Пирроло[2,3-d]пиримидины
Номер патента: 5852
Опубликовано: 30.06.2005
Авторы: Ченджелиан Пол Стивен, Блюменкопф Тодд Эндрю, Флэнэган Марк Эдвард, Браун Мэттью Фрэнк






Формула / Реферат
1. Соединение формулы

или его фармацевтически приемлемая соль, где R1 представляет группу формулы

где y означает 0, 1 или 2;
R4 выбирают из группы, состоящей из водорода, (C1-C6)алкила или (C2-C6)алкенила, где алкильные и алкенильные группы необязательно замещены гидроксильной группой, аминогруппой, (C1-C4)алкокси или (C1-C6)ациламиногруппой; или R4 является (C3-C10)циклоалкилом, где циклоалкильная группа необязательно замещена дейтерием, гидроксильной группой, аминогруппой, трифторметилом, (C1-C6)ацилокси, (C1-C6)ациламино, (C1-C6)алкиламино, ((C1-C6)алкил)2амино, циано, циано(C1-C6)алкилом, трифторметил(C1-C6)алкилом, нитро, нитро(C1-C6)алкилом или (C1-C6)ациламиногруппой;
R5 выбирают из группы, состоящей из (C3-C10)циклоалкила, где циклоалкильная группа необязательно замещена от одного до пяти заместителями, выбранными из гидрокси, (C1-C6)алкила, (C2-C6)алкенила; или R5 представляет и R13CO(C3-C10)циклоалкил, где R13 является R20O, где R20 представляет (C1-C6)алкил; или R5 является группой формулы

где w равно 1;
x равно 3;
или R5 является группой формулы

где g равно 1;
h равно 1 или 2;
j равно 0 или 1;
F, K и P представляют каждый CR7R8;
R7 и R8 каждый выбирают независимо из группы, включающей водород и (C1-C6)алкил;
R2 и R3 выбирают каждый независимо из группы, включающей водород, (C1-C6)алкил и (C6-C10)арил, где алкильные или арильные группы необязательно замещены от одного до трех галогенами;
при условии, что sp2- и sp-атомы углерода алкенила не могут быть замещены гидроксильной группой или аминогруппой.
2. Соединение по п.1, где R1 является NR4R5.
3. Соединение или его фармацевтически приемлемая соль, выбранные из группы, включающей
2-{4-метил-3-[метил-(7H-пирроло[2,3-d]пиримидин-4-ил)амино]циклогексил}пропан-2-ол;
2-{3-[(2-гидроксиэтил)-(7H-пирроло[2,3-d]пиримидин-4-ил)амино]-4-метилциклогексил}пропан-2-ол;
2-[(5-изопропенил-2-метилциклогексил)-(7H-пирроло[2,3-d]пиримидин-4-ил)амино]этанол;
(5-изопропенил-2-метилциклогексил)-(7H-пирроло[2,3-d]пиримидин-4-ил)-(2,2,2-трифторэтил)амин;
2-{4-метил-3-[(7H-пирроло[2,3-d]пиримидин-4-ил)-(2,2,2-трифторэтил)амино]циклогексил}пропан-2-ол;
(5-фтор-7H-пирроло[2,3-d]пиримидин-4-ил)-(5-изопренил-2-метилциклогексил)метиламин;
2-{3-[(5-фтор-7H-пирроло[2,3-d]пиримидин-4-ил)метиламино]-4-метилциклогексил}пропан-2-ол;
(2-этил-4-изопропенилциклопентил)метил-(7H-пирроло[2,3-d]пиримидин-4-ил)амин;
2-{3-этил-4-[метил-(7H-пирроло[2,3-d]пиримидин-4-ил)амино]циклопентил}пропан-2-ол;
2-{3-этил-4-[(2-гидроксиэтил)-(7H-пирроло[2,3-d]пиримидин-4-ил)амино]циклопентил}пропан-2-ол;
2-[(2-этил-4-изопропенилциклопентил)-(7H-пирроло[2,3-d]пиримидин-4-ил)амино]этанол;
(5-(S)-изопропенил-2-метилциклогексил)метил-(7H-пирроло[2,3-d]пиримидин-4-ил)амин;
2-[циклогептил-(7H-пирроло[2,3-d]пиримидин-4-ил)амино]этанол;
2-[циклооктил-(7H-пирроло[2,3-d]пиримидин-4-ил)амино]этанол и
бицикло[2,2,1]гепт-2-ил-метил-(7H-пирроло[2,3-d]пиримидин-4-ил)амин.
4. Фармацевтическая композиция для (а) лечения или предупреждения заболевания или состояния, выбранных из группы, включающей отторжение трансплантированного органа, волчанку, рассеянный склероз, ревматоидный артрит, псориаз, диабет типа I и осложнения, связанные с диабетом, рак, астму, атопический дерматит, аутоиммунные заболевания щитовидной железы, язвенный колит, болезнь Крона, болезнь Альцгеймера, лейкоз и другие аутоиммунные заболевания, или для (б) ингибирования протеинтирозинкиназ или Janus-киназы 3 (JAK3) у млекопитающего, включая человека, включающая количество соединения по п.1 или его фармацевтически приемлемой соли, эффективных при таких заболеваниях или состояниях, одного или в комбинации с одним или несколькими дополнительными модулирующими иммунную систему млекопитающего средствами или с противовоспалительными средствами и фармацевтически приемлемый носитель.
5. Способ ингибирования протеинтирозинкиназ или Janus-киназы 3 (JAK3) у млекопитающего, включая человека, включающий введение указанному млекопитающему эффективного количества соединения по п.1 или его фармацевтически приемлемой соли, одного или в комбинации с одним или несколькими дополнительными средствами, которые модулируют иммунную систему млекопитающего, или с противовоспалительными средствами.
6. Способ лечения или предупреждения заболевания или состояния, выбранных из группы, включающей отторжение трансплантированного органа, волчанку, рассеянный склероз, ревматоидный артрит, псориаз, диабет типа I или осложнения, связанные с диабетом, рак, астму, атопический дерматит, аутоиммунные заболевания щитовидной железы, язвенный колит, болезнь Крона, болезнь Альцгеймера, лейкоз и другие аутоиммунные заболевания млекопитающего, включая человека, включающий введение указанному млекопитающему количества соединения по п.1 или его фармацевтически приемлемой соли, эффективных при лечении такого состояния, одного или в комбинации с одним или несколькими дополнительными средствами, которые модулируют иммунную систему млекопитающего, или с противовоспалительными средствами.
Текст
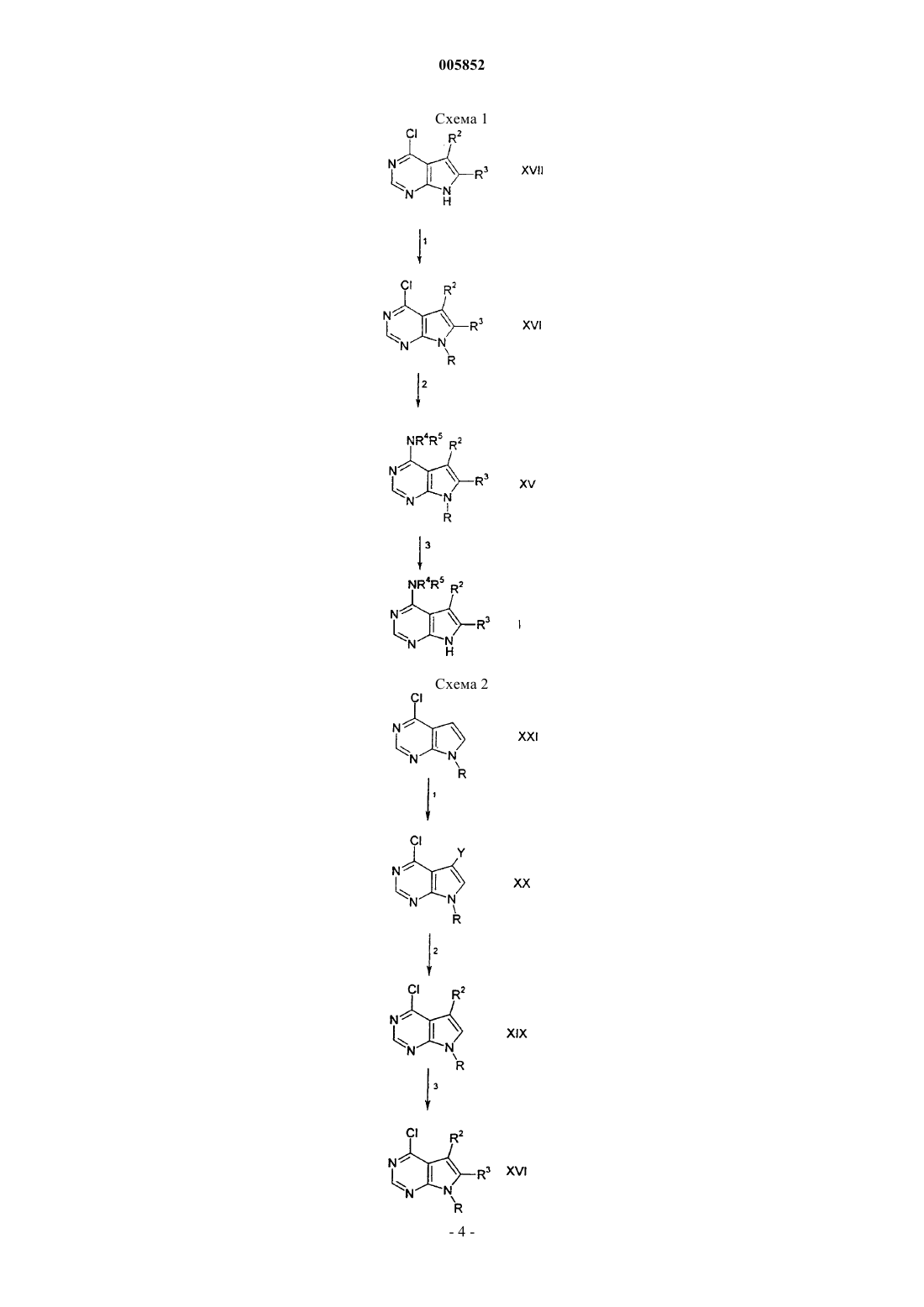
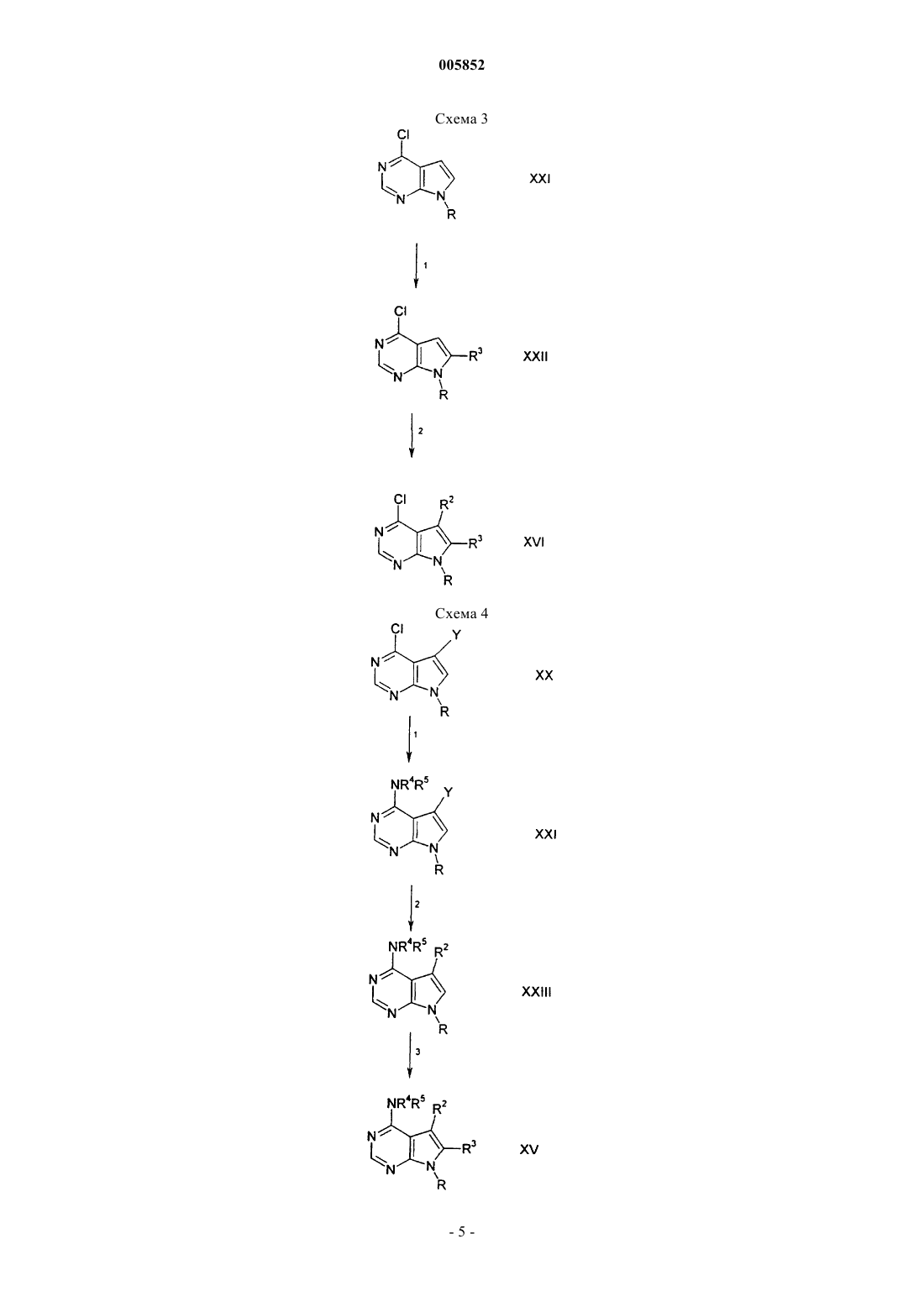
005852 Предпосылки создания изобретения Настоящее изобретение относится к пирроло[2,3-d]пиримидинам, которые являются ингибиторами протеинтирозинкиназ, таких как, например, фермент Janus-киназа З (в дальнейшем также упоминается как JAKЗ), и которые используются в терапии в качестве иммунодепрессивных средств при пересадке органов, для лечения волчанки, рассеянного склероза, ревматоидного артрита, псориаза, диабета типа I и осложнений от диабета, рака, астмы, атопического дерматита, аутоиммунных нарушений щитовидной железы, язвенного колита, болезни Крона, болезни Альцгеймера, лейкоза и при других показаниях, когда было бы желательно подавление иммунитета. Изобретение также относится к способу использования таких соединений для лечения вышеуказанных показаний у млекопитающих, особенно у людей, и к полезным для этого фармацевтическим композициям.JAKЗ является членом семейства Janus протеинтирозинкиназ. Хотя другие члены этого семейства экспрессируются, по существу, всеми тканями, экспрессия JAKЗ ограничивается кроветворными клетками. Это находится в соответствии с ее существенной ролью, заключающейся в передаче сигнала через рецепторы для интерлейкинов IL-2, IL-4, IL-7, IL-9 и IL-15 посредством нековалентного связыванияJAKЗ с гамма-цепью, общей для этих мультицепных рецепторов. Группы больных XSCID характеризуются сильно пониженными уровнями белка JAK3 или генетическими дефектами относительно общей гамма-цепи, что предполагает, что подавление иммунитета должно быть результатом блокирования передачи сигнала посредством JAKЗ. Из исследований на животных следует, что JAKЗ не только играет важную роль в созревании В- и Т-лимфоцитов, но JAKЗ, по существу, требуется для поддержания функции Т-клетки. Модуляция иммунной активности по этому новому механизму может оказаться полезной при лечении заболеваний, связанных с пролиферацией Т-клетки, таких как, например, отторжение трансплантата и аутоиммунные заболевания. Краткое описание изобретения Настоящее изобретение относится к соединению формулы или его фармацевтически приемлемой соли, где R1 представляет группу формулыR4 выбирают из группы, состоящей из водорода, (C1-C6)алкила или (C2-C6)алкенила, где алкильные и алкенильные группы необязательно замещены гидроксильной группой, аминогруппой, (C1-C4)алкокси или (C1-C6)ациламиногруппой; или R4 является (C3-C10)циклоалкилом, где циклоалкильная группа необязательно замещена дейтерием, гидроксильной группой, аминогруппой, трифторметилом, (C1-C6)ацилокси, (C1-C6)ациламино, (C1-C6)алкиламино, C1-C6)алкил)2 амино, циано, циано(C1-C6)алкилом, трифторметил(C1-C6)алкилом, нитро, нитро(C1-C6)алкилом или (C1-C6)ациламиногруппой;R5 выбирают из группы, состоящей из (C3-C10)циклоалкила, где циклоалкильная группа необязательно замещена от одного до пяти заместителями, выбранными из гидрокси, (C1-C6)алкила, (C2-C6)алкенила; или R5 представляет и R13 СO(С 3-С 10)циклоалкил, где R13 является R20O, где R20 представляет (C1C6)алкил; или R5 является группой формулыR7 и R8 каждый выбирают независимо из группы, включающей водород и (C1-C6)алкил;R2 и R3 выбирают каждый независимо из группы, включающей водород, (C1-C6)алкил и (C6C10)арил, где алкильные или арильные группы необязательно замещены от одного до трех галогенами; при условии, что sp2- и sp-атомы углерода алкенила не могут быть замещены гидроксильной группой или аминогруппой. Настоящее изобретение также относится к фармацевтически приемлемым кислотно-аддитивным солям соединений формулы I. Кислотами, которые применяются для получения фармацевтически приемлемых кислотно-аддитивных солей вышеупомянутых основных соединений данного изобретения, являются такие кислоты, которые образуют нетоксичные кислотно-аддитивные соли, то есть соли, содержащие фармакологически приемлемые анионы, такие как, например, гидрохлорид, гидроиодид, гидробромид, нитрат, сульфат, бисульфат, фосфат, кислый фосфат, ацетат, лактат, цитрат, кислый цитрат, тартрат, битартрат, сукцинат, малеат, фумарат, глюконат, сахарат, бензоат, метансульфонат, этансульфонат,бензолсульфонат, п-толуолсульфонат и памоат [то есть 1,1'-метилен-бис-(2-гидрокси-3-нафтоат)]. Изобретение также относится к основно-аддитивным солям соединений формулы I. Химическими основаниями, которые могут быть использованы в качестве реагентов для получения фармацевтически приемлемых основно-аддитивных солей соединений формулы I, которые являются кислотными по природе, являются такие, которые образуют нетоксичные основные соли с такими соединениями. Такие нетоксичные соли с основаниями включают, но не ограничиваются теми, которые получаются из таких фармакологически приемлемых катионов как, например, катионы щелочных металлов (например, калия и натрия) и катионы щелочно-земельных металлов (например, кальция и магния), аммониевые соли или водорастворимые соли, получаемые в результате присоединения амина, такие как, например, N-метилглюкамин-(меглюмин), и (низший)алканол-аммониевые соли, и другие основные соли фармацевтически приемлемых органических аминов. Термин "алкил", используемый здесь, если не указано иначе, включает насыщенные одновалентные углеводородные радикалы с прямой или разветвленной цепью, или циклические части, или их комбинации. Термин "алкокси", используемый здесь, включает O-алкильные группы, в которых "алкил" является таким, как определено выше. Термин "галоген", используемый здесь, если не указано иначе, включает фтор, хлор, бром или иод. Соединения по изобретению могут содержать двойные связи. Когда имеются такие связи, соединения по изобретению существуют в цис- и транс-конфигурациях и в виде их смесей. Если не указано иначе, алкильные и алкенильные группы, которые здесь упоминаются, а также алкильные части других упомянутых здесь групп (например, алкокси) могут быть линейными или разветвленными, а также они могут быть циклическими (например, циклопропил, циклобутил, циклопентил, циклогексил или циклогептил) или могут быть линейными или разветвленными и содержать циклические части. Если не указано иначе, галоген включает фтор, хлор, бром и иод. Термин (C3-C10)циклоалкил относится к циклоалкильным группам с уровнями ненасыщенности от 0 до 2, как, например, циклопропил, циклобутил, циклопентил, циклопентенил, циклогексил, циклогексенил, 1,3-циклогексадиен, циклогептил, циклогептенил, бицикло[3,2,1]октан, норборнанил и т.д. Термин (С 6-С 10)арил относится к фенилу или нафтилу. Соединения формулы (I) могут быть введены в фармацевтически приемлемой форме либо отдельно, либо в комбинации с одним или несколькими дополнительными средствами, которые являются модуляторами иммунной системы млекопитающего, или с противовоспалительными средствами. Эти средства могут включать, но не ограничиваются ими, циклоспорин А (например, Сандиммун или Неорал,рапамицин, FK-506 (такролимус), лефлуномид, дезоксиспергуалин, микофенолят (например, Селлсепт),азатиоприн (например, Имуран), даклизумаб (например, Зенапакс), ОКТЗ (например, Ортоклон), AtGam,аспирин, ацетаминофен, ибупрофен, напроксен, пироксикам и противовоспалительные стероиды (например, преднизолон или дексаметазон). Эти средства могут быть введены как часть той же самой дозированной лекарственной формы или как отдельная дозированная лекарственная форма одинаковыми или различными способами введения c использованием одинаковых или различных схем введения в соответствии со стандартной фармацевтической практикой. Соединения по изобретению включают все конформационные изомеры (например, цис- и транс-изомеры) и все оптические изомеры соединений формулы I(например, энантиомеры и диастереомеры), а также рацемические, диастереомерные и другие смеси таких изомеров. Предпочтительные соединения формулы I включают такие, в которых R1 означает NR4R5. Наиболее предпочтительные соединения формулы I включают следующие: 2-4-метил-3-[метил-(7 Н-пирроло[2,3-d]пиримидин-4-ил)амино]циклогексилпропан-2-ол; 2-3-[(2-гидроксиэтил)-(7 Н-пирроло[2,3-d]пиримидин-4-ил)амино]-4-метилциклогексилпропан-2-ол; 2-[(5-изопропенил-2-метилциклогексил)-(7 Н-пирроло[2,3-d]пиримидин-4-ил)амино]этанол;(5-(S)-изопропенил-2-метилциклогексил)метил-(7 Н-пирроло[2,3-d]пиримидин-4-ил)амин; 2-[циклогептил-(7 Н-пирроло[2,3-d]пиримидин-4-ил)амино]этанол; 2-[циклооктил-(7 Н-пирроло[2,3-d]пиримидин-4-ил)амино]этанол; бицикло[2,2,1]гепт-2-ил-метил-(7 Н-пирроло[2,3-d]пиримидин-4-ил)амин. Настоящее изобретение также относится к фармацевтической композиции для (а) лечения или предупреждения заболевания или состояния, выбранных из группы, включающей отторжение трансплантированного органа, волчанку, рассеянный склероз, ревматоидный артрит, псориаз, диабет типа I и осложнения от диабета, рак, астму, атопический дерматит, аутоиммунные нарушения щитовидной железы, язвенный колит, болезнь Крона, болезнь Альцгеймера, лейкоз и другие аутоиммунные заболевания, или для (б) ингибирования протеинтирозинкиназ или Janus-киназы 3 (JAK3) у млекопитающего, включая человека, включающей количество соединения формулы I или его фармацевтически приемлемой соли,эффективное при таких заболеваниях или состояниях, и фармацевтически приемлемый носитель. Настоящее изобретение также относится к фармацевтической композиции для (а) лечения или предупреждения заболевания или состояния, выбранных из группы, включающей отторжение трансплантированного органа, волчанку, рассеянный склероз, ревматоидный артрит, псориаз, диабет типа I и осложнения от диабета, рак, астму, атопический дерматит, аутоиммунные заболевания щитовидной железы,язвенный колит, болезнь Крона, болезнь Альцгеймера, лейкоз и другие аутоиммунные заболевания, или для (б) ингибирования протеинтирозинкиназ или Janus-киназы 3 (JAK3) у млекопитающего, включая человека, включающей количество соединения формулы I или его фармацевтически приемлемой соли,эффективного при таких нарушениях или состояниях, одного или в комбинации с Т-клеточным иммунодепрессантом или с противовоспалительными средствами, и фармацевтически приемлемого носителя. Настоящее изобретение также относится к способу ингибирования протеинтирозинкиназ или Janusкиназы 3 (JAK3) у млекопитающего, включая человека, включающему введение указанному млекопитающему эффективного количества соединения формулы I или его фармацевтически приемлемой соли. Настоящее изобретение относится к способу лечения или предупреждения заболевания или состояния, выбранных из группы, включающей отторжение трансплантированного органа, волчанку, рассеянный склероз, ревматоидный артрит, псориаз, диабет типа I и осложнения от диабета, рак, астму, атопический дерматит, аутоиммунные нарушения щитовидной железы, язвенный колит, болезнь Крона, болезнь Альцгеймера, лейкоз и другие аутоиммунные заболевания млекопитающего, включая человека, включающему введение указанному млекопитающему количества соединения формулы I или его фармацевтически приемлемой соли, эффективного для лечения такого состояния. Настоящее изобретение также относится к способу ингибирования протеинтирозинкиназ или Janusкиназы 3 (JAK3) у млекопитающего, включая человека, включающему введение указанному млекопитающему эффективного количества соединения формулы I или его фармацевтически приемлемой соли,одного или в комбинации с Т-клеточными иммунодепрессантами или с противовоспалительными средствами. Настоящее изобретение также относится к способу лечения или предупреждения заболевания или состояния, выбранных из группы, включающей отторжение трансплантированного органа, волчанку,рассеянный склероз, ревматоидный артрит, псориаз, диабет типа I или осложнения от диабета, рак, астму, атопический дерматит, аутоиммунные нарушения щитовидной железы, язвенный колит, болезнь Крона, болезнь Альцгеймера, лейкоз и другие аутоиммунные заболевания млекопитающего, включая человека, включающему введение указанному млекопитающему количества соединения формулы I или его фармацевтически приемлемой соли, эффективного для лечения такого состояния, одного или в комбинации с Т-клеточными иммунодепрессантами или противовоспалительными средствами. Подробное описание изобретения Следующие схемы реакций иллюстрируют получение соединений данного изобретения. Если не указано иначе, R1, R2, R3 и R9 в схемах реакций и при последующем обсуждении имеют указанные выше значения. В реакции 1 по схеме 1 производное 4-хлорпирроло[2,3-d]пиримидина формулы XVII превращают в соответствующее соединение формулы XVI, в котором R является бензолсульфонилом или бензилом,обработкой XVII бензолсульфонилхлоридом, хлористым бензилом или бромистым бензилом в присутствии основания, такого как, например, гидрид натрия или карбонат калия, и полярного апротонного растворителя, такого как, например, диметилформамид или тетрагидрофуран. Реакционную смесь перемешивают при температуре примерно от 0 до 70 С, предпочтительно около 30 С, в течение периода времени примерно от 1 до примерно 3 ч, предпочтительно около 2 ч. В реакции 2 по схеме 1 производное 4-хлорпирроло[2,3-d]пиримидина формулы XVI превращают в производное 4-аминопирроло[2,3-d]пиримидина формулы XV сочетанием соединения XVI с амином формулы HNR4R5. Реакцию проводят в спиртовом растворителе, например трет-бутаноле, метаноле или этаноле, или в других высококипящих органических растворителях, таких как, например, диметилформамид, 1,4-диоксан или 1,2-дихлорэтан, при температуре примерно между 60 и 120 С, предпочтительно около 80 С. Обычно продолжительность реакции составляет примерно от 2 до 48 ч, предпочтительно около 16 ч. В реакции 3 по схеме 1 удаление защитной группы у соединения формулы XV, где R означает бензолсульфонил, для получения соответствующего соединения формулы I осуществляют обработкой XV гидроокисью щелочного металла, такого как, например, гидроокись натрия или гидроокись калия, в спиртовом растворителе, таком как, например, метанол или этанол, или в смесях растворителей, как, например, спирт/тетрагидрофуран или спирт/вода. Реакцию проводят при комнатной температуре в тече-6 005852 ние времени от 15 мин до 1 ч, предпочтительно 30 мин. Удаление защитной группы у соединения формулы XV, где R является бензилом, проводят обработкой XV натрием в аммиаке при температуре около-78 С в течение времени примерно от 15 мин до около 1 ч. В реакции 1 по схеме 2 производное 4-хлорпирроло[2,3-d]пиримидина формулы XXI, где R означает водород или бензолсульфонат, превращают в производное 4-хлор-5-галоидпирроло[2,3-d]пиримидина формулы XX, где Y означает хлор, бром или иод, реакцией XXI с N-хлорсукцинимидом, N-бромсукцинимидом или N-иодсукцинимидом. Реакционную смесь в хлороформе кипятят с обратным холодильником примерно от 1 до 3 ч, предпочтительно около 1 ч. Альтернативно, в реакции 1 по схеме 2 4 хлорпирроло[2,3-d]пиримидин формулы XXI, где R означает водород, превращают в соответствующий 4-хлор-5-нитропирроло[2,3-d]пиримидин формулы XX, где Y является нитрогруппой, реакцией XXI с азотной кислотой в серной кислоте при температуре примерно от -10 до 10 С, предпочтительно около 0 С, в течение времени примерно от 5 до около 15 мин, предпочтительно около 10 мин. Соединение формулы XXI, где Y является нитрогруппой, превращают в соответствующий 4-хлор-5-аминопирроло[2,3-d]пиримидин формулы XX, где Y представляет аминогруппу, реакцией XXI в различных условиях,известных специалисту в данной области, как, например, гидрогенолиз в присутствии палладия или четыреххлористого олова и соляной кислоты. В реакции 2 по схеме 2 4-хлор-5-галоидпирроло[2,3-d]пиримидин формулы XX, где R означает водород, превращают в соответствующее соединение формулы XIX, где R2 представляет собой (C1-C6)алкил или бензил, обработкой XX N-бутиллитием при температуре около -78 С и реакцией образованного таким образом дианионного промежуточного соединения с алкилгалогенидом или бензилгалогенидом при температуре примерно от -78 С до комнатной температуры, предпочтительно при комнатной температуре. Альтернативно, образованный таким образом дианион реагирует с молекулярным кислородом с образованием соответствующего 4-хлор-5-гидроксипирроло[2,3-d]пиримидина формулы XIX, где R2 является гидроксильной группой. Соединение формулы XX, где Y представляет собой бром или иод и R является бензолсульфонатом, превращают в соединение формулы XIX, где R2 является (C6-C12)арилом или винилом,обработкой XX N-бутиллитием при температуре около -78 С с последующим присоединением хлористого цинка при температуре около -78 С. Образованное таким образом соответствующее цинкорганическое промежуточное соединение затем подвергают реакции с арилиодидом или винилиодидом в присутствии каталитического количества палладия. Реакционную смесь перемешивают при температуре около 5080 С, предпочтительно около 70 С в течение времени примерно от 1 до 3 ч, предпочтительно около 1 ч. В реакции 3 в схеме 2 соединение формулы XIX превращают в соответствующее соединение формулы XVI обработкой XIX N-бутиллитием, литийдиизопропиламином или гидридом натрия при температуре около -78 С в присутствии полярного апротонного растворителя, как, например, тетрагидрофуран. Образованное при этом анионное промежуточное соединение далее подвергают реакции с (а) алкилгалогенидом или бензилгалогенидом при температуре в пределах примерно от -78 С до комнатной температуры, предпочтительно при -78 С, когда R3 представляет алкил или бензил; (б) альдегидом или кетоном при температуре в пределах примерно от -78 С до комнатной температуры, предпочтительно при-78 С, когда R3 является алкоксигруппой; и (в) хлористым цинком при температуре примерно от -78 С до комнатной температуры, предпочтительно при -78 С, и образующееся при этом соответствующее промежуточное цинкорганическое соединения затем подвергают реакции с арилиодидом или винилиодидом в присутствии каталитического количества палладия. Полученную реакционную смесь перемешивают при температуре примерно от 50 до 80 С, предпочтительно около 70 С, в течение примерно от 1 до 3 ч, предпочтительно около 1 ч. Альтернативно, образованный таким образом анион подвергают реакции с молекулярным кислородом для образования соответствующего производного 4-хлор-6-гидроксипирроло[2,3-d]пиримидина формулы XVI, где R3 является гидроксильной группой. В реакции 1 по схеме 3 производное 4-хлорпирроло[2,3-d]пиримидина формулы XXI превращают в соответствующее соединение формулы XXII в соответствии с методикой, описанной выше для реакции 3 схемы 2. В реакции 2 схемы 3 соединение формулы XXII превращают в соответствующее соединение формулы XVI в соответствии с методиками, описанными выше для реакций 1 и 2 схемы 3. В реакции 1 схемы 4 производное 4-хлорпирроло[2,3-d]пиримидина формулы XX превращают в соответствующее производное 4-аминопирроло[2,3-d]пиримидина формулы XXIV в соответствии с методикой, описанной выше для реакции 2 схемы 1. В реакции 2 схемы 4 производное 4-амино-5-галоидпирроло[2,3-d]пиримидина формулы XXIV, гдеR является бензолсульфонатом и Z является бромом или иодом, превращают в соответствующее соединение формулы XXIII реакцией XXIV с (а) арилборной кислотой, когда R2 означает арил, в апротонном растворителе, таком как, например, тетрагидрофуран или диоксан, в присутствии каталитического количества палладия (0) при температуре примерно от 50 до 100 С, предпочтительно около 70 С, в течение времени примерно от 2 до 48 ч, предпочтительно около 12 ч; (б) алкинами, когда R2 означает алкинил, в присутствии каталитического количества иодида меди (I) и палладия (0) и полярного растворителя, такого как, например, диметилформамид, при комнатной температуре в течение времени примерно от 1 до 5 ч,предпочтительно около 3 ч; и (в) алкенами или стиролами, когда R2 означает винил или стирил, в присутствии каталитического количества палладия в диметилформамиде, диоксане или тетрагидрофуране-7 005852 при температуре примерно от 80 до 100 С, предпочтительно около 100 С, в течение времени примерно от 2 до 48 ч, предпочтительно около 48 ч. В реакции 3 схемы 4 соединение формулы XXIII превращают в соответствующее соединение формулы XV в соответствии с методикой, описанной выше для реакции 3 схемы 2. В реакции 1 схемы 5 производное 4-хлорпирроло[2,3-d]пиримидина формулы XVII превращают в соответствующее соединение формулы XVI, где R определен выше, согласно методике, описанной выше для реакции 1 схемы 1. В реакции 2 схемы 5 производное 4-хлорпирроло[2,3-d]пиримидина формулы XVI превращают в соответствующее соединение формулы XXV сочетанием XVI с соединением формулы R9 ОH в присутствии гидроокиси натрия. Реакцию проводят в полярном апротонном растворителе, таком как, например,тетрагидрофуран, при кипячении в течение примерно от 2 до 4 ч, предпочтительно около 3 ч. Удаление защитной группы осуществляют в соответствии с методикой, описанной выше для реакции 3 схемы 1. В реакции 1 схемы 6 производное 4-хлорпирроло[2,3-d]пиримидина формулы XVII превращают в соответствующее соединение формулы XXVI сочетанием XVII с соединением формулы SR9 в присутствии трет-бутоксида калия и полярного апротонного растворителя, такого как, например, тетрагидрофуран. Полученную в результате реакционную смесь кипятят в течение примерно от 2,5 до 5 ч, предпочтительно около 3,5 ч. Соединение формулы XXVI может быть далее подвергнуто реакции с окислителем,известным обычному специалисту в этой области, таким как перекись водорода, пероксимоносульфат калия, 3-хлорпероксибензойная кислота или трет-бутилпероксид, для образования соответствующего 4R9-cyльфинилпиppoлo[2,3-d]пиримидина или 4-R9-сульфонилпиррольных соединений. Соединения настоящего изобретения, которые являются основными по природе, способны образовывать множество различных солей с неорганическими и органическими кислотами. Хотя такие соли должны быть фармацевтически приемлемыми для введения животным, часто желательно на практике выделить сначала соединение настоящего изобретения из реакционной смеси в виде фармацевтически неприемлемой соли и затем просто превратить последнюю в свободное основание обработкой щелочным реагентом, и затем превратить это свободное основание в фармацевтически приемлемую кислотноаддитивную соль. Образующиеся в результате кислотно-аддитивные соли основных соединений по этому изобретению легко получают обработкой основного соединения по существу эквивалентным количеством выбранной минеральной или органической кислоты в среде водного растворителя или в подходящем органическом растворителе, таком как, например, метанол или этанол. При тщательном выпаривании растворителя легко получается желаемая твердая соль. Желаемая кислотная соль может также быть осаждена из раствора свободного основания в органическом растворителе добавлением к раствору подходящей минеральной или органической кислоты. Те соединения настоящего изобретения, которые являются кислотными по природе, способны образовывать соли с основаниями с различными фармакологически приемлемыми катионами. Примеры таких солей включают соли щелочных или щелочно-земельных металлов, и в частности натриевые и калиевые соли. Эти соли получают с помощью обычных методик. Химические основания, которые используются в качестве реагентов для получения фармацевтически приемлемых солей с основаниями по этому изобретению, являются такими, которые образуют в результате их присоединения нетоксичные основные соли с кислотными соединениями по настоящему изобретению. Такие нетоксичные основно-аддитивные соли включают производные таких фармакологически приемлемых катионов, как катионы натрия, калия,кальция и магния и т.д. Эти соли легко могут быть получены обработкой соответствующих кислотных соединений водным раствором, содержащим желаемые фармакологически приемлемые катионы, и затем упариванием полученного раствора досуха, предпочтительно при пониженном давлении. Альтернативно,они также могут быть получены смешиванием вместе спиртовых (низшие спирты жирного ряда) растворов кислотных соединений и желаемых алкоксидов щелочных металлов и затем упариванием полученного раствора досуха таким же образом, как описано выше. В каждом случае предпочтительно используют стехиометрические количества реагентов для обеспечения полноты прохождения реакции и максимальных выходов желаемого конечного продукта. Композиции по настоящему изобретению могут быть составлены общепринятым способом с использованием одного или нескольких фармацевтически приемлемых носителей. Таким образом, активные соединения по изобретению могут быть приготовлены в виде препаративных форм для перорального, буккального, внутриносового, парентерального (например, внутривенного, внутримышечного или подкожного) или ректального введения или в форме, подходящей для введения ингаляцией или инсуффляцией. Активные соединения по изобретению могут также быть получены в виде препаративной формы для пролонгированной доставки. Для перорального введения фармацевтические композиции могут иметь, например, форму таблеток или капсул, приготовленных общепринятыми способами с фармацевтически приемлемыми наполнителями,такими как, например, связывающие средства (например, прежелатинированный маисовый крахмал, поливинилпирролидон или гидроксипропилметилцеллюлоза); наполнители (например, лактоза, микрокристаллическая целлюлоза или фосфат кальция), смазывающие вещества (например, стеарат магния, тальк или двуокись кремния); разрыхлители (например, картофельный крахмал или модифицированный глико-8 005852 лятом натрия крахмал); или смачивающие средства (например, лаурилсульфат натрия). На таблетки может быть нанесено покрытие способами, хорошо известными в данной области. Жидкие препаративные формы для перорального введения могут быть, например, в виде растворов, сиропов или суспензий, или они могут быть представлены в виде сухого продукта для непосредственного получения перед использованием формы с применением воды или другого подходящего растворителя. Такие жидкие препаративные формы могут быть получены обычными способами с фармацевтически приемлемыми добавками, такими как, например, суспендирующие средства (например, сироп сорбита, метилцеллюлоза или гидрированные пищевые жиры); эмульгирующие средства (например, лецитин или гуммиарабик); неводные растворители (например, миндальное масло, маслянистые сложные эфиры или этиловый спирт) и консерванты(например, метиловый или пропиловый эфиры п-гидроксибензойной кислоты или сорбиновая кислота). Для буккального введения композиция может быть в виде таблеток или лепешек, приготовленных общепринятым способом. Из активных соединении по изобретению могут быть получены препаративные формы для парентерального введения путем инъекции, включая использование обычных методик катетеризации, или инфузии. Препаративные формы для инъекции могут быть представлены в виде единичной дозированной формы,например в ампулах или в упаковках для многократного приема с добавленным консервантом. Композиции могут быть в виде суспензий, растворов или эмульсий в масляных или водных растворителях и могут содержать дополнительные агенты, такие как, например, суспендирующие, стабилизирующие и/или диспергирующие агенты. Альтернативно, активный ингредиент может быть в виде порошка для реконституции перед использованием подходящим растворителем, например стерильной апирогенной водой. Из активных соединений по изобретению могут быть приготовлены ректальные композиции, такие как, например, суппозитории или удерживающие клизмы, например, содержащие обычные основы для суппозиториев, как, например, масло какао или другие глицериды. Для внутриносового введения или введения путем ингаляции активные соединения по изобретению обычно подаются в виде раствора или суспензии из распыляющего контейнера под давлением, который нажимается или накачивается больным, или в виде аэрозольной распыляемой формы из находящегося под давлением контейнера или распылителя с использованием подходящего пропеллента, например дихлордифторметана, трихлорфторметана, дихлортетрафторэтана, двуокиси углерода или другого подходящего газа. В случае находящегося под давлением аэрозоля единичная дозированная форма может быть обеспечена установкой клапана для подачи отмеренного количества. Находящийся под давлением контейнер или распылитель может содержать раствор или суспензию активного соединения. Капсулы и картриджи (сделанные, например, из желатина) для использования в ингаляторе или инсуффляторе могут быть приготовлены содержащими порошкообразную смесь соединения по изобретению и подходящую порошкообразную основу, такую как, например, лактоза или крахмал. Предлагаемая доза активных соединений по изобретению для перорального, парентерального или буккального введения для среднего взрослого человека для лечения состояний, которые упоминались выше (например, ревматоидный артрит), составляет от 0,1 до 1000 мг активного ингредиента на стандартную дозу, которая может вводиться, например, от 1 до 4 раз в день. Аэрозольные препаративные формы для лечения состояний, на которые ссылаются выше (например,астмы), для среднего взрослого человека предпочтительно готовятся так, чтобы каждая отмеренная доза или каждая порция аэрозоля содержала от 20 до 1000 мкг соединения по изобретению. Общая дневная доза в случае использования аэрозоля находится в пределах от 0,1 до 1000 мг. Введение может осуществляться несколько раз в день, например 2, 3, 4 или 8 раз, причем каждый раз вводится, например, 1, 2 или 3 дозы. Соединение формулы (I) вводится в фармацевтически приемлемой форме либо одно, либо в комбинации с одним или несколькими дополнительными средствами, которые модулируют иммунную систему млекопитающего, или с противовоспалительными средствами, которые включают, но не ограничиваются ими, циклоспорин А (например, Сандиммун или Неорал, рапамицин, FK-506 (такролимус), лефлуномид, дезоксиспергуалин, микофенолят (например, Селлсепт), азатиоприн (например, Имуран), даклизумаб (например, Зенапакс), ОКТЗ (например, Ортоколон), AtGam, аспирин, акктаминофен, ибупрофен, напроксен, пироксикам и противовоспалительные стероиды (например, преднизолон или дексаметазон); и такие средства могут быть введены как часть той же лекарственной формы или в виде отдельных дозированных форм такими же или другими способами введения и по такой же или по другим схемам введения в соответствии со стандартной фармацевтической практикой.FK-506 (такролимус) вводится перорально в дозе 0,10-0,15 мг/кг массы тела каждые 12 ч в пределах первых 48 послеоперационных часов. Доза контролируется по уровням такролимуса в сывортке. Циклоспорин А (Сандиммун в виде препаративной формы для перорального или внутривенного введения или Неорал, раствор для перорального введения или капсулы) вводится перорально в дозе 5 мг/кг массы тела каждые 12 ч в течение 48 послеоперационных часов. Доза контролируется по уровням циклоспорина А в крови. Могут быть получены готовые препаративные формы из активных веществ для пролонгированной доставки согласно методам, хорошо известным специалистам в данной области. Примеры таких препаративных форм есть в патентах США 3538214,4060598,4173626,3119742 и 3492397.-9 005852 Способность соединений формулы I или их фармацевтически приемлемых солей ингибироватьJanus-киназу 3, что, следовательно, демонстрирует их эффективность для лечения заболеваний или состояний, характеризуемых Janus-киназой 3, показана в следующих тестах in vitro. Биологические испытания Ферментативный метод анализа JAK3 (JH1:GST) В тесте киназы JAK3 используют белок, экспресированный в инфицированных бакуловирусом клетках SF9 (белок слияния GST и каталитического домена человеческой JAK3), очищенный с помощью аффинной хроматографии на глутатионсефарозе. Субстратом реакции является полиглутаминовая кислота-тирозин (PGT (4:1), Sigma catalog P0275), нанесенный на планшеты Nunc Maxi Sorp E концентрации 100 мкг/мл в течение ночи при 37 С. На утро после покрытия планшеты трижды промывают и добавляют JAK3 в ячейки, содержащие 100 мкл буфера для киназы (50 мМ N-2-гидроксиэтилпиперазин-N'2-этансульфоновая кислота (HEPES), рН 7,3, 125 мМ NaCl, 24 мМ MgCl2+0,2 мкМ АТФ+1 мМ ортованадат натрия). Реакция протекает в течение 30 мин при комнатной температуре, и планшеты промывают еще 3 раза. Уровень фосфорилированного тирозина в определенной ячейке количественно оценивают с помощью стандартного ферментного иммуносорбентного анализа (ELISA), используя антитело антифосфотирозина (ICN PY20, cat.69-151-1). Определение ингибирующей активности в отношении JAK3 с использованием DND 39/IL-4 клеточного анализаDND 39/IL-4 анализ предназначается для определения ингибиторов активности киназы JAK3, которые были бы первыми кандидатами для подавления иммунитета и/или аллергии. В анализе используют линию В-клеток, называемую DND39, которая имеет ген люциферазы, управляемый промотором IgE зародышевой линии, стабильно интегрированным в одну из хромосом. Когда эти клетки стимулируютсяIL-4, киназа JAK3, которая связана с рецептором IL-4, фосфорилирует преобразователь сигнала STAT6. Затем STAT6 присоединяется к промотору IgE зародышевой линии и начинает транскрипцию люциферазного гена. Люцифераза измеряется в лизате этих клеток при использовании системы реагентов Promega для анализа люциферазы. Клетки DND39 выращивают в среде RPMI 1640, дополненной 13% инактивированной при нагревании околоплодной сывороткой теленка, 2 мМ L-глутамином и 100 ед./мл пенициллина (Реn.)./ стрептомицина (Strep.). Поддерживают концентрацию клеток от 1 х 105 до 1 х 106 клеток/мл. Разведенные до концентрации 1 х 105 в пятницу, клетки находятся в концентрации около 1x106 в понедельник. Затем разведение составляет 1:2 в течение недели при сохранении, как требуется, объема 200 мл в сосуде. Помещают 3 х 105 клеток DND39 в 100 мкл среды RPMI 1640, дополненной 1% инактивированной при нагревании околоплодной сывороткой теленка, 2 мМ L-глутамином и 100 ед./мл Реn./Strep., в 96-ячеечный планшет с V-образным дном (Nunc). Соединения подвергают серийному разведению 1:2 в диметилсульфоксиде, начиная с 4 мМ до 1,9 мкМ, в 96-ячеечном полипропиленовом планшете, меняя пипетки после каждого разведения. Затем 5 мкл каждого разведения прибавляют в 500 мкл среды RPMI/1% сыворотки в штатив с 96 пробирками. По 125 мкл разведения соединений добавляют к клеткам и инкубируют при 37 С в атмосфере, содержащей 5% CO2, в течение 1 ч. Через час добавляют к клеткам 25 мкл IL-4 в концентрации 25 нг/мл и перемешивают. Конечная концентрация IL-4 составляет 2,5 нг/мл, и конечная концентрация соединения составляет от 20 мкМ до 156 нМ. Клетки затем инкубируют в течение ночи 16-18 ч. Планшет затем центрифугируют при 2500-3000 об./мин в настольной центрифуге в течение 5 мин. Культуральную надосадочную жидкость осторожно удаляют путем отсасывания с помощью отсасывающего устройства с 8 отверстиями. К осажденным при центрифугировании клеткам добавляют 100 мкл забуференного фосфатом физиологического раствора (ЗФР) с кальцием и магнием. Клетки повторно суспендируют в ЗФР и переносят в белый планшет OptiPlate (Packard). Добавляют 100 мкл Packard's LucLite-реагента в ячейки планшета OptiPlate. Следующие примеры иллюстрируют получение соединений по настоящему изобретению, не ограничивая его. Температуры плавления не скорректированы. Данные ЯМР-спектров указываются в миллионных доляхи приводятся относительно сигнала растворителя по дейтериевому локу (дейтерохлoроформ, если не указано иначе). Промышленные реагенты использовались без дополнительной очистки. ТГФ означает тетрагидрофуран. ДМФА N,N-диметилформамид. Масс-спектры низкого разрешения(LRMS) были сняты либо на приборе Hewlett Packard 59890 с использованием химической ионизации(аммоний), либо на приборе фирмы Fisons (или Micro Mass) с химической ионизацией при атмосферном давлении (ХИАД), смесь 50/50 ацетонитрил/вода с 0,1% муравьиной кислоты используется в качестве ионизирующего агента. Комнатная температура или температура окружающей среды означает 20-25 С. Пример 1. Циклогексилметил-(7 Н-пирроло[2,3-d]пиримидин-4-ил)амин. Метод А. Циклогексилметиламин. К раствору циклогексанона (98 мг/1 ммоль) и уксусной кислоты (120 мг/2 ммоль), растворенных в 2,0 мл 1,2-дихлорэтана, добавляли 2,0 мл 2 М раствора метиламина в метаноле и полученную в результате смесь перемешивали при комнатной температуре 4 ч. Добавляли боргидрид на полимерном носителе(1 г/2,5 ммоль) и смесь перемешивали при комнатной температуре в течение 1 ч, затем фильтровали и упаривали досуха в вакууме, получали 66 мг (40%) указанного в названии соединения в виде соли с ук- 10005852 сусной кислотой. 1H-ЯМР-спектр (400 МГц) (СD3 ОD) : 1,17-1,37 (м, 5 Н), 1,67 (уш. д, 1 Н, J=12,5 Гц),1,83 (уш. д, 2 Н, J=18,7 Гц), 1,86 (с, 3 Н), 2,04 (уш. д, 2 Н, J=10,2 Гц), 2,60 (с, 3 Н), 2,86-2,92 (м, 1 Н). Метод В. Циклогексилметил-(7 Н-пирроло[2,3-d]пиримидин-4-ил)амин. Смесь 200 мг (1,30 ммоль) 4-хлор-7 Н-пирроло[2,3-d]пиримидина (приготовленного по методу Davoll, J. Am. Chem. Soc., (1960), 82, 131), продукта, полученного методом А (539 мг/5,21 ммоль), и 3 мл трет-бутанола перемешивали в запаянной трубке при 100 С в течение 24 ч. Реакционную смесь добавляли в воду, подкисляли до рН 1 1 н. соляной кислотой (водной), дважды промывали диэтиловым эфиром и подщелачивали до рН 14 с помощью 1 н. гидроокиси натрия (NaOH). Выпавший в результате осадок отфильтровывали и сушили в вакууме, получали 263 мг (88%) указанного в названии соединения, т.пл. 177-180 С. 1 Н-ЯМР-спектр (400 МГц, СDСl3):1,11-1,22 (м, 1 Н), 1,43-1,63 (м, 4 Н), 1,73 (уш. д, 1 Н,J=13,3 Гц), 1,83-1,90 (м, 4 Н), 3,23 (с, 3 Н), 4,69 (уш., 1 Н), 6,53 (д, 1 Н, J=3,5 Гц), 7,03 (д, 1 Н, J=3,5 Гц), 8,30(с, 1 Н), 10,6 (уш., 1 Н). LRMS: 231 (М+1). Указанные в названиях примеров 2-84 соединения были получены методом, аналогичным описанному в примере 1. Пример 2. Бензилэтил-(7 Н-пирроло[2,3-d]пиримидин-4-ил)амин. Бензилэтиламин. Т.пл. 170-172 С; LRMS: 252,3. Пример 3. Метил-[(S)-1-фенилэтил]-(7 Н-пирроло[2,3-d]пиримидин-4-ил)амин. Метил[(S)-1-фенилэтил]амин. Т.пл. 131 С; LRMS: 253. Пример 4. Циклопентилметил-(7 Н-пирроло[2,3-d]пиримидин-4-ил)амин. Циклопентилметиламин. LRMS: 217,3. Пример 5. Аллилциклогексил-(7 Н-пирроло[2,3-d]пиримидин-4-ил)амин. Аллилциклогексиламин. LRMS: 257. Пример 6. Аллилциклопентил-(7 Н-пирроло[2,3-d]пиримидин-4-ил)амин. Аллилциклопентиламин. Т.пл. 173-175 С; LRMS: 243. Пример 7. Циклогексилэтил-(7 Н-пирроло[2,3-d]пиримидин-4-ил)амин. Циклогексилэтиламин. LRMS: 245,3. Пример 8. (1-Циклoгeксилэтил)метил-(7 Н-пирроло[2,3-d]пиримидин-4-ил)амин.- 13005852 Метод С. 7-Бензолсульфонил-4-хлор-7 Н-пирроло[2,3-d]пиримидин. В высушенную над пламенем колбу в атмосфере азота прибавляли 780 мг 60%-го гидрида натрия(19,5 ммоль) в минеральном масле к 30 мл диметилформамида (ДМФА) и полученную смесь охлаждали до 0 С. Медленно прибавляли в течение 5 мин раствор 2,0 г (13,0 ммоль) 4-хлор-7 Н-пирроло[2,3d]пиримидина в 10 мл ДМФА. Реакционную смесь перемешивали в течение 10 мин, за это время прекращалось генерирование водорода. Прибавляли бензолсульфонилхлорид (1,7 мл/13,0 ммоль), реакционную смесь нагревали до комнатной температуры и перемешивали в течение 1 ч. Прибавляли воду и отфильтровывали выпавший осадок, сушили в вакууме, получали 3,4 г (89%) указанного в заглавии соединения в виде твердого кристаллического вещества, т.пл. 163-167 С. Метод D. 7-Бензолсульфонил-4-хлор-6-фенил-7 Н-пирроло[2,3-d]пиримидин. В высушенной над пламенем колбе в атмосфере азота растворяли 0,53 мл (3,79 ммоль) диизопропиламина в 5 мл тетрагидрофурана (ТГФ) и раствор охлаждали до -78 С. Прибавляли н-бутиллитий (3,75 ммоль) в виде 2,5 М раствора в гексане и температуру полученной смеси доводили до 0 С при постоянном перемешивании в течение 10 мин. Реакционную смесь повторно охлаждали до -78 С и к смеси прибавляли раствор 1,0 г (3,40 ммоль) продукта, полученного по методу С, в 10 мл ТГФ в течение 10 мин. Реакционную смесь перемешивали в течение 1 ч при -78 С, в это время прибавляли 8,2 мл (4,10 ммоль) 0,5 М раствора хлористого цинка в ТГФ, температуру реакционной смеси доводили до комнатной и перемешивали 1 ч. Прибавляли иодбензол (0,46 мл/4,11 ммоль) и суспензию 197 мг тетракис(трифенилфосфин)палладия в 2 мл ТГФ. Полученную смесь перемешивали при кипячении с обратным холодильником в течение 3 ч,охлаждали до комнатной температуры и распределяли между дихлорметаном и водой. Водный слой подкисляли 1 н. соляной кислотой и экстрагировали дважды дихлорметаном. Дихлорметановые слои объединяли, промывали 1 н. соляной кислотой и раствором соли, сушили над сульфатом магния (MgSO4),фильтровали и упаривали в вакууме, получали указанное в заглавии соединение. LRMS: 370, 372 (М+2). Метод Е. 4-Хлор-6-фенил-7 Н-пирроло[2,3-d]пиримидин. Полученный методом D продукт растворяли в 10 мл ТГФ и к этому раствору прибавляли 5,0 мл метанола и 1,0 г едкого натра. Реакционную смесь перемешивали 15 мин, концентрировали в вакууме и распределяли между насыщенным водным раствором хлористого аммония (NН 4 Сl) и этилацетатoм. Полученный водный слoй дважды экстрагировали этилацетатом. Этилацетатные слои объединяли, промывали раствором соли, сушили над MgSO4, фильтровали и концентрировали в вакууме. Технический продукт очищали хроматографией на силикагеле (1:5 этилацетат/гексан), получали 0,59 г (76%) указанного в заглавии соединения в виде бледно-желтого твердого вещества, т.пл. 145 С (разл.). LRMS: 230, 232 (М+2). Метод F. Циклогексилметил-(6-фенил-7 Н-пирроло[2,3-d]пиримидин-4-ил)амин. Полученный по методу Е продукт (50 мг/0,218 ммоль) подвергали реакции с 0,12 мл N-метилциклогексиламина (0,920 ммоль), как описано в методе В. Реакционную смесь концентрировали в вакууме,прибавляли метанол и выпавший осадок отфильтровывали, получали 7 мг (10%) указанного в заглавии соединения в виде желтого твердого вещества. 1H-ЯМР-спектр (400 МГц, СDСl3) : 1,18-1,25 (м, 1 Н),1,47-1,66 (м, 4 Н), 1,75-1,90 (м, 5 Н), 3,30 (с, 3 Н), 4,74 (уш., 1 Н), 6,79 (с, 1 Н), 7,32-7,36 (м, 1 Н), 7,47-7,51 (м,2 Н), 7,77 (д, 2 Н, J=7,9 Гц), 8,33 (с, 1 Н). LRMS: 307 (М+1). Указанное в заглавии примера 86 соединение было получено методом, аналогичным описанному в примере 85. Пример 86. (1 Н-Индол-5-ил)-(6-фенил-7 Н-пирроло[2,3-d]пиримидин-4-ил)амин. 1H-Индоламин. LRMS: 326,4. Пример 87. Циклогексилметил-(6-метил-7 Н-пирроло[2,3-d]пиримидин-4-ил)амин. Циклогексилметиламин. Метод G. 7-Бензолсульфонил-4-хлор-6-метил-7 Н-пирроло[2,3-d]пиримидин. В высушенную над пламенем колбу загружали в атмосфере азота 0,57 мл (4,07 ммоль) диизопропиламина и 5,0 мл безводного ТГФ. Раствор охлаждали до -78 С и прибавляли 1,63 мл (4,08 ммоль) 2,5 М раствора н-бутиллития в гексане. Температуру полученной смеси доводили до 0 С и перемешивали 10 мин. После охлаждения смеси опять до -78 С прибавляли раствор 1,0 г (3,4 ммоль) технического продукта,полученного методом С, в 10 мл безводного ТГФ в течение 10 мин. Полученную смесь перемешивали в течение 1 ч, в это время прибавляли 0,28 мл (4,50 ммоль) иодистого метила. Реакционную смесь перемешивали в течение 2 ч, реакцию останавливали прибавлением насыщенного раствора NH4Cl и нагревали до комнатной температуры. Смесь перемешивали 5 мин, разбавляли водой и трижды экстрагировали этилацетатом. Объединенные экстракты промывали раствором соли, сушили над МgSO4, фильтровали и упаривали в вакууме, получали указанное в заглавии соединение. LRMS: 308, 310 (М+2). Метод Н. 4-Хлор-6-метил-7 Н-пирроло[2,3-d]пиримидин. У продукта, полученного по методу G, удаляли защитную группу, как описано в методе Е. Технический продукт очищали при растирании с гексаном и дихлорметаном, получали 250 мг (44%) указанного в заглавии соединения в виде твердого желтого вещества. Т.пл. 205 С (разл.). LRMS: 168, 170 (М+2). Метод I. Циклогексилметил-(6-метил-7 Н-пирроло[2,3-d]пиримидин-4-ил)амин. Полученный методом Н продукт (50 мг/0,298 ммоль) подвергали реакции с 100 мг (0,883 ммоль) Nметилциклогексиламина, как описано в методе В. Реакционную смесь обрабатывали, как в методе В, за- 14005852 исключением того, что использовали этилацетат вместо эфира. Указанное в заглавии соединение (42 мг,выход 58%) было получено в виде твердого вещества белого цвета. Т.пл. 221 С (разл.). 1H-ЯМР-спектр(М+1). Соединение, указанное в заглавии примера 88, было получено методом, аналогичным описанному в примере 87. Пример 88. Циклогексил-(6-метил-7 Н-пирроло[2,3-d]пиримидин-4-ил)амин. Циклогексиламин. LRMS: 231,3. Пример 89. 4-Циклогексилокси-7 Н-пирроло[2,3-d]пиримидин. Метод L. 7-Бензил-4-хлор-7 Н-пирроло[2,3-d]пиримидин. К перемешиваемому раствору 4-хлор-7 Н-пирроло[2,3-d]пиримидина (250 мг/1,63 ммоль) в 12 мл ДМФА прибавляли 676 мг (4,89 ммоль) карбоната калия и полученную смесь перемешивали 20 мин при комнатной температуре. Прибавляли хлористый бензил (310 мг/2,45 ммоль) и смесь перемешивали в течение 24 ч при комнатной температуре, затем фильтровали, концентрировали и остаток очищали хроматографией на силикагеле (3:1 гексан/этилацетат), получали 318 мг (80%) указанного в заглавии соединения. LRMS: 244,1 (М+1). Метод М. 7-Бензил-4-циклогексилокси-7 Н-пирроло[2,3-d]пиримидин. В высушенную над пламенем колбу в атмосфере азота загружали 84 мг (2,10 ммоль) 60%-ного гидрида натрия в минеральном масле и 3,0 мл ТГФ и смесь охлаждали до 0 С. Прибавляли циклогексанол(0,13 мл/1,70 ммоль) и реакционную смесь перемешивали в течение 5 мин. Добавляли раствор 102 мг(0,419 ммоль) полученного методом L продукта в 1,0 мл ТГФ и смесь кипятили с обратным холодильником в течение 3 ч. После охлаждения до комнатной температуры реакционную смесь подкисляли до рН 1 2 н. соляной кислотой и концентрировали в вакууме. Полученный остаток затем суспендировали в этилацетате, фильтровали и фильтрат концентрировали в вакууме, получали 76 мг (59%) указанного в заглавии соединения в виде масла. LRMS: 308 (М+1). Метод N. 4-Циклогексилокси-7 Н-пирроло[2,3-d]пиримидин. К жидкому аммиаку (6,0 мл) при -78 С прибавляли 33 мг (1,43 ммоль) металлического натрия и полученный в результате темно-синий раствор перемешивали в течение 10 мин при -78C. Прибавляли по каплям в течение 5 мин раствор 75 мг (0,244 ммоль) продукта, полученного по методу М, в 3,0 мл эфира. Полученный раствор перемешивали при -78 С в течение 1 ч, затем реакцию прекращали при добавлении 500 мг твердого хлористого аммония. После упаривания при комнатной температуре твердый остаток растирали в течение 1 ч с 25 мл этилацетата, содержащего 1 мл уксусной кислоты. Фильтрование и концентрирование в вакууме давали неочищенный продукт, который очищали с помощью препаративной тонкослойной хроматографии (силикагель, 2:1 этилацетат/гексан), получая 5 мг указанного в заглавии соединения. 1 Н-ЯМР-спектр (400 МГц, СDСl3) : 1,27-1,35 (м, 6 Н), 1,62-1,67 (м, 4 Н), 5,30-5,36 (м, 1 Н),6,55 (д, 1 Н, J=3,2 Гц), 7,11 (д, 1 Н, J=3,2 Гц), 8,37 (уш. с, 1 Н). LRMS: 218,2 (М+1). Пример 90. Метод О. 4-Циклогексилсульфанил-7 Н-пирроло[2,3-d]пиримидин. К раствору 100 мг (0,651 ммоль) 4-хлор-7 Н-пирроло[2,3-d]пиримидина в 3,0 мл ТГФ прибавляли 0,10 мл (0,818 ммоль) циклогексилмеркаптана и 100 мг (0,847 ммоль) 95%-ного трет-бутилата натрия и полученную смесь кипятили с обратным холодильником в течение 3,5 ч. После охлаждения до комнатной температуры реакционную смесь подкисляли до рН 1 2 н. соляной кислотой и концентрировали в вакууме. Остаток затем распределяли между этилацетатом и 1 н. соляной кислотой. Водный слой экстрагировали этилацетатом, этилацетатные слои объединяли, промывали солевым раствором, сушили над серно-кислым магнием, фильтровали и упаривали в вакууме. Технический продукт очищали хроматографией на силикагеле (1:3 этилацетат/гексан), получали 34 мг (22%) указанного в заглавии соединения в виде твердого белого вещества. Т.пл. 162-163 С. 1 Н-ЯМР-спектр (400 МГц) : 1,22-1,36 (м, 1 Н), 1,45-1,64(м, 5 Н), 1,75-1,79 (м, 2 Н), 2,12-2,14 (м, 2 Н), 4,18-4,20 (м, 1 Н), 6,50 (д, 1 Н, J=3,7 Гц), 7,19 (д, 1 Н, J=3,5 Гц),8,61 (с, 1 Н), 10,0 (уш. с, 1 Н). LRMS: 234 (M+1). Пример 91. 5-Хлор-4-пиперидин-1-ил-7 Н-пирроло[2,3-d]пиримидин. Метод R. 4,5-Дихлор-7 Н-пирродо[2,3-d]пиримидин. 4-Хлор-7 Н-пирроло[2,3-d]пиримидин (154 мг, 1,0 ммоль) суспендировали в 6,0 мл безводного дихлорметана в высушенной над пламенем колбе и к смеси добавляли в один прием N-хлорсукцинимид(147 мг, 1,1 ммоль). Полученную в результате смесь перемешивали 18 ч при комнатной температуре, за это время растворитель удаляли в вакууме. Остаток растирали с водой и отделяли фильтрованием, получая 137 мг (72%) указанного в заглавии соединения в виде твердого вещества серого цвета, т.пл. 224227 С (разл). LRMS: 188(М+1). Метод S. 5-Хлор-4-пиперидин-1-ил-7 Н-пирроло[2,3-d]пиримидин. Полученный методом R продукт (57 мг, 0,3 ммоль) суспендировали в 3,0 мл трет-бутанола, и к этому раствору добавляли пиперидин (90 мкл, 0,9 ммоль), и полученную смесь кипятили с обратным холо- 15005852 дильником в течение 1 ч. Реакционную смесь охлаждали до комнатной температуры и добавляли воду(4,0 мл). Доводили рН раствора до 1 1 н. НСl и затем промывали эфиром. Водный слой удаляли и доводили рН до 12 с помощью 2 н. едкого натра. Раствор затем экстрагировали 2 х 15 мл дихлорметаном и объединенные органические фракции промывали водой, затем раствором соли и сушили над MgSО 4. Выпаривание растворителя давало 45 мг твердого вещества желтого цвета, которое очищали хроматографией на силикагеле (3:1 этилацетат/гексан), получая 23 мг (32%) указанного в заглавии соединения в виде твердого вещества желтого цвета. Т.пл. 170-172 С. 1 Н-ЯМР-спектр (400 МГц, СDСl3) : 1,67-1,74(м, 6 Н), 3,65-3,67 (м, 4 Н), 7,10 (с, 1 Н), 8,31 (с, 1 Н). LRMS: 237 (М+1). Указанные в заглавиях примеров 92-94 соединения были получены методом, аналогичным описанному в примере 91. Пример 92. (5-Хлор-7 Н-пирроло[2,3-d]пиримидин-4-ил)-(3-этинилфенил)амин.(3-Этинилфенил)амин. Т.пл. 250 С. Пример 93. (5-Хлор-7 Н-пирроло[2,3-d]пиримидин-4-ил)циклогептилметиламин. Циклогептилметиламин. Т.пл. 152-153 С; LRMS: 279,8. Пример 94. (5-Хлор-7 Н-пирроло[2,3-d]пиримидин-4-ил)циклооктилметиламин. Циклооктилметиламин. Т.пл. 151-153 С; LRMS: 293,8. Пример 95. 5-Фенил-4-пиперидин-1-ил-7 Н-пирроло[2,3-d]пиримидин. Метод Т. 5-Бром-4-хлор-7 Н-пирроло[2,3-d]пиримидин. К перемешиваемому раствору 4-хлор-7 Н-пирроло[2,3-d]пиримидина (30 г/0,02 моль) в 75 мл хлороформа добавляли 3,5 г (0,02 моль) N-бромсукцинамида и полученную смесь кипятили с обратным холодильником в течение 1 ч. После охлаждения до комнатной температуры осадок отфильтровывали и сушили в вакууме, получали 4,1 г (89%) указанного в заглавии соединения. 1 Н-ЯМР-спектр (400 МГц,СDСl3) : 7,93 (д, 1 Н, J=2,8 Гц), 8,60 (с, 1 Н). Метод U. 7-Бензолсульфонил-5-бром-4-хлор-7 Н-пирроло[2,3-d]пиримидин. К суспензии полученного по методу Т продукта (4,1 г/0,018 моль) в ДМФА (15 мл), охлажденной до 0 С, прибавляли 1,0 г (0,025 моль) 60% гидрида натрия в минеральном масле и полученную смесь перемешивали в течение 15 мин при 0 С. Прибавляли бензолсульфонилхлорид (3,2 г/0,018 моль), реакционную смесь нагревали до комнатной температуры и перемешивали в течение 2 ч. Затем добавляли воду(15 мл) и полученное твердое вещество отделяли фильтрованием и сушили в вакууме, получали 5,9 г(89%) указанного в заглавии соединения. Метод V. 7-Бензолсульфонил-5-бром-4-пиперидин-1-ил-7 Н-пирроло[2,3-d]пиримидин. Смесь 2,0 г (5,37 ммоль) полученного по методу U продукта и 1,1 г (13,4 ммоль) пиперидина в 10 мл трет-бутанола нагревали при перемешивании в течение 2 ч при 60 С. После охлаждения до комнатной температуры реакционную смесь распределяли между дихлорметаном (25 мл) и водой (25 мл). Дихлорметановый слой сушили над сульфатом натрия (Na2SO4) и упаривали досуха в вакууме, получая 2,2 г(м, 4 Н), 7,53 (т, 2 Н, J=2,0 Гц), 7,60 (с, 1 Н), 7,61 (т, 1 Н, J=2,0 Гц), 8,17-8,20 (м, 2 Н), 8,43 (с, 1 Н). LRMS: 422,7, 420,7 (М+1). Метод W. 5-Фенил-4-пиперидин-1-ил-7 Н-пирроло[2,3-d]пиримидин. К перемешиваемому раствору полученного по методу V продукта (100 мг/0,237 ммоль) в 1,0 мл диоксана прибавляли 32 мг (0,261 ммоль) фенилборной кислоты и 75 мг (0,365 ммоль) трехосновного фосфата калия, затем 7 мг (0,006 ммоль) тетракис(трифенилфосфин)палладия. Полученную смесь дегазировали азотом и перемешивали 48 ч при 100 С. После охлаждения до комнатной температуры добавляли 1,0 мл метанола, затем 50 мг едкого натра и полученную смесь перемешивали 1 ч при комнатной температуре. Полученную в результате смесь затем распределяли между дихлорметаном и водой, дихлорметановый слой сушили над сульфатом магния и упаривали досуха в вакууме. Технический продукт очищали хроматографией на силикагеле (2:1 этилацетат/гексан), получали 13 мг (20%) указанного в заглавии соединения. 1 Н-ЯМР-спектр (400 МГц, СDСl3) : 1,33-1,34 (м, 4 Н), 1,43-1,44 (м, 2 Н), 3,26-3,28 (м, 4 Н) , 7,12 (с,1 Н), 7,27 (т, 1 Н, J=7,2 Гц), 7,38 (т, 2 Н, J=8,0 Гц), 7,45 (д, 2 Н, J=0,8 Гц), 8,42 (с, 1 Н). LRMS: 279,2 (М+1). Указанные в заглавиях примеров 96-99 соединения были получены методом, аналогичным описанному в примере 95. Пример 96. Циклогексилметил-(5-фенил-7 Н-пирроло[2,3-d]пиримидин-4-ил)амин. Циклогексилметиламин. Т.пл. 200 С; LRMS: 307,4. Пример 97. Циклогексил-[5-(4-фторфенил)-7 Н-пирроло[2,3-d]пиримидин-4-ил)метиламин. Циклогексилметиламин. Т.пл. 220 С; LRMS: 325,4. Пример 98. Бицикло[2,2,1]гепт-2-ил-(5-фенил-7 Н-пирроло[2,3-d]пиримидин-4-ил)амин. Бицикло[2,2,1]гепт-2-ил-амин. LRMS: 305,4. Пример 99. [5-(3-Хлорфенил)-7 Н-пирроло[2,3-d]пиримидин-4-ил]циклогексилметиламин. Циклогексилметиламин. LRMS: 455,9. Пример 100. Метод X. 4-Пиперидин-1-ил-7 Н-пирроло[2,3-d]пиримидин-5-карбонитрил.(приготовленного по методу Townsend и др., J. Am. Chem. Soc., 1969, 91, 2102) в 3,0 мл трет-бутанола добавляли пиперидин (59 мкл/0,60 ммоль). Полученную смесь затем кипятили с обратным холодильником в течение 2,5 ч и после охлаждения до комнатной температуры переносили в делительную воронку и разбавляли эфиром (20 мл). Раствор экстрагировали 2 х 10 мл 1 н. соляной кислотой, доводили рН 7 объединенных водных слоев с помощью 2 н. раствора гидроокиси калия (КОН), при этом образовывался осадок, который отфильтровывали, промывали водой и сушили в вакууме, получая 29 мг (42%) указанного в заглавии соединения в виде бесцветного твердого вещества. Т.пл. 209-211 С; 1 Н-ЯМР-спектр (400 МГц) (ацетон-d6) : 1,72-1,74 (м, 6 Н), 3,72-3,79 (м, 4 Н), 8,12(с, 1 Н), 8,29 (с, 1 Н). LRMS: 228 (М+1). Пример 101. 5-Этинил-4-пиперидин-1-ил-7 Н-пирроло[2,3-d]пиримидин. Метод Y. 4-Хлор-5-иод-7 Н-пирроло[2,3-d]пиримидин. К перемешиваемому раствору 4-хлор-7 Н-пирроло[2,3-d]примидина (30 г/0,02 моль) в 80 мл хлороформа добавляли 4,5 г (0,02 моль) N-иодсукцинимида и полученную смесь кипятили с обратным холодильником в течение 1 ч. После охлаждения до комнатной температуры осадок отфильтровывали и сушили в вакууме, получая 4,6 г (82%) указанного в заглавии соединения. Метод Z. 7-Бензолсульфонил-4-хлор-5-иод-7 Н-пирроло[2,3-d]пиримидин. Это соединение было получено, как ранее описано в методе U, с использованием полученного по методу Х продукта, получали 5,4 г (80%) вещества. LRMS: 419,6 (М+1), 279,7. Метод АА. 7-Бензолсульфонил-5-иод-4-пиперидин-1-ил-7 Н-пирроло[2,3-d]пиримидин. Это соединение было получено по методике, описанной в методе V, с использованием полученного по методу Z продукта для синтеза указанного в заглавии соединения. LRMS: 469 (М+1) 329,1. Метод ВВ. 7-Бензолсульфонил-4-пиперидин-1-ил-5-триэтилсиланилэтинил-7 Н-пирроло[2,3-d]пиримидин. В высушенную над пламенем колбу загружали в атмосфере азота 211 мг (0,5 ммоль) полученного по методу АА продукта, 19 мг (0,1 ммоль) иодида меди (I) и 58 мг (0,05 ммоль) тетракис(трифенилфосфин)палладия. Затем к этой смеси прибавляли 0,14 мл (1,0 ммоль) триэтиламина и 0,27 мл (1,5 ммоль) триэтилсилилацетилена в виде раствора в 1,5 мл безводного ДМФА. Полученную смесь перемешивали 3 ч при комнатной температуре, в это время прибавили 5,0 мл воды и смесь экстрагировали этилацетатом. Этилацетатный экстракт сушили над сульфатом магния и концентрировали в вакууме. Полученный технический продукт затем очищали хроматографией на силикагеле (7:1 гексан/этилацетат), получая 194 мг(89%) указанного в заглавии соединения. LRMS: 481 (М+1), 341. Метод СС. 5-Этинил-4-пиперидин-1-ил-7 Н-пирроло[2,3-d]пиримидин. К перемешиваемому раствору полученного методом ВВ продукта (194 мг/0,40 ммоль) в 2,0 мл безводного ТГФ прибавляли по каплям 0,4 мл (0,4 ммоль) 1 М раствора фтористого тетрабутиламмония в ТГФ. Полученную смесь перемешивали при комнатной температуре в течение 10 мин, затем переносили в раствор метанола (3,0 мл), содержащий 1 г гидроокиси калия, полученную при этом новую смесь перемешивали при комнатной температуре 15 мин и концентрировали в вакууме. Осадок распределяли между водой и этилацетатом, этилацетатный слой промывали водой и раствором соли, сушили над МgSO4 и упаривали досуха в вакууме. Технический продукт очищали хроматографией на силикагеле (2:1 этилацетат/гексан), получали 72 мг (64%) указанного в заглавии соединения в виде твердого кристаллического вещества белого цвета. Т.пл. 179-181 С. 1H-ЯМР-спектр (400 МГц, СDСl3) : 1,72 (уш. с, 6 Н), 3,20 (с,1 Н), 3,82-3,83 (м, 4 Н), 7,47 (с, 1 Н; 8,35 (с, 1 Н). LRMS: 227 (М+1). ФОРМУЛА ИЗОБРЕТЕНИЯ 1. Соединение формулы или его фармацевтически приемлемая соль, где R1 представляет группу формулыR4 выбирают из группы, состоящей из водорода, (C1-С 6)алкила или (C2-С 6)алкенила, где алкильные и алкенильные группы необязательно замещены гидроксильной группой, аминогруппой, (C1-С 4)алкокси или (C1-С 6)ациламиногруппой; или R4 является (C3-С 10)циклоалкилом, где циклоалкильная группа необязательно замещена дейтерием, гидроксильной группой, аминогруппой, трифторметилом, (C1-С 6)ацилок- 17005852 си, (C1-С 6)ациламино, (C1-С 6)алкиламино, C1-С 6)алкил)2 амино, циано, циано(C1-С 6)алкилом, трифторметил(C1-С 6)алкилом, нитро, нитро(C1-С 6)алкилом или (C1-С 6)ациламиногруппой;R5 выбирают из группы, состоящей из (С 3-С 10)циклоалкила, где циклоалкильная группа необязательно замещена от одного до пяти заместителями, выбранными из гидрокси, (C1-С 6)алкила, (С 2-С 6)алкенила; или R5 представляет и R13CO(С 3-С 10)циклоалкил, где R13 является R20O, где R20 представляет (C1 С 6)алкил; или R5 является группой формулыR7 и R8 каждый выбирают независимо из группы, включающей водород и (C1-С 6)алкил;R2 и R3 выбирают каждый независимо из группы, включающей водород, (C1-С 6)алкил и (C6-С 10) арил, где алкильные или арильные группы необязательно замещены от одного до трех галогенами; при условии, что sp2- и sp-атомы углерода алкенила не могут быть замещены гидроксильной группой или аминогруппой. 2. Соединение по п.1, где R1 является NR4R5. 3. Соединение или его фармацевтически приемлемая соль, выбранные из группы, включающей 2-4-метил-3-[метил-(7 Н-пирроло[2,3-d]пиримидин-4-ил)амино]циклогексилпропан-2-ол; 2-3-[(2-гидроксиэтил)-(7 Н-пирроло[2,3-d]пиримидин-4-иламино]-4-метилциклогексилпропан-2-ол; 2-[(5-изопропенил-2-метилциклогексил)-(7 Н-пирроло[2,3-d]пиримидин-4-ил)амино]этанол;(5-(S)-изопропенил-2-метилциклогексил)метил-(7 Н-пирроло[2,3-d]пиримидин-4-ил)амин; 2-[циклогептил-(7 Н-пирроло[2,3-d]пиримидин-4-ил)амино]этанол; 2-[циклооктил-(7 Н-пирроло[2,3-d]пиримидин-4-ил)амино]этанол и бицикло[2,2,1]гепт-2-ил-метил-(7 Н-пирроло[2,3-d]-пиримидин-4-ил)амин. 4. Фармацевтическая композиция для (а) лечения или предупреждения заболевания или состояния,выбранных из группы, включающей отторжение трансплантированного органа, волчанку, рассеянный склероз, ревматоидный артрит, псориаз, диабет типа I и осложнения, связанные с диабетом, рак, астму,атопический дерматит, аутоиммунные заболевания щитовидной железы, язвенный колит, болезнь Крона,болезнь Альцгеймера, лейкоз и другие аутоиммунные заболевания, или для (б) ингибирования протеинтирозинкиназ или Janus-киназы З (JAKЗ) у млекопитающего, включая человека, включающая количество соединения по п.1 или его фармацевтически приемлемой соли, эффективных при таких заболеваниях или состояниях, одного или в комбинации с одним или несколькими дополнительными модулирующими иммунную систему млекопитающего средствами или с противовоспалительными средствами и фармацевтически приемлемый носитель. 5. Способ ингибирования протеинтирозинкиназ или Janus-киназы З (JAKЗ) у млекопитающего,включая человека, включающий введение указанному млекопитающему эффективного количества соединения по п.1 или его фармацевтически приемлемой соли, одного или в комбинации с одним или несколькими дополнительными средствами, которые модулируют иммунную систему млекопитающего,или с противовоспалительными средствами.- 18005852 6. Способ лечения или предупреждения заболевания или состояния, выбранных из группы, включающей отторжение трансплантированного органа, волчанку, рассеянный склероз, ревматоидный артрит,псориаз, диабет типа I или осложнения, связанные с диабетом, рак, астму, атопический дерматит, аутоиммунные заболевания щитовидной железы, язвенный колит, болезнь Крона, болезнь Алдьцгеймера,лейкоз и другие аутоиммунные заболевания млекопитающего, включая человека, включающий введение указанному млекопитающему количества соединения по п.1 или его фармацевтически приемлемой соли,эффективных при лечении такого состояния, одного или в комбинации с одним или несколькими дополнительными средствами, которые модулируют иммунную систему млекопитающего, или с противовоспалительными средствами.
МПК / Метки
МПК: C07D 487/04, A61K 31/519, A61P 37/06
Метки: пирроло[2,3-d]пиримидины
Код ссылки
<a href="https://eas.patents.su/20-5852-pirrolo23-dpirimidiny.html" rel="bookmark" title="База патентов Евразийского Союза">Пирроло[2,3-d]пиримидины</a>
Способ синтеза (3as)-5,5-диоксо-2,3,3а,4-тетрагидро-1h-пирроло[2,1-c][1,2,4]бензотиадиазина

Номер патента: 4927
Опубликовано: 28.10.2004
Авторы: Фуше Эльза, Лекув Жан-Пьер, Томино Жилль, Рамсден Джеймс Эндрью, Кобли Кристофер Джеймс
МПК: C07D 513/04
Метки: синтеза, 3as)-5,5-диоксо-2,3,3а,4-тетрагидро-1h-пирроло[2,1-c][1,2,4]бензотиадиазина, способ
Формула / Реферат:
1. Способ синтеза (3aS)-5,5-диоксо-2,3,3a,4-тетрагидро-1H-пирроло[2,1-c][1,2,4]бензотиадиазина формулы (I) отличающийся тем, что 5,5-диоксо-2,3-дигидро-1H-пирроло[2,1-c][1,2,4]бензотиадиазин формулы (II) гидрогенизуют в присутствии катализатора (R)-BINAP RuCl2 (R,R)-DPEN формулы (III) в количестве от 0,4 до 2 ммоль на моль соединения формулы (II) в смеси толуола и изопропанола, в которой доля толуола составляет от 10 до 90 об.%, при давлении...
Пиразоло [3,4 - d] пиримидины с противоконвульсивным, противоаллергическим / противоастматическим действием

Номер патента: 2768
Опубликовано: 29.08.2002
Авторы: Тобер Кристине, Унферферт Клаус, Арнольд Томас, Менцер Манфред, Ланкау Ханс-Йоахим, Росток Ангелика
МПК: A61K 31/519, A61P 25/08, C07D 487/04...
Метки: противоастматическим, действием, противоаллергическим, пиразоло, пиримидины, противоконвульсивным
Формула / Реферат:
1. Соединения общей формулы 1 или их таутомеры, где Х обозначает кислород или серу и Y обозначает галоген, С1-С4-алкил, С1-С4-алкокси, трифторметил или трифторметокси, m равно 1 или 2, за исключением 2-бензил-4,5-дигидро-4-оксо-пиразоло[3,4d]пиримидина. 2. Соединения общей формулы 1, представляющие собой 2-(2-фторбензил)-пиразоло[3,4-d]пиримидин-4(5Н)-он, 2-(2-хлорбензил)-пиразоло[3,4-d]пиримидин-4(5Н)-он, ...
Композиции на основе пирроло [2,3d] пиримидинов и их применение

Номер патента: 3604
Опубликовано: 26.06.2003
Авторы: Кастелано Арлиндо Л., Маккиббен Брайан, Уиттер Дэвид Дж.
МПК: A61K 31/505, A61P 9/00, C07D 487/04...
Метки: применение, композиции, 2,3d, пирроло, основе, пиримидинов
Формула / Реферат:
1. Соединение, имеющее формулу I где каждый из R1 и R2 независимо представляет собой атом водорода, замещенный прямой (C1-C30)алкил, замещенный разветвленный (C3-C30)алкил, замещенный (C4-C10)циклоалкил, замещенный циклопропил или замещенный или незамещенный арил; где только один из R1 и R2 может быть водородом; где, когда алкил представляет собой (C1)алкил или (C2)алкил, тогда любой заместитель фенила, если он имеется, замещен; или R1 и R2...
Новая кристаллическая форма n-[4- [2- ( 2-амино-4,7-дигидро-4-оксо-3h-пирроло[ 2,3-d]пиримидин-5-ил) этил] бензоил] -l-глутаминовой кислоты и способ ее получения

Номер патента: 4684
Опубликовано: 24.06.2004
Авторы: Ройтцель-Эденс Сюзн Мари, Челиус Эрик Кристофер, Снорек Шэрон Ван Ден Берг
МПК: A61P 35/00, A61K 31/519, C07D 487/04...
Метки: новая, 2,3-d]пиримидин-5-ил, кислоты, n-[4, l-глутаминовой, бензоил, способ, получения, этил, 2-амино-4,7-дигидро-4-оксо-3h-пирроло, форма, кристаллическая
Формула / Реферат:
1. Гидратная кристаллическая форма динатриевой соли N-[4-[2-(2-амино-4,7-дигидро-4-оксо-3H-пирроло[2,3-d]пиримидин-5-ил)этил]бензоил]-L-глутаминовой кислоты ("гептагидратная кристаллическая форма"), характеризующаяся спектром дифракции рентгеновских лучей, который включает максимум, соответствующий межплоскостному расстоянию d: 7,78+ 0,04 Е, полученным измерением при 22+2шC и 20-80% относительной влажности с использованием медного источника...
Пиримидины, ингибирующие репликацию вич

Номер патента: 4049
Опубликовано: 25.12.2003
Авторы: Хо Чих Юнг, Андрис Конрад Йозеф Лодевийк Марсель, Каваш Роберт В., Лудовики Дональд Вилльям, Жанссен Поль Адриан Ян, Хэрес Ян, Кукла Майкл Джозеф, Де Жонж Марк Рене, Ван Акен Кун Жанн Альфонс, Де Корт Барт, Койманс Люсьен Мария Хенрикус
МПК: C07D 239/48, A61K 31/505, C07D 239/46...
Метки: пиримидины, ингибирующие, вич, репликацию
Формула / Реферат:
1. Соединение, имеющее формулу его N-оксид, аддитивная соль, четвертичный амин или стереохимически изомерная форма, где R1 представляет водород; арил; формил; C1-6алкилкарбонил; C1-6алкил, C1-6алкилоксикарбонил, C1-6алкил, замещенный формилом, C1-6-алкилкарбонилом, C1-6алкилоксикарбонилом, C1-6алкилкарбонилокси; C1-6алкилоксиC1-6алкилкарбонил, замещенный C1-6алкилоксикарбонилом; R2a представляет циано или аминокарбонил; L представляет...