Промежуточное соединение для получения макроциклических ингибиторов репликации вируса гепатита с и способ его синтеза
Номер патента: 19888
Опубликовано: 30.07.2014
Авторы: Лион Майкл, Мартин Пьер, Каус Роберт, Йанг Ютонг, Никольс Пол, Кеннеди Эприл, Керчер Тимоти, Сейверт Скот Д., Ванг Бин, Барнет Брэдли Р., Лью Вейдонг, Саммакиа Тэрек, Иэри С.Тод, Блат Лоренс М., Шумахер Андреас, Эндрюс Стивен В.




Формула / Реферат
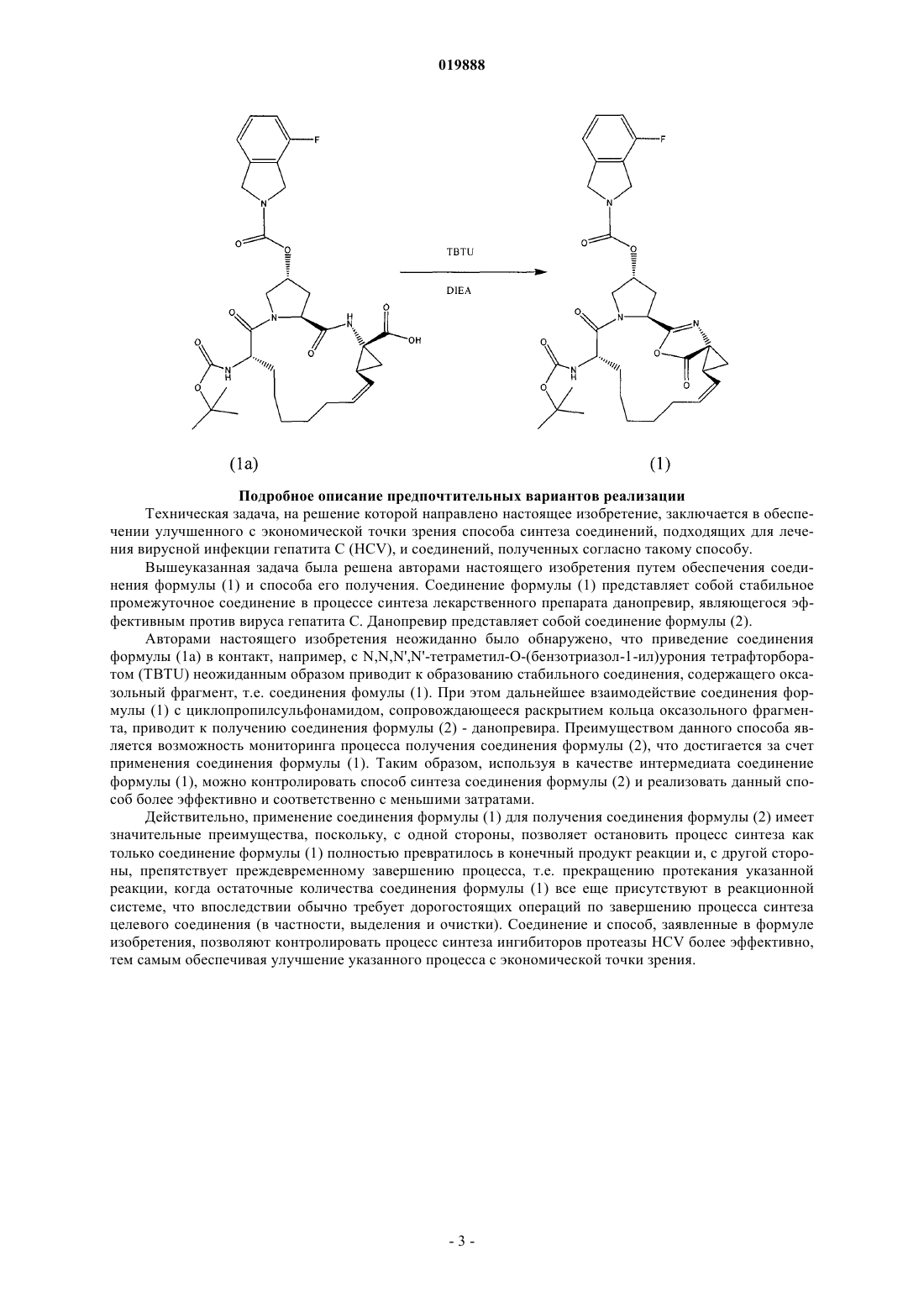
1. Соединение формулы (1)

2. Способ получения соединения по п.1, включающий смешивание соединения (1а) c TBTU и DIEA

Текст
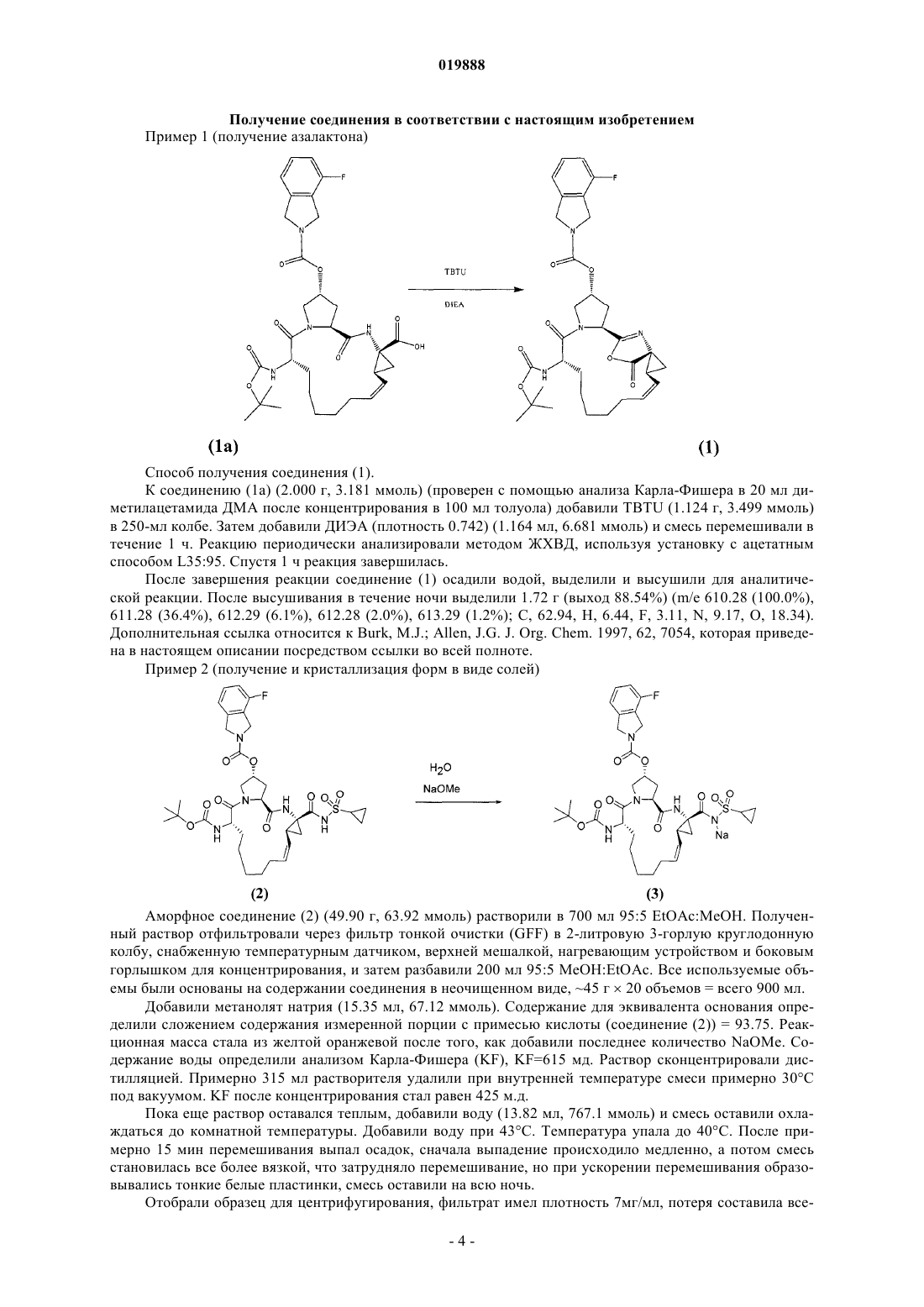
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ ДЛЯ ПОЛУЧЕНИЯ МАКРОЦИКЛИЧЕСКИХ ИНГИБИТОРОВ РЕПЛИКАЦИИ ВИРУСА ГЕПАТИТА С И СПОСОБ ЕГО СИНТЕЗА Настоящее изобретение предлагает соединение формулы (1) и способ его получения Предпосылки создания настоящего изобретения Область техники изобретения Настоящее изобретение относится к соединению формулы (1) и способу его синтеза. Описание уровня техники Вирус гепатита С (ВГС) в США является самой распространенной хронической инфекцией, передаваемой через кровь. Хотя число новых заражений снизилось, количество хронических больных является значительным, и по данным Центров по контролю за заболеваниями в США насчитывает приблизительно 3,9 млн (1,8%) инфицированных человек. Хроническое заболевание печени является десятой в списке лидирующих причин смертности среди взрослых в США, и насчитывает приблизительно 25000 смертей ежегодно или приблизительно 1% от всех смертей. Исследования показывают, что 40% хронических заболеваний печени связаны с вирусом гепатита С, в результате это оценивается приблизительно в 800010000 смертей каждый год. Связанная с вирусом гепатита С конечная стадия заболевания печени является самой частой причиной пересадки печени среди взрослых. За последнюю декаду антивирусная терапия хронического гепатита С динамично развивалась, показывая значительные улучшения в эффективности лечения. Тем не менее, даже при комбинированном лечении, используя интерферон-, конъюгированный с ПЭГ, в сочетании с рибавирином, у 40-50% пациентов лечение не оказывало действия, т.е. у пациентов отмечалось отсутствие лечебного эффекта или рецидив заболевания. Обычно для таких пациентов отсутствовала эффективная альтернатива лечения. В частности, пациенты с запущенным фиброзом или циррозом печени, выявляемые при биопсии, имеют значительный риск развития осложнений запущенных заболеваний печени, включая асцит, желтуху, варикозное кровотечение, энцефалопатию и прогрессирующее нарушение функции печени, также заметно возрастает риск гепатоцеллюлярной карциномы. Широкая распространенность хронической формы вирусной инфекции гепатита С несет серьезные последствия для общественного здравоохранения как отягощение хроническими заболеваниями печени в США в будущем. Данные, полученные в ходе Национального обзора по здоровью и исследованию питания (NHANES III), указывают, что значительное увеличение числа заражений пришлось на конец 60 начало 80 годов, в частности, среди людей в возрасте от 20 до 40 лет. Подсчитано, что количество людей с застарелой формой вирусной инфекции ГС 20 лет и более может увеличиться в четыре раза за промежуток времени с 1990 по 2015 годы с 750000 до свыше 3 млн. Пропорциональное увеличение числа людей, инфицированных в течение 30 или 40 лет, могло быть даже больше. Поскольку риск хронических заболеваний печени, связанных с ВГС, имеет отношение к продолжительности заражения, риск цирроза постепенно увеличивается для людей, инфицированных более чем 20 лет, это приведет в результате к значительному увеличению заболеваний, связанных с циррозом, и смертности среди пациентов, инфицированных между 1965-1985 г. ВГС представляет собой вирус семейства флавивирусов с линейной молекулой РНК положительной полярности. Геном ВГС представлен одноцепочечной молекулой РНК протяженностью около 9500 нуклеотидов и содержит единственную открытую рамку считывания (ORF), несущую информацию о единственном большом полипротеине размером около 3000 аминокислотных остатков. В зараженных клетках этот полипротеин расщепляется на многочисленные участки клеточными и вирусными протеазами с образованием структурных и неструктурных (NS) протеинов вируса. В случае ВГС образование зрелых неструктурных протеинов (NS2, NS3, NS4, NS4A, NS4B, NS5A и NS5B) осуществляется двумя вирусными протеазами. Первая вирусная протеаза расщепляет связь NS2-NS3 полипротеина. Вторая вирусная протеаза представляет собой сериновую протеазу, содержащую N-концевой участок NS3 (далее именуемую как "NS3 протеаза"). NS3 протеаза опосредует все последующие реакции расщепления на участках ниже положения NS3 в полипротеине (т.е. участках, расположенных между С-окончанием NS3 и Сокончанием полипротеина). NS3 протеаза проявляет активность как в цис-положении при расщеплении участка NS3-NS4, так и в транс-положении для остальных NS4A-NS4B, NS4B-NS5A, и NS5A-NS5B участков. Считается, что белок NS4A является многофункциональным полипротеином, способствует активности NS3 протеазы и возможно участвует в мембранной локализации NS3 и других вирусных репликазных компонентов. Очевидно,образование комплекса между NS3 и NS4A необходимо для процессов,опосредуемых NS3, и улучшает протеолитическую эффективность на всех участках, распознаваемых(1996), J. Hepatol. 24: 555-563; US Patent No. 5082659; Европейская патентная заявка ЕР 294160; патент США 4806347; Balish et al. (1992), J. Infect. Diseases 166: 1401-1403; Katayama et al. (2001), J. Viralal. (1992), Br. J. Haematol. 81:516-519; Европейская патентная заявка 294160; патент Канады 1321348; Европейская патентная заявка 276120; Wandl et al. (1992), Sem. Oncol. 19: 88-94; Balish etal. (1992), J. Infectious Diseases 166: 1401-1403; Van Dijk et al. (1994), Int. J. Cancer 56: 262-268; Sundmacher et al. (1987) Current Eye Res. 6: 273-276; патенты США 6172046; 6245740; 5824784; 5372808; 5980884; опубликованные международные патентные заявки WO 96/21468; WO 96/11953;Therap. 303: 540-548; Sheppard et al. (2003), Nat. Immunol. 4: 63-68; Chang et al. (1999), Nat. Biotechnol. 17: 793-797; Adolf (1995), Multiple Sclerosis 1 Suppl. 1: S44-S47; Chu et al., Tet. Lett. (1996), 7229-7232; Девятая Конференция по антивирусным исследованиям (Ninth Conference on Antiviral Research), Урабандай,Фукишима, Япония (1996) (Antiviral Research (1996), 30: 1, A23 (реферат 19; Steinkuhler et al., Biochem.,37: 8899-8905; Ingallinella et al., Biochem., 37: 8906-8914. Краткое описание изобретения Настоящее изобретение предлагает соединение формулы (1) Предпочтительный вариант реализации изобретения предлагает способ получения соединения формулы (1), включающий перемешивание соединения (1 а) с О-бензотриазол-1-ил-N,N,N',N'тетраметилурониум тетрафторборатом (TBTU) и N,N-диизопропилэтиламином (DIEA) Подробное описание предпочтительных вариантов реализации Техническая задача, на решение которой направлено настоящее изобретение, заключается в обеспечении улучшенного с экономической точки зрения способа синтеза соединений, подходящих для лечения вирусной инфекции гепатита С (HCV), и соединений, полученных согласно такому способу. Вышеуказанная задача была решена авторами настоящего изобретения путем обеспечения соединения формулы (1) и способа его получения. Соединение формулы (1) представляет собой стабильное промежуточное соединение в процессе синтеза лекарственного препарата данопревир, являющегося эффективным против вируса гепатита С. Данопревир представляет собой соединение формулы (2). Авторами настоящего изобретения неожиданно было обнаружено, что приведение соединения формулы (1 а) в контакт, например, с N,N,N',N'-тетраметил-O-(бензотриазол-1-ил)урония тетрафторборатом (TBTU) неожиданным образом приводит к образованию стабильного соединения, содержащего оксазольный фрагмент, т.е. соединения фомулы (1). При этом дальнейшее взаимодействие соединения формулы (1) с циклопропилсульфонамидом, сопровождающееся раскрытием кольца оксазольного фрагмента, приводит к получению соединения формулы (2) - данопревира. Преимуществом данного способа является возможность мониторинга процесса получения соединения формулы (2), что достигается за счет применения соединения формулы (1). Таким образом, используя в качестве интермедиата соединение формулы (1), можно контролировать способ синтеза соединения формулы (2) и реализовать данный способ более эффективно и соответственно с меньшими затратами. Действительно, применение соединения формулы (1) для получения соединения формулы (2) имеет значительные преимущества, поскольку, с одной стороны, позволяет остановить процесс синтеза как только соединение формулы (1) полностью превратилось в конечный продукт реакции и, с другой стороны, препятствует преждевременному завершению процесса, т.е. прекращению протекания указанной реакции, когда остаточные количества соединения формулы (1) все еще присутствуют в реакционной системе, что впоследствии обычно требует дорогостоящих операций по завершению процесса синтеза целевого соединения (в частности, выделения и очистки). Соединение и способ, заявленные в формуле изобретения, позволяют контролировать процесс синтеза ингибиторов протеазы HCV более эффективно,тем самым обеспечивая улучшение указанного процесса с экономической точки зрения. Получение соединения в соответствии с настоящим изобретением Пример 1 (получение азалактона) Способ получения соединения (1). К соединению (1 а) (2.000 г, 3.181 ммоль) (проверен с помощью анализа Карла-Фишера в 20 мл диметилацетамида ДМА после концентрирования в 100 мл толуола) добавили TBTU (1.124 г, 3.499 ммоль) в 250-мл колбе. Затем добавили ДИЭА (плотность 0.742) (1.164 мл, 6.681 ммоль) и смесь перемешивали в течение 1 ч. Реакцию периодически анализировали методом ЖХВД, используя установку с ацетатным способом L35:95. Спустя 1 ч реакция завершилась. После завершения реакции соединение (1) осадили водой, выделили и высушили для аналитической реакции. После высушивания в течение ночи выделили 1.72 г (выход 88.54%) (m/е 610.28 (100.0%),611.28 (36.4%), 612.29 (6.1%), 612.28 (2.0%), 613.29 (1.2%); С, 62.94, Н, 6.44, F, 3.11, N, 9.17, О, 18.34). Дополнительная ссылка относится к Burk, M.J.; Allen, J.G. J. Org. Chem. 1997, 62, 7054, которая приведена в настоящем описании посредством ссылки во всей полноте. Пример 2 (получение и кристаллизация форм в виде солей) Аморфное соединение (2) (49.90 г, 63.92 ммоль) растворили в 700 мл 95:5 EtOAc:MeOH. Полученный раствор отфильтровали через фильтр тонкой очистки (GFF) в 2-литровую 3-горлую круглодонную колбу, снабженную температурным датчиком, верхней мешалкой, нагревающим устройством и боковым горлышком для концентрирования, и затем разбавили 200 мл 95:5 MeOH:EtOAc. Все используемые объемы были основаны на содержании соединения в неочищенном виде, 45 г 20 объемов = всего 900 мл. Добавили метанолят натрия (15.35 мл, 67.12 ммоль). Содержание для эквивалента основания определили сложением содержания измеренной порции с примесью кислоты (соединение (2 = 93.75. Реакционная масса стала из желтой оранжевой после того, как добавили последнее количество NaOMe. Содержание воды определили анализом Карла-Фишера (KF), KF=615 мд. Раствор сконцентрировали дистилляцией. Примерно 315 мл растворителя удалили при внутренней температуре смеси примерно 30 С под вакуумом. KF после концентрирования стал равен 425 м.д. Пока еще раствор оставался теплым, добавили воду (13.82 мл, 767.1 ммоль) и смесь оставили охлаждаться до комнатной температуры. Добавили воду при 43 С. Температура упала до 40 С. После примерно 15 мин перемешивания выпал осадок, сначала выпадение происходило медленно, а потом смесь становилась все более вязкой, что затрудняло перемешивание, но при ускорении перемешивания образовывались тонкие белые пластинки, смесь оставили на всю ночь. Отобрали образец для центрифугирования, фильтрат имел плотность 7 мг/мл, потеря составила все-4 019888 го 4 г. Твердый осадок промыли этилацетатом EtOAc, высушили и проанализировали методом ЖХВД. Содержание кислоты было равно 0.97. Твердое вещество отфильтровали через полипропиленовый фильтр, промыли двумя объемами этилацетата EtOAc (90 мл). После высушивания на воздухе в течение 10 мин осадок перенесли на сушильную пластину и поместили в вакуумную печь на выходные дни при температуре 57 С. Дистиллят проанализировали, потери составили 4.02 г. Вещество было 98.8% чистоты с содержанием кислоты 0.83% (чистота диапазоном %). 4 дня спустя вещество (41.9 грамм) удалили из печи, теоретический выход составил 46.65 г (в пересчете на исходное вещество), выход по массе был равен 90%. Дополнительное количество 2-3% было потеряно при дистилляции из-за встряхивания. Анализ методом ЯМР-спектроскопии показал, что вещество содержало следы этилацетата EtOAc, остаточный анализ растворителя газовой хроматографией выявил этилацетат EtOAc на 3742 мд и этилового спирта EtOH на 1279 мд. Поскольку настоящее изобретение описано со ссылкой на конкретные варианты осуществления изобретения, специалисту в данной области техники будет очевидно, что могут быть выполнены различные изменения и могут быть использованы эквиваленты без отступления от истинной сущности и объема данного изобретения. Кроме того, многие модификации могут быть выполнены для того, чтобы применить данное изобретение к конкретной ситуации, материалу, смеси химически связанных веществ, процессу, стадии или стадиям процесса в целях, сущности и объеме настоящего изобретения. Подразумевается, что все такие модификации входят в объем прилагаемой формулы изобретения. ФОРМУЛА ИЗОБРЕТЕНИЯ 1. Соединение формулы (1) 2. Способ получения соединения по п.1, включающий смешивание соединения (1 а) c TBTU и DIEA
МПК / Метки
МПК: C07C 231/02, C07K 5/12, A61K 38/00, C07C 269/00, A61P 31/14, C07D 207/16, C07D 209/44
Метки: репликации, соединение, получения, вируса, макроциклических, способ, синтеза, промежуточное, гепатита, ингибиторов
Код ссылки
<a href="https://eas.patents.su/6-19888-promezhutochnoe-soedinenie-dlya-polucheniya-makrociklicheskih-ingibitorov-replikacii-virusa-gepatita-s-i-sposob-ego-sinteza.html" rel="bookmark" title="База патентов Евразийского Союза">Промежуточное соединение для получения макроциклических ингибиторов репликации вируса гепатита с и способ его синтеза</a>
Макроциклические карбоновые кислоты и ацилсульфонамиды в качестве ингибиторов репликации вируса гепатита с

Номер патента: 11857
Опубликовано: 30.06.2009
Авторы: Джианг Ютонг, Стенджел Питер Джон, Кеннеди Эйприл Лайн, Венгловски Стивен Марк, Кондроски Кевин Рональд, Догерти Джордж Эндрю, Вуддард Бенджамин Т., Джоси Джон Энтони, Эндрьюс Стивен Вейд, Блэтт Лоренс М., Маддуру Мачендер Р.
МПК: C08H 1/00, A61K 38/00
Метки: гепатита, макроциклические, вируса, качестве, ингибиторов, репликации, кислоты, ацилсульфонамиды, карбоновые
Формула / Реферат:
1. Макроциклическая карбоновая кислота или ацилсульфонамид, имеющие формулу I, VIII или IX где (a) R1 и R2, каждый независимо, представляют собой H, галоген, циано, гидрокси, С1-6алкил, С1-6алкокси, C1-6алкил, возможно замещенный не более чем тремя атомами фтора, тиазолил, C(O)NR6R7, NR6R7, C(O)OR8, NHC(O)R8, OCHnNR6R7 или OCHnR9a; где R9a представляет собой имидазолил или пиразолил; указанный тиазолил в определении R1 и R2 является...
Новые макроциклические ингибиторы репликации вируса гепатита с

Номер патента: 15752
Опубликовано: 30.12.2011
Авторы: Кеннеди Эприл, Ванг Бин, Блат Лоренс М., Йанг Ютонг, Лью Вейдонг, Мартин Пьер, Шумахер Андреас, Сейверт Скот Д., Эндрюс Стивен В., Саммакиа Тэрек, Лион Майкл, Каус Роберт, Керчер Тимати, Иэри С.Тод, Барнет Брэдли Р., Никольс Пол
МПК: C07C 231/02, A61P 31/14, A61K 38/00...
Метки: гепатита, ингибиторы, вируса, новые, репликации, макроциклические
Формула / Реферат:
1. Соединение общей формулы (Ia) или (Ib)или их фармацевтически приемлемая соль, гдеR1 представляет собой H, , где p равен 1, или OC(=O)-R1a, где R1a представляет собой возможно замещенный гетероарил, выбранный из пиридила, пиримидинила, пирролила, оксазолила, и R2 представляет собой NHR5;R3 выбирают из группы, состоящей из H, CH2R6, COR6, CO2R7, CSNH2, возможно замещенного 2-тиазола и R4 представляет собой водород или циклопропилметил;R5...
Промежуточное соединение для использования в синтезе доцетаксела и способ его получения

Номер патента: 1385
Опубликовано: 26.02.2001
Авторы: Систи Николас Дж., Свинделл Чарльз С.
МПК: C07D 305/14
Метки: использования, доцетаксела, синтезе, способ, получения, соединение, промежуточное
Формула / Реферат:
1. Химическое соединение, имеющее формулу 2. Способ получения соединения, имеющего формулу включающий стадию ацилирования 10-деацетилбаккатина III, по крайней мере, 1,5 эквивалентами н-бутиллития и, по крайней мере, 1,5 эквивалентами бензилхлорформиата в тетрагидрофуране. 3. Способ по п.2, по которому 10-деацетилбаккатин III сначала растворяют в вышеуказанном тетрагидрофуране с образованием раствора, затем добавляют н-бутиллитий с...
Промежуточное соединение для получения паклитаксела и способ получения промежуточного соединения

Номер патента: 678
Опубликовано: 28.02.2000
Авторы: Систи Николас Дж., Чандер Мадхави С., Свинделл Чарльз С.
МПК: C07D 305/14
Метки: промежуточное, получения, соединения, паклитаксела, способ, промежуточного, соединение
Формула / Реферат:
1. Промежуточное соединение для получения паклитаксела, где промежуточное соединение имеет общую формулу где P1 представляет гидрирующуюся бензильную защитную группу. 2. Промежуточное соединение паклитаксела по п.1, где гидрирующуюся бензильную защитную группу выбирают из группы, включающей бензилоксиметил и бензил. 3. Способ получения промежуточного соединения для получения паклитаксела, где промежуточное соединение имеет общую формулу ...
Производные пиримидина в качестве ингибиторов репликации вируса иммунодефицита человека

Номер патента: 2973
Опубликовано: 26.12.2002
Авторы: Янссен Поль Адриан Жан, Койманс Люсьен Мария Хенрикус, Янссен Марсель Огюст Констант, Де Корт Барт, Де Жонж Марк Рене, Кукла Майкл Джозеф, Андрис Кунрад Йозеф Лодевейк Марсель, Ван Акен Кун Жанн Альфонс, Хэрес Ян, Лудовики Дональд Уилльям, Хо Чих Юнг
МПК: A61K 31/505, A61P 31/18, C07D 239/50...
Метки: человека, пиримидина, производные, ингибиторов, качестве, вируса, иммунодефицита, репликации
Формула / Реферат:
1. Применение соединения формулы его N-оксида, фармацевтически приемлемой соли присоединения или стереохимически изомерной формы, где А является СН или N; n равен 0, 1, 2, 3 или 4; Q является водородом или -NR1R2; R1 и R2, каждый независимо, выбран из водорода, гидрокси, C1-12aлкила, С1-12алкилокси, С1-12алкилкарбонила, С1-12алкилоксикарбонила, где каждая из указанных С1-12алкильных групп необязательно и каждая независимо может быть замещена...