури и пригодные органические растворители или смеси растворителей. Для идентификации или характеризации различных комбинаций доз активного соединения в состав покрытия для таблетки или драже добавляют красители или пигменты. Фармацевтические препараты, которые можно вводить перорально, включают твердые капсулы, состоящие из двух частей, изготовленные из желатина, а также мягкие закрытые капсулы, изготовленные из желатина и пластификатора, такого как глицерин или сорбит. Твердые капсулы из двух частей включают активные ингредиенты в смеси с наполнителем, таким как лактоза, связующими, такими как крахмалы, и/или замасливателями, такими как тальк или стеарат магния, и, необязательно, стабилизаторами. Мягкие капсулы заполнены раствором или суспензией активных соединений в пригодных жидкостях,таких как жирные масла, вазелин, или жидкие полиэтиленгликоли. Кроме того, добавляют стабилизаторы. Для буккального или сублингвального введения композиции перерабатывают стандартным способом и получают таблетки, лепешки или гели. Парентеральные инъекции включают струйное вливание или непрерывное вливание. Фармацевтическую композицию соединения по настоящему изобретению перерабатывают и получают форму, пригодную для парентеральной инъекции, такую как стерильные суспензии, растворы или эмульсии в масляных или водных носителях, и указанная форма включает агенты для получения составов, такие как суспендирующие агенты, стабилизаторы и/или диспергирующие агенты. Фармацевтические составы для парентерального введения включают водные растворы активных соединений в водорастворимой форме. Кроме того, суспензии активных соединений получают в виде соответствующих масляных суспензий для инъекций. Пригодные липофильные растворители или носители включают жирные масла, такие как кунжутное масло, или сложные эфиры синтетических жирных кислот, такие как этилолеат, или триглицериды, или липосомы. Водные суспензии для инъекций содержат соединения, которые повышают вязкость суспензии, такие как натриевая соль карбоксиметилцеллюлозы, сорбит или декстран. Суспензия необязательно также включает пригодные стабилизаторы или агенты, которые повышают растворимость соединений, что позволяет получать высококонцентрированные растворы. В другом варианте активный ингредиент представляет собой порошок, который перед введением смешивают с пригодным носителем, например стерильной апирогенной водой. Соединения по настоящему изобретению вводят местным способом и получают в виде различных композиций, предназначенных для введения местным способом, таких как растворы, суспензии, лосьоны, гели, пасты, лечебные губные помады, бальзамы, кремы или мази. Указанные фармацевтические соединения содержат солюбилизаторы, стабилизаторы, агенты, повышающие тоничность, буферные вещества и консерванты. В составах, пригодных для чрескожного введения, используются устройства и пластыри для чрескожной доставки, и указанные составы представляют собой липофильные эмульсии или буферные водные растворы, растворенные и/или диспергированные в полимере или адгезиве. Конструкция указанных пластырей обеспечивает непрерывную, периодическую доставку фармацевтических агентов или доставку в требуемый момент времени. Кроме того, чрескожную доставку соединений по настоящему изобретению осуществляют с использованием ионтофоретических пластырей и т.п. Кроме того, чрескожные пластыри обеспечивают контролируемую доставку соединений по настоящему изобретению. Скорость абсорбции уменьшают за счет использования мембран, контролирующих скорость высвобождения, или за счет удерживания соединения в полимерной матрице или геле. С другой стороны, можно использовать ускорители абсорбции для повышения скорости абсорбции. Агент или носитель, повышающий скорость абсорбции, включает абсорбируемые фармацевтически приемлемые растворители, которые повышают проницаемость активного агента через кожу. Например, устройства для чрескожной доставки представляют собой повязку, включающую основу, резервуар, содержащий соединения необязательно в смеси с носителями, необязательно барьер, контролирующий скорость доставки соединения на кожу пациента и обеспечивающий контролируемую предварительно определенную скорость в течение продолжительного периода времени, и средство для закрепления устройства на коже. Для введения ингаляцией соединения по настоящему изобретению перерабатывают и получают аэрозоль, спрей или порошок. Фармацевтические композиции соединений по настоящему изобретению можно доставлять в форме аэрозоля из емкости или распылителя под давлением в смеси с пригодным пропеллентом, например дихлордифторметаном, трихлорфторметаном, дихлортетрафторэтаном, диоксидом углерода или другим пригодным газом. В случае аэрозоля под давлением доставку стандартной дозы- 20017392 осуществляют с использованием клапана, отмеряющего определенное количество аэрозоля. Капсулы и картриджи, например, из желатина, предназначенные для использования в ингаляторе или инсуффляторе, заполняют порошкообразной смесью соединения и пригодной порошкообразной основы, такой как лактоза или крахмал. Соединения по настоящему изобретению также перерабатывают в форму ректальных композиций,такую как клизмы, ректальные гели, ректальные пены, ректальные аэрозоли, суппозитории, желеобразные суппозитории, или удерживающие клизмы, содержащие стандартные основы для суппозиториев,такие как кокосовое масло или другие глицериды, а также синтетические полимеры, такие как поливинилпирролидон, ПЭГ и т.п. В суппозиториях, содержащих композиции, сначала плавится низкоплавкий воск, такой как, не ограничиваясь только ими, смесь глицеридов жирных кислот, необязательно в комбинации с кокосовым маслом. Фармацевтические композиции перерабатывают стандартным способом, используя один или более физиологически приемлемый носитель, включая эксципиенты и вспомогательные вещества, которые ускоряют переработку активных соединений в фармацевтические препараты. Соответствующий состав зависит от выбранного способа введения. Для получения составов можно использовать любые методики,носители и эксципиенты, известные в данной области техники. Фармацевтические композиции, включающие соединение по настоящему изобретению, можно получать стандартным способом, например при стандартном смешивании, растворении, грануляции, получении драже, растирании, эмульгировании,инкапсулировании, включении в матрицу или прессовании. Фармацевтические композиции включают по крайней мере один фармацевтически приемлемый носитель, разбавитель или эксципиент и соединение формулы (1), (2 А) или (2 В), описанное в данном контексте, в качестве активного ингредиента в форме свободной кислоты, или в форме свободного основания, или в форме его фармацевтически приемлемой соли. Кроме того, способы и фармацевтические композиции, описанные в данном контексте, включают применение N-оксидов, кристаллических форм (известных как полиморфные формы), а также активных метаболитов указанных соединений, характеризующихся активностью аналогичного типа. В некоторых вариантах соединения могут существовать в виде таутомеров. Все таутомеры включены в качестве соединений в объем настоящего изобретения. Кроме того, соединения, описанные в данном контексте, могут существовать в несольватированной, а также в сольватированной формах, включающих фармацевтически приемлемые растворители, такие как вода, этанол и т.п. Сольватированные формы соединений, представленных в настоящем описании, также включены в объем настоящего изобретения. Кроме того, фармацевтические композиции включают другие лекарственные препараты или фармацевтические агенты, носители, адъюванты, такие как консерванты, стабилизаторы, смачивающие или эмульгирующие агенты, агенты, повышающие растворимость, соли для регуляции осмотического давления, и/или буферные вещества. Кроме того, фармацевтические композиции содержат другие терапевтически ценные соединения. Способы получения композиций, включающих соединения, описанные в данном контексте, включают переработку соединений в смеси с одним или более инертных, фармацевтические приемлемых эксципиентов или носителей, при этом получают твердые, полутвердые или жидкие композиции. Твердые композиции включают, не ограничиваясь только ими, порошки, таблетки, диспергируемые гранулы, капсулы, пакетики и суппозитории. Жидкие композиции включают растворы, в которых растворено соединение, эмульсии, включающие соединение, или раствор, содержащий липосомы, мицеллы или наночастицы, включающие соединение, описанное в данном контексте. Полутвердые композиции включают, не ограничиваясь только ими, гели, суспензии и кремы. Композиции представляют собой жидкие растворы или суспензии, твердые формы, пригодные для растворения или суспендирования перед введением, или эмульсии. Указанные композиции включают также незначительные количества нетоксичных вспомогательных веществ, таких как смачивающие или эмульгирующие агенты, буферные вещества (стабилизирующие рН) и т.п. Краткое описание фармацевтических композиций, описанных в данном контексте, приведено, например, в книге Remington, The Science and Practice of Pharmacy, 19 изд. (Easton, Pa., Mack PublishingDecker, Нью-Йорк, N.Y. (1980), а также в книге Pharmaceutical Dosage Forms and Drug Delivery Systems,7oe изд. (Lippincott WilliamsWilkins (1999, включенных в настоящее описание в полном объеме в качестве ссылок. Способы введения и способы лечения. Композиции, включающие соединение(я), описанное(ые) в данном контексте, можно вводить для профилактики и/или лечения. В терапевтических целях композиции вводят пациенту, который уже страдает от заболевания или состояния, в количестве, достаточном для лечения или, по крайней мере, частичной остановки развития симптомов заболевания или состояния. Специалист в данной области определяет указанные терапевтически эффективные количества стандартными методами (включая, не ограничиваясь только ими, клинические испытания с повышением дозы).- 21017392 Соединения по настоящему изобретению можно использовать в комбинации со вторым терапевтическим агентом. Например, если при введении одного из соединений, описанных в данном контексте,одним из побочных действий, испытываемых пациентом, является воспаление, соединения по настоящему изобретению можно вводить в комбинации с противовоспалительным агентом. Терапевтическую эффективность соединения, описанного в данном контексте, можно также повысить при введении адъюванта. При введении соединений по настоящему изобретению совместно с другими терапевтическими агентами дозировки совместно вводимых соединений изменяются в зависимости от типа совместного лекарственного средства, специфичности применяемого лекарственного средства, заболевания или состояния, подлежащего лечению, и т.п. Кроме того, при совместном введении с одним или более биологически активных агентов соединения по настоящему изобретению можно вводить одновременно с биологически активным агентом(ами) или последовательно. При введении соединения по настоящему изобретению в комбинации с вторым терапевтическим агентом можно наблюдать аддитивное или синергетическое действие. В некоторых примерах соединения по настоящему изобретению можно использовать в комбинации с противофиброзным агентом, например с агентом, который препятствует развитию фиброзного заболевания (или модулирует его), такого как склеродермия, легочный фиброз, идиопатический легочный фиброз, первичная легочная гипертензия, первичная легочная артериальная гипертензия, идиопатическая артериальная гипертензия, фиброз печени, фиброз почек и фиброз сердца. Примеры противофиброзных агентов, которые можно использовать в комбинации с соединением по настоящему изобретению, включают, не ограничиваясь только ими, пирфенидон (Nakazoto и др., Eur. J. Pharmacol., 446, сс. 177-185(SMP-534,Sugaru и др., Am. J. Nephrology, 26, сс. 50-58 (2006. Соединения по настоящему изобретению можно также использовать в комбинации с противовоспалительным агентом, включая, не ограничиваясь только ими, кортикостероиды и кромолины, антагонисты лейкотриена и блокаторы IgE, такие как омализумаб. В других примерах соединения по настоящему изобретению можно использовать в комбинации с лекарственным средством, предназначенным для лечения астмы, например с бронхолитическими средствами,такими как 2-агонисты, с ксантинами (например, метилксантины) и антихолинергическими средствами,а также с противовоспалительным агентом, как описано выше. Соединения по настоящему изобретению можно также использовать в комбинации с химиотерапевтическим агентом, предназначенным для лечения нарушения клеточной пролиферации, включая, не ограничиваясь только ими, лимфому, остеосаркому, меланому, или опухоль молочной железы, почек,предстательной железы, колоректальную опухоль, опухоль щитовидной железы, яичников, поджелудочной железы, нейрональную опухоль, опухоль легких, матки или опухоль желудочно-кишечного тракта. Примеры химиотерапевтических агентов, которые можно использовать в композициях и способах по настоящему изобретению, включают, не ограничиваясь только ими, антрациклины, алкилирующие агенты (например, митомицин С), алкилсульфонаты, азиридины, этиленимины, метилмеламины, азотистый иприт, нитрозомочевины, антибиотики, антиметаболиты, аналоги фолиевой кислоты (например, ингибиторы дигидрофолатредуктазы, такие как метотрексат), аналоги пурина, аналоги пиримидина, ферменты,подофиллотоксины, агенты, содержащие платину, интерфероны и интерлейкины. Конкретные примеры известных химиотерапевтических агентов, которые можно использовать в композициях и способах по настоящему изобретению, включают, не ограничиваясь только ими, бусульфан, ипросульфан, пипосульфан, бензодепу, карбохинон, метуридепу, уридепу, алтретамин, триэтиленмеламин, триэтиленфосфорамид, триэтилентиофосфорамид, триметилолмеламин, хлорамбуцил, хлорнафазин, циклофосфамид, эстрамустин, ифосфамид, мехлоретамин, гидрохлорид оксида мехлоретамина, мелфалан, новембихин, фенестерин, преднимустин, трофосфамид, урамустин, кармустин, хлорозотоцин, фотемустин, ломустин, нимустин, ранимустин, дакарбазин, манномустин, митобронитол, митолактол, пипоброман, аклациномицины, актиномицин F(l), антрамицин, азасерин, блеомицин, кактиномицин, карубицин, карзинофилин, хромомицин, дактиномицин, даунорубицин, дауномицин, 6-диазо-5-оксо-1-норлейцин, доксорубицин, эпирубицин, митомицин С, микофеноловая кислота, ногаламицин, оливомицин, пепломицин, пликамицин,порфиромицин, пуромицин, стрептонигрин, стрептозоцин, туберсидин, убенимекс, зиностатин, зорубицин, деноптерин, метотрексат, птероптерин, триметрексат, флударабин, 6-меркаптопурин, тиамиприн,тиогуанин, анцитабин, азацитидин, 6-азауридин, кармофур, цитарабин, дидезоксиуридин, доксифлуридин, эноцитабин, флоксуридин, фторурацил, тегафур, L-аспарагиназу, пульмозим, ацеглатон, альдофосфамидгликозид, аминолевулиновую кислоту, амзакрин, бестрабуцил, бисантрен, карбоплатин, цисплатин, дефофамид, демеколцин, диазихинон, элфорнитин, ацетат эллиптиния, этоглуцид, этопозид, флутамид, нитрат галлия, гидроксимочевину, -интерферон, -интерферон, -интерферон, интерлейкин 2, лентинан, лонидамин, митогуазон, митоксантрон, мопидамол, нитракрин, пентостатин, фенамет, пирарубицин, подофиллиновую кислоту, 2-этилгидразид, прокарбазин, разоксан, сизофиран, спирогерманий, паклитаксел, тамоксифен, тенипозид, тенуазоновую кислоту, триазихинон, 2,2',2"-трихлортриэтиламин, уретан, винбластин, винкристин и виндезин.- 22017392 В основном, соединения по настоящему изобретению вводят в терапевтически эффективных количествах любым известным стандартным и приемлемым способом, в отдельности или в комбинации с одним или более терапевтических агентов. Терапевтически эффективное количество изменяется в широких пределах в зависимости от тяжести заболевания, возраста и относительного состояния здоровья субъекта, эффективности используемого соединения и других факторов. В основном установлено, что удовлетворительные результаты достигаются при системном введении в суточных дозах от приблизительно 0,03 до 2,5 мг/кг массы тела. Указанная суточная дозировка для крупного млекопитающего, например человека, находится в диапазоне от приблизительно 0,5 до приблизительно 100 мг, которую обычно вводят, например, в разделенных дозах вплоть до четырех раз в сутки или в форме с замедленным высвобождением. Пригодные стандартные лекарственные формы для перорального введения включают от приблизительно 1 до 50 мг активного ингредиента. Токсичность и терапевтическую эффективность в ходе указанных курсов лечения определяют стандартными фармацевтическими методами с использованием культур клеток или экспериментальных животных, включая, не ограничиваясь только ими, определение LD50 (летальную дозу для 50% популяции) и ED50 (терапевтически эффективную дозу для 50% популяции). Терапевтический индекс определяют по соотношению дозы, оказывающей токсическое действие, к дозе, оказывающей терапевтическое действие, т.е. терапевтический индекс равен LD50/ED50. Данные, полученные при проведении анализов с использованием клеточных культур и испытаний на моделях животных, можно использовать для определения диапазона доз для введения человеку. Дозировка указанных соединений предпочтительно находится в диапазоне концентраций в кровотоке, который включает ED50 с минимальной токсичностью. Дозировку изменяют в пределах указанного диапазона в зависимости от применяемой лекарственной формы и используемого способа введения. Способы получения соединений по настоящему изобретению. Общие методики получения соединений по настоящему изобретению приведены ниже в разделе Примеры. Если в составе конечного продукта присутствуют реакционноспособные функциональные группы, например гидроксигруппы, аминогруппы, иминогруппы, тиогруппы или карбоксигруппы, то при проведении указанных реакций указанные группы защищают для предотвращения их нежелательного участия в реакциях. Стандартные защитные группы используют в соответствии с общепринятой практикой, например, см. Greene T.W. и Wuts P.G.M. в книге Protective Groups in Organic Chemistry, JohnWiley and Sons (1991). Соединение по настоящему изобретению получают в виде фармацевтически приемлемой кислотноаддитивной соли при взаимодействии соединения в форме свободного основания с фармацевтически приемлемой неорганической или органической кислотой. В другом варианте фармацевтически приемлемую основно-аддитивную соль соединения по настоящему изобретению получают при взаимодействии соединения в форме свободной кислоты с фармацевтически приемлемым неорганическим или органическим основанием. В другом варианте солевые формы соединений по настоящему изобретению получают с использованием солей исходных или промежуточных соединений. Формы свободной кислоты или свободного основания соединений по настоящему изобретению можно получить из соответствующей основно-аддитивной соли или кислотно-аддитивной соли, соответственно. Например, соединение по настоящему изобретению в форме кислотно-аддитивной соли превращают в соответствующее свободное основание при обработке пригодным основанием (например,раствором гидроксида аммония, гидроксида натрия и т.п.). Соединение по настоящему изобретению в форме основно-аддитивной соли превращают в соответствующую свободную кислоту при обработке пригодной кислотой (например, соляной кислотой и т.д.). Соединения по настоящему изобретению в неокисленной форме можно получить из N-оксидов соединений по настоящему изобретению при обработке восстанавливающим агентом (например, серой,диоксидом серы, трифенилфосфином, боргидридом лития, боргидридом натрия, трихлоридом фосфора,трибромидом фосфора или т.п.) в пригодном инертном органическом растворителе (например, ацетонитриле, этаноле, водном растворе диоксана или т.п.) при температуре от 0 до 80C. Пролекарства соединений по настоящему изобретению получают известными способами (например, более подробная информация приведена в статье Saulnier и др., Bioorganic and Medicinal ChemistryLetters, т. 4, с. 1985 (1994. Например, соответствующие пролекарства получают при взаимодействии немодифицированного соединения по настоящему изобретению с пригодным карбамилирующим агентом (например, 1,1-ацилоксиалкилкарбанохлоридатом, паранитрофенилкарбонатом или т.п.). Защищенные производные соединений по настоящему изобретению получают известными способами. Подробное описание методик, используемых для введения защитных групп и их удаления, приведено в книге Greene T.W. Protecting Groups in Organic Chemistry, 3 е изд., John Wiley and Sons, Inc.(1999). Соединения по настоящему изобретению получают стандартным способом по настоящему изобретению или способом по изобретению в виде сольватов (например, гидратов). Гидраты соединений по настоящему изобретению получают стандартным способом при перекристаллизации из водно- 23017392 органических смесей растворителей с использованием органических растворителей, таких как диоксан,тетрагидрофуран или метанол. Соединения по настоящему изобретению можно получить в виде их индивидуальных стереоизомеров при взаимодействии рацемической смеси соединения с оптически активным разделяющим агентом,при этом получают пару диастереоизомерных соединений, разделяют диастереомеры и выделяют оптически чистые энантиомеры. Разделение энантиомеров проводят с использованием ковалентных диастереомерных производных соединений по настоящему изобретению, или с использованием диссоциирующих комплексов (например, кристаллических диастереомерных солей). Диастереомеры характеризуются различными физическими свойствами (например, температурами плавления, температурами кипения,растворимостью, реакционной способностью и т.п.), и их разделяют на основе указанных различий. Диастереомеры разделяют с использованием хроматографии или методик разделения, основанных на различиях в растворимости. Затем из смеси выделяют оптически чистый энантиомер и разделяющий агент любыми известными способами, которые не приводят к рацемизации. Более подробное описание методик, используемых для разделения стереоизомеров соединений из их рацемической смеси, приведено в книге Jacques J., Collet A., Samuel H.W. Enantiomers, Racemates and Resolutions, John Wiley And Sons,Inc. (1981). В основном, соединения формулы (1), (2 А), (2 В) или (2 С) получают способом, который включает следующие стадии:(a) общие методики, как описано ниже в разделе Примеры;(b) необязательное превращение соединения по настоящему изобретению в его фармацевтически приемлемую соль;(c) необязательное превращение солевой формы соединения по настоящему изобретению в несолевую форму;(d) необязательное превращение неокисленной формы соединения по настоящему изобретению в фармацевтически приемлемый N-оксид;(e) необязательное превращение соединения по настоящему изобретению в форме N-оксида в его неокисленную форму;(f) необязательное выделение индивидуального изомера соединения по настоящему изобретению из смеси изомеров;(g) необязательное превращение немодифицированного соединения по настоящему изобретению в фармацевтически приемлемое пролекарство и(h) необязательное превращение пролекарства соединения по настоящему изобретению в его немодифицированную форму. Получение исходных соединений подробно не описано, поскольку соединения являются известными, или их можно получить по известным методикам, или как описано ниже в примерах. Специалисту в данной области представляется очевидным, что приведенные выше превращения представлены только в качестве типичных примеров способов получения соединений по настоящему изобретению и что можно использовать другие известные способы. Следующие примеры представлены только для иллюстрации настоящего изобретения и не ограничивают его объем. Получение промежуточных соединений Получение 5-[5-(4-метоксифенил)пиримидин-2-иламино]пиридин-2-карбоновой кислоты 5. 5-Аминопиридин-2-карбоновую кислоту 1 (5,4 ммоль) растворяли в смеси бензол/МеОН (3:1, 40 мл) и охлаждали на ледяной бане до 0C. В раствор медленно добавляли 2 М раствор TMSCH2N2 в Et2O(6,5 ммоль). После завершения добавления реакционную смесь нагревали до КТ и перемешивали в течение 6 ч. После удаления растворителя в вакууме получали метиловый эфир 5-аминопиколиновой кислоты 2, который использовали на следующей стадии без дополнительной очистки. МС m/z: 153,1 (М+1)+. В флакон помещали метиловый эфир 5-аминопиколиновой кислоты 2 (0,83 ммоль), 2-хлор-5-(4 метоксифенил)пиримидин 3 (0,83 ммоль), Pd(OAc)2 (0,124 ммоль), реагент xanthphos (0,124 ммоль),- 24017392Cs2CO3 (0,83 ммоль) и безводный 1,4-диоксан (5 мл). Флакон дважды вакуумировали и заполняли азотом,смесь нагревали в микроволновом реакторе при 130C в течение 20 мин. Флакон охлаждали до КТ и реакционную смесь разбавляли ДХМ, промывали 10% раствором NH4Cl, солевым раствором и сушили надNa2SO4. Растворитель удаляли в вакууме и неочищенный остаток растирали в EtOAc, при этом получали метиловый эфир 5-[5-(4-метоксифенил)пиримидин-2-иламино]пиридин-2-карбоновой кислоты 4 в виде твердого вещества светло-коричневого цвета. 1H ЯМР (400 МГц, d6-ДМСО):10,62 (s, 1H), 8,93 (s, 2 Н), 8,83 (ушир. s, 1 Н), 8,46 (d, J=8,0 Гц, 1H,8,27 (d, J=8,0 Гц, 1H), 7,72-7,70 (m, 2 Н), 7,08-7,06 (m, 2 Н), 3,86 (s, 3H), 3,81 (s, 3H). МС m/z: 337,1 (М+1)+. В суспензию метилового эфира 5-[5-(4-метоксифенил)пиримидин-2-иламино]пиридин-2 карбоновой кислоты 4 (0,3 ммоль) в смеси ТГФ/MeOH/H2O (3:2:1, 5 мл) добавляли 6 М LiOH (1,8 ммоль). Реакционную смесь перемешивали при КТ в течение 12 ч. Затем растворитель удаляли в вакууме, остаток разбавляли H2O (6 мл) и рН доводили до нейтральных значений добавлением 6 М HCl. Твердое вещество, которое выпадало в осадок, собирали, промывали водой и сушили в вакуум-сушильном шкафу в течение 12 ч, при этом получали 5-[5-(4-метоксифенил)пиримидин-2-иламино]пиридин-2-карбоновую кислоту 5 в виде твердого вещества желтого цвета, которую использовали на следующей стадии без дополнительной очистки. МС m/z: 323,2 (М+1)+. 5-[5-(4-Метоксифенил)пиримидин-2-иламино]-2-метилникотиновая кислота 10. В раствор этилового эфира 2-метил-5-нитроникотиновой кислоты 7 (1,43 ммоль) в MeOH (15 мл) добавляли Pd (5% на угле, влажность 50%, 10 мас.%). После продувки реакционную смесь перемешивали в атмосфере водорода в течение 3 ч. Растворитель фильтровали через целит и осадок на целите промывали MeOH. Растворитель удаляли в вакууме, при этом получали этиловый эфир 5-амино-2 метилникотиновой кислоты 8 в виде твердого вещества желтого цвета, которое использовали без дополнительной очистки. 1 Н ЯМР (400 МГц, d6-ДМСО)7,19 (d, J=4,0 Гц, 1 Н), 6,78 (d, J=4,0 Гц, 2 Н), 4,10 (s, 2 Н), 3,56 (q,J=8,0 Гц, 2 Н), 1,80 (s, 3H), 0,59 (t, J=8,0 Гц, 3H). МС m/z: 182,0 (M+1)+. В флакон помещали этиловый эфир 5-амино-2-метилникотиновой кислоты 8 (1,25 ммоль), 2-хлор-5(4-метоксифенил)пиримидин 3 (1,25 ммоль), Pd(OAc)2 (0,187 ммоль), реагент xanthphos (0,187 ммоль),Cs2CO3 (1,25 ммоль) и безводный 1,4-диоксан (8 мл). Флакон вакуумировали и заполняли азотом (указанные процедуры повторяли два раза), реакционную смесь нагревали на масляной бане при 100C в течение 2 ч. Флакон охлаждали до КТ и реакционную смесь разбавляли ДХМ, промывали 10% растворомNH4Cl, солевым раствором и сушили над Na2SO4. Растворитель удаляли в вакууме и неочищенный остаток очищали хроматографией на коротком слое силикагеля (элюент: ДХМ/MeCN/MeOH, 8:2,5:0,5), при этом получали этиловый эфир 5-[5-(4-метоксифенил)пиримидин-2-иламино]-2-метилникотиновой кислоты 9 в виде твердого вещества светло-желтого цвета. 1 Н ЯМР (400 МГц, d6-ДМСО):10,13 (s, 1 Н), 9,10 (d, J=4,0 Гц, 1H), 8,94 (s, 2H), 8,78 (d, J=4,0 Гц,1H), 7,79-7,77 (m, 2H), 7,16-7,14 (m, 2 Н), 4,45 (q, J=8,0 Гц, 2 Н), 3,90 (s, 3H), 2,75 (s, 3H), 1,45 (t, J=8,0 Гц,3H).MC m/z: 365,1 (М+1)+. В суспензию этилового эфира 5-[5-(4-метоксифенил)пиримидин-2-иламино]-2-метилникотиновой кислоты 9 (0,9 ммоль) в смеси ТГФ/MeOH/H2O, 3:2:1 (7 мл) добавляли 6 М LiOH (2,7 ммоль). Реакционную смесь перемешивали при КТ в течение 12 ч. Затем растворитель удаляли в вакууме, остаток разбавляли H2O (6 мл) и рН доводили до нейтральных значений добавлением 6 М HCl. Полученное твердое вещество отделяли фильтрованием, промывали водой и сушили в вакуум-сушильном шкафу в течение 12 ч,при этом получали 5-[5-(4-метоксифенил)пиримидин-2-иламино]-2-метилникотиновую кислоту 10 в виде твердого вещества грязно-белого цвета, которую использовали на следующей стадии без дополнительной очистки. МС m/z: 337,2 (М+1)+. В суспензию 5-[5-(4-метоксифенил)пиримидин-2-иламино]-2-метилникотиновой кислоты 10 (0,59 ммоль) в безводном трет-BuOH (15 мл) добавляли ТЭА (8,3 ммоль). Реакционную смесь перемешивали при КТ в течение 30 мин, затем по каплям добавляли DPPA (8,3 ммоль). Затем смесь в колбе кипятили с обратным холодильником в течение 4 ч. Растворитель удаляли при пониженном давлении и вязкий маслообразный остаток очищали препаративной ЖХВР (элюент: градиент MeCN 20-90%), при этом получали трет-бутиловый эфир 5-[5-(4-метоксифенил)пиримидин-2-иламино]-2-метилпиридин-3-илкарбаминовой кислоты 11. 1 Н ЯМР (400 МГц, d6-ДМСО):10,22 (ушир. s, 1H), 9,17 (ушир. s, 1H), 8,87 (s, 2H), 8,50 (ушир. s,1H), 7,70-7,68 (m, 2H), 7,07-7,05 (m, 2 Н), 3,81 (s, 3H), 2,46 (s, 3H), 1,58 (s, 9H). МС m/z: 408,1 (М+1)+. Трет-бутиловый эфир 5-[5-(4-метоксифенил)пиримидин-2-иламино]-2-метилпиридин-3-илкарбаминовой кислоты 11 (0,33 ммоль) растворяли в ДХМ (2 мл) и обрабатывали ТФУ (3 мл). Реакционную смесь перемешивали при КТ в течение 2 ч. Растворитель удаляли, при этом получали твердый остаток грязно-белого цвета. Остаток переносили в воду, рН доводили до нейтральных значений добавлением 5% Na2CO3 и экстрагировали смесью ДХМ/IPA, 3:1 (340 мл). Органический слой промывали солевым раствором и сушили над Na2SO4. После упаривания органического растворителя при пониженном давлении получали N5-[5-(4-метоксифенил)пиримидин-2-ил]-2-метилпиридин-3,5-диамин 12, который использовали без дополнительной очистки. 1 Н ЯМР (400 МГц, d6-ДМСО):10,37 (s, 1H), 8,90 (s, 2H), 8,47 (ушир. s, 1H), 7,79 (ушир. s, 1H),7,71-7,69 (m, 2 Н), 7,08-7,06 (m, 2 Н), 6,36 (ушир. s, 2H), 3,81 (s, 3H), 2,43 (s, 3H). МС m/z: 308,2 (М+1)+. 2-Хлор-5-(4-(дифторметокси)фенил)пиримидин 17. Бром-4-(дифторметокси)бензол 13 (10 ммоль), ацетат калия (30,0 ммоль), 4,4,5,5-тетраметил-2(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1,3,2-диоксаборолан 14 (11,0 ммоль) и Pd(PPh3)4 (0,5 ммоль) добавляли в колбу Шленка объемом 40 мл, снабженную перемешивающим стержнем. Колбу вакуумировали и заполняли азотом (указанные процедуры повторяли несколько раз). 1,4-Диоксан (10 мл) добавляли шприцом. Колбу Шленка закрывали и нагревали при 150C в течение 20 мин в микроволновом реакторе. После завершения реакции растворитель удаляли в вакууме. Остаток растворяли в ДХМ (200 мл) и промывали водой. Органическую фазу сушили над безводным Na2SO4, фильтровали и концентрировали, при этом получали неочищенный продукт. После очистки колоночной хроматографией на силикагеле (элюент: EtOAc, гексан, градиент от 0 до 20%) получали 2-(4-(дифторметокси)фенил)-4,4,5,5 тетраметил-1,3,2-диоксаборолан 15. 1(s, 12H). МС m/z: 271,1 (M+1)+. В раствор 5-бром-2-хлорпиримидина 16 (7,7 ммоль) в 1,4-диоксане (1,5 мл) добавляли 2-(4(дифторметокси)фенил)-4,4,5,5-тетраметил-1,3,2-диоксаборолан 15 (8,9 ммоль), 1,8 М K2CO3 (16,2 ммоль) и Pd(PPh3)4 (0,38 ммоль). Реакционную смесь вакуумировали и колбу заполняли азотом (процедуры повторяли два раза), затем нагревали при 150C в течение 10 мин в микроволновом реакторе. Реакционную смесь разбавляли насыщенным раствором NH4Cl и экстрагировали ДХМ (350 мл). Органический слой промывали солевым раствором, сушили над Na2SO4 и концентрировали. После очистки колоночной хроматографией на силикагеле (элюент: гексан/EtOAc, 3:1) получали 2-хлор-5-(4-(дифторметокси)фенил) пиримидин 17. 1 Н ЯМР (400 МГц, CDCl3):8,73 (s, 2 Н), 7,47-7,52 (m, 2 Н), 7,20-7,24 (m, 2 Н), 6,52 (t, J=72 Гц, 1H). МС m/z: 257,0 (M+1)+.- 26017392 Получение конечных соединений Соединения типа А. В раствор 5-[5-(4-метоксифенил)пиримидин-2-иламино]пиридин-2-карбоновой кислоты 5 (0,12 ммоль) в ДМФА (0,5 мл) добавляли HATU (0,13 ммоль) и DIEA (0,36 ммоль) при КТ. Полученную смесь перемешивали в течение 2 мин и затем добавляли раствор трет-бутилового эфира пиперазин-1 карбоновой кислоты (0,12 ммоль) в ДМФА (1 мл). Реакционную смесь перемешивали при КТ в течение 4 ч и напрямую очищали препаративной ЖХ-МС, при этом получали трет-бутиловый эфир 4-5-[5-(4 метоксифенил)пиримидин-2-иламино]пиридин-2-карбонилпиперазин-1-карбоновой кислоты 6 в виде твердого вещества белого цвета. 1 Н ЯМР (400 МГц, d6-ДМСО):10,32 (s, 1H), 8,89 (s, 2H), 8,39-8,37 (m, 2H), 7,86 (dd, J=4,0 и 8,0 Гц,1 Н), 7,71-7,69 (m, 2 Н), 7,08-7,06 (m, 2 Н), 3,81 (s, 3H), 3,55-3,51 (ушир. m, 4 Н), 3,96-3,94 (ушир. s, 4H), 1,42(s, 9H). МС m/z: 491,1 (М+1)+. Трет-бутиловый эфир 4-5-[5-(4-метоксифенил)пиримидин-2-иламино]пиридин-2-карбонилпиперазин-1-карбоновой кислоты 6 (0,03 ммоль) растворяли в ДХМ (2 мл) и обрабатывали ТФУ (2 мл). Реакционную смесь перемешивали при КТ в течение 2 ч. Растворитель удаляли, при этом получали твердый остаток грязно-белого цвета, который промывали несколько раз EtOAc, при этом получали требуемый 5[5-(4-метоксифенил)пиримидин-2-иламино]пиридин-2-илпиперазин-1-илметанон А 1 в виде твердого вещества белого цвета. 1 Н ЯМР (400 МГц, d6-ДМСО):10,30 (s, 1H), 9,13 (ушир. s, 2H), 8,98 (d, J=4,0 Гц, 2 Н), 8,88 (s, 1 Н),8,43 (dd, J=4,0 и 8,0 Гц, 1 Н), 7,73 (d, J=8,0 Гц, 1H), 7,71-7,69 (m, 2 Н), 7,08-7,06 (m, 2 Н), 3,81 (s, 3H), 3,73 В раствор 5-[5-(4-метоксифенил)пиримидин-2-иламино]пиридин-2-карбоновой кислоты 5 (0,05 ммоль) в ДМФА (1 мл) добавляли HATU (0,06 ммоль) и DIEA (0,14 ммоль) при КТ. Полученную смесь перемешивали в течение 2 мин и затем добавляли пиперазин-2-он (0,05 ммоль). Реакционную смесь перемешивали при КТ в течение 12 ч и напрямую очищали препаративной ЖХ-МС, при этом получали 45-[5-(4-метоксифенил)пиримидин-2-иламино]пиридин-2-карбонилпиперазин-2-он А 2 в виде твердого вещества белого цвета. 1 Указанное в заголовке соединение получали аналогично тому, как описано для соединения А 2. Н ЯМР (400 МГц, d4-CH3OH):9,00 (d, J=4,0 Гц, 1 Н), 8,77 (s, 2H), 8,52 (dd, J=4,0 и 8,0 Гц, 1H), 7,68 В раствор 5-[5-(4-метоксифенил)пиримидин-2-иламино]-2-метилникотиновой кислоты 10 (0,05 ммоль) в ДМФА (1 мл) добавляли HATU (0,06 ммоль) и DIEA (0,14 ммоль) при КТ. Полученную смесь перемешивали в течение 2 мин, затем добавляли 1-метансульфонилпиперазин (0,05 ммоль). Реакционную смесь перемешивали при КТ в течение 4 ч и затем напрямую очищали препаративной ЖХ-МС, при этом получали (4-метансульфонилпиперазин-1-ил)5-[5-(4-метоксифенил)пиримидин-2-иламино]-2 метилпиридин-3-илметанон В 1. 1 Н ЯМР (400 МГц, d4-CH3OH):9,30 (d, J=4,0 Гц, 1H), 8,81 (s, 2H), 8,50 (d, J=4,0 Гц, 1H), 7,60-7,58 Указанное в заголовке соединение получали аналогично тому, как описано для соединения В 1. Н ЯМР (400 МГц, d4-CH3OH):9,44 (ушир. s, 1H), 8,84 (s, 2 Н), 8,58 (ушир. s, 1 Н), 7,60-7,58 (m, 2 Н),7,08-7,05 (m, 2 Н), 3,85 (s, 3H), 3,86 (t, J=4,0 Гц, 2 Н), 3,38 (t, J=4,0 Гц, 2 Н), 2,66 (s, 3H), 2,06-2,00 (m, 4 Н). МС m/z: 490,1 (М+1)+. В флакон помещали 5-аминоникотиновую кислоту 18 (1,45 ммоль), 2-хлор-5-(4 дифторметоксифенил)пиримидин 17 (1,45 ммоль), Pd(OAc)2 (0,22 ммоль), реагент xanthphos (0,22 ммоль),Cs2CO3 (1,45 ммоль) и безводный 1,4-диоксан (5 мл). Флакон дважды вакуумировали и заполняли азотом,смесь нагревали на масляной бане при 100C в течение 3 ч. Флакон охлаждали до КТ, реакционную смесь разбавляли водой (20 мл) и рН доводили до нейтральных значений добавлением 10% Na2CO3. Полученную суспензию экстрагировали смесью ДХМ/IPA (3:1, 350 мл). Органический слой промывали солевым раствором и сушили над Na2SO4. Растворитель удаляли в вакууме и неочищенный продукт оранжевого цвета очищали препаративной ЖХВР (элюент: градиент MeCN от 20 до 70%), при этом получали 5-(5-(4-(дифторметокси)фенил)пиримидин-2-иламино)никотиновую кислоту 19. МС m/z: 359,1 (М+1)+. В раствор 5-(5-(4-(дифторметокси)фенил)пиримидин-2-иламино)никотиновой кислоты 19 (0,028 ммоль) в ДМФА (1 мл) добавляли HATU (0,033 ммоль) и DIEA (0,084 ммоль) при КТ. Полученную смесь перемешивали в течение 2 мин, затем добавляли N-boc-пиперазин (0,03 ммоль). Реакционную смесь перемешивали при КТ в течение 12 ч и напрямую очищали препаративной ЖХ-МС, при этом получали трет-бутиловый эфир 4-(5-(5-(4-(дифторметокси)фенил)пиримидин-2-иламино)никотиноил)пиперазин-1 карбоновой кислоты. Остаток обрабатывали ТФУ (1 мл) при КТ в течение 1 ч, концентрировали в вакууме и напрямую очищали препаративной ЖХ-МС, при этом получали (5-(5-(4-(дифторметокси)фенил) пиримидин-2-иламино)пиридин-3-ил)(пиперазин-1-ил)метанон В 3. 1 Н ЯМР (400 МГц, d3-CH3CN):9,32 (ушир. s, 1H), 8,86 (s, 2 Н), 8,67 (ушир. s, 1 Н), 8,41 (ушир. s,1H), 7,75 (d, J=12,0 Гц, 2 Н), 7,33 (d, J=12,0 Гц, 2 Н), 6,87 (t, J=73 Гц, 1H), 4,20 (ушир. s, 4H), 3,30 (ушир. s,4H). МС m/z: 427,1 (М+1)+. Соединения типа С. В раствор 1-трет-бутилового эфира пиперидин-1,2-дикарбоновой кислоты (0,04 ммоль) в ДМФА (1 мл) добавляли HATU (0,05 ммоль) и DIEA (20,12 ммоль). Полученную смесь перемешивали при КТ в течение 2 мин и затем добавляли N5-[5-(4-метоксифенил)пиримидин-2-ил]-2-метилпиридин-3,5-диамин
МПК / Метки
МПК: C07D 409/14, C07D 401/14, A61K 31/506, C07D 405/14, A61P 35/00, C07D 401/12
Метки: качестве, производные, ингибиторов, 2-гетероариламинопиримидина, киназ
Код ссылки
<a href="https://eas.patents.su/30-17392-proizvodnye-2-geteroarilaminopirimidina-v-kachestve-ingibitorov-kinaz.html" rel="bookmark" title="База патентов Евразийского Союза">Производные 2-гетероариламинопиримидина в качестве ингибиторов киназ</a>
Производные 2-бифениламино-4-аминопиримидина в качестве ингибиторов киназ

Номер патента: 17252
Опубликовано: 30.11.2012
Авторы: Бурсалая Бадри, Чэнь Бэй, Грей Натанаэл С., Хе Сяосюй, Марсилдже Томас Х., Ли Кристиан Чо-Хуа, Лю Вэньшу
МПК: A61K 31/506, A61P 31/00, A61P 35/00...
Метки: киназ, производные, 2-бифениламино-4-аминопиримидина, ингибиторов, качестве
Формула / Реферат:
1. Соединение формулы (1)или его фармацевтически приемлемая соль, где R1a обозначает галоген, С1-С6алкил или галогензамещенный С1-С6алкил;R1b обозначает Н;R2 обозначает С6-С10карбоциклическое или 5-10-членное гетероарильное или 5-7-членное гетероциклическое кольцо, каждое из которых содержит от 1 до 3 гетероатомов, выбранных из N, О и S, каждый из которых необязательно замещен 1-2 С1-С6алкилом, -L-Y, -L-C(O)O0-1-(CR2)q-R8 или -L-C(O)-NRR8;R3...
Конденсированные производные тиазола в качестве ингибиторов киназ

Номер патента: 17187
Опубликовано: 30.10.2012
Авторы: Франклин Ричард Джереми, Крепи Карен-Вивьян-Люсиль, Александер Рикки Питер, Ауджла Павандип Сингх, Фоли Анне Мари
МПК: A61K 31/519, A61K 31/437, C07D 513/04...
Метки: ингибиторов, тиазола, качестве, конденсированные, производные, киназ
Формула / Реферат:
1. Соединение формулы (I) или его фармацевтически приемлемая соль или сольватгде R11 обозначает С1-С6алкил,R12 обозначает С1-С6алкил,Т обозначает кислород или N-R25,R23 обозначает водород, цианогруппу, карбоксигруппу, С2-С6алкоксикарбонил, ди(C1-C6)алкиламинокарбонил, (С1-С6алкил)(циано(C1-C6)алкил)аминокарбонил, (С1-С6алкокси(С1-C6)алкил)(C1-С6алкил)аминокарбонил или азетидинилкарбонил иR25 обозначает С1-С6алкил.2. Соединение по п.1, где Т...
Производные пиразолохиназолина: способ получения и применение в качестве ингибиторов киназ

Номер патента: 10904
Опубликовано: 30.12.2008
Авторы: Фергюсон Рон, Получчи Паоло, Ролетто Фульвия, Фанчелли Даниеле, Певарелло Паоло, Вианелло Паола, Квартьери Франческа, Тракванди Габриелла, Браска Мария Габриелла, Д`алессио Роберто, Панцери Акилле, Вульпетти Анна
МПК: A61P 35/00, A61K 31/519, C07D 487/04...
Метки: качестве, пиразолохиназолина, киназ, производные, ингибиторов, применение, получения, способ
Формула / Реферат:
1. Производное пиразолохиназолина, представленное формулой (Ia) или (Ib) или его фармацевтически приемлемая соль, где R обозначает водород или группу, выбранную из амино, линейного или разветвленного C1-C6алкила, C3-C10циклоалкила, арила, арилC1-C6алкила, гетероциклила или гетероциклилC1-C6алкила; X обозначает простую связь или двухвалентный радикал, выбранный из -NR'-, -CONR'-, -NH-CO-NH-, -О- или -S-, где R' обозначает водород или группу,...
Пиридил- и пиримидинилзамещенные производные пиррола, тиофена и фурана в качестве ингибиторов киназ

Номер патента: 15126
Опубликовано: 30.06.2011
Авторы: Ванотти Эрмес, Чирла Алессандра, Форте Барбара, Пиллан Антонио, Сколаро Алессандра, Эрмоли Антонелла, Меничинчери Мария, Кальдарелли Марина
МПК: A61K 31/435, A61K 31/495, A61P 35/00...
Метки: пиримидинилзамещенные, киназ, пиридил, производные, ингибиторов, фурана, качестве, пиррола, тиофена
Формула / Реферат:
1. Соединение формулы (I)где G представляет собой СН или атом азота;W представляет собой атом кислорода, NR1или S(O)n;где n равно 0;R1 представляет собой атом водорода или C1-С6-алкил;R2 представляет собойатом водорода;атом галогена;арильную группу, где арильная группа представляет собой одновалентную ароматическую карбоциклическую группу с 6-14 атомами углерода, имеющую одно кольцо или несколько конденсированных колец, где конденсированные...
Производные аминоиндазола, активные в качестве ингибиторов киназ, способ их получения и содержащая их фармацевтическая композиция

Номер патента: 7430
Опубликовано: 27.10.2006
Авторы: Салом Барбара, Мартина Катия, Вульпетти Анна, Амичи Раффаэлла, Д'анелло Маттео
МПК: A61P 35/00, A61K 31/416, C07D 231/56...
Метки: качестве, способ, активные, содержащая, ингибиторов, производные, композиция, аминоиндазола, киназ, фармацевтическая, получения
Формула / Реферат:
1. Способ лечения заболеваний, обусловленных и/или связанных с измененной (нарушенной) активностью протеинкиназ, включающий введение нуждающемуся в этом млекопитающему эффективного количества производного аминоиндазола, представленного формулой (I) где R выбирают из группы, состоящей из -NHCOR', -NHCONHR', -NHCONR'R", -NHSO2R' или -NHCOOR', где R' и R" каждый независимо выбран из прямого или разветвленного C1-С6алкила, С2-С6 алкенила или...
Предыдущий патент: Способ получения минеральных волокон
Следующий патент: Производные тиадиазинона
Случайный патент: Сельскохозяйственный пресс-подборщик