Конденсированные производные пиримидина и их композиции, применимые в качестве модуляторов cxcr3 рецепторов для профилактики и лечения воспалительных и иммунорегуляторных расстройств и заболеваний
Номер патента: 11439
Опубликовано: 27.02.2009
Авторы: Фу Цзыцэ, Маркус Эндрю П., Медина Хулио К., Гастин Дарин, Дукветт Джейсон А., Джонсон Майкл Дж., Чэнь Сяоци, Ли Ань-Жун, Дейгнан Джеффри, Лю Цзивень, Михалик Джеффри Т., Бергерон Филипп, Ду Сяохой



















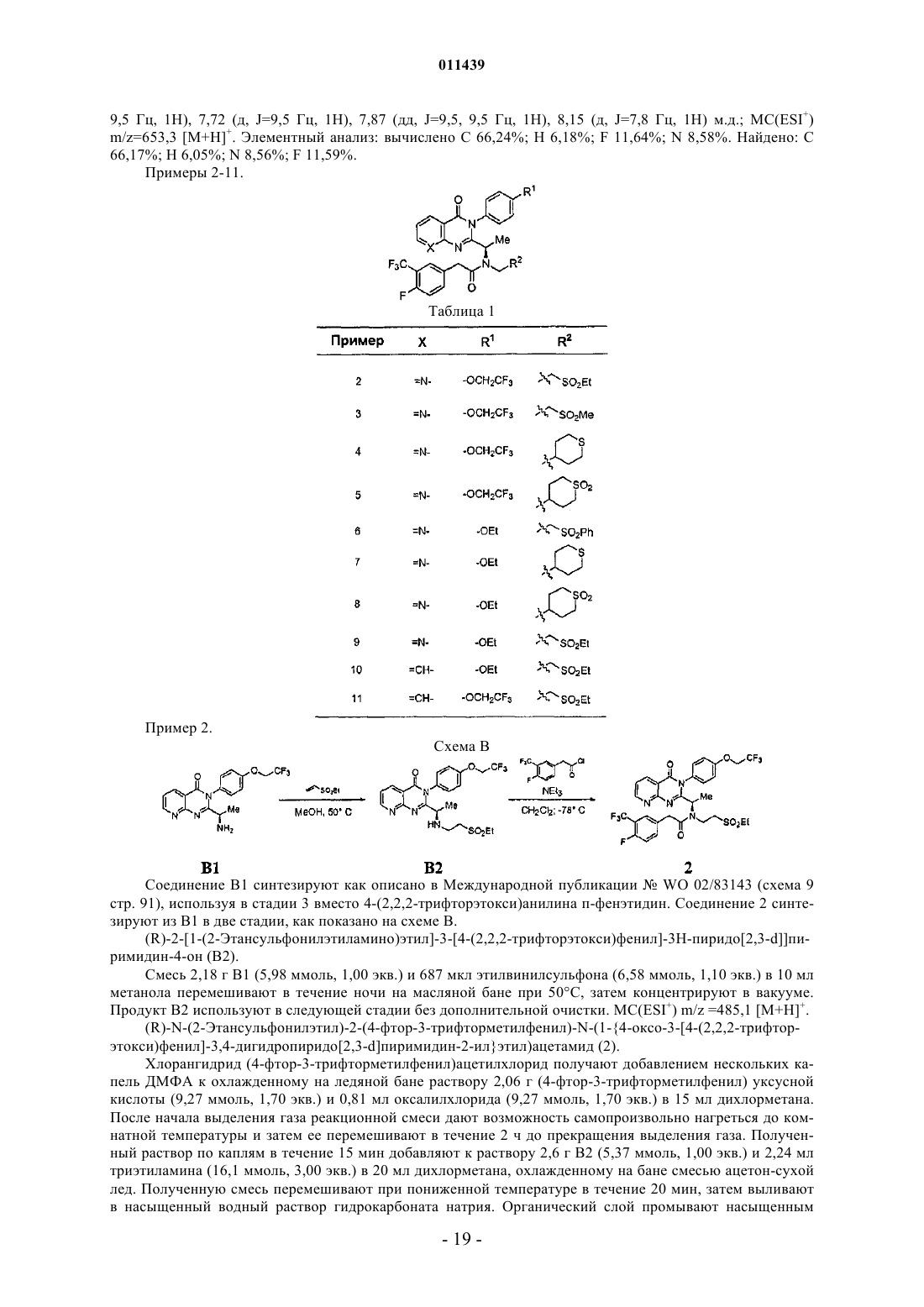
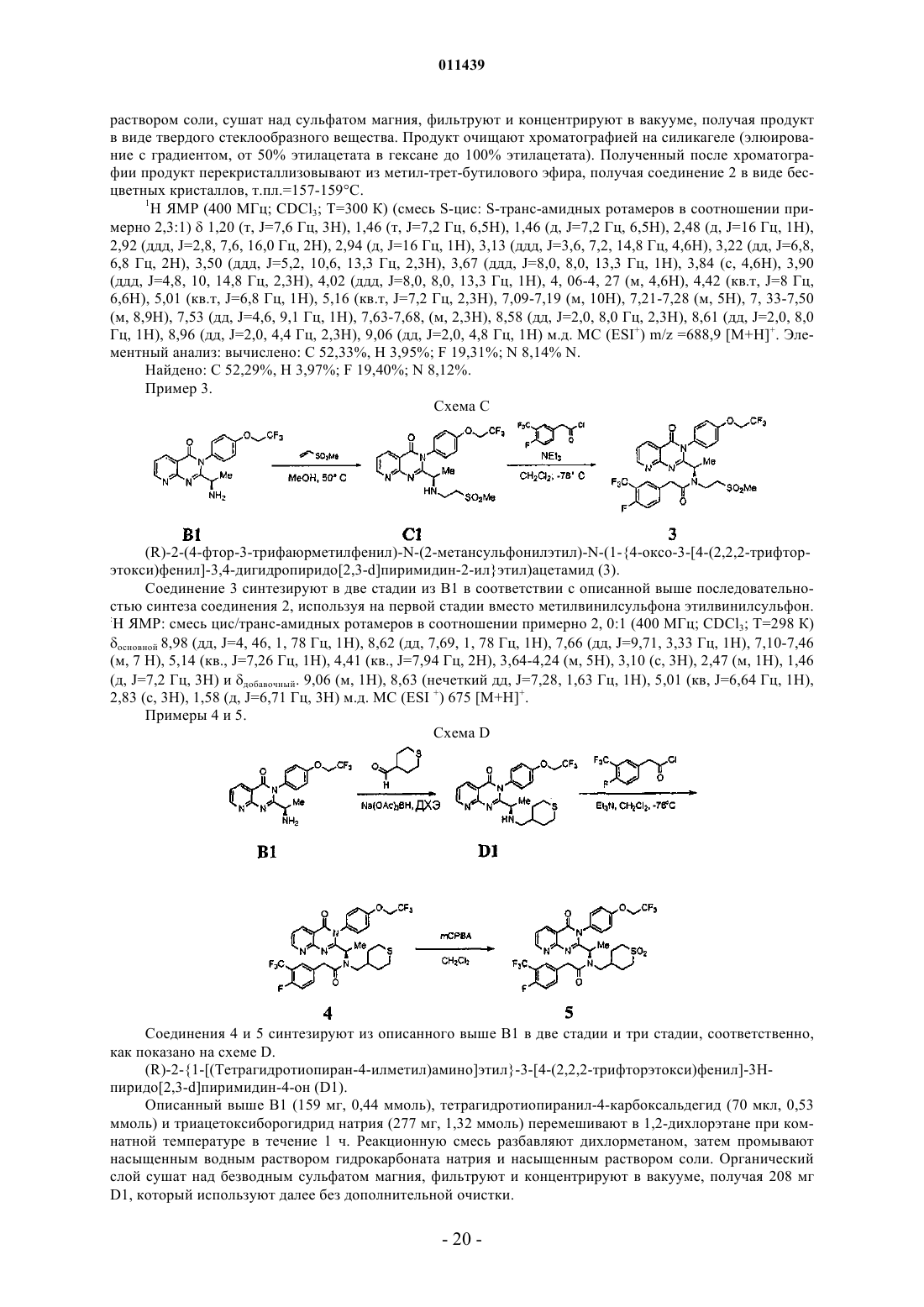








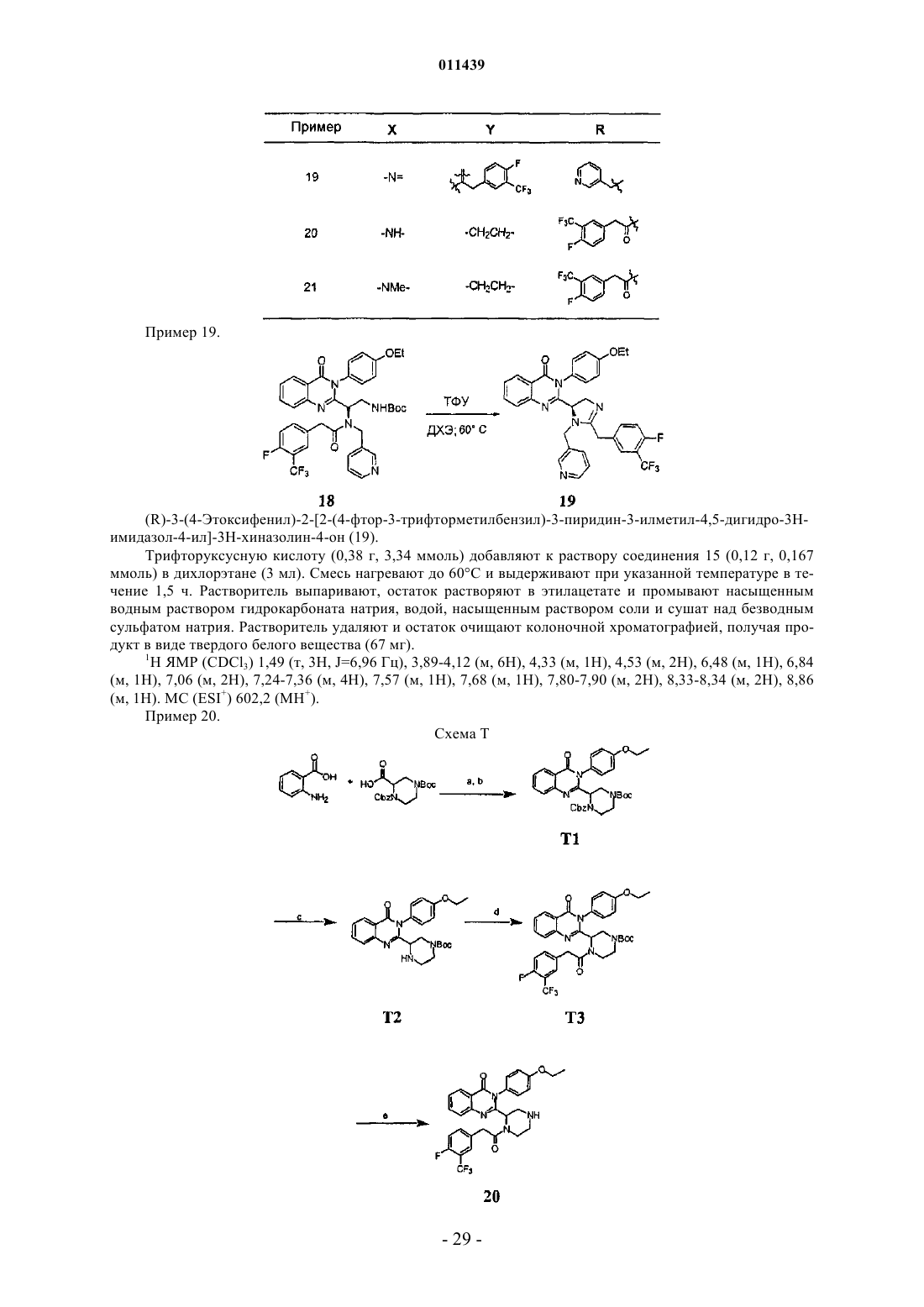
Формула / Реферат
1. Соединение формулы (I)

или его фармацевтически приемлемая соль или пролекарство,
где А1 представляет собой СН и А4 представляют собой СН или N;
Q представляет собой связь, -ОС(О)- и -СН2СО;
L представляет собой (СН2)m, где подстрочный индекс m равен 0, 1 или 2;
X представляет собой -С(О)-;
R1 представляет собой замещенный фенил, где заместитель выбиран из группы, состоящей из -OEt, -OCH2CF3, -CN и I;
R2 представляет собой -СН3, CH2NHBoc или, необязательно, R2 вместе с L и R3 могут образовывать 5- или 6-членный цикл, содержащий два атома азота;
R3 выбран из группы, включающей -Н, -SO2CH3, -SO2CH2CH3, -CH2SO2CH2CH3, -CH2SO2CH3,
-C(CH3)3, -CH-(OCH3)2, CH(CH3)2SO2CH2CH3, -СН(CH3)2SCH2CH3, -S-CH(CH3)2, SO2CH(CH3)2,
-SO2C(CH3)3, -CH(CH2OCH3)2,


или, необязательно, R3 вместе с R2 могут образовывать 5- или 6-членный цикл, содержащий два атома азота;
R4 представляет собой трет-бутил или замещенный фенил, где заместители выбраны из группы, состоящей из F, -CF3 и OCF3;
2. Соединение по п.1, где R3 выбран из группы, включающей -Н, -SO2CH3, -SO2CH2CH3,


3. Соединение по п.1 формулы (1а)

или его фармацевтически приемлемая соль или пролекарство.
4. Соединение по п.1 или 3, где А4 представляет собой N.
5. Соединение по п.1 или 3, где А4 представляет собой СН.
6. Соединение по п.1 или 3, где R1 представляет собой паразамещенный фенил, где заместитель выбран из группы, состоящей из OEt, -OCH2CF3, CN и I.
7. Соединение по п.1 или 3, где R2 представляет собой -СН3.
8. Соединение по п.1 или 3, где Q представляет собой -СН2С(О)-.
9. Соединение по п.1 или 3, где L представляет собой связь, -СН2- или -СН2СН2-.
10. Соединение по п.1 или 3, где -L-R3 вместе представляют собой 
11. Соединение по п.1 или 3, где Q-R4 представляет собой

12. Соединение по п.1 или 3, где R1 представляет собой парацианофенил.
13. Соединение по п.1 или 3 формулы (II)

или его фармацевтически приемлемая соль или пролекарство, где R11 представляет собой-СН2СН3 или -CH2CF3.
14. Соединение по п.13, где R2 представляет собой метил и -L-R3 представляет собой 
15. Соединение по п.13 или 14, где R3 выбран из группы, включающей -SO2CH3, -SO2CH2CH3,

16. Соединение по п.13 или 14, где -Q-R4 представляет собой

17. Соединение по п.16, где R3 выбран из группы, включающей -SO2CH3, -SO2CH2CH3,

18. Соединение формулы (III)

или его фармацевтически приемлемая соль или пролекарство,
где А1 представляет собой СН и А4 представляют собой СН или N;
Q представляет собой связь и -СН2СО-;
W и Y, каждый независимо, представляет собой атом углерода или гетероатом и W и Y соединены вместе с образованием 5- или 6-членного циклогетероалкильного кольца, где 5- или 6-членное кольцо является незамещенным или замещено метилом;
X представляет собой -С(О)-;
R1 представляет собой замещенный фенил, где заместитель выбран из группы, состоящей из -OEt, -OCH2CF3, -CN и I;
R3 выбран из группы, включающей -Н, 
R4 представляет собой замещенный фенил, где заместитель выбирают из группы, состоящей из F, -CF3 и OCF3.
19. Соединение по п.18, где А1 представляет собой СН.
20. Соединение по п.18, где А4 представляет собой N.
21. Соединение по п.18, где R1 представляет собой парацианофенил.
22. Соединение формулы (IV)

или его фармацевтически приемлемая соль или пролекарство,
где W1 отсутствует или представляет собой -СН2-;
W2 и Y независимо представляют собой -СН2-, -СН=, -NH-, N(СН3)- или -N=;
W3 отсутствует или представляет собой -СН2- или -N=;
А1 представляет собой СН и А4 представляют собой СН или N;
Q представляет собой связь и -CH2CO-;
X представляет собой -С(О)-;
R1 представляет собой замещенный фенил, где заместитель выбран из группы, состоящей из -OEt, -OCH2CF3, -CN и I;
R4 представляет собой замещенный фенил, где заместитель выбран из группы, состоящей из F, -CF3 и OCF3.
23. Соединение по п.22, где А1 представляет собой СН.
24. Соединение по п.23, где А4 представляет собой N.
25. Соединение по п.23, где А4 представляет собой СН.
26. Соединение по п.25, где R1 представляет собой паразамещенный фенил, где заместитель выбран из группы, состоящей из -OEt, -OCH2CF3, -CN и I.
27. Соединение по п.25, где R1 представляет собой

28. Соединение по п.25, где R представляет собой парацианофенил.
29. Соединение по п.25, где Q представляет собой -СН2С(O)-.
30. Соединение по п.25, где -Q-R4 представляет собой

31. Соединение по п.25, где W1 представляет собой -СН2-.
32. Соединение по п.25 или 31, где W2 представляет собой -N=, -NH- или -N(CH3)-.
33. Соединение по п.25, где W1, W3 и Y каждый представляет собой -СН2-.
34. Соединение по п.25, где W1 представляет собой -СН2-, W2 представляет собой -N=, W3 отсутствует и Y представляет собой -СН=.
35. Соединение по п.34, где W2 представляет собой -N(CH3)- или -NH-.
36. Соединение по п.35, где -Q-R4 представляет собой

37. Соединение по п.36 где А4 представляет собой -СН-.
38. Соединение по п.1, где соединение выбрано из группы, включающей

39. Фармацевтическая композиция, включающая соединение по п.1 или 18 и фармацевтически приемлемый носитель, разбавитель или наполнитель.
Текст
011439 Данная заявка на изобретение утверждает преимущество предварительной Заявки на изобретение США 60/583901, заявленной 28 июня 2004 г., содержание которой полностью введено в настоящее описание в виде ссылки. Настоящее изобретение относится к новым модуляторам CXCR3 рецепторов, композициям, включающим новые соединения, и способам их применения для лечения, например, воспалительных и иммунорегуляторных расстройств и заболеваний, включая астму и аллергические заболевания, а также аутоиммунных патологий, таких как ревматоидный артрит, рассеянный склероз, воспалительные заболевания кишечника, псориаз и атеросклероз. Хемокины представляют собой хемотаксические цитокины, которые вырабатываются самыми разными клетками для привлечения макрофагов, Т лимфоцитов, эозинофильных гранулоцитов, базофильных лейкоцитов и нейтрофильных гранулоцитов к сайтам воспаления (обзор свойств см. в публикацияхImmun., 12:593-633 (1994. Помимо стимулирования хемотасиса хемокины могут селективно вызывать другие изменения в чувствительных клетках, включая изменения формы клеток, временного возрастания концентрации внутриклеточных свободных ионов кальция ([Ca2+])i, экзоцитоз гранул, повышение целостной функциональной активности (integrin upregulation), образование биоактивных липидов (например,лейкотриенов) и респираторный бурст, связанный с активацией лейкоцитов. Таким образом, хемокины представляют собой механизмы раннего приведения в действие ответа на воспаление, вызывающие высвобождение медиатора воспаления, хемотаксис и транссудацию к сайтам заражения или воспаления. Существуют четыре класса хемокинов: СХС, СС), С, и СХ 3 С, которые различаются в зависимости от того, разделены ли первые два цистеина единственной аминокислотой (С-Х-С), являются ли они соседними (С-С), имеет ли место отсутствие цистеиновой пары (С) или они разделены тремя аминокислотами (СХ 3 С). -Хемокины, такие как интерлейкин-8 (IL-8), белок с активностью стимулирования роста меланомы (melanoma growth stimulatory activity protein - MGSA) и фактор, полученный из стромальных клеток (stromal cell derived factor - SDF-1), являются хемотаксическими, главным образом,для нейрофильных гранулоцитов и лимфоцитов, в то время как (-хемокины, такие как RANTES, MIP1, MIP-1, моноцитарный хемотаксический белок-1 (МСР-1), МСР-2, МСР-3 и эотаксин (eotaxin), являются хемотоксичными для макрофагов, Т лимфоцитов, эозинофилов и базофилов (Deng et al., Nature,381:661-666 (1996. С хемокиновый лимфотаксин проявляет специфичность в отношении лимфоцитов(Kelner et al., Science, 266:1395-1399 (1994, в то время как СХ 3 С хемокиновый фракталкин демонстрирует специфичность в отношении лимфоцитов и моноцитов (Bazan et al., Nature, 385:640-644 (1997. Хемокины связывают специфические рецепторы поверхностей клеток, принадлежащие к семействуG-протеин-связанных белков семи трансмембранных доменов (обзор свойств см. в публикации Horuk,Trends Pharm. Sci., 15:159-165 (1994, которые называют хемокиновыми рецепторами. Хемокиновые рецепторы при связывании с лигандами, обладающими к ним сродством, преобразуют внутриклеточный сигнал через ассоциированный гетеротримерный G-белок, приводя к быстрому повышению внутриклеточной концентрации кальция. Существует по меньшей мере двенадцать хемокиновых рецепторов человека, которые связывают -хемокины с последующей характеристической картиной или чувствительны к(1999, D6 MIP-1, RANTES и MCP-3 (Nibbs, et al., J. Biol. Chem., 272:32078-32083 (1997 и RANTES антиген группы крови Даффи, МСР-1 (Chaudhun, et al., J. Biol. Chem., 269:7835-7838 (1994. Считается, что хемокиновые рецепторы, такие как CCR1, CCR2, CCR2A, CCR2B, CCR3, CCR4,CCR5, CCR6, CCR7, CCR8, CCR9, CXCR1, CXCR2, CXCR3, CXCR4, CXCR5, CX3CR1 и XCR1, являются важными медиаторами воспаления и иммунорегуляторных расстройств и заболеваний, в том числе астмы и аллергических заболеваний, а также аутоиммунных патологий, таких как ревматоидный артрит и атеросклероз.CXCR3 хемокиновый рецептор экспрессирован, главным образом, в Т лимфоцитах, и его функциональная активность может количественно определяться повышением содержания цитозольного кальция или хемотаксисом. Ранее рецептор назывался GPR9 или CKR-L2. Среди хемокиновых рецепторов он выделяется хромосомным местоположеним, поскольку локализован в Xql3. Идентифицированными лигандами, которые являются селективными и обладают высоким сродством по отношению к нему, являются СХС хемокины, IP10, MIG и ITAC. Высокоселективная экспрессия CXCR3 делает его идеальной мишенью для вмешательства с целью-1 011439 прерывания аномального направления миграции Т лимфоцитов. Клиническими показаниями для такого вмешательства являются аутоиммунные заболевания, проводимые Т лимфоцитами, такие как рассеянный склероз, ревматоидный артрит и сахарный диабет I типа. Аномальная инфильтрация Т лимфоцитов также имеет место при псориазе и других патогенных воспалительных состояниях кожи, хотя заболевания могут не являться истинными аутоиммунными расстройствам. Тогда общим признаком кожных иммунопатологии служит повышение регуляции IP-10 экспрессии в кератиноцитах. Поэтому ингибированиеCXCR3 может быть полезно для ослабления функции отторжения при трансплантации органов. Эктопическая экспрессия CXCR3 в некоторых опухолях, особенно в подмножествах злокачественных новообразований В клеток, показывает, что селективные ингибиторы CXCR3 будут иметь значения при иммунотерапии опухолей, особенно для ослабления метастаза. Ввиду клинической важности CXCR3 - соединения, которые модулируют CXCR3 функцию, могут использоваться для разработки новых терапевтических средств. Такие соединения представлены в настоящем изобретении. Настоящее изобретение предоставляет соединения, пригодные для лечения или профилактики некоторых воспалительных и иммунорегуляторных расстройств и заболеваний, включая астму, псориаз,воспалительные заболевания кишечника и аллергические заболевания, а также аутоиммунных патологий, таких как ревматоидный артрит и рассеянный склероз. В соответствии с одним аспектом, предоставлены соединения общей формулы (I) где А 1 и А 4 независимо представляют собой C(Rb) или N; Q выбран из группы, включающей связь,(C1-C8)алкилен, (С 2-С 8) гетероалкилен, -С(О)-, -ОС(О)-, -СН 2 СО-, -CH2SO- и -CH2SO2-; L представляет собой связь или (С 1-С 5) алкилен; X представляет собой -СН 2-, SO2 или -С(О)-; Ra выбран из группы,включающей -OR', =O, =NR', =N-OR', -NR'R", -SR', -галоген, -SiR' R"R , -OC(O)R', -C(O)R', -CO2R', CONR'R", -OC(O)NR'R", -NR"C(O)R', -NR1-C(O)NR"R, -NR"C(O)2R', -NH-C(NH2)=NH, -NR'C(NH2) =NR' , -R', -S(O)R', -S(O)2R', -S(O)2NR'R", -CN и -NO2; подстрочный индекс n равен 0, 1, 2 или 3; каждый из R', R" и R независимо представляет собой Н, незамещенный (C1-C8)алкил, гетероалкил, незамещенный арил или замещенный арил; R1 представляет собой гетероарил или арил; R2 выбран из группы, включающей водород, галоген, (C1-С 10)алкил, (С 2-С 10)гетероалкил, гетеро(С 1-С 10)циклоалкил, (C1 С 10)алкиларил и (С 2-С 10)гетероалкиларил или, необязательно, R2 вместе с L могут образовывать 5-, 6-, 7 или 8-членный цикл, содержащий от 1 до 3 гетероатомов, выбранных из группы, включающей атомы N,O и S; R3 отсутствует или выбран из группы, включающей -Н, -CHR6R7, S(O)mR5, -S(О)mN(R8)R9,-S(O)mN(R8)CH2R6, -N(R8)SO2R5, -N(R6)CH2R10(C1-С 8)алкил, (С 2-С 8)гетероалкил, арил и гетероарил; R6 и R7 независимо представляют собой водород,(C1-С 8) алкил или (С 2-С 8)гетероалкил; R8 представляет собой водород, (C1-С 8)алкил, (С 2-С 8)гетероалкил,гетероарил или арил, R9 представляет собой (C1-С 8)алкил; R10 представляет собой арил; Z представляет собой СН или N; X1 представляет собой связь, (C1-С 6)алкилен или (C1-C6)гетероалкилен; Y1 представляет собой (C1-C6)алкилен; и подстрочный индекс m равен 0, 1 или 2. В соответствии с другим аспектом, изобретение предоставляет соединения формулы (III) где А 1 и А 4 независимо представляют собой C(Rb) или N; Q выбран из группы, включающей связь,(C1-С 8)алкилен, (С 2-С 8) гетероалкилен, -С(О)-, -ОС(О)-, -CH2CO-, -CH2SO- и -CH2SO2-; каждый из W и Y независимо представляет собой атом углерода или гетероатом, и W и Y, соединенные вместе, образуют 5- или 6-членное циклоалкильное или циклогетероалкильное кольцо, где 5- или 6-членный цикл является незамещенным или замещен галогеном, NH2, NO2, (C1-С 20) алкилом, (C1-С 20)гетероалкилом, гетероарилом, арилом, гетероарил (C1-С 6)алкилом, гетероарил (C2-С 6)гетероалкилом, арил (C2-С 6)алкилом и арил-OR',=O,=NR',=N-OR', -NR'R", -SR', -галоген, -SiR'R"R, -OC(O)R', -C(O)R', -CO2R', -CONR'R", OC(O)NR'R", -NR"C(O)R', -NR'-C(O)NR"R, -NR"C(O)2R', -NH-C(NH2)=NH, -NR'C(NH2)=NH, -NHC(NH2)=NR', -S(O)R', -S(O)2R' -S(O)2NR'R", -CN и -NO2; подстрочный индекс n равен 0, 1, 2 или 3; каждый R', R" и R независимо представляет собой Н, незамещенный (C2-С 8)алкил, гетероалкил, незамещенный арил или замещенный арил; R1 представляет собой гетероарил или арил; R3 отсутствует или выбран из группы, включающей -Н, -CHR6R7, S(O)mR5, -S(O)mN(R8)R9, -S(O)mN(R8)CH2R6, -N(R8)SO2R5, N(R8) CH2R10(C2-С 6)алкил, гетероарил(C2-С 6)гетероалкил, арил (C2-С 6)алкил и арил(C2-С 6)гетероалкил; R5 выбран из группы, включающей (C2-С 8)алкил, (C2-С 8)гетероалкил, арил и гетероарил; R6 и R7 независимо представляют собой водород, (C2-С 8)алкил или (C2-С 8)гетероалкил; R8 представляет собой водород, (C2-С 8)алкил,(С 2-С 8)гетероалкил, гетероарил или арил, R9 представляет собой (C2-С 8)алкил; и R10 представляет собой арил. Соединения согласно изобретению включают фармацевтически приемлемые соли, сольваты или пролекарства. В соответствии с другим аспектом, настоящее изобретение предоставляет фармацевтические композиции, включающие соединение формулы (I) или (III) и фармацевтически приемлемый наполнитель или носитель. В соответствии с еще одним аспектом, настоящее изобретение предоставляет способы лечения или профилактики воспалительного или иммунного заболевания или расстройства, включающие введение субъекту, нуждающемуся в таком лечении или профилактике, терапевтически эффективного количества соединения формулы (I) или (III). Предпочтительными субъектами для способов настоящего изобретения являются млекопитающие, например человек. Настоящее изобретение предоставляет также способы лечения или профилактики состояния или расстройства, проводимого CXCR3 хемокиновым рецептором, указанные способы включают введение субъекту, нуждающемуся в таком лечении или профилактике, терапевтически эффективного количества соединения формулы (I) или (III). Настоящее изобретение представляет также способы модуляции CXCR3, включающие контактирование клетки с соединением формулы (I) или (III). Настоящее изобретение предоставляет также способы модуляции CXCR3, включающие контактирование CXCR3 белка с соединением формулы (I) или (III). Кроме того, настоящее изобретение предоставляет способы получения соединений формулы (I) или(III). На фиг. 1 представлена общая схема синтеза соединений согласно настоящему изобретению; на фиг. 2 - общая схема синтеза соединений согласно настоящему изобретению; на фиг. 3 - общая схема синтеза соединений согласно настоящему изобретению. Термин алкил сам по себе или как часть другого заместителя означает, за исключением особо оговоренных случаев, прямой, разветвленный или циклический углеводородный радикал или сочетание указанных углеводородных радикалов, который(ые) может(гут) быть полностью насыщенным(и), моно-3 011439 или полиненасыщенным(и), может(гут) включать ди- и многовалентные радикалы и содержит указанное количество атомов углерода (т.е. C1-С 10 означает от одного до десяти атомов углерода). Примеры насыщенных углеводородных радикалов включают такие группы, как метил, этил, н-пропил, изопропил, нбутил, трет-бутил, изобутил, втор-бутил, циклогексил, (циклогексил)метил, циклопропилметил, их гомологи и изомеры, например, н-пентил, н-гексил, н-гептил, н-октил и т.п. Ненасыщенная алкильная группа представляют собой группу, содержащую одну или несколько двойных или тройных связей. Примеры ненасыщенных алкильных групп включают винил, 2-пропенил, кротил, 2-изопентил, 2-(бутадиенил), 2,4 пентадиенил, 3-(1,4-пентадиенил), этинил, 1- и 3-пропинил, 3-бутинил и их высшие гомологи и изомеры. Термин алкилен сам по себе или как часть другого заместителя означает, за исключением особо оговоренных случаев, двухвалентный радикал, полученный из алкана, например, -СН 2 СН 2 СН 2 СН 2-, а также группы, определенные ниже термином гетероалкилен. Обычно алкильная (или алкиленовая) группа будет содержать от 1 до 24 атомов углерода, причем группы, содержащие 10 или менее атомов углерода, предпочтительны согласно настоящему изобретению. Термин низший алкил или низший алкилен означает алкильную или алкиленовую группу с более короткой цепью, содержащей обычно восемь атомов углерода или менее. Термины алкокси, алкиламино и алкилтио (или тиоалкокси), используются в настоящем описании в их обычном значении и относятся к алкильным группам, присоединенным к остальной части молекулы через атом кислорода, аминогруппу или атом серы, соответственно. Аналогично, термин диалкиламино относится к аминогруппе, содержащей две присоединенные алкильные группы, которые могут быть одинаковыми или разными. Термин гетероалкил сам по себе или как часть другого заместителя означает, за исключением особо оговоренных случаев, стабильный прямой, разветвленный или циклический углеводородный радикал или сочетание указанных углеводородных радикалов, включающий(ее) указанное количество атомов углерода и от одного или до трех гетероатомов, выбранных из группы, включающей атомы О, N, Si иS, где атомы азота и серы могут быть необязательно окисленными и азотный гетероатом может необязательно быть четвертичным. Гетероатом(ы) О, N и S могут располагаться в любом положении внутри гетероалкильной группы. Гетероатом Si может находиться в любом положении гетероалкильной группы,включая положение, в котором алкильная группа присоединена к остальной части молекулы. Примеры гетероалкила включают -СН 2-СН 2-О-СН 3, -CH2-CH2-NH-CH3, -CH2-CH2-N(CH3-)-СН 3, -CH2-S-CH2-СН 3,-СН 2-СН 2, -S(O)-CH3, -СН 2-СН 2-S(O)2-СН 3, -СН=СН-O-СН 3, -Si(CH3)3, -CH2-CH=N-OCH3 и -CH=CH-N(СН 3)-СН 3. Последовательно соединенными могут быть не более двух гетероатомов, например -СН 2-NHOCH3 и -CH2-O-Si(СН 3)3. Когда в определении гетероалкильной группы используется подстрочный индекс, например, (С 2-С 8), количество атомов углерода (в данном случае 2-8) включает также гетероатомы. Например, подразумевается, что С 2-гетероалкильная группа включает в частности, -СН 2 ОН (один атом углерода и один гетероатом, замещающий атом углерода) и -CH2SH. Термин гетероалкилен сам по себе или как часть другого заместителя означает, за исключением особо оговоренных случаев, двухвалентный радикал, полученный из гетероалкила, например -CH2-CH2-S-CH2CH2- и -CH2-S-CH2-CH2-NHCH2-. В гетероалкиленовых группах гетероатомы также могут занимать одно или оба концевых положения в цепи (например, алкиленокси, алкилендиокси, алкиленамино, алкилендиамино и т.п.). Кроме того,определение алкиленовых и гетероалкиленовых связующих групп не предполагает определения точного расположения связующей группы. Термин циклоалкил или гетероциклоалкил сам по себе или как часть другого заместителя означает, за исключением особо оговоренных случаев, циклические разновидности алкила или гетероалкила, соответственно. Кроме того, в гетероциклоалкиле гетероатом может занимать положение, в котором гетероцикл присоединяется к остальной части молекулы. Примеры циклоалкила включают циклопентил, циклогексил, 1-циклогексенил, 3-циклогексенил, циклогептил и т.п. Примеры гетероциклоалкила включают 1-(1,2,5,6-тетрагидропиридил), 1-пиперидинил, 2-пиперидинил, 3-пиперидинил, 4 морфолинил, 3-морфолинил, тетрагидрофуран-2-ил, тетрагидрофуран-3-ил, тетрагидротиен-2-ил, тетрагидротиен-3-ил, 1-пиперазинил, 2-пиперазинил и т.п. Термин галоген сам по себе или как часть другого заместителя означает, за исключением особо оговоренных случаев, атом фтора, хлора, брома или йода. Кроме того, значение термина галогеналкил включает моногалогеналкил и полигалогеналкил. Например, подразумевается, что термин галоген(C1C4)алкил включает трифторметил, 2,2,2-трифторэтил, 4-хлорбутил, 3-бромпропил и т.п. Термин арил сам по себе или как часть другого заместителя означает, за исключением особо оговоренных случаев, полиненасыщенный, как правило, ароматический, углеводородный заместитель, который может представлять собой единственное кольцо или состоять из нескольких колец (до трех колец),конденсированных или соединенных ковалентно. Термин гетероарил относится к арильным группам(или циклам), которые содержат от нуля до четырех гетероатомов, выбранных из атомов N, О и S, где атомы азота и серы необязательно являются окисленными и атом(ы) азота необязательно является(ются) четвертичными. Гетероарильная группа может присоединяться к остальной части молекулы через гетероатом. Неограничивающие примеры арильных и гетероарильных групп включают фенил, 1-нафтил, 2 нафтил, 4-бифенил, 1-пирролил, 2-пирролил, 3-пирролил, 3-пиразолил, 2-имидазолил, 4-имидазолил, пи-4 011439 разинил, 2-оксазолил, 4-оксазолил, 2-фенил-4-оксазолил, 5-оксазолил, 3-изоксазолил, 4-изоксазолил, 5 изоксазолил, 2-тиазолил, 4-тиазолил, 5-тиазолил, 2-фурил, 3-фурил, 2-тиенил, 3-тиенил, 2-пиридил, 3 пиридил, 4-пиридил, 2-пиримидил, 4-пиримидил, 5-бензотиазолил, пуринил, 2-бензимидазолил, 5 индолил, 1-изохинолил, 5-изохинолил, 2-хиноксалинил, 5-хиноксалинил, 3-хинолил и 6-хинолил. Заместители для каждой из перечисленных выше арильных или гетероарильных кольцевых систем выбраны из группы приемлемых заместителей, описанных ниже. Для краткости, значение термина арил при использовании его в сочетании с другими терминами(например, арилокси, арилтиокси, арилалкил), значение включает арильные и гетероарильные циклы,которые определены выше. Таким образом, подразумевается, что термин арилалкил включает радикалы, в которых арильная группа присоединена к алкильной группе (например, бензил, фенэтил, пиридилметил и т.п.), включая алкильные группы, в которых атом углерода (например, метиленовая группа) замещен, например, атомом кислорода (например, феноксиметил, 2-пиридилоксиметил, 3-(1 нафтилокси)пропил и т.п.). Подразумевается, что каждый из определенных выше терминов (например, алкил, гетероалкил,арил и гетероарил) включает как замещенные, так и незамещенные формы указанного радикала. Предпочтительные заместители для каждого типа радикала представлены ниже. Заместители для алкильных и гетероалкильных радикалов (включая группы, определяемые как алкилен, алкенил, гетероалкилен, гетероалкенил, алкинил, циклоалкил, гетероциклоалкил, циклоалкенил и гетероциклоалкенил) могут представлять собой различные группы, выбранные из -OR', =O, =NR' , =NOR', -NR'R", -SR', -галоген, -SiR'R"R, -OC(O)R', -C(O)R', -CO2R', -CONR'R", -OC(O)NR'R", -NR"C(O)R',-NR'-C(O)NR"R, -NR"C(O)2R', -NH-C(NH2)=NH, -NR'С (NH2)=NH, -NH-C(NH2)=NR', -S(O)R', -S(O)2R',-S(O)2NR'R", -CN и -NO2 и взятые в количестве в интервале от нуля до (2m+1), где m равно общему количеству атомов углерода в таком радикале. R', R" и R каждый независимо представляет собой Н, незамещенный (C1-C8)алкил и гетероалкил, незамещенный арил, арил, замещенный 1-3 галогенами, алкокси или тиоалкоксигруппами или арил-(С 1-C4)алкильными группами. Когда R' и R" присоединены к одному атому азота, они могут объединяться с атомом азота с образованием 5-, 6- или 7-членного цикла. Например подразумевается, что -NR'R" включает 1-пирролидинил и 4-морфолинил. Подразумевается также,что из приведенного выше обсуждения заместителей специалист данной области техники может представить, что термин алкил в наиболее широком смысле включает такие группы, как галогеналкил (например, -CF3 и -CH2CF3) и ацил (например, -C(O)CH3, -C(O)CF3, -С(O)СН 2 ОСН 3 и т.п.). Предпочтительно, за исключением особо оговоренных случаев, алкильные группы будут содержать 0-3 заместителя,более предпочтительно, 0, 1 или 2 заместителя. Аналогично, заместители для арильных и гетероарильных групп являются различными, выбраны из галогена, -OR', OC(O)R', -NR'R", -SR', -R', -CN, -NO2, -CO2R', -CONR'R", -C(O)R', -ОС(O)NR'R", NR"C(O)R', -NR"C(O)2R', -NR'-C(O)NR"R, -NH-C(NH2)=NH, -NR'C(NH2)=NH, -NH-C(NH2)=NR', -S(O)R',-S(O)2R', -S(O)2NR'R", -N3, -CH(Ph)2, перфтор (C1-C4) алкокси и перфтор(C1-C4)алкила и присутствуют в количестве в интервале от нуля до общего количества открытых валентностей на ароматической циклической системе; где R', R" и R независимо выбраны из Н, (C1-C8)алкила и гетероалкила, незамещенного арила и гетероарила, (незамещенного арил)-(C1-C4)алкила и (незамещенного арил)окси-(С 1-С 4)алкила. Два заместителя на соседних атомах арильного или гетероарильного цикла могут необязательно замещаться заместителем формулы -Т-С(О)-(СН 2)q-U-, где Т и U независимо представляют собой -NH-, -O-, -СН 2 или одинарную связь, и q представляет собой целое число от 0 до 2. Альтернативно, два из заместителей на соседних атомах арильного или гетероарильного цикла необязательно могут замещаться заместителем формулы -А-(СН 2)r-В-, где А и В независимо представляют собой -СН 2-, -O-, -NH-, -S-, -S(O)-, -S(O)2-, -S(O)2NR'или одинарную связь, и r представляет собой целое число от 1 до 3. Одна из одинарных связей новых циклов,образованных таким образом, необязательно может быть замещена двойной связью. Альтернативно, два из заместителей на соседних атомах арильного или гетероарильного цикла необязательно могут быть замещены заместителем формулы -(СН 2)S-X-(СН 2)t- где s и t независимо представляют собой целые числа от 0 до 3, и X представляет собой -O-, -NR'-, -S-, -S(O)-, -S(O)2- или -S(O)2NR'-. Заместитель R' в -NR'- и S(O)2NR'- выбран из водорода или незамещенного (C1-С 6)алкила. Подразумевается, что термин гетероатом, когда используется в настоящем описании, означает атом кислорода (О), азота (N), серы (S) и кремния (Si). Аббревиатура "Me", когда используется в настоящем описании, означает метил (т.е. -СН 3), аббревиатура Et, когда используется в настоящем описании, означает этил, и аббревиатура Ph, когда используется в описании, означает фенил. Подразумевается, что термин фармацевтически приемлемые соли включает соли соединений, которые получены с относительно нетоксичными кислотами или основаниями, в зависимости от конкретных заместителей, находящихся на соединениях согласно настоящему изобретению. Когда соединения согласно настоящему изобретению содержат относительно кислотные функциональности, основноаддитивные соли могут быть получены контактированием нейтральной формы таких соединений с достаточным количеством нужного основания в чистом виде или в подходящем инертном растворителе. Примеры фармацевтически приемлемых основно-аддитивных солей включают соль натрия, калия, каль-5 011439 ция, аммония, органическую аминную или магниевую соль и т.п. Когда соединения согласно настоящему изобретению содержат относительно основные функциональности, кислотно-аддитивные соли могут быть получены контактированием нейтральной формы таких солей с достаточным количеством нужной кислоты в чистом виде или в подходящем инертном растворителе. Примеры фармацевтически приемлемых кислотно-аддитивных солей включают соли, полученные из неорганических кислот, таких как соляная, бромистоводородная, азотная, угольная, гидрокарбонат, форсфорная (ортофосфорная) , моногидрофосфат, дигидрофосфат, серная, моногидросульфат, йодисто-водородная или фосфористая и т.п. кислоты, а также соли, полученные из относительно нетоксичных органических кислот, таких как уксусная,пропионовая, изомасляная, малеиновая, малоновая, бензойная, янтарная, субериновая, фумаровая, миндальная, фталевая, бензолсульфоновая, п-толуолсульфоновая, лимонная, винная, метансульфоновая и т.п. кислоты. Они также включают соли аминокислот, таких как аргинат и т.п., и соли органических кислот, таких как глюкуроновая или галактуноровая (galactunoric) кислоты и т.п. (см., например, Berge, etal. (1977) J. Pharm. Sci 66:1-19). Некоторые конкретные соединения согласно настоящему изобретению содержат как основные, так и кислотные функциональности, которые позволяют превращать эти соединения как в основно-, так и в кислотно-аддитивные соли. Нейтральные формы соединений могут регенерироваться контактированием соли с основанием или кислотой и выделением исходного соединения удобным образом. Исходная форма соединения отличается от различных солевых форм определенными физическими свойствами, такими как растворимость в полярных растворителях, но что касается целей настоящего изобретения, то в остальном соли эквивалентны исходной форме соединений согласно настоящему изобретению. Помимо солевых форм, настоящее изобретение предоставляет соединения, которые являются пролекарственной формой. Пролекарства активных соединений, описанных в настоящем изобретении, являются неактивными соединениями, которые легко подвергаются химическим превращениям в физиологических условиях с получением активных соединений согласно настоящему изобретению. Кроме того,пролекарства могут подвергаться превращению в активные соединения согласно настоящему изобретению химическими или биохимическими методами ex vivo. Например, пролекарства могут подвергаться медленному превращению в активные соединения согласно настоящему изобретению при размещении их в резервуаре трансдермальной заплаты с подходящим ферментом или химическим реагентом. Зачастую применение пролекарств обусловлено тем, что в некоторых случаях они могут вводиться легче, чем активное соединение. Они могут, например, быть биологически доступными посредством перорального введения, тогда как активное соединение - нет. Пролекарство может также обладать лучшей растворимостью в фармакологических композициях, чем активное соединение. В данной области техники известен широкий спектр пролекарственных производных, таких как пролекарства, расщепление с активацией которых происходит в результате гидролиза или окисления. Например, но без ограничения,пролекарством могло бы быть соединение согласно настоящему изобретению, которое вводится в форме сложного эфира (пролекарство), но затем подвергается метаболитическому гидролизу до карбоновой кислоты - активного ингредиента. Дополнительные примеры включают пептидил-производные активного соединения согласно настоящему изобретению. Некоторые соединения согласно настоящему изобретению могут существовать как в несольватированных формах, так и в сольватированных формах, включая гидратные формы. Обычно сольватированные формы эквиваленты несольватированным формам, и подразумевается, что они включены в область настоящего изобретения. Некоторые соединения согласно настоящему изобретению могут существовать во множестве кристаллических или аморфных форм. Обычно все физические формы эквивалентны для областей применения, предусмотренных настоящим изобретением, и подразумевается, что они включены в область настоящего изобретения. Некоторые соединения согласно настоящему изобретению содержат асимметричные атомы углерода (оптические центры) или двойные связи; и подразумевается, что все рацематы, энантиомеры, диастереомеры, геометрические изомеры и индивидуальные изомеры включены в область настоящего изобретения. За исключением особо оговоренных случаев, термин стереизомер или стереомерно чистый, когда используется в настоящем описании, означает один стереоизомер соединения, который, по существу,свободен от других стереоизомеров данного соединения. Например, стереомерно чистое соединение,содержащее один хиральный центр, будет, по существу, свободно от противоположного энантиомера данного соединения. Стереомерно чистое соединение, содержащее два хиральных центра, будет, по существу, свободно от других диастереомеров соединения. Обычно стереомерно чистое соединение состоит из более чем примерно 80 мас.% одного стереоизомера соединения и менее чем примерно 20 мас.% других стереоизомеров соединения, более предпочтительно, из более чем примерно 90 мас.% одного стереоизомера соединения и менее чем примерно 10 мас.% других стереоизомеров соединения, еще более предпочтительно, более чем примерно 95 мас.% одного стереоизомера соединения и менее чем примерно 5 мас.% других стереоизомеров соединения, и наиболее предпочтительно, более чем примерно 97 мас.% одного стереоизомера соединения и менее чем примерно 3 мас.% других стереоизомеров соединения. Следует отметить, что если стереохимия структуры или части структуры не указана, например,-6 011439 жирной или пунктирной линиями, то подразумевается, что структура или часть структуры включает все ее стереоизомеры. Различные соединения согласно настоящему изобретению содержат один или несколько хиральных центров и могут существовать в виде рацемических смесей энантиомеров, смесей диастереомеров или в виде энантиомерно или оптических чистых соединений. Данное изобретение включает применение стереомерно чистых форм таких соединений, а также применение смесей этих форм. Например, смеси,включающие в себя равные или неравные количества энантиомеров конкретного соединения согласно настоящему изобретению, могут применяться в способах и композициях согласно настоящему изобретению. Такие изомеры могут быть синтезированы способами ассиметрического синтеза или могут разделяться с использованием стандартных методов, таких как применение хиральных колонок или хиральных разделяющих агентов (см., например, Jacques, J., et al., Enantiomers, Racemates и Resolutions (WileyInterscience, New York, 1981); Wilen, S.H., et al., Tetrahedron 33:2725 (1977); Eliel, E.L., Stereochemistry ofp. 268 (E.L. Eliel, Ed., Univ. of Notre Dame Press, Notre Dame, IN, 1972). Соединения согласно настоящему изобретению могут также содержать необычные доли атомных изотопов одного или нескольких атомов, которые составляют такие соединения. Например, соединение может содержать радиоизотопную метку, такую как, например, тритий (3 Н), йод-125 (125I) или углерод-14(14 С). Меченые радиоизотопом соединения применимы в качестве терапевтических средств, например терапевтических средств против рака, реагентов, используемых при исследованиях, например, в качестве реагентов, использующихся для исследования связывания, и в качестве средств диагностики, например радиофармацевтических средств для анализа в условиях in vivo. Подразумевается, что все изотопные разновидности соединений согласно настоящему изобретению, независимо, являются они радиоактивными или нет, включены в область настоящего изобретения. Термин активный, когда используется в настоящем описании, означает эффективный для осуществления модуляции, например, для ингибирования, функции CXCR3. Термин лечение (treat, treating или treatment), когда используется в настоящем описании,относится к способу облегчения или устранения заболевания и/или сопровождающих его симптомов. Термин профилактика (prevent, preventing или prevention), когда используется в настоящем описании, относится к способу защиты субъекта от заболевания. Настоящее изобретение относится к соединениям, композициям и способам, применимым для модуляции активности хемокинового рецептора, в частности CXCR3. Соединения согласно изобретению применимы для лечения, например, воспалительных и иммунорегуляторных расстройств и могут вводиться непосредственно субъектам, например человеку, в виде фармацевтических препаратов. Соединения согласно настоящему изобретению применимы также для идентификации и/или разработки соединений, которые модулируют функцию CXCR3, например, антагонистов CXCR3, и соединений, которые превращаются в одно или несколько соединений, модулирующих функцию CXCR3 в физиологических условиях. Соединения согласно настоящему изобретения представляют собой соединения, которые ингибируют по меньшей мере одну функцию или характеристику CXCR3 белка млекопитающих, напримерCXCR3 белка человека. Способность соединения ингибировать такую функцию может быть показана при исследовании связывания (например, связывания лиганда или связывания агониста), исследовании сигнала (например, активации G белка млекопитающих, индуцирования быстрого и преходящего повышения концентрации свободного цитозольного кальция) и/или функции клеточного ответа (например,стимуляции хемотаксиса, эксоцитоза или высвобождения медиатора воспаления лейкоцитами). Примеры таких исследований описаны в Публикациях Заявок на Патент США 2002/0169159 А 1 и 2003/0055054 А 1, содержание которых введено во всей полноте в данное описание в виде ссылок. Настоящее изобретение предоставляет соединения, которые применимы в качестве антагонистовCXCR3, пригодных, в частности, для лечения или профилактики воспалительных или иммунных состояний или расстройств. В соответствии с одним аспектом, настоящее изобретение предоставляет соединение формулы (I) где Ra, R1, R2, R3, R4, A1, A4, L, Q, X и подстрочный индекс n принимают значения, определенные ниже. Соединения приведенной выше формулы включают, за исключением особо оговоренных случаев,фармацевтически приемлемые соли, сольваты или их пролекарства. А 1 и А 4 независимо представляют собой C(Rb) или N. В некоторых примерах осуществления настоящего изобретения А 4 представляет собой N.-7 011439 В некоторых примерах осуществления настоящего изобретения А 4 представляет собой C(Rb). В некоторых примерах осуществления настоящего изобретения А 1 представляет собой СН. В некоторых примерах осуществления настоящего изобретения А 4 представляет собой СН.Q выбран из группы, включающей связь, (С 1-С 8)алкилен, (С 2-С 8)гетероалкилен, -С(О)-, -ОС(О)-,-СН 2 СО-, -CH2SO- и -CH2SO2-. В некоторых примерах осуществления настоящего изобретения Q представляет собой -С(О)-.L представляет собой связь или (С 1-С 5)алкилен. В некоторых примерах осуществления настоящего изобретения L представляет собой связь, -СН 2 или -СН 2 СН 2-.X представляет собой -СН 2-, SO2 или -С(О)-. В некоторых примерах осуществления изобретения X представляет собой -С(О)-.Rb выбран из группы, включающей -OR', =O, =NR', =N-OR', -NR'R", -SR', -галоген, -SiR'R"R'",-OC(O)R', -C(O)R', -CO2R', -CONR'R", -OC(O)NR'R", -NR"C(O)R', -NR'-C(O)NR"R, -NR"C(O)2R', -NHC(NH2)=NH, -NR'С (NH2)=NH, -NH-C(NH2)=NR', -R', -S(O)R', S(O)2R', -S(O)2NR'R", -CN и -NO2. Подстрочный индекс n равен 0, 1, 2 или 3. Каждый R', R" и R'" независимо представляет собой Н, незамещенный (C1-C8)алкил, гетероалкил,незамещенный арил или замещенный арил.R1 представляет собой гетероарил или арил. В некоторых вариантах осуществления настоящего изобретения R1 представляет собой незамещенный или мета- или паразамещенный фенил, где заместитель представляет собой галоген, циано, (С 2-С 8) алкил, (С 2-С 8) гетероалкил, (С 2-С 8) алкокси или (С 2-С 8) гетероалкокси. В некоторых вариантах осуществления настоящего изобретения R1 представляет собой парацианофенил.R2 выбран из группы, включающей водород, галоген, (С 1-С 10)алкил, (С 2-С 10)гетероалкил, гетеро (С 1 С 10)циклоалкил, (С 1-С 10)алкиларил и (С 2-С 10)гетероалкиларил или необязательно R2 вместе с L образуют 5-, 6-, 7- или 8-членный цикл, содержащий от 1 до 3 гетероатомов, выбранных из группы, включающей атомы N, О и S. В некоторых вариантах осуществления настоящего изобретения R2 выбран из группы, включающейR3 отсутствует или выбран из группы, включающей Н, -CHR6R7, -S(O)mR5, -S(O)mN(R8)R9, S(O)mN(R8)CH2R6, -N(R8)SO2R5, -N(R8)CH2R10, или,необязательно, R3 вместе с R2 могут образовывать 4-, 5-, 6-, 7-или 8-членный цикл, содержащий от 1 до 3 гетероатомов, выбранных из группы, включающей атомы N, О и S. В некоторых вариантах осуществления настоящего изобретения R3 выбран из группы, включающей-Н, -SO2CH3, -SO2CH2CH3, СН(СН 3)СН 3, В некоторых вариантах осуществления настоящего изобретения -L-R3 вместе представляют собой(C1-C6)алкил, гетероарил (С 2-С 6)гетероалкил, арил(C1-C6)алкил и арил(С 2-С 6)гетероалкил. В некоторых вариантах осуществления настоящего изобретения -Q-R4 представляет собойR6 и R7 независимо представляют собой водород, (C1-C8)алкил или (С 2-С 8)гетероалкил.Z представляет собой СН или N.Y1 представляет собой (C1-C6) алкилен. Подстрочный индекс m равен 0, 1 или 2. В некоторых вариантах осуществления настоящего изобретения соединение соответствует формуле где Ra, R2, R3, R4, A1, A4, L, Q, X и подстрочный индекс n принимают значения, описанные выше для формулы I, R11 представляет собой водород, (C1-C8)алкил или (С 2-С 8)гетероалкил. В некоторых вариантах осуществления настоящего изобретения R11 представляет собой -Н, -СН 3, СН 2 СН 3 или -CH2CF3. В некоторых вариантах осуществления настоящего изобретения R2 представляет собой метил и -L3 В некоторых вариантах осуществления настоящего изобретения R3 выбран из группы, включающей В некоторых вариантах осуществления настоящего изобретения соединение согласно настоящему изобретению соответствует формуле (III) где Ra, R1, R3, R4, A1, A4, Y, Q, X и подстрочный индекс п принимают значения, определенные ниже. Соединения представленной выше формулы включают, за исключением особо оговоренных случаев, фармацевтически приемлемые соли, сольваты или их пролекарства. А 1 и А 4 независимо представляют собой C(Rb) или N. В некоторых вариантах осуществления настоящего изобретения А 4 представляет собой N. В некоторых вариантах осуществления настоящего изобретения А 1 представляет собой C(Rb).-9 011439 В некоторых вариантах осуществления настоящего изобретения А 4 представляет собой C(Rb). В некоторых вариантах осуществления настоящего изобретения А 4 представляет собой -СН 2-.Q выбран из группы, включающей связь, (C1-C8)алкилен, (С 2-С 8)гетероалкилен, -С(О)-, -ОС(О)-,-СН 2 СО-, -CH2SO- и -CH2SO2-. В некоторых вариантах осуществления настоящего изобретения Q представляет собой -С(О)-.W и Y каждый независимо представляет собой атом углерода или гетероатом и W и Y, соединенные вместе, образуют 5- или 6-членное циклоалкильное или циклогетероалкильное кольцо, где 5- или 6 членное кольцо является незамещенным или замещено галогеном, NH2, NO2, (С 1-С 20)алкилом, (С 2-С 20) гетероалкилом, гетероарилом, арилом, гетероарил (С 1-С 6)алкилом, гетероарил (С 2-С 6)гетероалкилом,арил(C2-С 6)алкилом и арил(C2-С 6)гетероалкилом.X представляет собой -СН 2-, SO2 или -С(О)-. В некоторых вариантах осуществления настоящего изобретения X представляет собой -С(О)-.R', R" и R'" каждый независимо представляет собой Н, незамещенный (C1-C8) алкил, гетероалкил,незамещенный арил или замещенный арил.R1 представляет собой гетероарил или арил. В некоторых вариантах осуществления настоящего изобретения R1 представляет собой незамещенный или мета- или пара-замещенный фенил, где заместитель представляет собой галоген, циано, (C1-C8) алкил, (C1-C8)гетероалкил, (C1-C8)алкокси или (C1-C8)гетероалкокси. В некоторых вариантах осуществления настоящего изобретения R1 представляет собой В некоторых осуществления настоящего изобретения R1 представляет собой пара-цианофенил.R3 отсутствует или выбран из группы, включающей -Н, -CHR6R7, -S(O)mR5, -S(О)mN(R8)R9, S(O)mN(R8) CH2R6, -N(R8)SO2R5, -N(R8)CH2R10,(C1-C6)алкил, гетероарил (С 2-C6)гетероалкил, арил(C1-C6)алкил и арил(С 2-С 6)гетероалкил. В некоторых вариантах осуществления настоящего изобретения -Q-R4 представляет собойR6 и R7 независимо представляют собой водород, (C1-C8)алкил или (С 2-С 8)гетероалкил.R10 представляет собой арил. В некоторых вариантах осуществления настоящего изобретения соединение соответствует формулеW1 отсутствует или выбран из группы, включающей -O-, -S-, -S(O)-, -S(O)2-, -С(О)-, -СН 2- или NR -. В некоторых вариантах осуществления настоящего изобретения W1 представляет собой -СН 2-.W2 и Y независимо представляют собой -СН 2-, -CHR12-, -СН=, -CR12=, -NH-, -N= или -NR12-. В некоторых вариантах осуществления настоящего изобретения W2 представляет собой -N=, -NHили -N(СН 3)-.W3 отсутствует или представляет собой -O-, -S-, -S(O)-, -S(O)2-, -СН 2-, -CHR13-, -СН=, -CR13=, -NH-,-N= или -NR13-. В некоторых вариантах осуществления настоящего изобретения W1, W3 и Y каждый представляет собой -СН 2-. В некоторых вариантах осуществления настоящего изобретения W1 представляет собой -СН 2-, W2 представляет собой -N=, W3 отсутствует и Y представляет собой -СН= или -CR12-. В некоторых вариантах осуществления настоящего изобретения W2 представляет собой -N(CH3)или -NH-. А 1 и А 4 независимо представляют собой C(Rb) или N.R12 и R13 независимо представляют собой (C1-C20)алкил, (С 2-С 20)гетероалкил, гетероарил, арил, гетероарил (C1-C6)алкил, гетероарил(С 2-С 6)гетероалкил, арил(C1-C6)алкил и арил(С 2-С 6)гетероалкил. В некоторых вариантах осуществления настоящего изобретения R1 представляет собой парацианофенил. В некоторых вариантах осуществления настоящего изобретения соединение соответствует формуле где Ra, Rb, R1, R3, R4, Y, Q, W2, W3 и подстрочный индекс n принимают значения, определенные выше. Легко представить, что соединения согласно настоящему изобретению существуют в виде стереоизомеров. В некоторых вариантах осуществления настоящего изобретения соединение формулы (I) представляет собой рацемическое соединение. В некоторых вариантах осуществления настоящего изобретения соединение формулы (I) включает смесь (S) и (R) энантиомеров. В некоторых вариантах осуществления настоящего изобретения соединение соответствует формуле где Ra, R1, R2, R3, R4, A1, A4, L, Q, X и подстрочный индекс n принимают значения, определенные выше для формулы I. В других вариантах осуществления настоящего изобретения соединение соответствует формуле (Ib) где Ra, R1, R2, R3, R4, A1, A4, L, Q, X и подстрочный индекс n принимают значения, определенные выше для формулы I. В дополнительных вариантах осуществления настоящее изобретение предоставляет рацемическую смесь соединений Ia и Ib. В некоторых вариантах осуществления настоящего изобретения соединение соответствует формуле где Ra, R2, R3, R4, R11, A1, A4, L, Q, X и подстрочный индекс n принимают значения, определенные выше для формулы II. В других вариантах осуществления настоящего изобретения соединение соответствует формуле где Ra, R2, R3, R4, R11, A1, A4, L, Q, X и подстрочный индекс n принимают значения, определенные выше для формулы II. В дополнительных вариантах осуществления настоящее изобретение предоставляет рацемическую смесь соединений На и IIb. В некоторых вариантах осуществления настоящего изобретения соединение согласно настоящему изобретению представляет собой твердое вещество. Например, в некоторых вариантах осуществления изобретения соединение согласно настоящему изобретению имеет кристаллическую форму. В некоторых вариантах осуществления настоящего изобретения соединение имеет аморфную форму. В некоторых вариантах осуществления изобретения соединение согласно настоящему изобретению в кристаллической форме имеет чистоту по меньшей мере 80%, по меньшей мере 90%, по меньшей мере 92%, по меньшей мере 95%, по меньшей мере 97% или по меньшей мере 98%. Соединения согласно настоящему изобретению могут быть получены различными синтетическими или полусинтетическими способами. На фиг. 1-3 и ниже в примерах представлены различные пути синтеза соединений согласно настоящему изобретению. Синтез подходящих исходных материалов может быть осуществлен в соответствии с методиками, известными или очевидными для специалиста данной области техники, или исходные вещества могут быть коммерчески доступными. Например, такие вещества могут быть получены в соответствии со способами, описанными в заявках на Патент США 2002/0160159 А 1 и 2003/0055054 А 1 и в Международной ПубликацииWO 02/83143, содержание которых введено в данное описание во всех полноте. Например, соединения согласно настоящему изобретению могут быть получены в соответствии с представленной ниже схемой или ее вариантами, очевидными специалисту данной области техники: На представленной выше схеме продукт с может быть получен из исходного вещества а, полученного,например, в соответствии с методикой публикации WO 02/83143 с использованием методик и реагентов, очевидных для специалиста данной области техники. Например, для получения промежуточного продукта b исходное вещество а может подвергаться взаимодействию с активированным производным (-Y-R3). В свою очередь, для получения продукта с промежуточный продукт b может подвергаться взаимодействию с активированным производным (Q-R4). Многочисленные примеры синтезов соединений согласно настоящему изобретению представлены в примерах ниже, включая исходные вещества, реагенты, условия реакций, промежуточные продукты и конечные продукты. Дополнительные способы получения соединений согласно изобретению будут понятны из приведенной выше схемы, вариантов схем, приведенных в примерах ниже, и представленных описаний. Специалисту данной области техники будет понятно, что заместители могут добавляться или изменяться перед, в процессе или после получения гетероциклических каркасов, и специалист сможет подходящим образом подобрать условия (например, температуры, растворители и т.д.).Кроме того, специалисту данной области техники понятно, что для получения некоторых соединений могут быть необходимы защитные группы, и он будет представлять условия, совместимые с выбранной защитной группой. Типичные способы и примеры, описанные в изобретении, являются иллюстративными примерами- 12011439 настоящего изобретения и не должны рассматриваться как ограничивающие его область. В соответствии с другим аспектом, настоящее изобретение предоставляет фармацевтические композиции для модулирования активности хемокиновых рецепторов у людей и животных. Композиции включают в себя соединение согласно настоящему изобретению с фармацевтически приемлемым носителем или разбавителем. Подразумевается, что термин модуляция или модулирование активности хемокиновых рецепторов, когда используется в описании в различных формах, включает антагонизм, агонизм, частичный антагонизм и/или частичный агонизм активности, ассоциированный с конкретным хемокиновым рецептором, предпочтительно CXCR3 рецептором. Подразумевается, что термин композиция, когда используется в данном описании, включает изделие, содержащее перечисленные ингредиенты (и в конкретных количествах, если они указаны), а также любой продукт, который является непосредственно или опосредовано результатом сочетания конкретных ингредиентов в конкретных количествах. Термин фармацевтически приемлемый означает носитель, разбавитель или наполнитель, который должен быть совместимым с другими ингредиентами препарата и не оказывать неблагоприятного воздействия на реципиента. Фармацевтические композиции для введения соединений согласно настоящему изобретению могут удобно быть представлены в лекарственной форме стандартной дозы и могут быть получены любым из способов, хорошо известным в области фармакопеи. Все способы включают стадию объединения активного ингредиента с носителем, который составляет один или несколько вспомогательных ингредиентов. Обычно фармацевтические композиции получают тщательным однородным смешением активного ингредиента с жидким носителем или тонко измельченным твердым носителем или обоими указанными носителями и затем, если это необходимо, формованием изделия в целевой препарат. В фармацевтическую композицию соединение вводится в количестве, достаточном для получения желаемого воздействия на течение или состояние болезни. Фармацевтические композиции, содержащие активный ингредиент, могут быть представлены в форме, подходящей для перорального применения, например в виде таблеток, лепешек, пастилок, водных или масляных суспензий, диспергируемых порошков или гранул, эмульсий, твердых или мягких капсул, сиропов или эликсиров. Композиции, предназначенные для перорального применения, могут быть получены в соответствии с любым способом, известным в области производства фармацевтических композиций, и такие композиции могут содержать одно или несколько вспомогательных добавок, выбранных из группы, включающей, подслащивающие средства, вкусовые агенты, красители и консерванты, для обеспечения фармацевтически изысканных и приятных на вкус препаратов. Таблетки содержат активный ингредиент в смеси с нетоксичными фармацевтически приемлемыми наполнителями, которые подходят для производства таблеток. Такими наполнителями могут быть, например, инертные разбавители, такие как карбонат кальция, карбонат натрия, лактоза, фосфат кальция или фосфат натрия, гранулирующие добавки и дезинтегранты, например, кукурузный крахмал или альгиновая кислота; связующие добавки, например крахмал, желатин или акасия, и добавки, повышающие скольжение, например, стеарат магния, стеариновая кислота или тальк. Таблетки могут не иметь покрытия или могут покрываться оболочкой известными методами для замедления дезинтеграции и абсорбции в желудочно-кишечном тракте и, таким образом, обеспечения прологнированного действия в течение более продолжительного периода времени. Например, может применяться материал с более длительным разложением, такой как глицерилмоностеарат или глицерилдистеарат. Они могут также покрываться оболочками способами,описанными в Патентах США 4256108; 4166452 и 4265874, для получения осмотических терапевтических таблеток для контролируемого высвобождения действующего вещества. Препараты для перорального применения также могут быть представлены в виде твердых желатиновых капсул, в которых активный ингредиент смешан с твердым инертным разбавителем, например карбонатом кальция, фосфатом кальция или каолином, или в виде мягких желатиновых капсул, где активный ингредиент смешан с водой или масляной средой, например арахисовым маслом, жидким парафином или оливковым маслом. Водные суспензии содержат активные ингредиенты в смеси с наполнителями, подходящими для производства водных суспензий. Такие наполнители представляют собой суспензаторы, такие как натрийкарбоксиметилцеллюлоза, метилцеллюлоза, гидроксипропилметилцеллюлоза, альгинат натрия, поливинилпирролидон, трагакантова смола и гумарабик; диспергирующие или смачивающие агенты могут представлять собой природный фосфатид, например лецитин, продукты конденсации алкиленоксида с жирными кислотами, например полиоксиэтиленстеарат, продукты конденсации этиленоксида и алифатических спиртов с длинными углеродными цепями, например гептадекаэтиленоксицетанол, продукты конденсации этиленоксида с неполными сложноэфирными производными жирных кислот с гекситом,например, полиоксиэтиленсорбитолмоноолеат, или продукты конденсации этиленоксида с неполными сложными эфирами, полученными из жирных кислот и гекситовых ангидридов, например полиэтиленсорбитанмоноолеат. Водные суспензии могут также содержать один или несколько консервантов, например, этил- или н-пропил-, п-гидроксибензоат, один или несколько красителей, одну или несколько вкусовых добавок и одно или несколько подслащивающих веществ, такие как сахароза или сахарин.- 13011439 Масляные суспензии могут быть получены суспендированием активного ингредиента в растительном масле, например арахисовом масле, оливковом масле, кунжутном масле или кокосовом масле, или в минеральном масле, таком как жидкий парафин. Масляные суспензии могут содержать загуститель, например пчелиный воск, твердый парафин или цетиловый спирт. Подслащивающие вещества, например,упомянутые выше, и вкусовые добавки могут добавляться для получения приятного на вкус препарата для перорального введения. Такие композиции могут быть защищены добавлением антиоксиданта, такого как аскорбиновая кислота. Диспергируемые порошки и гранулы, подходящие для получения водной суспензии при добавлении воды, включают в себя активный ингредиент в смеси с дисперсантом или смачивающим агентом,суспензатором и одним или несколькими консервантами. Подходящие примеры дисперсантов или смачивающих агентов и суспензаторов упомянуты выше. Могут также присутствовать дополнительные наполнители, например, подслащивающие вещества, вкусовые добавки и красители. Фармацевтические композиции согласно настоящему изобретению также могут быть представлены в форме эмульсий типа масло-в-воде. Масляная фаза может представлять собой растительное масло,например оливковое масло или арахисовое масло, или минеральное масло, например, жидкий парафин,или их смеси. Подходящие эмульгаторы могут представлять собой природные смолы, например гуммиарабик или трагакант, природные фосфатиды, например соевые бобы, лецитин, или сложные эфиры или неполные эфирные производные жирных кислот и гекситовых ангидридов, например сорбитанмоноолеат, а также продукты конденсации указанных неполных сложных эфиров с этиленоксидом, например полиоксиэтиленсорбитанмоноолеат. Эмульсии могут также содержать подслащивающие вещества и вкусовые добавки. Сиропы и эликсиры могут быть получены с подслащивающими веществами, например глицерином,пропиленгликолем, сорбитом или сахарозой. Такие препараты могут также содержать эмульгатор, консервант и вкусовые добавки и красители. Фармацевтические композиции могут быть представлены в форме стерильной водной или масляной суспензии для инъекции. Такая суспензия может быть получена в соответствии со способами, широко известными в данной области техники, с использованием подходящих дисперсантов или смачивающих агентов и суспензаторов, которые были перечислены выше. Стерильный препарат для инъекции может также представлять собой раствор или суспензию для инъекции в нетоксичном парентерально приемлемом разбавителе или растворителе, например в виде раствора в 1,3-бутандиоле. Подходящими носителями и растворителями, которые могут применяться, являются вода, раствор Рингера и изотонический раствор хлорида натрия. Кроме того, для этой цели может применяться любое стерильное нелетучее масло в качестве растворителя или суспендирующей среды. Для этих целей могут применяться любое мягкое нелетучее масло, включая синтетические моно- или диглицериды. Кроме того, для получения препаратов для введения в виде инъекций нашли применения жирные кислоты, такие как олеиновая кислота. Соединения согласно настоящему изобретению могут также применяться в форме свечей для ректального введения лекарственного средства. Такие композиции могут быть получены смешением лекарственного средства с подходящим, не вызывающим раздражения наполнителем, который является твердым при комнатных температурах, но жидким при температуре прямой кишки и, следовательно, будет плавиться в прямой кишке с высвобождением лекарственного средства. Такие вещества включают, но без ограничения, масло какао и полиэтиленгликоли. Для местного применения применяются кремы, мази, гели, растворы или суспензии и т.п., содержащие соединения согласно настоящему изобретению. Термин местное применение, когда используется в настоящем описании, означает применение промывных растворов и полосканий для ротовой полости. Фармацевтическая композиция и способ согласно настоящему изобретению могут дополнительно включать другие терапевтически эффективные соединения, которые указаны в описании и которые обычно применяются для лечения или профилактики перечисленных выше патологических состояний. В соответствии с другим аспектом, настоящее изобретение предоставляет способы лечения CXCR3 опосредуемых состояний или заболеваний путем введения субъекту, страдающему таким заболеванием или состоянием, терапевтически эффективного количества соединения или композиции согласно настоящему изобретению. Термин субъект, упомянутый в описании, включает животных, таких как млекопитающие, включая, но без ограничения, приматов (например, человека), крупный рогатый скот, овец, коз, лошадей, свиней, собак, кошек, кроликов, крыс, мышей и т.п. Фраза CXCR3-опосредуемое состояние или заболевание и производные от нее фразы и термины относятся к состоянию, характеризующемуся необычной, например меньшей или большей по сравнению с нормальной, активностью CXCR3. Необычная активность CXCR3 может возникать в результате экспрессии CXCR3 в клетках, которые обычно не экспрессируют CXCR3, повышенной экспрессии CXCR3(приводящей, например, к воспалительным и иммунорегуляторным расстройствам и заболеваниям), или пониженной CXCR3 экспрессии (приводящей, например, к некоторым видам рака и ангиогенным расстройствам и расстройствам, связанным с развитием кровеносных сосудов). Необычная функциональная- 14011439 активность CXCR3 может возникать в результате экспрессии CXCR3 в клетках, которые обычно не экспрессируют CXCR3, повышенной CXCR3 экспрессии (приводящей, например, к воспалительным и иммунорегуляторным расстройствам и заболеваниям) или сниженной CXCR3 экспрессии. Необычная функциональная активность CXCR3 может также возникать как результат секреции хемокина клетками,которые обычно не секретируют СХС хемокин, повышенной экспрессии хемокина (приводящей, например, к воспалительным и иммунорегуляторным расстройствам и заболеваниям) или пониженной экспрессии хемокина. CXCR3-опосредуемое состояние или заболевание может полностью или частично проводиться необычной функциональной активностью CXCR3. Однако CXCR3-опосредуемое состояние или заболевание представляет собой заболевание, в котором модуляция CXCR3 приводит к некоторому влиянию на рассматриваемое состояние или заболевание (например, антагонист CXCR3 приводит к некоторому улучшению здоровья, по меньшей мере, у некоторых пациентов). Термин терапевтически эффективное количество означает количество рассматриваемого соединения, вызывающее биологический или медицинский ответ ткани, системы, животного или человека,которого добивался исследователь, ветеринар, доктор или другой врач или который является достаточным для предотвращения развития или облегчения до некоторой степени одного или нескольких симптомов заболевания, подлежащего лечению. Заболевания или состояния, связанные с воспалением, инфекцией или раком, могут лечиться соединениями и композициями согласно настоящему изобретению. В одной группе вариантов осуществления изобретения заболевания или состояния, включая хронические заболевания, людей или других видов могут лечиться с помощью ингибиторов функции CXCR3. Эти заболевания или состояния включают (1) воспалительные или аллергические заболевания, такие как системные анафилактический или гипертензивный ответы, аллергические реакции на лекарственные средства, аллергические реакции на укусы насекомых и пищевые аллергии; воспалительные заболевания кишечника, такие как болезнь Крона, язвенный колит, илеит и энтерит; вагинит; псориаз и воспалительные дерматозы, такие как дерматит, экзема,атопический дерматит, аллергический контактный дерматит, крапивница; васкулит; спордилоартропатия; склеродерма; астма и респираторные аллергические заболевания, такие как аллергический ринит,аллергические заболевания легких и т.п., (2) аутоиммунные заболевания, такие как артрит (ревматоидный и псориатический), рассеянный склероз, системная красная волчанка, сахарный диабет I типа, гломеруллонефрит и т.п., (3) отторжение трансплантата (включая отторжение аллотрансплантата и реакцию трансплантат против хозяина) и состояния, связанные с ним, (4) другие заболевания, при которых нежелательные воспалительные ответы необходимо ингибировать, например, атеросклероз, миозит, нейродегенеративные заболевания (например, болезнь Альцгеймера), энцефалит, менингит, гепатит, нефрит,сепсис, саркоидоз, конъюнктивит, отит, хроническая обструктивная болезнь легких, синусит и синдром Бехчета. В другой группе вариантов осуществления настоящего изобретения заболевания или состояния лечатся с помощью агонистов функции CXCR3. Примеры заболеваний, подлежащих лечению агонистами функции CXCR3, включают раковые опухоли, заболевания, в которых ангиогенез или неоваскуляризация играет важную роль (неопластические заболевания, ретинопатия и дегенерация желтого пятна),инфекционные заболевания и иммунодепрессивные заболевания. Предпочтительно, представленные способы относятся к лечению или профилактике заболеваний или состояний, выбранных из нейродегенеративных заболеваний (например, болезни Альцгеймера), рассеянного склероза, системной красной волчанки, ревматоидного артрита, атеросклероза, энцефалита,менингита, гепатита, нефрита, сепсиса, саркоидоза, псориаза, экземы, крапивницы, сахарного диабета I типа, астмы, конъюнктивита, отита, аллергического ринита, хронической обструктивной болезни легких,синусита, дерматита, воспалительного заболевания толстого кишечника, язвенного колита, болезни Крона, синдрома Бехчета, подагры, рака, вирусных инфекций (например, ВИЧ), бактериальных инфекций и состояний, связанных с трансплантацией органов или трансплантацией кожи. Подразумевается, что термин состояния, связанные с трансплантацией органов, включает состояния, связанные с трансплантацией костного мозга и трансплантацией цельных органов (например, почки, печени, легкого, сердца,поджелудочной железы или их сочетаний). Заболевания или состояния, которые могут лечиться с помощью соединений и композиций согласно настоящему изобретению, обычно ассоциированы с (1) воспалительными или аллергические заболеваниями, (2) аутоиммунными заболеваниями, (3) отторжением трансплантата и (4) другими заболеваниями, при которых нежелательные воспалительные ответы должны быть ингибированы, как описано выше. Например, рестеноз после такой операции, как пластическая операция на сосудах, обычно связан с атеросклерозом и может лечиться соединениями и композициями согласно настоящему изобретению. В зависимости от заболевания, подлежащего лечению, и состояния субъекта соединения согласно настоящему изобретению могут вводиться пероральным, парентеральным (например, внутримышечно,интраперитонеально, внутривенно, ICV, интрацистернальным введением или вливанием, подкожной инъекцией или имплантированием), ингаляцией спрея, назальным, вагинальным, ректальным, подъязычным или местным способами введения, и препараты на основе соединений согласно настоящему изобретению могут содержать единственное активное соединение или смесь активных соединений и представлять собой подходящие лекарственные препараты стандартной дозы, содержащие традиционные неток- 15011439 сичные фармацевтически приемлемые носители, адъюванты и разбавители, подходящие для каждого способа введения. При лечении или профилактике состояний, которые требуют модуляции хемокинового рецептора,подходящая дозировка будет обычно находиться в интервале примерно от 0,001 до 100 мг на кг массы тела пациента в день и может вводиться в разовой дозе или в виде нескольких доз. Предпочтительно уровень дозировки будет составлять от примерно 0,01 до примерно 25 мг/кг в день; более предпочтительно, от примерно 0,05 до примерно 10 мг/кг в день. Подходящий уровень дозировки может составлять примерно от 0,01 до 25 мг/кг в день, примерно от 0,05 до 10 мг/кг в день или примерно от 0,1 до 5 мг/кг в день. В пределах этого интервала дозировка может составлять от 0,005 до 0,05, от 0,05 до 0,5 или от 0,5 до 5,0 мг/кг в день. Для перорального введения композиции предпочтительно предоставляют в форме таблеток, содержащих от 1,0 до 1000 мг активного ингредиента, предпочтительно 1,0, 5,0, 10,0, 15,0, 20,0,25,0, 50,0, 75,0, 100,0, 150,0, 200,0, 250,0, 300,0, 400,0, 500,0, 600,0, 750,0, 800,0, 900,0 и 1000,0 мг активного ингредиента для симптоматического регулирования дозировки, которая вводится пациенту, подлежащему лечению. Соединения могут вводиться в режиме от 1 до 4 раз в день, предпочтительно один или два раза в день. Однако очевидно, что конкретный уровень дозировки и частота введения дозы для любого конкретного пациента может изменяться и будет зависеть от различных факторов, включая активность конкретного применяемого соединения, метаболической стабильности и продолжительности действия данного соединения, возраста, массы тела, общего состояния здоровья, пола, рациона питания, способа и времени введения, скорости выведения из организма, сочетания лекарственных средств, тяжести конкретного состояния и организма-хозяина, подлежащего терапевтическому лечению. Соединения согласно настоящему изобретению могут объединяться с другими соединениями, применимыми для лечения или профилактики воспалительных и иммунных расстройств и заболеваний,включая астму и аллергические заболевания, а также аутоиммунных патологий, таких как ревматоидный артрит и атеросклероз, и патологий, перечисленные выше. Во многих примерах композиции, которые включают соединение согласно настоящему изобретению и альтернативное или второе терапевтическое лекарственное средство, при введении обладают дополнительными или синергическими эффектами. Например, при лечении или профилактике воспаления соединения согласно настоящему изобретению могут применяться в сочетании или в комбинации с противовоспалительным средством или анальгетиком, таким как агонист опиата, ингибитором липоксигеназы, например, ингибитором 5 липоксигеназы, ингибитором циклооксигеназы, например, ингибитором циклооксигеназы-2, ингибитором интерлейкина, например ингибитором интерлейкина-1, антагонистом NMDA, ингибитором оксида азота или ингибитором синтеза оксида азота, нестероидным противовоспалительным средством или цитокин-супрессирующим противовоспалительным средством, например, с таким соединением, как ацетаминофен, аспирин, кодеин, фентанил, ибупрофен, индометацин, кеторолак, морфин, напроксен, фенацетин, пироксикам, стероидный анальгетик, суфентанил, сунлиндак, тенидап и т.п. Аналогично, рассматриваемые соединения могут вводиться с соединением, облегчающим боль; потенцирующим средством,таким как кофеин, Н 2-антагонист, симетикон, гидроксид алюминия или магния; противозастойным средством, таким как фенилэфрин, фенилпропаноламин, псевдофедрин, оксиметазолин, эфинэфрин, нафазолин, ксилометазолин, пропилгекседрин или леводезоксиэфедрин; противокашлевым средством, таким как кодеин, гидрокодон, карамифен, карбетапентан или декстрометорфан; диуретиком; и седативным или неседативным антигистамином. Аналогично, соединения согласно настоящему изобретению могут применяться в сочетании с другими лекарственными средствами, которые применяются для лечения/профилактики/подавления или облегчения заболеваний или состояний, для лечения которых применимы соединения согласно настоящему изобретению. Такие дополнительные лекарственные средства могут вводиться способом и в количестве, обычно используемым при его применении, одновременно или последовательно с соединением согласно настоящему изобретению. Когда соединение согласно настоящему изобретению применяется одновременно с одним или несколькими дополнительными лекарственными средствами, фармацевтическая композиция, включающая в себя помимо соединений согласно настоящему изобретению такие дополнительные лекарственные средства, является предпочтительной. Соответственно, фармацевтические композиции согласно настоящему изобретению включают фармацевтические композиции, которые также содержат один или несколько дополнительных активных ингредиентов помимо соединения согласно настоящему изобретению. Примеры дополнительных активных ингредиентов, которые могут объединяться с соединением согласно настоящему изобретению и вводиться отдельно или в одних и тех же фармацевтических композициях, включают, но без ограничения, (а) антагонисты VLA-4, (b) стероиды, такие как беклометазон, метилпреднизолон, бетаметазон, преднизон,дексаметазон и гидрокортизон; (с) иммунодепрессанты, такие как циклоспорин (циклоспорин А,Sandimmune, Neoral), такролим (FK-506, Prograf), рапамицин (сиролим, Rapamune) и другие иммунодепрессанты, относящиеся к иммунодепрессантам FK-506 типа, и микофенолят, например микофенолят-мофетил (CellCept); (d) антигистамины (антагонисты Н 1-гистамина), такие как бромофенирамин,хлорфенирамин, дексхлорфенирамин, трипролидин, клемастин, дифенилгидрамин, дифенилпиралин,- 16011439 трипеленнамин, гидроксизин, метдилазин, прометазин, тримепразин, азатадин, ципрогептадин, антазолин, фенирамин-пириламин, астемизол, терфенадин, лоратадин, цетиризин, фексофенадин, дескарбэтоксилоратадин и т.п.; (е) нестероидные противоастматические средства, такие как (2-агонист (тербуталин,метапротеренол, фенотерол, изоэтарин, албутерол, битолтерол и пирбутерол), теофиллин, кромолиннатрия, атропин, ипратропиум бромид, антагонисты лейкотриена (зафирлукаст, монтелукаст, пранлукаст,иралукаст, побилукаст, SKB-106203), ингибиторы биосинтеза лейкотриена (зилеутон, BAY-1005); (f) нестероидные противовоспалительные средства (non-steroidal anti-inflammatory agents - NSAID), такие как производные пропионовой кислоты (алминопрофен, беноксапрофен, буклоксиновая кислота, карпрофен, фенбуфен, фенопрофен, флупрофен, флурбипрофен, ибупрофен, индопрофен, кетопрофен, миропрофен, напроксен, оксапрозин, пирпрофен, пранопрофен, супрофен, тиапрофеновая кислота и тиоксапрофен) , производные уксусной кислоты (индометацин, ацеметацин, аклофенак, клиданак, диклофенак, фенклофенак, фенклозиновая кислота, фентиазак, фурофенак, ибуфенак, изоксепак, оспинак, сулиндак, тиопинак, толметин, зидометацин и зомепирак); производные фенаминовой кислоты (флуфенаминовая кислота, меклофенаминовая кислота, мефенаминовая кислота, нифлуминовая кислота и толфенаминовая кислота), производные бифенилкарбоновой кислоты (дифлунизал и флуфенизал), оксикамы (изоксикам, пироксикам, судоксикам и теноксикан), салицилаты (ацетилсалициловая кислота, сульфасалазин) и пиразолоны (апазон, безпиперилон, фепразон, мофебутазон, оксифенбутазон, фенилбутазон); (g) ингибиторы циклооксигеназы-2 (СОХ-2), такие как целекоксиб (Celebrex) и рофекоксиб (Vioxx); ингибиторы фосфодиэстеразы IV типа (PDE-IV); (i) соединения золота, такие как ауранофин и ауротиоглюкоза; (j) ингибиторы фосфодиэстеразы IV типа (PDE-IV); (k) другие антагонисты хемокиновых рецепторов, в частности CCR1, CCR2, CCR3, CCR5, CCR6, CCR8 и CCR10/ (1) средства, снижающие содержание холестерина, такие как ингибиторы HMG-CoA редуктазы (ловастатин, симвастатин и правастатин,флувастатин, аторвастатин и другие статины), секвесторы (холестирамин и колестипол), никотиновая кислота, производные фенофибриновой кислоты (гемфиброзил, клофибрат, фенофибрат и бензафибрат) и пробукол; (m) противодиабетические средства, такие как инсулин, сульфонилмочевины, бигуамиды(метформин), ингибиторы -глюкозидазы (акарбоза) и глитазоны (троглитазоны и пиоглитазон); (n) препараты бэта-интерферона (интерферон (-1, интерферон (-1; (о) этанерцепт (Enbrel), (р) терапевтические антитела, такие как ортоклон (ОКТЗ), даклизумаб (Zenapax), инфликсимаб (Remicade), басиликсимаб (Simulect) и антитела к анти-СD40-лигандам (например, MRP-1); и (q) другие соединения,такие как 5-аминосалициловая кислота и ее пролекарства, гидроксихлороквин, D-пеницилламин, антиметаболиты, такие как азатиопрен и 6-меркаптопурин, и химиотерапевтические средства, цитотоксические по отношению к раковым клеткам. Массовое соотношение соединения согласно настоящему изобретению ко второму активному ингредиенту могут изменяться и будут зависеть от эффективной дозы каждого ингредиента. Обычно будет применяться эффективная доза каждого активного ингредиента. Таким образом, например, когда соединение согласно настоящему изобретению объединяется с NSAID, массовое соотношение соединения согласно настоящему изобретению и NSAID обычно будет находиться в интервале от примерно 1000:1 до примерно 1:1000, предпочтительно от примерно 200:1 до примерно 1:200. Сочетания соединения согласно настоящему изобретению и дополнительных активных ингредиентов обычно будут также находиться в вышеуказанном интервале значений, но в каждом случае должна использоваться эффективная доза каждого активного ингредиента. Иммунодепрессанты, включенные в область настоящего изобретения, дополнительно включают, но без ограничения, лефлуномид, RAD001, ERL080, FTY720, CTLA-4, терапевтические антитела, такие как ортоклон (ОКТЗ), даклизумаб (Zenapax) и басиликсимаб (Simulect), и антитимоцитарные глобулины,такие как тимоглобулины. В особенно предпочтительных вариантах осуществления изобретения представленные способы направлены на лечение или профилактику множественного склероза с использованием соединения согласно настоящему изобретению самого по себе или в сочетании со вторым терапевтическим средством, выбранным из бетасерона, авонекса, азатиопрена (Imurek, Imuran), капоксона, преднизолона и циклофосфамида. При комбинированном применении лечащий врач может назначить сочетание терапевтических средств или введение может быть последовательным. В дополнительных особенно предпочтительных вариантах осуществления изобретения представленные способы относятся к лечению или профилактике ревматоидного артрита, где соединение согласно настоящему изобретению вводится само по себе или в сочетании со вторым терапевтическим средством, выбранным из группы, включающей метотрексат, сульфасалазин, гидроксихлороквин, циклоспорин А,D-пеницилламин, инфликсимаб (Remicade), этанерцепт (Enbrel), ауранофин и ауротиоглюкозу. В еще одних особенно предпочтительных вариантах осуществления изобретения представленные способы относятся к лечению или профилактике состояния органических трансплантатов, где соединение согласно изобретению применяется само по себе или в сочетании со вторым терапевтическим средством, выбранным из группы, включающей циклоспорин A, FK-506, рапамицин, микофенолат, преднизолон, азатиопрен, циклофосфамид и глобулин антилимфоцита.- 17011439 Примеры Реагенты и растворители, используемые в примерах, представленных ниже, могут быть получены из коммерческих источников, таких как Aldrich Chemical Co. (Milwaukee, Wis., USA). 1H ЯМР спектры записаны на спектрометре Bruker 500 MHZ NMR. Пики, имеющие значение, приведены в таблице в следующем порядке: число протонов, множественность (с - синглет; д - дуплет; т - триплет; кв. - квартет; м мультиплет; уш.с - уширенный синглет) и связанная(ые) константа(ы) в Герцах (Гц). Массспектрометрический анализ методом электрораспылительной ионизации (ESI) проводят на электрораспылительном массспектрометре Hewlett-Packard 1100 MSD с использованием НР 1 100 HPLC (ВЭЖХ) для доставки образца. Результаты масс-спектрометрического анализа записывают в виде отношения массы к заряду. Каждое соединение растворяют в метаноле с получением концентрации 0,1 мг/мл и 1 мкл раствора вводят с растворителем доставки в масс-спектрометр, который сканирует от 100 до 1500 Да. Каждое соединение может анализироваться в положительном режиме ESI с использованием смеси ацетонитрил/вода (1:1) с 1% уксусной кислотой в качестве растворителя доставки. Каждое соединение может анализироваться в отрицательном режиме с использованием 2 мМ раствора NH4OAc в смеси ацетонитрил/вода в качестве растворителя доставки. Пример 1. Схема А Соединение 1 синтезируют из А 1 в две стадии, как представлено на схеме А. Соединение А 1 синтезируют в соответствии с методикой, описанной в Международной ПубликацииWO 02/83143 (схема 3 стр. 77), которая полностью введена в описании в виде ссылки.(А 2). К раствору 18,7 г 1-изопропилпиперидина-4-карбальдегида (0,12 моль, 1,00 экв.), растворенному в 300 мл 1,2-дихлорэтана, охлажденному до температуры -45 С, тремя равными порциями с интервалом 5 мин добавляют 37,3 г А 1 (0,12 моль, 1,00 экв.). Смесь перемешивают при -45 С в течение 15 мин, после чего тремя равными порциями с интервалом 5 мин добавляют 39 г триацетоксиборогидрида натрия (0,18 моль, 1,50 экв.). Реакционной смеси дают возможность медленно нагреться от -45 С до комнатной температуры в течение ночи. К смеси добавляют насыщенный водный раствор гидрокарбоната натрия до достижения в водном слое рН=9-10. Отделенный водный слой дважды экстрагируют дихлорметаном. Объединенные органические слои сушат над сульфатом магния, фильтруют и концентрируют в вакууме,получая А 2 в виде твердого стеклообразного вещества, которое используют далее без дополнительной очистки. 1 Н ЯМР (400 МГц; CDCl3; Т=298,1 К)1,01 (д, J=6,6 Гц, 6 Н), 1,11-1,29 (м, 2 Н), 1,23 (д, J=6, 6 Гц,3 Н), 1,11-1,41 (м, 1 Н), 1,46 (т, J=6,9 Гц, 3 Н), 1,72 (дд, J=14,2, 33,2 Гц, 2 Н), 1, 98-2,20 (м, 3 Н), 2,20-2, 37 (м,2 Н), 2,58-2,73 (м, 1 Н), 2,78-2,92 (м, 2 Н), 3,43 (дд, J=7,6, 7,6 Гц, 1 Н), 4,10 (кв.т, J=6,9 Гц, 2 Н), 6,97-7, 08 (м,2 Н), 7,08-7,18 (м, 2 Н), 7,46 (дд, J=8,5, 8,5 Гц, 1 Н), 7,69 (д, J=8,5 Гц, 1 Н), 8,27 (д, J=8,5 Гц, 1 Н) м.д.(0,13 моль, 1,20 экв.) в 120 мл N,N-диметилформамида (ДМФА) и 40 мл дихлорметана при комнатной температуре добавляют 12 мл N-метилморфолина (NMM) (0,11 моль, 1,00 экв.) и 14,9 г 1 гидроксибензотриазола (НОВТ) (0,11 моль, 1,00 экв.). К раствору двумя равными порциями с интервалом 5 мин добавляют гидрохлорид 1-(3-диметиламинопропил)-3-этилкарбодиимида (EDC) (0,22 моль, 2,00 экв.). Реакционную смесь перемешивают при комнатной температуре в течение 16 ч, затем переносят в 800 мл этилацетата и шесть раз последовательно промывают насыщенным водным раствором гидрокарбоната натрия и насыщенным водным раствором соли. Органический экстракт сушат над сульфатом магния, фильтруют и концентрируют в вакууме, получая твердый стеклообразный продукт. Продукт очищают перекристаллизацией из диэтилового эфира, получая 41 г 1 в виде твердого бесцветного вещества. 1 Н ЯМР (400 МГц; d6-ДМСО; Т=393 К):0,92 (д, J=6,6 Гц, 6 Н), 0,95-1,20 (м, 2 Н), 1,35 (т, J=7,0 Гц,3 Н), 1, 32-1,50 (м, 2 Н), 1,50 (д, J=6,6 Гц, 3 Н), 1,58 (д, J=11,0 Гц, 1 Н), 1,85 (дд, J=10,3, 10,3 Гц, 1H), 1,99 Соединение В 1 синтезируют как описано в Международной публикацииWO 02/83143 (схема 9 стр. 91), используя в стадии 3 вместо 4-(2,2,2-трифторэтокси)анилина п-фенэтидин. Соединение 2 синтезируют из В 1 в две стадии, как показано на схеме В.(R)-2-[1-(2-Этансульфонилэтиламино)этил]-3-[4-(2,2,2-трифторэтокси)фенил]-3 Н-пиридо[2,3-dпиримидин-4-он (В 2). Смесь 2,18 г В 1 (5,98 ммоль, 1,00 экв.) и 687 мкл этилвинилсульфона (6,58 ммоль, 1,10 экв.) в 10 мл метанола перемешивают в течение ночи на масляной бане при 50 С, затем концентрируют в вакууме. Продукт В 2 используют в следующей стадии без дополнительной очистки. MC(ESI+) m/z =485,1 [М+Н]+.(R)-N-(2-Этансульфонилэтил)-2-(4-фтор-3-трифторметилфенил)-N-(1-4-оксо-3-[4-(2,2,2-трифторэтокси)фенил]-3,4-дигидропиридо[2,3-d]пиримидин-2-илэтил)ацетамид (2). Хлорангидрид (4-фтор-3-трифторметилфенил)ацетилхлорид получают добавлением нескольких капель ДМФА к охлажденному на ледяной бане раствору 2,06 г (4-фтор-3-трифторметилфенил) уксусной кислоты (9,27 ммоль, 1,70 экв.) и 0,81 мл оксалилхлорида (9,27 ммоль, 1,70 экв.) в 15 мл дихлорметана. После начала выделения газа реакционной смеси дают возможность самопроизвольно нагреться до комнатной температуры и затем ее перемешивают в течение 2 ч до прекращения выделения газа. Полученный раствор по каплям в течение 15 мин добавляют к раствору 2,6 г В 2 (5,37 ммоль, 1,00 экв.) и 2,24 мл триэтиламина (16,1 ммоль, 3,00 экв.) в 20 мл дихлорметана, охлажденному на бане смесью ацетон-сухой лед. Полученную смесь перемешивают при пониженной температуре в течение 20 мин, затем выливают в насыщенный водный раствор гидрокарбоната натрия. Органический слой промывают насыщенным- 19011439 раствором соли, сушат над сульфатом магния, фильтруют и концентрируют в вакууме, получая продукт в виде твердого стеклообразного вещества. Продукт очищают хроматографией на силикагеле (элюирование с градиентом, от 50% этилацетата в гексане до 100% этилацетата). Полученный после хроматографии продукт перекристаллизовывают из метил-трет-бутилового эфира, получая соединение 2 в виде бесцветных кристаллов, т.пл.=157-159 С. 1 Н ЯМР (400 МГц; CDCl3; Т=300 К) (смесь S-цис: S-транс-амидных ротамеров в соотношении примерно 2,3:1)1,20 (т, J=7,6 Гц, 3 Н), 1,46 (т, J=7,2 Гц, 6,5 Н), 1,46 (д, J=7,2 Гц, 6,5 Н), 2,48 (д, J=16 Гц, 1 Н),2,92 (ддд, J=2,8, 7,6, 16,0 Гц, 2 Н), 2,94 (д, J=16 Гц, 1 Н), 3,13 (ддд, J=3,6, 7,2, 14,8 Гц, 4,6 Н), 3,22 (дд, J=6,8,6,8 Гц, 2 Н), 3,50 (ддд, J=5,2, 10,6, 13,3 Гц, 2,3 Н), 3,67 (ддд, J=8,0, 8,0, 13,3 Гц, 1 Н), 3,84 (с, 4,6 Н), 3,90(R)-2-(4-фтор-3-трифаюрметилфенил)-N-(2-метансульфонилэтил)-N-(1-4-оксо-3-[4-(2,2,2-трифторэтокси)фенил]-3,4-дигидропиридо[2,3-d]пиримидин-2-илэтил)ацетамид (3). Соединение 3 синтезируют в две стадии из В 1 в соответствии с описанной выше последовательностью синтеза соединения 2, используя на первой стадии вместо метилвинилсульфона этилвинилсульфон. Соединения 4 и 5 синтезируют из описанного выше В 1 в две стадии и три стадии, соответственно,как показано на схеме D.(R)-2-1-[(Тетрагидротиопиран-4-илметил)амино]этил-3-[4-(2,2,2-трифторэтокси)фенил]-3 Нпиридо[2,3-d]пиримидин-4-он (D1). Описанный выше В 1 (159 мг, 0,44 ммоль), тетрагидротиопиранил-4-карбоксальдегид (70 мкл, 0,53 ммоль) и триацетоксиборогидрид натрия (277 мг, 1,32 ммоль) перемешивают в 1,2-дихлорэтане при комнатной температуре в течение 1 ч. Реакционную смесь разбавляют дихлорметаном, затем промывают насыщенным водным раствором гидрокарбоната натрия и насыщенным раствором соли. Органический слой сушат над безводным сульфатом магния, фильтруют и концентрируют в вакууме, получая 208 мгD1, который используют далее без дополнительной очистки.(R)-2-(4-Фтор-3-трифторметилфенил)-N-(1-4-оксо-3-[4-(2,2,2-трифторэтокси)фенил]-3,4-дигидропиридо[2,3-d]пиримидин-2-илэтил)-N-(тетрагидротиопиран-4-илметил)ацетамид (4). 4-Фтор-3-трифторметилфенилуксусную кислоту (164 мг, 0,74 ммоль) растворяют в дихлорметане и охлаждают до 0 С. Добавляют оксалилхлорид (64 мкл, 0,74 ммоль), спустя 5 мин добавляют N,Nдиметилформамид (5,7 мкл, 0,07 ммоль). Реакционную смесь перемешивают при 0 С в течение 30 мин и при комнатной температуре в течение 1 ч. Полученный раствор медленно добавляют к смеси D1 (208 мг,0,43 ммоль) и триэтиламина (182 мкл, 1,30 ммоль) в дихлорметане при -78 С. Смесь перемешивают в течение 30 мин, затем промывают насыщенным водным раствором гидрокарбоната натрия и насыщенным раствором соли. Органический экстракт сушат над безводным сульфатом магния, фильтруют и концентрируют в вакууме. Полученный продукт очищают хроматографией (2% метанола и 0,1% гидроксида аммония в дихлорметане), получая 265 мг соединения 4.(R)-N-(1,1-Диоксогексагидро-16-тиопиран-4-илметил)-2-(4-фтор-3-трифторметилфенил)-N-(1-4 оксо-3-[4-(2,2,2-трифторэтокси)фенил]-3,4-дигидропиридо[2,3-d]пиримидин-2-илэтил)ацетамид (5). Соединение 4 (265 мг, 0,39 ммоль) растворяют в дихлорметане и охлаждают до 0 С. К полученному раствору добавляют 3-хлороксибензойную кислоту (77%, 174 мг, 0,78 ммоль) и смеси дают возможность нагреться до комнатной температуры в течение ночи. Реакционную смесь промывают дважды 10% водным раствором тиосульфата натрия и один раз насыщенным раствором соли. Затем органический слой сушат над безводным сульфатом магния, фильтруют и концентрируют в вакууме. Продукт очищают хроматографией (3% метанола и 0,1% гидроксида аммония в дихлорметане), получая 136 мг соединения 5. МС (МН+): 688,2. 1 Н ЯМР (500 МГц; CDCl3; Т=298 К):8,30 (т, 1 Н, J=7,9 Гц), 7,86 (м, 1 Н), 7,69-7,35 (м, 4 Н), 7,25 (м,2 Н), 7,13 (м, 3 Н), 5,23-4,93 (дкв., 1 Н, J=7,2, 143 Гц), 4,43 (квинт., 2 Н, J=8,0 Гц), 4,22-3,58 (м, 2 Н), 3,26 (м,1 Н), 3, 06-2, 97 (м, 3 Н), 1,55 (д, 2 Н, J=6,7 Гц), 1,43 (дт, 3 Н, J=7,6, 2,0 Гц), 1,32 (т, 3 Н, J=7,5 Гц) м.д. Пример 6. Схема Е(R)-N-(2-Бензолсульфонилэтил)-N-1-[3-(4-этоксифенил)-4-оксо-3,4-дигидропиридо[2,3-d]пиримидин-2-ил]этил-2-(4-фтор-3-трифторметилфенил)ацетамид (6). Соединение 6 синтезируют в две стадии из соединения Е 1 в соответствии с последовательностью синтеза соединения 2, но используя на стадии 1 вместо фенилвинилсульфона этилвинилсульфон. 1 Н ЯМР: смесь цис/транс-амидных ротамеров в соотношении 2,0:1 (400 МГц, CDCl3; Т=25 С) основной 8,98 (дд, J=4,63, 1,94 Гц, 1 Н), 8,61 (д, J=7,97, 1,80 Гц, 1 Н), 7,98 (д, J=7,43 Гц, 1 Н), 7,01-7,75 (м,12 Н), 5,14 (кв., J=7,20 Гц, 1 Н), 4,07 (м, 2 Н), 2,86-3,90 (м, 6 Н), 1,45 (нечеткий т, J=6,93 Гц, 3 Н), 1,34 (д,J=7,23 Гц, 3 Н) и дополн. 9,09 (дд, J=4,3, 1,84 Гц, 1 Н), 8,64 (дд, J=7,88, 1,87 Гц, 1 Н), 1,54 (д, J=6,73 Гц, 3 Н),1,44 (нечеткий т, J=6,74 Гц, 3 Н). МС (ESI +) 683 [М+Н]+. Соединения 7 и 8 синтезируют в две и три стадии, соответственно, как описано ранее для Е 1.(R)-3-(4-Этоксифенил)-2-1-[(тетрагидротиопиран-4-илметил)амино]этил-3 Н-пиридо[2,3-d]пиримидин-4-он (F1). К смеси Е 1 (531 мг, 1,71 ммоль) и тетрагидротиопиран-4-карбальдегида (234 мг, 1,79 ммоль) в 9 мл 1,2-дихлорэтана добавляют 1,09 г (5,13 ммоль) триацетоксиборогидрида натрия при комнатной температуре. Полученную смесь перемешивают при комнатной температуре в течение 3 ч. После этого добавляют 25 мл насыщенного водного раствора гидрокарбоната натрия и полученную смесь экстрагируют дихлорметаном (30 мл х 3). Объединенные экстракты промывают водой и насыщенным раствором соли,сушат над безводным сульфатом натрия и концентрируют в вакууме. Остаток сушат в вакууме, получая 550 мг продукта F1, который используют в следующей стадии без дополнительной очистки. МС (ESI+) 425 [М+Н]+.(R)-N-1-[3-(4-Этоксифенил)-4-оксо-3,4-дигидропиридо[2,3-d]пиримидин-2-ил]этил)-2-(4-фтор-3 трифторметилфенил)-N-(тетрагидротиопиран-4-илметил)ацетамид (7). Смесь F1 (550 мг, 1,29 ммоль), 4-фтор-3-трифторметилфенилуксусной кислоты (374 мг, 1,68 ммоль), EDC (371 мг, 1,93 ммоль), НОВТ (198 мг, 1,29 ммоль) и 300 мкл (2,58 ммоль) NMM в 13 мл дихлорметана перемешивают при комнатной температуре в течение 3 дней. После этого смесь концентрируют и добавляют 15 мл воды. Полученную смесь экстрагируют этилацетатом (30 мл 3). Объединенные экстракты промывают водой и насыщенным раствором соли, сушат над безводным сульфатом натрия и концентрируют. Полученный остаток очищают колоночной хроматографией (силикагель, элюирование: смесь этилацетат/гексан, последовательно 50%, 80%), получая 531 мг продукта 7. 1 Н ЯМР для смеси цис/транс-амидных ротамеров в соотношении примерно 2:1 (400 МГц, CDCl3; Т=25 С) основной 8,96 (дд, J=4,58, 2,01 Гц, 1 Н), 8,56 (дд, J=7,87, 2,02 Гц, 1 Н), 7,0-7,55 (м, 8 Н), 5,16 (кв,J=7,21 Гц, 1 Н), 4,05 (кв., J=6,92 Гц, 2 Н), 1,44 (нечеткий д, J=7,19 Гц, 3 Н), 1,43 (нечеткий т, J=6,96 Гц, 3 Н) и дополн. 9,06 (дд, J=4,56, 1,96 Гц, 1 Н), 8,63 (дд, J=7,90, 1,96 Гц, 1 Н), 5,02 (кв, J=6,81 Гц, 1 Н), 1,58 (д,J=6,74 Гц, 3 Н). МС (ESI +) 629 [М+Н]+.(R)-N-(1,1-Диоксогексагидро-16-тиопиран-4-илметил)-N-1-[3-(4-этоксифенил)-4-оксо-3,4-дигидропиридо[2,3-d]пиримидин-2-ил]этил-2-(4-фтор-3-трифторметилфенил)ацетамид (8). К раствору соединения 7 (531 мг, 0,845 ммоль) в 8 мл дихлорметана медленно добавляют 379 мг(1,69 ммоль) 3-хлороксибензойной кислоты (77%). Полученную смесь перемешивают при комнатной температуре в течение 3 ч. После этого добавляют 30 мл насыщенного водного раствора тиосульфата натрия и полученную смесь экстрагируют дихлорметаном (40 мл 3). Объединенные экстракты промывают насыщенным водным раствором тиосульфата натрия и водой, сушат над безводным сульфатом натрия и концентрируют в вакууме. Продукт очищают колоночной хроматографией (элюирование: этилацетат/гексан, последовательно 50 и 100%), получая 484 мг соединения 8. 1 Н ЯМР: смесь цис/транс-амидных ротамеров в соотношении примерно 4:1 (400 МГц, CDCl3; Т=25 С) основной 3,60 (д, J=3,60 Гц, 1 Н), 8,85 (д, J=6,64 Гц, 1 Н), 7,04-7,68 (м, 8 Н), 5,13 (кв, J=7,29 Гц, 1 Н),4,10 (кв, J=6,97 Гц, 2 Н), 3,90 (ABd, JAB=16,3 Гц, 1 Н), 3,59-3,75 (м, 3 Н), 2,85-3,17 (м, 4 Н), 1,80-2,35 (м, 5 Н),1,46 (J=6,95 Гц, 3 Н), 1,43 (д, J=7,43 Гц, 3 Н) и дополн. 9,10 (м, 1 Н), 8,78 (м, 1 Н), 1,59 (д, J=6,74 Гц, 3 Н). МС(R)-N-(2-Этансульфонилэтил)-N-1-[3-(4-этоксифенил)-4-оксо-3,4-дигидропиридо[2,3-]пиримидин 2-ил]этил-2-(4-фтор-3-трифторметилфенил)ацетамид (9). Соединение 9 синтезируют в две стадии из Е 1 в соответствии с последовательностью синтеза соединения 2. 1 Н ЯМР соединения 9 (смесь цис/транс-амидных ротамеров в соотношении примерно 1,2:1) (400 МГц, CDCl3; Т=25 С) основной 8,94 (дд, J=4,60, 2,00 Гц, 1 Н), 8,58 (дд, J=7,87, 1,99 Гц, 1 Н), 7,02-7,55 (м,8 Н), 5,21 (м, 1 Н), 3,82-4,19 (м, 5 Н), 3,48 (м, 1 Н), 3,09-3,21 (м, 2 Н), 2,88-2,93 (м, 2 Н), 1,41-1,47 (м,9 Н) и дополн. 9,05 (дд, J=4,60, 2,02 Гц, 1 Н), 8,63 (дд, J=7,89, 1,98 Гц, 1 Н), 5,05 (м, 1 Н), 3,66 (м, 1 Н), 2,43 (ABd,JAB=16,3 Гц, 1 Н), 1,58 (д, J=6,89 Гц, 3 Н), 1,22 (т, J=7,45 Гц, 3 Н). МС (ESI+) 635 [М+Н]+. Пример 10. Схема Н Соединение примера 10 синтезируют в две стадии из описанного ранее соединения А 1.(R)-2-[1-(2-Этансульфонилэтиламино)этил]-3-(4-этоксифенил)-3 Н-хиназолин-4-он (H1). Смесь 0,2 г соединения А 1 (0,647 ммоль, 1,00 экв.) и этилвинилсульфона (0,809 ммоль, 1,25 экв.) в 5 мл метанола перемешивают при 70 С в течение 5 ч, затем раствор концентрируют в вакууме. Продукт используют в следующей стадии без дополнительной очистки. МС (ESI+) 430,1 (МН+).EDC (18 4 мг, 0,96 ммоль) добавляют к смеси соединения H1 (137 мг, 0,32 ммоль), 4-фтор-3 трифторметилфенилуксусной кислоты (108 мг, 0,49 ммоль), HOBt (22 мг, 0,16 ммоль) и NMM (97 мг,0,96 ммоль) в 5 мл ДМФА. Смесь перемешивают при комнатной температуре в течение ночи и затем разбавляют этилацетатом. Раствор промывают насыщенным водным раствором гидрокарбоната натрия,водой и насыщенным раствором соли, сушат над безводным сульфатом натрия и концентрируют. Остаток очищают хроматографией, получая продукт в виде твердого белого вещества (160 мг). 1 Н ЯМР (CDCl3) (смесь цис-транс-амидных ротамеров в соотношении примерно 2,57:1) 1,31 (т,2,16 Н, J=7,5 Гц), 1,46 (м, 4,68 Н), 1,56 (д, 2,16 Н, J=7 Гц), 2,5 (д, 0,72 Н, J=16,5 Гц), 2,98 (м, 3 Н), 3,28 (м,1 Н), 3,55 (м, 0,28 Н), 3,72 (м, 1 Н), 3,82 (м, 0,56 Н), 3,95 (м, 0,72 Н), 4,11 (м, 2,72 Н), 5,00 (кв., 0,72 Н, J=7 Гц),5,28 (кв., 0,28 Н, J=7 Гц), 7,05 (м, 0,72 Н), 7,16 (м, 5,28 Н), 7,29 (м, 1 Н), 7,49-7,61 (м, 2 Н), 7,80 (м, 0,28 Н),7,86 (м, 0,72 Н), 8,31 (д, 1 Н, J=7,5 Гц), MC (ESI +) 634,1 (МН+). Пример 11. Схема I Соединение II синтезируют в соответствии с методикой, описанной в Международной публикацииWO 02/83143 (схема 3, стр. 77) с тем отличием, что 4-(2,2,2-трифторэтокси)анилин используют вместо п-фенэтидина в стадии b. Соединение 11 синтезируют из описанного выше I1 в две стадии, как показано на схеме I.- 23011439 Описанный выше I1 (158 мг, 0,43 ммоль) и этилвинилсульфон (68 мкл, 0,65 ммоль) растворяют в метаноле и перемешивают при 60 С в течение 3 ч. Метанол удаляют в вакууме, получая 210 мг соединения 12, которое используют в следующей стадии синтеза без дополнительной очистки. МС (ESI+): m/z=484,l [М+Н]+.(R)-N-(2-Этансульфонилэтил)-2-(4-фтор-3-трифторметилфенил)-N-(1-4-оксо-3-[4-(2,2,2-трифторэтокси)фенил]-3,4-дигидрохиназолин-2-илэтил)ацетамид (11). 4-Фтор-3-трифторметилфенилуксусную кислоту (164 мг, 0,74 ммоль) растворяют в дихлорметане и охлаждают до 0 С. К полученной смеси добавляют оксалилхлорид (64 мкл, 0,74 ммоль), спустя 5 мин добавляют N,N-диметилформамид (5,7 мкл, 0,07 ммоль). Реакционную смесь перемешивают при 0 С в течение 30 мин и при комнатной температуре в течение 1 ч. Полученный раствор медленно добавляют к смеси соединения 12 (210 мг, 0,43 ммоль) и триэтиламина (182 мкл, 1,30 ммоль) в дихлорметане при -78 С. Полученную смесь перемешивают в течение 30 мин, затем промывают насыщенным водным раствором гидрокарбоната натрия и насыщенным раствором соли. Смесь сушат над сульфатом магния, фильтруют и концентрируют в вакууме. Остаток очищают ВЭЖХ с обращенной фазой, получая 256 мг 11 в виде твердого бесцветного вещества. МС (ESI+): m/z=715,2 [М+Н]+. 1 Н ЯМР (500 МГц; CDCl3; Т=298 К):9,10-8,95 (м, 1 Н), 8,66-8,58 (м, 1 Н), 7,78-7,57 (м, 1 Н), 7, 47-7,36 (м, 3 Н), 7,24 (м, 1 Н), 7,13 (м, 3 Н), 5,03 (кв, 1 Н, J=7,4 Гц), 4,43 (квинт., 2 Н, J=8,0 Гц), 3,77 (с, 1 Н), 3,71 Промежуточный продукт J3 синтезируют из коммерчески доступного J1 в две стадии, как показано на схеме J. 2,5-(Диметил-2 Н-пиразол-3-ил)метанол (J2). Раствор алюмогидрида лития (1,0 М в тетрагидрофуране, 1,7 8 мл) в течение 5 мин при комнатной температуре добавляют к раствору коммерчески доступного этил-1,3-диметил-1 Н-пиразол-5-карбоксилата (J1, 300 мг, 1,78 ммоль) в тетрагидрофуране. Реакционную смесь перемешивают в течение ночи. После этого добавляют воду (67 мкл), 15% водный раствор гидроксида натрия (202 мкл) и снова воду (67 мкл). Полученный твердый продукт отфильтровывают и сушат в вакууме, затем очищают хроматографией (элюирование с градиентом: метанола в дихлорметана - от 3 до 7%), получая 175 мг J2 в виде твердого бесцветного вещества. 5-Бромметил-1,3-диметил-1 Н-пиразол (J3). Бром (229 мг, 1,4 4 ммоль) в дихлорметане добавляют к раствору трифенилфосфина (376 мг, 1,44 ммоль) в дихлорметане, охлажденному на бане с ледяной водой. Реакционную смесь перемешивают в течение 10 мин, затем одной порцией добавляют соединение J2 (170 мг, 1,34 ммоль), после чего раствору дают возможность нагреться до комнатной температуры в течение 2 ч. Реакционную смесь гасят водой,водный слой экстрагируют дихлорметаном (2x25 мл). Объединенный органический слой промывают- 24011439 последовательно 10% водным раствором тиосульфата натрия и насыщенным раствором соли. Органический слой сушат над сульфатом магния, фильтруют и концентрируют в вакууме. Остаток очищают хроматографией (элюирование: 1:1 гексан:этилацетат), получая 195 мг J3 в виде твердого бесцветного вещества. 1 Н ЯМР (400 МГц; CDCl3; Т=298 К):6,07 м.д. (с, 1 Н), 4,44 (с, 2 Н), 3,83 (с, 3 Н), 2,23 (с, 3 Н) м.д. Схема K Соединение 12 синтезируют из соединения Е 1 в две стадии, как показано на схеме К. СоединенияE1, J3 и карбонат калия подвергают взаимодействию в диметилформамиде, продукт очищают с получением соединения К 5. К 5 подвергают взаимодействию с 4-фтор-3-трифторметилфенилуксусной кислотой и оксалилхлоридом в триэтиламине, дихлорметане и ДМФА. Соединение 14 фильтруют и концентрируют. Схема L Соединение 13 синтезируют из описанного ранее соединения Е 1 в четыре стадии, как показано на схеме L. Е 1 подвергают взаимодействию с метилвинилкетоном и полученный продукт выделяют с получением соединения L1. L1 подвергают реакции с 4-фтор-3-трифторметилфенилуксусной кислотой и полученный продукт выделяют с получением соединения L2. L2 подвергают взаимодействию с циклобутиламином и триацетоксиборогидридом натрия и полученный продукт выделяют с получением соединения Соединение 14 синтезируют из описанного выше соединения Е 1, как показано на схеме М. Соединение Е 1 подвергают взаимодействию с 1-изопропил-4-пиперидоном и триацетоксиборогидридом на- 25011439 трия, после чего выделяют полученный продукт M1. M1 подвергают взаимодействию с 4-фтор-3 трифторметилфенилуксусной кислотой и оксалилхлоридом, выделяя в качестве продукта соединение 14. Пример 15. Схема N(с) 1) 4-йоданилин, CH2Cl2, от -10 до 15 С, 12 ч; (d) NMM, IBCF, CH2Cl2, -25 С, 12 ч (общий выход 13%). Соединение 15 синтезируют из коммерчески доступных веществ, как показано на схеме N.(температура реакционной смеси). К смеси последовательно добавляют N-метилморфолин (NMM) (13,8 мл, 125 ммоль) и изобутилхлорформиат (IBCF) (13,5 мл, 104 ммоль) с такой скоростью, чтобы температура реакционной смеси оставалась ниже -25 С. Спустя 1,5 ч смесь переносят с помощью канюли в трехгорлую колбу объемом 250 мл, снабженную термометром и содержащую сухую 2-аминоникотиновую кислоту (7,28 г, 52,7 ммоль). После завершения добавления (примерно 10 мин) внутреннюю температуру смеси доводят до -10 С. Реакционной смеси дают возможность нагреться в течение 17 ч при энергичном перемешивании до температуры 15 С. Смесь охлаждают до 0 С и промывают охлажденной льдом INHCl (2x100 мл), насыщенным раствором соли (100 мл) и сушат над Na2SO4. Полученный раствор 2 загружают в трехгорлую колбу объемом 250 мл, охлаждают до -25 С и обрабатывают твердым 4 йоданилином (11,61 г, 53 ммоль). Полученной темной смеси дают возможность нагреться до 15 С и перемешивают смесь в течение 12 ч. Раствор промывают IN HCl (2100 мл), насыщенным раствором NaHCO3 (2100 мл), насыщенным раствором соли (100 мл) и сушат над Na2SO4. Смесь охлаждают до -25 С и обрабатывают NMM (6,8 мл, 61,8 ммоль), затем IBCF (6,7 мл, 56,1), поддерживая температуру реакционной смеси ниже -25 С. После перемешивания в течение 12 ч реакционную смесь промывают IN HCl(2x100 мл), насыщенным раствором NaHCO3 (2100 мл), насыщенным раствором соли (100 мл), сушат над Na2SO4 и концентрируют. Очистка остатка колоночной хроматографией (колонка 50x400 мм, силикагель; элюирование с градиентом: ацетон/CH2Cl2 от 5 до 25%) приводит к получению соединения 15 (3,41 г, 13%; чистота 96% AUC). Rf = 0,37 (15% ацетон/CH2Cl2). Пример 16. Схема О(R)-трет-Бутил-1-(3-(4-цианофенил)-4-оксо-3,4-дигидропиридо[2,3-d]пиримидин-2-ил)этилкарбамат (16). Соединение 15 (2,02 г, 4,10 ммоль), полученное в соответствии с методикой, приведенной выше,объединяют с (Ph3P)4Pd (439 мг, 0,38 ммоль), Cul (157 мг, 0,82 ммоль) и NaCN (406 мг, 8,29 ммоль) в колбе грушевидной формы объемом 25 мл, снабженной обратным холодильником. Смесь сушат в высоком вакууме в атмосфере сухого N2 три раза. После этого добавляют ацетонитрил (6 мл), полученную суспензию нагревают до 70 С и выдерживают при указанной температуре в течение 30 мин, после чего анализ ТСХ и ВЭЖХ показывает почти полный расход соединения 15. Смесь разбавляют EtOAc (100 мл) и фильтруют через рыхлый слой целита. Фильтрат промывают насыщенным водным раствором NaHCO3(100 мл). Водный слой экстрагируют дополнительным количеством EtOAc (250 мл). Объединенные экстракты сушат над Na2SO4 и концентрируют. Очистка остатка колоночной хроматографией (колонка 50400 мм; силикагель; элюирование с градиентом: от 80% EtOAc/гексаны до 100% EtOAc) приводит к получению соединения 16 (1,42 г, 89%; чистота 96% AUC; 92% (хиральная ВЭЖХ. Rf = 0,35 (80%(R)-4-(2-(1-аминоэтил)-4-оксопиридо[2,3-d]пиримидин-3(4H)-ил)бенэонитрил (P1). Соединение 16 (1,05 г, 2,68 ммоль), полученное как описано выше, растворяют в CH2Cl2 (40 мл) и обрабатывают ТФУК (40 мл). Полученную смесь перемешивают в течение 1,5 ч затем концентрируют в вакууме. Концентрат снова растворяют в CH2Cl2 (100 мл) и промывают насыщенным NaHCO3 (100 мл). Водные слои экстрагируют дополнительными порциями CH2Cl2 (350 мл). Объединенные экстракты сушат над Na2SO4, концентрируют и сушат в высоком вакууме в течение 17 ч, получая продукт Р 1 (767 мг, 98%), который используют далее без дополнительной очистки.(R)-N-(1-(3-(4-цианофенил)-4-оксо-3,4-дигидропиридо[2,3-d]пиримидин-2-ил)этил)-N-(2-(этилсульфонил)этил)-2-(4-фтор-3-(трифторметил)фенил)ацетамид (17). Сырой Р 1 (567 мг, 1,95 ммоль) и этилвинилсульфон (0,26 мл, 2,49 ммоль) объединяют в безводном МеОН (6,5 мл). Смесь нагревают 50 С (внутренняя температура) при перемешивании и выдерживают при этой температуре в течение 17 ч, после чего ЖХ-МС анализ реакционной смеси показывает полное расходование исходного вещества. Реакционную смесь распределяют между EtOAc (100 мл) и водой (50 мл). Этилацетатный слой промывают водой (250 мл). Объединенные промывные слои экстрагируют EtOAc (250 мл). Объединенные экстракты сушат над Na2SO4 и концентрируют. Остаток объединяют с 4-фтор-3-трифторметилфенилуксусной кислотой(680 мг, 2,93 ммоль), EDCI (2,98 ммоль) и HOBt (376 мг, 2,78 ммоль) в ДМФА (5 мл). Полученную смесь обрабатывают основанием Ханига (Hunig's base) (1,35 мл, 7,75 ммоль) и перемешивают при комнатной температуре в течение 17 ч. Реакционную смесь разбавляют EtOAc (200 мл) и промывают 1N HCl (2100 мл). Объединенные промывные воды экстрагируют EtOAc (2100 мл). Объединенные экстракты промывают насыщенным NaHCO3 (200 мл), водой (3100 мл) и насыщенным раствором соли (100 мл), затем сушат над Na2SO4 и концентрируют. Остаток очищают хроматографией (силикагель; элюирование с градиентом: ТГФ/CH2Cl2 от 10 до 20%, затем до 100% ТГФ), получая 17 (780 мг, 1,27 ммоль, 65%) в виде твердого аморфного вещества белого цвета (чистота 97% AUC при 254 нМ). Rf 0,2 (15% ТГФ/CH2Cl2).DSC показывает единственный эндотермический переход при 179 С. МС (ESI +) 616,6 (МН+). Соединение 18 синтезируют из коммерчески доступных исходных веществ в четыре стадии, как показано на схеме Q. Бензиловый эфир (R)-2-трет-бутоксикарбониламино-1-[3-(4-этоксифенил)-4-оксо-3,4-дигидрохиназолин-2-ил]этилкарбаминовой кислоты (Q1). К раствору антраниловой кислоты (1,86 г, 13,59 ммоль) и R-2-бензилоксикарбониламино-3-третбутоксикарбониламинопропионовой кислоты (4,6 г, 13,59 ммоль) в 25 мл безводного пиридина добавляют трифенилфосфит (8,43 г, 27,18 ммоль) при комнатной температуре. Полученный раствор перемешивают при 60 С в течение 15 ч. С помощью шприца добавляют п-фенетидин (1,86 г, 13,59 ммоль). Реакционную смесь перемешивают в течение дополнительных 3 ч при 60 С, охлаждают до комнатной температуры и упаривают в вакууме для удаления большей части пиридина. Остаток в виде раствора в простом эфире промывают последовательно 1N HCl, 1N NaOH, водой и насыщенным раствором соли. Органический слой сушат над безводным сульфатом натрия и упаривают в вакууме до сухого остатка коричневого цвета, который очищают хроматографией, получая соединение Q1 в виде твердого белого вещества (4,48 г). MC (ESI+) 559,2 (МН+). Трет-бутиловый эфир(R)-2-амино-2-[3-(4-этоксифенил)-4-оксо-3,4-дигидрохиназолин-2 ил]этилкарбаминовой кислоты (Q2). К раствору Q1 (1 г, 1,79 ммоль) и циклогекса-1,3-диена (2,87 г, 35,84 ммоль) в 30 мл этанола добавляют 10% палладий на активированном угле (0,19 г, 0,18 ммоль). Полученный раствор кипятят с обратным холодильником в течение 3 ч. Смесь фильтруют и упаривают в вакууме, получая продукт в виде твердого белого вещества (0,53 г). 1 Н ЯМР (CDCl3) 1,35 (с, 9 Н), 1,48 (т, 3 Н, J=7 Гц), 3,42 (м, 2 Н), 3,80 (с, 1 Н), 4,11 (кв, 2 Н, J=7 Гц), 5,09(м, 1 Н), 7,06 (м, 2 Н), 7,22 (д, 1 Н, J=8,5 Гц), 7,29 (м, 1 Н), 7,50 (т, 1 Н, J=7,5 Гц), 7,72 (м, 1 Н), 7,79 (м, 1 Н),8,30 (д, 1 Н, J=8,0 Гц) м.д. МС (ESI +) 425,2 (МН+). трет-Бутиловый эфир (R)-2-[3-(4-этоксифенил)-4-оксо-3,4-дигидрохиназолин-2-ил]-2-[(пиридин-3 илметил)амино]этилкарбаминовой кислоты (18). Пиридин-3-карбальдегид (0,13 г, 1,2 ммоль) добавляют к раствору соединения Q2 (0, 424 г, 1 ммоль) в дихлорэтане (10 мл) при -10 С с последующим добавлением триацетоксиборогидрида натрия(0,32 г, 1,5 ммоль). Смесь выдерживают при указанной температуре в течение 1,5 ч, затем дают ей возможность нагреться до комнатной температуры и перемешивают в течение ночи. Раствор разбавляют дихлорметаном, промывают насыщенным водным раствором гидрокарбоната натрия, водой, насыщенным раствором соли и сушат над безводным сульфатом натрия. Растворитель выпаривают с получением соединения 18 в виде твердого вещества желтого цвета (0,50 г). MC (ESI+) 516,2 (МН+). Примеры 19-21.(R)-3-(4-Этоксифенил)-2-[2-(4-фтор-3-трифторметилбензил)-3-пиридин-3-илметил-4,5-дигидро-3 Нимидазол-4-ил]-3 Н-хиназолин-4-он (19). Трифторуксусную кислоту (0,38 г, 3,34 ммоль) добавляют к раствору соединения 15 (0,12 г, 0,167 ммоль) в дихлорэтане (3 мл). Смесь нагревают до 60 С и выдерживают при указанной температуре в течение 1,5 ч. Растворитель выпаривают, остаток растворяют в этилацетате и промывают насыщенным водным раствором гидрокарбоната натрия, водой, насыщенным раствором соли и сушат над безводным сульфатом натрия. Растворитель удаляют и остаток очищают колоночной хроматографией, получая продукт в виде твердого белого вещества (67 мг). 1 Н ЯМР (CDCl3) 1,49 (т, 3 Н, J=6,96 Гц), 3,89-4,12 (м, 6 Н), 4,33 (м, 1 Н), 4,53 (м, 2 Н), 6,48 (м, 1 Н), 6,84- 29011439 Соединение 20 синтезируют из коммерчески доступных исходных веществ в пять стадий, как показано на схеме Т. 4-трет-Бутиловый эфир 1-бензиловый эфир (R)-2-[3-(4-этоксифенил)-4-оксо-3,4-дигидрохиназолин 2-ил]пиперазин-1,4-дикарбоновой кислоты (Т 1). К раствору антраниловой кислоты (1,91 г, 13,96 ммоль) и 1-бензилового эфира 4-третбутилового эфира пиперазин-1,2,4-трикарбоновой кислоты (5,08 г, 13,96 ммоль) в 25 мл безводного пиридина при комнатной температуре добавляют трифенилфосфит (8,66 г,27,92 ммоль). Полученный раствор перемешивают при 70 С в течение 15 ч. С помощью шприца добавляют п-фенетидин (1,91 г, 13,96 ммоль). Реакционную смесь перемешивают в течение еще 3 ч при 60 С,охлаждают до комнатной температуры и упаривают в вакууме для удаления большей части пиридина. Остаток в эфире промывают последовательно водным 1N раствором соляной кислоты, водным 1N раствором гидроксида натрия, водой и насыщенным раствором соли. Органический слой сушат над безводным сульфатом натрия и упаривают в вакууме до остатка коричневого цвета, который очищают хроматографией, получая продукт в виде твердого белого вещества(2,7 г). MC(ESI +) 585,2 (МН+). трет-Бутиловый эфир (R)-3-[3-(4-этоксифенил)-4-оксо-3,4-дигидрохиназолин-2-ил]пиперазин-1 карболовой кислоты (Т 2). К раствору соединения Т 1 (2,5 г, 4,28 ммоль) и циклогекса-1,3-диена (6,85 г,85, 62 ммоль) в 60 мл этанола добавляют 10% палладий на активированном угле (0,45 г, 0,43 ммоль). Полученный раствор кипятят с обратным холодильником в течение 3 ч. Смесь фильтруют и упаривают в вакууме, получая продукт в виде твердого белого вещества (1,85 г). 1 Н ЯМР (CDCl3) 1,43 (с, 9 Н), 1,49 (т, 3 Н, J=7 Гц), 2,70 (м, 1 Н), 2,85 (уш., 1 Н), 2,98 (м, 1 Н), 3,15 (д,1 Н, J=8 Гц), 3,59 (д, 1 Н, J=8 Гц), 3,95 (уш., 1 Н), 4,13 (м, 3 Н), 7,08 (м, 2 Н), 7,24-7,33 (м, 2 Н), 7,52 (м, 1 Н),7,76 (м, 2 Н), 8,30 (д, 1 Н, J=7,5 Гц). MC(ESI +) 451,2 (МН+). трет-Бутиловый эфир (R)-3-[3-(4-этоксифенил)-4-оксо-3,4-дигидрохиназолин-2-ил]-4-[(4-фтор-3 трифторметилфенил) ацетил] пиперазин-1 -карбоновой кислоты (ТЗ). EDC (1,53 г, 8,01 ммоль) добавляют к смеси соединения Т 2 (1,2 г, 2,67 ммоль), 4-фтор-3-трифторметилфенилуксусной кислоты (0,71 г,3,20 ммоль), HOBt (0,18 г, 1,34 ммоль) и NMM (0,81 г, 8,01 ммоль) в 15 мл ДМФА. Полученную смесь перемешивают при комнатной температуре в течение ночи и затем разбавляют этилацетатом. Полученный раствор промывают насыщенным водным раствором гидрокарбоната натрия, водой и насыщенным раствором соли, сушат над безводным сульфатом натрия и концентрируют. Остаток очищают хроматографией, получая продукт в виде твердого белого вещества (1,44 г). 1 Н ЯМР (CDCl3) 1,08 (с, 9 Н), 1,47 (т, 3 Н, J=7 Гц), 3,05 (м, 2 Н), 3,67 (м, 1 Н), 3,81 (м, 2 Н), 4,10 (м, 3 Н),4,24 (м, 1 Н), 4,49 (м, 1 Н), 5,19 (м, 1 Н), 7,11 (м, 3 Н), 7,29 (м, 1 Н), 7,51 (м, 5 Н), 7,75 (м, 1 Н), 8,30 (д, 1 Н,J=7,65 Гц) MC(ESI +) 655,2 (МН+).(R)-3-(4-Этоксифенил)-2-1-[(4-фтор-3-трифторметилфенил)ацетил]пиперазин-2-ил-3 Нхиназолин-4-он (20). Трифторуксусную кислоту (5,7 г, 50,15 ммоль) добавляют к раствору соединение Т 3 (1,64 г, 2,51 ммоль) в дихлорметане (90 мл). Полученную смесь перемешивают при комнатной температуре в течение 3 ч. Растворитель выпаривают, остаток растворяют в этилацетате и промывают насыщенным водным раствором гидрокарбоната натрия, водой, насыщенным раствором соли и сушат над безводным сульфатом натрия. Растворитель удаляют и остаток очищают колоночной флэш-хроматографией, получая продукт в виде твердого белого вещества (1,28 г). 1 Н ЯМР (CDCl3) 1,42 (т, 3 Н, J=7,04 Гц), 2, 69-2, 79 (м, 2 Н), 3,15 (м, 2 Н), 3,58 (м, 1 Н), 3,72 (м, 2 Н),4,04 (кв, 2 Н, J=7,04 Гц), 4,20 (м, 1 Н), 5,14 (м, 1 Н), 6,97-7,15 (м, 4 Н), 7,34-7,50 (м, 5 Н), 7,72 (м, 1 Н), 8,24 (д,1 Н, J=7,76 Гц). MC (ESI+) 555,1 (МН+). Пример 21.(R)-3-(4-Этоксифенил)-2-1-[(4-фтор-3-трифторметилфенил)ацетил]-4-метилпиперазин-2-ил-3 Нхиназолин-4-он (21). Формальдегид (37% в воде) (0,015 г, 0,18 ммоль) при комнатной температуре добавляют к раствору соединения 20 (0,05 г, 0,09 ммоль) в дихлорэтане (5 мл) с последующим добавлением Na(OAC)3BH(0,057 г, 0,27 ммоль). Смесь перемешивают в течение ночи. Полученный раствор разбавляют дихлорметаном, промывают насыщенным раствором гидрокарбоната натрия, водой, насыщенным раствором соли и сушат над безводным сульфатом натрия. Растворитель выпаривают и остаток очищают колоночной флэш-хроматографией, получая продукт в виде твердого белого вещества (40 мг).
МПК / Метки
МПК: A61P 29/00, A61P 37/00, A61K 31/519, C07D 239/91, C07D 471/04
Метки: применимые, модуляторов, иммунорегуляторных, пиримидина, рецепторов, cxcr3, заболеваний, воспалительных, композиции, расстройств, производные, профилактики, лечения, конденсированные, качестве
Код ссылки
<a href="https://eas.patents.su/30-11439-kondensirovannye-proizvodnye-pirimidina-i-ih-kompozicii-primenimye-v-kachestve-modulyatorov-cxcr3-receptorov-dlya-profilaktiki-i-lecheniya-vospalitelnyh-i-immunoregulyatornyh-rasst.html" rel="bookmark" title="База патентов Евразийского Союза">Конденсированные производные пиримидина и их композиции, применимые в качестве модуляторов cxcr3 рецепторов для профилактики и лечения воспалительных и иммунорегуляторных расстройств и заболеваний</a>
Конденсированные арильные и гетероарильные производные в качестве модуляторов метаболизма и для профилактики и лечения расстройств, связанных с нарушением метаболизма

Номер патента: 10023
Опубликовано: 30.06.2008
Авторы: Чои Цзинь Сунь Кэролайн, Сэмпл Грэем, Джоунз Роберт М., Фьораванти Беатриц, Сюн Ифэн, Рен Альберт С., Сейдж Карлтон Р., Шин Янг-Дзун, Кальдерон Имельда
МПК: A61K 31/4375, A61K 31/47, A61K 31/519...
Метки: гетероарильные, метаболизма, арильные, качестве, связанных, нарушением, производные, конденсированные, лечения, модуляторов, расстройств, профилактики
Формула / Реферат:
1. Соединение формулы (I) или его фармацевтически приемлемая соль, гидрат или сольват; где А и В, каждый независимо, означает C1-3 алкилен, возможно замещенный 1-4 заместителями, выбранными из группы, состоящей из C1-3 алкила, C1-4 алкокси, карбокси, циано, C1-3 галогеналкила и галогена; D является CR1R2 или N-R2, где R1 выбран из группы, состоящей из Н, C1-8 алкила, C1-4 алкокси, галогена и гидроксила; Е является N, С или CR3, где R3 означает...
Диарильные и арилгетероарильные производные мочевины в качестве модуляторов 5-ht2a-рецептора серотонина, пригодные для профилактики и лечения связанных с ним заболеваний

Номер патента: 9732
Опубликовано: 28.02.2008
Авторы: Страх-Плейнет Соня, Йаякумар Хоннаппа, Тигарден Бредли, Доса Петер Ян, Ли Хунмэй
МПК: C07D 231/16, A61K 31/415
Метки: заболеваний, диарильные, производные, серотонина, лечения, мочевины, связанных, арилгетероарильные, профилактики, пригодные, качестве, ним, модуляторов, 5-ht2a-рецептора
Формула / Реферат:
1. Соединение формулы (I) или его фармацевтически приемлемая соль, гидрат или сольват; где i) R1 означает арил, который необязательно замещен R9, R10, R11, R12, R13, R14 и R15, каждый из которых независимо выбран из группы, включающей C1-6ацил, C1-6ацилокси, С2-6алкенил, C1-6алкокси, C1-6алкил, C1-6алкилкарбоксамид, С2-6алкинил, C1-6алкилсульфонамид, C1-6алкилсульфинил, C1-6алкилсульфонил, C1-6алкилтио, C1-6алкилуреил, амино, C1-6алкиламино,...
Производные 2-(пурин-9-ил)-тетрагидрофуран-3,4-диола, способы их получения, фармацевтическая композиция и способ лечения или профилактики воспалительных заболеваний с их применением

Номер патента: 3194
Опубликовано: 27.02.2003
Авторы: Чен Чуэн, Кокс Брайан, Казинз Ричард Питер Чарлз
МПК: A61P 29/00, A61K 31/52, C07D 473/32...
Метки: воспалительных, композиция, получения, фармацевтическая, 2-(пурин-9-ил)-тетрагидрофуран-3,4-диола, заболеваний, профилактики, производные, способ, применением, способы, лечения
Формула / Реферат:
1. Производное 2-(пурин-9-ил)-тетрагидрофуран-3,4-диола формулы (I) где R1 и R2 независимо представляют собой группу, выбранную из (1) C3-8циклоалкил-; (2) водород; (3) арил2CHCH2-; (4) C3-8циклоалкилC1-6алкил-; (5) C1-8алкил-; (6) арилC1-6алкил-; (7) R4R5N-C1-6алкил-; (8) C1-6алкил-CH(CH2OH)-; (9) арилC1-5алкил-CH(CH2OH)-; (10) C3-8циклоалкил, независимо замещенный одной или более чем одной группой -(CH2)pR6; (11) H2NC(=NH)NHC1-6алкил-; (12)...
Производные 3-(3,5-диоксо-4,5-дигидро-3н-(1,2,4)триазин-2-ил)бензамида в качестве ингибиторов р2х7 для лечения воспалительных заболеваний

Номер патента: 8591
Опубликовано: 29.06.2007
Авторы: Дюплантьер Аллен Джекоб, Домброуски Марк Энтони
МПК: C07D 253/06, C07D 401/06, A61K 31/53...
Метки: воспалительных, заболеваний, качестве, ингибиторов, производные, лечения, р2х7, 3-(3,5-диоксо-4,5-дигидро-3н-(1,2,4)триазин-2-ил)бензамида
Формула / Реферат:
1. Соединение формулы где R1 представляет собой (С1-С6)алкил, возможно замещенный (С3-С10)циклоалкилом или (С6-С10)арилом, причем каждый указанный (С1-С6)алкил, (С3-С10)циклоалкил или (С6-С10)арил возможно замещен одной-тремя подходящими группировками, независимо выбранными из группы, состоящей из гидрокси, галогена, CN-, (С1-С6)алкила, НО(С1-С6)алкила, фенила или п-толила; R2 представляет собой галоген и (С1-С6)алкил; R3 представляет собой...
Производные полигидроксифенола, содержащие их лекарственные композиции и применение указанных веществ для профилактики и лечения заболеваний костей и хрящей

Номер патента: 1172
Опубликовано: 30.10.2000
Авторы: Сакай Куниказу, Китамура Казуюки, Сатох Юсуке, Дой Казуюки
МПК: A61K 31/12, C07C 49/713
Метки: лекарственные, лечения, производные, хрящей, профилактики, веществ, композиции, указанных, полигидроксифенола, костей, содержащие, применение, заболеваний
Формула / Реферат:
1. Соединение общей формулы (I) или его соль, где R1 обозначает алкильную группу с разветвленной или прямой цепью, имеющую 1-15 атомов углерода (при условии, что метильная группа исключается), замещенную или незамещенную бензильную группу либо замещенную или незамещенную арильную группу; R2 обозначает атом водорода, алкильную группу с разветвленной или прямой цепью, имеющую 1-15 атомов углерода, алкенильную группу с разветвленной или прямой...
Предыдущий патент: Аспарагиназа, полученная из aspergillus niger, полинуклеотид, вектор, клетка-хозяин, способ получения пищевого продукта и пищевой продукт.
Следующий патент: Новые стронциевые соли сульфоновых кислот, способ их получения и фармацевтические композиции, содержащие их
Случайный патент: Многофазный препарат для контрацепции на основе натурального эстрогена