Амидофеноксииндазолы в качестве ингибиторов c-met
Номер патента: 18385
Опубликовано: 30.07.2013
Авторы: Побанс Марк Эндрю, Ян Вэй Дженнифер, Ли Течао, Ших Чуан, Чжун Боюй, У Чжипэй
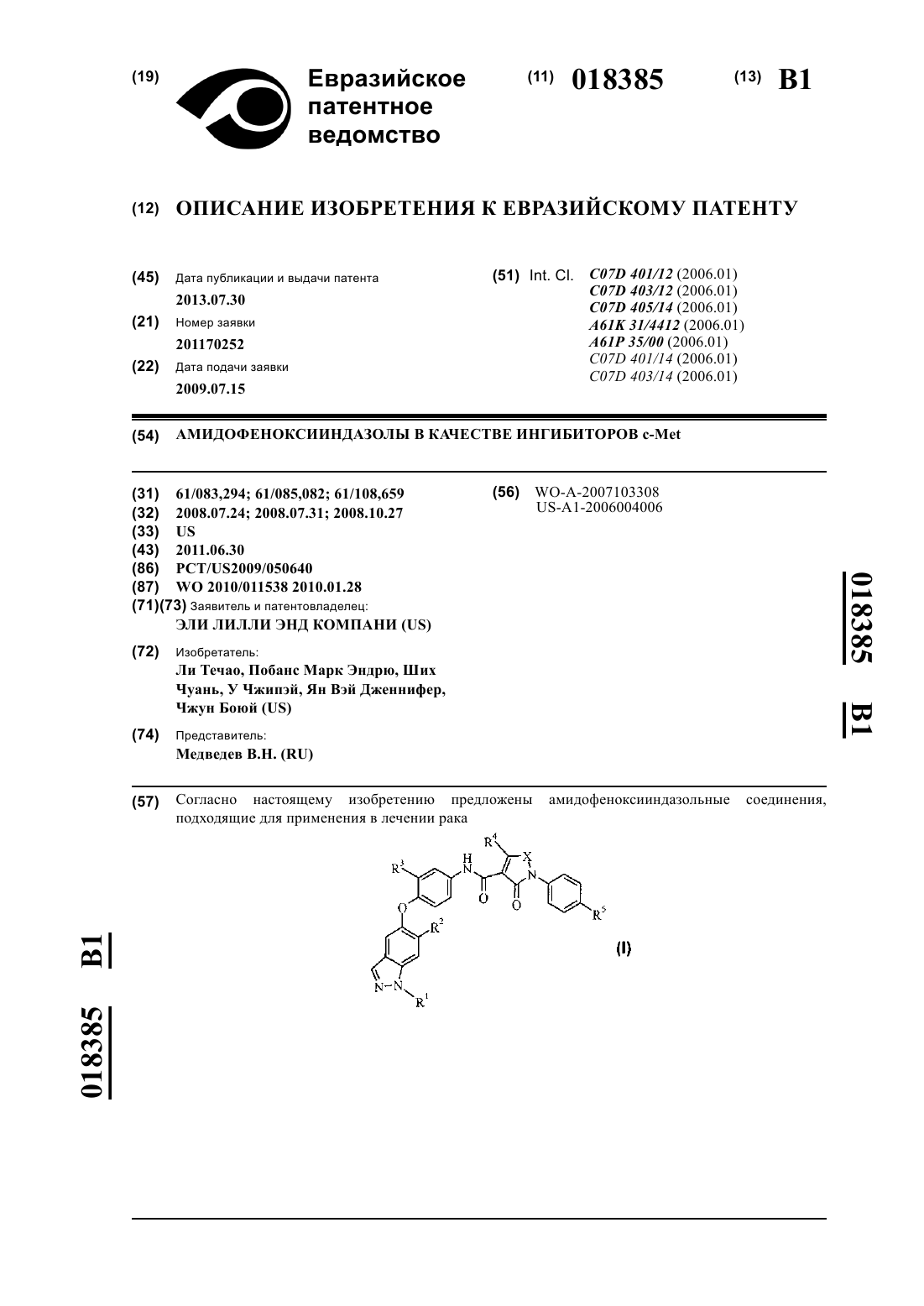






Формула / Реферат
1. Соединение формулы

где R1 представляет собой Н или метил;
R2 представляет собой амино, диметиламино, фтор, циклопропил, пиридил, необязательно замещенный амино или 1-2 метильными группами, пиразолил, необязательно замещенный двумя метильными группами, 2-метоксипиримидин-5-ил, 4-метилсульфонилфенил, тетрагидро-2Н-пиран-4-иламино, (тетрагидро-2Н-пиран-4-ил)аминокарбонил или морфолин-4-ил:

где Ra, Rb и Rc независимо выбраны из Н или метила;
R3 представляет собой Н или F;
R4 представляет собой Н, метил, пиперидин-1-илметил, морфолин-4-илметил или пиразол-1-илметил;
R5 представляет собой Н или F;
X представляет собой CH=N, CH=CH, СН=С(СН3), С(СН3)=СН, C(CH3)=N, N(CH3) или С(морфолин-4-илметил)=СН;
или его фармацевтически приемлемая соль.
2. Соединение по п.1 или его фармацевтически приемлемая соль, в котором R2 представляет собой амино, диметиламино, циклопропил, пиридил, необязательно замещенный амино или 1-2 метильными группами, пиразол-4-ил или морфолин-4-ил.
3. Соединение по п.1 или его фармацевтически приемлемая соль, в котором R2 представляет собой амино, диметиламино, пиразол-4-ил или морфолин-4-ил.
4. Соединение по п.1 или его фармацевтически приемлемая соль, в котором R2 представляет собой пиразол-4-ил.
5. Соединение по пп.1-4 или его фармацевтически приемлемая соль, в котором R4 представляет собой Н, метил или морфолин-4-илметил.
6. Соединение по пп.1-4 или его фармацевтически приемлемая соль, в котором R4 представляет собой Н.
7. Соединение по пп.1-6 или его фармацевтически приемлемая соль, в котором X представляет собой СН=СН или СН=С(СН3).
8. Соединение по п.1, представляющее собой N-(3-фтор-4-(1-метил-6-(1H-пиразол-4-ил)-1H-индазол-5-илокси)фенил)-1-(4-фторфенил)-6-метил-2-оксо-1,2-дигидропиридин-3-карбоксамид или его фармацевтически приемлемую соль.
9. Соединение по п.1, представляющее собой N-(3-фтор-4-(1-метил-6-(1H-пиразол-4-ил)-1H-индазол-5-илокси)фенил)-6-метил-2-оксо-1-фенил-1,2-дигидропиридин-3-карбоксамид или его фармацевтически приемлемую соль.
10. Соединение по пп.1-9, отличающееся тем, что фармацевтически приемлемая соль представляет собой метансульфонатную соль.
11. Фармацевтическая композиция, содержащая соединение по пп.1-10 или фармацевтически приемлемую соль указанного соединения и фармацевтически приемлемый носитель, разбавитель или наполнитель.
12. Способ лечения рака, выбранного из группы, включающей рак легкого, рак молочной железы, рак ободочной и прямой кишки, рак почки, рак поджелудочной железы, рак головы, рак шеи, наследственную папиллярную почечно-клеточную карциному, печеночно-клеточную карциному у детей и рак желудка у млекопитающего, включающий введение млекопитающему, нуждающемуся в таком лечении, эффективного количества соединения по любому из пп.1-10 или его фармацевтически приемлемой соли.
13. Применение соединения по любому из пп.1-10 или фармацевтически приемлемой соли указанного соединения в качестве лекарственного средства для лечения рака.
14. Применение соединения или фармацевтически приемлемой соли указанного соединения по любому из пп.1-10 для лечения рака.
Текст
Согласно настоящему изобретению предложены подходящие для применения в лечении рака(71)(73) Заявитель и патентовладелец: ЭЛИ ЛИЛЛИ ЭНД КОМПАНИ (US)c-Met является членом семейства тирозинкиназных рецепторов фактора роста. Экспрессия c-Met происходит в клетках эндотелия, эпителия и мезенхимы. Связывание эндогенного лиганда, фактора роста гепатоцитов (HGF), с c-Met способствует миграции, пролиферации и инвазии клеток.c-Met вовлечен в прогрессирование некоторых опухолей. Сверхэкспрессия c-Met была обнаружена в многочисленных видах опухолей, включая опухоли толстой кишки, молочной железы, почки, легкого,гемангиомы, плоскоклеточный миелоидный лейкоз, меланомы, глиобластомы и астроцитомы. Активация рецепторов с-Met клеток опухоли усиливает пролиферацию, инвазию/метастазирование и устойчивость клеток опухоли к апоптозу и цитотоксическим видам терапии. Существуют данные о различных амидофеноксигетероарильных ингибиторах c-Met. См., например,US 2005/0288290, US 2006/0211695, US 2006/0004006 и WO 2007103308. Однако по-прежнему существует необходимость в разработке дополнительных соединений, ингибирующих c-Met. Согласно настоящему изобретению предложены новые амидофеноксииндазольные соединения, которые, как считают, находят клиническое применение для лечения рака путем ингибирования c-Met. Также считают, что предпочтительные соединения согласно настоящему изобретению обеспечивают повышение активности по сравнению с некоторыми другими соединениями-ингибиторами cMet, известными из уровня техники. Согласно настоящему изобретению предложено соединение(я) формулы IR2 представляет собой амино, диметиламино, фтор, циклопропил, пиридил, необязательно замещенный аминозаместителем или 1-2 метильными заместителями, пиразолил, необязательно замещенный двумя метильными заместителями, 2-метоксипиримидин-5-ил, 4-метилсульфонилфенил, тетрагидро-2 Нпиран-4-иламино, (тетрагидро-2 Н-пиран-4-ил)аминокарбонил или морфолин-4-ил: где Ra, Rb и Rc независимо выбраны из Н или метила;X представляет собой CH=N, CH=CH, СН=С(СН 3), С(СН 3)=СН, C(CH3)=N, N(CH3) или С (морфолин-4-илметил)=СН; или фармацевтически приемлемая соль указанного соединения. Согласно настоящему изобретению предложен способ лечения рака, выбранного из группы, включающей рак легкого, рак молочной железы, рак ободочной и прямой кишки, рак почки, рак поджелудочной железы, рак головы, рак шеи, наследственную папиллярную почечно-клеточную карциному, печеночно-клеточную карциному у детей и рак желудка у млекопитающего, при этом указанный способ включает введение млекопитающему, нуждающемуся в таком лечении, эффективного количества соединения формулы I или фармацевтически приемлемой соли указанного соединения. Согласно настоящему изобретению также предложена фармацевтическая композиция, содержащая соединение формулы I или фармацевтически приемлемую соль указанного соединения и фармацевтически приемлемый носитель, разбавитель или наполнитель. Согласно настоящему изобретению также предложено соединение формулы I или фармацевтически приемлемая соль указанного соединения для применения в качестве лекарственного средства. Кроме того, согласно настоящему изобретению предложено применение соединения формулы I или фармацевтически приемлемой соли указанного соединения для приготовления лекарственного средства для лечения рака. В частности, данные раковые опухоли выбраны из группы, включающей рак легкого, рак молочной железы, рак ободочной и прямой кишки, рак почки, рак поджелудочной железы, рак головы, рак шеи, наследственную папиллярную почечно-клеточную карциному, печеночно-клеточную карциному у детей и рак желудка. Кроме того, согласно настоящему изобретению предложена фармацевтическая композиция для лечения рака, выбранного из группы, включающей рак легкого, рак молочной железы,рак ободочной и прямой кишки, рак почки, рак поджелудочной железы, рак головы, рак шеи, наследственную папиллярную почечно-клеточную карциному, печеночно-клеточную карциному у детей и рак желудка, при этом указанная композиция содержит в качестве активного компонента соединение формулы I или фармацевтически приемлемую соль указанного соединения. Для специалиста очевидно, что большинство или все соединения согласно настоящему изобретению способны образовывать соли. Соединения согласно настоящему изобретению представляют собой амины и соответственно реагируют с любой из неорганических и органических кислот с образованием фармацевтически приемлемых кислотно-аддитивных солей. Указанные фармацевтически приемлемые кислотно-аддитивные соли и общая методика их получения хорошо известны в данной области техники. См., например, P. Stahl, et al., HANDBOOK OF PHARMACEUTICAL SALTS: PROPERTIES, SELECTIONAND USE (VCHA/Wiley-VCH, 2002); L.D. Bighley, S.M. Berge, D.C. Monkhouse, in "Encyclopedia of Pharmaceutical Technology". Eds. J. Swarbrick and J.C. Boylan, Vol. 13, Marcel Dekker, Inc., New York, Basel,Hong Kong 1995, с. 453-499; S.M. Berge, et al., "Pharmaceutical Salts", Journal of Pharmaceutical Sciences,Vol. 66, No. 1, January 1977. Предпочтительные фармацевтически приемлемые соли соединений согласно настоящему изобретению представляют собой метансульфонатные соли. Предпочтительные соединения представляют собой соединения формулы I, где:(f) R1 представляет собой метил и R2 представляет собой пиразол-4-ил;(u) R1 представляет собой метил, R2 представляет собой пиразол-4-ил, R3 представляет собой F, R4 представляет собой Н, R5 представляет собой F и X представляет собой СН=С(СН 3). Соединения согласно настоящему изобретению могут быть получены в соответствии со следующими схемами синтеза, способами, хорошо известными и очевидными в данной области техники. Подходящие условия реакций для стадий данных схем хорошо известны в данной области техники, а подходящие замены растворителей и дополнительных реагентов находятся в рамках компетенции специалиста в данной области. Для специалистов в данной области техники также очевидно, что промежуточные соединения синтеза могут при необходимости или желании быть выделены и/или очищены различными хорошо известными способами и что часто возможно напрямую использовать различные промежуточные соединения на следующих стадиях синтеза после незначительной очистки или без очистки. Кроме того, специалисту в данной области техники очевидно, что в некоторых случаях порядок введения групп не является определяющим. Конкретный порядок стадий, необходимый для получения соединений согласно настоящему изобретению, зависит от конкретного синтезируемого соединения, исходного соединения и относительной лабильности замещаемых групп, что очевидно квалифицированному химику. Все заместители, если не указано иное, являются такими, как определено выше, и все реагенты хорошо известны и очевидны в данной области техники. Соединения согласно настоящему изобретению могут быть синтезированы, как показано на следующей схеме I, где X, R3, R4 и R5 являются такими, как определено ранее, и R1' и R2' соответствуют или являются предшественниками R1 и R2. Схема I Соединения согласно настоящему изобретению могут быть получены в стандартных условиях сочетания пептидов, хорошо известных специалисту. Замещенный подходящим образом индазол анилин формулы (а) подвергают взаимодействию с кислым соединением формулы (b) в присутствии реагента для сочетания пептидов, такого как 1-(3-диметиламинопропил)-3-этилкарбодиимида гидрохлорид(EDCI), 1-гидроксибензотриазола гидрат (HOBt), подходящего основания, такого как N-метилморфолин,N,N-диизопропилэтиламин (DIPEA) или триэтиламин (ТЭА), в подходящем растворителе, таком как дихлорметан (ДХМ), N,N-диметилформамид (ДМФ) или тетрагидрофуран (ТГФ) с получением необходимого амида согласно настоящему изобретению. В случае если R1' представляет собой подходящую защитную группу азота, такую как тетрагидропиран (ТГП), или если R2' представляет собой функциональную группу, защищенную защитной группой азота, требуется стадия снятия защиты для получения необходимого соединения согласно настоящему изобретению. Схема II Соединение формулы (а) может быть получено, как показано на схеме II, где R1, R2 и R3 являются такими, как определено ранее. Соединение формулы (f) может реагировать с замещенным подходящим образом соединением формулы (g) при повышенной температуре в присутствии подходящего основания, такого как бикарбонат натрия, в подходящем растворителе, таком как ДМФ, с получением соединения формулы (е). Соединение формулы (е) может быть метилировано подходящим метилирующим реагентом, таким как метилиодид, в подходящем растворителе, таком как ТГФ, с подходящим основанием, таким как третбутоксид калия, с получением соединения формулы (d), где R1' представляет собой метил. Соединение формулы (е) также может быть защищено подходящим защитным реагентом, таким как 2,3 дигидропиран (DHP), в подходящем растворителе, таком как ТГФ, в присутствии кислоты, такой какCH3SO3H, с получением соединения формулы (d), где R1 представляет собой защитную группу, такую как 2-тетрагидропиранил [Способы введения или снятия защитных групп хорошо известны в данной области техники; см., например, Greene and Wuts, Protective Groups in Organic Synthesis, 3rd Ed., John Wileyand Sons, New York, (1999)]. Соединение формулы (d) может реагировать в различных облегчаемых переходными металлами условиях сочетания с получением необходимого соединения формулы (с) с замещенным подходящим образом R2', как определено выше. В частности, в случае если это сочетание углерод-углерод, соединение формулы (с) может быть получено в условиях сочетания, таких как сочетание Сузуки, с подходящей бороновой кислотой, подходящим катализатором, таким как хлорид 1,1'-бис-(дифенилфосфино)ферроцен палладия(II) (PdCl2(dppf, и подходящим основанием, таким как CsF, в подходящем растворителе, таком как 1,4-диоксан при повышенной температуре. В случае если это сочетание углерод-азот, соединение формулы (с) может быть получено в условиях сочетания, таких как сочетание Бухвальда-Хартвига, с подходящим азотсодержащим реагентом, таким как морфолин, подходящим лигандом, таким как 2-дитрет-бутилфосфино-2'-метилбифенил, основанием, таким как KOH, и катализатором, таким как трис(дибензилиденацетон)дипалладий(0) (Pd2(dba)3), в подходящем растворителе, таком как трет-бутанол,при повышенной температуре. Реакцию сочетания можно проводить в традиционных условиях теплового режима или в условиях микроволновых реакций, хорошо известных специалисту в данной области техники. Соединение формулы (с) может быть восстановлено до аминосоединения в различных условиях восстановления, хорошо известных специалисту в данной области техники. В частности, соединение формулы (с) может реагировать с хлоридом олова в подходящем растворителе, таком как этилацетат(EtOAc)/этанол (EtOH), при повышенной температуре с получением необходимого соединения формулы(а). В случае если R1' представляет собой защитную группу азота, такую как 2-тетрагидропиранил, данная защитная группа будет расщеплена. R1' будет представлять собой протон после восстановления. Соединение формулы (с) также может быть восстановлено путем взаимодействия с N,Nдиметилгидразином и FeCl3 в подходящем растворителе, таком как метанол (МеОН), или Н 2/палладиемна-углероде (Pd/C) в подходящем растворителе, таком как EtOH. Оба условия восстановления только восстановят нитрогруппу до аминогруппы и не затронут защитную группу азота, R1'. Специалисту в данной области техники хорошо известно, что реакции по атому азота некоторых азотсодержащих гетероциклов, таких как индазольные соединения, могут приводить к образованию таутомеров. Соотношение таутомеров, по существу, зависит от способа проведения реакций и разных условий. Промежуточные соединения, описанные согласно настоящему изобретению, с защитными группами азота, такими как ТНР, могут существовать в виде региоизомеров. В соединениях согласно настоящему изобретению, когда R1 представляет собой протон, соединение может существовать в виде пары быстро меняющихся таутомеров, как показано на схеме III. Схема IIIN-(4-Метокси-2-метилфенил)ацетамид. К раствору 4-метокси-2-метиланилина (120 г, 0,88 моль) в ДХМ (400 мл) по каплям добавляют ангидрид уксусной кислоты (120 мл, 1,2 моль). Реакционную смесь перемешивают при комнатной температуре (RT) в течение 3 ч. После добавления петролейного эфира (ПЭ) (1,6 л) суспензию перемешивают в течение еще одного часа. Осадок собирают путем фильтрования и промывают ПЭ с получением продукта в виде розового твердого вещества (130 г, выход 82%).N-(5-Бром-4-метокси-2-метилфенил)ацетамид. К раствору N-(4-метокси-2-метилфенил)ацетамида (130 г, 0,73 моль) в уксусной кислоте (800 мл) по каплям добавляют бром (42 мл, 0,8 моль) при 0 С. После добавления полученную смесь перемешивают при комнатной температуре в течение ночи. Добавляют насыщенный водный NaHSO3 до получения прозрачного раствора, а затем добавляют большое количество воды (2,0 л) с образованием осадка. Осажденное твердое вещество собирают и промывают водой с получением неочищенного продукта (растворитель для тонкослойной хроматографии (ТСХ):ДХМ:EtOAc=10:1). Твердое вещество растворяют в минималь-4 018385 ном количестве нагреваемого в колбе с обратным холодильником ДХМ, а затем добавляют ПЭ (около 10-20% объема используемого ДХМ). Полученный раствор охлаждают до комнатной температуры. Через 2 ч осажденное твердое вещество собирают и промывают ПЭ с получением белого продукта (108 г, выход 57%).MS (m/z): 260,0 (М+Н). Пример получения 3. 5-Бром-4-метокси-2-метиланилин. К раствору N-(5-бром-4-метокси-2-метилфенил)ацетамида (108 г, 0,42 моль) в МеОН (400 мл) добавляют концентрированную HCl (160 мл, 2 моль). После нагревания в колбе с обратным холодильником в течение ночи смесь нейтрализуют водным NaHCO3 и экстрагируют EtOAc (1200 мл). Органическую фазу сушат и концентрируют с получением продукта (76 г, выход 84%).(280 мл) добавляют раствор NaNO2 (27,6 г, 0,4 моль) в Н 2 О (60 мл) при 0 С. По завершении добавления реакционную смесь перемешивают при 0 С в течение 30 мин. Осадок фильтруют, промывают несколько раз небольшим количеством ледяной воды и сушат с получением продукта (110 г, выход 99%).MS (m/z): 229,0 (М+Н). Пример получения 5. 6-Бром-5-метокси-1H-индазол. Тетрафторборат 1-(5-бром-4-метокси-2-метилфенил)диазония (110 г, 0,35 моль) одной порцией добавляют к перемешиваемой смеси KOAc (140,5 г, 1,43 моль) и 18-краун-6-эфира (9,3 г, 0,035 моль) вCHCl3 (800 мл) и перемешивают при комнатной температуре в течение ночи. Смесь фильтруют и твердое вещество промывают ДХМ. Смешанный фильтрат концентрируют и остаток очищают путем колоночной хроматографии на силикагеле (ДХМ:МеОН = 50:1) с получением продукта (10 г). Осадок добавляют в ТГФ (1600 мл) и смесь перемешивают при комнатной температуре в течение 1 ч. Смесь фильтруют и фильтрат концентрируют с получением 10 г неочищенного продукта (общий выход 88%).BBr3 (105 г, 0,42 моль) в ДХМ (200 мл) при 0 С. Реакционную смесь нагревают до комнатной температуры и перемешивают в течение ночи. Затем реакционный раствор гасят МеОН при 0 С. Растворитель удаляют под вакуумом и остаток нейтрализуют твердым NaHCO3. Смесь разделяют водой (1500 мл) иEtOAc (1500 мл). Водную фазу два раза экстрагируют EtOAc (1500 мл). Смешанные органические фазы сушат и концентрируют с получением неочищенного продукта, который очищают путем колоночной хроматографии на силикагеле (ПЭ:ТГФ = 2:1) с получением необходимого продукта (42,5 г, выход 75%).MS (m/z): 215,0 (М+Н). Пример получения 7. 6-Бром-5-(2-фтор-4-нитрофенокси)-1H-индазол. Смесь 6-бром-5-гидрокси-1H-индазола (7,0 г, 33,0 ммоль), 3,4-дифторнитробензола (4,98 мг,31,0 ммоль), NaHCO3 (2,5 г, 31,0 ммоль) в ДМФ (100 мл) перемешивают при 80 С в течение 4 ч. Затем добавляют LiCl (10% водный раствор) и раствор экстрагируют EtOAc. Органическую фазу сушат над безводным MgSO4, фильтруют и концентрируют. Остаток очищают путем колоночной хроматографии на силикагеле, элюируя ПЭ:ДХМ (1:1), с получением необходимого продукта (5,5 г, выход 46,6%).MS (m/z): 354,0 (M+H). Следующее соединение получают по существу по способу из примера получения 7. Пример получения 9. 6-Бром-5-(2-фтор-4-нитрофенокси)-1-(тетрагидро-2H-пиран-2-ил)-1H-индазол. К раствору 6-бром-5-(2-фтор-4-нитрофенокси)-1H-индазола (5,0 г, 14,2 ммоль) в ТГФ (50 мл) добавляют DHP (2,4 г, 28,5 ммоль) и CH3SO3H (1,0 г, 10,4 ммоль). Полученную смесь перемешивают при комнатной температуре в течение 1 ч. Затем добавляют раствор EtOAc и водный раствор NaHCO3. Отделяют органическую фазу, сушат над безводным MgSO4, фильтруют и концентрируют. Остаток очищают путем колоночной хроматографии на силикагеле, элюируя ПЭ:EtOAc (5:1), с получением необходимогоMS (m/z): 436,0 (M+H). Следующее соединение получают по существу по способу из примера получения 9. Пример получения 11. 6-Бром-1-метил-5-(4-нитрофенокси)-1H-индазол. К смеси 6-бром-5-(4-нитрофенокси)-1H-индазола (2,68 г, 8,02 ммоль) и KOH (600 мг, 10,69 ммоль) в ацетоне (100 мл), охлажденной с помощью ледяной бани, по каплям добавляют метилиодид (0,55 мл,8,83 ммоль). После перемешивания реакционной смеси при 0 С в течение 2 ч смесь перемешивают при комнатной температуре в течение ночи. Удаляют растворитель и остаток очищают путем колоночной хроматографии на силикагеле, элюируя ПЭ:EtOAc (3:1), с получением продукта (1,50 г, выход 53,7%).MS (m/z): 348,0 (М+Н). Следующее соединение получают по существу по способу из примера получения 11. В качестве альтернативы 6-бром-5-(2-фтор-4-нитрофенокси)-1-метил-1H-индазол (пример получения 12) также может быть получен следующим 3-стадийным способом. 2,4-Дибром-5-гидроксибензальдегид (50 г, 178,6 ммоль), 1,2-дифтор-4-нитробензол (28,4 г, 178,6 ммоль) и карбонат калия (49,0 г, 357,2 ммоль) перемешивают в ДМФ (500 мл) при 60 С в течение 2 ч. Реакцию гасят водой (1000 мл) и экстрагируют метил-трет-бутиловым эфиром (МТБЭ) (2500 мл). Органическую фазу промывают насыщенным водным раствором хлорида натрия (2500 мл), сушат безводным сульфатом натрия и концентрируют с получением желтого твердого вещества. Неочищенное твердое вещество перекристаллизовывают из EtOAc/ПЭ (1:5) с получением 2,4-дибром-5-(2-фтор-4 нитрофенокси)бензальдегида в виде желтого твердого вещества (52,8 г, выход 70,6%). 2,4-Дибром-5-(2-фтор-4-нитрофенокси)бензальдегид (13,0 г, 31,0 ммоль) нагревают в колбе с обратным холодильником с метилгидразином (4,3 г, 93 ммоль) в ТГФ (130 мл) в течение 2 ч. Реакцию гасят водой (150 мл) и экстрагируют МТБЭ (2150 мл). Органическую фазу промывают насыщенным водным раствором хлорида натрия (2150 мл), сушат безводным сульфатом натрия и концентрируют с получением твердого вещества. Неочищенное твердое вещество перекристаллизовывают из смеси EtOAc/гексан с получением 1-(2,4-дибром-5-(2-фтор-4-нитрофенокси)бензилиден)-2-метилгидразина в виде желтого твердого вещества (10,6 г, выход 76,7%).MS (m/z): 447,9 (М+Н). 1-(2,4-Дибром-5-(2-фтор-4-нитрофенокси)бензилиден)-2-метилгидразин (1,0 г, 2,2 ммоль), хлорид меди (22 мг, 0,2 ммоль), карбонат калия (0,638 г, 7,2 ммоль), ДМФ (10 мл) и смешивают магнитной мешалкой в пробирке под давлением в атмосфере азота. Герметически закрывают пробкой и пробирку помещают в масляную баню при перемешивании. Баню нагревают до 100 С в течение 30 мин и поддерживают при 100 С в течение 6 ч, а затем охлаждают до комнатной температуры. Реакционную смесь вливают в делительную воронку, содержащую МТБЭ (10 мл) и воду (10 мл). Фазы разделяют и водную фазу экстрагируют дополнительной порцией МТБЭ (10 мл). Органические фазы смешивают и промывают водой, а затем насыщенным водным раствором хлорида натрия. Органическую фазу сушат над сульфатом магния и концентрируют с получением 0,70 г неочищенного продукта. Неочищенное твердое вещество растворяют в ДХМ и гептане, а затем концентрируют с удалением ДХМ, что приводит к образованию суспензии. Продукт собирают путем фильтрования и сушат под вакуумом с получением 6-бром-5(2-фтор-4-нитрофенокси)-1-метил-1H-индазола в виде твердого вещества (0,65 г, выход 79%). Пример получения 13. 6-Бром-2-(тетрагидро-2H-пиран-2-ил)-2H-индазол-5-ол. К раствору 6-бром-5-гидрокси-1H-индазола (725,0 г, 3,4 моль) в ТГФ (10,2 л) добавляют DHP (336,5 мл, 3,57 моль) и CH3SO3H (65,4 г, 0,68 моль) при комнатной температуре. Полученную смесь перемешивают при комнатной температуре в течение 22 ч. Реакционную смесь гасят дистиллированной водой (6 л) и экстрагируют EtOAc (6 л). Добавляют насыщенный водный раствор NaHCO3 (1100 мл), в результате чего доводят значение рН до 8. После разделения фаз органическую фазу промывают водой (4 л), а затем насыщенным водным хлоридом натрия (3 л), сушат над безводным Na2SO4, фильтруют и концентрируют с получением продукта в виде твердого вещества (1,64 кг, выход 99,3%).(5,3 л) добавляют 3,4-дифторнитробензол (1,18 кг, 7,45 моль) и NaHCO3 (625,5 г, 7,45 моль) при комнатной температуре. Реакционную смесь нагревают при 70 С в течение 10 ч, а затем охлаждают до менее 20 С. К раствору медленно добавляют деионизированную (DI) воду (7,9 л) и полученную суспензию перемешивают при 15 С в течение 1 ч. Твердое вещество собирают путем фильтрования под вакуумом и твердый осадок промывают водой (8 л). Затем его сушат на воздухе под вакуумом на фильтре и растирают с метил МТБЭ (18,6 л) в условиях нагревания в колбе с обратным холодильником в течение 2 ч. Затем суспензию охлаждают до комнатной температуры и твердое вещество собирают путем фильтрования под вакуумом. Твердый осадок промывают МТБЭ (4 л 2) и сушат в вакуумной печи при 35 С с получением необходимого продукта (1,405 кг, выход 64,9%).NH4Cl и EtOAc. Органическую фазу отделяют, сушат над безводным MgSO4 и концентрируют. Остаток очищают путем колоночной хроматографии на силикагеле, элюируя ПЭ:EtOAc (3:1), с получением необходимого продукта (2,8 г, выход 56%).MS (m/z): 435,1 (М+Н). Следующие соединения получают по существу по способу из примера получения 15.: Реакцию проводят в микроволновых условиях при 95 С в течение примерно 90 мин. Пример получения 28. 6-(2,6-Диметилпиридин-4-ил)-5-(2-фтор-4-нитрофенокси)-1-(тетрагидропиран-2-ил)-1H-индазол. К раствору 6-бром-5-(2-фтор-4-нитрофенокси)-1-(тетрагидропиран-2-ил)-1H-индазола (1,31 г,3,0 ммоль) в 1,4-диоксане (20 мл) добавляют бис-(пинаколато)дибор (0,91 г, 3,6 ммоль), Pd2(dba)3 (0,14 г,0,2 ммоль), трициклогексилфосфин (0,1 г, 0,4 ммоль), ацетат калия (KOAc) (0,59 г, 6,0 ммоль) в атмосфере N2. После нагревания реакционной смеси в колбе с обратным холодильником в течение 3 ч в атмосфере N2 смесь охлаждают до комнатной температуры. Добавляют гидробромид 4-бром-2,6 диметилпиридина (0,8 г, 3,0 ммоль), PdCl2(dppf) (0,16 г, 0,2 ммоль), CsF (1,38 г, 9,0 ммоль) в атмосфереN2. Полученную смесь нагревают в колбе с обратным холодильником в течение ночи в атмосфере N2. Твердое вещество отфильтровывают и фильтрат концентрируют. Остаток очищают путем колоночнойMS (m/z): 463,1 (М+Н). Пример получения 29. 4-(5-(2-Фтор-4-нитрофенокси)-1-(тетрагидро-2H-пиран-2-ил)-1 Н-индазол-6-ил)морфолин. К раствору 6-бром-5-(2-фтор-4-нитрофенокси)-1-(тетрагидропиран-2-ил)-1H-индазола (8,0 г,18 ммоль) в трет-бутаноле (150 мл) и Н 2 О (3,2 мл) добавляют Pd2(dba)3 (320 мг, 0,35 ммоль), 2-ди-третбутилфосфино-2'-метилбифенил (480 мг, 1,54 ммоль), KOH (3,2 г, 57 ммоль) и морфолин (3,2 г, 36,7 ммоль) в атмосфере N2. После перемешивания смеси при 70 С в течение 2 ч в атмосфере N2 смесь фильтруют и фильтрат концентрируют. Остаток разделяют EtOAc (100 мл) и насыщенным воднымNH4Cl (30 мл). Органическую фазу отделяют, сушат над MgSO4 и концентрируют. Остаток очищают путем колоночной хроматографии на силикагеле, элюируя ПЭ:EtOAc (3:1), с получением продукта (4,5 г,выход 56,5%).MS (m/z): 443,1 (М+Н). Следующие соединения получают по существу по способу из примера получения 29.(рацемический). В 25-мл пробирку для микроволновой печи добавляют 6-бром-5-(2-фтор-4-нитрофенокси)-2(тетрагидро-2H-пиран-2-ил)-2H-индазол(12 мл, 113 ммоль), а затем добавляют гидрохлорид 2-метилморфолина (278 мг, 2,02 ммоль) и третбутоксид натрия (454 мг, 4,58 ммоль). Реакционную смесь нагревают при 150 С в течение 25 мин в микроволновом реакторе. Затем реакционную смесь концентрируют и остаток очищают на колонке с силикагелем, элюируя гексанами (А) и EtOAc (В), градиент от 90% (А):10% (В) до 60% (А):40% (В) в течение 60 мин, затем до 50% (А):50% (В) в течение 20 мин, с получением оранжевого твердого вещества в качестве необходимого продукта (410 мг, выход 39%).MS (m/z): 457 (М+Н). Следующие соединения получают по существу способом из примера получения 32. ние 10 мин. К указанной смеси добавляют гидрохлорид 2,2-диметилморфолина (1,04 г, 6,9 ммоль) и раствор KOH (617 мг, 11,0 ммоль), растворенного в 0,3 мл дистиллированной воды. Реакционную смесь нагревают до 80 С в течение 2 ч, а затем охлаждают до комнатной температуры. Далее смесь вливают вEtOAc (300 мл), промывают дистиллированной водой (1100 мл), а затем насыщенным водным хлоридом натрия (100 мл). Смешанные водные фазы экстрагируют EtOAc (100 мл). Смешанный органический раствор сушат Na2SO4, фильтруют и концентрируют досуха с получением твердого вещества золотистого цвета. Твердое вещество очищают на колонке с силикагелем, элюируя гексаном (А) и EtOAc (В), градиент 95% (А):5% (В) - 75% (А):25% (В) в течение 70 мин с получением светло-желтого твердого вещества в качестве необходимого продукта (410 мг, выход 25%).MS (m/z): 470,8 (М+Н). Пример получения 36. 5-(2-Фтор-4-нитрофенокси)-6-(пиридин-4-ил)-1H-индазол. К раствору 5-(2-фтор-4-нитрофенокси)-6-(пиридин-4-ил)-1-(тетрагидро-2H-пиран-2-ил)-1Hиндазола (2,8 г, 6,45 ммоль) в EtOH (30 мл) добавляют HCl (концентрированная, 5,0 мл). Смесь перемешивают при 80 С в течение 2 ч. Затем добавляют водный NaHCO3 и EtOAc. Органическую фазу сушат над безводным MgSO4 и концентрируют. Остаток очищают путем колоночной хроматографии на силикагеле, элюируя ПЭ:EtOAc (3:1), с получением необходимого продукта (1,95 г, выход 86,0%).(10,2 г, 45,3 ммоль) в EtOAc/EtOH (200 мл/10 мл) перемешивают при 80 С в течение 2 ч. Затем добавляют водный NaHCO3 и EtOAc. Органическую фазу сушат над безводным MgSO4 и концентрируют. Остаток очищают путем колоночной хроматографии на силикагеле, элюируя ПЭ:EtOAc (2:1), с получением необходимого продукта (1,10 г, выход 61,0%).MS (m/z): 321,0 (М+Н). Пример получения 38. 3-Фтор-4-(6-(4-(метилсульфонил)фенил)-1-(тетрагидро-2H-пиран-2-ил)-1H-индазол-5 илокси)анилин. К раствору 5-(2-фтор-4-нитрофенокси)-6-(4-(метилсульфонил)фенил)-1-(тетрагидро-2H-пиран-2 ил)-1H-индазола (320 мг, 0,63 ммоль) в EtOAc (50 мл) добавляют Pd/C (10%, 100 мг). Полученную смесь дегазируют путем вакуумирования и снова наполняют Н 2 три раза. Смесь перемешивают в атмосфере Н 2 при комнатной температуре в течение ночи. Твердое вещество удаляют путем фильтрования и фильтрат концентрируют. Остаток очищают путем колоночной хроматографии на силикагеле, элюируя ДХМ:МеОН (20:1), с получением необходимого продукта (250 мг, выход 83%).MS (m/z): 482,1 (М+Н). Следующие соединения получают по существу по способу из примера получения 38.(900 мг, 2,13 ммоль) и трет-бутилового эфира 4-[5-(2-фтор-4-нитрофенокси)-1-(тетрагидропиран-2-ил)1H-индазол-6-ил]пиразол-1-карбоновой кислоты (3,1 г, 5,92 ммоль) в EtOAc/EtOH (75 мл/75 мл) добавляют дигидрат хлорида олова (10 г, 52,74 ммоль). Реакционную смесь перемешивают в колбе с обратным холодильником в течение ночи. После охлаждения и подщелачивания насыщенным водным NaHCO3 до рН 8-9 смесь экстрагируют EtOAc (100 мл). Органическую фазу отделяют, сушат над MgSO4 и концентрируют. Остаток очищают путем колоночной хроматографии на силикагеле, элюируя сначала ПЭ:EtOAc (1:1), а затем EtOAc, с получением продукта (1,75 г, выход 71,0%). Следующие соединения получают по существу по способу из примера получения 41. Пример получения 53. 4-(6-Циклопропил-2-(тетрагидро-2 Н-пиран-2-ил)-2H-индазол-5-илокси)-3-фторанилин. В 25-мл стеклянную пробирку добавляют 6-циклопропил-5-(2-фтор-4-нитрофенокси)-2(тетрагидро-2H-пиран-2-ил)-2H-индазол (395 мг, 994 мкмоль) и МеОН (20 мл, 494 ммоль). К суспензии добавляют N,N-диметилгидразин (756 мкл, 9,94 ммоль) и FeCl3 (163 мг, 994 мкмоль). Пробирку закрывают пробкой, нагревают до 60 С и перемешивают в течение 2 ч, а затем перемешивают при 50 С в течение ночи. Смесь фильтруют через воронку Бюхнера и твердое вещество промывают МеОН (100 мл). Фильтрат собирают и концентрируют с получением коричневого/оранжевого твердого вещества. Остаток очищают на колонке с силикагелем, элюируя ДХМ (А) и 10% МеОН в растворе ДХМ (В), градиент 100%(А) - 85% (А):15% (В) в течение 50 мин, с получением желтого твердого вещества в качестве соединения,указанного в названии (358 мг, выход 98%).MS (m/z): 368 (М+Н). Следующие соединения получают по существу по способу из примера получения 53.N-(Дифенилметилен)-5-(2-фтор-4-нитрофенокси)-1-метил-1H-индазол-6-амин. В 10-мл пробирку для микроволновой печи добавляют 6-бром-5-(2-фтор-4-нитрофенокси)-1-метил 1 Н-индазол (500 мг, 1,37 ммоль), 4,5-бис-(дифенилфосфино)-9,9-диметилксантен (73 мг, 123 мкмоль) иPd2(dba)3 (38 мг, 41 мкмоль). Смесь суспендируют в толуоле (5 мл, 4 7 ммоль) и добавляют бензофенонимин (272 мг, 1,5 ммоль) и трет-бутоксид натрия (203 мг, 2,05 ммоль). Смесь нагревают при 150 С в течение 20 мин в микроволновом реакторе. После охлаждения реакционный раствор концентрируют с получением коричневого масла, которое растворяют в ДХМ (150 мл) и промывают насыщенным водным хлоридом натрия (250 мл). Водные фазы смешивают и экстрагируют ДХМ (150 мл), сушат Na2SO4,фильтруют и концентрируют с получением коричневого остатка. Остаток очищают на колонке с силикагелем, элюируя гексанами (А) и EtOAc (В), градиент 85% (А):15% (В) - 50% (А):50% (В) в течение 50 мин, с получением желтого твердого вещества в качестве соединения, указанного в названии (574 мг,выход 90,1%).MS (m/z): 467,2 (М+Н). Пример получения 60. 5-(4-Амино-2-фторфенокси)-N-бензгидрил-1-метил-1H-индазол-6-амин. В 100-мл круглодонную колбу добавляют N-(дифенилметилен)-5-(2-фтор-4-нитрофенокси)-1 метил-1H-индазол-6-амин (424 мг, 909 мкмоль) и EtOH (35 мл, 601 ммоль). К смеси добавляют формиат аммония (1000 мг, 15,9 ммоль) с последующим добавлением 10% Pd/C (300 мг, 141 мкмоль), реакционную смесь перемешивают при комнатной температуре в течение 1 ч и нагревают при 45 С в течение 15 мин. Растворитель удаляют и остаток растворяют в ДХМ (150 мл) и дистиллированной воде (100 мл). Органическую фазу промывают дистиллированной водой (1100 мл) и смешанные водные фазы экстрагируют ДХМ (150 мл). Смешанный органический раствор сушат Na2SO4, фильтруют и концентрируют. Неочищенное вещество очищают на колонке с силикагелем, элюируя гексанами (А) и EtOAc (В), градиент 90% (А):10% (В) - 40% (А):60% (В) в течение 90 мин с получением почти белого твердого вещества в качестве соединения, указанного в названии (204 мг, выход 51%).MS (m/z): 438,8 (М+Н). Пример получения 61. 1-Ацетил-6-бром-1H-индазол-5-ил ацетат. Смесь 6-бром-5-гидрокси-1H-индазола (25 г, 117 ммоль) в ангидриде уксусной кислоты (75 мл) нагревают при 110 С при перемешивании в течение 2 ч. После охлаждения добавляют диэтиловый эфир(100 мл). Осадок собирают путем фильтрования, промывают диэтиловым эфиром (30 мл) и сушат под вакуумом с получением продукта (34 г, выход 98%).(дифенилфосфино)ферроцен (dppf) (4,7 г, 8,4 ммоль) и EtOH (500 мл), и перемешивают при 130 С в атмосфере СО в течение 8 ч. Реакционную смесь концентрируют и остаток очищают путем колоночной хроматографии на силикагеле с получением продукта (15,5 г, выход 90%).(100 мл), содержащему метансульфоновую кислоту (721 мг, 7,5 ммоль), по каплям добавляют раствор 3,4-дигидро-2 Н-пирана (6,6 г, 79 ммоль) в ДХМ (20 мл) при комнатной температуре. Полученный раствор перемешивают при комнатной температуре в течение ночи. После удаления растворителя добавляют EtOAc (500 мл), насыщенный водный хлорид натрия (500 мл) и NaHCO3 (5 г). Органическую фазу отделяют, сушат и концентрируют. Остаток очищают путем колоночной хроматографии на силикагеле с получением продукта (16,8 г, выход 77%).MS (m/z): 291,1 (М+Н). Пример получения 64. Этил-5-(2-фтор-4-нитрофенокси)-1-(тетрагидро-2H-пиран-2-ил)-1H-индазол-6-карбоксилат. Смесь этил-5-гидрокси-1-(тетрагидро-2H-пиран-2-ил)-1H-индазол-6-карбоксилата (10,8 г, 37 ммоль), 1,2-дифтор-4-нитробензола (7,1 г, 44 ммоль), NaHCO3 (7,4 г, 88 ммоль) и ДМФ (100 мл) перемешивают при 80 С в течение 2 ч. После охлаждения до комнатной температуры добавляют воду (400 мл) и ПЭ (50 мл), и смесь перемешивают в течение 20 мин. Осадок собирают и сушат под вакуумом при 50 С с получением продукта (15 г, выход 95%).MS (m/z): 430,1 (М+Н). Пример получения 65. 5-(2-Фтор-4-нитрофенокси)-1-(тетрагидро-2 Н-пиран-2-ил)-1H-индазол-6-карбоновая кислота. К раствору этил-5-(2-фтор-4-нитрофенокси)-1-(тетрагидро-2 Н-пиран-2-ил)-1 Н-индазол-6 карбоксилата (22 г, 51,2 ммоль) в ТГФ (60 мл) и МеОН (60 мл), и Н 2 О (10 мл) добавляют LiOH (3 г,125 ммоль). Полученный раствор перемешивают при комнатной температуре в течение 1,5 ч и удаляют органические растворители. Водный остаток подкисляют холодной 6 н. HCl до рН, равного 2, и экстрагируют EtOAc (150 мл). Органическую фазу отделяют, сушат и концентрируют. Остаток очищают путем колоночной хроматографии на силикагеле, элюируя смесью ПЭ:EtOAc:уксусная кислота (100:100:1), с получением продукта (19,5 г, выход 95%).(590 мг, 1,47 ммоль), дифенилфосфорилазида (485 мг, 1,76 ммоль), ТЭА (179 мг, 1,76 ммоль), молекулярных сит 4(3 г) в трет-бутаноле (40 мл) перемешивают при 80 С в течение ночи. Смесь фильтруют и фильтрат концентрируют. Остаток очищают путем колоночной хроматографии на силикагеле, элюируя ПЭ:EtOAc (10:1) с получением необходимого продукта (400 мг, выход 65%).MS (m/z): 473,1 (М+Н). Пример получения 67. трет-Бутил-5-(4-амино-4-фторфенокси)-1-(тетрагидро-2H-пиран-2-ил)-1H-индазол-6-илкарбамат. К раствору трет-бутил-5-(2-фтор-4-нитрофенокси)-1-(тетрагидро-2H-пиран-2-ил)-1H-индазол-6 илкарбамата (400 мг, 0,85 ммоль) в EtOAc (50 мл) добавляют Pd/C (10%, 50 мг). Полученную смесь дегазируют путем вакуумирования и снова наполняют Н 2 три раза. Смесь перемешивают в атмосфере Н 2 при комнатной температуре в течение ночи. Твердое вещество удаляют путем фильтрования и фильтрат концентрируют. Остаток очищают путем колоночной хроматографии на силикагеле, элюируя ДХМ:МеОН (10:1), с получением необходимого продукта (300 мг, выход 80%).(0,17 г, 1,25 ммоль), N-метилморфолина (0,5 мл) в ДМФ (5 мл) перемешивают при комнатной температуре в течение ночи. Затем реакционную смесь разделяют насыщенным водным NH4Cl (20 мл) и EtOAc(50 мл). Органическую фазу отделяют, сушат над MgSO4 и концентрируют. Остаток очищают путем колоночной хроматографии на силикагеле, элюируя ПЭ:EtOAc (1:1), с получением продукта (0,49 г, выход 81,2%). Пример получения 69. 5-(4-Амино-2-фторфенокси)-1-(тетрагидро-2H-пиран-2-ил)-N-(тетрагидро-2H-пиран-4-ил)-1Hиндазол-6-карбоксамид. К раствору 5-(2-фтор-4-нитрофенокси)-1-(тетрагидро-2H-пиран-2-ил)-N-(тетрагидро-2H-пиран-4 ил)-1H-индазол-6-карбоксамида (0,49 г, 1,0 ммоль) в МеОН (50 мл) добавляют Pd/C (100 мг, 10 мас.%) в атмосфере N2. Полученную смесь дегазируют путем вакуумирования и снова наполняют азотом. Затем реакционную смесь перемешивают при комнатной температуре в атмосфере Н 2 в течение ночи. Твердое вещество удаляют путем фильтрования и фильтрат концентрируют с получением неочищенного продукта (0,42 г, выход 91,4%).MS (m/z): 455,1 (М+Н). Пример получения 70. 5-(Бензилокси)-6-бром-1H-индазол. К раствору 6-бром-5-гидрокси-1H-индазола (5 г, 23,5 ммоль) в ТГФ (50 мл) добавляют бензиловый спирт (3,05 г, 28,2 ммоль), PPh3 (7,39 г, 28,2 ммоль) и диэтилазодикарбоксилат (4,46 мл, 2 8,2 ммоль). После перемешивания реакционной смеси при комнатной температуре в течение ночи добавляют EtOAc(50 мл) и насыщенный водный NH4Cl (30 мл). Органическую фазу отделяют, сушат над MgSO4 и концентрируют. Остаток очищают путем колоночной хроматографии на силикагеле, элюируя ПЭ:EtOAc (4:1), с получением продукта (5,0 г, выход 70,2%).MS (m/z): 303,0 (М+Н). Пример получения 71. 5-(Бензилокси)-6-бром-1-(тетрагидро-2H-пиран-2-ил)-1H-индазол. К раствору 5-(бензилокси)-6-бром-1H-индазола (5 г, 16,5 ммоль) в ТГФ (30 мл) и ДХМ (30 мл) добавляют 3, 4-дигидро-2H-пиран (3 мл, 32,8 ммоль) и метансульфоновую кислоту (1 мл, 15,3 ммоль). После перемешивания реакционной смеси при комнатной температуре в течение 2 ч добавляют EtOAc (50 мл) и насыщенный водный NaHCO3. Органическую фазу отделяют, сушат над MgSO4 и концентрируют. Остаток очищают путем колоночной хроматографии на силикагеле, элюируя ПЭ:EtOAc (4:1), с получением необходимого продукта (2,7 г, выход 42,2%). MS(m/z): 387,0 (М+Н). Пример получения 72. 5-(Бензилокси)-1-(тетрагидро-2H-пиран-2-ил)-N-(тетрагидро-2H-пиран-4-ил)-1H-индазол-6-амин. К раствору 5-(бензилокси)-6-бром-1-(тетрагидро-2H-пиран-2-ил)-1H-индазола (2 г, 5,16 ммоль) в толуоле (30 мл) добавляют тетрагидропиран-4-иламин (800 мг, 7,91 ммоль), ацетат палладия (60 мг,267 мкмоль), 2-дициклогексилфосфино-2'-(N,N-диметиламино)бифенил (200 мг, 508 мкмоль) и третбутоксид натрия (700 мг, 7,28 ммоль) в атмосфере N2. После перемешивания реакционной смеси при 100 С в атмосфере N2 в течение 1 ч добавляют EtOAc (30 мл) и реакционную смесь фильтруют. Фильтрат концентрируют и остаток очищают путем колоночной хроматографии на силикагеле, элюируя сначала EtOAc:ПЭ (1/1), а затем EtOAc, с получением необходимого продукта (1,87 г, выход 88,8%).K2CO3 (760 мг, 5,50 ммоль). После нагревания реакционной смеси в колбе с обратным холодильником в течение 30 мин добавляют Н 2 О (20 мл) и EtOAc (30 мл). Органическую фазу отделяют, промывают насыщенным водным NH4Cl, сушат и концентрируют с получением продукта (1,7 г, выход 82,4%).N-(5-Гидрокси-1-(тетрагидро-2H-пиран-2-ил)-1H-индазол-6-ил)-N-(тетрагидро-2H-пиран-4 ил)ацетамид. К раствору N-(5-(бензилокси)-1-(тетрагидро-2H-пиран-2-ил)-1H-индазол-6-ил)-N-(тетрагидро-2Hпиран-4-ил)ацетамида(1,7 г, 3,78 ммоль) в EtOH (100 мл) и EtOAc (100 мл) добавляют Pd/C (1 г,10 мас.%). После перемешивания реакционной смеси при комнатной температуре в атмосфере Н 2 в течение ночи смесь фильтруют и фильтрат концентрируют. Остаток очищают путем колоночной хроматографии на силикагеле, элюируя сначала ПЭ:EtOAc (1:1), а затем EtOAc, с получением продукта (1,2 г,выход 88,2%). 4,02 ммоль) и Cs2CO3 (1,63 г, 5,00 ммоль). После перемешивания реакционной смеси при 100 С в течение 2 ч добавляют EtOAc (60 мл) и насыщенный водный NH4Cl. Органическую фазу отделяют, сушат надMgSO4 и концентрируют. Остаток очищают путем колоночной хроматографии на силикагеле, элюируя сначала ПЭ:EtOAc (1:1), а затем EtOAc, с получением продукта (1,44 г, выход 86,5%).MS (m/z): 499,2 (M+H). Пример получения 76. 5-(4-Амино-2-фторфенокси)-N-(тетрагидро-2H-пиран-4-ил)-1H-индазол-6-амин. Соединение, указанное в названии, получают по существу по способу из примера получения 41.MS (m/z): 343,1 (M+H). Пример получения 77. 5-(Бензилокси)-6-(6-метилпиридин-3-ил)-1-(тетрагидро-2H-пиран-2-ил)-1H-индазол. К раствору 5-(бензилокси)-6-бром-1-(тетрагидро-2H-пиран-2-ил)-1H-индазола (4 г, 10,3 ммоль) в 1,4-диоксане (25 мл) добавляют бис-(пинаколато)дибор (3 г, 11,8 ммоль), dppf (0,3 г, 541 мкмоль),PdCl2(dppf) (0,42 г, 514 мкмоль) и KOAc (2 г, 20,4 ммоль) в атмосфере N2. После перемешивания реакционной смеси в колбе с обратным холодильником в течение 5 ч добавляют 5-бром-2-метилпиридин (2,1 г,12,2 ммоль), тетракис-(трифенилфосфин)палладий (Pd(PPh3)4) (0,6 г, 519 мкмоль) и CS2CO3 (4 г,12,3 ммоль) и смесь перемешивают в колбе с обратным холодильником в течение ночи. Реакционную смесь охлаждают и фильтруют, и фильтрат концентрируют. Остаток очищают путем колоночной хроматографии на силикагеле, элюируя ПЭ:EtOAc (2:1), с получением продукта (1,9 г, выход 46,0%).(1,44 г, 3,60 ммоль) в EtOH (50 мл) и EtOAc (50 мл) добавляют Pd/C (500 мг, 10 мас.%) в атмосфере N2. Полученную смесь дегазируют путем вакуумирования и снова наполняют азотом. Затем реакционную смесь перемешивают при комнатной температуре в атмосфере Н 2 в течение ночи. Реакционную смесь фильтруют. Фильтрат концентрируют с получением продукта (982 мг, выход 88,0%).MS (m/z): 310,1 (M+H). Пример получения 79. 5-(2-Фтор-4-нитрофенокси)-6-(6-метилпиридин-3-ил)-1-(тетрагидро-2H-пиран-2-ил)-1H-индазол. Соединение, указанное в названии, получают по существу по способу из примера получения 75.MS (m/z): 449,2 (М+Н). Пример получения 80. 3-Фтор-4-(6-(6-метилпиридин-3-ил)-1H-индазол-5-илокси)анилин. Соединение, указанное в названии, получают по существу по способу из примера получения 41.MS (m/z): 335,1 (М+Н). Пример получения 81. 1-(4-Бром-2-фторфенокси)-2-фтор-5-метил-4-нитробензол. Соединение, указанное в названии, получают по существу по способу из примера получения 75. 1 Н ЯМР (d6-диметилсульфоксид (ДМСО 8,20 (д, 1 Н), 7,80 (д, 1 Н), 7,42 (д, 1 Н), 7,30 (д, 1 Н), 7,10(д, 1 Н). Пример получения 82. 4-(4-Бром-2-фторфенокси)-5-фтор-2-метиланилин. Соединение, указанное в названии, получают по существу по способу из примера получения 53.MS (m/z): 315,8 (М+Н). Пример получения 83. 5-(4-Бром-2-фторфенокси)-6-фтор-1H-индазол. К суспензии 4-(4-бром-2-фторфенокси)-5-фтор-2-метиланилина (5,2 г, 16,6 ммоль) в воде (100 мл) добавляют фторборную кислоту (6,06 г, 33,1 ммоль) при комнатной температуре. Раствор охлаждают до 0 С и добавляют NaNO2 (1,71 г, 24,8 ммоль) в воде (2 мл). Реакционную смесь перемешивают при 0 С в течение 40 мин. Добавляют CHCl3 (200 мл). Органическую фазу отделяют, сушат над Na2SO4 и фильтруют. К фильтрату добавляют KOAc (7,31 г, 74,5 ммоль) и 18-краун-6-эфир (0,218 г, 0,82 ммоль). Реакционную смесь перемешивают при комнатной температуре в течение 1 ч. Реакционную смесь промывают насыщенным водным хлоридом натрия, сушат над Na2SO4, фильтруют и выпаривают. Остаток промывают ДХМ и фильтруют с получением необходимого продукта в виде светло-желтого твердого вещества. Фильтрат очищают путем хроматографии, элюируя ДХМ, а затем смесью EtOAc:гексан (1:1), с получением дополнительного количества необходимого продукта. Обе порции твердого вещества смешивают с получением необходимого продукта (3,2 г, выход 59%). Пример получения 84. 5-(4-Бром-2-фторфенокси)-6-фтор-1-(тетрагидро-2H-пиран-2-ил)-1H-индазол. Соединение, указанное в названии, получают по существу способом из примера получения 9.(0,10 г, 0,075 моль), CS2CO3 (1,16 г, 3,56 ммоль), Pd2(dba)3 (0,11 г, 0,12 ммоль) в 1,4-диоксане (20 мл) продувают азотом, нагревают до 100 С и перемешивают в течение ночи. Реакционную смесь охлаждают до комнатной температуры, экстрагируют EtOAc, промывают насыщенным водным хлоридом натрия,сушат над Na2SO4, фильтруют и концентрируют (1,45 г).MS (m/z): 510 (М+Н). Пример получения 86. 3-Фтор-4-(6-фтор-1-(тетрагидро-2H-пиран-2-ил)-1H-индазол-5-илокси)анилин. К раствору N-(дифенилметилен)-3-фтор-4-(6-фтор-1-(тетрагидро-2H-пиран-2-ил)-1H-индазол-5 илокси)анилина (1,45 г, 2,85 ммоль) в ТГФ (30 мл) и воде (5 мл) добавляют 1 н. водную HCl (5,69 мл,5,69 ммоль). Реакционную смесь перемешивают при комнатной температуре в течение 2 ч. ДобавляютEtOAc и смесь промывают насыщенным водным раствором NaHCO3, насыщенным водным хлоридом натрия, сушат над Na2SO4, фильтруют и выпаривают. Остаток очищают путем хроматографии, элюируя ДХМ, а затем смесью EtOAc:гексан (1:1), с получением необходимого продукта в виде светлого геляMS (m/z): 346 (М+Н). Пример получения 87. Этил-5-(2-фтор-4-нитрофенокси)-1-(тетрагидро-2H-пиран-2-ил)-1H-индазол-6-карбоксилат. Соединение, указанное в названии, получают по существу тем же способом из примера получения 75.MS (m/z): 430,1 (М+Н). Пример получения 88. Этил-5-(2-фтор-4-нитрофенокси)-1 Н-индазол-6-карбоксилат. К раствору этил-5-(2-фтор-4-нитрофенокси)-1-(тетрагидро-2H-пиран-2-ил)-1H-индазол-6 карбоксилата (15 г, 35 ммоль) в ДХМ (300 мл) добавляют трифторуксусную кислоту (ТФУ) (30 мл) и перемешивают при комнатной температуре в течение ночи. Выпаривают ДХМ. Остаток разделяют между EtOAc (800 мл) и 5% водным NaHCO3 (400 мл). Органическую фазу промывают насыщенным водным хлоридом натрия, сушат над MgSO4 и концентрируют. Остаток очищают путем колоночной хроматографии на силикагеле, элюируя смесью ПЭ:ацетон (5:1), с получением продукта (10,4 г, выход 86%).MS (m/z): 346,1 (М+Н). Пример получения 89. Этил-5-(2-фтор-4-нитрофенокси)-1-метил-1H-индазол-6-карбоксилат. Соединение, указанное в названии, получают по существу по способу из примера получения 11.MS (m/z): 360,1 (М+Н). Пример получения 90. 5-(2-Фтор-4-нитрофенокси)-1-метил-1H-индазол-6-карбоновая кислота. К раствору этил-5-(2-фтор-4-нитрофенокси)-1-метил-1H-индазол-6-карбоксилата (1,19 г, 3,5 ммоль) в ТГФ/Н 2 О (15 мл/5 мл) добавляют LiOH (0,2 г, 8,3 ммоль). После перемешивания реакционной смеси при комнатной температуре в течение 2 ч растворители удаляют. Остаток подкисляют 2 н. HCl до рН,равного 5, и экстрагируют EtOAc (50 мл). Органическую фазу промывают насыщенным водным хлоридом натрия, сушат и концентрируют с получением продукта (1,1 г, выход 100%).(2,5 г) в трет-бутаноле (50 мл) нагревают в колбе с обратным холодильником в течение ночи. Затем твердое вещество отфильтровывают и фильтрат концентрируют. Остаток разделяют между насыщенным водным хлоридом натрия (40 мл) и EtOAc (100 мл). Органическую фазу отделяют, сушат и концентрируют, и остаток очищают путем колоночной хроматографии на силикагеле, элюируя смесью ПЭ:ацетон Пример получения 92. трет-Бутил-5-(4-амино-2-фторфенокси)-1-метил-1H-индазол-6-илкарбамат. Соединение, указанное в названии, получают по существу по способу из примера получения 38.MS (m/z): 373,1 (М+Н). Пример получения 93. 5-(5-(Бензилокси)-1-(тетрагидро-2H-пиран-2-ил)-1H-индазол-6-ил)пиридин-2-амин. К раствору 5-(бензилокси)-6-бром-1-(тетрагидро-2H-пиран-2-ил)-1H-индазола (1,5 г, 3,87 ммоль) в 1,4-диоксане (50 мл) добавляют 5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил) пиридин-2-амин (1 г,4,54 ммоль), Pd(PPh3)4 (200 мг, 173 мкмоль) и CS2CO3 (5 г, 10,6 ммоль). После перемешивания реакционной смеси в колбе с обратным холодильником в течение ночи смесь разделяют между EtOAc (100 мл) и насыщенным водным NH4Cl (50 мл). Органическую фазу отделяют, сушат и концентрируют. Остаток очищают путем колоночной хроматографии на силикагеле, элюируя ДХМ:МеОН (15:1), с получением продукта (0,80 г, выход 51,5%).(1 мл, 7,17 ммоль). После перемешивания реакционной смеси при комнатной температуре в течение 3 ч смесь разделяют между EtOAc (50 мл) и насыщенным водным NH4Cl (20 мл). Органическую фазу отделяют, сушат и концентрируют. Остаток очищают путем колоночной хроматографии на силикагеле,элюируя ПЭ:EtOAc (3:1), с получением продукта (260 мг, выход 67,1%).MS (m/z): 501,2 (М+Н). Пример получения 95. трет-Бутил-5-(5-гидрокси-1-(тетрагидро-2H-пиран-2-ил)-1H-индазол-6-ил)пиридин-2-илкарбамат. К раствору трет-бутил-5-(5-(бензилокси)-1-(тетрагидро-2H-пиран-2-ил)-1 Н-индазол-6-ил)пиридин 2-илкарбамата (260 мг, 519 мкмоль) в EtOH (25 мл) и EtOAc (25 мл) добавляют Pd/C (100 мг, 10 мас.%) в атмосфере N2. Полученную смесь дегазируют путем вакуумирования и снова наполняют азотом. Затем реакционную смесь перемешивают при комнатной температуре в атмосфере Н 2 в течение 6 ч. Реакционную смесь фильтруют. Фильтрат концентрируют с получением продукта (200 мг, выход 93,8%).MS (m/z): 411,1 (М+Н). Пример получения 96. трет-Бутил-5-(5-(2-фтор-4-нитрофенокси)-1-(тетрагидро-2H-пиран-2-ил)-1H-индазол-6-ил)пиридин 2-илкарбамат. Соединение, указанное в названии, получают по существу по способу из примера получения 75.MS (m/z): 550,2 (М+Н). Пример получения 97. трет-Бутил-5-(5-(4-амино-2-фторфенокси)-1-(тетрагидро-2H-пиран-2-ил)-1H-индазол-6-ил)пиридин 2-илкарбамат. Соединение, указанное в названии, получают по существу способом из примера получения 38.(15,6 мл) добавляют ди-трет-бутилдикарбонат (1,09 мл, 4,83 ммоль). Смесь перемешивают в течение 30 мин. ВЭЖХ демонстрирует завершение реакции. Растворитель удаляют при пониженном давлении. Затем добавляют МТБЭ (5 мл). Раствор охлаждают с помощью бани с ледяной водой и перемешивают в течение 15 мин, затем фильтруют с получением соединения, указанного в названии, в виде твердого вещества (0,764 г, 48,9%). Пример получения 99. 6-Бром-5-(2-фтор-4-нитрофенокси)-1H-индазол. В 3000-мл колбу добавляют 6-бром-5-(2-фтор-4-нитрофенокси)-2-(тетрагидро-2H-пиран-2-ил)-2Hиндазол (250 г, 573,1 ммоль) с последующим добавлением МеОН (1000 мл) и MeSO3H (112,71 мл, 1,72 моль) и нагревают до 50 С в течение 14 ч. Твердое вещество выкристаллизовывается из раствора. Затем смесь охлаждают до комнатной температуры. Реакционную смесь концентрируют под вакуумом при 45 С досуха с получением твердого вещества. Твердое вещество разбавляют ДХМ (2000 мл) и действуют на него водой (1000 мл) и 5 н. NaOH (примерно 350 мл) до значения рН, равного 8-9. Органическую фазу отделяют и растворитель удаляют с последующей азеотропной смесью с толуолом с получением 195,6 г желтого твердого вещества. Пример получения 100. 6-Бром-5-(2-фтор-4-нитрофенокси)-1-метил-1H-индазол. В 4000-мл колбу добавляют 6-бром-5-(2-фтор-4-нитрофенокси)-1H-индазол (230 г, 653,2 ммоль),ДМФ (3000 мл) и K2CO3 (135,41 г, 979,78 ммоль). Реакционную смесь охлаждают до 4,5 С с помощью ледяной бани. К смеси добавляют метилиодид (92,71 г, 653,19 ммоль) и реакционную смесь оставляют перемешиваться в течение 20 мин при той же температуре. Реакционную смесь оставляют нагреваться до комнатной температуры и перемешиваться в течение 13 ч. За реакционной смесью наблюдают с помощью жидкостной хромато-масс-спектрометрии. Добавляют дополнительное количество метилиодида(50 г) и NaHCO3 (33 г) и реакционную смесь перемешивают при комнатной температуре до завершения реакции. Реакционную смесь делят на две порции, обозначаемые (А) и (В), и каждую порцию гасят водой (1 л) с последующей экстракцией EtOAc (1 л), вызывающей образование некоторого количества твердых веществ. Смешанные твердые вещества из (А) и (В) собирают путем фильтрования под вакуумом с получением 70 г неочищенной массы, содержащей около 90% изомера 6-бром-5-(2-фтор-4-нитрофенокси)-1 метил-1 Н-индазола (предпочтительный изомер) и 8% изомера 6-бром-5-(2-фтор-4-нитрофенокси)-2 метил-2 Н-индазола. Затем твердое вещество растирают с 1000 мл ДХМ и 750 мл МеОН в течение ночи(около 14 ч). Твердое вещество собирают путем фильтрования и промывают свежим ДХМ (100 мл) с получением 55,5 г необходимого вещества, обозначаемого (С). Маточный раствор концентрируют и обозначают (D). Органическую фазу из (А) и (В) отделяют и концентрируют. Остатки из (А) и (В) смешивают с получением 185 г неочищенного вещества в виде оранжевого твердого вещества, обозначаемого (Е). Неочищенное соединение (Е) растворяют в МеОН и ДХМ, а затем раствор делят на 3 порции. 600 г силикагеля смешивают с каждой порцией раствора и затем смесь силикагеля загружают в семь 270 г пустых картриджей. Используют семь 1,5 кг колонок ISCO с системой растворителей, начиная с 25%EtOAc в гексанах, затем ее меняют на 50% EtOAc в гексанах с получением 62 г необходимого соединения в виде желтого порошка, обозначаемого (F). После 7 прохождений существует несколько смешанных фракций изомеров 6-бром-5-(2-фтор-4-нитрофенокси)-1-метил-1 Н-индазола и 6-бром-5-(2-фтор-4 нитрофенокси)-2-метил-2 Н-индазола. Указанные фракции объединяют и смешивают с маточным раствором (D), полученным в результате процедуры, описанной выше, с получением 17 г смеси. Смесь растворяют в МеОН и ДХМ, смешивают с 170 г силикагеля с получением раствора и пропускают через 1,5 кг колонку с получением 10 г желтого порошка в качестве необходимого продукта, обозначаемого (G). Общий выход (55,5 г (С), 62 г (F) и 10 г (G составляет 127,5 г. Пример получения 101. трет-Бутил-4-(5-(2-фтор-4-нитрофенокси)-1-метил-1H-индазол-6-ил)-1H-пиразол-1-карбоксилат. В 12-л круглодонную колбу, оснащенную мешалкой, устанавливаемую в колбу сверху, термопарой,колбонагревателем, конденсатором и устройством для барботирования азотом под поверхностью, добавляют 1,4-диоксан (7,44 л) и воду (1,67 л). Раствор продувают N2 (подводящая трубка). Затем добавляют 6-бром-5-(2-фтор-4-нитрофенокси)-1-метил-1H-индазол (595 г, 1,63 моль) и раствор снова продувают N2. Добавляют трет-бутил-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-пиразол-1-карбоксилат(717,02 г, 2,44 моль), N-гидрат трехосновного фосфата калия (689,8 г, 3,25 моль) и 1,l'-бис-(ди-третбутилфосфино)ферроцен (7,71 г, 16,25 ммоль). Наконец, добавляют Pd2(dba)3 (7,44 г, 8,13 ммоль). Раствор продувают в течение 15 мин и затем нагревают при 60 С в течение 12 ч. Реакцию не завершают и добавляют больше Pd2(dba)3 (7,44 г, 8,13 ммоль). Раствор снова нагревают при 60 С в течение еще 3 ч и завершают реакцию. Затем удаляют 1,4-диоксан (баня Buchi, температура 60 С) и остаток снова растворяют в 10 объемах (6 л) ДХМ. Добавляют воду (3 л) и затем разделяют фазы. Органический раствор сушат над Na2SO4, фильтруют и концентрируют до масла темного цвета (835 г). Вещество не очищают и используют далее на следующей стадии. Указанное вещество представляет собой около 60% необходимого продукта и около 40% 5-(2-фтор-4-нитрофенокси)-1-метил-6-(1 Н-пиразол-4-ил)-1 Н-индазола. Полученное неочищенное вещество снова защищают на следующей стадии. Считают, что 50% неочищенного продукта представляет собой 5-(2-фтор-4-нитрофенокси)-1-метил-6-(1 Н-пиразол-4-ил)-1 Н-индазол. В 22-л круглодонную колбу, оснащенную мешалкой, устанавливаемую в колбу сверху, термопарой,1-л дополнительной воронкой, устройством для продувки N2 и охлаждающей баней, добавляют раствор неочищенного 5-(2-фтор-4-нитрофенокси)-1-метил-6-(1 Н-пиразол-4-ил)-1 Н-индазола (835 г, 1,18 моль) в ДХМ (6 л). В дополнительную воронку добавляют ди-трет-бутилдикарбонат (283,69 г, 1,30 моль), растворенный в ДХМ (350 мл). Данный раствор по каплям добавляют в течение 48 мин. После завершения реакции ДХМ удаляют путем ротационного выпаривания с получением масла темного цвета. К данному маслу темного цвета добавляют МТБЭ (2,5 л) и масляный раствор охлаждают до примерно 0-5 С. В качестве затравки в раствор вводят вещество, полученное в примере получения 98. После внесения затравки наблюдают кристаллизацию и полученную суспензию перемешивают в течение 30-40 мин. Светложелтую суспензию фильтруют через полипропиленовую прокладку и осадок промывают холодным (05 С) МТБЭ (1,5 л). Твердые вещества сушат в вакуумной печи с температурой 40 С в течение ночи с получением необходимого продукта (443 г, выход неочищенного вещества 60%).MS (m/z): 354,0 (М+Н). Как показано, чистота вещества составляет 93-95% по данным ВЭЖХ и, следовательно, его используют далее. Пример получения 102. трет-Бутил-4-(5-(4-амино-2-фторфенокси)-1-метил-1H-индазол-6-ил)-1H-пиразол-1-карбоксилат. В емкость объемом 3 галлона добавляют трет-бутил 4-(5-(2-фтор-4-нитрофенокси)-1-метил-1 Ниндазол-6-ил)-1 Н-пиразол-1-карбоксилат (433,0 г, 952,8 ммоль) с последующим добавлением ТГФ (6,5 л) и Pd/C (21,65 г, 10% Pd/C и 21,65 г, 5% Pd/C). Смесь нагревают до 35 С в атмосфере газообразного водорода в течение 2 ч. Затем реакционную смесь охлаждают до комнатной температуры и оставляют перемешиваться в атмосфере газообразного водорода в течение ночи, затем нагревают до 40 С в атмосфере газообразного водорода в течение 7 ч. Добавляют дополнительные 2 г Pd/C (1 г, 10% Pd/C и 1 г, 5% Pd/C) и перемешивают в атмосфере газообразного водорода в течение еще одного часа, и охлаждают до комнатной температуры. Смесь снова перемешивают в атмосфере газообразного водорода в течение ночи и завершают реакцию. Смесь фильтруют через комбинацию бумаги GFF и Watman. Фильтрат концентрируют до получения твердого вещества слегка желтого цвета (446 г, выход 110%). Пример получения 103. Метил-2-оксо-1,2-дигидропиридин-3-карбоксилат. В колбу добавляют 2-гидроксиникотиновую кислоту (10 г, 72 ммоль), концентрированную H2SO4(2 мл) и МеОН (400 мл) с последующим добавлением толуола (80 мл). Реакционную смесь нагревают в колбе с обратным холодильником в течение ночи с ловушкой Дина-Старка. После охлаждения до комнатной температуры смесь фильтруют и фильтрат концентрируют. Остаток растворяют в ДХМ (200 мл),нейтрализуют насыщенным водным NaHCO3 до рН, равного 7, и три раза экстрагируют ДХМ (200 мл). Смешанные органические фазы сушат и концентрируют с получением необходимого продукта (8,2 г,выход 74%).(6,0 г,39 ммоль),4 фторфенилбороновой кислоты (16,3 г, 116 ммоль), ацетата меди (14 г, 78 ммоль) и пиридина (12 г, 0,156 моль) в ДХМ (300 мл) добавляют молекулярные сита 4(5 г). Реакционную смесь перемешивают при комнатной температуре в течение ночи с доступом воздуха. После фильтрации и промывки твердых веществ водой фильтрат экстрагируют ДХМ (100 мл). Органическую фазу сушат над MgSO4 и концентрируют. Остаток очищают путем колоночной хроматографии на силикагеле, элюируя ПЭ:ДХМ (2:1-0:1), с получением продукта (9,6 г, выход 100%).DIPEA (6 мл, 34,40 ммоль), этилхлорформиат (1,2 мл, 12,55 ммоль) и 4-диметиламинопиридин (0,16 г,1,31 ммоль). Смесь перемешивают при комнатной температуре в течение 4 ч. Реакционную смесь разбавляют водой и экстрагируют EtOAc, органическую фазу промывают насыщенным водным хлоридом натрия, сушат над Na2SO4 и концентрируют. Остаток очищают путем колоночной хроматографии на силикагеле, элюируя ДХМ, затем ДХМ:МеОН (80:1), с получением соединения, указанного в названииMS (m/z): 199 (М+Н). Пример получения 106. 1-(4-Фторфенил)-2-метилгидразин. К смеси LiAlH4 (1,1 г) в ТГФ (15 мл) по каплям добавляют раствор этил-2-(4 фторфенил)гидразинкарбоксилата (1,9 г, 9,59 ммоль) в ТГФ (10 мл) в атмосфере азота на ледосоляной бане. После добавления ледяную баню убирают и смесь нагревают до 60 С в течение ночи. Реакционную смесь охлаждают и гасят водой. Осадок фильтруют и промывают EtOAc. Органическую фазу отделяют,сушат над Na2SO4 и концентрируют. Остаток очищают путем флэш-хроматографии, элюируя ПЭ:EtOAcMS (m/z): 141 (М+Н). Пример получения 107. Этил-2-(4-фторфенил)-1,5-диметил-3-оксо-2,3-дигидро-1H-пиразол-4-карбоксилат. Смесь 1-(4-фторфенил)-2-метилгидразина (0,6 г, 4,28 ммоль), диэтил-2-ацетилмалоната (0,96 г,4,75 ммоль) и уксусной кислоты (0,51 г, 8,49 ммоль) в воде (15 мл) нагревают при 115 С в течение 3 ч. Реакционную смесь охлаждают и дважды экстрагируют EtOAc. Органическую фазу промывают насыщенным водным хлоридом натрия, сушат над Na2SO4 и концентрируют. Остаток очищают путем колоночной хроматографии на силикагеле, элюируя ДХМ:МеОН (150:1-60:1), с получением соединения, указанного в названии (0,59 г, выход 49,5%).(Е)-2-(2-(4-Фторфенил)гидразоно)ацетальдегид. На раствор гидрохлорида 4-фторфенилгидразина (1 г, 6 ммоль) в ТГФ (20 мл) действуют DIPEA(3 мл, 18 ммоль). Затем по каплям добавляют глиоксаль (40% в воде, 0,89 г, 6,0 ммоль) в ТГФ (10 мл) на ледяной бане. После добавления смесь перемешивают при комнатной температуре в течение 30 мин, гасят водой и экстрагируют EtOAc. Органическую фазу промывают насыщенным водным хлоридом натрия, сушат над Na2SO4 и концентрируют с получением неочищенного соединения, указанного в названии (0,97 г, выход 97%).EtOH (50 мл) и добавляют диэтилмалонат (0,93 г, 5,8 ммоль) и пиперидин (0,2 мл, 2,0 ммоль). Смесь нагревают в колбе с обратным холодильником в течение ночи. Затем реакционную смесь охлаждают и концентрируют. Остаток разбавляют водой и дважды экстрагируют EtOAc. Смешанные органические экстракты сушат над Na2SO4 и концентрируют. Остаток очищают путем колоночной хроматографии на силикагеле, элюируя ПЭ:EtOAc (3:1-1:1), с получением соединения, указанного в названии (0,26 г, выход двух стадий 17%).MS (m/z): 263 (М+Н). Пример получения 110. Метил-3-(4-фторфениламино)-3-оксопропаноат. К раствору 4-фторанилина (4,4 мл, 45 ммоль) в ацетоне добавляют ТЭА (8,5 мл, 67 ммоль) и метилмалонилхлорид (7,2 мл, 67 ммоль). Смесь перемешивают при комнатной температуре в течение 5 ч, а затем концентрируют. После добавления воды (20 мл) остаток подкисляют концентрированной HCl до рН, равного 3. Осадок собирают путем фильтрования и промывают ПЭ с получением продукта (14,1 г,выход 100%).MS (m/z): 212,1 (М+Н). Следующие соединения получают по существу по способу из примера получения 110. Пример получения 113. Этил-1-(4-фторфенил)-6-метил-2-оксо-1,2-дигидропиридин-3-карбоксилат. К раствору метил-3-(4-фторфениламино)-3-оксопропаноата (10,1 г, 48 ммоль) в EtOH (100 мл) добавляют транс-4-метокси-3-бутен-2-он (5,7 г, 57 ммоль) и CH3ONa (3,1 г, 57 ммоль). Смесь нагревают в колбе с обратным холодильником в течение ночи и затем концентрируют. Остаток очищают путем колоночной хроматографии на силикагеле, элюируя ПЭ:EtOAc (5:1-2:1), с получением продукта (2,1 г, выход 16%).MS (m/z): 276,1 (М+Н). Следующие соединения получают по существу по способу из примера получения 113.(Z)-4-Аминопент-3-ен-2-он. К раствору 2,4-пентандиона (20,0 г, 0,2 моль) в воде (20 мл) по каплям добавляют 25% водный раствор аммиака (13,2 мл, 0,2 моль) при комнатной температуре. Смесь перемешивают при комнатной температуре в течение 2 ч. Затем удаляют растворитель с получением продукта (20,0 г, выход 100%) в виде(ушир.с, 1 Н). Пример получения 117. Этил-4,6-диметил-2-оксо-1,2-дигидропиридин-3-карбоксилат. Раствор (Z)-4-аминопент-3-ен-2-она (20,0 г, 0,2 моль) в сухом ТГФ (20 мл) добавляют к раствору этилцианоацетата (22,6 г, 0,2 моль) и ТЭА (20,2 г, 0,2 моль) в сухом ТГФ (200 мл). Реакционную смесь нагревают в колбе с обратным холодильником в течение 56 ч и затем концентрируют. Остаток оставляют при комнатной температуре на 5 ч, осадок фильтруют и промывают EtOAc с получением соединения,указанного в названии (7,3 г, выход 19,9%).MS (m/z): 196,0 (М+Н). Пример получения 118. Этил-1-(4-фторфенил)-4,6-диметил-2-оксо-1,2-дигидропиридин-3-карбоксилат. Суспензию этил-4,6-диметил-2-оксо-1,2-дигидропиридин-3-карбоксилата (5,0 г, 0,025 моль), 4 фторфенилбороновой кислоты (10,7 г, 0,077 моль), ацетата меди (1,33 г, 0,007 моль), пиридина (6,86 мл) и молекулярных сит 4(5,0 г) в 1,4-диоксане (80 мл) нагревают при 80 С в течение 68 ч. Твердые вещества отфильтровывают и фильтрат концентрируют. Остаток разделяют EtOAc (50 мл) и 2 н. HCl (70 мл). Осажденное твердое вещество собирают путем фильтрования и промывают EtOAc с получением продукта, указанного в названии (1,2 г). Фильтрат EtOAc отделяют, сушат и концентрируют. Остаток очищают путем колоночной хроматографии на силикагеле, элюируя ПЭ:EtOAc:МеОН (40:20:1), с получением другой порции продукта (0,12 г), (общее количество 1,32 г, выход 17,8%).MS (m/z): 290,1 (М+Н). Пример получения 119. Этил-4,6-диметил-2-оксо-1-фенил-1,2-дигидропиридин-3-карбоксилат. Суспензию этил-4,6-диметил-2-оксо-1,2-дигидропиридин-3-карбоксилата (7,5 г, 0,038 моль), фенилбороновой кислоты (14,1 г, 0,115 моль), ацетата меди (1,38 г, 0,008 моль), пиридина (7,5 мл) в 1,4 диоксане (80 мл) нагревают при 80 С в течение 96 ч. Твердое вещество отфильтровывают и фильтрат концентрируют. Остаток разделяют EtOAc (50 мл) и 2 н. HCl (70 мл), осадок собирают путем фильтрования и промывают EtOAc с получением продукта, указанного в названии (2,95 г). Раствор EtOAc отделяют, сушат и концентрируют. Остаток очищают путем колоночной хроматографии на силикагеле, элюируя смесью ПЭ:ацетон (2:1), с получением другой порции продукта (0,3 г). (3,25 г, выход 31,2%).(1,2 г, 4,3 ммоль) и азобисизобутиронитрила (AIBN) (0,09 г, 0,06 ммоль) в ДХМ тремя порциями добавляют N-бромсукцинимид (NBS) (0,9 г, 4,3 ммоль) при комнатной температуре. Полученную реакционную смесь перемешивают при комнатной температуре в течение ночи. Смесь концентрируют и остаток очищают путем колоночной хроматографии на силикагеле, элюируя ПЭ:EtOAc (4:1), с получением продукта в виде белого твердого вещества (1,3 г, выход 84%).MS (m/z) 357,0 (М+Н). Пример получения 121. Этил-5-1 Н-пиразол-1-ил)метил)-2-(4-фторфенил)-1-метил-3-оксо-2,3-дигидро-1H-пиразол-4 карбоксилат. К раствору этил-5-(бромметил)-2-(4-фторфенил)-1-метил-3-оксо-2,3-дигидро-1H-пиразол-4 карбоксилата (1,3 г, 3,6 ммоль) и 1H-пиразола (0,25 г, 3,6 ммоль) в ТГФ (50 мл) и Н 2 О (1 мл), охлажденному на бане с ледяной водой, тремя порциями добавляют твердый KOH (0,2 г, 3,6 ммоль). После перемешивания реакционной смеси при комнатной температуре в течение ночи растворитель удаляют. Остаток очищают путем колоночной хроматографии на силикагеле, элюируя ПЭ:EtOAc (4:1), с получением продукта (0,87 г, выход 70%). Следующие соединения получают по существу по способу из примера получения 121. Пример получения 124. Этил-1-(4-фторфенил)-5-(морфолинометил)-2-оксо-1,2-дигидропиридин-3-карбоксилат. Смесь этил-1-(4-фторфенил)-5-метил-2-оксо-1,2-дигидропиридин-3-карбоксилата (0,275 г, 1 ммоль),NBS (0,2 г, 1,1 ммоль), AIBN (40 мг, 0,2 ммоль) в CCl4 (20 мл) нагревают в колбе с обратным холодильником в течение 3 ч. После охлаждения реакционной смеси добавляют морфолин (0,088 мл, 1 ммоль) иK2CO3 (0,14 г, 1 ммоль). Реакционную смесь перемешивают при комнатной температуре в течение еще 2 ч. Растворитель удаляют, остаток разбавляют водой (10 мл) и дважды экстрагируют EtOAc (20 мл). Смешанные органические фазы промывают насыщенным водным хлоридом натрия, сушат и концентрируют. Остаток очищают путем колоночной хроматографии на силикагеле, элюируя смесью ДХМ:МеОНMS (m/z): 361,1 (М+Н). Пример получения 125. Этил-2-(4-фторфенилкарбамоил)-3-метилбут-2-еноат. К раствору 4-фторанилина (2 г, 18 ммоль) в диэтилизопропилиденмалонате (10 г, 50 ммоль) добавляют 1 Н-имидазол (0,25 г, 3,67 ммоль). Реакционную смесь перемешивают при 200 С в атмосфере N2 в течение 3 ч. После охлаждения реакционной смеси добавляют силикагель. Смесь загружают в колонку с силикагелем и элюируют сначала ПЭ, а затем ПЭ:EtOAc (3:1) с получением продукта (1,85 г, выход 39%).N,N-диметилформамида (5 мл, 37,45 ммоль) перемешивают при 90 С в течение 90 мин. После охлаждения реакционной смеси добавляют EtOAc (30 мл) и насыщенный раствор NH4Cl (20 мл). Органическую фазу отделяют, сушат и концентрируют. Остаток очищают путем колоночной хроматографии на силикагеле, элюируя ДХМ:МеОН (10:1). Фракции, содержащие продукт, концентрируют и остаток смешивают с эфиром с получением 1,2 г желтого твердого вещества, которое фильтруют и отбрасывают. Фильтрат концентрируют и снова очищают путем колоночной хроматографии на силикагеле, элюируя ДХМ:МеОНMS (m/z): 276,1 (М+Н). Пример получения 127. Этил-2-(4-фторфенил)-6-метил-3-оксо-2,3-дигидропиридазин-4-карбоксилат. К суспензии 1-трифенилфосфоранилиден-2-пропанона (3,18 г, 9,99 ммоль) в ТГФ (50 мл) добавляют диэтилкетомалонат (1,74 г, 9,99 ммоль). Полученную смесь перемешивают при комнатной температуре в течение 30 мин с получением прозрачного раствора. Затем данный раствор добавляют к раствору гидрохлорида 4-фторфенилгидразина (1,62 г, 9,96 ммоль) в EtOH:H2O (30 мл:30 мл). После нагревания полученной смеси в колбе с обратным холодильником в течение 1 ч добавляют ТЭА (3 мл) и смесь перемешивают в колбе с обратным холодильником в течение еще 1 ч. Затем смесь концентрируют и остаток разделяют насыщенным водным NH4Cl (30 мл) и EtOAc (30 мл). Органическую фазу отделяют, сушат и концентрируют. Остаток очищают путем колоночной хроматографии на силикагеле, элюируя ПЭ:EtOAcMS (m/z): 277,1 (М+Н). Пример получения 128. 1-(4-Фторфенил)-2-оксо-1,2-дигидропиридин-3-карбоновая кислота. К раствору метил-1-(4-фторфенил)-2-оксо-1,2-дигидропиридин-3-карбоксилата (10,0 г, 0,04 моль) в МеОН (150 мл) и воде (50 мл) добавляют LiOH (1,9 г, 0,08 моль). Полученную реакционную смесь перемешивают при комнатной температуре в течение 0,5 ч. После удаления большей части МеОН смесь под- 22018385 кисляют концентрированной водной HCl до осаждения белого твердого вещества. Твердое вещество собирают путем фильтрования и промывают водой (5 мл) с получением необходимого продукта (8,7 г, выход 93%).MS (m/z): 234,0 (М+Н). Следующие соединения получают по существу способом из примера получения 128. Пример получения 142. 6-Метил-2-оксо-1-фенил-1,2-дигидропиридин-3-карбоновая кислота. К раствору этоксида натрия в EtOH (полученному в результате растворения натрия (0,5 г,21,75 ммоль) в EtOH (40 мл добавляют этил-3-оксо-3-(фениламино)пропаноат (4 г, 19,30 ммоль) и транс-4-метокси-3-бутен-2-он (2 г, 19,98 ммоль). После перемешивания реакционной смеси в колбе с обратным холодильником в течение ночи смесь концентрируют. Остаток разделяют между Н 2 О (50 мл) иEtOAc (50 мл). Водную фазу подкисляют концентрированной HCl до рН, равного 34, и затем экстрагируют EtOAc. Органические экстракты сушат и концентрируют. Остаток очищают путем колоночной хроматографии на силикагеле, элюируя ДХМ:МеОН (25:1), с получением продукта (1,20 г, выход 27%).MS (m/z): 230,0 (М+Н). Пример получения 143. 1,5-Диметил-3-оксо-2-фенил-2,3-дигидро-1H-пиразол-4-карбоновая кислота. К смеси 1,5-диметил-3-оксо-2-фенил-2,3-дигидро-1H-пиразол-4-карбальдегида (2 г, 9,25 ммоль) в воде (100 мл) при 75 С медленно добавляют раствор KMnO4 (1,5 г, 9,49 ммоль) в воде (200 мл). После завершения добавления реакционную смесь перемешивают при 75 С в течение еще 1 ч. Добавляют твердый KOH, в результате чего раствор становится щелочным, и смесь фильтруют, пока она горячая. К фильтрату добавляют EtOH (10 мл) и EtOAc (50 мл). Органическую фазу отделяют и отбрасывают. Водную фазу подкисляют концентрированной HCl до рН, равного 5, и экстрагируют EtOAc (60 мл) и ДХМMS (m/z): 233,1 (М+Н). Пример получения 144. Этил-3-(4-фторфениламино)-3-оксопропаноат. В 4-горлую 10-л круглодонную колбу добавляют ДХМ (5000 мл), 4-фторанилин (111 г, 1,0 моль) и ТЭА (166 мл, 1,2 моль). По каплям добавляют этилмалонилхлорид (196 г, 1,3 моль) в ДХМ (500 мл) в атмосфере N2 при 0-5 С (на бане с ледяной водой) в течение 4 ч. После добавления смесь перемешивают в течение 30 мин при данной температуре. К смеси добавляют воду (2 л) при перемешивании при данной температуре. Органическую фазу промывают насыщенным NaHCO3, насыщенным водным хлоридом натрия и сушат над безводным Na2SO4. Водную фазу экстрагируют EtOAc (21 л). Органическую фазу промывают насыщенным водным хлоридом натрия и сушат над безводным Na2SO4. После фильтрации смешанный органический раствор концентрируют досуха при пониженном давлении. Остаток промывают ПЭ (1 л) с получением этил-3-(4-фторфениламино)-3-оксопропаноата (230 г, выход неочищенного вещества 102%) в виде желтого твердого вещества.MS (m/z): 226,1 (М+Н). Пример получения 145. 1-(4-Фторфенил)-6-метил-2-оксо-1,2-дигидропиридин-3-карбоновая кислота. В 4-горлую 3-л круглодонную колбу добавляют EtOH (1500 мл), этил-3-(4-фторфениламино)-3 оксопропаноат (139 г, 617 ммоль), этоксид натрия (65,6 г, 925 ммоль) и 4-метокси-3-бутен-2-он (103 г,925 ммоль). Смесь нагревают в колбе с обратным холодильником в течение 3 ч. После охлаждения раствора до комнатной температуры реакционную смесь концентрируют при пониженном давлении с удалением EtOH. Остаток разбавляют ДХМ (700 мл) и 1 М HCl (1500 мл). Водную фазу экстрагируют ДХМ(3500 мл). Смешанные органические экстракты промывают насыщенным водным хлоридом натрия(1000 мл), сушат над безводным Na2SO4 и концентрируют при пониженном давлении. Остаток растирают с EtOAc (150 мл) при комнатной температуре (10-15 С) в течение 1 ч. После фильтрации и промывкиEtOAc (50 мл) получают 1-(4-фторфенил)-6-метил-2-оксо-1,2-дигидропиридин-3-карбоновую кислоту в виде желтого твердого вещества (85,3 г, выход 56%). В 100-мл круглодонную колбу добавили трет-бутил 4-(5-(4-амино-2-фторфенокси)-1-метил-1Hиндазол-6-ил)-1H-пиразол-1-карбоксилат (1,43 г, 3,38 ммоль), 1-(4-фторфенил)-6-метил-2-оксо-1,2 дигидропиридин-3-карбоновую кислоту (1,25 г, 5,07 ммоль), EDCI (1,48 г, 7,6 ммоль) и HOBt (776 мг,- 24018385 5,07 ммоль) с последующим добавлением ДМФ (15 мл, 193,99 ммоль), а затем DIPEA (1,47 мл,8,44 ммоль). Смесь оставили перемешиваться при комнатной температуре в течение ночи. Реакционную смесь разбавили EtOAc (300 мл) и промыли насыщенным водным хлоридом натрия (5100 мл). Смешанный водный раствор экстрагировали EtOAc (1100 мл) и далее смешанные органические растворы сушили над безводным Na2SO4, фильтровали и концентрировали досуха. Твердое вещество очищали на колонке с силикагелем, элюируя ДХМ (А) и 10% МеОН в растворе ДХМ (В), градиент 100% (А) 80% (А):20% (В) в течение 70 мин, с получением трет-бутил 4-(5-(2-фтор-4-(1-(4-фторфенил)-6-метил-2 оксо-1,2-дигидропиридин-3-карбоксамидо)фенокси)-1-метил-1H-индазол-6-ил)-1H-пиразол-1 карбоксилата в виде твердого вещества золотистого цвета (2,20 г, выход 87%).MS (m/z): 653 (М+Н), 675 (M+Na). В круглодонную колбу добавили трет-бутил 4-(5-(2-фтор-4-(1-(4-фторфенил)-6-метил-2-оксо-1,2 дигидропиридин-3-карбоксамидо)фенокси)-1-метил-1 Н-индазол-6-ил)-1H-пиразол-1-карбоксилат (1,92 г,2,94 ммоль) и ДХМ (50 мл) с последующим добавлением триэтилсилана (1,88 мл, 11,77 ммоль) и ТФУ(17,8 мл, 235,35 ммоль). Реакционную смесь оставили перемешиваться при комнатной температуре в течение 1,5 ч. Растворитель удалили и разбавили ДХМ (150 мл), и промыли насыщенным водным раствором NaHCO3 (2100 мл). Органический раствор сушили Na2SO4 и концентрировали при пониженном давлении с получением твердого вещества. Твердое вещество очищали на колонке с силикагелем, элюируя ДХМ (А) и 10% МеОН в растворе ДХМ (В), градиент 100% (А) - 75% (А):25% (В) в течение 70 мин,поддерживали в данном соотношении 75:25 в течение 15 мин с получением соединения, указанного в названии, в виде почти белого твердого вещества. Твердое вещество растворили в горячем EtOH (50 мл) с последующим добавлением дистиллированной воды (250 мл) порциями, что вызвало осаждение белого твердого вещества. Твердое вещество фильтровали через воронку Бюхнера и промыли дистиллированной водой (315 мл), сушили на воздухе и под вакуумом при 60 С в течение 15 ч с получением соединения, указанного в названии, в виде почти белого твердого вещества (1,27 г, выход 78%). 12-л круглодонную колбу оснастили мешалкой, устанавливаемой в колбу сверху, термопарой и устройством для продувки N2. трет-Бутил 4-(5-(4-амино-2-фторфенокси)-1-метил-1H-индазол-6-ил)-1Hпиразол-1-карбоксилат (404 г, 954,08 ммоль) растворили в ДМФ (2 л) и поместили в указанную колбу. ДМФ (1 л) использовали для ополаскивания колбы. Добавили 1-(4-фторфенил)-6-метил-2-оксо-1,2 дигидропиридин-3-карбоновую кислоту (259,46 г, 1,05 моль) и EDCI (228,63 г, 1,19 моль) и промыли ДМФ (500 мл). Затем добавили HOBt (189,94 г, 1,24 моль) и снова промыли ДМФ (500 мл). Наконец,медленно добавили DIPEA (184,97 г, 1,43 моль). Затем раствор темного цвета перемешивали при комнатной температуре в течение выходных. В 20-л колбу с нижним выходным отверстием добавили DI воду (3 л) и ДХМ (5 л). Влили в нее реакционную смесь и промывали ДХМ (1 л). Органическую фазу отделили, промыли DI водой (33 л), сушили над Na2SO4, фильтровали, промывали твердые вещества ДХМ и концентрировали фильтрат. К остатку добавили EtOAc (2 л) и раствор перемешивали в течение 1 ч. Продукт выкристаллизовался. Смесь концентрировали. Добавили другую порцию EtOAc (2 л) и концентрируют с удалением всего ДХМ. К остатку добавляют EtOAc (650 мл) и МТБЭ (3 л) и раствор перемешивали на ледяной бане в течение 1 ч. Желто-коричневую суспензию фильтровали с использованием полипропиленовой прокладки. Осадок промыли МТБЭ (2500 мл). Светло-желто-коричневое твердое вещество сушили в течение ночи в вакуумной печи при 40 С с получением неочищенного продукта (553 г). Неочищенный продут очищали путем колоночной хроматографии на силикагеле, элюируя смесью (50%EtOAc (50%):35% ДХМ (35%):н-гептан (15%, с получением чистого необходимого продукта, третбутил 4-(5-(2-фтор-4-(1-(4-фторфенил)-6-метил-2-оксо-1,2-дигидропиридин-3-карбоксамидо)фенокси)-1 метил-1H-индазол-6-ил)-1H-пиразол-1-карбоксилата (424 г, 68%).MS (m/z): 651,0 (М-Н). трет-Бутил-4-(5-(2-фтор-4-(1-(4-фторфенил)-6-метил-2-оксо-1,2-дигидропиридин-3 карбоксамидо)фенокси)-1-метил-1H-индазол-6-ил)-1H-пиразол-1-карбоксилат (423,9 г, 649,50 ммоль) растворили в ДХМ (4,24 л). Добавили HCl в МеОН (5,74 Н, 799,99 мл, 4,59 моль) и раствор нагревали при 30 С в течение 1 ч. Затем реакционную смесь нагрели до 45 С и добавили ДХМ (1,5 л). Через 2 ч раствор нагрели до 50 С и добавили ДХМ (2). Через 3 ч добавили ДХМ (2 л) с последующим добавлением HCl в МеОН (4,5 Н, 721,67 мл, 3,25 моль). Через еще 45 мин добавили ДХМ (1 л), HCl в МеОН (4,5 Н,288,67 мл, 1,30 моль) и МеОН (1,5 л). Затем реакционный раствор нагрели до 60 С. Через 4 ч добавили МеОН (2 л) и 10 мин спустя добавили ДХМ (1 л) с последующим добавлением HCl в МеОН (4,5 Н,200 мл). Через 5 ч реакцию завершили. Реакционную смесь концентрировали до примерно 1/3 объема. Добавили МеОН (2 л) и раствор концентрировали до получения густой суспензии. Снова добавили МеОН (2 л) и смесь концентрировали до получения густой суспензии. Суспензию охладили до примерно 1015 С и затем фильтровали. Твердые вещества промыли МеОН. Твердые вещества помещали в вакуумную печь с температурой 55 С на 2 дня с получением необходимого продукта, гидрохлорида N-(3-фтор 4-(1-метил-6-(1H-пиразол-4-ил)-1H-индазол-5-илокси)фенил)-1-(4-фторфенил)-6-метил-2-оксо-1,2 дигидропиридин-3-карбоксамида (377 г, 92,8%).MS (m/z): 551,0 (М-Н). В 22-л круглодонную колбу, оснащенную механической мешалкой, в атмосфере азота добавили гидрохлорид N-(3-фтор-4-(1-метил-6-(1H-пиразол-4-ил)-1H-индазол-5-илокси)фенил)-1-(4-фторфенил)-6 метил-2-оксо-1,2-дигидропиридин-3-карбоксамида (367 г, 0,62 моль) с последующим добавлением ДХМ(7,34 л) и воды (7,34 л). Добавили Na2CO3 (181,6 г, 1,71 моль) и смесь перемешивали при комнатной температуре в течение 30 мин. Проверили значение рН и обнаружили, что оно составляет около 9,4. Смесь фильтровали через полипропилен. Твердые вещества собирали и помещали в 5-л круглодонную колбу. Добавили 20% раствор вода/МеОН (2,6 л) и суспензию перемешивали в течение 30 мин. Суспензию фильтровали и твердые вещества промыли 20% раствором вода/МеОН (600 мл). Твердые вещества помещали в вакуумную печь при 35 С на ночь. Первое определение массы показало 394 г (теоретический выход 324,8 г, выход по массе примерно 121%). Данные ТГА (термогравиметрический анализ)/ДСК(дифференциальная сканирующая калориметрия) показали, что примерно 17 мас.%, составляла свободная вода и 10-11 мас.%, составляли потери летучих веществ при плавке. Твердые вещества сушили при 55 С в вакуумной печи с продуванием N2 в течение 3,5 ч (354,7 г, выход по массе примерно 109%, данные ЯМР демонстрировали примерно 9,3 мас.% ДХМ). Согласно ТГА/ДСК свободная вода отсутствовала. Вещество отправляли на измельчение. Вихревую мельницу (Aljet 0101) в перчаточном боксе собрали во встроенной вытяжке и подсоединили к N2 к 100-фунтовой насадке. Входной наконечник толкателя настроили на максимальную подачу и ввели максимальный поток азота в мельницу. Фиксировали следующие показатели давления: 90 фунт/кв.дюйм на наконечнике толкателя и 85 фунт/кв.дюйм на обоих размалывающих наконечниках. Исходное вещество (353,4 г) медленно подавали во входное отверстие мельницы, останавливаясь, чтобы освободить мешок приемника по мере необходимости. Общая продолжительность размола составила 22 мин и 25 с. Рассчитанная скорость подачи составила 15,8 г/мин (353,4 г, разделенные на 22,42 мин). Измельченное вещество (335,7 г, 95%) получили с 17,7-граммовой потерей. Результат анализа размера частиц измельченного вещества покзал d90 4,6 мкм. Данные ТГА/ДСК показывали, что примерно 11,4 мас.%, составляли летучие вещества при плавке и данные ЯМР (ДМСО) демонстрировали присутствие примерно 9,3 мас.% ДХМ. 1 Н ЯМР (ДМСО)12,94 (ушир.с, 1 Н), 11,88 (с, 1 Н), 8,44 (д, J=7,47 Гц, 1 Н), 8,12 (ушир.с, 1 Н), 8,00LC/MS: (М + Н) 553,1. Получение безводной кристаллической формы. К 10 мл EtOH добавили 120 мг вышеуказанного соединения в 20 мл пробирку. Образец нагрели до 70 С при перемешивании. Сначала твердые вещества начинали растворяться, а затем образовалась суспензия с последующим белым осадком. Образец охладили до комнатной температуры при перемешивании. Небольшой образец суспензии отобрали пипеткой и оставили сушиться на воздухе. Данное вещество было высококристаллическим и по данным ТГА оказалось этанольным сольватом. К оставшейся суспензии добавили 10 мл гептана и затем нагрели до кипения. Измеренную температуру наблюдали при 70,8 С до уменьшения объема до 10 мл. Когда температура начала повышаться, нагревание прекратили и суспензию перемешивали при комнатной температуре в течение ночи. Твердое вещество выделили путем фильтрования под вакуумом и сушили в вакуумной печи при 45 С в течение 3 ч, что привело к 77% выходу. Кристаллическая форма теряла в массе 0,17%, при нагревании от 25 до 238 С при ТГА. Начало плавления данной формы составило 247,8 С.N-метилморфолин (0,5 мл). После перемешивания реакционной смеси при 60 С в течение ночи смесь разделили между EtOAc (50 мл) и насыщенным водным NH4Cl (30 мл). Органическую фазу отделили,сушили и концентрировали. Остаток растерли с EtOAc (5 мл) и твердые вещества собрали путем фильтрования с получением N-(3-фтор-4-(1-метил-6-(1H-пиразол-4-ил)-1H-индазол-5-илокси)фенил)-1-(4 фторфенил)-4,6-диметил-2-оксо-1,2-дигидропиридин-3-карбоксамида в качестве необходимого продукта(10 мл) добавили MeSO3H (39 мг, 0,406 ммоль). После перемешивания реакционной смеси при комнатной температуре в течение 0,5 ч смесь концентрировали. Остаток промыли эфиром и сушили с получением метансульфоната N-(3-фтор-4-(1-метил-6-(1H-пиразол-4-ил)-1H-индазол-5-илокси)фенил)-1-(4 фторфенил)-4,6-диметил-2-оксо-1,2-дигидропиридин-3-карбоксамида (250 мг, выход 92,9%).MS (m/z): 567,2 (M+H). Следующие соединения получают по существу по способу примера 3 за исключением того, что в некоторых случаях получают свободное основание, а не соль.
МПК / Метки
МПК: C07D 403/12, C07D 401/12, C07D 405/14, A61P 35/00, A61K 31/4412
Метки: амидофеноксииндазолы, c-met, качестве, ингибиторов
Код ссылки
<a href="https://eas.patents.su/30-18385-amidofenoksiindazoly-v-kachestve-ingibitorov-c-met.html" rel="bookmark" title="База патентов Евразийского Союза">Амидофеноксииндазолы в качестве ингибиторов c-met</a>
Гетероариламины в качестве ингибиторов гликогенсинтаза-киназы 3-бета (ингибиторов gsk3)

Номер патента: 7298
Опубликовано: 25.08.2006
Авторы: Эмбрехтс Вернер Констант Йохан, Ван Акен Кун Жанн Альфонс, Дильс Гастон Станислас Марселла, Жанссен Поль Адриан Ян, Лав Кристофер Джон, Койманс Люсьен Мария Хенрикус, Виллемс Марк, Винкерс Хендрик Мартен, Леви Паулус Йоаннес, Хэрес Ян, Фрейн Эдди Жан Эдгар, Бейнстерс Петер Якобус Йоханнес Антониус, Де Жонж Марк Рене
МПК: A61K 31/50, A61K 31/435, A61K 31/505...
Метки: 3-бета, гетероариламины, гликогенсинтаза-киназы, качестве, gsk3, ингибиторов
Формула / Реферат:
1. Соединение формулы его N-оксид, фармацевтически приемлемая аддитивная соль, четвертичный амин и стереохимически изомерная форма, где R1 обозначает водород; X обозначает -O-; -О-С1-4алкил- или прямую связь; Z обозначает прямую связь, -NH-, -С1-4алкил-NH- или -С(=O)-; R2 обозначает водород или фенил; R3 обозначает водород, С1-4алкил, полигалогенC1-6алкил или циано; R4 обозначает моноциклический или бициклический насыщенный гетероцикл;...
Сульфонамидные производные в качестве ингибиторов рассасывания костной ткани и ингибиторов адгезии клеток,способ их получения, применение и фармацевтическая композиция

Номер патента: 3102
Опубликовано: 26.12.2002
Авторы: Вилл Дэвид Вильям, Шойнеманн Карлхайнц, Катбертсон Роберт Эндрю, Карниато Дени, Гурвест Жан-Франсуа, Макдауэлл Роберт, Пейман Ануширван, Гадек Томас, Бодари Сара Кэтрин, Кнолле Йохен
МПК: A61P 19/10, A61K 31/505, C07D 239/42...
Метки: производные, получения, ингибиторов, фармацевтическая, ткани, рассасывания, сульфонамидные, применение, адгезии, качестве, костной, клеток,способ, композиция
Формула / Реферат:
1. Сульфонамидные производные общей формулы I где R1 и R2 вместе образуют двухвалентный (С2-С3)алкиленовый радикал; R4 является Н или (С1-С6)алкилом; R5 является (С1-С10)алкилом, (С6-С14)арилом, (C5-C14)гетероарилом или (С6-С14)арил(С1-С10)алкильной группой, где арил, гетероарил или алкил возможно замещены R3, или 2-оксобицикло[2.2.1]гепт-1-илметильной группой; R3 является (C1-C4)алкилом, (C1-C4)алкилокси, галогеном, трифторметилом, циано,...
Гетероариламины-производные пиримидина и пиридазина в качестве ингибиторов гликогенсинтаза-киназы 3-бета (ингибиторов gskз)

Номер патента: 10859
Опубликовано: 30.12.2008
Авторы: Бейнстерс Петер Якобус Йоханнес Антониус, Дильс Гастон Станислас Марселла, Койманс Люсьен Мария Хенрикус, Жанссен Поль Адриан Ян, Леви Паулус Йоаннес, Эмбрехтс Вернер Констант Йохан, Лав Кристофер Джон, Хэрес Ян, Ван Акен Кун Жанн Альфонс, Винкерс Хендрик Мартен, Де Жонж Марк Рене, Фрейн Эдди Жан Эдгар, Виллемс Марк
МПК: A61K 31/505, A61K 31/50, A61K 31/435...
Метки: ингибиторов, пиридазина, гликогенсинтаза-киназы, gskз, гетероариламины-производные, пиримидина, 3-бета, качестве
Формула / Реферат:
1. Соединение формулы его N-оксид, фармацевтически приемлемая аддитивная соль, четвертичный амин и стереохимически изомерная форма, где кольцо А является R1 обозначает водород; X обозначает -O-; -O-C1-6алкил- или прямую связь; Z обозначает прямую связь, -С(=O)- или -NR1-C1-6алкил-; R обозначает водород или R20; R3 обозначает водород; галоген; циано; полигалогенС1-6алкил; R4 обозначает моноциклический, насыщенный или частично насыщенный или...
Триамид-замещённые индолы, бензофураны и бензотиофены в качестве ингибиторов микросомального белка, переносящего триглицериды, (мтр) и/или ингибиторов секреции аполипопротеина в (аро в)

Номер патента: 7008
Опубликовано: 30.06.2006
Авторы: Ченг Хенгмиао, Бронк Брайан Скотт, Блайз Алан Элвуд, Мейсон Клайв Филип, Хуатан Хип, Бертинато Питер, Ли Дзин
МПК: A61K 31/4439, A61K 31/404, A61K 31/343...
Метки: триамид-замещённые, переносящего, секреции, мтр, бензофураны, ингибиторов, бензотиофены, индолы, аро, аполипопротеина, белка, качестве, микросомального, триглицериды
Формула / Реферат:
1. Соединение формулы 1b или его фармацевтически приемлемая соль, где R1 представляет собой заместитель в 5 или 6 положении формулы 1b и имеет структуру или, когда R7 представляет собой фенил, пиридил, фенил-Z1-или пиридил-Z1-, необязательно замещенные от одного до пяти, независимо, выбранными R12, то R1 представляет собой (C1-С6)алкил, (С3-С8)циклоалкил, (С5-С10)бициклоалкил, -(CRaRb)tO(C1-C6 алкил), -(СRaRb)tS(C1-C6 алкил), -(CRaRb)rC(O)R15,...
Арил и гетероарил мочевины в качестве ингибиторов снк1 для использования в качестве радиосенсибилизаторов и химиосенсибилизаторов

Номер патента: 14954
Опубликовано: 29.04.2011
Авторы: Кук Адам Уэйд, Кесицки Эдвард А., Годино Джон Джозеф, Кауэн Скотт Даглас, Киган Кэтлин С., Бэрджесс Лоренс Эдуард
МПК: A61K 31/4965, A61P 35/00, A61K 31/498...
Метки: качестве, снк1, использования, ингибиторов, мочевины, химиосенсибилизаторов, арил, гетероарил, радиосенсибилизаторов
Формула / Реферат:
1. Способ ингибирования киназы 1 точек контроля в клетке, включающий стадию контакта клетки с терапевтически эффективным количеством соединения формулыгде X1представляет -O-, -S-, -СН2- или -N(R1)-;X2 представляет -O-, -S- или -N(R1)-;Y представляет О или S;W представляет Z представляет где Z необязательно замещен 1-4 заместителями, представленными R2, a W необязательно замещен 1-3 заместителями, представленными R5;R1 выбран из группы,...
Предыдущий патент: Композиция дигидрата 3-(2,2,2-триметилгидразиниум) пропионата для наружного местного применения
Следующий патент: Пуриновые соединения
Случайный патент: Композиция, содержащая жидкий жирный компонент.