Соединения пирроло[2,3-d]пиримидина в качестве иммунодепрессантов
Номер патента: 6153
Опубликовано: 27.10.2005
Авторы: Флэнэган Марк Эдвард, Манчхоф Майкл Джон, Блуменкопф Тодд Эндрю







Формула / Реферат
1. Соединение формулы

или его фармацевтически приемлемая соль;
где R1 представляет собой группу формулы

где R4 представляет собой (C1-C6)алкил;
R5 представляет собой пиперидинил, который должен быть замещен группами в количестве от одной до пяти, состоящими из (C1-C6)алкила, циано(C1-C6)алкила, [(циано(C1-C6)алкил)((C1-C6)алкил)амино]ацила, гидрокси(C1-C6)алкилацила, нитро(C1-C6)алкилацила, (C1-C6)алкилSO2HN(C1-C6)алкилацила, (C1-C6)алкоксиацила, циано(C1-C6)алкил-O-C(=O), (C1-C6)алкил-O-C(=O), циано(C1-C6)алкил-HN-C(=O), циано(C1-C6)алкил-(C1-C6)алкилN-C(=O), (C1-C6)алкилсульфонил(C1-C6)алкилацила, трифторметил(C1-C6)алкилсульфонила, (C3-C10)циклоалкил(C1-C6)алкил-C(=O)NH(C1-C6)алкилсульфонила, (C3-C10)циклоалкил(C1-C6)алкила, (C1-C6)алкилацил(C1-C6)алкила, (C1-C6)алкил-(C(=O))2, (C1-C6)алкокси-(C(=O))2, [((C1-C6)алкил)2амино]ацил(C1-C6)алкила; и группы формулы II

где a равен 0, 1, 2, 3 или 4;
b, c, e, f и g, каждый независимо, равен 0 или 1;
d равен 0, 1, 2 или 3;
X представляет собой S(O)2, кислород или карбонил;
Y представляет собой S(O)2 или карбонил; и
Z представляет собой карбонил, C(O)O-, C(O)NR-, где R представляет собой водород или (C1-C6)алкил; или Z представляет собой S(O)2;
R6, R7, R8, R9, R10 и R11, каждый независимо, выбран из группы, состоящей из водорода или (C1-C6)алкила, возможно, замещенного дейтерием, гидрокси, амино, трифторметилом, (C1-C6)ацилокси, (C1-C6)ациламино, (C1-C6)алкиламино, ((C1-C6)алкил)2амино, циано, циано(C1-C6)алкилом, трифторметил(C1-C6)алкилом, нитро, нитро(C1-C6)алкилом или (C1-C6)ациламино;
R12 представляет собой (C6-C10)арил, (C2-C9)гетероарил, (C3-C10)циклоалкил или (C2-C9)гетероциклоалкил, где арильная, гетероарильная, циклоалкильная и гетероциклоалкильная группы, возможно, замещены группами в количестве от одной до четырех, состоящими из водорода, (C1-C6)алкокси, галогено, оксо, (C1-C6)алкоксиацила, (C1-C6)алкилацила, нитро, циано, (C1-C6)алкила, гидрокси(C1-C6)алкила, трифторметила, карбокси, гидрокси, (C1-C6)алкоксиацилHN-, аминоацила, (C1-C6)алкилHNацила, ((C1-C6)алкил)2Nацила, карбокси(C1-C6)алкила, амино, (C1-C6)алкилHN, ((C1-C6)алкил)2N, амино(C1-C6)алкилHN, ((C1-C6)алкил)2N(C1-C6)алкилHN, аминосульфонила, (C1-C6)алкоксиацил(C1-C6)алкила, (C2-C9)гетероциклоалкил(C1-C6)алкилHN, амино(C1-C6)алкокси, (C1-C6)алкоксиацилHN(C1-C6)алкокси, (C1-C6)алкилсульфонила, ((C1-C6)алкил)2N(C1-C6)алкила, (C2-C9)гетероциклоалкила, (C6-C10)арил(C1-C6)алкиламино, ((C1-C6)алкил)2N(C1-C6)алкила, (C1-C6)алкилациламино, ((C1-C6)алкил)2N((C1-C6)алкил)HNсульфонила, (C2-C9)гетероариламиносульфонила, (C2-C9)гетероциклоалкилацила, (C2-C9)гетероарил(C1-C6)алкилацила, (C1-C6)алкиламиноацила, (C2-C9)гетероциклоалкилсульфонила, (C1-C6)алкиламиносульфонила, гидрокси(C1-C6)алкиламиносульфонила и галогеносульфонила;
R2 и R3, каждый независимо, представляет собой водород;
при условии, что R5 должен быть замещен группой формулы II.
2. Соединение по п.1, где указанное соединение выбрано из группы, состоящей из
4-{4-метил-3-[метил-(7H-пирроло[2,3-d]пиримидин-4-ил)амино]пиперидин-1-илметил}бензолсульфонамида;
(4-сульфамоилфенил)амида 4-метил-3-[метил-(7H-пирроло[2,3-d]пиримидин-4-ил)амино]пиперидин-1-карбоновой кислоты;
(4-нитрофенил)амида 4-метил-3-[метил-(7H-пирроло[2,3-d]пиримидин-4-ил)амино]пиперидин-1-карбоновой кислоты;
1-{4-метил-3-[метил-(7H-пирроло[2,3-d]пиримидин-4-ил)амино]пиперидин-1-ил}-2-тетразол-1-ил-этанона;
(4-метилсульфамоилфенил)амида 4-метил-3-[метил-(7H-пирроло[2,3-d]пиримидин-4-ил)амино]пиперидин-1-карбоновой кислоты;
(3-гидроксипирролидин-1-ил)-{4-метил-3-[метил-(7H-пирроло[2,3-d]пиримидин-4-ил)амино]пиперидин-1-ил}метанона;
[2-({4-метил-3-[метил-(7H-пирроло[2,3-d]пиримидин-4-ил)амино]пиперидин-1-карбонил}амино)тиазол-4-ил]уксусной кислоты;
5-(2-{4-метил-3-[метил-(7H-пирроло[2,3-d]пиримидин-4-ил)амино]пиперидин-1-ил}-2-оксоэтил)тиазолидин-2,4-диона;
метил[4-метил-1-(5-нитротиазол-2-ил)пиперидин-3-ил]-(7H-пирроло[2,3-d]пиримидин-4-ил)амина;
этилового эфира [2-({4-метил-3-[метил-(7H-пирроло[2,3-d]пиримидин-4-ил)амино]пиперидин-1-карбонил}амино)тиазол-4-ил]уксусной кислоты;
(4-метансульфонилфенил)амида 4-метил-3-[метил-(7H-пирроло[2,3-d]пиримидин-4-ил)амино]пиперидин-1-карбоновой кислоты;
тиазол-2-иламида 4-метил-3-[метил-(7H-пирроло[2,3-d]пиримидин-4-ил)амино]пиперидин-1-карбоновой кислоты;
(4-цианофенил)амида 4-метил-3-[метил-(7H-пирроло[2,3-d]пиримидин-4-ил)амино]пиперидин-1-карбоновой кислоты;
{4-метил-3-[метил-(7H-пирроло[2,3-d]пиримидин-4-ил)амино]пиперидин-1-ил}пирролидин-1-ил-метанона;
(2-{4-метил-3-[метил-(7H-пирроло[2,3-d]пиримидин-4-ил)амино]пиперидин-1-сульфонил}этил)амида фуран-2-карбоновой кислоты;
{4-метил-3-[метил-(7H-пирроло[2,3-d]пиримидин-4-ил)амино]пиперидин-1-ил}(тетрагидрофуран-3-ил)метанона;
изоксазол-3-иламида 4-метил-3-[метил-(7H-пирроло[2,3-d]пиримидин-4-ил)амино]пиперидин-1-карбоновой кислоты;
(6-цианопиридин-3-ил)амида 4-метил-3-[метил-(7H-пирроло[2,3-d]пиримидин-4-ил)амино]пиперидин-1-карбоновой кислоты;
(4-метилтиазол-2-ил)амида 4-метил-3-[метил-(7H-пирроло[2,3-d]пиримидин-4-ил)амино]пиперидин-1-карбоновой кислоты;
2-циклопропил-1-{4-метил-3-[метил-(7H-пирроло[2,3-d]пиримидин-4-ил)амино]пиперидин-1-ил}этанона;
циклопентил-{4-метил-3-[метил-(7H-пирроло[2,3-d]пиримидин-4-ил)амино]пиперидин-1-ил}метанона;
(3-метилизоксазол-4-ил)амида 4-метил-3-[метил-(7H-пирроло[2,3-d]пиримидин-4-ил)амино]пиперидин-1-карбоновой кислоты;
[4-({4-метил-3-[метил-(7H-пирроло[2,3-d]пиримидин-4-ил)амино]пиперидин-1-карбонил}амино)фенил]уксусной кислоты;
[1-(5-аминотиазол-2-ил)-4-метилпиперидин-3-ил]метил-(7H-пирролю[2,3-d]пиримидин-4-ил)амина;
(3-метилизотиазол-5-ил)амида 4-метил-3-[метил-(7H-пирроло[2,3-d]пиримидин-4-ил)амино]пиперидин-1-карбоновой кислоты и
3-{4-метил-3-[метил-(7H-пирроло[2,3-d]пиримидин-4-ил)амино]пиперидин-1-карбонил}циклопентанона.
3. Фармацевтическая композиция для (а) лечения или предупреждения расстройства или состояния, выбранного из отторжения трансплантата органа, ксенотрансплантации, волчанки, рассеянного склероза, ревматоидного артрита, псориаза, диабета I типа и осложнений диабета, рака, астмы, атопического дерматита, аутоиммунных расстройств щитовидной железы, неспецифического язвенного колита, болезни Крона, болезни Альцгеймера, лейкемии и других аутоиммунных заболеваний, или (б) ингибирования протеинкиназ, в частности Janus Киназы 3 (JAK3), у млекопитающего, включая человека, содержащая количество соединения по п.1 или его фармацевтически приемлемой соли, отдельно или в комбинации с одним или более чем одним дополнительным агентом, который модулирует иммунную систему млекопитающего, или с противовоспалительными агентами, эффективное при таких расстройствах или состояниях, и фармацевтически приемлемый носитель.
4. Способ ингибирования протеинкиназ, в частности Janus Киназы 3 (JAK3), у млекопитающего, включая человека, при котором указанному млекопитающему вводят эффективное количество соединения по п.1 или его фармацевтически приемлемой соли, отдельно или в комбинации с одним или более чем одним дополнительным агентом, который модулирует иммунную систему млекопитающего, или с противовоспалительными агентами.
5. Способ лечения или предупреждения расстройства или состояния, выбранного из отторжения трансплантата органа, ксенотрансплантации, волчанки, рассеянного склероза, ревматоидного артрита, псориаза, диабета I типа и осложнений диабета, рака, астмы, атопического дерматита, аутоиммунных расстройств щитовидной железы, неспецифического язвенного колита, болезни Крона, болезни Альцгеймера, лейкемии и других аутоиммунных заболеваний у млекопитающего, включая человека, при котором указанному млекопитающему вводят количество соединения по п.1 или его фармацевтически приемлемой соли, отдельно или в комбинации с одним или более чем одним дополнительным агентом, который модулирует иммунную систему млекопитающего, или с противовоспалительными агентами, эффективное при лечении такого состояния.
Текст
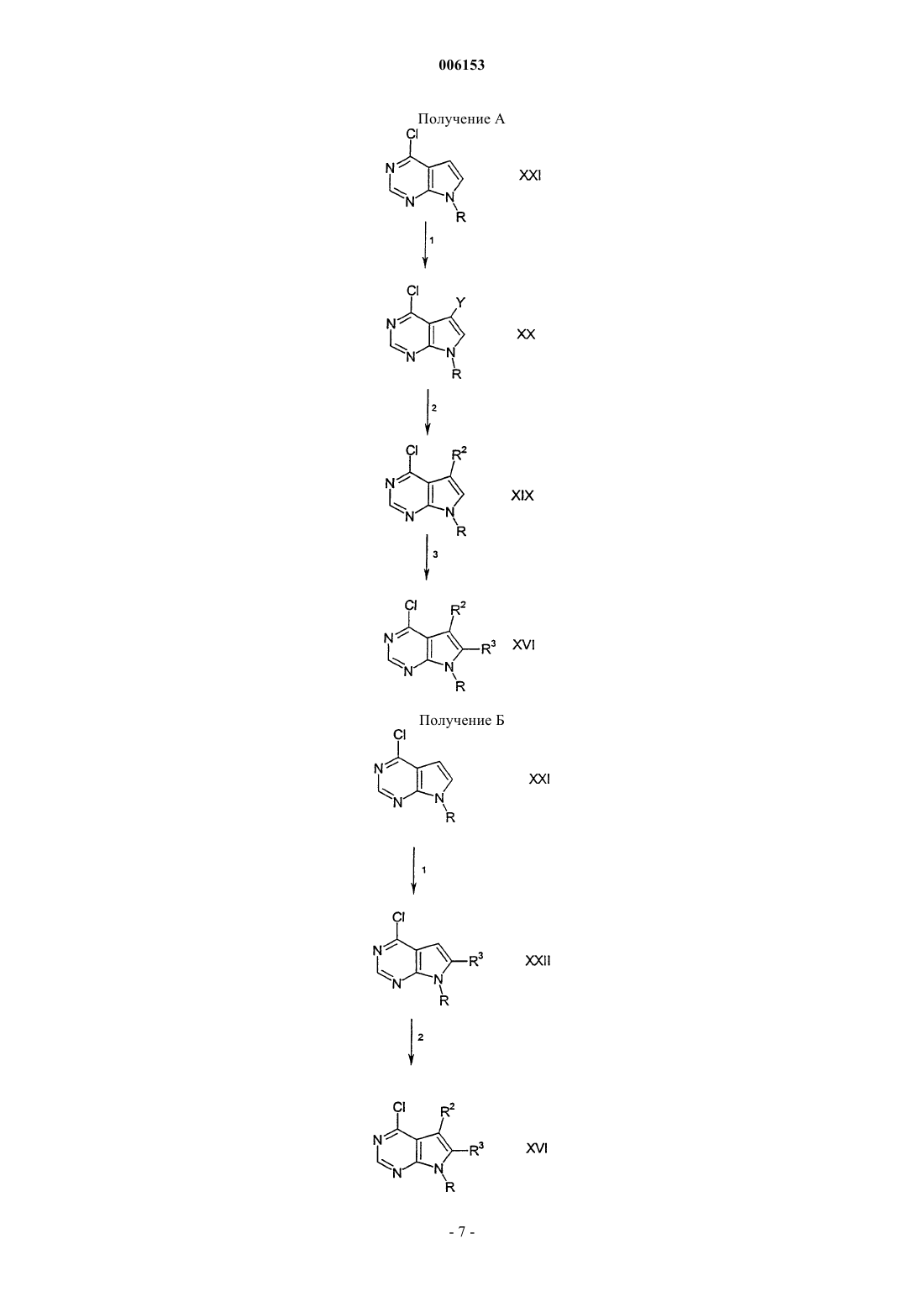
006153 Предшествующий уровень техники Настоящее изобретение относится к соединениям пирроло[2,3-d]пиримидина, которые являются ингибиторами протеинкиназ, таких как фермент Janus киназа 3 (в дальнейшем также называемая JAK3),и как таковые полезны в качестве иммунодепрессантов для трансплантатов органов, ксенотрансплантации, в терапии волчанки, рассеянного склероза, ревматоидного артрита, псориаза, диабета I типа и осложнений диабета, рака, астмы, атопического дерматита, аутоиммунных расстройств щитовидной железы, неспецифического язвенного колита, болезни Крона, болезни Альцгеймера, лейкемии и других показаний, при которых желательна иммунодепрессия. Данное изобретение также относится к способу применения таких соединений в лечении упомянутых выше показаний у млекопитающих, в особенности у людей, и фармацевтическим композициям, полезным для этого.JAK3 является членом семейства Janus протеинкиназ. Несмотря на то, что другие члены этого семейства экспрессируются, по существу, во всех тканях, экспрессия JAK3 ограничивается гемопоэтическими клетками. Это согласуется с ее важной ролью в передаче сигналов через рецепторы для интерлейкинов ИЛ-2, ИЛ-4, ИЛ-7, ИЛ-9 и ИЛ-15 посредством нековалентного связывания JAK3 с гамма-цепью,общей для этих многоцепочечных рецепторов. Были выявлены XSCID (Х-сцепленная форма тяжелой комбинированной иммунной недостаточности) популяции пациентов со значительно пониженными уровнями белка JAK3 или с генетическими дефектами в отношении общей гамма-цепи, означающие, что иммунодепрессия должна являться результатом блокирования передачи сигналов через JAK3 путь метаболизма. Эксперименты на животных дали основание предположить, что JAK3 не только играет решающую роль в созревании В- и Т-лимфоцитов, но также и то, что JAK3 является неотъемлемым требованием для поддержания функции Т-клеток. Модуляция иммунной активности посредством этого нового механизма может быть полезной в лечении нарушений пролиферации Т-клеток, таких как отторжение трансплантата и аутоиммунные болезни. Краткое изложение сущности изобретения Настоящее изобретение относится к соединению формулы или его фармацевтически приемлемой соли; где R1 представляет собой группу формулыR5 представляет собой (С 2-С 9)гетероциклоалкил, где гетероциклоалкильные группы должны быть замещены группами в количестве от одной до пяти, состоящими из карбокси, циано, амино, дейтерия,гидрокси, (С 1-С 6)алкила, (С 1-С 6)алкокси, галогено, (С 1-С 6)ацила, (С 1-С 6)алкиламино, амино(С 1-С 6)алкила,(С 1-С 6)алкокси-СО-NН, (С 1-С 6)алкиламино-СО-, (C2-С 6)алкенила, (С 2-С 6)алкинила, (С 1-С 6)алкиламино,амино(С 1-С 6)алкила, гидрокси(С 1-С 6)алкила, (С 1-С 6)алкокси(С 1-С 6)алкила, (С 1-С 6)ацилокси(С 1-С 6)алкила,нитро, циано(С 1-С 6)алкила, галогено(С 1-С 6)алкила, нитро(С 1-С 6)алкила, трифторметила, трифторметил(С 1-С 6) алкила, (С 1-С 6)ациламино, (С 1-С 6)ациламино(С 1-С 6)алкила, (С 1-С 6)алкокси(С 1-С 6)ациламино, амино(С 1 С 6)ацила, амино(С 1-С 6)ацил(С 1-С 6)алкила, (С 1-С 6)алкиламино(С 1-С 6)ацила, С 1-С 6)алкил)2 амино(С 1-С 6) ацила, R15R16N-CO-O-, R15R16N-CO-(С 1-С 6)aлкилa, (С 1-С 6)алкил-S(O)m, R15R16NS(O)m, R15R16NS(O)m(С 1 С 6)алкила, R15S(O)mR16N, R15S(O)mR16N(С 1-С 6)aлкилa, где m равен 0, 1 или 2 и R15 и R16, каждый независимо, выбран из водорода или (С 1-С 6)алкила; и группы формулыZ представляет собой карбонил, С(O)O-, C(O)NR-, где R представляет собой водород или (С 1-С 6)алкил; или Z представляет собой S(O)n, где n равен 0, 1 или 2;R6, R7, R8, R9, R10 и R11, каждый независимо, выбран из группы, состоящей из водорода или (С 1-С 6) алкила, возможно, замещенного дейтерием, гидрокси, амино, трифторметилом, (С 1-С 6)ацилокси, (С 1-С 6) ациламино, (С 1-С 6)алкиламино, С 1-С 6)алкил)2 амино, циано, циано(С 1-С 6)алкилом, трифторметил(С 1-С 6) алкилом, нитро, нитро(С 1-С 6)алкилом или (С 1-С 6)ациламино;R12 представляет собой (С 6-С 10)арил, (С 2-С 9)гетероарил, (С 3-С 10)циклоалкил или (С 2-С 9)гетероциклоалкил, где арильная, гетероарильная, циклоалкильная и гетероциклоалкильная группы, возможно, замещены группами в количестве от одной до четырех, состоящими из водорода, дейтерия, амино, галогено, оксо, гидрокси, нитро, карбокси, (С 2-С 6)алкенила, (С 2-С 6)алкинила, трифторметила, трифторметокси,(С 1-С 6)алкила, (С 1-С 6)алкокси, (С 3-С 10)циклоалкила, (С 1-С 6)алкил-СО-NН-, (С 1-С 6)алкокси-СО-NH-, (С 1 С 6)алкил-СО-NН-(С 1-С 6)алкила, (С 1-С 6)алкокси-СО-NН-(С 1-С 6)алкила, (С 1-С 6)алкокси-СО-NН-(С 1-С 6)алкокси, карбокси, карбокси(С 1-С 6)алкила, карбокси(С 1-С 6)алкокси, бензилоксикарбонил(С 1-С 6)алкокси,(С 1-С 6)алкоксикарбонил(С 1-С 6)алкокси, (С 6-С 10)арила, амино, амино(С 1-С 6)алкила, (С 1-С 6)алкоксикарбониламино, (С 6-С 10)арил(С 1-С 6)алкоксикарбониламино, (С 1-С 6)алкиламино, С 1-С 6)алкил)2 амино, (С 1-С 6) алкиламино(С 1-С 6)алкила, С 1-С 6)алкил)2 амино(С 1-С 6)алкила, гидрокси, (С 1-С 6)алкокси, карбокси, карбокси(С 1-С 6)алкила, (С 1-С 6)алкоксикарбонила, (С 1-С 6)алкоксикарбонил(С 1-С 6)алкила, (С 1-С 6)алкоксиСО-NH-, (С 1-С 6)алкил-СО-NН-, циано, (С 5-С 9)гетероциклоалкила, амино-CO-NH-, (С 1-С 6)алкиламиноСО-NН-, С 1-С 6)алкил)2 амино-СО-NН-, (С 6-С 10)ариламино-CO-NH-, (С 5-С 9)гетероариламино-СО-NН-,(С 1-С 6)алкиламино-СО-NН-(С 1-С 6)алкила, С 1-С 6)алкил)2 амино-СО-NН-(С 1-С 6)алкила, (С 6-С 10)ариламино-СО-NН-(С 1-С 6)алкила, (С 5-С 9)гетероариламино-СО-NН-(С 1-С 6)алкила, (С 1-С 6)алкилциано, (С 1-С 6)алкилкарбокси(С 1-С 6)алкокси, (С 1-С 6)алкилкарбокси, сульфониламино, аминосульфонила, сульфониламино(С 1-С 6)алкила, сульфониламинокарбокси(С 1-С 6)алкила, (С 1-С 6)алкилсульфонила, (С 1-С 6)алкилсульфониламино, (С 1-С 6)алкилсульфониламино(С 1-С 6)алкила, (С 6-С 10)арилсульфонила, (С 6-С 10)арилсульфониламино, (С 6-С 10)арилсульфониламино(С 1-С 6)алкила, (С 1-С 6)алкилсульфониламино, (С 1-С 6)алкилсульфониламино(С 1-С 6)алкила, (С 3-С 10)циклоалкила, (С 3-С 10)циклоалкокси, (С 1-С 6)алкиламино, С 1-С 6)алкил)2 амино, (С 6-С 10)ариламино, (С 1-С 6)алкилтио, (С 6-С 10)арилтио, (С 1-С 6)алкилсульфинила, (С 6-С 10)арилсульфинила, (С 1-С 6)алкилсульфонила, (С 6-С 10)арилсульфонила, (С 1-С 6)ацила, (С 1-С 6)алкокси-СО-NН-,(С 1-С 6)алкиламино-СО-, (С 5-С 9)гетероарила, (С 2-С 9)гетероциклоалкила или (С 6-С 10)арила, где гетероарильная, гетероциклоалкильная и арильная группы, которые являются, возможно, замещенными по R12, могут быть дополнительно замещены группами в количестве от одной до трех, состоящими из галогено, (С 1-С 6) алкила, (С 1-С 6)алкил-СО-NH-, (С 1-С 6)алкокси-СО-NН-, (С 1-С 6)алкил-СО-NН-(С 1-С 6)алкила, (С 1-С 6)алкоксиСО-NН-(С 1-С 6)алкила, (С 1-С 6)алкокси-СО-NН-(С 1-С 6)алкокси, карбокси, карбокси(С 1-С 6)алкила, карбоксиR2 и R3, каждый независимо, выбран из группы, состоящей из водорода, дейтерия, амино, галогено,гидрокси, нитро, карбокси, (С 2-С 6)алкенила, (С 2-С 6)алкинила, трифторметила, трифторметокси, (С 1-С 6) алкила, (С 1-С 6)алкокси, (С 3-С 10)циклоалкила, где алкильная, алкоксильная или циклоалкильная группы,-2 006153 возможно, замещены группами в количестве от одной до трех, выбранными из галогено, гидрокси, карбокси, амино(С 1-С 6)алкилтио, (С 1-С 6)алкиламино, С 1-С 6)алкил)2 амино, (С 5-С 9)гетероарила, (С 2-С 9)гетероциклоалкила, (С 3-С 9)циклоалкила или (С 6-С 10)арила; или R2 и R3, каждый независимо, представляет собой (С 3-С 10)циклоалкил, (С 3-С 10)циклоалкокси, (С 1-С 6)алкиламино, С 1-С 6)алкил)2 амино, (С 6-С 10)ариламино, (С 1-С 6)алкилтио, (С 6-С 10)арилтио, (С 1-С 6)алкилсульфинил, (С 6-С 10)арилсульфинил, (С 1-С 6)алкилсульфонил, (С 6-С 10)арилсульфонил, (С 1-С 6)ацил, (С 1-С 6)алкокси-СО-NH-, (С 1-С 6)алкиламино-СО-, (С 5 С 9)гетероарил, (С 2-С 9)гетероциклоалкил или (С 6-С 10)арил, где гетероарильная, гетероциклоалкильная и арильная группы, возможно, замещены заместителями в количестве от одного до трех галогено, (С 1-С 6) алкилом, (С 1-С 6)алкил-СО-NН-, (С 1-С 6)алкокси-СО-NН-, (С 1-С 6)алкил-СО-NН-(С 1-С 6)алкилом, (С 1-С 6)алкокси-СО-NН-(С 1-С 6)алкилом, (С 1-С 6)алкокси-СО-NН-(С 1-С 6)алкокси, карбокси, карбокси(С 1-С 6)алкилом,карбокси(С 1-С 6)алкокси, бензилоксикарбонил(С 1-С 6)алкокси, (С 1-С 6)алкоксикарбонил(С 1-С 6)алкокси, (С 6-С 10) арилом, амино, амино(С 1-С 6)алкилом, (С 1-С 6)алкоксикарбониламино, (С 6-С 10)арил(С 1-С 6)алкоксикарбониламино, (С 1-С 6)алкиламино, С 1-С 6)алкил)2 амино, (С 1-С 6)алкиламино(С 1-С 6)алкилом, С 1-С 6)алкил)2 амино(С 1-С 6)алкилом, гидрокси, (С 1-С 6)алкокси, карбокси, карбокси(С 1-С 6)алкилом, (С 1-С 6)алкоксикарбонилом, (С 1-С 6)алкоксикарбонил(С 1-С 6)алкилом, (С 1-С 6)алкокси-CO-NH-, (С 1-С 6)алкил-СО-NН-, циано,(С 5-С 9)гетероциклоалкилом, амино-СО-NH-, (С 1-С 6)алкиламино-СО-NН-, С 1-С 6)алкил)2 амино-СО-NН-,(С 6-С 10)ариламино-СО-NН-, (С 5-С 9)гетероариламино-СО-NН-, (С 1-С 6)алкиламино-СО-NН-(С 1-С 6)алкилом,С 1-С 6)алкил)2 амино-СО-NН-(С 1-С 6)алкилом, (С 6-С 10)ариламино-СО-NН-(С 1-С 6)алкилом, (С 5-С 9)гетероариламино-СО-NН-(С 1-С 6)алкилом, (С 1-С 6)алкилсульфонилом, (С 1-С 6)алкилсульфониламино, (С 1-С 6)алкилсульфониламино(С 1-С 6)алкилом, (С 6-С 10)арилсульфонилом, (С 6-С 10)арилсульфониламино, (С 6-С 10) арилсульфониламино(С 1-С 6)алкилом, (С 1-С 6)алкилсульфониламино, (С 1-С 6)алкилсульфониламино(С 1-С 6) алкилом, (C5-С 9)гетероарилом или (С 2-С 9)гетероциклоалкилом; при условии, что R5 должен быть замещен группой формулы II. Настоящее изобретение также относится к фармацевтически приемлемым солям присоединения кислоты соединений формулы I. Кислотами, которые используют для получения фармацевтически приемлемых солей присоединения кислоты вышеупомянутых основных соединений по данному изобретению,являются кислоты, которые образуют нетоксичные соли присоединения кислоты, то есть соли, содержащие фармакологически приемлемые анионы, такие как соли гидрохлорид, гидробромид, гидройодид,нитрат, сульфат, бисульфат, фосфат, гидрофосфат, ацетат, лактат, цитрат, кислый цитрат, тартрат, битартрат, сукцинат, малеат, фумарат, глюконат, сахарат, бензоат, метансульфонат, этансульфонат, бензолсульфонат, паратолуолсульфонат и памоат (то есть 1,1'-метилен-бис-(2-гидрокси-3-нафтоат. Данное изобретение также относится к солям присоединения оснований формулы I. Химические основания, которые можно использовать в качестве реагентов для получения фармацевтически приемлемых основных солей тех соединений формулы I, которые являются кислыми по своей природе, представляют собой основания, которые образуют нетоксичные основные соли с такими соединениями. Такие нетоксичные основные соли включают в себя, но не ограничиваются ими, соли, производные от таких фармакологически приемлемых катионов, как катионы щелочных металлов (например, калия и натрия) и катионы щелочно-земельных металлов (например, кальция и магния), аммониевые или растворимые в воде соли присоединения аминов, таких как N-метилглюкамин-(меглумин), и низший алканоламмоний, и другие основные соли фармацевтически приемлемых органических аминов. Термин "Охоnе" является названием моноперсульфатного соединения, применяемого в данном изобретении, имеющего формулу 2KHSO5KHSO4K2SO4, от Aldrich Chemical Company, P.O. Box 2060,Milwaukee, WI 53201, USA. Термин "алкил", как он использован здесь, если не указано иначе, включает в себя насыщенные моновалентные углеводородные радикалы, имеющие группировки с прямой или разветвленной цепью или их комбинации. Термин "алкокси", как он использован здесь, включает в себя O-алкильные группы, где "алкил" является таким, как определено выше. Термин "галогено", как он использован здесь, если не указано иначе, включает в себя фторо, хлоро,бромо или йодо. Соединения по настоящему изобретению могут содержать двойные связи. Когда такие связи присутствуют, то соединения по данному изобретению существуют в виде цис- и транс-конфигураций или в виде их смесей. Если не указано иначе, алкильные и алкенильные группы, упоминаемые здесь, а также алкильные группировки других групп, упоминаемых здесь (например, алкокси), могут быть линейными или разветвленными, а также они могут быть циклическими (например, циклопропил, циклобутил, циклопентил, циклогексил или циклогептил) или быть линейными или разветвленными и содержать циклические группировки. Если не указано иначе, галоген включает в себя фтор, хлор, бром и йод.(С 2-С 9)гетероциклоалкил, как он использован здесь, относится к пирролидинилу, тетрагидрофуранилу, дигидрофуранилу, тетрагидропиранилу, пиранилу, тиопиранилу, азиридинилу, оксиранилу, метилендиоксилу, хроменилу, изоксазолидинилу, 1,3-оксазолидин-3-илу, изотиазолидинилу, 1,3-тиазолидин 3-илу, 1,2-пиразолидин-2-илу, 1,3-пиразолидин-1-илу, пиперидинилу, тиоморфолинилу, 1,2-тетрагидро-3 006153 тиазин-2-илу, 1,3-тетрагидротиазин-3-илу, тетрагидротиадиазинилу, морфолинилу, 1,2-тетрагидродиазин-2-илу, 1,3-тетрагидродиазин-1-илу, тетрагидроазепинилу, пиперазинилу, хроманилу и т.д. Специалисту в данной области техники понятно, что связывание указанных (С 2-С 9)гeтероциклоалкильных колец осуществляется через атом углерода или sр 3-гибридизованный гетероатом азота.(С 2-С 9)гетероарил, как он использован здесь, относится к фурилу, тиенилу, тиазолилу, пиразолилу,изотиазолилу, оксазолилу, изоксазолилу, пирролилу, триазолилу, тетразолилу, имидазолилу, 1,3,5-оксадиазолилу, 1,2,4-оксадиазолилу, 1,2,3-оксадиазолилу, 1,3,5-тиадиазолилу, 1,2,3-тиадиазолилу, 1,2,4-тиадиазолилу, пиридилу, пиримидилу, пиразинилу, пиридазинилу, 1,2,4-триазинилу, 1,2,3-триазинилу, 1,3,5 триазинилу, пиразоло[3,4-b]пиридинилу, циннолинилу, птеридинилу, пуринилу, 6,7-дигидро-5 Н-[1]пириндинилу, бензо[b]тиофенилу, 5,6,7,8-тетрагидрохинолин-3-илу, бензоксазолилу, бензотиазолилу, бензизотиазолилу, бензизоксазолилу, бензимидазолилу, тианафтенилу, изотианафтенилу, бензофуранилу,изобензофуранилу, изоиндолилу, индолилу, индолизинилу, индазолилу, изохинолилу, хинолилу, фталазинилу, хиноксалинилу, хиназолинилу, бензоксазинилу и т.д. Специалисту в данной области техники понятно, что связывание указанных (С 2-С 9)гетероциклоалкильных колец осуществляется через атом углерода или sp3-гибридизованный гетероатом азота.(С 6-С 10)арил, как он использован здесь, относится к фенилу или нафтилу. Соединения формулы I можно вводить в фармацевтически приемлемой форме или отдельно, или в комбинации с одним или более чем одним дополнительным агентом, который модулирует иммунную систему млекопитающего, или с противовоспалительными агентами. Эти агенты могут включать в себя циклоспорин А (например, Sandimmune или Neoral), рапамицин, FK-506 (такролимус), лефлуномид,деоксиспергуалин, микофенолат (например, Cellcept), азатиоприн (например, Imuran), даклизумаб (например, Zenapax), ОКТЗ (например, Orthoclone), AtGam, аспирин, ацетаминофен, ибупрофен, напроксен, пироксикам и противовоспалительные стероиды (например, преднизолон или дексаметазон), но не ограничиваются ими. Эти агенты можно вводить в виде составной части той же самой либо отдельной лекарственной формы посредством одинаковых или разных путей введения и по одинаковым или разным схемам введения в соответствии со стандартной фармацевтической практикой. Соединения по данному изобретению включают в себя все конформационные изомеры (например,цис- и транс-изомеры). Соединения по настоящему изобретению имеют ассиметрические центры и, следовательно, существуют в разных энантиомерных и диастереомерных формах. Данное изобретение относится к применению всех оптических изомеров и стереоизомеров соединений по настоящему изобретению и их смесей и ко всем фармацевтическим композициям и способам лечения, в которых они могут применяться или которые могут их содержать. В этом отношении данное изобретение включает в себя как Е-, так и Z-конфигурации. Соединения формулы I также могут существовать в виде таутомеров. Данное изобретение относится к применению всех таких таутомеров и их смесей. Данное изобретение также включает в себя фармацевтические композиции, содержащие пролекарства соединений формулы I. Данное изобретение также включает в себя способы лечения или предупреждения расстройств, которые можно лечить или предупреждать ингибированием протеинкиназ, таких как фермент Janus киназа 3, при которых вводят пролекарства соединений формулы I. Соединения формулы I, имеющие свободные амино-, амидо-, гидрокси- или карбоксильные группы, можно превращать в пролекарстсва. Пролекарства включают в себя соединения, где аминокислотный остаток или полипептидная цепь из двух или более чем двух (например, двух, трех или четырех) аминокислотных остатков ковалентно связаны пептидными связями со свободными амино-, гидрокси- или карбоксильными группами соединений формулы I. Аминокислотные остатки включают в себя остатки 20 существующих в природе аминокислот, обычно обозначаемых тремя буквенными символами, а также включают 4-гидроксипролин, гидроксилизин, демозин, изодемозин, 3-метилгистидин, норвалин, бета-аланин, гамма-аминомасляную кислоту, цитруллин, гомоцистеин, гомосерин, орнитин и метионинсульфон. Пролекарства также включают в себя соединения, где карбонаты, карбаматы, амиды и сложные алкиловые эфиры ковалентно связаны с упомянутыми выше заместителями формулы I по карбонильной углеродной боковой цепи пролекарства. Предпочтительные соединения формулы I включают в себя соединения, где R5 представляет собой(С 2-С 9)гетероциклоалкил, возможно, замещенный группами в количестве от одной до трех, выбранными из дейтерия, гидрокси, (С 1-С 6)алкила, галогено, (С 1-С 6)алкокси и группы формулы II. Другие предпочтительные соединения формулы I включают в себя соединения, где а равен 0; b равен 1; X представляет собой карбонил; с равен 0; d равен 0; е равен 0; f равен 0; и g равен 0. Другие предпочтительные соединения формулы I включают в себя соединения, где а равен 0; b равен 1; Х представляет собой карбонил; с равен 0; d равен 1; е равен 0; f равен 0; и g равен 0. Другие предпочтительные соединения формулы I включают в себя соединения, где а равен 0; b равен 1; Х представляет собой карбонил; с равен 1; d равен 0; е равен 0; f равен 0; и g равен 0. Другие предпочтительные соединения формулы I включают в себя соединения, где а равен 0; b равен 1; Х представляет собой -С(=N=циано)-; с равен 1; d равен 0; е равен 0; f равен 0; и g равен 0. Другие предпочтительные соединения формулы I включают в себя соединения, где а равен 0; b равен 0; с равен 0; d равен 0; е равен 0; f равен 0; g равен 1; и Z представляет собой -С(O)-O-.-4 006153 Другие предпочтительные соединения формулы I включают в себя соединения, где а равен 0; b равен 1; Х представляет собой S(O)n; n равен 2; с равен 0; d равен 0; е равен 0; f равен 0; и g равен 0. Другие предпочтительные соединения формулы I включают в себя соединения, где а равен 0; b равен 1; Х представляет собой S(O)n; n равен 2; с равен 0; d равен 2; е равен 0; f равен 1; g равен 1; и Z представляет собой карбонил. Другие предпочтительные соединения формулы I включают в себя соединения, где а равен 0; b равен 1; X представляет собой S(O)n; n равен 2; с равен 0; d равен 2; е равен 0; f равен 1; и g равен 0. Другие предпочтительные соединения формулы I включают в себя соединения, где а равен 0; b равен 1; Х представляет собой карбонил; с равен 1; d равен 0; е равен 1; Y представляет собой S(O)n; n равен 2; f равен 0; и g равен 0. Другие предпочтительные соединения формулы I включают в себя соединения, где а равен 0; b равен 1; Х представляет собой S(O)n; n равен 2; с равен 1; d равен 0; е равен 0; f равен 0; и g равен 0. Другие предпочтительные соединения формулы I включают в себя соединения, где а равен 1; b равен 1; Х представляет собой карбонил; с равен 1; d равен 0; е равен 0; f равен 0; и g равен 0. Другие предпочтительные соединения формулы I включают в себя соединения, где а равен 0; b равен 1; Х представляет собой S(O)n; с равен 0; d равен 1; е равен 1; Y представляет собой S(O)n; n равен 2;f равен 0; и g равен 0. Другие предпочтительные соединения формулы I включают в себя соединения, где а равен 0; b равен 1; Х представляет собой S(O)n; с равен 0; d равен 1; е равен 1; Y представляет собой S(O)n; n равен 2;f равен 1; и g равен 0. Другие предпочтительные соединения формулы I включают в себя соединения, где а равен 0; b равен 1; Х представляет собой кислород; с равен 0; d равен 1; е равен 1; Y представляет собой S(O)n; n равен 2; f равен 1; и g равен 0. Другие предпочтительные соединения формулы I включают в себя соединения, где а равен 0; b равен 1; Х представляет собой кислород; с равен 0; d равен 1; е равен 1; Y представляет собой S(O)n; n равен 2; f равен 0; и g равен 0. Другие предпочтительные соединения формулы I включают в себя соединения, где а равен 0; b равен 1; Х представляет собой карбонил; с равен 1; d равен 1; е равен 1; Y представляет собой S(O)n; f равен 0; и g равен 0. Другие предпочтительные соединения формулы I включают в себя соединения, где а равен 0; b равен 1; X представляет собой карбонил; с равен 1; d равен 1; е равен 1; Y представляет собой S(O)n; n равен 2; f равен 1; и g равен 0. Другие предпочтительные соединения формулы I включают в себя соединения, где R12 представляет собой (С 6-С 10)арил или (С 2-С 9)гетероарил, где арильная или гетероарильная группа, возможно, замещена группами в количестве от одной до четырех, состоящими из водорода, галогено, гидрокси, карбокси, трифторметила, (С 1-С 6)алкила, (С 1-С 6)алкокси, (С 1-С 6)алкил-СО-NН-, амино, амино(С 1-С 6)алкила,(С 1-С 6)алкиламино, С 1-С 6)алкил)2 амино, циано, амино-CO-NH-, (С 1-С 6)алкиламино-СО-NН-, С 1-С 6)алкил)2 амино-СО-NН-, (C5-С 9)гетероариламино-СО-NН-, (С 1-С 6)алкилсульфонила, (C1-С 6)алкилсульфониламино, (С 6-С 10)арилсульфониламино, (С 1-С 6)алкилсульфониламино и (С 1-С 6)алкокси-СО-NН-. Конкретные предпочтительные соединения формулы I включают в себя соединения, выбранные из группы, состоящей из 4-4-метил-3-[метил-(7 Н-пирроло[2,3-d]пиримидин-4-ил)амино]пиперидин-1-илметилбензолсульфонамида;(3-метилизотиазол-5-ил)амида 4-метил-3-[метил-(7 Н-пирроло[2,3-d]пиримидин-4-ил)амино]пиперидин-1-карбоновой кислоты; 3-4-метил-3-[метил-(7 Н-пирроло[2,3-d]пиримидин-4-ил)амино]пиперидин-1-карбонилциклопентанона; бензилметиламида 4-метил-3-[метил-(7 Н-пирроло[2,3-d]пиримидин-4-ил)амино]пиперидин-1-карбоновой кислоты; и диметиламида 4-метил-3-[метил-(7 Н-пирроло[2,3-d]пиримидин-4-ил)амино]пиперидин-1-карбоновой кислоты. Настоящее изобретение также относится к фармацевтической композиции для (а) лечения или предупреждения расстройства или состояния, выбранного из отторжения трансплантата органа, ксенотрансплантации, волчанки, рассеянного склероза, ревматоидного артрита, псориаза, диабета I типа и осложнений диабета, рака, астмы, атопического дерматита, аутоиммунных расстройств щитовидной железы, неспецифического язвенного колита, болезни Крона, болезни Альцгеймера, лейкемии и других аутоиммунных заболеваний, или (б) ингибирования протеинкиназ или Janus киназы 3 (JAK3) у млекопитающего,включая человека, содержащей количество соединения формулы I или его фармацевтически приемлемой соли, эффективное при таких расстройствах или состояниях, и фармацевтически приемлемый носитель. Настоящее изобретение также относится к способу ингибирования протеинтирозинкиназ или Janus киназы 3 (JAK3) у млекопитающего, включая человека, при котором указанному млекопитающему вводят эффективное количество соединения формулы I или его фармацевтически приемлемой соли. Настоящее изобретение также относится к способу лечения или предупреждения расстройства или состояния, выбранного из отторжения трансплантата органа, ксенотрансплантации, волчанки, рассеянного склероза, ревматоидного артрита, псориаза, диабета I типа и осложнений диабета, рака, астмы, атопического дерматита, аутоиммунных расстройств щитовидной железы, неспецифического язвенного колита, болезни Крона, болезни Альцгеймера, лейкемии и других аутоиммунных заболеваний у млекопитающего, включая человека, при котором указанному млекопитающему вводят количество соединения формулы I или его фармацевтически приемлемой соли, эффективное при лечении такого состояния. Подробное описание изобретения Следующие cхемы реакций иллюстрируют получение соединений по настоящему изобретению. Если не указано иначе, R2, R3, R4 и R5 в cхемах реакций и обсуждении, которое следует ниже, являются такими, как определено выше. В реакции 1 получения А соединение 4-хлорпирроло[2,3-d]пиримидина формулы XXI, где R представляет собой водород или защитную группу, такую как бензолсульфонил или бензил, превращают в соединение 4-хлор-5-галогенпирроло[2,3-d]пиримидина формулы XX, где Y представляет собой хлоро,бромо или йодо, путем осуществления взаимодействия XXI с N-хлорсукцинимидом, N-бромсукцинимидом или N-йодсукцинимидом. Реакционную смесь нагревают до температуры флегмообразования в хлороформе в течение периода времени от приблизительно 1 ч до приблизительно 3 ч, предпочтительно приблизительно 1 ч. Альтернативно, в реакции 1 получения А 4-хлорпирроло[2,3-d]пиримидин формулыXXI, где R представляет собой водород, превращают в соответствующий 4-хлор-5-нитропирроло[2,3-d] пиримидин формулы XX, где Y представляет собой нитро, путем осуществления взаимодействия XXI с азотной кислотой в серной кислоте при температуре от приблизительно -10 до приблизительно 10 С,предпочтительно при приблизительно 0 С, в течение периода времени от приблизительно 5 до прибли-9 006153 зительно 15 мин, предпочтительно приблизительно 10 мин. Соединение формулы XXI, где Y представляет собой нитро, превращают в соответствующий 4-хлор-5-аминопирроло[2,3-d]пиримидин формулыXX, где Y представляет собой амино, путем осуществления взаимодействия XXI при ряде условий, известных специалистам в данной области техники, таких как гидрогенолиз с использованием палладия или с хлоридом олова (IV) и соляной кислотой. В реакции 2 получения А соединение 4-хлор-5-галогенпирроло[2,3-d]пиримидина формулы XX, гдеR представляет собой водород, превращают в соответствующее соединение формулы XIX, где R2 представляет (С 1-С 6)алкил или бензил, путем обработки XX N-бутиллитием при температуре приблизительно-78 С и осуществления взаимодействия полученного таким образом промежуточного дианиона с алкилгалогенидом или бензилгалогенидом при температуре от приблизительно -78 С до комнатной температуры, предпочтительно при комнатной температуре. Альтернативно, полученный таким образом дианион подвергают взаимодействию с молекулярным кислородом с образованием соответствующего соединения 4-хлор-5-гидроксипирроло[2,3-d]пиримидина формулы XIX, где R2 представляет собой гидрокси. Соединение формулы XX, где Y представляет собой бром или йод и R представляет собой бензолсульфонат, превращают в соединение формулы XIX, где R2 представляет собой (С 6-С 12)арил или винил, путем обработки XX N-бутиллитием при температуре приблизительно -78 С, с последующим добавлением хлорида цинка при температуре приблизительно -78 С. Полученное таким образом соответствующее цинкорганическое промежуточное соединение затем подвергают взаимодействию с арилйодидом или винилйодидом в присутствии каталитического количества палладия. Реакционную смесь перемешивают при температуре от приблизительно 50 до приблизительно 80 С, предпочтительно при приблизительно 70 С, в течение периода времени от приблизительно 1 до приблизительно 3 ч, предпочтительно приблизительно 1 ч. В реакции 3 получения А соединение формулы XIX превращают в соответствующее соединение формулы XVI путем обработки XIX N-бутиллитием, диизопропиламином лития или гидридом натрия при температуре приблизительно -78 С в присутствии полярного апротонного растворителя, такого как тетрагидрофуран. Полученное таким образом анионное промежуточное соединение затем подвергают взаимодействию с (а) алкилгалогенидом или бензилгалогенидом при температуре от приблизительно-78 С до комнатной температуры, предпочтительно при -78 С, когда R3 представляет собой алкил или бензил; (б) альдегидом или кетоном при температуре от приблизительно -78 С до комнатной температуры, предпочтительно при -78 С, когда R3 представляет собой алкокси; и в) хлоридом цинка при температуре от приблизительно -78 С до комнатной температуры, предпочтительно при -78 С, и полученное таким образом соответствующее цинкорганическое промежуточное соединение затем подвергают взаимодействию с арилйодидом или винилйодидом в присутствии каталитического количества палладия. Полученную реакционную смесь перемешивают при температуре от приблизительно 50 до приблизительно 80 С, предпочтительно при приблизительно 70 С, в течение периода времени от приблизительно 1 до приблизительно 3 ч, предпочтительно приблизительно 1 ч. Альтернативно, полученный таким образом анион подвергают взаимодействию с молекулярным кислородом с образованием соответствующего соединения 4-хлор-6-гидроксипирроло[2,3-d]пиримидина формулы XVI, где R3 представляет собой гидрокси. В реакции 1 получения Б соединение 4-хлорпирроло[2,3-d]пиримидина формулы XXI превращают в соответствующее соединение формулы XXII по методике, аналогичной описанной выше в реакции 3 получения А. В реакции 2 получения Б соединение формулы XXII превращают в соответствующее соединение формулы XVI по методике, аналогичной описанной выше в реакциях 1 и 2 получения А. В реакции 1 получения В соединение 4-метилпиридина формулы XXXI превращают в соответствующее соединение формулы XXX путем алкилирования XXXI на первой стадии бензилхлоридом в присутствии полярного апротонного растворителя, такого как ацетон. Реакционную смесь перемешивают при температуре от приблизительно 40 до приблизительно 80 С в течение периода времени от приблизительно 4 до приблизительно 24 ч. Полученное таким образом пиридиниевое промежуточное соединение затем восстанавливают восстановителем, таким как борогидрид натрия, в присутствии полярного протонного растворителя, такого как метанол, этанол, вода или их смеси. Реакционную смесь перемешивают при температуре от приблизительно 0 С до приблизительно комнатной температуры в течение периода времени от приблизительно 18 до приблизительно 24 ч. В реакции 2 получения В соединение формулы XXX превращают в соответствующее соединение формулы XXIX путем обработки XXX эфиратом трифторида бора в присутствии восстановителя и апротонного растворителя, такого как тетрагидрофуран. Реакционную смесь перемешивают при температуре от приблизительно 0 С до комнатной температуры в течение периода времени от приблизительно 1 до приблизительно 3 ч. Полученное таким образом комплексное промежуточное соединение затем подщелачивают водным раствором гидроксида натрия и затем обрабатывают окислителем, таким как пероксид водорода или Oхоnе, при температуре от приблизительно 0 С до комнатной температуры в течение периода времени от приблизительно 12 до приблизительно 24 ч.- 10006153 В реакции 3 получения В соединение формулы XXIX обрабатывают окислителем, таким как оксид хрома или диметилсульфоксид, оксалилхлорид или комплексом SО 3-пиридин в течение периода времени от приблизительно 1 до приблизительно 3 ч при температуре окружающей среды. Полученный таким образом промежуточный кетон затем обрабатывают амином (R4-NH2) в присутствии кислоты, такой как уксусная кислота, при приблизительно комнатной температуре в течение периода времени от приблизительно 2 до приблизительно 24 ч в органическом растворителе, таком как метанол, этанол или тетрагидрофуран. Полученный таким образом соответствующий промежуточный имин затем обрабатывают восстановителем, таким как борогидрид натрия, цианоборогидрид натрия или триацетоксиборогидрид натрия, при температуре окружающей среды в течение периода времени от приблизительно 2 до приблизительно 24 ч. В реакции 1 схемы 1 соединение 4-хлорпирроло[2,3-d]пиримидина формулы XVII превращают в соответствующее соединение формулы XVI, где R представляет собой бензолсульфонил или бензил, путем обработки XVII бензолсульфонилхлоридом, бензилхлоридом или бензилбромидом в присутствии основания, такого как гидрид натрия или карбонат калия, и полярного апротонного растворителя, такого как диметилформамид или тетрагидрофуран. Реакционную смесь перемешивают при температуре от приблизительно 0 до приблизительно 70 С, предпочтительно при приблизительно 30 С, в течение периода времени от приблизительно 1 до приблизительно 3 ч, предпочтительно приблизительно 2 ч. В реакции 2 схемы 1 соединение 4-хлорпирроло[2,3-d]пиримидина формулы XVI превращают в соответствующее соединение 4-аминопирроло[2,3-d]пиримидина формулы XV путем реакции сочетанияXVI с амином формулы HNR4R5. Реакцию проводят в спиртовом растворителе, таком как трет-бутанол,метанол или этанол, или других высококипящих органических растворителях, таких как диметилформамид,триэтиламин, 1,4-диоксан или 1,2-дихлорэтан, при температуре от приблизительно 60 до приблизительно 120 С, предпочтительно при приблизительно 80 С. Типичная продолжительность реакции находится в интервале от приблизительно 2 до приблизительно 48 ч, предпочтительно приблизительно 16 ч. Когда R5 представляет собой азотсодержащую гетероциклоалкильную группу, то каждый атом азота должен быть защищен защитной группой, такой как бензил. Удаление защитой группы с R5 проводят в условиях, подходящих для конкретной защитной группы, которые не будут оказывать влияния на R защитную группу пирроло[2,3-d]пиримидинового кольца. Удаление защитной группы с R5 в случае бензила проводят в спиртовом растворителе, таком как этанол, в присутствии водорода и катализатора, такого как гидроксид палладия на угле. Полученную таким образом R5 азотсодержащую гетероциклоалкильную группу затем подвергают взаимодействию с рядом различных электрофилов формулы II. Для образования мочевины электрофилы формулы II, такие как изоцианаты, карбаматы и карбамоилхлориды, подвергают взаимодействию с азотом R5 гетероалкильной группы в растворителе, таком как ацетонитрил или диметилформамид, в присутствии основания, такого как карбонат натрия или калия, при температуре от приблизительно 20 до приблизительно 100 С в течение периода времени от приблизительно 24 до приблизительно 72 ч. Для образования амида и сульфонамида электрофилы формулы II, такие как ацилхлориды и сульфонилхлориды, подвергают взаимодействию с азотом R5 гетероалкильной группы в растворителе, таком как метиленхлорид, в присутствии основания, такого как пиридин, при температуре окружающей среды в течение периода времени от приблизительно 12 до приблизительно 24 ч. Образование амида также проводят взаимодействием карбоновой кислоты с гетероалкильной группой в присутствии карбодиимида, такого как 1-(3-диметиламинопропил)-3-этилкарбодиимид, в растворителе, таком как метиленхлорид, при температуре окружающей среды в течение 12-24 ч. Для образования алкила электрофилы формулы II, такие как ,-ненасыщенные амиды, кислоты, нитрилы, сложные эфиры и галогенамиды, подвергают взаимодействию с азотом R5 гетероалкильной группы в растворителе, таком как метанол, при температуре окружающей среды в течение периода времени от приблизительно 12 до приблизительно 18 ч. Образование алкила также можно проводить взаимодействием альдегидов с гетероалкильной группой в присутствии восстановителя, такого как цианоборогидрид натрия, в растворителе, таком как метанол, при температуре окружающей среды в течение периода времени от приблизительно 12 до приблизительно 18 ч. В реакции 3 схемы 1 удаление защитной группы с соединения формулы XV, где R представляет собой бензолсульфонил, с образованием соответствующего соединения формулы I, проводят путем обработки XV основанием, таким как гидроксид натрия или гидроксид калия, в спиртовом растворителе, таком как метанол или этанол, или смешанных растворителях, таких как спирт/тетрагидрофуран или спирт/вода. Реакцию проводят при комнатной температуре в течение периода времени от приблизительно 15 мин до приблизительно 1 ч, предпочтительно 30 мин. Удаление защитной группы с соединения формулы XV, где R представляет собой бензил, проводят путем обработки XV натрием в аммиаке при температуре приблизительно -78 С в течение периода времени от приблизительно 15 мин до приблизительно 1 ч. В реакции 1 схемы 2 соединение 4-хлорпирроло[2,3-d]пиримидина формулы XX превращают в соответствующее соединение 4-аминопирроло[2,3-а]пиримидина формулы XXIV по методике, аналогичной описанной выше в реакции 2 схемы 1.R представляет собой бензолсульфонат и Z представляет собой бром или йод, превращают в соответствующее соединение формулы XXIII путем осуществления взаимодействия XXIV с (а) арилбороновой кислотой, где R2 представляет собой арил, в апротонном растворителе, таком как тетрагидрофуран или диоксан, в присутствии каталитического количества палладия (0) при температуре от приблизительно 50 до приблизительно 100 С, предпочтительно при приблизительно 70 С, в течение периода времени от приблизительно 2 до приблизительно 48 ч, предпочтительно приблизительно 12 ч; (б) алкинами, где R2 представляет собой алкинил, в присутствии каталитического количества йодида меди (I) и палладия (0),и полярного растворителя, такого как диметилформамид, при комнатной температуре в течение периода времени от приблизительно 1 до приблизительно 5 ч, предпочтительно приблизительно 3 ч; и(в) алкенами или стиролами, где R2 представляет собой винил или стирил, в присутствии каталитического количества палладия в диметилформамиде, диоксане или тетрагидрофуране при температуре от приблизительно 80 до приблизительно 100 С, предпочтительно при приблизительно 100 С, в течение периода времени от приблизительно 2 до приблизительно 48 ч, предпочтительно приблизительно 48 ч. В реакции 3 схемы 2 соединение формулы XXIII превращают в соответствующее соединение формулы XV по методике, аналогичной описанной выше в реакции 3 получения А. В реакции 1 схемы 3 соединение формулы XVII превращают в соответствующее соединение формулы I по методике, аналогичной описанной выше в реакции 2 схемы 1. Соединения по настоящему изобретению, которые являются основными по своей природе, способны к образованию широкого ряда различных солей с различными неорганическими и органическими кислотами. Хотя такие соли должны быть фармацевтически приемлемыми для введения животным, часто на практике является желательным первоначальное выделение соединения по настоящему изобретению из реакционной смеси в виде фармацевтически неприемлемой соли и затем простое превращение последней обратно в свободное основание путем обработки щелочным реагентом с последующим превращением последнего свободного основания в фармацевтически приемлемую соль присоединения кислоты. Соли присоединения кислоты основных соединений по данному изобретению легко получают путем обработки основного соединения, по существу, эквивалентным количеством выбранной минеральной или органической кислоты в среде водного растворителя или в подходящем органическом растворителе, таком как метанол или этанол. После осторожного выпаривания растворителя легко получают желаемую твердую соль. Желаемую кислую соль также осаждают из раствора свободного основания в органическом растворителе, добавляя к этому раствору подходящую минеральную или органическую кислоту. Соединения по настоящему изобретению, которые являются кислыми по своей природе, способны к образованию основных солей с различными фармакологически приемлемыми катионами. Примеры таких солей включают в себя соли щелочных металлов или щелочно-земельных металлов, и особенно соли натрия и калия. Все эти соли получают по традиционным методикам. Химические основания, которые применяют в качестве реагентов для получения фармацевтически приемлемых основных солей по данному изобретению, представляют собой такие основания, которые образуют нетоксичные основные соли с кислыми соединениями по настоящему изобретению. Такие нетоксичные основные соли включают в себя соли, которые являются производными таких фармакологически приемлемых катионов, как натрий, калий, кальций и магний и т.д. Эти соли могут быть легко получены путем обработки соответствующих кислых соединений водным раствором, содержащим желаемые фармакологически приемлемые катионы, и затем упаривания полученного раствора до сухого состояния, предпочтительно при пониженном давлении. Альтернативно, они также могут быть получены путем смешивания низших спиртовых растворов кислых соединений вместе с желаемым алкоголятом щелочного металла и затем упаривания полученного раствора до сухого состояния, как описано выше. В любом случае, для обеспечения полноты протекания реакции и максимальных выходов желаемого конечного продукта предпочтительно использовать стехиометрические количества реагентов. Композиции по настоящему изобретению могут быть приготовлены традиционным способом с применением одного или более чем одного фармацевтически приемлемого носителя. Таким образом,активные соединения по данному изобретению могут быть приготовлены в виде препаратов для перорального, буккального, интраназального, парентерального (например, внутривенного, внутримышечного или подкожного) или ректального введения либо в форме, подходящей для введения путем ингаляции или инсуффляции. Активные соединения по данному изобретению также могут быть приготовлены в виде препарата для продолжительной доставки. Для перорального введения фармацевтические композиции могут быть в форме, например, таблеток или капсул, приготавливаемых традиционными способами с фармацевтически приемлемыми эксципиентами, такими как связывающие агенты (например, предварительно желатинизированный кукурузный крахмал, поливинилпирролидон или гидроксипропилметилцеллюлоза); наполнители (например,лактоза, микрокристаллическая целлюлоза или фосфат кальция); смазывающие агенты (например, стеарат магния, тальк или диоксид кремния); разрыхлители (например, картофельный крахмал или натрия крахмал гликолят); или смачивающие агенты (например, лаурилсульфат натрия). Таблетки могут быть- 12006153 покрыты оболочкой с использованием хорошо известных способов. Жидкие препараты для перорального введения могут быть в форме, например, растворов, сиропов или суспензий или могут быть представлены в виде сухого продукта для разведения водой или другим подходящим растворителем перед применением. Такие жидкие препараты могут быть получены традиционными способами с фармацевтически приемлемыми добавками, такими как суспендирующие агенты (например, сироп сорбита, метилцеллюлоза или гидрогенизированные пищевые жиры); эмульгаторы (например, лецитин или гуммиарабик); безводные носители (например, миндальное масло, жирные сложные эфиры или этиловый спирт); и консерванты (например, метиловый или пропиловый эфиры парагидроксибензойной кислоты или сорбиновая кислота). Для буккального введения композиция может быть в форме таблеток или лепешек, приготовленных традиционным способом. Активные соединения по данному изобретению могут быть приготовлены в виде препаратов для парентерального введения путем инъекции, в том числе с использованием традиционных методик катетеризации, или инфузии. Препараты для инъекции могут быть представлены в стандартной лекарственной форме, например в ампулах или контейнерах для многократного дозирования, с добавленными консервантами. Композиции могут быть в таких формах как суспензии, растворы или эмульсии в масляных или водных растворителях, и могут содержать такие агенты для препаратов, как суспендирующие агенты, стабилизаторы и/или диспергирующие агенты. Альтернативно, активный ингредиент может быть в виде порошка для разведения подходящим растворителем, например апирогенной стерильной водой,перед применением. Активные соединения по данному изобретению также могут быть приготовлены в форме композиций для ректального введения, таких как суппозитории или удерживающие клизмы, например, содержащие традиционные основы для суппозиториев, такие как какао-масло или другие глицериды. Для интраназального введения или введения путем ингаляции активные соединения по данному изобретению удобно доставлять в форме раствора или суспензии из нагнетательного контейнера с аэрозолем, который сжимается или накачивается пациентом, или в виде аэрозоля, находящегося под давлением в контейнере или распылителе, с использованием подходящего пропеллента, например дихлордифторметана, трихлорфторметана, дихлортетрафторэтана, диоксида углерода или другого подходящего газа. В случае аэрозоля, находящегося под давлением, стандартную дозу можно установить при помощи клапана для доставки отмеренного количества. Находящийся под давлением контейнер или распылитель может содержать раствор или суспензию активного соединения. Капсулы и картриджи (сделанные, например,из желатина) для применения в ингаляторе или инсуффляторе могут быть приготовлены в виде препарата, содержащего смесь порошка соединения по данному изобретению и порошка подходящей основы,такой как лактоза или крахмал. Предлагаемая доза активных соединений по данному изобретению для перорального, парентерального или буккального введения среднему взрослому человеку для лечения состояний, упомянутых выше(например, ревматоидного артрита), составляет 0,1-1000 мг активного ингредиента в стандартной дозе,которую следует вводить, например, 1-4 раза в день. Аэрозольные препараты для лечения состояний, упомянутых выше (например, астмы), для среднего взрослого человека предпочтительно приготавливают таким образом, чтобы каждая отмеренная доза или"пшик" аэрозоля содержал от 20 до 1000 мкг соединения по данному изобретению. Общая суточная доза при применении аэрозоля будет находиться в пределах от 0,1 до 1000 мг. Введение можно осуществлять несколько раз в сутки, например 2, 3, 4 или 8 раз, с введением каждый раз, например, 1, 2 или 3 доз. Соединение формулы I вводят в фармацевтически приемлемой форме или отдельно, или в комбинации с одним или более чем одним дополнительным агентом, который модулирует иммунную систему млекопитающего, или с противовоспалительными агентами, которые могут включать в себя циклоспорин А (например, Sandimmune или Neoral), рапамицин, FK-506 (такролимус), лефлуномид, деоксиспергуалин, микофенолат (например, Cellcept), азатиоприн (например, Imuran), даклизумаб (например,Zenapax), ОКТЗ (например, Orthoclone), AtGam, аспирин, ацетаминофен, ибупрофен, напроксен, пироксикам и противовоспалительные стероиды (например, преднизолон или дексаметазон), но не ограничиваются ими; и такие агенты можно вводить в виде составной части той же самой или отдельной формы дозы одинаковым или разным путями введения и по одинаковой или разной схемам введения в соответствии со стандартной фармацевтической практикой.FK-506 (такролимус) принимают перорально в количестве 0,10-0,15 мг/кг массы тела каждые 12 ч в течение первых 48 ч после операции. Дозу контролируют по уровням такролимуса в сыворотке. Циклоспорин A (Sandimmune, пероральный или внутривенный препарат, или Neoral, пероральный раствор или капсулы) принимают перорально в количестве 5 мг/кг массы тела каждые 12 ч в течение 48 ч после операции. Дозу контролируют по уровням циклоспорина А в крови. Активные агенты могут быть приготовлены в виде препарата для продолжительной доставки в соответствии со способами, хорошо известными специалистам в данной области техники. Примеры таких препаратов можно найти в патентах США 3538214, 4060598, 4173626, 3119742 и 3492397.- 13006153 Способность соединений формулы I или их фармацевтически приемлемых солей ингибировать Janus киназу 3 и, вследствие этого, проявлять свою эффективность для лечения расстройств или состояний,определяемых Janus киназой 3, показана следующими анализами in vitro. Биологический анализ Ферментативный анализ JAK3 (JH1:GST) При анализе JAK3 киназы используют белок, экспрессируемый в инфицированных бакуловирусом клетках SF9 (белок слияния GST и каталитического домена человеческой JAK3), очищенный аффинной хроматографией на глутатион-сефарозе. Субстратом для реакции является полиглутаминовая кислотатирозин (РGТ (4:1), каталог Sigma P0275), нанесенный на планшеты Nunc Maxi Sorp в течение ночи при 37 С в концентрации 100 мкг/мл. Утром, после нанесения, планшеты промывают 3 раза и к лункам, содержащим 100 мкл буфера для киназы (50 мМ HEPES, рН 7,3, 125 мМ NaCl, 24 мМ MgCl2+0,2 мкМ АТФ+1 мМ ортованадата Na), добавляют JAK3. Реакция продолжается в течение 30 мин при комнатной температуре, и планшеты промывают еще 3 раза. Уровень фосфорилированного тирозина в данной лунке вычисляют стандартным анализом ELISA, используя антифосфотирозин антитело (ICN PY20, Cat.69151-1). Ингибирование пролиферации ИЛ-2 зависимых бластных Т-клеток человека Этот метод исследования измеряет ингибирующее влияние соединений на пролиферацию ИЛ-2 зависимых бластных Т-клеток in vitro. Поскольку для передачи сигналов через ИЛ-2 рецептор необходимаJAK-3, то активные клеточные ингибиторы JAK-3 должны ингибировать пролиферацию ИЛ-2 зависимых бластных Т-клеток. Клетки для этого анализа выделяют из свежей человеческой крови. После отделения мононуклеарных клеток с использованием Accuspin System Histopaque-1077 (SigmaA7054), исходные человеческие Т-клетки выделяют отрицательной селекцией с использованием Lympho-Kwik T (One Lambda, Inc., Cat.LK-50T). Т-клетки культивируют в количестве 1-2 х 106/мл в питательной среде (RPMI + 10% фетальной телячьей сыворотки, инактивированной тепловой обработкой (Hyclone Cat.A-1111-L) + 1% пенициллин/ стрептомицин (Gibco и индуцируют пролиферацию добавлением 10 мкг/мл PHA (Murex Diagnostics,Cat.НА 16). Через 3 дня при 37 С в 5% СO2 клетки промывают 3 раза средой, ресуспендируют до концентрации 1-2 х 106 клеток/мл в среде + 100 Единиц/мл человеческого рекомбинантного ИЛ-2 (RD Systems,Cat.202-IL). Через 1 неделю клетки становятся ИЛ-2 зависимыми и могут сохраняться до 3 недель подпиткой равными объемами среды + 100 Единиц/мл ИЛ-2 2 раза в неделю. Для того чтобы проанализировать способность тестируемых соединений ингибировать пролиферацию ИЛ-2 зависимых Т-клеток, ИЛ-2 зависимые клетки промывают 3 раза, ресуспендируют в питательной среде и затем помещают в плоскодонный 96-луночный титрационный микропланшет (Falcon353075)(50000 клеток/лунка/0,1 мл). Последовательные 2-кратные разведения соединения, начиная с 10 мкМ,выполняемые из исходного 10 мМ раствора тестируемого соединения в ДМСО, добавляют в лунки в трех повторностях. Через 1 ч к каждой анализируемой лунке добавляют 10 Единиц/мл ИЛ-2. Затем планшеты инкубируют при 37 С 5% СO2 в течение 72 ч. Затем в планшеты вводят 3H-тимидин (0,5 мкКюри/ лунка) (NEN CatNET-027A) и инкубируют в течение дополнительных 18 ч. Затем культуральные планшеты собирают с использованием коллектора для 96-луночных планшетов и определяют количество 3H-тимидина, включенного в пролиферирующие клетки, с помощью сцинтилляционного счетчика PackardTop Counter. Данные анализируют при помощи графика зависимости % ингибирования пролиферации от концентрации тестируемого соединения. Из этого графика определяют величину ИK50 (мкМ). Следующие примеры иллюстрируют получение соединений по настоящему изобретению, которое не ограничивается их подробным описанием. Коммерческие реагенты применяли без дополнительной очистки. ТГФ относится к тетрагидрофурану. ДМФА относится к N,N-диметилформамиду. Масс-спектры низкого разрешения (LRMS) регистрировали либо на Hewlett Packard 5989 с химической ионизацией(аммоний), либо на Fisons (или Micro Mass) Platform с химической ионизацией при атмосферном давлении(APCI) и использованием смеси ацетонитрил/вода (50/50) с 0,1%-ной муравьиной кислотой в качестве ионизирующего агента. Комнатная температура или температура окружающей среды относится к 20-25 С. Пример 1. Фуран-2-ил-4-метил-3-[метил-(7 Н-пирроло[2,3-d]пиримидин-4-ил)амино]пиперидин-1 илметанон. Способ А. 1-Бензил-4-метилпиридиний хлорид. К перемешиваемому раствору 4-метилпиридина (26 мл/0,268 моль) в 70 мл ацетона добавляли 31 мл(0,268 моль) бензилхлорида. Полученную смесь перемешивали при 50 С в течение 18 ч. После охлаждения до комнатной температуры реакционную смесь фильтровали, промывали ацетоном и сушили при пониженном давлении с получением 38 г указанного в заголовке соединения. Фильтрат концентрировали при пониженном давлении с образованием дополнительных 5,6 г указанного в заголовке соединения(суммарный выход 74%). LRMS: 184. Способ Б. 1-Бензил-4-метил-1,2,3,6-тетрагидропиридин. К перемешиваемому раствору продукта, полученного cпособом А (38 г/0,171 моль), растворенного в 140 мл смеси этанол/вода (10:1) при 0 С, добавляли порциями в течение 25 мин 16 г (0,427 моль) борогидрида натрия. Полученную смесь перемешивали при комнатной температуре в течение 18 ч, после че- 14006153 го реакцию гасили, добавляя 100 мл воды. Реакционную смесь фильтровали, осадок на фильтре промывали водой и этилацетатом и для удаления органики объединенные фильтраты концентрировали при пониженном давлении. Остаток разбавляли водой (100 мл) и экстрагировали этилацетатом 3 раза по 150 мл. Объединенные этилацетатные экстракты сушили над Na2SO4 и концентрировали под вакуумом до сухого состояния, с получением 32 г (100%) указанного в заголовке соединения в виде желтого масла. LRMS: 188 (М+1). Способ В. 1-Бензил-4-метилпиперидин-3-ол. К раствору продукта, полученного способом Б (72,45 г/0,387 моль), растворенного в 240 мл ТГФ,добавляли 21,4 г NaBH4 и смесь охлаждали до 0 С. Затем по каплям в течение 1,5 ч добавляли раствор эфирата трифторида бора (109,4 мл, растворенного в 200 мл ТГФ). После добавления реакционную смесь доводили до комнатной температуры и перемешивали в течение 2 ч. Реакционную смесь снова охлаждали до 0 С и по каплям в течение 15 мин добавляли 29,3 мл воды, а затем по каплям в течение 20 мин 2 н гидроксид натрия (97,5 мл). Полученную смесь перемешивали при 0 С в течение 40 мин и затем доводили до комнатной температуры. В реакционную смесь по каплям со скоростью, при которой температура не превышала 50 С, добавляли пероксид водорода (30%, 97,5 мл, приблизительно 30 мин). После окончания добавления реакционную смесь перемешивали в течение 10 мин, затем охлаждали до 0 С. В течение 5 мин добавляли концентрированную соляную кислоту (97,5 мл), под вакуумом уменьшали объем реакционной смеси в 3 раза и устанавливали рН до 9-10 6 н гидроксидом натрия (водн.). Полученную смесь 3 раза экстрагировали эфиром, объединенные эфирные слои сушили над MgSО 4 и под вакуумом упаривали до сухого состояния, получая 65,32 г (79%) указанного в заголовке соединения в виде желтого масла. LRMS: 206,1 (М+1). Альтернативный способ. К раствору продукта, полученного способом Б (18,7 г/0,1 моль), в ТГФ (150 мл) добавляли NaBH4(6,5 г/0,170 моль) при комнатной температуре в атмосфере N2. Суспензию охлаждали до 0 С и через капельную воронку медленно добавляли BF3OEt2 (15 мл, 16,8 г/0,118 моль) в ТГФ (25 мл). Добавление осуществляли медленно, для поддержания температуры реакционной смеси ниже 0 С. После добавления реакционную смесь перемешивали при 0 С в течение 1 ч и при комнатной температуре в течение 1,5 ч. Реакционную смесь снова охлаждали до 0 С и медленно, для погашения избытка борана, добавляли воду(50 мл). Реакционную смесь перемешивали при комнатной температуре в течение 2 ч, а затем при 0 С добавляли Охоnе (110 г/0,343 моль) в воде (500 мл). Реакционную смесь оставляли нагреваться до комнатной температуры и перемешивали в течение ночи. Реакцию останавливали, добавляя твердыйNaHSO3 до полного погашения избытка окислителя. (KI/крахмал индикаторная бумага). рН реакционной смеси составил 1-2. Затем реакционную смесь экстрагировали этилацетатом 3 раза по 50 мл, устанавливали рН водного слоя 12 с помощью 6 н гидроксида натрия и экстрагировали этилацетатом (4 раза по 100 мл). Органический слой промывали рассолом, сушили над Na2SO4 и концентрировали под вакуумом, получая 19,0 г (92%) указанного в заголовке соединения в виде масла. LRMS: 206,1 (М+1). Способ Г. Соль 1-бензил-4-метилпиперидин-3-ол-толуол-4-сульфокислоты. К перемешиваемому раствору продукта, полученного cпособом В (65,32 г/0,318 моль), растворенного в 175 мл ацетона и охлажденного до 0 С, добавляли раствор моногидрата паратолуолсульфокислоты в 350 мл ацетона (по каплям) в течение 2 ч и полученную смесь перемешивали при 0 С в течение 1,5 ч. Осадок отфильтровывали, остаток на фильтре промывали 90 мл диизопропилового эфира. Затем твердый продукт сушили под вакуумом, получая 58,55 г (100%) указанного в заголовке соединения в виде белого твердого вещества. LRMS: 378,5 (М+1). Способ Д. 1-Бензил-4-метилпиперидин-3-он. К раствору продукта, полученного cпособом Г (9,8 г/0,026 моль), и 31,7 мл диизопропилэтиламина,растворенного в 250 мл дихлорметана и охлажденного до 0 С, добавляли (по каплям) в течение 40 минутного периода 12,4 г комплекса SО 3 пиридин, растворенного в 153 мл диметилсульфоксида. Сразу после добавления реакционную смесь перемешивали в течение 1,5 ч при комнатной температуре и затем гасили, добавляя 200 мл насыщенного NaHCO3 (водн.). Под вакуумом удаляли дихлорметан и оставшуюся водную часть экстрагировали 4 раза диизопропиловым эфиром (150 мл). Объединенные эфирные слои промывали 4 раза водой (100 мл), сушили над Na2SO4 и под вакуумом концентрировали до сухого состояния, получая 3,81 г (72,97%) указанного в заголовке соединения в виде желтого масла. LRMS: 204(М+1). Способ Е. (1-Бензил-4-метилпиперидин-3-ил)метиламин. К перемешиваемому раствору продукта, полученного cпособом Д (3,81 г/0,019 моль), и 38 мл 2,0 М метиламина в ТГФ добавляли 2,2 мл уксусной кислоты и полученную смесь перемешивали при комнатной температуре в течение 1,5 ч. Добавляли твердый триацетоксиборогидрид натрия (NаВ(ОАс)3 Н) (7,94 г/ 0,038 моль) и новую смесь перемешивали при комнатной температуре в течение 18 ч. Реакцию гасили 2 н соляной кислотой и устанавливали рН 1. Реакционную смесь промывали 2 раза эфиром, затем 6 н гидроксидом натрия (водн.) устанавливали рН водного слоя 12 и 3 раза экстрагировали дихлорметаном. Объединенные дихлорметановые слои сушили над Na2SO4, фильтровали и под вакуумом упаривали до сухогоChem. Soc., (1960), 82, 131, продукта, полученного cпособом Е (1,7 г, 7,95 ммоль), и 10 мл триэтиламина нагревали в герметичной пробирке при 100 С в течение 4 дней. После охлаждения до комнатной температуры и концентрирования при пониженном давлении остаток очищали флэш-хроматографией (диоксид кремния; 3%-ный метанол в дихлорметане), получая 1,0 г (38%) указанного в заголовке соединения в виде бесцветного масла. LRMS: 336,1 (М+1). Способ З. Метил-(4-метилпиперидин-3-ил)-(7 Н-пирроло[2,3-d]пиримидин-4-ил)амин. К продукту, полученному способом Ж (0,7 г, 2,19 ммоль), растворенному в 15 мл этанола, добавляли 0,5 г 20% гидроксида палладия на угле (50% воды) (Aldrich) и полученную смесь встряхивали в аппарате Парра в атмосфере водорода (345 кПа) при комнатной температуре в течение 2 дней. После фильтрования на целите реакционную смесь под вакуумом концентрировали до сухого состояния и остаток очищали флэш-хроматографией (диоксид кремния; 5%-ный метанол в дихлорметане), получая 0,48 г(90%) указанного в заголовке соединения. LRMS: 246,1 (М+1). Способ И. [1-(4-Метоксибензолсульфонил)-4-метилпиперидин-3-ил]метил-(7 Н-пирроло[2,3-d]пиримидин-4-ил)амин. К перемешиваемому раствору 1 мл пиридина и 9 мл дихлорметана добавляли 40 мг (0,163 ммоль) продукта, полученного способом З, и 20 мкл 4-метоксибензолсульфонилхлорида и полученную смесь перемешивали при комнатной температуре в течение 18 ч. Затем реакцию гасили, добавляя насыщенныйNаНСО 3 (водн.), удаляли органический слой и экстрагировали водный слой дихлорметаном. Дихлорметановый слой сушили над Na2SO4 и под вакуумом концентрировали до сухого состояния. Остаток очищали PTLC (диоксид кремния; дихлорметан/метанол (10:1, получая 22 мг (32%) указанного в заголовке соединения в виде светло-желтого твердого вещества. LRMS: 416,5 (М+1). Соединения, указанные в заголовках примеров 2-297, получали способом, аналогичным способу,описанному в примере 1. Пример 2. [1-(4-Метоксибензолсульфонил)-4-метилпиперидин-3-ил]метил-(7 Н-пирроло[2,3-d]пиримидин-4-ил)амин.LRMS: 285. Пример 298. [1-(2-Фторбензил)-4-метилпиперидин-3-ил]метил-(7 Н-пирроло[2,3-d]пиримидин-4-ил) амин. Способ К. К раствору продукта, полученного cпособом 3 (50 мг, 0,204 ммоль), растворенного в 5 мл метанола,добавляли 154 мкл 2-фторбензальдегида. Полученную смесь перемешивали при комнатной температуре в течение 4 ч, после чего добавляли 51 мг (0,816 ммоль) цианоборогидрида натрия и новую смесь перемешивали при комнатной температуре в течение 18 ч. Реакцию гасили добавлением 2 капель 1 н NaOH(водн.) и для удаления метанола смесь концентрировали при пониженном давлении. Остаток растворяли в хлороформе и промывали водой. Водный слой 3 раза промывали хлороформом, объединенные хлороформные экстракты сушили над МgSO4 и концентрировали под вакуумом до сухого состояния. Затем неочищенный продукт очищали флэш-хроматографией (диоксид кремния; 2,5%-ный метанол в хлороформе), получая 36 мг (47,5%) указанного в заголовке соединения в виде белого твердого вещества.
МПК / Метки
МПК: C07D 487/04, A61P 17/06, A61K 31/505
Метки: пирроло[2,3-d]пиримидина, качестве, иммунодепрессантов, соединения
Код ссылки
<a href="https://eas.patents.su/30-6153-soedineniya-pirrolo23-dpirimidina-v-kachestve-immunodepressantov.html" rel="bookmark" title="База патентов Евразийского Союза">Соединения пирроло[2,3-d]пиримидина в качестве иммунодепрессантов</a>
Пирроло[2,3-d]пиримидиновые соединения

Номер патента: 6034
Опубликовано: 25.08.2005
Авторы: Блуменкопф Тодд Эндрю, Чанджелиэн Пол Стивен, Браун Маттью Фрэнк, Фланаган Марк Эдуард
МПК: A61P 35/02, A61K 31/505, C07D 487/04...
Метки: соединения, пирроло[2,3-d]пиримидиновые
Формула / Реферат:
1. Соединение формулы или его фармацевтически приемлемая соль, где R1 представляет собой группу формулы где пунктирная линия обозначает возможные двойные связи; m равняется 0, 1 или 2; n равняется 1 или 2, X представляет собой кислород, NR6 или CR7R8; каждый из B и D представляет собой CR7R8; каждый из A и E представляет собой CR7R8; и R6 выбран из группы, состоящей из водорода, (C1-C6)алкила; каждый из R7 и R8 независимо выбран из группы,...
Производные пиримидина в качестве ингибиторов репликации вируса иммунодефицита человека

Номер патента: 2973
Опубликовано: 26.12.2002
Авторы: Кукла Майкл Джозеф, Де Корт Барт, Хо Чих Юнг, Койманс Люсьен Мария Хенрикус, Ван Акен Кун Жанн Альфонс, Лудовики Дональд Уилльям, Де Жонж Марк Рене, Янссен Марсель Огюст Констант, Янссен Поль Адриан Жан, Андрис Кунрад Йозеф Лодевейк Марсель, Хэрес Ян
МПК: A61P 31/18, A61K 31/505, C07D 239/50...
Метки: пиримидина, репликации, ингибиторов, производные, вируса, качестве, иммунодефицита, человека
Формула / Реферат:
1. Применение соединения формулы его N-оксида, фармацевтически приемлемой соли присоединения или стереохимически изомерной формы, где А является СН или N; n равен 0, 1, 2, 3 или 4; Q является водородом или -NR1R2; R1 и R2, каждый независимо, выбран из водорода, гидрокси, C1-12aлкила, С1-12алкилокси, С1-12алкилкарбонила, С1-12алкилоксикарбонила, где каждая из указанных С1-12алкильных групп необязательно и каждая независимо может быть замещена...
Производные пиримидина в качестве антагонистов 5-нт2с- рецептора

Номер патента: 1311
Опубликовано: 26.02.2001
Авторы: Витерхед Габриэль, Флиппин Ли
МПК: C07D 239/74, A61K 31/517, A61P 25/00...
Метки: рецептора, производные, пиримидина, антагонистов, 5-нт2с, качестве
Формула / Реферат:
1. Соединение формулы в котором R1, R2, R3, R4 и R5 независимо друг от друга обозначают водород, C1-С8алкил, C1-С8алкокси, галоген или трифторметил, Х обозначает кислород, серу, NR7 или СН2, Y обозначает NR7; где R7 обозначает водород или C1-С8алкил, и R6 обозначает C1-С8алкил или необязательно замещенный арил, а именно, моноциклическое или бициклическое ароматическое кольцо, такое как фенил, нафтил, тиофен, фуран, имидазол, пиридин,...
Азабициклические соединения, способ их получения и их применение в качестве лекарственных средств, в частности, в качестве антибактериальных средств

Номер патента: 4920
Опубликовано: 28.10.2004
Авторы: Роулендс Дэвид Ален, Асзоди Жозеф, Лампила Максим, Фроментэн Клод
МПК: A61P 31/04, A61K 31/529, C07D 487/08...
Метки: средств, частности, азабициклические, соединения, лекарственных, антибактериальных, способ, качестве, получения, применение
Формула / Реферат:
1. Соединение общей формулы (I) или одна из его солей с основанием или с кислотой в которой R1 означает атом водорода, радикал COOH, CN, COOR, CONR6R7, (CH2)n'R5 или радикал где R выбирают из группы, состоящей из алкила с 1-6 атомами углерода, возможно замещенного пиридилом или карбамоилом; -CH2-алкенила, содержащего в целом 3-9 атомов углерода; арила с 6-10 атомами углерода или аралкила с 7-11 атомами углерода, причем арильное или...
Трициклические соединения, обладающие активностью в отношении интегринов, в частности, в отношении интегрина альфаvбета 3, способ их получения и промежуточные соединения, используемые в этом способе,их применение в качестве медикаментов и содержащие их фармацевтические композиции.

Номер патента: 2271
Опубликовано: 28.02.2002
Авторы: Кнолле Йохен, Питти Роберт М., Бернар Серж, Тетш Жан-Жорж, Гурвест Жан-Франсуа, Карниато Дени, Бодари Сара С., Макдауэлл Роберт С., Венер Фолькмар, Штильц Ханс-Ульрих, Гадек Томас Р.
МПК: C07C 281/12, A61K 31/19, A61P 9/10...
Метки: альфаvбета, используемые, содержащие, активностью, интегринов, частности, фармацевтические, способ, отношении, трициклические, соединения, способе,их, применение, композиции, обладающие, качестве, интегрина, этом, медикаментов, получения, промежуточные
Формула / Реферат:
1. Соединения общей формулы (I) в которой R1 обозначает группу -О-[А]-[В]-COR6, в которой R6 обозначает -ОН, C1-С6алкокси, -О-СН2-СН(ОН)-СН2OН, [A] обозначает группу C1-С6алкилен, возможно замещенный оксогруппой, [B] обозначает радикал -CH(Z)- или простую связь, Z обозначает группу -NHCO2Rc, или -NHSO2Rc, где Rc обозначает радикал фенил(C1-С4)алкил-, хинолинил или пиридинилимидазолил(C1-С4)алкил-, R2 и R3, одинаковые или разные, обозначают атом...
Предыдущий патент: Способ транспортировки и упаковки пищевых продуктов
Следующий патент: N-фенпропилциклопентил-замещенные производные глутарамида в качестве nep ингибиторов при fsad
Случайный патент: Чайная композиция (варианты)