Антагонисты рецептора cgrp
Номер патента: 20409
Опубликовано: 30.10.2014
Авторы: Мэйкор Джон Е., Луо Гуанглин, Дубовчик Джин М.



























Формула / Реферат
1. Соединение формулы I

где R1 представляет собой водород;
R2 представляет собой

или пиперидинил, замещенный

R3 представляет собой водород;
R4 представляет собой водород;
R5 представляет собой водород или гидроксигруппу;
R6 представляет собой водород;
R7 представляет собой водород;
R8 представляет собой водород;
R9 представляет собой водород или гидроксигруппу;
R10 представляет собой водород;
R11 представляет собой водород, гидрокси-, азидо-, амино-, алкиламино-, диалкиламиногруппу или алкоксикарбонил;
или R10 и R11, взятые вместе, представляют собой О или N-OH;
Ar1 представляет собой фенил, замещенный 0-2 заместителями, выбранными из галогена; и
X представляет собой О, СН2 или NH;
или его фармацевтически приемлемая соль.
2. Соединение по п.1, в котором
R1 представляет собой водород;
R2 представляет собой пиперидинил, замещенный

R3 представляет собой водород;
R4 представляет собой водород;
R5 представляет собой водород или гидроксигруппу;
R6 представляет собой водород;
R7 представляет собой водород;
R8 представляет собой водород;
R9 представляет собой водород или гидроксигруппу;
R10 представляет собой водород;
R11 представляет собой водород, гидрокси-, азидо-, амино-, алкиламино- или диалкиламиногруппу;
или R10 и R11, взятые вместе, представляют собой оксогруппу;
Ar1 представляет собой фенил, замещенный 0-2 заместителями, выбранными из галогена; и
X представляет собой О, СН2 или NH;
или его фармацевтически приемлемая соль.
3. Соединение по п.1 с приведенной ниже стереохимией

4. Соединение по п.1, в котором R5 представляет собой водород, R6 представляет собой водород, R7 представляет собой водород, R8 представляет собой водород, R9 представляет собой водород или гидроксигруппу и R10 и R11, взятые вместе, представляют собой оксогруппу; или в котором R5 представляет собой водород, R6 представляет собой водород, R7 представляет собой водород, R8 представляет собой водород, R9 представляет собой гидроксигруппу, R10 представляет собой водород и R11 представляет собой водород; или в котором R5 представляет собой гидроксигруппу, R6 представляет собой водород, R7 представляет собой водород, R8 представляет собой водород, R9 представляет собой водород, R10 представляет собой водород и R11 представляет собой водород.
5. Соединение по п.1, в котором Ar1 представляет собой фенил, замещенный двумя галогеновыми заместителями.
6. Соединение по п.5, в котором Ar1 представляет собой 2,3-дифторфенил.
7. Соединение по п.1, в котором X представляет собой О.
8. Соединение по п.1, выбранное из группы, состоящей из
(6R,9R)-6-(2,3-дифторфенил)-6-гидрокси-5-оксо-6,7,8,9-тетрагидро-5H-циклогепта[b]пиридин-9-ил 4-(2-оксо-2,3-дигидро-1Н-имидазо[4,5-b]пиридин-1-ил)пиперидин-1-карбоксилат;
(9R)-6-(2,3-дифторфенил)-5-оксо-6,7,8,9-тетрагидро-5H-циклогепта[b]пиридин-9-ил 4-(2-оксо-2,3-дигидро-1H-имидазо[4,5-b]пиридин-1-ил)пиперидин-1-карбоксилат;
(5S,6R,9R)-6-(2,3-дифторфенил)-5,6-дигидрокси-6,7,8,9-тетрагидро-5Н-циклогепта[b]пиридин-9-ил 4-(2-оксо-2,3-дигидро-1H-имидазо14,5-b]пиридин-1-ил)пиперидин-1-карбоксилат;
(5S,6S,9R)-6-(2,3-дифторфенил)-5-гидрокси-6,7,8,9-тетрагидро-5Н-циклогепта[b]пиридин-9-ил 4-(2-оксо-2,3-дигидро-1Н-имидазо[4,5-b]пиридин-1-ил)пиперидин-1-карбоксилат;
(5R,6S,9R)-6-(2,3-дифторфенил)-5-гидрокси-6,7,8,9-тетрагидро-5H-циклогепта[b]пиридин-9-ил 4-(2-оксо-2,3-дигидро-1Н-имидазо[4,5-b]пиридин-1-ил)пиперидин-1-карбоксилат;
(5S,6S,9R)-6-(2,3-дифторфенил)-5-гидрокси-6,7,8,9-тетрагидро-5Н-циклогепта[b]пиридин-9-ил 4-(2-оксо-2,3-дигидро-1Н-имидазо[4,5-b]пиридин-1-ил)пиперидин-1-карбоксилат;
(5S,6S,9R)-5-азидо-6-(2,3-дифторфенил)-6,7,8,9-тетрагидро-5Н-циклогепта[b]пиридин-9-ил 4-(2-оксо-2,3-дигидро-1Н-имидазо[4,5-b]пиридин-1-ил)пиперидин-1-карбоксилат;
(5S,6S,9R)-5-амино-6-(2,3-дифторфенил)-6,7,8,9-тетрагидро-5Н-циклогепта[b]пиридин-9-ил 4-(2-оксо-2,3-дигидро-1Н-имидазо[4,5-b]пиридин-1-ил)пиперидин-1-карбоксилат;
(6S,9R)-6-(2,3-дифторфенил)-6-гидрокси-6,7,8,9-тетрагидро-5Н-циклогепта[b]пиридин-9-ил 4-(2-оксо-2,3-дигидро-1Н-имидазо[4,5-b]пиридин-1-ил)пиперидин-1-карбоксилат;
(5S,6S,9R)-5-амино-6-(3,5-дифторфенил)-6,7,8,9-тетрагидро-5Н-циклогепта[b]пиридин-9-ил 4-(2-оксо-2,3-дигидро-1Н-имидазо[4,5-b]пиридин-1-ил)пиперидин-1-карбоксилат;
(5S,6S,9R)-6-(3,5-дифторфенил)-5-гидрокси-6,7,8,9-тетрагидро-5Н-циклогепта[b]пиридин-9-ил 4-(2-оксо-2,3-дигидро-1Н-имидазо[4,5-b]пиридин-1-ил)пиперидин-1-карбоксилат;
(6S,8R,9S)-6-(2,3-дифторфенил)-8-гидрокси-6,7,8,9-тетрагидро-5Н-циклогепта[b]пиридин-9-ил 4-(2-оксо-2,3-дигидро-1H-имидазо[4,5-b]пиридин-1-ил)пиперидин-1-карбоксилат;
(5S,6S,9R)-6-(2,3-дифторфенил)-5-(метиламино)-6,7,8,9-тетрагидро-5Н-циклогепта[b]пиридин-9-ил 4-(2-оксо-2,3-дигидро-1Н-имидазо[4,5-b]пиридин-1-ил)пиперидин-1-карбоксилат;
(5S,6S,9R)-6-(2,3-дифторфенил)-5-(диметиламино)-6,7,8,9-тетрагидро-5Н-циклогепта[b]пиридин-9-ил 4-(2-оксо-2,3-дигидро-1Н-имидазо[4,5-b]пиридин-1-ил)пиперидин-1-карбоксилат;
(6S,9R,Z)-6-(2,3-дифторфенил)-5-(гидроксиимино)-6,7,8,9-тетрагидро-5Н-циклогепта[b]пиридин-9-ил 4-(2-оксо-2,3-дигидро-1Н-имидазо[4,5-b]пиридин-1-ил)пиперидин-1-карбоксилат;
(6S,9R,Е)-6-(2,3-дифторфенил)-5-(гидроксиимино)-6,7,8,9-тетрагидро-5Н-циклогепта[b]пиридин-9-ил 4-(2-оксо-2,3-дигидро-1Н-имидазо[4,5-b]пиридин-1-ил)пиперидин-1-карбоксилат;
(5S,6S,9R)-5-амино-6-(2,3-дифторфенил)-6,7,8,9-тетрагидро-5Н-циклогепта[b]пиридин-9-ил 2'-оксо-1,1',2',3-тетрагидроспиро[инден-2,3'-пирроло[2,3-b]пиридин]-5-илкарбамат;
трет-бутил (5S,6S,9R)-9-амино-6-(2,3-дифторфенил)-6,7,8,9-тетрагидро-5Н-циклогепта[b]пиридин-5-илкарбамат;
трет-бутил (5S,6S,9S)-6-(2,3-дифторфенил)-9-(2-оксо-2-(4-(2-оксо-2,3-дигидро-1Н-имидазо[4,5-b]пиридин-1-ил)пиперидин-1-ил)этил)-6,7,8,9-тетрагидро-5Н-циклогепта[b]пиридин-5-илкарбамат;
трет-бутил (5S,6S,9R)-6-(2,3-дифторфенил)-9-(2-оксо-2-(4-(2-оксо-2,3-дигидро-1Н-имидазо[4,5-b]пиридин-1-ил)пиперидин-1-ил)этил)-6,7,8,9-тетрагидро-5Н-циклогепта[b]пиридин-5-илкарбамат и
1-(1-(2-((5S,6S,9R)-5-амино-6-(2,3-дифторфенил)-6,7,8,9-тетрагидро-5Н-циклогепта[b]пиридин-9-ил)ацетил)пиперидин-4-ил)-1Н-имидазо[4,5-b]пиридин-2(3Н)-он;
или его фармацевтически приемлемая соль.
9. Соединение по п.1, которое представляет собой (5S,6S,9R)-5-амино-6-(2,3-дифторфенил)-6,7,8,9-тетрагидро-5Н-циклогепта[b]пиридин-9-ил 4-(2-оксо-2,3-дигадро-1Н-имидазо[4,5-b]пиридин-1-ил)пиперидин-1-карбоксилат

или его фармацевтически приемлемая соль.
10. Композиция для лечения состояния, связанного с аберрантными уровнями CGRP, содержащая соединение по п.1 или его фармацевтически приемлемую соль и фармацевтически приемлемый носитель.
11. Композиция по п.10, в которой соединением 1 является (5S,6S,9R)-5-амино-6-(2,3-дифторфенил)-6,7,8,9-тетрагидро-5Н-циклогепта[b]пиридин-9-ил 4-(2-оксо-2,3-дигидро-1Н-имидазо[4,5-b]пиридин-1-ил)пиперидин-1-карбоксилат.
12. Способ лечения состояния, связанного с аберрантными уровнями CGRP, включающий введение пациенту терапевтически эффективного количества соединения по п.1 или его фармацевтически приемлемой соли.
13. Способ по п.12, где состоянием является мигрень.
14. Способ по п.12, где соединением по п.1 является (5S,6S,9R)-5-амино-6-(2,3-дифторфенил)-6,7,8,9-тетрагидро-5Н-циклогепта[b]пиридин-9-ил 4-(2-оксо-2,3-дигидро-1Н-имидазо[4,5-b]пиридин-1-ил)пиперидин-1-карбоксилат.
15. Способ по п.13, где соединением по п.1 является (5S,6S,9R)-5-амино-6-(2,3-дифторфенил)-6,7,8,9-тетрагидро-5Н-циклогепта[b]пиридин-9-ил 4-(2-оксо-2,3-дигидро-1Н-имидазо[4,5-b]пиридин-1-ил)пиперидин-1-карбоксилат.
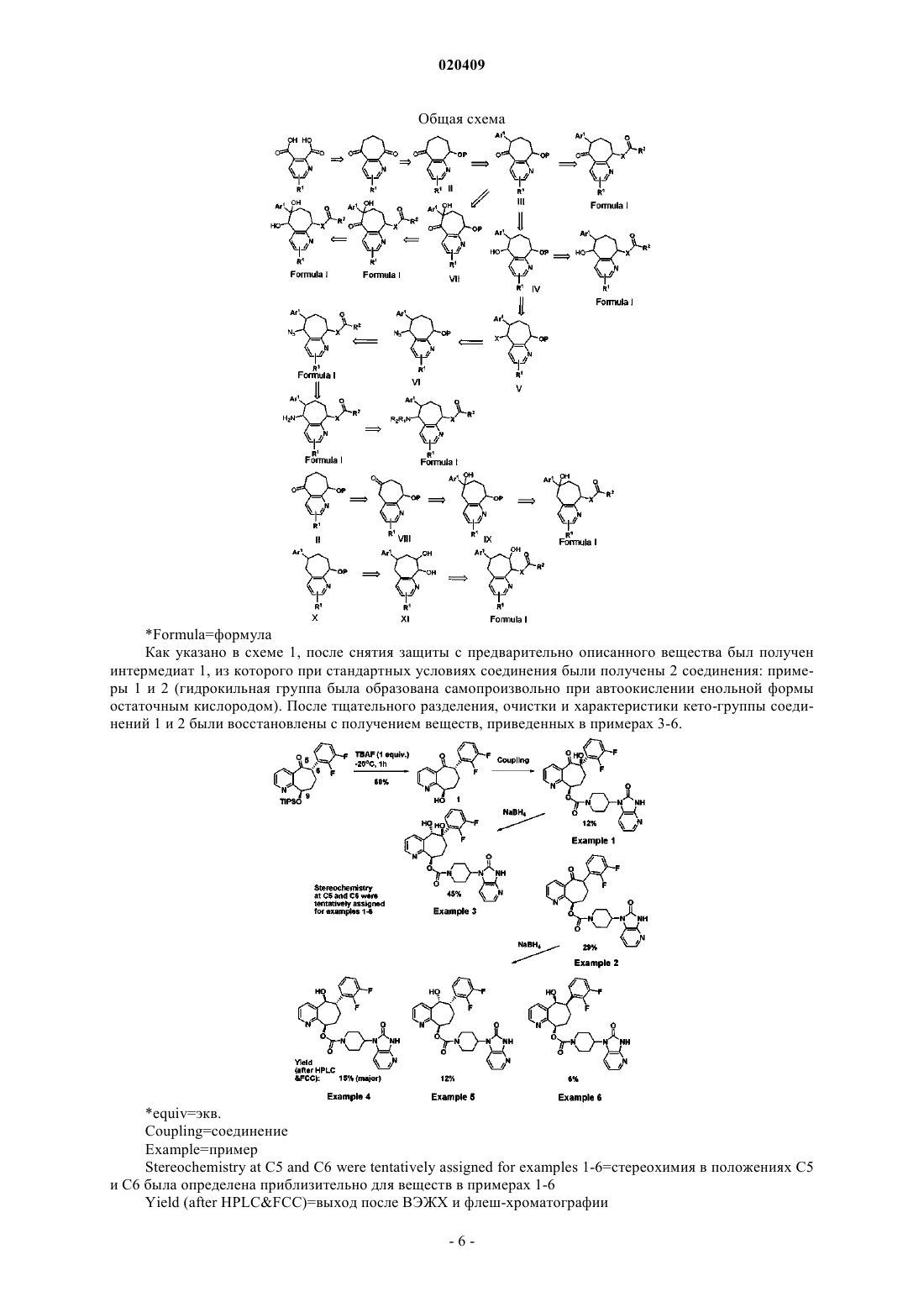
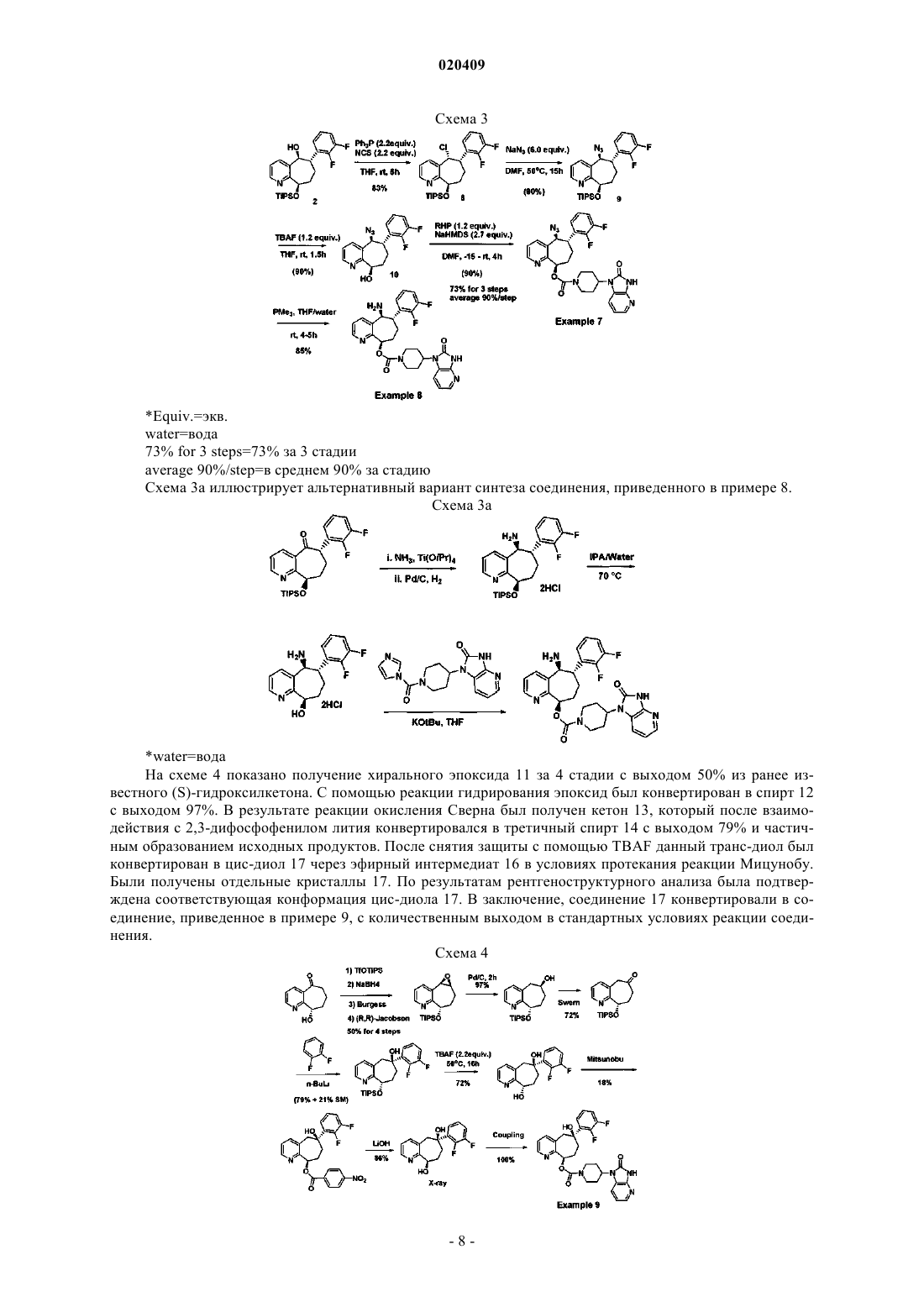
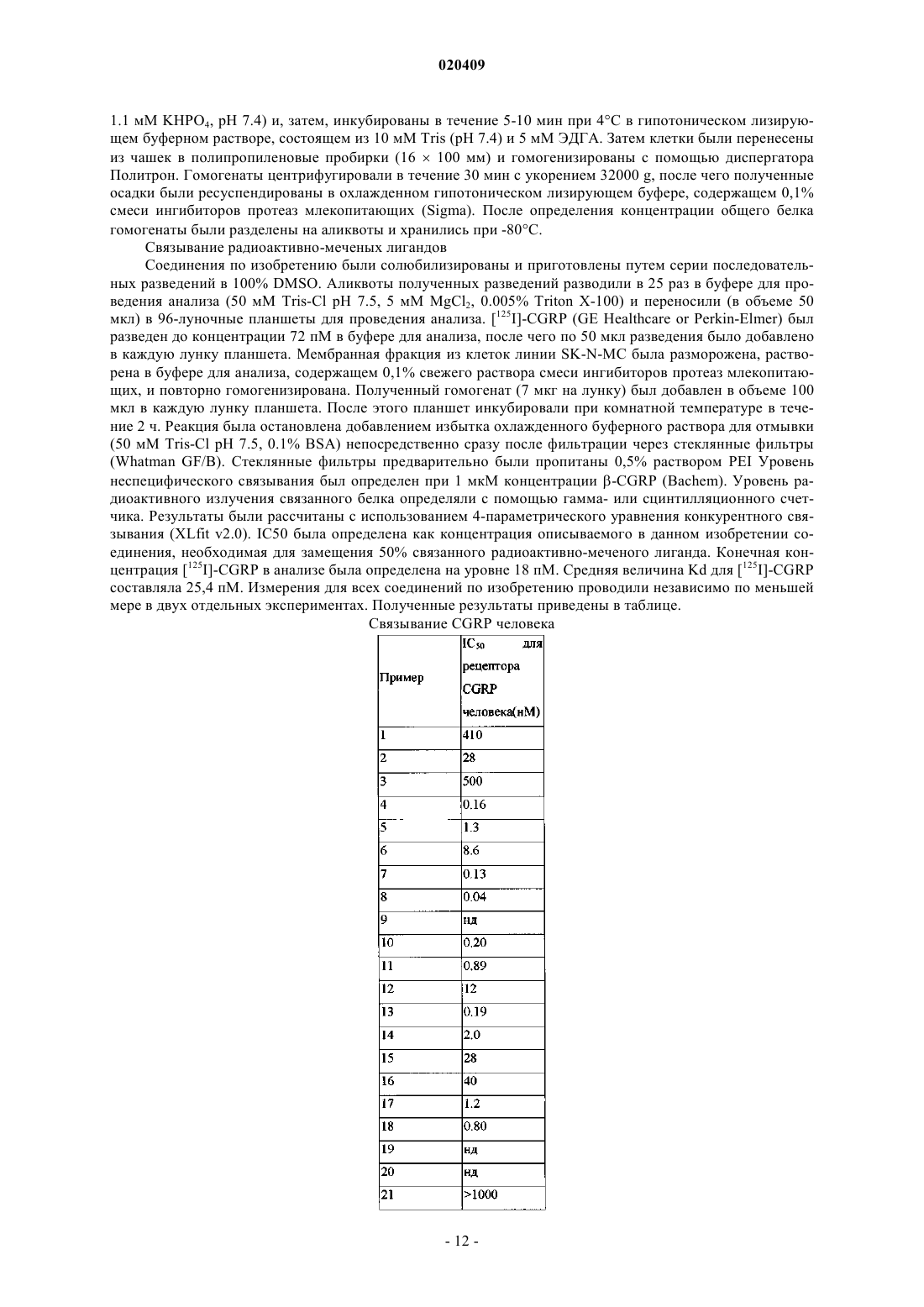
Текст
Изобретение главным образом относится к новым соединениям формулы I, включая их фармацевтически приемлемые соли, которые являются антагонистами рецептора CGRP. Изобретение также относится к фармацевтическим композициям и способам использования соединений при лечении нарушений, связанных с CGRP, включая мигрень и другие виды головной боли, нейрогенную вазодилатацию, нейрогенное воспаление, термический ожог, циркуляторный шок, приливы, вызванные менопаузой, воспалительные заболевания дыхательных путей, такие как астма и хроническая обструктивная болезнь легких (COPD). Родственные заявки По настоящей заявке испрашивается приоритет предварительной заявки США 61/251477, поданной 14 октября 2009 г. Уровень техники Изобретение главным образом относится к новым компонентам общей формулы I, включая фармацевтически приемлемые соли, являющимся антагонистами рецептора CGRP. Также описание относится к фармакологическим композициям и способам применения этих веществ при лечении заболеваний, связанных с изменениями в функционировании рецептора CGRP, включая мигрень, нейрогенную вазодилатацию, нейрогенное воспаление, термический ожог, циркуляторный шок, приливы, вызванные менопаузой, воспалительные заболевания дыхательных путей, такие как астма и хроническая обструктивная болезнь легких (ХОБЛ). Пептид, связанный с геном кальцитонина (CGRP) - природный пептид, состоящий из 37 аминокислотных остатков, впервые идентифицирован в 1982 г. (Amara S.G. et al., Science 1982, 298, 240-244). Обнаружены две экспрессирующиеся формы данного пептида (CGRP и CGRP), различающиеся одним и тремя аминокислотными остатками у крысы и у человека, соответственно. Данный пептид широко распространен в тканях периферической (ПНС) и центральной нервной системы (ЦНС) и, в основном, локализуется в афферентных и центральных нейронах. Пептид вызывает ряд биологических эффектов, включая вазодилатацию. При высвобождении из клетки CGRP связывается со специфическими рецепторами клеточной поверхности, сопряженными с G-белками, и осуществляет свои функции преимущественно за счет активации внутриклеточной аденилатциклазы (Poyner D.R. et al., Br J Pharmacol 1992, 105, 441-7; Van Valen F. etal., Neurosci Lett 1990, 119, 195-8). Предполагается существование двух классов рецепторов CGRP:CGRP1 и CGRP2. Данные предположения основаны на антагонистических свойствах пептидного фрагмента CGRP (8-37) и на способности линейных аналогов CGRP актировать рецепторы CGRP2 (Juaneda С.et al. TiPS 2000, 21, 432-438). Однако непосредственно на молекулярном уровне рецепторы CGRP2 не найдены (Brain S.D. et al., TiPS 2002, 23, 51-53). Рецептор CGRP1 состоит из трех частей: (i) семи трансмембранных доменов, подобных рецептору кальцитонина (CRLR); (ii) одного домена белка первого типа,модифицирующего активность трансмембранных рецепторов (RAMP1); (iii) и внутриклеточного домена рецептора (RCP) (Evans В.N. et al., J Biol Chem. 2000, 275, 31438-43). RAMP1 необходим для транспортаCRLR на поверхность клеточной мембраны, а также принимает участие в связывании лиганда с рецептором CGRP (McLatchie L.M. et al., Nature 1998, 393, 333-339). RCP необходим для передачи сигнала в клетку (Evans В.N. et al., J Biol Chem. 2000, 275, 31438-43). Существуют видоспецифические отличия в связывании низкомолекулярных антагонистов с рецептором CGRP. Обычно, большая аффинность наблюдается при связывании с рецептором человека, по сравнению с рецепторами из других организмов(Brain S.D. et al., TiPS 2002, 23, 51-53). Видоспецифичность связана с отличиями в аминокислотной последовательности RAMP1, в частности, аминокислотный остаток Trp74 ответственен за описанный выше фенотип рецептора человека (Mallee et al. J Biol Chem 2002, 277, 14294-8). Предполагается, что ингибиторы CGRP на уровне рецептора применимы при таких патофизиологических состояниях, при которых наблюдается избыточная активация рецептора CGRP. Некоторые из них включают нейрогенную вазодилатацию, нейрогенное воспаление, мигрень, кластерные головные боли и головные боли других типов, термические ожоги, циркуляторный шок, приливы, вызванные менопаузой,астму. Есть основания предполагать, что активация рецептора CGRP вовлечена в патогенез мигрени (Edvinsson L. CNS Drugs 2001; 15(10):745-53; Williamson D.J. Microsc. Res. Tech. 2001, 53, 167-178; GrantA.D. Brit. J. Pharmacol. 2002, 135, 356-362). Показано, что сывороточный уровень CGRP повышен при приступе мигрени (Goadsby P.J. et al. Ann Neurol 1990; 28:183-7). Прием противомигреневых препаратов снижает уровни CGRP до нормальных одновременно с облегчением головной боли (Gallai V. et al. Cephalalgia 1995; 15:384-90). У пациентов, страдающих мигренями, показан более высокий базальный уровеньCGRP по сравнению с представителями контрольной группы (Ashina M. et al., Pain 2000, 86(1-2): 1338.2000). Также, внутривенное введение CGRP провоцирует продолжительную головную боль у лиц,страдающих мигренями (Lassen L.H., et al. Cephalalgia 2002 Feb; 22(l):54-61). В доклинических испытаниях на собаках и крысах показано, что системная блокада CGRP пептидным антагонистом CGRP (8-37a.o.) не приводит к изменению системной гемодинамики в покое, а также не вызывает регионарных изменений тока крови (Shen Y-T. et al., J Pharmacol Exp Ther 2001, 298, 551-8). Таким образом, антагонисты рецептора CGRP, возможно, являются новым средством лечения мигрени, не вызывающим склонности к акивной вазоконстрикции, в отличие от неселективных агонистов 5-HT1B/1D - триптанов (например,Суматриптан). Эффективность антагонистов CGRP продемонстрирована в ряде клинических исследований (DavisA.M.; Neurology 2008 70:1304. Epub 2007 Oct 3). Антагонисты рецептора CGRP раскрыты в публикациях РСТ WO 2004/092166, 2004/092168 и 2007/120590. Данное изобретение обеспечивает технические преимущества, например, те, что соединения являются новыми и ингибируют CGRP. Дополнительно, соединения имеют преимущества при фармацевтическом применении, например, в отношении одного или нескольких из их механизма действия, связывания, эффективности ингибирования, специфичности к мишеням, растворимости, профилей безопасности или биодоступности. Описание изобретения Изобретение охватывает серию соединений-антагонистов CGRP, включающую фармацевтически приемлемые соли, композиции, способы их получения, а также способы использования их при терапевтическом лечении. Одним из объектов изобретения является соединение формулы IR5 представляет собой водород или гидроксигруппу;R9 представляет собой водород или гидроксигруппу;X представляет собой О, СН 2 или NH; или его фармацевтически приемлемая соль. Другим объектом изобретения является соединение формулы I, гдеR5 представляет собой водород или гидроксигруппу;R9 представляет собой водород или гидроксигруппу;X представляет собой О, СН 2 или NH; или его фармацевтически приемлемая соль. Другим объектом изобретения является соединение формулы I с приведенной ниже стереохимией. Другим объектом изобретения является соединение формулы I, гдеR5 представляет собой водород или гидроксигруппу;R9 представляет собой водород или гидроксигруппу;X представляет собой О, СН 2 или NH; или его фармацевтически приемлемая соль. Другим объектом изобретения является соединение формулы I, где R1 представляет собой водород; 2R5 представляет собой водород или гидроксигруппу;R9 представляет собой водород или гидроксигруппу;Ar1 представляет собой фенил или дифторфенил; иX представляет собой О, СН 2 или NH; или его фармацевтически приемлемая соль.-3 020409 Другим аспектом изобретения является соединение формулы I, где R1 представляет собой водород. Другим объектом изобретения является соединение формулы I, где R2 представляет собой Другим объектом изобретения является соединение формулы I, где R5 представляет собой водород,R представляет собой водород, R7 представляет собой водород, R8 представляет собой водород, R9 представляет собой водород или гидроксигруппу, R10 и R11, взятые вместе, являются оксогруппой; или где R5 представляет собой водород, R6 представляет собой водород, R7 представляет собой водород, R8 представляет собой водород, R9 представляет собой гидроксигруппу, R10 представляет собой водород, R11 представляет собой водород; или где R5 представляет собой гидроксигруппу, R6 представляет собой водород, R7 представляет собой водород, R8 представляет собой водород, R9 представляет собой водород,R10 представляет водород, R11 представляет собой водород. Другим объектом изобретения является соединение формулы I, где Ar1 представляет собой замещенный двумя галогенозаместителями фенил. Другим объектом изобретения является соединение формулы I, где Ar1 представляет собой 2,3 дифторфенил. Другим объектом изобретения является соединение формулы I, где X представляет собой О. Сочетание любых вариантов переменных групп заместителей, включая R1, R2, R3, R4, R5, R6, R7, R9,10R , R11, Ar1 и X, может быть независимо использовано с сочетанием любых других вариантов групп заместителей. В связи с этим, изобретение включает различные комбинации вышеупомянутых групп. Если не указано иное, перечисленные ниже термины имеют следующие значения. Алкил означает линейную или разветвленную алкильную группу, состоящую из 1-6 атомов углерода (преимущественно 1-3 атома). Алкенил означает линейную или разветвленную группу из 2-6 атомов углерода с по меньшей мере одной двойной связью. Циклоалкил означает моноциклическую кольцевую систему из 3-7 атомов углерода. Гидроксиалкил, алкокси и другие термины с замещенной алкильной частью включают линейные и разветвленные изомеры, состоящие из 1-6 атомов углерода для алкильной части. Галогеналкил или галогеналкокси включают все галогенированные изомеры, от однозамещенного галогеналкила до пергалогенозамещенного алкила. Арил объединяет карбоциклические и гетероциклические системы ароматических колец. Амино объединяет первичные, вторичные и третичные аминогруппы. Карбонил означает СО. Окси означает -О-. Аминокарбонил означает-N(R)C(=O)-. Оксикарбонил означает -ОС(=О)-. Метиленкарбонил означает CHYDROGENC(=O)-. Амино(циано)иминометил означает -NHC(=NCN)-. Заключение названий в скобки (или несколько скобок) применяется для уточнения связей в сложных химических структурах, что приемлемо для специалистов в данной области. Например, такой термин, как R)алкил), означает алкильный заместитель, дполнительно замещенный заместителем R. Настоящее изобретение включает в себя также все формы фармацевтически приемлемых солей соединений. Фармацевтически приемлемыми солями являются соли, противоионы которых не вносят значимый вклад в их физиологическую активность или токсичность соединений и, фактически, являются фармакологическими эквивалентами. Такие соли могут быть получены с помощью обычных методов органической химии из коммерчески доступных реактивов. Некоторые формы анионных солей включают: ацетат, ацистрат, бесилат, бромид, хлорид, цитрат, фумарат, глюкоуранат, гидробромид, гидрохлорид, гидройодид, йодид, лактат, малеат, мезилат, нитрат, памоат, фосфат, сукцинат, сульфат, тартрат,тозилат и ксинафоат. Некоторые формы катионных солей включают соли аммония, алюминия, бензатина, висмута, кальция, холина, диэтиламина, диэтаноламина, лития, магния, меглюмина, 4 фенилциклогексиламина, пиперазина, калия, натрия, трометамина и цинка. Некоторые соединения по изобретению могут существовать в виде стереоизомеров, пример одного из которых приведен ниже. Данное изобретение включает все стерео- и таутомерные формы описанных веществ. 6 Данное изобретение подразумевает включение всех возможных изотопов атомов, присутствующих в описанных соединениях. Под изотопами подразумеваются атомы, имеющие тот же атомный номер,однако отличающиеся по атомным массам. В качестве общего примера, не предназначенного для ограничения, изотопы водорода включают дейтерий и тритий. Изотопы углерода включают 13 С и 14 С. Меченные изотопами соединения по изобретению могут быть получены в общем случае с помощью традиционных методик, известных специалистам в данной области, или с помощью способов, аналогичных описанным далее, при использовании соответствующих изотопно-меченых реактивов вместо немеченых. Такие соединения потенциально могут быть использованы для различных целей, например, в качестве стандартных веществ и реагентов при определении биологической активности. В случае использования стабильных изотопов, такие соединения, потенциально, могут обладать лучшими биологическими, фармакологическими или фармакокинетическими свойствами. Методы синтеза Соединения могут быть получены известными в соответствующей области способами, включая те,которые описаны ниже, а также включая варианты способов, разработанные специалистами в данной области. Некоторые реактивы и интермедиаты известны в соответствующей сфере. Другие реактивы могут быть получены известными способами с использованием легко доступных исходных материалов. Приводимые ниже методы иллюстрируют, но не ограничивают методы синтеза. Специалисты в данной области должны принять во внимание, что существует ряд способов для синтеза описываемых соединений и что подход к их получению не ограничивается способами, приведенными ниже в качестве примеров. Также, помимо описанных, существуют другие методики получения описываемых соединений и их производных. Условные обозначения, входящие в общие структурные формулы и особенности схем синтеза, следует считать отличными и рассматривать отдельно от использованных в "формуле изобретения" или в остальной части данного документа. Данные условные обозначения приведены только в качестве иллюстрации способов получения некоторых соединений по изобретению. Используемые в данных схемах аббревиатуры, в основном, традиционны для данной области. Химические аббревиатуры, используемые в описании и примерах, определяются следующим образом:"ВОС", "DMSO" - диметилсульфоксид; "h" - час; "rt" - комнатная температура или время удержания (в зависимости от контекста); "min" - минута; "EtOAc" - этилацетат; "THF" - тетрагидрофуран; "EDTA" этилендиаминтетраацетат; "Et2O" - диэтиловый эфир; "DMAP" - 4-диметиламинопиридин; "DCE" - 1,2 дихлорэтан; "ACN" - ацетонитрил; "DME" - 1,2-диметоксиэтан; "HOBt" - 1-гидроксибензотриазол гидрат;"DIEA" - диизопропилэтиламин, "Nf" - CF3(CF2)3SO2-; "TMOF" - триметилортоформиат. Некоторые соединения формулы I могут быть получены в соответствии с нижеследующими общими схемами. Предварительно известная структура II может быть арилирована с помощью различных арил-бромидов с образованием структуры III. Далее защита может быть удалена и структруа III переведена в кето-аналоги веществ с формулой I. Кето-группа структуры III может быть гидроксилирована по альфа-атому с образованием интермедиата VII, который затем может быть превращен в гидроксикетон и диольные производные вещества с формулой I. Альтернативный способ заключается в том, что кетогруппа интермедиата III может быть восстановлена до спиртового интермедиата IV, который затем может быть либо напрямую превращен в гидрокси-аналоги вещества, описываемого формулой I, либо превращен в галогенированные аналоги V. Интермедиат V может быть превращен в галогенированные интермедиаты V. Из азидных интермедиатов VI могут быть получены различные азидные или аминные производные. Кетогруппа преварительно известной структуры II может быть перенесена на кетоинтермедиаты VIII. Далее, для получения интермедиатов структуры IX, могут быть добавлены арильные группы. Затем интермедиаты IX могут быть превращены в гидрокси-аналоги вещества с формулой I. Предварительно известная структура X может быть дегидрирована и ди-дегидратирована для получения интермедиатов XI, которые могут быть превращены в позиционные ОН-аналоги вещества с формулой I.Formula=формула Как указано в схеме 1, после снятия защиты с предварительно описанного вещества был получен интермедиат 1, из которого при стандартных условиях соединения были получены 2 соединения: примеры 1 и 2 (гидрокильная группа была образована самопроизвольно при автоокислении енольной формы остаточным кислородом). После тщательного разделения, очистки и характеристики кето-группы соединений 1 и 2 были восстановлены с получением веществ, приведенных в примерах 3-6.Stereochemistry at C5 and C6 were tentatively assigned for examples 1-6=стереохимия в положениях С 5 и С 6 была определена приблизительно для веществ в примерах 1-6Major=основное вещество. Стереохимия примера 4 была подтверждена. Стереоспецифический синтез данного вещества был проведен экспериментально в соответствии со схемой 2. После простого восстановления кетона борогидридом натрия были получены соединения 2 и 3. С помощью обработки этой смеси TBAF при комнатной температуре соединение 2, основной компонент смеси, при удалении блокирующей группы было конвертировано в соединение 4, которое было легко отделено от соединения 3. С помощью рентгеноструктурного анализа были проанализированы чистые кристаллы соединения 4 и подтверждено присутствие диольной группы в цис-положении. Соединение 3 также было обработано TBAF при повышенной температуре. Был получен транс-диол 5, структура которого также была подтверждена с помощью РСА. После диастереоселективного восстановления кето-группы, добавления ацетатной защитной группы и снятия защитной группы TIPS интермедиат 7 может быть конвертирован в соединение в примере 4. Стереоспецифические свойства такого соединения совпадают с таковыми для соединения, полученного с помощью нестереоспецифического синтеза, проводившегося ранее. Схема 2Previous coupling=ранее описанные условия реакции соединенияExample=пример Как показано на схеме 3, соединение 2 может быть конвертировано в хлорид 8 при обработке триенилфосфином и NCS с выходом 83%. Хлорид 8 может быть превращен в азидный интермедиат 9. Соединения 8 и 9 представляют собой стереоизомеры, т.о. наиболее вероятно, что реакции проходили по пути двойной инверсии вокруг атома углерода, несущего гидроксильный заместитель. Соединение 10 было получено после снятия блокирующей группы с помощью TBAF. Затем, в результате стандартной реакции соединения, с высоким выходом было получено соединение, показанное в примере 7. Обработка этого соединения трифенилфосфином в присутствии THF привела к образованию аминоаналога - пример 8.average 90%/step=в среднем 90% за стадию Схема 3 а иллюстрирует альтернативный вариант синтеза соединения, приведенного в примере 8. Схема 3 аwater=вода На схеме 4 показано получение хирального эпоксида 11 за 4 стадии с выходом 50% из ранее известного (S)-гидроксилкетона. С помощью реакции гидрирования эпоксид был конвертирован в спирт 12 с выходом 97%. В результате реакции окисления Сверна был получен кетон 13, который после взаимодействия с 2,3-дифосфофенилом лития конвертировался в третичный спирт 14 с выходом 79% и частичным образованием исходных продуктов. После снятия защиты с помощью TBAF данный транс-диол был конвертирован в цис-диол 17 через эфирный интермедиат 16 в условиях протекания реакции Мицунобу. Были получены отдельные кристаллы 17. По результатам рентгеноструктурного анализа была подтверждена соответствующая конформация цис-диола 17. В заключение, соединение 17 конвертировали в соединение, приведенное в примере 9, с количественным выходом в стандартных условиях реакции соединения. Схема 4Example=пример Варианты арильных групп приведены на схеме 5. Интермедиат 18 был получен при описанных выше условиях. Синтез соединений, приведенных в примерах 10 и 11, был проведен в соответствии с методиками, описанными для примеров 8 и 4 соответственно. Данные методики подробно описаны в экспериментальной части. Схема 5for example 4/8=для вещества пример 4/8 example=пример Как показано на схеме 6, соединение 19 было получено из соответствующего спирта путем обработки реактивом Бургесса. В результате обычного ди-гидроксилирования были получены два разделяемых дистереомерных диола. Затем, менее полярный транс-диол (вещество 20) был конвертирован в соединение, приведенное в примере 12. Схема 6Example=пример Как показано на схеме 7, моно- и бис-метилендиаминовые аналоги были получены простой обработкой аминоаналогов примера 8 формальдегидом и NaBH3CN.Example=пример Как показано на схеме 8, оксимные продукты из примера 2 были получены с помощью обработки гидроксиламином. Схема 8Example=пример Азидная группа интермедиата 9 может быть восстановлена до амина 21 и защищена группой Вос(схема 9). После снятия защиты спиртовая группа может взаимодействовать с изоцианатами, такими как 24, которые были получены в одну стадию из известного анилина 26. После взаимодействия спиртовой группы с изоцианатами образуется карбаматный интермедиат 25. После снятия защиты может быть получено соединение, приведенное как пример 17. Схема 9Example=пример Как показано на схеме 10, интермедиат 23 был превращен в интермедиат 27 с помощью реакции Мицунобу. После второй реакции Мицунобу хиральный центр спирта менял направление, давая интермедиат 28, который после обработки гидразином превращался в моно-защищенный диамин 29. Затем, в заранее известных условиях при реакции с известным реактивом 30, было получено соединение 31. После снятия защитной группы Вос было получено соединение, показанное как пример 18.Example=пример Как показано на схеме 11, интермедиат 23 также может быть превращен в кетонный интермедиат 32 с помощью реакции окисления Сверна. Данный кетон был переведен в форму ненасыщенного эфира 33 с помощью реакции Виттига. Гидрирование интермедиата 33 привело к образованию двух разделяемых изомеров 34 и 35, которые затем были гидролизованы водным раствором LiOH и получены кислотные интермедиаты 36 и 37, соответственно. С помощью стандартной реакции соединения интермедиаты 36 и 37 были конвертированы в соединения, приведенные в примерах 19 и 20, соответственно. После обработки соединения, приведенного как пример 20, TFA, было получено соединение, приведенное в примере 21. Схема 11Example=пример Биологические методы Фармакология in vitro Культура ткани Клетки линии SK-N-MC культивировали в виде монослоя при 37 С и 5% СО 2 в среде MEM, кондиционированной солями Эрла и L-глутамином (Invitrogen) и содержащей 10% эмбриональной бычьей сыворотки (Invitrogen). Выделение мембранной фракции Мембранная фракция была получена из клеток линии SK-N-MC, экспрессирующих рецепторыCGRP. Клетки были отмыты дважды фосфатным буферным раствором (155 мМ NaCl, 3.3 мМ Na2HPO4,- 11020409 1.1 мМ KHPO4, рН 7.4) и, затем, инкубированы в течение 5-10 мин при 4 С в гипотоническом лизирующем буферном растворе, состоящем из 10 мМ Tris (рН 7.4) и 5 мМ ЭДГА. Затем клетки были перенесены из чашек в полипропиленовые пробирки (16100 мм) и гомогенизированы с помощью диспергатора Политрон. Гомогенаты центрифугировали в течение 30 мин с укорением 32000 g, после чего полученные осадки были ресуспендированы в охлажденном гипотоническом лизирующем буфере, содержащем 0,1% смеси ингибиторов протеаз млекопитающих (Sigma). После определения концентрации общего белка гомогенаты были разделены на аликвоты и хранились при -80 С. Связывание радиоактивно-меченых лигандов Соединения по изобретению были солюбилизированы и приготовлены путем серии последовательных разведений в 100% DMSO. Аликвоты полученных разведений разводили в 25 раз в буфере для проведения анализа (50 мМ Tris-Cl pH 7.5, 5 мМ MgCl2, 0.005% Triton Х-100) и переносили (в объеме 50 мкл) в 96-луночные планшеты для проведения анализа. [125I]-CGRP (GE Healthcare or Perkin-Elmer) был разведен до концентрации 72 пМ в буфере для анализа, после чего по 50 мкл разведения было добавлено в каждую лунку планшета. Мембранная фракция из клеток линии SK-N-MC была разморожена, растворена в буфере для анализа, содержащем 0,1% свежего раствора смеси ингибиторов протеаз млекопитающих, и повторно гомогенизирована. Полученный гомогенат (7 мкг на лунку) был добавлен в объеме 100 мкл в каждую лунку планшета. После этого планшет инкубировали при комнатной температуре в течение 2 ч. Реакция была остановлена добавлением избытка охлажденного буферного раствора для отмывки(50 мМ Tris-Cl pH 7.5, 0.1% BSA) непосредственно сразу после фильтрации через стеклянные фильтры(Whatman GF/B). Стеклянные фильтры предварительно были пропитаны 0,5% раствором PEI Уровень неспецифического связывания был определен при 1 мкМ концентрации -CGRP (Bachem). Уровень радиоактивного излучения связанного белка определяли с помощью гамма- или сцинтилляционного счетчика. Результаты были рассчитаны с использованием 4-параметрического уравнения конкурентного связывания (XLfit v2.0). IC50 была определена как концентрация описываемого в данном изобретении соединения, необходимая для замещения 50% связанного радиоактивно-меченого лиганда. Конечная концентрация [125I]-CGRP в анализе была определена на уровне 18 пМ. Средняя величина Kd для [125I]-CGRP составляла 25,4 пМ. Измерения для всех соединений по изобретению проводили независимо по меньшей мере в двух отдельных экспериментах. Полученные результаты приведены в таблице. Связывание CGRP человека Фармацевтические композиции и способы лечения Соединения формулы I ингибируют рецептор CGRP. Таким образом, они могут использоваться при лечения состояний и нарушений, связанных с аберрантными (измененными) уровнями экспрессии CGRP,а также при тех состояниях, когда модуляция уровней CGRP может иметь терапевтический эффект. В соответствии с вышесказанным, другим объектом изобретения является фармацевтическая композиция, содержащая соединение формулы I и фармацевтически приемлемый адъювант, носитель или растворитель. Соединения главным образом включают в состав фармацевтических композиций, содержащих терапевтически эффективное количество соединения формулы I или фармацевтиически приемлемую соль,а также фармацевтически приемлемый носитель, и могут содержать обычные наполнители. Терапевтически эффективным количеством является то количество, которое необходимо для значимого положительного эффекта при терапевтическом применении, который определяется специалистами в данной области. Фармацевтически приемлемыми носителями являются традиционно используемые носители, которые имеют определенные и приемлемые профили безопасности. Композиции включают все общие тврдые и жидкие формы, включая капсулы, таблетки, пастилки и порошки, а также жидкие суспензии, сиропы,эликсиры и растворы. Твердые композиции могут быть включены в составы отложенного или замедленного высвобождения. Композиции изготавливают с помощью обычных технологий приготовления лекарственных средств и традиционных наполнителей (таких как связывающие и увлажняющие агенты) и разбавителей (таких как вода и спирты). Твердые композиции обычно изготавливают с помощью дозирующих устройств, которые позволяют изменять количество активного компонента в дозе от примерно 1 до примерно 1000 мг. Некоторые примеры дозировки, которую обычно получают, используя традиционные устройства: 0.1, 1, 10, 100, 500 и 1000 мг. Для жидких композиций обычно используется дозировка в диапазоне 1-100 мг/мл. Некоторые примеры дозировки жидких композиций: 0.1, 1, 10, 25, 50 и 100 мг/мл. Настоящее изобретение охватывает все традиционные способы введения медицинских препаратов,включая пероральный, парентеральный, интраназальный, сублингвальный и трансдермальный способы. Обычно суточная доза составит 0,01-100 мг/кг веса в день. Как правило, при пероральном способе введения требуется большее количество вещества, тогда как при парентеральном - меньшее. Точный режим приема вещества должен определяться врачом с учетом результатов тщательного обследования. Считается, что ингибиторы CGRP на уровне рецептора эффективны при терапии патофизиологических состояний, при которых наблюдается избыточная активация рецептора CGRP. Некоторые из таких состояний включают нейрогенную вазодилатацию, нейрогенное воспаление, мигрень, кластерные и другие головные боли, термический ожог, циркуляторный шок, приливы, вызванные менопаузой, астму. Предполагается, что активация рецептора CGRP вовлечена в патогенез мигрени (Edvinsson L. CNS Drugs 2001, 15(10), 745-53; Williamson D.J. Microsc. Res. Tech. 2001, 53, 167-178; Grant A.D. Brit. J. Pharmacol. 2002, 135, 356-362). Показано, что уровень CGRP в плазме повышается во время приступа мигрени(Goadsby P.J. et al. Ann. Neurol. 1990, 28, 183-7). При этом терапия противомигреневыми препаратами снижает сывороточный уровень CGRP до нормального, что сопровождается облегчением головной боли(Gallai V. et al. Cephalalgia 1995, 15, 384-90). У лиц, страдающих мигренями, показан повышенный базальный уровень CGRP по сравнению с контрольной группой (Ashina M. et al., Pain 2000, 86(1-2), 133-8). Также, внутривенное введение CGRP провоцирует продолжительную головную боль у страдающих мигренями (Lassen L.H. et al. Cephalalgia 2002 Feb; 22(1):54-61). В доклинических испытаниях на собаках и крысах показано, что системная блокада CGRP пептидным антагонистом (8-37 а.о.) не приводит к изменению системной гемодинамики в покое, а также не вызывает регионарных изменений тока крови (Shen,Y-T. et al., J Pharmacol Exp Ther 2001, 298, 551-8). Таким образом, антагонисты рецептора CGRP, возможно, являются новым средством терапии при мигренях, не вызывающим склонности к активной вазоконстрикции, в отличие от неселективных агонистов 5-HT1B/1D - триптанов (например, Суматриптан). Другим объектом изобретения является способ ингибирования рецептора CGRP, включающий контактирование рецептора CGRP с соединением формулы I или его фармацевтически примелемой солью. Другим объектом изобретения является способ лечения состояний, ассоциированных с аберрантными (измененными) уровнями CGRP, включающий введение пациенту терапевтически эффективного количества соединения общей формулы I. Другим объектом изобретения является способ лечения мигрени или головной боли. Другой объект изобретения относится к способу лечения воспаления (в частности, нейрогенного воспаления), боли, термического ожога, циркуляторного шока, диабета, Синдрома Рейно, заболевания периферических артерий, субарахноидальной/краниальной геморрагии, опухолевого роста, приливов,ассоциированных с менопаузой, и других состояний, эффективность лечения которых может быть повышена подавлением функционирования рецептора CGRP путем приема фармацевтических композиций,содержащих соединения формулы I, описанные в настоящем документе. Другой объект изобретения относится к способам, выбираемым из следующей группы: (а) иммунная регуляция микрофлоры в слизистой оболочке кишечника, (б) протективный эффект при анафилактическом поражении сердца, (в) стимуляция или блокирование интерлейкин-1b-зависимой костной резорп- 13020409 ции, (г) изменение уровня экспрессии рецептора NK-1 у нейронов спинного мозга и (д) респираторные заболевания и хроническая обструктивная болезнь легких, включая астму (см. (a) Calcitonin ReceptorLike Receptor Is Expressed on Gastrointestinal Immune Cells. Hagner, Stefanie; Knauer, Jens; Haberberger,Rainer; Goeke, Burkhard; Voigt, Karlheinz; McGregor, Gerard Patrick. Institute of Physiology, Philipps University, Marburg, Germany. Digestion (2002), 66(4), 197-203; (6) Protective effects of calcitonin gene-related peptide-mediated evodiamine on guinea-pig cardiac anaphylaxis. Rang, Wei-Qing; Du, Yan-Hua; Hu, Chang-Ping;Chemistry: Anti-InflammatoryAnti-Allergy Agents (2003), 2(2), 191-218). Мигрень, головная боль и др. соответствующие термины в данном документе имеют те же значения, что и в медицинской литературе. Термин мигрень включает в себя все типы мигрени, в т.ч. общую, классическую, кластерную, стреляющую, гемиплегическую, офтальмоплегическую и офтальмологическую. Термин терапевтически эффективный означает, что на пациента оказывается значимое положительное воздействие по заключению врача. Термин пациент означает субъекта, терапевтическое воздействие на которого может привести к положительному итогу по заключению врача. Описание предпочтительных вариантов осуществления изобретения Используемые аббревиатуры в основном соответствуют традиционно используемым в данной области. Химические аббревиатуры, использованные в описании и в примерах определяются следующим образом: "NaHMDS" - бис(триметилсилил)амид натрия, "DMFE" - N,N-диметилформамид, "МеОН" - метанол, "NBS" - N-бромсукцинимид, "TFA" - трифторуксусная кислота, "LAH" - алюмогидрид лития,"ВОС" и "DMSO" - диметилсульфоксид, "h" - час, "rt" - комнатная температура или время удержания (в зависимости от контекста), "min" - минута, "EtOAc" - этилацетат, "THF" - тетрагидрофуран, "EDTA" этилендиаминтетрацетат, "Et2O" - диэтиловый эфир, "DMAP" - 4-диметиламинопиридин, "DCE" - 1,2 дихлорэтан, "ACN" - ацетонитрил, "DME" - 1,2-диметоксиэтан, "HOBt" - 1-гидроксибензотриазолгидрат,"DIEA" - диизопропилэтиламин, "Nf" - CF3(CF2)3SO2-, и "TMOF" - триметиловый эфир ортомуравьиной кислоты. Используемые в данном документе аббревиатуры определяются следующим образом: 1 - однократно, 2 - дважды, 3 - трижды, С - градус Цельсия, eq - эквиваленты или эквивалент, г - грамм или граммы, мг - миллиграмм или миллиграммы, л - литр или литры, мл - миллилитр или миллилитры,мкл - микролитр или микролитры, Н - нормальность, М - молярность, ммоль - милимоль или милимоли, мин - минута или минуты, h - час или часы, rt - комнатная температура, tR - время удержания,атм - атмосфера, psi - фунтов на квадратный дюйм, конц - концентрированный, нас или насыщ - насыщенный, MW - молекулярный вес, тп - точка плавления, ее - энантиомерный избыток, MS - массспектрометрия, LCMS - жидкостная хроматография и масс-спектрометрия, ВЭЖХ - высокоэффективная жидкостная хроматография, ОФ ВЭЖХ - обратнофазная высокоэффективная жидкостная хроматография,ТСХ или тех - тонкослойная хроматография, ЯМР - спектроскопия ядерного магнитного резонанса,1 Н - протон,- дельта, с - синглетный, д - дублет, т - триплет, к - квартет, м - мультиплет, шширокий, Гц - Герц, и , , "R", "S", "E" and "Z" - стереохимические обозначения, известные специалисту в данной области. Спектры протонного магнитного резонанса (1 Н ЯМР) были получены на установках Bruker AC 300 и АС 500. Все спектры были получены в указанных растворителях. Химические сдвиги приводятся в единицах с меньшей частотой относительно тетраметилсилана в качестве внутреннего стандарта. Константы спин-спиннового взаимодействия приводятся в Герцах (Гц). Паттерны обозначены следующим образом: с - синглет, д - дублет, т - триплет, к - квартет, м - мультиплет, ш - широкий пик. Масс-спектры низкого разрешения и различимые молекулярные спектры (МН+) или (М-Н)+ были получены на платформе Micromass. Элементный анализ приводится как процент-вес. Продукты были выделены с помощью препаративной ВЭЖХ на колонке YMC S5 ODS (30100 мм) при скорости потока 40 мл/мин и градиенте продолжительностью 8,0 мин. Стартовый буфер содержал 40% метанола, 60% воды и 0,1%TFA. Конечный буфер состоял из 95% метанола, 5% воды и 0,1 TFA. Полученные продукты анализировали с помощью ВЭЖХ на колонке XTERA (3,050 мм S7) при градиенте от буфера А (10% метанола,90% воды и 0,1% TFA) до буфера Б (10% воды, 90% метанола и 0.1% TFA) в течение 2 мин, при скорости потока 5 мл/мин. Время удержания для продукта определялось по уровню поглощения фракции при длине волны 220 нм. Интермедиат 1(6S,9R)-6-(2,3-Дифторфенил)-9-гидрокси-6,7,8,9-тетрагидро-5 Н-циклогепта[b]пиридин-5-он. 0,218 г (0,49 ммоль) (9R)-6-(2,3-дифторфенил)-9-гидрокси-6,7,8,9-тетрагидро-5 Н-циклогепта[b]пиридин 5-он растворяли в 5 мл тетрагидрофурана в круглодонной колбе объемом 250 мл до получения бесцветного раствора. После охлаждения до -15 С (на лед/метанольной бане) под азотом добавляли 0,490 мл (0,490 ммоль) TBAF и перемешивали полученный ярко-желтый раствор при -15 С в течение 1 ч (12:00). Реакцию останавливали добавлением раствора бикарбоната натрия и разбавляли этилацетатом. После разделения слоев водный слой экстрагировали этилацетатом. Объединенные органические слои были промыты солевым раствором, обезвожены и сконцентрированы с получением масла желто-коричневого цвета. С помощью флэш-хроматографии (25 г силикагеля в колонке, градиент до 100% этилацетата в гексане) был выделен целевой продукт (112 мг, 62%). 1(6R,9R)-6-(2,3-Дифторфенил)-6-гидрокси-5-оксо-6,7,8,9-тетрагидро-5 Н-циклогепта[b]пиридин-9-ил 4-(2 оксо-2,3-дигидро-1 Н-имидазо[4,5-b]пиридин-1-ил)пиперидин-1-карбоксилат (пример 1) и (9R)-6-(2,3 дифторфенил)-5-оксо-6,7,8,9-тетрагидро-5 Н-циклогепта[b]пиридин-9-ил 4-(2-оксо-2,3-дигидро-1 Нимидазо[4,5-b]пиридин-1-ил)пиперидин-1-карбоксилат (пример 2). В абсолютно сухой круглодонной колбе объемом 100 мл была приготовлена суспензия (6S,9R)-6(2,3-дифторфенил)-9-гидрокси-6,7,8,9-тетрагидро-5 Н-циклогепта[b]пиридин-5-она (112.45 мг, 0.389 ммоль) (в азеотропной смеси с безводным бензолом) и 4-нитрофенил 4-(2-оксо-2,3-дигидро-1 Нимидазо[4,5-b]пиридин-1-ил)пиперидин-1-карбоксилата (224 мг, 0.583 ммоль) в диметилформамиде (3 мл). После охлаждения до -15 С на лед/метанольной бане к смеси по каплям добавляли NaHMDS (1,555 мл, 1,555 ммоль) (10:30). Полученный раствор желтого цвета перемешивали в атмосфере азота при -15 С в течение 1 ч (нагретый до 10 С раствор/суспензия окрашивался в темно-красный цвет). По прошествии еще 30 мин (и нагрева раствора до -5 С) реакцию останавливали раствором бикарбоната натрия и разбавляли этилацетатом. Получившиеся слои разделяли. Экстракцию водорастворимых веществ этилацетатом проводили дважды. Объединенные органические фазы были промыты солевым раствором, обезвожены над сульфатом натрия и сконцентрированы с получением масла желтого цвета. С помощью флеш-хроматографии при градиенте до 10% метанола в метиленхлориде был выделен целевой продукт(пример 2, 60 мг, 29%, в виде смеси диастереоизомеров), а также его окисленная форма (пример 1, 25,5 мг, 12%, один диастереоизомер) в виде осадка белого цвета. Пример 2.(5S,6R,9R)-6-(2,3-Дифторфенил)-5,6-дигидрокси-6,7,8,9-тетрагидро-5 Н-циклогепта[b]пиридин-9-ил 4-(2-оксо-2,3-дигидро-1 Н-имидазо[4,5-b]пиридин-1-ил)пиперидин-1-карбоксилат. В круглодонной колбе объемом 100 мл растворяли (6S,9R)-6-(2,3-дифторфенил)-6-гидрокси-5-оксо 6,7,8,9-тетрагидро-5 Н-циклогепта[b]пиридин-9-ил 4-(2-оксо-2,3-дигидро-1 Н-имидазо[4,5-b]пиридин-1 ил)пиперидин-1-карбоксилат (пример 1, 25.5 мг, 0.046 ммоль) в метаноле (1 мл) до получения бесцветного раствора. После добавления бикарбоната натрия (3,51 мг, 0,093 ммоль) смесь перемешивали при комнатной температуре в течение 30 мин. По результатам LCMS конверсия до более полярного вещества прошла полностью. Смесь концентрировали и очищали с помощью препаративной ВЭЖХ. Для защелачивания раствора к смеси добавляли насыщенный раствор бикарбоната натрия. Летучие компоненты удаляли при глубоком вакууме. Оставшиеся твердые частицы были повторно промыты метиленхлоридом и отфильтрованы. Затем раствор концентрировали с получением осадка белого цвета (12,2 мг, 45%). Полученное вещество представляло собой один диастереоизомер. Однако, соответствующее определение стереохимических показателей не проводилось.(5S,6S,9R)-6-(2,3-Дифторфенил)-5-гидрокси-6,7,8,9-тетрагидро-5H-циклогепта[b]пиридин-9-ил 4-(2 оксо-2,3-дигидро-1 Н-имидазо[4,5-b]пиридин-1-ил)пиперидин-1-карбоксилат (пример 4); (5R,6S,9R)-6(2,3-дифторфенил)-5-гидрокси-6,7,8,9-тетрагидро-5 Н-циклогепта[b]пиридин-9-ил-4-(2-оксо-2,3-дигидро 1 Н-имидазо[4,5-b]пиридин-1-ил)пиперидин-1-карбоксилат (пример 5) и (5S,6R,9R)-6-(2,3-дифторфенил)5-гидрокси-6,7,8,9-тетрагидро-5 Н-циклогепта[b]пиридин-9-ил 4-(2-оксо-2,3-дигидро-1 Н-имидазо[4,5b]пиридин-1-ил)пиперидин-1-карбоксилат. В круглодонной колбе объемом 100 мл готовили бесцветный раствор (9R)-6-(2,3-дифторфенил)-5 оксо-6,7,8,9-тетрагидро-5 Н-циклогепта[b]пиридин-9-ил 4-(2-оксо-2,3-дигидро-1 Н-имидазо [4,5-b]пиридн 1-ил)пиперидин-1-карбоксилат (44.4 мг, 0.083 ммоль) в метаноле (1 мл). Затем в раствор добавляли бикарбонат натрия (6,30 мг, 0,166 ммоль) и полученную смесь перемешивали при комнатной температуре в течение 30 мин. Анализ с помощью LCMS показал, что вещество полностью перешло в три компонента(предположительно являющихся диастереоизомерами), каждый из которых имел целевую молекулярную массу (М+Н=536). Полученную смесь концентрировали и разделяли с помощью препаративной ВЭЖХ(0,1% TFA-метанол-вода) с получением трех веществ (порядок элюции: пример 456; были собраны только фракции, содержащие чистое вещество). Прямое концентрирование (кислого раствора) при глубоком вакууме привело к некоторому распаду (по данным LCMS и ЯМР спектроскопии). Каждую фрак- 16020409 цию отдельно смешивали с бикарбонатом натрия и концентрировали досуха, после чего несколько раз промывали метиленхлоридом с получением трех оснований. Затем каждое основание отделяли от примесей с помощью флэш-хроматографии (при градиентной элюции до 10% метанола в метиленхлориде) с получением целевых продуктов, обозначенных как пример 4 (6,7 мг, 14%), пример 5 (5,5 мг, 12%) и пример 6 (3,0 мг, 6%) в виде осадков белого цвета. Соответствующий стереохимический анализ не требовался. Пример 4. 1 Н ЯМР (400 МГц, ХЛОРОФОРМ-d)млн-1 10.21 (ш. с., 1 Н), 8.52 (д, J=3.53 Гц, 1 Н), 7.97-8.16 (м,2 Н), 7.47 (ш. с., 1 Н), 7.27-7.37 (м, 1 Н), 6.90-7.22 (м, 4 Н), 5.97 (д, J=10.32 Гц, 1 Н), 5.32 (д, J=10.4 Гц, 1 Н),4.26-4.74 (м, 3 Н), 2.55-3.29 (м, 3 Н), 2.18-2.49 (м, 4 Н), 2.07-2.17 (м, 1 Н), 1.59-2.02 (м, 4 Н); 19F ЯМР (376 МГц, ХЛОРОФОРМ-d)млн-1 -137.26136.84 (м, 1F) -142.46142.13 (м, 1F). Пример 5. 1H ЯМР (400 МГц, ХЛОРОФОРМ-d)млн-1 10.04 (ш. с., 1 Н), 8.60 (дд, J=4.78, 1.26 Гц, 1 Н), 8.05 (ш. с., 1 Н), 7.66 (д, J=6.55 Гц, 1 Н), 7.31 (дд, J=7.43, 4.91 Гц, 3 Н), 7.03-7.17 (м, 5H), 6.91-7.03 (м, 1 Н), 6.25 (д,J=5.79 Гц, 1 Н), 4.80 (д, J=8.56 Гц, 1 Н), 4.18-4.66 (м, 3 Н), 3.38-3.58 (м, 5H), 3.02 (д, J=6.29 Гц, 5H), 2.68 (д,J=13.60 Гц, 5H), 2.05-2.45 (м, 3 Н), 1.93 (ш. с., 3 H); 19F ЯМР (376 МГц, ХЛОРОФОРМ-d)млн-1 -138.28(5S,6S,9R)-6-(2,3-Дифторфенил)-9-(тршзопропилсилилокси)-6,7,8,9-тетрагидро-5 Н-циклогепта[b]пиридин-5-ол и (5R,6S,9R)-6-(2,3-дифторфенил)-9-(триизопропилсилилокси)-6,7,8,9-тетрагидро-5 Н-циклогепта[b]пиридин-5-ол. В круглодонной колбе объемом 100 мл в метаноле (5 мл) растворяли (9R)-6-(2,3-дифторфенил)-9(триизопропилсилилокси)-6,7,8,9-тетрагидро-5 Н-циклогепта[b]пиридин-5-он (510 мг, 1.144 ммоль) (в основном транс-изомер) до получения бесцветного раствора. Затем к раствору добавляли борогидрид натрия (87 мг, 2,29 ммоль) и перемешивали полученную смесь при комнатной температуре в течение 1 ч. По результатам LCMS реакция прошла полностью. Далее из смеси под вакуумом удаляли метанол, а остаток разделяли на водную и этилацетатную фазы. Органическую фазу промывали солевым раствором,обезвоживали и концентрировали с получением масляной фракции светло-желтого цвета (492 мг, 96%). Интермедиат 4(5S,6S,9R)-6-(2,3-Дифторфенил)-6,7,8,9-тетрагидро-5 Н-циклогепта[b]пиридин-5,9-диол. В круглодонной колбе объемом 100 мл растворяли (9R)-6-(2,3-дифторфенил)-9(триизопропилсилилокси)-6,7,8,9-тетрагидро-5 Н-циклогепта[b]пиридин-5-ол (смесь интермедиатов 2 и 3, 224,3 мг, 0,501 ммоль) в тетрагидрофуране (4 мл) до получения бесцветного раствора. Затем к раствору добавляли TBAF (0,752 мл, 0,752 ммоль) и перемешивали смесь при комнатной температуре в течение 2 ч. По результатам LCMS основной компонент смеси полностью конвертировался, тогда как минорный компонент не претерпел изменений. После этого удаляли из смеси тетрагидрофуран под вакуумом и разделяли остаток на водную и этилацетатную фазы. Водную фазу дополнительно экстрагировали этилацетатом. Органические фракции объединяли, промывали солевым раствором, осушали над сульфатом натрия и концентрировали с получением масла желто-коричневого цвета. С помощью флешхроматографии при элюции 50%-й смесью этилацетат/гексан были выделены не измененный интермедиат 3 (38 мг, 17%) в виде белого кристаллического осадка и интермедиат 4 (95 мг, 65%) в виде бесцветного масла (затвердевающего при отстаивании). В дальнейшем были получены кристаллы чистого интер- 17020409 медиата 4, стереохимия которых была подтверждена с помощью РСА. Интермедиат 3 5R,6S,9R)-6-(2,3-Дифторфенил)-6,7,8,9-тетрагидро-5 Н-циклогепта[b]пиридин-5,9-диол. В круглодоннной колбе объемом 100 мл растворяли (5R,6S,9R)-6-(2,3-дифторфенил)-9(триизопропилсилилокси)-6,7,8,9-тетрагидро-5 Н-циклогепта[b]пиридин-5-ол (91 мг, 0.203 ммоль) (интермедиат 3) в тетрагидорфуране (2 мл) с получением бесцветного раствора. Затем к раствору добавлялиTBAF (0,407 мл, 0,407 ммоль) и инкубировали полученную смесь при 50 С в течение 16 ч. По результатам LCMS реакция прошла полностью. Разбавляли смесь этилацетатом и водой, разделяли полученные слои и экстрагировали водный слой этилацетатом. Смешанную органическую фазу промывали солевым раствором, осушали над сульфатом натрия и концентрировали с получением масла желто-коричневого цвета. С помощью флеш-хроматографии выделяли целевой продукт (55,5 мг, 94%) в виде белого кристаллического осадка при градиентной элюции смесью этилацетат/гексан до 50%. В дальнейшем были получены кристаллы чистого интермедиата 5, стереохимия которых была подтверждена с помощью РСА.(5S,6S,9R)-6-(2,3-Дифторфенил)-9-(триизопропилсилилокси)-6,7,8,9-тетрагидро-5 Н-циклогепта[b]пиридин-5-ил ацетат. В круглодонной колбе объемом 250 мл растворяли (5S,6S,9R)-6-(2,3-дифторфенил)-9(триизопропилсилилокси)-6,7,8,9-тетрагидро-5 Н-циклогепта[b]пиридин-5-ол (1.004 г, 2.243 ммоль) в метиленхлориде (20 мл) до получения бесцветного раствора. Затем к раствору приливали уксусный ангидрид (0,423 мл, 4,49 ммоль) и триэтиламин (0,938 мл, 6,73 ммоль) с последующим добавлением DMAP(0,055 г, 0,449 ммоль). Смесь перемешивали при комнатной температуре в атмосфере азота в течение 2 ч. По результатам LCMS реакция прошла полностью. Процесс останавливали добавлением бикарбоната натрия и разбавляли раствор этилацеататом. После разделения слоев отделяли органическую фазу, про- 18020409 мывали раствором солей, осушали и концентрировали с получением бесцветного масла (100%). Полученное масло использовали непосредственно для следующей реакции без очистки и характеристики.(5S,6S,9R)-6-(2,3-Дифторфенил)-9-гидрокси-6,7,8,9-тетрагидро-5 Н-циклогепта[b]пиридин-5-ил ацетат. В круглодонной колбе объемом 100 мл растворяли (5S,6S,9R)-6-(2,3-дифторфенил)-9(триизопропилсилилокси)-6,7,8,9-тетрагидро-5 Н-циклогепта[b]пиридин-5-ил ацетат (1098 мг, 2,243 ммоль) (азеотропная смесь с безводным бензолом) в тетрагидрофуране (20 мл) до получения бесцветного раствора. После добавления TBAF (2,69 мл, 2,69 ммоль) полученный раствор светло-желтого цвета перемешивали при комнатной температуре в течение 2 ч. (8:30). По результатам LCMS реакция прошла полностью. Тетрагидрофуран из смеси удаляли под вакуумом, а к остатку добавляли этилацетат и воду. После разделения слоев органическую фазу промывали солевым раствором, осушали и концентрировали с получением бесцветного масла. Целевой продукт в виде бесцветного масла (648 мг, 87% за две стадии) выделяли с помощью флеш-хроматографии при градиентной элюции с содержанием этилацетата в смеси этилацетат/гексан до 70%.(5S,6S,9R)-6-(2,3-Дифторфенил)-5-гидрокси-6,7,8,9-тетрагидро-5H-циклогепта[b]пиридин-9-ил 4-(2 оксо-2,3-дигидро-1 Н-имидазо[4,5-b]пиридин-1-ил)пиперидин-1-карбоксилат (пример 4). В абсолютно сухой круглодонной колбе объемом 100 мл готовили суспензию (5S,6S,9R)-6-(2,3 дифторфенил)-9-гидрокси-6,7,8,9-тетрагидро-5 Н-циклогепта[b]пиридин-5-ил ацетата (96.7 мг, 0.290 ммоль) (в азеотропной смеси с безводным бензолом) и 4-нитрофенил 4-(2-оксо-2,3-дигидро-1Hимидазо[4,5-b]пиридин-1-ил)пиперидин-1-карбоксилата (167 мг, 0.435 ммоль) в диметилформамиде (3 мл) в атмосфере азота. После охлаждения до -15 С (на лед/метанольной бане) к суспензии добавляли по каплям NaHMDS (0,870 мл, 0,870 ммоль). Полученный раствор темно-красного цвета перемешивали при температуре -15 С 0 С в течение 1 ч в атмосфере азота. По результатам LCMS в смеси присутствал целевой продукт, а также некоторое количество продукта избыточного гидролиза. После дополнительного перемешивания в течение 1 ч полный гидролиз так и не был достигнут. Процесс останавливали добавлением бикарбоната натрия, после чего удаляли летучие фракции. Затем к смеси приливали этилацетат и воду и после разделения слоев экстрагировали водный слой дважды этилацетатом. Объединенную органическую фазу промывали солевым раствором, осушали над сульфатом натрия и концентрировали с получением масла желтого цвета. Ацетат-блокированный продукт (2-й пик, 51 мг, 30%, с примесями), а также целевой спирт (3-й пик, 20 мг, 13%) выделяли с помощью флеш-хроматографии при градиентной элюции с содержанием метанола в смеси метанол/метиленхлорид до 10%. В круглодонной колбе объемом 250 мл готовили бесцветный раствор (5S,6S,9R)-5-ацетокси-6-(2,3-дифторфенил)-6,7,8,9-тетрагидро 5 Н-циклогепта[b]пиридин-9-ил 4-(2-оксо-2,3-дигидро-1H-имидазо[4,5-b]пиридин-1-ил)пиперидин-1 карбоксилата (51 мг, 0.088 ммоль) (ацетат-блокированный продукт, полученный на предыдущей стадии) в метаноле (1 мл). К раствору добавляли карбонат калия (122 мг, 0,883 ммоль) и перемешивали полученную смесь при комнатной температуре в течение 1 ч. По результатам LCMS реакция прошла полностью. Затем удаляли метанол под вакуумом. Остаток раствора был разделен на водную и этилацетатную фазы с последующим разделением слоев (анализ водной фазы с помощью LCMS показал отсутствие продукта). Органическую фазу промывали солевым растовром, осушали и концентрировали с получением осадка белого цвета. Целевой продукт (28 мг, 56%) в виде желтого осадка выделяли с помощью флешхроматографии при градиентной элюции с содержанием метанола в смеси метанол/метиленхлорид до 10%. Далее были получены спектры ЯМР 1 Н и 19F, совпавшие с таковыми для вещества, приведенного в примере 4. Интермедиат 8(5R,6S,9R)-5-Хлоро-6-(2,3-дифторфенил)-9-(триизопропилсилилокси)-6,7,8,9-тетрагидро-5 Н-циклогепта[b]пиридин. В абсолютно сухой круглодонной колбе объемом 250 мл готовили суспензию NCS (0,751 г, 5,62 ммоль) в тетрагидрофуране (15 мл). Затем к суспензии добавляли трифенилфосфин (1,475 г, 5,62 ммоль). После перемешивания в атмосфере азота в течение 5 мин к полученной суспензии серого цвета в один прием добавляли (5S,6S,9R)-6-(2,3-дифторфенил)-9-(триизопропилсилилокси)-6,7,8,9-тетрагидро-5 Нциклогепта[b]пиридин-5-ол (1.007 г, 2.250 ммоль). Полученную красноватую суспензию перемешивали при комнатной температуре, постепенно растворяя до получения раствора желто-коричневого цвета. После этого под вакуумом удаляли тетрагидрофуран и полученное масло красного цвета подвергали очистке с помощью ISCO (240 г кремния на колонку), при содержании этилацетата в смеси этилацетат/гексан до 60%. Чистый этилацетат элюировал неполярный компонент раствора. Продукт элюировали 10% метанолом в метиленхлориде (при концентрации NH4OH, равной 2,0 М). Полученные фракции продукта объединяли и проводили еще один раунд очистки с помощью флеш-хроматографии при содержании этилацетата в смеси этилацетат/гексан до 50%. Целевой продукт в виде бесцветного масла был выделен в количестве 869 мг (83%).(5S,6S,9R)-5-азидо-6-(2,3-Дифторфенил)-9-(триизопропилсилилокси)-6,7,8,9-тетрагидро-5 Н-циклогепта[b]пиридин. В курглодонной колбе объемом 100 мл был готовили бесцветный раствор (5R,6S,9R)-5-хлор-6-(2,3 дифторфенил)-9-(триизопропилсилилокси)-6,7,8,9-тетрагидро-5 Н-циклогепта[b]пиридина (566 мг, 1.214 ммоль) в диметилформамиде (5 мл). К раствору добавляли азид натрия (474 мг, 7,29 ммоль) и перемешивали полученную смесь при комнатной температуре в атмосфере азота в течение 2,5 ч. По результатамLCMS реакция прошла только частично. Смесь нагревали до 50 С и инкубировали в течение 15 ч. Анализ с помощью LCMS показал полную конверсию вещества, а также частичное разрушение продукта. Затем смесь разбавляли водой и этилацетатом, разделяли получившиеся слои и промывали ограническую фазу раствором солей, осушали и концентрировали с получением бесцветного масла. Полученный неочищенный продукт был использован непосредственно для последующей реакции без очистки и характеристики. Аналитический образец был получен после менее масштабной процедуры выделения.(5S,6S,9R)-5-азидо-6-(2,3-Дифторфенил)-6,7,8,9-тетрагидро-5 Н-циклогепта[b]пиридин-9-ол. В круглодонной колбе объемом 100 мл готовили бесцветный раствор (5S,6S,9R)-5-азидо-6-(2,3 дифторфенил)-9-(триизопропилсилилокси)-6,7,8,9-тетрагидро-5 Н-циклогепта[b]пиридина (0,732 г, 1,549 ммоль, неочищенный) в тетрагидрофуране (8 мл). К раствору добавляли TBAF (1,859 мл, 1,859 ммоль) и полученный раствор светло-желтого цвета перемешивали при комнатной температуре в течение 1,5 ч. По результатам LCMS реакция прошла полностью. Затем удаляли тетрагидрофуран под вакуумом и к оставшемуся раствору приливали воду и этилацетат. После разделения слоев органическую фазу промывали раствором солей, осушали и концентрировали до получения масла светло-желтого цвета. Далее проводили очистку с помощью флеш-хроматографии при градиенте этилацетата в смеси этилацетат/гексан до 60%, что позволило получить целевой продукт в виде бесцветного масла (приблизительный вес: 480 мг). Аналитический образец был получен после менее масштабной процедуры выделения:(5S,6S,9R)-5-азидо-6-(2,3-Дифторфенил)-6,7,8,9-тетрагидро-5 Н-циклогепта[b]пиридин-9-ил 4-(2 оксо-2,3-дигидро-1 Н-имидазо[4,5-b]пиридин-1-ил)пиперидин-1-карбоксилат. В круглодонной колбе объемом 100 мл в диметилформамиде (8 мл) в атмосфере азота готовили суспензию светло-желтого цвета, содержавшую (5S,6S,9R)-5-азидо-6-(2,3-дифторфенил)-6,7,8,9 тетрагидро-5 Н-циклогепта[b]пиридин-9-ол (0.490 г, 1.549 ммоль) (в азеотропной смеси с безводным бензолом) и 4-нитрофенил 4-(2-оксо-2,3-дигидро-1 Н-имидазо[4,5-b]пиридин-1-ил)пиперидин-1-карбоксилат(0.713 г, 1.859 ммоль). После охлаждения до -15 С (на лед/метанольной бане) к суспензии по каплям добавляли NaHMDS (4,18 мл, 4,18 ммоль). Полученный раствор желто-коричневого цвета перемешивали при -100 С в течение 2 ч и затем еще в течение 2 ч при комнатной температуре. По результатам LCMS реакция прошла полностью. Затем реакцию останавливали добавлением раствора бикарбоната натрия и разбавляли смесь этилацетатом. После разделения слоев водный слой экстрагировали этилацетатом. Полученную объединенную органическую фазу промывали солевым раствором, осушали над сульфатом натрия и концентрировали с получением масла желто-коричневого цвета Далее проводили очистку с помощью флеш-колоночной хроматографии при содержании метанола в смеси метанол/метиленхлорид до 8%, что позволило выделить целевой продукт (основной пик, 632 мг, 73% за три шага) в виде пены светло-желтого цвета.(5S,6S,9R)-5-амино-6-(2,3-Дифторфенил)-6,7,8,9-тетрагидро-5 Н-циклогепта[b]пиридин-9-ил 4-(2 оксо-2,3-дигидро-1 Н-имидазо[4,5-b]пиридин-1-ил)пиперидин-1-карбоксилат. В круглодонной колбе объемом 100 мл готовили бесцветный раствор (5S,6S,9R)-5-азидо-6-(2,3 дифторфенил)-6,7,8,9-тетрагидро-5 Н-циклогепта[b]пиридин-9-ил 4-(2-оксо-2,3-дигидро-1 Н-имидазо[4,5b]пиридин-1-ил)пиперидин-1-карбоксилат (620 мг, 1.106 ммоль) (пример 7) в тетрагидрофуране (5 мл). Далее к раствору добавляли триметилфосфин (3,32 мл, 3,32 ммоль, 1,0 М раствор в толуоле) и перемешивали смесь при комнатной температуре. Через 2 ч по результатам LCMS в системе не осталось исходных компонентов реакционной смеси. После этого добавляли 0,080 мл (4,42 ммоль) воды и смесь перемешивали еще 3 ч. По результатам LCMS реакция прошла полностью. Летучие компоненты удаляли из смеси под вакуумом, а из оставшегося раствора выделяли целевой продукт в виде белого осадка (510 мг,85%) с помощью флеш-колоночной хроматографии при содержании метанола в метиленхлориде до 10%. Эпоксид 1. В абсолютно сухой круглодонной колбе объемом 250 мл готовили раствор желто-коричневого цвета 5(S)-9-гидрокси-6,7,8,9-тетрагидро-5 Н-циклогепта[b]пиридин-5-она (3.16 г, 17.83 ммоль) в метиленхлориде (50 мл). После охлаждения до 0 С к раствору с помощью шприца добавляли TIPS-OTf (4,84 мл, 17,83 ммоль) и триэтиламин (4,97 мл, 35,7 ммоль), после чего смесь перемешивали при 0 С в течение 1 ч. По результатам LCMS реакция прошла полностью. Летучие компоненты удаляли с помощью вакуума, а к остатку приливали раствор бикарбоната натрия и этилацетат. После разделения слоев слой, содержащий органическую фазу, промывали солевым раствором, осушали и концентрировали с получением масла желто-коричневого цвета. Такой продукт без даьнейшей очистки был использован для дальнейших реакций. MS(ESI)[M+H+]=334.28. 2. В круглодонной колбе объемом 250 мл готовили раствор желто-коричневого цвета (S)-9(триизопропилсилилокси)-6,7,8,9-тетрагидро-5 Н-циклогепта[b]пиридин-5-она (5.95 г, 17.83 ммоль) (неочищенный) в метаноле (50 мл). К полученному раствору добалвляи борогидрид натрия (0,675 г, 17,83 ммоль) и перемешивали смесь в течение 1 ч при комнатной температуре. По результатам LCMS реакция прошла полностью. Затем удаляли метанол под вакуумом, а к остатку раствора приливали воду и этилацетат. После разделения слоев органическую фазу промывали солевым раствором, осушали над сульфатом натрия и концентрировали с получением масла желто-коричневого цвета. Продукт использовали непосредственно для дальнейших реакций без дополнительной очистки и характеристики.MS(ESI)[M+H+]=336.28 (по результатам LCMS продукт состоял из двух диастереоизомеров). 3. В круглодонной колбе объемом 250 мл была приготовлена суспензия (9S)-9(триизопропилсилилокси)-6,7,8,9-тетрагидро-5 Н-циклогепта[b]пиридин-5-ола (5.98 г, 17.83 ммоль) и внутренней соли (метоксикарбонилсульфамоил)триэтиламмония гидроксид (6.37 г, 26.7 ммоль) в бензоле (100 мл). Смесь нагревали с обратным холодильником в течение 5 ч при перемешивании в атмосфере азота на предварительно нагретой масляной бане при температуре 85 С. По результатам LCMS реакция прошла полностью. Затем удаляли летучие компоненты под вакуумом, а к остатку приливали воду и этилацетат. После разделения слоев органическую фазу промывали солевым раствором, осушали и концентрировали до получения масла желто-коричневого цвета (6,3 г), которое использовали непосредственно для дальнейших реакций без дополнительной очистки и характеристики MS(ESI)[M+H+]=318.32. 4. В круглодонной колбе объемом 2 л смешивали гипохлорит натрия (658 мл, 574 ммоль) и двухосновный фосфат натрия (3,04 г, 2140 ммоль). После охлаждения до 0 С к смеси добавляли (S,Z)-9(триизопропилсилилокси)-8,9-дигидро-7 Н-циклогепта[b]пиридин (5.66 г, 17 83 ммоль) (неочищенный) и марганец(III) 6,6'-(1 Е,1'Е)-(1R,2R)-циклогексан-1,2-диилбис(азан-1-ил-1-илиден)бис(метан-1-ил-1- 22020409 илиден)бис(2,4-ди-трет-бутилфенолят)хлорид (1.359 г, 2.140 ммоль), растворенные в метиленхлориде (140 мл), по каплям на протяжении 1 ч. После этого реакционной смеси темного цвета позволяли достичь комнатной температуры и перемешивали в течение 20 ч. При анализе с помощью LCMS был получен пик продукта реакции в отсутствие пика исходного вещества Смесь разбавляли водой и эфиром. После разделения слоев водную фазу экстрагировали эфиром дважды. Объединенную ограническую фазу промывали водой и солевым раствором, осушали над цеолитом, фильтровали и концентрировали с получением масла темного цвета. После очистки с помощью флеш-хроматографии при содержании этилацетата в смеси этилацетат/гексан до 50% был получен целевой продукт в виде масла светло-желтого цвета (2,98 г, 50% за 4 стадии). 1 Н ЯМР (400 МГц, ХЛОРОФОРМ-d)млн-1 8.25-8.44 (м, 1 Н), 7.81 (д, J=8.31 Гц, 1 Н), 7.13 (тд,J=7.05, 3.53 Гц, 1 Н), 4.94-5.16 (м, 1 Н), 3.88-4.04 (м, 1 Н), 3.25-3.48 (м, 1 Н), 2.18-2.38 (м, 1 Н), 1.89-2.11 (м,5H), 1.11-1.29 (м, 1 Н), 0.62-1.10 (м, 21 Н). Интермедиат 12(6S,9S)-9-(Триизопропилсилилокси)-6,7,8,9-тетрагидро-5 Н-циклогепта[b]пиридин-6-ол. В круглодонной колбе объемом 500 мл готовили светло-желтого цвета раствор интермедиата 11(2,98 г, 8,93 ммоль) в метаноле (60 мл), к которому добавляли Pd/C (10%, 0,475 г, 0,447 ммоль). Полученную смесь перемешивали в атмосфере азота (1 атм) при комнатной температуре в течение 2 ч. По результатам LCMS реакция прошла в достаточной степени. По прошествии еще 1 ч смесь фильтровали и промывали метанолом. Объединенную органическую фазу концентрировали с получением масла светложелтого цвета. Затем масло было высушено на протяжении 3 дней до получения светло-желтого осадка(2,91 г, 97%), который был использован для следующей стадии без очистки и характеристики.(S)-9-(триизопропилсилилокси)-8,9-дигидро-5 Н-циклогепта[b]пиридин-6(7 Н)-он. В круглодонной колбе объемом 250 мл готовили бесцветный раствор оксалил-хлорида (9,54 мл,19,08 ммоль) в метиленхлориде при -55 С в атмосфере азота. Медленно, в течение 2-х мин, к полученному раствору по каплям добавляли DMSO (2,71 мл, 38,2 ммоль), после чего раствор дополнительно перемешивали в течение 30 мин. Затем к раствору через канюлю в течение 5 мин приливали (6S,9S)-9(триизопропилсилилокси)-6,7,8,9-тетрагидро-5 Н-циклогепта[b]пиридин-6-ол (2.91 г, 8.67 ммоль) (неочищенный, азеотропная смесь с сухим бензолом), растворенный в 8 мл метиленхлорида (плюс 8 мл для ополаскивания). Затем реакционную смесь дополнительно перемешивали в течение 40 мин при -5055 С и вносили триэтиламин (6,04 мл, 43,4 ммоль) через шприц при -50 С. После чего реакционную смесь постепенно нагревали до -20 С за 30 мин. Анализ с помощью ТСХ показал, что конверсия вещества прошла полностью. После добавления воды и этилацетата разделяли слои и, экстрагировали водный слой этилацетатом. Объединенную органическую фракцию осушали над сульфатом натрия и концентрировали с получением желто-коричневого масла. Дальнейшая очистка при использовании флешхроматографии с содержанием этилацетата в смеси этилацетат/гексан до 50% позволила выделить целевой продукт в виде масла светло-желтого цвета (2,08 мг, 72%). В абсолютно сухой круглодонной колбе объемом 250 мл готовили раствор 1,2-дифторбензола(0,680 мл, 6,90 ммоль) в тетрагидрофуране (12 мл) в атмосфере азота. После охлаждения раствора до 65 С через шприц по каплям добавляли n-BuLi (1M в гексане, 2,208 мл, 5,52 ммоль) После этого смесь перемешивали в течение 30 мин при температуре -6560 С, с последующим охлаждением до -78 С. Затем к раствору через шприц добавляли раствор (S)-9-(триизопропилсилилокси)-8,9-дигидро-5 Нциклогепта[b]пиридин-6(7 Н)-она (920.5 мг, 2.76 ммоль) (80304-043) в тетрагидрофуране (4 мл раствора и 4 мл для ополаскивания) (результирующий раствор стал желтым) и перемешивали при -78 С в течение 1 ч (раствор имел желтый цвет), а затем при комнатной температуре в течение 30 мин (раствор приобрел красный цвет). По результатам LCMS реакция прошла в достаточной степени. Реакцию останавливали добавлением перенасыщенного раствора NH4Cl. Затем тетрагидрофуран удаляли и к остатку приливали воду и этилацетат. После разделения фаз органическую фазу промывали солевым раствором, осушали над сульфатом натрия и концентрировали с получением масла желто-коричневого цвета. Дальнейшая очистка с помощью флеш-хроматографии при содержании этилацетата в смеси этилацетат/гексан до 80% позволила получить восстановленный SM (197 мг, 21%) в виде масла желтого цвета, а также целевые продукты (977 мг, 79%) в виде бесцветного масла.(6S,9S)-6-(2,3-Дифторфенил)-6,7,8,9-тетрагидро-5 Н-циклогепта[b]пиридин-6,9-диол. В круглодонной колбе объемом 250 мл готовили бесцветный раствор (6R,9S)-6-(2,3-дифторфенил)9-(триизопропилсилилокси)-6,7,8,9-тетрагидро-5 Н-циклогепта[b]пиридин-6-ола (977 мг, 2.183 ммоль) в тетрагидрофуране (10 мл). К полученному раствору добавляли TBAF (4,80 мл, 4,80 ммоль) и перемешивали смесь в течение 16 ч при 50 С. По результатам LCMS реакция прошла в достаточной степени с некоторым смещением SM влево. После этого к раствору добавляли еще 0,2 эквивалента TBAF и инкубировали смесь при 50 С еще 2 ч. Затем из системы удаляли тетрагидрофуран, а к остатку раствора добавляли воду и этилацетат. После разделения слоев и экстракции водной фазы этилацетатом объединенную органическую фазу промывали солевым раствором, сушили над сульфатом натрия и концентрировали до получения масла желто-коричневого цвета. Целевой продукт в виде белого осадка был выделен с помощью флеш-колоночной хроматографии при содержании метанола в смеси метанол/метиленхлорид до 10% (458 мг, 72%).(6S,9R)-6-(2,3-Дифторфенил)-6-гидрокси-6,7,8,9-тетрагидро-5 Н-циклогепта[b]пиридин-9-ил 4 нитробензоат. В круглодонной колбе объемом 250 мл готовили раствор (6S,9S)-6-(2,3-дифторфенил)-6,7,8,9 тетрагидро-5 Н-циклогепта[b]пиридин-6,9-диола (458 мг, 1.572 ммоль) (азеотропная смесь с безводным бензолом) в тетрагидрофуране (8 мл) светло-желтого цвета. Затем к раствору в атмосфере азота добавляли 4-нитробезойную кислоту (394 мг, 2,359 ммоль) и трифенилфосфин (619 мг, 2,358 ммоль), а также по каплям диизопропилазодикарбоксилат (0,464 мл, 2,358 ммоль). Полученную смесь перемешивали 15 ч,после чего по результатам LCMS реакция прошла полностью, однако целевой продукт являлся минорным компонентом смеси. Смесь концентрировали до масла светло-желтого цвета и выделяли целевой продукт в виде белого осадка (125 мг, 18%) при помощи флеш-хроматографии (при градиенте этилацетата в смеси этилацетат/гексан от 5 до 100%).(6S,9R)-6-(2,3-Дифторфенил)-6,7,8,9-тетрагидро-5 Н-циклогепта[b]пиридин-6,9-диол. В круглодонной колбе объемом 25 о мл был приготовлен бесцветный раствор (6S,9R)-6-(2,3 дифторфенил)-6-гидрокси-6,7,8,9-тетрагидро-5 Н-циклогепта[b]пиридин-9-ил 4-нитробензоат (125 мг,0.284 ммоль) в тетрагидрофуране (2 мл). К раствору добавили хлорид лития (0,568 ммоль) и перемешивали полученную смесь при комнатной температуре в течение 2 ч. По результатам LCMS реакция прошла полностью, после чего к смеси добавили воду и этилацетат. После разделения слоев водная фаза была экстрагирована этилацетатом и полученная объединенная органическая фаза была промыта солевым раствором, высушена и сконцентрирована до получения осадка белого цвета. Целевой продукт в виде белого кристаллического осадка (71 мг, 86%) был выделен с помощью колоночной флешхроматографии с содержанием смеси метано/метиленхлорид до 6%. Несколько кристаллов были использованы для рентгеноструктурного анализа, чтобы подтвердить цис-диольную конформацию продукта.(6S,9R)-6-(2,3-Дифторфенил)-6-гидрокси-6,7,8,9-тетрагидро-5 Н-циклогепта[b]пиридин-9-ил 4-(2 оксо-2,3-дигидро-1 Н-имидазо[4,5-b]пиридин-1-ил)пиперидин-1-карбоксилат. В абсолютно сухой круглодонной колбе объемом 100 мл готовили суспензию светло-желтого цвета (6S,9R)-6-(2,3-дифторфенил)-6,7,8,9-тетрагидро-5 Н-циклогепта[b]пиридин-6,9-диола (71 мг,0.244 ммоль) (азеотропная смесь с безводным бензолом) и 4-нитрофенил 4-(2-оксо-2,3-дигидро-1Hимидазо[4,5-b]пиридин-1-ил)пиперидин-1-карбоксилата (121 мг, 0.317 ммоль) в диметилформамиде (2 мл) в атмосфере азота. Затем к суспензии по каплям добавляли NaHMDS (0,926 мл, 0,926 ммоль). Полученную суспензию перемешивали в атмосфере азота при комнатной температуре в течение 3,5 ч. По результатам LCMS реакция прошла полностью. Реакцию останавливали насыщенным раствором бикарбоната натрия и разбавляли смесь этилацетатом. После разделения слоев водную фазу экстрагировали этилацетатом (в водной фазе не обнаруживался остаток продукта по результатам анализа с помощьюLCMS). Объединенную органическую фазу промывали солевым раствором, осушали над сульфатом натрия и концентрировали с получением масла желтого цвета. Целевой продукт был выделен в виде порошка белого цвета (131 мг, 100%) с помощью колоночной флеш-хроматографии при содержании метанола в смеси метанол/метиленхлорид до 10%. По результатам анализа с помощью хроматограф-массспектрометрии и ВЭЖХ чистота продукта достигала более 99%.(0.881 мл, 7.65 ммоль) в толуоле (24 мл, дегазированного перед использованием) нагревали в атмосфере азота в течение 18 ч. Затем из системы удаляли большую часть растворителя под вакуумом и разбавляли смесь этилацетатом. После разделения слоев фазу с этилацетатом промывали водой 3 раза, осушали над сульфатом натрия, фильтровали и концентрировали. Целевой арилированный продукт (51%) выделяли с помощью флеш-хроматоргафии при содержании этилацетата в смеси этилацетат/гексан 0-35-50%. tR при ВЭЖХ составило 3,55 мин.(6S,9R)-6-(3,5-дифторфенил)-9-(триизопропилсилилокси)-6,7,8,9-тетрагидро-5 Нциклогепта[b]пиридин-5-она (1.4496 г, 3.25 ммоль) в циклопентилметиловом эфире (15 мл) при 0 С в атмосфере азота был добавлен борогидрид лития (0,283 г, 13,01 ммоль). Реакцию проводили при перемешивании при 0 С в течение 6 ч при комнатной температруе, после чего останавливали добавлением метанола и перемешивали еще в течение 0,5 ч. Затем из смеси под вакуумом удаляли растворитель и приливали к остатку этилацетат, трижды промытый водой. Целевой продукт выделяли с помощью флешхроматографии (56%) при содержании этилацетата в гексане от 0 до 10%. tR при ВЭЖХ составило 3,05 мин.(5S,6S,9R)-5-амино-6-(3,5-Дифторфенил)-6,7,8,9-тетрагидро-5 Н-циклогепта[b]пиридин-9-ил 4-(2 оксо-2,3-дигидро-1 Н-имидазо[4,5-b]пиридин-1-ил)пиперидин-1-карбоксилат. В круглодонной колбе объемом 100 мл готовили суспензию NCS (324 мг, 2,423 ммоль) в тетрагидрофуране (4 мл). К суспензии в один прием добавляли трифенилфосфин (636 мг, 2,423 ммоль) и перемешивали в атмосфере азота в течение 5 мин. После этого к полученной суспензии серого цвета добавляли раствор(5S,6S,9R)-6-(3,5-дифторфенил)-9-(триизопропилсилилокси)-6,7,8,9-тетрагидро-5 Нциклогепта[b]пиридин-5-ола (493 мг, 1.101 ммоль) в 1 мл тетрагидрофурана (дополнительно 1 мл для ополаскивания) через канюлю. Получившуюся суспензию сероватого цвета перемешивали при комнатной температуре 5 ч. После этого смесь анализировали на хроматограф-масс-спектрометре. Через 5 ч инкубации конверсия наблюдалась в слабой степени. Реакцию продолжали при 40 С в течение 16 ч, после чего по результатам LCMS конверсия прошла полностью. Реакцию останавливали раствором бикарбоната натрия и разбавляли смесь этилацетатом. После разделения слоев органическую фазу промывали солевым раствором, осушали и концентрирована с получением масла темного цвета. Целевой продукт в виде бесцветного масла (386 мг, 75%, загрязненный веществом с очень близкой подвижностью на колонке) выделяли с помощью флеш-хроматографии при содержании этилацетата в смеси этилацетат/гексан до 20%. Основной пик (tR=3,39 мин) представлял собой продукт элиминации, тогда как минорный пик(tR=3,29) представляет собой хлорид (MS(ESI)[M+H+]=466.22). 2. В круглодонной колбе объемом 100 мл был приготовлен бесцветный раствор (5R,6S,9R)-5-хлор 6-(3,5-дифторфенил)-9-(триизопропилсилилокси)-6,7,8,9-тетрагидро-5 Н-циклогепта[b]пиридина (386 мг,0.828 ммоль) в диметилформамиде (4 мл). К раствору добавляли азид натрия (323 мг, 4,97 ммоль), после чего перемешивали смесь при 50 С в атмосфере азота в течение 20 ч. По результатам ТСХ (4/1 смесь гексан/этилацетат) обнаружено два близких пика: менее полярный компонент представлял собой продукт элиминации стартовых веществ, более полярный пик являлся азидом продукта. Смесь разбавляли водой и этилацетатом. После разделения слоев органическую фазу промывали солевым раствором, осушали и концентрировали с получением бесцветного масла. После очистки с помощью колоночной флешхроматографии, при элюции градиентом этилацетата в смеси этилацетат/гексан до 20%, был выделен целевой продукт (второй минорный пик) (70 мг, 18%, 13% за 2 этапа). ХЛОРОФОРМ-dмлн-1 8.59 (дд,J=4.91, 1.64 Гц, 1 Н), 7.64-7.72 (м, 1 Н), 7.26-7.36 (м, 1 Н), 6.64-6.82 (м, 3 Н), 5.27 (т, J=4.41 Гц, 1 Н), 4.71 (д,J=8.31 Гц, 1 Н), 3.57-3.71 (м, 1 Н), 2.10 (д, J=4.53 Гц, 3 Н), 1.72-1.87 (м, 1 Н), 1.14-1.27 (м, 3 Н), 0.99-1.12 (м,18 Н); 19F ЯМР (376 МГц, ХЛОРОФОРМ-d)млн-1-109.44109.27 (м, 2F). 3. В круглодонной колбе объемом 100 мл готовили бесцветный раствор (5S,6S,9R)-5-азидо-6-(3,5 диффторфенил)-9-(триизопропилсилилокси)-6,7,8,9-тетрагидро-5 Н-циклогепта[b]пиридина (70 мг, 0.148 ммоль) в тетрагидрофуране (1 мл). К раствору добавляли TBAF и перемешивали бесцветный раствор при комнатной температуре в течение 1 ч. По результатам LCMS реакция прошла полностью. После этого тетрагидрофуран удаляли под вакуумом, а остаток разбавляли водой и этилацетатом. После разделения слоев органическую фазу промывали солевым раствором, осушали и концетрировади с получением бесцветного масла. Целевой продукт в виде бесцветного масла (46, 2 мг, 99%) выделяли с помощью флешхроматографии при содержании этилацетата в смеси этилацетат/гексан до 60%.(с, 2 С), 50.08 (с, 1 С), 35.24 (с, 2 С), 34.74 (с, 2 С). 4. В круглодонной колбе объемом 100 мл готовили суспензию (5S,6S,9R)-5-азидо-6-(3,5 дифторфенил)-6,7,8,9-тетрагидро-5 Н-циклогепта[b]пиридин-9-ола (46 мг, 0.145 ммоль) (в азеотропной смеси с безводным бензолом) и 4-нитрофенил 4-(2-оксо-2,3-дигидро-1 Н-имидазо[4,5-b]пиридин-1 ил)пиперидин-1-карбоксилата (66.9 мг, 0.175 ммоль) в диметилформамиде (1 мл) в атмосфере азота. Суспензию охлаждали до -15 С (на лед/метанольной бане) и приливали NaHMDS (0,393 мл, 0,393 ммоль) по каплям. Полученный раствор перемешивали в атмосфере азота в течение 2 ч при -100 С и 2 ч при комнатной температуре. По результатам LCMS реакция прошла в достаточной степени. После этого останавливали реакцию раствором бикарбоната натрия и разбавляли полученный раствор этилацетатом. После разделения слоев водную фазу экстрагировали этилацетатом. Объединенную органическую фазу пормывали водой, осушали над сульфатом натрия и концентрировали до получения желто-коричневого масла. Целевой продукт в виде белого осадка (62 мг, 76%) был выделен с помощью флеш-хроматографии при содержании метанола в смеси метанол/метиленхлорид до 8%.(т, J=9.4 Гц, 2F). 5. В круглодонной колбе объемом 100 мл готовили бесцветный раствор (5S,6S,9R)-5-азидо-6-(3,5 дифторфенил)-6,7,8,9-тетрагидро-5 Н-циклогепта[b]пиридин-9-ил 4-(2-оксо-2,3-дигидро-1 Н-имидазо[4,5b]пиридин-1-ил)пиперидин-1-карбоксилата (62 мг, 0.11 ммоль) в тетрагидрофуране (1 мл). К раствору добавляли триметилфосфин (0,332 мл, 0,332 ммоль) и перемешивали полученную смесь при комнатной температуре. Через 2 ч добавляли воду (7,97 мкл, 0,442 ммоль) и перемешивали полученную смесь при комнатной температуре в течение ночи. По результатам LCMS реакция прошла полностью с образованием целевого продукта. После удаления летучих компонентов смеси под вакуумом, из остатка, с помощью флеш-хроматографии при содержании метанола в смеси метанол в метиленхлориде до 10%, выделяли целевой продукт в виде белого осадка. После очистки целевой продукт сушили под вакуумом в течение трех дней (56 мг, 90%).(5S,6S,9R)-6-(3,5-дифторфенил)-5-гидрокси-6,7,8,9-тетрагидро-5 Н-циклогепта[b]пиридин-9-ил 4-(2 оксо-2,3-дигидро-1 Н-имидазо[4,5-b]пиридин-1-ил)пиперидин-1-карбоксилат. 1. К раствору (5S,6S,9R)-6(3,5-дифторфенил)-9-(триизопропилсилилокси)-6,7,8,9-тетрагидро-5 Н-циклогепта[b]пиридин-5-ол (0.492 г, 1.099 ммоль) в метиленхлориде (10 мл) в атмосфере азота при комнатной температуре приливали уксусный ангидрид (0,207 мл, 2,198 ммоль), триэтиламин (0,460 мл. 3,30 ммоль) и DMAP (0,027 г, 0,220 ммоль). Реакционную смесь перемешивали в течение 2 ч и разбавляли метиленхлоридом, после чего промывали насыщенным раствором карбоната натрия. Отделяли фазу метиленхлорида, осушали над сульфатом натрия, фильтровали и концентрировали с получением неочищенного целевого продукта в виде масла светло-желтого цвета (0,538 г, 100%). ВЭЖХ tR=3,19 мин, MS(ESI)[M+H+]=490.26; 1H ЯМР(400 МГц, ХЛОРОФОРМ-d)млн-1 8.56 (дд, J=4.78, 1.51 Гц, 1 Н), 7.72 (дд, J=7.81, 1.26 Гц, 1 Н), 7.23 (дд,J=7.81, 4.78 Гц, 1 Н), 6.62-6.76 (м, 3 Н), 6.19 (д, J=9.07 Гц, 1 Н), 5.23-5.30 (м, 1 Н), 3.48-3.57 (м, 1 Н), 3.153.18 (м, 1 Н), 2.08-2.20 (м, 5H), 1.92-1.98 (м, 1 Н), 1.17-1.25 (м, 3 Н), 1.07-1.14 (м, 9 Н), 1.04 (д, J=7.30 Гц,9 Н); 19F ЯМР (376 МГц, ХЛОРОФОРМ-d)млн-1 -109.71 (т, J=8.62 Гц, 2F). 2. К раствору (5S,6S,9R)-6-(3,5-дифторфенил)-9-(триизопропилсилилокси)-6,7,8,9-тетрагидро-5 Нциклогепта[b]пиридин-5-ил ацетата (0.538 г, 1.099 ммоль) в тетрагидрофуране (6 мл) при комнатной температуре в атмосфере азота приливали TBAF (1,319 мл, 1,319 ммоль) и перемешивали смесь в течение 1 ч. По результатам LCMS реакция прошла полностью. Затем удаляли ратворитель под вакуумом, а оставшуюся неочищенную смесь фракционировали между этилацетатом и солевым раствором. Отделяли фазу этилацетата, осушали над сульфатом натрия, фильтровали и концентрировали. Очистку целевого продукта (0,2786 г, 75%) (Rf ca. 0.86 при 50% этилацетата в гексане) проводили с помощью колоночной флеш-хроматографии при содержании этилацетата в гексане от 0 до 50%. ВЭЖХ tR=1,88 мин.(1,839 мл, 1,839 ммоль) в атмосфере азота при -20 С. Реакционную смесь перемешивали при -20 С в течение 3 ч, после чего приливали 0,1 экв 4-нитрофенил 4-(2-оксо-2,3-дигидро-1 Н-имидазо [4,5-b]пиридин 1-ил)пиперидин-1-карбоксилата и 0,15 мл NaHMDS. Затем реакционную смесь перемешивали еще в течение 1 ч, после чего поднимали температуру до -10 С. По результатам LCMS в смеси обнаруживался целевой продукт, а также гидролизованный спирт (без альдегидной группы). Реакцию останавливали водой с последующим добавлением этилацетата. Слой этилацетата трижды промывали водой до разделения, после которого вещество обезвоживали над сульфатом натрия, фильтровали и концентрировали с получением неочищенного целевого продукта. ВЭЖХ tR=2,58 мин, MS(ESI)[M+H+]=578.26. К раствору(5 мл) добавляли карбонат калия (785 мг, 5,68 ммоль) при комнатной температуре, после чего перемешивали реакционную смесь в течение 1 ч перед удалением растворителя. Неочищенную смесь фракционировали водой и этилацетатом. Отделенный слой этилацетата промывали водой еще раз, после чего обезвоживали над сульфатом натрия, фильтровали и концентрировали. Целевой продукт был выделен (135,3 мг, 42%) с помощью колоночной флеш-хроматографии при содержании метанола в метиленхлориде от 0 до 10%. ВЭЖХ tR=2,17 мин.(R,Z)-6-(2,3-Дифторфенил)-6,7-дигидро-5 Н-циклогепта[b]пиридин. В круглодонной колбе объемом 250 мл готовили суспензию (6R,9R)-6-(2,3-дифторфенил)-6,7,8,9 тетрагидро-5 Н-циклогепта[b]пиридин-9-ола (1.93 г, 7.01 ммоль) и внутренней соли (метоксикарбонилсульфамоил)триэтиламмоний гидроксида (2,005 г, 8,41 ммоль) в бензоле (60 мл). Суспензию инкубировали при 85 С при перемешивании в атмосфере азота в течение 1 ч (суспензия быстро окрасилась в темно-красный цвет). Анализ с помощью LCMS показал наличие продукта с целевым молекулярным весом. По результатам ТСХ (смесь 1/1 этилацетат/гексан) в смеси обнаруживались следы SM со сдвигом влево,а также мажорный более полярный компонент. Затем из смеси удаляли бензол под вакуумом, а к остатку приливали воду и этилацетат. После разделения водный слой был экстрагирован этилацетатом. Объединенную органическую фазу промывали солевым раствором, обезвоживали и концентрировали до получения желто-коричневого масла. Целевой продукт был выделен в виде масла светло-желтого цвета (0,81 г, 45%) с помощью колоночной флеш-хроматографии при содержании смеси этилацетат/гексан до 80%.(6S,8R,9S)-6-(2,3-Дифторфенил)-6,7,8,9-тетрагидро-5 Н-циклогепта[b]пиридин-8,9-диол. В круглодонной колбе объемом 50 мл готовили раствор желто-коричневого цвета (R,Z)-6-(2,3 дифторфенил)-6,7-дигидро-5 Н-циклогепта[b]пиридина (110 мг, 0.428 ммоль) и NMO (110 мг, 0,941 ммоль) в ацетоне (2 мл) и воде (0,04 мл). Затем к раствору добавляли тетраоксид осмия (0,021 мл, 1,710 мкмоль) (2,5 вес.% раствор в 2-метил-2-пропаноле). Цвет раствора немедленно менялся до очень светложелтого. Смесь перемешивали при комнатной температуре в течение 1 ч. При этом конверсия достигала менее 5%. После чего добавляли еще 0,021 мл OsO4 и инкубировали смесь 22 ч. При этом конверсия достигла 1/3. После этого приливали еще 0,021 мл раствора OsO4 и инкубировали смесь еще 28 ч (суммарно 50 ч). Затем добавляли к смеси 1,2 г бисульфита натрия и продолжали перемешивать в течение 30 мин. После чего удаляли ацетон под вауумом и экстрагировали остаток этилацетатом три раза. Объединенную органическую фазу отмывали солевым раствором, обезвоживали и концентрировали до получения осадка грязно-белого цвета. С помощью флеш-хроматографии при содержании метанола в смеси метанол/метиленхлорид до 10% были выделены два продукта в виде белых осадков: наиболее вероятный целевой менее полярный продукт в транс-форме (61 мг, 49%) и более полярный продукт в цис-форме (47,1 мг, 38%).
МПК / Метки
МПК: A61K 31/4704, A61K 31/4745, C07D 471/20, C07D 401/12, A61P 25/06, A61P 11/06, A61K 31/473, A61K 31/551, A61K 31/519, C07D 471/10, A61K 31/4747, C07D 471/04, A61K 31/517
Метки: антагонисты, рецептора
Код ссылки
<a href="https://eas.patents.su/30-20409-antagonisty-receptora-cgrp.html" rel="bookmark" title="База патентов Евразийского Союза">Антагонисты рецептора cgrp</a>
Выбранные cgrp – антагонисты, способ их получения, а также их применение в качестве лекарственных средств

Номер патента: 9984
Опубликовано: 28.04.2008
Авторы: Рудольф Клаус, Шиндлер Маркус, Дрейер Александер, Бауер Эккхард, Додс Хенри, Арндт Кирстен, Штенкамп Дирк, Лустенбергер Филипп, Мюллер Штефан Георг
МПК: C07D 401/14
Метки: выбранные, способ, лекарственных, качестве, получения, средств, применение, также, антагонисты
Формула / Реферат:
1. CGRP-антагонисты общей формулы в которой А обозначает атом кислорода, цианиминогруппу или фенилсульфонилиминогруппу, X обозначает атом кислорода или серы, необязательно замещенную C1-C6алкильной группой иминогруппу или необязательно замещенную C1-C6алкильной группой метиленовую группу, Y и Z независимо друг от друга обозначают неразветвленную или разветвленную C1-C6алкильную группу, в которой каждая метиленовая группа может быть замещена 1-2...
Производные индола как антагонисты рецептора 5-нт

Номер патента: 304
Опубликовано: 29.04.1999
Авторы: Дакворт Дэвид Малькольм, Вайман Пол Эдриан, Малхоллэнд Кит Раймонд, Гэстер Лэрэми Мэри, Дэвис Дэвид Томас, Джоунз Грэхэм Элджин, Форбес Ян Томсон
МПК: A61K 31/44, C07D 401/12
Метки: 5-нт, индола, антагонисты, производные, рецептора
Формула / Реферат:
1. Соединение формулы (I) или его соль где Р1 и Р2 независимо представляет собой фенил, ароматические или частично насыщенные моноциклические или бициклические гетероциклические кольца, содержащие до трех гетероатомов, выбранных из азота, кислорода или серы; А представляет собой связь, цепь из 1-5 атомов, необязательно замещенных С1-6алкилом, или А представляет собой необязательно замещенный фенил или необязательно замещенное 5-7-членное...
Антагонисты рецептора 5-нт7

Номер патента: 16787
Опубликовано: 30.07.2012
Авторы: Филла Сандра Энн, Галлахер Питер Таддеуш, Бадеску Валентина О., Ваттон Мария Энн
МПК: A61P 25/00, C07D 401/14, A61K 31/497...
Метки: 5-нт7, рецептора, антагонисты
Формула / Реферат:
1. Соединение формулыгде А представляет собой -С(Н)= или -N=;R1 представляет собой заместитель, выбранный из группы, состоящей из i) водорода, ii) метила, iii) этила, iv) гидроксиметила, v) гидроксиэтила, vi) фенила, необязательно содержащего в качестве заместителей от 1 до 3 фторгрупп, vii) бензила, необязательно содержащего в качестве заместителей от 1 до 3 фторгрупп, и viii) пиридила;R2 представляет собой водород, метил или этил;R3...
Производные имидазола, как антагонисты глутаматного рецептора

Номер патента: 10577
Опубликовано: 30.10.2008
Авторы: Бюттельманн Бернд, Йэшке Георг, Фиайра Эрик, Чеккарелли Симона-Мария, Кольчевски Забине, Портер Ричард Хью Филлип
МПК: A61K 31/4439, A61P 25/28, A61K 31/444...
Метки: рецептора, производные, глутаматного, имидазола, антагонисты
Формула / Реферат:
1. Соединение общей формулы в которой R1 обозначает галоген или цианогруппу; R2 обозначает C1-C6-алкил; R3 обозначает фенил или 5- или 6-членный гетероарил, которые необязательно замещены 1, 2 или 3 заместителями, выбранными из группы, включающей галоген, C1-C6-алкил, C3-C6-циклоалкил, C1-C6-алкилгалоген, цианогруппу, C1-C6-алкоксигруппу, NR'R"; R', R" независимо друг от друга обозначают водород или C1-C6-алкил; n равно 1 или 2; R4 обозначает...
Антагонисты рецептора интегрина

Номер патента: 2822
Опубликовано: 31.10.2002
Авторы: Коулман Пол Дж., Халщенко Василь, Даггэн Марк Е., Пэйтан Майкл А., Аскью Бен С., Мейсснер Роберт С., Хатчинсон Джон Х., Хартман Джордж Д., Ванг Дзиабинг, Хант Сесилия, Смит Гарри Р.
МПК: A61K 31/4375, C07D 471/04, A61P 19/00...
Метки: антагонисты, рецептора, интегрина
Формула / Реферат:
1. Соединение формулы где Х выбирают из группы, состоящей из Y выбирают из группы, состоящей из (СН2)m, (СН2)m-S-(СН2)n и (СН2)m-NR4-(СН2)n, где каждый атом углерода метиленовой группы (СН2) в группировке Y в отличие от R4 может быть замещен одним или двумя заместителями R3, при условии, что когда Y обозначает -(СН2)m-NR4-(СН2)n- и n=1, то заместитель R3 на атоме углерода метиленовой группы в -(СН2)m-, соседнем с атомом азота, не может...
Предыдущий патент: Электрод для электролитических применений
Следующий патент: Способ и устройство для прокладки и ремонта изолированного трубопровода
Случайный патент: Получение блоков галогенидов редкоземельных металлов