Производные пиримидина для лечения патологического роста клеток






Формула / Реферат
1. Производное пиримидина формулы 1

или его фармацевтически приемлемая соль, сольват или гидрат,
где n означает целое число от 1 до 3;
каждый R1означает заместитель, независимо выбранный из группы, состоящей из водорода, -(C1-C6)алкила, который может быть замещен гидроксигруппой, -(C3-C7)циклоалкила и -О(C1-C6)алкила;
каждый R2означает заместитель, независимо выбранный из группы, состоящей из водорода, -(C1-C6)алкила, который может быть замещен гидроксигруппой, и -(C3-C7)циклоалкила;
R1 и R2, взятые вместе с атомом, с которым они связаны, могут образовывать -(C3-C10)циклоалкил, который может быть замещен гидроксигруппой или -SO2R7 и который необязательно прерван -SO2 или -О-;
R3 означает заместитель, выбранный из группы, состоящей из:
(a) водорода;
(b) фенила или -(C1-C9)гетероарила, содержащего 1-3 гетероатома (азота, серы или кислорода), которые необязательно могут быть замещены 1-3 заместителями, выбранными из группы, состоящей из галогена, гидрокси, -(C1-C6)алкила, -(C1-C6)алкил-Р(О)(О(C1-C6)алкила)2, -(C3-C10)циклоалкила, фенила, -NR5R6,
-NHSO2-(C1-C6)алкила, -N((C1-C6)алкил)-SO2-(C1-C6)алкила), -O(C1-C6)алкила, -O-SO2-(C1-C6)алкила, -(CO)-O-(C1-C6)алкила, -SO2-(C1-C6)алкила, SO2CF3, SO2NH2и =O;
(c) -(C3-C10)циклоалкила или -(C2-C9)гетероциклила, содержащего 1-2 гетероатома (азота, серы или кислорода) или группу -SO2, который необязательно может быть замещен 1-2 заместителями, выбранными из группы, состоящей из гидрокси, -(C1-C6)алкила, -О(C1-C6)алкила, (C1-C9)гетероарила, содержащего 1-2 гетероатома азота в качестве гетероатома, -С(О)(C1-C6)алкила, -C(O)CF3, -С(O)-O-(C1-C6)алкила, -С(О)(C3-C10)циклоалкила, -С(О)(C1-C6)алкил-O(C1-C6)алкила, -SO2-(C1-C6)алкила и =O; и
(d) -(C1-C6)алкила, необязательно замещенного 1-2 заместителями, выбранными из группы, состоящей из гидрокси, -(C1-C6)алкила, -(C3-C10)циклоалкила, -(C1-C9)гетероарила, содержащего 2 атома азота в качестве гетероатома, -NR5R6, -NHSO2-(C1-C6)алкила, -N((C1-C6)алкил)-SO2-(C1-C6)алкила, -О(C1-C6)алкила, -С(О)(C1-C6)алкила и -SO2-(C1-C6)алкила;
R4 означает заместитель, выбранный из группы, состоящей из водорода, (C1-C6)алкила; и
каждый из R5 и R6означает заместители, независимо выбранные из группы, состоящей из водорода или
-(C1-C6)алкила;
R7 означает заместитель, выбранный из группы, состоящей из -(C1-C6)алкила и -(C2-C9)гетероциклила, содержащего в качестве гетероатома 2 атома азота.
2. Соединение по п.1, в котором R1 выбран из водорода и -(C1-C6)алкила, необязательно замещенного гидроксигруппой; R2 является водородом или -(C1-C6)алкилом, необязательно замещенным гидроксигруппой; и n равно 1.
3. Соединение по любому из предшествующих пунктов, в котором R3 означает фенил или -(C1-C9)гетероарил, содержащий 1-3 гетероатома (азота, серы или кислорода), необязательно замещенный 1-3 заместителями, независимо выбранными из группы, состоящей из галогена, гидрокси, -(C1-C6)алкила, -(C1-C6)алкил-Р(О)(O(C1-C6)алкила)2, -(C3-C10)циклоалкила, фенила, -NR5R6, -NHSO2-(C1-C6)алкила, -N((C1-C6)алкил)-(SO2-(C1-C6)алкила), -О(C1-C6)алкила, -O-SO2-(C1-C6)алкила, -(CO)-O-(C1-C6)алкила, -SO2-(C1-C6)алкила.
4. Соединение по п.1 или 2, в котором R3 выбран из -(C3-C10)циклоалкила и -(C2-C9)гетероциклила, необязательно замещенного 1-2 заместителями, независимо выбранными из группы, состоящей из гидрокси, -(C1-C6)алкила, -(C1-C9)гетероарила, содержащего 1-2 гетероатома азота в качестве гетероатома, -О(C1-C6)алкила, -С(О)(C1-C6)алкила, -(CO)CF3, -С(О)(C3-C10)циклоалкила, -С(О)О(C1-C6)алкила, -С(О)О(C3-C10)циклоалкила, -С(О)(C1-C6)алкил-O(C1-C6)алкила, -SO2-(C1-C6)алкила.
5. Соединение по п.1 или 2, в котором R3 означает -(C1-C6)алкил, необязательно замещенный 1-2 заместителями, выбранными из группы, состоящей из гидрокси, -(C1-C6)алкила и -(C3-C10)циклоалкила.
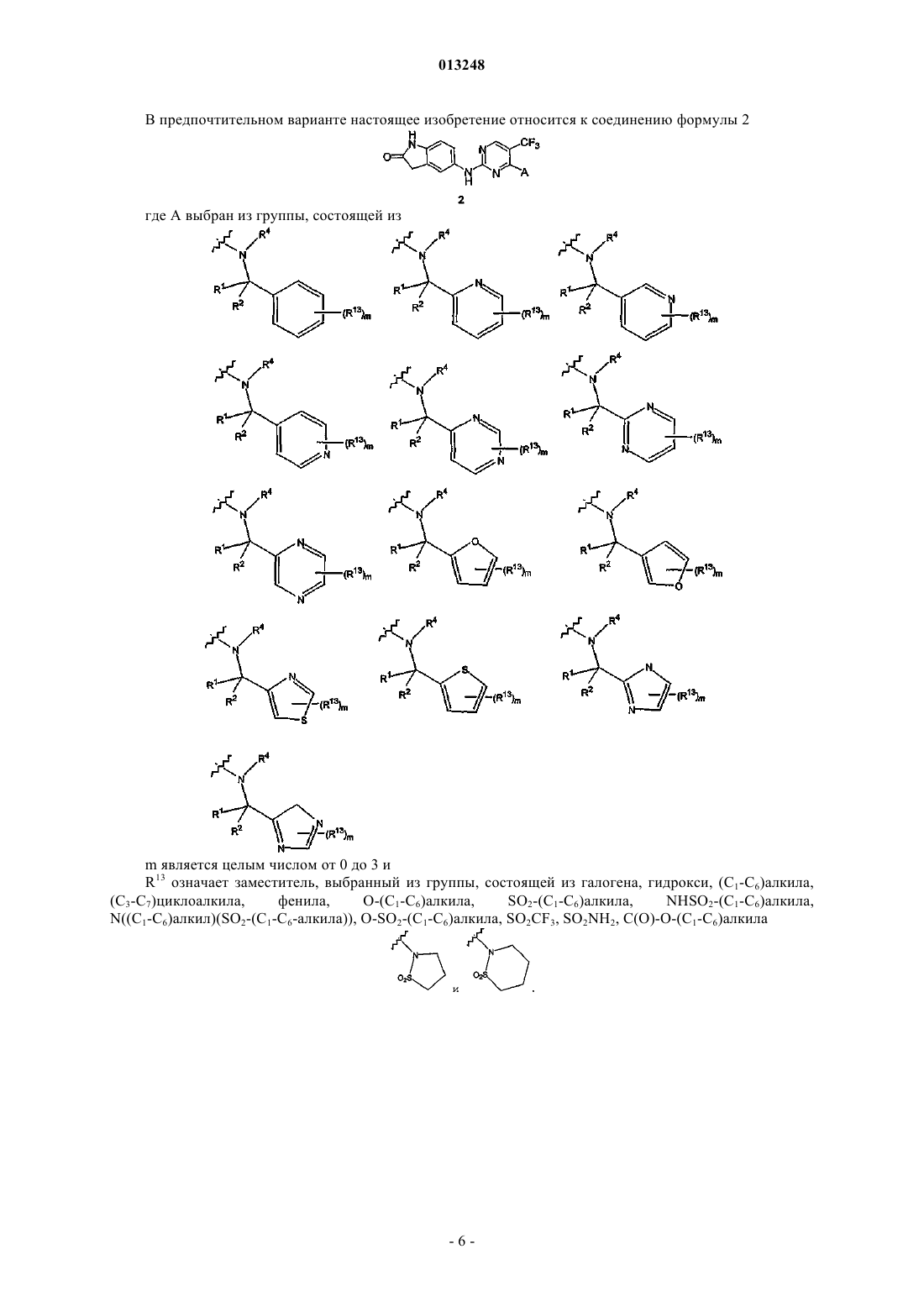
6. Соединение по п.1 формулы 2

где А выбран из группы, состоящей из

где R1, R2 и R4определены в п.1;
m является целым числом от 0 до 3 и
R13 означает заместитель, выбранный из группы, состоящей из галогена, гидрокси, (C1-C6)алкила, (C3-C7)циклоалкила, фенила, О-(C1-C6)алкила, SO2-(C1-C6)алкила, NHSO2-(C1-C6)алкила, N((C1-C6)алкил)(SO2-(C1-C6)алкила), OSO2-(C1-C6)алкила, SO2CF3, SO2NH2, С(О)-О-(C1-C6)алкила,

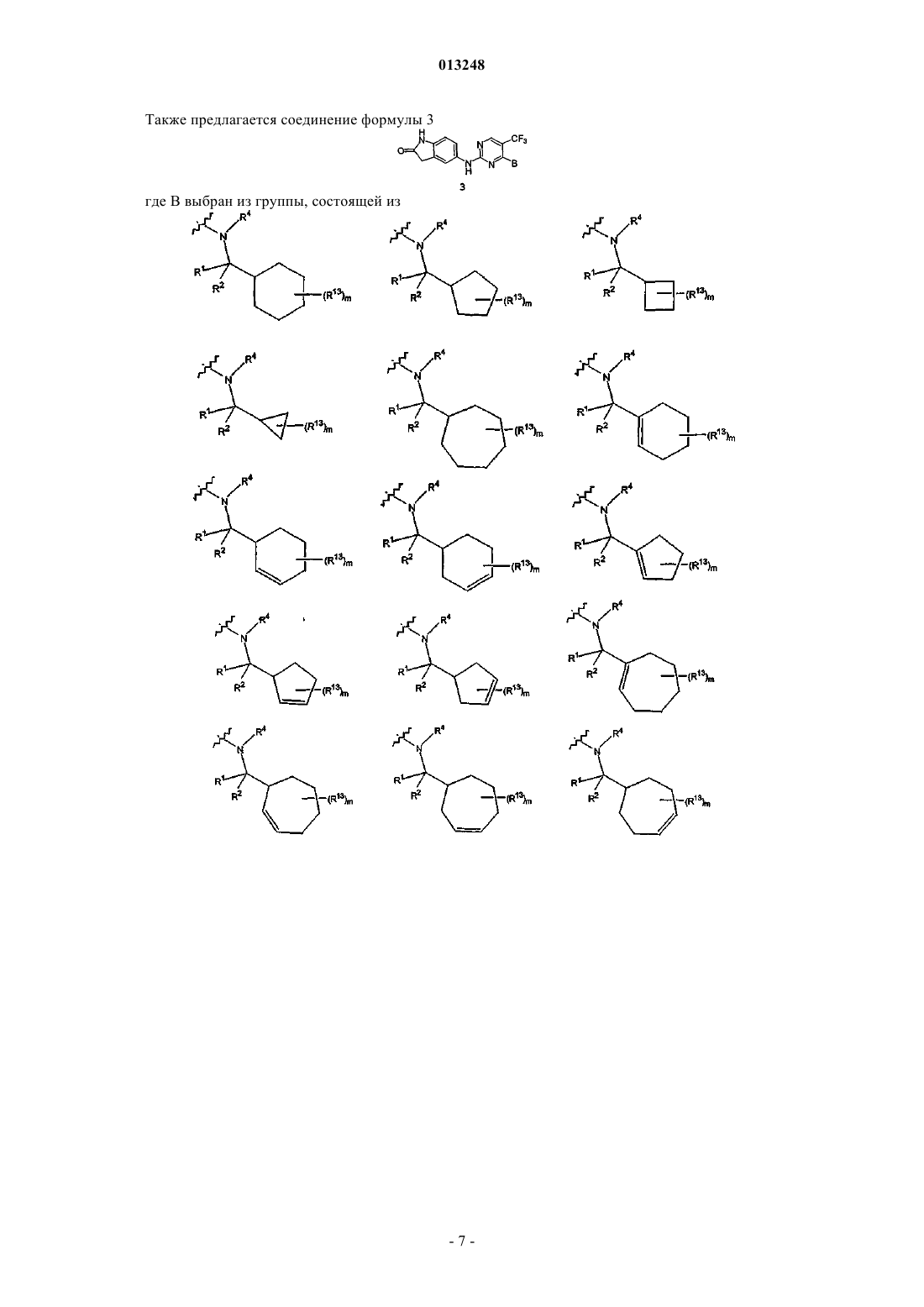
7. Соединение по п.1 формулы 3

где В выбран из группы, состоящей из

где m является целым числом от 0 до 3,
R13 означает заместитель, выбранный из группы, состоящей из галогена, гидрокси, (C1-C6)алкила, (C3-C7)циклоалкила, фенила, О-(C1-C6)алкила, SO2-(C1-C6)алкила, NHSO2-(C1-C6)алкила, N((C1-C6)алкил)(SO2-(C1-C6)-алкила), O-SO2-(C1-C6)алкила, SO2CF3, SO2NH2, С(О)-О-(C1-C6)алкила,

R1, R2и R4 определены в п.1.
8. Соединение по п.1 формулы 4

где D выбран из группы, состоящей из


где q означает целое число от 1 до 2;
m является целым числом от 0 до 3;
R13 означает заместитель, выбранный из группы, состоящей из галогена, гидрокси, (C1-C6)алкила, (C3-C7)циклоалкила, фенила, О-(C1-C6)алкила, SO2-(C1-C6)алкила, NHSO2-(C1-C6)алкила, N((C1-C6)алкил)(SO2-(C1-C6)алкила), OSO2-(C1-C6)алкила, SO2CF3, SO2NH2, С(О)-О-(C1-C6)алкила,

R1, R2и R4 определены в п.1.
9. Соединение по п.1 формулы 5

где Е выбран из группы, состоящей из

где R14 выбран из группы, состоящей из (C1-C6)алкила и (C3-C6)циклоалкила;
R15 выбран из группы, состоящей из водорода, (C1-C6)алкила и (C3-C7)циклоалкила;
q означает целое число от 1 до 2 и
R1, R2и R4 определены в п.1.
10. Соединение, выбранное из группы, состоящей из
5-[4-(3-метансульфонилбензиламино)-5-трифторметилпиримидин-2-иламино]-1,3-дигидроиндол-2-она;
метил-{3-[2-(2-оксо-2,3-дигидро-1Н-индол-5-иламино)-5-трифторметилпиримидин-4-иламино]пропил} амида этансульфоновой кислоты;
5-{4-[(изохроман-1-илметил)амино]-5-трифторметилпиримидин-2-иламино}-1,3-дигидроиндол-2-она;
5-{4-[2-(пиридин-3-илокси)пропиламино]-5-трифторметилпиримидин-2-иламино}-1,3-дигидроиндол-2-она;
3-{[2-(2-оксо-2,3-дигидро-1Н-индол-5-иламино)-5-трифторметилпиримидин-4-иламино]метил}бензолсульфонамида;
5-{4-[(1-метансульфонилпиперидин-3-илметил)амино]-5-трифторметилпиримидин-2-иламино}-1,3-дигидроиндол-2-она;
N-(3-{[2-(2-оксо-2,3-дигидро-1Н-индол-5-иламино)-5-трифторметилпиримидин-4-иламино]метил} фенил)метансульфонамида;
N-метил-N-{2-[2-(2-оксо-2,3-дигидро-1Н-индол-5-иламино)-5-трифторметилпиримидин-4-иламино]этил} метансульфонамида;
5-{4-[(4-метансульфонилморфолин-2-илметил)амино]-5-трифторметилпиримидин-2-иламино}-1,3-дигидроиндол-2-она;
5-[4-(3-метансульфонилметилбензиламино)-5-трифторметилпиримидин-2-иламино]-1,3-дигидроиндол-2-она;
5-{4-[(1-метансульфонилпирролидин-3-илметил)амино]-5-трифторметилпиримидин-2-иламино}-1,3-дигидроиндол-2-она;
N-метил-N-{3-[2-(2-оксо-2,3-дигидро-1Н-индол-5-иламино)-5-трифторметилпиримидин-4-иламино]пропил}метансульфонамида;
5-{4-[2-(1-метансульфонилпиперидин-2-ил)этиламино]-5-трифторметилпиримидин-2-иламино}-1,3-дигидроиндол-2-она;
5-{4-[(4-метансульфонилпиридин-2-илметил)амино]-5-трифторметилпиримидин-2-иламино}-1,3-дигидроиндол-2-она;
5-[4-(3-изопропоксипропиламино)-5-трифторметилпиримидин-2-иламино]-1,3-дигидроиндол-2-она;
5-{4-[(5-метилфуран-2-илметил)амино]-5-трифторметилпиримидин-2-иламино}-1,3-дигидроиндол-2-она;
5-{4-[(бицикло[2.2.1]гепт-5-ен-2-илметил)амино]-5-трифторметилпиримидин-2-иламино}-1,3-дигидроиндол-2-она;
N-(4-фтор-3-{[2-(2-оксо-2,3-дигидро-1Н-индол-5-иламино)-5-трифторметилпиримидин-4-иламино]метил} фенил)-N-метилметансульфонамида;
5-{4-[(6-метансульфонилпиридин-2-илметил)амино]-5-трифторметилпиримидин-2-иламино}-1,3-дигидроиндол-2-она;
5-{4-[(5-метансульфонилпиридин-3-илметил)амино]-5-трифторметилпиримидин-2-иламино}-1,3-дигидроиндол-2-она;
5-[4-(2-метансульфонилбензиламино)-5-трифторметилпиримидин-2-иламино]-1,3-дигидроиндол-2-она;
5-{4-[(1-пиримидин-2-илпиперидин-3-илметил)амино]-5-трифторметилпиримидин-2-иламино}-1,3-дигидроиндол-2-она;
5-{4-[2-(1-метансульфонилпиперидин-2-ил)этиламино]-5-трифторметилпиримидин-2-иламино}-1,3-дигидроиндол-2-она;
N-(2-{[2-(2-оксо-2,3-дигидро-1Н-индол-5-иламино)-5-трифторметилпиримидин-4-иламино]метил}фенил) метансульфонамида;
5-{4-[(1-метансульфонилпирролидин-2-илметил)амино]-5-трифторметилпиримидин-2-иламино}-1,3-дигидроиндол-2-она;
N-метил-N-(2-{[2-(2-оксо-2,3-дигидро-1Н-индол-5-иламино)-5-трифторметилпиримидин-4-иламино]метил} фенил)метансульфонамида;
N-метил-N-(2-метил-6-{[2-(2-оксо-2,3-дигидро-1Н-индол-5-иламино)-5-трифторметилпиримидин-4-иламино]метил}фенил)метансульфонамида;
5-[4-(2-гидроксииндан-1-иламино)-5-трифторметилпиримидин-2-иламино]-1,3-дигидроиндол-2-она;
5-{4-[(1-гидроксициклопентилметил)амино]-5-трифторметилпиримидин-2-иламино}-1,3-дигидроиндол-2-она;
5-{4-[2-гидрокси-2-(1-метансульфонилпиперидин-2-ил)этиламино]-5-трифторметилпиримидин-2-иламино}-1,3-дигидроиндол-2-она и
N-метил-N-(3-{[2-(2-оксо-2,3-дигидро-1Н-индол-5-иламино)-5-трифторметилпиримидин-4-иламино]метил} пиридин-2-ил)метансульфонамида.
11. Фармацевтическая композиция, содержащая эффективное количество соединения по п.1 и фармацевтически приемлемый носитель.
12. Соединение, представляющее собой N-метил-N-(3-{[2-(2-оксо-2,3-дигидро-1Н-индол-5-иламино)-5-трифторметилпиримидин-4-иламино]метил}пиридин-2-ил)метансульфонамид или его фармацевтически приемлемую соль.
13. Фармацевтическая композиция, включающая фармацевтически приемлемый носитель и N-метил-N-(3-{[2-(2-оксо-2,3-дигидро-1Н-индол-5-иламино)-5-трифторметилпиримидин-4-иламино]метил}пиридин-2-ил)метансульфонамид или его фармацевтически приемлемую соль.
14. Применение N-метил-N-(3-{[2-(2-оксо-2,3-дигидро-1Н-индол-5-иламино)-5-трифторметилпиримидин-4-иламино]метил}пиридин-2-ил)метансульфонамида или его фармацевтически приемлемой соли для получения лекарственного средства для лечения рака у млекопитающих.
Текст
013248 Данное изобретение относится к новым производным пиримидина, которые применимы для лечения патологического роста клеток, такого как в случае злокачественной опухоли, у млекопитающих. Данное изобретение также относится к способу применения таких соединений для лечения патологического роста клеток у млекопитающих, в частности у человека, и к фармацевтическим композициям, содержащим такие соединения. Известно, что клетка может стать канцерогенной в силу трансформации части ее ДНК в онкоген(т.е. ген, который при активации приводит к образованию злокачественных опухолевых клеток). Многие онкогены кодируют белки, которые являются атипичными тирозинкиназами, способными вызывать трансформацию клеток. Альтернативно, сверхэкспрессия нормальной протоонкогенной тирозинкиназы также может приводить к пролиферативным расстройствам, иногда приводя к злокачественному фенотипу. Рецепторные тирозинкиназы являются ферментами, которые пронизывают клеточную мембрану и имеют внеклеточный связывающий домен для факторов роста, таких как эпидермальный фактор роста,трансмембранный домен, и внутриклеточную часть, которая функционирует в качестве киназы, фосфорилируя специфичные остатки тирозина в белках и поэтому влияя на пролиферацию клеток. Другие рецепторные тирозинкиназы включают c-verbB-2, c-met, tie-2, PDGFr, FGFr и VEGFR. Известно, что такие киназы часто аномально экспрессируются в распространенных злокачественных опухолях человека, таких как рак молочной железы, рак желудочно-кишечного тракта, такой как рак ободочной кишки, прямой кишки или желудка, лейкоз и рак яичника, бронхов или поджелудочной железы. Также показано,что рецептор эпидермального фактора роста (EGFR), который обладает тирозинкиназной активностью,мутирован и/или сверхэкспрессируется во многих злокачественных опухолях человека, таких как опухоли головного мозга, легких, сквамозных клеток, мочевого пузыря, желудка, молочной железы, головы и шеи, пищевода, гинекологические опухоли и опухоли щитовидной железы. Соответственно, было выяснено, что ингибиторы рецепторных тирозинкиназ применимы в качестве избирательных ингибиторов роста злокачественных клеток млекопитающих. Например, эрбстатин, ингибитор тирозинкиназы, у бестимусных мышей nude избирательно ослабляет рост трансплантированной карциномы молочной железы человека, которая экспрессирует тирозинкиназу рецептора эпидермального фактора роста, но не влияет на рост другой карциномы, которая не экспрессирует рецептор EGF. Таким образом, избирательные ингибиторы некоторых рецепторных тирозинкиназ применимы для лечения патологического роста клеток, в частности злокачественной опухоли, у млекопитающих. Кроме рецепторных тирозинкиназ, избирательные ингибиторы некоторых нерецепторных тирозинкиназ, таких как FAK(киназа фокальной адгезии), Ick, src, abl, или серин/треониновых киназ (например, циклинзависимые киназы) применимы для лечения патологического роста клеток, в частности злокачественной опухоли, у млекопитающих. FAK также известна как тирозиновая протеинкиназа 2, PTK2. Убедительное доказательство свидетельствует, что FAK, цитоплазматическая нерецепторная тирозинкиназа, играет важную роль в путях сигнальной трансдукции клетка-матрикс (Clark и Brugge 1995,Science 268: 233-239), и ее аномальная активация связана с увеличением метастатического потенциала опухолей (Owens et al., 1995, Cancer Research, 55: 2752-2755). FAK исходно идентифицирована как белок с молекулярной массой 125 кДа, высокофосфорилированный по тирозину в клетках, трансформированных v-Src. Впоследствии обнаружили, что FAK является тирозинкиназой, которая локализована у фокальных адгезий, которые являются точками контакта между культивируемыми клетками и подлежащим субстратом и местами интенсивного фосфорилирования тирозина. FAK фосфорилируется и таким образом активируется в ответ на связывание внеклеточного матрикса (ЕСМ) с интегринами. Недавно исследования показали, что увеличение уровней мРНК FAK сопровождалось инвазивной трансформацией опухолей, а ослабление экспрессии FAK (посредством применения антисмысловых олигонуклеотидов) индуцирует апоптоз опухолевых клеток (Xu et al., 1996, Cell Growth and Diff. 7: 413-418). Кроме того чтоFAK экспрессируется в большинстве типов тканей, обнаружены повышенные уровни FAK в большинстве злокачественных опухолей человека, в частности в высокоинвазивных метастазах. Также показано, что различные соединения, такие как производные стирола, обладают свойствами ингибиторов тирозинкиназ. Пять публикаций Европейских патентов, а именно ЕР 0566226 А 1(опубликованный 30 декабря 1992 г.), относятся к некоторым бициклическим производным, в частности производным хиназолина, обладающим противоопухолевыми свойствами, которые являются результатом их ингибирующих свойств в отношении тирозинкиназы. Также международная заявка на выдачу патента WO 92/20642 (опубликованная 26 ноября 1992 г.) относится к некоторым бис-моно- и бициклическим арильным и гетероарильным соединениям в качестве ингибиторов тирозинкиназы, которые применимы для ингибирования аномальной пролиферации клеток. Международные заявки на выдачу патентов WO 96/16960 (опубликованная 6 июня 1996 г.), WO 96/09294-1 013248 производным в качестве ингибиторов тирозинкиназы, которые применимы для тех же целей. Заявка на выдачу патента США с регистрационным 60/435670, поданная 20 декабря 2002 г.( дела патентного поверенного РС 25339), относится к широкому классу новых производных пиримидина, которые являются избирательными ингибиторами FAK. Как таковые указанные соединения применимы для лечения патологического роста клеток. Таким образом, существует необходимость в дополнительных избирательных ингибиторах некоторых рецепторных и нерецепторных тирозинкиназ, применимых для лечения патологического роста клеток, такого как злокачественная опухоль, у млекопитающих. Настоящее изобретение относится к новым производным пиримидина, которые являются избирательными ингибиторами нерецепторной тирозинкиназы FAK и применимы для лечения патологического роста клеток. Настоящее изобретение относится к соединению формулы 1 или его фармацевтически приемлемой соли, сольвату или гидрату,где n означает целое число от 1 до 3; каждый R1 означает заместитель, независимо выбранный из группы, состоящей из водорода,-(C1-C6)алкила, который может быть замещен гидроксигруппой; -(C3-C7)циклоалкила и -O-(C1-C6)алкила; каждый R2 означает заместитель, независимо выбранный из группы, состоящей из водорода,-(C1-C6)алкила, который может быть замещен гидроксигруппой; и -(C3-C7)циклоалкила;R1 и R2, вместе взятые с атомом, с которым они связаны, могут образовывать -(C3-C10)циклоалкил,который может быть замещен гидроксигруппой или -SO2R7 и который необязательно прерван -SO2 или(b) фенила или -(C1-C9)гетероарила, содержащего 1-3 гетероатома (азота, серы или кислорода),которые необязательно могут быть замещены 1-3 заместителями, выбранными из группы, состоящей из галогена, гидрокси, -(C1-C6)алкила, -(C1-C6)алкил-Р(O)(-O-(C1-C6)алкила)2, -(C3-C10)циклоалкила,фенила, -NR5R6, -NHSO2-(C1-C6)алкила, -NC1-C6)алкил)(SO2-(C1-C6)алкила), -O-(C1-C6)алкила,-O-SO2-(C1-C6)алкила, -(CO)-O-(C1-C6)алкила, -SO2-(C1-C6)алкила, SO2CF3, SO2NH2 и =O;(c) -(C3-C10)циклоалкила или -(C2-C9)гетероциклила, содержащего 1-2 гетероатома (азота, серы или кислорода), или группу -SO2, который необязательно может быть замещен 1-2 заместителями, выбранными из группы, состоящей из гидрокси,-(C1-C6)алкила, -O(C1-C6)алкила, (C1-C9)гетероарила, содержащего 1-2 гетероатома азота в качестве гетероатома, -C(O)(C1-C6)алкила, -C(O)CF3, -С(O)-O-(C1-C6)алкила,-С(O)(C3-C10)циклоалкила, -С(O)(C1-C6)алкил-O(C1-C6)алкила, -SO2-(C1-C6)алкила и =O; иR4 означает заместитель, выбранный из группы, состоящей из водорода, (C1-C6)алкила; и каждый из R5 и R6 означают заместители, независимо выбранные из группы, состоящей из водорода или -(C1-C6)алкила;-(C2-C9)гетероциклила, содержащего в качестве гетероатома 2 атома азота. Настоящее изобретение также включает меченные изотопами соединения, которые идентичны соединениям, указанным в формуле 1, не считая того факта, что один или несколько атомов заменены атомом, имеющим массу атома и массовое число, отличное от массы атома и массового числа, обычно встречаемого в природе. Примерами изотопов, которые могут быть включены в соединения согласно изобретению, являются изотопы водорода, углерода, азота, кислорода, фосфора, фтора и хлора, такие как 2H, 3H, 13C, 14C, 15N, 18O, 17O, 31P, 32P, 35S, 18F и 36Cl соответственно. Соединения согласно настоящему изобретению и фармацевтически приемлемые соли указанных соединений, которые содержат указанные выше изотопы и/или другие изотопы других атомов, входят в объем данного изобретения. Некоторые меченные изотопами соединения согласно настоящему изобретению, например соединения, в которые включены такие радиоактивные изотопы, как 3 Н и 14 С, применимы в анализах тканевого распределения лекарственного средства и/или субстрата. Изотопы трития, т.е. 3 Н, и углерода-14, т.е. 14 С, особенно предпочтительны из-за простоты их получения и возможностей регистрации. Кроме того, замещение более тяжелыми изотопами, такими как дейтерий, т.е. 2 Н, может давать некоторые терапевтические преимущества в результате более высокой метаболической стабильности, например увеличенного времени полужизни in vivo, или из-за потребности в более низких дозах, и, следовательно, может быть предпочти-2 013248 тельным в некоторых случаях. Меченные изотопами соединения формулы 1 согласно данному изобретению в общем могут быть получены при осуществлении способов, описанных ниже на схемах и/или в примерах и получениях, путем замены немеченных изотопами реагентов легко доступными меченными изотопами реагентами. Настоящее изобретение также относится к фармацевтически приемлемым кислотно-аддитивным солям соединений формулы 1. Кислотами, которые используются для получения фармацевтически приемлемых кислотно-аддитивных солей указанных выше основных соединений согласно данному изобретению, являются кислоты, которые образуют нетоксичные кислотно-аддитивные соли, т.е. соли, содержащие фармакологически приемлемые анионы, такие соли, как хлорид, бромид, йодид, нитрат, сульфат,бисульфат, фосфат, кислый фосфат, ацетат, лактат, цитрат, кислый цитрат, тартрат, битартрат, сукцинат,малеат, фумарат, глюконат, сахарат, бензоат, метансульфонат, этансульфонат, бензолсульфонат, паратолуолсульфонат и памоат [т.е. 1,1'-метилен-бис-(2-гидрокси-3-нафтоат)]. Изобретение также относится к основно-аддитивным солям формулы 1. Химическими основаниями, которые могут быть использованы в качестве реагентов для получения фармацевтически приемлемых солей оснований указанных соединений формулы 1, которые являются кислотными по своей природе, являются основания, которые образуют нетоксичные соли оснований таких соединений. Такие нетоксичные соли оснований включают, но не ограничены указанным, соли, полученные из таких фармакологически приемлемых катионов, как катионы щелочных металлов (например, калия и натрия) и катионы щелочно-земельных металлов (например, кальция и магния), аммония или водорастворимые соли присоединения аминов, таких как N-метилглюкамин (меглюмин), и соли низшего алканоламмония и другие соли оснований фармацевтически приемлемых органических аминов. Фраза фармакологически приемлемая соль(и) в используемом в данном описании смысле, если не оговорено особо, включает соли кислотных и основных групп, которые могут быть образованы с соединениями согласно настоящему изобретению. Соединения согласно настоящему изобретению, которые являются основаниями по своей природе, способны образовывать множество солей с различными неорганическими и органическими кислотами. Кислотами, которые могут быть использованы для получения фармацевтически приемлемых кислотно-аддитивных солей таких основных соединений, являются кислоты, которые образуют нетоксичные кислотно-аддитивные соли, т.е. соли, содержащие фармакологически приемлемые анионы, такие соли, как гидрохлорид, гидробромид, гидройодид, нитрат, сульфат,бисульфат, фосфат, кислый фосфат, изоникотинат, ацетат, лактат, салицилат, цитрат, кислый цитрат,тартрат, пантотенат, битартрат, аскорбат, сукцинат, малеат, гентизинат, фумарат, глюконат, глюкуронат,сахарат, формиат, бензоат, глютамат, метансульфонат, этансульфонат, бензолсульфонат, паратолуолсульфонат и памоат [т.е. 1,1'-метилен-бис-(2-гидрокси-3-нафтоат)]. Соединения согласно настоящему изобретению, которые содержат остаток основания, такой как аминогруппа, могут образовывать фармацевтически приемлемые соли с различными аминокислотами, кроме кислот, указанных выше. Данное изобретение также охватывает фармацевтические композиции, содержащие соединения формулы 1. Данное изобретение также охватывает соединения формулы 1, содержащие защитные группы. Специалисту в данной области также будет понятно, что соединения согласно изобретению также можно получить с некоторыми защитными группами, которые применимы для очистки или хранения и могут быть удалены перед введением пациенту. Защита и удаление защиты функциональных групп описаны вOrganic Synthesis, 3rd edition, T.W. Greene и P.G.M. Wuts, Wiley-Interscience (1999). Соединения согласно данному изобретению включают все стереоизомеры (например, цис- и трансизомеры) и все оптические изомеры соединений формулы 1 (например, R- и S-энантиомеры), а также рацемические, диастереомерные и другие смеси таких изомеров. Соединения, соли согласно настоящему изобретению могут существовать в нескольких таутомерных формах, включая енольную форму и иминную форму и форму кетона и енамина, а также их геометрические изомеры и смеси. Все такие таутомерные формы включены в объем настоящего изобретения. В растворе таутомеры существуют в виде смесей набора таутомеров. В твердой форме обычно преобладает один таутомер. Даже если может быть описан один таутомер, настоящее изобретение включает все таутомеры данных соединений. Настоящее изобретение также включает атропизомеры согласно настоящему изобретению. Атропизомеры относятся к соединениям формулы 1, которые могут быть разделены в поворотные изомеры. Соединения согласно данному изобретению могут содержать олефиноподобные двойные связи. В том случае, когда присутствуют такие связи, соединения согласно изобретению существуют в цис- и транс-конфигурациях и в виде их смесей. Подразумевается, что подходящий заместитель означает химически и фармацевтически приемлемую функциональную группу, т.е. остаток, который не оказывает негативного действия на биологическую активность предлагаемых в изобретении соединений. Такие подходящие заместители обычно могут быть выбраны специалистами в данной области. Иллюстративные примеры подходящих заместителей включают, но не ограничены указанным, группы галогена, перфторалкильные группы, перфторалкокси-3 013248 группы, алкильные группы, алкенильные группы, алкинильные группы, гидрокси, оксогруппы, меркаптогруппы, алкилтиогруппы, алкоксигруппы, арильные или гетероарильные группы, арилоксигруппы или гетероарилоксигруппы, аралкильные или гетероаралкильные группы, аралкокси- или гетероаралкоксигруппы, HO-(C=O)-группы, аминогруппы, алкил- и диалкиламиногруппы, карбамоильные группы, алкилкарбонильные группы, алкоксикарбонильные группы, алкиламинокарбонильные группы, диалкиламинокарбнильные группы, арилкарбонильные группы, арилоксикарбонильные группы, алкилсульфонильные группы, арилсульфонильные группы и т.п. Специалистам в данной области будет понятно, что многие заместители могут быть замещены дополнительными заместителями. Следующие примеры подходящих заместителей включают заместители, перечисленные в определении соединений формулы 1,включая определенные выше R1-R11. Термин прерван относится к соединениям, в которых атом углерода в цикле заменен элементом,выбранным из группы, состоящей из -(С=O), -SO2, -S-, -O-, -N-, -NH- и -NR5. Например, если R7 означает то цикл может быть прерван или может быть произведена замена гетероатомом азота с образованием следующего цикла: так, что атом углерода в цикле заменяется гетероатомом азота. Соединения согласно изобретению могут вмещать до трех таких замен или прерываний. В используемом в данном описании смысле термин алкил, а также алкильные остатки других групп, указанных в данном описании (например, алкоксигрупп), может означать неразветвленные или разветвленные (такие как метил, этил, н-пропил, изопропил, н-бутил, изобутил, вторичный бутил, третичный бутил); необязательно замещенный 1-3 подходящими заместителями, которые определены выше,такими как фтор, хлор, трифторметил, (C1-C6)алкоксигруппа, (C6-C10)арилоксигруппа, трифторметоксигруппа, дифторметоксигруппа или (C1-C6)алкил. Фраза каждый из указанных алкилов в используемом в данном описании смысле относится к любому из упомянутых выше алкильных остатков в группе, такому как алкоксигруппа, алкенил или алкиламиногруппа. Предпочтительные алкилы включают (C1-C6)алкил, более предпочтительным является(C1-C4)алкил и наиболее предпочтительными являются метил и этил. В используемом в данном описании смысле термин циклоалкил относится к моноциклическому,бициклическому или трициклическому карбоциклическому кольцу (например, циклопропилу, циклобутилу, циклопентилу, циклогексилу, циклогептилу, циклооктилу, циклононилу, циклопентенилу, циклогексенилу, бицикло[2.2.1]гептанилу, бицикло[3.2.1]октанилу и бицикло[5.2.0]нонанилу, и т.д.), необязательно содержащему 1 или 2 двойных связи и необязательно замещенному 1-3 подходящими заместителями, которые определены выше, такими как фтор, хлор, трифторметил, (C1-C6)алкоксигруппа,(C6-C10)арилоксигруппа, трифторметоксигруппа, дифторметоксигруппа или (C1-C6)алкил. В используемом в данном описании смысле термин галоген включает фтор, хлор, бром или йод или фторид, хлорид, бромид или йодид. В используемом в данном описании смысле термин алкенил означает ненасыщенные радикалы с прямой или разветвленной цепью из 2-6 атомов углерода, включая, но не ограничиваясь указанным,этенил, 1-пропенил, 2-пропенил (аллил), изопропенил, 2-метил-1-пропенил, 1-бутенил, 2-бутенил и т.п.,необязательно замещенные 1-3 подходящими заместителями, которые определены выше, такими как фтор, хлор, трифторметил, (C1-C6)алкоксигруппа, (C6-C10)арилоксигруппа, трифторметоксигруппа, дифторметоксигруппа или (C1-C6)алкил. В используемом в данном описании смысле термин алкинил означает радикалы с прямой или разветвленной углеводородной цепью, имеющие одну тройную связь, включая, но не ограничиваясь указанным, этинил, пропинил, бутинил и т.п.; необязательно замещенные 1-3 подходящими заместителями,которые определены выше, такими как фтор, хлор, трифторметил, (C1-C6)алкоксигруппа,(C6-C10)арилоксигруппа, трифторметоксигруппа, дифторметоксигруппа или (C1-C6)алкил. В используемом в данном описании смысле термин карбонил или (С=O) (который используют в таких фразах, как алкилкарбонил, алкил-(С=O)- или алкоксикарбонил) относится к присоединению остатка С=O ко второму остатку, такому как алкил или аминогруппа (т.е. амидогруппа). Алкоксикарбониламиногруппа (т.е. алкокси(С=O)-NH-) относится к группе алкилкарбамата. Карбонильная группа также эквивалентна группе, определяемой в данной описании, как (С=O). Алкилкарбониламиногруппа относится к таким группам, как ацетамид.-4 013248 В используемом в данном описании смысле термин арил означает ароматические радикалы, такие как фенил, нафтил, тетрагидронафтил, инданил и т.п.; необязательно замещенные 1-3 подходящими заместителями, которые определены выше. В используемом в данном описании смысле термин гетероарил относится к ароматической гетероциклической группе обычно с одним гетероатомом в цикле, выбранным из O, S и N. Кроме указанного гетероатома, ароматическая группа необязательно может иметь до 4 атомов N в цикле. Например, к гетероарильным группам относятся пиридил, пиразинил, пиримидинил, пиридазинил, тиенил, фурил, имидазолил, пирролил, оксазолил (например, 1,3-оксазолил, 1,2-оксазолил), тиазолил (например, 1,2-тиазолил,1,3-тиазолил), пиразолил, тетразолил, триазолил (например, 1,2,3-триазолил, 1,2,4-триазолил), оксадиазолил (например, 1,2,3-оксадиазолил), тиадиазолил (например, 1,3,4-тиадиазолил), хинолил, изохинолил,бензотиенил, бензофурил, индолил и т.п.; необязательно замещенные 1-3 подходящими заместителями,которые определены выше, такими как фтор, хлор, трифторметил, (C1-C6)алкоксигруппа,(C6-C10)арилоксигруппа, трифторметоксигруппа, дифторметоксигруппа или (C1-C6)алкил. Термин гетероциклическая в используемом в данном описании смысле относится к циклической группе, содержащей 1-9 атомов углерода и от 1 до 4 гетероатомов, выбранных из N, O, S(O)n или NR. Примеры таких циклов включают азетидинил, тетрагидрофуранил, имидазолидинил, пирролидинил, пиперидинил, пиперазинил, оксазолидинил, тиазолидинил, пиразолидинил, тиоморфолинил, тетрагидротиазинил, тетрагидротиадиазинил, морфолинил, оксетанил, тетрагидродиазинил, оксазинил, оксатиазинил, индолинил, изоиндолинил, хинуклидинил, хроманил, изохроманил, бензоксазинил и т.п. Примерами указанных моноциклических насыщенных или частично насыщенных кольцевых систем являются тетрагидрофуран-2-ил, тетрагидрофуран-3-ил, имидазолидин-1-ил, имидазолидин-2-ил, имидазолидин-4-ил,пирролидин-1-ил, пирролидин-2-ил, пирролидин-3-ил, пиперидин-1-ил, пиперидин-2-ил, пиперидин-3 ил, пиперазин-1-ил, пиперазин-2-ил, пиперазин-3-ил, 1,3-оксазолидин-3-ил, изотиазолидин,1,3-тиазолидин-3-ил, 1,2-пиразолидин-2-ил, 1,3-пиразолидин-1-ил, тиоморфолинил, 1,2-тетрагидротиазин-2-ил, 1,3-тетрагидротиазин-3-ил, тетрагидротиадиазинил, морфолинил, 1,2-тетрагидродиазин-2 ил, 1,3-тетрагидродиазин-1-ил, 1,4-оксазин-2-ил, 1,2,5-оксатиазин-4-ил и т.п.; необязательно содержащие 1 или 2 двойных связи и необязательно замещенные 1-3 подходящими заместителями, которые определены выше, такими как фтор, хлор, трифторметил, (C1-C6)алкоксигруппа, (C6-C10)арилоксигруппа,трифторметоксигруппа, дифторметоксигруппа или (C1-C6)алкил. Гетероатомы азота в используемом в данном описании смысле относятся к N=, N и -NH; где -N= относится к двойной связи азота; N относится к атому азота, имеющему связывания по двум связям, и-N относится к атому азота, содержащему одну связь. Вариант в используемом в данном описании смысле относится к конкретному группированию соединений или применений в отдельные подгруппы. Такие подгруппы можно различать по одному конкретному заместителю, такому как конкретная группа R1 или R3. Другие подгруппы различают по комбинациям различных заместителей, таких как все соединения, в которых R2 является водородом и R1 является (C1-C6)алкилом. Таким образом, настоящее изобретение относится к соединению формулы 1, в которомR1 выбран из водорода и -(C1-C6)алкила, необязательно замещенного гидроксигруппой;R2 является водородом или -(C1-C6)алкилом, необязательно замещенным гидроксигруппой; иn равно 1. Настоящее изобретение, кроме того, относится к соединению формулы 1, в котором R3 означает фенил или -(C1-C9)гетероарил, содержащий 1-3 гетероатома (азота, серы или кислорода), необязательно замещенный 1-3 заместителями, независимо выбранными из группы, состоящей из галогена,гидрокси, -(C1-C6)алкила, -(C1-C6)алкил-Р(O)(O(C1-C6)алкила)2, -(C3-C10)циклоалкила, фенила, -NR5R6,-NHSO2-(C1-C6)алкила, -NC1-C6)алкил)(SO2-(C1-C6)алкила), -O(C1-C6)алкила, -O-SO2-(C1-C6)алкила,-(CO)-O-(C1-C6)алкила, -SO2-(C1-C6)алкила. Настоящее изобретение также относится к соединению формулы 1, в котором R3 выбран из-(C3-C10)циклоалкила и -(C2-C9)гетероциклила, необязательно замещенного 1-2 заместителями,независимо выбранными из группы, состоящей из гидрокси, -(C1-C6)алкила, -(C1-C9)гетероарила, содержащего 1-2 гетероатома азота в качестве гетероатома, -O(C1-C6)алкила, -С(O)(C1-C6)алкила, -(CO)CF3,-С(O)(C3-C10)циклоалкила, -С(O)-O-(C1-C6)алкила, -С(O)-O-(C3-C10)циклоалкила, -С(O)(C1-C6)алкилO(C1-C6)алкила, -SO2-(C1-C6)алкила. Изобретение также охватывает соединения формулы 1, в которых R3 означает -(C1-C6)алкил, необязательно замещенный 1-2 заместителями, выбранными из группы, состоящей из гидрокси, -(C1-C6)алкила и -(C3-C10)циклоалкила. Настоящее изобретение также относится к соединению формулы 1, в котором R3 является водородом.-5 013248 В предпочтительном варианте настоящее изобретение относится к соединению формулы 2m является целым числом от 0 до 3 и-6 013248 Также предлагается соединение формулы 3-7 013248 Настоящее изобретение также относится к соединению формулы 4 где q означает целое число от 1 до 2.-8 013248 Кроме того, настоящее изобретение относится к соединению формулы 5q означает целое число от 1 до 2. Конкретными вариантами настоящего изобретения являются соединения, выбранные из 5-[4-(3-метансульфонилбензиламино)-5-трифторметилпиримидин-2-иламино]-1,3-дигидроиндол-2 она; метил-3-[2-(2-оксо-2,3-дигидро-1 Н-индол-5-иламино)-5-трифторметилпиримидин-4-иламино]пропиламида этансульфоновой кислоты; 5-4-[(изохроман-1-илметил)амино]-5-трифторметилпиримидин-2-иламино-1,3-дигидроиндол-2 она; 5-4-[2-(пиридин-3-илокси)пропиламино]-5-трифторметилпиримидин-2-иламино-1,3-дигидроиндол-2-она; 3-[2-(2-оксо-2,3-дигидро-1 Н-индол-5-иламино)-5-трифторметилпиримидин-4-иламино]метилбензолсульфонамида; 5-4-[(1-метансульфонилпиперидин-3-илметил)амино]-5-трифторметилпиримидин-2-иламино-1,3 дигидроиндол-2-она;N-метил-N-(3-[2-(2-оксо-2,3-дигидро-1 Н-индол-5-иламино)-5-трифторметилпиримидин-4 иламино]метилпиридин-2-ил)метансульфонамида. Данное изобретение также относится к фармацевтической композиции для лечения патологического роста клеток у млекопитающего, включая человека, содержащей определенное количество соединения формулы 1, которое определено выше, или его фармацевтически приемлемой соли, сольвата, которое является эффективным при лечении патологического роста клеток, и фармацевтически приемлемому носителю. В одном варианте указанной композиции указанным патологическим ростом клеток является злокачественная опухоль, включая без ограничения рак легкого, рак костей, рак поджелудочной железы,рак кожи, злокачественную опухоль головы или шеи, кожную или внутриглазную меланому, рак матки,рак яичника, рак прямой кишки, злокачественную опухоль анальной области, рак желудка, рак ободочной кишки, рак молочной железы, рак матки, карциному фаллопиевых труб, карциному эндометрия,карциному шейки матки, вагинальную карциному, карциному вульвы, болезнь Ходжкина, рак пищевода,рак тонкого кишечника, злокачественную опухоль эндокринной системы, рак щитовидной железы, рак паращитовидной железы, рак надпочечника, саркому мягкой ткани, рак уретры, рак пениса, рак простаты, хронический и острый лейкоз, лимфоцитарные лимфомы, рак мочевого пузыря, рак почки или мочеточника, почечноклеточную карциному, карциному почечной лоханки, неоплазмы центральной нервной системы (ЦНС), первичную лимфому ЦНС, опухоли спинного мозга, глиому ствола головного мозга,аденому гипофиза или комбинацию одной или нескольких указанных выше злокачественных опухолей. В одном варианте указанной фармацевтической композиции указанный патологический рост клеток является доброкачественным пролиферативным заболеванием, включая, но не ораничиваясь указанным,псориаз, доброкачественную гипертрофию простаты или рестеноз. Изобретение также относится к фармацевтической композиции для лечения патологического роста клеток у млекопитающего, включая человека, которая содержит определенное количество соединения формулы 1, которое определено выше, или его фармацевтически приемлемой соли, сольвата или пролекарства, которое является эффективным при лечении патологического роста клеток, в комбинации с фармацевтически приемлемым носителем и противоопухолевым агентом, выбранным из группы, состоящей из ингибиторов митоза, алкилирующих агентов, антиметаболитов, интеркалирующих антибиотиков, ингибиторов фактора роста, ингибиторов клеточного цикла, ферментов, ингибиторов топоизомеразы, модификаторов биологического ответа, антигормонов и антиандрогенов. Данная фармацевтическая композиция может быть предназначена для лечения патологического роста клеток у млекопитающего, которая содержит количество соединения формулы 1 или его фармацевтически приемлемой соли, сольвата и количество одного или нескольких веществ, выбранных из средств против ангиогенеза, ингибиторов сигнальной трансдукции и антипролиферативных средств, и данные количества вместе являются эффективными при лечении указанного патологического роста клеток. Средства против ангиогенеза, такие как ингибиторы ММР-2 (металлопротеиназа 2 матрикса), ингибиторы ММР-9 (металлопротеиназа 9 матрикса) и ингибиторы ЦОГ-II (циклооксигеназа II) могут быть использованы вместе с соединением формулы 1 в фармацевтических композициях, приведенных в данном описании. Примеры применимых ингибиторов ЦОГ-11 включают CELEBREX (алекоксиб), валдекоксиб и рофекоксиб. Примеры применимых ингибиторов металлопротеиназ матрикса описаны в WO 96/33172(опубликованной 24 октября 1996 г.), WO 96/27583 (опубликованной 7 марта 1996 г.), европейской заявке на выдачу патента 97304971.1 (поданной 8 июля 1997 г.), европейской заявке на выдачу патента 99308617.2 (поданной 29 октября 1999 г.), WO 98/07697 (опубликованной 26 февраля 1998 г.),WO 98/03516 (опубликованной 29 января 1998 г.), WO 98/34918 (опубликованной 13 августа 1998 г.),WO 98/34915 (опубликованной 13 августа 1998 г.), WO 98/33768 (опубликованной 6 августа 1998 г.),WO 98/30566 (опубликованной 16 июля 1998 г.), публикации европейского патента 606046 (опубликованной 13 июля 1994 г.), публикации европейского патента 931788 (опубликованной 28 июля 1999 г.),WO 90/05719 (опубликованной 31 марта 1990 г.), WO 99/52910 (опубликованной 21 октября 1999 г.),WO 99/52889 (опубликованной 21 октября 1999 г.), WO 99/29667 (опубликованной 17 июня 1999 г.), ме- 10013248 ждународной заявке РСТPCT/IB98/01113 (поданной 21 июля 1998 г.), европейской заявке на выдачу патента 99302232.1 (поданной 25 марта 1999 г.), заявке на выдачу патента Великобритании номер 9912961.1 (поданной 3 июня 1999 г.), предварительной заявке на выдачу патента США 60/148464 (поданной 12 августа 1999 г.), патенте США 5863949 (опубликованном 26 января 1999 г.), патенте США 5861510 (опубликованном 19 января 1999 г.) и в публикации патента Европы 780386 (опубликованной 25 июня 1997 г.), все публикации включены в данное описание в виде ссылки в полном объеме. Предпочтительными ингибиторами ММР-2 и ММР-9 являются ингибиторы, которые обладают низкой активностью или не имеют активности, ингибирующей ММР-1. Более предпочтительными являются ингибиторы, которые избирательно ингибируют ММР-2 и/или ММР-9 по сравнению с другими металлопротеиназами матрикса (т.е. ММР-1, ММР-3, ММР-4, ММР-5, ММР-6, ММР-7, ММР-8, ММР-10, ММР-11,ММР-12 и ММР-13). Некоторыми конкретными примерами ингибиторов ММР, применимых в комбинации с соединениями согласно настоящему изобретению, являются AG-3340, RO 32-3555, RS 13-0830 и следующие соединения: 3-4-(4-фторфенокси)бензолсульфонил]-(1-гидроксикарбамоилциклопентил)амино]пропионовая кислота; гидроксиамид 3-экзо-3-[4-(4-фторфенокси)бензолсульфониламино]-8-оксабицикло[3.2.1]октан-3 карбоновой кислоты; гидроксиамид(2R,3R)-1-[4-(4-фтор-2-метилбензилокси)бензолсульфонил]-3-гидрокси-3-метилпиперидин-2-карбоновой кислоты; 3-4-(4-фторфенокси)бензолсульфонил]-(1-гидроксикарбамоил-1-метилэтил)амино]пропионовая кислота; 3-4-(4-фторфенокси)бензолсульфонил]-(4-гидроксикарбамоилтетрагидропиран-4-ил)амино]пропионовая кислота; гидроксиамид 3-экзо-3-[4-(4-хлорфенокси)бензолсульфониламино]-8-оксабицикло[3.2.1]октан-3 карбоновой кислоты; гидроксиамид 3-эндо-3-[4-(4-фторфенокси)бензолсульфониламино]-8-оксабицикло[3.2.1]октан-3 карбоновой кислоты и гидроксиамид 3-[4-(4-фторфенокси)бензолсульфониламино]тетрагидрофуран-3-карбоновой кислоты; и фармацевтически приемлемые соли, сольваты указанных соединений. Соединения формулы 1 и их фармацевтически приемлемые соли, сольваты также могут быть использованы в комбинации с ингибиторами сигнальной трансдукции, такими как агенты, которые могут ингибировать ответы EGFR (рецептора эпидермального фактора роста), такие как EGFR-антитела, EGFантитела и молекулы, которые являются ингибиторами EGFR; ингибиторы VEGF (фактора роста эндотелия сосудов); и ингибиторы рецептора erbB2, такие как органические молекулы или антитела, которые связываются с рецептором erbB2, например HERCEPTIN (Genentech, Inc. of South San Francisco,California, USA). Ингибиторы EGFR описаны, например, в WO 95/19970 (опубликованной 27 июля 1995 г.),WO 98/14451 (опубликованной 9 апреля 1998 г.), WO 98/02434 (опубликованной 22 января 1998 г.) и в патенте США 5747498 (опубликованном 5 мая 1998 г.).EGFR-ингибирующие агенты включают, но не ограничены указанным, CI-1033 (Pfizer Inc.), моноклональные антитела С 225 и анти-EGFR 22-Mab (ImClone Systems Incorporated of New York, New York,USA), соединения ZD-1839 (AstraZeneca), BIBX-1382 (Boehringer Ingelheim), MDX-447 (Medarex Inc. ofSouth San Francisco, California, USA), также можно комбинировать с соединением формулы 1. Ингибиторы VEGF описаны, например, в WO 99/24440 (опубликованной 20 мая 1999 г.), международной заявке РСТ PCT/IB99/00797 (поданной 3 мая 1999 г.), в WO 95/21613 (опубликованной 17 августа 1995 г.), WO 99/61422 (опубликованной 2 декабря 1999 г.), патенте США 5834504 (опубликованном 10 ноября 1998 г.), WO 98/50356 (опубликованной 12 ноября 1998 г.), патенте США 5883113- 11013248 22 января 1998 г.), WO 99/16755 (опубликованной 8 апреля 1999 г.) и WO 98/02437 (опубликованной 22 января 1998 г.), все из которых включены в данное описание в виде ссылки в полном объеме. Другими примерами некоторых конкретных ингибиторов VEGF являются IM862 (Cytran Inc. ofKirkland, Washington, USA); моноклональное антитело против VEGF Genentech, Inc. of South SanFrancisco, California; и ангиозим, синтетический рибозим из Ribozyme (Boulder, Colorado) и Chiron(Chiron), могут быть введены в комбинации с соединением формулы 1. Такие ингибиторы erbB2 включают ингибиторы, описанные в WO 98/02434 (опубликованной 22 января 1998 г.), WO 99/35146 (опубликованной 15 июля 1999 г.), WO 99/35132 (опубликованной 15 июля 1999 г.), WO 98/02437 (опубликованной 22 января 1998 г.), WO 97/13760 (опубликованной 17 апреля 1997 г.), WO 95/19970 (опубликованной 27 июля 1995 г.), патенте США 5587458 (опубликованном 24 декабря 1996 г.) и патенте США 5877305 (опубликованном 2 марта 1999 г.), каждая публикация включена в данное описание в виде ссылки в полном объеме. Ингибиторы рецептора erbB2, применимые в настоящем изобретении, также описаны в предварительной заявке на выдачу патента США 60/117341, поданной 27 января 1999 г., и в предварительной заявке на выдачу патента США 60/117346, поданной 17 января 1999 г., обе заявки включены в данное описание в виде ссылки в полном объеме. Другие антипролиферативные средства, которые могут быть использованы с соединениями согласно настоящему изобретению, включают ингибиторы HDI (CI-994, Pfizer Inc.), MEK (CI-1040, Pfizer Inc.),фермент фарнезилпротеинтрансферазу и рецепторную тирозинкиназу PDGFr, включая соединения, описанные и заявленные в следующих заявках на выдачу патентов США: 09/221946 (поданной 28 декабря 1998 г.); 09/454058 (поданной 2 декабря 1999 г.); 09/501163 (поданной 9 февраля 2000 г.); 09/539930 (поданной 31 марта 2000 г.); 09/202796 (поданной 22 мая 1997 г.); 09/384339 (поданной 26 августа 1999 г.) и 09/383755 (поданной 26 августа 1999 г.); и соединения, описанные и заявленные в следующих предварительных заявках на выдачу патента США: 60/168207 (поданной 30 ноября 1999 г.); 60/170119 (поданной 10 декабря 1999 г.); 60/177718 (поданной 21 января 2000 г.); 60/168217 (поданной 30 ноября 1999 г.) и 60/200834 (поданной 1 мая 2000 г.). Соединения согласно изобретению также могут быть использованы в комбинации с ингибиторами топоизомеразы I, например, иринотеканом (Camptosar) и эдотекарином. Каждая из указанных выше заявок на выдачу патента и предварительных заявок на выдачу патента включена в данное описание в виде ссылки в полном объеме. Соединение формулы 1 также может быть использовано с другими агентами, применимыми при лечении патологического роста клеток или злокачественной опухоли, включая, но не ограничиваясь указанным, агенты, способные усиливать противоопухолевые иммунные ответы, такие как антитела противCTLA4 (антиген 4 цитотоксических лимфоцитов) и другие агенты, способные блокировать CTLA4; и антипролиферативные агенты, такие как другие ингибиторы фарнезилпротеинтрансферазы, например,ингибиторы фарнезилпротеинтрансферазы, описанные в публикациях, цитированных в разделе Уровень техники выше. Конкретные CTLA4-антитела, которые могут быть использованы в настоящем изобретении, включают антитела, описанные в предварительной заявке на выдачу патента США 60/113647 (поданной 23 декабря 1998 г.), которая включена в данное описание в виде ссылки в полном объеме. Патологический рост клеток в используемом в данном описании смысле, если не оговорено особо, относится к росту клеток, который не зависит от нормальных регуляторных механизмов (например,отсутствие контактного ингибирования). Термин включает патологический рост:(1) опухолевых клеток (опухолей), которые пролиферируют при экспрессии мутантной тирозиновой киназы или сверхэкспрессии рецепторной тирозинкиназы;(2) доброкачественных и злокачественных клеток при других пролиферативных заболеваниях, при которых происходит активация атипичной тирозинкиназы;(3) любых опухолей, которые пролиферируют при действии рецепторных тирозинкиназ;(4) любых опухолей, которые пролиферируют при активации атипичной сериновой/треониновой киназы; и(5) доброкачественных и злокачественных клеток при других пролиферативных заболеваниях, при которых происходит активация атипичной сериновой/треониновой киназы.- 12013248 Соединения согласно настоящему изобретению являются эффективными ингибиторами тирозиновых протеинкиназ FAK и, следовательно, все подходят для терапевтического применения в качестве антипролиферативных средств (например, противораковых), противоопухолевых средств (например, эффективных против солидных опухолей), средств против ангиогенеза (например, для остановки или предотвращения пролиферации кровеносных сосудов) у млекопитающих, в частности у человека. Соединения согласно настоящему изобретению, в частности, применимы для профилактики и лечения множества гиперпролиферативных расстройств у человека, таких как злокачественные и доброкачественные опухоли печени, почек, мочевого пузыря, молочной железы, желудка, яичника, прямой и ободочной кишки,простаты, поджелудочной железы, легкого, вульвы, щитовидной железы, карциномы печени, саркомы,глиобластомы, опухоли головы и шеи и другие гиперпластические состояния, такие как доброкачественная гиперплазия кожи (например, псориаз) и доброкачественная гиперплазия простаты (например, ВРН). Кроме того, предполагается, что соединение согласно настоящему изобретению может обладать активностью против ряда лейкозов и лимфоидных злокачественных новообразований. Настоящее изобретение также относится к соединению, представляющему собойN-метил-N-(3-[2-(2-оксо-2,3-дигидро-1 Н-индол-5-иламино)-5-трифторметилпиримидин-4-иламино]метилпиридин-2-ил)метансульфонамид или его фармацевтически приемлемую соль. Кроме того, настоящее изобретение обеспечивает фармацевтическую композицию, включающую фармацевтически приемлемый носитель и N-метил-N-(3-[2-(2-оксо-2,3-дигидро-1 Н-индол-5-иламино)5-трифторметилпиримидин-4-иламино]метилпиридин-2-ил)метансульфонамид или его фармацевтически приемлемую соль. Также настоящее изобретение относится к применению N-метил-N-(3-[2-(2-оксо-2,3-дигидро-1 Ниндол-5-иламино)-5-трифторметилпиримидин-4-иламино]метилпиридин-2-ил)метансульфонамида или его фармацевтически приемлемой соли для получения лекарственного средства для лечения рака у млекопитающих. В одном предпочтительном варианте согласно настоящему изобретению злокачественная опухоль выбрана из рака легкого, рака костей, рака поджелудочной железы, рака кожи, злокачественной опухоли головы или шеи, кожной или внутриглазной меланомы, рака матки, рака яичника, злокачественной опухоли гинекологических органов, рака прямой кишки, злокачественной опухоли анальной области, рака желудка, рака ободочной кишки, рака молочной железы, рака матки, карциномы фаллопиевых труб, карциномы эндометрия, карциномы шейки матки, вагинальной карциномы, карциномы вульвы, болезни Ходжкина, рака пищевода, рака тонкого кишечника, злокачественной опухоли эндокринной системы,рака щитовидной железы, рака паращитовидной железы, рака надпочечника, саркомы мягкой ткани, рака уретры, рака пениса, рака сквамозных клеток, рака простаты, хронического или острого лейкоза, лимфоцитарных лимфом, рака мочевого пузыря, рака почки или мочеточника, почечно-клеточной карциномы,карциномы почечной лоханки, неоплазм центральной нервной системы (ЦНС), первичной лимфомы ЦНС, опухолей спинного мозга, опухолей головного мозга, аденомы гипофиза или комбинации одной или нескольких указанных выше злокачественных опухолей. В более предпочтительном варианте злокачественная опухоль выбрана из солидной опухоли, такой как без ограничения опухоль молочной железы, легкого, ободочной кишки, головного мозга, простаты,желудка, поджелудочной железы, яичника, кожи (меланома), эндокринной системы, матки, семенника и мочевого пузыря. Соединения согласно настоящему изобретению также могут быть применимы при лечении дополнительных расстройств, в которые вовлечены аномальная экспрессия, взаимодействия лиганд/рецептор или события активации или передачи сигнала, связанные с различными тирозиновыми протеинкиназами. Такие расстройства могут включать расстройства, происходящие в нервных клетках, глие, астроцитах,гипоталамусе и других железах, макрофагах, эпителии, строме и бластоцеле, в которые вовлечены аномальная функция, экспрессия, активация или передача сигнала тирозинкиназ erbB. Кроме того, соединения согласно настоящему изобретению могут иметь терапевтическую применимость при воспалительных, ангиогенных и иммунологических расстройствах, в которые вовлечены как идентифицированные,так и пока неидентифицированные тирозинкиназы, которые ингибируются соединениями согласно настоящему изобретению. Термин лечение в используемом в данном описании смысле, если не оговорено особо, означает отмену, ослабление, ингибирование прогрессирования или предотвращение расстройства или состояния,к которому применяется такой термин, или одного или нескольких симптомов такого расстройства или состояния. Термин лечение в используемом в данном описании смысле, если не оговорено особо, относится к действию лечения в том виде, как лечение определено непосредственно выше. Настоящее изобретение также относится к фармацевтической композиции, содержащей соединение формулы 1 или его фармацевтически приемлемую соль или сольват, которые определены в данном описании выше, в ассоциации с фармацевтически приемлемым адъювантом, разбавителем или носителем. Фармацевтическую композицию согласно изобретению получают смешиванием соединения формулы 1 или его фармацевтически приемлемой соли или сольвата, которые определены в данном описании выше, с фармацевтически приемлемым адъювантом, разбавителем или носителем.- 13013248 Для указанных выше терапевтических применений вводимая доза, конечно, будет варьировать в зависимости от используемого соединения, способа введения, требуемого лечения и показанного расстройства. Суточная доза соединения формулы 1/соли/сольвата (активного ингредиента) может быть в пределах от 1 мг до 1 г, предпочтительно от 1 до 250 мг, более предпочтительно от 10 до 100 мг. Настоящее изобретение также предусматривает композиции длительного высвобождения. Подробное описание изобретения Соединения формулы 1 могут быть получены с использованием пути синтеза, изображенного на схеме 1. Заместители на схеме 1 имеют такое же значение, как и заместители, определенные для формулы 1. Схема 1 Соединения формулы 1 могут быть получены, исходя из 5-аминооксиндола (2) и пиримидина (3). Объединение 3 с эквимолярным количеством кислоты Льюиса при температурах в диапазоне от -15 до 45 С в течение периода времени, равного 10-60 мин, в инертном растворителе (или смеси растворителей) с последующим добавлением 2 и подходящего основания, в результате через 1-24 ч образуется промежуточный продукт 4-хлорпиримидин (4) с высокими выходами. Примеры инертных растворителей включают без ограничения ТГФ, 1,4-диоксан, н-BuOH, изо-PrOH, дихлорметан и 1,2-дихлорэтан. Примеры подходящих используемых оснований могут включать без ограничения (i) ненуклеофильные органические основания, например триэтиламин или диизопропилэтиламин, (ii) неорганические основания, такие как карбонат натрия или карбонат цезия, или (iii) связанные со смолой основания, такие как МР-карбонат. Примеры кислот Льюиса включают без ограничения галогенидные соли магния, меди, цинка, олова или титана. В следующей реакции промежуточный продукт 4 подвергают взаимодействию с амином формулы 5 либо неразбавленным, либо в присутствии инертного растворителя (или смеси растворителей) при температурах в диапазоне от 0 до 150 С, получая соединения формулы 1. Необязательно данную реакцию можно проводить в присутствии подходящего основания. Примеры подходящих растворителей для данной реакции включают без ограничения ТГФ, 1,4-диоксан, ДМФА, N-метилпирролидинон,EtOH, н-BuOH, изо-PrOH, дихлорметан, 1,2-дихлорэтан, ДМСО или ацетонитрил. Подходящие основания указаны выше. Соединения согласно настоящему изобретению могут быть искусственно превращены в другие соединения согласно изобретению способами, известными специалистам в данной области. Только в иллюстративных целях и без ограничения такие способы включают:a) удаление защитной группы способами, указанными в T.W. Greene and P.G.M. Wuts, Protectiveb) замену удаляемой группы (галогенида, мезилата, тозилата и т.д.) функциональными группами,такими как без ограничения первичный или вторичный амин, тиол или спирт, с образованием вторичного или третичного амина, тиоэфира или эфира соответственно;c) обработку фенил- (или замещенный фенил) карбаматов первичными или вторичными аминами с образованием соответствующих мочевин, как в Thavonekham, В et. al. Synthesis (1997), 10, р. 1189;d) восстановление пропаргилового или гомопропаргилового спирта или N-Boc-защищенных первичных аминов до соответствующих Е-аллильных или Е-гомоаллильных производных обработкой бис(2-метоксиэтокси)алюмогидридом натрия (Red-AI), как в Denmark, S.E.; Jones, T.K.J. Org. Chem. (1982),47, 4595-4597 или van Benthem, R.A.T.M.; Miches, J.J.; Speckamp, W.N. Synlett (1994), 368-370;e) восстановление алкинов до соответствующих производных Z-алкена обработкой водородным газом и Pd-катализатором как в Tomassy, В. et. al. Synth. Commun. (1998), 28, p. 1201;f) обработку первичных и вторичных аминов изоцианатом, хлорангидридом кислоты (или другим активированным производным карбоновой кислоты), алкиларилхлорформиатом или сульфонилхлоридом с получением соответствующей мочевины, амида, карбамата или сульфонамида;g) восстановительное аминирование первичного или вторичного амина с использованием альдегида или кетона и подходящего восстанавливающего реагента;h) обработку спиртов изоцианатом, хлорангидридом кислоты (или другим активированным производным карбоновой кислоты), алкил/арилхлорформиатом или сульфонилхлоридом с получением соответствующего карбамата, сложного эфира, карбоната или сложного эфира сульфоновой кислоты. Амины формулы 5 могут быть приобретены и использованы непосредственно или, альтернативно,могут быть получены специалистом в данной области с использованием обычных химически превращений. Например, арилалкиламины или гетероарилалкиламины могут быть получены из соответствующего нитрила каталитическим гидрированием с использованием катализаторов, таких как Pd/C или никель Ренея, или восстановлением алюмогидридом лития (см. Rylander, Catalytic Hydrogenation in Organic Исходные вещества, нитрилы, могут быть либо приобретены, либо получены из соответствующего арил/гетероарилбромида, йодида или трифторметансульфоната и Zn(CN)2 с использованием условий сочетания в присутствии Pd, описанных в Tschaen, D.M., et al. Synthetic Communications (1994), 24, 6,p. 887-890 Альтернативно, бензиламины или гетероарилметиламины могут быть получены при взаимодействии подходящего арилалкил- или гетероарилалкилгалогенида и калиевой соли (Boc)2NH и последующем удалении Boc-групп с использованием кислоты Амины, защищенные формы аминов, предшественники аминов и предшественники защищенных форм аминов формулы 5 могут быть получены при объединении подходящего алкина или алкенилстаннана, алкенилборана, алкенилбороновой кислоты, сложного эфира бороновой кислоты с подходящим арил- или гетероарилбромидом, йодидом или трифторметансульфонатом с использованием условий сочетания в присутствии Pd, которые описаны в Tsuji, J., Palladium Reagents and Catalysis, John Wiley and Соответствующим образом защищенные амины формулы 5 могут быть превращены в различные амины формулы 5 согласно способам, известным специалистам в данной области, например, но не ораничиваясь указанным:(а) окислением тиоэфира до сульфоксида или сульфона(b) N-алкилирование сульфанилида можно осуществить посредством фазового перехода с использованием условий, описанных в Brehme, R. Synthesis (1976), р. 113-114. Как понятно специалистам в данной области, химическое превращение для того, чтобы превратить арилгалогенид или трифторметансульфонат, или гетероарилгалогенид, или трифторметансульфонат в ароматический или гетероароматический амин, можно осуществить с использованием условий, описанных в настоящее время в литературе, см. Hartwig, J.F.: Angew. Chem. Int. Ed. (1998), 37, p. 2046-2067,Wolfe, J.P.; Wagaw, S.; Marcoux, J.F.; Buchwald, S.L.; Acc. Chem. Res., (1998), 31, p. 805-818, Wolfe, J.P.;Chemistry (2002), p. 131-209, и цитированных в указанной работе ссылках. Кроме того, как понятно специалистам в данной области, такие же химические превращения арил- или гетероариламинирования,альтернативно, можно осуществить, используя предшественники в виде нитрила (или первичного амида), которые дают амины формулы 5 после восстановления нитрила (или амида). Защищенные амины формулы 5, кроме того, можно превратить в другие амины формулы 5 согласно способам, известным специалистам в данной области Активность соединений формулы 1 in vitro может быть определена следующим способом. Более конкретно, следующий анализ обеспечивает способ определения того, ингибируют ли соединения формулы 1 тирозинкиназную активность каталитической конструкции FAK (410-689). Анализ осуществляют в форме, основанной на ELISA, измеряя ингибирование поли-glu-tyr-фосфорилирования под действиемFAK (410-689). Протокол анализа состоит из трех частей.Ni-NTA-агароза (Qiagen); колонка XK-16 (Amersham-Pharmacia); 300 мМ имидазола; препаративная колонка Superdex 200 HiLoad 16/60 (Amersham Biotech.); антитело: антифосфотирозиновое антитело Ру 20, конъюгированное с HRP (Transduction labs);FAKcd: очищенная и активированная на месте; субстрат пероксидазы ТМВ Microwell (Oncogene Research ProductsCL07); БСА: SigmaА 3294; Твин-20: SigmaР 1379; ДМСО: SigmaD-5879;D-PBS: Gibco14190-037. Реагенты для очистки: буфер А: 50 мМ HEPES, рН 7,0,500 мМ NaCl,0,1 мМ ТСЕР,таблетки полной смеси ингибиторов протеаз ТМ (Roche); буфер В: 25 мМ HEPES, рН 7,0,400 мМ NaCl,0,1 мМ ТСЕР; буфер С: 10 мМ HEPES рН 7,5,200 мМ сульфата аммония,0,1 мМ ТСЕР. Реагенты для активации:FAK(410-689): 3 пробирки замороженных аликвот по 150 мкл/пробирку, всего 450 мкл с концентрацией 1,48 мг/мл (660 мкг),His-Src(249-524): 0,74 мг/мл маточного раствора в 10 мМ HEPES, 200 мМ (NH4)2SO4; буфер для реакции Src (Upstate Biotech): 100 мМ трис-HCl рН 7,2,125 мМ MgCl2, 25 мМ MnCl2, 2 мМ ЭДТА, 250 мкМ Na3VO4, 2 мМ DTT; смесь Mn2+/АТФ (Upstate Biotech) 75 мМ MnCl2,500 мкМ АТФ,20 мМ MOPS, рН 7,2,1 мМ Na3VO4,25 мМ -глицеринфосфата,5 мМ EGTA,1 мМ DTT; АТФ: 150 мМ исходного раствора;DTT: 1 М исходного раствора. Реагенты для ELISA киназы FAKcd: буфер для фосфорилирования: 50 мМ HEPES, рН 7,5,125 мМ NaCl, 48 мМ MgCl2; буфер для промывки: TBS + 0,1% Твин-20; блокирующий буфер: Трис-солевой буфер, 3% БСА; 0,05% Твин-20, профильтрованный; буфер для покрывания планшета,50 мг/мл поли-Glu-Tyr (SigmaР 0275) в фосфатно-солевом буфере (DPBS);- 16013248 АТФ: 0,1 М АТФ в H2O или HEPES, рН 7. Примечание: буфер для анализа АТФ готовят в виде 75 мкМ АТФ в PBS так, чтобы 80 мкл в 120 мкл реакционного раствора = конечной концентрации АТФ 50 мкМ.I. Очистка His-FAKcd410-689. 1. Ресуспендируют 130 г массы клеток, инфицированных бакуловирусами, содержащей сверхэкспрессированный рекомбинантный белок His-FAKcd410-689, в 3 объемах (400 мл) буфера А. 2. Клетки лизируют, пропуская один раз в микрофлюидизаторе. 3. Остатки клеток удаляют центрифугированием при 4 С в течение 35 мин при 14000 об/мин в роторе Sorval SLA-1500. 4. Надосадок переносят в чистую пробирку и добавляют 6,0 мл Ni-NTA-агарозы (Qiagen). 5. Суспензию инкубируют при осторожном встряхивании при 4 С в течение 1 ч. 6. Суспензию центрифугируют при 700g в бакет-роторе. 7. Надосадок отбрасывают и агарозные шарики ресуспендируют в 20,0 мл буфера А. 8. Шарики переносят на колонку XK-16 (Amersham-Pharmacia), соединенную с FPLCTM. 9. Агарозные шарики промывают 5 объемами колонки буфера А и элюируют с колонки ступенчатым градиентом буфера А, содержащим 300 мМ имидазола. 10. Осуществляют замену буфера в элюированных фракциях на буфер В. 11. После замены буфера фракции объединяют и добавляют тромбин в соотношении 1:300(мас./мас.) и инкубируют в течение ночи при 13 С, чтобы удалить N-концевую His-метку (His-FAK410698FAK410-689 (также называемая FAKcd. 12. Реакционную смесь снова наносят на колонку Ni-NTA, уравновешенную буфером А, и собирают проходящий поток. 13. Проходящий поток концентрируют до 1,7 мл и наносят непосредственно на препаративную колонку Superdex 200 HiLoad 16/60, уравновешенную буфером С. Требуемый белок элюируется от 85 до 95 мл. 14. Белок FAKcd делят на аликвоты и хранят замороженными при -80 С.II. Активация FAK. 1. К 450 мкл FAK410-689 в концентрации 1,48 мг/мл (660 мкг) добавляют следующее: 30 мкл 0,037 мг/мл (1 мкМ) His-Src(249-524); 30 мкл 7,5 мМ АТФ; 12 мкл 20 мМ MgCl2; 10 мкл смеси Mn2+/АТФ (UpState Biotech.); 4 мкл 6,7 мМ DTT; 60 мкл буфера реакции Src (UpState Biotech.). 2. Реакционную смесь инкубируют по меньшей мере в течение 3 ч при комнатной температуре. Во временной точке t0 почти вся FAK410-689 однократно фосфорилирована. Второе фосфорилирование протекает медленно. В точке t120 (t=120 мин) добавляют 10 мкл 150 мМ АТФ. Т 0 = (начало) 90% однократно фосфорилированной FAK410-689 (1 PO4),Т 43 = (43 мин) 65% однократно фосфорилированной (1 PO4),35% дважды фосфорилированной (2 PO4),Т 90 = (90 мин) 45% 1 PO4, 55% 2 PO4,T150 = 15% 1 PO4, 85% 2 PO4,Т 210 = 10% 1 PO4, 90% 2 PO4, обессоленный образец. 3. Добавляют аликвоту объемом 180 мкл обессоленного вещества на центрифужную колонкуNi-NTA и инкубируют на центрифужной колонке. 4. Центрифугируют при 10000 об/мин (микроцентрифуга) в течение 5 мин, чтобы выделить и собрать проходящий поток (активированная FAK410-689) и удалить His-Src (улавливаемую на колонке).III. ELISA киназы FAKcd. 1. 96-луночный планшет Nunc MaxiSorp покрывают поли-glu-tyr (pGT) по 10 мкг/лунку: готовят 10 мкг/мл pGT в PBS и вносят аликвоты 100 мкл/лунку. Планшеты инкубируют при 37 С в течение ночи,отбирают надосадок, планшеты 3 раза промывают буфером для промывки и стряхивают, чтобы подсушить перед хранением при 4 С. 2. Готовят исходные растворы соединений 2,5 мМ в 100% ДМСО. Затем исходные растворы разбавляют до 60 конечной концентрации в 100% ДМСО и разбавляют 1:5 в буфере для киназного фосфорилирования. 3. Готовят 75 мкМ рабочего раствора АТФ в буфере для киназного фосфорилирования. В каждую лунку добавляют 80 мкл до конечной концентрации АТФ 50 мкМ. 4. Переносят 10 мкл разбавленных соединений (серийные разведения 0,5 log) в каждую лунку планшета для анализа pGT, внося каждое соединение в трех повторах на одном и том же планшете. 5. На льду разбавляют белок FAKcd до 1:1000 в буфере для киназного фосфорилирования. Разносят по 30 мкл на лунку.- 17013248 6. Примечание: Линейность и подходящее разведение необходимо определить предварительно для каждой партии белка. Выбранная концентрация фермента должна быть такой, чтобы количественная оценка сигнала в анализе составляла примерно 0,8-1,0 при OD450 и была в линейном диапазоне скорости реакции. 7. Готовят как контроль без АТФ (шума), так и контроль без соединения (сигнала). 8. (Шум). В один пустой ряд лунок вносят 10 мкл соединений, разведенных 1:5 в ДМСО, 80 мкл буфера для фосфорилирования (минус АТФ) и 30 мкл раствора FAKcd. 9. (Сигнал). В контрольные лунки вносят 10 мкл ДМСО, разведенного 1:5 (минус соединение) в буфере для киназного фосфорилирования, 80 мкл 75 мкМ АТФ и 30 мкл фермента FAKcd в разведении 1:1000. 10. Реакционную смесь инкубируют при комнатной температуре в течение 15 мин при осторожном встряхивании на встряхивающем устройстве для планшетов. 11. Реакцию останавливают отсасыванием реакционной смеси и промыванием 3 раза буфером для промывки. 12. Разбавляют антитело против фосфотирозина, конъюгированное с HRP (pY20HRP) до 0,250 мкг/мл (1:1000 исходного раствора) в блокирующем буфере. Разносят по 100 мкл на лунку и инкубируют при встряхивании в течение 30 мин при комнатной температуре. 13. Надосадок отсасывают и планшет промывают 3 раза буфером для промывки. 14. Добавляют 100 мкл на лунку раствора ТМВ комнатной температуры, чтобы инициировать развитие окраски. Развитие окраски останавливают примерно через 15-30 с добавлением 100 мкл 0,09 МH2SO4 на лунку. 15. Сигнал оценивают количественно посредством измерения оптической плотности при 450 нм на считывающем устройстве для микропланшетов BioRad или считывающем устройстве для микропланшетов, способном регистрировать при OD450. 16. Результатом ингибирования тирозинкиназной активности будет уменьшенный сигнал оптической плотности. Сигнал обычно составляет 0,8-1,0 единиц OD. Значения представлены в виде концентрации IC50 в мкМ. Основанный на клетках ELISA индуцированной FAK: конечный протокол. Материалы: 96-луночные планшеты с антикроличьим антителом козы Reacti-Bind (Pierce Product15135ZZ,115,00 долларов США),поликлональное антитело кролика FAKpY397 (Biosource44624, 315,00 долларов США),IgG кролика, полная молекула ChromePure (Jackson Laboratories001-000-003, 60 долларов США 25 мг),мышиное моноклональное антитело 2 А 7 клона UBI FAK (Upstate05-182, 289,00 долларов США),антимышиный IgG козы, конъюгированный с пероксидазой AffiniPure (Jackson Labs115-035-146,95 долларов США 1,5 мл),TBS SuperBlock (Pierce Product37535ZZ, 99 долларов США),бычий сывороточный альбумин (SigmaА-9647, 117,95 долларов США 100 г),субстрат пероксидазы ТМВ (Oncogene Research ProductsCL07, 100 мл 40 долларов США),ортованадат натрия Na3VO4 (SigmaS6508, 43,95 долларов США 50 г),субстрат МТТ (SigmaМ-2128, 25,95 долларов США 500 мг). Среда роста: DMEM + 10% FBS, P/S, Glu, 750 мкг/мл зеоцина и 50 мкг/мл гигромицина (ZeocinInVitrogen250-05, 725 долларов США и гигромицин InVitrogenR220-05, 150 долларов США). Мифепристон InVitrogenH110-01, 125 долларов США. Таблетка не содержащих ЭДТА полной смеси ингибиторов протеаз Boehringer Mannheim1873580. Основанный на клетках протокол анализа FAK в отношении избирательности зависимой от киназыphosphoFAKY397. Способ. Разрабатывали основанный на клетках анализ индуцируемой FAK в форме ELISA для скрининга химического вещества, чтобы идентифицировать специфичные ингибиторы тирозинкиназы. В основанном на клетках анализе используют механизм системы GeneSwitch (InVitrogen), чтобы экзогенно регулировать экспрессию и фосфорилирование FAK и зависимого от киназы сайта автофосфорилирования остатка Y397. Ингибирование зависимого от киназы автофосфорилирования Y397 приводит к уменьшенному сигналу оптической плотности при OD450. Сигнал обычно составляет от 0,9 до 1,5 единиц OD450 с шумом в диапазоне от 0,08 до 0,1 единицы OD450. Значения приведены в виде концентрации IC50 в мкМ. В 1-й день выращивают A431FAKwt во флаконах Т 175. За 1 день до выполнения основанного на клетках анализа FAK клетки A431FAKwt высевают в среду роста в 96-луночные планшеты сU-образным дном. Клетки оставляют при 37 С, 5% CO2 на 6-8 ч перед индукцией FAK. Готовят исходный раствор мифепристона в концентрации 10 мкМ в 100% этаноле. Затем исходный раствор разбавляют до 10 конечной концентрации в среде роста. В каждую лунку переносят по 10 мкл указанного разведения (конечная концентрация 0,1 нМ мифепристона). Клетки оставляют при 37 С, 5% CO2 в течение ночи(12-16 ч). Также готовят контрольные лунки без индукции мифепристоном экспрессии и фосфорилирования FAK. На 2-й день планшет(ы) с антикроличьим антителом козы покрывают поликлональным антителом,специфичным к фосфату FAKpY397, приготовленным в буфере TBS SuperBlock, и обеспечивают возможность встряхивания планшета(тов) на встряхивающем устройстве при комнатной температуре в течение 2 ч. Необязательно контрольные лунки могут быть покрыты 3,5 мкг/мл контрольного антитела для захвата (полные молекулы кроличьего IgG), приготовленного в TBS SuperBlock. 3 раза отмывают от избытка антитела FAKpY397, используя буфер. Блокируют покрытый анти-PAKpY397 планшет(ы) 200 мкл на лунку блокирующего буфера с 3% БСА/0,5% Твин в течение 1 ч при комнатной температуре на устройстве для встряхивания планшетов. Во время блокирования планшета(ов) готовят исходные растворы соединений в концентрации 5 мМ в 100% ДМСО. Затем исходные растворы серийно разводят до 100 конечной концентрации в 100% ДМСО. Делают разведение 1:10, используя 100 раствор, в среде роста и переносят по 10 мкл соответствующих разведений соединения в каждую лунку, содержащую либо индуцированные по FAK, либо неиндуцированные контрольные клетки А 431, на 30 мин при 37 С, 5% CO2. Готовят лизирующий буфер RIPA (50 мМ трис-HCl, pH 7,4, 1% NP-40, 0,25% Na-дезоксихолат, 150 мМNaCl, 1 мМ ЭДТА, 1 мМ Na3VO4, 1 мМ NaF и одна таблетка полной смеси ингибиторов протеазы, не содержащей ЭДТА, на 50 мл раствора). В конце 30-минутной обработки соединением отмывают от соединения 3 раза, используя буфер для промывки TBS-T. Клетки лизируют 100 мкл/лунку буфера RIPA. Из покрытых планшетов удаляют блокирующий буфер и 3 раза промывают, используя буфер для промывки TBS-T. Используя 96-луночный автоматизированный микродиспенсер, переносят по 100 мкл целого клеточного лизата (со стадии 6) в планшет(ы), покрытый антителом козы против FAKpY397 кролика, чтобы захватить белки phosphoFAKY397. Встряхивают при комнатной температуре в течение 2 ч. Отмывают несвязанные белки 3 раза, используя буфер для промывки TBS-T. Готовят 0,5 мкг/мл (разведение 1:2000) регистрирующего антитела UBI FAK в блокирующем буфере 3% БСА/0,5% Твин. Распределяют по 100 мкл раствора UBI FAK на лунку и встряхивают в течение 30 мин при комнатной температуре. Отмывают избыток антитела UBI FAK 3 раза, используя буфер для промывки TBS-T. Готовят 0,08 мкг/мл (разведение 1:5000) второго антимышиного конъюгированного с пероксидазой антитела (анти-2MHRP). Распределяют по 100 мкл на лунку раствора анти-2MHRP и встряхивают в течение 30 мин при комнатной температуре. Отмывают от избытка анти-2MHRP-антитела 3 раза, используя буфер для промывки TBS-T. Добавляют по 100 мкл на лунку раствора субстрата ТМВ комнатной температуры,обеспечивая возможность развития окраски. Реакцию ТМВ останавливают, используя 100 мкл на лунку останавливающего раствора для ТМВ (0,09 М H2SO4) и количественно оценивают сигнал, измеряя оптическую плотность при 450 нм на считывающем устройстве для микропланшетов BioRad. Дополнительные клеточные анализы FAK включены в данное описание в виде ссылки на заявкуPfizer сдела патентного поверенного PC11699, озаглавленной Inducible focal adhesion kinase cell assay. В предпочтительном варианте соединения согласно настоящему изобретению обладают активностью in vivo, составляющей менее 100 нМ, как определено в анализе киназы, например, таком, который указан в данном описании. Предпочтительно соединения имеют IC50 менее 25 нМ в анализе киназы, и более предпочтительно менее 10 нМ. В следующем предпочтительном варианте соединения имеют IC50 в основанном на клетках анализе FAK, например, таком как анализ, приведенный в данном описании, менее 1 мкМ, более предпочтительно менее 100 нМ и наиболее предпочтительно менее 25 нМ. Введение соединений согласно настоящему изобретению (в дальнейшем активного соединения(ий можно осуществить любым способом, который делает возможным доставку соединений к месту действия. Такие способы включают пероральные пути, интрадуодеальные пути, парентеральную инъекцию (включая внутривенную, подкожную, внутримышечную, внутрисосудистую инъекцию или инфузию), местное и ректальное введение. Количество вводимого активного соединения будет зависеть от субъекта, находящегося на лечении,тяжести расстройства или состояния, скорости введения, распределения соединения и решения назначающего врача. Однако эффективная доза находится в пределах примерно от 0,001 до 100 мг на кг массы тела в сутки, предпочтительно примерно от 1 до 35 мг/кг/сутки, в виде однократных или дробных доз. Для человека массой 70 кг такое количество может составлять примерно от 0,05 до 7 г/сутки, предпочтительно примерно от 0,2 до 2,5 г/сутки. В некоторых случаях уровни доз ниже нижнего предела указанного выше диапазона могут быть более чем адекватными, тогда как в других случаях могут быть использованы еще более высокие дозы, не вызывая какого-либо опасного побочного эффекта, при условии, что такие более высокие дозы сначала делят на несколько небольших доз для введения на протяжении суток.- 19013248 Активное соединение может применяться в качестве единственной терапии, или могут быть включены одно или несколько других противоопухолевых веществ, например, веществ, выбранных из ингибиторов митоза, например, винбластина; алкилирующих агентов, например, цис-платина, карбоплатина и циклофосфамида; антиметаболитов, например 5-фторурацила, цитозинарабинозида и гидроксимочевины,или, например, одного из предпочтительных антиметаболитов, описанных в Европейской заявке на выдачу патента 239362, такого как N-(5-[N-(3,4-дигидро-2-метил-4-оксохиназолин-6-илметил)-Nметиламино]-2-теноил)-L-глутаминовая кислота; ингибиторов фактора роста; ингибиторов клеточного цикла; интеркалирующих антибиотиков, например, адриамицина и блеомицина; ферментов, например интерферона; и антигормонов, например антиэстрогенов, таких как нолвадекс (тамоксифен), или, например антиандрогенов, таких как казодекс (4'-циано-3-(4-фторфенилсульфонил)-2-гидрокси-2-метил-3'(трифторметил)пропионанилид). Такое совместное лечение может быть осуществлено путем одновременного, последовательного или раздельного дозирования отдельных компонентов лечения. Фармацевтическая композиция, например, может быть в форме, подходящей для перорального введения в виде таблетки, капсулы, пилюли, порошка, препаратов длительного высвобождения, раствора,суспензии, для парентеральной инъекции в виде стерильного раствора, суспензии или эмульсии, для местного введения в виде мази или крема, или для ректального введения в виде суппозитория. Фармацевтическая композиция может быть в дозированных лекарственных формах, подходящих для однократного введения точных доз. Фармацевтическая композиция будет содержать обычный фармацевтический носитель или эксципиент и соединение согласно изобретению в виде активного ингредиента. Кроме того, она может содержать другие медицинские и фармацевтические агенты, носители, адъюванты и т.д. Примеры форм для парентерального введения включают растворы или суспензии активных соединений в стерильных водных растворах, например, водных растворах пропиленгликоля или декстрозы. При желании такие дозированные формы могут быть соответствующим образом забуферены. Подходящие фармацевтические носители включают инертные разбавители или наполнители, воду и различные органические растворители. Фармацевтические композиции при желании могут включать дополнительные ингредиенты, такие как корригенты, связывающие вещества, эксципиенты и т.п. Таким образом, для перорального введения таблетки, содержащие различные эксципиенты, такие как лимонная кислота, могут быть использованы вместе с различными дезинтегрирующими агентами, такими как крахмал, альгиновая кислота и некоторые комплексные силикаты, и со связывающими агентами, такими как сахароза, желатин и акациевая камедь. Кроме того, скользящие агенты, такие как стеарат магния,лаурилсульфат натрия и тальк, часто применимы в целях таблетирования. Твердые композиции сходного типа также могут быть использованы в мягких и твердых наполняемых желатиновых капсулах. Предпочтительные для этого вещества включают лактозу или молочный сахар и высокомолекулярные полиэтиленгликоли. В том случае, когда требуются водные суспензии или эликсиры для перорального введения активного соединения, в них соединения можно комбинировать с различными подсластителями или корригентами, красящими веществами или красителями, и при желании с эмульгаторами или суспендирующими агентами, вместе с такими разбавителями, как вода, этанол, пропиленгликоль, глицерин или их комбинации. Способы получения различных фармацевтических композиций с конкретным количеством активного соединения известны или будут известны специалистам в данной области. Например, см. Remington'sPharmaceutical Sciences, Mack Publishing Company, Easter, Pa., 15th Edition (1975). Примеры и получения, приведенные ниже, дополнительно иллюстрируют и поясняют на примерах соединения согласно настоящему изобретению и способы получения таких соединений. Следует понимать, что объем настоящего изобретения никоим образом не ограничен объемом следующих примеров и получений. В следующих примерах молекулы с одним хиральным центром, если не оговорено особо,существуют в виде рацемической смеси. Такие молекулы с двумя или более хиральными центрами, если не оговорено особо, существуют в виде рацемической смеси диастереомеров. Отдельные энантиомеры/диастереомеры могут быть получены способами, известными в данной области. В том случае, когда упоминают ВЭЖХ-хроматографию, применительно к приведенным ниже получениям и примерам, общими используемыми условиями, если не оговорено особо, являются следующие условия. Использую колонку ZORBAX RXC18 (производства Hewlett Packard) длиной 150 мм и внутренним диаметром 4,6 мм. Образцы анализируют на системе Hewlett Packard-1100. Используют способ на основе градиента растворителей от буфера 100% ацетата аммония/уксусная кислота (0,2 М) до 100% ацетонитрила в течение 10 мин. Затем система переходит к циклу промывки 100-процентным ацетонитрилом в течение 1,5 мин и затем 100-процентным буферным раствором в течение 3 мин. Скорость потока в течение указанного периода постоянна и составляет 3 мл/мин. В следующих примерах и описаниях получения Et означает этил, Ас означает ацетил, Me означает метил и Bu означает бутил.- 20013248 Примеры Общие способы. Получение 5-нитрооксиндола. К раствору оксиндола (26 г) в 100 мл концентрированной серной кислоты при -15 С по каплям добавляли дымящую азотную кислоту (8,4 мл). Особое внимание обращали на то, чтобы поддерживать температуру реакционной смеси при -15 С. После завершения добавления реакционную смесь перемешивали в течение 30 мин и затем вливали в ледяную воду. Образовывался желтый осадок, который выделяли фильтрованием, получая 34 г (98%) 5-нитрооксиндола. Получение 5-аминооксиндола (2). К раствору 5-нитрооксиндола (25 г) в 120 мл диметилацетамида в сосуде Парра добавляли 10%Pd/C (0,5 г). Смесь гидрировали (40 фунтов/кв. дюйм Н 2) в течение 16 ч. Катализатор удаляли фильтрованием и фильтрат разбавляли эфиром (2 л), получая 5-аминооксиндол (10,5 г; 50%). Получение 2,4-дихлор-5-трифторметилпиримидина (3). 5-Трифторметилурацил (250 г, 1,39 моль) и оксихлорид фосфора (655 мл, 6,94 моль, 5 экв.) загружали в 4-горлую колбу объемом 3 л, оборудованную верхней мешалкой, обратным холодильником, капельной воронкой и внутренней термопарой. Содержимое поддерживали в атмосфере азота по мере того,как к взвеси частями добавляли концентрированную фосфорную кислоту (85 мас.%, 9,5 мл, 0,1 экв.), получая умеренный экзотермический эффект. Затем по каплям добавляли диизопропилэтиламин (245 мл,1,39 моль, 1 экв.) в течение 15 мин с такой скоростью, чтобы внутренняя температура реакции достигала 85-90 С к концу добавления. К концу добавления амина реакционная смесь представляла собой гомогенный светло-оранжевый раствор. Начинали нагревание и оранжевый раствор поддерживали при 100 С в течение 20 ч, в этой временной точке анализ ВЭЖХ реакционной смеси показал, что исходное вещество было истрачено. Наружное нагревание убирали и содержимое колбы охлаждали до 40 С и затем по каплям добавляли к охлажденной смеси 3 н. HCl (5 л, 10 экв.) и диэтиловый эфир (2 л), поддерживая температуру чаши для гашения от 10 до 15 С. Слои разделяли и водный слой один раз экстрагировали эфиром(1 л). Органические слои объединяли. Промывали водой до тех пор, пока промывные воды становились нейтральными (промывки 51,5 л), сушили, используя MgSO4, и концентрировали, получая 288 г (выход 95%) светло-оранжевого масла с чистотой 96% (ВЭЖХ). Полученный продукт можно дополнительно очистить дистилляцией (т.кип. 109 С при 79 мм Hg). Получение 5-(4-хлор-5-трифторметилпиримидин-2-иламино)-1,3-дигидроиндол-2-она (4). К раствору 5-трифторметил-2,4-дихлорпиримидина (214,8 г; 0,921 моль) в смеси 1:1 DCE/третBuOH (1,240 л) добавляли 1 М раствора хлорида цинка в эфире (1 экв.; 0,921 л). Через 0,5 ч добавляли 5-аминооксиндол (124 г; 0,837 моль), затем триэтиламин (129,4 мл; 0,921 моль), поддерживая температуру при 25 С. Реакционной смеси давали возможность перемешиваться при комнатной температуре в течение ночи, затем концентрировали и продукт растирали в метаноле, получая в виде желтого твердого вещества (224,3 г; 82%). 1(R)(+)-альфа-фенэтиламин (0,071 мл; 0,547 ммоль) и диизопропилэтиламин (0,081 мл, 0,456 ммоль). Полученный в результате раствор перемешивали в атмосфере азота и нагревали до 80 С в течение 16 ч. Реакционную смесь охлаждали до комнатной температуры, разбавляли 10 мл смеси 1:1 дихлорметана и метанола с последующим добавлением 0,5 г МР-карбоната. Полученную в результате смесь перемешивали, фильтровали, концентрировали и очищали хроматографией на силикагеле (соотношение 97:2,8:0,3 хлороформ/метанол/концентрированный гидроксид аммония). Требуемое указанное в заголовке соединение получали в виде белого твердого вещества (0,021 г; 11%).LRMS (M+) 413,4. Следующие соединения согласно изобретению получали нагреванием хлорпиримидина (4) с соответствующим амином, как в примере 1. Амины, используемые в данных реакциях, либо получали коммерчески и использовали в полученном виде, либо, альтернативно, их получали обычными для аминов способами синтеза, известными специалистам в данной области. Если не оговорено особо, соединения,имеющие хиральные центры, получали в виде рацемических смесей. Таблица 1 Соединения, полученные способом согласно примеру 1
МПК / Метки
МПК: C07D 405/14, C07D 413/14, C07D 401/14, C07D 417/14, C07D 513/04, A61K 31/505, C07D 403/14, A61P 35/00, C07D 409/14, C07D 403/12, C07D 239/00
Метки: патологического, пиримидина, лечения, клеток, роста, производные
Код ссылки
<a href="https://eas.patents.su/30-13248-proizvodnye-pirimidina-dlya-lecheniya-patologicheskogo-rosta-kletok.html" rel="bookmark" title="База патентов Евразийского Союза">Производные пиримидина для лечения патологического роста клеток</a>
Замещенные бициклические производные для лечения аномального роста клеток

Номер патента: 5525
Опубликовано: 28.04.2005
Авторы: Бхаттачариа Самит Кумар, Моррис Джоэл, Кат Джон Чарльз
МПК: A61K 31/517, A61P 35/00, C07D 401/12...
Метки: производные, лечения, клеток, бициклические, роста, аномального, замещенные
Формула / Реферат:
1. Соединение формулы 1 или его фармацевтически приемлемая соль, где m равно целому числу от 0 до 3; p равно целому числу от 0 до 4; каждый из R1 и R2 независимо выбран из H и C1-C6-алкила; R3 выбран из где упомянутые выше R3-группы необязательно замещены 1-3 R8-группами; R4 представляет -(CR16R17)m-Cу C-(CR16R17)kR13 или -(CR16R17)m-C=C-(CR16R17)kR13, где каждое из k равно целому числу от 1 до 3 и каждое из m равно целому числу от 0 до...
Конденсированные производные пиримидина и их композиции, применимые в качестве модуляторов cxcr3 рецепторов для профилактики и лечения воспалительных и иммунорегуляторных расстройств и заболеваний

Номер патента: 11439
Опубликовано: 27.02.2009
Авторы: Чэнь Сяоци, Маркус Эндрю П., Фу Цзыцэ, Ду Сяохой, Бергерон Филипп, Лю Цзивень, Дукветт Джейсон А., Джонсон Майкл Дж., Медина Хулио К., Дейгнан Джеффри, Ли Ань-Жун, Гастин Дарин, Михалик Джеффри Т.
МПК: A61P 29/00, A61K 31/519, A61P 37/00...
Метки: воспалительных, пиримидина, рецепторов, модуляторов, конденсированные, иммунорегуляторных, качестве, композиции, лечения, производные, применимые, заболеваний, cxcr3, профилактики, расстройств
Формула / Реферат:
1. Соединение формулы (I) или его фармацевтически приемлемая соль или пролекарство, где А1 представляет собой СН и А4 представляют собой СН или N; Q представляет собой связь, -ОС(О)- и -СН2СО; L представляет собой (СН2)m, где подстрочный индекс m равен 0, 1 или 2; X представляет собой -С(О)-; R1 представляет собой замещенный фенил, где заместитель выбиран из группы, состоящей из -OEt, -OCH2CF3, -CN и I; R2 представляет собой -СН3, CH2NHBoc...
Производные пиридо[2,3-d]пиримидина и их фармацевтически приемлемые соли, фармацевтическая композиция, обладающая ингибирующей протеин-тирозин-киназу активностью, способ лечения заболеваний, опосредованных клеточной пролиферацией, и способ лечения рака, атеросклероза, псориаза или рестеноза

Номер патента: 897
Опубликовано: 26.06.2000
Авторы: Пэнек Роберт, Клачко Сильвестер, Блэнкли Клифтон Джон, Хэмби Джэймс Марино, Босчелли Дайянэ Хэррис, Доэрти Эннет Мэриэн
МПК: A61P 35/00, C07D 471/04, A61K 31/519...
Метки: соли, производные, фармацевтически, способ, активностью, атеросклероза, пролиферацией, лечения, рестеноза, фармацевтическая, приемлемые, заболеваний, опосредованных, рака, протеин-тирозин-киназу, клеточной, композиция, псориаза, обладающая, ингибирующей, пиридо[2,3-d]пиримидина
Формула / Реферат:
1. Производные пиридо[2,3-d]пиримидина общей формулы (I) где X означает иминогруппу, N-ацил, кислород или серу; R1 означает группы NR3R4S(O)mR3, где m является 0,1 или 2, или ОR3; R2, R3 и R4 независимо означают водород, группу (СН2)nPh, где Ph является фенилом, не замещенным или замещенным 1-3 остатками из группы, включающей галоген, гидроксил, карбоксил, алкил с 1-6 атомами углерода, не замещенный или замещенный гидроксилом,...
Лекарственное средство и способ лечения патологического синдрома

Номер патента: 8106
Опубликовано: 27.04.2007
Авторы: Дыгай Александр Михайлович, Гольдберг Евгений Данилович, Эпштейн Олег Ильич
МПК: A61K 39/395, A61P 31/12
Метки: лечения, синдрома, патологического, лекарственное, средство, способ
Формула / Реферат:
1. Лекарственное средство на основе антител, характеризующееся тем, что содержит активированную форму моноклональных или поликлональных антител к интерферону в малых или сверхмалых дозах, приготовленную путем многократного последовательного разведения и внешнего воздействия по гомеопатической технологии. 2. Лекарственное средство по п.1, характеризующееся тем, что для получения антител используют человеческий или гетерологичный интерферон альфа,...
Способ продуцирования гормона роста человека и применение среды, не содержащей сыворотки, для культивирования клеток млекопитающих, экспрессирующих гормон роста человека

Номер патента: 11749
Опубликовано: 30.06.2009
Авторы: Касаторрес Эрнандес Хосе, Мартин Пьера Карлос
МПК: C07K 14/825, C12N 5/02, C07K 14/61...
Метки: сыворотки, млекопитающих, гормон, продуцирования, клеток, применение, гормона, содержащей, человека, среды, роста, культивирования, экспрессирующих, способ
Формула / Реферат:
1. Способ продуцирования гормона роста человека, включающий стадию культивирования клеточной линии, экспрессирующей гормон роста, в культуральной среде, не содержащей компонентов, полученных из сыворотки животных, причем среда содержит цинк в концентрации в интервале от 0,2 до 1,75 мкМ, медь в концентрации в интервале от 10 до 75 нМ и ионы железа в концентрации в интервале от 1 до 10 мкМ, где клетки представляют собой мышиные клетки С127 и среда...
Предыдущий патент: Монорельсовая транспортная система
Следующий патент: Производные тиазола
Случайный патент: Кремнийсодержащее соединение в качестве ингибитора коррозии в полиолефиновых композициях