Соединения и композиции в качестве ингибиторов протеазы, активирующей каналы
Номер патента: 16327
Опубликовано: 30.04.2012
Авторы: Ван Чживэй, Чаттерджи Арнаб К., Талли Дейвид С.





















Формула / Реферат
1. Соединение формулы (1)

или его фармацевтически приемлемые соли, где
В представляет собой
или (CR2)kR5;
Y представляет собой -SO2- или -О-С(=О)-;
J представляет собой OH, OCH3, NH-CH(фенил)2, NH(C0-C6-алкилен)-О(фенил), NH(C0-C6-алкилен)-SO2(морфолинил) или NH-(CR2)lR6, где
R6 представляет собой C1-6алкил, фенил, не замещенный или замещенный группами C1-6алкокси, SO2NR2, галогеном, фенилом, бензилокси, CONH(CR2)2(фенил) или CONR8R9; пиридил, циклопропил, циклогексил, бензотиазолил, 2,3-дигидро-1Н-инденил, морфолинил, имидазолил, тетрагидропиранил, пиперидинил, тиофенил, 2,3-дигидробензо[b][1,4]диоксинил или бензотиофенил;
R1 представляет собой -(CH2)m-NH2, -(CH2)m-NHC(=NH)-NH2 или -(CH2)m-X, где
X представляет собой пиперидинил, C3-7циклоалкил или фенил;
R2 является заместителем в любом положении кольца А и представляет собой -O-(CR2)p-R7 или -OC(O)-NR8R9;
R3 представляет собой C1-6алкил или -(CR2)l-R7;
R4 представляет собой С2-6алкенил или -CR=CR-R6, где
R6 представляет собой C1-6алкил или фенил, замещенный C1-6алкоксигруппой;
или R4 представляет собой фенил, пиперидинил или
, где
кольцо Е представляет собой циклогексил, необязательно замещенный NR2, или
кольцо Е представляет собой пиперидинил;
R5 представляет собой гетероарил, замещенный C1-6алкилом, NR8R9 или обеими группами;
R7 представляет собой 5-7-членное карбоциклическое кольцо или фенил, необязательно замещенный галогеном;
R8 и R9 вместе с атомом N могут образовывать 5-7-членное гетероциклическое кольцо;
каждый R представляет собой Н или C1-6алкил;
l имеет значение 0-4;
m имеет значение 1-4;
n имеет значение 1 или 2;
k и p равны 1.
2. Соединение по п.1, где R2 представляет собой -O-(CH2)-R7 и R7 представляет собой галогензамещенный фенил.
3. Соединение по п.1, где указанное соединение имеет формулу (2)

или его фармацевтически приемлемые соли, где
R8 и R9 вместе образуют пиперидинил.
4. Соединение по п.1, где указанное соединение имеет формулу (3)

или его фармацевтически приемлемые соли, где
q равно 1 и
R10 представляет собой галоген.
5. Соединение по п.4, где R1 представляет собой -(CH2)m-NH2, -(CH2)m-NHC(=NH)-NH2 или (CH2)m-X, где X представляет собой пиперидинил.
6. Соединение по п.4, где R3 представляет собой C1-6алкил, циклогексил или бензил.
7. Соединение по п.4, где R4 представляет собой пиперидинил.
8. Соединение по п.4, где R1 представляет собой (CR2)m-X, где X представляет собой С3-7циклоалкил или фенил; R4 представляет собой пиперидинил; Y представляет собой SO2.
9. Соединение по п.1, где указанное соединение имеет формулу (4)

или его фармацевтически приемлемые соли, где
q равно 1 и
R10 представляет собой галоген.
10. Соединение по п.9, где R5 представляет собой тиазолил, необязательно замещенный NR8R9.
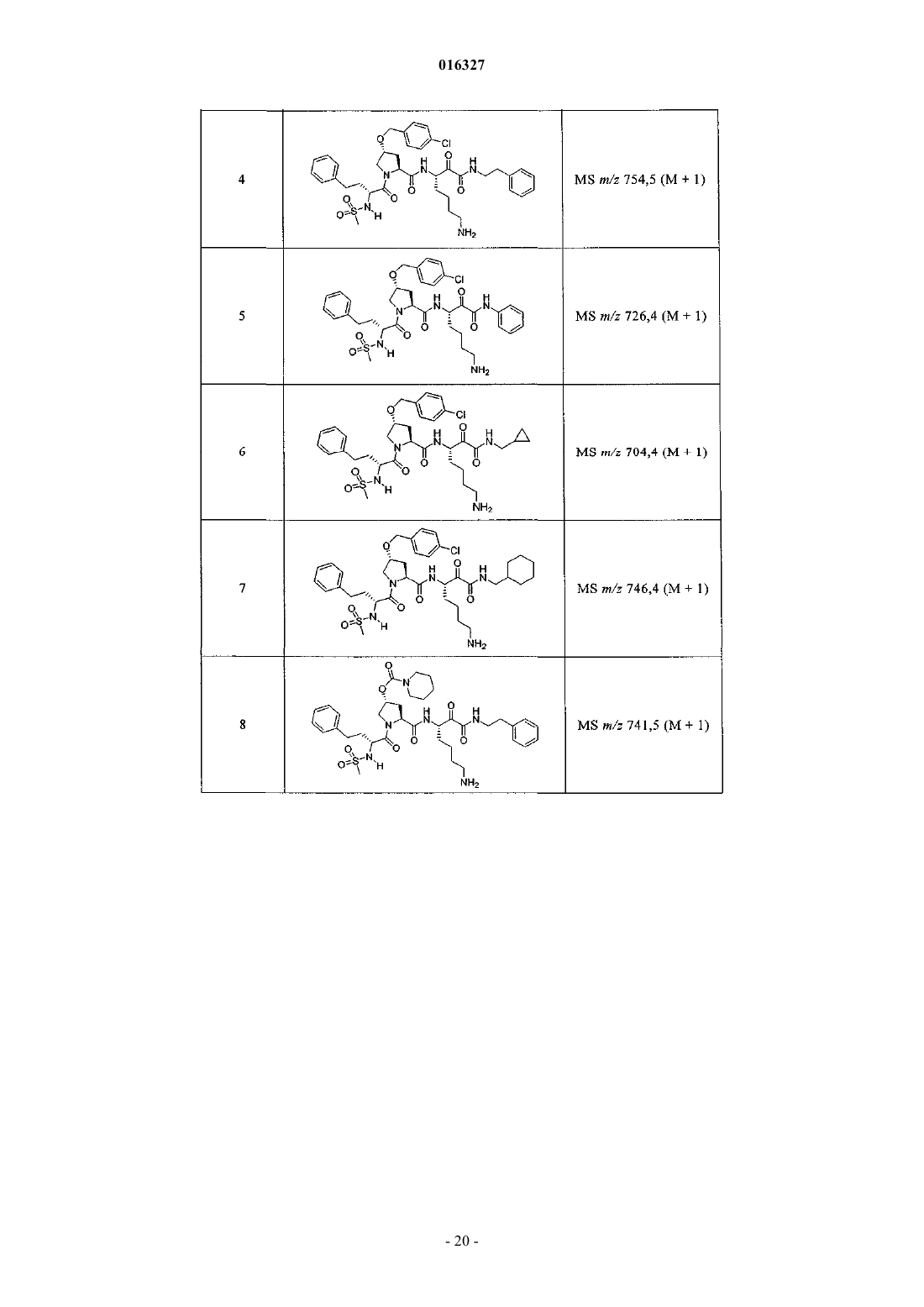
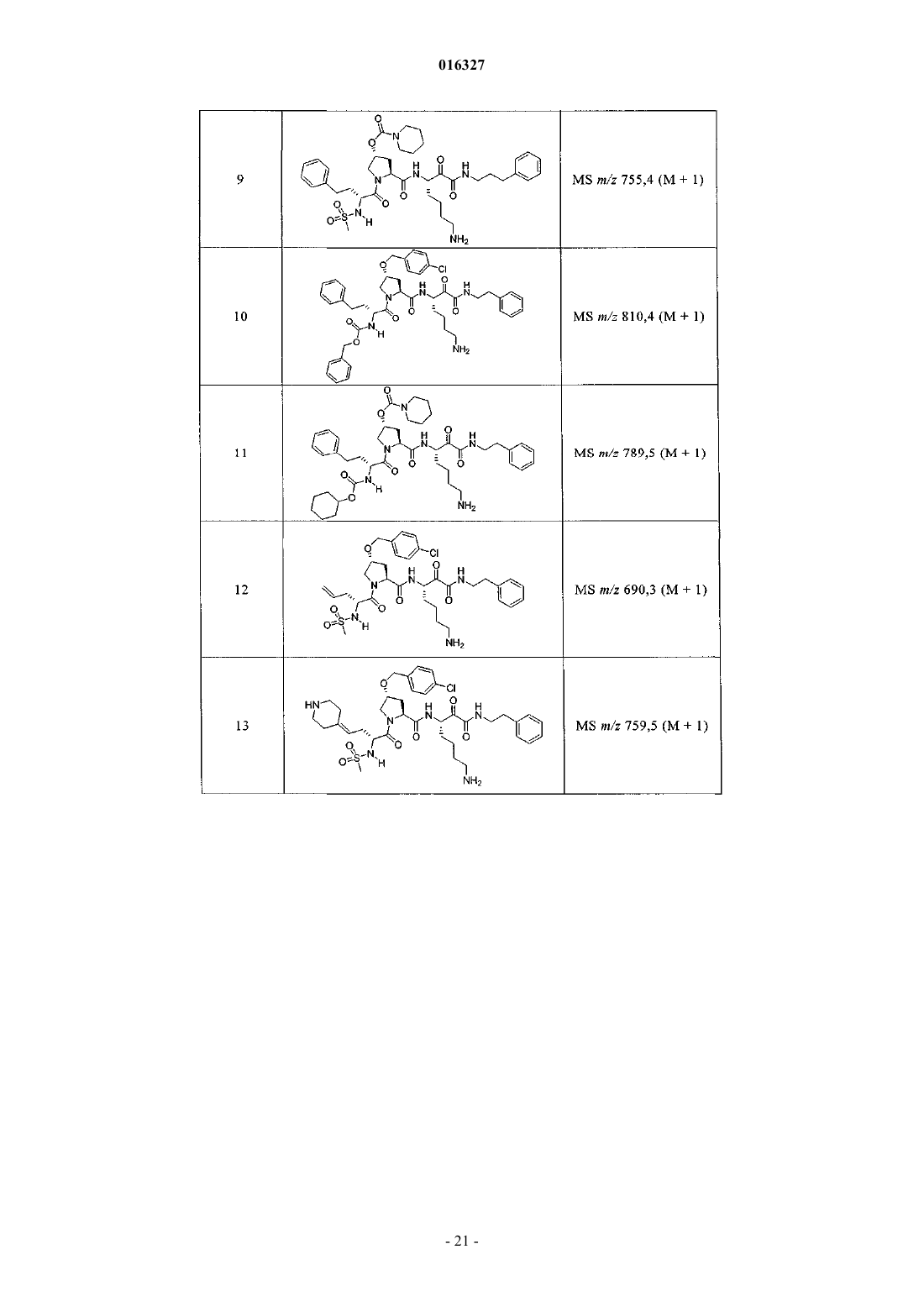
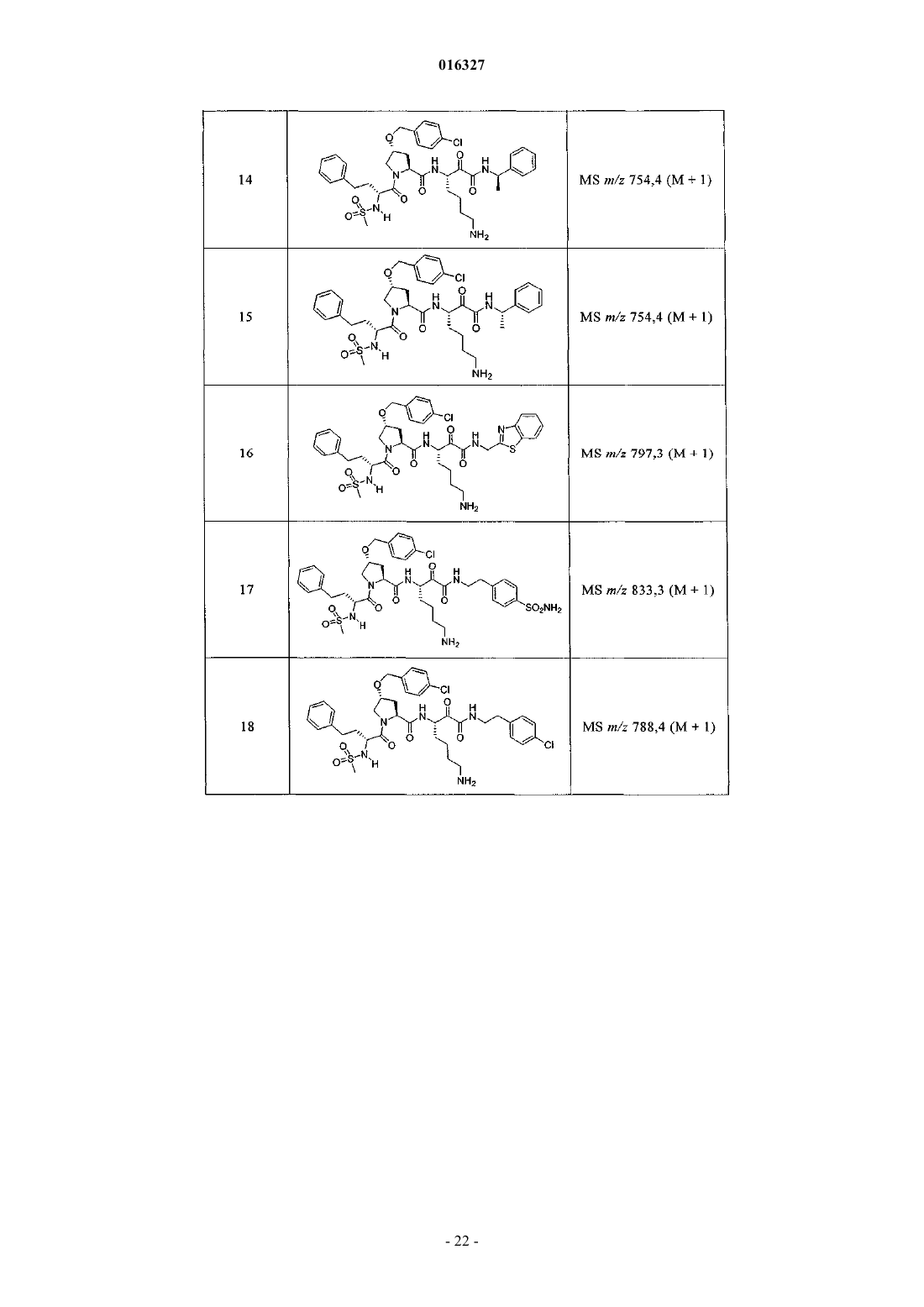
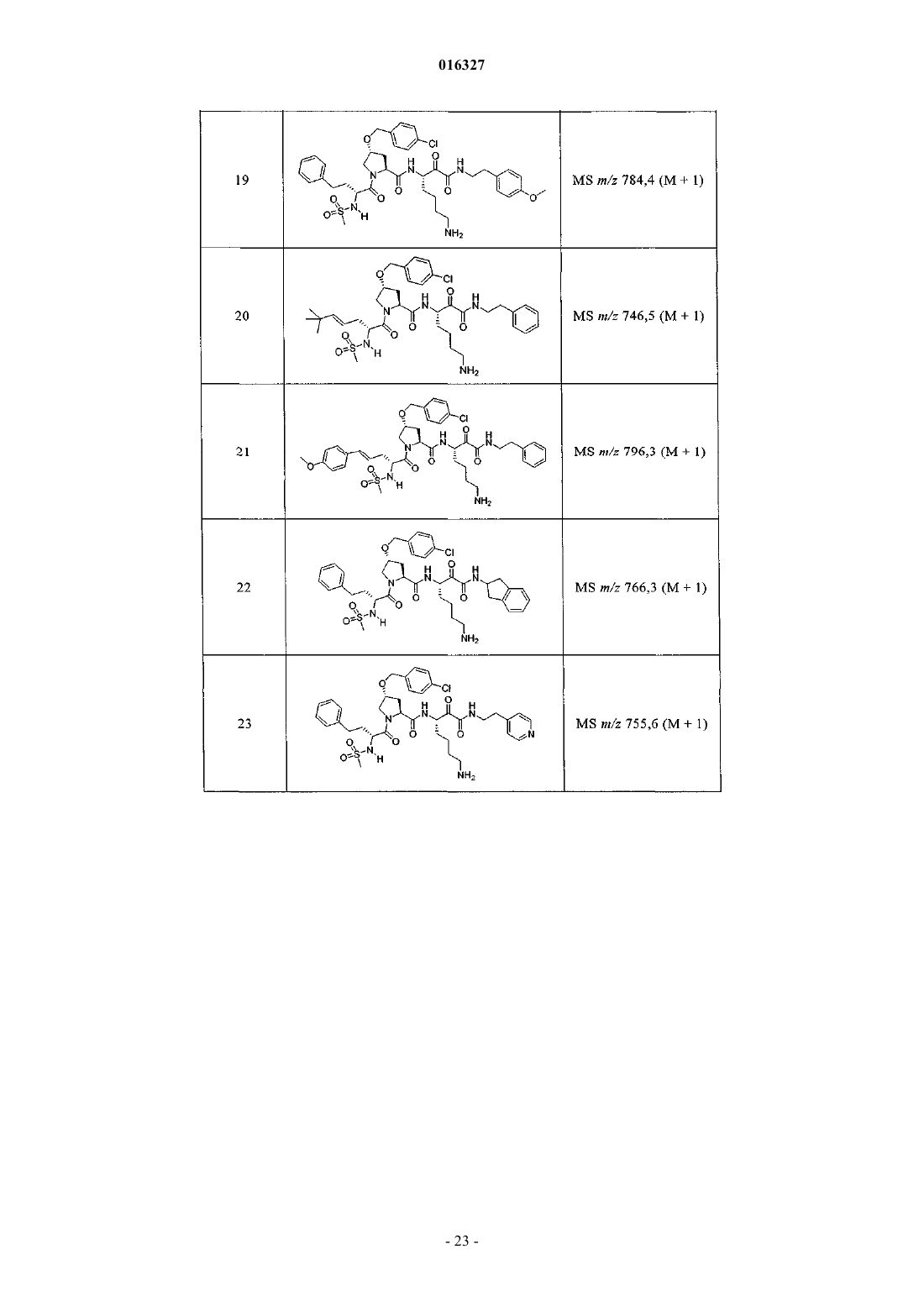
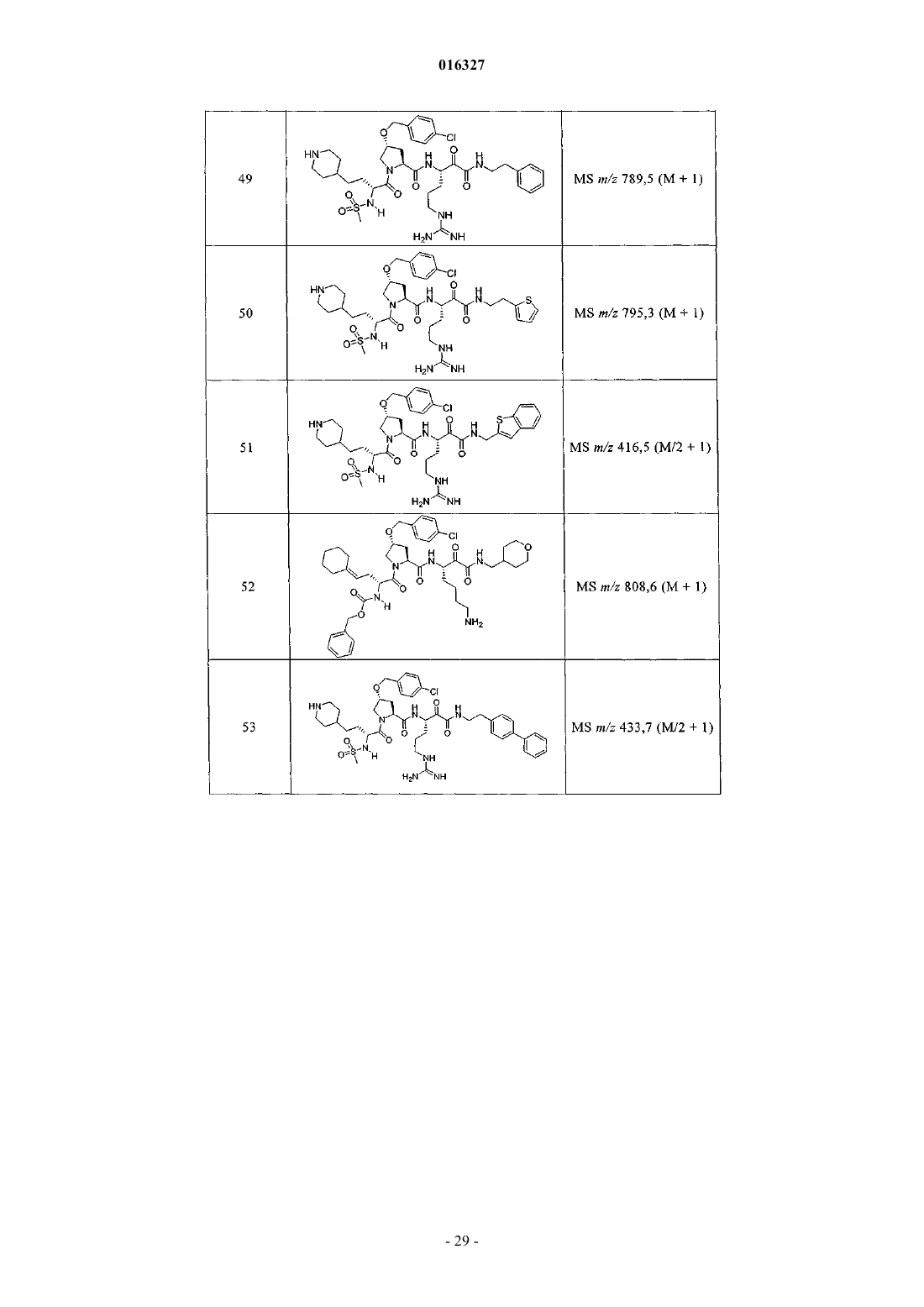
11. Соединение по п.1, выбранное из группы, состоящей из следующих соединений:








или его фармацевтически приемлемые соли.
12. Соединение формулы

или его фармацевтически приемлемые соли.
13. Фармацевтическая композиция, содержащая терапевтически эффективное количество соединения по любому из пп.1-12.
14. Применение соединения по любому из пп.1-12 для ингибирования протеазы, активирующей каналы, в системе клеток или тканей или у млекопитающего, где указанная протеаза, активирующая каналы, представляет собой простазин, матриптазу (MTSP-1) или трипсин.
15. Применение соединения по любому из пп.1-12, и необязательно в комбинации со вторым терапевтическим агентом, для изготовления лекарственного средства для лечения состояния, опосредованного протеазой, активирующей каналы, в системе клеток или тканей или у млекопитающего, где указанная протеаза, активирующая каналы, представляет собой простазин, матриптазу (MTSP-1) или трипсин.
16. Применение по п.15, в котором указанное состояние связано с движением жидкости через ионотранспортирующий эпителий, или с накоплением слизи и мокроты в тканях дыхательных путей, или их комбинацией.
17. Применение по п.15, в котором указанное состояние представляет собой циститный фиброз, первичную цилиарную дискинезию, легочную карциному, хронический бронхит, хроническое обструктивное заболевание легких, астму или инфекцию дыхательных путей.
18. Применение по п.15, в котором указанный второй терапевтический агент представляет собой противовоспалительный агент, бронхорасширяющий агент, антигистаминный агент, противокашлевый агент, антибиотик или ДНазу и его вводят до, одновременно или после соединения по любому из пп.1-12.
19. Применение по п.14 или 15, в котором указанная протеаза, активирующая каналы, представляет собой простазин.
20. Применение по п.14 или 15, в котором указанная система клеток или тканей включает бронхиальные эпителиальные клетки.
Текст
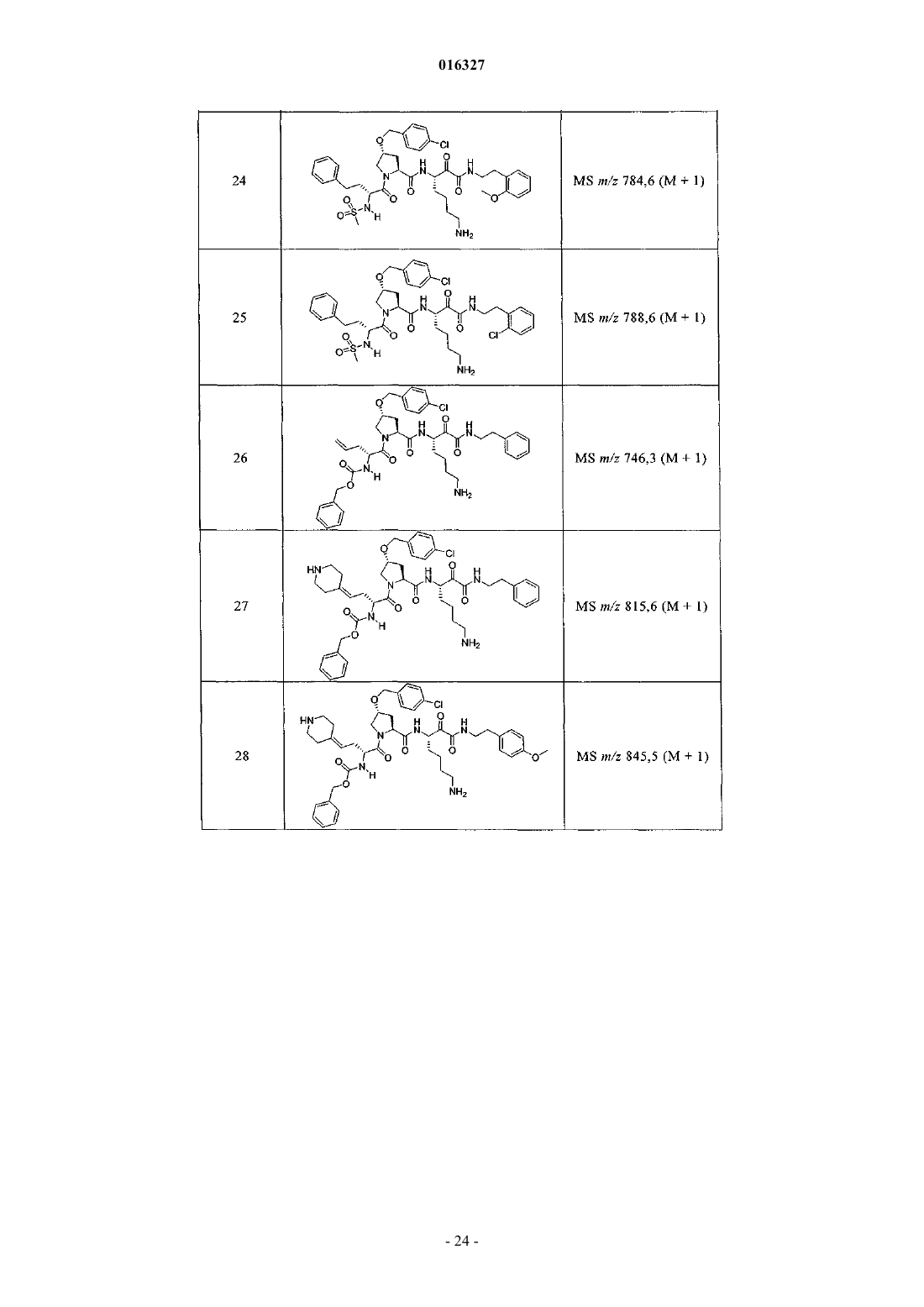
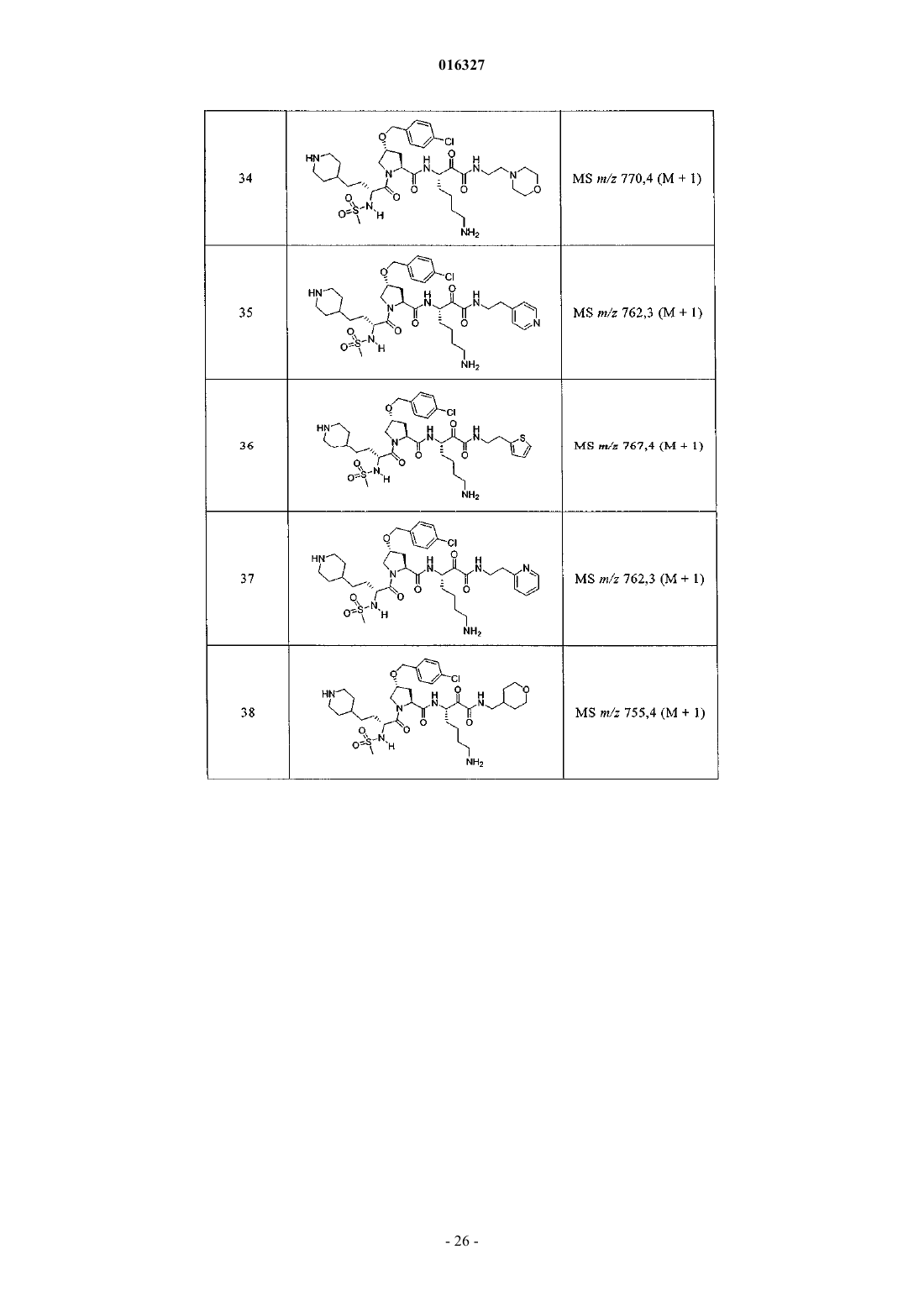
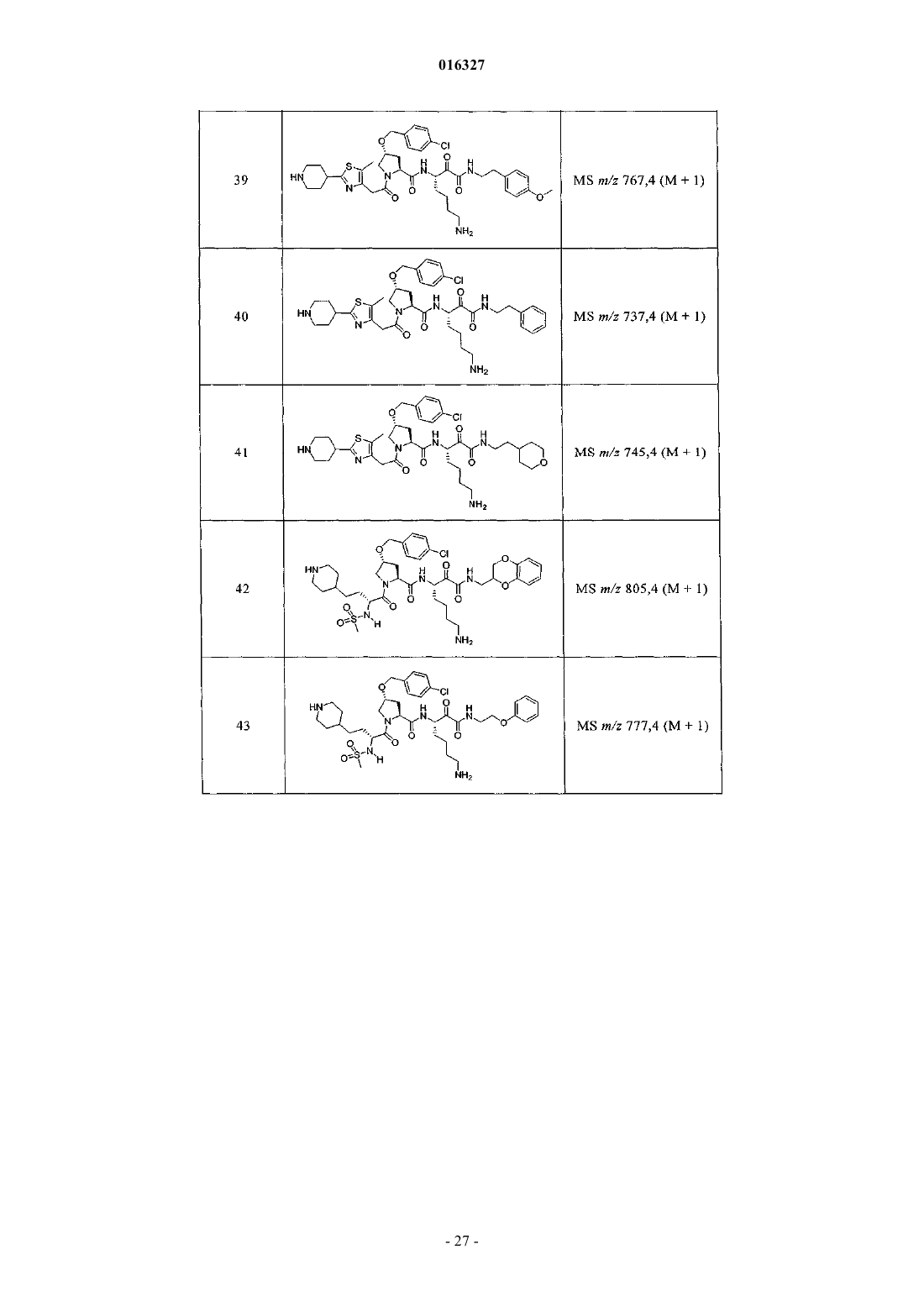
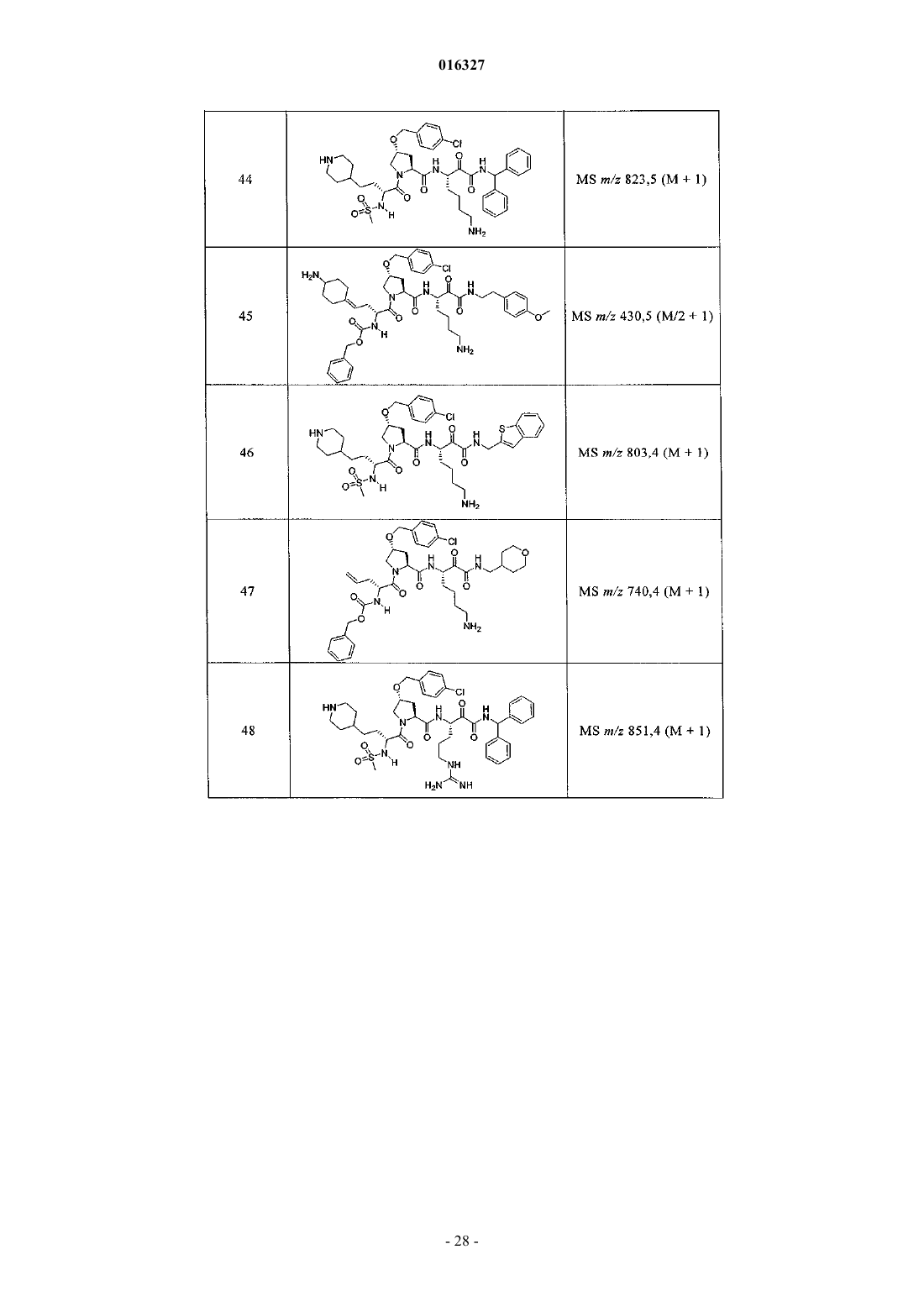
Настоящее изобретение относится к соединениям формулы (1) и содержащим их фармацевтическим композициям, которые полезны для модулирования протеаз,активирующих каналы, и к способам применения таких соединений для лечения, облегчения или профилактики состояния, связанного с протеазой, активирующей каналы, включая, но не ограничиваясь ими, простазин, PRSS22, TMPRSS11 (например, TMPRSS11B, TMPRSS11E),TMPRSS2, TMPRSS3, TMPRSS4 (MTSP-2), матриптазу (MTSP-1), CAP2, САР 3, трипсин, катепсин А или нейтрофилэластазу. 016327 Ссылки на заявки-аналоги Заявка на данный патент является продолжением заявки US 60/889018, поданной 9.02.2007 г., которая приведена здесь полностью в качестве ссылки. Область техники Настоящее изобретение в целом относится к ингибиторам протеазы, активирующей каналы (САР). Уровень техники Простазин является трипсиноподобной сериновой протеазой, которая присутствует в различных тканях млекопитающего. Она является мембранозакрепленной протеазой, которая экспрессируется на внешней клеточной мембране клеток, но также может секретироваться в жидкостях организма, таких как сперма, моча и жидкость поверхности дыхательных путей. Простазин (PRSS8) вместе с протеазами, такими как матрипатаза, САР 2, САР 3, трипсин, PRSS22, TMPRSS11, катепсин А и нейтрофилэластаза, могут стимулировать активность амилоридчувствительного эпителиального натриевого канала (ENaC). Ингибирование этих ферментов может вызывать изменения в эпителиальном ионном транспорте и, следовательно, гомеостазе жидкости через эпителиальные мембраны. Например, ингибирование САР в почках, как полагают, промотирует диурез, тогда как ингибирование САР в легочных путях промотирует выведение слизи и мокроты из легких. Ингибирование САР в почках, следовательно, может использоваться терапевтически для лечения гипертензии. Ингибирование САР в легочных путях предотвращает застой секреции дыхательных путей, который делает пациентов незащищенными по отношению ко вторичным бактериальным инфекциям. Описание изобретения Настоящее изобретение относится к соединениям, фармацевтическим композициям и способам применения таких соединений для моделирования протеаз, активирующих каналы (САР). Например,соединения и композиции по изобретению могут использоваться для модулирования простазина,PRSS22, TMPRSS11 (например, TMPRSS11B, TMPRSS11E), TMPRSS2, TMPRSS3, TMPRSS4 (MTSP-2),матриптазы (MTSP-1), CAP2, САР 3, трипсина, катепсина А и нейтрофилэластазы. В одном варианте осуществления настоящее изобретение относится к соединениям формулы (1) или их фармацевтически приемлемым солям, гдеR6 представляет собой C1-6 алкил, фенил, не замещенный или замещенный группами C1-6 алкокси,SO2NR2, галогеном, фенилом, бензилокси, CONH(CR2)2(фенил) или CONR8R9; пиридил, циклопропил,циклогексил, бензотиазолил, 2,3-дигидро-1H-инденил, морфолинил, имидазолил, тетрагидропиранил,пиперидинил, тиофенил, 2,3-дигидробензо[b][1,4]диоксинил или бензотиофенил;R2 является заместителем в любом положении кольца А и представляет собой -O-(CR2)p-R7 илиR6 представляет собой C1-6 алкил или фенил, замещенный C1-6 алкоксигруппой; или R4 представляет собой фенил, пиперидинил или где кольцо Е представляет собой циклогексил, необязательно замещенный NR2; или кольцо Е представляет собой пиперидинил;R7 представляет собой 5-7-членное карбоциклическое кольцо или фенил, необязательно замещенный галогеном;k и p равны 1. В описанной выше формуле (1) R2 представляет собой -O-(CH2)-R7 и R7 представляет собой галогензамещенный фенил. В одном варианте осуществления настоящее изобретение относится к соединениям формулы (2) или их фармацевтически приемлемым солям, гдеR8 и R9 вместе образуют пиперидинил. В другом варианте осуществления настоящее изобретение относится к соединениям формулы (3) или их фармацевтически приемлемым солям, гдеR10 представляет собой галоген. В указанных выше формулах (2) и (3) R1 представляет собой -(CH2)m-NH2, -(CH2)m-NHC(=NH)-NH2 или (CH2)m-X, где X представляет собой пиперидинил. В других примерах R3 представляет собой C1-6 алкил, циклогексил или бензил. В других примерах R4 представляет собой пиперидинил. В некоторых вариантах осуществления настоящее изобретение относится к соединениям формулыR4 представляет собой пиперидинил иY представляет собой SO2. В другом варианте осуществления настоящее изобретение относится к соединениям формулы (4) или их фармацевтически приемлемым солям, гдеR10 представляет собой галоген. В описанной выше формуле (4) R5 представляет собой тиазолил, необязательно замещенный NR8R9. В описанных выше формулах (1)-(4) J может представлять собой ОН, ОСН 3, NH-СН(фенил)2,NH(C0-C6-алкилен)-О(фенил), NH(C0-C6-алкилен)-SO2(морфолинил) или NH-(CR2)lR6, где R6 представляет собой C1-6 алкил, фенил, не замещенный или замещенный группами C1-6 алкокси, SO2NR2, галогеном,фенилом, бензилокси, CONH(CR2)2(фенил) или CONR8R9; пиридил, циклопропил, циклогексил, бензотиазолил, 2,3-дигидро-1 Н-инденил, морфолинил, имидазолил, тетрагидропиранил, пиперидинил, тиофенил, 2,3-дигидробензо[b][1,4]диоксинил или бензотиофенил. В другом варианте осуществления настоящее изобретение относится к фармацевтическим композициям, содержащим соединение формул (1)-(3) или (4) и фармацевтически приемлемый эксципиент. Настоящее изобретение также относится к способам модулирования протеазы, активирующей каналы, включающим введение в систему или субъекту, нуждающемуся в этом, терапевтически эффективного количества соединения формул (1)-(3) или (4), или его фармацевтически приемлемых солей, или-2 016327 содержащих его фармацевтических композиций, модулируя указанную протеазу, активирующую каналы. В одном варианте осуществления настоящее изобретение относится к способу ингибирования протеазы, активирующей каналы, включающему введение в систему клеток или тканей или млекопитающему терапевтически эффективного количества соединения формул (1)-(3) или (4), или его фармацевтически приемлемых солей, или его фармацевтических композиций; где указанная протеаза, активирующая каналы, представляет собой простазин, PRSS22, TMPRSS11 (например, TMPRSS11B, TMPRSS11E),TMPRSS2, TMPRSS3, TMPRSS4 (MTSP-2), матриптазу (MTSP-1), CAP2, САР 3, трипсин, катепсин А или нейтрофилэластазу, ингибируя указанную протеазу, активирующую каналы. В конкретных примерах настоящее изобретение относится к способу ингибирования простазина. В другом варианте осуществления настоящее изобретение относится к способу облегчения или лечения состояния, опосредованного протеазой, активирующей каналы, включающему введение в систему клеток или тканей или млекопитающему терапевтически эффективного количества соединения формул(1)-(3) или (4), или его фармацевтически приемлемых солей, или его фармацевтических композиций, и необязательно в комбинации со вторым терапевтическим агентом; где указанная протеаза, активирующая каналы, представляет собой простазин, PRSS22, TMPRSS11 (например, TMPRSS11B, TMPRSS11E),TMPRSS2, TMPRSS3, TMPRSS4 (MTSP-2), матриптазу (MTSP-1), САР 2, САР 3, трипсин, катепсин А или нейтрофилэластазу, излечивая указанное состояние. Кроме того, настоящее изобретение относится к соединениям формул (1)-(3) или (4) для применения для лечения состояния, опосредованного протеазой, активирующей каналы. Настоящее изобретение также относится к применению соединения формул (1)-(3) или (4) и необязательно в комбинации со вторым терапевтическим агентом для изготовления лекарственного средства для лечения состояния, опосредованного протеазой, активирующей каналы. В конкретных примерах соединения по изобретению могут использоваться для лечения состояния,опосредованного простатином. В одном варианте осуществления второй терапевтический агент может представлять собой противовоспалительное средство, бронхорасширяющее средство, антигистаминовое средство, противокашлевое средство, антибиотик или ДНКазу и вводиться до, одновременно или после соединения формул (1)-(3) или (4). В некоторых примерах соединения по изобретению вводят в бронхиальные эпителиальные клетки, особенно в бронхиальные эпителиальные клетки человека. Примеры состояний, которые могут облегчаться или излечиваться соединениями по изобретению,включают, но не ограничиваются ими, состояние, связанное с движением жидкости через ионотранспортирующий эпителий или накоплением слизи и мокроты в тканях дыхательных путей, или их комбинация. В некоторых примерах состояние, которое может излечиваться соединениями по изобретению, представляет собой циститный фиброз, первичную цилиарную дискинезию, карциному легких, хронический бронхит, хроническое обструктивное заболевание легких, астму или инфекции дыхательного тракта. Определения."Алкил" как группа и в качестве структурного элемента других групп, например галогензамещенного алкила и алкокси, может быть линейным или разветвленным. Необязательно замещенный алкил,алкенил или алкенил, как здесь используется, может быть необязательно галогенирован (например, CF3) или может содержать один или несколько атомов углерода, которые являются замещенными или заменены гетероатомом, таким как NR, О или S (например, -ОСН 2 СН 2 О-, алкилтиолы, тиоалкокси, алкиламины и т.д.)."Арил" обозначает моноциклическое или конденсированное бициклическое ароматическое кольцо,содержащее атомы углерода. Например, арил может представлять собой фенил или нафтил. "Арилен" обозначает двухвалентный радикал, образованный от арильной группы."Гетероарил", как здесь используется, является таким, как определено выше для арила, где один или несколько членов кольца представляют собой гетероатом. Примеры гетероарилов включают, но не ограничиваются ими, пиридил, индолил, индазолил, хиноксалинил, хинолинил, бензофуранил, бензопиранил,бензотиопиранил, бензо[1,3]диоксол, имидазолил, бензоимидазолил, пиримидинил, фуранил, оксазолил,изоксазолил, триазолил, тетразолил, пиразолил, тиенил и т.д."Карбоциклическое кольцо", как здесь используется, обозначает насыщенное или частично ненасыщенное, моноциклическое, конденсированное бициклическое или мостиковое полициклическое кольцо, содержащее атомы углерода, которые необязательно могут быть замещены, например, с помощью=O. Примеры карбоциклических колец включают, но не ограничиваются ими, циклопропил, циклобутил,циклопентил, циклогексил, циклопропилен, циклогексанон и т.д."Гетероциклическое кольцо", как здесь используется, является таким, как определено выше для карбоциклического кольца, где один или несколько кольцевых атомов углерода являются гетероатомами. Например, гетероциклическое кольцо может содержать N, О, S, -N=, -S-, -S(O), -S(O)2- или -NR-, где R может представлять собой водород, C1-6 алкил или защитную группу. Примеры гетероциклических колец включают, но не ограничиваются ими, морфолино, пирролидинил, пирролидинил-2-он, пиперазинил,пиперидинил, пиперидинилон, 1,4-диокса-8-азаспиро[4.5]дец-8-ил и т.д. Если не указано иное, когда заместитель определен как "необязательно замещенный", подразумева-3 016327 ется, что заместитель представляет собой группу, которая может быть замещена одной или несколькими группами, отдельно и независимо выбранными, например, из необязательно галогенированного алкила,алкенила, алкинила, алкокси, алкиламина, алкилтио, алкинила, амида, амино, включая моно- и дизамещенные аминогруппы, арила, арилокси, арилтио, карбонила, карбоцикла, циано, циклоалкила, галогена,гетероалкила, гетероалкенила, гетероалкинила, гетероарила, гетероцикла, гидрокси, изоцианато, изотиоцианато, меркапто, нитро, О-карбамила, N-карбамила, О-тиокарбамила, N-тиокарбамила, С-амидо, Nамидо, S-сульфонамидо, N-сульфонамидо, С-карбокси, О-карбокси, пергалогеналкила, перфторалкила,силила, сульфонила, тиокарбонила, тиоцианато, тиогалогенметансульфонила и их защищенных соединений. Защитные группы, которые могут образовывать защищенные соединения в указанных выше заместителях, известны специалисту в данной области техники и могут быть обнаружены в источниках, таких как Greene и Wuts, Protective Groups in Organic Synthesis, 3-e издание, John WileySons, New York, NY,1999 и Kocienski, Protective Groups, Thieme Verlag, New York, NY, 1994, которые приведены здесь полностью в качестве ссылки. Термины "совместное введение" или "объединенное введение" или им подобные, как здесь используется, обозначают осуществление введения выбранных терапевтических агентов отдельному пациенту и включают режимы лечения, при которых агенты необязательно вводят одним способом введения или в одно время. Термин "фармацевтическая комбинация", как здесь используется, обозначает продукт, полученный смешением или объединением активных ингредиентов, и включает фиксированные и нефиксированные комбинации активных ингредиентов. Термин "фиксированная комбинация" обозначает, что активные ингредиенты, например соединение формулы (1) и совместно вводимый агент, оба, вводятся пациенту одновременно в форме одного целого или одной дозировки. Термин "нефиксированная комбинация" обозначает, что активные ингредиенты, например, соединение формулы (1) и совместно вводимый агент,оба, вводятся пациенту раздельными частями одновременно, параллельно или последовательно без особых временных интервалов, где такое введение обеспечивает терапевтически эффективные уровни активных ингредиентов в организме пациента. Последнее также относится к коктейльной терапии, например введению трех или более активных ингредиентов. Термин "терапевтически эффективное количество" обозначает количество соединения, которое вызывает биологический или медицинский отклик в клетке, ткани, органе, системе, животном или человеке, которое понятно исследователю, ветеринару, медицинскому доктору или другому врачу. Термин "введение" соединения следует понимать как обеспечение соединения по изобретению,включая пролекарство соединения по изобретению, пациенту, нуждающемуся в таком лечении. Как здесь используется, термины "лечить", "излечение" и "лечение" относятся к способу облегчения или ослабления заболевания и/или сопровождающих его симптомов. Термин "простазин" также может обозначаться как протеаза, активирующая каналы человека(hCAP); протеаза, активирующая каналы-1; и PRSS8, MERPOPS ID S01,159. Способы осуществления изобретения Настоящее изобретение относится к соединениям, фармацевтическим композициям и способам применения таких соединений для модулирования протеаз, активирующих каналы (САР). В одном варианте осуществления настоящее изобретение относится к соединениям формулы (1) или их фармацевтически приемлемым солям, гдеR1 представляет собой -(CR2)m-NR2, -(CR2)m-NRC(=NR)-NR2 или -(CR2)m-C(=NR)-NR2, или необязательно замещенное 5-7-членное азотсодержащее гетероциклическое кольцо; илиX представляет собой C3-7 циклоалкил или арил, каждый из которых необязательно замещен-(CR2)mNR2, -(CR2)m-NRC(=NR)-NR2 или -(CR2)m-C(=NR)-NR2;R2 представляет собой заместитель в любом положении кольца А и представляет собой -O-(CR2)p-R7 или -L-NR8R9, гдеR4 представляет собой Н, C1-6 алкил, C2-6 алкенил, -CR=CR-R6, C2-6 алкинил или необязательно замещенное 5-12-членное карбоциклическое кольцо, гетероциклическое кольцо, арил или гетероарил; или, где кольцо Е представляет собой необязательно замещенное 5-12-членное моноциклическое или конденсированное карбоциклическое или гетероциклическое кольцо;R5 и R7 независимо представляют собой необязательно замещенное 5-7-членное карбоциклическое кольцо, гетероциклическое кольцо, арил или гетероарил;R6 представляет собой C1-6 алкил, C2-6 алкенил, C2-6 алкинил или необязательно замещенное 5-12 членное карбоциклическое кольцо, гетероциклическое кольцо, арил или гетероарил;R8 и R9 независимо представляют собой Н, C1-6 алкил, C2-6 алкенил, C2-6 алкинил или -(CR2)l-R7; илиR8 и R9 вместе с атомом N могут образовывать необязательно замещенное 5-7-членное гетероциклическое кольцо; каждый R представляет собой Н или C1-6 алкил, C2-6 алкенил или C2-6 алкинил;k, l, m, n и р независимо имеют значение 0-6. В конкретных примерах Y представляет собой -SO2-, -NHCO-, -СО- или -О-С(=О)-;k, m, n и р независимо имеют значения 1-6. В других примерах заместитель, соответствующий R2, может находиться в 3-положении кольца А. В одном варианте осуществления настоящее изобретение относится к соединению формулы (2) где R8 и R9 вместе образуют необязательно замещенное 5-7-членное азотсодержащее гетероциклическое кольцо;R, R1, R3, R4, Y, J и n являются такими, как определено в формуле (1). В другом варианте осуществления настоящее изобретение относится к соединению формулы (3)R, R1, R3, R4, Y, J и n являются такими, как определено в формуле (1). В другом варианте осуществления настоящее изобретение относится к соединениям формулы (4)R, R1, R5, J и k являются такими, как определено в формуле (1). В описанных выше формулах (1)-(4) каждая необязательно замещенная группа может быть замеще-5 016327 на галогеном, =O, C1-6 алкокси, амино, C1-6 алкилом, C2-6 алкенилом или C2-6 алкинилом, каждый из которых необязательно может быть галогенирован или необязательно может содержать атом углерода, который может быть замещен или замещен N, О или S; CO2R11, О-(CR2)l-C(O)-R11; -(CR2)l-R11, -(CR2)l-C(O)-R11 или -(CR2)l-SO2-R11; или их комбинациями, где каждый R11 представляет собой Н, амино, C1-6 алкил или необязательно замещенное карбоциклическое кольцо, гетероциклическое кольцо, арил или гетероарил. Например, J может представлять собой фенил, замещенный одним или несколькими атомами галогена,C1-6 алкокси, SO2NH2 или -(CR2)l-C(O)-R11, где R11 представляет собой 5-7-членное гетероциклическое кольцо, такое как морфолинил. В другом примере R3 может представлять собой тиазолил, замещенный одним или несколькими C1-6 алкилами, или 5-7-членное гетероциклическое кольцо, такой как пиперидинил. В других примерах R4 может представлять собой фенил, замещенный C1-6 алкокси, или метиленциклогексан, замещенный амино. Настоящее изобретение также включает все подходящие изотопные варианты соединений по изобретению или их фармацевтически приемлемых солей. Изотопный вариант соединения по изобретению или его фармацевтически приемлемой соли обозначает соединение, в котором по крайней мере один атом заменен на атом, имеющий тот же атомный номер, но атомная масса которого отличается от атомной массы, обычно встречающейся в природе. Примеры изотопов, которые могут быть включены в соединения по изобретению, и их фармацевтически приемлемые соли включают, но не ограничиваются ими, изотопы водорода, углерода, азота и кислорода, такие как 2 Н, 3 Н, 11C, 13 С, 14 С, 15N, 17O, 18O,35S, 18F,36Cl и 123I. Некоторые изотопные варианты соединений по изобретению и их фармацевтически приемлемых солей, например те, в которые включен радиоактивный изотоп, такой как 3H или 14C, являются полезными в качестве лекарственного средства и/или вещества для исследований распределения в ткани. В конкретных примерах изотопы 3 Н и 14 С могут использоваться для их более легкого получения и определения. В других примерах замещение изотопов, таких как 2 Н, может иметь некоторые терапевтические преимущества, вытекающие из большей метаболической стабильности, такие как повышенное время полураспада in vivo или необходимости пониженной дозировки. Изотопные варианты соединений по изобретению или их фармацевтически приемлемых солей обычно могут быть получены обычными методиками, используя подходящие изотопные варианты подходящих реагентов. Соединения и композиции по изобретению могут использоваться для модулирования протеазы, активирующей каналы. Примеры протеаз, активирующих каналы, которые могут модулироваться соединениями и композициями по изобретению, включают, но не ограничиваются ими, простазин, PRSS22,TMPRSS11 (например, TMPRSS11B, TMPRSS11E), TMPRSS2, TMPRSS3, TMPRSS4 (MTSP-2), матриптазу (MTSP-1), CAP2, САР 3, трипсин, катепсин А или нейтрофилэластазу. Новые соединения настоящего изобретения также могут ингибировать активность протеазы, которые стимулируют активность ионных каналов, таких как эпителиальные натриевые каналы, и могут использоваться для лечения САРсвязанных заболеваний. Фармакология и полезность. Соединения по изобретению модулируют активность протеазы, активирующей каналы, например,трипсиноподобных сериновых протеаз, таких как простазин, и как таковые являются полезными для лечения заболеваний или нарушений, при которых простазин участвует в патологии и/или симптоматике заболевания. Заболевания, опосредованные ингибированием протеазы, активирующей каналы, например, трипсиноподобной сериновой протеазы, такой как простазин, включают заболевания, связанные с регулированием объемов жидкости через эпителиальные мембраны. Например, объем жидкости поверхности дыхательных путей является ключевым регулятором мукоцилиарного клиренса и поддержания здоровья легких. Ингибирование протеазы, активирующей каналы, вызывает накопление жидкости на слизистой стороне эпителия дыхательных путей, тем самым вызывая мукоцилиарный клиренс и предотвращение накопления слизи и мокроты в респираторных тканях (включая легочные пути). Такие заболевания включают респираторные заболевания, такие как циститный фиброз, первичную цилиарную дискинезию,хронический бронхит, хроническое обструктивное заболевание легких (COPD), астму, инфекции дыхательных путей (острые и хронические; вирусные и бактериальные) и легочную карциному. Заболевания,опосредованные ингибированием протеаз, активирующих каналы, также включают заболевания, отличные от респираторных заболеваний, которые связаны с нарушением регулирования потока через эпителий, вероятно включающим нарушенную физиологию жидкостей защитных поверхностей на их поверхности, например сухость слизистой оболочки рта (сухой рот) или сухой кератоконъюнктивит (сухие глаза). Кроме того, регулирование CAP ENaC в почках могли бы использоваться для защиты диуреза и тем самым вызывать гипотензивный эффект. Хроническое обструктивное легочное заболевание включает хронический бронхит или связанную с ним одышку, эмфизему, а также обострение гиперреактивности легочных путей вследствие терапии другими лекарственными средствами, в частности терапии другими ингалируемыми лекарственными препаратами. Настоящее изобретение также включает лечение бронхита любого типа или генеза, включая, например, острый, арахиновый, катаральный, фибринозный, хронический или фтиноидный бронхит. Астма включает врожденную (неаллергическую) астму и приобретенную (аллергическую) астму,-6 016327 легкую астму, астму средней степени, тяжелую астму, бронхиальную астму, вызванную физическими упражнениями астму, профессиональную астму и астму, вызванную бактериальной инфекцией. Астма также включает состояние, обозначенное как "синдром свистящих младенцев", который включает субъектов возраста менее 4 или 5 лет, проявляющих свистящие симптомы и диагностированные как "свистящие младенцы", категория пациентов основного медицинского назначения и сейчас идентифицируемая как начальная или астматики ранней фазы. Приспособленность ингибитора протеазы, активирующей каналы, такого как ингибитор простазина, для лечения заболевания, опосредованного ингибированием протеазы, активирующей каналы, может тестироваться путем определения ингибирующего эффекта ингибитора протеазы, активирующей каналы,в соответствии с анализами, описанными далее, и используя способы, известные из предшествующего уровня техники. В соответствии с вышеизложенным настоящее изобретение также относится к способу профилактики или лечения любого заболевания или нарушения, описанного выше, у субъекта, нуждающегося в таком лечении, который включает введение указанному субъекту терапевтически эффективного количества соединения формул (1)-(3) или (4) или его фармацевтически приемлемой соли. Для любого из описанных выше применений необходимая дозировка будет зависеть от способа введения, конкретного излечиваемого состояния и желаемого эффекта (см. раздел "Введение и фармацевтические композиции"). Введение и фармацевтические композиции. Обычно соединения по изобретению вводят в терапевтически эффективных количествах любым обычным и приемлемым способом, известным из предшествующего уровня техники, отдельно или в комбинации с одним или несколькими терапевтическими агентами. Ингибиторы протеазы, ингибирующей каналы, по изобретению также полезны в качестве совместно вводимых терапевтических агентов для применения в комбинации с другим терапевтическим агентом. Например, ингибитор протеазы, ингибирующей каналы, может использоваться в комбинации с противовоспалительным, бронхорасширяющим, антигистаминным или противокашлевым терапевтическим агентом, антибиотиком или ДНазы. Ингибитор протеазы, ингибирующей каналы, и другой терапевтический агент может находиться в одной или различных фармацевтических композициях. Ингибитор протеазы,ингибирующей каналы, может быть смешан с другим терапевтическим агентом в фиксированной фармацевтической композиции, или он может вводиться раздельно, перед, одновременно или после другого терапевтического агента. Комбинация может быть полезна, в частности, для лечения циститного фиброза или обструктивных или воспалительных заболеваний легочных путей, таких как заболевания, указанные выше, например, в качестве вещества, усиливающего терапевтическую активность таких лекарственных препаратов, или в качестве средств для снижения необходимой дозировки или возможных побочных эффектов таких лекарственных препаратов. Такие противовоспалительные лекарственные вещества включают стероиды, в частности глюкокортикостероиды, такие как будесонид, бекламетазон, флутиказон, циклесонид или мометазон, или стероиды, описанные в документах WO 02/88167, WO 02/12266, WO 02/100879 или WO 02/00679 (особенно соединения примеров 3, 11, 14, 17, 19, 26, 34, 37, 39, 51, 60, 67, 72, 73, 90, 99 и 101), WO 03/35668,WO 03/48181, WO 03/62259, WO 03/64445, WO 03/72592, WO 04/39827 и WO 04/66920; нестероидные агонисты, такие как соединения, описанные в документах DE 10261874, WO 00/00531, WO 02/10143,WO 03/82280, WO 03/82787, WO 03/86294, WO 03/104195, WO 03/101932, WO 04/05229, WO 04/18429,WO 04/19935 и WO 04/26248; антагонисты LTD4, такие как монтелукаст и зафирлукаст; ингибиторыPDE4, такие как циломиласт (ARIFLO GlaxoSmithKline), ROFLUMILAST (Byk Gulden), V-11294A(Napp), BAY19-8004 (Bayer), SCH-351591 (Schering-Plough), AROFYLLINE (Almirall Prodesfarma),PD189659/PD168787 (Parke-Davis), AWD-12-281 (Asta Medica), CDC-801 (Celgene), SelCID(TM) CC10004 (Celgene), VM554/UM565 (Vernalis), T-440 (Tanabe), KW-4490 (Kyowa Hakko Kogyo), и соединения, описанные в документах WO 92/19594, WO 93/19749, WO 93/19750, WO 93/19751, WO 98/18796,WO 99/16766, WO 01/13953, WO 03/104204, WO 03/104205, WO 03/39544, WO 04/000814, WO 04/000839,WO 04/005258, WO 04/018450, WO 04/018451, WO 04/018457, WO 04/018465, WO 04/018431,WO 04/018449, WO 04/018450, WO 04/018451, WO 04/018457, WO 04/018465, WO 04/019944,WO 04/019945, WO 04/045607 и WO 04/037805; и антагонисты рецептора аденозина A2B, такие как соединения, описанные в международной заявке на патент WO 02/42298, каждый из которых приведен здесь полностью в качестве ссылки. Подходящие бронхорасширяющие терапевтические агенты включают агонисты адренорецептора бета-2, такие как албутерол (салбутамол), метапротеренол, тербуталин, салметерол, фенотерол, прокатерол, формотерол, кармотерол или их фармацевтически приемлемые соли, и соединения (в свободной форме или форме соли или сольвата) формулы (1), описанные в международной заявке на патентWO 00/75114, каждый из которых приведен здесь полностью в качестве ссылки, такой как соединение формулы и его фармацевтически приемлемый соли, а также соединения (в свободной форме или форме соли или сольвата) формулы I документа WO 04/16601, а также соединения документов ЕР 1440966, JP 05025045,WO 93/18007, WO 99/64035, US 2002/0055651, WO 01/42193, WO 01/83462, WO 02/66422, WO 02/70490,WO 02/76933, WO 03/24439, WO 03/42160, WO 03/42164, WO 03/72539, WO 03/91204, WO 03/99764,WO 04/16578, WO 04/22547, WO 04/32921, WO 04/33412, WO 04/37768, WO 04/37773, WO 04/37807,WO 04/39762, WO 04/39766, WO 04/45618 WO 04/46083 и WO 04/80964, каждый из которых приведен здесь полностью в качестве ссылки. Подходящие бронхорасширяющие терапевтические агенты также включают антихолинергические или антимускариновые агенты, в частности бромид ипратропия, бромид окситропия, соли тиотропия иCHF 4226 (Chiesi), и гликопирролят, а также соединения, описанные в документах ЕР 424021,US 3714357, US 5171744, WO 01/04118, WO 02/00652, WO 02/51841, WO 02/53564, WO 03/00840,WO 03/33495, WO 03/53966, WO 03/87094, WO 04/018422 и WO 04/05285, каждый из которых приведен здесь полностью в качестве ссылки. Подходящие двойные противовоспалительные и бронхорасширяющие терапевтические агенты включают двойные агонисты адренорецептора бета-2; антагонисты мускаринового рецептора, такие как агенты, описанные в документах US 2004/0167167, WO 04/74246 и WO 04/74812. Подходящие антигистаминные терапевтические агенты включают гидрохлорид цетиризина, ацетаминофен, фумарат клемастина, прометазин, лоратидин, деслоратидин, дифенгидрамин и гидрохлорид фексофенадина, активастин, астемизол, азеластин, эбастин, эпинастин, мизоластин и тефенадин, а также соединения, описанные в документах JP 2004107299, WO 03/099807 и WO 04/026841, каждый из которых приведен здесь полностью в качестве ссылки. Подходящие антибиотики включают макролидные антибиотики, например тобрамицин (TOBI). Подходящие терапевтические агенты ДНазы включают дорназу альфа (PULMOZYME), высокоочищенный раствор рекомбинантной дезоксирибонуклеазы I человека (rhDNase), которая селективно расщепляет ДНК. Дорназу альфа используют для лечения циститного фиброза. Другими полезными комбинациями ингибиторов протеазы, активирующей каналы, с противовоспалительными терапевтическими агентами являются комбинации с антагонистами хемокиновых рецепторов, например антагонисты CCR-1, CCR-2, CCR-3, CCR-4, CCR-5, CCR-6, CCR-7, CCR-8, CCR-9 иCCR10, CXCR1, CXCR2, CXCR3, CXCR4, CXCR5, особенно CCR-5, такие как антагонисты ШерингаПлуга SC-351125, SCH-55700 и SCH-D, антагонисты Такеда, такие как хлорид N-4-6,7-дигидро-2-(4 метилфенил)-5 Н-бензоциклогептен-8-ил]карбонил]амино]фенил]метил]тетрагидро-N,N-диметил-2 Нпиран-4-аминия (TAK-770), и антагонисты CCR-5, описанные в документах US 6166037, WO 00/66558,WO 00/66559, WO 04/018425 и WO 04/026873, каждый из которых приведен здесь полностью в качестве ссылки. Для лечения заболевания, опосредованного ингибированием простазина в соответствии с изобретением, ингибитор протеазы, активирующей каналы, по изобретению в свободной форме или в форме фармацевтически приемлемой соли может вводиться любым подходящим способом, например перорально, например в форме таблетки, капсулы или в жидкой форме; парентерально, например в форме инъекционного раствора или суспензии; интраназально, например в форме аэрозоля или другого измельченного состава, используя подходящее интраназальное устройство для доставки, например назальный спрей,такой как спрей, известные из предшествующего уровня техники; или ингаляцией, такой как с помощью аэрозольного устройства. Ингибитор протеазы, активирующей каналы, может вводиться в фармацевтической композиции вместе с фармацевтически приемлемым разбавителем или носителем. Такие композиции могут представлять собой, например, сухие порошки, таблетки, капсулы и жидкости, а также инъекционные растворы, инфузионные растворы или ингаляционные суспензии, которые могут быть получены, используя другие ингредиенты состава и методики, известные из предшествующего уровня техники. Дозировка ингибитора протеазы, активирующей каналы, в свободной форме или в форме фармацевтически приемлемой соли может зависеть от различных факторов, таких как активность и длительность действия активного ингредиента, степени тяжести излечиваемого состояния, способа введения,типа, пола, этнического происхождения, возраста и веса субъекта и/или от его индивидуального состояния. В обычном случае суточная дозировка для введения, например перорального введения теплокровному животному, конкретно человеку, весом около 75 кг, составляет приблизительно от 0,7 приблизи-8 016327 тельно до 1400 мг; или в некоторых примерах приблизительно от 5 приблизительно до 200 мг. Эта дозировка может вводиться одной дозой или несколькими частичными дозами, например, от 5 до 200 мг. Когда композиция включает аэрозольный состав, она может содержать газ-вытеснитель гидрофторалкана (HFA), такой как HFA134a или HFA227 или их смесь; один или несколько сорастворителей, известных из уровня техники, таких как этанол (вплоть до 20 вес.%); одно или несколько поверхностноактивных веществ, таких как олеиновая кислота или триолеат сорбита; и/или один или несколько наполнителей, таких как лактоза. Когда композиция включает состав в виде сухого порошка, она предпочтительно содержит, например, ингибитор протеазы, активирующей каналы, с диаметром частиц вплоть до 10 мкм, необязательно вместе с разбавителем или носителем, таким как лактоза, желаемого распределения размера частиц, и соединением, которое помогает защитить продукт от появления повреждения вследствие влаги (например, стеарат магния). Когда композиция включает аэрозольный состав, она может содержать, например, ингибитор протеазы, активирующей каналы, в растворенной или суспендированной форме, в носителе, содержащем воду, сорастворитель, такой как этанол или пропиленгликоль, и стабилизатор, которым может быть поверхностно-активное вещество. В конкретных вариантах осуществления настоящее изобретение относится к соединениям формул(1)-(4) в ингалируемой форме, например в форме аэрозоля или другой распыляемой композиции, или в форме ингалируемых частиц, например в микронозированной форме. Настоящее изобретение также относится к ингалируемому лекарственному средству, содержащему соединения по изобретению в ингалируемой форме; фармацевтическому продукту, включающему соединения по изобретению в ингалируемой форме вместе с устройством для ингаляции; и устройству для ингаляции, включающему соединения по изобретению в ингалируемой форме. Способ получения соединений по изобретению Соединения по изобретению могут быть получены методиками, описанными в примерах. В описанных реакциях реакционные функциональные группы, при необходимости в конечном продукте (например, гидрокси, амино, имино, тио или карбоксигруппы), могут быть защищены, используя защитные группы, известные из предшествующего уровня техники, для устранения их нежелательного участия в реакциях. Обычные защитные группы могут использоваться в соответствии со стандартной практикой, например, см. книгу T.W. Greene и P.G.M. Wuts, "Protective Groups in Organic Chemistry", JohnWiley and Sons, 1991. Соединения по изобретению также могут быть получены в форме фармацевтически приемлемой кислотной аддитивной соли реакцией формы свободного основания соединения с фармацевтически приемлемой неорганической или органической кислотой. Альтернативно, фармацевтически приемлемая основная аддитивная соль соединения по изобретению может быть получена реакцией формы свободной кислоты соединения с фармацевтически приемлемым неорганическим или органическим основанием. Альтернативно, формы соли соединений по изобретению могут быть получены, используя соли исходных материалов или промежуточных соединений. Формы свободной кислоты или свободного основания соединений по изобретению могут быть получены из соответствующей формы основной аддитивной соли или кислотной аддитивной соли соответственно. Например, соединение по изобретению в форме кислотной аддитивной соли может быть превращено в соответствующее свободное основание обработкой подходящим основанием (например, раствором гидроксида аммония, гидроксида натрия и им подобными). Соединение по изобретению в форме основной аддитивной соли может быть превращено в соответствующую свободную кислоту обработкой подходящей кислотой (например, хлористо-водородной кислотой и т.д.). Соединения по изобретению в неокисленной форме могут быть получены из N-оксидов соединений по изобретению обработкой восстанавливающим агентом (например, сера, диоксид серы, трифенилфосфин, боргидрид лития, натрийборгидрид, трихлорид фосфора, трибромид или им подобные) в подходящем инертном органическом растворителе (например, ацетонитрил, этанол, водный диоксан или им подобные) при температуре от 0 до 80 С. Пролекарственные производные соединений по изобретению могут быть получены способами, известными специалисту в данной области техники (например, более подробно см. статью Saulnier и др.,Bioorganic and Medicinal Chemistry Letters, 1994, т. 4, с. 1985). Например, подходящие пролекарства могут быть получены реакцией незамещенного соединения по изобретению с подходящим карбамилирующим агентом (например, 1,1-ацилоксиалкилкарбанохлоридатом, пара-нитрофенилкарбонатом или им подобными). Защищенные производные соединений по изобретению могут быть получены способами, известными специалисту в данной области техники. Подробное описание методик, использующихся для создания защитных групп, и их удаление может быть обнаружено в книге Т.W. Greene, "Protecting Groups inOrganic Chemistry", 3-e издание, John Wiley and Sons, Inc., 1999. Соединения настоящего изобретения могут быть получены обычным образом, или в ходе способа по изобретению, в виде сольватов (например, гидратов). Гидраты соединений настоящего изобретения могут быть получены обычным образом перекристаллизацией из смеси водный/органический растворитель, используя органические растворители, такие как диоксин, тетрагидрофуран или метанол.-9 016327 Соединения по изобретению могут быть получены в виде их индивидуальных стереоизомеров реакцией рацемической смеси соединения с оптически активным расщепляющим агентом с образованием пары диастереоизомерных соединений, разделением диастереомеров и выделением оптически чистых энантиомеров. Расщепление энантиомеров может осуществляется, используя ковалентные диастереомерные производные соединений по изобретению или используя диссоциируемые комплексы (например,кристаллические диастереомерные соли). Диастереомеры имеют различные физические свойства (например, точки плавления, точки кипения, растворимости, реакционоспособность и т.д.) и могут быть легко разделены на основании этих различий. Диастереомеры могут быть разделены хроматографией,или методиками разделения/расщепления на основании различий в растворимости. Оптически чистый энантиомер затем извлекают, вместе с расщепляющим агентом, любыми обычными способами, которые не приводят к рацемизации. Более подробное описание методик, использующихся для выделения стереоизомеров соединений из их рацемической смеси, может быть обнаружено в книге Jean Jacques, AndreCollet, Samuel H. Wilen, "Enantiomers, Racemates and Resolutions", John Wiley And Sons, Inc., 1981. В целом соединения по изобретению могут быть получены способом, описанным в примерах, и соединения формул (1)-(4) могут быть получены способом, который включает:(а) необязательно превращение соединения по изобретению в фармацевтически приемлемую соль;(б) необязательно превращение формы соли соединения по изобретению в форму, отличную от соли;(в) необязательно превращение неокисленной формы соединения по изобретению в фармацевтически приемлемый N-оксид;(г) необязательно превращение формы N-оксида соединения по изобретению в его неокисленную форму;(д) необязательно выделение индивидуального изомера соединения по изобретению из смеси изомеров;(е) необязательно превращение свободного соединения по изобретению в фармацевтически приемлемое пролекарственное производное; и(ж) необязательно превращение пролекарственного производного соединения по изобретению в его свободную форму. Несмотря на то что получение исходных материалов конкретно не описано, соединения являются известными или могут быть получены аналогично способам, известным из предшествующего уровня техники, или как описано далее в примерах. Специалисту в данной области техники ясно, что описанные выше превращения только иллюстрируют способы получения соединений настоящего изобретения и что другие хорошо известные способы могут аналогично использоваться. Настоящее изобретение далее представлено примерами, но не ограничивается ими, следующих промежуточных соединений (ссылочные соединения) и примеров, которые иллюстрируют получение соединений по изобретению. В одном варианте осуществления описаны соединения по изобретению формулы (1), где R1 представляет собой -(CR2)m-NR2, -(CR2)m-NRC(=NR)-NR2 или -(CR2)m-C(=NR)-NR2, или необязательно замещенный 5-7-членное азотсодержащее гетероциклическое кольцо; или R1 представляет собой (CR2)m-X,где X представляет собой C3-7 циклоалкил или арил, который замещен -(CR2)m-NR2, -(CR2)m-NRC(=NR)NR2 или -(CR2)m-C(=NR)-NR2. Конкретные соединения могут быть получены способом, описанным для ссылочных соединений и в примерах. Ссылочное соединение 1. Гидрохлорид этиловый эфир D-гомофенилаланин (5,00 г, 20,5 ммоль) и DIPEA (8,7 мл,51,25 ммоль) растворяли в ТГФ (100 мл) и перемешивали при комнатной температуре. По каплям добавляли мезилхлорид (1,67 мл, 21,52 ммоль) и реакционную смесь перемешивали в течение 6 ч при комнатной температуре. ТГФ упаривали и сырой продукт растворяли в EtOAc (100 мл) и промывали водой(100 мл), 1 N HCl (2100 мл) и насыщенным раствором хлорида натрия (100 мл), сушили (MgSO4). Растворитель удаляли в вакууме и сырой материал очищали ускоренной хроматографией (гексан:EtOAc) с получением этилового эфира. Этиловый эфир растворяли в диоксане (50 мл) и перемешивали при комнатной температуре. Добавляли LiOHH2O (1,00 мг, 24 ммоль), растворенный в воде (20 мл), и реакционную смесь перемешивали до исчезновения этилового эфира (по данным ТСХ и LCMS). Растворитель удаляли в вакууме и сырой материал разделяли с помощью EtOAc (50 мл) и 1 N HCl (50 мл). Водный слой экстрагировали EtOAc (250 мл) и объединенные органические фазы промывали 1 М NaHSO4 (250 мл) и насыщенным раствором хлорида натрия (50 мл) и сушили MgSO4. Растворитель упаривали и сырой материал очищали ускоренной хроматографией (градиент EtOAc:гексан) с получением ссылочного со- 10016327 единения 1 в виде белого порошка. Ссылочное соединение 2. На реакционной схеме для получения ссылочного соединения 2 реагенты и условия являются следующими: (a) TBTU/CH2Cl2/CH3OH, Et3N, 23 С; (б) DIBAL-H, CH2Cl2, -78 С; (в) NaCN, ТЕВАС (кат.),Ас 2 О, CH2Cl2/H2O -20 С; (г) 3 М HCl (газ) в Et2O, CH3OH, 4 С, затем Н 2 О, рН 11, 0 С; (д) Вос 2 О, ДМФА,Et3N, 23 С; (е) Н 2, МеОН, Pd/C 10%, 23 С, 12 ч. 2-В. Раствор Cbz-Lys(Boc)-OH (28 г, 73,60 ммоль) и TBTU (23,5 г, 73,60 ммоль) в 500 мл смесиCH2Cl2:MeOH (9:1) доводили до рН 8,0 добавлением Et3N. Реакционную смесь перемешивали при комнатной температуре в течение 2 ч. Смесь промывали последовательно холодным 1 N раствором HCl, водой, 5% раствором NaHCO3 и водой, сушили Na2SO4. Фильтрат упаривали и остаток хроматографировали на силикагеле (гептан:EtOAc, 4:1) с получением соединения 2-В. ТСХ (EtOAc:гептан, 3:1) Rf=0,85. 2-С. К раствору соединения 2-В (10,5 г, 26,6 ммоль) в CH2Cl2 (350 мл) добавляли DIBAL-H (80 мл,1M в растворе гексана) при -78 С, раствор оставляли перемешиваться при этой температуре в течение 1 ч. Добавляли лимонную кислоту (5% водный раствор) для погашения реакции и оставляли перемешиваться при комнатной температуре в течение 10 мин. CH2Cl2 слой отделяли и водный слой промывалиCH2Cl2 (2200 мл). Объединенные органические слои промывали водой, сушили Na2SO4 и отфильтровывали. Фильтрат упаривали и остаток соединения 2-С использовали немедленно на следующей стадии. 2-D. Сырой раствор соединения 2-С (9,6 г, 26,6 ммоль) в CH2Cl2 (350 мл) охлаждали до -20 С, добавляли раствор NaCN (15 г, 306 ммоль), хлорида триэтилбензиламмония (1,75 г, 7,67 ммоль) в 245 мл воды. По каплям добавляли ангидрид уксусной кислоты (7,7 мл, 81,6 ммоль) в течение 10 мин при интенсивном перемешивании. Реакционную смесь перемешивали в течение 10 мин и затем CH2Cl2 слой отделяли. Водный слой экстрагировали CH2Cl2. Объединенные органические слои промывали водой,сушили Na2SO4 и отфильтровывали. Фильтрат упаривали и остаток соединения 2-D очищали автоматической хроматографией на силикагеле (30-40% этилацетат в гексане). Собранные фракции, содержащие целевой материал, относятся к соединению 2-D. ТСХ (CH2Cl2:EtOAc, 7:3) Rf=0,6. 2-Е. Промежуточное соединение 2-D растворяли в безводном МеОН (80 мл) и диэтиловом эфире(200 мл) и охлаждали до -20 С. Газообразный HCl пропускали через раствор до повышения веса до 36 г. Температуру поддерживали ниже 0 С в течение ночи, затем добавляли воду, поддерживая температуру реакции при 0 С. Реакционную смесь оставляли перемешиваться при комнатной температуре в течение 16 ч, наблюдая за ходом реакции по данным LC/MS. Эфир удаляли в вакууме и значение рН доводили до 11,0, поддерживая температуру реакции при 0 С, добавлением водного раствора NaOH. Реакционную смесь экстрагировали этилацетатом, сушили Na2SO4 и отфильтровывали. Сырой материал 2-Е использовали непосредственно на следующей стадии. LC/MS (рассчит. 325,2 (М+1), обнаруж. 325,1). 2-F. Промежуточное соединение 2-Е (4,94 г, 15,2 ммоль) растворяли в ДМФА (250 мл). Добавляли триэтиламин (6,5 мл, 45 ммоль) для доведения значения рН до 8,0. Добавляли ди-трет-бутилдикарбонат(5,0 г, 23 ммоль) одной порцией и реакционную смесь оставляли перемешиваться в течение ночи при комнатной температуре. Растворитель удаляли в вакууме и остаток растворяли в этилацетате, промывали водой и насыщенным раствором хлорида натрия, сушили Na2SO4, отфильтровывали и упаривали. Остаток соединения 2-F очищали автоматической хроматографией на силикагеле (40% этилацетата в гексане). Собранные фракции, содержащие целевой материал, относятся к соединению 2-F. Ссылочное соединение 2. Раствор соединения 2-F (3,7 г, 8,73 ммоль) растворяли в этаноле (50 мл). Добавляли Pd/C (10%, сырой, типа Degussa) и колбу помещали в аппарат Парра в течение ночи и обрабатывали водородом при 40 пси. Катализатор отфильтровывали через целит и растворитель удаляли в вакууме. Растворитель удаляли в вакууме с получением целевого ссылочного соединения 2 в виде прозрачного масла. LC/MS (рассчит. 291,2 (М+Н), обнаруж. 291,4). На реакционной схеме для ссылочного соединения 3 реагенты и условия являются следующими: (a)(60 мл) и воду (20 мл). Смесь перемешивали при комнатной температуре и добавляли Et3N (11,6 мл,70,2 ммоль), реакционную смесь перемешивали в течение ночи при комнатной температуре. Прозрачный раствор разбавляли EtOAc (200 мл) и промывали 1 N HCl (3100 мл) и насыщенным раствором хлорида натрия (1100 мл) и сушили MgSO4. Растворитель упаривали в вакууме с получением целевого продукта в виде прозрачного масла, которое использовали без дополнительной очистки. MS m/z 280,1 (М+1). 3-С. 4-Нитрофенилхлороформиат (1,514 г, 7,51 ммоль) добавляли к раствору соединения 3-В (1,92 г,6,83 ммоль) и пиридина (663 мкл, 8,19 ммоль) в CH2Cl2 (100 мл). Реакционную смесь перемешивали в течение ночи. Смесь промывали тремя порциями NaHSO4 1 M и двумя порциями насыщенного раствора хлорида натрия; сушили (MgSO4) и концентрировали в вакууме с получением соединения 3-С в виде желтого масла. MS m/z 445,1 (М+1). 3-D. Пиперидин (320 мг, 3,76 ммоль) добавляли к раствору соединения 3-С (1,4 г, 3,14 ммоль) в ДХМ (100 мл) и растворенную смесь перемешивали при комнатной температуре в течение 3 ч. Смесь затем промывали тремя порциями водного 1 М раствора NaHSO4, тремя порциями насыщенного водного раствора NaHCO3 и двумя порциями насыщенного раствора хлорида натрия. Органический слой сушили(MgSO4) и концентрировали в вакууме. Остаток очищали автоматически на силикагеле (EtOAc/гексан, от 0 до 100%) с получением соединения 3-D в виде масла. MS m/z 391,3 (М+1). Ссылочное соединение 3. Раствор соединения 3-D (883 мг, 2,26 ммоль) растворяли в 4:1 смеси третBuOH и воды (50 мл). Добавляли Pd/C (10%, сырой, типа Degussa) и колбу помещали в аппарат Парра в течение ночи, обрабатывали водородом при 40 пси. Катализатор отфильтровывали через целит и растворитель удаляли в вакууме. Ссылочное соединение 3 выделяли в виде прозрачного масла. LC/MS (рассчит. 257,2 (М+Н), обнаруж. 257,4). Ссылочное соединение 4. Реагенты и условия для приведенной реакции являются следующими: (а) Cbz-OSu, Et3N, ТГФ, вода,(4,49 г, 18,0 ммоль) добавляли в круглодонную колбу, содержащую ТГФ (60 мл) и воду (20 мл). Смесь перемешивали при комнатной температуре и добавляли Et3N (10,1 мл, 72,0 ммоль) и реакционную смесь перемешивали в течение ночи при комнатной температуре. Прозрачный раствор разбавляли EtOAc(200 мл) и промывали 1 N HCl (3100 мл) и насыщенным раствором хлорида натрия (1100 мл), сушилиMgSO4. Растворитель упаривали в вакууме с получением целевого продукта в виде белого твердого вещества, которое использовали без дополнительной очистки. Ссылочное соединение 5 получали в условиях, аналогичных описанным для получения ссылочного соединения 4, используя циклогексилкарбонил-OSu вместо Cbz-OSu. Ссылочное соединение 6. Ссылочное соединение 6 получали в условиях, аналогичных описанным для получения ссылочного соединения 1, используя гидрохлорид метилового эфира D-аллилглицина. Ссылочное соединение 7. Ссылочное соединение 7 получали в условиях, аналогичных описанным для получения ссылочного соединения 4, используя D-аллилглицин в качестве реагента. Ссылочное соединение 8.(5,6 мл, 40 ммоль) и раствор охлаждали до 0 С. Добавляли Boc2O (9,59 г, 44 ммоль) и реакционную смесь перемешивали в течение ночи при комнатной температуре. Растворитель удаляли в вакууме и сырой остаток растворяли в этилацетате (120 мл). Раствор промывали 0,1 N HCl (3100 мл) и насыщенным раствором хлорида натрия (1100 мл); сушили MgSO4; отфильтровывали и растворитель упаривали в вакууме с получением соединения 8-В в виде прозрачного масла. 8-С. Трихлоризоциануровую кислоту (2,66 г, 11,46 ммоль) добавляли к раствору спирта (2,39 г,10,42 ммоль) в CH2Cl2, раствор перемешивали и поддерживали при 0 С, затем добавляли каталитическое количество TEMPO. После добавления смесь нагревали при комнатной температуре и перемешивали в течение часа и затем отфильтровывали через целит. Органическую фазу промывали насыщенным водным раствором Na2CO3, затем 1 N HCl и насыщенным раствором хлорида натрия. Органический слой сушили (MgSO4) и растворитель упаривали с получением соединения 8-С. 1 Н ЯМР (CDCl3, 400 МГц)9,72 (1 Н, s), 4,07-4,01 (2H, m), 2,70-2,57 (2H, m), 2,35-2,31 (2H, m), 2,051,94 (1H, m), 1,64-1,46 (2H, m), 1,39 (9H, s), 1,30-1,02 (2H, m). 8-D. К раствору триметилового эфира Cbzфосфоноглицина (2,8 г, 8,45 ммоль) в ТГФ при -78 С добавляли 1,1,3,3-тетраметилгуанидин (1,022 мл, 8,14 ммоль). Через 10 мин добавляли альдегид 8-С(1,76 г, 7,76 ммоль). Раствор затем помещали в ледяную баню при 0 С на 1 ч и затем оставляли нагре- 13016327 ваться до комнатной температуры и перемешивали в течение часа. Раствор разбавляли EtOAc, промывали 1 М NaHSO4, сушили (MgSO4) и концентрировали в вакууме. Остаток очищали автоматической хроматографией на силикагеле с помощью EtOAc/гексан (0-100%) с получением соединения 8-D в виде прозрачного масла. MS m/z 333,2 (М+1). 1 Н ЯМР (CDCl3, 400 МГц)7,35-7,33 (5 Н, m), 6,63 (1H, t, J=8 Гц), 6,30 (1H, bs), 5,12 (2 Н, s), 4,104,04 (2 Н, m), 3,73 (3 Н, s), 2,67-2,62 (2 Н, m), 2,14 (2 Н, t, J=6,8 Гц), 1,63-1,46 (3 Н, m), 1,43 (9 Н, s), 1,14-1,06(2 Н, m). 8-Е. В аппарат Парра загружали соединение 8-D (1 г, 2,31 ммоль) и МеОН (100 мл) в атмосфере азота. Раствор подвергали трем циклам вакуумирования и насыщения азота и добавляли катализатор(R,R)-этим-DuPHOS-Rh(COD) трифлат (30 мг, 0,04 ммоль). Смесь помещали в атмосферу газообразного водорода при 60 пси при комнатной температуре в течение 24 ч. Превращение в соединение 8-Е завершалось через 24 ч с 99% е.е., растворитель удаляли в вакууме и сырой продукт очищали хроматографией на силикагеле (гексан/EtOAc). 8-F. Промежуточное соединение 8-Е растворяли в МеОН, раствор насыщали азотом и добавлялиPd/Carbon (5% сырой, Degussa). Смесь помещали при 50 пси водорода при комнатной температуре и перемешивали в течение 24 ч. Смесь насыщали азотом и отфильтровывали через целит. Слой промывали МеОН и объединенные органические растворы концентрировали в вакууме. Добавляли гексан и затем упаривали до азеотропы оставшегося метанола с получением соединения 8-F в виде масла, которое затем использовали на следующей стадии без дополнительной очистки. 8-G. Сырое промежуточное соединение 8-F (0,6 г, 1,99 ммоль) растворяли в ТГФ (10 мл), к раствору добавляли 2,4,6-коллидин (315 мг, 2,38 ммоль) и метансульфонилхлорид (0,170 мл, 2,19 ммоль) и перемешивали в течение 2 ч. Реакционную смесь разбавляли EtOAc (50 мл) и раствор промывали 1 М NaHSO4 (225 мл) и насыщенным раствором хлорида натрия (25 мл) и сушили (MgSO4). Растворитель удаляли в вакууме и сырой остаток очищали ускоренной хроматографией, используя градиент гексана иEtOAc с получением целевого продукта 8-G. Ссылочное соединение 8. Соединение 8-G (0,70 г, 1,84 ммоль) растворяли в диоксане (7 мл) и добавляли LiOHH2O (232 мг,5,55 ммоль), растворенный в воде (4 мл). Реакционную смесь перемешивали в течение 1 ч при 23 С. Растворитель упаривали, остаток разбавляли EtOAc (25 мл) и промывали 1 N NaHSO4 (25 мл) и насыщенным раствором хлорида натрия (25 мл), сушили MgSO4 и отфильтровывали. Растворитель удаляли в вакууме и сырой продукт очищали хроматографией на силикагеле (градиент гексан/EtOAc) с получением целевого продукта ссылочного соединения 8 в виде белого твердого вещества. Ссылочное соединение 9. Ссылочное соединение 9 получали олефинированием соответствующего циклогекасанона. Раствор бромида метилтрифенилфосфония (156 мг, 0,44 ммоль) и KHMDS (880 мкл 0,5 М раствора в толуоле,0,44 ммоль) в ТГФ (10 мл) при 23 С оставляли перемешиваться в течение 1 ч. Добавляли раствор в ТГФ 4-N-Вос-амино-1-циклогексанона (75 мг, 0,35 ммоль) в 5 мл ТГФ и реакционную смесь перемешивали при 23 С до окончания реакции по данным LC/MS. Реакцию гасили водой; ТГФ упаривали и растворяли в EtOAc и слои разделяли. Водную фазу промывали 210 мл EtOAc и объединенные органические слои промывали водой, насыщенным NaCl, сушили MgSO4 и отфильтровывали. Растворитель удаляли в вакууме и сырой продукт очищали хроматографией на силикагеле (градиент гексан/EtOAc) с получением целевого продукта ссылочного соединения 9 в виде прозрачного масла. LC/MS обнаруж. 212,3 [М+Н]. Ссылочное соединение 10.- 14016327 В описанной выше реакционной схеме использовали следующие реагенты и условия:(г) HgCl2, HgO, MeOH, H2O, 23 С; (д) Н 2, МеОН, Pd/C 10%, 23 С, 12 ч. Промежуточные соединения 10-В и 10-С получали, следуя методикам, аналогичным описанным для получения ссылочного соединения 2, используя N-бис-Вос аргинин (10-А) и промежуточное соединение 10-В в качестве реагентов соответственно. 10-D. н-BuLi (6,4 мл 2,5 М раствора в гексан, 15,94 ммоль) добавляли в течение 10 мин к перемешиваемому раствору трис-(метилтио)метана (2,24 мл, 16,74 ммоль) в ТГФ (45 мл) при -65 С. Через 20 мин при -65 С добавляли раствор альдегида 10-С (1,83 г, 3,71 ммоль) в ТГФ (20 мл) в течение 30 мин. Раствор оставляли перемешиваться при -65 С в течение 5 ч. Реакционную смесь гасили добавлением 400 мл насыщенного раствора NH4Cl и CH2Cl2 (1:12). Слои разделяли и водный слой промывали CH2Cl2 (3100 мл). Объединенные органические слои промывали Н 2 О, насыщенным раствором хлорида натрия, сушилиMgSO4, отфильтровывали и упаривали досуха. Автоматическая хроматография на силикагеле 0-10%EtOAc в CH2Cl2 приводит к получению соединения 10-D в виде прозрачного масла. LC/MS обнаруж. 647,3 [М+Н]. 10-Е. К раствору соединения 10-D (1,0 г, 1,55 ммоль), растворенного в МеОН (30 мл) и Н 2 О (2,2 мл),добавляли хлорид ртути(II) (1,42 г, 5,23 ммоль) и оксид ртути(II) (422 мг, 1,95 ммоль) при интенсивном перемешивании при 23 С в течение 72 ч. Реакционную смесь отфильтровывали через целит и остаток промывали CH2Cl2 (75 мл), МеОН (10 мл) и водой (10 мл). Фильтрат разделяли и водный слой экстрагировали CH2Cl2 (50 мл). Объединенные органические слои промывали насыщенным водным растворомNH4OAc (350 мл) и насыщенным водным раствором NH4Cl (275 мл), сушили Na2SO4, отфильтровывали и упаривали с получением сырого соединения 10-Е. Автоматическая хроматография на силикагеле 0100% EtOAc в гексане приводила к получению соединения 10-Е в виде прозрачного масла. LC/MS обнаруж. 553,3 [М+Н]. Ссылочное соединение 10 получали в условиях, аналогичных описанным для получения ссылочного соединения 2, используя промежуточное соединение 10-Е в качестве реагента. Пример 1. В примере 1 использовали следующие реагенты и условия: (а) KOH, 4-хлорбензилхлорид, ДМСО,от 0 до 23 С; (б) TMS-CHN2, CH2Cl2/MeOH, 23 С; (в) ТФУК/CH2Cl2 (50:50), 23 С;(к) ТФУК/CH2Cl2 (20:80), 23 С, затем ВЭЖХ очистка с массовым распределением, 23 С. 1-В. Тонкоизмельченный порошок KOH (19,4 г, 0,346 моль) растворяли в ДМСО и перемешивали при комнатной температуре в течение 20 мин и затем охлаждали до 0 С. N-Вос-транс-4-гидрокси-Lпролин (Вос-Нур-ОН, 1-А) (10 г, 43,3 ммоль) растворяли в ДМСО (10 мл) и добавляли к смеси и реакционную смесь перемешивали в течение 10 мин при 0 С. Затем добавляли 4-хлорбензилхлорид (33 г, 0,204 моль) и реакционную смесь перемешивали при 0 С в течение 15 мин, после чего ледяную баню удаляли и реакционную смесь оставляли нагреваться до комнатной температуры и перемешивали в течение 4 ч. Реакционную смесь выливали в воду (300 мл) и реакционный сосуд промывали дополнительным количеством воды (300 мл). Объединенные водные слои экстрагировали диэтиловым эфиром (2300 мл) и выгружали. Водный слой подкисляли 87% Н 3 РО 4 до значения рН 2,3 и затем экстрагировали диэтиловым эфиром (3300 мл). Объединенные эфирные экстракты промывали водой (2400 мл) и насыщенным раствором хлорида натрия (2400 мл) и затем сушили MgSO4, отфильтровывали и концентрировали в вакууме. Остаток очищали хроматографией на силикагеле с помощью смеси EtOAc/гексан (градиент от 0 до 100%) с получением соединения 1-В в виде прозрачного масла. MS m/z 256,1 (М+1-Boc). 1 Н ЯМР (ДМСО-D6, 400 МГц)7,39-7,31 (4 Н, m), 4,52-4,40 (2 Н, m), 4,16-4,10 (2 Н, m), 3,48-3,41 (2 Н,m), 2,40-2,30 (1 Н, m), 2,03-1,94 (1H, m), 1,39-1,34 (9 Н, m). 1-С. Раствор (триметилсилил)диазометана (2 М в диэтиловом эфире) (4,7 мл, 9,45 ммоль) добавляли к карбоновой кислоте 1-В (2,4 г, 8,6 ммоль), растворенной в смеси CH2Cl2/MeOH 5:1 (25 мл) при комнатной температуре. Когда исходный материал исчезал по данным LC/MS, реакционную смесь гасили уксусной кислотой, концентрировали в вакууме и сырой остаток очищали ускоренной хроматографией(градиент EtOAc:гексан) с получением метилового эфира 1-С в виде прозрачного масла. 1-D. В круглодонную колбу помещали мешалку и соединение 1-С (1,03 г, 2,80 ммоль). Добавляли ТФУК (50%) в CH2Cl2 (6 мл) и раствор перемешивали в течение 1 ч при комнатной температуре. Растворитель удаляли в вакууме, добавляли гексан и затем снова упаривали в вакууме досуха и повторяли при необходимости до образования азеотропной смеси ТФУК/гексан. Сырой материал использовали непосредственно на следующей стадии без дополнительной очистки. 1-Е. Сырой материал 1-D растворяли в CH2Cl2 (30 мл), добавляли ссылочное соединение 1 (1,02 г,2,80 ммоль) и HATU (1,12 г, 2,94 ммоль), раствор перемешивали при комнатной температуре в течение 10 мин. Пипеткой добавляли DIPEA (1,5 мл, 8,4 ммоль) и реакционную смесь оставляли перемешиваться в течение ночи при комнатной температуре. Растворитель удаляли в вакууме и сырой материал непосредственно очищали ускоренной хроматографией (40 г силикагеля, градиент гексан/EtOAc). Растворитель удаляли в вакууме с получением соединения 1-Е в виде маслянистого полутвердого вещества. 1-F. Метиловый эфир 1-Е (1,15 г, 1,86 ммоль) растворяли в диоксане (15 мл). Моногидрат гидроксида лития (120 мг, 2,00 ммоль) растворяли в воде (15 мл) и по каплям добавляли к раствору метилового эфира 1-Е и оставляли перемешиваться в течение 3 ч при комнатной температуре. Реакционную смесь концентрировали для удаления диоксана и затем подкисляли с помощью 1 М NaHSO4. Смесь экстрагировали EtOAc и объединенные органические слои промывали насыщенным раствором хлорида натрия и сушили MgSO4. Растворитель удаляли в вакууме с получением карбоновой кислоты 1-F в виде воскоподобного твердого вещества. 1-G. Карбоновую кислоту 1-F (385 мг, 0,78 ммоль) растворяли в CH2Cl2 (10 мл). Добавляли ссылочное соединение 2 (151 мг, 0,52 ммоль) и HATU (356 мг, 0,94 ммоль) и смесь перемешивали в течение 10 мин при комнатной температуре. Пипеткой добавляли DIPEA (0,27 мл, 1,56 ммоль) и реакционную смесь оставляли перемешиваться в течение 3 ч при комнатной температуре. Растворитель удаляли в вакууме, сырой продукт растворяли в EtOAc (50 мл) и промывали 1 М HCl (225 мл), затем насыщенным водным раствором NaHCO3 (225 мл) и насыщенным раствором хлорида натрия (25 мл), сушили безводным Na2SO4. Растворитель удаляли в вакууме и сырой материал непосредственно очищали ускоренной хроматографией (40 г силикагеля, градиент CH2Cl2/МеОН). Растворитель удаляли в вакууме с получением соединения 1-G в виде маслянистого полутвердого вещества. 1-Н. Метиловый эфир 1-G (300 мг, 0,39 ммоль) растворяли в диоксане (18 мл). Гидроксид лития(12 мг, 0,47 ммоль) растворяли в воде (7,5 мл) и добавляли по каплям к раствору метилового эфира 1-G и оставляли перемешиваться в течение 3 ч при комнатной температуре. Реакционную смесь концентрировали в вакууме для удаления диоксана и затем подкисляли 1 М NaHSO4. Продукт экстрагировали EtOAc и объединенные органические слои промывали насыщенным раствором хлорида натрия и сушилиMgSO4. Растворитель удаляли в вакууме с получением карбоновой кислоты 1-Н в виде воскоподобного твердого вещества. 1-I. Карбоновую кислоту 1-Н (72 мг, 0,10 ммоль) растворяли в CH2Cl2 (2 мл). Добавляли 4 аминометилпиридин (13 мг, 0,11 ммоль) и HATU (54 мг, 0,14 ммоль), смесь перемешивали в течение 10 мин при комнатной температуре. Пипеткой добавляли DIPEA (50 мкл, 0,29 ммоль) и реакционную смесь оставляли перемешиваться в течение 3 ч при комнатной температуре. Растворитель удаляли в вакууме, сырой материал растворяли в EtOAc (50 мл) и промывали 1 М HCl (225 мл), затем насыщенным- 16016327 водным раствором NaHCO3 (225 мл) и насыщенным раствором хлорида натрия (25 мл) и сушили безводным Na2SO4. Растворитель удаляли в вакууме и сырой материал 1-I использовали непосредственно в следующей реакции. 1-J. Сырой спирт 1-1 (80 мг, 0,10 ммоль) растворяли в CH2Cl2 (2 мл) и добавляли периодинан ДессаМартина (65 мг, 0,15 ммоль). Реакционную смесь перемешивали в течение 2 ч при комнатной температуре. Растворитель удаляли в вакууме, сырой продукт растворяли в EtOAc (50 мл) и промывали насыщенным раствором Na2S2O3 (225 мл), затем насыщенным водным раствором NaHCO3 (225 мл) и насыщенным раствором хлорида натрия (25 мл) и сушили безводным Na2SO4. Сырой материал очищали ускоренной хроматографией (колонка с 40 г силикагеля), используя градиент CH2Cl2:MeOH с получением кетона 1-J в виде белой пены. Пример 1. Промежуточное соединение 1-J (42 мг, 0,05 ммоль) растворяли в 20% ТФУК в CH2Cl2(3 мл). Реакционную смесь перемешивали при комнатной температуре в течение 2 ч и растворитель удаляли в вакууме. Сырой материал очищали ВЭЖХ с обратной фазой и растворитель лиофилизовали с получением соединения 1 в виде белого порошка в виде моно-ТФУК соли. Примеры 2-9. Примеры 2-7 получали способами, аналогичными описанным для синтеза соединения примера 1,используя подходящие реагенты на стадии (з): пример 2 - используя метиламин; пример 3 - используя бензиламин; пример 4 - используя фенэтиламин; пример 5 - используя анилин; пример 6 - используя циклопропилметиламин и пример 7 - используя циклогексилметиламин. Примеры 8 и 9 получали способами, аналогичными описанным в примере 1, используя ссылочное соединение 1 и ссылочное соединение 3 в качестве реагентов. Для стадии (з) фенэтиламин и 3 фенилпропиламин использовали в примерах 8 и 9 соответственно. Пример 10. В примере 10 использовали следующие реагенты и условия: (а) HATU/DIPEA/CH2Cl2, ссылочное соединение 2, 23 С; (б) LiOH, диоксан/вода (4:1), 23 С; (в) LiOH, диоксан/вода (4:1), 23 С; (г)HATU/DIPEA/CH2Cl2, фенэтиламин, 23 С; (д) Н 2, Pd/C, EtOH, 23C; (e) HATU/DIPEA/CH2Cl2, 23C. Промежуточные соединения 10-A-10-D получали способами, аналогичными описанным в примере 1, стадиях (г), (д), (ж) и (з), используя ссылочное соединение 4, 10-А и 10-В в качестве реагентов для стадий (г), (д) и (ж) соответственно. На стадии (з) промежуточное соединение 10-С использовали в качестве кислотного компонента и фенэтиламин - в качестве аминокомпонента. Промежуточное соединение 10-Е получали способами, аналогичными описанным в примере 2, стадии (f), используя соединение 10-D в качестве субстрата для снятия защитных групп. Промежуточное соединение 10-F получали способами, аналогичными описанным в примере 1, стадии (з), используя соединение 10-В в качестве кислотного компонента и промежуточное соединение 10-Е в качестве амино- 17016327 компонента. Окисление Десса-Мартина и снятие Вос-защит осуществляли аналогично примеру 1, стадиям (и) и (к). Примеры 11, 12. Соединения примеров 11 и 12 получали способами, аналогичными описанным в примере 10, используя ссылочное соединение 5 и ссылочное соединение 6 в качестве кислотного компонента соответственно. Пример 13. В примере 13 использовали следующие реагенты и условия: (а) катализатор метатезиса ГовейдаГраббса, 4-метилен-N-Вос-пиперидин, соединение 13-А, CH2Cl2, 40 С; 65%; (б) ТФУК:CH2Cl2 (1:1),23 С, ВЭЖХ очистка, 30%. 13-А. Это соединение получали как промежуточное соединение при синтезе примера 12. Синтез соединения 13-А являлся аналогичным примеру 1, стадии (и). 13-В. Безводный дихлорметан (5 мл) добавляли пипеткой к соединению 13-А (108 мг, 0,137 ммоль,1,0 экв.), катализатору метатезиса Говейда-Груббса второго поколения [1,3-бис-(2,4,6-триметилфенил)-2(имидазолидинилиден)дихлор (о-изопропоксифенилметилен)рутения(II) дихлорид] (10 мг, 0,013 ммоль,10 мол.%) в атмосфере азота. Пипеткой добавляли N-Boc-4-метиленпиперидин (100 мкл, 0,507 ммоль,3,5 экв.) и реакцию оснащали обратным холодильником и нагревали при 40 С в течение 12 ч. После окончания реакции по данным LC/MS реакционную смесь непосредственно очищали автоматической очисткой на силикагеле (80-100% этилацетата в гексане) с получением соединения 13-В в виде темнозеленого масла. MS m/z 859,5 (М-Вос+1). Пример 13. Это соединение получали способами, аналогичными описанным в примере 1, стадии(к), используя промежуточное соединение 13-В в качестве реагента. Пример 14-57. Соединения примеров 14-19, 22-25, 29-30 получали способами, аналогичными описанным в примере 1, используя подходящие аминокомпоненты на стадии (з), например используя (R)-метилбензиламин,(S)-метилбензиламин и 2-аминометилбензотиазол в примерах 14-16 соответственно. Соединения примеров 20-21, 27-28, 45 и 52 получали способами, аналогичными описанным в примере 13, используя подходящие олефиновые соединения перекрестного метатезиса на стадии (а), например: пример 20, используя трет-бутилэтилен; пример 21, используя 4-виниланизол; пример 27, используя соответствующее промежуточное соединение из примера 26; пример 28, используя соответствующее промежуточное соединение из примера 26, где фенэтиламин замещен n-метоксифенэтиламином; пример 45, используя ссылочное соединение 9 и пример 52, используя 4-метиленциклогексен. Соединение примера 26 получали способами, аналогичными описанным в примере 10, используя ссылочное соединение 7 в качестве кислотного компонента на стадии (б). Соединения примеров 31-44 и 46 получали способами, аналогичными описанным в примере 1, используя подходящие кислотные компоненты на стадии (г) и подходящие аминокомпоненты на стадиях(е) и (з). Соединение примера 47 получали способами, аналогичными описанным в примере 26, за исключением аминокомпонента на стадии (г). Соединения примеров 48-51 и 53-54 получали способами, аналогичными описанным в примере 1,- 18016327 используя ссылочное соединение 10 и подходящий аминокомпонент на стадиях (е) и (з) соответственно. Соединения примеров 55 и 56 а получали способами, аналогичными описанным в примере 1, за исключением осуществления стадий (и) и (к) непосредственно с промежуточными соединениями 1-Н и 1-G соответственно. Соединение примера 57 получали способами, аналогичными описанным в примере 1, используя ссылочное соединение 3 в качестве реагента. Примеры 58-60 получали способами, аналогичными описанным в примере 1, используя подходящий кислотный компонент на стадии (г) и подходящие аминокомпоненты на стадиях (е) и (з). В табл. 1 показаны соединения формулы (1), как описано в примерах 1-60. Таблица 1
МПК / Метки
МПК: C07K 5/08, A61K 31/4025, A61P 11/00, A61K 31/454, A61K 31/4178, A61K 31/4015, A61K 31/4545, C07K 5/06, A61K 31/4439, A61K 31/428
Метки: композиции, соединения, ингибиторов, активирующей, протеазы, каналы, качестве
Код ссылки
<a href="https://eas.patents.su/30-16327-soedineniya-i-kompozicii-v-kachestve-ingibitorov-proteazy-aktiviruyushhejj-kanaly.html" rel="bookmark" title="База патентов Евразийского Союза">Соединения и композиции в качестве ингибиторов протеазы, активирующей каналы</a>
Соединения и композиции как ингибиторы активирующей канал протеазы

Номер патента: 16199
Опубликовано: 30.03.2012
Авторы: Спраггон Глен, Чаттерджи Арнаб К., Бурсулая Бадри, Талли Дейвид С., Видаль Аньес
МПК: A61K 31/425, A61K 31/444, A61K 31/401...
Метки: соединения, протеазы, канал, активирующей, ингибиторы, композиции
Формула / Реферат:
1. Соединение формулы (1)или его фармацевтически приемлемые соли,в которой O-(CR2)p-R2 обозначает заместитель в любом положении кольца А;J обозначает, пиридил или пиперидинил;где один или более из Z1, Z2, Z3, Z4, Z5, Z6 и Z7 обозначает гетероатом, выбранный из N, NR, О или S, а другие атомы Z1-Z7 обозначают СН при условии, что J не обозначает триазолил;В обозначаетили (CR2)k-R5;Y обозначает связь, -SO2-, -NHCO- или -O-(CO)-;R1 обозначает...
Новые соединения и композиции в качестве ингибиторов протеазы

Номер патента: 5369
Опубликовано: 24.02.2005
Авторы: Бьюнин Бэрри А., Пэттерсон Джон У., Крейнэк Эрика А., Брайант Клиффорд М.
МПК: A61K 31/275, C07C 271/22, A61P 11/00...
Метки: новые, протеазы, качестве, ингибиторов, соединения, композиции
Формула / Реферат:
1. Соединение формулы (I) где R1 представляет собой группу формулы (a) где X1 представляет собой -C(O)- или -CH2S(O)2-; R5 представляет собой водород, R7 представляет собой водород; R9 представляет собой (C1-6)алкил, замещенный -(O)R14 или SR14, и где R14 представляет собой (C3-6) циклоалкил(C0-6)алкил, фенил(C0-6)алкил, бифенилил(C0-6)алкил или гетеро(C5-6)арил(C0-6)алкил, где в R9 любая присутствующая алициклическая или ароматическая...
Соединения и композиции в качестве ингибиторов катепсина

Номер патента: 7334
Опубликовано: 25.08.2006
Авторы: Ципфель Шила, Паттерсон Джон В.
МПК: C07C 255/42, A61K 31/277
Метки: качестве, ингибиторов, композиции, соединения, катепсина
Формула / Реферат:
1. Соединение формулы I где X1 является -NHC(R1)(R2)2; X2 является циано, -C(R7)(R8)X3; R7 и R8 вместе образуют оксогруппу; X3 содержит гетеромоноциклическое кольцо, имеющее от 4 до 6 атомов, являющихся членами кольца или конденсированную гетеробициклическую кольцевую систему, содержащую от 8 до 14 атомов, являющихся членами кольца и любой карбоциклический кетон, иминокетон или его тиокетоновое производное; R1 является водородом и R2 является...
Соединения и композиции в качестве ингибиторов протеинкиназ

Номер патента: 15751
Опубликовано: 30.12.2011
Авторы: Пан Шифенг, Ван Син, Пун Дэниел, Чжан Чэньчжи, Ренхауэ Пол, Чжан Цюн, Лю Цзошэн, Ван Ся, Дин Цян, Жэнь Пинда, Албо Памела, Чопиук Грегори С., Чжан Гобао, Сендцик Мартин, Се Юнпин, Хуан Шэньлинь
МПК: A61P 35/00, C07D 401/14, A61K 31/506...
Метки: композиции, ингибиторов, протеинкиназ, соединения, качестве
Формула / Реферат:
1. Соединение формулы Iгде А представляет собой пиразол, необязательно замещенный одним или двумя радикалами R3, где R3выбирают из водорода, C1-6алкила, необязательно замещенного 1-3 атомами F, -XOR4a, -XCN, -XC(O)OR4a, -XC(O)R4b, -XNR4aR4a, -XNR4aXNR4aR4a, -XC(O)NR4aXNR4aR4a, -XC(O)NR4aXR4b, -XNR4aXOR4a, -XC(O)NR4aXOR4a, -XNR4aXCF3, XC(O)NR4aR4a и -XR4b; где каждый X независимо выбирают из связи и C1-3 алкилена; R4a выбирают из водорода и...
Производные аминофенилсульфонамида в качестве ингибиторов протеазы вич

Номер патента: 16060
Опубликовано: 30.01.2012
Авторы: Ласт Стефаан Жюльен, Сюрлеро Доминик Луи Нестор Гилейн, Де Кок Херман Аугустинус, Йонкерс Тим Хьюго Мария, Бонантс Пол Йозеф Габриел Мария, Вигеринк Пит Том Берт Поль
МПК: C07D 401/04, A61K 31/34, A61P 31/18...
Метки: качестве, вич, аминофенилсульфонамида, протеазы, производные, ингибиторов
Формула / Реферат:
1. Соединение, имеющее формулуили его N-оксид, соль, стереоизомерная форма, рацемическая смесь или сложный эфир,где заместитель -NR1R2 присоединен к фенильному кольцу в 3 или 5 положении указанного фенильного кольца;R1 представляет собой водород;R2 представляет собой Het1, Het2 или Y илиR1 и R2 образуют кольцо, указанное кольцо является 3-7-членным и может быть насыщенным или ненасыщенным, в котором члены кольца необязательно замещены гидрокси;Y...
Предыдущий патент: Вакцина
Следующий патент: Способ магнитной сепарации и устройство для его осуществления
Случайный патент: Устройство и способ фиксации окна