4-(8-метокси-1-((1-метоксипропан-2-ил)-2-(тетрагидро-2н-пиран-4-ил)-1н-имидазо[4,5-c]хинолин-7-ил)-3,5-диметилизоксазол и его применение в качестве ингибитора бромодомена
Номер патента: 23374
Опубликовано: 31.05.2016
Авторы: Ватсон Роберт Дж., Демон Эммануэль Юбер, Джонс Кэтрин Луис























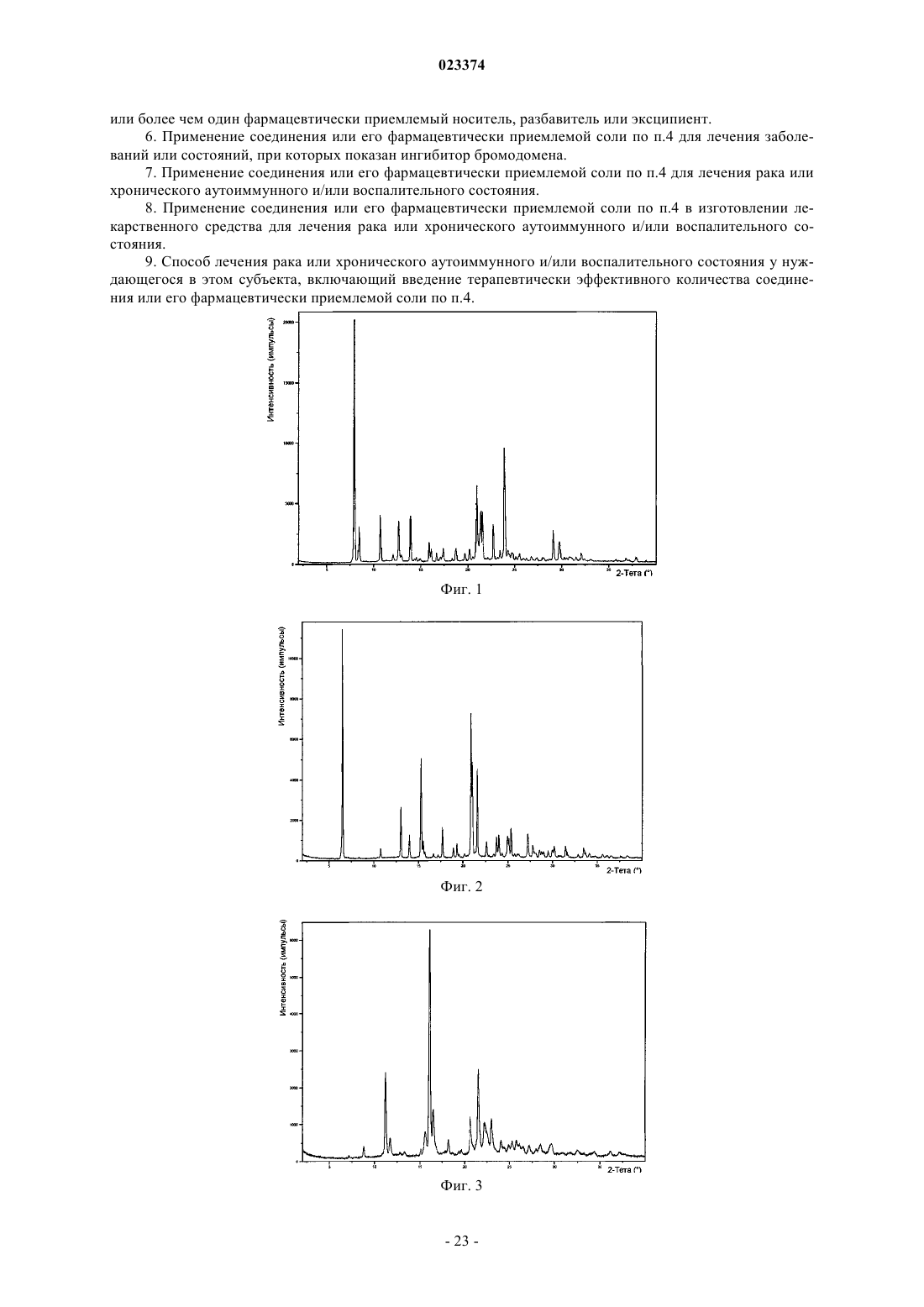
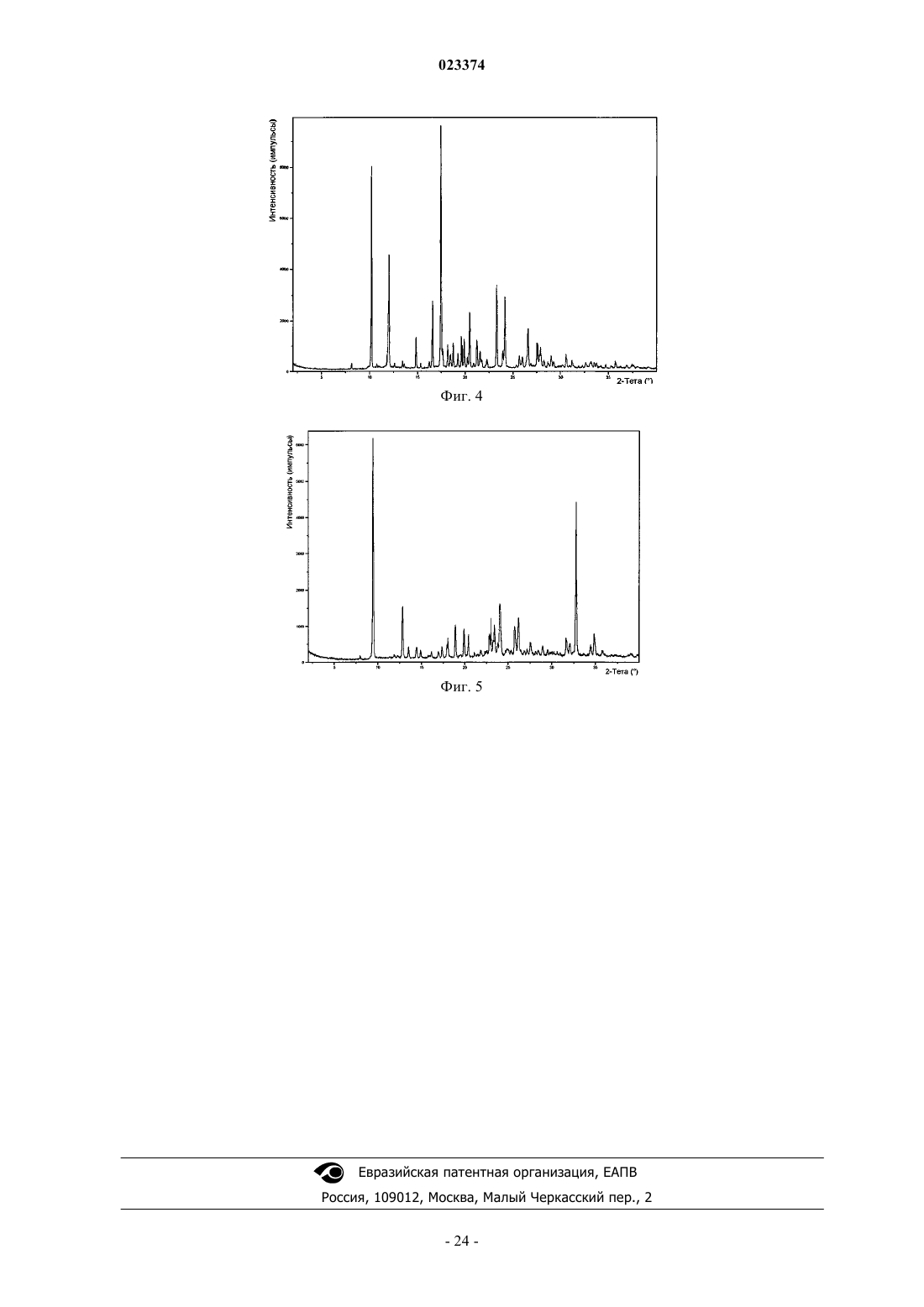
Формула / Реферат
1. Соединение формулы (I), представляющее собой 4-(8-метокси-1-(1-метоксипропан-2-ил)-2-(тетрагидро-2Н-пиран-4-ил)-1Н-имидазо[4,5-с]хинолин-7-ил)-3,5-диметилизоксазол

или его соль.
2. Соединение формулы (IA) по п.1, представляющее собой 4-(8-метокси-1-((R)-1-метоксипропан-2-ил)-2-(тетрагидро-2Н-пиран-4-ил)-1Н-имидазо[4,5-с]хинолин-7-ил)-3,5-диметилизоксазол

или его соль.
3. Соединение формулы (IB) по п.1, представляющее собой 4-(8-метокси-1-((S)-1-метоксипропан-2-ил)-2-(тетрагидро-2Н-пиран-4-ил)-1Н-имидазо[4,5-с]хинолин-7-ил)-3,5-диметилизоксазол

или его соль.
4. Соединение по любому из пп.1-3 или его фармацевтически приемлемая соль.
5. Фармацевтическая композиция для лечения заболеваний или состояний, при которых показан ингибитор бромодомена, содержащая соединение или его фармацевтически приемлемую соль по п.4 и один или более чем один фармацевтически приемлемый носитель, разбавитель или эксципиент.
6. Применение соединения или его фармацевтически приемлемой соли по п.4 для лечения заболеваний или состояний, при которых показан ингибитор бромодомена.
7. Применение соединения или его фармацевтически приемлемой соли по п.4 для лечения рака или хронического аутоиммунного и/или воспалительного состояния.
8. Применение соединения или его фармацевтически приемлемой соли по п.4 в изготовлении лекарственного средства для лечения рака или хронического аутоиммунного и/или воспалительного состояния.
9. Способ лечения рака или хронического аутоиммунного и/или воспалительного состояния у нуждающегося в этом субъекта, включающий введение терапевтически эффективного количества соединения или его фармацевтически приемлемой соли по п.4.
Текст
Новые хинолиновые соединения формулы (I), фармацевтические композиции, содержащие такие соединения, и их применение в терапии в качестве ингибиторов бромодомена. Область изобретения Настоящее изобретение относится к новым соединениям, фармацевтическим композициям, содержащим такие соединения, и к их применению в терапии. Предшествующий уровень техники Геномы эукариотических организмов сильно организованы внутри ядра клетки. Длинные нити дуплексной ДНК обернуты вокруг октамера гистоновых белков (обычно содержащего по две копии гистонов Н 2 А, Н 2 В, H3 и Н 4) с образованием нуклеосомы. Потом эта основная единица дополнительно уплотняется посредством агрегации и сворачивания нуклеосом с образованием сильно конденсированной хроматиновой структуры. Возможен ряд различных состояний конденсации, и плотность этой структуры варьирует в течение клеточного цикла, становясь наиболее компактной во время процесса деления клетки. Структура хроматина играет важную роль в регулировании транскрипции генов, которая не может протекать эффективно из сильно конденсированного хроматина. Структура хроматина регулируется серией посттрансляционных модификаций гистоновых белков, особенно гистонов H3 и Н 4, и наиболее часто в пределах гистоновых хвостов, которые протягиваются за пределы центральной нуклеосомной структуры. Эти модификации включают ацетилирование, метилирование, фосфорилирование, убиквитинирование, сумоилирование. Эти эпигенетические метки вносятся и стираются специфическими ферментами, которые помещают метки на специфических остатках в гистоновом хвосте, формируя посредством этого эпигенетический код, который затем интерпретируется клеткой, обеспечивая возможность генспецифичной регуляции структуры хроматина и, посредством этого, транскрипции. Ацетилирование гистонов наиболее часто ассоциировано с активацией транскрипции генов, так как эта модификация ослабляет взаимодействие ДНК и гистонового октамера за счет изменения электростатики. В дополнение к этому физическому изменению специфические белки связываются с ацетилированными остатками лизина в гистонах для считывания эпигенетического кода. Бромодомены представляют собой небольшие (110 аминокислот) особые домены в белках, которые связываются с ацетилированными остатками лизина, как правило, но не всегда, в контексте гистонов. Известно, что существует семейство из приблизительно 50 белков, которые содержат бромодомены, и они имеют ряд функций в клетке. Семейство BET бромодоменсодержащих белков включает 4 белка (BRD2, BRD3, BRD4 и BRD-t),которые содержат тандемные бромодомены, способные связываться с двумя ацетилированными остатками лизина, расположенными в непосредственной близости, увеличивая специфичность взаимодействия. Сообщается, что BRD2 и BRD3 ассоциированы с гистонами в активно транскрибируемых генах и могут быть вовлечены в облегчение транскрипционной элонгации (Leroy et al., Mol. Cell. 2008, 30(1): 5160), в то время как BRD4, по-видимому, вовлечен в рекрутинг комплекса pTEF- в индуцибельных генах,вызывая фосфорилирование РНК-полимеразы и увеличение транскрипционного выхода (Hargreaves etal., Cell, 2009, 138(1): 129-145). Также сообщалось, что BRD4 или BRD3 могут сливаться с NUT (ядерный белок в яичках), образуя новые слитые онкогены, BRD4-NUT или BRD3-NUT, при очень злокачественной форме эпителиальной неоплазии (French et al. Cancer Research, 2003, 63, 304-307 и French et al. Journal of Clinical Oncology, 2004, 22 (20), 4135-4139). Данные указывают на то, что слитые белки BRD-NUT содействуют канцерогенезу (Oncogene, 2008, 27, 2237-2242). BRD-t экспрессируется исключительно в яичках и яичниках. Сообщалось, что все члены семейства обладают некоторой функцией в аспектах контроля или исполнения клеточного цикла, и было показано, что они остаются в комплексе с хромосомами во время клеточного деления, что предполагает роль в поддержании эпигенетической памяти. Кроме того, некоторые вирусы используют эти белки для привязывания своих геномов к хроматину клеткихозяина как часть процесса репликации вируса (You et al. Cell, 2004, 117(3): 349-60). В японской патентной заявке JP 2008-156311 раскрыто производное бензимидазола, которое, как утверждается, представляет собой связывающий бромодомен BRD2 агент, который является применимым в отношении вирусной инфекции/пролиферации. В патентной заявке WO 2009084693A1 раскрыт ряд производных тиенотриазолодиазепина, которые, как утверждается, ингибируют связывание между ацетилированным гистоном и бромодоменсодержащим белком, которые, как утверждается, полезны в качестве противораковых агентов. В патентной заявке WO 2011/054846 раскрыт ряд производных хинолина, которые ингибируют связывание бромодоменов семейства BET с ацетилированными остатками лизина. Были обнаружены новые соединения, которые ингибируют связывание бромодоменов с когнатными им ацетилированными белками, более конкретно, класс соединений, которые ингибируют связывание бромодоменов семейства BET с ацетилированными остатками лизина. Такие соединения в дальнейшем называют "ингибиторами бромодомена". Краткое изложение сущности изобретения В первом аспекте настоящего изобретения предложено соединение формулы (I) или его соль, более конкретно соединение формулы (I) или его фармацевтически приемлемая соль Во втором аспекте настоящего изобретения предложена фармацевтическая композиция, содержащая соединение формулы (I) или его фармацевтически приемлемую соль и один или более чем один фармацевтически приемлемый носитель, разбавитель или эксципиент. В третьем аспекте настоящего изобретения предложено соединение формулы (I) или его фармацевтически приемлемая соль для применения в терапии, в частности в лечении заболеваний или состояний,для которых показан ингибитор бромодомена. В четвертом аспекте настоящего изобретения предложен способ лечения заболеваний или состояний, для которых показан ингибитор бромодомена, у нуждающегося в этом субъекта, включающий введение терапевтически эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли. В пятом аспекте настоящего изобретения предложено применение соединения формулы (I) или его фармацевтически приемлемой соли в изготовлении лекарственного средства для лечения заболеваний или состояний, для которых показан ингибитор бромодомена. Краткое описание графических материалов На фиг. 1 показана картина XRPD кристаллической формы 4-(8-метокси-1-R)-1-метоксипропан-2 ил)-2-(тетрагидро-2 Н-пиран-4-ил)-1 Н-имидазо[4,5-с]хинолин-7-ил)-3,5-диметилизоксазола(безводная форма 3). На фиг. 4 показана картина XRPD кристаллической формы 4-(8-метокси-1-R)-1-метоксипропан-2 ил)-2-(тетрагидро-2 Н-пиран-4-ил)-1 Н-имидазо[4,5-с]хинолин-7-ил)-3,5-диметилизоксазола (гидрат). На фиг. 5 показана картина XRPD кристаллической формы гидрохлорида 4-(8-метокси-1-R)-1 метоксипропан-2-ил)-2-(тетрагидро-2 Н-пиран-4-ил)-1 Н-имидазо[4,5-с]хинолин-7-ил)-3,5 диметилизоксазола (гидрохлорид). Подробное описание изобретения Настоящее изобретение относится к соединению формулы (I), представляющему собой 4-(8 метокси-1-(1-метоксипропан-2-ил)-2-(тетрагидро-2 Н-пиран-4-ил)-1 Н-имидазо[4,5-с]хинолин-7-ил)-3,5 диметилизоксазол или его соли. Соединение формулы (I) содержит хиральный атом, так что могут образовываться оптические изомеры, например энантиомеры. Таким образом, настоящее изобретение охватывает все изомеры соединения формулы (I), как в виде отдельных изомеров, выделенных так, чтобы они были, по существу, свободны от другого изомера (т.е. чистых), так и в виде смесей (т.е. рацематов и рацемических смесей). Отдельный изомер, выделенный так, чтобы он был, по существу, свободен от другого изомера (т.е. чистый),может быть выделен так, что он будет присутствовать с менее чем 10%, конкретно менее чем примерно 1%, например менее чем примерно 0,1% другого изомера. Разделение изомеров может быть выполнено посредством традиционных методик, известных специалисту в данной области техники, например фракционной кристаллизацией, хроматографией илиHPLC (высокоэффективной жидкостной хроматографией). В одном воплощении предложено соединение формулы (IA), представляющее собой 4-(8-метокси 1-R)-1-метоксипропан-2-ил)-2-(тетрагидро-2 Н-пиран-4-ил)-1 Н-имидазо[4,5-с]хинолин-7-ил)-3,5-диме-2 023374 или его соль. В еще одном воплощении предложено соединение формулы (IB), представляющее собой 4-(8 метокси-1-S)-1-метоксипропан-2-ил)-2-(тетрагидро-2 Н-пиран-4-ил)-1 Н-имидазо[4,5-с]хинолин-7-ил)3,5-диметилизоксазол или его соль. Понятно, что соединения формулы (IA) и формулы (IB) находятся в пределах объема соединения формулы (I). При использовании и в данном описании изобретения, если не указано иное, ссылка на соединение формулы (I) также включает ссылку на соединение формулы (IA) и соединение формулы (IB). Понятно, что настоящее изобретение охватывает соединения формулы (I) в виде свободного основания и в виде его солей, например в виде его фармацевтически приемлемой соли. В одном воплощении изобретение относится к соединениям формулы (I) в форме свободного основания. В одном воплощении изобретение относится к соединениям формулы (I) или его фармацевтически приемлемой соли. Вследствие их возможного применения в медицине, желательно, чтобы соли соединений формулы(I) являлись фармацевтически приемлемыми. Подходящие фармацевтически приемлемые соли могут включать соли присоединения кислоты (для обзора подходящих фармацевтически приемлемых солей см.Berge et al., J. Pharm. Sci., 66: 1-19, 1977). Обычно, фармацевтически приемлемая соль может быть легко получена посредством использования требуемых кислоты или основания в зависимости от обстоятельств. Полученная соль может осаждаться из раствора и быть собрана фильтрацией или может быть извлечена выпариванием растворителя. Фармацевтически приемлемая соль присоединения кислоты может быть образована в результате взаимодействия соединения формулы (I) с подходящей неорганической или органической кислотой (такой как бромисто-водородная, соляная, серная, азотная, фосфорная, янтарная, малеиновая, уксусная,пропионовая, фумаровая, лимонная, винная, молочная, бензойная, салициловая, глутаминовая, аспарагиновая, п-толуолсульфоновая, бензолсульфоновая, метансульфоновая, этансульфоновая, нафталинсульфоновая, такая как 2-нафталинсульфоновая, или гексановая кислота), возможно в подходящем растворителе, таком как органический растворитель, с получением соли, которую обычно выделяют, например,кристаллизацией и фильтрацией или упариванием с последующим растиранием. Фармацевтически приемлемая соль присоединения кислоты соединения формулы (I) может включать или представлять собой,например, соль гидробромид, гидрохлорид, сульфат, нитрат, фосфат, сукцинат, малеат, ацетат, пропионат, фумарат, цитрат, тартрат, лактат, бензоат, салицилат, глутамат, аспартат, п-толуолсульфонат, бензолсульфонат, метансульфонат, этансульфонат, нафталинсульфонат (например, 2-нафталинсульфонат) или гексаноат. В одном воплощении фармацевтически приемлемая соль присоединения кислоты соединения формулы (I) может включать или представлять собой соль гидрохлорид, сульфат, малеат, фумарат,цитрат, п-толуолсульфонат, бензолсульфонат или метансульфонат. В конкретном воплощении предложено соединение формулы (IA) в форме соли гидрохлорида. Другие фармацевтически неприемлемые соли, например формиаты, оксалаты или трифторацетаты,могут быть использованы, например, в выделении соединений формулы (I), и они включены в объем данного изобретения. В объем изобретения включены все возможные стехиометрические и нестехиометрические формы солей соединений формулы (I). Понятно, что многие органические соединения могут образовывать комплексы с растворителями, в которых они взаимодействуют или из которых они осаждаются или кристаллизуются. Эти комплексы известны как "сольваты". Например, комплекс с водой известен как "гидрат". Растворители с высокими точками кипения и/или способные к образованию водородных связей, такие как вода, ксилол, Nметилпирролидон, метанол и этанол, можно использовать для образования сольватов. Способы идентификации сольватов включают, но не ограничиваются этим, ЯМР (ядерный магнитный резонанс) и мик-3 023374 роанализ. Сольваты соединений формулы (I) входят в объем данного изобретения. В объем изобретения включены все возможные стехиометрические и нестехиометрические формы сольватов соединений формулы (I). Изобретение охватывает все пролекарства соединения формулы (I) или его фармацевтически приемлемой соли, которые при введении реципиенту способны давать (прямо или опосредованно) соединение формулы (I) или его фармацевтически приемлемую соль или их активный метаболит или остаток. Такие производные могут быть распознаны специалистом в данной области техники без излишнего экспериментирования (тем не менее, сделана ссылка на руководство Burger's Medicinal Chemistry and DrugDiscovery, 5th Edition, vol. 1: Principles and Practice, которое включено в данное описание изобретения посредством ссылки в объеме описания таких производных). Соединения формулы (I) могут находиться в кристаллической или аморфной форме. Кроме того,некоторые из кристаллических форм соединений формулы (I) могут существовать в виде полиморфов,которые включены в объем настоящего изобретения. Полиморфные формы соединений формулы (I) могут быть охарактеризованы и дифференцированы, используя ряд традиционных аналитических методик,включая картины дифракции рентгеновских лучей на порошке (XRPD), инфракрасные (IR) спектры,спектры комбинационного рассеяния, дифференциальную сканирующую калориметрию (DSC), термогравиметрический анализ (TGA) и твердотельный ядерный магнитный резонанс (ТТЯМР), но не ограничиваясь этим. Было обнаружено, что соединение формулы (IA), то есть 4-(8-метокси-1-R)-1-метоксипропан-2 ил)-2-(тетрагидро-2 Н-пиран-4-ил)-1 Н-имидазо[4,5-с]хинолин-7-ил)-3,5-диметилизоксазол, в форме свободного основания существует в ряде различных кристаллических форм, а именно безводных форм 1, 2 и 3 и гидратированной формы 1. Такие кристаллические формы могут быть получены способами, описанными в данном описании изобретения. Таким образом, в одном воплощении предложена кристаллическая форма 4-(8-метокси-1-R)-1 метоксипропан-2-ил)-2-(тетрагидро-2 Н-пиран-4-ил)-1 Н-имидазо[4,5-с]хинолин-7-ил)-3,5-диметилизоксазола (безводная форма 1), характеризующаяся, по существу, картиной дифракции рентгеновских лучей на порошке (XRPD), как показано на фиг. 1, где картина XRPD выражена в единицах углов 2-тета и получена на дифрактометре с использованием K-излучения меди, используя методы, описанные в данном описании изобретения. Картина XRPD безводной формы 1 демонстрирует характеристические пики при углах 2-тета, перечисленных в примере 9 в табл. 1. В еще одном воплощении предложена кристаллическая форма 4-(8-метокси-1-R)-1-метоксипропан-2-ил)-2-(тетрагидро-2 Н-пиран-4-ил)-1 Н-имидазо[4,5-с]хинолин-7-ил)-3,5-диметилизоксазола(безводная форма 2), характеризующаяся, по существу, картиной дифракции рентгеновских лучей на порошке (XRPD), как показано на фиг. 2, где картина XRPD выражена в единицах углов 2-тета и получена на дифрактометре с использованием K-излучения меди, используя описанные методы. КартинаXRPD безводной формы 2 показывает характеристические пики при углах 2-тета, перечисленных в примере 9 в табл. 1. В еще одном воплощении предложена кристаллическая форма 4-(8-метокси-1-R)-1-метоксипропан-2-ил)-2-(тетрагидро-2 Н-пиран-4-ил)-1 Н-имидазо[4,5-с]хинолин-7-ил)-3,5-диметилизоксазола(безводная форма 3), характеризующаяся, по существу, картиной дифракции рентгеновских лучей на порошке (XRPD), как показано на фиг. 3, где картина XRPD выражена в единицах углов 2-тета и получена на дифрактометре с использованием K-излучения меди, используя методы, описанные в данном описании изобретения. Картина XRPD безводной формы 3 демонстрирует характеристические пики при углах 2-тета, перечисленных в примере 9 в табл. 1. В еще одном воплощении предложена кристаллическая форма 4-(8-метокси-1-R)-1-метоксипропан-2-ил)-2-(тетрагидро-2 Н-пиран-4-ил)-1 Н-имидазо[4,5-с]хинолин-7-ил)-3,5-диметилизоксазола(гидрат), характеризующаяся, по существу, картиной дифракции рентгеновских лучей на порошке(XRPD), как показано на фиг. 4, где картина XRPD выражена в единицах углов 2-тета и получена на дифрактометре с использованием K-излучения меди, используя методы, описанные в данном описании изобретения. Картина XRPD гидрата демонстрирует характеристические пики при углах 2-тета, перечисленных в примере 9 в табл. 1. В изобретении также предложен гидрохлорид соединения формулы (IA), то есть 4-(8-метокси-1R)-1-метоксипропан-2-ил)-2-(тетрагидро-2 Н-пиран-4-ил)-1 Н-имидазо[4,5-с]хинолин-7-ил)-3,5-диметилизоксазола, в кристаллической форме. Такая кристаллическая форма может быть получена способами,описанными в данном описании изобретения. В еще одном воплощении предложена кристаллическая форма гидрохлорида 4-(8-метокси-1-R)-1 метоксипропан-2-ил)-2-(тетрагидро-2 Н-пиран-4-ил)-1 Н-имидазо[4,5-с]хинолин-7-ил)-3,5-диметилизоксазола (форма 1 гидрохлорида), характеризующаяся, по существу, картиной дифракции рентгеновских лучей на порошке (XRPD), как показано на фиг. 5, где картина XRPD выражена в единицах углов 2-тета и получена на дифрактометре с использованием K-излучения меди, используя методы, описанные в данном описании изобретения. Картина XRPD этой формы демонстрирует характеристические пики при углах 2-тета, перечисленных в примере 9 в табл. 1. Из предыдущего понятно, что сольваты, изомеры и полиморфные формы соединения формулы (I) и его соли включены в объем данного изобретения. Соединения формулы (I) или их соли могут быть получены различными способами, включая стандартные химические способы. Иллюстративные общие способы синтеза изложены ниже, а получение конкретных соединений формулы (I) и их фармацевтически приемлемых солей описано далее в рабочих примерах. Соединение формулы (I) может быть получено из соединения формулы (II), например, нагреванием соединения формулы (II) в уксусной кислоте или п-толуолсульфоновой кислоте в толуоле Соединение формулы (II) может быть получено приведением соединения формулы (III) во взаимодействие с соединением формулы (IV) или его активированным производным Подходящим активированным производным соединения формулы (IV) является хлорангидрид кислоты. Взаимодействие между соединением формулы (III) и соединением формулы (IV) может быть осуществлено в подходящем органическом растворителе, возможно в присутствии основания. Когда соединение формулы (I) представляет собой смесь изомеров, соединения формулы (IA) и формулы (IB) могут быть получены из соединения формулы (I), используя подходящие методики разделения, которые хорошо известны специалисту в данной области техники, такие как методики, описанные в данном описании изобретения. Альтернативно соединения формулы (IA) и (IB) могут быть получены способом хирального синтеза. В качестве иллюстрации соединение формулы (IA) может быть получено посредством способа, представленного на следующей схеме: Специалисту в данной области техники понятно, что может быть выгодным защитить одну или более чем одну функциональную группу соединений, описанных выше. Примеры защитных групп и способы для их удаления можно найти в Т.W. Greene "Protective Groups in Organic Synthesis" (4th edition, J.Wiley and Sons, 2006). Подходящие аминозащитные группы включают ацил (например ацетил, карбамат(например бензил), которые могут быть удалены гидролизом (например, используя кислоту, такую как соляная кислота в диоксане или трифторуксусная кислота в дихлорметане) или восстановлением (например гидрогенолизом бензильной или бензилоксикарбонильной группы или восстановительным удалением 2',2',2'-трихлорэтоксикарбонильной группы, используя цинк в уксусной кислоте) в зависимости от обстоятельств. Другие подходящие аминозащитные группы включают трифторацетил (-COCF3), который может быть удален гидролизом, катализируемым основанием. Понятно, что в любом из путей, описанных выше, точный порядок стадий синтеза, посредством которых различные группы и группировки вводят в молекулу, можно варьировать. Обеспечение того, чтобы группы или группировки, введенные на одной стадии процесса, не подвергались воздействию последующих трансформаций и взаимодействий, и, таким образом, выбор порядка стадий синтеза находятся в рамках компетенции практикующего специалиста в данной области техники. Некоторые описанные выше промежуточные соединения образуют еще один аспект изобретения. Соединения формулы (I) и их соли представляют собой ингибиторы бромодомена, и, таким образом, предполагается, что они обладают потенциальной пользой в лечении заболеваний или состояний,для которых показан ингибитор бромодомена. Кроме того, соединения формулы (I) и их фармацевтически приемлемые соли (такие как соединение формулы (IA) или его фармацевтически приемлемая соль) могут обладать одним или более чем одним свойством ADMET (абсорбция, распределение, метаболизм,выделение и токсичность), что делает их особенно подходящими. Таким образом, в настоящем изобретении предложено соединение формулы (I) или его фармацевтически приемлемая соль для применения в терапии. В одном воплощении предложено соединение формулы (IA) или его фармацевтически приемлемая соль для применения в терапии. Соединение формулы (I) или его фармацевтически приемлемую соль можно применять в лечении заболеваний или состояний, для которых показан ингибитор бромодомена. Таким образом, в настоящем изобретении предложено соединение формулы (I) или его фармацевтически приемлемая соль для применения в лечении любых заболеваний или состояний, для которых показан ингибитор бромодомена. В одном воплощении предложено соединение формулы (IA) или его фармацевтически приемлемая соль для применения в лечении любых заболеваний или состояний, для которых показан ингибитор бромодомена. В другом воплощении предложено соединение формулы (I) (такое как соединение формулы (IA или его фармацевтически приемлемая соль для применения в лечении хронических аутоиммунных и/или воспалительных состояний. В еще одном воплощении предложено соединение формулы (I) (такое как соединение формулы (IA или его фармацевтически приемлемая соль для применения в лечении рака. Также предложено применение соединения формулы (I) или его фармацевтически приемлемой соли в изготовлении лекарственного средства для лечения заболеваний или состояний, для которых показан ингибитор бромодомена. В одном воплощении предложено применение соединения формулы (IA) или его фармацевтически приемлемой соли в изготовлении лекарственного средства для лечения заболеваний или состояний, для которых показан ингибитор бромодомена. Также предложен способ лечения заболеваний или состояний, для которых показан ингибитор бромодомена, у нуждающегося в этом субъекта, включающий введение терапевтически эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли. В одном воплощении предложен способ лечения заболеваний или состояний, для которых показан ингибитор бромодомена, у нуждающегося в этом субъекта, включающий введение терапевтически эффективного количества соединения формулы (IA) или его фармацевтически приемлемой соли. Подходящим образом, нуждающимся в этом субъектом является млекопитающее, в частности человек. При использовании в данном описании изобретения термин "эффективное количество" означает количество лекарственного средства или фармацевтического агента, вызывающее биологический или медицинский ответ ткани, системы или субъекта (например человека), которого добивается, например,исследователь или клиницист. Кроме того, термин "терапевтически эффективное количество" означает любое количество, которое, по сравнению с соответствующим субъектом, не получившим такого количества, приводит к улучшению лечения, излечению, предупреждению или облегчению симптомов заболевания, расстройства, или уменьшению побочного эффекта, или снижению скорости развития заболевания или расстройства. В объем данного термина также включены количества, эффективные для улучшения нормальной физиологической функции. Считается, что ингибиторы бромодомена полезны в лечении различных заболеваний или состояний, связанных с системным воспалением или воспалением ткани, воспалительными ответами на инфекцию или гипоксию, активацией и пролиферацией клеток, метаболизмом липидов, фиброзом, и в предупреждении и лечении вирусных инфекций. Ингибиторы бромодомена могут быть полезны в лечении широкого ряда хронических аутоиммунных и воспалительных состояний, таких как ревматоидный артрит, остеоартрит, острая подагра, псориаз,системная красная волчанка, рассеянный склероз, воспалительное заболевание кишечника (болезнь Крона и язвенный колит), астма, хроническое обструктивное заболевание дыхательных путей, пневмонит,миокардит, перикардит, миозит, экзема, дерматит (включая атопический дерматит), алопеция, витилиго,буллезные заболевания кожи, нефрит, васкулит, атеросклероз, болезнь Альцгеймера, депрессия, синдром Шегрена, сиаладенит, окклюзия центральной вены сетчатки, окклюзия ветвей вены сетчатки, синдром Ирвина-Гасса (после катаракты и послеоперационный), пигментный ретинит, парс планит (pars plantis),дробьевидная (birdshot) хориоретинопатия, эпиретинальная мембрана, кистозный макулярный отек, парафовеальная телеангиэктазия, тракционные макулопатии, витреомакулярные тракционные синдромы,отслоение сетчатки, нейроретинит, идиопатический макулярный отек, ретинит, сухой глаз (сухой кератоконъюнктивит), весенний кератоконъюнктивит, атопический кератоконъюнктивит, увеит (такой как передний увеит, панувеит, задний увеит, ассоциированный с увеитом макулярный отек), склерит, диабетическая ретинопатия, диабетический макулярный отек, возрастная макулярная дистрофия, гепатит, панкреатит, первичный билиарный цирроз, склерозирующий холангит, болезнь Аддисона, гипофизит, тиреоидит, диабет I типа и острое отторжение трансплантированных органов. Ингибиторы бромодомена могут быть полезны в лечении широкого ряда острых воспалительных состояний, таких как острая подагра, гигантоклеточный артериит, нефрит, включая волчаночный нефрит,васкулит с вовлечением органов, такой как гломерулонефрит, васкулит, включая гигантоклеточный артериит, гранулематоз Вегенера, узелковый полиартериит, болезнь Бехчета, болезнь Кавасаки, артериит Такаясу, гангренозная пиодермия, васкулит с вовлечением органов и острое отторжение трансплантированных органов. Ингибиторы бромодомена могут быть полезны в лечении заболеваний или состояний, которые вовлекают воспалительные ответы на инфицирования бактериями, вирусами, грибами, паразитами или их токсины, таких как сепсис, септический синдром, септический шок, эндотоксемия, синдром системного воспалительного ответа (SIRS), синдром мультиорганной дисфункции, синдром токсического шока, острое повреждение легких, ARDS (респираторный дистресс-синдром взрослых), острая почечная недостаточность, фульминантный гепатит, ожоги, острый панкреатит, послеоперационные синдромы, саркоидоз,реакции Герксгеймера, энцефалит, миелит, менингит, малярия и SIRS, ассоциированный с вирусными инфекциями, такими как грипп, опоясывающий герпес, простой герпес и коронавирусная инфекция. Ингибиторы бромодомена могут быть полезны в лечении состояний, ассоциированных с ишемически-реперфузионным повреждением, таких как инфаркт миокарда, цереброваскулярная ишемия (инсульт), острые коронарные синдромы, реперфузионное повреждение почек, трансплантация органа, аортокоронарное шунтирование, процедуры искусственного кровообращения, легочная, почечная, печеночная, желудочно-кишечная эмболия или периферическая эмболия конечности. Ингибиторы бромодомена могут быть полезны в лечении расстройств метаболизма липидов посредством регуляции АРО-А 1 (аполипопротеинов-А 1), таких как гиперхолестеринемия, атеросклероз и болезнь Альцгеймера. Ингибиторы бромодомена могут быть полезны в лечении фиброзных состояний, таких как идиопатический фиброз легких, фиброз почек, послеоперационная стриктура, образование келоидных рубцов,склеродермия (включая кольцевидную склеродермию) и фиброз сердца. Ингибиторы бромодомена могут быть полезны в лечении вирусных инфекций, таких как инфекция вирусом герпеса, вирусом папилломы человека, аденовирусом, поксвирусом и другими ДНК-вирусами. Ингибиторы бромодомена могут быть полезны в лечении рака, включая гематологический (такой как лейкоз, лимфома и множественная миелома), эпителиальный, включая карциномы легкого, молочной железы и ободочной кишки, срединные карциномы, мезенхимальные опухоли, опухоли печени, почки и неврологические опухоли. Ингибиторы бромодомена могут быть полезны в лечении одного или более чем одного вида рака,выбранного из рака головного мозга (глиомы), глиобластом, синдрома Баньяна-Зонана, болезни Коудена,болезни Лермитта-Дюкло, рака молочной железы, воспалительного рака молочной железы, колоректального рака, опухоли Вильмса, саркомы Юинга, рабдомиосаркомы, эпендимомы, медуллобластомы, рака ободочной кишки, рак головы и шеи, рака почки, рака легкого, рака печени, меланомы, плоскоклеточной карциномы, рака яичника, рака поджелудочной железы, рака предстательной железы, саркомы, рака, остеосаркомы, гигантоклеточной опухоли кости, рака щитовидной железы, лимфобластного Т-клеточного лейкоза, хронического миелогенного лейкоза, хронического лимфоцитарного лейкоза, волосатоклеточного лейкоза, острого лимфобластного лейкоза, острого миелогенного лейкоза, хронического нейтрофильного лейкоза, острого лимфобластного Т-клеточного лейкоза, плазмоцитомы, иммунобластного крупноклеточного лейкоза, мантийноклеточного лейкоза, множественной миеломы, мегакариобластного лейкоза, острого мегакариоцитарного лейкоза, промиелоцитарного лейкоза, лейкоза смешанного происхождения, эритролейкоза, злокачественной лимфомы, ходжкинской лимфомы, неходжкинской лимфомы,лимфобластной Т-клеточной лимфомы, лимфомы Беркитта, фолликулярной лимфомы, нейробластомы,рака мочевого пузыря, уротелиального рака, рака вульвы, рака шейки матки, рака эндометрия, почечного рака, мезотелиомы, рака пищевода, рака слюнной железы, гепатоцеллюлярного рака, рака желудка, рака носоглотки, рака щеки, рака ротовой полости, GIST (желудочно-кишечной стромальной опухоли) и рака яичка. В одном воплощении рак представляет собой лейкоз, например лейкоз, выбранный из острого моноцитарного лейкоза, острого миелогенного лейкоза, хронического миелогенного лейкоза, хронического лимфоцитарного лейкоза и лейкоза смешанного происхождения (MLL). В другом воплощении рак представляет собой срединную NUT-карциному. В другом воплощении рак представляет собой множественную миелому. В другом воплощении рак представляет собой рак легкого, такой как мелкоклеточный рак легкого (SCLC). В другом воплощении рак представляет собой нейробластому. В другом воплощении рак представляет собой лимфому Беркитта. В другом воплощении рак представляет собой рак шейки матки. В другом воплощении рак представляет собой рак пищевода. В другом воплощении рак представляет собой рак яичника. В другом воплощении рак представляет собой рак молочной железы. В другом воплощении рак представляет собой колоректальный рак. В одном воплощении заболевание или состояние, для которых показан ингибитор бромодомена,выбирают из заболеваний, ассоциированных с синдромом системного воспалительного ответа, таких как сепсис, ожоги, панкреатит, обширная травма, кровотечение и ишемия. В этом воплощении ингибитор бромодомена предполагают вводить в момент диагностики для снижения инцидентности: SIRS, возникновения шока, синдрома мультиорганной дисфункции, которые включают возникновение острого повреждения легких, ARDS, острого повреждения почек, печени, сердца или желудочно-кишечного тракта и смертности. В другом воплощении ингибитор бромодомена предполагают вводить до хирургических или других процедур, ассоциированных с высоким риском сепсиса, кровотечения, обширного поражения ткани, SIRS или MODS (синдрома мультиорганной дисфункции). В конкретном воплощении заболевание или состояние, для которых показан ингибитор бромодомена, представляет собой сепсис, септический синдром, септический шок и эндотоксемию. В другом воплощении ингибитор бромодомена показан для лечения острого или хронического панкреатита. В другом воплощении ингибитор бромодомена показан для лечения ожогов. В одном воплощении заболевание или состояние, для которых показан ингибитор бромодомена,выбирают из инфекций и реактиваций простого герпеса, герпетических лихорадок, инфекций и реактиваций опоясывающего герпеса; ветряной оспы, опоясывающего лишая, вируса папилломы человека, вируса иммунодефицита человека (ВИЧ), неоплазии шейки матки, аденовирусных инфекций, включая острое респираторное заболевание, поксвирусных инфекций, таких как коровья оспа и натуральная оспа, и инфекции вирусом африканской лихорадки свиней. В одном конкретном воплощении ингибитор бромодомена показан для лечения кожи или эпителия шейки матки от инфекций вирусом папилломы человека. В одном воплощении ингибитор бромодомена показан для лечения латентной ВИЧ-инфекции. При использовании в данном описании изобретения ссылка на "лечение" конкретного заболевания или состояния включает предупреждение или профилактику такого заболевания или состояния. Термин "заболевания или состояния, для которых показан ингибитор бромодомена" подразумевает включение каждого из указанных выше заболеваний или состояний или их всех. В изобретения также предложен способ ингибирования бромодомена, включающий приведение бромодомена в контакт с соединением формулы (I) или его фармацевтически приемлемой солью. Хотя возможно, что для применения в терапии соединение формулы (I), а также его фармацевтически приемлемые соли можно вводить в виде необработанного химического вещества, обычно активный ингредиент представляют в виде фармацевтической композиции. По этой причине в еще одном аспекте настоящего изобретения предложена фармацевтическая композиция, содержащая соединение формулы (I) или фармацевтически приемлемую соль и один или более чем один фармацевтически приемлемый носитель, разбавитель или эксципиент. В одном воплощении предложена фармацевтическая композиция, содержащая соединение формулы (IA) или фармацевтически приемлемую соль и один или более чем один фармацевтически приемлемый носитель, разбавитель или эксципиент. Соединения формулы (I) и фармацевтически приемлемые соли являются такими, как описано выше. Носитель(и), разбавитель(и) или эксципиент(ы) должны быть приемлемыми в смысле совместимости с другими ингредиентами композиции и не вредными для ее реципиента. В соответствии с другим аспектом изобретения также предложен способ получения фармацевтической композиции, включающий смешивание соединения формулы (I) или его фармацевтически приемлемой соли с одним или более чем одним фармацевтически приемлемым носителем, разбавителем или эксципиентом. Фармацевтическую композицию можно применять в лечении любого из состояний, описанных в данном описании изобретения. Так как соединения формулы (I) предназначены для применения в фармацевтических композициях,легко понять, что каждое из них предпочтительно предложено, по существу, в чистой форме, например, с чистотой по меньшей мере 85%, особенно с чистотой по меньшей мере 98% (проценты даны в виде мас.% в пересчете на общую массу). Фармацевтические композиции могут быть представлены в стандартных лекарственных формах,содержащих предопределенное количество активного ингредиента на стандартную дозу. Предпочти-8 023374 тельными стандартными лекарственными композициями являются композиции, содержащие суточную дозу или субдозу активного ингредиента, или ее подходящую часть. Такие стандартные дозы, следовательно, можно вводить более одного раза в сутки. Предпочтительными стандартными лекарственными композициями являются композиции, содержащие суточную дозу или субдозу (для введения более одного раза в сутки) активного ингредиента, как изложено выше в данном описании изобретения, или ее подходящую часть. Фармацевтические композиции могут быть адаптированы для введения любым подходящим путем,например пероральным (включая трансбуккальный или сублингвальный), ректальным, ингаляционным,интраназальным, местным (включая трансбуккальный, сублингвальный или чрескожный), глазным(включая местный, внутриглазный, субконъюнктивальный, эписклеральный, субтеноновый), вагинальным или парентеральным (включая подкожный, внутримышечный, внутривенный или внутрикожный) путем. Такие композиции могут быть получены любым способом, известным в области фармации, например путем объединения активного ингредиента с носителем(ями) или эксципиентом(ами). В одном воплощении фармацевтическая композиция адаптирована для парентерального введения, в частности для внутривенного введения. В одном воплощении фармацевтическая композиция адаптирована для перорального введения. В одном воплощении фармацевтическая композиция адаптирована для местного введения. Фармацевтические композиции, адаптированные для парентерального введения, включают водные и неводные стерильные растворы для инъекций, которые могут содержать антиоксиданты, буферы, бактериостаты и растворенные вещества, которые приводят композицию в изотоническое состояние с кровью предполагаемого реципиента; и водные и неводные стерильные суспензии, которые могут содержать суспендирующие агенты и загустители. Композиции могут быть представлены в однодозовых или многодозовых контейнерах, например в запаянных ампулах и флаконах, и могут храниться в сублимированном (лиофилизированном) состоянии, требующем только добавления стерильного жидкого носителя,например воды для инъекций, непосредственно перед применением. Приготовленные для немедленного приема растворы и суспензии для инъекций могут быть получены из стерильных порошков, гранул и таблеток. Фармацевтические композиции, адаптированные для перорального введения, могут быть представлены в виде дискретных единиц, таких как капсулы или таблетки; порошки или гранулы; растворы или суспензии в водных или неводных жидкостях; съедобные пены или муссы; или жидкие эмульсии типа масло-в-воде или жидкие эмульсии типа вода-в-масле. Например, для перорального введения в форме таблетки или капсулы активный компонент лекарственного средства может быть объединен с пероральным, нетоксичным фармацевтически приемлемым инертным носителем, таким как этанол, глицерин, вода и тому подобное. Порошки, подходящие для включения в таблетки или капсулы, могут быть приготовлены доведением соединения до подходящего мелкодисперсного размера (например, микронизацией) и смешиванием с приготовленным аналогичным образом фармацевтическим носителем, таким как пищевой углевод, например крахмал или маннит. Также могут присутствовать корригент, консервант, диспергирующий агент и краситель. Капсулы могут быть изготовлены путем приготовления порошковой смеси, как описано выше, и заполнением формованных желатиновых капсул. Глиданты и смазывающие вещества, такие как коллоидный диоксид кремния, тальк, стеарат магния, стеарат кальция или твердый полиэтиленгликоль, могут быть добавлены к порошковой смеси перед операцией заполнения. Разрыхляющий или солюбилизирующий агент, такой как агар-агар, карбонат кальция или карбонат натрия, также могут быть добавлены для улучшения доступности лекарственного средства при проглатывании капсулы. Кроме того, при желании или необходимости, в смесь также могут быть введены подходящие связующие вещества, глиданты, смазывающие вещества, подсластители, корригенты, разрыхляющие агенты и красители. Подходящие связующие вещества включают крахмал, желатин, природные сахара, такие как глюкоза или бета-лактоза, кукурузные подсластители, природные и синтетические смолы, такие как гуммиарабик, трагакант или альгинат натрия, карбоксиметилцеллюлозу, полиэтиленгликоль, воски и тому подобное. Смазывающие вещества, используемые в этих лекарственных формах, включают олеат натрия, стеарат натрия, стеарат магния, бензоат натрия, ацетат натрия, хлорид натрия и тому подобное. Разрыхляющие агенты включают крахмал, метилцеллюлозу, агар, бентонит, ксантановую смолу и тому подобное. Таблетки получают, например, путем приготовления порошковой смеси, гранулирования или комкования, добавления смазывающего вещества и разрыхляющего агента и прессования в таблетки. Порошковую смесь получают смешением подходящим образом измельченного соединения с разбавителем или основой, как описано выше, и возможно, со связующим веществом, таким как карбоксиметилцеллюлоза, альгинат, желатин или поливинилпирролидон, замедлителем растворения, таким как парафин, ускорителем ресорбции, таким как четвертичная соль, и/или агентом абсорбции, таким как бентонит, каолин или дикальцийфосфат. Порошковую смесь можно подвергнуть влажному гранулированию со связующим веществом, таким как сироп, крахмальная паста, клейкое вещество акации или растворами целлюлозных или полимерных веществ, и пропустить через сито. В качестве альтернативы гранулированию порошковую смесь можно пропустить через таблетирующую машину и в результате получать не-9 023374 идеально сформованные комки, разломанные на гранулы. Гранулы можно смазать для предупреждения прилипания к образующим таблетки пуансонам посредством добавления стеариновой кислоты, стеаратной соли, талька или минерального масла. Затем смазанную смесь прессуют в таблетки. Соединения формулы (I) и их фармацевтически приемлемые соли можно также объединять с легкосыпучим инертным носителем и спрессовывать в таблетки непосредственно без проведения стадий гранулирования или комкования. Может быть выполнено прозрачное или непрозрачное защитное покрытие, состоящее из изолирующего покрытия шеллаком, покрытия сахаром или полимерным веществом и полирующего воскового покрытия. К этим покрытиям могут быть добавлены красящие вещества, чтобы различать разные стандартные дозировки. Пероральные жидкости, такие как раствор, сиропы и эликсиры, могут быть приготовлены в стандартной лекарственной форме, так что заданное количество содержит предопределенное количество соединения. Сиропы могут быть приготовлены растворением соединения в подходящим образом ароматизированном водном растворе, в то время как эликсиры приготавливают посредством использования нетоксичного спиртового разбавителя. Суспензии могут быть приготовлены диспергированием соединения в нетоксичном разбавителе. Также можно добавлять солюбилизаторы и эмульгаторы, такие как этоксилированные изостеариловые спирты и полиоксиэтилен-сорбитные эфиры, консерванты, ароматизирующие добавки, такие как масло перечной мяты или природные подсластители, или сахарин или другие искусственные подсластители и тому подобное. Композиции для введения (например, перорального введения) могут быть предназначены для обеспечения модифицированного профиля высвобождения для замедления или иным образом контроля высвобождения терапевтически активного агента. Модифицированный профиль высвобождения терапевтически активного агента можно получать посредством конструирования полимерных матриц, включающих биоразлагаемые/биоразрушаемые полимеры с разным ассортиментом и свойствами (например, поли(этиленвинил)ацетат (EVA), супергидролизованный PVA (поливинилацетат, гидроксиалкилцеллюлоза (НРС), метилцеллюлоза (МС), гидроксипропилметилцеллюлоза (НРМС), поликапролактон, поли(гликолевая) кислота, поли(молочная) кислота, полиангидрид, молекулярных масс полимера, кристалличности полимера, соотношений в сополимере, условий обработки, поверхностной обработки, геометрии, добавления эксципиента и полимерных покрытий, которые улучшают диффузию, эрозию, растворение и осмос лекарственного средства. Когда это целесообразно, композиции со стандартной дозировкой для перорального введения могут быть микроинкапсулированными. Можно получать композицию для пролонгированного или замедленного высвобождения, например, посредством покрытия или включения частиц вещества в полимеры,воск или тому подобное. Соединения формулы (I) и их фармацевтически приемлемые соли также можно вводить в форме липосомных систем доставки, таких как небольшие однослойные везикулы, крупные однослойные везикулы и многослойные везикулы. Липосомы могут быть образованы из различных фосфолипидов, таких как холестерин, стеариламин или фосфатидилхолины. Фармацевтические композиции, адаптированные для местного введения, могут быть приготовлены в виде мазей, кремов, суспензий, эмульсий, лосьонов, порошков, растворов, паст, гелей, пен, спреев, аэрозолей или масел. Такие фармацевтические композиции могут включать традиционные добавки, которые включают, консерванты, растворители для содействия проникновению лекарственного средства,сорастворители, смягчающие вещества, пропелленты, модифицирующие вязкость агенты (гелеобразующие агенты), поверхностно-активные вещества и носители, но не ограничиваются этим. В одном воплощении предложена фармацевтическая композиция, адаптированная для местного введения, содержащая от 0,01 до 10% или от 0,01 до 1% соединения формулы (I) или его фармацевтически приемлемой соли от массы композиции. Для лечения глаза или других наружных тканей, например ротовой полости и кожи, композиции предпочтительно наносят в виде местной мази, крема, геля, спрея или пены. При приготовлении мази активный ингредиент может быть использован либо с парафиновой, либо с водорастворимой основой мази. Альтернативно, активный ингредиент может быть приготовлен в виде крема с основой для крема типа масло-в-воде или с основой типа вода-в-масле. Фармацевтические композиции, адаптированные для местных введений в глаз, включают глазные капли, где активный ингредиент растворен или суспендирован в подходящем носителе, особенно в водном растворителе. Композиции, предназначенные для введения в глаз, имеют офтальмологически совместимые рН и осмоляльность. В композицию по изобретению могут быть включены один или более чем один офтальмологически приемлемый регулирующий рН агент и/или буферизующий агент, включая кислоты, такие как уксусная, борная, лимонная, молочная, фосфорная и соляная кислоты; основания,такие как гидроксид натрия, фосфат натрия, борат натрия, цитрат натрия, ацетат натрия и лактат натрия; и буферы, такие как цитрат/декстроза, бикарбонат натрия и хлорид аммония. Такие кислоты, основания и буферы могут быть включены в количестве, требующемся для поддерживания рН композиции в офтальмологически приемлемом диапазоне. Одна или более чем одна офтальмологически приемлемая соль может быть включена в композицию в количестве, достаточном для доведения осмоляльности композиции до офтальмологически приемлемого диапазона. Такие соли включают соли, содержащие катионы натрия,калия или аммония и анионы хлорид, цитрат, аскорбат, борат, фосфат, бикарбонат, сульфат, тиосульфат или бисульфит. Устройство для глазной доставки может быть сконструировано для контролируемого высвобождения одного или более чем одного терапевтического агента с различными определенными скоростями высвобождения и замедленной кинетикой доз и проницаемостью. Фармацевтические композиции для глазной доставки также включают гелеобразующую in situ водную композицию. Такая композиция содержит гелеобразующий агент в концентрации, эффективной для содействия гелеобразованию при контакте с глазом или со слезной жидкостью. Подходящие гелеобразующие агенты включают термореактивные полимеры, но не ограничиваются ими. Термин "гелеобразующий in situ" при использовании в данном описании изобретения включает не только жидкости с низкой вязкостью, которые образуют гели при контакте с глазом или со слезной жидкостью, но также включает более вязкие жидкости, такие как полужидкие и тиксотропные гели, которые проявляют существенно повышенную вязкость или прочность геля при введении в глаз (см., например, Ludwig (2005) Adv.Drug Deliv. Rev. 3; 57: 1595-639, включенную в данное описание изобретения посредством ссылки в части описания примеров полимеров для применения в глазной доставке лекарственного средства). Лекарственные формы для назального или ингаляционного введения могут быть легко приготовлены в виде аэрозолей, растворов, суспензий, гелей или сухих порошков. Для композиций, подходящих и/или адаптированных для ингаляционного введения, предпочтительно, чтобы соединение формулы (I) или его фармацевтически приемлемая соль находились в форме измельченных частиц, полученной, например, посредством микронизации. Предпочтительный размер частиц измельченного (например микронизированного) соединения или соли определяют по величинеD50 от примерно 0,5 до примерно 10 микрон (как, например, при измерении с использованием лазерной дифракции). Аэрозольные композиции, например, для ингаляционного введения, могут содержать раствор или тонкую суспензию активного вещества в фармацевтически приемлемом водном или неводном растворителе. Аэрозольные композиции могут быть представлены в одно- или многодозовых количествах в стерильной форме в герметизированном контейнере, который может иметь форму картриджа или модуля для повторного заполнения для использования с распылительным устройством или ингалятором. Альтернативно, герметизированный контейнер может представлять собой одноразовое дозирующее устройство, которое выбрасывают сразу после использования его содержимого, такое как однодозовый назальный ингалятор или аэрозольный дозатор, оборудованный мерным клапаном (ингалятор отмеренной дозы). Когда лекарственная форма содержит аэрозольный дозатор, он предпочтительно содержит подходящий пропеллент под давлением, такой как сжатый воздух, диоксид углерода или органический пропеллент, такой как гидрофтороуглерод (HFC). Подходящие пропелленты HFC включают 1,1,1,2,3,3,3 гептафторпропан и 1,1,1,2-тетрафторэтан. Аэрозольные лекарственные формы могут также представлять собой нагнетательный распылитель. Аэрозоль под давлением может содержать раствор или суспензию активного соединения. Это может потребовать включения дополнительных эксципиентов, например сорастворителей и/или поверхностно-активных веществ, для улучшения дисперсионных характеристик и гомогенности суспензионных композиций. Композиции раствора также могут требовать добавления сорастворителей, таких как этанол. Для фармацевтических композиций, подходящих и/или адаптированных для ингаляционного введения, фармацевтическая композиция может представлять собой сухую порошковую ингаляционную композицию. Такая композиция может содержать порошковую основу, такую как лактоза, глюкоза, трегалоза, маннит или крахмал, соединение формулы (I) или его фармацевтически приемлемую соль (предпочтительно в измельченной форме, например в микронизированной форме) и, возможно, модификатор характеристик, такой как L-лейцин или другая аминокислота и/или металлическая соль стеариновой кислоты, такая как стеарат магния или кальция. Предпочтительно сухая порошковая ингаляционная композиция содержит сухую порошковую смесь лактозы, например моногидрата лактозы, и соединения формулы (I) или его соли. Такие композиции могут быть введены пациенту при помощи подходящего устройства, такого как устройство DISKUS, поставляемое GlaxoSmithKline, которое описано, например, вGB 2242134 А. Соединения формулы (I) и их фармацевтически приемлемые соли могут быть приготовлены в виде жидкой композиции для доставки из жидкостного дозатора, например жидкостного дозатора, имеющего дозирующую насадку или дозирующее отверстие, через которые отмеренная доза жидкой композиции дозируется при приложении пользователем силы к нагнетающему механизму жидкостного дозатора. Такие жидкостные дозаторы обычно снабжены резервуаром для многократных отмериваемых доз жидкой композиции, причем дозы дозируются при последовательных приведениях насоса в действие. Дозирующая насадка или отверстие могут быть сконструированы для введения в ноздри пользователя для дозирования жидкой композиции в носовую полость в виде спрея. Жидкостный дозатор вышеупомянутого типа описан и проиллюстрирован в WO-A-2005/044354. Терапевтически эффективное количество соединения формулы (I) или его фармацевтически приемлемой соли будет зависеть от ряда факторов, включая, например, возраст и массу субъекта, точное состояние, требующее лечения, и его тяжесть, природу композиции и путь введения, и, в конечном счете,будет определяться лечащим врачом или ветеринаром. В фармацевтической композиции каждая дозировочная единица для перорального или парентерального введения предпочтительно содержит от 0,01 до 3000 мг, более предпочтительно от 0,5 до 1000 мг соединения формулы (I) или его фармацевтически приемлемой соли в расчете на свободное основание. Каждая дозировочная единица для назального или ингаляционного введения предпочтительно содержит от 0,001 до 50 мг, более предпочтительно от 0,01 до 5 мг соединения формулы (I) или его фармацевтически приемлемой соли в расчете на свободное основание. Фармацевтически приемлемые соединения формулы (I) и их фармацевтически приемлемые соли могут быть введены в суточной дозе (для взрослого пациента), например в пероральной или парентеральной дозе от 0,01 до 3000 мг в сутки, от 0,5 до 1000 мг в сутки или от 100 до 2500 мг в сутки или в назальной или ингаляционной дозе от 0,001 до 50 мг в сутки или от 0,01 до 5 мг в сутки соединения формулы (I) или его фармацевтически приемлемой соли в расчете на свободное основание. Это количество можно вводить однократной дозой один раз в сутки или чаще несколькими (например, двумя, тремя, четырьмя, пятью или шестью) субдозами в сутки, но так, чтобы суммарная суточная доза была той же. Эффективное количество его соли может быть определено как пропорциональное эффективному количеству самого соединения формулы (I). Соединения формулы (I) и их фармацевтически приемлемые соли можно использовать сами по себе или в комбинации с другими терапевтическими агентами. Комбинированные терапии по настоящему изобретению включают, таким образом, введение по меньшей мере одного соединения формулы (I) или его фармацевтически приемлемой соли и использование по меньшей мере одного другого терапевтически активного агента. Предпочтительно комбинированные терапии по настоящему изобретению включают введение по меньшей мере одного соединения формулы (I) или его фармацевтически приемлемой соли и по меньшей мере одного другого терапевтически активного агента. Соединение(я) формулы (I) и его(их) фармацевтически приемлемая(ые) соль(и) и другой(ие) терапевтически активный(ые) агент(ы) могут быть введены вместе в единой фармацевтической композиции или раздельно, и при введении раздельно введение можно проводить одновременно или последовательно в любом порядке. Количества соединения(ий) формулы (I) и его(их) фармацевтически приемлемой(ых) соли(ей) и другого(их) терапевтически активного(ых) агента(ов) и относительный хронометраж введения выбирают так, чтобы достичь желаемого комбинированного терапевтического эффекта. Таким образом, в еще одном аспекте предложена комбинация, содержащая соединение формулы (I) или его фармацевтически приемлемую соль и по меньшей мере один другой терапевтически активный агент. Таким образом, в одном аспекте соединение формулы (I) или его фармацевтически приемлемая соль и фармацевтические композиции, содержащие соединение формулы (I) или его фармацевтически приемлемую соль, согласно изобретению могут быть использованы в комбинации с одним или более чем одним другим терапевтическим агентом, например, выбранным из антибиотиков, противовирусных средств, глюкокортикостероидов, мускариновых антагонистов, агонистов бета-2 и аналогов витаминаD3, или включать их. В еще одном воплощении соединение формулы (I) или его фармацевтически приемлемая соль могут быть использованы в комбинации с дополнительным терапевтическим агентом, подходящим для лечения рака. Примеры таких дополнительных терапевтических агентов описаны в CancerPrinciples and Practice of Oncology by V.T. Devita and S. Hellman (editors), 6th edition (2001), Lippincott WilliamsWilkins Publishers. Средний специалист в данной области техники смог бы понять, какие комбинации агентов будут полезны, на основе конкретных характеристик лекарственных средств и вовлеченного рака. Дополнительные терапевтические агенты для применения в комбинации с соединением формулы (I) или его фармацевтически приемлемой солью включают антимикротрубочковые агенты (такие как дитерпеноиды и алкалоиды барвинка); платиновые координационные комплексы; алкилирующие агенты (такие как азотистые аналоги иприта, оксазафосфорины, алкилсульфонаты, нитрозомочевины и триазены); антибиотические агенты (такие как антрациклины, актиномицины и блеомицины); ингибиторы топоизомеразы II (такие как эпиподофиллотоксины); антиметаболиты (такие как аналоги пурина и пиримидина и антифолатные соединения); ингибиторы топоизомеразы I (такие как камптотецины; гормоны и аналоги гормонов); ингибиторы пути сигнальной трансдукции (такие как ингибиторы тирозинового рецептора); ингибиторы ангиогенеза, опосредованного нерецепторными тирозинкиназами; иммунотерапевтические агента; проапоптозные агенты; эпигенетические или транскрипционные модуляторы(такие как ингибиторы гистондеацетилазы) и ингибиторы передачи сигнала в клеточном цикле, но не ограничиваются ими. Понятно, что если соединение формулы (I) или его фармацевтически приемлемую соль вводят в комбинации с другими терапевтическими агентами, обычно вводимыми посредством ингаляционного,внутривенного, перорального или интраназального пути, то получаемую фармацевтическую композицию можно вводить теми же самыми путями. Альтернативно, отдельные компоненты композиции можно вводить различными путями. Одно воплощение изобретения охватывает комбинации, содержащие один или два других терапевтических агента. Специалисту в данной области техники будет ясно, что, когда это целесообразно, другой(ие) терапевтический(ие) ингредиент(ы) может(гут) быть использован(ы) в виде солей, например в виде солей щелочных металлов или аминов или в виде солей присоединения кислоты, или пролекарств, или в виде сложных эфиров, например эфиров низших алкилов, или в виде сольватов, например гидратов, для оптимизации активности и/или стабильности и/или физических характеристик терапевтического ингредиента, таких как растворимость. Также ясно, что, когда это целесообразно, терапевтические ингредиенты можно использовать в оптически чистой форме. Комбинации, указанные выше, можно легко представлять для применения в форме фармацевтической композиции, и, таким образом, фармацевтические композиции, содержащие комбинацию, как определено выше, совместно с фармацевтически приемлемым разбавителем или носителем, представляют собой еще один аспект изобретения. Соединения формулы (I) и их фармацевтически приемлемые соли можно получать способами, описанными ниже или аналогичными способами. Таким образом, следующие промежуточные соединения и примеры служат для иллюстрации получения соединений формулы (I) и их фармацевтически приемлемых солей и не должны рассматриваться как ограничивающие объем изобретения каким-либо образом. Общие подробности экспериментов Все указанные значения температур представлены в С. Названия следующих соединений получали, используя программу присвоения названий соединениям "ACD Name Pro 6 02" или Chem Draw Ultra 12.0. Сокращения Методика LCMS Способ с формиатом Условия для LC Анализ UPLC выполняли на колонке Acquity UPLC ВЕН С 18 (50 мм 2,1 мм в.д. (внутренний диаметр) 1,7 мкм диаметр частиц наполнителя) при 40 С. Использованные растворители А = 0,1% об./об. раствор муравьиной кислоты в воде; В = 0,1% об./об. раствор муравьиной кислоты в ацетонитриле. УФ-обнаружение представляло собой суммарный сигнал с длиной волны от 210 до 350 нм. Условия для MS ЯМР Спектры прогоняли на аппарате для ЯМР, действующем при 400 МГц, либо при 302 K, либо для спектров VT при 392-393 K. Промежуточное соединение 1 7-(3,5-Диметилизоксазол-4-ил)-6-метокси-N-(1-метоксипропан-2-ил)-3-нитрохинолин-4-амин(20 г, 59,9 ммоль) в 1,4-диоксане (200 мл) добавляли 1-метоксипропан-2-амин (31,6 мл, 300 ммоль) и реакционную смесь нагревали при 70 С в течение 1,5 ч. Растворитель удаляли при пониженном давлении и полученное твердое вещество распределяли между этилацетатом (3750 мл) и водой (750 мл). Органические слои объединяли, сушили (гидрофобная фритта) и упаривали при пониженном давлении с получением 7-(3,5-диметилизоксазол-4-ил)-6-метокси-N-(1-метоксипропан-2-ил)-3-нитрохинолин-4-амина(25,8 г), который использовали без дополнительной очистки на следующей стадии. ЯМР (D6-DMSO): H 9.03 (s, 1H), 8.63 (d, 1H), 7.83 (s, 1H), 7.81 (s, 1H), 4.39 (m, 1H), 3.97 (s, 3H),3.54 (m, 2H), 3.27 (s, 3H), 2.34 (s, 3H), 2.15 (s, 3H), 1.41 (d, 3H). 7-(3,5-Диметилизоксазол-4-ил)-6-метокси-N-(1-метоксипропан-2-ил)-3-нитрохинолин-4-амин (для получения см. промежуточное соединение 1) (25,8 г, 66,8 ммоль) растворяли в смеси этилацетата (1000 мл) и DMSO (50 мл) и раствор гидрировали, используя проточный аппарат для гидрирования (H-cube)CatCart 70. Картридж с катализатором заменяли всякий раз, когда он засорялся. Реакционную смесь упаривали при пониженном давлении и распределяли между этилацетатом (750 мл) и водой (3750 мл). Водные слои объединяли и экстрагировали этилацетатом (750 мл). Органические слои объединяли, сушили (гидрофобная фритта) и упаривали при пониженном давлении. Остаток растворяли в DCM и наносили на картридж с диоксидом кремния (100 г). Картридж элюировали 2 М аммиака в градиенте метанол/DCM (0-4%). Соответствующие фракции объединяли и упаривали при пониженном давлении с получением 7-(3,5-диметилизоксазол-4-ил)-6-метокси-N4-(1-метоксипропан-2-ил)хинолин-3,4-диамина и извлеченного 7-(3,5-диметилизоксазол-4-ил)-6-метокси-N-(1-метоксипропан-2-ил)-3-нитрохинолин-4 амина (6,5 г). Извлеченное исходное вещество растворяли в этилацетате (250 мл) и раствор гидрировали, используя проточный аппарат для гидрирования (H-cube) (параметры: 20 С, 1 бар (0,1 МПа), скорость потока 1 мл/мин) и картридж с катализатором 10% Pd/C CatCart 70. Реакционную смесь упаривали при пониженном давлении и остаток растворяли в DCM и наносили на картридж с диоксидом кремния (100 г). Картридж элюировали с помощью 2 М аммиака в градиенте метанол/DCM (0-4%). Соответствующие фракции объединяли и упаривали при пониженном давлении с получением 7-(3,5-диметилизоксазол-4 ил)-6-метокси-N4-(1-метоксипропан-2-ил)хинолин-3,4-диамина. Порции продукта объединяли с получением 7-(3,5-диметилизоксазол-4-ил)-6-метокси-N4-(1-метоксипропан-2-ил)хинолин-3,4-диамина (15,6 г, 43,8 ммоль, выход 65,6%) в виде липкой темно-коричневой смолы. ЯМР (D6-DMSO): Н 8.29 (s, 1 Н), 7.55 (s, 1H), 7.45 (s, 1H), 5.13 (s, 2H), 4.48 (d, 1 Н), 3.88 (s, 3H), 3.54(для получения см. промежуточное соединение 2) (9,1 г) в DCM (300 мл) обрабатывали пиридином (30 мл) и тетрагидро-2 Н-пиран-4-карбонилхлоридом (5,0 мл) и раствор перемешивали в атмосфере азота при температуре окружающей среды в течение 3,5 ч и потом оставляли стоять в течение ночи (температура окружающей среды, атмосфера азота). Летучие компоненты выпаривали в вакууме и остаток распределяли между DCM и водой. Органическую фазу дважды промывали водой и объединенную водную фазу экстрагировали с помощью DCM. Объединенные органические фазы промывали рассолом, сушили (гидрофобная фритта) и упаривали досуха в вакууме. Объединенную водную фазу, включая промывку рассолом (рН 4), подщелачивали твердым гидрокарбонатом натрия и водную фазу экстрагировали с помощью DCM 2. Органические фракции сушили (гидрофобная фритта), объединяли с предыдущим веществом и упаривали досуха в вакууме с получением коричневой пены. Эту пену дополнительно сушили в вакууме, растирали с диэтиловым эфиром, суспензию охлаждали (баня лед/вода) и твердое вещество выделяли фильтрацией. Твердое вещество промывали небольшим количеством диэтилового эфира и сушили на воздухе с получением N-(7-(3,5-диметилизоксазол-4-ил)-6-метокси-4-1-метоксипропан-2-ил)амино)хинолин-3-ил)тетрагидро-2 Н-пиран-4-карбоксамида в виде бежевого твердого вещества (11,95 г,100%). ЯМР (D6-DMSO): H 9.49 (s, 1H), 8.38 (s, 1 Н), 7.70 (s, 1H), 7.63 (s, 1H), 5.33 (d, 1 Н), 3.96-3.90 (m,6 Н), 3.43-3.28 (m частично скрыт водой, 7 Н), 2.69 (m, 1H), 2.32 (s, 3H), 2.12 (s, 3H), 1.81-1.67 (m, 4H), 1.19 Суспензию N-(7-(3,5-диметилизоксазол-4-ил)-6-метокси-4-1-метоксипропан-2-ил)амино)хинолин 3-ил)тетрагидро-2 Н-пиран-4-карбоксамида (для получения см. промежуточное соединение 3) (29,4 г, 62,7 ммоль) в уксусной кислоте (250 мл, 62,7 ммоль) нагревали при 120 С в течение 2 ч. Добавляли молекулярные сита 3(20 г) и нагревание продолжали в течение 3,5 ч. Дополнительно добавляли молекулярные сита 3(20 г, высушенные в печи) и продолжали нагревание в течение ночи. Дополнительно добавляли молекулярные сита 3(20 г, высушенные в печи) и нагревание продолжали в течение следующих 24 ч. Реакционную смесь оставляли охлаждаться до комнатной температуры и твердое вещество удаляли фильтрацией. Остаток и фильтрат объединяли и упаривали при пониженном давлении. Добавляли воду(3 л) и полученную суспензию нейтрализовали медленным добавлением твердого гидрокарбоната натрия. Водную суспензию экстрагировали с помощью DCM (31 л) и органические фазы объединяли,- 15023374 сушили (гидрофобная фритта) и упаривали при пониженном давлении с получением коричневой смолы. Смолу растворяли при нагревании и обрабатывании ультразвуком в минимальном количествеDCM. Раствор наносили на колонку с диоксидом кремния (750 г), который предварительно был смоченDCM. Колонку элюировали с градиентом (2 М аммиака в метаноле, метанол (3:1/DCM (0-8%). Фракции продукта объединяли и упаривали досуха при пониженном давлении с получением кремовой пены/стекла (13,027 г). Смешанные фракции продукта объединяли и упаривали досуха при пониженном давлении с получением густого желтого масла. Это масло растворяли в диэтиловом эфире и летучие компоненты выпаривали в вакууме с получением желтого твердого вещества. Твердое вещество растирали с диэтиловым эфиром, твердое вещество выделяли фильтрацией и промывали диэтиловым эфиром ( 2) с получением 4-(8-метокси-1-(1-метоксипропан-2-ил)-2-(тетрагидро-2 Н-пиран-4-ил)-1 Н-имидазо[4,5-с]хинолин-7-ил)3,5-диметилизоксазола. Кремовую пену/стекло растирали с диэтиловым эфиром/этилацетатом, смесь упаривали досуха при пониженном давлении и повторяли растирание с диэтиловым эфиром. Фракцию предыдущего продукта добавляли в суспензию и смесь выдерживали в течение ночи. Твердое вещество выделяли фильтрацией и промывали диэтиловым эфиром с получением 4-(8-метокси-1-(1-метоксипропан-2-ил)-2-(тетрагидро-2 Нпиран-4-ил)-1 Н-имидазо[4,5-с]хинолин-7-ил)-3,5-диметилизоксазола в виде кремового твердого вещества. Твердое вещество сушили в вакууме и повторно растирали с диэтиловым эфиром при перемешивании в течение 30 мин. Твердое вещество выделяли фильтрацией и промывали диэтиловым эфиром. Твердое вещество сушили в вакууме с получением 4-(8-метокси-1-(1-метоксипропан-2-ил)-2-(тетрагидро-2 Нпиран-4-ил)-1 Н-имидазо[4,5-с]хинолин-7-ил)-3,5-диметилизоксазола в виде белого твердого веществаN-(7-(3,5-диметилизоксазол-4-ил)-6-метокси-4-1-метоксипропан-2-ил)амино)хинолин-3 ил)тетрагидро-2 Н-пиран-4-карбоксамида (для получения см. промежуточное соединение 3) (11,95 г, 25,5 ммоль) и п-толуолсульфоновой кислоты (1,2 г, 25,5 ммоль) в толуоле (250 мл) нагревали в атмосфере азота с обратным холодильником, используя аппарат Дина-Старка в течение 3 суток. Добавляли дополнительно п-толуолсульфоновую кислоту (0,2 г, 4,3 ммоль) и нагревание продолжали в течение ночи. Добавляли воду (750 мл) и смесь подщелачивали до рН 8, используя насыщенный водный раствор гидрокарбоната натрия. Водную фракцию экстрагировали этилацетатом (3750 мл), органические слои объединяли, сушили (гидрофобная фритта) и упаривали при пониженном давлении. Полученное твердое вещество растирали в эфире (200 мл), обрабатывали ультразвуком в течение короткого периода времени. Большая часть твердого вещества представляла собой мелкодисперсный порошок. Мелкодисперсный порошок, суспендированный в эфире, декантировали, твердое вещество выделяли фильтрацией и сушили в вакуумной печи с получением неочищенного 4-(8-метокси-1-(1-метоксипропан-2-ил)-2-(тетрагидро 2 Н-пиран-4-ил)-1 Н-имидазо[4,5-с]хинолин-7-ил)-3,5-диметилизоксазола (6,3 г). Это вещество растирали в эфире (100 мл), обрабатывали ультразвуком в течение короткого периода времени и оставляли в течение ночи при комнатной температуре. Твердое вещество выделяли фильтрацией и сушили в вакуумной печи с получением 4-(8-метокси-1-(1-метоксипропан-2-ил)-2-(тетрагидро-2 Н-пиран-4-ил)-1 Н-имидазо[4,5-с]хинолин-7-ил)-3,5-диметилизоксазола (4,9 г, 10,88 ммоль, выход 42,6%). LCMS (формиат): Rt 0,75 мин, МН+451. Скомкованное твердое вещество, оставшееся от декантирования, растирали в эфире (100 мл), обрабатывали ультразвуком в течение 15 мин и оставляли при комнатной температуре в течение 3 суток. Твердое вещество выделяли фильтрацией и сушили в вакуумной печи с получением 4-(8-метокси-1-(1 метоксипропан-2-ил)-2-(тетрагидро-2 Н-пиран-4-ил)-1 Н-имидазо[4,5-с]хинолин-7-ил)-3,5-диметилизоксазола (3,6 г, 7,99 ммоль, выход 31,3%). LCMS (формиат): Rt 0,76 мин, МН+ 451. Пример 2 4-(8-Метокси-1-R)-1-метоксипропан-2-ил)-2-(тетрагидро-2 Н-пиран-4-ил)-1 Н-имидазо[4,5-с]хинолин-7-ил)-3,5-диметилизоксазол имидазо[4,5-с]хинолин-7-ил)-3,5-диметилизоксазола выполняли, используя следующие условия: колонка: Chiralpak AD-H (25030 мм, 5 микрон) [ADH10029-01]; скорость потока: 45 мл/мин; обнаружение: УФ DAD (диодно-матричный детектор) (300 нм (ширина полосы пропускания 180 нм, эталон 550 нм (ширина полосы пропускания 100 нм; подвижная фаза А: н-гексан (10 мл изопропиламина на Winchester (2,5 л; подвижная фаза В: этанол (10 мл изопропиламина на Winchester (2,5 л; изократическая система - 85:15 подвижная фаза А:В; время прогона - приблизительно 35 мин. 4-(8-Метокси-1-(1-метоксипропан-2-ил)-2-(тетрагидро-2 Н-пиран-4-ил)-1 Н-имидазо[4,5-с]хинолин 7-ил)-3,5-диметилизоксазол (500 мг) суспендировали в этаноле и этиленгликоле (10 мл:5 мл) и потом обрабатывали ультразвуком и нагревали для облегчения растворения. Потом добавляли изопропиламин(1 мл). Этот рабочий раствор нагревали на плите (60 С) при сохранении продолжающейся очистки в растворе. Ввод пробы (1,5 мл) выполняли, используя автоматический пробоотборник. Фракции собирали на основе заранее установленного времени между 28 и 35 мин, используя коллектор фракций с воронкой. Растворы объединенных фракций упаривали досуха, используя роторный испаритель (температура бани 30 С) и остаток переносили в тарированный 20-мл стеклянный флакон, используя этанол (приблизительно 12 мл). Этанол выпаривали в потоке газа азота (комнатная температура). 4-(8-Метокси-1-R)-1-метоксипропан-2-ил)-2-(тетрагидро-2 Н-пиран-4-ил)-1 Н-имидазо[4,5-с]хинолин-7-ил)-3,5-диметилизоксазол (1,16 г), выделенный, используя указанный выше способ, растирали с диэтиловым эфиром (3 мл) в течение 4 ч при температуре окружающей среды. Твердое вещество выделяли фильтрацией, промывали диэтиловым эфиром и сушили в вакууме с получением 4-(8-метокси-1R)-1-метоксипропан-2-ил)-2-(тетрагидро-2 Н-пиран-4-ил)-1 Н-имидазо[4,5-с]хинолин-7-ил)-3,5-диметилизоксазола в виде кремового твердого вещества (0,96 г). ЯМР VT (D6-DMSO): H 9.03 (s, 1 Н), 7.98 (s, 1 Н), 7.74 (s, 1H), 5.43 (m, 1H), 4.13 (m, 1 Н), 4.04-4.00 Анализ хиральности 4-(8-метокси-1-(1-метоксипропан-2-ил)-2-(тетрагидро-2 Н-пиран-4-ил)-1 Нимидазо[4,5-с]хинолин-7-ил)-3,5-диметилизоксазола выполняли, используя следующие условия: колонка: Chiralpak AD-H (25030 мм, 5 микрон) [ADH10029-01]; скорость потока: 45 мл/мин; обнаружение: УФ DAD (300 нм (ширина полосы пропускания 180 нм, эталон 550 нм (ширина полосы пропускания 100 нм; подвижная фаза А: гептан; подвижная фаза В: этанол; изократическая система - 85:15 подвижная фаза А:В; время прогона - приблизительно 35 мин. 4-(8-Метокси-1-(1-метоксипропан-2-ил)-2-(тетрагидро-2 Н-пиран-4-ил)-1 Н-имидазо[4,5-с]хинолин 7-ил)-3,5-диметилизоксазол (85 мг) растворяли в этаноле (приблизительно 4 мл) при нагревании и обрабатывании ультразвуком. Ввод пробы (400 мкл) выполняли посредством пластмассового шприца. Собирали фракции между 24 и 26,5 мин и растворы объединенных фракций упаривали досуха, используя роторный испаритель (температура бани 30 С). Остаток переносили в тарированный 20-мл стеклянный флакон, используя этанол (приблизительно 12 мл). Потом растворитель удаляли выпариванием в потоке газа азота (комнатная температура) с получением 4-(8-метокси-1-S)-1-метоксипропан-2-ил)-2-(тетрагидро-2 Н-пиран-4-ил)-1 Н-имидазо[4,5-с]хинолин-7-ил)-3,5-диметилизоксазола (38 мг). ЯМР VT (D6-DMSO): H 9.00 (s, 1H), 7.95 (s, 1H), 7.72 (s, 1H), 5.41 (m, 1H), 4.10 (m, 1 Н), 4.03-3.97 Получение кристаллической формы 4-(8-метокси-1-R)-1-метоксипропан-2-ил)-2-(тетрагидро-2 Нпиран-4-ил)-1 Н-имидазо[4,5-с]хинолин-7-ил)-3,5-диметилизоксазола (безводная форма 1) 4-(8-Метокси-1-R)-1-метоксипропан-2-ил)-2-(тетрагидро-2 Н-пиран-4-ил)-1 Н-имидазо[4,5-с]хинолин-7-ил)-3,5-диметилизоксазол (для получения см. пример 2 или реакционную схему 1; 18,87 г) растворяли в изопропилацетате (95 мл) с обратным холодильником и потом оставляли охлаждаться до 20 С в течение 1 ч. Потом добавляли циклогексан (190 мл) в течение 1 ч и полученную суспензию выдерживали в течение 1 ч. Затем суспензию фильтровали и промывали при помощи смеси 2:1 циклогексан:изопропилацетат (30 мл) и затем циклогексана (30 мл) перед сушкой под тягой. Затем осадок сушили в печи в течение ночи при 40 С в вакууме. XRPD этого вещества была зарегистрирована (см. пример 9). Пример 5 Получение кристаллической формы 4-(8-метокси-1-R)-1-метоксипропан-2-ил)-2-(тетрагидро-2 Нпиран-4-ил)-1 Н-имидазо[4,5-с]хинолин-7-ил)-3,5-диметилизоксазола (безводная форма 2) Предварительно смешанный раствор изопропилацетата (211 мл) и циклогексана (422 мл) добавляли к 4-(8-метокси-1-(R)-1-метоксипропан-2-ил)-2-(тетрагидро-2 Н-пиран-4-ил)-1 Н-имидазо[4,5-с]хинолин-7 ил)-3,5-диметилизоксазолу (для получения см. пример 2 или реакционную схему 1, 42,19 г, 94 ммоль). Полученную суспензию перемешивали в течение 24 ч, фильтровали, полученное твердое вещество промывали (1 циклогексан:изопропилацетат (2:1, 180 мл), 1 циклогексан (180 мл), 1 ТВМЕ (180 мл,сушили под тягой досуха и сушили в вакууме при 40 С до постоянной массы, получая почти белое твердое вещество (выход 90%). XRPD этого вещества была зарегистрирована (см. пример 9). Пример 6 Получение кристаллической формы 4-(8-метокси-1-R)-1-метоксипропан-2-ил)-2-(тетрагидро-2 Нпиран-4-ил)-1 Н-имидазо[4,5-с]хинолин-7-ил)-3,5-диметилизоксазола (гидрат) Предварительно смешанный раствор этанола (2,000 мл) и воды (8,00 мл) добавляли к 4-(8-метокси 1-R)-1-метоксипропан-2-ил)-2-(тетрагидро-2 Н-пиран-4-ил)-1 Н-имидазо[4,5-с]хинолин-7-ил)-3,5-диметилизоксазолу (для получения см. пример 2 или реакционную схему 1, 1,00 г, 2,220 ммоль) и полученную суспензию перемешивали в течение ночи. Суспензию фильтровали, промывали (2 вода, 2 ТВМЕ),сушили под тягой досуха и сушили в вакууме при 40 С с получением белого порошка. XRPD этого вещества была зарегистрирована (см. пример 9). Пример 7 Получение кристаллической формы 4-(8-метокси-1-R)-1-метоксипропан-2-ил)-2-(тетрагидро-2 Нпиран-4-ил)-1 Н-имидазо[4,5-с]хинолин-7-ил)-3,5-диметилизоксазола (безводная форма 3) Гидрат 4-(8-метокси-1-R)-1-метоксипропан-2-ил)-2-(тетрагидро-2 Н-пиран-4-ил)-1 Н-имидазо[4,5 с]хинолин-7-ил)-3,5-диметилизоксазола (для получения см. пример 6; 860 мг, 1,835 ммоль) нагревали при 135 С при 9 мбар (0,9 кПа) в течение ночи. XRPD этого вещества была зарегистрирована (см. пример 9). Пример 8 Получение гидрохлорида 4-(8-метокси-1-R)-1-метоксипропан-2-ил)-2-(тетрагидро-2 Н-пиран-4 ил)-1 Н-имидазо[4,5-с]хинолин-7-ил)-3,5-диметилизоксазола (гидрохлорид) В первом реакторе раствор 7-(3,5-диметилизоксазол-4-ил)-6-метокси-4-(N-R)-1-метоксипропан-2 ил)тетрагидро-2 Н-пиран-4-карбоксамидо)хинолин-3-карбоновой кислоты (для получения см. схему 1; 23,3 кг) в DCM (113 кг) и DIPEA (3,8 кг) объединяли с ацетонитрилом (61,6 кг) и диизопропиламином(12,8 кг), смесь охлаждали до температуры от -10 до -3 С. Добавляли дифенилфосфорилазид (19,0 кг),поддерживая температуру от -10 до -3 С, и реакционную смесь перемешивали при этой температуре. Добавляли дополнительно дифенилфосфорилазид (2,4 кг), поддерживая температуру от -10 до -3 С, и реакционную смесь перемешивали при этой температуре с образованием первого раствора. Во втором реакторе диметилформамид (144 кг) и воду (72 кг) нагревали до 90-100 С. Добавляли первый раствор, поддерживая температуру 85-100 С, первый реактор ополаскивали ацетонитрилом (7 кг) и промывной раствор добавляли во второй реактор, поддерживая температуру 85-100 С. Смесь перемешивали при этой температуре и потом охлаждали до 20-30 С. Регулировали рН до 10 при помощи 30 мас.% водного раствора гидроксида натрия (10,4 кг), поддерживая температуру 20-30 С. Загружали дихлорметан (315 кг) и воду (351 кг), слои разделяли и водный слой экстрагировали дихлорметаном (315 кг). Объединенные органические слои промывали водой (234 кг). Загружали воду (234 кг) к органическому слою, смесь перемешивали, добавляли хлорид натрия (80 кг) и слои разделялись. Органический слой концентрировали при пониженном давлении и температуре ниже 30 С, загружали метанол (201 кг) и раствор концентрировали при пониженном давлении и температуре ниже 50 С. Загружали дополнительно метанол (199 кг) и 4 М соляную кислоту в метаноле (68,5 кг), поддерживая температуру 20-30 С,и реакционную смесь перемешивали при этой температуре. Смесь концентрировали при пониженном давлении и температуре ниже 50 С и загружали ацетонитрил (104 кг). Смесь концентрировали при пониженном давлении и температуре ниже 50 С и загружали дополнительно ацетонитрил (97 кг). Смесь концентрировали при пониженном давлении и температуре ниже 50 С и загружали дополнительно ацетонитрил (116 кг). Смесь упаривали при пониженном давлении и температуре ниже 50 С и загружали дополнительно ацетонитрил (103,0 кг). Смесь охлаждали до температуры -5-0 С и перемешивали при этой температуре. Суспензию центрифугировали двумя порциями, каждую порцию промывали ацетонитрилом (9 кг) с получением указанного в заголовке соединения (33,40 кг, чистота 99,3%) в виде влажного осадка. Пример 9 Исследования дифракции рентгеновских лучей на порошке (XRPD) кристаллических форм 4-(8 метокси-1-R)-1-метоксипропан-2-ил)-2-(тетрагидро-2 Н-пиран-4-ил)-1 Н-имидазо[4,5-с]хинолин-7-ил)3,5-диметилизоксазола в виде свободного основания и в виде соли гидрохлорида Данные получали на порошковом дифрактометре PANalytical X'Pert Pro, модель PW3040/60, используя детектор X'Celerator. Условиями сбора данных были: излучение: Cu K, напряжение генератора: 40 кВ, электрический ток генератора: 45 мА, начальный угол: 2,0 2, конечный угол: 40,0 2, величина шага: 0,0167 2, время на стадию: 31,75 с. Образец получали посредством установления нескольких миллиграммов образца на планшет из кремниевой пластинки (нулевой уровень), получая тонкий слой порошка. Положения пиков измеряли, используя программное обеспечение Highscore. Предел погрешности равен приблизительно 0,1 2 для каждого из распределений пиков. Интенсивности пиков могут варьировать от образца к образцу вследствие преимущественной ориентации. В табл. 1 показаны положения характеристических пиков XRPD и межатомные расстояния для кристаллической формы соединения 4-(8-метокси-1-R)-1-метоксипропан-2-ил)-2-(тетрагидро-2 Н-пиран-4 ил)-1 Н-имидазо[4,5-с]хинолин-7-ил)-3,5-диметилизоксазола в виде свободного основания и в виде соли гидрохлорида. Таблица 1 Методики биологических тестирований Соединения формулы (I) могут быть испытаны в одном или более чем одном из следующих анализов. Анализ резонансного переноса энергии флюоресценции с временным разрешением (TR-FRET) Связывание соединений формулы (I) с бромодоменами BRD2, BRD3 и BRD4 оценивали, используя анализ резонансного переноса энергии флюоресценции с временным разрешением, в котором измеряется связывание ацетилированного гистонового пептида с бромодоменовым белком. Бромодоменовый белок, гистоновый пептид и испытываемое соединение с изменяемой концентрацией инкубируют вместе до достижения термодинамического равновесия. Анализ конфигурируют так,что в отсутствие испытываемого соединения бромодомен и пептид связываются существенно (30%), а в присутствии достаточной концентрации сильного ингибитора это взаимодействие разрушается, приводя к измеримому снижению резонансного переноса энергии флюоресценции. Гистоновый пептид Защищенный пептид собирали на твердофазном синтезаторе, используя предварительно загруженную смолу Ванга и применяя стандартные протоколы Fmoc-синтеза. С-концевой лизин защищали посредством гиперкислотолабильной группы, допускающей ее селективное удаление в конце сборки и присоединение биотина. Неочищенный пептид получали после отщепления от смолы при помощи смеси трифторуксусная кислота (TFA), триизопропилсилан и вода (95:2,5:2,5) в течение 3 ч при комнатной температуре и затем очищали, применяя обращенно-фазовую колонку С 18 с использованием градиента 0,1% TFA-буферизованная вода/ацетонитрил. Полученные фракции анализировали и фракции, которые имели чистоту более 95% согласно аналитической HPLC и показывающие правильную ММ (молекулярную массу) (посредством масс-спектроскопии MALDiTOF), объединяли и лиофилизировали. Конечное вещество анализировали посредством HPLC для подтверждения чистоты. Продуцирование белка: Рекомбинантные бромодомены человека (BRD2 (1-473), BRD3 (1-435) иBRD4 (1-477 экспрессировали в клетки E.coli (в вектор рЕТ 15b) при помощи 6(His)-метки на N-конце. Бромодомен с His-меткой извлекали из клеток E.coli, применяя обработку ультразвуком, и очищали, используя колонку 6FF с никелем на сефарозе, белки отмывали и потом элюировали при помощи 50 мМ трис-HCl рН 8,0; 300 мМ NaCl, 1 мМ -меркаптоэтанола и 20 мМ имидазола. Дополнительную очистку выполняли посредством аффинной хроматографии на колонке HisTRAP HP, элюируя при помощи линейного градиента 0-500 мМ хлорида натрия, посредством 20 объемов колонки. Конечную очистку заканчивали посредством препаративной колонки для вытеснительной хроматографии с разделением частиц по размерам Superdex 200. Очищенный белок хранили при -80 С в 20 мМ HEPES (4-(2 гидроксиэтил)-1-пиперазинэтансульфоновая кислота) рН 7,5 и 100 мМ NaCl. Идентичность белка подтверждали способом идентификации белков по массовым "отпечаткам пальцев" и расчетную молекулярную массу подтверждали масс-спектрометрией. Протокол для анализов бромодомена BRD 2, 3 и 4: все компоненты для анализа растворяли в буферной композиции 50 мМ HEPES рН 7,4; 50 мМ NaCl и 0,5 мМ CHAPS (3-3-холамидопропил)диметиламмоний)-1-пропансульфоновая кислота). Конечная концентрация бромодоменовых белков составляла 100 нМ, а концентрация гистонового пептида составляла 300 нМ; эти компоненты предварительно смешивали и оставляли уравновешиваться в течение 1 ч в темноте. Эту реакционную смесь добавляли в объеме 8 мкл во все лунки, содержащие в различных концентрациях 50 нл испытываемого соединения или растворителя DMSO (0,5% конечная), в 384-луночных черных малообъемных планшетахGreiner для микротитрования и инкубировали в темноте в течение 60 мин при комнатной температуре. Смесь для обнаружения в объеме 2 мкл, содержащую меченые антитела против 6His XL665 и стрептавидин, меченный криптатом европия, добавляли во все лунки и выполняли дополнительное инкубирование в темноте в течение по меньшей мере 30 мин. Затем планшеты считывали на планшет-ридере Envision,(ex (длина волны возбуждения) = 317 нм, донорная EM (длина волны испускания) = 615 нм; акцепторная EM = 665 нм; дихроичный двойной LANCE). Измерения интенсивности флюоресценции с временным разрешением выполняли при обеих длинах волн испускания и вычисляли соотношение акцептор/донор и использовали для анализа данных. Все данные нормализовали к среднему по 16 высшим и 16 низшим контрольным лункам на каждом планшете. Затем применяли четырехпараметрическую кривую приближения следующего вида: где "а" равен минимуму, "b" означает наклон Хилла, "с" равен pIC50 и "d" равен максимуму. Соединения примеров 1-3 испытывали во всех анализах BRD2, BRD3 и BRD4, описанных выше, и было обнаружено, что они имеют pIC50 в диапазоне 6,5-7,5 в каждом анализе. Измерение LPS-индуцированной секреции IL-6 из цельной крови Активация моноцитарных клеток агонистами toll-подобных рецепторов, таких как бактериальный липополисахарид (LPS), приводит к продуцированию ключевых медиаторов воспаления, включая IL-6(интерлейкин-6). Широко распространено мнение, что такие пути являются главными в патофизиологии ряда аутоиммунных и воспалительных расстройств. Соединения, предназначенные для испытания, разбавляли с получением диапазона соответствующих концентраций, от которых по 1 мкл разбавленных исходных растворов добавляют на 96-луночный планшет. После добавления цельной крови (130 мкл) планшеты инкубируют при 37 С (5% СО 2) в течение 30 мин перед добавлением 10 мкл 2,8 мкг/мл LPS, разбавленного в полной RPMI 1640 (конечная концентрация составляет 200 нг/мл), с получением общего объема 140 мкл на лунку. После дополнительной инкубации в течение 24 ч при 37 С добавляют 140 мкл PBS (забуференного фосфатом физиологического раствора) в каждую лунку. Планшеты герметизируют, встряхивают в течение 10 мин и затем центрифугируют (2500 об/мин 10 мин). Извлекают 100 мкл супернатанта и анализируют уровни IL-6 посредством иммунологического анализа (обычно посредством методики MesoScale Discovery) или незамедлительно или после хранения при -20 С. Кривые концентрация-ответ для каждого соединения строили по данным и вычисляли значение IC50. Соединения примеров 1 и 2 испытывали в этом анализе и обнаружили, что они имеют pIC50 в диапазоне 6,0-7,0. Эти данные демонстрируют, что ингибиторы бромодомена, испытываемые в указанном выше анализе цельной крови, ингибировали продуцирование ключевого медиатора воспаления IL-6. Мышиная модель in vivo для демонстрирования модуляции провоспалительного ответа Самец мышей CD1 (25-30 г, n=4 на группу) получал одну внутривенную болюсную инъекцию LPS(100 мкг/кг) в хвостовую вену через 1 ч после предварительной обработки одним пероральным введени- 20023374 ем либо растворителя (1% (мас./об.) метилцеллюлозы в стерильной воде), либо испытываемого соединения (3, 10 и 30 мг/кг). Серийные образцы крови для анализа концентраций IL-6 и испытываемого соединения посредством Meso Scale Discovery (MSD) и LC-MS/MS, соответственно, получали из хвостовой вены посредством прямой венопункции в различные моменты времени вплоть до 5 ч после введенияLPS. У получающих растворитель контрольных мышей внутривенное введение LPS индуцировало зависящее от времени увеличение концентраций IL-6 в сыворотке крови, в то время как мыши, которым предварительно вводили перорально соединение из примера 2 (3, 10 и 30 мг/кг), имели максимальные уровни (Смакс) IL-6, пониженные на величину 46, 79, 73% соответственно, и суммарные воздействия(AUC) IL-6, пониженные на величину 35, 70, 63% соответственно. Это понижение в отношении IL-6 является сравнимым с максимальным понижением, наблюдаемым у положительного контроля с дексаметазоном, т.е. понижение Смакс и AUC в отношении IL-6 на 73 и 70% соответственно. Эти данные демонстрируют, что соединение из примера 2, испытанное в указанном выше анализе,понижало LPS-индуцированные системные уровни IL-6 у мыши после одного перорального введения и,следовательно, может быть полезным в качестве противовоспалительного агента. Анализ роста онкологических клеток Клеточные линии человека (n = 80, содержащие клеточные линии, описанные ниже в табл. 2) культивировали в среде RPMI-1640, содержащей 10% фетальной бычьей сыворотки, высевали 1000 жизнеспособных клеток на лунку с плоским дном в 384-луночных черных полистиреновых планшетах (Greiner 781086) в 48 мкл культуральных сред. Все планшеты оставляли при 5% СО 2, 37 С в течение ночи. На следующие сутки с одного планшета собирали клетки при помощи CellTiter-Glo (CTG, Promega G7573) в течение времени, равного 0 измерению (Т 0) и добавляли соединение (20 точек титрования от 14,7 мкМ до 7 пМ) в остальные планшеты. Конечная концентрация DMSO во всех лунках составляла 0,15%. Клетки инкубировали в течение 72 ч или указанного времени и каждый планшет проявляли реагентомCellTiter-Glo, используя объем, равный объему клеточной культуры в лунках. Планшеты встряхивали в течение приблизительно 2 мин и считывали хемилюминесцентный сигнал на планшет-ридере Analyst GT(Molecular Devices) или EnVision (Perkin Elmer). Результаты выражали в виде процента от Т 0 и наносили на график против концентрации соединения. Значение Т 0 нормализовали до 100%, оно представляет количество клеток во время добавления соединения, и данные концентрация-ответ приводили в соответствие с 4-параметрической кривой приближения, применяя программное обеспечение XLfit (model 205). Концентрация, которая ингибировала клеточный рост на 50% (gIC50), представляет собой среднюю точку "окна роста" (между Т 0 и контролемDMSO). Значение Yмин - Т 0 определяют вычитанием значения Т 0 (100%) из значения Yмин (%), определенного из приближения кривой концентрация-ответ. Значения от лунок без клеток вычитали из всех образцов для коррекции фона. Соединение из примера 2 испытывали в соответствии с указанным выше способом и обнаружили,что оно имеет gIC50, как показано в табл. 2. Таблица 2 Соединение из примера 2 также испытывали в соответствии с аналогичным способом, применяя 96 луночные планшеты с соответствующими модификациями для объемов и концентраций, которые будут очевидны для специалиста в данной области техники. Соединение из примера 2 испытывали и обнаружили, что оно имеет gIC50, как показано в табл. 3. Эти данные демонстрируют, что соединение из примера 2, испытанное в указанном выше анализе,ингибировало клеточный рост на панели онкологических клеточных линий и по этой причине может быть полезным в лечении одного или более чем одного вида рака. Все публикации, включая, без ограничения, патенты и патентные заявки, цитированные в этом описании, включены в данное описание посредством ссылки, как если бы каждую отдельную публикацию указали специально и отдельно для включения посредством ссылки в данное описание, как если бы она была приведена целиком. ФОРМУЛА ИЗОБРЕТЕНИЯ 1. Соединение формулы (I), представляющее собой 4-(8-метокси-1-(1-метоксипропан-2-ил)-2(тетрагидро-2 Н-пиран-4-ил)-1 Н-имидазо[4,5-с]хинолин-7-ил)-3,5-диметилизоксазол или его соль. 4. Соединение по любому из пп.1-3 или его фармацевтически приемлемая соль. 5. Фармацевтическая композиция для лечения заболеваний или состояний, при которых показан ингибитор бромодомена, содержащая соединение или его фармацевтически приемлемую соль по п.4 и один или более чем один фармацевтически приемлемый носитель, разбавитель или эксципиент. 6. Применение соединения или его фармацевтически приемлемой соли по п.4 для лечения заболеваний или состояний, при которых показан ингибитор бромодомена. 7. Применение соединения или его фармацевтически приемлемой соли по п.4 для лечения рака или хронического аутоиммунного и/или воспалительного состояния. 8. Применение соединения или его фармацевтически приемлемой соли по п.4 в изготовлении лекарственного средства для лечения рака или хронического аутоиммунного и/или воспалительного состояния. 9. Способ лечения рака или хронического аутоиммунного и/или воспалительного состояния у нуждающегося в этом субъекта, включающий введение терапевтически эффективного количества соединения или его фармацевтически приемлемой соли по п.4.
МПК / Метки
МПК: A61P 29/00, C07D 471/04, A61K 31/4745, A61P 37/00
Метки: 4-(8-метокси-1-((1-метоксипропан-2-ил)-2-(тетрагидро-2н-пиран-4-ил)-1н-имидазо[4,5-c]хинолин-7-ил)-3,5-диметилизоксазол, применение, качестве, бромодомена, ингибитора
Код ссылки
<a href="https://eas.patents.su/25-23374-4-8-metoksi-1-1-metoksipropan-2-il-2-tetragidro-2n-piran-4-il-1n-imidazo45-chinolin-7-il-35-dimetilizoksazol-i-ego-primenenie-v-kachestve-ingibitora-bromodomena.html" rel="bookmark" title="База патентов Евразийского Союза">4-(8-метокси-1-((1-метоксипропан-2-ил)-2-(тетрагидро-2н-пиран-4-ил)-1н-имидазо[4,5-c]хинолин-7-ил)-3,5-диметилизоксазол и его применение в качестве ингибитора бромодомена</a>
Гидраты и полиморфы 4-[[(7r)-8-циклопентил-7-этил-5,6,7,8-тетрагидро-5-метил-6-оксо-2-птеридинил]амино]-3-метокси-n-(1-метил-4-пиперидинил)бензамида, способы их получения и их применение в качестве лекарственных средств

Номер патента: 11407
Опубликовано: 27.02.2009
Авторы: Шмид Рольф, Хоффманн Маттиас, Хертер Рольф, Зигер Петер, Крэмер Герд Ф., Линц Гюнтер, Ралль Вернер
МПК: C07D 211/58, A61K 31/519, A61P 35/00...
Метки: получения, лекарственных, применение, гидраты, 4-[[(7r)-8-циклопентил-7-этил-5,6,7,8-тетрагидро-5-метил-6-оксо-2-птеридинил]амино]-3-метокси-n-(1-метил-4-пиперидинил)бензамида, качестве, средств, полиморфы, способы
Формула / Реферат:
1. Гидраты соединения формулы (I) 2. Гидрат соединения формулы (I) по п.1, отличающийся тем, что он представляет собой моногидрат соединения (I). 3. Гидрат соединения формулы (I) по п.1, отличающийся тем, что он представляет собой тригидрат соединения (I). 4. Соединение формулы (I) где соединение это ангидрат формы II или формы III. 5. Ангидрат по п.4, отличающийся тем, что он представлен в виде безводной формы II соединения формулы (I),...
8-метокси-9н-изотиазоло[5,4-b]хинолин-3,4-дионы и родственные соединения в качестве противоинфекционных средств

Номер патента: 13244
Опубликовано: 30.04.2010
Авторы: Хасимото Акихиро, Брэдбери Бартон Джеймс, Уайлс Джейсон Аллан, Ванг Циупинг, Ким Ха Юнг, Дешпенде Майлинд, Пуччи Майкл Джон, Лусиен Эдлэйн, Пейс Годвин Клэренс Гилрой
МПК: A61K 31/4365, A61P 33/00, C07D 513/04...
Метки: противоинфекционных, родственные, соединения, качестве, 8-метокси-9н-изотиазоло[5,4-b]хинолин-3,4-дионы, средств
Формула / Реферат:
1. Соединение формулы Аили таутомер формулы Вили фармацевтически приемлемая соль формулы А или формулы В, гдеR3 представляет собой водород, C1-С6алкил или C2-С6алканоил;R5 представляет собой водород, гидрокси, амино, С1-С2алкил, С1-С2алкокси, моно- или ди(С1-С4алкил)амино или моно- или ди(С1-С4алкил)гидразинил;R6 представляет собой водород, галоген или амино;R7 представляет собой присоединенную по азоту гетероциклоалкильную группу, которая...
Производные пиридазино [4,5-b] хинолин-5-оксида или их фармацевтически приемлемые соли, их применение в качестве антагонистов глицина, фармацевтическая композиция, способ получения производных пиридазино [ 4,5-b] хинолин-5-оксида или их соли холина

Номер патента: 1711
Опубликовано: 25.06.2001
Авторы: Калвиньш Иварс, Пискунова Ирина, Парсонс Кристофер Грэхам Рафаэль, Гольд Маркус, Даниш Войцех, Рожков Евгений
МПК: C07D 471/04, A61P 25/26, A61K 31/5025...
Метки: производных, фармацевтически, антагонистов, хинолин-5-оксида, качестве, 4,5-b, соли, глицина, приемлемые, пиридазино, фармацевтическая, применение, производные, холина, композиция, получения, способ
Формула / Реферат:
1. Производные пиридазино[4,5-b]хинолин-5-оксида общей формулы где R1 и R2 выбирают из группы, включающей водород, галоген и метокси, или где R1 и R2 вместе образуют метилендиокси, или их фармацевтически приемлемые соли. 2. Соединение по п.1, где соль выбирают из соли холина и соли 4-тетраметиламмония. 3. Соединение по п.1, которое выбирают из группы, включающей 4-гидрокси-1-оксо-1,2-дигидропиридазино[4,5-b]хинолин-5-оксид,...
Применение комбинации 7-(2,5-дигидро-4-имидазо[1,2-а]пиридин-3-ил-2,5-диоксо-1н-пиррол-3-ил)-9-фтор-1,2,3,4-тетрагидро-2-(1-пиперидинилкарбонил)пирроло[3,2,1-jk][1,4]бензодиазепина с химиотерапевтическим агентом для лечения злокачественных опухолей

Номер патента: 18447
Опубликовано: 30.08.2013
Авторы: Чедид Марсио, Абуруб Актхам, Васудеван Венкатрагхаван, Энглер Томас Альберт
МПК: A61K 31/24, A61K 31/4745, A61K 31/4985...
Метки: опухолей, применение, 7-(2,5-дигидро-4-имидазо[1,2-а]пиридин-3-ил-2,5-диоксо-1н-пиррол-3-ил)-9-фтор-1,2,3,4-тетрагидро-2-(1-пиперидинилкарбонил)пирроло[3,2,1-jk][1,4]бензодиазепина, злокачественных, химиотерапевтическим, лечения, агентом, комбинации
Формула / Реферат:
1. Применение комбинации 7-(2,5-дигидро-4-имидазо[1,2-a]пиридин-3-ил-2,5-диоксо-1H-пиррол-3-ил)-9-фтор-1,2,3,4-тетрагидро-2-(1-пиперидинилкарбонил)пирроло[3,2,1-jk][1,4]бензодиазепина или фармацевтически приемлемой соли или сольвата указанного соединения с химиотерапевтическим агентом, выбранным из группы, состоящей из СРТ-11, пеметрекседа, гемцитабина, этопозида, доксорубицина, цисплатина и карбоплатина, для получения лекарственного средства...
Производные имидазо[1,2-b]пиридазина и их применение в качестве ингибиторов pde10

Номер патента: 20847
Опубликовано: 27.02.2015
Авторы: Вертс Йохан Эрвин Эдмонд, Макдональд Грегор Джеймс, Конде-Сейде Сусана, Суинни Келли Энн, Мартин-Мартин Мария Лус, Ванхоф Грета Констанция Петер, Дельгадо-Гонсалес Оскар, Пастор-Фернандес Хоакин, Алонсо-Де Диего Серхио-Алвар, Ван Гол Михиль Люк Мария, Бартоломе-Небреда Хосе Мануэль, Лейс Карина
МПК: A61P 3/00, A61K 31/5025, A61P 25/00...
Метки: имидазо[1,2-b]пиридазина, ингибиторов, применение, производные, качестве, pde10
Формула / Реферат:
1. Соединение формулы (I)или его стереоизомерная форма,где R1 представляет собой пиридинил; пиридинил, необязательно замещенный галогеном, С1-4алкилом, трифторметилом или C1-4алкилоксигруппой; тетрагидропиранил или NR6R7;R2 представляет собой водород, С1-4алкил, трифторметил, С3-8циклоалкил или С1-4алкилоксигруппу;R3 представляет собой водород, хлор, С1-4алкил, трифторметил или С3-8циклоалкил;Het представляет собой 5- или 6-членное...