Фенилацетамиды и их применение в качестве модуляторов глюкокиназы
Номер патента: 12700
Опубликовано: 30.12.2009
Авторы: Ясуда Косуки, Навано Масао, Скофилд Карен Лесли, Файф Мэттью Колин Тор, Шах Виласбен Канджи, Проктер Мартин Джеймс, Разамизон Кристель Мари, Гарднер Лайза Сара







Формула / Реферат
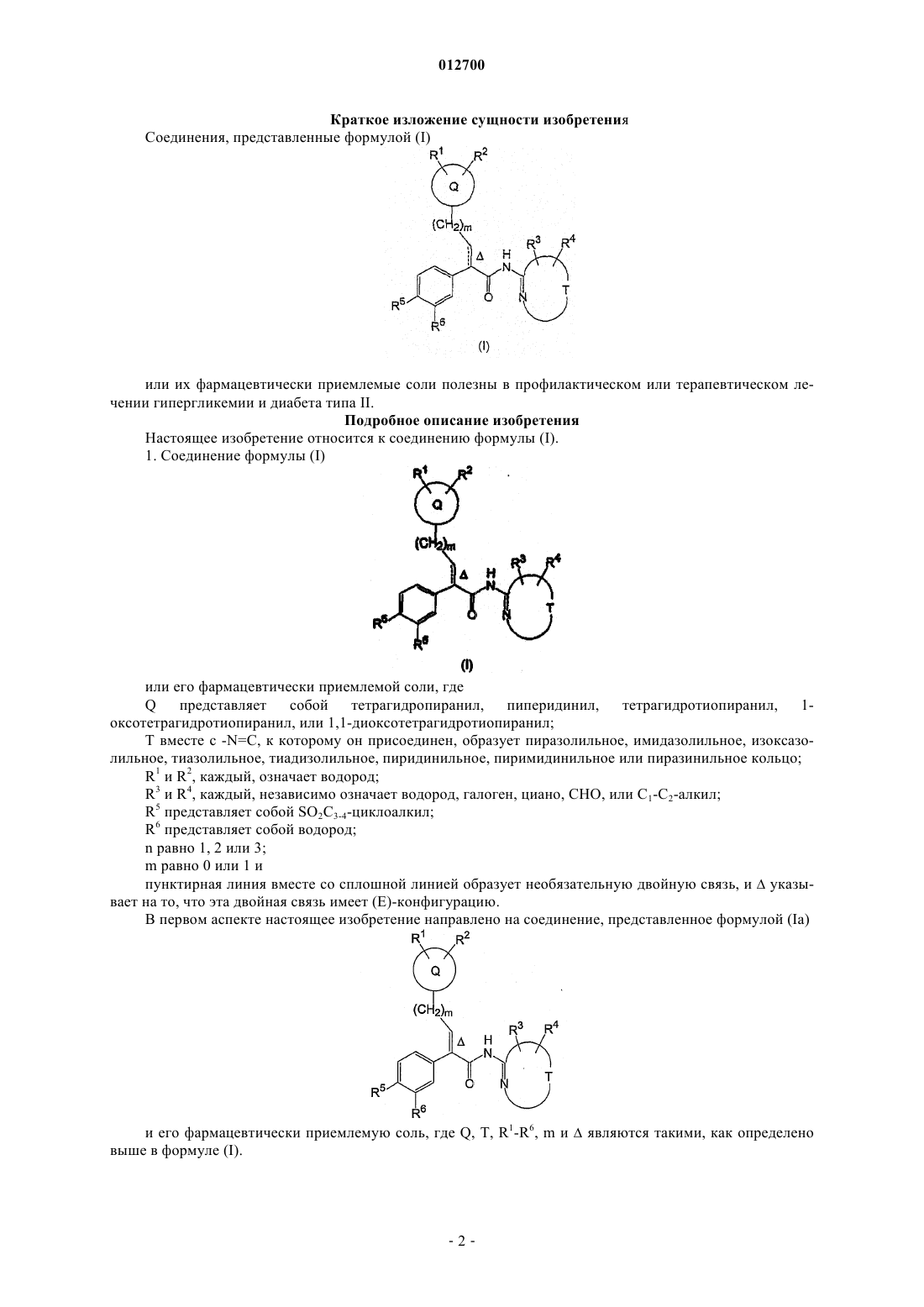
1. Соединение формулы (I)

или его фармацевтически приемлемая соль, где
Q представляет собой тетрагидропиранил, пиперидинил, тетрагидротиопиранил, 1-оксотетрагидротиопиранил или 1,1-диоксотетрагидротиопиранил;
Т вместе с -N=C, к которому он присоединен, образует пиразолильное, имидазолильное, изоксазолильное, тиазолильное, тиадизолильное, пиридинильное, пиримидинильное или пиразинильное кольцо;
R1 и R2, каждый, означает водород;
R3 и R4, каждый независимо, означает водород, галоген, циано, СНО, или С1-С2-алкил;
R5 представляет собой SO2C3-4-циклоалкил;
R6 представляет собой водород;
n равно 1, 2 или 3;
m равно 0 или 1;
пунктирная линия вместе со сплошной линией образует необязательную двойную связь, и D указывает на то, что эта двойная связь имеет (E)-конфигурацию.
2. Соединение по п.1 или его фармацевтически приемлемая соль, где пунктирная линия вместе со сплошной линией образует двойную связь.
3. Соединение по п.1 или его фармацевтически приемлемая соль, где пунктирная линия вместе со сплошной линией образует простую связь.
4. Соединение по п.3 или его фармацевтически приемлемая соль, где пунктирная линия вместе со сплошной линией образует простую связь и абсолютная конфигурация при асимметрическом центре в положении a относительно карбонильного атома углерода амида представляет собой (R)-конфигурацию.
5. Соединение по любому из предшествующих пунктов, где m равно 0.
6. Соединение по любому из предшествующих пунктов или его фармацевтически приемлемая соль, где Q представляет собой 4-тетрагидропиранил.
7. Соединение по любому из предшествующих пунктов или его фармацевтически приемлемая соль, где группа формулы

представляет собой тиазолил, тиадиазолил, изоксазолил, пиримидинил, пиразинил или пиридинил.
8. Соединение по любому из предшествующих пунктов или его фармацевтически приемлемая соль, где группа формулы

представляет собой 2-пиразинил или 2-тиазолил.
9. Соединение по любому из предшествующих пунктов или его фармацевтически приемлемая соль, где R3 и R4 независимо выбраны из водорода, галогена и метила.
10. Соединение, выбранное из
2-(4-циклопропансульфонилфенил)-3-(тетрагидропиран-4-ил)-N-тиазол-2-илпропионамида,
2-(4-циклопропансульфонилфенил)-N-(5-фтортиазол-2-ил)-3-(тетрагидропиран-4-ил)пропионамида,
2-(4-циклопропансульфонилфенил)-N-(3-метил-[1,2,4]тиадиазол-5-ил)-3-(тетрагидропиран-4-ил)пропионамида,
2-(4-циклопропансульфонилфенил)-N-пиразин-2-ил-3-(тетрагидропиран-4-ил)пропионамида,
2-(4-циклопропансульфонилфенил)-3-(тетрагидропиран-4-ил)-N-[1,2,4]тиадиазол-5-илпропионамида,
(Е)-2-(4-циклопропансульфонилфенил)-3-(тетрагидропиран-4-ил)-N-тиазол-2-илакриламида,
2-(4-циклопропансульфонилфенил)-N-(5-формилтиазол-2-ил)-3-(тетрагидропиран-4-ил)пропионамида,
(2R)-2-(4-циклопропансульфонилфенил)-N-(5-фтортиазол-2-ил)-3-(тетрагидропиран-4-ил)пропионамида,
(2R)-2-(4-циклопропансульфонилфенил)-3-(тетрагидропиран-4-ил)-N-[1,2,4]тиадиазол-5-илпропионамида,
(2R)-2-(4-циклопропансульфонилфенил)-N-пиразин-2-ил-3-(тетрагидропиран-4-ил)пропионамида,
(2R)-2-(4-циклопропансульфонилфенил)-N-(5-фторпиридин-2-ил)-3-(тетрагидропиран-4-ил)пропионамида,
(2R)-2-(4-циклопропансульфонилфенил)-3-(тетрагидропиран-4-ил)-N-тиазол-2-илпропионамида,
(2R)-2-(4-циклопропансульфонилфенил)-N-(3-метил-[1,2,4]тиадиазол-5-ил)-3-(тетрагидропиран-4-ил)пропионамида,
(2R)-2-(4-циклобутансульфонилфенил)-N-пиразин-2-ил-3-(тетрагидропиран-4-ил)пропионамида,
(2R)-2-(4-циклобутансульфонилфенил)-N-пиримидин-4-ил-3-(тетрагидропиран-4-ил)пропионамида,
(2R)-2-(4-циклобутансульфонилфенил)-N-изоксазол-3-ил-3-(тетрагидропиран-4-ил)пропионамида,
(2R)-2-(4-циклобутансульфонилфенил)-N-(1-метил-1H-пиразол-3-ил)-3-(тетрагидропиран-4-ил)пропионамида,
(2R)-2-(4-циклобутансульфонилфенил)-N-(5-фтортиазол-2-ил)-3-(тетрагидропиран-4-ил)пропионамида,
(Е)-2-(4-циклопропансульфонилфенил)-N-(5-фторпиридин-2-ил)-3-(тетрагидропиран-4-ил)акриламида,
(Е)-2-(4-циклопропансульфонилфенил)-N-(5-фтортиазол-2-ил)-3-(тетрагидропиран-4-ил)акриламида,
N-(5-цианотиазол-2-ил)-2-(4-циклопропансульфонилфенил)-3-(тетрагидропиран-4-ил)пропионамида и
2-(4-циклобутансульфонилфенил)-3-(тетрагидропиран-4-ил)-N-тиазол-2-илпропионамида
или их фармацевтически приемлемой соли.
11. Соединение, выбранное из
2-(4-циклопропансульфонилфенил)-3-(тетрагидропиран-4-ил)-N-тиазол-2-илпропионамида,
2-(4-циклопропансульфонилфенил)-N-(5-фтортиазол-2-ил)-3-(тетрагидропиран-4-ил)пропионамида,
2-(4-циклопропансульфонилфенил)-N-(3-метил-[1,2,4]тиадиазол-5-ил)-3-(тетрагидропиран-4-ил)пропионамида,
2-(4-циклопропансульфонилфенил)-N-пиразин-2-ил-3-(тетрагидропиран-4-ил)пропионамида,
2-(4-циклопропансульфонилфенил)-3-(тетрагидропиран-4-ил)-N-[1,2,4]тиадиазол-5-илпропионамида,
(Е)-2-(4-циклопропансульфонилфенил)-3-(тетрагидропиран-4-ил)-N-тиазол-2-илакриламида,
(2R)-2-(4-циклопропансульфонилфенил)-N-(5-фтортиазол-2-ил)-3-(тетрагидропиран-4-ил)пропионамида,
(2R)-2-(4-циклопропансульфонилфенил)-3-(тетрагидропиран-4-ил)-N-[1,2,4]тиадиазол-5-илпропионамида,
(2R)-2-(4-циклопропансульфонилфенил)-N-пиразин-2-ил-3-(тетрагидропиран-4-ил)пропионамида,
(2R)-2-(4-циклопропансульфонилфенил)-N-(5-фторпиридин-2-ил)-3-(тетрагидропиран-4-ил)пропионамида,
(2R)-2-(4-циклопропансульфонилфенил)-3-(тетрагидропиран-4-ил)-N-тиазол-2-илпропионамида,
(2R)-2-(4-циклопропансульфонилфенил)-N-(3-метил-[1,2,4]тиадиазол-5-ил)-3-(тетрагидропиран-4-ил)пропионамида,
(2R)-2-(4-циклобутансульфонилфенил)-N-пиразин-2-ил-3-(тетрагидропиран-4-ил)пропионамида,
(2R)-2-(4-циклобутансульфонилфенил)-N-(5-фтортиазол-2-ил)-3-(тетрагидропиран-4-ил)пропионамида,
(Е)-2-(4-циклопропансульфонилфенил)-N-(5-фтортиазол-2-ил)-3-(тетрагидропиран-4-ил)акриламида,
2-(4-циклопропилметансульфонилфенил)-3-(тетрагидропиран-4-ил)-N-тиазол-2-илпропионамида
или их фармацевтически приемлемой соли.
12. Соединение, выбранное из
(2R)-2-(4-циклобутансульфонилфенил)-N-пиразин-2-ил-3-(тетрагидропиран-4-ил)пропионамида,
(2R)-2-(4-циклобутансульфонилфенил)-N-(5-фтортиазол-2-ил)-3-(тетрагидропиран-4-ил)пропионамида,
(2R)-2-(4-циклопропансульфонилфенил)-N-(5-фтортиазол-2-ил)-3-(тетрагидропиран-4-ил)пропионамида,
(2R)-2-(4-циклопропансульфонилфенил)-N-пиразин-2-ил-3-(тетрагидропиран-4-ил)пропионамида
или их фармацевтически приемлемой соли.
13. Соединение формулы (I)

или его фармацевтически приемлемая соль, где
Q представляет собой 4-тетрагидропиранил;
Т вместе с -N=C, к которому он присоединен, образует 2-пиразинильное или 2-тиазолильное кольцо;
R1 и R2, каждый, означает водород;
R3 означает водород;
R4 означает водород или фтор;
R5 представляет собой SO2C3-4-циклоалкил;
R6 представляет собой водород;
m равно 0;
пунктирная линия вместе со сплошной линией образует необязательную двойную связь и D указывает на то, что эта двойная связь имеет (E)-конфигурацию.
14. Фармацевтическая композиция, содержащая соединение по любому из пп.1-13 или его фармацевтически приемлемую соль и фармацевтически приемлемый носитель.
15. Способ профилактического или терапевтического лечения состояния, где желательна активация глюкокиназы (GK), включающий стадию введеэшя эффективного количества соединения по любому из пп.1-13 или его фармацевтически приемлемой соли.
16. Способ профилактического или терапевтического лечения гипергликемии или диабета, включающий стадию введения эффективного количества соединения по любому из пп.1-13 или его фармацевтически приемлемой соли.
17. Способ по п.16, где соединение по любому из пп.1-13 вводят в комбинации с одним или более чем одним гипогликемическим агентом или противодиабетическим агентом.
18. Способ предупреждения диабета у человека, проявляющего преддиабетическую гипергликемию или нарушенную толерантность к глюкозе, включающий стадию введения эффективного количества соединения по любому из пп.1-13 или его фармацевтически приемлемой соли.
19. Способ получения соединения формулы (Ia)

включающий стадию конденсации соединения формулы (I+V)

с соединением формулы (V)

где Q, T, R1-R6, m и D являются такими, как определено в п.1.
20. Способ получения соединения формулы (Ib)

включающий стадию конденсации соединения формулы (VIII)

с соединением формулы (V)

где Q, T, R1-R6 и m являются такими, как определено в п.1.
21. Соединение формулы (IV)

где Q представляет собой тетрагидропиранил;
R1 и R2 представляют собой водород;
R5 представляет собой SO2C3-4-циклоалкил;
R6 представляет собой водород;
m равно нулю и
D указывает на то, что эта двойная связь имеет (E)-конфигурацию.
22. Соединение формулы (IV), выбранное из
(Е)-2-(4-циклопропансульфонилфенил)-3-(тетрагидропиран-4-ил)акриловой кислоты,
(Е)-2-(4-циклопропансульфинилфенил)-3-(тетрагидропиран-4-ил)акриловой кислоты.
23. Соединение формулы (VIII)

где Q представляет собой тетрагидропиранил;
R1 и R2 представляют водород;
R5 представляет собой SO2C3-4-циклоалкил;
R6 представляет собой водород;
m равно 0.
24. Соединение формулы (VIII), выбранное из
2-(4-циклопропансульфонилфенил)-3-(тетрагидропиран-4-ил)пропионовая кислота;
2-(4-циклобутансульфонилфенил)-3-(тетрагидропиран-4-ил)пропионовая кислота;
(2R)-2-(4-циклопропансульфонилфенил)-3-(тетрагидропиран-4-ил)пропионовая кислота и
(2R)-2-(4-циклобутансульфонилфенил)-3-(тетрагидропиран-4-ил)пропионовая кислота.
Текст
012700 Предшествующий уровень техники Настоящее изобретение относится к три(цикло)замещенным амидным соединениям. В частности,настоящее изобретение относится к амидным соединениям, замещенным 1) по карбонильному атому углерода этилом/этенилом, присоединенным к фенильному кольцу и арильному/гетероарильному/гетероциклическому кольцу, и 2) по амино гетероарильным кольцом, несущим атом азота, которые являются модуляторами глюкокиназы и полезны в профилактическом и терапевтическом лечении гипергликемии и диабета типа II. Считают, что глюкокиназа (GK) важна в организме при регуляции уровня глюкозы в плазме. GK,обнаруживаемая, главным образом, в печени и поджелудочной железе, представляет собой одну из четырех гексокиназ, которые катализируют первоначальный метаболизм глюкозы. Путь GK насыщается при более высоких уровнях глюкозы, чем другие гексокиназные пути (см. R.L. Printz et al., Annu. Rev. Nutr.,13: 463-496 (1993. GK является критическим фактором при поддержании баланса глюкозы у млекопитающих. Животные, у которых GK не экспрессируется, умирают от диабета вскоре после рождения, тогда как животные, у которых наблюдается повышенная экспрессия GK, обладают улучшенной толерантностью к глюкозе. Активация GK может привести к гиперинсулинемической гипогликемии (см., например, Н.В.Т. Christesen et al., Diabetes, 51: 1240-1246 (2002. Кроме того, диабет взрослых типа II у молодых людей вызван потерей функциональных мутаций в гене GK, что позволяет предположить, что GK действует в качестве сенсора глюкозы у людей (Y. Liang et al., Biochem. J. 309: 167-173 (1995. Таким образом, соединения, которые активируют GK, повышают чувствительность сенсорной системы GK и будут полезны в лечении гипергликемии, в частности гипергликемии, ассоциированной с диабетом типаII. Поэтому желательна разработка новых соединений, которые активируют GK, для лечения диабета. В международной патентной публикацииWO 2001044216 и в патенте США 6353111 описаны(E)-2,3-дизамещенные-N-гетероарилакриламиды в качестве активаторов GK. В международной патентной публикацииWO 2002014312 и в патентах США 6369232, 6388088 и 6441180 описаны тетразолилфенилацетамидные активаторы GK. В международной патентной публикацииWO 2000058293, в заявке на европейский патентЕР 1169312 и в патенте США 6320050 описаны арилциклоалкилпропионамидные активаторы GK. В международной патентной публикацииWO 2002008209 и в патенте США 6486184 описаны активаторы GK, представляющие собой альфа-ацил- и альфагетероатомзамещенный бензолацетамид, в качестве противодиабетических агентов. В международной патентной публикацииWO 2001083478 описаны гидантоинсодержащие активаторы GK. В международной патентной публикацииWO 2001083465 и в патенте США 6388071 описаны алкинилфенилгетероароматические активаторы GK. В международной патентной публикацииWO 2001085707 и в патенте США 6489485 описаны парааминзамещенные фениламидные активаторы GK. В международной патентной публикацииWO 2002046173 и в патентах США 6433188, 6441184 и 6448399 описаны конденсированные гетероароматические активаторы GK. В международной патентной публикацииWO 2002048106 и в патенте США 6482951 описаны активаторы GK, представляющие собой изоиндолин-1-оны. В международной патентной публикацииWO 2001085706 описаны замещенные фенилацетамидные активаторы GK для лечения диабета типа II. В патенте США 6384220 описаны активаторы GK, представляющие собой параарил- или гетероарилзамещенный фенил. Во французском патенте 2834295 описаны способы очистки и кристаллическая структура GK человека. В международной патентной публикацииWO 2003095438, опубликованной после даты приоритета настоящей заявки, описаныN-гетероарилфенилацетамиды и родственные соединения в качестве активаторов GK для лечения диабета типа II. В патенте США 6610846 описано получение циклоалкилгетероарилпропионамидов в качестве активаторов GK. В международной патентной публикацииWO 2003000262 описаны винилфениловые активаторы GK. В международной патентной публикацииWO 2003000267 описаны производные аминоникотината в качестве модуляторов GK. В международной патентной публикацииWO 2003015774, опубликованной после даты приоритета настоящей заявки, описаны соединения в качестве модуляторов GK. В международной патентной публикацииWO 2003047626, опубликованной после даты приоритета настоящей заявки, описано применение активатора GK в комбинации с антагонистом глюкагона для лечения диабета типа II. В международной патентной публикацииWO 2003055482, опубликованной после даты приоритета настоящей заявки, описаны амидные производные в качестве активаторов GK. В международной патентной публикацииWO 2003080585, опубликованной после даты приоритета настоящей заявки, описаны производные аминобензамида сGK-активностью для лечения диабета и ожирения. В международной патентной публикацииWO 2003097824, опубликованной после даты приоритета настоящей заявки, описаны кристаллы GK печени человека и их применение для разработки лекарств на основе структуры. В международной патентной публикацииWO 2004002481, опубликованной после даты приоритета настоящей заявки, раскрыты арилкарбонильные производные в качестве активаторов GK.-1 012700 Краткое изложение сущности изобретения Соединения, представленные формулой (I) или их фармацевтически приемлемые соли полезны в профилактическом или терапевтическом лечении гипергликемии и диабета типа II. Подробное описание изобретения Настоящее изобретение относится к соединению формулы (I). 1. Соединение формулы (I) или его фармацевтически приемлемой соли, гдеQ представляет собой тетрагидропиранил, пиперидинил, тетрагидротиопиранил, 1 оксотетрагидротиопиранил, или 1,1-диоксотетрагидротиопиранил; Т вместе c -N=C, к которому он присоединен, образует пиразолильное, имидазолильное, изоксазолильное, тиазолильное, тиадизолильное, пиридинильное, пиримидинильное или пиразинильное кольцо;m равно 0 или 1 и пунктирная линия вместе со сплошной линией образует необязательную двойную связь, иуказывает на то, что эта двойная связь имеет (E)-конфигурацию. В первом аспекте настоящее изобретение направлено на соединение, представленное формулой (Ia) и его фармацевтически приемлемую соль, где Q, T, R1-R6, m иявляются такими, как определено выше в формуле (I).-2 012700 Во втором аспекте настоящее изобретение направлено на соединение, представленное формулой и его фармацевтически приемлемую соль, где Q, T, R1-R6 и m являются такими, как определено выше в формуле (I). Молекулярная масса соединений формулы (I) предпочтительно меньше 800, более предпочтительно меньше 600, наиболее предпочтительно меньше 500. В настоящем изобретении пунктирная линия вместе со сплошной линией образует двойную связь. В другом воплощении пунктирная линия вместе со сплошной линией образует простую связь. В одном из воплощений пунктирная линия вместе со сплошной линией образует простую связь, и абсолютная конфигурация при асимметрическом центре в положенииотносительно карбонильного атома углерода амида представляет собой (R)-конфигурацию. В настоящем изобретении m предпочтительно равно 0. В настоящем изобретении Q предпочтительно представляет собой 4-тетрагидропиранил. В настоящем изобретении группа формулы предпочтительно представляет собой тиазолил, тиадиазолил, изоксазолил, пиримидинил, пиразинил или пиридинил. В одном из воплощений группа формулы предпочтительно представляет собой 2-пиразинил или 2-тиазолил. В настоящем изобретении R3 и R4 предпочтительно независимо выбраны из водорода, галогена и метила. Конкретные соединения по изобретению, которые могут быть упомянуты, представляют собой те соединения, которые описаны в примерах, в частности в примерах 1-201, и их фармацевтически приемлемые соли. В одном из воплощений настоящего изобретения конкретные соединения, которые могут быть упомянуты, выбраны из 2-(4-циклопропансульфонилфенил)-3-(тетрагидропиран-4-ил)-N-тиазол-2-илпропионамида,2-(4-циклопропансульфонилфенил)-N-(5-фтортиазол-2-ил)-3-(тетрагидропиран-4-ил)пропионамида,2-(4-циклопропансульфонилфенил)-N-(3-метил-[1,2,4]тиадиазол-5-ил)-3-(тетрагидропиран-4 ил)пропионамида,2-(4-циклопропансульфонилфенил)-N-пиразин-2-ил-3-(тетрагидропиран-4-ил)пропионамида,2-(4-циклопропансульфонилфенил)-3-(тетрагидропиран-4-ил)-N-[1,2,4]тиадиазол-5 илпропионамида,(Е)-2-(4-циклопропансульфонилфенил)-3-(тетрагидропиран-4-ил)-N-тиазол-2-илакриламида,2-(4-циклопропансульфонилфенил)-N-(5-формилтиазол-2-ил)-3-(тетрагидропиран-4 ил)пропионамида,(2R)-2-(4-циклопропансульфонилфенил)-N-(5-фтортиазол-2-ил)-3-(тетрагидропиран-4 ил)пропионамида,(2R)-2-(4-циклопропансульфонилфенил)-3-(тетрагидропиран-4-ил)-N-[1,2,4]тиадиазол-5 илпропионамида,(2R)-2-(4-циклопропансульфонилфенил)-N-пиразин-2-ил-3-(тетрагидропиран-4-ил)пропионамида,(2R)-2-(4-циклопропансульфонилфенил)-N-(5-фторпиридин-2-ил)-3-(тетрагидропиран-4-3 012700 ил)пропионамида,(2R)-2-(4-циклопропансульфонилфенил)-3-(тетрагидропиран-4-ил)-N-тиазол-2-илпропионамида,(2R)-2-(4-циклопропансульфонилфенил)-N-(3-метил-[1,2,4]тиадиазол-5-ил)-3-(тетрагидропиран-4 ил)пропионамида,(2R)-2-(4-циклобутансульфонилфенил)-N-пиразин-2-ил-3-(тетрагидропиран-4-ил)пропионамида,(2R)-2-(4-циклобутансульфонилфенил)-N-пиримидин-4-ил-3-(тетрагидропиран-4-ил)пропионамида,(2R)-2-(4-циклобутансульфонилфенил)-N-изоксазол-3-ил-3-(тетрагидропиран-4-ил)пропионамида,(2R)-2-(4-циклобутансульфонилфенил)-N-(1-метил-1H-пиразол-3-ил)-3-(тетрагидропиран-4 ил)пропионамида,(2R)-2-(4-циклобутансульфонилфенил)-N-(5-фтортиазол-2-ил)-3-(тетрагидропиран-4 ил)пропионамида,(Е)-2-(4-циклопропансульфонилфенил)-N-(5-фторпиридин-2-ил)-3-(тетрагидропиран-4-ил)акриламида,(Е)-2-(4-циклопропансульфонилфенил)-N-(5-фтортиазол-2-ил)-3-(тетрагидропиран-4-ил)акриламидаN-(5-цианотиазол-2-ил)-2-(4-циклопропансульфонилфенил)-3-(тетрагидропиран-4-ил)пропионамида и 2-(4-циклобутансульфонилфенил)-3-(тетрагидропиран-4-ил)-N-тиазол-2-илпропионамида или их фармацевтически приемлемой соли. В другом воплощении настоящего изобретения соединение выбрано из 2-(4-циклопропансульфонилфенил)-3-(тетрагидропиран-4-ил)-N-тиазол-2-илпропионамида,2-(4-циклопропансульфонилфенил)-N-(5-фтортиазол-2-ил)-3-(тетрагидропиран-4-ил)пропионамида,2-(4-циклопропансульфонилфенил)-N-(3-метил-[1,2,4]тиадиазол-5-ил)-3-(тетрагидропиран-4 ил)пропионамида,2-(4-циклопропансульфонилфенил)-N-пиразин-2-ил-3-(тетрагидропиран-4-ил)пропионамида,2-(4-циклопропансульфонилфенил)-3-(тетрагидропиран-4-ил)-N-[1,2,4]тиадиазол-5 илпропионамида,(Е)-2-(4-циклопропансульфонилфенил)-3-(тетрагидропиран-4-ил)-N-тиазол-2-илакриламида,(2R)-2-(4-циклопропансульфонилфенил)-N-(5-фтортиазол-2-ил)-3-(тетрагидропиран-4-ил)пропионамида,(2R)-2-(4-циклопропансульфонилфенил)-3-(тетрагидропиран-4-ил)-N-[1,2,4]тиадиазол-5 илпропионамида,(2R)-2-(4-циклопропансульфонилфенил)-N-пиразин-2-ил-3-(тетрагидропиран-4-ил)пропионамида,(2R)-2-(4-циклопропансульфонилфенил)-N-(5-фторпиридин-2-ил)-3-(тетрагидропиран-4 ил)пропионамида,(2R)-2-(4-циклопропансульфонилфенил)-3-(тетрагидропиран-4-ил)-N-тиазол-2-илпропионамида,(2R)-2-(4-циклопропансульфонилфенил)-N-(3-метил-[1,2,4]тиадиазол-5-ил)-3-(тетрагидропиран-4 ил)пропионамида,(2R)-2-(4-циклобутансульфонилфенил)-N-пиразин-2-ил-3-(тетрагидропиран-4-ил)пропионамида,(2R)-2-(4-циклобутансульфонилфенил)-N-(5-фтортиазол-2-ил)-3-(тетрагидропиран-4-ил)пропионамида,(Е)-2-(4-циклопропансульфонилфенил)-N-(5-фтортиазол-2-ил)-3-(тетрагидропиран-4 ил)акриламида,2-(4-циклопропилметансульфонилфенил)-3-(тетрагидропиран-4-ил)-N-тиазол-2-илпропионамида или их фармацевтически приемлемой соли. В другом воплощении настоящего изобретения соединение выбрано из(2R)-2-(4-циклобутансульфонилфенил)-N-пиразин-2-ил-3-(тетрагидропиран-4-ил)пропионамида,(2R)-2-(4-циклобутансульфонилфенил)-N-(5-фтортиазол-2-ил)-3-(тетрагидропиран-4-ил)пропионамида,(2R)-2-(4-циклопропансульфонилфенил)-N-(5-фтортиазол-2-ил)-3-(тетрагидропиран-4-ил)пропионамида,(2R)-2-(4-циклопропансульфонилфенил)-N-пиразин-2-ил-3-(тетрагидропиран-4-ил)пропионамида или их фармацевтически приемлемой соли. В одном из предпочтительных вариантов осуществления изобретения соединение представляет собой соединение формулы (I) или его фармацевтически приемлемую соль, гдеR4 означает водород или фтор;m равно 0 и пунктирная линия вместе со сплошной линией образует необязательную двойную связь, иуказывает на то, что эта двойная связь имеет (E)-конфигурацию. Хотя предпочтительные группы для каждой переменной в целом перечислены выше отдельно для каждой переменной, предпочтительные соединения по данному изобретению включают те, в которых некоторые переменные или каждая переменная в формуле (I) выбраны из предпочтительных, более предпочтительных или конкретно перечисленных групп для каждой переменной. Следовательно, в данное изобретение нужно включать все комбинации предпочтительных, более предпочтительных, наиболее предпочтительных, особенно или конкретно перечисленных групп. При использовании здесь, если не указано иное, алки, а также другие группы, имеющие префикс алк, такие как, например, алкокси, алканил, алкенил, алкинил и тому подобное, означает углеродные цепи, которые могут быть линейными или разветвленными, или их комбинации. Примеры алкильных групп включают метил, этил, пропил, изопропил, бутил, втор-бутил, трет-бутил, пентил, гексил, гептил и тому подобное. Алкенил, алкинил и другие подобные термины включают углеродные цепи, имеющие по меньшей мере одну ненасыщенную углерод-углеродную связь. При использовании здесь, например, С 0-4-алкил используют для обозначения алкила, имеющего 0-4 атома углерода, то есть 0, 1, 2, 3 или 4 атома углерода в неразветвленной или разветвленной конфигурации. Алкил, не имеющий атомов углерода, представляет собой водород, когда алкил представляет собой концевую группу. Алкил, не имеющий атомов углерода, представляет собой прямую связь, когда алкил представляет собой мостиковую (связывающую) группу. Термины циклоалкил и карбоциклическое кольцо означают карбоциклы, не содержащие гетероатомов, и включают моно-, би- и трициклические насыщенные карбоциклы, а также конденсированные и мостиковые системы. Такие конденсированные кольцевые системы могут включать одно кольцо,которое является частично или полностью ненасыщенным, такое как бензольное кольцо, с образованием конденсированных кольцевых систем, таких как бензоконденсированные карбоциклы. Циклоалкил включает такие конденсированные кольцевые системы, как спироконденсированные кольцевые системы. Примеры циклоалкильных и карбоциклических колец включают С 3-8-циклоалкил, такой как циклопропил, циклобутил, циклопентил, циклогексил и декагидронафталин, адамантан, инданил, 1,2,3,4 тетрагидронафталин и тому подобное. Термин галоген включает атомы фтора, хлора, брома и йода. Термин арил включает, например, фенил и нафтил. Если не указано иное, термин гетероциклическое кольцо включает 4-8-членные насыщенные кольца, содержащие один или два гетероатома, выбранные из кислорода, серы и азота. Гетероатомы непосредственно не присоединены друг к другу. Примеры гетероциклических колец включают оксетан,тетрагидрофуран, тетрагидропиран, оксепан, оксокан, тиетан, тетрагидротиофен, тетрагидротиопиран,тиепан, тиокан, азетидин, пирролидин, пиперидин, азепан, азокан, [1,3]диоксан, оксазолидин, пиперазин и тому подобное. Другие примеры гетероциклических колец включают окисленные формы серосодержащих колец. Таким образом, тетрагидротиофен-1-оксид, тетрагидротиофен-1,1-диоксид, тетрагидротиопиран-1-оксид и тетрагидротиопиран-1,1-диоксид также считают гетероциклическими кольцами. Если не указано иное, термин гетероарил включает 5- или 6-членные гетероарильные кольца, содержащие 1-4 гетероатома, выбранные из кислорода, серы и азота. Примерами таких гетероарильных колец являются фурил, тиенил, пирролил, пиразолил, имидазолил, оксазолил, изоксазолил, тиазолил,изотиазолил, триазолил, оксадиазолил, тиадиазолил, тетразолил, пиридинил, пиридазинил, пиримидинил,-5 012700 пиразинил и триазинил. Приведенные выше формулы показаны без определенной стереохимии некоторых положений. Настоящее изобретение включает все стереоизомеры (например, геометрические изомеры, оптические изомеры, диастереоизомеры и так далее) и их фармацевтически приемлемые соли, за исключением того, что конкретно изображено или указано иначе. Кроме того, включены также как смеси стереоизомеров, так и отдельные конкретные стереоизомеры, за исключением того, что конкретно изображено или указано иначе. В процессе методик синтеза, используемых для получения таких соединений, или при использовании методик рацемизации или эпимеризации, известных специалистам в данной области техники, продукты таких методик могут представлять собой смесь стереоизомеров. Когда существует таутомер соединения приведенных выше формул, в настоящее изобретение включены все возможные таутомеры и их фармацевтически приемлемые соли, а также их смеси, за исключением того, что конкретно изображено или указано иначе. Когда соединение приведенных выше формул и его фармацевтически приемлемые соли существуют в форме сольватов или полиморфных форм, в настоящее изобретение включены все возможные сольваты и полиморфные формы. Тип растворителя, который образует сольват, конкретно не ограничен, при условии, что этот растворитель является фармакологически приемлемым. Например,можно использовать воду, этанол, пропанол, ацетон или тому подобное. Поскольку соединения формулы (I) предназначены для фармацевтического применения, они предпочтительно представлены, по существу, в чистой форме, например по меньшей мере на 60% чистой,более пригодно по меньшей мере на 75% чистой, особенно по меньшей мере на 98% чистой (мас./мас.% на основании массы). В изобретение также включена фармацевтическая композиция, которая содержит соединение формулы (I) или его фармацевтически приемлемую соль в комбинации с фармацевтически приемлемым носителем. Предпочтительно эта композиция содержит фармацевтически приемлемый носитель и нетоксичное терапевтически эффективное количество соединения формулы (I), как описано выше, или его фармацевтически приемлемой соли. Кроме того, в объем данного предпочтительного воплощения изобретения включена фармацевтическая композиция для профилактики или лечения гипергликемии и диабета посредством активации GK,содержащая фармацевтически приемлемый носитель и нетоксичное терапевтически эффективное количество соединения формулы (I), как описано выше, или его фармацевтически приемлемой соли. В изобретении также предложено применение соединения формулы (I) или его фармацевтически приемлемой соли в качестве фармацевтического агента. Соединения и композиции по настоящему изобретению эффективны для лечения гипергликемии у млекопитающих, таких как, например, люди. В изобретении также предложен способ профилактического или терапевтического лечения состояния, где желательна активация GK, включающий стадию введения эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли. В изобретении также предложен способ профилактического или терапевтического лечения гипергликемии или диабета, включающий стадию введения эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли. В изобретении также предложен способ предупреждения диабета у человека, проявляющего преддиабетическую гипергликемию или нарушенную толерантность к глюкозе, включающий стадию введения эффективного профилактического количества соединения формулы (I) или его фармацевтически приемлемой соли. В изобретении также предложено применение соединения формулы (I) или его фармацевтически приемлемой соли в качестве активатора GK. В изобретении также предложено применение соединения формулы (I) или его фармацевтически приемлемой соли для профилактического или терапевтического лечения гипергликемии или диабета. В изобретении также предложено применение соединения формулы (I) или его фармацевтически приемлемой соли для предупреждения диабета у человека, проявляющего преддиабетическую гипергликемию или нарушенную толерантность к глюкозе. В изобретении также предложено применение соединения формулы (I) или его фармацевтически приемлемой соли в производстве лекарства для активации GK. В изобретении также предложено применение соединения формулы (I) или его фармацевтически приемлемой соли в производстве лекарства для профилактического или терапевтического лечения гипергликемии или диабета. В изобретении также предложено применение соединения формулы (I) или его фармацевтически приемлемой соли в производстве лекарства для предупреждения диабета у человека, проявляющего преддиабетическую гипергликемию или нарушенную толерантность к глюкозе. Соединения и композиции по настоящему изобретению возможно можно использовать в комбинации с одним или более чем одним другим противодиабетическим агентом или антигипергликемическим агентом, которые включают, например, сульфонилмочевины (например, глибурид, глимепирид, глипи-6 012700 рид, глипизид, хлорпропамид, гликлазид, глизоксепид, ацетогексамид, глиборнурид, толбутамид, толазамид, карбутамид, гликвидон, глигексамид, фенбутамид, толцикламид и так далее), бигуаниды (например, метформин, фенформин, буформин и так далее), антагонисты глюкагона (например, пептидный или непептидный антагонист глюкагона), ингибиторы глюкозидазы (например, акарбоза, миглитол и так далее), вещества, способствующие секреции инсулина, сенсибилизаторы инсулина (например, троглитазон,розиглитазон, пиоглитазон и так далее) и тому подобное, либо агенты против ожирения (например, сибутрамин, орлистат и так далее) и тому подобное. Соединения и композиции по настоящему изобретению и другие противодиабетические агенты или антигипергликемические агенты можно вводить одновременно, последовательно или по отдельности. Термин фармацевтически приемлемые соли относится к солям, полученным из фармацевтически приемлемых нетоксичных оснований или кислот. Когда соединение по настоящему изобретению является кислым, его соответствующую соль можно удобно получить из фармацевтически приемлемых нетоксичных оснований, включая неорганические основания и органические основания. Соли, полученные из таких неорганических оснований, включают алюминиевые, аммониевые, кальциевые, содержащие двухвалентную медь, содержащие одновалентную медь, содержащие трехвалентное железо, содержащие двухвалентное железо, литиевые, магниевые, содержащие трехвалентный марганец, содержащие двухвалентный марганец, калийные, натриевые, цинковые и подобные соли. Особенно предпочтительными являются аммониевые, кальциевые, магниевые, калиевые и натриевые соли. Соли, образованные из фармацевтически приемлемых органических нетоксичных оснований, включают соли первичных, вторичных и третичных аминов, а также циклических аминов и замещенных аминов, таких как природные или синтетические амины. Другие фармацевтически приемлемые органические нетоксичные основания, из которых можно образовать соли, включают, например, аргинин, бетаин, кофеин, холин, N',N'дибензилэтилендиамин, диэтиламин, 2-диэтиламиноэтанол, 2-диметиламиноэтанол, этаноламин, этилендиамин, N-этилморфолин, N-этилпиперидин, глюкамин, глюкозамин, гистидин, изопропиламин, лизин,метилглюкамин, морфолин, пиперазин, пиперидин, полиаминные смолы, прокаин, пурины, теобромин,триэтиламин, триметиламин, трипропиламин, трометамин и тому подобное. Когда соединение по настоящему изобретению является основным, его соответствующую соль можно удобно получить из фармацевтически приемлемых нетоксичных кислот, включая неорганические и органические кислоты. Такие кислоты включают, например, уксусную, бензолсульфоновую, бензойную, камфарсульфоновую, лимонную, этансульфоновую, фумаровую, глюконовую, глутаминовую, бромоводородную, соляную, изэтионовую, молочную, малеиновую, яблочную, миндальную, метансульфоновую, слизевую, азотную, памовую, пантотеновую, фосфорную, янтарную, серную, винную, паратолуолсульфоновую кислоту и тому подобное. Особенно предпочтительны лимонная, бромоводородная, соляная, малеиновая, фосфорная, серная, метансульфоновая и винная кислоты. Фармацевтические композиции по настоящему изобретению содержат соединение формулы (I) или его фармацевтически приемлемую соль в качестве активного ингредиента, фармацевтически приемлемый носитель и возможно другие терапевтические ингредиенты или адъюванты. Эти композиции включают композиции, пригодные для перорального, ректального, местного и парентерального (включая подкожное, внутримышечное и внутривенное) введения, а также для введения посредством ингаляции, хотя наиболее подходящий путь в любом данном случае будет зависеть от конкретного хозяина, а также от природы и тяжести состояний, для которых вводят активный ингредиент. Фармацевтические композиции могут быть удобно представлены в виде стандартной лекарственной формы и изготовлены любым из способов, известных в области фармацевтики. Фармацевтические композиции по изобретению предпочтительно адаптированы для перорального введения. На практике соединения формулы (I) или их фармацевтически приемлемые соли по данному изобретению можно объединять в качестве активного ингредиента в гомогенной смеси с фармацевтическим носителем в соответствии с общепринятыми фармацевтическими методиками смешивания. Носитель может принимать широкое разнообразие форм в зависимости от формы препарата, желательной для введения, например перорального или парентерального (включая внутривенный). Таким образом, фармацевтические композиции по настоящему изобретению могут быть представлены в виде отдельных единиц, пригодных для перорального введения, таких как капсулы, крахмальные облатки или таблетки, каждая из которых содержит предварительно определенное количество активного ингредиента. Кроме того, эти композиции могут быть представлены в виде порошка, в виде гранул, в виде раствора, в виде суспензии в водной жидкости, в виде неводной жидкости, в виде эмульсии "масло в воде" или в виде жидкой эмульсии "вода в масле". В дополнение к распространенным лекарственным формам, изложенным выше, соединение, представленное формулой (I), или его фармацевтически приемлемую соль можно также вводить с помощью средств с регулируемым высвобождением и/или устройств для доставки. Эти композиции можно готовить любыми способами фармацевтики. В целом такие способы включают стадию приведения в контакт активного ингредиента с носителем, который содержит один или более чем один необходимый ингредиент. В целом композиции готовят путем однородного и тесного смешивания активного ингредиента с жидкими носителями, либо тонкоизмельченными твердыми носителями, либо с-7 012700 теми и с другими. Затем продукту можно придать удобную форму в желаемом виде. Таким образом, фармацевтические композиции по данному изобретению могут включать фармацевтически приемлемый носитель и соединение формулы (I) или его фармацевтически приемлемую соль. Соединения формулы (I) или их фармацевтически приемлемые соли могут быть также включены в фармацевтические композиции в комбинации с одним или более чем одним другим терапевтически активным соединением. Фармацевтические композиции по данному изобретению включают фармацевтически приемлемую липосомную композицию, содержащую соединение формулы (I) или его фармацевтически приемлемую соль. Используемый фармацевтический носитель может представлять собой, например, твердое вещество, жидкость или газ. Примеры твердых носителей включают лактозу, каолин, сахарозу, тальк, желатин,агар, пектин, аравийскую камедь, стеарат магния и стеариновую кислоту. Примерами жидких носителей являются сахарный сироп, арахисовое масло, оливковое масло и вода. Примеры газообразных носителей включают диоксид углерода и азот. При изготовлении композиций для пероральной лекарственной формы можно использовать любые удобные фармацевтические среды. Например, воду, гликоли, масла, спирты, корригенты, консерванты,красящие агенты и тому подобное можно использовать для образования пероральных жидких препаратов, таких как суспензии, эликсиры и растворы; тогда как носители, такие как крахмалы, сахара, микрокристаллическую целлюлозу, разбавители, агенты для гранулирования, смазывающие агенты, связывающие агенты, разрыхляющие агенты и тому подобное можно использовать для образования пероральных твердых препаратов, таких как порошки, капсулы и таблетки. В связи с легкостью введения таблетки и капсулы являются предпочтительными пероральными лекарственными формами, где используют твердые фармацевтические носители. Возможно, таблетки могут быть покрыты с помощью стандартных водных или неводных методик. Таблетка, содержащая композицию по данному изобретению, может быть получена путем прессования или формования, возможно, с одним или более чем одним вспомогательным ингредиентом или адъювантом. Прессованные таблетки могут быть получены путем прессования на подходящей машине активного ингредиента в сыпучей форме, такой как порошок или гранулы, возможно, смешанного со связывающим агентом, смазывающим агентом, инертным разбавителем, поверхностно-активным или диспергирующим агентом либо другим таким эксципиентом. Эти эксципиенты могут представлять собой, например, инертные разбавители, такие как карбонат кальция, карбонат натрия, лактозу, фосфат кальция или фосфат натрия; агенты для гранулирования и разрыхляющие агенты, например кукурузный крахмал или альгиновую кислоту; связывающие агенты, например крахмал, желатин или аравийскую камедь, и смазывающие агенты, например стеарат магния, стеариновую кислоту или тальк. Таблетки могут быть непокрытыми либо они могут быть покрыты с помощью известных методик для замедления распадаемости и всасывания в желудочно-кишечном тракте и обеспечивать тем самым пролонгированное действие в течение длительного времени. Например, можно использовать вещество для замедления времени, такое как глицерилмоностеарат или глицерилдистеарат. В твердых желатиновых капсулах активный ингредиент смешан с инертным твердым разбавителем,например карбонатом кальция, фосфатом кальция или каолином. В мягких желатиновых капсулах активный ингредиент смешан с водой или масляной средой, например арахисовым маслом, вазелиновым маслом или оливковым маслом. Формованные таблетки могут быть получены путем формования на подходящем аппарате смеси порошкообразного соединения, увлажненного инертным жидким разбавителем. Каждая таблетка предпочтительно содержит от примерно 0,05 мг до примерно 5 г активного ингредиента, и каждая крахмальная облатка или капсула предпочтительно содержат от примерно 0,05 мг до примерно 5 г активного ингредиента. Например, композиция, предназначенная для перорального введения людям, может содержать от примерно 0,5 мг до примерно 5 г активного агента, смешанного с подходящим и удобным количеством вещества-носителя, которое может варьировать от примерно 5 до примерно 95% от всей композиции. Стандартные лекарственные формы будут, как правило, содержать между примерно 1 мг до примерно 2 г активного ингредиента, типично 25, 50, 100, 200, 300, 400, 500, 600, 800 или 1000 мг. Фармацевтические композиции по настоящему изобретению, пригодные для парентерального введения, можно готовить в виде растворов или суспензий активных соединений в воде. Можно включать подходящий сурфактант, такой как, например, гидроксипропилцеллюлоза. Дисперсии можно также готовить в глицерине, жидких полиэтиленгликолях и их смесях в маслах. Кроме того, можно включать консервант для предотвращения вредного роста микроорганизмов. Фармацевтические композиции по настоящему изобретению, пригодные для инъекционного применения, включают стерильные водные растворы или дисперсии. Кроме того, композиции могут находиться в форме стерильных порошков для приготовления таких стерильных инъекционных растворов или дисперсий непосредственно перед применением. Во всех случаях готовая инъекционная форма должна быть стерильной и должна быть эффективно текучей для легкого набора в шприц. Эти фармацевтические композиции должны быть стабильными в условиях производства и хранения; таким образом,-8 012700 они предпочтительно должны быть защищены от загрязняющего действия микроорганизмов, таких как бактерии и грибы. Носитель может представлять собой растворитель или дисперсионную среду, содержащие, например, воду, этанол, полиол (например, глицерин, пропиленгликоль и жидкий полиэтиленгликоль), растительные масла и их подходящие смеси. Фармацевтические композиции по настоящему изобретению могут находиться в форме, пригодной для местного применения, такой как, например, аэрозоль, крем, мазь, лосьон, присыпка или тому подобное. Кроме того, композиции могут находиться в форме, пригодной для применения в чрескожных устройствах. Эти препараты можно готовить, используя соединение формулы (I) или его фармацевтически приемлемую соль, посредством способов общепринятой обработки. Например, крем или мазь готовят путем смешивания гидрофильного вещества и воды вместе с соединением в количестве от примерно 5 до примерно 10 мас./мас.% с получением крема или мази, имеющих желаемую консистенцию. Фармацевтические композиции по настоящему изобретению могут находиться в форме, пригодной для ректального введения, где носитель представляет собой твердое вещество. Предпочтительно, чтобы смесь образовала стандартную лекарственную форму суппозитории. Пригодные носители включают масло какао и другие вещества, обычно используемые в данной области техники. Суппозитории можно удобно образовать сначала путем смешивания композиции с размягченным(и) или расплавленным(и) носителем(ями) с последующим охлаждением и приданием формы. Фармацевтические композиции по настоящему изобретению могут находиться в форме, пригодной для ингаляционного введения. Такое введение может осуществляться в формах и использовать носители,описанные, например, в 1) Particulate Interactions in Dry Powder Formulations for Inhalation, Xian Zeng etDekker, 3) Respiratory Drug Delivery, 1990, Editor: P.R. Byron, CRC Press. В дополнение к вышеупомянутым ингредиентам-носителям фармацевтические композиции, описанные выше, могут включать, как пригодно, один или более чем один дополнительный ингредиентноситель, такой как разбавители, буферы, корригенты, связывающие агенты, поверхностно-активные агенты, загустители, смазывающие агенты, консерванты (включая антиоксиданты) и тому подобное. Кроме того, можно включать другие адъюванты, чтобы сделать препарат изотоническим с кровью предназначенного реципиента. Композиции, содержащие соединение формулы (I) или его фармацевтически приемлемые соли, можно также готовить в порошкообразной или жидкой концентрированной форме. Как правило, уровни дозировки порядка от примерно 0,01 до примерно 150 мг/кг массы тела в сутки полезны при лечении вышеуказанных состояний либо, альтернативно, от примерно 0,5 мг до примерно 10 г на пациента в сутки. Например, диабет можно эффективно лечить путем введения от примерно 0,01 до 100 мг соединения на килограмм массы тела в сутки или альтернативно от 0,5 мг до примерно 7 г на пациента в сутки. Должно быть понятно, однако, что конкретный уровень дозировки для каждого конкретного пациента будет зависеть от ряда факторов, включая возраст, массу тела, общее состояние здоровья, пол, режим питания, время введения, путь введения, скорость выведения, комбинацию лекарств и тяжесть заболевания у конкретного пациента с диабетом, подлежащего лечению. Кроме того, должно быть понятно,что соединения и их соли по данному изобретению можно вводить в субтерапевтических уровнях профилактически при предупреждении гипергликемического состояния. Соединения формулы (I) могут проявлять полезные свойства по сравнению с известными активаторами глюкокиназы, например как проиллюстрировано в анализах, описанных здесь. Конкретные соединения по изобретению могут проявлять улучшенные значения для Km, Vmax, ЕС 50, максимальной активации (концентрация глюкозы = 5 мМ) и/или максимального снижения глюкозы в крови на базальных уровнях глюкозы (например, у мышей C57BL/6J) или другие полезные фармакологические свойства по сравнению с известными активаторами GK. Экспериментальная часть В соответствии с данным изобретением соединения формулы (Ia) могут быть получены, следуя протоколу, проиллюстрированному ниже на схеме 1 Схема 1 где Q, Т, R1-R6, m иявляются такими, как описано выше, a R11 представляет собой С 0-4-алкил. Альдегиды II и фенилуксусные кислоты или сложные эфиры III имеются в продаже или могут быть легко получены, используя известные методики. Когда Q представляет собой ароматическое или гетероароматическое кольцо, IV можно получить с помощью реакции Перкина (G. Karminski-Zamola et al., Tetrahedron 1982, 38, 1329-1335). В данной реакции II конденсируют с фенилуксусной кислотой III(R11 = С 0-алкил) в присутствии ангидрида карбоновой кислоты, например уксусного ангидрида, и третичного аминного основания, например триэтиламина, при температуре дефлегмации с получением акриловой кислоты IV. Альтернативно, IV может быть получена посредством конденсации II и III(R11 = С 0-алкил) под действием аминного основания, такого как пиперидин, в толуоле при кипячении с обратным холодильником (D. Deschenes et al., WO 01/46151). Когда Q представляет собой гетероциклическое кольцо, -карбанион фенилуксусного эфира III (R11 = С 1-4-алкил), образованный при -78 С, например, в тетрагидрофуране сильным основанием, например диизопропиламидом лития, можно конденсировать со II с получением ,-ненасышенного эфира (Т. Severin et al. Chem. Ber. 1985, 118, 4760-4773),который можно подвергнуть омылению, используя, например, гидроксид натрия (W.L. Corbett et al.,WO 01/44216), с получением IV.,-Ненасыщенные карбоновые кислоты IV можно конденсировать с гетероароматическими аминами V, многие из которых имеются в продаже, используя разнообразные условия сочетания, например карбодиимид-1-гидроксибензотриазол на полимерной подложке в N,N-диметилформамиде при 20 С (репрезентативные методики см. http://www.argotech.com/PDF/resins/ps carbodiimide.pdf, и доступны от Argonaut Technologies, Inc., Foster City, California), с получением (la). Соединения формулы (lb) могут быть получены путем, изображенным ниже на схеме 2 Схема 2CO2CH2Ph, a X представляет собой хлоро, бромо, йодо или -OSO2R12; где R11 является таким, как описано выше, a R12 представляет собой С 1-4-алкил, возможно замещенный одним или более чем одним атомом фтора, или возможно замещенный арил. Галогениды и сульфонатные эфиры VI имеются в продаже или могут быть легко получены, используя известные методики. Эти алкилирующие агенты можно подвергнуть взаимодействию с дианионами фенилуксусных кислот VII, образованными при -78 С в тетрагидрофуране с 2 эквивалентами сильного основания, такого как диизопропиламид лития, с образованием VIII непосредственно (F.Т. Bizzarro et al.,WO 00/58293). Альтернативно, -карбанион фенилуксусного эфира VII, образованный при -78 С в тетрагидрофуране сильным основанием, таким как бис(триметилсилил)амид лития (L. Snyder et al., J. Org.Chem., 1994, 59, 7033-7037), можно алкилировать VI с получением -замещенных эфиров. Омыление этих эфиров с использованием, например, гидроксида натрия в водном метаноле при температуре от 20 С до температуры дефлегмации приводит к образованию карбоновых кислот VIII. Карбоновые кислоты VIII можно конденсировать с гетероароматическими аминами V, используя разнообразные условия сочетания, например карбодиимид-1-гидроксибензотриазол на полимерной подложке в N,Nдиметилформамиде при 20 С (репрезентативные методики см. http://www.arqotech.com/PDF/resins/ps carbodiimide.pdf, и доступны от Argonaut Technologies, Inc., Foster City, California) с получением амидов (Ib). Соединение формулы (Ib) имеет асимметрический атом углерода, который связывает между собой карбонильный атом углерода амида, арильное кольцо и Q-содержащую боковую цепь. В соответствии с данным изобретением предпочтительной стереоконфигурацией при этом асимметрическом центре является (R). Если желательно выделить чистые (R)- или (S)-стереоизомеры соединения формулы (Ib), возможно разделить рацемическую смесь хирального карбоново-кислотного предшественника VIII любыми общепринятыми химическими методами, а затем конденсировать энантиомерночистые карбоновые кислоты с амином формулы V, используя реагент, который вызывает пренебрежимо малую рацемизацию. Для иллюстрации рацемическое соединение VIII можно конденсировать с хиральным производным оксазолидинона (см., например, F.Т. Bizzarro et al. WO 00/58293) с образованием смеси диастереоизомерных имидов, которую можно разделить любым общепринятым способом, например колоночной хроматографией. В результате гидролиза чистых имидов образуется стереомерночистые (R)- и (S)-карбоновые кислоты,которые можно затем конденсировать с гетероциклическими аминами V, используя реагент, который минимизирует рацемизацию хирального центра, например гексафторфосфат бензотриазол-1 илокситрис(пирролидино)фосфония (J. Coste et al. Tetrahedron Lett. 1990, 31, 205-208), с образованием энантиомерночистых (R)- или (S)-амидов формулы (Ib). Альтернативно, рацемическую смесь амидов формулы (Ib) можно разделить с помощью хиральной высокоэффективной жидкостной хроматографии,используя хиральную стационарную фазу, которую можно приобрести, например, у Daicel Chemical Industries, Ltd, Tokyo, Japan. Дополнительные подробности получения соединений формулы (I) находятся в примерах.- 10012700 Соединения формулы (I) можно получать по отдельности или в виде библиотек соединений, содержащих по меньшей мере 2, например от 5 до 1000, соединений и более предпочтительно от 10 до 100 соединений формулы (I). Библиотеки соединений могут быть получены с помощью комбинаторного подхода "разделения и смешивания" либо путем множественного параллельного синтеза с использованием либо химии растворов, либо твердофазной химии с использованием методик, известных специалистам в данной области техники. Во время синтеза соединений формулы (I) лабильные функциональные группы в промежуточных соединениях, например гидрокси-, карбокси- и аминогруппы, можно защитить. Защитные группы могут быть удалены на любой стадии синтеза соединений формулы (I) или могут присутствовать на конечном соединении формулы (I). Всестороннее обсуждение путей, при которых различные лабильные функциональные группы можно защитить, и способов расщепления полученных в результате защищенных производных приведено, например, в Protective Groups in Organic Chemistry, T.W. Greene and P.G.M. Wuts(1991) Wiley-lnterscience, New York, 2nd edition. Любые новые промежуточные соединения, как определено выше, также включены в объем изобретения. Согласно другому аспекту изобретения предложено соединение формулы (IV) и применение таких соединений в синтезе активаторов GKm равно 0 иуказывает на то, что эта двойная связь имеет (E)-конфигурацию. Согласно предпочительному варианту осуществления изобретения соединение формулы (IV) выбрано из(Е)-2-(4-циклопропансульфонилфенил)-3-(тетрагидропиран-4-ил)акриловой кислоты,(Е)-2-(4-циклопропансульфинилфенил)-3-(тетрагидропиран-4-ил)акриловой кислоты. Согласно еще одному аспекту изобретения предложено соединение формулы (VIII) и применение таких соединений в синтезе активаторе GKm равно 0. Согласно предпочительному варианту осуществления изобретения соединение формулы (VIII) выбрано из 2-(4-циклопропансульфонилфенил)-3-(тетрагидропиран-4-ил)пропионовой кислоты; 2-(4-циклобутансульфонилфенил)-3-(тетрагидропиран-4-ил)пропионовой кислоты;(2R)-2-(4-циклобутансульфонилфенил)-3-(тетрагидропиран-4-ил)пропионовой кислоты. Конкретные соединения формул (IV) и (VIII) включают соединения, описанные в подготовительных примерах. Согласно другому аспекту изобретения также предложен 5-фтортиазол-2-иламин либо его амид,или соль присоединения кислоты. В частности, в изобретении предложены амиды и соли присоединения кислоты данного соединения. Подходящие соли присоединения кислоты включают соли, образованные с неорганическими и органическими кислотами. Такие кислоты включают, например, уксусную, трифторуксусную, бензолсульфоновую, бензойную, камфарсульфоновую, лимонную, этансульфоновую, фумаровую, глюконовую, глутаминовую, бромоводородную, соляную, фтороводородную, изэтионовую, молочную, малеиновую, яблочную, миндальную, метансульфоновую, слизевую, азотную, памовую, пантотеновую, фосфорную, янтарную, серную, винную, паратолуолсульфоновую кислоту, трифторметансульфоновую и тому подобное. Особенно предпочтительны гидрогалогенидные соли, особенно гидрохлорид. Амиды и соли присоединения кислоты 5-фтортиазол-2-иламина могут быть полезны в качестве промежуточных соединений для синтеза соединений формулы (I) или могут сами по себе действовать в качестве активаторов GK и, следовательно, иметь применение в профилактическом или терапевтическом лечении гипергликемии и диабета типа II. Все публикации, включая патенты и патентные заявки, цитируемые в данном описании, но не ограничиваясь ими, включены здесь путем ссылки, так что каждая индивидуальная публикация конкретно и индивидуально указана как включенная здесь путем ссылки. Материалы и методы Микроволновые реакции проводили в системе СЕМ Explorer при 100 Вт. Колоночную хроматографию проводили на SiO2 (40-63 меш), если не указано иное. Данные LCMS (жидкостной хроматографии масс-спектрометрии) получали, используя два способа: способ А: колонка Waters Symmetry C18 3,5(2,130,0 мм, скорость потока = 0,8 мл/мин), элюция раствором (5% MeCN в H2O)-MeCN, содержащим 0,1% HCO2H, в течение 6 мин и УФ детекция при 220 нм. Информация о градиенте: 0,0-1,2 мин: 100%(5% MeCN в H2O); 1,2-3,8 мин: повышали до 10% (5% MeCN в Н 2 О)-90% MeCN; 3,8-4,4 мин: держали при 10% (5% MeCN в Н 2 О)-90% MeCN; 4,4-5,5 мин: повышали до 100% MeCN; 5,5-6,0 мин: возвращались к 100% (5% MeCN в H2O). Способ В: колонка Phenomenex Mercury Luna C18 3(2,010,0 мм, скорость потока = 1,5 мл/мин), элюция раствором (5% MeCN в H2O)-MeCN (от 4:1 до 1:4), содержащим 0,1% HCO2H, в течение 2,95 мин и использовали детекцию с диодной матрицей. Масс-спектры для обоих способов А и В получали, используя источник ионизации электрораспылением в режиме либо положительного иона (ES+), либо отрицательного иона (ES-). Спектры химической ионизации при атмосферном давлении (APCI) получали на приборе FumiganMat SSQ 7000C. Синтез приведенных ниже соединений описан ранее: 2-амино-5-хлор-4-метилтиазол; S. Kyoichi et al. ЕР 412404; 2-амино-5-формилтиазол: М.D.(3R)-3-(тозилокси)тетрагидрофуран: А. Bouzide et al. Tetrahedron Lett. 2001, 42, 8781-8783; (3S)-3(тозилокси)тетрагидрофуран: F.J.A. Hundscheid et al. Tetrahedron 1987, 43, 5073-5088; 3(тозилокси)оксетан: K. Baum et al. J. Org. Chem. 1983, 48, 2953-2956. (Е)-2-Фенил-3-тиофен-2 илакриловую кислоту покупали у Maybridge (Tintagel, UK). Сокращения и акронимы: Ас: ацетил; i-Am: изопентил; АТФ: аденозин-5'-трифосфат; ВОР: гексафторфосфат бензотриазол-1-илокситрис(диметиламино)фосфония; n-Bu: н-бутил; t-Bu: трет-бутил; Bz: бензоил; dba: дибензилиденацетон; DIPEA: N,N-диизопропилэтиламин; DMAc: N,N-диметилацетамид; ДМЭ: 1,2-диметоксиэтан; ДМФ: N,N-диметилформамид; DMPU: 1,3-диметил-3,4,5,6-тетрагидро-2(1 Н)пиримидинон; ДМСО: диметилсульфоксид; DPEPhos: бис(2-дифенилфосфинофенил)эфир; EDCI: гидрохлорид 1-(3-диметиламинопропил)-3-этилкарбодиимида; Et: этил; FA: кратность активации; GK: глюкокиназа; Glc: глюкоза; G6P: глюкозо-6-фосфат; G6PDH: глюкозо-6-фосфатдегидрогеназа; GST-GK: слитый белок глутатион-S-трансфераза-глюкокиназа; HATU: гексафторфосфат O-(7-азабензотриазол-1-ил)N,N,N',N'-тетраметилурония; HOBt: 1-гидроксибензотриазол; IH: изогексан; i-Pr: изопропил; LDA: диизопропиламид лития; LHMDS: бис(триметилсилил)амид лития; mCPBA: 3-хлорпероксибензойная кислота; Me: метил; т. пл.: точка плавления; НАДФ(Н): -никотинамидадениндинуклеотидфосфат (восстановленный); NBS: N-бромсукцинимид; Ph: фенил; PS: на полимерной подложке; Rf: фактор удерживания;RT: время удерживания; RTA: время удерживания при способе A; RTB: время удерживания при способе В; ОФ-ВЭЖХ: высокоэффективная жидкостная хроматография с обращенной фазой; ТВА-ОХ: тетрабутиламмонийоксон; ТФУ: трифторуксусная кислота; ТФА: трифторуксусный ангидрид; TFFH: гексафторфосфат фтор-N,N,N',N'-тетраметилформамидиния; ТГФ: тетрагидрофуран. Подготовительный пример 1. (E)-2-(4-Метансульфонилфенил)-3-тиофен-3-илакриловая кислота Смесь 3-тиофенкарбоксальдегида (1,40 мл, 15,0 ммоль), (4-метансульфонилфенил)уксусной кислоты (3,23 г, 15,0 ммоль) и пиперидина (0,45 мл, 4,5 ммоль) в PhMe (21 мл) нагревали с обратным холодильником при перемешивании в течение 16 ч. После охлаждения PhMe декантировали с маслянистого твердого вещества, которое осело на дне реакционного сосуда. Это твердое вещество распределяли между 1 М HCl (60 мл) и EtOAc (400 мл), затем водную фазу дополнительно экстрагировали EtOAc (60 мл). Объединенные органические слои промывали H2O (60 мл), предварительно встряхнув с насыщенным водным Na2CO3 (100 мл). Полученную в результате эмульсию фильтровали через целит. Органический слой дополнительно экстрагировали насыщенным водным Na2CO3 (2100 мл). Объединенные водные слои промывали Et2O (80 мл), предварительно профильтровав через целит и осторожно подкислив АсОН до доведения рН до 4. Образовавшийся беловатый осадок собирали, тщательно промывали H2O и сушили на воздухе с получением соединения, указанного в заголовке: m/z (ES+) = 634,2 [2 М+ NH4]+. Несколько других акриловых кислот были получены (табл. 1) катализируемой пиперидином конденсацией (4-метансульфонилфенил)уксусной кислоты с соответствующим гетероароматическим альдегидом, как описано в подготовительном примере 1. Таблица 1 Смесь 4-бромфенилуксусной кислоты (12,90 г, 60,0 ммоль), 2-фуранкарбоксальдегида (6,0 мл,72,0 ммоль), NEt3 (12,0 мл, 86,4 ммоль) и Ac2O (12,0 мл, 127,2 ммоль) нагревали при 140 С (баня) при перемешивании в течение 13/4 ч. Эту реакционную смесь охлаждали на ледяной бане, предварительно обработав 2 М HCl (30 мл) для доведения рН до 1. Твердое вещество выпадало в осадок из раствора. Это твердое вещество экстрагировали Et2O (500 мл). Слой Et2O промывали H2O (100 мл), предварительно экстрагировав 5 мас./об.% водным раствором Na2CO3 (5100 мл). Водные экстракты промывали Et2O (2- 1301270050 мл), предварительно осторожно подкислив АсОН до рН 6. Образовавшееся твердое вещество кремового цвета собирали, промывали Н 2 О и перекристаллизовали из MeOH-H2O с получением соединения,указанного в заголовке: m/z (ES+) = 604,0 [2M+NH4]+. Конденсацию Перкина, используя NEt3 и Ac2O, использовали для получения других акриловых кислот (табл. 2) из соответствующей арилуксусной кислоты и (гетеро)ароматического альдегида, как описано в подготовительном примере 9. Таблица 2SOCl2 (8,2 мл, 112,0 ммоль) добавляли к перемешиваемой суспензии (4-метансульфонилфенил)уксусной кислоты (20,00 г, 93,3 ммоль) в EtOH (80 мл) при -10 С. Этой смеси давали нагреться до 20 С в течение 16 ч, затем растворители удаляли при пониженном давлении. Остаток растворяли вEtOAc, затем полученный в результате раствор промывали H2O до тех пор, пока рН водной фазы не стал нейтральным. Раствор EtOAc дополнительно промывали насыщенным водным Na2CO3, предварительно просушив (MgSO4). В результате фильтрования и выпаривания растворителя получили соединение, указанное в заголовке: m/z (ES+) = 284,1 [M+MeCN+H]+. Подготовительный пример 16. Этил-(4-метилсульфанилметилфенил)ацетат(4-Метилсульфанилметилфенил)уксусную кислоту (2,00 г, 10,2 ммоль) этерифицировали, как описано выше в подготовительном примере 15, с получением соединения, указанного в заголовке: m/z (ES+)(3-Фтор-4-метилсульфанилфенил)уксусную кислоту (7,54 г, 37,7 ммоль) этерифицировали, как описано выше в подготовительном примере 15, с получением соединения, указанного в заголовке: RTA = 3,62 мин. Подготовительный пример 18. Этил-(4-метансульфинилфенил)ацетат(метилсульфанилфенил)ацетата (4,66 г, 22,2 ммоль) в CH2Cl2 (70 мл) при охлаждении на бане лед-H2O. Эту смесь перемешивали в течение 4 суток при 20 С, после чего гасили насыщенным водным Na2CO3. Органический слой отделяли, промывали насыщенным водным NaHCO3 и сушили (MgSO4). В результате фильтрования, выпаривания растворителя и флэш-хроматографии (IH-EtOAc, от 1:1 до 0:1) получили соединение, указанное в заголовке: m/z (ES+) = 227,0 [М+Н]+. Подготовительный пример 19. Этил-(4-этансульфонилфенил)ацетат В результате алкилирования этил-(4-меркаптофенил)ацетата (20 г, 102 ммоль) Etl (9,8 мл, 122 ммоль) с использованием методики, подобной описанной в подготовительном примере 39, получили этил-(4-этилсульфанилфенил)ацетат: m/z (ES+) = 225,2 [М+Н]+. В результате окисления этого соединения Перемешиваемый раствор 4-йодметилтетрагидропирана (3,43 г, 15,2 ммоль) и PPh3 (3,98 г, 15,2 ммоль) в безводном MeCN (10 мл) нагревали с обратным холодильником в течение 19 ч. После охлаждения до 20 С добавляли Et2O (50 мл). Образовавшийся осадок собирали, промывали Et2O (150 мл) и перекристаллизовали (MeCN) с получением соединения, указанного в заголовке: m/z (ES+) = 361,2 [М]+. Подготовительный пример 22. 2-(4-Циклопропансульфонилфенил)-3-(тетрагидропиран-4 ил)пропионовая кислота Перемешиваемую суспензию AlCl3 (12,90 г, 96,8 ммоль) в безводном CH2Cl2 (135 мл) обрабатывали порциями при 0 С этилхлороксоацетатом (8,5 мл, 76,0 ммоль). К этой смеси добавляли по каплям циклопропилфенилсульфид (10,0 мл, 70,0 ммоль) в течение 1 ч, поддерживая в это время температуру реакционной смеси ниже 10 С. Смеси давали нагреться до 20 С, после чего перемешивали в течение дополнительных 70 мин. Охлажденную во льду H2O (35 мл) добавляли после охлаждения до 0 С, затем смесь перемешивали дополнительно в течение 10 мин. Слой CH2Cl2 отделяли, затем водный слой экстрагировали дополнительным количеством CH2Cl2 (250 мл). Объединенные органические слои сушили(MgSO4), фильтровали и концентрировали с получением этил-(4-циклопропилсульфанилфенил)оксоацетата: RTB = 1,74 мин. LHMDS (3,7 мл 1,0 М раствора в ТГФ, 3,7 ммоль) добавляли к перемешиваемой суспензии йодида трифенил(тетрагидропиран-4-илметил)фосфония (подготовительный пример 21, 1,82 г, 3,7 ммоль) в безводном ТГФ (5,6 мл) при 0 С. Через 1 ч раствор этил-(4-циклопропилсульфанилфенил)-оксоацетата (0,78 г, 3,1 ммоль) в безводном ТГФ (4 мл) добавляли в течение 5 мин. Эту смесь перемешивали при 0 С в течение 1 ч, после чего давали нагреться до 20 С в течение 16 ч. H2O(7 мл) добавляли после охлаждения до 0 С. 1 М HCl добавляли для доведения рН до 6, затем смесь перемешивали в течение 1 ч при 20 С. ТГФ удаляли в вакууме, затем добавляли Et2O (35 мл). Эту смесь перемешивали в течение 30 мин и фильтровали, промывая Et2O. Водный слой отделяли и экстрагировалиEt2O (310 мл). Объединенные органические экстракты промывали рассолом (20 мл), сушили, фильтровали и концентрировали. В результате флэш-хроматографии (IH-CH2Cl2, от 2:1 до 1:1, затем ТГФ-CH2Cl2,1:99) получили этил-2-(4-циклопропилсульфанилфенил)-3-(тетрагидропиран-4-ил)акрилат: m/z (ES+) = 333,2 [М+Н]+. Перемешиваемый раствор этого тиоэфира (609 мг, 1,83 ммоль) в CH2Cl2 (35 мл) обрабатывали раствором mCPBA (992 мг с чистотой 65%, 3,74 ммоль) в CH2Cl2 (15 мл). Через 16 ч добавляли насыщенный водный NaHCO3 (25 мл), затем перемешивание продолжали в течение 5 мин. Слои разделяли,затем водную фазу экстрагировали CH2Cl2(20 мл). Объединенные органические слои промывали насыщенным водным NaHCO3 (25 мл), H2O (25 мл) и рассолом (25 мл), а затем сушили (MgSO4). В результате- 15012700 фильтрования и выпаривания растворителя получили этил-2-(4-циклопропансульфонилфенил)-3(тетрагидропиран-4-ил)акрилат: m/z (ES+) = 382,2 [M+NH4]+. Раствор этого соединения (667 мг, 1,83 ммоль) в EtOAc (60 мл) обрабатывали Pd (10% на С, 424 мг, 0,39 ммоль). Эту реакционную смесь перемешивали в атмосфере Н 2 в течение 3 суток, после чего фильтровали через целит. Целит промывалиEtOAc (100 мл), затем объединенные фильтраты концентрировали с получением этил-2-(4 циклопропансульфонилфенил)-3-(тетрагидропиран-4-ил)пропионата: RF (CH2Cl2-ТГФ, 30:1) = 0,56. Раствор этого эфира (664 мг, 1,81 ммоль) в ТГФ-H2O (3:1, 20 мл) перемешивали с LiOHH2O (168 мг, 4,00 ммоль) в течение 23 ч. ТГФ выпаривали при пониженном давлении, затем остаток разбавляли H2O(10 мл). Эту смесь промывали Et2O (220 мл), после чего подкисляли 2 M HCl (5 мл) до рН 1. Остаток экстрагировали EtOAc (320 мл). Объединенные органические экстракты промывали рассолом (20 мл),сушили (MgSO4), фильтровали и выпаривали с получением соединения, указанного в заголовке: m/z В результате частичного окисления этил-2-(4-циклопропилсульфанилфенил)-3-(тетрагидропиран-4 ил)акрилата (см. подготовительный пример 22, 3,14 г, 9,44 ммоль) mCPBA с использованием протокола,описанного выше в подготовительном примере 18, получили этил-2-(4-циклопропансульфинилфенил)-3(тетрагидропиран-4-ил)акрилат: m/z (ES+) = 349,2 [M+H]+. В результате омыления этого эфира (1,15 г,3,3 ммоль) с использованием методики, описанной в подготовительном примере 25, получили соединение, указанное в заголовке: m/z (ES+) = 641,4 [2M+H]+. Подготовительный пример 25. (Е)-2-(4-Метансульфонилфенил)-3-(тетрагидропиран-4-ил)акриловая кислотаLDA (24 мл 1,8 М растворе в н-C7H16-ТГФ-PhEt, 43,3 ммоль) добавляли по каплям к перемешиваемому раствору DMPU (19 мл, 153,0 ммоль) в безводном ТГФ (100 мл) при -78 С. Через 30 мин добавляли по каплям раствор этил-(4-метансульфонилфенил)-ацетата (подготовительный пример 15, 5,00 г, 20,6 ммоль) в безводном ТГФ (42 мл). Эту смесь дополнительно перемешивали в течение 1 ч, после чего обрабатывали по каплям раствором тетрагидропиран-4-карбоксальдегида (2,36 г, 20,6 ммоль) в безводном ТГФ (25 мл). После того как реакционной смеси давали нагреться до 20 С в течение 16 ч, ее гасили насыщенным водным NH4Cl (210 мл). ТГФ удаляли при пониженном давлении, затем остаток экстрагировали EtOAc (3250 мл). Объединенные экстракты EtOAc сушили (MgSO4) фильтровали и концентрировали. В результате колоночной хроматографии (IH-EtOAc, 7:3) получили (Е)-этил-2-(4 метансульфонилфенил)-3-(тетрагидропиран-4-ил)акрилат: m/z (ES+) = 356,2 [M+NH4]+. Раствор этого эфира (6,46 г, 19,1 ммоль) в МеОН (30 мл) и 1 М NaOH (40 мл, 40,0 ммоль) нагревали с обратным холодильником в течение 1 ч. После охлаждения смесь промывали EtOAc. Водную фазу подкисляли 1 М HCl,после чего экстрагировали EtOAc. Объединенные органические экстракты сушили (MgSO4). В результате фильтрования и выпаривания растворителя получили соединение, указанное в заголовке: m/z (ES+) = 621,3 [2 М+Н]+. Акриловые кислоты, перечисленные в табл. 3, синтезировали, используя способы, подобные опи- 16012700 санным в подготовительном примере 25. Таблица 3NaOEt (0,63 мл 0,5 М раствора в EtOH, 0,32 ммоль) добавляли по каплям к перемешиваемому раствору этил-(4-[1,2,3]триазол-1-илфенил)ацетата (730 мг, 3,16 ммоль) и тетрагидропиран-4 карбоксальдегида (396 мг, 3,47 ммоль) в безводном ДМСО (3 мл). Эту смесь нагревали при 80 С в течение 16 ч, после чего обрабатывали АсОН для доведения рН до 7. Добавляли EtOAc (30 мл), затем раствор промывали H2O (210 мл) и рассолом (10 мл), после чего сушили (MgSO4). В результате фильтрования,выпаривания растворителя и колоночной хроматографии (IH-EtOAc, 1:1) получили этил-3(тетрагидропиран-4-ил)-2-(4-[1,2,3]триазол-1-илфенил)акрилат: m/z (ES+) = 328,2 [М+Н]+. Этот эфир(404 мг, 1,23 ммоль) подвергали омылению, как описано выше в подготовительном примере 25: m/z (ES+)= 300,2 [М+Н]+. Способ, подробно описанный в подготовительном примере 30, включающий конденсацию фенилуксусного эфира с соответствующим альдегидом с последующим омылением промежуточного,-ненасыщенного эфира, использовали для получения акриловых кислот, перечисленных в табл. 4. Таблица 4DMPU (50 мл, 413 ммоль) добавляли к раствору LDA (65 мл 1,8 М раствора в н-С 17 Н 16-ТГФ-PhEt,117 ммоль) в безводном ТГФ (250 мл) при -78 С. Эту смесь перемешивали в течение 1 ч с образованием осадка кремового цвета. Раствор (4-метансульфонилфенил)уксусной кислоты (12,00 г, 56 ммоль) в безводном ТГФ (120 мл) добавляли в течение 20 мин. Добавляли дополнительное количество безводного ТГФ (30 мл), затем густую желтую суспензию перемешивали в течение 1 ч. Эту смесь обрабатывали раствором 2-хлорметилтиофена (7,50 г, 57 ммоль) и PhMe (5,20 г, 57 ммоль) в безводном ТГФ (20 мл), затем перемешивание продолжали при -78 С в течение 20 мин. Затем реакционной смеси давали нагреться до 20 С в течение 16 ч, после чего гасили H2O (500 мл). ТГФ удаляли при пониженном давлении, затем добавляли 12 M HCl до доведения рН до 2. Смесь экстрагировали EtOAc (2300 мл), затем экстракты промывали Н 2 О (2200 мл) и рассолом (2100 мл), после чего сушили (MgSO4). В результате фильтрования, выпаривания растворителя и колоночной хроматографии (IH-EtOAc, 3:2, содержащий 0,5% АсОН) получили соединение, указанное в заголовке: m/z (ES+) = 638,3 [2M+NH4]+. Подготовительный пример 39. Этил-[4-(тетрагидропиран-4-илсульфанил)фенил]ацетатNEt3 (1,3 мл, 9,0 ммоль) и 4-йодтетрагидропиран (1,93 г, 9,0 ммоль) добавляли к перемешиваемому раствору этил-(4-меркаптофенил)ацетата (1,21 г, 6,0 ммоль) в безводном ДМФ (10 мл) при 0 С. Этой смеси давали нагреться до комнатной температуры в течение 3 суток, затем растворители удаляли при пониженном давлении. Остаток распределяли между Et2O (100 мл) и насыщенным водным NH4Cl(50 мл), водную фазу дополнительно экстрагировали Et2O (45 мл). Объединенные эфирные экстракты промывали Н 2 О (50 мл), Н 2 О-насыщенным водным Na2CO3 (1:1, 50 мл) и рассолом (50 мл), после чего сушили (MgSO4). В результате фильтрования, выпаривания растворителя и колоночной хроматографии(59,3 мл, 490,3 ммоль) в безводном ТГФ (150 мл) при -78 С. Эту смесь перемешивали в течение 30 мин,после чего обрабатывали по каплям раствором этил-(4-метансульфонилфенил)ацетата (подготовительный пример 15, 16,97 г, 70,0 ммоль) в безводном ТГФ (50 мл). Перемешивание продолжали при -78 С в течение 45 мин, затем добавляли раствор 4-йодметилтетрагидропирана (19,00 г, 84,0 ммоль) в безводном ТГФ (40 мл). Этой смеси давали нагреться до 20 С в течение 16 ч, после чего гасили 1 М HCl (70 мл). ТГФ удаляли при пониженном давлении, затем добавляли дополнительное количество Н 2 О (40 мл), и остаток экстрагировали EtOAc (2250 мл). Экстракты EtOAc сушили (MgSO4). В результате фильтрования, выпаривания растворителя и колоночной хроматографии (IH-EtOAc, от 9:1 до 1:1) получили этил-2(4-метансульфонилфенил)-3-(тетрагидропиран-4-ил)пропионат: m/z (ES+) = 698,3 [2M+NH4]+. Раствор этого соединения (20,27 г, 59,6 ммоль) в МеОН (100 мл) и 2 М NaOH (62,5 мл, 125,0 ммоль) нагревали с обратным холодильником в течение 1 ч. Растворители удаляли при пониженном давлении, затем остаточное твердое вещество растирали с Et2O (5x100 мл), после чего растворяли в Н 2 О (100 мл). Этот водный раствор промывали EtOAc (50 мл), подкисляли 2 М HCl до рН 1 и экстрагировали EtOAc (21 л). После сушки (MgSO4), фильтрования и выпаривания растворителя получили соединение, указанное в заголовке: m/z (ES+) = 642,3 [2M+NH4]+. Подходы, подобные раскрытым в подготовительном примере 41, включающие алкилирование соответствующего эфира 4-йодметилтетрагидропираном с последующим гидролизом продукта, использовали для получения карбоновых кислот, показанных в табл. 5. Таблица 5mCPBA (1,15 г с чистотой 60%, 4,0 ммоль). Через 16 ч этот раствор фильтровали, затем фильтрат очищали колоночной хроматографией (IH-EtOAc-АсОН, от 320:80:1 до 80:320:1) с получением соединения,указанного в заголовке: m/z (ES+) = 678,3 [2M+NH4]+. Подготовительный пример 51. 2-(4-Этилсульфамилфенил)-3-(тетрагидропиран-4-ил)пропионовая кислота(32,4 г, 143,4 ммоль) согласно протоколу, описанному в подготовительном примере 41, с получением этил-2-(4-нитрофенил)-3-(тетрагидропиран-4-ил)пропионата: н (CDCl3): 1,21 (3 Н, t), 1,25-1,45 (3 Н, m),1,55-1,65 (2 Н, m), 1,70-1,80 (1 Н, m), 2,05-2,15 (1 Н, m), 3,25-3,35 (2 Н, m), 3,79 (1 Н, t), 3,90-3,95 (2 Н, m),4,10-4,20 (2 Н, m), 7,49 (2 Н, d), 8,19 (2 Н, d). Нитрогруппу этого соединения (6,55 г, 18,1 ммоль) восстанавливали, используя методику, описанную в примере 145, с получением этил-2-(4-аминофенил)-3(тетрагидропиран-4-ил)пропионата: m/z (ES+) = 278,2 [М+Н]+. Это соединение (30,5 г, 110 ммоль) превращали в этил-2-(4-хлорсульфонилфенил)-3-(тетрагидропиран-4-ил)пропионат, используя протокол,описанный в подготовительном примере 59. Раствор этого сульфонилхлорида (33,6 г, 93,2 ммоль) в безводном ТГФ (100 мл) добавляли в течение 30 мин при 0C к EtNH2 (116,5 мл 2,0 М раствора в ТГФ,- 20012700 233,0 ммоль). Эту смесь нагревали до 20 С, после чего перемешивали в течение 16 ч. Суспензию фильтровали через слой целита, который промывали ТГФ (350 мл). Объединенные растворы ТГФ концентрировали с получением этил-2-(4-этилсульфамилфенил)-3-(тетрагидропиран-4-ил)пропионата: m/z (ES+)= 370,2 [М+Н]+. В результате гидролиза этого эфира (33,7 г, 91,2 ммоль) с использованием методики,изложенной в подготовительном примере 41, с последующей очисткой путем ОФ-ВЭЖХ получили соединение, указанное в заголовке: m/z (ES+) = 342,2 [М+Н]+. Подготовительный пример 52. 2-(4-Циклобутансульфонилфенил)-3-(тетрагидропиран-4-ил)пропионовая кислота В результате алкилирования этил-(4-меркаптофенил)ацетата (9,5 г, 48,4 ммоль) c-BuBr (7,84 г,58,1 ммоль) с использованием методики, подобной описанной в примере 161, получили этил-(4 циклобутилсульфанилфенил)ацетат: RTA = 4,17 мин. В результате окисления этого соединения (18,5 г,73,9 ммоль) nCPBA (222 ммоль) с использованием протокола, подобного описанному в подготовительном примере 22, получили этил-(4-циклобутансульфонилфенил)ацетат: m/z (ES+) = 283,2 [М+Н]+. В результате конденсации этого соединения (18,84 г, 66,7 ммоль) с тетрагидропиран-4-карбоксальдегидом(8,38 г, 73,4 ммоль) с использованием методики, описанной в подготовительном примере 30, получили этил-2-(4-циклобутансульфонилфенил)-3-(тетрагидропиран-4-ил)акрилат: m/z (ES+) = 396,2 [M+NH4]+. В результате восстановления этого ,-ненасыщенного эфира (13,00 г, 34,4 ммоль) с использованием протокола, описанного в подготовительном примере 22, получили этил-2-(4-циклобутансульфонилфенил)-3(тетрагидропиран-4-ил)пропионат: m/z (ES+) = 381,2 [М+Н]+. Этот эфир подвергали гидролизу, используя методику, описанную в подготовительном примере 22, с получением соединения, указанного в заголовке: m/z (ES+) = 370,2 [M+NH4]+. Подготовительный пример 53. (2R)-2-(4-Метансульфонилфенил)-3-(тетрагидропиран-4-ил)пропионовая кислотаNEt3 (15,4 мл, 110 ммоль) добавляли к перемешиваемой суспензии 2-(4-метансульфонилфенил)-3(тетрагидропиран-4-ил)пропановой кислоты (подготовительный пример 41, 30,0 г, 96,0 ммоль) в безводном ТГФ (300 мл) при 0 С. Через 10 мин добавляли по каплям пивалоилхлорид (13,6 мл, 110 ммоль) в течение 20 мин, и смесь перемешивали при 0 С в течение 2 ч. В это же время n-BuLi (45,3 мл 2,5 М раствора в гексанах, 115 ммоль) добавляли к раствору (R)-(+)-4-бензил-2-оксазолидинона (20,4 г, 115 ммоль) в безводном ТГФ (300 мл) при -78 С. Эту смесь перемешивали при температуре от -78 до 20 С в течение 2 ч. Полученный таким образом раствор добавляли по каплям к вышеупомянутому раствору смешанного ангидрида при -78 С. Эту реакционную смесь перемешивали при -78 С в течение 1 ч, а затем при 20 С в течение 4 ч, после чего обрабатывали H2O (300 мл). ТГФ удаляли в вакууме, затем остаток экстрагировали EtOAc (3300 мл). Объединенные органические слои промывали H2O, сушили (Na2SO4), фильтровали и концентрировали в вакууме. В результате хроматографического разделения (EtOAc - n-C6H14, от 1:2 до 1:1) получили два продукта: (1) (4R)-4-бензил-3-[(2R)-2-[4-(метилсульфонил)фенил]-3-(тетрагидро 2R-пиран-4-ил)пропаноил]-1,3-оксазолидин-2-он: т. пл. 139-141 С (из Et2O-ТГФ); (2) (4R)-4-бензил-3[(2S)-2-[4-(метилсульфонил)фенил]-3-(тетрагидро-2H-пиран-4-ил)пропаноил]-1,3-оксазолидин-2-он: m/z(400 мл) добавляли по каплям в течение 40 мин к перемешиваемому раствору (4R)-4-бензил-3-[(2S)-2-[4(метилсульфонил)фенил]-3-(тетрагидро-2 Н-пиран-4-ил)пропаноил]-1,3-оксазолидин-2-она (15,1 г, 10,9 ммоль) в ТГФ-H2O (3:1, 1,6 л) при 0 С. Эту реакционную смесь перемешивали при 0 С в течение 1,5 ч,затем остаточный окислитель разрушали 10% водным Na2SO3. Смесь промывали Et2O (4300 мл), подкисляли 10% водной HCl и экстрагировали EtOAc (3200 мл). Объединенные органические слои промывали рассолом, сушили (Na2SO4), фильтровали и концентрировали в вакууме. В результате растирания с Et2O-гексанами получили соединение, указанное в заголовке: т. пл. 217 С; абсолютную конфигурацию определяли с использованием кристаллографического анализа с помощью рентгеновских лучей.(4-Хлор-3-нитрофенил)уксусную кислоту (10,00 г, 46,4 ммоль) этерифицировали, как описано выше в подготовительном примере 15, с получением этил-(4-хлор-3-нитрофенил)ацетата: m/z (ES+) = 285,2[М+MeCN+Н]+. В результате алкилирования этого эфира (10,50 г, 43,1 ммоль) с использованием протокола, описанного в подготовительном примере 41, получили этил-2-(4-хлор-3-нитрофенил)-3(тетрагидропиран-4-ил)пропионат: m/z (ES+) = 342,1 [М+Н]. Раствор этого соединения (7,42 г,19,7 ммоль) в ДМСО (50 мл) обрабатывали NaSMe (1,52 г, 21,6 ммоль). Эту смесь перемешивали при 20 С в течение 5,5 ч, а затем при 50 С в течение 2 ч, после чего ее наливали на колотый лед (500 мл). После того как лед полностью растаял, смесь распределяли между EtOAc (250 мл) и Н 2 О (100 мл). Водный слой дополнительно экстрагировали EtOAc (4200 мл), затем объединенные органические экстракты промывали рассолом и сушили (MgSO4). В результате фильтрования, выпаривания растворителя и колоночной хроматографии (IH-EtOAc, 7:3) получили этил-2-(4-метилсульфанил-3-нитрофенил)-3(тетрагидропиран-4-ил)пропионат: m/z (ES+) = 371,0 [M+NH4]+. Этот эфир (7,48 г, 19,2 ммоль) подвергали гидролизу LiOH-Н 2 О, как описано выше в подготовительном примере 22, с получением соединения,указанного в заголовке: m/z (ES+) = 343,3 [M+NH4]+.(10 мл) и H2O (1 мл). Эту смесь перемешивали в атмосфере Н 2 в течение 24 ч, после чего фильтровали через целит. Целит промывали EtOAc (550 мл), затем объединенные фильтраты выпаривали с получением этил-2-(3-аминофенил)-3-(тетрагидлропиран-4-ил)пропионата: m/z (ES+) = 278,2 [М+Н]+. Раствор этого соединения (2,77 г, 10,0 ммоль) в ДМЭ (10 мл) добавляли в течение 30 мин к перемешиваемой смеси i-AmONO (2,0 мл, 15,0 ммоль) и MeSSMe (9,9 мл, 110,0 ммоль). Температуру повышали до 45 С в течение 0,5 ч, а затем до 85 С в течение 1,5 ч. После охлаждения растворители удаляли при пониженном давлении, затем остаток растворяли в EtOAc (60 мл). Раствор EtOAc промывали 1 М HCl (220 мл), H2O(20 мл) и рассолом (20 мл). В результате фильтрования, выпаривания растворителя и колоночной хроматографии (CH2Cl2-Et2O, от 1:0 до 99:1) получили этил-2-(3-метилсульфанил-фенил)-3-(тетрагидропиран 4-ил)пропионат: m/z (ES+) = 309,2 [М+Н]+. В результате омыления этого эфира LiOH-H2O по протоколу,описанному в подготовительном примере 22, получили соединение, указанное в заголовке: m/z (ES+) = 561,3 [2 М+Н]+. Подготовительный пример 59. 4-[2-(Тетрагидропиран-4-ил)-1-(тиазол-2-илкарбамоил)этил]-бензолсульфонилхлорид[М+Н]+. Нитрогруппу этого соединения (6,55 г, 18,1 ммоль) восстанавливали, используя методику, описанную в примере 145, с получением 2-(4-аминофенил)-3-(тетрагидропиран-4-ил)-N-тиазол-2 илпропионамида: m/z (ES+) = 332,1 [М+Н]+. Раствор NaNO2 (2,11 г, 30,5 ммоль) в H2O (20 мл) медленно добавляли к перемешиваемой смеси вышеописанного анилина (9,40 г, 28,4 ммоль), 12 М HCl (30 мл) иH2O (30 мл) при 0 С. Через 1 ч полученный в результате раствор диазониевой соли добавляли в течение 15 мин к смеси CuCl22H2O (1,29 г, 7,6 ммоль) в АсОН (64,5 мл) и H2O (3,2 мл), которая была предварительно насыщена SO2. Эту смесь перемешивали в течение 1,5 ч, обрабатывали H2O (200 мл) и экстрагировали EtOAc (300 + 150 мл). Объединенные экстракты EtOAc промывали H2O (2200 мл), фильтровали и сушили (MgSO4). В результате фильтрования и выпаривания растворителя получили соединение, указанное в заголовке: m/z (ES+) = 466,1 [М+MeCN+Н]+. Подготовительный пример 60. 4-[1-(5-Хлортиазол-2-илкарбамоил)-2-(тетрагидропиран-4 ил)этил]бензолсульфонилхлоридNEt3 (63,4 мл, 455 ммоль) добавляли к перемешиваемой суспензии гидробромида 5-бромтиазол-2 иламина (102,7 г, 379 ммоль) в CH2Cl2 (1,5 л). Через 1 ч добавляли по каплям ТФА (64,2 мл, 455 ммоль) при 0 С в течение 15 мин. Смеси давали нагреться до 20 С в течение 1 ч, после чего ее перемешивали в- 23012700 течение дополнительных 2 ч. Добавляли H2O (600 мл), и полученный в результате осадок собирали. Водный слой фильтрата отделяли и экстрагировали CHCl3 (3300 мл). Объединенные органические экстракты промывали рассолом, сушили (Na2SO4), фильтровали и концентрировали. Собранный осадок и остаточное твердое вещество объединяли и растирали с EtOAc-n-C6H14 с получением N-(5-бромтиазол-2 ил)-2,2,2-трифторацетамида: н (CDCl3): 7,45 (1 Н, s), 13,05 (1 Н, br). n-BuLi (253 мл 1,58 М раствора в гексанах, 403 ммоль) добавляли по каплям в течение 50 мин к перемешиваемому раствору вышеуказанного амида (50,0 г, 183 ммоль) в безводном ТГФ (1,3 л) при -78 С. Через 1,5 ч добавляли раствор Nфторбензолсульфонимида (86,0 г, 275 ммоль) в безводном ТГФ (250 мл) по каплям в течение 30 мин. Эту смесь перемешивали в течение 3 ч, после чего нагревали до -30 С. Добавляли H2O (300 мл), и смесь фильтровали через слой целита. Твердое вещество собирали, и целит промывали Et2O (400 мл) и H2O(400 мл). Органический слой фильтрата отделяли и экстрагировали водой (2400 мл). Объединенные водные слои промывали Et2O (400 мл), после чего подкисляли до рН 6,5 2 М HCl и экстрагировалиEtOAc (2400 мл). Объединенные органические экстракты промывали Н 2 О (2400 мл) и рассолом, после чего сушили (MgSO4), фильтровали и концентрировали. В результате колоночной хроматографии(1 Н, d). AcCl (12,6 мл, 175 ммоль) добавляли по каплям к перемешиваемому раствору этого амида (15,7 г,73 ммоль) в МеОН (300 мл) при 0 С. Смесь перемешивали при 20 С в течение 30 мин, нагревали с обратным холодильником в течение 1 ч и, наконец, концентрировали в вакууме. Остаточное твердое вещество растирали с ТГФ с получением соединения, указанного в заголовке: H (D2O): 7,00 (1 Н, d). Подготовительный пример 62. 4-[2-(Тетрагидропиран-4-ил)-1-(тиазол-2-илкарбамоил)этил]бензойная кислота Метил-4-трет-бутоксикарбонилметилбензоат (1,71 г, 6,84 ммоль) алкилировали 4-йодметилтетрагидропираном (1,86 г, 8,21 ммоль), используя способ, описанный в подготовительном примере 41, с получением метил-4-[1-трет-бутоксикарбонил-2-(тетрагидропиран-4-ил)этил]бензоата: RTA = 3,86 мин. Раствор этого соединения (1,37 г, 3,94 ммоль) в CH2Cl2 (5 мл) обрабатывали ТФУ-CH2Cl2 (2:1, 15 мл) при 0 С в течение 10 мин. Смесь перемешивали при 20 С в течение 3 ч, после чего концентрировали в вакууме. Остаток обрабатывали PhMe, и растворители выпаривали при пониженном давлении. Этот процесс повторяли дважды с получением сырого метил-4-[1-карбокси-2-(тетрагидропиран-4 ил)этил]бензоата. Эту карбоновую кислоту конденсировали с тиазол-2-иламином, используя протокол,описанный в примере 65, с получением метил-4-[2-(тетрагидропиран-4-ил)-1-(тиазол-2-илкарбамоил)этил]бензоата: m/z (ES+) = 375,2 [М+Н]+. Этот эфир (1,20 г, 3,21 ммоль) подвергали омылению LiOHH2O, используя методику, описанную в подготовительном примере 22 с получением соединения, указанного в заголовке: m/z (ES-) = 359,2 [М-Н-]. Пример 1. (E)-2-(4-Метансульфонилфенил)-N-тиазол-2-ил-3-тиофен-3-илакриламид Суспензию PS-карбодиимида (688 мг, нагрузка 1,34 мкмоль мг-1, 922 мкмоль), (E)-2-(4 метансульфонилфенил)-3-тиофен-3-илакриловой кислоты (подготовительный пример 1, 139 мг, 450 мкмоль) и HOBt (84 мг, 622 мкмоль) в безводном ДМФ перемешивали в течение 15 мин при 20 С. Добавляли тиазол-2-иламин (32 мг, 320 мкмоль), затем эту смесь перемешивали в течение 40 ч при 20 С,после чего фильтровали через целит. Фильтровальный кек промывали ДМФ (10 мл), EtOAc (20 мл) иCH2Cl2 (20 мл). Эти растворы объединяли, растворители удаляли при пониженном давлении, и остаток растворяли в EtOAc (50 мл). Раствор EtOAc промывали насыщенным водным Na2CO3 (320 мл), H2O(20 мл) и рассолом (20 мл), после чего сушили (Na2SO4). В результате фильтрования, выпаривания растворителя и флэш-хроматографии (IH-EtOAc, от 3:1 до 1:3) получили соединение, указанное в заголовке:RTA = 3,43 мин; m/z (ES+) = 391,0 [М+Н]+. Опосредованная PS-карбодиимидом-HOBt конденсация соответствующей карбоновой кислоты с тиазол-2-иламином, как описано в примере 1, была также использована для синтеза амидов, перечисленных ниже в табл. 6. Раствор (Е)-2-(4-метансульфонилфенил)-3-тиофен-2-илакриловой кислоты (подготовительный пример 2, 309 мг, 1,0 ммоль), HATU (813 мг, 2,1 ммоль), гидрохлорида 5-хлортиазол-2-иламина (258 мг, 1,5 ммоль) и DIPEA (0,71 мл, 4,0 ммоль) в безводном ДМФ (5 мл) нагревали при микроволновом облучении при перемешивании при 60 С в течение 2 мин. Растворители выпаривали при пониженном давлении,затем остаток распределяли между CH2Cl2 (60 мл) и 1 М HCl (60 мл). Органический слой отделяли и промывали 1 М HCl (60 мл), Н 2 О (60 мл), насыщенным водным Na2CO3 (260 мл), H2O (60 мл) и рассолом(60 мл), после чего сушили (Na2SO4). В результате фильтрования, выпаривания растворителя и флэшхроматографии (IH-EtOAc, от 4:1 до 2:3) получили соединение, указанное в заголовке: RTA = 3,77 мин;m/z (ES+) = 466,1 [M+MeCN+H]+. Опосредованную микроволновым облучением конденсацию соответствующей карбоновой кислоты- 28012700 с тиазол-2-иламинами, изложенную в примере 36, также использовали для получения амидов, перечисленных ниже в табл. 7. Таблица 7 Суспензию (E)-2-(4-бромфенил)-3-фуран-2-илакриловой кислоты (подготовительный пример 9,4,10 г, 14,0 ммоль) и оксалилхлорида (2,5 мл, 28,0 ммоль) в безводном CH2Cl2 (100 мл) обрабатывали каталитическим количеством безводного ДМФ (25 мкл). Полученный в результате раствор перемешивали при 20 С в течение 4 ч, затем растворители удаляли при пониженном давлении. К остатку добавлялиCH2Cl2 (50 мл), затем растворители выпаривали при пониженном давлении с получением 2-(4 бромфенил)-3-фуран-2-илакрилоилхлорида в виде коричневого твердого вещества. Раствор этого хлорангидрида (343 мг, 1,1 ммоль) в безводном ТГФ (1 мл) добавляли к раствору гидрохлорида 5-хлортиазол-2-иламина (171 мг, 1,0 ммоль) и NEt3 (0,56 мл, 4,0 ммоль) в безводном ТГФ (1 мл). Эту суспензию перемешивали в течение 16 ч при 20 С, затем растворители удаляли при пониженном давлении. Остаток распределяли между CH2Cl2 (10 мл) и насыщенным водным NaHCO3 (5 мл). Органический слой промывали Н 2 О (5 мл) и рассолом (5 мл), после чего концентрировали. Полученное в результате твердое вещество перекристаллизовали из МеОН с получением соединения, указанного в заголовке: RTA = 4,39 мин;m/z (ES+) = 410,9 [М+Н]+. Некоторые другие енамиды получили путем конденсации соответствующего хлорангидрида с гетероароматическими аминами, как представлено примером 44. Эти соединения перечислены ниже в табл. 8.
МПК / Метки
МПК: A61P 3/10, A61K 31/454, A61K 31/495, C07D 413/12, A61K 31/427, C07D 277/46, C07D 417/14, C07D 491/08, C07D 417/12, C07D 405/12, A61K 31/4439
Метки: глюкокиназы, фенилацетамиды, применение, качестве, модуляторов
Код ссылки
<a href="https://eas.patents.su/30-12700-fenilacetamidy-i-ih-primenenie-v-kachestve-modulyatorov-glyukokinazy.html" rel="bookmark" title="База патентов Евразийского Союза">Фенилацетамиды и их применение в качестве модуляторов глюкокиназы</a>
Замещённые фенилацетамиды и их применение в качестве активаторов глюкокиназы

Номер патента: 11297
Опубликовано: 27.02.2009
Авторы: Хейнс Нэнси-Эллен, Мэхэней Пейж Эрин, Корбетт Уэнди Ли, Гримсби Джозеф Самьюэл, Сарабу Рамакант, Кестер Роберт Франсис, Рача Ягдиш Кумар, Ван Ка
МПК: C07C 275/52, A61K 31/38, A61K 31/335...
Метки: качестве, активаторов, применение, замещённые, глюкокиназы, фенилацетамиды
Формула / Реферат:
1. Соединение формулы где R1 и R2 независимо означают водород, галоген, перфтор(низш.)алкил или (низш.)алкилсульфонил, комбинация R3 и атома углерода, к которому присоединен R3, означает незамещенный тетрагидрофуранил, незамещенный тетрагидропиранил, тетрагидротиопиранил, в котором атом S необязательно замещен оксогруппой, или пяти- или шестичленный циклоалкил, который монозамещен по одному атому углерода в цепи одним остатком, выбранным из...
Производные бензазола и их применение в качестве модуляторов jnk

Номер патента: 7152
Опубликовано: 25.08.2006
Авторы: Готтелан Жан-Пьер, Гайяр Паскаль, Алази Серж, Черч Деннис, Кампс Монсеррат
МПК: A61K 31/506, A61P 25/00, C07D 403/06...
Метки: применение, модуляторов, производные, качестве, бензазола
Формула / Реферат:
1. Производные бензазола формулы I а также их таутомеры, их геометрические изомеры, их оптически активные формы в виде энантиомеров, диастереомеров и их рацематов, а также фармацевтически приемлемые соли или N-оксиды, где G представляет собой пиримидинильную группу, замещенную по меньшей мере одним заместителем, выбранным из группы, включающей галоген, гидрокси, гидразино, нитро, алкилсульфанил, необязательно замещенный алкил, необязательно...
Производные пиримидин-4-она и их применение в качестве модуляторов киназы p38

Номер патента: 9743
Опубликовано: 28.04.2008
Авторы: Дивадас Балекудру, Хикори Брайан, Мэдсен Хизер, Дерли Ричард, Селнесс Шон, Палмквист Катерин
МПК: C07D 239/46, A61K 31/513, C07D 239/52...
Метки: модуляторов, применение, пиримидин-4-она, киназы, производные, качестве
Формула / Реферат:
1. Соединение формулы или его фармацевтически приемлемая соль, где L и M независимо выбраны из -О-, -СН2-; R5 представляет собой или где Х1, Х2, Xa, Xb, Xc, Xd и Xe независимо выбраны из -C(O)NR6R7, -(С1-С4алкил)-C(O)NR6R7, -NR6R7, гидрокси(С1-С4)алкила, С1-С4дигидроксиалкила, Н, галогена, C1-С6галогеноалкила, C1-С6алкила, C1-С6галогеноалкокси, R6R7N-(С1-С6алкил)-, -СО2-(С1-С6)алкила, -N(R)C(O)NR6R7, -N(R)C(O)-(C1-C6)алкокси,...
Производные бензоксазина в качестве модуляторов 5-нт-6, способ их получения, фармацевтические композиции, содержащие эти производные, и их применение

Номер патента: 9982
Опубликовано: 28.04.2008
Авторы: Чжао Шухай, Бергер Якоб, Кларк Робин Дуглас
МПК: A61P 25/18, C07D 265/36, A61K 31/536...
Метки: фармацевтические, способ, модуляторов, бензоксазина, производные, эти, 5-нт-6, применение, композиции, качестве, содержащие, получения
Формула / Реферат:
1. Соединение формулы его фармацевтически приемлемая соль или пролекарство, где m означает целое число от 0 до 3; n означает 2 или 3; р означает 2; Y означает -S(O2)-; Z1 означает N; R1 и R2 означают водород; R3 означает фенил или нафтил, которые необязательно замещены галогеном, алкилом, алкокси, гидрокси, циано, пиперазинилом, метилсульфониламино, метилсульфонилом, аминокарбонилом, или означает хинолинил, изохинолинил, тиофенил,...
Производные пропионамида, полезные в качестве модуляторов андрогенных рецепторов

Номер патента: 12430
Опубликовано: 30.10.2009
Авторы: Вольфарт Герд, Мойланен Ану, Каллио Пекка, Хухтала Пааво, Термекангас Олли, Карьялайнен Арья, Ратилайнен Яри
МПК: A61K 31/395, A61K 31/167, A61K 31/277...
Метки: производные, модуляторов, полезные, качестве, пропионамида, рецепторов, андрогенных
Формула / Реферат:
1. Производные пропионамида формулы (I) в которой R1 представляет (С1-С7)алкил, гидрокси(С1-С7)алкил или -СНО; R2 представляет нитро, циано или атом галогена; R3 представляет (С1-С7)алкил; R4 представляет атом водорода; X представляет О или NH; А является группой, выбранной из в которых R5, R6, R7, R8 и R9 независимо представляют атом водорода, атом галогена, нитро, циано, (C1-C7)алкил, галоген (С1-С7)алкил, циано (С1-С7)алкил,...
Предыдущий патент: Композиция на основе замещенных производных 1,3-дифенилпроп-2-ен-1-она и ее применение
Следующий патент: Фармацевтическая композиция, содержащая ингибитор фермента дипептидилпептидазы
Случайный патент: Составы, содержащие фунгицидный штамм и активное соединение