Азабициклопроизводные в качестве антагонистов мускариновых рецепторов
Номер патента: 7932
Опубликовано: 27.02.2007
Авторы: Кумар Нареш, Салман Мохаммад, Дхармараджан Санкаранарейянан, Шетти Шанкар Джайрам, Чагх Анита, Силамкоти Арундутт Висванатан, Сарма Пакала Кумара Савитру, Мехта Анита


















Формула / Реферат
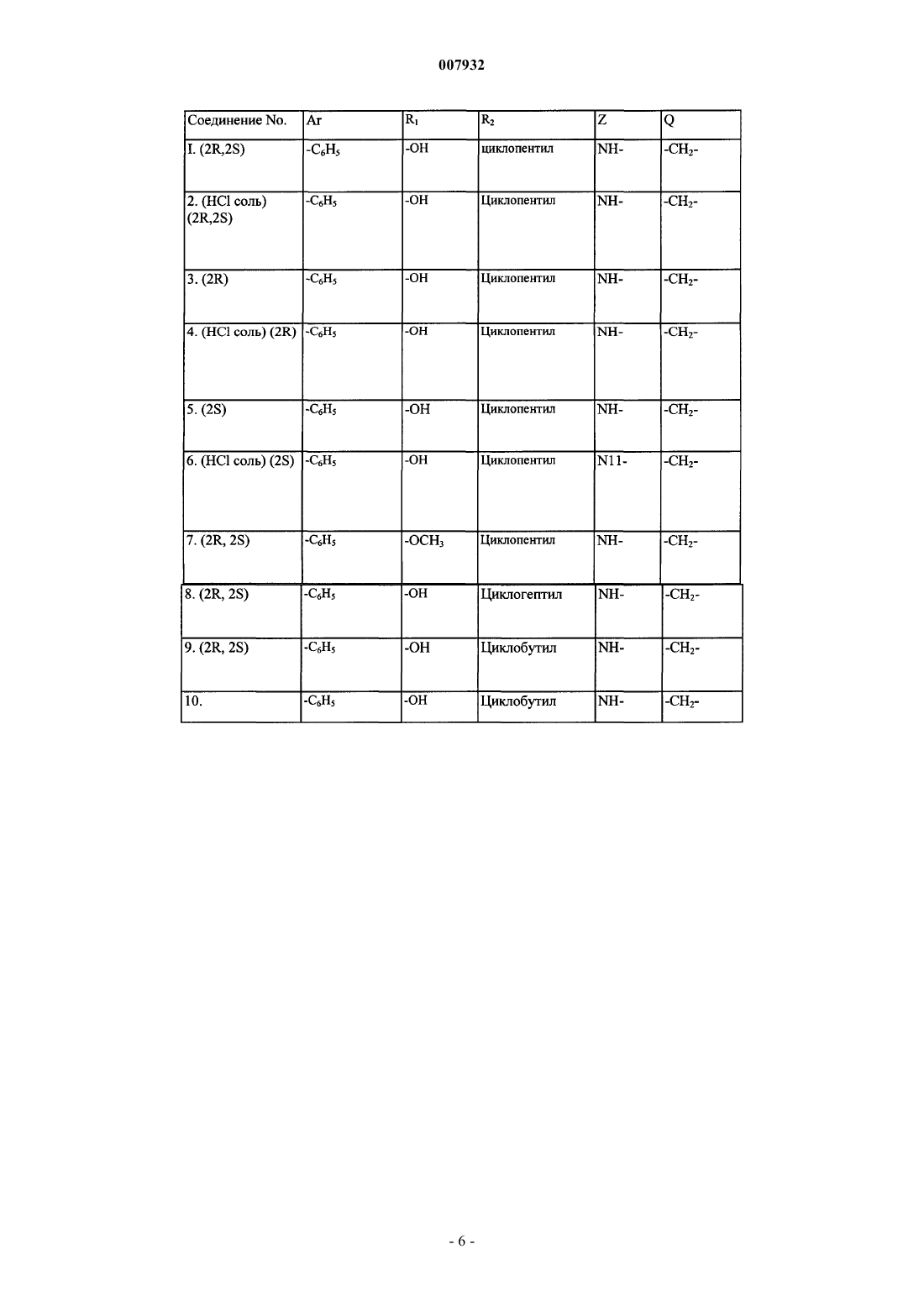
1. Соединения, имеющие структурную формулу I

формула I
и их фармацевтически приемлемые соли, фармацевтически приемлемые сольваты, эфиры, энантиомеры, диастереоизомеры,
где Ar представляет фенил;
R1 представляет ОН, МеО и MeSO3;
R2 представляет фенил, С4-С7 циклоалкил, 3,3-дифторциклопентил, 3-фторциклопентил;
W представляет простую связь;
X представляет NH, Nme, О или отсутствие атома;
Y представляет простую связь;
Z представляет NH, Nme, кислород или простую связь;
Q представляет СН2 и
R6=R7=H.
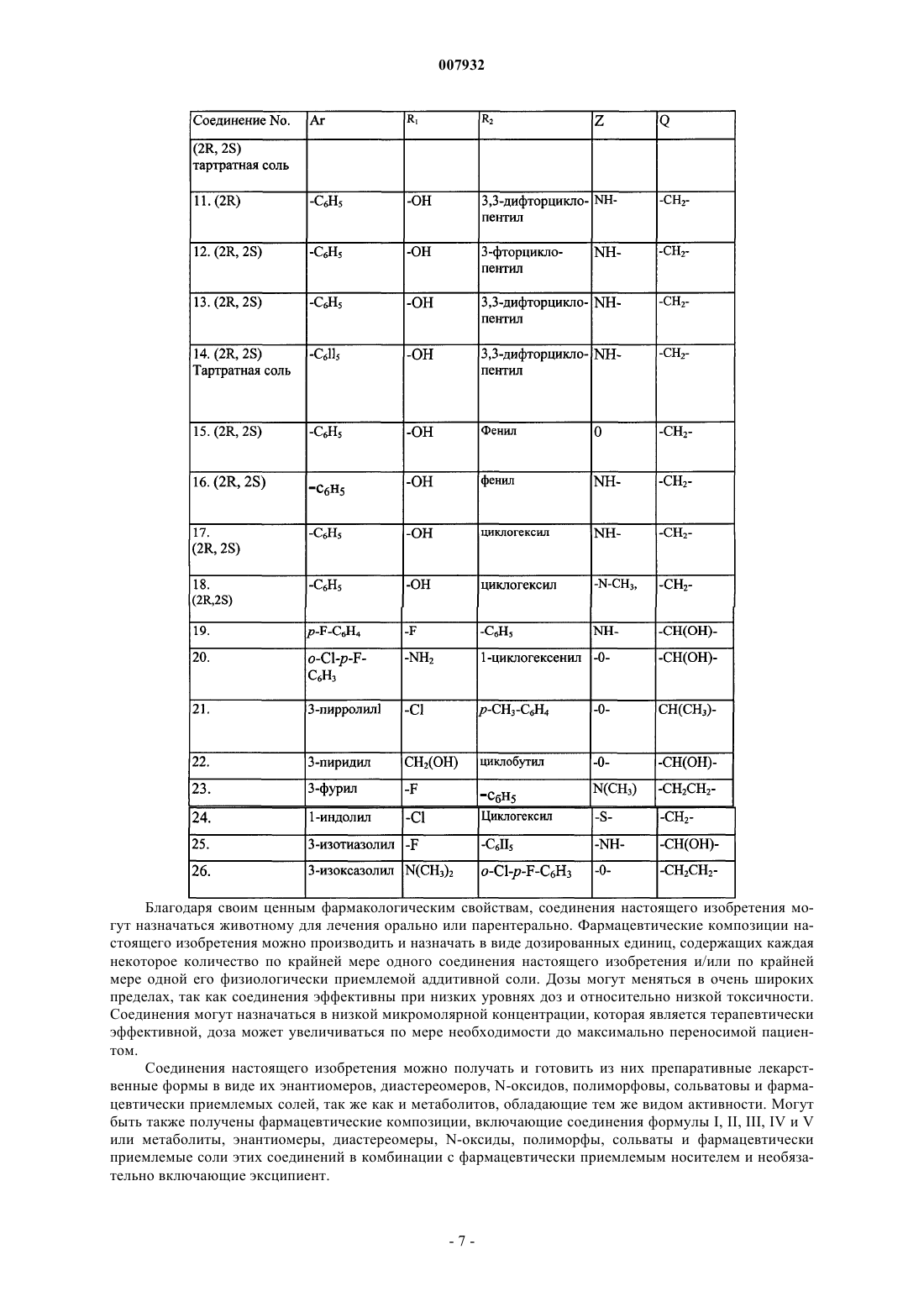
2. Соединения по п.1, имеющие структурную формулу II, и их фармацевтически приемлемые соли, фармацевтически приемлемые сольваты, эфиры, энантиомеры, диастереоизомеры

формула II
3. Соединения по п.1, имеющие структурную формулу III, и их фармацевтически приемлемые соли, фармацевтически приемлемые сольваты, эфиры, энантиомеры, диастереоизомеры

формула III
4. Соединения по п.1, имеющие структурную формулу IV, и их фармацевтически приемлемые соли, фармацевтически приемлемые сольваты, эфиры, энантиомеры, диастереоизомеры, где r представляет от 1 до 4

формула IV
5. Соединения по п.1, имеющие структурную формулу V, и их фармацевтически приемлемые соли, эфиры, энантиомеры, где s представляет от 1 до 2

формула V
6. Соединение, выбранное из группы, состоящей из
(2R,2S)-(1a,5a,6a)-N-[3-азабицикло[3.1.0]гекс-6-илметил]-2-гидрокси-2-циклопентил-2-фенилацетамида (соединение 1);
(2R,2S)-(1a,5a,6a)-N-[3-азабицикло[3.1.0]гексил-6-(аминометил)ил]-2-гидрокси-2-циклопентил-2-фенилацетамида, хлористо-водородная соль (соединение 2);
(2R)-(1a,5a,6a)-N-[3-азабицикло[3.1.0]гексил-6-(аминометил)ил]-2-гидрокси-2-циклопентил-2-фенилацетамида (соединение 3);
(2R)-(1a,5a,6a)-N-[3-азабицикло[3.1.0]гексил-6-(аминометил)ил]-2-гидрокси-2-циклопентил-2-фенилацетамида, хлористо-водородная соль (соединение 4);
(2S)-(1a,5a,6a)-N-[3-азабицикло[3.1.0]гексил-6-(аминометил)ил]-2-гидрокси-2-циклопентил-2-фенилацетамида (соединение 5);
(2S)-(1a,5a,6a)-N-[3-азабицикло[3.1.0]гексил-6-(аминометил)ил]-2-гидрокси-2-циклопентил-2-фенилацетамида, хлористо-водородная соль (соединение 6);
(2R,2S)-(1a,5a,6a)-N-[3-азабицикло[3.1.0]гексил-6-(аминометил)ил]-2-метокси-2-циклопентил-2-фенилацетамида (соединение 7);
(2R,2S)-(1a,5a,6a)-N-[3-азабицикло[3.1.0]гексил-6-(аминометил)ил]-2-гидрокси-2-циклогексил-2-фенилацетамида (соединение 7);
(2R,2S)-(1a,5a,6a)-N-[3-(2-(2,3-дигидробензофуран-5-ил)этил)-3-азабицикло[3.1.0]гексил-6-(аминометил)ил]-2-гидрокси-2-циклогептил-2-фенилацетамида (соединение 8);
(2R,2S)-(1a,5a,6a)-N-[3-азабицикло[3.1.0]гексил-6-(аминометил)ил]-2-гидрокси-2-циклобутил-2-фенилацетамида (соединение 9);
(2R,2S)-(1a,5a,6a)-N-[3-азабицикло[3.1.0]гексил-6-(аминометил)ил]-2-гидрокси-2-циклобутил-2-фенилацетамида, тартратная соль (соединение 10);
(2R)-(1a,5a,6a)-N-[3-азабицикло[3.1.0]гексил-6-(аминометил)ил]-2-гидрокси-2-(3,3-дифторциклопентил)-2-фенилацетамида (соединение 11);
(2R,2S)-(1a,5a,6a)-N-[3-азабицикло[3.1.0]гексил-6-(аминометил)ил]-2-гидрокси-2-(3-фторциклопентил)-2-фенилацетамида (соединение 12);
(2R,2S)-(1a,5a,6a)-N-[3-азабицикло[3.1.0]гексил-6-(аминометил)ил]-2-гидрокси-2-(3,3-дифторциклопентил-2-фенилацетамида (соединение 13);
(2R,2S)-(1a,5a,6a)-N-[3-азабицикло[3.1.0]гексил-6-(аминометил)ил]-2-гидрокси-2-(3,3-дифторциклопентил)-2-фенилацетамида, тартратная соль (соединение 14);
(2R,2S)-(1a,5a,6a)-N-[3-азабицикло[3.1.0]гексил-6-(метил)ил]-2-гидрокси-2,2-дифенилацетата (соединение 15);
(2R,2S)-(1a,5a,6a)-N-[3-азабицикло[3.1.0]гексил-6-(аминометил)ил]-2-гидрокси-2,2-дифенилацетамида (соединение 16);
(2R,2S)-(1a,5a,6a)-N-[3-азабицикло[3.1.0]гексил-6-(аминометил)ил]-2-гидрокси-2-циклогексил-2-фенилацетамида (соединение 17) и
(2R,2S)-(1a,5a,6a)-N-[3-азабицикло[3.1.0]гек-6-силметил]-2-циклопентил-2-гидрокси-N-метил-2-фенилацетамида (соединение 18).
7. Фармацевтическая композиция, включающая терапевтически эффективное количество соединения по пп.1, 2, 3, 4, 5 вместе с фармацевтически приемлемым носителем, эксципиентом или разбавителем.
8. Способ лечения или профилактики животных или человека, страдающих от болезни или расстройств респираторной, мочевой и желудочно-кишечной систем, где эти болезни или расстройства опосредуются мускариновыми рецепторами, включающий назначение указанному животному или человеку терапевтически эффективного количества соединения, имеющего структурную формулу I

формула I
их фармацевтически приемлемых солей, фармацевтически приемлемых сольватов, эфиров, энантиомеров, диастереоизомеров,
где Ar представляет фенил;
R1 представляет ОН, МеО и MeSO3;
R2 представляет фенил, С4-С7 циклоалкил, 3,3-дифторциклопентил, 3-фторциклопентил;
W представляет простую связь;
X представляет NH, Nme, О или отсутствие атома;
Y представляет простую связь;
Z представляет NH, Nme, кислород или простую связь;
Q представляет СН2 и
R6=R7=H.
9. Способ по п.8 для лечения или профилактики животных или человека, страдающих от болезни или расстройств респираторной, мочевой и желудочно-кишечной систем, где эти болезни или расстройства опосредуются мускариновыми рецепторами, включающий назначение указанному животному или человеку терапевтически эффективного количества соединения, имеющего структурную формулу II, их фармацевтически приемлемых солей, фармацевтически приемлемых сольватов, эфиров, энантиомеров, диастереоизомеров

формула II
10. Способ по п.8 для лечения или профилактики животных или человека, страдающих от болезни или расстройств респираторной, мочевой и желудочно-кишечной систем, где эти болезни или расстройства опосредуются мускариновыми рецепторами, включающий назначение указанному животному или человеку терапевтически эффективного количества соединения, имеющего структурную формулу III, их фармацевтически приемлемых солей, фармацевтически приемлемых сольватов, эфиров, энантиомеров, диастереоизомеров

формула III
11. Способ по п.8 для лечения или профилактики животных или человека, страдающих от болезни или расстройств респираторной, мочевой и желудочно-кишечной систем, где эти болезни или расстройства опосредуются мускариновыми рецепторами, включающий назначение указанному животному или человеку терапевтически эффективного количества соединения, имеющего структурную формулу IV, их фармацевтически приемлемых солей, фармацевтически приемлемых сольватов, эфиров, энантиомеров, диастереоизомеров, где r означает от 1 до 4

формула IV
12. Способ по п.8 для лечения или профилактики животных или человека, страдающих от болезни или расстройств респираторной, мочевой и желудочно-кишечной систем, где эти болезни или расстройства опосредуются мускариновыми рецепторами, включающий назначение указанному животному или человеку терапевтически эффективного количества соединения, имеющего структурную формулу V, их фармацевтически приемлемых солей, фармацевтически приемлемых сольватов, эфиров, энантиомеров, диастереоизомеров, где s представляет от 1 до 2

формула V
13. Способ по п.8, где заболеваниями или расстройствами являются недержание мочи, расстройство нижних мочевых путей (LUTS), бронхиальная астма, хроническое обструктивное заболевание легких (COPD), фиброз легких, синдром раздраженной толстой кишки, ожирение, диабет или желудочно-кишечная гиперкинезия.
14. Способ по п.9, где заболеваниями или расстройствами являются недержание мочи, симптомы нижних мочевых путей (LUTS), бронхиальная астма, хроническое обструктивное заболевание легких (COPD), фиброз легких, синдром раздраженной толстой кишки, ожирение, диабет или желудочно-кишечная гиперкинезия.
15. Способ по п.10, где заболеваниями или расстройствами являются недержание мочи, симптомы нижних мочевых путей (LUTS), бронхиальная астма, хроническое обструктивное заболевание легких (COPD), фиброз легких, синдром раздраженной толстой кишки, ожирение, диабет или желудочно-кишечная гиперкинезия.
16. Способ по п.11, где заболеваниями или расстройствами являются недержание мочи, симптомы нижних мочевых путей (LUTS), бронхиальная астма, хроническое обструктивное заболевание легких (COPD), фиброз легких, синдром раздраженной толстой кишки, ожирение, диабет или желудочно-кишечная гиперкинезия.
17. Способ по п.12, где заболеваниями или расстройствами являются недержание мочи, симптомы нижних мочевых путей (LUTS), бронхиальная астма, хроническое обструктивное заболевание легких (COPD), фиброз легких, синдром раздраженной толстой кишки, ожирение, диабет или желудочно-кишечная гиперкинезия.
18. Способ лечения или профилактики животных или человека, страдающих от болезни или расстройств респираторной, мочевой и желудочно-кишечной систем, где эти болезни или расстройства опосредуются мускариновыми рецепторами, включающий назначение указанному животному или человеку терапевтически эффективного количества фармацевтической композиции по п.7.
19. Способ по п.18, где заболеваниями или расстройствами являются недержание мочи, симптомы нижних мочевых путей (LUTS), бронхиальная астма, хроническое обструктивное заболевание легких (COPD), фиброз легких, синдром раздраженной толстой кишки, ожирение, диабет или желудочно-кишечная гиперкинезия.
20. Способ получения соединений формулы I

формула I
и их фармацевтически приемлемых солей, фармацевтически приемлемых сольватов, эфиров, энантиомеров, диастереоизомеров,
где Ar представляет фенил;
R1 представляет ОН, МеО и MeSO3;
R2 представляет фенил, С4-С7 циклоалкил, 3-3-дифторциклопентил, 3-фторциклопентил;
W представляет простую связь;
X представляет NH, Nme, О или простую связь;
Y представляет простую связь;
Z представляет NH, Nme, кислород или простую связь;
Q представляет СН2 и
R6=R7=H;
включающий:
(а) реакцию соединения формулы VII с соединением формулы VI

формула VII

формула VII
с получением защищенного соединения формулы VIII, где Ar, R1, R2, W, X, Y, Z и Q, как указано выше, и Р представляет собой защитную для аминогруппы группу

формула VIII
(b) снятие защиты с соединения формулы формула VIII в присутствии агента для снятия защиты с получением соединения формулы I, где Ar, R1, R2, W, X, Y, Z и Q, как указано выше,

формула I
21. Способ по п.20, где Р представляет собой любую защитную для аминогруппы группу, выбранную из группы, состоящей из бензила и t-бутоксикарбонила.
22. Способ по п.20, где реакцию соединения формулы VI с соединением формулы VII с получением соединения формулы VIII проводят в присутствии конденсирующего агента, который выбирают из группы, состоящей из 1-(3-диметиламинопропил)-3-этил карбодиимид гидрохлорида (ЭДК) и 1,8-диазабицикло[5.4.0]ундец-7-ена (ДБУ).
23. Способ по п.20, где реакцию соединения формулы VI с соединением формулы VII ведут в полярном апротонном растворителе, выбранном из группы, состоящей из N,N-диметилформамида, диметилсульфоксида, толуола и ксилола.
24. Способ по п.20, где реакцию соединения формулы VI с соединением формулы VII ведут при 0-140шС.
25. Способ по п.20, где снятие защиты с соединения формулы VIII ведут агентом для снятия защиты, который выбирают из группы, состоящей из палладия на угле, трифторуксусной кислоты (ТФК) и хлористо-водородной кислоты.
26. Способ по п.20, где снятие защиты с соединения формулы VIII с получением соединения формулы I ведут в подходящем органическом растворителе, который выбирают из группы, состоящей из метанола, этанола, тетрагидрофурана и ацетонитрила.
Текст
007932 Область изобретения Настоящее изобретение относится к антагонистам мускариновых рецепторов, которые полезны,между прочим, при лечении различных расстройств респираторной, мочевой и желудочно-кишечной систем, опосредованных мускариновыми рецепторами. В частности изобретение относится к производным азабициклосоединений, включая, например, 6-замещенные азабицикло[3.1.0]гексаны, 2,6- и 4,6 дизамещенные производные, и 2,4,6-тризамещенные производные, а также к фармацевтическим композициям, содержащим эти соединения, и способам лечения заболеваний, опосредованных мускариновыми рецепторами. Предпосылки создания изобретения Мускариновые рецепторы как члены рецепторов, сопряженных с протеином G (GPCRs), составляют семейство из 5 рецепторных подтипов (M1, М 2, М 3, М 4 и М 5) и активируются нейромедиатором ацетилхолином. Эти рецепторы широко распределены во многих органах и тканях и являются решающими в поддержании центральной и периферической холинергической нейротрансмиссии. Подтверждено расположение этих рецепторных подтипов в головном мозге и других органах. Например, подтип M1 локализован преимущественно в нейронных тканях, таких как кора головного мозга и вегетативный ганглий,подтип М 2 в основном расположен в сердце, где он опосредует холинергически индуцированную брадикардию, и подтип М 3 доминирует в гладких мышцах и слюнных железах (Nature, 323, р.411 (1986; Sciences, 237, р.527 (1987. Обзор Eglen и др., представленный в Current Opinions in Chemical Biology, 3, p.426 (1999), а также вTrends in Pharmacological Sciences, 22, p.409 (2001), описывает биологические возможности модулирования подтипов мускариновых рецепторов лигандами при различных болезненных состояниях, таких как болезнь Альцгеймера, боль, уренарные расстройства, хроническое обструктивное заболевание легких и тому подобные. Обзор Felder et al. в J. Med. Chem., 43, p. 4333 (2000) описывает терапевтические возможности мускариновых рецепторов в центральной нервной системе и конкретизирует структуру и функции мускариновых рецепторов, фармакологию и их терапевтическое применение. В Molecules, 6, p. 142 (2001) представлены фармакологические и медицинские аспекты класса мускариновых агонистов и антагонистов ацетилхолина.Birdsall et al. в Trends in Pharmacological Sciences, 22, p.215 (2001) суммируют последние исследования роли различных подтипов мускариновых рецепторов, используя различные мускариновые рецепторы нокаут мышей. Мускариновые агонисты, такие как мускарин и пилокарпин, и антагонисты, такие как атропин, известны более ста лет, но в открытии соединений, селективных в отношении рецепторных подтипов, достигнут незначительный прогресс, что затрудняет оценку специфических функций индивидуальных рецепторов. Хотя классические мускариновые антагонисты, такие как атропин, являются сильнодействующими бронходилататорами, их клиническое использование ограничено ввиду значительных побочных эффектов периферического и центрального действия, таких как тахикардия, пелена перед глазами,сухость во рту, запор, слабоумие и т.д. Разработанные впоследствии четвертичные производные атропина лучше переносятся, чем парентерально вводимые средства, но большинство из них не являются идеальными антихолинергическими бронходилататорами благодаря низкой селективности в отношении подтипов мускариновых рецепторов. Существующие соединения обладают ограниченной терапевтической эффективностью из-за их низкой селективности, приводящей к необходимости ограничения дозы, чтобы снизить побочные эффекты, такие как жажда, тошнота и сопровождающие сердечные расстройства, такие как опосредованная М 2 рецептором тахикардия. В Annual review of Pharmacological Toxicol., 41, p. 691 (2001) описывается фармакология инфекций нижнего мочевого тракта. Хотя антимускариновые агенты, такие как оксибутинин и толтеродин, которые действуют не селективно на мускариновые рецепторы, многие годы использовались для лечения гиперфункции мочевого пузыря, клиническая эффективность этих агентов ограничивается побочными эффектами, такими как сухость во рту, пелена перед глазами и запор. Обсуждается лучшая переносимость организмом толтеродина, чем оксибутинина. (W.D.Steers et al. in Curr. Opin. Invest. Drugs, 2, 268; Chapple et.Louis, MO; Mosby, 3rd edition (1996. Остается необходимость разработки новых высоко эффективных мукариновых антагонистов, которые могут взаимодействовать с определенными подтипами таким образом, чтобы избежать побочных эффектов. Соединения, обладающие антагонистической активностью по отношению к мускариновым рецепторам, описаны в патентных заявках Японии 92921/1994 и 135958/1994; WO 93/16048; патенте США 3176019; GB 940540; ЕР 325571; WO 98/29402; ЕР 0801067; ЕР 0388054; WO 9109013; патентах США 5281601, 6174900, 6130232, 5948792; и WO 97/45414 и представляют собой производные 1,4 дизамещенных пиперидинов; WO 98/05641 описывает фторированные производные 1,4-дизамещенных пиперидинов; WO 93/16018 и WO 96/33973 представляют собой остальные близкие ссылки.-1 007932 В отчете в Med. Chem., 44, p. 984 (2002) описаны производные циклогексилметил пиперидинил трифенилпропионамида в качестве селективных М 3 антагонистов в отличие от других рецепторных подтипов. Изложение сущности изобретения Предложены азабициклопроизводные,включая,например,6-замещенные азабицикло[3.1.0]гексаны, 2,6- и 4,6-дизамещенные производные, и 2,4,6-тризамещенные производные в качестве антагонистов мускариновых рецепторов, которые могут быть полезны, как безопасные и эффективные терапевтические или профилактические агенты для лечения различных заболеваний респираторной, мочевой и желудочно-кишечной систем. Предложены также способы синтеза этих соединений. Изобретение также относится к фармацевтическим композициям, содержащим эти соединения вместе с пригодными носителями, эксципиентами или разбавителями, которые могут быть полезны для лечения различных заболеваний респираторной, мочевой и желудочно-кишечной систем. Предложены также энантиомеры, диастереоизомеры, фармацевтически приемлемые соли и фармацевтически приемлемые сольваты этих соединений, а также фармацевтические композиции, включающие эти соединения, энантиомеры, диастереоизомеры, сольваты или их фармацевтически приемлемые соли в комбинации с фармацевтически приемлемым носителем и, необязательно, эксципиенты. Другие аспекты будут представлены далее в описании и частично будут очевидны из описания или могут следовать из практического использования изобретения. В соответствии с первым объектом настоящего изобретения описываются соединения, имеющие структурную формулу I формула 1 и их фармацевтически приемлемые соли, фармацевтически приемлемые сольваты, эфиры, энантиомеры,диастереоизомеры, гдеZ представляет NH, Nme, кислород или простую связь;R6 = R7 = H. В соответствии со вторым объектом настоящего изобретения предложены соединения структурной формулы II (формула I, где R6 и R7=H) и их фармацевтически приемлемые соли, фармацевтически приемлемые сольваты, эфиры, энантиомеры, диастереоизомеры, где Ar, R1, R2, W, X, Y, Z и Q, как указано для формулы I формула II В соответствии с третьим объектом настоящего изобретения предложены соединения, имеющие общую структурную формулу III (формула I, где W представляет (СН 2)p, где р=0, X означает отсутствие атома и Y представляет (CH2)q, где q=0, R6=H, R7=H) и их фармацевтически приемлемые соли, фармацевтически приемлемые сольваты, эфиры, энантиомеры, диастереоизомеры, где Ar, R1, R2, Z и Q, как указано для формулы I.-2 007932 В соответствии с четвертым объектом настоящего изобретения предложены соединения, имеющие общую структурную формулу IV (формула I, где W представляет (СН 2)p, где р=0, X означает отсутствие атома и Y представляет (СН 2)q, где q=0, R6=H, R7=H, и их фармацевтически приемлемые соли, фармацевтически приемлемые сольваты, эфиры, энантиомеры, диастереоизомеры, где Ar, R1, Z и Q, как указано для формулы I и r представляет от 1 до 4. формула IV В соответствии с пятым объектом настоящего изобретения предложены соединения, имеющие общую структурную формулу V (формула I, где W представляет (СН 2)p, где р=0, X означает отсутствие атома и Y представляет (СН 2)q, где q=0, R6=H, R7=H,R1 представляет гидрокси, Ar представляет фенил), и их фармацевтически приемлемые соли, фармацевтически приемлемые сольваты, эфиры, энантиомеры, диастереоизомеры, где Z и Q как указано для формулы I, s представляет от 1 до 2. формула V В соответствии с шестым объектом настоящего изобретения предложен способ лечения или профилактики животных или человека, страдающих от болезни или расстройств респираторной, мочевой и желудочнокишечной систем, где эти болезни или расстройства опосредуются мускариновыми рецепторами. Способ включает назначение по крайней мере одного соединения, имеющего структуру формулы I. В соответствии с седьмым объектом настоящего изобретения предложен способ лечения или профилактики животных или человека, страдающих от болезни или расстройств, ассоциированных с мускариновыми рецепторами, включающий назначение пациенту, нуждающемуся в этом, эффективного количества соединения-антагониста мускариновых рецепторов, как указано выше. В соответствии с восьмым объектом настоящего изобретения предложен способ лечения или профилактики животных или человека, страдающих от болезни или расстройств респираторной системы,таких как бронхиальная астма, хроническое обструктивное заболевание легких (COPD), фиброз легких и т.д.; мочевой системы, которые индуцируют такие расстройства мочевой системы, как недержание мочи,расстройства нижних мочевых путей (LUTS) и т.д.; и желудочно-кишечной системы, таких как синдром раздраженной толстой кишки, ожирение, диабет и желудочно-кишечная гиперкинезия, указанными выше соединениями, где эти болезни или расстройства ассоциированы с мускариновыми рецепторами. В соответствии с девятым объектом настоящего изобретения предложены способы получения указанных выше соединений. Описываемые соединения проявляют значительную активность, которая определялась in vitro в тестах на связывание и функционирование рецептора и в экспериментах in vivo с использованием наркотизированных кроликов. Соединения, для которых была установлена активность in vitro, тестировались invivo. Некоторые из соединений являются сильнодействующими антагонистами мускариновых рецепторов с высоким сродством к М 3 рецепторам. Предложены также фармацевтические композиции для лечения заболеваний и расстройств, ассоциированных с мускариновыми рецепторами. В добавление к этому соединения настоящего изобретения могут назначаться орально или парентерально. Детальное описание изобретения Соединения настоящего изобретения могут быть получены с помощью представленной ниже последовательности реакций: Формула I Соединения формулы I могут быть получены с помощью последовательности реакций, приведенной в схеме I. Способ получения включает реакцию соединения формулы VII с соединением формулыZ представляет NH, Nme, кислород или простую связь;R6=R7 = Н. Р представляет любую защитную для аминогруппы группу, например, бензильную или третбутилоксикарбонильную группу. Реакцию соединения формулы VII с соединением формулы VI можно проводить в присутствии конденсирующего агента (например 1-(3-диметиламинопропил)-3-этилкарбодиимид гидрохлорида (ЭДК) или 1,8-диазабицикло[5.4.0]ундец-7-ена (ДБУ), в растворителе (таком как N,N-диметилформамид, диметилсульфоксид, толуол или ксилол при температуре около 0-140 С) с получением защищенного соединения формулы VIII, с которого снимают защиту в присутствии агента для снятия защиты (например,палладия на угле, трифторуксуной кислоты (ТФК) или хлористо-водородной кислоты), в органическом растворителе (например, метанол, этанол, тетрагидрофуран или ацетонитрил, при температуре около 1050 С) с получением соединения формулы I. В приведенной выше схеме, где указаны специфические основания, конденсирующие агенты, защитные группы, агенты для снятия защиты, растворители, катализаторы, температуры и т.д., могут использоваться другие, известные специалисту в данной области основания, конденсирующие агенты, защитные группы, агенты для снятия защиты, растворители, катализаторы, температуры и т.д. Аналогично температура реакции и продолжительность могут регулироваться в соответствии с необходимостью. Получают растворимые в водной среде соли соединений формулы I, чтобы они были пригодны для биологических испытаний, совместимы с лекарственными формами а также способствовали биодоступности соединений. Примерами таких солей являются фармакологически приемлемые соли, такие как соли неорганических кислот (например, гидрохлориды, гидробромиды, сульфаты, нитраты и фосфаты),-4 007932 соли органических кислот (например, ацетаты, тартраты, цитраты, фумараты, малеаты, толуолсульфонаты и метансульфонаты). Когда соединение формулы I включает карбоксильную группу в качестве заместителя, то оно может быть в виде соли щелочного металла (например, натрия, калия, кальция, магния и тому подобное). Эти соли могут быть получены обычными известными методами, такими как обработка соединения эквивалентным количеством неорганической или органической кислоты или основания в подходящем растворителе. Предпочтительные соединения приведены ниже:(Формула I, где W=(CH2)p, где р=0, X представляет отсутствие атома и Y=(CH2)q, где q=0, R6=R7=H) Благодаря своим ценным фармакологическим свойствам, соединения настоящего изобретения могут назначаться животному для лечения орально или парентерально. Фармацевтические композиции настоящего изобретения можно производить и назначать в виде дозированных единиц, содержащих каждая некоторое количество по крайней мере одного соединения настоящего изобретения и/или по крайней мере одной его физиологически приемлемой аддитивной соли. Дозы могут меняться в очень широких пределах, так как соединения эффективны при низких уровнях доз и относительно низкой токсичности. Соединения могут назначаться в низкой микромолярной концентрации, которая является терапевтически эффективной, доза может увеличиваться по мере необходимости до максимально переносимой пациентом. Соединения настоящего изобретения можно получать и готовить из них препаративные лекарственные формы в виде их энантиомеров, диастереомеров, N-оксидов, полиморфовы, сольватовы и фармацевтически приемлемых солей, так же как и метаболитов, обладающие тем же видом активности. Могут быть также получены фармацевтические композиции, включающие соединения формулы I, II, III, IV и V или метаболиты, энантиомеры, диастереомеры, N-оксиды, полиморфы, сольваты и фармацевтически приемлемые соли этих соединений в комбинации с фармацевтически приемлемым носителем и необязательно включающие эксципиент.-7 007932 Соединения, такие как описанные, например, в табл. I, могут быть получены из соответствующей уксусной кислоты по аналогии со специальными приводимыми ниже примерами. Способы получения таких уксусных кислот хорошо известны среднему специалисту в данной области и о них можно узнать,ссылаясь на цитированные специальные примеры, приводимые ниже. Другие соединения, входящие в объем настоящего изобретения, такие как имеющие R6 и/или R7 означающие метил, карбоновая кислота,амид, аминео или метиламино, могут быть получены по аналогии со специальными процедурами, указанными в специальных приводимых ниже примерах, используя соответствующие азабицикло[3.1.0]гексаны, которые синтезируются известными методами. Другие соединения, входящие в объем настоящего изобретения, такие как имеющие X означающий кислород, сера или вторичный или третичный амин, могут быть получены по аналогии со специальными процедурами, указанными в специальных приводимых ниже примерах, используя соответствующие эфиры, тиосоединения или амиды, которые могут быть получены известными среднему специалисту в данной области методами. Подобно этому соединения, входящие в объем настоящего изобретения, такие как имеющие Y означающий CHR5CO, гдеR5 водород или метил, могут быть получены по аналогии со специальными процедурами, указанными в специальных приводимых ниже примерах, используя соответствующие ангидриды, имиды или тиоангидриды, которые могут быть получены известными среднему специалисту в данной области методами. Другие соединения, входящие в объем настоящего изобретения, такие как имеющие Z означающий кислород, серу или вторичный или третичный амин, могут быть получены по аналогии со специальными процедурами, указанными в специальных приводимых ниже примерах, используя соответствующие исходные материалы, которые могут быть получены известными среднему специалисту в данной области методами. Приводимые ниже примеры демонстрируют основные способы синтеза, так же как и специальные приготовления предпочтительных соединений. Примеры предназначены для иллюстрации деталей настоящего изобретения и не должны рассматриваться, как ограничивающие объем притязаний в соответствии с изобретением. Примеры Различные растворители, такие как ацетон, метанол, пиридин, диэтиловый эфир, тетрагидрофуран,гексан и дихлорметан, осушают с помощью различных известных осушители используя описанные в литературе методы. Инфракрасные спектры (ИК) в минеральном масле (нуджол) или в тонкой пленке снимали на приборе Perkin Elmer Paragon, спектр Ядерного Магнитного Резонанса (ЯМР) снимали наVarian XL-300 MHz, используя тетраметилсилан в качестве внутреннего стандарта. Пример 1. Получение (2R,2S)-(1,5,6)-N-[3-азабицикло[3.1.0]гекс-6-илметил)-2-гидрокси-2 циклопентил-2-фенилацетамида (соединение 1) Стадия а: Синтез (2R,2S)-(1,5,6)-N-[3-бензил-3-азабицикло[3.1.0]гексил-6-(аминометил)-ил]-2 гидрокси-2-циклопентил-2-фенилацетамида. К раствору (1,5,6)-6-аминометил-3-бензил-3-азабицикло[3.1.0]гексана (полученному как описано в ЕР 0413455 А 2) (29,9 ммол, 6,05 г) в диметилформамиде (100 мл) добавляют 2-(R,S)-гидрокси-2 циклопентил-2-фенилуксусную кислоту (получена, следуя J.Am.Chem.Soc, 1953; 75:2654) (27,2 ммол, 6,0 г) и охлаждают до 0 С. Реакционную смесь обрабатывают гидроксибензотриазолом (29,9 ммол, 4,0 г) с последующей обработкой N-метилморфолином (54,4 ммол, 5,2 г) и перемешивают при 0 С 0,5 ч. Добавляют (1-[3(-(диметиламино)пропил]-3-этилкарбодиимид гидрохлорид (ЭДК) (29,9 ммол, 5,7 г) и реакционную смесь перемешивают при 0 С 1 ч и при комнатной температуре в течение ночи. Затем реакционную смесь вливают в насыщенный бикарбонат натрия и эктрагируют этилацетатом. Органические слои промывают водой, сушат над сульфатом натрия и концентрируют при пониженном давлении. Остаток очищают колоночной хроматографией (силикагель 100-200 меш), элюируя соединение 93-95% чистоты. Для получения более высокой чистоты (около 99%) соединение растирают с толуолом и фильтруют. Стадия b: Получение (2R,2S)-(1,5,6)-N-[3-азабицикло[3.1.0]гексил-6-(аминометил)-ил]-2 гидрокси-2-циклопентил-2-фенилацетамида. К раствору(2R,2S)-(1,5,6)-N-[3-бензил-3-азабицикло[3.1.0]гексил-6-(аминометил)-ил]-2 гидрокси-2-циклопентил-2-фенилацетамида из примера 1, стадия а, (1,0 г, 2,48 ммол) в метаноле (25,0 мл) добавляют в атмосфере азота 5% Pd-C (0,2 г) (50 вес.%). Затем при перемешивании добавляют муравьинокислый аммоний (0,8 г, 12,38 ммол) и реакционную смесь нагревают с обратным холодильником в течение получаса в атмосфере азота. Смесь охлаждают до комнатной температуры и реакционную смесь фильтруют через слой Hyflo. Hyflo слой промывают метанолом (75,0 мл), этилацетатом (25,0 мл) и водой (25,0 мл). Фильтрат концентрируют под вакуумом. Остаток разводят водой и доводят рН полученного раствора до рН 14 с помощью 1N NaOH. Раствор экстрагируют этилацетатом (2x50 мл) и этилацетатный слой промывают водой и соляным раствором. Слой сушат над безводным Na2SO4 и концентрируют с получением указанного в заголовке соединения в виде твердого остатка с выходом 96,2% (0,75 г,2,39 ммол) и чистотой 98% (HPLC) (жидкостная хроматография высокого разрешения). Соединение имеет точку плавления 149-151 С и ИК (KBr) 3410, 2951,5, 2868,3 и 1652,5 см-1.(m, 4H, включая ОН), l,51-l,71(m, 8H), 1.19-1.27(m, 4 Н), 0.70-0.72 (m, 1 Н). Масс спектры показывают пики при m/е 315(МН+), 297(М-ОН). Пример 2. Получение (2R,2S)-(1,5,6)-N-[3-азабицикло[3.1.0]гексил-6-(аминометил)-ил]-2 гидрокси-2-циклопентил-2-фенилацетамида, хлористо-водородная соль (соединение 2) К раствору (2R,2S)-(l,5,6)-N-[3-азабицикло[3.1,0]гексил-6-(аминометил)-ил]-2-гидрокси-2 циклопентил-2-фенилацетамида (0,2 г, 0,637 ммол) (полученному в примере 1) в дихлорметане (4,0 мл) добавляют этанольный HCl (1,45 N, 0,5 мл, 0,725 ммол) при комнатной температуре и перемешивают 10 мин. При той же температуре к реакционной смеси добавляют диэтиловый эфир (100 мл), перемешивают 5 мин и концентрируют под вакуумом без нагрева. Остаток растирают с эфиром до получения твердого материала. Эфирный слой декантируют и твердый остаток сушат под вакуумом до получения указанного в заголовке соединения в виде гигроскопичного твердого вещества с выходом 94% (0,21 г, 0,6 ммол) и чистотой 98% (HPLC). Пример 3. Получение(2R)-(1,5,6)-N-[3-азабицикло[3.1.0]гексил-6-(аминометил)-ил]-2 гидрокси-2-циклопентил-2-фенилацетамида (соединение 3) Стадия а: Синтез (2R)-(l,5,6)-N-[3-бензил-3-азабицикло[3.1.0]гексил-6-(аминометил)-ил]-2 гидрокси-2-циклопентил-2-фенилацетамида. Это соединение получено, следуя методике примера 1, стадия а, используя (2R)-2-гидрокси-2 циклопентил-2-фенилуксусную кислоту (синтезированную, как указано Grover et al., J.Org. Chem., 2000; 65:6283-6287) вместо 2-гидрокси-2-циклопентил-2-фенилуксусной кислоты. Стадия b: Синтез (2R)-(1,5,6)-N-[3-азабицикло[3.1.0]гексил-6-(аминометил)-ил]-2-гидрокси-2 циклопентил-2-фенилацетамида. Это соединение синтезировано, следуя методике примера 1, стадия b, используя (2R)-(1,5,6)-N[3-бензил-3-азабицикло[3.1.0]гексил-6-(аминометил)-ил]-2-гидрокси-2-циклопентил-2-фенилацетамид вместо(R) изомеров. S-изомер элюируется приблизительно 11,11 мин и R-изомер элюируется приблизительно 11,81 мин. Оптическая чистота 99%. Соединение имеет точку плавления 150,2 С. ИК (DCM): 1653,8 см-1. 1 Н-ЯМР (CDCl3):7.61 (d, J=9Hz, 2H), 7.30-7.38 (m, 2H), 6.70 (s, 1H), 3.61-3.68 (m, 2H), 3.08-3.28 (m,5H), l,49-l,68(m, 10H), 1.11-1.26(m, 2H), 0.75-0.85 (m, 1H). Пример 4. Получение хлористо-водородной соли (2R)-(l,5,6)-N-[3-азабицикло[3.1.0]гексил-6(аминометил)-ил]-2-гидрокси-2-циклопентил-2-фенилацетамида (соединение 4) Хлористо-водородная соль была синтезирована, следуя примеру 2 с использованием (2R)(1,5,6)-N-[3-азабицикло[3.1.0]гексил-6-(аминометил)-ил]-2-гидрокси-2-циклопентил-2-фенилацетамида вместо(2R,2S)-(1,5,6)-N-[3-бензил-3-азабицикло[3.1.0]гексил-6-(аминометил)-ил]-2-гидрокси-2-циклопентил-2-фенилацетамида. Энантимерный избыток (ее) определяют с помощью HPLC (Chinacel OD, подвижная фаза 90% гексан/10% этанол/0,1% трифторуксусная кислота), наблюдая (S) и (R) изомеры. Sизомер элюируется приблизительно 11,11 мин. R-изомер элюируется приблизительно 11,81 мин. Оптическая чистота 99%. Соединение имеет точку плавления 62,6-63,3 С. ИК (KBr): 1653,7 см-1. 1 Н-ЯМР (CDCl3):7.59-7.62 (m, 2H), 7.29-7.37 (m, 3 Н), 3.58-3.65 (m, 2H), 3.02-3.24 (m, 4H), 1,111,34 (m, 11 Н), 0.75-0.95 (m, 1H).-9 007932 Пример 6. Получение хлористо-водородной соли (2S)-(l,5,6)-N-[3-азабицикло[3.1.0]гексил-6(аминометил)-ил]-2-гидрокси-2-циклопентил-2-фенилацетамида (соединение 6). Хлористо-водородная соль была синтезирована с 90% выходом, следуя примеру 2, с использованием(2R,2S)-(1,5,6)-N-[3-азабицикло[3.1.0]гексил-6-(аминометил)-ил]-2 гидрокси-2-циклопентил-2-фенилацетамида. Пример 7. Получение (2R,2S)-(1,5,6)-N-[3-азабицикло[3.1.0]гексил-6-(аминометил)-ил]-2 метокси-2-циклопентил-2-фенилацетамида (соединение 7) Стадия а: Получение этилового эфира (2R,2S)-2-метокси-2-циклопентил-2-фенилуксусной кислоты К охлажденному раствору этилового эфира (2R,2S)-2-гидрокси-2-циклопентил-2-фенилуксусной кислоты (синтезирован, как указано в J.Am.Chem. Soc, 1953; 75:2654) (4,5 ммол) в диметилформамиде порциями добавляют гидрид натрия (9,08 ммол) при 0 С и перемешивают при комнатной температуре 1 ч. Реакционную смесь охлаждают до 0 С и добавляют иодометан (18,0 ммол). Затем реакционную смесь перемешивают при комнатной температуре 2 ч. TLC (тонкослойная жидкостная хроматография) показывает отсутствие исходного материала. К реакционной смеси добавляют воду и экстрагируют этилацетатом. Органический слой сушат над безводным сульфатом натрия и концентрируют. Сырое соединение очищают с помощью колоночной хроматографии, элюируя требуемый продукт 2% EtOAc/гексан. 1 Н-ЯМР: (CDCl3) -величины: 7.47-7.36 (5 Н, m), 4.31 (2 Н, q, 3.26 (3 Н, s), 2.43 (1 Н, m), 1.66-1.46(11 Н, m). Стадия b: Синтез (2R,2S) 2-метокси-2-циклопентил-2-фенилуксусной кислоты К раствору этилового эфира (2R,2S) 2-метокси-2-циклопентил-2-фенилуксусной кислоты (1,8 ммол) в метаноле добавляют гидроксид калия (КОН) (2,2 ммол) и реакционную смесь нагревают с обратным холодильником 7 ч. TLC показывает наличие исходного материала, затем добавляют 3 мольэквивалента КОН и реакционную смесь нагревают с обратным холодильником 3 ч. TLC показывает отсутствие исходных материалов. Реакционную смесь концентрируют, остаток разводят водой, нейтрализуют концентрированной соляной кислотой и экстрагируют этилацетатом. Органический слой промывают водой,рассолом, сушат над безводным сульфатом натрия и концентрируют при пониженном давлении, получая желаемый продукт. 1 Н-ЯМР: (CDCl3) -величины: 7.48-7.35 (5 Н, m), 3.20 (3 Н, s), 2.94-2.86 (1 Н, m), 1.86-1.50 (8 Н, m) Стадия с: Получение (2R,2S)-(1,5,6)-N-[3-бензил-3-азабицикло[3.1.0]гексил-6-(аминометил)ил]-2-метокси-2-циклопентил-2-фенилацетамида. Получен, следуя методике примера 1, стадия а, используя 2-метокси-2-циклопентил-2 фенилуксусную кислоту вместо 2-гидрокси-2-циклопентил-2-фенилуксусной кислоты. Стадия d: Получение (2R,2S)-(1,5,6)-N-[3-азабицикло[3.1.0]гексил-6-(аминометил)-ил]-2 метокси-2-циклопентил-2-фенилацетамида Получен, следуя методике примера 1, стадия b, используя (2R,2S)-(1,5,6)-N-[3 азабицикло[3.1.0]гексил-6-(аминометил)-ил]-2-метокси-2-циклопентил-2-фенилацетамид вместо (2R,2S)(1,5,6)-N-[3-бензил-3-азабицикло[3.1.0]гексил-6-(аминометил)-ил]-2-гидрокси-2-циклопентил-2 фенилацетамида. 1 Н-ЯМР: (CDCl3) -величины: 7.45-7.30 (5 Н, m), 7.03 (1 Н, m), 3.25-3.02 (9 Н, m), 2.00-0.86 (12 Н, m). Пример 8. Получение (2R,2S)-(1,5,6)-N-[3-азабицикло[3.1.0]гексил-6-(аминометил)-ил]-2 гидрокси-2-циклопентил-2-фенилацетамида (соединение 8) Стадия а: Получение (2R,2S)-(l,5,6)-N-[3-бензил-3-азабицикло[3.1.0]гексил-6-(аминометил)-ил]2-гидрокси-2-циклопентил-2-фенилацетамида. Это соединение синтезировано, следуя примеру 1, стадия а, используя (2R,2S)-2-гидpoкcи-2 циклoпeнтил-2-фeнилyкcycнyю кислоту (синтезирована, как указано в Grover et all, J.Org.Chem., 2000; 65:6283-6287) вместо 2-гидрокси-2-циклопентил-2-фенилуксусной кислоты. Стадия b: Синтез (2R,2S) (1,5,6)-N-[3-азабицикло[3.1.0]гексил-6-(аминометил)-ил]-2-гидрокси 2-циклогептил-2-фенилацетамида. Это соединение синтезировано, следуя примеру 1, стадия b, используя (2R,2S)(1,5,6)-N-[3 бензил-3-азабицикло[3.1.0]гексил-6-(аминометил)-ил]-2-гидрокси-2-циклогептил-2-фенилацетамид вместо (2R,2S)(1,5,6)-N-[3-бензил-3-азабицикло[3.1.0]гексил-6-(аминометил)-ил]-2-гидрокси-2-циклопентил-2-фенилацетамида, с выходом 90%. 1 Н-ЯМР (CDCl3):7.59-7.61 (m, 2H), 7.13-7.36 (m, 3 Н), 6.76 (brs, 1H), 3.00-3.20 (m, 2H), 2,80-2,92(m, 2H), 2.50-2.80 (m, 1H), 2.40 (brs, 2H), 1.28-1.73 (m, 12H), l,00-l,20(m, 2H), 0.80-0.90 (m, 1H). ИК(DCM): 1655,7 см-1. Масс-спектр имеет пики при m/e: 343 (МН+). Пример 9. Получение (2R,2S)(1,5,6)-N-[3-азабицикло[3.1.0]гексил-6-(аминометил)-ил]-2 гидрокси-2-циклобутил-2-фенилацетамида (соединение 9) Стадия а: Синтез (2R,2S)(1,5,6)-N-[3-бензил-3-азабицикло[3.1.0]гексил-6-(аминометил)-ил]-2 гидрокси-2-циклобутил-2-фенилацетамида.(6 Н, m), 1.45 (1 Н, m), 1,00 (1 Н, m). Пример 10. Получение тартратной соли (2R,2S)-(l,5,6)-N-[3-азабицикло[3.1.0]гексил-6(аминометил)-ил]-2-гидрокси-2-циклобутил-2-фенилацетамида (соединение 10) К раствору соединения 9 в этаноле добавляют твердую винную кислоту, раствор перемешивают 1 ч при комнатной температуре и упаривают растворитель. Добавляют этиловый эфир до высаживания соли. 4 раза промывают эфиром, декантируя надосадочную жидкость с получением соли в виде порошка 95,66% чистоты (HPLC). 1(2R)-(l,5,6)-N-[3-азабицило[3.1.0]гексил-6-(аминометил)-ил]-2 гидрокси-2-(3,3-дифторциклопентил)-2-фенилацетамида (соединение 11) Стадия а: Получение (2R,5R)-2-третбутил-5-фенил-1,3-диоксолан-4-она Это соединение получено, следуя процедуре, описанной в J.Org. Chem., 2000; 65:6283-6287. Стадия b: Получение (2R,5R)-2-третбутил-5-[(1R или 1S)-3-оксоциклопентил]-5-фенил-1,3 диоксолан-4-она. К суспензии соединения со стадии а (1,36 ммол) в тетрагидрофуране (ТГФ) (12 мл) по каплям добавляют литийдиизопропиламид (ЛДА) в ТГФ (1,5 мл) при -78 С в атмосфере азота. Реакционную смесь перемешивают при этой же температуре 2 ч. К реакцонной смеси добавляют по каплям раствор 2 циклопентен-1-она (1,52 мл) в ТГФ (2 мл) и еще перемешивают 3 ч. Реакционную смесь резко охлаждают путем добавления холодного насыщенного водного раствора хлорида аммония и экстрагируют этилацетатом. Органический слой сушат и полученный после удаления в вакууме растворителя остаток очищают колоночной хроматографией (100-200 меш, силикагель). Продукт элюируют 10% смесью ЕtOАс-гексан. 1 Н-ЯМР (CDCl3):7.70-7.26 (m, 5 Н, ArH), 5.43-5.37 (d, 1H), 2.91-2.88 (m, 1 Н), 3.37-1.77 (m, 6 Н),0.92 (s, 9H). Стадия с: Получение (2R,5R)-2-третбутил-5-[(1R или 1S)-3,3-дифторциклопентил]-5-фенил-1,3 диоксолан-4-она К раствору соединения со стадии b (1 ммол) в хлороформе при 0 С добавляют трехфтористую диэтиламиносеру (ДАСТ) (3,3 мл) в атмосфере азота. Реакционную смесь перемешивают при этой температуре 30 мин и затем 3 дня при комнатной температуре. После охлаждения до 0 С реакционную смесь(PC) сразу резко охлаждают добавлением холодной воды. Отделяют органический слой и водный слой экстрагируют хлороформом. Объединенные органические слои сушат и полученный после удаления растворителя остаток очищают колоночной хроматографией (100-200 меш, силикагель). Продукт элюируют 5% смесью EtOAc-гексан. 1H-ЯМР (CDCl3):7.73-7.35 (m, 5 Н, ArH), 5.49 (s,1H), 2.86-2.82 (m, 1H), 2.27-1.80 (m,6H), 0.98 (s,H) ИK(DCM): 1793 см-1 Стадия d: Получение (2R)-[(1S или 1R)-3,3-дифторциклопентил]-2-гидрокси-2-фенилуксусной кислоты Раствор соединения со стадии с (1 ммол) в МеОН (10 мл) перемешивают с 3N водным раствором гидроокиси натрия в течение ночи при комнатной температуре. Реакционную смесь концентрируют при пониженном давлении. Остаток разводят водой и экстрагируют дихлорметаном. Водный слой подкисляют концентрированной соляной кислотой и экстрагируют EtOAc. Органический слой сушат и концентрируют при пониженном давлении, получая требуемый продукт. Т.пл. 123 С. 1 Н-ЯМР (CDCl3):7.69-7.37 (m, 5H, ArH), 3.29-3.20 (m, 1 Н), 2.39-1.68 (m, 6 Н) Стадия е: Получение (1,5,6)-6-аминометил-3-бензил-3-азабицикло[3.1.0]гексана. Соединение синтезировано, следуя процедуре, описанной в ЕР 0413455 А 2. Стадия f: Получение (2R)-(1,5,6)-N-[3-бензил-3-азабицикло[3.1.0]гексил-6-(аминометил)-ил]-2[(1R или 1S)-3,3-дифторциклопентил]-2-гидрокси-2-фенилацетамида Это соединение получено, следуя методике примера 1, стадия а, используя кислоту, синтезированную на стадии d, вместо 2-гидрокси-2-циклопентил-2-фенилуксусной кислоты. Стадия g: Синтез (2R)-(1,5,6)-N-[3-азабицикло[3.1.0]гексил-6-(аминометил)-ил]-2-гидрокси-2(3,3-дифторциклопентил)-2-фенилацетамида- 11007932 Это соединение получено, следуя методике примера 1, стадия b, используя (2R)-(1,5,6)-N-[3 бензил-3-азабицикло[3.1.0]гексил-6-(аминометил)-ил]-2-гидрокси-2-(3,3-дифторциклопентил]-2-фенилацетамид вместо(2R,2S)(1,5,6)-N-[3-бензил-3-азабицикло[3.1.0]гексил-6-(аминометил)-ил]-2 гидрокси-2-циклопентил-2-фенилацетамида. Оптическая чистота 87,27% (HPLC). 1 Н-ЯМР (CDCl3):7.59-7.55 (2 Н, m), 7.35-7.31 (3 Н, m), 7.03 (1 Н, m), 3.18-3.11 (7 Н, m), 1,87-1.62(9 Н, m). Пример 12. Получение (2R,2S)(1,5,6)-N-[3-азабицикло[3.1.0]гексил-6-(аминометил)-ил]-2 гидрокси-2-(3-фторциклопентил)-2-фенилацетамида (соединение 12) Стадия а: Получение (2R,2S)-2-трет-бутил-5-[(1R или 1S)-3-гидроксициклопентил]-5-фенил-1,3 диоксолан-4-она. К охлажденному до 0 С раствору (2R,2S,5R)-2-трет-бутил-5-[(1R или 1S)-3-оксоциклопентил]-5 фенил-1,3-диоксалан-4-она (1 ммоль) в метаноле (10 мл) при перемешивании добавляют небольшими партиями борогидрид натрия. Реакционную смесь перемешивают при 0 С 1 ч. Концентрируют при пониженном давлении, остаток разбавляют водой и экстрагируют EtOAc. Органический слой сушат и полученный после удаления растворителя остаток очищают колоночной хроматографией (100-200 меш,силикагель), элюируя соединение 20% смесью EtOAc-гексан. 1 Н-ЯМР (CDCl3):7.68-7.29 (m, 5H, ArH), 5.45 (d, 1H), 4.30 (m, 1H), 3.25 (m, 1 Н), 2.65-2.63 (m, 1 Н),1.80-1.63 (m, 6 Н), 0.92 (s, 9H). Стадия b: Получение (2R,2S)-2-третбутил-5-[(1R или 1S,3R или 3S)-3-фторциклопентил]-5-фенил 1,3-диоксолан-4-она Раствор соединения со стадии а (1 ммол) в хлороформе (10 мл) охлаждают до 0 С и добавляют по каплям трехфтористую диэтиламиносеру (ДАСТ) (1,5 мл) в атмосфере азота. Реакционную смесь перемешивают при 0 С 30 мин и затем 3 дня при комнатной температуре. Реакционную смесь охлаждают и сразу резко охлаждают добавлением холодного раствора хлорида аммония. Отделяют органический слой и водный слой экстрагируют EtOАс. Объединенные органические слои сушат и полученный после удаления растворителя остаток очищают колоночной хроматографией (100-200 меш, силикагель), элюируя соединение 5% смесью EtOАс-гексан. 1 Н-ЯМР (CDCl3):7.68-7.28 (m, 5 Н, ArH), 5.49 (d,lH), 5.39 (m, 1H), 2.90 (m, 1H), 1.98-1.25 (m, 6H),0.93(s, 9H) Стадия с: Получение (2R,2S)-[(1R или 1S,3R или 3S)-3-фторциклопентил]-2-гидрокси-2 фенилуксусной кислоты Соединение синтезируют, следуя методике примера 11, стадия d, используя (2R,2S,5R)-2 третбутил-5-[(1R или 1S,3R или 3S)-3-фторциклопентил]-5-фенил-1,3-диоксалан-4-он вместо (2R,5R)-2 третбутил-5-[(1R или 1S)-3,3-дифторциклопентил]-5-фенил-1,3-диоксалан-4-она. 1 Н-ЯМР (CDCl3):7.66-7.27 (m, 5 Н, ArH), 5.30-5.00 (m, Н), 3.32-3.16 (m, 1 Н), 2.05-1.26 (m,6 Н) ИK(DCM): 1710 см-1 Стадия d: Получение (2R,2S)-(1,5,6)-N-[3-бензил-3-азабицикло[3.1.0]гексил-6-(аминометил)ил]-2-[(1R или 1S,3R или 3S)-3-фторциклопентил]-2-гидрокси-2-фенилацетамида. Это соединение получено, следуя методике примера 1, стадия а, используя кислоту, синтезированную на вышеуказанной стадии с, вместо 2-гидрокси-2-циклопентил-2-фенилуксусной кислоты. Стадия е: Получение (2R,2S)(1,5,6)-N-[3-азабицикло[3.1.0]гексил-6-(аминометил)-ил]-2 гидрокси-2-(3-фторциклопентил]-2-фенилацетамида. Это соединение получено, следуя методике примера 1, стадия b, используя (2R,2S)-(1,5,6)-N-[3 бензил-3-азабицикло[3.1.0]гексил-6-(аминометил)-ил]-2-[1R или 1S,3R или 3S]-3-фторциклопентил]-2 гидрокси-2-фенилацетамид вместо (2R,2S)-(l,5,6)-N-[3-бензил[-3-азабицикло[3.1.0]гексил-6-(аминометил)-ил]-2-гидрокси-2-циклопентил-2-фенилацетамида. Оптическая чистота 87,27% (HPLC). 1H-ЯМР (CDCl3):7.56 (2 Н, m), 7.35(3 Н, m), 6.08 (1 Н, m), 5.30-5.03 (1 Н, m), 3,27 (1 Н, m), 3.11 (2 Н,m), 2.91(4 Н, m), 2.04-1.48 (9 Н, m), 0.71 (1 Н, m). Пример 1. Получение (2R,2S)-(1,5,6)-N-[3-азабицикло[3.1.0]гексил-6-(аминометил)-ил]-2 гидрокси-2-(3,3-дифторциклопентил-2-фенилацетамида (соединение 13) Это соединение получают, следуя методике примера 11, используя (2R,2S)-[(1,5,6)-N-[3 бензил-3-азабицикло[3.1.0]гексил-6-(аминометил)-ил]-2-гидрокси-2-(3,3-дифторциклопентил)-2-фенилацетамид] вместоH-ЯМР: (CDCl3) -величины: 7.57-7.30 (5 Н, m), 6.49-6.44 (1H, m), 3.33 (1 Н, m), 3.10 (2 Н, m), 6,496.44 (1 Н, m), 3.33 (1 Н, m), 3.10 (2 Н, m), 2.87 (3 Н, m), 2.23-1.80 (8 Н, m), 1.79-1.20 (2 Н, m) ИК(КBr): 3410, 1654 см-1 Пример 14. Получение тартратной соли (2R,2S)-(l,5,6)-N-[3-азабицикло[3.1.0]гексил-6(аминометил)-ил]-2-гидрокси-2-(3,3-дифторциклопентил)-2-фенилацетамида (соединение 14)- 12007932 К раствору соединения 13 в этаноле добавляют винную кислоту и реакционную смесь греют при 60 С 12 ч. Затем реакционную смесь концентрируют при пониженном давлении, добавляют диэтиловый эфир и отделяют органический слой, получая светлокоричневый твердый остаток требуемого продукта. Оптическая чистота 98,214% (HPLC). 1(1,5,6)-3-бензил-6-(метансульфонилокси)метил-3-азабицикло[3.1.0]гексана: Раствор указанного в заголовке стадии (i) соединения (0,203 г, 1 ммол) и триэтиламина (0,21 г, 2 ммол) в этилацетате (25 мл) охлаждают до -10 С и обрабатывают метансульфонилхлоридом (0,17 г, 1,5 ммол). После перемешивания в течение 1 ч при -10 С реакционную смесь вливают в насыщенный водный раствор бикарбоната натрия. Органический слой сушат над сульфатом натрия, фильтруют и в вакууме удаляют растворитель, получая указанное в заголовке соединение в виде желтого масла, которое без очистки используют на следующей стадии. 1 Н-ЯМР: (CDCl3) -величины: 7.45 (m, 5 Н, аром.), 4.29 (s, 2 Н), 3.81 (m, 2 Н), 3.13 (m, 4 Н), 2,84 (s,3 Н), 1,38 (m, 3 Н) Стадия (iii): Получение (1,5,6)-[3-бензил-3-азабицикло[3.1.0]гексил-6-(метил)-ил]-2-гидрокси 2,2-дифенилацетата К раствору 2-гидрокси-2,2-дифенилуксусной кислоты (синтезирована, как описано в Vogel's textbook of "Practical Organic Chemistry", стр. 1046 (5th Ed); J. Am.Chem. Soc, 1953; 75: 2654 и ЕР 613232) (1 ммол, 0,228 г) в ксилоле добавляют (1,5,6)-[3-бензил-6-(метансульфонилокси)метил-3 азабицикло[3.1.0]гексан (0,28 г, 1 ммол), затем добавляют ДБУ (1,8-диазабицикло[5,4,0]ундец-7-ен (2 ммол, 0,305 г) и реакционную смесь нагревают с обратным холодильником 6 ч. Затем реакционную смесь промывают водой, рассолом и сушат над сульфатом натрия. Упаривают растворитель и полученное сырое соединение очищают колоночной хроматографией (силикагель, 100-200 меш), элюируя соединение 20-80, этилацетат гексан. 1 Н-ЯМР: (CDCl3) -величины: 7.46-7.22 (m, 15H, аром.), 4.24 (s, 1H), 4.11-4.09 (d, 2H), 3.56 (s, 2H),2,91-2/89 (d, 2H), 2,31-2.29 (d, 2H), 1.67-1.62 (m, 1H), 1.3 (s,2H) ИK(DCM): 1724 см-1 Стадия b: Синтез (2R,2S)-(1,5,6)-N-[3-азабицикло[3.1.0]гексил-6-(метил)-ил]-2-гидрокси-2,2 дифенилацетата Осуществляют дебензилирование, следуя процедуре примера 1, стадия b, получая указанное в заголовке соединение в выходом 60%. ИК(КBr): 1731.6 см-1 1(m, 2 Н), 0,88 (t, 1H) Масс-спектр показывает пики при m/е 324 (М+1) Пример 16. Получение (2R,2S)(la,5a,6a)-N-[3-азабицикло[3.1.0]гексил-6-(аминометил)-ил]-2 гидрокси-2,2-дифенилацетамида (соединение 16); Стадия а: Получение (2R,2S)(1,5,6)-N-[3-бензил-3-азабицикло[3.1.0]гексил-6-(аминометил)-ил]2-гидрокси-2,2-дифенилацетамида. Соединение получено следуя методике примера 1, стадия а, используя 2-гидрокси-2,2 дифенилуксусную кислоту (синтезирована, как описано в Vogel's textbook of "Practical OrganicChemistry", стр. 1046 (5th Ed); J. Am.Chem. Soc, 1953; 75:2654 и ЕР 613232) вместо 2-гидрокси-2 циклопентил-2-фенилуксусной кислоты. Стадия b: Получение (2R,2S)(1,5,6)-N-[3-азабицикло[3.1.0]гексил-6-(аминометил)-ил]-2-гидрокси-2-(3,3-дифторциклопентил)-2-фенилацетамида. Соединение получено, следуя методике примера 1, стадия b, используя (1,5,6)-N-[3-бензил-3 азабицикло[3.1.0]гексил-6-(аминометил)-ил]-2-гидрокси-2,2-дифенилацетамид вместо (2R,2S)(1,5,6)N-[3-бензил-3-азабицикло[3.1.0]гексил-6-(аминометил)-ил]-2-гидрокси-2-циклопентил-2-фенилацетамида, получая указанное в заголовке соединение с выходом 70%. ИК(КBr): 1658.0 см-1 1 Н-ЯМР: (CDCl3) -величины: 7.34-7.44 (m, 10H), 6.53 (s, 1H), 3.17-3.26 (m, 2 Н), 2.87-3.01 (m, 4 Н),1.38 (s, 2H), 0.88 (t, lH). Масс-спектр показывает пики при m/е 323 (М+1), 305 (М-ОН).- 13007932 Пример 17. Получение (2R,2S)-(l,5,6)-N-[3-азабицикло[3.1.0]гексил-6-(аминометил)-ил]-2 гидрокси-2-циклогексил-2-фенилацетамида (соединение 17) Стадия а: Получение (1,5,6)-N-[3-бензил-3-азабицикло[3.1.0]гексил-6-(аминометил)-ил]-2 гидрокси-2-циклогексил-2-фенилацетамида. Соединение получено, следуя методике примера 1, стадия b, используя 2-гидрокси-2-циклогексил 2-фенилуксусную кислоту (синтезирована, как описано в J. Am. Chem. Soc, 1953; 75: 2654 вместо 2 гидрокси-2-циклопентил-2-фенилуксусной кислоты. Стадия b: Получение (2R,2S)(1,5,6)-N-[3-азабицикло[3.1.0]гексил-6-(аминометил)-ил]-2 циклогексил-2-фенилацетамида Соединение получено, следуя методике примера 1, стадия b, используя (2R,2S)(1,5,6)-N-[3 бензил-3-азабицикло[3.1.0]гексил-6-(аминометил)-ил]-2-гидрокси-2-циклогексил-2-фенилацетамид вместо (2R,2S)(1,5,6)-N-[3-бензил-3-азабицикло[3.1.0]гексил-6-(аминометил)-ил]-2-гидрокси-2-циклопентил-2-фенилацетамида, получая указанное в заголовке соединение с выходом 80%. ИК(КBr): 1654.7 см-1 1 Н-ЯМР: (CDCI3) -величины: 7.59-7.62 (m, 2H), 7.29-7.37 (m, 3 Н), 6.71 (s, 1 Н), 3.03-3.14 (m, 2H),2.80-2.92 (m, 4H), 2.42 (m, 1H), 1.13-1.35 (m, 12H), 0.88 (m, 1H) Масс-спектр показывает пики при m/e 329 (M+l), 311 (М-ОН). Пример 18. Получение (2R,2S)-(1,5,6)-N-[3-азабицикло[3.1.0]гексил-6-метил]-2-циклопентил-2 гидрокси-N-метил-2-фенилацетамида (соединение 18) Стадия а: Получение (2R, 2S) (1,5,6)-N-[3-третбутилоксикарбонил-3-азабицикло[3.1.0]гексил-6 метил]-2-гидрокси-2-циклопентил-2-фенилацетамида К раствору (1,5,6)-3N-бензил-6-амино-3-азабицикло[3.1.0]гексана (синтезирован в соответствии с процедурой, описанной Braish T.F. et al., Synlett, 1996; 1100) (2,5 г, 7,96 мл) в (50,0 ммол) при 0 С добавляют триэтиламин (3,9 мл, 28 ммол) и ангидрид бутилоксикарбонила (Воc) (5,2 г, 23,9 ммол). Реакционную смесь перемешивают при 0 С 30 мин и 12 ч при комнатной температуре. Разбавляют дихлорметаном (50 мл) и промывают водой и рассолом. Сушат над безводным сульфатом натрия и концентрируют. Остаток очищают колоночной хроматографией, используя 25% этилацетат в гексане и получая указанное в заголовке соединение в виде твердого вещества с выходом 86% (2,85 г, 6,9 ммол). Вещество имеет температуру плавления 179,5-182,9 С. Стадия b: Получение (2R,2S)(1,5,6)-N-[3-третбутилоксикарбонил-3-азабицикло[3.1.0]гексил-6 метил]-2-(триметилсилилокси)-2-циклопентил-2-фенилацетамида К раствору производного бутилоксикарбонила (Воc) (2,0 г, 4,8 ммол) в диметилформамиде (10,0 мл) добавляют имидазол (1,2 г, 16,9 ммол) и триметилхлорсилан (1,54 мл, 12,0 ммол) и реакционную смесь перемешивают при комнатной температуре 12 ч. Разводят водой и экстрагируют этилацетатом. Этилацетатный слой промывают водой и рассолом. Сушат и концентрируют. Остаток очищают колоночной хроматографией, используя 15% этилацетат в гексане, получая указанное в заголовке соединение с выходом 85% (2,0 г, 4,1 ммол). Стадия с: Получение (2R,2S)(1,5,6)-N-[3-третбутилоксикарбонил-3-азабицикло[3.1.0]гексил-6 метил]-2-циклопентил-2-гидрокси-N-метил-2-фенилацетамида К раствору триметилсилильного производного (2,0 г, 4,1 ммол) и n-тетрабутиламмонийиодида (0,11 г, 0,3 ммол) в сухом тетрагидрофуране (ТГФ) (20,9 мл) при 0 С порциями добавляют гидрид натрия (0,6 г, 12,3 ммол), полученный раствор перемешивают при 0 С 15 мин и дают остыть до комнатной температуры и перемешивают 1 ч при комнатной температуре. Опять охлаждают до 0 С и по каплям добавляют метилиодид (2,3 мл, 36,8 ммол) в сухом ТГФ (2,0 мл). Перемешивают при комнатной температуре 12 ч. Добавляют насыщенный водный раствор NаНСО 3 (10,0 мл), отделяют органический слой и сушат над безводным сульфатом натрия. Концентрируют и остаток очищают колоночной хроматографией, используя 15% этилацетат в гексане, получая указанное в заголовке соединение в полутвердом виде с выходом 61% (1,25 г, 2,49 ммол). Стадия d: Получение (2R,2S)(1,5,6)-N-[3-азабицикло[3.1.0]гек-6-силметил]-2-циклопентил-2 гидрокси-N-метил-2-фенилацетамида К раствору соединения с предыдущей стадии (0,2 г, 0,4 ммол) в этаноле (5,0 мл) по каплям добавляют концентрированный HCl так, чтобы рН реакционной смеси было 2. Реакционную смесь перемешивают при комнатной температуре 24 ч. Нейтрализуют насыщенным водным раствором бикарбоната натрия. Концентрируют в вакууме, остаток поглощают дихлорметаном (10,0 мл) и промывают водой и рассолом. Органический слой сушат над безводным сульфатом натрия и концентрируют, получая указанное в заголовке соединение в твердом виде с выходом 54% (0,07 г, 0,21 ммол) Т.пл. 91,5 С 1 Н-ЯМР: (CDCl3) -величины: 7.29-7.42 (m, 5 Н), 5.39 (m, 1H), 2.81-3.52 (m, 10 Н), 1.11-1.82 (m, 12 Н) ИK(DCM): 1621.9 см-1 Масс-спектр показывает пик 329 (МН+).- 14007932 Биологическая активность Исследование на связывание с использованием радиоактивного лиганда. Сродство тестируемых соединений к М 2 и M1 подтипам мускариновых рецепторов определяли, исследуя связывание [3H]-N-метилскополамина с использованием сердечной мышцы и подчелюстной железы крысы соответственно, как описано Moriya et al., (Life Sci, 1999, 64(25):2351-2358) со следующими небольшими изменениями: низкоскоростное центрифугирование ведут при 500 g в течение 10 мин при 4 С; буфер 20 mM HEPES, 10 mМ EDTA при рН 7,4; высокоскоростное центрифугирование ведут при 40000 g и перед каждым центрифугированием гомогенизат пропускают через стандартный фильтр. Условия исследования модифицированы следующим образом: исследуемый объем составлял 250 мкл; время инкубации составляло 2 ч; концентрация РЕ 0,1%; использовали фильтр GF/B от Wallac; сцинтиллятор Supermix от Wallac; количество сцинциллятора 500 мкл/лунку; сцинтилляционный счетчик 1450 microbeta PLUS от Wallac. Подготовка мембран: Сразу после умерщвления извлекают подчелюстные железы и сердце крысы и помещают в охлаждаемый льдом гомогенизирующий буфер (HEPES 20 мМ, 10 мМ EDTA, рН 7,4). Ткани гомогенизирут в 10 объемах гомогенизирующего буфера, гомогенизат фильтруют через два слоя влажной марли и фильтрат центрифугируют при 500 g 10 мин. Затем супернатант центрифугируют при 40000g 20 мин. Осадок в пробирке после центрифугирования ресуспендируют в том же количестве аналитического буфера (HEPES 20 мМ, EDTA 5 мМ, рН 7,4) и хранят при -70 С вплоть до исследования. Исследование на связывание с помощью лиганда: Соединения растворяют и разводят в диметилсульфоксиде. Гомогенизаты мембран (150-250 мкг протеина) инкубируют в 250 мкл аналитического буфера (HEPES 20 мМ, рН 7,4) при 24-25 С 3 ч. Неспецифическое связывание определяют в присутствии 1 мкМ атропина. Инкубацию прерывают вакуумной фильтрацией через стекловолоконные фильтры GF/B(Wallas). Затем фильтры промывают 50 мМ охлажденного льдом Трис HCI буфера (рН 7,4). Фильтровальные слои сушат и подсчитывают удержанную на фильтрах предельную радиоактивность. IC50 и Kd определяют используя программу построения нелинейной кривой с помощью программного обеспечения G Pad Prism. Рассчитывают коэффициент ингибирования Ki, изучая конкурентное связывание с использованием уравнения Cheng и Prusoff (Biochem Pharmacol, 1973, 22:3099-3108) Ki=IC50/(1+L/Kd), гдеL представляет собой концентрацию [3 Н] NMS, используемую в конкретном эксперименте. pKi = -[lg Ki] Экспериментальные примеры с использованием извлеченного мочевого пузыря крысы Методика Животных умерщвляют сверхдозой уретана, отделяют целый мочевой пузырь, извлекают и быстро помещают в охлаждаемый льдом Tyrode буфер следующего состава (мМол/л) NaCl 137; KCl 2,7; CaCl2 1,8; MgCl2 0,1; NaHCO3 11,9; NaH2PO4 0,4; глюкоза 5,55 и непрерывное насыщение 95% O2 или 5% СO2. Мочевой пузырь разрезают на продольные стрипы (3 мм шириной и 5-6 мм длиной) и помещают в 10 мл ванну при 30 С, одним концом соединяют с основанием держателя ткани и другим концом соединяют с полиграфом через преобразователь силовой подачи. Каждую ткань поддерживают при постоянном основном натяжении 2 г и в течение 1 ч дают возможность достичь равновесия, в течение которого меняют PSS каждые 15 мин. В конце периода уравновешивания 2-3 раза последовательно оценивают стабилизацию ответной сократительной реакции ткани с помощью 1 мкмол/л карбахола (Carbachol). Получают кривую суммарного ответа на карбохол (10-9 мол/л до 3x10-5 мол/л). После нескольких промывок получают базисную линию, кривую суммарного ответа получают в присутствии NCE (NCE добавляют за 20 мин до второго CRC). Результаты сократительной способности выражают как % от контрольного Е макс. Величину ED50 расчитывают строя нелинейную кривую регрессии или возврата в прежнее состояние (Graph Pad Prism). Величину рКВ рассчитывают по формуле pKB=-log [(мольная концентрация антагониста(дозовый коэффициент], где дозовый коэффициент = ED50 в присутствии антагониста/ ED50 в отсутствии антагониста. Эксперименты in vivo на анестезированных кроликах Действие исследуемых соединений изучали по изменениям индуцированного карбахолом давления на мочевом пузыре, частоты сердечных сокращений и слюноотделения. Самцов кроликов весом 1,2-3 кг анестезируют уретаном 1,5 г/кг путем медленного внутривенного вливания через краевую ушную вену. Трахею катетеризируют для поддержания раскрытого состояния дыхательных путей. Кровяное давление регистрируют на бедренной артерии с помощью преобразователя давления Statham P10 EZ, соединенного с полиграфом модели Grass 7D. Частоту сердечных сокращений регистрируют по тахографии, вызываемой пульсовой волной кровяного давления. Вторую бедренную артерию катетеризируют для введения карбахола. Тестируемое соединение и физиологический раствор вливают внутривенно через бедренную вену. Обнажают мочевой пузырь путем лапаротомии, распознают оба мочеточника, осторожно отделяют и перевязывают. Мочеточники проксимально рассекают,позволяя моче свободно вытекать из почки наружу. Осторожно поддерживают шейку мочевого пузыря,различают уретру и отделяют от примыкающих тканей. В мочевой пузырь вводят РЕ канюлю и перевязывают. Мочевой пузырь дренируют и затем заполняют 15 мл теплого физиологического раствора(37 С). Другой конец внутрипузырного катетра соединяют с полиграфом Grass 7D через преобразователь- 15007932 давления Statham P10 EZ для регистрации давления на мочевом пузыре. Следят за поддержанием в зоне влаги и тепла. В течение 30-60 мин дают стабилизироваться параметрам, являющимся следствием хирургического вмешательства. После введения карбахола оценивают слюнный ответ, помещая предварительно взвешенную абсорбирующую хлопчатобумажную марлю в полость рта на 2 мин. Наблюдают действие соединения на индуцированные карбахолом (1,5 мкг/кг, внутриартериально) изменения кровяного давления, частоты сердечных сокращений и давления на мочевом пузыре. Получают по крайней мере два стабильных ответа. Эти ответы рассматривают как 100%. Затем исследуют эффект увеличения дозы тестируемого соединения или наполнителя (за 12-15 мин до введения карбохола). Изменения давления на мочевом пузыре, слюноотделения и индуцированную агонистом брадикардию выражают как % изменения от предварительной контрольной величины. Величину ID50 (доза, необходимая для 50% ингибирования ответа) рассчитывают из нелинейной кривой, построенной для сигмовидной кривой ответа на дозу с помощью программного обеспечения Grafh Pad Prism и выражают в мкг/ кг. Результаты исследований in-vitro и in-vivo приведены в табл. II и III. Таблица II ФОРМУЛА ИЗОБРЕТЕНИЯ 1. Соединения, имеющие структурную формулу I формула I и их фармацевтически приемлемые соли, фармацевтически приемлемые сольваты, эфиры, энантиомеры,диастереоизомеры,где Ar представляет фенил;Z представляет NH, Nme, кислород или простую связь;R6=R7=H. 2. Соединения по п.1, имеющие структурную формулу II, и их фармацевтически приемлемые соли,фармацевтически приемлемые сольваты, эфиры, энантиомеры, диастереоизомеры формула II 3. Соединения по п.1, имеющие структурную формулу III, и их фармацевтически приемлемые соли,фармацевтически приемлемые сольваты, эфиры, энантиомеры, диастереоизомеры формула III 4. Соединения по п.1, имеющие структурную формулу IV, и их фармацевтически приемлемые соли,фармацевтически приемлемые сольваты, эфиры, энантиомеры, диастереоизомеры, где r представляет от 1 до 4 формула IV 5. Соединения по п.1, имеющие структурную формулу V, и их фармацевтически приемлемые соли,эфиры, энантиомеры, где s представляет от 1 до 2(2R,2S)-(1,5,6)-N-[3-азабицикло[3.1.0]гек-6-силметил]-2-циклопентил-2-гидрокси-N-метил-2 фенилацетамида (соединение 18). 7. Фармацевтическая композиция, включающая терапевтически эффективное количество соединения по пп.1, 2, 3, 4, 5 вместе с фармацевтически приемлемым носителем, эксципиентом или разбавителем. 8. Способ лечения или профилактики животных или человека, страдающих от болезни или расстройств респираторной, мочевой и желудочно-кишечной систем, где эти болезни или расстройства опосредуются мускариновыми рецепторами, включающий назначение указанному животному или человеку терапевтически эффективного количества соединения, имеющего структурную формулу I формула I их фармацевтически приемлемых солей, фармацевтически приемлемых сольватов, эфиров, энантиомеров, диастереоизомеров,где Ar представляет фенил;Z представляет NH, Nme, кислород или простую связь;R6=R7=H. 9. Способ по п.8 для лечения или профилактики животных или человека, страдающих от болезни или расстройств респираторной, мочевой и желудочно-кишечной систем, где эти болезни или расстройства опосредуются мускариновыми рецепторами, включающий назначение указанному животному или- 18007932 человеку терапевтически эффективного количества соединения, имеющего структурную формулу II, их фармацевтически приемлемых солей, фармацевтически приемлемых сольватов, эфиров, энантиомеров,диастереоизомеров формула II 10. Способ по п.8 для лечения или профилактики животных или человека, страдающих от болезни или расстройств респираторной, мочевой и желудочно-кишечной систем, где эти болезни или расстройства опосредуются мускариновыми рецепторами, включающий назначение указанному животному или человеку терапевтически эффективного количества соединения, имеющего структурную формулу III, их фармацевтически приемлемых солей, фармацевтически приемлемых сольватов, эфиров, энантиомеров,диастереоизомеров формула III 11. Способ по п.8 для лечения или профилактики животных или человека, страдающих от болезни или расстройств респираторной, мочевой и желудочно-кишечной систем, где эти болезни или расстройства опосредуются мускариновыми рецепторами, включающий назначение указанному животному или человеку терапевтически эффективного количества соединения, имеющего структурную формулу IV, их фармацевтически приемлемых солей, фармацевтически приемлемых сольватов, эфиров, энантиомеров,диастереоизомеров, где r означает от 1 до 4 формула IV 12. Способ по п.8 для лечения или профилактики животных или человека, страдающих от болезни или расстройств респираторной, мочевой и желудочно-кишечной систем, где эти болезни или расстройства опосредуются мускариновыми рецепторами, включающий назначение указанному животному или человеку терапевтически эффективного количества соединения, имеющего структурную формулу V, их фармацевтически приемлемых солей, фармацевтически приемлемых сольватов, эфиров, энантиомеров,диастереоизомеров, где s представляет от 1 до 2 формула V 13. Способ по п.8, где заболеваниями или расстройствами являются недержание мочи, расстройство нижних мочевых путей (LUTS), бронхиальная астма, хроническое обструктивное заболевание легких(COPD), фиброз легких, синдром раздраженной толстой кишки, ожирение, диабет или желудочнокишечная гиперкинезия. 14. Способ по п.9, где заболеваниями или расстройствами являются недержание мочи, симптомы нижних мочевых путей (LUTS), бронхиальная астма, хроническое обструктивное заболевание легких(COPD), фиброз легких, синдром раздраженной толстой кишки, ожирение, диабет или желудочнокишечная гиперкинезия. 15. Способ по п.10, где заболеваниями или расстройствами являются недержание мочи, симптомы нижних мочевых путей (LUTS), бронхиальная астма, хроническое обструктивное заболевание легких(COPD), фиброз легких, синдром раздраженной толстой кишки, ожирение, диабет или желудочнокишечная гиперкинезия. 16. Способ по п.11, где заболеваниями или расстройствами являются недержание мочи, симптомы нижних мочевых путей (LUTS), бронхиальная астма, хроническое обструктивное заболевание легких(COPD), фиброз легких, синдром раздраженной толстой кишки, ожирение, диабет или желудочнокишечная гиперкинезия.- 19007932 17. Способ по п.12, где заболеваниями или расстройствами являются недержание мочи, симптомы нижних мочевых путей (LUTS), бронхиальная астма, хроническое обструктивное заболевание легких(COPD), фиброз легких, синдром раздраженной толстой кишки, ожирение, диабет или желудочнокишечная гиперкинезия. 18. Способ лечения или профилактики животных или человека, страдающих от болезни или расстройств респираторной, мочевой и желудочно-кишечной систем, где эти болезни или расстройства опосредуются мускариновыми рецепторами, включающий назначение указанному животному или человеку терапевтически эффективного количества фармацевтической композиции по п.7. 19. Способ по п.18, где заболеваниями или расстройствами являются недержание мочи, симптомы нижних мочевых путей (LUTS), бронхиальная астма, хроническое обструктивное заболевание легких(COPD), фиброз легких, синдром раздраженной толстой кишки, ожирение, диабет или желудочнокишечная гиперкинезия. 20. Способ получения соединений формулы I формула I и их фармацевтически приемлемых солей, фармацевтически приемлемых сольватов, эфиров, энантиомеров, диастереоизомеров,где Ar представляет фенил;Z представляет NH, Nme, кислород или простую связь;(а) реакцию соединения формулы VII с соединением формулы VI формула VII с получением защищенного соединения формулы VIII, где Ar, R1, R2, W, X, Y, Z и Q, как указано выше, и Р представляет собой защитную для аминогруппы группу(b) снятие защиты с соединения формулы формула VIII в присутствии агента для снятия защиты с получением соединения формулы I, где Ar, R1, R2, W, X, Y, Z и Q, как указано выше, формула I- 20007932 21. Способ по п.20, где Р представляет собой любую защитную для аминогруппы группу, выбранную из группы, состоящей из бензила и t-бутоксикарбонила. 22. Способ по п.20, где реакцию соединения формулы VI с соединением формулы VII с получением соединения формулы VIII проводят в присутствии конденсирующего агента, который выбирают из группы, состоящей из 1-(3-диметиламинопропил)-3-этил карбодиимид гидрохлорида (ЭДК) и 1,8 диазабицикло[5.4.0]ундец-7-ена (ДБУ). 23. Способ по п.20, где реакцию соединения формулы VI с соединением формулы VII ведут в полярном апротонном растворителе, выбранном из группы, состоящей из N,N-диметилформамида, диметилсульфоксида, толуола и ксилола. 24. Способ по п.20, где реакцию соединения формулы VI с соединением формулы VII ведут при 0140 С. 25. Способ по п.20, где снятие защиты с соединения формулы VIII ведут агентом для снятия защиты, который выбирают из группы, состоящей из палладия на угле, трифторуксусной кислоты (ТФК) и хлористо-водородной кислоты. 26. Способ по п.20, где снятие защиты с соединения формулы VIII с получением соединения формулы I ведут в подходящем органическом растворителе, который выбирают из группы, состоящей из метанола, этанола, тетрагидрофурана и ацетонитрила.
МПК / Метки
МПК: C07D 407/12, C07D 417/12, C07D 209/52, C07D 413/12, C07D 401/12, C07D 403/12, A61K 31/403
Метки: качестве, антагонистов, азабициклопроизводные, мускариновых, рецепторов
Код ссылки
<a href="https://eas.patents.su/22-7932-azabicikloproizvodnye-v-kachestve-antagonistov-muskarinovyh-receptorov.html" rel="bookmark" title="База патентов Евразийского Союза">Азабициклопроизводные в качестве антагонистов мускариновых рецепторов</a>
Соединения мочевины, обладающие активностью антагонистов мускариновых рецепторов

Номер патента: 6437
Опубликовано: 29.12.2005
Авторы: Оуэр Дэвид, Маммен Матай
МПК: C07D 211/58, A61P 13/10, A61K 31/4468...
Метки: мускариновых, мочевины, антагонистов, рецепторов, обладающие, активностью, соединения
Формула / Реферат:
1. Соединение формулы где R46 означает C1-10алкил, C3-20циклоалкил или C1-40гетероцикл; R47 означает C1-10алкил, C6-20арил, HC(O)-, C1-10алкил-C(O)-, C3-20циклоалкил-C(O)-, C4-20циклоалкенил-C(O)-, C6-20арил-C(O)-, C1-15гетероарил-C(O)-, C1-40гетероциклил-C(O)-, C1-40гетероцикл или -COOR50, в котором R50 означает C1-10алкил; или R46 и R47 вместе с атомом азота, к которому они присоединены, образуют C1-40гетероцикл; X означает C3-20алкилен, где...
Азабициклические, азатрициклические и азаспироциклические производные аминоциклогексана в качестве антагонистов рецепторов nmda, 5ht и нейронных никотиновых рецепторов

Номер патента: 7098
Опубликовано: 30.06.2006
Авторы: Ванейевс Максимс, Гольд Маркус, Йиргенсонс Айгарс, Каусс Валерьянс, Парсонс Кристофер Грахам Рафаэль, Калвиньш Ивар, Даныш Войцех, Хенрих Маркус
МПК: A61P 25/18, A61P 25/16, A61K 31/40...
Метки: никотиновых, nmda, качестве, аминоциклогексана, азатрициклические, нейронных, производные, антагонистов, азаспироциклические, азабициклические, рецепторов
Формула / Реферат:
1. Соединения формулы (1) где R и R1-R5, каждый независимо, выбран из C1-6-алкильных групп, С2-6-алкенильных групп, С2-6-алкинильных групп, С6-12-арил-С1-4-алкильных групп, необязательно замещенных С6-12-арильных групп и в случае R и R2-R5 - из атомов водорода, с условием, что по меньшей мере один из R2 и R3 и по меньшей мере один из R4 и R5 не является водородом, или R и R1 вместе представляют С3-5-алкиленовую или алкениленовую группу, причем...
Полициклоалкилпурины в качестве антагонистов аденозиновых рецепторов

Номер патента: 5211
Опубликовано: 30.12.2004
Авторы: Лин Ко-Чунг, Чан Хэ Си, Даулинг Джеймс Е., Кумаравел Гнанасамбандам, Кесман Уилльям Ф., Энсингер Кэрол Л., Петтер Расселл К.
МПК: A61K 31/52, A61P 9/00, C07D 493/08...
Метки: аденозиновых, качестве, полициклоалкилпурины, рецепторов, антагонистов
Формула / Реферат:
1. Соединение, имеющее формулу где R1 и R2 независимо выбраны из группы, состоящей из a) водорода; b) C1-C6алкила, незамещенного или замещенного C1-C6алкокси или галогеном; и c) замещенного арила или замещенного арил-C1-C6алкила; R3 выбран из группы, состоящей из (a) бициклической, трициклической или пентациклической группы, выбранной из группы, состоящей из где бициклическая или трициклическая группа является незамещенной или...
Полиморфные модификации кристаллического цитрата (2-бензгидрил-1-азабицикло[2.2.2]окт-3-ил)-(5-изопропил-2-метоксибензил)амина в качестве антагонистов рецепторов нейрокинина 1 (nk1)

Номер патента: 4206
Опубликовано: 26.02.2004
Авторы: Куоллич Джордж Джозеф, Роуз Питер Роберт, Массетт Стивен Сарджент, Уинт Льюин Теофилус
МПК: A61P 25/28, A61K 31/435, C07D 453/02...
Метки: рецепторов, 2-бензгидрил-1-азабицикло[2.2.2]окт-3-ил)-(5-изопропил-2-метоксибензил)амина, цитрата, кристаллического, качестве, антагонистов, полиморфные, нейрокинина-2, nk1, модификации
Формула / Реферат:
1. Кристаллические формы цитрата (2-бензгидрил-1-азабицикло[2.2.2]окт-3-ил)-(5-изопропил-2-метоксибензил)амина, имеющего формулу где указанная кристаллическая форма выбрана из группы, состоящей из (а) стабильной негигроскопичной безводной формы цитрата, имеющей картину дифракции рентгеновских лучей на порошке Пик ь Расстояние D 1 17,61 2 10,95 3 8,78 4 7,96 5 7,37 6 6,80 7 6,57 8 5,87 9 5,46 (б) моногидрата...
Производные бензамидопиперидина в качестве антагонистов рецепторов вещества р

Номер патента: 5409
Опубликовано: 24.02.2005
Авторы: Арнольд Эрик Платт, О'нэйлл Брайан Томас, Соболов-Джейнс Сьюзн Бет, Хуанг Дзианхуа, Винсент Лоренс Альберт, Нэйджел Артур Адам, Хамфри Джон Майкл, Чэппи Томас Аллен
МПК: A61P 1/00, A61K 31/5395, C07D 401/14...
Метки: вещества, производные, качестве, бензамидопиперидина, рецепторов, антагонистов
Формула / Реферат:
1. Соединение формулы где Q обозначает C=S или C=O; A обозначает CH, CH2, C(C1-C6)алкил, CH(C1-C6)алкил, C(CF3) или CH(CF3), при условии, что, когда B присутствует, A должен представлять либо CH, C(C1-C6)алкил, либо C(CF3); B отсутствует или обозначает метилен или этилен; Y обозначает N и Z обозначает CH или Y обозначает CH и Z обозначает N; G обозначает NH(CH2)q, S(CH2)q или O(CH2)q, где q равно 0 или 1; при условии, что, когда q равно 0,...
Предыдущий патент: Способ получения производных 4-тиоалкилбромбензола
Следующий патент: 2,4,5-тризамещенные производные тиазолила и их противовоспалительная активность
Случайный патент: Способ распределения функциональных молекул по поверхности носителя и носитель, получаемый в результате этого способа