Гидраты и полиморфы 4-[[(7r)-8-циклопентил-7-этил-5,6,7,8-тетрагидро-5-метил-6-оксо-2-птеридинил]амино]-3-метокси-n-(1-метил-4-пиперидинил)бензамида, способы их получения и их применение в качестве лекарственных средств
Номер патента: 11407
Опубликовано: 27.02.2009
Авторы: Хоффманн Маттиас, Крэмер Герд Ф., Ралль Вернер, Зигер Петер, Линц Гюнтер, Хертер Рольф, Шмид Рольф







Формула / Реферат
1. Гидраты соединения формулы (I)

2. Гидрат соединения формулы (I) по п.1, отличающийся тем, что он представляет собой моногидрат соединения (I).
3. Гидрат соединения формулы (I) по п.1, отличающийся тем, что он представляет собой тригидрат соединения (I).
4. Соединение формулы (I)

где соединение это ангидрат формы II или формы III.
5. Ангидрат по п.4, отличающийся тем, что он представлен в виде безводной формы II соединения формулы (I), идентифицируемой рентгеновской порошковой дифрактометрией и интенсивностью (нормированная) при нормальных комнатных условиях


6. Ангидрат по п.4, отличающийся тем, что он представлен в виде безводной формы III соединения формулы (I), идентифицируемой рентгеновской порошковой дифрактометрией и интенсивностью (нормированная) при 100шС


7. Фармацевтическая композиция, отличающаяся тем, что она содержит гидрат или ангидрат соединения формулы (I) по одному из пп.1-2, соответственно по одному из пп.4-6 в терапевтически эффективном количестве и одно или несколько фармацевтически приемлемых вспомогательных веществ.
8. Способ получения соединения формулы (I)

отличающийся тем, что соединение формулы 4

подвергают взаимодействию с соединением формулы 9

9. Способ получения соединения формулы 9

отличающийся тем, что соединение формулы 8

подвергают метилированию диметилкарбонатом.
10. Соединение формулы 3

11. Соединение формулы 4

12. Способ получения моногидрата соединения формулы (I) по п.2, заключающийся в том, что
(а) приготавливают раствор соединения формулы (I) в смеси растворителей из 1-пропанола или изопропанола и воды,
(б) из смеси растворителей кристаллизуют моногидрат соединения формулы (I) и
(в) выделяют моногидрат соединения формулы (I).
13. Способ по п.12, на стадии а) которого приготавливают раствор соединения формулы (I) в смеси растворителей из 1-пропанола и воды.
14. Способ получения безводной формы II соединения формулы (I) по п.5, заключающийся в том, что
(а) приготавливают раствор соединения формулы (I) в этилацетате,
(б) из этилацетата кристаллизуют безводную форму I соединения формулы (I) с последующим добавлением диэтилового эфира и
(в) выделяют безводную форму II соединения формулы (I).
Текст
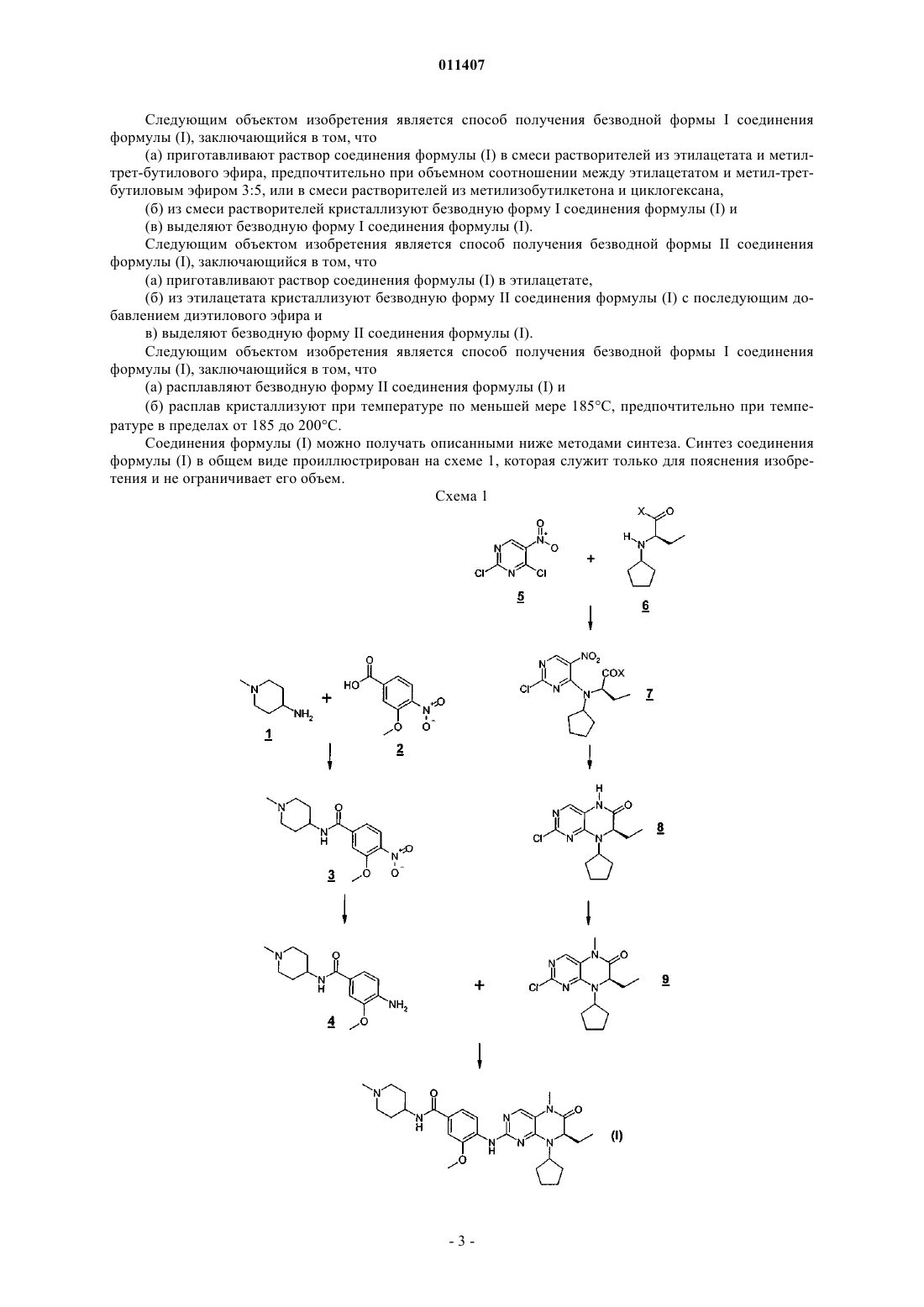
011407 Настоящее изобретение относится к новым гидратам и новым полиморфам 4-(7R)-8-циклопентил 7-этил-5,6,7,8-тетрагидро-5-метил-6-оксо-2-птеридинил]амино]-3-метокси-N-(1-метил-4-пиперидинил) бензамида, к способам их получения и к их применению в качестве лекарственных средств. Предпосылки создания изобретения Соединение 4-(7R)-8-циклопентил-7-этил-5,6,7,8-тетрагидро-5-метил-6-оксо-2-птеридинил]амино]-3-метокси-N-(1-метил-4-пиперидинил)бензамид играет важную роль в качестве ингибитора Poloподобной киназы (PLK, от англ. "Polo-like kinase") в регуляции клеточного цикла у эукариот. Соединение 4-(7R)-8-циклопентил-7-этил-5,6,7,8-тетрагидро-5-метил-6-оксо-2-птеридинил]амино]-3-метокси-N-(1-метил-4-пиперидинил)бензамид имеет структуру, соответствующую следующей формуле (I): Аналогичные дигидроптеридиноны описаны в WO 03/020722. Подробное описание изобретения В настоящем изобретении предлагаются новые гидраты и новые полиморфные формы соединения формулы (I) с антипролиферативным действием. В соответствии с этим изобретение относится к гидратам соединения формулы (I) Предпочтительным гидратом соединения формулы (I) является моногидрат. Другим предпочтительным гидратом соединения формулы (I) является тригидрат. Следующим объектом изобретения является ангидрат (безводная форма) соединения формулы (I). Равным образом предпочтителен ангидрат соединения формулы (I), представленный в виде безводной формы I. Предпочтителен также ангидрат соединения формулы (I), представленный в виде безводной формы II. Предпочтителен далее ангидрат соединения формулы (I), представленный в виде безводной формы III. Следующим объектом изобретения является фармацевтическая композиция, которая содержит один из описанных выше предлагаемых в изобретении гидратов и полиморфов соединения формулы (I) в терапевтически эффективном количестве и один или несколько фармацевтически приемлемых вспомогательных веществ. Следующим объектом изобретения являются предлагаемые в нем гидраты и полиморфы соединения формулы (I) для применения в качестве лекарственных средств с антипролиферативным действием. Следующим объектом изобретения является применение предлагаемых в нем гидратов и полиморфов соединения формулы (I) для получения лекарственного средства, предназначенного для лечения и/или профилактики рака, инфекционных, воспалительных и аутоиммунных заболеваний. Следующим объектом изобретения является применение предлагаемых в нем гидратов и полиморфов соединения формулы (I) для получения лекарственного средства, предназначенного для ингибирования Polo-подобной киназы. Предпочтительно применение предлагаемых в изобретении гидратов и полиморфов соединения формулы (I) для получения лекарственного средства, предназначенного для ингибирования Poloподобной киназы PLK-1. Особенно предпочтительно применение предлагаемых в изобретении гидратов и полиморфов соединения формулы (I), предусматривающее пероральное, энтеральное, внутривенное, перитонеальное введение действующего вещества в организм или введение действующего вещества в организм путем инъекции. Следующим объектом изобретения является способ получения соединения формулы (I), отличающийся тем, что соединение формулы 4 Следующим объектом изобретения является способ получения соединения формулы 9 путем метилирования диметилкарбонатом в присутствии основания при повышенной температуре (в интервале от 80 до 180 С), предпочтительно при температуре 130 С. Следующим объектом изобретения является соединение формулы 3 Следующим объектом изобретения является соединение формулы 4 Следующим объектом изобретения является способ получения моногидрата соединения формулы(а) приготавливают раствор соединения формулы (I) в смеси растворителей из изопропанола и воды или ацетона и воды,(б) из смеси растворителей кристаллизуют моногидрат соединения формулы (I) и(в) выделяют моногидрат соединения формулы (I). Следующим объектом изобретения является способ получения тригидрата соединения формулы (I),заключающийся в том, что моногидрат соединения формулы (I) подвергают выдержке в атмосфере с влажностью воздуха по меньшей мере 70%. Следующим объектом изобретения является способ получения безводной формы III соединения формулы (I), заключающийся в том, что(а) моногидрат соединения формулы (I) выдерживают в токе сухого азота или(б) моногидрат соединения формулы (I) выдерживают при температуре примерно 70 С, предпочтительно при температуре в пределах от 70 до 120 С, наиболее предпочтительно при температуре в пределах от 70 до 90 С. Следующим объектом изобретения является способ получения безводной формы I соединения формулы (I), заключающийся в том, что расплавляют безводную форму III соединения формулы (I) и затем расплав кристаллизуют при температуре по меньшей мере 140 С, предпочтительно при температуре в пределах от 140 до 160 С.-2 011407 Следующим объектом изобретения является способ получения безводной формы I соединения формулы (I), заключающийся в том, что(а) приготавливают раствор соединения формулы (I) в смеси растворителей из этилацетата и метилтрет-бутилового эфира, предпочтительно при объемном соотношении между этилацетатом и метил-третбутиловым эфиром 3:5, или в смеси растворителей из метилизобутилкетона и циклогексана,(б) из смеси растворителей кристаллизуют безводную форму I соединения формулы (I) и(в) выделяют безводную форму I соединения формулы (I). Следующим объектом изобретения является способ получения безводной формы II соединения формулы (I), заключающийся в том, что(а) приготавливают раствор соединения формулы (I) в этилацетате,(б) из этилацетата кристаллизуют безводную форму II соединения формулы (I) с последующим добавлением диэтилового эфира и в) выделяют безводную форму II соединения формулы (I). Следующим объектом изобретения является способ получения безводной формы I соединения формулы (I), заключающийся в том, что(а) расплавляют безводную форму II соединения формулы (I) и(б) расплав кристаллизуют при температуре по меньшей мере 185 С, предпочтительно при температуре в пределах от 185 до 200 С. Соединения формулы (I) можно получать описанными ниже методами синтеза. Синтез соединения формулы (I) в общем виде проиллюстрирован на схеме 1, которая служит только для пояснения изобретения и не ограничивает его объем. Схема 1-3 011407 Получение анилинового фрагмента 4. Получение соединения 3. Вариант 1 А. Суспензию 100 г (0,507 моль) 3-метокси-4-нитробензойной кислоты 2, 163 г (0,508 моль) тетрафторбората О-(бензотриазол-1-ил)-N,N,N',N'-тетраметилурония и 192 мл (1,1 моль) этилдиизопропиламина в 1,2 л дихлорметана перемешивают в течение часа при 25 С. К полученному раствору добавляют 58 г(0,508 моль) 1-метил-4-аминопиперидина 1 и перемешивают в течение 16 ч при 20 С. Затем раствор упаривают до объема 600 мл и органическую фазу пятикратно промывают 80 мл 1-молярного раствора аммиака. Органическую фазу упаривают и остаток хроматографируют на силикагеле, используя смесь из дихлорметана, метанола и конц. аммиака (в соотношении 15:1:0,1). Содержащие продукт фракции объединяют, выпаривают растворитель и продукт кристаллизуют из смеси этилацетата и метанола. Таким путем получают 123 г продукта 3. Вариант 1 Б. 4,00 кг (20,3 моль) 3-метокси-4-нитробензойной кислоты 2 добавляют в 54 л толуола. Далее при нормальном давлении отгоняют 16 л толуола. После этого смесь охлаждают до 105 С и добавляют 40 мл диметилформамида в 2 л толуола. При температуре рубашки 120 С в течение 30 мин приливают 2,90 кг(24,3 моль) тионилхлорида и промывают 4 л толуола. После этого реакционную смесь в течение 1 ч перемешивают при нагревании с обратным холодильником. Затем при нормальном давлении отгоняют 12 л толуола. Далее содержимое реактора охлаждают. Затем при 55-65 С приливают раствор 2,55 кг (22,3 моль) 1-метил-4-аминопиперидина 1 в 2 л толуола и 2,46 кг (24,3 моль) триэтиламина в 2 л толуола. После этого промывают 4 л толуола. Суспензию перемешивают в течение 1 ч. Затем приливают 20 л воды и при 35-40 С добавляют 3,08 кг (30,4 моль) конц. соляной кислоты (36%-ной). Далее промывают 2 л воды. При 35-40 С образуются две фазы. Органическую фазу отделяют, а содержащую продукт водную фазу вновь загружают в реактор. После этого промывают 4 л воды. Затем при пониженном давлении и при 50 С отгоняют 3,2 л воды. К оставшемуся раствору при 40 С приливают 4,87 кг (60,9 моль) раствора гидроксида натрия (50%-ного). После этого промывают 4 л воды. Содержащей продукт суспензии дают охладиться до 22 С и затем в течение 30 мин перемешивают при этой температуре. Суспензию фильтруют на нутче и осадок на фильтре промывают 40 л воды. Продукт сушат в вакуумном сушильном шкафу при 40 С. Таким путем получают 5,65 кг продукта. Получение соединения 4. Вариант 2 А. Раствор 145 г (0,494 моль) соединения 3 в 2 л метанола гидрируют в присутствии 2 г палладия на угле (10%-ного) при давлении 4 бара. После этого катализатор отфильтровывают и фильтрат упаривают. Таким путем получают 128 г продукта 4. Вариант 2 Б. К 5,00 кг (17,0 моль) соединения 3 и 600 г активированного угля (технического) добавляют 25 л деминерализованной воды. После этого добавляют 2,05 кг (34,1 моль) уксусной кислоты. Затем суспензию перемешивают в течение 15 мин при 22-25 С. Далее добавляют 500 г палладия на угле (10%-ного), взмученного в 3 л деминерализованной воды, и затем промывают 2 л деминерализованной воды. После этого содержимое реактора нагревают до 40 С и гидрируют при этой температуре до прекращения поглощения водорода. Затем реакционную смесь фильтруют и осадок на фильтре промывают 10 л деминерализованной воды. Для кристаллизации фильтрат переносят в реактор и транспортировочную емкость затем промывают 5 л деминерализованной воды. Содержимое реактора нагревают до 50 С. Далее добавляют смесь из 5,45 кг (68,2 моль) раствора гидроксида натрия (50%-ного, технического) и 7 л деминерализованной воды. После этого перемешивают в течение 10 мин при 45-50 С. Затем суспензию охлаждают до 20 С и-4 011407 перемешивают при этой температуре в течение 1-1,5 ч. Продукт отфильтровывают на нутче, промывают 30 л деминерализованной воды и сушат в вакуумном сушильном шкафу при 45 С. Таким путем получают 4,13 кг продукта 4. Получение дигидроптеридинонового фрагмента 9. Получение эфиров аминокислот 6a-6d. Метиловый эфир 6 а, этиловый эфир 6b и 2-пропиловый эфир 6 с получают известными из литературы методами, например, согласно WO 03/020722. трет-Бутиловый эфир 6d получают переэтерификацией трет-бутилового эфира уксусной кислоты в присутствии перхлорной кислоты (J. Med. Chem., т. 37,20,1994, cc. 3294-3302). В последующей реакции нуклеофильного замещения аминокислоты можно использовать в виде оснований или гидрохлоридов. Получение амидов аминокислот 6 е, 6f. Амид аминокислоты 6 е получают путем аминолиза метилового эфира 6 а 40%-ным водным раствором метиламина при комнатной температуре. Амид аминокислоты 6f получают путем образования амида свободной аминокислоты взаимодействием со взятым в пятикратном избытке 2-молярным раствором диметиламина в тетрагидрофуране в присутствии тетрафторбората О-(бензотриазол-1-ил)-N,N,N',N'тетраметилурония в качестве агента сочетания. Получение соединений 7. Получение метилового эфира 7 а. Суспензию 457 г (2,06 моль) метилового эфира аминокислоты 6 а. и 693 г (8,25 моль) измельченного в порошок гидрокарбоната натрия в 10 л циклогексана перемешивают в течение 15 мин при комнатной температуре. Затем добавляют 440 г (2,27 моль) 2,4-дихлор-5-нитропиримидина 5 и 1,5 л циклогексана и перемешивают в течение 3 дней при комнатной температуре. Протекание реакции контролируют с помощью жидкостной хроматографии высокого разрешения. Для перевода выкристаллизовавшегося продукта обратно в раствор к суспензии добавляют 4 л дихлорметана. После добавления 335 г сульфата магния суспензию фильтруют на нутче и неорганический осадок на фильтре затем промывают дихлорметаном. Фильтрат упаривают при пониженном давлении до массы 3,1 кг и полученную суспензию нагревают с обратным холодильником. После этого раствору дают медленно охладиться и затем перемешивают в течение часа при 10-15 С. Суспензию фильтруют на нутче и осадок на фильтре промывают циклогексаном. Продукт сушат в вакуумном сушильном шкафу при 40 С. Таким путем получают 582 г соединения 7 а (X обозначает ОСН 3) в виде темно-желтого твердого вещества. Для достижения высокой региоселективности при нуклеофильном замещении в соединении 5 наи-5 011407 более пригодны липофильные растворители, такие, например, как циклогексан, метилциклогексан, толуол и их смеси. Аналогично этой процедуре получают соединения 7b-7f. При превращении амидов аминокислот 7 е,7f для улучшения растворимости добавляют небольшое количество более полярного растворителя, например этилацетата или дихлорметана. Получение соединения 8. Свежеприготовленную суспензию 560 г (1,63 моль) соединения 7 а и 185 г никеля Ренея в 2,8 л уксусной кислоты гидрируют при 75 С. По завершении поглощения водорода катализатор отфильтровывают и раствор для гидрирования упаривают при пониженном давлении. К остатку добавляют 4 л деминерализованной воды и 4 л этилацетата. Между фазами образуется осадок, содержащий продукт. Водную фазу отделяют. К органической фазе добавляют 2 л этилацетата и осадок отфильтровывают на нутче. Осадок суспендируют в 600 мл деминерализованной воды, перемешивают в течение 1 ч при комнатной температуре, отфильтровывают на нутче и промывают деминерализованной водой. Таким путем получают 110 г влажного продукта А. Фильтрат трижды промывают раствором хлорида натрия. Органическую фазу упаривают. Таким путем получают 380 г красновато-коричневого остатка Б, который объединяют с влажным продуктом А. Объединенные сырые продукты А и Б при нагревании с обратным холодильником растворяют в 1,5 л этанола. После этого раствор фильтруют до прозрачного состояния и фильтр затем промывают 150 мл этанола. Далее при нагревании с обратным холодильником к раствору добавляют 550 мл деминерализованной воды. Затем раствору дают охладиться, после чего перемешивают сначала в течение 16 ч при комнатной температуре, а затем в течение 3 ч при 0-5 С. Осадок отфильтровывают на нутче и промывают сначала смесью из деминерализованной воды и метанола (в соотношении 1:1), а затем деминерализованной водой. Продукт сушат в вакуумном сушильном шкафу при 50 С. Таким путем получают 266 г продукта 8 в виде твердого вещества. Получение соединения 9 (вариант 3 А). К раствору 264 г (0,94 моль) соединения 8 и 161 г (1,13 моль) метилиодида в 2 л диметилацетамида при 4-10 С порциями в течение часа добавляют 38 г (0,95 моль) гидрида натрия (60%-ная дисперсия в минеральном масле). Затем охлаждающую баню удаляют и смеси в течение 2 ч дают нагреться до 20 С. После этого охлаждают до 10 С и добавляют еще 0,38 г (9,5 ммоль) гидрида натрия. Далее перемешивают в течение 4 ч при 10-15 С. Затем к реакционному раствору добавляют 100 мл этилацетата и 1 кг льда. Образовавшуюся суспензию разбавляют 3 л деминерализованной воды. После этого суспензию перемешивают в течение 2 ч, осадок отфильтровывают на нутче и осадок на фильтре промывают деминерализованной водой. Продукт сушат в вакуумном сушильном шкафу при 50 С. Таким путем получают 273 г продукта 9 в виде бесцветных кристаллов. Получение соединения 9 (вариант 3 Б). Суспензию 100 г (356 ммоль) соединения 8 и 73,8 г (534 ммоль) карбоната калия в 400 мл диметил-6 011407 карбоната нагревают в автоклаве до 130 С с последующей выдержкой при этой температуре в течение 6 ч. Затем смеси дают охладиться и при перемешивании добавляют 300 мл деминерализованной воды и 200 мл этилацетата. Водную фазу вместе с нерастворимыми солями отделяют. От органической фазы при давлении 180 мбар и при температуре нагревательной бани 70 С отгоняют 500 мл растворителя. К остатку добавляют 600 мл деминерализованной воды и при давлении 150 мбар и при температуре нагревательной бани 80 С отгоняют 100 мл растворителя. К суспензии добавляют 350 мл этанола и нагревают до 65 С. Затем раствору дают охладиться и добавляют затравку. После этого охлаждают до 10 С, осадок отфильтровывают на нутче и затем промывают смесью из деминерализованной воды и этанола (в соотношении 2,5:1). Продукт сушат в вакуумном сушильном шкафу при 50 С. Таким путем получают 95,5 г продукта 9. Получение соединения формулы (I). Суспензию 201 г (1,06 моль) гидрата паратолуолсульфоновой кислоты, 209 г (706 ммоль) соединения 9 и 183 г (695 ммоль) соединения 4 в 800 мл 2-метил-4-пентанола кипятят с обратным холодильником. При этом отгоняют 100 мл растворителя. После этого в течение 3 ч кипятят с обратным холодильником, добавляют 200 мл 2-метил-4-пентанола и отгоняют 120 мл растворителя. После 2-часового кипячения с обратным холодильником отгоняют еще 280 мл растворителя. Затем реакционному раствору дают охладиться до 100 С и добавляют к нему 1 л деминерализованной воды, а затем 0,5 л этилацетата. Органическую фазу отделяют, а водную фазу еще раз промывают 0,5 л этилацетата. К кислой водной фазе добавляют 1,5 л дихлорметана и 0,5 л этилацетата. Значение рН водной фазы устанавливают на 9,2 добавлением 260 мл 6 н. раствора едкого натра. Водную фазу отделяют и органические фазы трижды промывают 1 н. водным раствором гидрокарбоната натрия порциями по 1 л. Органическую фазу сушат над сульфатом натрия, фильтруют и растворитель выпаривают при пониженном давлении. Таким путем получают 406 г сырого продукта. Этот сырой продукт растворяют в 1,5 л этилацетата. Затем при температуре 50-55 С добавляют 2,5 л метил-трет-бутилового эфира. При 45 С добавляют затравку и перемешивают в течение 16 ч при охлаждении до комнатной температуры. Далее суспензию перемешивают в течение 3,5 ч при 0-5 С и осадок отфильтровывают на нутче. Осадок на фильтре после этого промывают смесью метил-трет-бутилового эфира и этилацетата (в соотношении 2:1), а затем метил-трет-бутиловым эфиром. Продукт сушат в вакуумном сушильном шкафу при 50 С. Таким путем получают 236 г кристаллического соединения формулы(I) в виде безводной формы I. Кристаллизация. 46,5 г описанной выше кристаллической безводной формы I соединения формулы (I) растворяют в 310 мл 1-пропанола и раствор фильтруют до прозрачного состояния. Далее нагревают до 70 С и добавляют 620 мл деминерализованной воды. Раствору дают охладиться до комнатной температуры, охлаждают до 0-10 С и добавляют затравочные кристаллы. Образовавшуюся суспензию перемешивают в течение 3 ч при 0-10 С. После этого фильтруют на нутче и промывают холодной смесью 1-пропанола и деминерализованной воды (в соотношении 1:2), а затем деминерализованной водой. Продукт сушат в вакуумном сушильном шкафу при 50 С. Таким путем получают 40,5 г кристаллического соединения формулы (I) в виде моногидрата. Сырой продукт, полученный при описанном выше химическом превращении, можно также непосредственно кристаллизовать в виде кристаллического моногидрата из смеси 1-пропанола и деминерализованной воды. Гидраты и ангидраты соединения формулы (I) можно получать описанными ниже методами. Эти методы проиллюстрированы на схеме 2 и служат только для пояснения изобретения без ограничения его объема.-7 011407 Схема 2 Полиморфные превращения соединения формулы (I) ОВ означает "относительная влажность" Характеристики предлагаемых в изобретении гидратов и ангидратов определяли с помощью ДСК/ТГ (дифференциальной сканирующей калориметрии/термогравиметрии) и с помощью рентгеновской порошковой дифрактометрии (с получением рентгеновских порошковых дифрактограмм) (табл. 1-5,фиг. 1 а-5 а). Все рентгеновские порошковые дифрактограммы получали известными из уровня техники методами, используя рентгеновский порошковый дифрактометр (Bruker D8 Advanced). Дифрактограммы получали при следующих условиях измерения: K-излучение меди ( = 1,5418 ), 40 кВ, 40 мА. Для ДСК/ТГ-измерений использовали приборы фирмы Mettler Toledo (DSC 821 и TGA 851). ДСКизмерения проводили на образцах массой от 2 до 10 мг, а ТГ-измерения проводили на образцах массой от 10 до 30 мг. Измерения проводили при скорости нагрева 10 K/мин в атмосфере инертного газа (в токе азота). Таблица 1 Дифракционные максимумы (рефлексы) и их интенсивность (нормированная),выявленные при анализе моногидрата соединения формулы (I) рентгеновской порошковой дифрактометрией при нормальных комнатных условиях-8 011407 Таблица 2 Дифракционные максимумы (рефлексы) и их интенсивность (нормированная), выявленные при анализе тригидрата соединения формулы (I) рентгеновской порошковой дифрактометрией при комнатной температуре и 90%-ной относительной влажности Таблица 3 Дифракционные максимумы (рефлексы) и их интенсивность (нормированная),выявленные при анализе безводной формы I соединения формулы (I) рентгеновской порошковой дифрактометрией при нормальных комнатных условиях Таблица 4 Дифракционные максимумы (рефлексы) и их интенсивность (нормированная),выявленные при анализе безводной формы II соединения формулы (I) рентгеновской порошковой дифрактометрией при нормальных комнатных условиях- 10011407 Таблица 5 Дифракционные максимумы (рефлексы) и их интенсивность (нормированная), выявленные при анализе безводной формы III соединения формулы (I) рентгеновской порошковой дифрактометрией при 100 С Предлагаемые в изобретении ангидрат I, ангидрат II и моногидрат соединения формулы (I) могут применяться индивидуально либо в комбинации с другими предлагаемыми в изобретении действующими веществами и необязательно также в комбинации с фармакологически активными действующими веществами иного типа. В качестве примера лекарственных форм, пригодных для введения в организм в их составе предлагаемых в изобретении соединений, можно назвать таблетки, капсулы, суппозитории, растворы, прежде всего растворы для инъекций (подкожных, внутривенных, внутримышечных) и инфузий, микстуры,эмульсии и диспергируемые порошки. В таких лекарственных формах на долю фармацевтически активного(ых) соединения(ий) должно приходиться в каждом случае от 0,01 до 90 мас.%, предпочтительно от 0,1 до 50 мас.% от общей массы препарата, т.е. в подобных лекарственных формах фармацевтически активное(ые) соединение(ия) должно(ы) содержаться в количествах, достаточных для его(их) введения в организм в дозировке, соответствующей указанному ниже интервалу значений. При необходимости действующее(ие) вещество(вещества) можно вводить в организм в указанных ниже дозах несколько раз в сутки. Соответствующие таблетки можно изготавливать, например, смешением действующего вещества или действующих веществ с известными вспомогательными веществами, например инертными разбавителями, такими как карбонат кальция, фосфат кальция или лактоза, разрыхлителями, такими как кукурузный крахмал или альгиновая кислота, связующими, такими как крахмал или желатин, смазывающими веществами, такими как стеарат магния или тальк, и/или средствами для обеспечения депо-эффекта, такими как карбоксиметилцеллюлоза, ацетофталат целлюлозы или поливинилацетат. Таблетки могут также состоять из нескольких слоев. Соответствующим образом можно изготавливать драже нанесением на полученные аналогично таблеткам ядра покрытий из обычно применяемых в этих целях материалов, например, коллидона или шеллака, гуммиарабика, талька, диоксида титана или сахара. Ядра драже для обеспечения депо-эффекта или во избежание несовместимости также можно изготавливать многослойными. Равным образом и оболочка драже также может состоять для обеспечения депо-эффекта из нескольких слоев, для чего можно использовать вспомогательные вещества, указанные выше для таблеток. В состав микстур с предлагаемыми в изобретении действующими веществами, соответственно комбинациями действующих веществ дополнительно могут входить также подслащивающее вещество, такое как сахарин, цикламат, глицерин или сахар, а также улучшитель вкуса, например ароматизатор, такой как ванилин или апельсиновый экстракт. Помимо этого микстуры могут содержать суспендирующие вспомогательные вещества или загустители, такие как натрийкарбоксиметилцеллюлоза, смачиватели,например продукты конденсации жирных спиртов с этиленоксидом, или защитные вещества (консерванты), такие как n-гидроксибензоаты. Растворы для инъекций и инфузий приготавливают по известной технологии, например, с добавлением придающих изотоничность агентов, консервантов, таких как n-гидроксибензоаты, или стабилизаторов, таких как соли этилендиаминтетрауксусной кислоты с щелочными металлами, и при необходимости с применением эмульгаторов и/или диспергаторов, при этом, например, при применении воды в качестве разбавителя при необходимости можно использовать органические растворители в качестве гидротропных солюбилизаторов, соответственно вспомогательных растворителей, и затем разливают по бутылкам для инъекций, ампулам или бутылкам для инфузий.- 11011407 Капсулы, содержащее одно или несколько действующих веществ, соответственно комбинации действующих веществ, можно изготавливать, например, смешением действующих веществ с инертными носителями, такими как лактоза или сорбит, и расфасовыванием полученной смеси в желатиновые капсулы. Соответствующие суппозитории можно изготавливать, например, смешением действующего вещества или действующих веществ с предусмотренными для этой цели носителями, такими как нейтральные жиры или полиэтиленгликоль, соответственно его производные. В качестве примера вспомогательных веществ, которые можно использовать при приготовлении лекарственных форм, можно назвать воду, фармацевтически приемлемые (безвредные) органические растворители, такие как парафины (например, фракции минерального масла), масла растительного происхождения (например, арахисовое или кунжутное масло), моно- или полифункциональные спирты (например, этанол или глицерин), носители, такие как природная мука горных пород (например, каолин,глиноземы, тальк, мел), синтетическая мука горных пород (например, высокодисперсная кремниевая кислота и силикаты), сахара (например, тростниковый, молочный и виноградный сахар), эмульгаторы (например, лигнин, сульфитный щелок, метилцеллюлоза, крахмал и поливинилпирролидон) и скользящие вещества (например, стеарат магния, тальк, стеариновая кислота и лаурилсульфат натрия). Предлагаемые в изобретении соединения можно вводить в организм обычным путем, предпочтительно перорально, путем инъекции или трансдермально. Для приема внутрь могут использоваться таблетки, в состав которых помимо вышеуказанных носителей можно, как очевидно, включать также такие добавки, как, например, цитрат натрия, карбонат кальция и дикальцийфосфат, совместно с различного рода наполнителями, такими как крахмал, предпочтительно картофельный крахмал, желатин и т.п. Помимо этого при производстве таблеток могут использоваться также скользящие вещества, такие как стеарат магния, лаурилсульфат натрия и тальк. В случае водных суспензий действующие вещества помимо вышеописанных вспомогательных веществ можно также смешивать с различного рода улучшителями вкуса или красителями. Для парентерального введения можно использовать растворы действующих веществ, получаемые с применением пригодных для этой цели жидких носителей. При внутривенном введении действующее вещество вводят в организм в дозе, которая составляет от 1 до 1000 мг/ч, предпочтительно от 5 до 500 мг/ч. Однако в некоторых случаях в зависимости от веса тела пациента, соответственно пути введения действующего вещества в организм, индивидуальной реакции на лекарственное средство, типа его лекарственной формы и времени, соответственно интервала времени введения лекарственного средства в организм, доза действующего вещества может отличаться от указанных выше количеств. Так, в частности, иногда может оказаться достаточным использовать действующее вещество или действующие вещества в дозе, меньшей указанного выше нижнего предела, тогда как в других случаях может потребоваться использовать действующее вещество или действующие вещества в дозе, превышающий вышеуказанный верхний предел. При необходимости введения действующего вещества или действующих веществ в больших дозах может оказаться целесообразным дробить их на несколько более мелких доз из расчета на несколько приемов в сутки. Ниже изобретение проиллюстрировано на примерах, в которых представлены составы некоторых лекарственных форм и которые не ограничивают его объем. Примеры лекарственных форм (фармацевтических композиций) Тонкоизмельченное действующее вещество смешивают с лактозой и частью от всего предусмотренного количества кукурузного крахмала. Полученную смесь просеивают, после чего ее увлажняют раствором поливинилпирролидона в воде, месят, гранулируют во влажном состоянии и сушат. Полученный гранулят совместно с остальным количеством кукурузного крахмала и стеаратом магния просеивают и смешивают между собой. Из полученной смеси прессуют таблетки требуемых формы и размеров. Тонкоизмельченное действующее вещество смешивают с частью от всего предусмотренного количества кукурузного крахмала, лактозой, микрокристаллической целлюлозой и поливинилпирролидоном,полученную смесь просеивают и совместно с остальным количеством кукурузного крахмала и водой перерабатывают в гранулят, который сушат и просеивают. Далее добавляют натрийкарбоксиметилкрахмал и стеарат магния, перемешивают и из полученной смеси прессуют таблетки требуемых размеров. Действующее вещество при его собственном значении рН или при необходимости при рН 5,5-6,5 растворяют в воде и смешивают с хлоридом натрия в качестве придающего изотоничность агента. От полученного раствора отфильтровывают пирогенные продукты и фильтрат в асептических условиях расфасовывают в ампулы, которые затем стерилизуют и запаивают. Такие ампулы могут содержать 5, 25 или 50 мг действующего вещества. ФОРМУЛА ИЗОБРЕТЕНИЯ 1. Гидраты соединения формулы (I) 2. Гидрат соединения формулы (I) по п.1, отличающийся тем, что он представляет собой моногидрат соединения (I). 3. Гидрат соединения формулы (I) по п.1, отличающийся тем, что он представляет собой тригидрат соединения (I). 4. Соединение формулы (I) где соединение это ангидрат формы II или формы III. 5. Ангидрат по п.4, отличающийся тем, что он представлен в виде безводной формы II соединения формулы (I), идентифицируемой рентгеновской порошковой дифрактометрией и интенсивностью (нормированная) при нормальных комнатных условиях 6. Ангидрат по п.4, отличающийся тем, что он представлен в виде безводной формы III соединения формулы (I), идентифицируемой рентгеновской порошковой дифрактометрией и интенсивностью (нормированная) при 100 С 7. Фармацевтическая композиция, отличающаяся тем, что она содержит гидрат или ангидрат соединения формулы (I) по одному из пп.1-2, соответственно по одному из пп.4-6 в терапевтически эффективном количестве и одно или несколько фармацевтически приемлемых вспомогательных веществ. 8. Способ получения соединения формулы (I) отличающийся тем, что соединение формулы 4 9. Способ получения соединения формулы 9 отличающийся тем, что соединение формулы 8 12. Способ получения моногидрата соединения формулы (I) по п.2, заключающийся в том, что(а) приготавливают раствор соединения формулы (I) в смеси растворителей из 1-пропанола или изопропанола и воды,(б) из смеси растворителей кристаллизуют моногидрат соединения формулы (I) и(в) выделяют моногидрат соединения формулы (I). 13. Способ по п.12, на стадии а) которого приготавливают раствор соединения формулы (I) в смеси растворителей из 1-пропанола и воды. 14. Способ получения безводной формы II соединения формулы (I) по п.5, заключающийся в том,что(а) приготавливают раствор соединения формулы (I) в этилацетате,(б) из этилацетата кристаллизуют безводную форму I соединения формулы (I) с последующим добавлением диэтилового эфира и(в) выделяют безводную форму II соединения формулы (I).
МПК / Метки
МПК: A61P 35/00, A61K 31/519, C07D 211/58, C07D 475/00
Метки: полиморфы, получения, 4-[[(7r)-8-циклопентил-7-этил-5,6,7,8-тетрагидро-5-метил-6-оксо-2-птеридинил]амино]-3-метокси-n-(1-метил-4-пиперидинил)бензамида, применение, средств, способы, лекарственных, гидраты, качестве
Код ссылки
<a href="https://eas.patents.su/19-11407-gidraty-i-polimorfy-4-7r-8-ciklopentil-7-etil-5678-tetragidro-5-metil-6-okso-2-pteridinilamino-3-metoksi-n-1-metil-4-piperidinilbenzamida-sposoby-ih-polucheniya-i-ih-primenenie-v-k.html" rel="bookmark" title="База патентов Евразийского Союза">Гидраты и полиморфы 4-[[(7r)-8-циклопентил-7-этил-5,6,7,8-тетрагидро-5-метил-6-оксо-2-птеридинил]амино]-3-метокси-n-(1-метил-4-пиперидинил)бензамида, способы их получения и их применение в качестве лекарственных средств</a>
Применение (+)-4-амино-5-хлор-n-(3-метокси-4-пиперидинил)-2-метоксибензамида для приготовления лекарственных препаратов для лечения заболеваний желудочно-кишечного тракта

Номер патента: 2362
Опубликовано: 25.04.2002
Авторы: Схююркес Йоаннес Адрианус Якобус, Хейкантс Йозеф Ян Питер, Мэлдерманс Виллем Эмиль Гюстаф, Мегенс Антониус Адрианус Хендрикус Петрус
МПК: A61P 1/14, A61K 31/4468
Метки: +)-4-амино-5-хлор-n-(3-метокси-4-пиперидинил)-2-метоксибензамида, лекарственных, тракта, лечения, желудочно-кишечного, препаратов, применение, заболеваний, приготовления
Формула / Реферат:
1. Применение (+)-4-амино-5-хлор-N-(3-метокси-4-пиперидинил)-2-метоксибензамида для получения лекарственного препарата для лечения гастроэзофагиального рефлюкса. 2. Применение (+)-4-амино-5-хлор-N-(3-метокси-4-пиперидинил)-2-метоксибензамида для получения лекарственного препарата для лечения синдрома воспаления пищеварительного тракта или синдрома воспаления пищеварительного тракта с преобладанием диареи. 3. Применение...
Новые дигидроптеридиноны, способы их получения и их применение в качестве лекарственных средств

Номер патента: 7062
Опубликовано: 30.06.2006
Авторы: Леманн-Линтц Торстен, Бауер Эккхарт, Шнапп Гизела, Айкмайер Кристиан, Грауэрт Маттиас, Поль Геральд, Редеманн Норберт, Штегмайер Мартин, Хоффманн Маттиас, Квант Йенс Юрген, Брайтфельдер Штеффен
МПК: A61P 31/18, A61P 35/00, A61K 31/505...
Метки: средств, качестве, новые, лекарственных, получения, применение, дигидроптеридиноны, способы
Формула / Реферат:
1. Соединения общей формулы (I) в которой R1 представляет собой остаток, выбранный из группы, включающей водород, NH2 и необязательно замещенную одним либо несколькими атомами галогена C1-С3алкильную группу, R2 представляет собой остаток, выбранный из группы, включающей водород или необязательно замещенную C1-С3алкильную группу, R3 и R4 имеют идентичные или разные значения и представляют собой остаток, выбранный из группы, включающей...
Азабициклические соединения, способ их получения и их применение в качестве лекарственных средств, в частности, в качестве антибактериальных средств

Номер патента: 4920
Опубликовано: 28.10.2004
Авторы: Лампила Максим, Фроментэн Клод, Асзоди Жозеф, Роулендс Дэвид Ален
МПК: A61K 31/529, A61P 31/04, C07D 487/08...
Метки: антибактериальных, соединения, получения, применение, качестве, способ, частности, азабициклические, лекарственных, средств
Формула / Реферат:
1. Соединение общей формулы (I) или одна из его солей с основанием или с кислотой в которой R1 означает атом водорода, радикал COOH, CN, COOR, CONR6R7, (CH2)n'R5 или радикал где R выбирают из группы, состоящей из алкила с 1-6 атомами углерода, возможно замещенного пиридилом или карбамоилом; -CH2-алкенила, содержащего в целом 3-9 атомов углерода; арила с 6-10 атомами углерода или аралкила с 7-11 атомами углерода, причем арильное или...
Производные 6-деоксиэритромицина, способ их получения и их применение в качестве лекарственных средств

Номер патента: 3776
Опубликовано: 28.08.2003
Авторы: Ожер Жан-Мишель, Дени Алексис
МПК: A61K 31/70, C07H 17/08
Метки: средств, качестве, лекарственных, применение, производные, получения, 6-деоксиэритромицина, способ
Формула / Реферат:
1. Соединения формулы (I) в которой X представляет собой радикал (NH)a, CH2 или SO2 или атом кислорода, тогда как символ а представляет собой число, равное 0 или 1, Y представляет собой радикал (CH2)m-(CH=CH)n-(CH2)o, причем m+n+o_ 8, а n=0 или 1, Ar представляет собой фенил или нафтил, или гетероарил, представляющий собой тиенил, фурил, пиролил, тиазолил, оксазолил, имидазолил, в частности, 4-(3-пиридинил)-1H-имидазолил, тиадиазолил,...
Новые пролекарства (n-2-пиридил-n-2-гидроксикарбонилэтил)амида 1 – метил-2-(4-амидинофениламинометил)бензимидазол-5-илкарбоновой кислоты, их получение и их применение в качестве лекарственных средств

Номер патента: 8249
Опубликовано: 27.04.2007
Авторы: Хауэль Норберт, Буш Ульрих, Кольбацки Флориан
МПК: A61P 7/02, A61K 31/245, C07D 401/12...
Метки: кислоты, пролекарства, получение, средств, метил-2-(4-амидинофениламинометил)бензимидазол-5-илкарбоновой, применение, n-2-пиридил-n-2-гидроксикарбонилэтил)амида, лекарственных, новые, качестве
Формула / Реферат:
1. Соединения общей формулы в которой а) R' обозначает атом водорода, a R обозначает метоксикарбонильную группу или б) R' обозначает атом водорода либо С1-С6алкильную группу, a R обозначает гидроксигруппу, при этом алкильные группы, содержащие более 2 атомов углерода, включают также их изомеры с разветвленной цепью, такие, например, как изопропильная, трет-бутильная и изобутильная группа, их таутомеры и их соли. 2. Соединения общей формулы I по...
Предыдущий патент: Производные пиридо-пиримидина, их получение, их применение в терапии
Следующий патент: Новый продукт, способ и промежуточные продукты получения производных азетидина
Случайный патент: Оберточный материал с противолежащими адгезивными средствами