2,4-диаминопиримидины как ингибиторы aurora
Номер патента: 16358
Опубликовано: 30.04.2012
Авторы: Мантоулидис Андреас, Тонч-Грунт Ульрике, Энгельхардт Харальд, Цан Штефан Карл, Трой Маттиас, Райзер Ульрих, Штадтмюллер Хайнц, Гюртлер Ульрих, Крэмер Оливер, Брюкнер Ральф, Бёмельт Гуидо, Золка Флавио, Шоп Андреас, Херфурт Ларс, Райтер Шарлотте




















Формула / Реферат
1. Соединения общей формулы (1)

в которой
R1 обозначает остаток, выбранный из группы, включающей циклопропил, циклобутил, циклопентил, циклогексил и циклогептил, замещенный группой -NHC(O)(С1-С4алкил) или -C(O)NRcRd, где Rc означает H или С1-С4алкил, Rd означает H, С1-С4алкил, галоген(С1-С4алкил), (C1-C4)алкокси(С1-С4алкил), гидрокси(C1-С4алкил), ди(С1-С4алкил)амино(С1-С4алкил), фенил, циклопропил, циклопентил, 1,1-диоксотетрагидротиенил, или NRcRd образует азетидинил, пирролидинил, пиперидинил;
R2 обозначает фенил, замещенный группой -C(O)NRaRb, -SO2NRaRb или -NRaRb, где Ra обозначает H, C1-С4алкил, Rb обозначает C0-С4алкил, или -NRaRb образует 5-7-членный гетероциклил, включающий 1-2 атома азота, необязательно замещенные 5-7-членным гетероциклилом, включающим 1-2 гетероатома, выбранные из O и N, фенилом или С1-С4алкилом,
R3 обозначает остаток, выбранный из группы, включающей хлор, бром, -CN, -NO2, циклопропилэтинил, метэтинил, фенилэтинил, -CF3, циклопропил;
или их фармацевтически активные соли.
2. Соединения по п.1, в которых R3 обозначает -CF3.
3. Соединения общей формулы (1A)

в которой
все Rc независимо друг от друга выбраны из группы, включающей водород, С1-С5алкил, необязательно замещенный с помощью -ORf, циклопропил, фенил, где Rf означает водород или метил;
R3 обозначает остаток, выбранный из группы, включающей хлор, бром, -CN, -NO2, циклопропилэтинил, метэтинил, фенилэтинил, -CF3, циклопропил;
R4 обозначает остаток, выбранный из группы, включающей -C(O)NRaRb, -SO2NRaRb или -NRaRb, где Ra обозначает H, C1-С4алкил, Rb обозначает C0-С4алкил, или -NRaRb образует 5-7-членный гетероциклил, включающий 1-2 атома азота, необязательно замещенные 5-7-членным гетероциклилом, включающим 1-2 гетероатома, выбранные из O и N, фенилом или C1-С4алкилом.
4. Соединения по п.3, в которых R3 обозначает -CF3.
5. Применение соединений или их фармацевтически активных солей по пп.1-4 для приготовления фармацевтической композиции.
6. Фармацевтические препараты, содержащие в качестве активного вещества одно или большее количество соединений общей формулы (1) или (1A) по одному из пп.1-4 или их физиологически приемлемые соли необязательно совместно с обычными инертными наполнителями и/или носителями.
7. Применение соединений общей формулы (1) или (1A) по пп.1-4 для приготовления фармацевтической композиции, предназначенной для лечения и/или предупреждения рака, инфекций, воспалительных и аутоиммунных заболеваний.
8. Фармацевтический препарат, включающий соединение общей формулы (1) или (1A) по пп.1-4 и по меньшей мере одно другое цитостатическое или цитотоксическое активное вещество, не описываемое формулой (1) или (1A), необязательно в виде их таутомеров, рацематов, энантиомеров, диастереоизомеров и смесей и необязательно их фармакологически приемлемых солей присоединения.
Текст
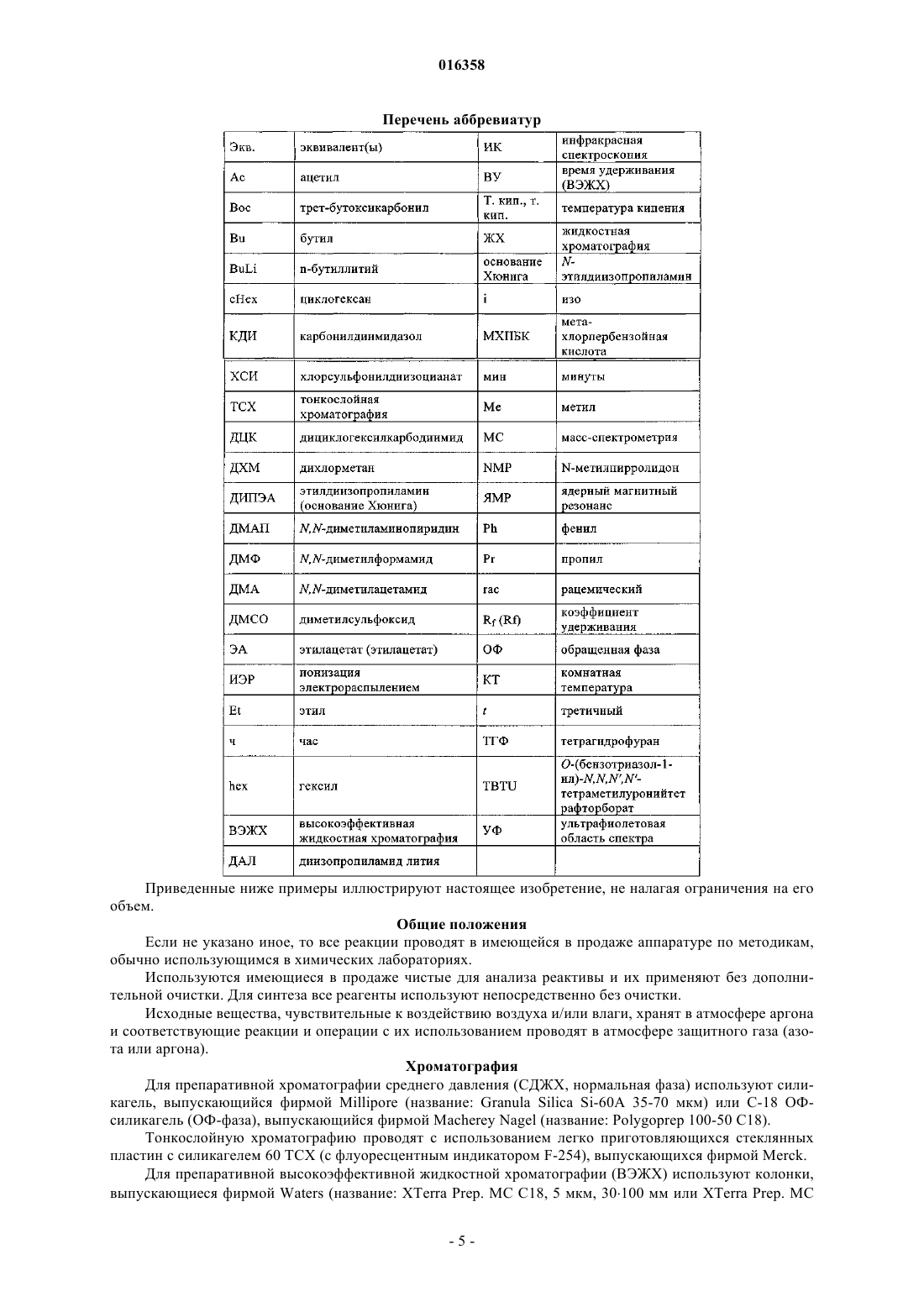
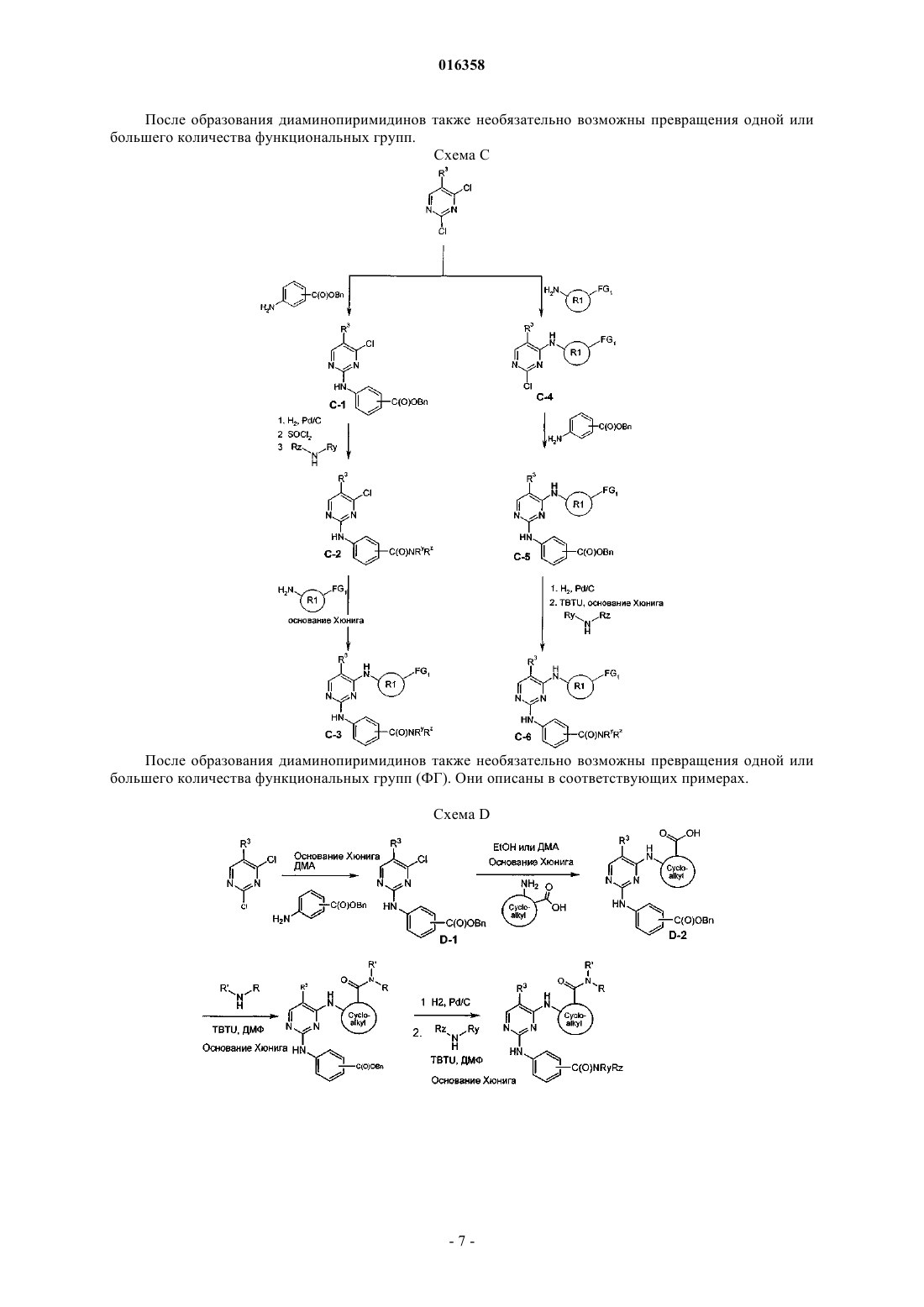
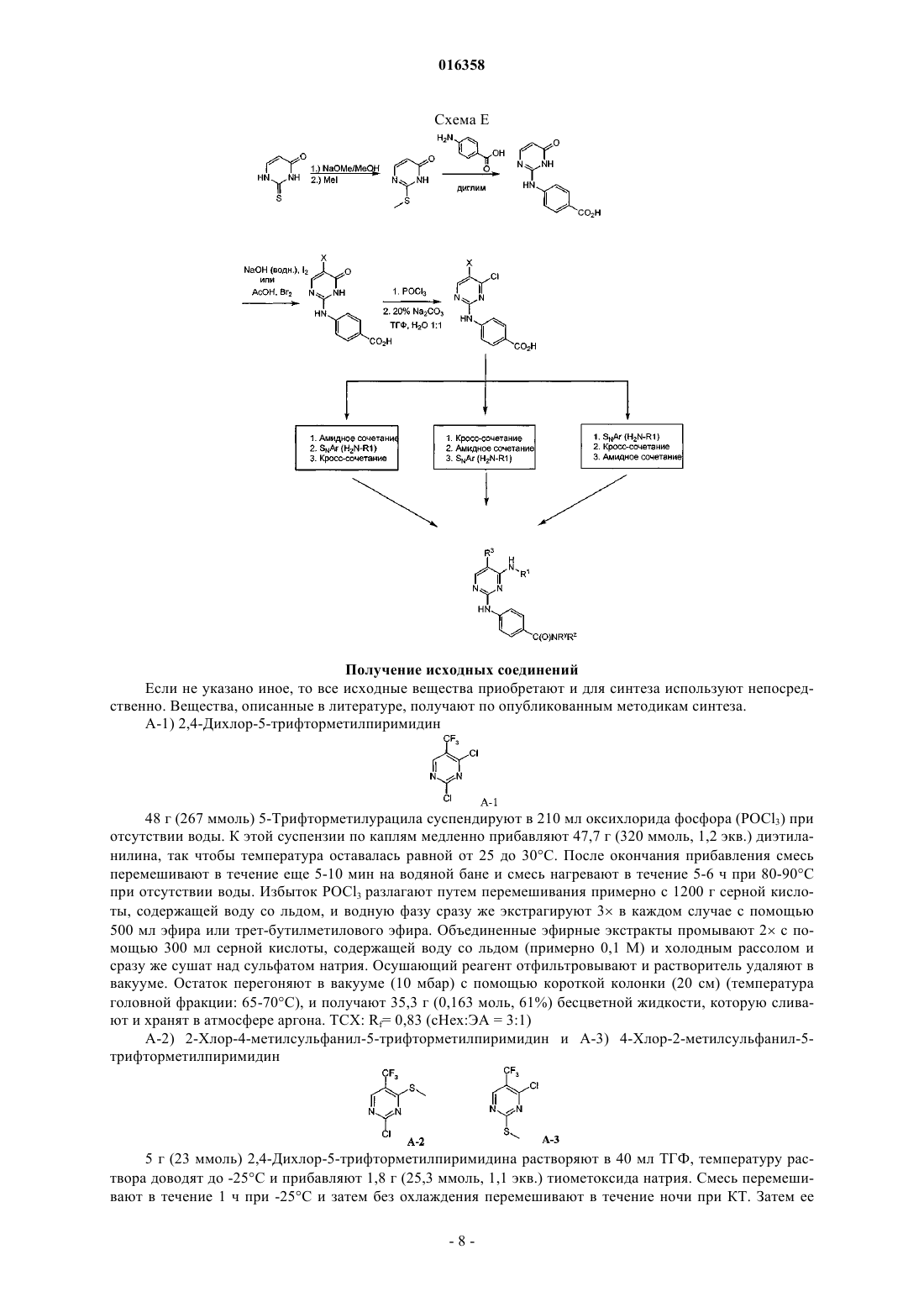
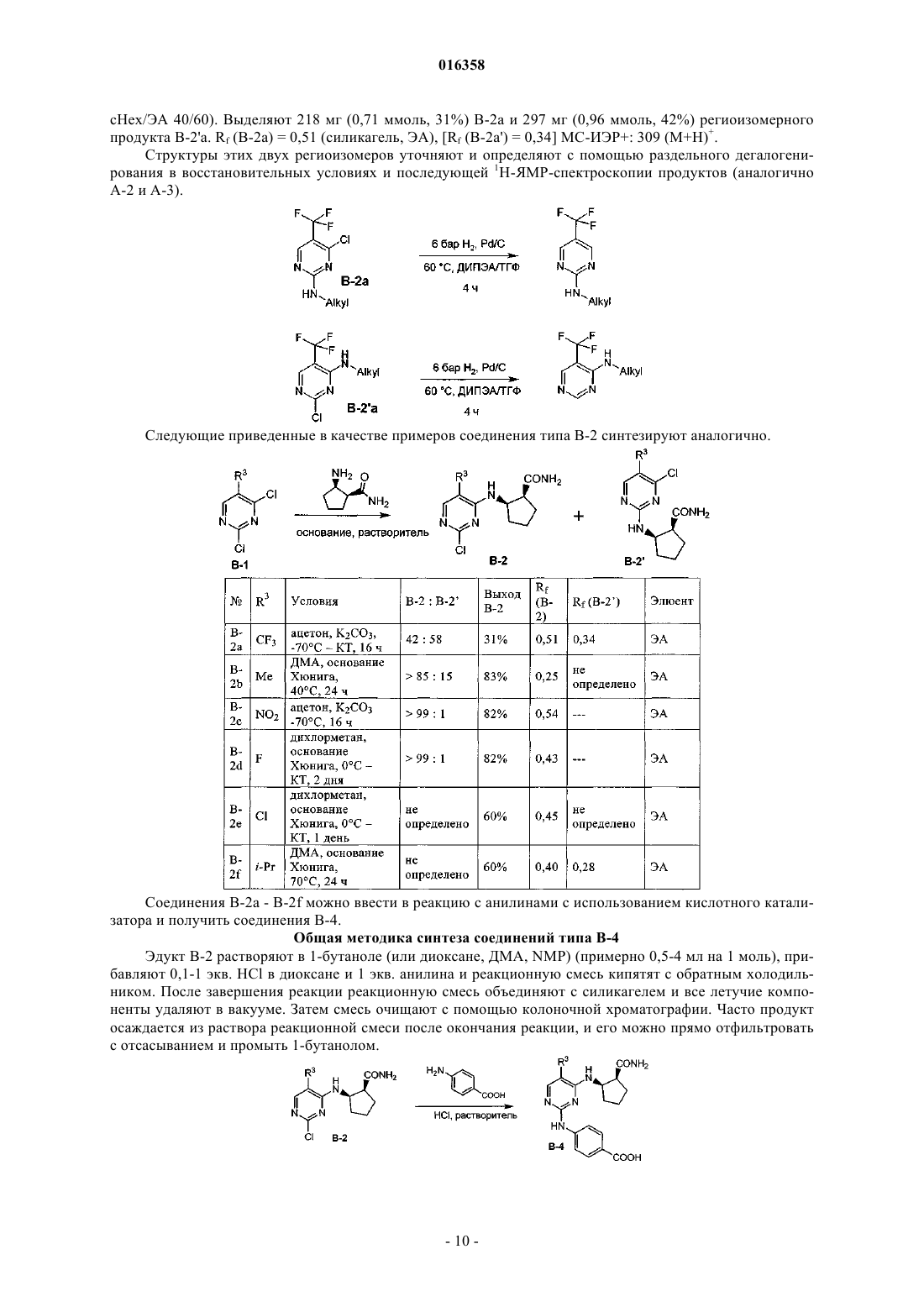
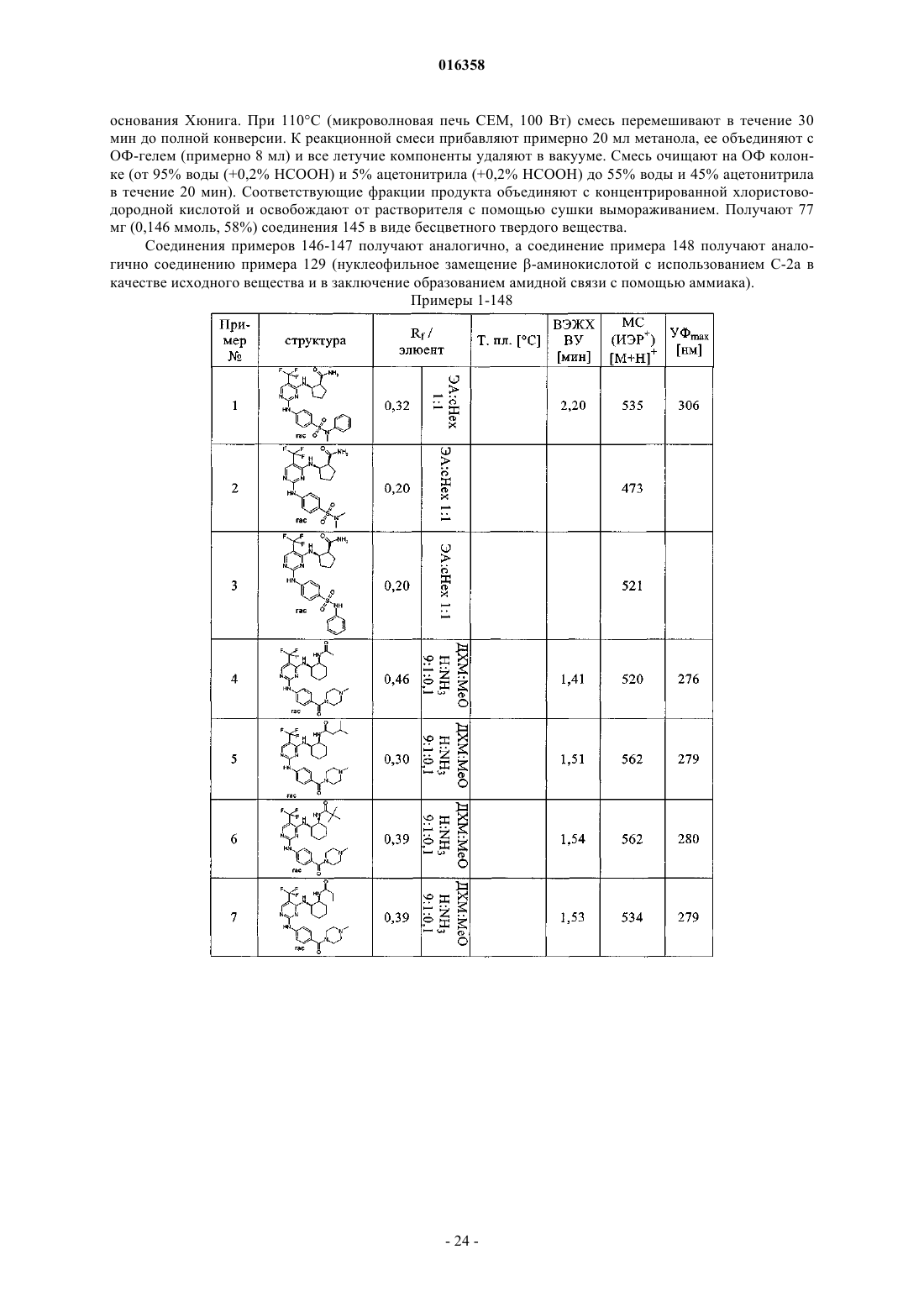
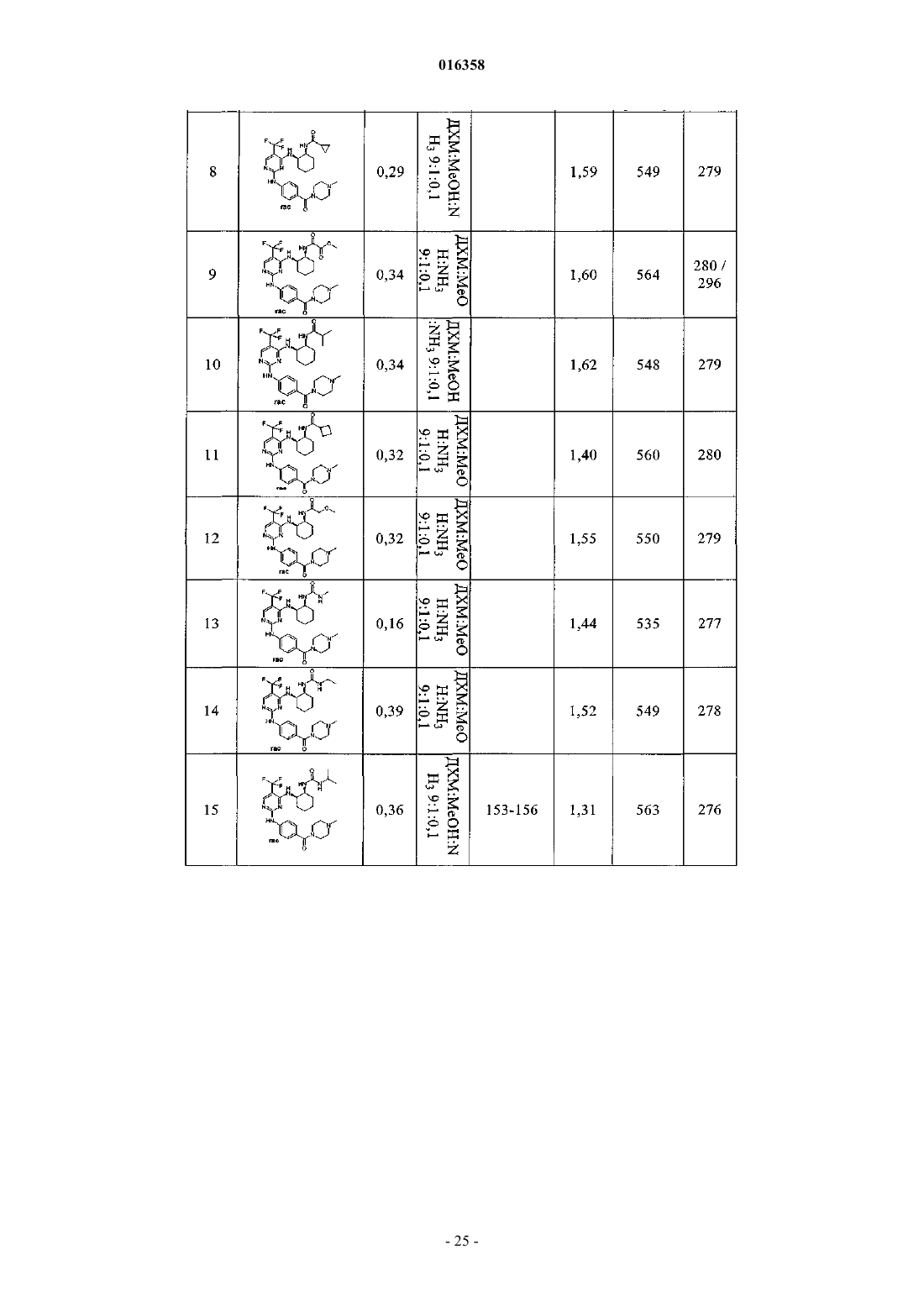
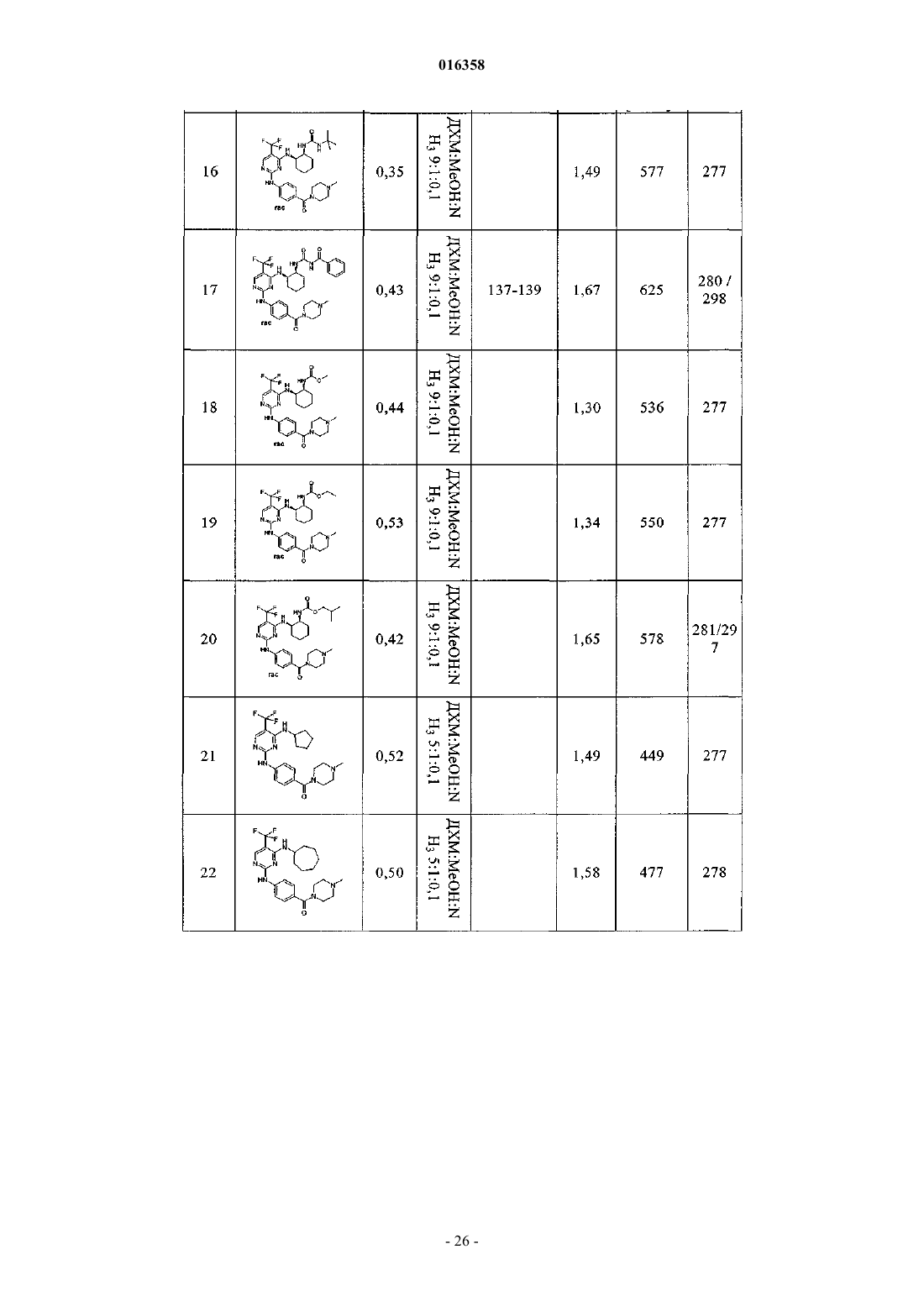
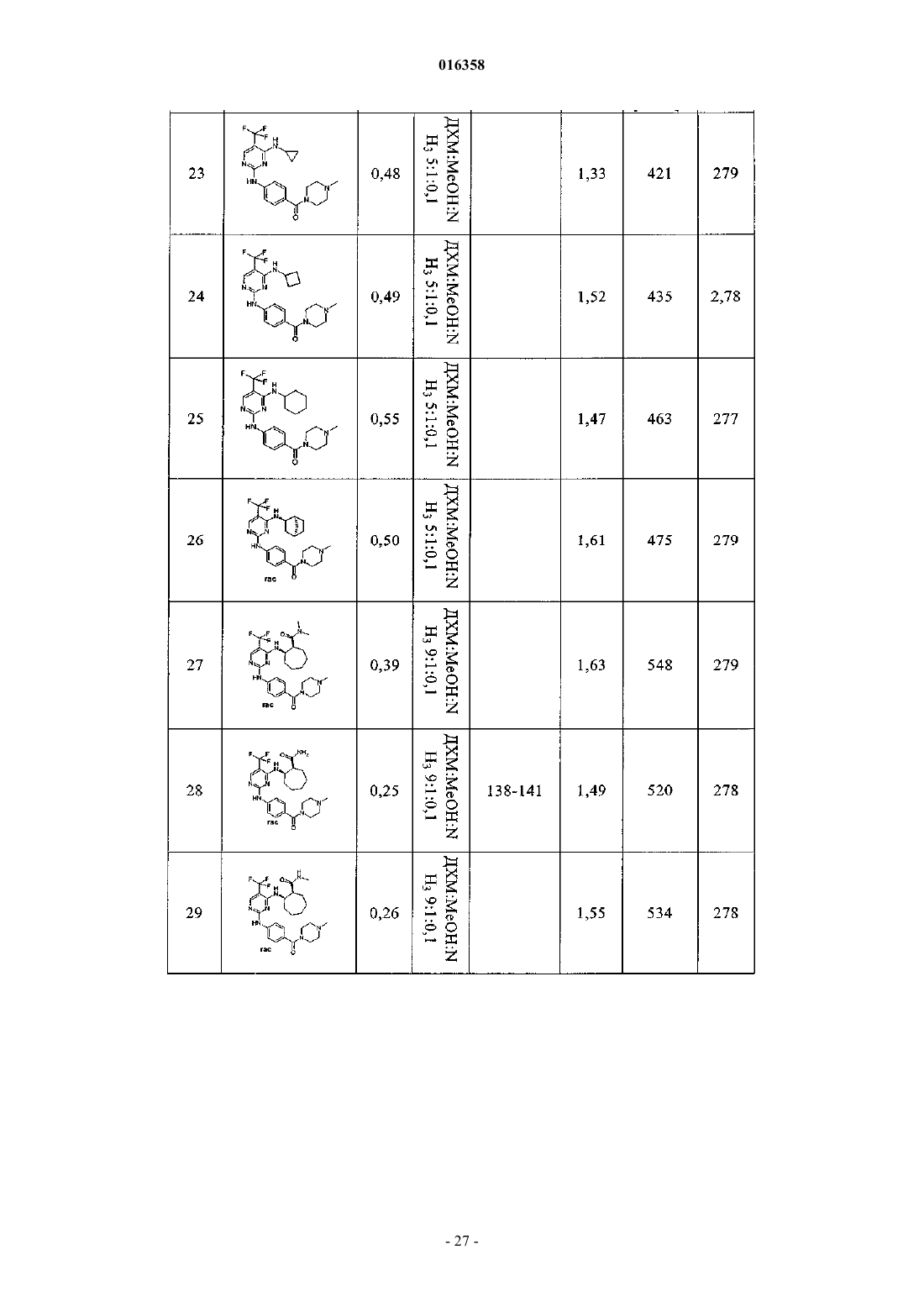
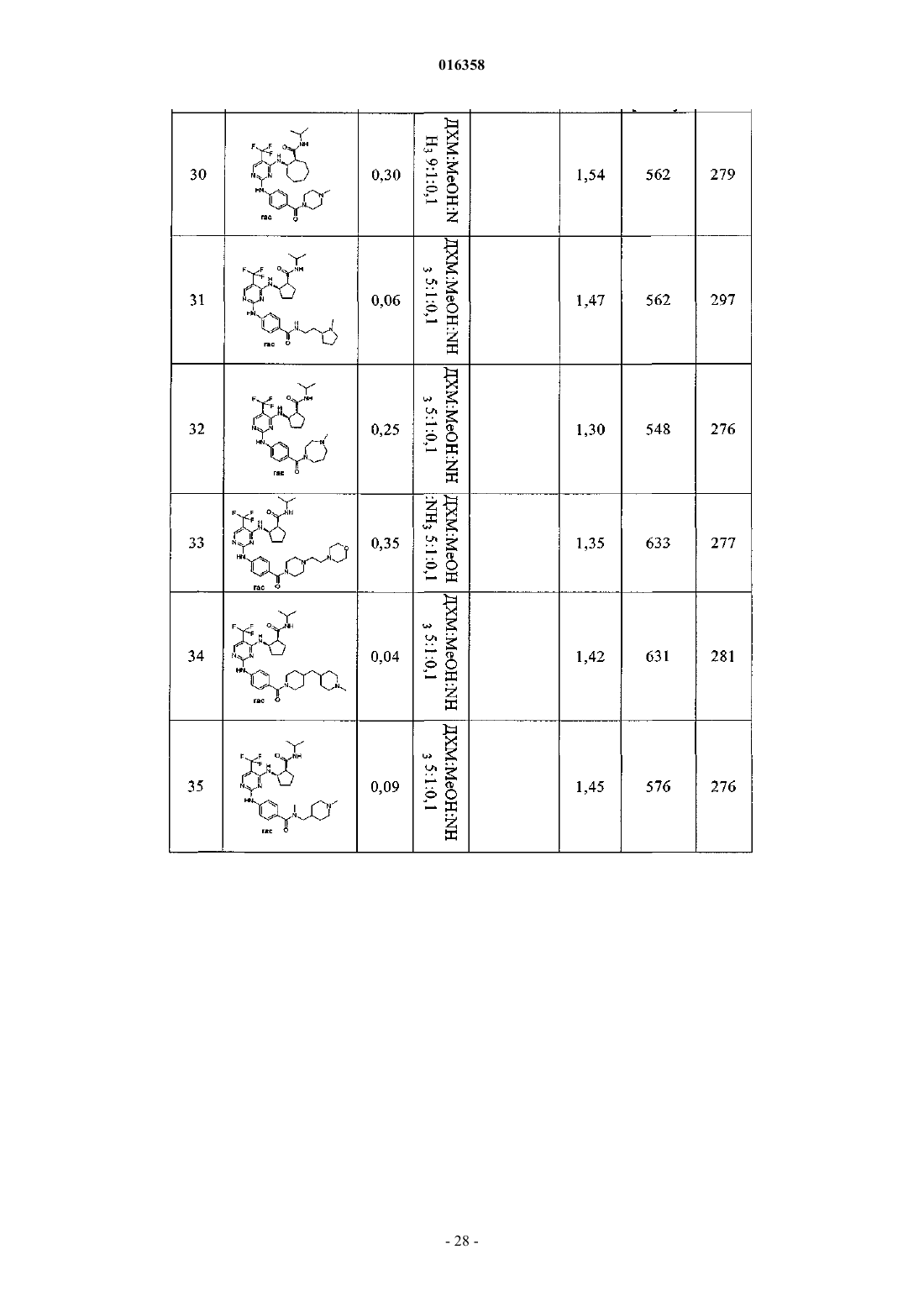
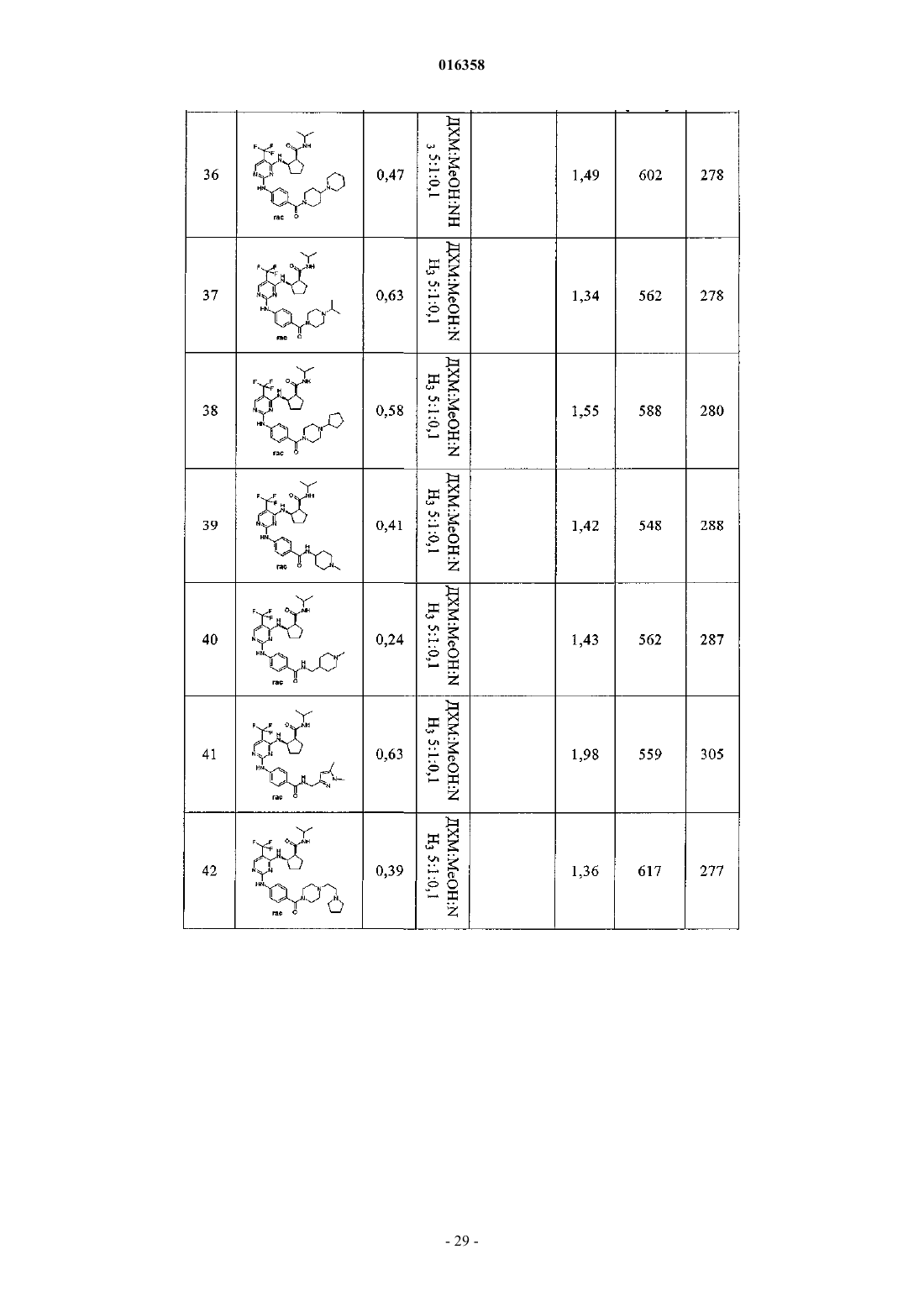
В патенте описаны соединения общей формулы (1) в которой R1-R3 являются такими, как определено в п.1, которые применимы для лечения заболеваний, характеризующихся чрезмерной или аномальной пролиферацией клеток, и их применение для приготовления фармацевтической композиции, обладающей указанными выше характеристиками.(71)(73) Заявитель и патентовладелец: БРИНГЕР ИНГЕЛЬХАЙМ ИНТЕРНАЦИОНАЛЬ ГМБХ (DE) в которой группы R1-R3 обладают значениями, указанными в формуле изобретения и описании, к их изомерам, к способам получения этих пиримидинов и к их применению в качестве фармацевтических композиций. Уровень техники Опухолевые клетки полностью или частично не подвергаются регулированию и управлению организмом и характеризуются неконтролируемым ростом. Это обусловлено, с одной стороны, потерей регуляторных белков, таких как, например, Rb, p16, p21 и p53, а с другой стороны, активацией так называемых ускорителей клеточного цикла, циклинзависимых киназ. Исследования на модельных микроорганизмах, таких как Schizosaccharomyces pombe, Drosophilamelanogaster или Xenopus, а также исследования на клетках человека показали, что переход от фазы G2 к митозу регулируется CDK1/циклин B киназой (Nurse, 1990). Эта киназа, которая также известна как"фактор стимулирования митоза" (ФСМ), фосфорилирует и регулирует множество белков, таких как,например, ядерные пластинки, кинезиноподобные двигательные белки, конденсины и матриксные белки Гольджи, которые играют важную роль в разрушении оболочки ядра, в разделении центросом, структуре аппарата митотического веретена, конденсации хромосом и разрушении аппарата Гольджи (Nigg, 2001). Воздействие на опухолевые клетки человека ингибиторами CDK1/циклина B, такими как, например, бутиролактон, приводит к остановке фазы G2/M и последующему апоптозу (Nishio, et al. 1996). В дополнение к циклинзависимым киназам так называемые polo-подобные серин/треонинкиназы(PLK-1, PLK-2, PLK-3 и PLK-4) играют важную роль в регуляции клеточного цикла эукариотов. В частности, обнаружено, что PLK-1 играет главную роль в регуляции фазы митоза. PLK-1 обеспечивает созревание центросом, активацию фосфатазы Cdc25C, а также активацию стимулирующего анафазу комплекса (Glover et al., 1998, Qian et al., 2001). Инъекция антител PLK-1 приводит к остановке G2 у неизмененных клеток, а для опухолевых клеток происходит остановка во время фазы митоза (Lane and Nigg, 1996). Кроме того, остановку на фазе G2/M также может быть инициирована ингибированием специальных двигательных белков, так называемых кинезинов, таких как, например, Eg5 (Mayer et al., 1999), или агентами, стабилизирующими или дестабилизирующими микротрубочки (например, колхицином, таксолом, этопозидом, винбластином, винкристином) (Schiff and Horwitz, 1980). Киназы Ser/Thr группы Aurora регулируют различные процессы деления клеток. К ним относятся конденсация хромосом, динамика веретена, взаимодействия кинетохор-микротрубочка, ориентирование хромосом, выравнивание метафазной пластинки и цитокинез (Meraldi et al., 2004; Carmena and Earnshaw,2003; Andrews et al., 2003). Для млекопитающих описаны три представителя этой группы - Aurora A, B иC. Киназы Aurora типа А и В также содержатся в Caenorhabditis elegans и Drosophila melanogaster, тогда как дрожжи содержат только один ген Aurora, известный под названием IPL1 (в S cerevisiae), или ARK1(в S pombe). Все белки Aurora обладают сходной общей структурой, которая включает различные Nконцы, хорошо сохраняющуюся центральную киназную область и короткий C-концевой участок. Несмотря на сходство последовательностей киназы группы Aurora характеризуются разным расположением внутри клеток, что обусловлено специализированными функциями. Таким образом, Aurora A должна в интерфазе обнаруживаться в центросомах, а во время митоза - и в центросомах, и в микротрубочке веретена вблизи от полюсов. В соответствии с этим - как подтверждено с помощью экспериментов по интерференции РНК - Aurora A важна для вхождения в фазу митоза,поскольку созревание и отделение центросом не может происходить при отсутствии Aurora А. Имеются различные активаторы Aurora А, такие как, например, TPX2, Ajuba и ингибитор протеинфосфатазы-2.TPX2, видимо, обеспечивает надлежащую по времени и пространству активацию Aurora A на микротрубочках веретена вблизи полюса (Hirota et al., 2003; Bayliss et al., 2003; Eyers and Mailer, 2004; Kufer et al.,2002; Satinover et al., 2004).Aurora B в начале профазы связывается с конденсирующимися хромосомами, в метафазе располагается на центросомах, затем перемещается в центральную зону центрального веретена и в заключение во время цитокинеза накапливается в так называемом флеминговом, или центральном тельце, в узкой области между дочерними клетками. Эти характерные пространственные изменения во время митоза оправдывают введенное для Aurora B название: белок-"пассажир хромосомы". Известны по меньшей мере три других белка-"пассажира хромосомы", которые образуют комплекс с Aurora B. Ими являются INCENP (внутренний центромерный белок), сурвивин и бореалин (Andrews et al., 2003; Carmena and Earnshaw, 2003; Meraldi et al., 2004). Важный центр взаимодействия между Aurora B и этим комплексом образует C-конец белка INCENP, так называемый "IN-box". "IN-box" является наиболее хорошо сохраняющимся участком INCENP. Он связывается с Aurora B и активирует ее и фосфорилируется этой киназойAurora C является наименее изученным представителем группы Aurora. Aurora C также связывается с INCENP и ведет себя, как белок-"пассажир хромосомы", хотя она характеризуется наибольшими уровнями экспрессирования после Aurora B. Aurora С, видимо, способна выполнять дополнительные по сравнению с Aurora B функции, так, например, обедненные полиядерным фенотипом Aurora B клетки могут быть нормализованы путем экспрессирования Aurora C (Sasai et al., 2004; Li et al., 2004).Aurora B фосфорилирует гистон H3 по остаткам Ser10 и Ser28. Хотя фосфорилирование совпадает с моментом конденсации хромосом, эффект этого явления проявляется только на более поздней стадии клеточного цикла. Это подтверждается тем фактом, что гистон H3 концентрируется в митотических хромосомах с фосфорилированным Ser10 и трижды метилированным Lys9 гетерохроматином вблизи от центромера. Таким образом, модифицированный гистон H3 предотвращает связывание белка 1 гетерохроматина (HP1) и разрешает комплексу белок-"пассажир хромосомы" доступ к центромерным кинетохорным регионам (Hirota T. et al., Manuscript in Preparation). Одна функция Aurora B, которая стала очевидной при ингибировании Aurora B, представляет собой комбинирование разных белков на кинетохоре во время метафазы (Ditchfield et al., 2003; Hauf et al., 2003;Murata-Hori and Wang, 2002; Vigneron et al., 2004). Aurora B играет главную роль в прохождении сигнала,который обнаруживает и корректирует синтелические (дефектные, поскольку они начинаются только с одного полюса веретена) присоединения микротрубочек к кинетохору (Andrews et al., 2003; Carmena andEarnshaw, 2003; Meraldi et al., 2004). Если это состояние присоединения не скорректировано, то происходят ошибки при сегрегации хромосом. Опосредуемое Aurora B фосфорилирование деполимеразы MCAK микротрубочек связанно с этим механизмом коррекции (Gorbsky, 2004).Aurora B также фосфорилирует белки, которые важны для образования репликативной формы и цитогенеза, такие как, например, MgcRacGAP, регулирующую восприятие света цепь миозина II, виментин,десмин, GFAP (глиальный фибриллярный кислый белок), а также кинезины MKLP1 и MKLP2, из которых MKLP2 предположительно обеспечивает завершение переноса комплекса белок-"пассажир хромосомы" от кинетохоров в центральную зону (Gruneberg et al., 2004). Вследствие различных функций Aurora B в клеточном цикле оказалось неожиданным установление того, что ингибирование Aurora B в опухолевых клетках приводит не к остановке митоза, а к продолжению клеточного цикла без цитокинеза (Hauf et al., 2003). Вследствие накопления синтелических присоединений микротрубочка-кинетохор и поэтому ошибочных сегрегации хромосом протекает массовая полиплодия, в результате приводящая к апоптозу. Даже одновременное ингибирование Aurora A не может повлиять на этот фенотип (Keen and Taylor, 2004). Сначала преимущественно приводили указания на онкогенную активность Aurora A (например,превращение мышиных фибробластов после сверхэкспрессирования), тогда как для Aurora B такие указания были только косвенными (Zhou et al., 1998; Bischoff et al., 1998; Katayama et al., 1999). Положение изменилось после обнаружения того, что сверхэкспрессирование Aurora B в эмбриональных клетках хомяка и их применение в экспериментах с ксенотрансплантатами непосредственно увеличивает частоту,размер и инвазивность опухолей. Соответствующие опухоли характеризуются хромосомной нестабильностью и усиленным фосфорилированием гистона H3 по Ser10 (Ota et al., 2002). Эти результаты подчеркивают важность Aurora B в генезе опухоли. Общеизвестно, что пиримидины являются ингибиторами киназ. Так, например, в заявках WO 02/096888 и WO 03/032997 замещенные пиримидины, содержащие неароматическую группу в положении 4, описаны, как активные компоненты, оказывающие противораковое воздействие. Задачей настоящего изобретения является выявление новых активных веществ, которые можно применять для предупреждения и/или лечения заболеваний, характеризующихся чрезмерной или аномальной пролиферацией клеток. Подробное описание изобретения Согласно изобретению неожиданно было установлено, что соединения общей формулы (1), в которой группы R1, R2 и R3 являются такими, как определено ниже в настоящем изобретении, действуют, как ингибиторы специфических киназ клеточного цикла. Таким образом, соединения, предлагаемые в настоящем изобретении, можно применять, например, для лечения заболеваний, связанных с активностью специфических киназ клеточного цикла и характеризующихся чрезмерной или аномальной пролиферацией клеток. Настоящее изобретение относится к соединениям общей формулы (1)H, С 1-С 4 алкил, Rb обозначает С 0-С 4 алкил, или -NRaRb образует 5-7-членный гетероциклил, включающий 1-2 атома азота, необязательно замещенные 5-7-членным гетероциклилом, включающим 1-2 гетероатома,выбранные из O и N, фенилом или С 1-С 4 алкилом,R3 обозначает остаток, выбранный из группы, включающей хлор, бром, -CN, -NO2, циклопропилэтинил, метэтинил, фенилэтинил, -CF3, циклопропил; или их фармацевтически активные соли. В другом варианте осуществления настоящее изобретение относится к соединениям общей формулы (1), в которой R3 обозначает -CF3. В другом варианте осуществления настоящее изобретение относится к соединениям общей формулы (1A), в которой все Rc независимо друг от друга выбраны из группы, включающей водород, С 1-С 5 алкил, необязательно замещенный с помощью -ORf, циклопропил, фенил, где Rf означает водород или метил;R обозначает H, С 1-С 4 алкил, Rb обозначает С 0-С 4 алкил, или -NRaRb образует 5-7-членный гетероциклил,включающий 1-2 атома азота, необязательно замещенные 5-7-членным гетероциклилом, включающим 12 гетероатома, выбранные из O и N, фенилом или С 1-С 4 алкилом. В другом варианте осуществления настоящее изобретение относится к соединениям общей формулы (1A), в которой R3 обозначает CF3. В другом варианте осуществления настоящее изобретение относится к применению соединений или их фармацевтически активных солей по пп.1-4 для приготовления фармацевтической композиции. В другом варианте осуществления настоящее изобретение относится к фармацевтическим препаратам, содержащим в качестве активного вещества одно или большее количество соединений общей формулы (1) или (1A) по одному из пп.1-4 или их физиологически приемлемые соли необязательно совместно с обычными инертными наполнителями и/или носителями. В другом варианте осуществления настоящее изобретение относится к применению соединений общей формулы (1) или (1A) по пп.1-4 для приготовления фармацевтической композиции, предназначенной для лечения и/или предупреждения рака, инфекций, воспалительных и аутоиммунных заболеваний. В другом варианте осуществления настоящее изобретение относится к фармацевтическому препарату, включающему соединение общей формулы (1) или (1A) по пп.1-4 и по меньшей мере одно другое цитостатическое или цитотоксическое активное вещество, не описываемое формулой (1) или (1A), необязательно в виде их таутомеров, рацематов, энантиомеров, диастереоизомеров и смесей и необязательно их фармакологически приемлемых солей присоединения. Определения При использовании в настоящем изобретении используются следующие определения, если не указано иное. Алкильные заместители в каждом случае означают насыщенные, ненасыщенные, обладающие линейной или разветвленной цепью углеводородные группы (алкильные группы), и это определение включает и насыщенные алкильные группы, и ненасыщенные алкенильные и алкинильные группы. Алкенильные в каждом случае означают обладающие линейной или разветвленной цепью ненасыщенные алкильные группы, которые содержат по меньшей мере одну двойную связь. Алкинильные в каждом случае означают обладающие линейной или разветвленной цепью ненасыщенные алкильные группы, которые содержат по меньшей мере одну тройную связь. Гетероалкил означает обладающие линейной или разветвленной цепью алифатические углеводородные группы, которые содержат от 1 до 3 гетероатомов, причем каждый доступный атом углерода или гетероатом гетероалкильной цепи необязательно может быть независимо друг от друга замещен и гетероатомы независимо друг от друга выбраны из группы, включающей O, N, P, PO, PO2, S, SO и SO2 (на-3 016358 пример, диметиламинометил, диметиламиноэтил, диметиламинопропил, диэтиламинометил, диэтиламиноэтил, диэтиламинопропил, 2-диизопропиламиноэтил, бис-2-метоксиэтиламиногруппа, [2-(диметиламиноэтил)этиламино]метил, 3-[2-(диметиламиноэтил)этиламино]пропил, гидроксиметил, 2-гидроксиэтил,3-гидрокмипропил, метоксигруппа, этоксигруппа, пропоксигруппа, метоксиметил, 2-меткосиэтил). Галогеналкил означает алкильные группы, в которых один или большее количество атомов водород замещены на атомы галогенов. Галогеналкил включает и насыщенные алкильные группы, и ненасыщенные алкенильные и алкинильные группы, такие как, например, -CF3, -CHF2, -CH2F, -CF2CF3, -CHFCF3,-CH2CF3, -CF2CH3, -CHFCH3, -CF2CF2CF3, -CF2CH2CH3, -CF=CF2, -CCl=СН 2, -CBr=CH2, -CJ=CH2, -CCF3,-CHFCH2CH3 и -CHFCH2CF3. Галоген означает атомы фтора, хлора, брома и/или йода. Циклоалкил означает моно- или полициклические кольца, в которых кольцевая система может представлять собой насыщенное кольцо, но может представлять собой и ненасыщенное, ароматическое кольцо или спиросоединение, которое также может необязательно содержать двойные связи, такие как,например, циклопропил, циклопропенил, циклобутил, циклобутенил, циклопентенил, циклогексил, циклогексенил, циклогептанил, циклогептенил, норборнил, норборненил, инданил, адамантил, спирогептанил и спиро[4.2]гептанил. Циклоалкилалкил включает нециклическую алкильную группу, в которой атом водорода, связанный с атомом углерода, заменен циклоалкильной группой. Арил означает моноциклические или бициклические кольца, содержащие 6-12 атомов углерода, такие как, например, фенил и нафтил. Арилалкил включает нециклическую алкильную группу, в которой атом водорода, связанный с атомом углерода, заменен арильной группой. Гетероарил означает моно- или полициклические кольца, которые вместо одного или большего количества атомов углерода содержат один или большее количество гетероатомов, которые могут быть одинаковыми или разными, такие как, например, атомы азота, серы или кислорода. Примеры включают фурил, тиенил, пирролил, оксазолил, тиазолил, изоксазолил, изотиазолил, пиразолил, имидазолил, триазолил, тетразолил, оксадиазолил, тиадиазолил, пиридил, пиримидил, пиридазинил, пиразинил триазинил. Примерами бициклических гетероарильных групп являются индолил, изоиндолил, бензофуранил, бензотиенил, бензоксазолил, бензотиазолил, бензизоксазолил, бензизотиазолил, бензимидазолил, индазолил,изохинолинил, хинолинил, хиноксалинил, циннолинил, фталазинил, хиназолинил и бензотриазинил, индолизинил, оксазолопиридинил, имидазопиридинил, нафтиридинил, индолинил, изохроманил, хроманил,тетрагидроизохинолинил, изоиндолинил, изобензотетрагидрофуранил, изобензотетрагидротиенил, изобензотиенил, бензоксазолил, пиридопиридинил, бензотетрагидрофуранил, бензотетрагидротиенил, пуринил, бензодиоксолил, триазинил, феноксазинил, фенотиазинил, претидинил, бензотиазолил, имидазопиридинил, имидазотиазолил, дигидробензизоксазолил, бензизоксазинил, дигидробензизотиазинил, бензопиранил, бензотиопиранил, кумаринил, изокумаринил, хромонил, хроманонил, пиридинил-N-оксид, тетрагидрохинолинил, дигидрохинолинил, дигидрохинолинонил, дигидроизохинолинонил, дигидрокумаринил, дигидроизокумаринил, изоиндолинил, бензодиоксанил, бензоксазолинил, пирролил-N-оксид, пиримидинил-N-оксид, пиридазинил-N-оксид, пиразинил-N-оксид, хинолинил-N-оксид, индолил-N-оксид,индолинил-N-оксид, изохинолинил-N-оксид, хиназолинил-N-оксид, хиноксалинил-N-оксид, фталазинилN-оксид, имидазолил-N-оксид, изоксазолил-N-оксид, оксазолил-N-оксид, тиазолил-N-оксид, индолизинил-N-оксид, индазолил-N-оксид, бензотиазолил-N-оксид, бензимидазолил-N-оксид, пирролил-N-оксид,оксадиазолил-N-оксид, тиадиазолил-N-оксид, триазолил-N-оксид, тетразолил-N-оксид, бензотиопиранил-S-оксид и бензотиопиранил-S,S-диоксид. Гетероарилалкил включает нециклическую алкильную группу, в которой атом водорода, связанный с атомом углерода, заменен гетероарильной группой. Гетероциклил означает насыщенные или ненасыщенные неароматические моно-, бициклические или мостиковые полициклические кольца или спиросоединения, содержащие 3-12 атомов углерода, которые вместо одного или большего количества атомов углерода содержат гетероатомы, такие как атомы азота, кислорода или серы. Примерами таких гетероциклильных групп являются тетрагидрофуранил,пирролидинил, пирролинил, имидазолидинил, имидазолинил, пиразолидинил, пиразолинил, пиперидинил, пиперазинил, индолинил, изоиндолинил, морфолинил, тиоморфолинил, гомоморфолинил, гомопиперидинил, гомопиперазинил, гомотиоморфолинил, тиоморфолинил-S-оксид, тиоморфолинил-S,Sдиоксид, тетрагидропиранил, тетрагидротиенил, гомотиоморфолияил-S,S-диоксид, оксазолидинонил,дигидропиразолил, дигидропирролил, дигидропиразинил, дигидропиридинил, дигидропиримидинил,дигидрофурил, дигидропиранил, тетрагидротиенил-S-оксид, тетрагидротиенил-S,S-диоксид, гомотиоморфолинил-S-оксид, 2-окса-5-азабицикло[2.2.1]гептан, 8-окса-3-азабицикло[3.2.1]октан, 3,8-диазабицикло[3.2.1]октан, 2,5-диазабицикло[2.2.1]гептан, 3,8-диазабицикло[3.2.1]октан, 3,9-диазабицикло[4.2.1]нонан и 2,6-диазабицикло[3.2.2]нонан. Гетероциклоалкилалкил означает нециклическую алкильную группу, в которой том водорода, связанный с атомом углерода, заменен гетероциклоалкильной группой. Приведенные ниже примеры иллюстрируют настоящее изобретение, не налагая ограничения на его объем. Общие положения Если не указано иное, то все реакции проводят в имеющейся в продаже аппаратуре по методикам,обычно использующимся в химических лабораториях. Используются имеющиеся в продаже чистые для анализа реактивы и их применяют без дополнительной очистки. Для синтеза все реагенты используют непосредственно без очистки. Исходные вещества, чувствительные к воздействию воздуха и/или влаги, хранят в атмосфере аргона и соответствующие реакции и операции с их использованием проводят в атмосфере защитного газа (азота или аргона). Хроматография Для препаративной хроматографии среднего давления (СДЖХ, нормальная фаза) используют силикагель, выпускающийся фирмой Millipore (название: Granula Silica Si-60A 35-70 мкм) или C-18 ОФсиликагель (ОФ-фаза), выпускающийся фирмой Macherey Nagel (название: Polygoprep 100-50 C18). Тонкослойную хроматографию проводят с использованием легко приготовляющихся стеклянных пластин с силикагелем 60 TCX (с флуоресцентным индикатором F-254), выпускающихся фирмой Merck. Для препаративной высокоэффективной жидкостной хроматографии (ВЭЖХ) используют колонки,выпускающиеся фирмой Waters (название: XTerra Prep. MC C18, 5 мкм, 30100 мм или XTerra Prep. MCZorbax SB-C8, 5 мкм, 21,250 мм). Для хиральной высокоэффективной жидкостной хроматографии (ВЭЖХ) используют колонки, выпускающиеся фирмой Daicel Chemical Industries, Ltd. (название: Chiralpak AD-H или Chiralpak AS, илиChiracel OD-RH, или Chiracel OD-H, или Chiracel OJ-H различных размеров с насадкой, обладающей частицами размером 5 мкм). Спектроскопия ядерного магнитного резонанса (ЯМР). Спектры ядерного магнитного резонанса снимают в дейтерированном диметилсульфоксиде-d6 в качестве растворителя. В случае использования других растворителей они явно указаны в примерах или методиках. Химические сдвиги указаны относительно стандарта, тетраметилсилана (= 0,00 ч./млн). Исследования проводят с помощью приборов Avance 400 (400 МГЦ ЯМР-спектрометр) или Avance 500 (500 МГЦ ЯМР спектрометр), выпускающихся фирмой Bruker Biospin GmbH. ВЭЖХ-масс-спектроскопия/УФ-спектрометрия. Времена удерживания в МС-ИЭР+ для характеризации соединений в примерах получают с помощью прибора ВЭЖХ-МС (аппарат для высокоэффективной жидкостной хроматографии с детектором по массе), выпускающегося фирмой Agilent. Конструкция прибора такова, что диодный матричный детектор (G1315B, выпускающийся фирмойAgilent) и детектор по массе (1100 LS-MSD SL; G1946D; Agilent) соединены последовательно и установлены после хроматографа (колонка: XTerra MC C18, 2,5 мкм, 2,130 мм, Waters или Synergi POLAR-ОФ 80 А; 4 мкм, Phenomenex). Прибор работает при скорости потока, равной 1,1 мл/мин. Для разделения градиентный режим проводят в течение 3,1 мин (начало градиентного режима: 95% воды и 5% ацетонитрила; конец градиентного режима: 5% воды и 95% ацетонитрила; в каждом случае к этим двум растворителям прибавляют 0,1% муравьиной кислоты). Температуры плавления Температуры плавления определяют с помощью прибора типа B-540, выпускающегося фирмойBchi, и не корректируют. В случаях, когда получение исходных соединений не описано, они имеются в продаже или их можно получить аналогично известным соединениям или по методикам, описанным в настоящем изобретении. Получение соединений, предлагаемых в настоящем изобретении. Соединения, предлагаемые в настоящем изобретении, можно получить по методикам синтеза, описанным ниже в настоящем изобретении, и заместители в общих формулах обладают указанными выше значениями. Эти методики предназначены для иллюстрации настоящего изобретения без ограничения его объекта и объема соединениями, указанными в текстах этих примеров. Схема A-6 016358 После образования диаминопиримидинов также необязательно возможны превращения одной или большего количества функциональных групп. Схема C После образования диаминопиримидинов также необязательно возможны превращения одной или большего количества функциональных групп (ФГ). Они описаны в соответствующих примерах. Схема D Получение исходных соединений Если не указано иное, то все исходные вещества приобретают и для синтеза используют непосредственно. Вещества, описанные в литературе, получают по опубликованным методикам синтеза. 48 г (267 ммоль) 5-Трифторметилурацила суспендируют в 210 мл оксихлорида фосфора (POCl3) при отсутствии воды. К этой суспензии по каплям медленно прибавляют 47,7 г (320 ммоль, 1,2 экв.) диэтиланилина, так чтобы температура оставалась равной от 25 до 30C. После окончания прибавления смесь перемешивают в течение еще 5-10 мин на водяной бане и смесь нагревают в течение 5-6 ч при 80-90C при отсутствии воды. Избыток POCl3 разлагают путем перемешивания примерно с 1200 г серной кислоты, содержащей воду со льдом, и водную фазу сразу же экстрагируют 3 в каждом случае с помощью 500 мл эфира или трет-бутилметилового эфира. Объединенные эфирные экстракты промывают 2 с помощью 300 мл серной кислоты, содержащей воду со льдом (примерно 0,1 М) и холодным рассолом и сразу же сушат над сульфатом натрия. Осушающий реагент отфильтровывают и растворитель удаляют в вакууме. Остаток перегоняют в вакууме (10 мбар) с помощью короткой колонки (20 см) (температура головной фракции: 65-70C), и получают 35,3 г (0,163 моль, 61%) бесцветной жидкости, которую сливают и хранят в атмосфере аргона. TCX: Rf= 0,83 (cHex:ЭА = 3:1) 5 г (23 ммоль) 2,4-Дихлор-5-трифторметилпиримидина растворяют в 40 мл ТГФ, температуру раствора доводят до -25C и прибавляют 1,8 г (25,3 ммоль, 1,1 экв.) тиометоксида натрия. Смесь перемешивают в течение 1 ч при -25C и затем без охлаждения перемешивают в течение ночи при КТ. Затем ее-8 016358 разбавляют дихлорметаном и промывают 3 с помощью 1 н. HCl. Органическую фазу сушат над сульфатом магния и выпаривают в вакууме. Неочищенный продукт очищают с помощью колоночной хроматографии (силикагель, циклогексан/дихлорметан; от 90/10 до 80/20% в течение примерно 20 мин). 1,56 г(6,8 ммоль, 30%) Продукта A-3 и 1,46 г (6,4 ммоль, 28%) продукта A-2 выделяют в виде бесцветных масел. Кроме того, 0,24 г (4%) 2,4-бис-метилсульфанил-5-трифторметилпиримидина можно выделить в виде бесцветного твердого вещества. Анализ структуры проводят путем получения производных и последующего исследования с помощью спектроскопии ЯМР. Для этого A-2 и A-3 сначала по отдельности полностью дегалогенируют в ТГФ при 100C, 5 бар H2, Pd/C и Pd(OH)2 в каждом случае при соотношении 1:1. Вследствие разной симметрии полученных продуктов можно точно определить степень регионизомерной чистоты. 4-Амино-N-метил-N-фенилбензолсульфонамид (эдукт в примере 1) 9,5 мл (85,7 ммоль, 98%) N-Метиланилина растворяют в 100 мл дихлорметана и при 0C по каплям прибавляют 20 г (85,7 ммоль, 95%) 4-нитробензосульфонилхлорида, растворенного в 150 мл дихлорметана, и смесь перемешивают в течение еще 1,5 ч. Органическую фазу промывают насыщенным водным раствором карбоната натрия и сушат над сульфатом натрия. В заключение ее фильтруют через силикагель и сразу же все летучие компоненты удаляют в вакууме и получают 24,6 г неочищенного N-метил-4 нитро-N-фенилбензолсульфонамида. 14,6 г (49,9 ммоль) Амида нитросульфоновой кислоты растворяют в 100 мл ТГФ/MeOH 1/1. После прибавления Pd/C (10%) смесь перемешивают в течение 16 ч при 50C и давлении H2, равном 5 бар. После прибавления молекулярного сита для связывания воды, последующего прибавления Pd/C и дополнительного перемешивании при условиях гидрирования (давление H2 5 бар, 60C) в течение 16 ч получают 13,1 г (48,9 ммоль, 100%) неочищенного A-4 а в виде бежевого твердого вещества. Этот неочищенный продукт используют для синтеза без какой-либо дополнительной очистки. 4-Амино-N-фенилбензолсульфонамид и 4-амино-N,N-диметилбензолсульфонамид получают аналогично (эдукты в примерах 2 и 3). Описанная методика в общем случае применима для получения замещенных или незамещенных амидов аминобензолсульфоновой кислоты из соответствующих хлорангидридов нитробензолсульфоновой кислоты. Общая методика синтеза соединений типа B-2 Соответствующий R3-замещенный 2,4-дихлорпиримидин B-1 (имеется в продаже или его можно получить путем хлорирования соответствующего урацила, как это в качестве примера описано для A-1) растворяют в ТГФ (или диоксане, ДМА, NMP, ацетоне) (примерно 2-5 мл/ммоль), прибавляют 1-1,6 экв. основание Хюнига (или триэтиламина, карбоната калия или другого подходящего основания) и устанавливают необходимую температуру реакционной смеси (-78C для очень реакционноспособных пиримидинов, КТ или повышенную температуру для мало реакционноспособных пиримидинов). Затем прибавляют примерно 0,75-1 экв. амина, растворенного в соответствующем растворителе (см. выше), и реакционную смесь перемешивают в течение заданного количества времени при соответствующей температуре или оттаивают, или нагревают в течение заданного количества времени, зависящего от реакционной способности использующегося пиримидина. После завершения реакции (за протеканием реакции следят с помощью ВЭЖХ или ТСХ) реакционную смесь объединяют с силикагелем и все летучие компоненты удаляют в вакууме. Очистка с помощью колоночной хроматографии дает искомые замещенные продукты. В зависимости от группы R3 пиримидина два возможных региоизомера получают в разных соотношениях. Обычно их можно разделить с помощью хроматографии. 500 мг (2,3 ммоль) A-1 и 636 мг (4,6 ммоль, 2 экв.) карбоната калия суспендируют в 11 мл ацетона,охлаждают до -70C, затем прибавляют цис(1S.2R)-2-аминоциклопентанкарбоксамид. Реакционную смесь оставляют оттаивать в течение ночи при перемешивании при КТ и затем перемешивают в течение еще 24 ч при температуре окружающей среды. Затем прибавляют 40 мл силикагеля и все летучие компоненты удаляют в вакууме. Эти два региоизомерных продукта разделяют с помощью колоночной хроматографии, при которой искомый региоизомер - это продукт, элюирующийся первым (силикагель,-9 016358cHex/ЭА 40/60). Выделяют 218 мг (0,71 ммоль, 31%) B-2a и 297 мг (0,96 ммоль, 42%) региоизомерного продукта В-2'а. Rf (B-2a) = 0,51 (силикагель, ЭА), [Rf (B-2a') = 0,34] MC-ИЭР+: 309 (M+H)+. Структуры этих двух региоизомеров уточняют и определяют с помощью раздельного дегалогенирования в восстановительных условиях и последующей 1H-ЯМР-спектроскопии продуктов (аналогично Следующие приведенные в качестве примеров соединения типа B-2 синтезируют аналогично. Соединения B-2a - B-2f можно ввести в реакцию с анилинами с использованием кислотного катализатора и получить соединения B-4. Общая методика синтеза соединений типа B-4 Эдукт B-2 растворяют в 1-бутаноле (или диоксане, ДМА, NMP) (примерно 0,5-4 мл на 1 моль), прибавляют 0,1-1 экв. HCl в диоксане и 1 экв. анилина и реакционную смесь кипятят с обратным холодильником. После завершения реакции реакционную смесь объединяют с силикагелем и все летучие компоненты удаляют в вакууме. Затем смесь очищают с помощью колоночной хроматографии. Часто продукт осаждается из раствора реакционной смеси после окончания реакции, и его можно прямо отфильтровать с отсасыванием и промыть 1-бутанолом. 1 мл (8,84 ммоль, 1,3 экв.) N-Метилпиперазина растворяют в 40 мл дихлорметана и этот раствор объединяют с 1,5 мл (8,84 ммоль, 1,3 экв.) основание Хюнига. Затем по каплям при охлаждении медленно прибавляют 1-5 г (6,82 моль, 1 экв.) 4-нитро-2-хлорбензоилхлорида, растворенного в 10 мл дихлорметана. Через 2 ч по каплям при перемешивании медленно прибавляют 9 мл насыщенного водного раствора бикарбоната натрия, органическую фазу отделяют и растворитель удаляют в вакууме. Продукт очищают с помощью колоночной хроматографии (силикагель, ДХМ/MeOH/NH3 9/1/01) и 1,83 г (6,45 ммоль,95%) и получают амида нитробензойной кислоты. Последний растворяют в 2 л ТГФ, прибавляют 300 мг никеля Ренея и смесь перемешивают в течение 16 ч при давлении H2, равном 3 бар, и при КТ. Затем никель Ренея отфильтровывают и летучие компоненты удаляют в вакууме и получают 1,2 г (4,73 ммоль,73%) (4-амино-2-хлорфенил)-(4-метилпиперазин-1-ил)-метанона. Rf = 0,38 (силикагель, ДХМ:MeOH:NH3= 9:1:0,1) MC-ИЭР+: 254 (M+H)+. Эта методика также применима для замещенных или незамещенных амидов аминобензойной кислоты, как это сделано, например, при синтезе соединений примеров 71-75. Соединения этих примеров получают аналогично соединению примера 70. При синтезе соединений примеров 106, 107 и 144 используют амиды м-аминобензойной кислоты, которые получают по такой же методике. Изопропиламид цис 2-аминоциклопентанкарбоновой кислоты. 55 мг (0,43 ммоль) цис 2-аминоциклопентанкарбоновой кислоты суспендируют в 900 мкл (25 экв.) изопропиламина и к этой суспензии прибавляют 205 мг (0,064 ммоль, 1,5 экв.) TBTU и 550 мкл ДМФ. Ее перемешивают в течение 16 ч и реакционную смесь растворяют в смеси ДХМ:MeOH:NH3 9:1:0,1 и объединяют с 7 мл силикагеля. После удаления всех летучих компонентов в вакууме смесь хроматографируют (силикагель ДХМ:MeOH:KH3 9:1:0,1). Получают 63 мг (0,37 ммоль, 86%) бесцветного твердого вещества. Rf = 0,33 (силикагель, ДХМ:MeOH:NH3 85:15:1,5).- 11016358 2 г (9,2 ммоль) A-1 и 1,8 мл (11,2 ммоль, 1,2 экв.) основания Хюнига растворяют в 60 мл ТГФ, смесь охлаждают до -78C, затем при -78C по каплям медленно прибавляют изопропиламид цис 2 аминоциклопентанкарбоновой кислоты, растворенный в 60 мл ТГФ. Реакционную смесь при перемешивании выдерживают для оттаивания при КТ в течение ночи. Затем прибавляют 40 мл силикагеля и все летучие компоненты удаляют в вакууме. Эти два региоизомерных продукта разделяют с помощью колоночной хроматографии, при которой искомый региоизомер - это продукт, элюирующийся первым (силикагель, cHex/ЭА от 85/15 до 80/20 в течение 30 мин). Выделяют 590 мг (1,68 ммоль, 24%) B-2g и 690 мг 2 г (12,6 ммоль) 3,4-Дифторнитробензола растворяют в 1,6 мл этанола, прибавляют 2,4 мл (15,1 ммоль, 1,2 экв.) основание Хюнига и затем при охлаждении льдом по каплям прибавляют 1,44 г (12,6 ммоль, 1 экв.) гексагидро-1-метил-1H-1,4-диазепина. Примерно через 12 ч перемешивания при КТ реакция завершается. Затем прибавляют метанол и 50 мл силикагеля, летучие компоненты удаляют в вакууме и смесь очищают с помощью колоночной хроматографии (ДХМ/MeOH от 97/3 до 85/15 в течение 35 мин). Получают 3 г (11,9 ммоль, 94%) нитросоединения.Rf = 0,39 (силикагель, ДХМ:MeOH:NH3 9:1:0,1). MC-ИЭР+: 253 (M+H)+. Нитросоединение растворяют в 600 мл ТГФ и объединяют с примерно 300 мг никеля Ренея. Смесь гидрируют в течение 3 ч при давлении H2, равном 3 бар. Никель Ренея отфильтровывают и раствор освобождают от всех летучих компонентов в вакууме. Получают 2,15 г (9,6 ммоль, 81%) 3-фтор-4-(4-метил[1.4] диазепан-1-ил)-фениламина. Rf = 0,48 (силикагель, ДХМ:MeOH:NH3 4:1:0,1) MC-ИЭР+: 224 (M+H)+ Анилины, которые используют в качестве эдуктов в примерах 142-143 получают аналогично. Бензил-4-аминобензоат. 10,01 г 4-Нитробензойной кислоты суспендируют в 500 мл ацетонитрила и затем объединяют с 15,03 г (108,7 ммоль, 1,2 экв.) карбоната калия. При перемешивании по каплям прибавляют 15,40 г (171,0 ммоль, 1 экв.) бензилбромида и затем реакционную смесь при перемешивании нагревают при 60C в течение 5 ч. Ее объединяют с 750 мл дистиллированной воды, экстрагируют 4 с помощью 250 мл ЭА и затем органические фазы объединяют, сушат над сульфатом натрия. После удаления всех летучих компонентов в вакууме неочищенный продукт последовательно суспендируют 2 в толуоле и все летучие компоненты удаляют в вакууме (удаление избыточного бензилбромида). 20,60 г (80,1 ммоль) Бензил-4 нитробензоат получают в виде бесцветного твердого вещества, которое используют на следующей стадии без дополнительной очистки. 20,6 г Бензил-4-нитробензоата растворяют в 350 мл диоксана и этот раствор объединяют с 6,9 г(49,9 ммоль, 0,61 экв.) никеля Ренея. Смесь гидрируют в течение 16 ч при перемешивании при давленииH2, равном 5 бар. Катализатор отфильтровывают, все летучие компоненты удаляют в вакууме. Получают 17,0 г (74,8 ммоль, 93%) бензил-4-аминобензоата в виде бесцветного твердого вещества. 10 г (44 ммоль) Бензил-4-аминобензоата растворяют в 200 мл ДМА, прибавляют 8 мл основание Хюнига (0,97 экв.) и при КТ к прозрачному раствору по каплям прибавляют 10,4 г (48,21 ммоль) 2,4 дихлор-5-трифторметилпиримидина, растворенного в 50 мл ДМА. Раствор реакционной смеси перемешивают в течение ночи при 60C, затем объединяют с 300 мл дихлорметана и экстрагируют дистиллированной водой (3 300 мл). Органическую фазу сушат над сульфатом натрия и растворитель удаляют в вакууме. Неочищенный продукт объединяют с 100 мл MeOH, кипятят и выдерживают в течение 2 ч. Затем смесь перемешивают в течение 10 мин, осадок отфильтровывают и промывают метанолом (метанольный фильтрат содержит нежелательный региоизомер, полученный посредством нуклеофильного замещения). В заключение неочищенный продукт еще раз суспендируют в метаноле, отфильтровывают,- 12016358 промывают небольшим количеством метанола и сушат при 60C в вакуумном сушильном устройстве. Получают 8,5 г (20,7 ммоль, 43%) C-1a в виде светло-желтого твердого вещества.(20% мас./мас. Pd, 2,14 ммоль, 0,32 экв.) и смесь перемешивают в течение 16 ч при давлении H2, равном 3 бар, и КТ. Реакционную смесь фильтруют через цеолит, растворитель удаляют в вакууме и получают 1,87 г (5,89 ммоль, 88%) 4-(4-хлор-5-трифторметилпиримидин-2-иламино)бензойной кислоты в виде бесцветного твердого вещества, которое используют без дополнительной очистки. 1,1 г (3,46 ммоль) Бензойной кислоты объединяют с 20 мл толуола и 301 мкл (4,16 ммоль, 1,2 экв.) тионилхлорида и кипятят с обратным холодильником в течение 1,5 ч. Все летучие компоненты удаляют в вакууме и неочищенный хлорангидрид бензойной кислоты непосредственно используют для последующей реакции. 536 мг (1,6 ммоль) этого вещества растворяют в 4 мл ТГФ и объединяют с 410 мкл (1,5 экв.) основания Хюнига. После прибавления 179 мкл (1 экв.) N-метилпиперазина раствор перемешивают в течение 16 ч при КТ. Реакционную смесь выливают примерно в 40 мл дистиллированной воды, перемешивают в течение 30 мин и водную фазу экстрагируют 3 с помощью 50 мл этилацетата. После сушки органической фазы над сульфатом магния, фильтрования и удаления летучих компонентов в вакууме получают 645 мг (1,5 ммоль, 94%) C-2a в виде твердого вещества. Rf = 0,69 (силикагель, CH2Cl2:MeOH:NH3 5:1:0,1)[3.3.1]нонана (9-ББН) растворяют в 8 мл ТГФ/NMP 1/1 и перемешивают в течение 45 мин при КТ. К немного мутному раствору прибавляют 2,4 мл (16,2 ммоль, 1 экв.) бензилхлорформиата (Cbz-хлорид). Примерно через 1 ч реакционную смесь объединяют дистиллированной водой и перемешивают в течение нескольких минут. Затем водный раствор объединяют с этилацетатом и водную фазу промывают 3 с помощью примерно 50 мл этилацетата. Весь продукт содержится в водной фазе, а загрязнения - в органической фазе. Водную фазу подщелачивают с помощью NaHCO3 (pH 8), смешивают с дихлорметаном,экстрагируют 3 с помощью 10 мл дихлорметана, объединенные органические фазы сушат над сульфатом магния и растворитель удаляют в вакууме. Получают 2,29 г (9,22 ммоль, 57%) бензил 1S,2R)2-аминоциклогексил)карбамата в виде бесцветной маслянистой жидкости. Rf = 0,45 (силикагель,CH2Cl2:MeOH:NH3 9:1:0,1) MC-ИЭР+: 249 (M+H)+.- 13016358 800 мг (2 ммоль) C-2a растворяют в 1 мл NMP, прибавляют 569 мг (2,4 ммоль, 1,2 экв.) бензил-1S,2R)-2-аминоциклогексил)карбамата и затем 521 мкл (3 ммоль, 1,5 экв.) основания Хюнига. Через 48 ч при 70C реакция останавливается. После удаления растворителя в вакууме неочищенный продукт очищают с помощью колоночной хроматографии (ДХМ/MeOH/NH3 от 19/1/0,1 до 9/1/0,1) и получают 826 мг (1,35 ммоль, 68%) продукта в виде бесцветной смолы. MC-ИЭР+: 612 (M+H)+. 112 мг (0,18 ммоль) C-3a растворяют в ДМФ (10 мл) и объединяют с дистиллированной водой (1 мл). Затем прибавляют еще 9 мл ДМФ, раствор переносят в аппарат для гидрирования и объединяют сPd/C (200 мг, 5% Pd). Раствор реакционной смеси перемешивают в течение 12 ч при давлении H2, равном 4 бар. Реакционную смесь растворяют в дихлорметан и объединяют с 10 мл ОФ-геля и все летучие компоненты удаляют в вакууме. Очистку проводят с помощью колоночной хроматографии (ОФ-фаза, ацетонитрил/вода от 5/95 до 95/5 в течение 20 мин). После объединения фракций продукта и сушки вымораживанием получают 27 мг (0,06 ммоль, 30%) искомого продукта в виде бесцветного твердого вещества. 440 мг (1,1 ммоль) C-2a растворяют в 500 мкл NMP и объединяют с 565 мкл основание Хюнига (3,3 ммоль, 3 экв.) и 256 мг цис-2-аминоциклогептанкарбоновой кислоты (рацемической). Реакционную смесь помещают на масляную баню, температура которой поддерживается равной 100C, и нагревают при этой температуре в течение 8 ч при перемешивании. После окончания реакции реакционную смесь растворяют в метаноле, объединяют с 20 мл ОФ-геля и все летучие компоненты удаляют в вакууме. Очистку проводят с использованием обращенной фазы (элюент: ацетонитрил/вода (от 15/85 до 35/65 в течение 15 мин). После объединения фракций продукта и сушки вымораживанием получают 160 мг (0,31 ммоль, 28%) искомого продукта в виде бесцветного твердого вещества. MC-ИЭР+: 521 (M+H)+. 563 мг (1,13 ммоль) C-2a растворяют в 5 мл 1-бутанола и к нему прибавляют 163 мг цис-2-амино-1 циклопентанкарбоновой кислоты (рацемической). После прибавления 540 мкл основание Хюнига смесь нагревают при 110C в течение примерно 60 мин (микроволновая печь СЕМ, 100 Вт). Реакционную смесь выпаривают в вакууме, перемешивают примерно с 100 мл воды и экстрагируют 3 с помощью 50 мл этилацетата. Объединенные органические фазы сушат над сульфатом магния и растворитель удаляют в вакууме. Получают 530 мг (1,08 ммоль, 96%) C-3d. MC-ИЭР+: 493 (M+H)+. 200 мг C-2b растворяют в 750 мкл ДМА и прибавляют 160 мкл (0,93 ммоль, 2 экв.) основание Хюнига. Затем прибавляют 72 мг (0,56 ммоль, 1,2 экв.) (1A,3R)-3-аминоциклопентанкарбоновой кислоты и реакционную смесь нагревают при 120C в течение 40 мин. Реакционную смесь объединяют с ОФ-гелем,летучие компоненты удаляют в вакууме и продукт очищают с помощью колоночной хроматографии на ОФ-фазе и выделяют (от 85% воды (+0,2% HCOOH) и 15% ацетонитрила (+0,2% HCOOH) до 76% воды и 24% ацетонитрила в течение 20 мин). Соответствующие фракции продукта объединяют, освобождают от растворителя с помощью сушки вымораживанием и получают 150 мг (0,29 ммоль, 62%) C-3f в виде бесцветной пленки. Это соединение получают по литературной методике (Csomos et al., 2002). 2,05 г (5 ммоль) C-1a и 1 г гидрохлорида (1S.2R)-(+)-2-амино-l-циклопентанкарбоновой кислоты (6 ммоль, 1,2 экв.) помещают в 18 мл этанола. 7,3 мл (42,5 ммоль, 3,4 экв.) прибавляют основание Хюнига и смесь перемешивают в течение 4 ч при 70C. Реакционную смесь перемешивают с 275 мл воды, фильтруют для удаления нерастворенного вещества, значение pH доводят до 2 насыщенным водным растворомKHSO4, перемешивают в течение 5 мин и образовавшийся осадок фильтруют с отсасыванием. Неочищенный продукт промывают водой, сушат в вакууме и получают 2,37 г (4,74 ммоль, 94%) D-2a в виде светло-бежевого твердого вещества. MC-ИЭР+: 501 (M+H)+. Синтез с использованием производного (1R,2S)-(-)-2-амино-l-циклопентанкарбоновой кислоты или 22,64 мл (0,26 моль, 0,95 экв.) ХСИ при -75C в атмосфере аргона по каплям прибавляют к 23 мл(0,273 моль, 1 экв.) циклопентена. Во время прибавления реакционной смеси температуру всегда поддерживают ниже -65C. Реакционной смеси дают нагреваться до КТ в течение 2 ч и дополнительно перемешивают в течение ночи. Восстановительную обработку проводят путем проводимого по каплям прибавления раствора реакционной смеси к смеси 600 мл лед/вода, к которой прибавлено 60 г сульфита натрия и 180 г NaHCO3. Водную фазу экстрагируют 4 с помощью 200 мл дихлорметана, органические фазы объединяют, сушат над сульфатом магния и все летучие компоненты удаляют в вакууме. Получают 25,75 г (85%) немного желтоватых кристаллов. Их растворяют в 400 мл диизопропилового эфира, 1,6 мл воды и прибавляют 20 г связанной со смолой липолазы (липазно-акриловая смола, полученная из Candida antartica, Sigma-Aldrich) и смесь встряхивают в течение 11 дней при 60C. Суспензию реакционной смеси фильтруют через цеолит, промывают- 15016358 диизопропиловым эфиром и фильтрат выпаривают досуха. Полученное желтоватое масло растворяют в 200 мл дихлорметана и промывают с помощью примерно 150 мл насыщенного раствора NaHCO3. Водную фазу экстрагируют 3 дихлорметаном, органические фазы объединяют и сушат над сульфатом магния. После удаления всех летучих компонентов в вакууме получают 8,93 г хирального лактама в виде желтоватого масла. Последний продукт растворяют в 10 мл воды и при охлаждении в бане со льдом и перемешивании прибавляют 10 мл 37% водного раствора HCl. После 10 мин перемешивания при 0C раствор реакционной смеси выдерживают в течение ночи при КТ. Осадившиеся кристаллы отфильтровывают, промывают небольшим количеством ацетонитрила и сушат в высоком вакууме. Маточный раствор выпаривают почти досуха, осадившиеся кристаллы отфильтровывают, промывают ацетонитрилом и также сушат в высоком вакууме. Получают 11,74 г (70,9 ммоль, 31% в пересчете на рацемический лактам) бесцветных кристаллов гидрохлорида (1S,2R)-2-аминоциклопентанкарбоновой кислоты. (Энантиомерная кислота осаждается на стадии кинетического разделения и содержится в осадке, который отделяют фильтрованием через цеолит). Последовательность синтеза описана в литературе (Forro and Fueloep, 2003). 2,59 г (4,9 ммоль) D-2a, 2,21 г (6,9 ммоль, 1,4 экв.) TBTU и 4,21 мл (24,6 ммоль, 5 экв.) основание Хюнига растворяют в 75 мл ДМФ и перемешивают в течение 20 мин при КТ. Затем прибавляют 0,63 мл(7,38 ммоль, 1,5 экв.) изопропиламина и смесь перемешивают в течение ночи при КТ. Ее фильтруют с отсасыванием через основной оксид алюминия, промывают с помощью ДМФ и маточный раствор перемешивают с 400 мл воды, перемешивают в течение еще 30 мин и осадок фильтруют с отсасыванием. Неочищенный продукт промывают водой и сушат в вакууме. Для очистки его перемешивают с 50 мл ацетонитрила в течение 30 мин при 5C, фильтруют с отсасыванием, промывают небольшим количеством холодного ацетонитрила и остаток сушат в вакууме. Получают 2,13 г (3,9 ммоль, 80%) D-3a в виде светлобежевого твердого вещества. Rf = 0,53 (силикагель, cHx:ЭА 1:1) MC-ИЭР+: 542 (M+H)+. 2,13 г (3,9 ммоль) D-3a растворяют в 150 мл ТГФ и прибавляют 250 мг катализатора гидроксид палладия/C (20 мас.% Pd на древесном угле). Смесь гидрируют в течение 16 ч при перемешивании при КТ при давлении H2, равном 6 бар. Затем прибавляют 30 мл метанола, катализатор фильтруют через кизельгур, промывают метанолом и фильтрат выпаривают. Остаток кипятят с 45 мл этанола, медленно охлаждают до 5C, перемешивают в течение еще 1 ч и затем фильтруют с отсасыванием и промывают холодным этанолом. Получают 2,46 г (3,2 ммоль, 82%) кислоты D-4a. Rf = 0,46 (силикагель, CH2Cl2:MeOH: 450 мг (1 ммоль) D-4c растворяют в 1,8 мл сухого толуола и последовательно прибавляют 222 мкл(1,3 ммоль, 1,3 экв.) основания Хюнига и 940 мкл трет-бутанола. Затем прибавляют 258 мкл дифенилфосфорилазида и смесь нагревают при 80C в течение 16 ч. Реакционную смесь объединяют с 20 мл этилацетата, промывают 2 с помощью 20 мл 0,5 М раствора NaOH и водную фазу промывают 2 с помощью 20 мл этилацетата. Объединенные органические фазы промывают насыщенным водным раствором хлорида натрия, нерастворимые компоненты отфильтровывают, фильтрат сушат над хлоридом магния и растворитель удаляют в вакууме. Получают 461 мг (0,88 ммоль, 89%) D-5c в виде желтоватого твердого вещества. MC-ИЭР+: 523 (M+H)+. 461 мг (0,88 ммоль) D-5c растворяют в 5 мл дихлорметана, прибавляют 2 мл трифторуксусной кислоты и смесь перемешивают в течение 1 ч при КТ. Реакционную смесь перемешивают с 50 мл воды и водную фазу промывают с помощью 50 мл этилацетата. Органическую фазу экстрагируют еще 2 с помощью 30 мл 10% хлористоводородной кислоты, водные фазы объединяют, значение pH доводят до 10 с помощью 10% раствора гидроксида натрия и экстрагируют 3 с помощью 50 мл этилацетата. Объединенные органические фазы сушат над сульфатом магния, летучие компоненты удаляют в вакууме и получают 243 мг (0,58 ммоль, 65%) D-6c в виде бесцветного твердого вещества. Rf = 0,08 (силикагель,cHex:ЭА 1:1) MC-ИЭР+: 423 (M+H)+. 20 г (153 ммоль) 2-Тиоурацила суспендируют в 250 мл метанола и затем прибавляют 8,7 г (152,9 ммоль, 1 экв.) метоксида натрия. Раствор перемешивают в течение 5 мин при КТ и затем по каплям прибавляют 12,4 мл (198,8 ммоль, 1,3 экв.) метилйодида. Реакционную смесь перемешивают в течение ночи,затем выливают в воду и экстрагируют 3 с помощью примерно 150 мл хлороформа. Объединенные органические фазы сушат над сульфатом магния, растворитель удаляют в вакууме и получают 16 г (121,5 ммоль, 74%) E-1 в виде бесцветного твердого вещества. 4,1 г (28,8 ммоль) E-1 растворяют в 10 мл диглима (диметиловый эфир диэтиленгликоля) и этот раствор объединяют с 4,79 г (34,6 ммоль, 1,2 экв.) 4-аминобензойной кислоты. Реакционную смесь кипятят с обратным холодильником в течение 16 ч После охлаждения до КТ осадок фильтруют с отсасыванием, промывают небольшим количеством диглима, затем диэтиловым эфиром и сушат в вакууме. Получают 5,27 г (22,8 ммоль, 79%) E-2 в виде бесцветного твердого вещества. MC-ИЭР+: 232 (M+H)+. 9 г (38,9 ммоль) E-2 помещают в 100 мл воды, прибавляют 2,18 г NaOH (54,5 ммоль, 1,4 экв.). Раствор объединяют с 11,9 г (46,7 моль, 1,2 экв.) йода и перемешивают в течение 3 ч при 65C. После охлаждения до 50C прибавляют пентагидрат тиосульфата натрия для удаления избытка йода, затем смесь перемешивают в течение еще 1 ч и охлаждают до КТ. Коричневатый осадок фильтруют с отсасыванием,промывают водой и сушат в вакууме. Получают 13,7 г (38,4 ммоль, 82%) E-3a. MC-ИЭР+: 358 (M+H)+. 9 г (38,9 ммоль) E-2 помещают в 10 мл уксусной кислоты и к нему по каплям прибавляют раствор 2,1 мл (40,9 ммоль 1,05 экв.) брома в 50 мл уксусной кислоты и смесь перемешивают в течение примерно 1 ч при КТ. Реакционную смесь перемешивают с 800 мл воды, осадок фильтруют с отсасыванием и полученный коричневатый осадок промывают водой и сушат в вакууме. Получают 11,5 г (37,1 ммоль, 95%)E-3b в виде бесцветного твердого вещества. 6,5 г (18,2 ммоль) Е-3 а суспендируют в 80 мл оксихлорида фосфора и в течение 3 ч при перемешивании смесь кипятят с обратным холодильником. Реакционную смесь при энергичном перемешивании по каплям прибавляют к 800 мл смеси воды со льдом, перемешивают в течение еще 30 мин и неочищенный хлорангидрид кислоты E-4a отфильтровывают. Его сушат в вакууме и затем используют без дополнительной очистки. Для получения кислоты неочищенный хлорангидрид кислоты растворяют в 200 мл ТГФ и прибавляют 200 мл 20% водного раствора NaHCaO3. Реакционную смесь перемешивают в течение 16 ч при КТ. ТГФ удаляют в вакууме, значение pH водной фазы доводят до 2 с помощью концентрированной HCl,перемешивают в течение 10 мин, полученный остаток фильтруют с отсасыванием и промывают водой. После сушки в вакууме получают 6,3 г (16,7 ммоль, 92%) E-5a в виде бесцветного твердого вещества. Rf Получают из E-3b аналогично получению производных E-4a и E-5a. 559 мг (1,6 ммоль) E-4b растворяют в 5 мл ТГФ и объединяют с 414 мкл (2,4 ммоль, 1,5 экв.) основания Хюнига. К этому раствору по каплям прибавляют 181 мкл (1,6 ммоль, 1 экв.) N-метилпиперазина и смесь перемешивают в течение 90 мин при КТ. Затем прибавляют 100 мл воды и смесь экстрагируют 3 с помощью 50 мл этилацетата. Объединенные органические фазы сушат над сульфатом магния и растворитель удаляют в вакууме. Получают 566 мг (1,4 ммоль, 86%) E-6b в виде бесцветной смолы. MC-ИЭР+: 410/412 (M+H)+ (1Br). 459 мг (1,1 ммоль) E-6b растворяют в 5 мл 1-бутанола и объединяют с 536 мкл (3,1 ммоль, 2,8 экв.) основания Хюнига. К раствору прибавляют 162 мг цис-2-аминоциклопентанкарбоновой кислоты (рацемической) и реакционную смесь перемешивают в течение 100 мин при 110C (микроволновая печь СЕМ,100 Вт). Реакционную смесь выпаривают, перемешивают примерно с 200 мл воды и экстрагируют 3 с помощью 50 мл этилацетата. Объединенные органические фазы сушат над сульфатом магния и растворитель удаляют в вакууме. Получают 321 мг (0,64 ммоль, 57%) E-7b в виде бесцветной смолы. MC-ИЭР+: 503/505 (M+H)+ (1Br).(рацемического) и реакционную смесь перемешивают в течение 60 мин при 120C. Реакционную смесь выпаривают, остаток растворяют в 5 мл 1-бутанола и осадок фильтруют с отсасыванием. После промывки с помощью 5 мл холодного 1-бутанола и сушки в вакууме получают 935 мг(2,2 ммоль, 73%) E-8b в виде бежевого твердого вещества. MC-ИЭР+: 420/422 (M+H)+ (1Br). Йодпроизводное Е-8 а получают аналогично из E-5a. Однако температура реакции равна 80C. 935 мг (2,23 ммоль) E-8b растворяют в 8 мл ДМФ и в атмосфере аргона прибавляют 403 мг (4,45 ммоль, 2 экв.) цианида меди(I). Желтый раствор объединяют с 80 мг (0,067 ммоль, 3 мол.%) тетракистрифенилфосфинпалладия и нагревают при 145C в течение 24 ч, и за это время в реакцию входит примерно 50% эдукта. Повторно прибавляют такое же количество катализатора, смесь нагревают в течение- 19016358 еще 5 ч и затем реакционную смесь обрабатывают. Реакционную смесь фильтруют через пористый фильтр с силикагелем (растворитель: ДМФ), фильтрат выпаривают примерно до 5 мл и выливают примерно в 400 мл дистиллированной воды. Образовавшийся осадок отфильтровывают, промывают с помощью 100 мл воды и растворяют в метаноле. Прибавляют ОФ-гель и растворитель удаляют в вакууме. Смесь очищают с помощью хроматографии с использованием обращенной фазы (от 5% ацетонитрилаHCOOH. Выделяют 160 мг (0,44 ммоль, 20%) E-9b в виде бежевого твердого вещества. Rf = 0,30 (силикагель, CH2Cl2:MeOH:AcOH 5:1:0,1) MC-ИЭР+: 367 (M+H)+. Пример 1.-(1S,2R)-2-2-[4-(Метилфенилсульфамоил)фениламино]-5-трифторметилпиримидин-4 иламино-циклопентанкарбонамид (схема синтеза A). 150 мг (0,6 ммоль) A-2, 519 мг (1,98 ммоль, 3 экв.) 4-Амино-N-метил-N-фенилбензолсульфонамида и 130 мкл (0,76 ммоль, 1,15 экв.) N-этилдиизопропиламина растворяют в 3 мл N,N-диметилацетамида и раствор перемешивают в течение 10 мин при 180C (при нагревании в микроволновой печи). Раствор перемешивают с 30 мл воды, значение pH доводят до 3 с помощью 0,1 н. HCl (водный раствор), экстрагируют 3 с помощью 10 мл этилацетата, сушат над сульфатом магния и летучие компоненты удаляют в вакууме. Остаток очищают с помощью колоночной хроматографии (циклогексан/этилацетат 2/1). Получают 92 мг (0,2 ммоль) N-метил-4-(4-метилсульфанил-5-трифторметилпиримидин-2-иламино)-Nфенилбензолсульфонамида в виде светло-коричневого твердого вещества. 85 мг (0,19 ммоль) этого промежуточного продукта растворяют в 7,5 мл дихлорметана, прибавляют 64 мг (0,285 ммоль, 1,5 экв., 77%) м-хлорпербензойной кислоты и смесь перемешивают в течение 3 ч при КТ. Органическую фазу промывают 3 с помощью 20 мл насыщенного водного раствора NaHCaO3 и таким образом удаляют 3-хлорбензойную кислоту. После сушки органической фазы над сульфатом натрия получают 83 мг (0,18 ммоль, 95%) 4-(4-метансульфинил-5-трифторметилпиримидин-2-иламино)-Nметил-N-фенилбензолсульфонамида (A-4a), который используют на следующей стадии без дополнительной очистки. 83 мг (0,18 ммоль) A-4a, 26 мг цис-2-амино-1-циклопентанкарбоксамида (0,2 ммоль, 1,1 экв., рацемический) и 35 мкл (0,2 ммоль, 1,1 экв.) основания Хюнига растворяют в 2 мл ДМА и перемешивают в течение 1 ч при 60C. Реакционную смесь перемешивают с 10 мл 0,1 н. HCl (водный раствор), смесь перемешивают в течение 30 мин, образовавшийся осадок фильтруют с отсасыванием, промывают водой и сушат. Заключительную очистку проводят с помощью колоночной хроматографии (cHex/ЭА от 60/40 до 50/50 в течение 20 мин). Получают 43 мг (0,08 ммоль, 45%) соединения 1 в виде бесцветного твердого вещества. Соединения примеров 2 и 3 получают аналогично. Пример 4.-N-(1S,2R)-2-2-[4-(4-Метилпиперазин-1-карбонил)фениламино]-5-трифторметилпиримидин 4-иламиноциклогексил)ацетамид (схема синтеза C). 38 мг (0,08 ммоль) C-3b растворяют в 50 мкл ДМА, прибавляют 25 мкл (0,16 моль, 2 экв.) основания Хюнига и растворяют в течение нескольких минут при КТ. 5 мкл Ацетилхлорида (1 экв.) растворяют в небольшом количестве ДМА и по каплям прибавляют к реакционной смеси. Примерно через 10 мин реакционную смесь растворяют в дихлорметане, объединяют с 10 мл ОФ-геля и все летучие компоненты удаляют в вакууме. Смесь очищают с помощью хроматографии с ОФ фазой (AcCN/вода от 5/95 до 95/5% в течение 20 мин). После объединения фракций продукта и сушки вымораживанием получают 18 мг(0,034 ммоль, 42%) соединения 4 в виде бесцветного твердого вещества. Соединения примеров 5-12 получают аналогично. Пример 13. -1-Метил-3-1S,2R)-2-2-[4-(4-метилпиперазин-1-карбонил)фениламино]-5-трифторметилпиримидин-4-иламиноциклогексил)мочевина (схема синтеза C). 50 мг (0,105 ммоль) C-3b растворяют в 50 мкл ДМФ и объединяют с 55 мкл (0,315 ммоль, 3 экв.) основания Хюнига. К этому раствору при КТ прибавляют 6 мкл метилизоцианата (1 экв.). Примерно через 10 мин реакционную смесь растворяют в дихлорметане, объединяют с 10 мл ОФ-геля и все летучие компоненты удаляют в вакууме. Смесь очищают с помощью хроматографии с ОФ фазой (AcCN/вода от 5/95 до 95/5% в течение 20 мин). После объединения фракций продукта и сушки вымораживанием получают 24 мг (0,045 ммоль, 43%) соединения 13 в виде бесцветного твердого вещества. Соединения примеров 14-17 получают аналогично. Пример 18. Метил-)-(1S,2R)-2-2-[4-(4-метилпиперазин-1-карбонил)фениламино]-5-трифторметилпиримидин-4-иламино-циклогексил)-карбамат (схема синтеза C). 30 мг (0,063 ммоль) C-3b растворяют в 50 мкл ДМФ и объединяют с 22 мкл (0,126 ммоль, 2 экв.) основания Хюнига. К этому раствору при КТ прибавляют 6 мкл метилхлорформиата (1,2 экв.). Примерно через 10 мин реакционную смесь растворяют в дихлорметане, объединяют с 10 мл ОФ-геля и все летучие компоненты удаляют в вакууме. Смесь очищают с помощью хроматографии с ОФ фазой (AcCN/вода от 5/95 до 95/5% в течение 20 мин). После объединения фракций продукта и сушки вымораживанием полу- 20016358 чают 13 мг (0,025 ммоль, 39%) соединения 13 в виде бесцветного твердого вещества. Соединения примеров 19 и 20 получают аналогично. Пример 21.[4-(4-Циклопентиламино-5-трифторметилпиримидин-2-иламино)фенил]-(4-метилпиперазин-1-ил)метанон (схема синтеза C). 88 мг (0,22 ммоль) C-2a растворяют в 290 мкл ДМА, прибавляют 26 мкл (0,26 ммоль, 1,2 экв.) циклопентиламина и 75 мкл (0,44 ммоль, 2 экв.) основания Хюнига и реакционную смесь нагревают при 120C. Примерно через 90 мин реакционную смесь выливают примерно в 10 мл дистиллированной воды и образовавшийся осадок отфильтровывают. Суспензию экстрагируют 3 с помощью 20 мл этилацетата,объединенные органические фазы сушат с помощью насыщенного водного раствора NaCl и сульфата магния, объединяют с 100 мкл раствора HCl в диоксане и все летучие компоненты удаляют в вакууме. Получают 106 мг (0,219 ммоль, 99%) соединения 21 в виде гидрохлорида. Соединения примеров 22-26 получают аналогично. Пример 27. Диметиламид -(1S,2R)-2-2-[4-(4-метилпиперазин-1-карбонил)фениламино]-5 трифторметилпиримидин-4-иламиноциклогептанкарбоновой кислоты (схема синтеза C). 35 мг (0,067 ммоль) C-3d растворяют в 250 мкл ДМФ, прибавляют 30 мкл (0,175 ммоль, 2,6 экв.) основание Хюнига и затем 35 мг (0,11 ммоль, 1,6 экв.) TBTU. Реакционную смесь перемешивают в течение 10 мин при КТ и затем объединяют с 118 мкл диметиламина (2 М раствор в ТГФ, 0,235 ммоль, 3,5 экв.). Смесь встряхивают в течение 4 ч при 35C, затем реакционную смесь растворяют в ацетонитриле и объединяют с 6 мл ОФ-геля и все летучие компоненты удаляют в вакууме. Очистку проводят с помощью колоночной хроматографии с ОФ фазой (ацетонитрил/вода от 12/88 до 40/60 в течение 12 мин). Фракции продукта сушат вымораживанием и получают 19 мг (0,035 ммоль, 52%) соединения 27. Соединения примеров 28-30 получают аналогично. Пример 31. -4-[4-1R,2S)-2-Изопропилкарбамоилпиклопентиламино)-5-трифторметилпиримидин-2-иламино]-N-[2-(1-метилпирролидин-2-ил)этил]бензамид (схема синтеза D). 80 мг (0,18 ммоль) D-4c растворяют в 2,4 мл ДМФ, прибавляют 179 мкл (1,03 моль, 1,5 экв.) основания Хюнига и раствор объединяют с 83 мг (0,25 ммоль, 1,4 экв.) TBTU. Раствор перемешивают в течение 40 мин при КТ, затем прибавляют 38,5 мкл (0,27 ммоль, 1,5 экв.) 2-(2-аминоэтил)-1-метилпирролидина и смесь перемешивают в течение 2 дня. Затем к реакционной смеси прибавляют силикагель и летучие компоненты удаляют в вакууме. Очистку проводят с помощью колоночной хроматографии с нормальной фазой (ДХМ/MeOH/NH3 (водный раствор) 5/1/0,1). Получают 70 мг (0,125 ммоль, 70%) соединения 31. Соединения примеров 32-58 получают аналогично. Пример 59. Изопропиламид -(1S,2R)-2-2-[4-(4-метилпиперазин-1-карбонил)фениламино]-5 трифторметилпиримидин-4-иламиноциклопентанкарбоновой кислоты (схема синтеза C). 88 мг (0,18 ммоль) C-3d растворяют в 2 мл ДМФ, прибавляют 153 мкл (0,90 ммоль, 5 экв.) основания Хюнига и раствор объединяют с 81 мг (0,25 ммоль, 1,4 экв.) TBTU. Раствор перемешивают в течение 20 мин при КТ, затем прибавляют 12 мкл (0,27 ммоль, 1,5 экв.) изопропиламина и смесь перемешивают в течение 16 ч. Затем ее фильтруют через основной оксид алюминия и промывают с помощью 20 мл метанола. К фильтрату прибавляют ОФ-гель и летучие компоненты удаляют в вакууме. Неочищенный продукт, иммобилизованный на ОФ-геле, очищают в режиме обращенной фазы (от 95% воды (+0,2%HCOOH) и 5% ацетонитрила (+0,2% HCOOH) до 55% воды и 45% ацетонитрила в течение 20 мин). Соответствующие фракции продукта объединяют с 1 экв. концентрированной хлористоводородной кислоты и освобождают от растворителя с помощью сушки вымораживанием. 14 мг (0,025 ммоль, 14%) Гидрохлорида соединения 59 остается в виде бесцветной пленки. Соединения примеров 60-69 получают аналогично. Соединения примеров 68 и 69 являются хиральными и их получают из C-2a, с использованием энантиомеров цис-2-аминоциклопентанкарбоновой кислоты с последующим получением изопропиламидов. Альтернативно, 68 и 69 также можно получить из 59 с помощью препаративной хиральной ВЭЖХ. Пример 70. Изопропиламид -(1S,2R)-2-2-[3-хлор-4-(4-метилпиперазин-1-карбонил)фениламино]-5-трифторметилпиримидин-4-иламиноциклопентанкарбоновой кислоты (схема синтеза B). 30 мг (85,5 ммоль) B-2a растворяют в 100 мкл NMP и объединяют с 35 мг (0,14 ммоль, 1,6 экв.) (4 амино-2-хлорфенил)-(4-метилпиперазин-1-ил)метанона. К этой реакционной смеси прибавляют 107 мкл 4 М HCl в диоксане (0,43 ммоль, 5 экв.) и ее перемешивают в течение 12 ч при 5C. Реакционную смесь растворяют в смеси ДХМ/MeOH/NH3 9/1/0,1 и объединяют с 6 мл ОФ-геля, летучие компоненты удаляют в вакууме и очищают с помощью хроматографии с ОФ фазой (от 5% ацетонитрила до 95% ацетонитрила в течение 10 мин). Соответствующие фракции продукта освобождают от растворителя с помощью сушки вымораживанием. Остается 35 мг (0,06 ммоль, 72%) соединения 70. Соединения примеров 71-75 получают аналогично. Примеры 76-105 (общая методика). 1 экв. Соединения B-4 (соединение Е-8b для примеров 98-101 и соединение E-8a для примеров 102105) растворяют в ДМФ (примерно 1-10 мл на 1 ммоль), прибавляют 4-6 экв. основания Хюнига и затем- 21016358 1,3-1,5 экв. TBTU. Реакционную смесь перемешивают в течение 10-30 мин при КТ и затем прибавляют 11,5 экв. амина или анилина. После окончания реакции реакционную смесь объединяют с силикагелем,все летучие компоненты удаляют в вакууме и продукт очищают с помощью колоночной хроматографии(нормальная или ОФ-фаза) и выделяют. Пример 106. -(3-[4-1R,2S)-2-Карбамоилциклопентиламино)-5-трифторметилпиримидин-2 иламино]-N-фенилбензамид (схема синтеза A). 700 мг (3,06 ммоль) A-3 растворяют в 6 мл ДМА. Прибавляют 800 мкл (4,6 ммоль, 1,5 экв.) основание Хюнига и по каплям прибавляют 440 мг цис-2-амино-1-циклопентанкарбоксамида, растворенного в 24 мл ДМА. Реакционную смесь перемешивают при КТ. Через 1 ч ее разбавляют с помощью 400 мл дихлорметана и экстрагируют 2 с помощью 200 мл разбавленного вдвое насыщенного раствора хлорида аммония, затем сушат над сульфатом магния, и растворитель удаляют в вакууме. 1,1 г Неочищенного-(1S,2R)-2-(2-метилсульфанил-5-трифторметилпиримидин-4-иламино)циклопентанкарбоксамид остается в виде бежевого твердого вещества. Его вводят в реакцию без дополнительной очистки. Для этого твердое вещество растворяют в 60 мл ТГФ, порциями прибавляют 1,31 г (5,5 ммоль, 77% 2 экв.) МХПБК и смесь перемешивают в течение 1 ч при КТ. Органическую фазу промывают 3 с помощью 20 мл насыщенного водного раствора бикарбоната натрия и таким образом удаляют 3 хлорбензойную кислоту. После сушки органической фазы над сульфатом магния получают 1,15 г неочищенного-(1S,2R)-2-(2-метансульфинил-5-трифторметилпиримидин-4-иламино)циклопентанкарбоксамида, который используют на следующей стадии без дополнительной очистки. 150 мг (0,45 ммоль) -(1S,2R)-2-(2-Метансульфинил-5-трифторметилпиримидин-4-иламино)циклопентанкарбоксамида растворяют в 500 мкл NMP и прибавляют 148 мг (0,68 ммоль, 1,5 экв.) маминобензанилида. К этому раствору прибавляют 34 мкл хлористоводородной кислоты (4 М раствор в диоксане, 0,3 экв.) и его перемешивают в течение 16 ч при 50C. Реакционную смесь перемешивают с 30 мл воды, значение pH доводят до 3 с помощью 10 мл 0,1 н. HCl и экстрагируют 3 с помощью 15 мл этилацетата. Объединенные органические фазы сушат над сульфатом магния, все летучие компоненты удаляют в вакууме и неочищенный продукт перемешивают со смесью циклогексан/этилацетат 60/40,осадок фильтруют с отсасыванием и промывают 2-пропанолом. Получают 15 мг (0,03 ммоль, 7%) соединения 106 в виде бесцветного твердого вещества. Соединения примеров 107-109 получают аналогично. В этом случае очистку проводят с помощью колоночной хроматографии (этилацетат/циклогексан, силикагель). Пример 110. Пиклопропиламид -(1S,2R)-2-5-бром-2-[4-(4-метилпиперазин-1-карбонил)фениламино]пиримидин-4-иламиноциклопентанкарбоновой кислоты (схема синтеза E). 39 мг (0,077 ммоль) E-7b растворяют в 500 мкл ДМФ, прибавляют 66 мкл (0,39 ммоль, 5 экв.) основания Хюнига и 35 мг (0,11 ммоль, 1,4 экв.) TBTU. Раствор перемешивают в течение 20 мин при КТ и затем прибавляют 8 мкл (0,116 ммоль, 1,5 экв.) циклопропиламина и смесь выдерживают в течение ночи при КТ. Ее фильтруют через основной оксид алюминия, промывают с помощью примерно 20 мл метанола и фильтрат объединяют с 8 мл ОФ-геля. После удаления летучих компонентов в вакууме смесь очищают в режиме обращенной фазы (от 95% воды (+0,2% HCOOH) и 5% ацетонитрила (+0,2% HCOOH) до 5% воды и 95% ацетонитрила в течение 20 мин). Соответствующие фракции продукта освобождают от растворителя с помощью сушки вымораживанием. Соединение 110 получают в виде бесцветной пленки,12 мг (0,021 ммоль, 27%). MC-ИЭР+: 542/544 (M+H)+ (1 Br). Соединения примеров 111-120 получают аналогично. Пример 121. N-Метил-N-(1-метилпиперидин-4-ил)-4-4-[-(1R,2S)-2-(пирролидин-1-карбонил) циклопентиламино]-5-трифторметилпиримидин-2-иламинобензамид (схема синтеза C). 80 мг (0,15 ммоль) С-3 е растворяют в 1,4 мл ДМФ, прибавляют 132 мкл (0,77 ммоль, 5 экв.) основания Хюнига и 69 мг (0,22 ммоль, 1,4 экв.) TBTU. Реакционную смесь перемешивают в течение 30 мин при КТ, затем прибавляют 119 мкл (0,144 ммоль, 9,4 экв.) пирролидина и смесь перемешивают в течение 16 ч при КТ. Ее фильтруют через основной оксид алюминия, промывают с помощью примерно 20 мл метанола и фильтрат объединяют с силикагелем. После удаления летучих компонентов в вакууме смесь очищают с помощью колоночной хроматографии. (ДХМ/MeOH/NH3 9/1/0,1). После сбора фракций продукта их смешивают со 100 мкл HCl (4 М раствор в диоксане) и растворитель удаляют в вакууме, получают гидрохлорид соединения 121 в виде бесцветной пленки, 29 мг (0,048 ммоль, 31%). MC-ИЭР+: 574(M+H)+ Соединения примеров 122-128 получают аналогично. Пример 129. 4-[4-1R,3S)-3-Карбамоилциклопентиламино)-5-трифторметилпиримидин-2-иламино]-N-метил-N-(1-метилпиперидин-4-ил)-бензамид (схема синтеза C). 75 мг (0,14 ммоль) C-3f растворяют в 1 мл ДМФ, прибавляют 123 мкл (0,7 ммоль, 5 экв.) основания Хюнига и реакционную смесь перемешивают в течение 30 мин. Затем прибавляют 14 мкл (0,22 ммоль,1,5 экв.) водного раствора аммиака (28%) и смесь перемешивают в течение 5 ч при КТ. Раствор объединяют с ОФ-гелем, все летучие компоненты удаляют в вакууме и смесь очищают с помощью колоночной хроматографии (от 10% ацетонитрила (+0,2% HCOOH) и 90% воды (+0,2% HCOOH) до 24% ацетонитри- 22016358 ла и 76% воды в течение 12 мин). Фракции продукта объединяют с 100 мкл раствора HCl в диоксане и все летучие компоненты удаляют с помощью сушки вымораживанием. Получают 35 мг (0,063 моль,44%) соединения 129 в виде гидрохлорида. Соединение примера 130 получают аналогично. Пример 131. Изопропиламид -(1S,2R)-2-[2-(4-ацетиламинофениламино)-5-трифторметилпиримидин-4-иламино]циклопентанкарбоновой кислоты (схема синтеза D). 22 мг D-6c растворяют в 1 мл ТГФ, объединяют с 14 мкл (0,075 ммоль, прибавляют 1,5 экв.) основания Хюнига и затем 3 мкл ацетилхлорида, растворенного в 500 мкл ТГФ. Примерно через 90 мин раствор реакционной смеси разбавляют с помощью 10 мл метанола и 8 мл ОФ-геля прибавляют. Хроматографическую очистку проводят с использованием обращенной фазы (от 78% воды (+0,2% HCOOH) и 22% ацетонитрила (+0,2% HCOOH) до 51% воды и 49% ацетонитрила в течение 15 мин). Соответствующие фракции продукта объединяют и растворитель удаляют с помощью сушки вымораживанием. Получают 14 мг (0,028 ммоль, 54%) соединения 131. Соединения примеров 132-133 получают аналогично. Пример 134. -(1S,2R)-2-5-Циано-2-[4-(4-метилпиперазин-1-карбонил)фениламино]пиримидин-4-иламиноциклопентанкарбоксамид (схема синтеза E). 40 мг (0,11 ммоль) E-9b растворяют в 1,5 мл ДМФ, прибавляют 110 мкл (0,63 ммоль, 5,8 экв.) основания Хюнига и реакционную смесь перемешивают в течение 40 мин. Затем прибавляют 18 мкл (0,16 ммоль, 1,5 экв.) N-метилпиперазина и смесь перемешивают в течение 48 ч при КТ. Раствор объединяют с силикагелем, все летучие компоненты удаляют в вакууме и смесь очищают с помощью колоночной хроматографии (ДХМ/MeOH 9/1). Получают 33 мг (0,07 моль, 67%) соединения 134. Соединения примеров 135-136 получают аналогично. Пример 137. -(1S,2R)-2-5-Циклопропилэтинил-2-[4-(4-метилпиперазин-1-карбонил)фениламино]пиримидин-4-иламиноциклопентанкарбоксамид (схема синтеза E). 50 мг (0,09 ммоль) соединения 105 растворяют в 220 мкл ДМФ и затем прибавляют 15 мг дихлорбис(трифенилфосфин)палладия (0,021 ммоль, 23 мол.%) и 10 мг (0,03 ммоль, 0,58 экв.) йодида меди(I). Раствор объединяют с 320 мкл основания Хюнига и затем с 18 мг (0,27 ммоль, 3 экв.) этинилциклопропана. Реакционную смесь фильтруют через силикагель со смесью ДХМ/MeOH/NH3 4/1/0,1 и затем 6 прибавляют мл ОФ-геля. После удаления летучих компонентов очистку с помощью колоночной хроматографии проводят с использованием ОФ-фазы (от 95% воды (+0,2% HCOOH) и 5% ацетонитрила (+0,2%HCOOH) до 50% воды и 50% ацетонитрила в течение 20 мин). Соответствующие фракции продукта объединяют и растворитель удаляют с помощью сушки вымораживанием. Получают 32 мг (0,065 ммоль,71%) соединения 137. Соединения примеров 138-139 получают аналогично, а в примере 138 реакцию проводят в атмосфере пропина в колбе, заполненной азотом, при 40C. Пример 140. -4-[4-1R,2S)2-Карбамоилциклопентиламино)-5-циклопропилпиримидин-2-иламино]-N-(1-метилпиперидин-4-ил)бензамид (схема синтеза E). 100 мг (0,15 ммоль) соединения 104 суспендируют в 1,4 мл диоксана и прибавляют 13 мг (0,15 ммоль, 1 экв.) циклопропилборной кислоты. Раствор дегазируют в вакууме и в атмосфере аргона прибавляют 3,5 мг (0,004 ммоль, 3 мол.%) аддукта дихлор[1,1'-бис(дифенилфосфино)-ферроцен]палладий(II)дихлорметана с (PdCl2dppf ДХМ) и 2 мл раствора карбоната натрия (2M в воде). Эту двухфазную смесь нагревают при 130C в течение 5 мин (микроволновая печь СЕМ, 100 Вт). Органическую фазу отделяют,разбавляют метанолом и объединяют с 6 мл ОФ-геля. После удаления летучих компонентов очистку проводят с помощью колоночной хроматографии с использованием обращенной фазы (от 97% воды(+0,2% HCOOH) до 3% ацетонитрила (+0,2% HCOOH) до 70% воды и 30% ацетонитрила в течение 12 мин). Соответствующие фракции продукта объединяют и растворитель удаляют с помощью сушки вымораживанием. Получают 2 мг (0,003 ммоль, 2%) соединения 140. Пример 141. Изопропиламид -(1S,2R)-2-[2-(4-[1,4]диазепан-1-ил-3-фторфениламино)-5 трифторметилпиримидин-4-иламино]циклопентанкарбоновой кислоты (схема синтеза B). 23 мг (0,066 ммоль) B-2a растворяют в 100 мкл NMP, прибавляют 17 мг (0,079 ммоль, 1,2 экв.) 3 фтор-4-(4-метил-[1.4]диазепан-1-ил)-фениламина и в заключение 46 мкл HCl (0,18 ммоль, 2,8 экв., 4 М раствор в диоксане). Реакционную смесь нагревают при 90C в течение 12 ч, объединяют с 6 мл ОФ-геля и летучие компоненты удаляют в вакууме. Хроматографическую очистку проводят с использованием обращенной фазы (от 95% воды (+0,2% HCOOH) и 5% ацетонитрила (+0,2% HCOOH) до 55% воды и 45% ацетонитрила в течение 25 мин). Соответствующие фракции продукта объединяют и растворитель удаляют с помощью сушки вымораживанием. Получают 3 мг (0,005 ммоль, 8%) соединения 141. Соединения примеров 142-144 получают аналогично. Пример 145. -(1R,2R)-2-2-[4-(4-Метилпиперазин-1-карбонил)фениламино]-5-трифторметилпиримидин-4-иламиноциклопентанкарбоксамид (схема синтеза C). 100 мг (0,25 ммоль) C-2a растворяют в 1 мл 1-бутанола и этот раствор объединяют с 35 мг (0,275 ммоль, 1,1 экв.) рацемического транс-2-аминоциклопентанкарбоксамида и 60 мкл (0,35 ммоль, 1,4 экв.)- 23016358 основания Хюнига. При 110C (микроволновая печь СЕМ, 100 Вт) смесь перемешивают в течение 30 мин до полной конверсии. К реакционной смеси прибавляют примерно 20 мл метанола, ее объединяют с ОФ-гелем (примерно 8 мл) и все летучие компоненты удаляют в вакууме. Смесь очищают на ОФ колонке (от 95% воды (+0,2% HCOOH) и 5% ацетонитрила (+0,2% HCOOH) до 55% воды и 45% ацетонитрила в течение 20 мин). Соответствующие фракции продукта объединяют с концентрированной хлористоводородной кислотой и освобождают от растворителя с помощью сушки вымораживанием. Получают 77 мг (0,146 ммоль, 58%) соединения 145 в виде бесцветного твердого вещества. Соединения примеров 146-147 получают аналогично, а соединение примера 148 получают аналогично соединению примера 129 (нуклеофильное замещение -аминокислотой с использованием C-2a в качестве исходного вещества и в заключение образованием амидной связи с помощью аммиака). Примеры 1-148
МПК / Метки
МПК: C07D 239/48, A61K 31/505, A61P 35/00, A61P 31/00
Метки: ингибиторы, aurora, 2,4-диаминопиримидины
Код ссылки
<a href="https://eas.patents.su/30-16358-24-diaminopirimidiny-kak-ingibitory-aurora.html" rel="bookmark" title="База патентов Евразийского Союза">2,4-диаминопиримидины как ингибиторы aurora</a>
Диаминопиримидины в качестве фунгицидов

Номер патента: 15174
Опубликовано: 30.06.2011
Авторы: Фёрстэ Арнд, Хиллебранд Штефан, Дамен Петер, Гэртцен Оливер, Дункель Ральф, Маттес Амос, Гройль Йорг Нико, Вахендорфф-Нойманн Ульрике, Кокеро Пьер-Иф, Шрайер Петер
МПК: A01N 43/54, A01P 3/00, C07D 239/95...
Метки: качестве, диаминопиримидины, фунгицидов
Формула / Реферат:
1. Применение в качестве фунгицидов соединений формулы (I)в которой символы имеют следующие значения:X1 означает азот или CR3,X2 означает азот или CR4,A означает C(R14)2 или простую химическую связь,R1-R5независимо один от другого означают водород, галоид, циано-, гидрокси-, нитрогруппу, 3-8-членный, незамещенный или замещенный, насыщенный или ненасыщенный цикл, который не содержит ни одного или может содержать до четырех гетероатомов,...
Ингибиторы тромбина

Номер патента: 2767
Опубликовано: 29.08.2002
Авторы: Цирке Томас, Мак Хельмут, Зайтц Вернер, Хорнбергер Вильфрид, Бём Ханс-Йоахим, Козер Штефан, Хёффкен Ханс Вольфганг, Пфайффер Томас
МПК: C07K 5/06, A61P 7/02, A61K 38/55...
Метки: ингибиторы, тромбина
Формула / Реферат:
1. Ингибиторы тромбина формулы (I) где R1 - алкил с 1-20 атомами углерода, фенил- или нафтилалкил с 1-10 атомами углерода в алкильной части, группа R2OOC-(CH2), где R2 означает водород или алкил с 1-10 атомами углерода, А - остаток a-аминокислоты формулы (II) где R3 - водород, алкил с 1-8 атомами углерода, циклоалкил с 3-7 атомами углерода, фенил и нафтил, незамещенные или замещенные алкилом или алкоксилом, каждый с 1-4 атомами углерода, или...
Производные фениламинопиримидина как ингибиторы bcr-abl

Номер патента: 13328
Опубликовано: 30.04.2010
Авторы: Подили Кхадгапатхи, Венкайах Чоудари Наннапанени, Компелла Амала Кишан, Рачаконда Сринивас, Адибхатла Кали Сатьа Бхуджанга Рао
МПК: C07C 233/66, A61P 35/02, A61K 31/506...
Метки: bcr-abl, производные, фениламинопиримидина, ингибиторы
Формула / Реферат:
1. Фениламинопиридинилпиримидин общей формулы Iгде X представляет собой СН или N, n=1 или 2 и R представляет собой Н или СН3, или его фармацевтически приемлемые соли.2. Соединение по п.1, где X представляет собой N, n=1 и R представляет собой Н или СН3, или его фармацевтически приемлемые соли.3. Соединение по п.1, где X представляет собой СН, n=1 или 2 и R представляет собой Н или СН3, или его фармацевтически приемлемые соли.4. Соединение по...
Ингибиторы киназы

Номер патента: 15189
Опубликовано: 30.06.2011
Авторы: Побанс Марк Эндрю, Де Дьес Альфонсо, Чжун Боюй, Лопес Де Уралде-Гармениа Беатрис, Худзиак Кевин Джон, Ли Течао, Блас Де Блас Хесус Андрес, Бастиан Джолие Анне, Ших Чуан, Мэйдер Мэри Маргарет, Майерс Майкл Рэй
МПК: A61K 31/44, A61K 31/497, C07D 401/14...
Метки: ингибиторы, киназы
Формула / Реферат:
1. Соединение формулы Iгде Z выбран из группы, которая включаетX выбран из группы, которая включаетR1 представляет собой С1-С7алкил, необязательно замещенный от одного до шести заместителями, выбранными из группы, которая включает галоген и C1-C4алкилгалоген; С3-С6циклоалкил, необязательно замещенный одним или двумя заместителями, выбранными из группы, которая включает C1-C4алкил и трифторметил; или триметилсилил;R2 представляет собой фенил,...
Замещенные амидазо [1, 2а] азины как селективные сох-2 ингибиторы

Номер патента: 3399
Опубликовано: 24.04.2003
Авторы: Лагунас Арналь Кармен, Хименес Гвасч Ферран, Фарреронс Гальеми Карлес, Микель Боно Игнасио-Хосе, Фернандес Гарсия Андрес, Фернандес Серрат Ана Мария, Монсеррат Видаль Карлос
МПК: A61K 31/53, C07D 471/04, A61P 35/00...
Метки: ингибиторы, замещенные, селективные, амидазо, азины, сох-2
Формула / Реферат:
1. Замещенный имидазо[1,2a]азин формулы (I) или его фармацевтически приемлемый сольват и его соль присоединения кислоты, где A и B независимо выбирают из N и CH, при условии, что если A представляет N, тогда B также представляет N; R1 выбирают из группы, состоящей из CH3 и NH2; R2 и R3 выбирают из группы, состоящей из H, CH3, Cl, Br, COCH3 и OCH3; и R4, R5 и R6, каждый независимо, выбирают из группы, состоящей из H, F, Cl, Br, (C1-C3)алкила,...
Предыдущий патент: Антитела, направленные против бета-амилоидного пептида, и способы их применения
Следующий патент: Безопасная личная карточка и способ ее использования
Случайный патент: Разветвленные производные 3-фенилпропионовой кислоты и их применение