Производные пиразолоспирокетонов для применения в качестве ингибиторов ацетил-коа-карбоксилаз
Номер патента: 22375
Опубликовано: 30.12.2015
Авторы: Гриффит Дэвид Эндрю, Саутерс Джеймс Альфред, Эдмондс Дейвид Джеймс, Доу Роберт Ли




























Формула / Реферат
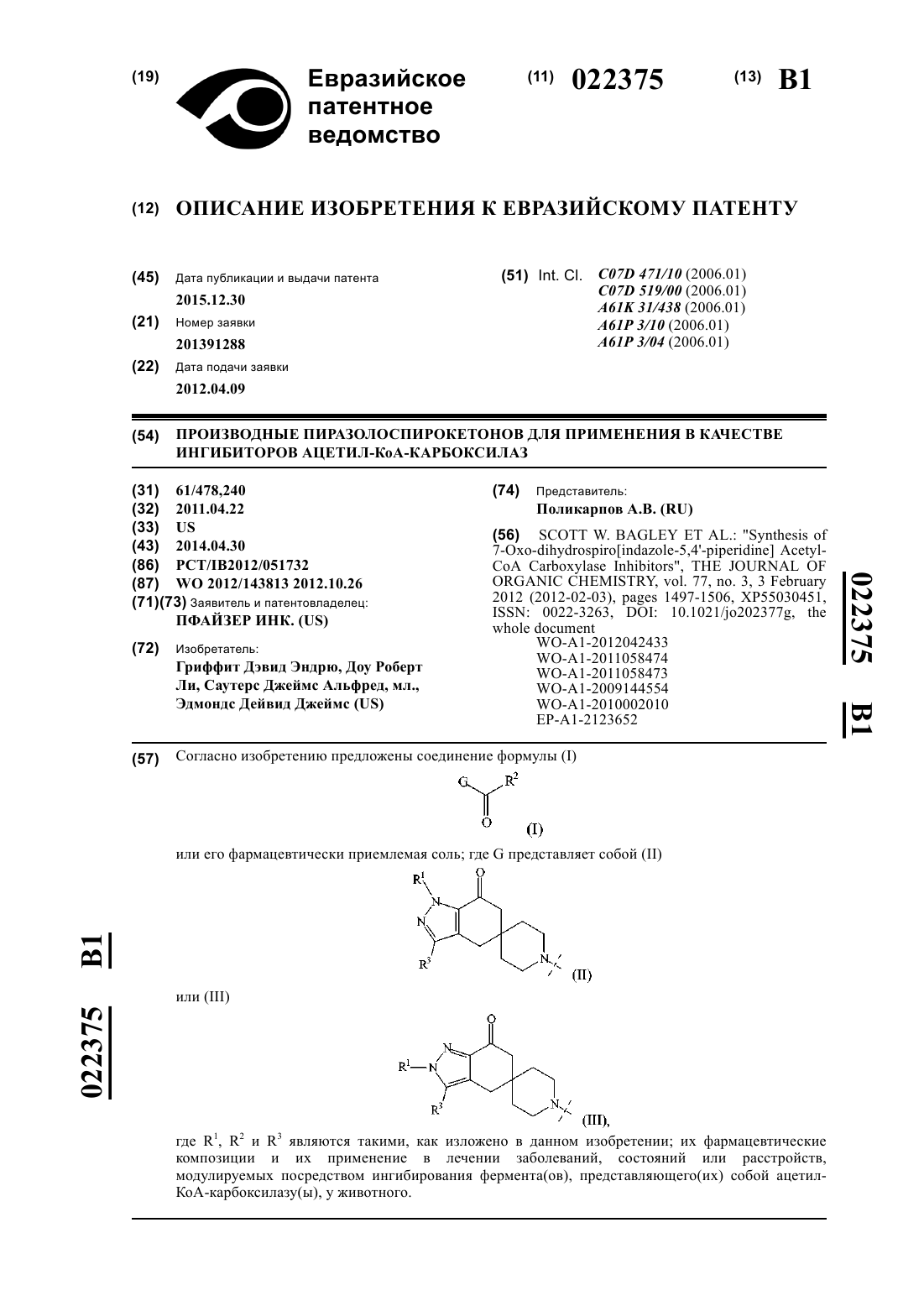
1. Соединение структуры

или его фармацевтически приемлемая соль.
2. Соединение структуры

или его фармацевтически приемлемая соль.
3. Соединение структуры

или его фармацевтически приемлемая соль.
4. Соединение структуры

или его фармацевтически приемлемая соль.
5. Фармацевтическая композиция для лечения заболеваний, опосредованных активностью ацетил-КоА-карбоксилазы, содержащая соединение по любому из пп.1-4 или его фармацевтически приемлемую соль и фармацевтически приемлемый эксципиент, разбавитель или носитель.
6. Применение соединения по любому из пп.1-4 в изготовлении лекарственного средства для лечения заболевания, состояния или расстройства, модулируемого посредством ингибирования фермента(ов), представляющего(их) собой ацетил-КоА-карбоксилазу(ы).
7. Применение по п.6, где заболевание, состояние или расстройство представляет собой диабет 2 типа, связанные с диабетом расстройства, неалкогольную жировую болезнь печени или печеночную инсулинорезистентность.
8. Применение по п.6, где заболевание, состояние или расстройство представляют собой диабет 2 типа.
Текст
ПРОИЗВОДНЫЕ ПИРАЗОЛОСПИРОКЕТОНОВ ДЛЯ ПРИМЕНЕНИЯ В КАЧЕСТВЕ ИНГИБИТОРОВ АЦЕТИЛ-КоА-КАРБОКСИЛАЗ Согласно изобретению предложены соединение формулы (I) или его фармацевтически приемлемая соль; где G представляет собой (II) где R1, R2 и R3 являются такими, как изложено в данном изобретении; их фармацевтические композиции и их применение в лечении заболеваний, состояний или расстройств,модулируемых посредством ингибирования фермента(ов), представляющего(их) собой ацетилКоА-карбоксилазу(ы), у животного. Область изобретения Данное изобретение относится к замещенным соединениям пиразолоспирокетонов, действующим в качестве ингибиторов ацетил-КоА-карбоксилаз(ы), и их применению в лечении заболеваний, состояний или расстройств, модулируемых посредством ингибирования фермента(ов), представляющего(их) собой ацетил-КоА-карбоксилазу(ы). Предшествующий уровень техники Ацетил-КоА-карбоксилазы (АСС) представляют собой семейство ферментов, обнаруженных у большинства видов, и ассоциированы с синтезом и метаболизмом жирных кислот через катализ продукции малонил-КоА из ацетил-КоА. У млекопитающих идентифицированы две изоформы фермента АСС. АСС 1, которая экспрессируется на высоких уровнях в липогенных тканях, таких как жировая ткань и печень, регулирует первую стадию, осуществляемую в биосинтезе длинноцепочечных жирных кислот. Если ацетил-КоА не карбоксилируется с образованием малонил-КоА, то он метаболизируется через цикл Кребса. АСС 2, минорный компонент печеночной АСС, но преобладающая изоформа в сердечной и скелетной мышце, катализирует продукцию малонил-КоА на цитозольной поверхности митохондрий и регулирует, в какой степени жирные кислоты утилизируются при -окислении, путем ингибирования карнитинпальмитоилтрансферазы. Таким образом, путем увеличения утилизации жирных кислот и предотвращения увеличения синтеза жирных кислот de novo постоянное введение ингибитора АСС (ACC-I) также может истощать запасы триглицеридов (TG) в печени и жировой ткани у субъектов, страдающих ожирением, придерживающихся диеты с высоким или низким содержанием жиров, что приводит к избирательной потере жира в организме. Исследования, проведенные Abu-Etheiga и др., указывают на то, что АСС 2 играет существенную роль в регуляции окисления жирных кислот и как таковая может представлять собой мишень в терапии против ожирения и связанных с ожирением заболеваний, таких как диабет 2 типа. См. Abu-Etheiga L., etal., "Acetyl-CoA carboxylase 2 mutant mice are protected against obesity and diabetes induced by high-fat/highcarbohydrate diets", PNAS, 100(18), 10207-10212 (2003). См. также Choi C.S., et al., "Continuous fat oxidation in acetyl-CoA carboxylase 2 knockout mice increases total energy expenditure, reduces fat mass, and improves insulin sensitivity", PNAS, 104(42), 16480-16485 (2007). Становится все более ясно, что накопление печеночного жира вызывает печеночную инсулинорезистентность и вносит вклад в патогенез диабета 2 типа. Salvage и др. продемонстрировали, что оба фермента АСС 1 и АСС 2 вовлечены в регуляцию окисления жира в гепатоцитах, в то время как АСС 1, преобладающая изоформа в печени крыс, представляет собой единственный регулятор синтеза жирных кислот. Кроме того, в их модели совместное уменьшение обеих изоформ необходимо, чтобы значительно снизить уровни малонил-КоА в печени, усилить окисление жиров в сытом состоянии, уменьшить накопление липидов и улучшить действие инсулина in vivo. Таким образом, показано, что ингибиторы печеночных АСС 1 и АСС 2 могут быть полезны в лечении неалкогольной жировой болезни печени (NAFLD) и печеночной инсулинорезистентности. См. Savage D.В. et al., "Reversal of diet-induced hepatic steatosisacetyl-CoA carboxylase 2 knockout mice", PNAS, 102(5), 1384-1389 (2005). Таким образом, существует потребность в лекарственных средствах, содержащих ингибиторы АСС 1 и/или АСС 2, для лечения ожирения и связанных с ожирением заболеваний (таких как NAFLD и диабет 2 типа) путем ингибирования синтеза жирных кислот и путем усиления окисления жирных кислот. Краткое изложение сущности изобретения Первым воплощением настоящего изобретения является соединение структуры или его фармацевтически приемлемая соль. Другим воплощением настоящего изобретения является соединение структуры-1 022375 или его фармацевтически приемлемая соль. Другим воплощением настоящего изобретения является соединение структуры или его фармацевтически приемлемая соль. Другим воплощением настоящего изобретения является соединение структуры или его фармацевтически приемлемая соль. Другим аспектом настоящего изобретения является фармацевтическая композиция для лечения заболеваний, опосредованных активностью ацетил-КоА-карбоксилазы, содержащая количество соединения по настоящему изобретению, как описано в любом из воплощений, или его фармацевтически приемлемую соль и фармацевтически приемлемый эксципиент, разбавитель или носитель. Предпочтительно композиция содержит терапевтически эффективное количество соединения по настоящему изобретению. Композиция также может содержать по меньшей мере один дополнительный фармацевтический агент. Предпочтительные агенты включают антидиабетические агенты и/или агенты против ожирения. В настоящем изобретении также раскрыт способ лечения заболевания, состояния или расстройства,опосредованного ингибированием фермента(ов),представляющего(их) собой ацетил-КоАкарбоксилазу(ы), у млекопитающего, включающий стадию введения млекопитающему, предпочтительно человеку, нуждающемуся в таком лечении, терапевтически эффективного количества соединения по настоящему изобретению, или его фармацевтически приемлемой соли, или их фармацевтической композиции. Заболевания, расстройства или состояния, опосредованные ингибиторами ацетил-КоА-карбоксилаз,включают диабет 2 типа и связанные с диабетом заболевания, такие как неалкогольная жировая болезнь печени (NAFLD), печеночная инсулинорезистентность, гипергликемия, метаболический синдром, нарушенная толерантность к глюкозе, диабетическая невропатия, диабетическая нефропатия, диабетическая ретинопатия, ожирение, дислипидемия, гипертензия, гиперинсулинемия и синдром инсулинорезистентности. Предпочтительные заболевания, расстройства или состояния включают диабет 2 типа, неалкогольную жировую болезнь печени (NAFLD), печеночную инсулинорезистентность, гипергликемию, нарушенную толерантность к глюкозе, ожирение и синдром инсулинорезистентности. Более предпочтительными являются диабет 2 типа, неалкогольная жировая болезнь печени (NAFLD), печеночная инсулинорезистентность, гипергликемия и ожирение. Наиболее предпочтительным заболеванием является диабет 2 типа. В настоящем изобретении также раскрыт способ лечения (например, замедления прогрессирования или возникновения) диабета 2 типа и связанных с диабетом расстройств у животных, включающий стадию введения животному, нуждающемуся в таком лечении, терапевтически эффективного количества соединения по настоящему изобретению, или его фармацевтически приемлемой соли, или их композиции. В настоящем изобретении также раскрыт способ лечения или замедления прогрессирования или возникновения диабета 2 типа и связанных с диабетом расстройств у человека, включающий стадию введения человеку, нуждающемуся в таком лечении, терапевтически эффективного количества соединения по настоящему изобретению, или его фармацевтически приемлемой соли, или их композиции. В настоящем изобретении также раскрыт способ лечения или замедления прогрессирования или возникновения диабета 2 типа у человека, включающий стадию введения человеку, нуждающемуся в таком лечении, терапевтически эффективного количества соединения по настоящему изобретению, или его фармацевтически приемлемой соли, или их композиции. В настоящем изобретении также раскрыт способ лечения ожирения и связанных с ожирением расстройств у животных, включающий стадию введения животному, нуждающемуся в таком лечении, терапевтически эффективного количества соединения по настоящему изобретению, или его фармацевтически приемлемой соли, или их композиции.-2 022375 В настоящем изобретении также раскрыт способ лечения ожирения и связанных с ожирением расстройств у человека, включающий стадию введения человеку, нуждающемуся в таком лечении, терапевтически эффективного количества соединения по настоящему изобретению, или его фармацевтически приемлемой соли, или их композиции. В настоящем изобретении также раскрыт способ лечения неалкогольной жировой болезни печени(NAFLD) или печеночной инсулинорезистентности у животных, включающий стадию введения животному, в частности человеку, нуждающемуся в таком лечении, терапевтически эффективного количества соединения по настоящему изобретению, или его фармацевтически приемлемой соли, или их композиции. В настоящем изобретении также раскрыт способ лечения неалкогольной жировой болезни печени(NAFLD) или печеночной инсулинорезистентности у человека, включающий стадию введения человеку,нуждающемуся в таком лечении, терапевтически эффективного количества соединения по настоящему изобретению, или его фармацевтически приемлемой соли, или их композиции. Соединения по настоящему изобретению могут быть введены в комбинации с другими фармацевтическими агентами (в частности, агентами против ожирения и противодиабетическими агентами, описанными ниже в данном изобретении). Комбинированную терапию можно вводить в виде (а) единой фармацевтической композиции, содержащей соединение по настоящему изобретению, по меньшей мере один дополнительный фармацевтический агент, описанный в данном изобретении, и фармацевтически приемлемый эксципиент, разбавитель или носитель; или (б) двух отдельных фармацевтических композиций, содержащих (1) первую композицию, содержащую соединение по настоящему изобретению и фармацевтически приемлемый эксципиент, разбавитель или носитель, и (2) вторую композицию, содержащую по меньшей мере один дополнительный фармацевтический агент, описанный в данном изобретении, и фармацевтически приемлемый эксципиент, разбавитель или носитель. Фармацевтические композиции можно вводить одновременно или последовательно и в любом порядке. Другим воплощением является применение соединения по настоящему изобретению в изготовлении лекарственного средства для лечения заболевания, состояния или расстройства, модулируемого посредством ингибирования фермента(ов), представляющего(их) собой ацетил-КоА-карбоксилазу(ы). Другим воплощением является применение соединения по настоящему изобретению в изготовлении лекарственного средства для лечения заболевания, состояния или расстройства, модулируемого посредством ингибирования фермента(ов), представляющего(их) собой ацетил-КоА-карбоксилазу(ы), где заболевание, состояние или расстройство представляет собой диабет 2 типа, связанные с диабетом расстройства, неалкогольную жировую болезнь печени (NAFLD) или печеночную инсулинорезистентность. Другим воплощением является применение соединения по настоящему изобретению в изготовлении лекарственного средства для лечения заболевания, состояния или расстройства, модулируемого посредством ингибирования фермента(ов), представляющего(их) собой ацетил-КоА-карбоксилазу(ы), где заболевание, состояние или расстройство представляет собой диабет 2 типа. Другим воплощением является применение соединения примера 6, 14 или 25 или 1'-(2-(третбутиламино)хинолин-7-карбонил)-1-изопропил-4,6-дигидроспиро[индазол-5,4'-пиперидин]-7(1H)-она в изготовлении лекарственного средства для лечения заболевания, состояния или расстройства, модулируемого посредством ингибирования фермента(ов), представляющего(их) собой ацетил-КоАкарбоксилазу(ы), где заболевание, состояние или расстройство представляет собой диабет 2 типа, связанные с диабетом расстройства, неалкогольную жировую болезнь печени (NAFLD) или печеночную инсулинорезистентность. Другим воплощением является применение соединения примера 6, 14 или 25 в изготовлении лекарственного средства для лечения заболевания, состояния или расстройства, модулируемого посредством ингибирования фермента(ов), представляющего(их) собой ацетил-КоА-карбоксилазу(ы), где заболевание, состояние или расстройство представляет собой диабет 2 типа. Другим воплощением является применение соединения примера 6 в изготовлении лекарственного средства для лечения заболевания, состояния или расстройства, модулируемого посредством ингибирования фермента(ов), представляющего(их) собой ацетил-КоА-карбоксилазу(ы), где заболевание, состояние или расстройство представляет собой диабет 2 типа. Другим воплощением является применение соединения примера 14 в изготовлении лекарственного средства для лечения заболевания, состояния или расстройства, модулируемого посредством ингибирования фермента(ов), представляющего(их) собой ацетил-КоА-карбоксилазу(ы), где заболевание, состояние или расстройство представляет собой диабет 2 типа. Другим воплощением является применение соединения примера 25 в изготовлении лекарственного средства для лечения заболевания, состояния или расстройства, модулируемого посредством ингибирования фермента(ов), представляющего(их) собой ацетил-КоА-карбоксилазу(ы), где заболевание, состояние или расстройство представляет собой диабет 2 типа. Подробное описание изобретения Определения. Фраза "терапевтически эффективное количество" означает количество соединения по настоящему изобретению или его фармацевтически приемлемой соли, которое (1) лечит или предупреждает конкретное заболевание, состояние или расстройство, (2) ослабляет, смягчает или устраняет один или более чем один симптом конкретного заболевания, состояния или расстройства или (3) предупреждает или замедляет возникновение одного или более чем одного симптома конкретного заболевания, состояния или расстройства, описанного в данном изобретении. Термин "животное" относится к людям (мужского или женского пола), домашним животным (например, собакам, кошкам и лошадям), животным-источникам пищи, животным зоопарков, морским животным, птицам и другим подобным видам животных. Термин "годные в пищу животные" относится к животным-источникам пищи, таким как коровы, свиньи, овцы и домашняя птица. Фраза "фармацевтически приемлемый" указывает на то, что вещество или композиция должны быть химически и/или токсикологически совместимы с другими ингредиентами, составляющими препарат, и/или млекопитающим, которое ими. Термины "процесс лечения", "лечить" или "лечение" охватывают как превентивное, т.е. профилактическое, так и паллиативное лечение. Термины "модулируемый", или "модулирующий", или "модулируют(ет)", как использовано в данном изобретении, если не указано иное, относятся к ингибированию фермента(ов), представляющего(их) собой ацетил-КоА-карбоксилазу(ы) (АСС), соединениями по настоящему изобретению. Термины "опосредованный", или "опосредующий", или "опосредуют(ет)", как использовано в данном изобретении, если не указано иное, относится к (1) лечению или предупреждению конкретного заболевания, состояния или расстройства, (2) ослаблению, смягчению или устранению одного или более чем одного симптома конкретного заболевания, состояния или расстройства или (3) замедлению возникновения одного или более чем одного симптома конкретного заболевания, состояния или расстройства, описанного в данном изобретении, путем ингибирования фермента(ов), представляющего(их) собой ацетилКоА-карбоксилазу(ы) (АСС). Термин "соединения по настоящему изобретению" (если специально не указано иное) относится к указанным выше соединениям и любым фармацевтически приемлемым солям этих соединений, а также всем стереоизомерам (включая диастереоизомеры и энантиомеры), таутомерам, конформационным изомерам и меченым изотопами соединениям. Гидраты и сольваты соединений по настоящему изобретению рассматривают как композиции по настоящему изобретению, где соединение находится в ассоциации с водой или растворителем соответственно. Термины "(C1-C6)алкил" и "(C1-C3)алкил" означают алкильные группы с определенным числом атомов углерода, от одного до шести или от одного до трех атомов углерода, соответственно, которые могут быть либо с прямой цепью, либо разветвленными. Например, термин "(C1-C3)алкил" имеет от одного до трех атомов углерода и состоит из метила, этила, н-пропила и изопропила. Алкоксигруппы с определенным числом атомов углерода названы аналогичным образом. Термин "(C1-C3)алкилен" означает бирадикальные (C1-C3)алкильные группы, имеющие от одного до трех атомов углерода, которые могут быть либо с прямой цепью, либо разветвленными. Типичные примеры термина "(C1-C3)алкилен" включают -СН 2-, -СН 2 СН 2-, -СН(СН 3)СН 2-, -СН 2 СН 2(СН 3)- или-СН 2 СН 2 СН 2-, но не ограничиваются ими. Термин "(C3-C7)циклоалкил" означает циклоалкильную группу с атомами углерода в количестве от трех до семи и состоит из циклопропила, циклобутила, циклопентила, циклогексила и циклогептила или может представлять собой бициклическую кольцевую систему, такую как бицикло[1.1.1]пентанил. Термин "галогено" означает фторо, хлоро, бромо или йодо. Термин "4-7-членный гетероциклил" означает радикал 4-7-членного неароматического гетероцикла. Место присоединения может проходить через атом углерода или атом азота. Его неограничивающие примеры включают оксетанил, тетрагидрофуранил, морфолинил, азетидинил, пирролидинил, пиперидинил, пиперазинил и т.п. Термины "индолил", "индазолил", "пирролопиридинил", "пиразолопиридинил", "хинолинил" или"бензимидазолил" означают радикалы приведенных ниже групп, и местом присоединения является атом углерода этой группы. Соединения по настоящему изобретению могут быть синтезированы с помощью путей синтеза,включающих способы, аналогичные хорошо известным в области химии, в частности, в свете описания,приведенного в данном изобретении. Исходные вещества обычно доступны у коммерческих источников,таких как Aldrich Chemicals (Милуоки, Висконсин (WI, или могут быть легко получены с использованием способов, хорошо известных специалистам в данной области (например, получены способами, в целом описанными в Louis F. Fieser and Mary Fieser, Reagents for Organic Synthesis, v. 1-19, Wiley, NewYork (1967-1999 ed.) или Beilsteins Handbuch der organischen Chemie, 4, Aufl. ed. Springer-Verlag, Berlin,включая приложения (также доступны через онлайновую базу данных Бейльштейна. В иллюстративных целях на приведенных ниже реакционных схемах представлены возможные пути синтеза соединений по настоящему изобретению, а также ключевых промежуточных соединений. Более подробное описание отдельных реакционных стадий см. в разделе "Примеры", приведенном ниже. Специалистам в данной области техники будет понятно, что для синтеза соединений по изобретению можно использовать другие пути синтеза. Хотя на схемах приведены и ниже обсуждаются конкретные исходные вещества и реагенты, они легко могут быть заменены на другие исходные вещества и реагенты для обеспечения разнообразия производных и/или реакционных условий. Кроме того, многие из соединений, полученных описанными ниже способами, могут быть дополнительно модифицированы в свете данного описания с использованием методов традиционной химии, хорошо известных специалистам в данной области. При получении соединений по настоящему изобретению может быть необходима защита отдаленной функциональной группы (например, первичного или вторичного амина) промежуточных соединений. Необходимость в такой защите будет варьировать в зависимости от природы отдаленной функциональной группы и условий способов получения. Подходящие аминозащитные группы (NH-Pg) включают ацетил, трифторацетил, трет-бутоксикарбонил (ВОС), бензилоксикарбонил (CBz) и 9 флуоренилметиленоксикарбонил (Fmoc). Аналогично, "гидроксизащитная группа" относится к заместителю гидроксигруппы, который блокирует или защищает функциональную группу гидрокси. Подходящие гидроксизащитные группы (O-Pg) включают, например, аллил, ацетил, силил, бензил, параметоксибензил, тритил и т.п. Специалист в данной области легко определяет необходимость в такой защите. Общее описание защитных групп и их применение см. в Т.W. Greene, Protective Groups in OrganicSynthesis, John WileySons, New York, 1991. На следующих далее реакционных схемах, от реакционной схемы I до реакционной схемы III, приведены типичные процедуры, которые используют для получения соединений по настоящему изобретению. Следует понимать, что эти реакционные схемы не должны истолковываться как ограничивающие и что для получения соединений по настоящему изобретению можно использовать разумные вариации представленных способов. На реакционной схеме I приведены общие процедуры, которые могут быть использованы для получения N1 АСС-ингибирующих соединений по настоящему изобретению, имеющих формулу Ia, где R1 представляет собой (C1-C6)алкил или (С 3-С 7)циклоалкил; R2 представляет собой индолил, индазолил,пирролопиридинил, пиразолопиридинил, хинолинил или бензимидазолил; где каждая группа R2 возможно замещена заместителями в количестве от одного до двух, независимо выбранными из циано, -LC(O)NR4R5, -L-NR4R5, (С 1-С 3)алкила, (С 1-С 3)алкокси и галогено; R3 представляет собой водород; L представляет собой прямую связь или -Х(C1-C3)алкилен; X представляет собой прямую связь, О или S; каждый из R4 и R5 независимо представляет собой водород, (C1-C3)алкил, (C3-C7)циклоалкил или 4-7 членный гетероциклил, где указанный (C1-C3)алкил, (C3-C7)циклоалкил или 4-7-членный гетероциклил возможно замещен фторо или (C1-C3)алкокси в количестве от одного до трех. Согласно схеме I соединение формулы VIIa может быть образовано путем приведения соединения формулы VIIIa, где Pg представляет собой соответствующую амино-защитную группу, во взаимодействие с трис(диметиламино)метаном в соответствующем растворителе. Реакция может быть осуществлена в соответствующем растворителе, таком как толуол, при повышенной температуре, такой как температура дефлегмации, в течение 1-24 ч с получением соединения формулы VIIa. Соединение формулы VIa может быть образовано путем приведения соединения формулы VIIa во взаимодействие с соответствующим алкил- или циклоалкилгидразином (R1NHNH2, таким как трет-бутилгидразин, изопропилгидразин или бицикло[1.1.1]пентанилгидразин) в соответствующем растворителе, таком как этанол. Например,соединение формулы VIa может быть образовано путем приведения соединения формулы VIIa во взаимодействие с соответствующим алкилгидразином (R1NHNH2), возможно в присутствии основания, такого как карбонат калия ("K2CO3"), в этаноле при температуре дефлегмации с получением желаемого циклизованного соединения, при температуре от примерно 20 до примерно 80 С в течение примерно 2-24 ч. Соединение формулы Va может быть образовано путем превращения соединения формулы VIa в соответствующее гидроксибромидное производное путем взаимодействия с соответствующим бромирующим реагентом и водой в соответствующем растворителе. Например, соединение формулы Va может быть образовано путем приведения соединения формулы VIa во взаимодействие с N-бромсукцинимидом(NBS) и водой в тетрагидрофуране при комнатной температуре в течение 1 ч с получением соответствующего гидроксибромидного производного формулы Va. Затем может быть образовано соединение формулы IVa путем окисления соединения формулы Va соответствующим окисляющим агентом в соответствующем растворителе. Например, окисление соединения формулы Va может быть осуществлено путем обработки реагентом Джонса в ацетоне при температуре от 0 С до комнатной температуры в течение периода времени от 15 мин до 4 ч с последующей экстракцией. Затем можно осуществить дебромирование соединения формулы IVa путем обработки водным хлоридом аммония и металлическим цинком в соответствующем растворителе, таком как тетрагидрофуран, в течение периода времени от 15 мин до 4 ч обычно при комнатной температуре. Затем с соединения формулы IIIa можно снять защиту с получением свободного спиропиперидинового производного формулы IIa, используя стандартные способы в зависимости от того, какая защитная группа Pg была применена. Например, когда Pg представляет собой ВОС, то для удаления группы ВОС можно использовать стандартные условия снятия защиты с применением сильной кислоты, такой как 4 н. соляная кислота в диоксане или трифторуксусная кислота в соответствующем растворителе, таком как дихлорметан. Когда Pg представляет собой Cbz, для снятия защиты можно использовать гидрирование над палладием на угле в этаноле или обработку источником водорода, таким как формиат аммония или 1-метил-1,4-циклогексадиен, в присутствии палладия на угле в этаноле или этилацетате. Затем, чтобы получить соединение формулы Ia, спиропиперидиновое производное формулы IIa можно ацилировать путем применения стандартных способов. Например, в таком случае соединение (Ia) может быть образовано с использованием стандартной реакции пептидного сочетания с желаемой карбоновой кислотой (R2CO2H). Например, сочетание спиропиперидинового промежуточного соединения (IIa) и карбоновой кислоты (R2CO2H) может быть осуществлено путем образования активированного сложного эфира карбоновой кислоты, например, путем приведения карбоновой кислоты (R2CO2H) в контакт с реагентом пептидного сочетания, таким как гексафторфосфат О-(7-азабензотриазол-1-ил)-N,N,N',N'тетраметилурония ("HATU") или гидрохлорид 1-этил-3-(3-диметиламинопропил)карбодиимида("HOBt"), и в присутствии подходящего основания, такого как N,N-диизопропилэтиламин ("DIEA"), триэтиламин или N-метилморфолин ("NMM"), в подходящем растворителе, таком как THF (тетрагидрофуран) и/или DMF (диметилформамид), диметилацетамид ("DMA") или дихлорметан, и затем приведения этого активированного эфира карбоновой кислоты в контакт со спиропиперидиновым производным IIa с образованием соединения формулы Ia. Соединения по настоящему изобретению могут быть выделены и использованы per se или в форме их фармацевтически приемлемых солей. В соответствии с настоящим изобретением соединения с много-6 022375 численными основными атомами азота могут образовывать соли с варьирующим числом эквивалентов("экв.") кислоты. Практикующим специалистам будет понятно, что все такие соли находятся в пределах объема настоящего изобретения. Фармацевтически приемлемые соли, как использовано в данном изобретении в отношении соединений по настоящему изобретению, включают фармацевтически приемлемые неорганические и органические соли соединения. Эти соли могут быть получены in situ в процессе конечного выделения и очистки соединения или путем отдельного взаимодействия соединения с подходящей органической или неорганической кислотой и выделения соли, образованной таким образом. Типичные соли включают соли гидробромид, гидрохлорид, гидройодид, сульфат, бисульфат, нитрат, ацетат, трифторацетат, оксалат,безилат, пальмитат, памоат, малонат, стеарат, лаурат, малат, борат, бензоат, лактат, фосфат, гексафторфосфат, бензолсульфонат, тозилат, формиат, цитрат, малеат, фумарат, сукцинат, тартрат, нафтилат, мезилат, глюкогептонат, лактобионат и лаурилсульфонат и тому подобное, но не ограничиваются ими. Они также могут включать катионы на основе щелочных и щелочноземельных металлов, таких как натрий,литий, калий, кальций, магний и тому подобное, а также нетоксичные катионы аммония, четвертичного аммония и амина, включая аммоний, тетраметиламмоний, тетраэтиламмоний, метиламмоний, диметиламмоний, триметиламмоний, триэтиламмоний, этиламмоний и т.п., но не ограничиваясь ими. Дополнительные примеры см., например, в Berge, et al., J. Pharm. Sci., 66, 1-19 (1977). Соединения по настоящему изобретению могут существовать в более чем одной кристаллической форме. Полиморфы соединений по настоящему изобретению и их солей (включая сольваты и гидраты) составляют часть данного изобретения и могут быть получены путем кристаллизации соединения по настоящему изобретению в различных условиях. Например, используя для перекристаллизации различные растворители или различные смеси растворителей; кристаллизацию при различных температурах; различные режимы охлаждения, варьирующие от очень быстрого до очень медленного охлаждения во время кристаллизации. Полиморфы также могут быть получены путем нагревания или плавления соединения по настоящему изобретению с последующим постепенным или быстрым охлаждением. Присутствие полиморфов может быть определено посредством спектроскопии ядерного магнитного резонанса (ЯМР) твердого образца, инфракрасной (ИК) спектроскопии, дифференциальной сканирующей калориметрии,дифракции рентгеновских лучей на порошке или других таких методов. Данное изобретение также включает меченые изотопами соединения, которые идентичны соединениям по настоящему изобретению, но с учетом того, что один или более чем один атом заменен атомом,имеющим атомную массу или массовое число, отличные от атомной массы или массового числа, обычно встречающихся в природе. Примеры изотопов, которые могут быть включены в соединения по изобретению, включают изотопы водорода, углерода, азота, кислорода, серы и фтора, такие как 2 Н, 3 Н, 13 С, 14 С,15N, 18O, 17O, 35S, 36Cl, 125I, 129I и 18F соответственно. Некоторые меченые изотопами соединения по настоящему изобретению, например те, в которые включены радиоактивные изотопы, такие как 3 Н и 14 С,полезны в анализах распределения лекарственного средства и/или субстрата в тканях. Изотопы трития(т.е. 3 Н) и углерода-14 (т.е. 14 С) особенно предпочтительны ввиду простоты их получения и детектирования. Кроме того, замещение на более тяжелые изотопы, такие как дейтерий (т.е. 2 Н), может давать некоторые терапевтические преимущества, являющиеся результатом большей метаболической стабильности,например, увеличенный in vivo период полувыведения или уменьшенные требования к дозировкам, и поэтому может быть предпочтительным в некоторых обстоятельствах. Меченые изотопами соединения по настоящему изобретению обычно могут быть получены путем осуществления процедур, описанных ниже на схемах и/или в примерах, путем замены не меченого изотопом реагента на легко доступный меченый изотопом реагент. Соединения по настоящему изобретению могут содержать стереогенные центры. Эти соединения могут существовать в виде смесей энантиомеров или в виде чистых энантиомеров. Когда соединение включает стереогенный центр, тогда соединения могут быть разделены на чистые энантиомеры способами, известными специалистам в данной области, например, путем образования диастереоизомерных солей, которые могут быть разделены, например, посредством кристаллизации; образования стереоизомерных производных или комплексов, которые могут быть разделены, например, посредством кристаллизации, газожидкостной или жидкостной хроматографии; селективного взаимодействия одного энантиомера с энантиомер-специфическим реагентом, например, ферментативной этерификации; или газожидкостной или жидкостной хроматографии в хиральном окружении, например на хиральном носителе, например диоксиде кремния со связанным хиральным лигандом, или в присутствии хирального растворителя. Следует понимать, что когда желаемый стереоизомер превращают в другую химическую сущность с помощью одной из процедур разделения, описанных выше, тогда требуется дополнительная стадия для высвобождения желаемой энантиомерной формы. Альтернативно, конкретные стереоизомеры можно синтезировать с использованием оптически активного исходного вещества, применяя асимметрический синтез с использованием оптически активных реагентов, субстратов, катализаторов или растворителей или превращая один стереоизомер в другой путем асимметрической трансформации. Соединения по настоящему изобретению могут существовать в различных стабильных конформационных формах, которые могут поддаваться разделению. Торсионная асимметрия вследствие ограни-7 022375 ченного вращения вокруг асимметричной одинарной связи, например ввиду стерических затруднений или напряжения кольца, может обеспечить разделение различных конформеров. Соединения по настоящему изобретению также включают каждый конформационный изомер соединений по настоящему изобретению и их смеси. Соединения по настоящему изобретению полезны для лечения заболеваний, состояний и/или расстройств, модулируемых посредством ингибирования фермента(ов), представляющего(их) собой ацетилКоА-карбоксилазу(ы) (в частности, АСС 1 и АСС 2). Другим воплощением настоящего изобретения является фармацевтическая композиция, содержащая терапевтически эффективное количество соединения по настоящему изобретению и фармацевтически приемлемый эксципиент, разбавитель или носитель. Соединения по настоящему изобретению (включая композиции и способы, используемые в данном изобретении) также могут быть применены в изготовлении лекарственного средства для терапевтических применений, описанных в данном изобретении. Типичную композицию готовят путем смешивания соединения по настоящему изобретению и носителя, разбавителя или эксципиента. Подходящие носители, разбавители и эксципиенты хорошо известны специалистам в данной области техники и включают такие вещества, как углеводы, воски, растворимые и/или набухаемые полимеры, гидрофильные или гидрофобные вещества, желатин, масла, растворители, воду и т.п. Конкретный используемый носитель, разбавитель или эксципиент будет зависеть от способа и назначения, для которых соединение по настоящему изобретению будут применять. Растворители обычно выбирают, основываясь на растворителях, признанных специалистами в данной области безопасными (GRAS) для введения млекопитающему. В общем случае растворители представляют собой нетоксичные водные растворители, такие как вода и другие нетоксичные растворители, которые растворимы в воде или смешиваются с водой. Подходящие водные растворители включают воду, этанол, пропиленгликоль, полиэтиленгликоли (например, ПЭГ 400, ПЭГ 300) и т.д. и их смеси. Композиции также могут включать один или более чем один буфер, стабилизирующий агент, поверхностно-активное вещество, увлажняющий агент, смазывающее вещество эмульгатор, суспендирующий агент, консервант, антиоксидант, замутняющий агент, глидант, технологическую добавку, краситель, подсластитель, ароматизатор, корригент и другие известные добавки для обеспечения наилучшего представления лекарственного средства (т.е. соединения по настоящему изобретению или его фармацевтической композиции) или содействия изготовлению фармацевтического продукта (т.е. для применения в изготовлении лекарственного средства). Композиции могут быть приготовлены с использованием традиционных процедур растворения и смешивания. Например, нефасованное лекарственное вещество (т.е. соединение по настоящему изобретению или стабилизированную форму соединения (например, комплекс с циклодекстриновым производным или другим известным агентом комплексообразования растворяют в подходящем растворителе в присутствии одного или более чем одного эксципиента, описанного выше. Скорость растворения малорастворимых в воде соединений можно повысить путем применения высушенной распылением дисперсии, как например описано Takeuchi H. и др. в "Enhancement of the dissolution rate of a poorly water-solubledrug (tolbutamide) by a spray-drying solvent deposition method and disintegrants", J. Pharm. Pharmacol., 39,769-773 (1987); и в ЕР 0901786 B1 (US2002/009494), которые включены в данную заявку посредством ссылки. Соединение по настоящему изобретению типично изготавливают в виде фармацевтических лекарственных форм для того, чтобы обеспечить легко контролируемую дозировку лекарственного средства и предоставить пациенту прекрасный и простой в применении продукт. Фармацевтические композиции также включают сольваты и гидраты соединений по настоящему изобретению. Термин "сольват" относится к молекулярному комплексу соединения по настоящему изобретению (включая его фармацевтически приемлемые соли), с одной или более чем одной молекулой растворителя. Такие молекулы растворителя представляют собой молекулы растворителей, обычно используемых в области фармацевтики, которые известны как безвредные для реципиента, например вода,этанол, этиленгликоль и т.п. Термин "гидрат" относится к комплексу, в котором молекулой растворителя является вода. Сольваты и/или гидраты предпочтительно существуют в кристаллической форме. В качестве промежуточных сольватов при получении более желаемых сольватов могут быть использованы другие растворители, такие как метанол, метил-трет-бутиловый эфир, этилацетат, метилацетат, (S)пропиленгликоль, (R)-пропиленгликоль, 1,4-бутиндиол и т.п. Фармацевтическая композиция (или препарат) для применения может быть упакована различным образом в зависимости от способа, используемого для введения лекарственного средства. В общем случае, изделие для распространения включает контейнер с помещенной внутрь него фармацевтической композицией в соответствующей форме. Подходящие контейнеры хорошо известны специалистам в данной области и включают такие материалы, как бутылки (пластиковые и стеклянные), саше, ампулы, пластиковые пакеты, металлические цилиндры и т.п. Контейнер также может включать защищающую от неумелого обращения систему для предупреждения неосторожного доступа к содержимому упаковки. Помимо этого, контейнер имеет помещенную на него этикетку, описывающую содержимое контейнера. Этикетка также может содержать соответствующие предостережения. В настоящем изобретении также раскрыт способ лечения заболеваний, состояний и/или рас-8 022375 стройств, модулируемых посредством ингибирования фермента(ов), представляющего(их) собой ацетилКоА-карбоксилазу(ы), у животного, включающий введение животному, нуждающемуся в таком лечении,терапевтически эффективного количества соединения по настоящему изобретению или фармацевтической композиции, содержащей эффективное количество соединения по настоящему изобретению и фармацевтически приемлемый эксципиент, разбавитель или носитель. Способ особенно полезен для лечения заболеваний, состояний и/или расстройств, при которых благоприятно ингибирование фермента(ов),представляющего(их) собой ацетил-КоА-карбоксилазу(ы). Одним из аспектов настоящего изобретения является лечение ожирения и связанных с ожирением расстройств (например, избыточная масса тела, увеличение массы тела или поддержание массы тела). Ожирение и избыточную массу тела обычно определяют по индексу массы тела (BMI), который коррелирует с общим содержанием жира в организме и позволяет оценить относительный риск заболевания. BMI рассчитывают как массу в килограммах, деленную на рост в метрах в квадрате (кг/м 2). Избыточную массу тела типично определяют, когда BMI составляет 25-29,9 кг/м 2, а ожирение типично определяют, когда BMI составляет 30 кг/м 2. См., например, National Heart, Lung, and Blood Institute, ClinicalReport, Washington, DC: U.S. Department of Health and Human Services, NIH publication no. 98-4083 (1998). Другим аспектом настоящего изобретения является лечение (например, замедление прогрессирования или возникновения) диабета или связанных с диабетом расстройств, включая диабет 1 типа (инсулинозависимый сахарный диабет, также обозначаемый как "IDDM") и 2 типа (инсулинонезависимый сахарный диабет, также обозначаемый как "NIDDM"), нарушенную толерантность к глюкозе, инсулинорезистентность, гипергликемию и диабетические осложнения (такие как атеросклероз, ишемическая болезнь сердца, инсульт, заболевание периферических сосудов, нефропатия, гипертензия, невропатия и ретинопатия). Еще одним аспектом настоящего изобретения является лечение сопутствующих ожирению заболеваний, таких как метаболический синдром. Метаболический синдром включает заболевания, состояния или расстройства, такие как дислипидемия, гипертензия, инсулинорезистентность, диабет (например,диабет 2 типа), ишемическая болезнь сердца и сердечная недостаточность. Для более подробной информации о метаболическом синдроме см., например, Zimmet P.Z., et al., "The Metabolic Syndrome: Perhaps anEndocrinology, 7(2), (2005); и Alberti K.G., et al., "The Metabolic Syndrome - A New Worldwide Definition",Lancet, 366, 1059-62 (2005). Предпочтительно, введение соединений по настоящему изобретению обеспечивает статистически значимое (р 0,05) уменьшение по меньшей мере одного фактора риска сердечнососудистого заболевания, такого как снижение содержания лептина, С-реактивного белка (CRP) и/или холестерина в плазме крови по сравнению с контролем-носителем, не содержащим лекарственного средства. Введение соединений по настоящему изобретению также может обеспечивать статистически значимое (р 0,05) снижение уровня глюкозы в сыворотке. Еще одним аспектом изобретения является лечение неалкогольной жировой болезни печени(NAFLD) и печеночной инсулинорезистентности. Для нормального взрослого человека, имеющего массу тела примерно 100 кг, дозировка в диапазоне от примерно 0,001 до примерно 10 мг на 1 кг массы тела обычно является достаточной, предпочтительно от примерно 0,01 до примерно 5,0 мг/кг, более предпочтительно от примерно 0,01 до примерно 1 мг/кг. Однако может потребоваться некоторая вариабельность в общем диапазоне дозировок в зависимости от возраста и массы субъекта, которого лечат, предполагаемого пути введения, конкретного вводимого соединения и тому подобного. Определение диапазонов дозировок и оптимальных дозировок для конкретного пациента находится в пределах компетенции обычного специалиста в данной области, получающего пользу от настоящего описания. Следует также отметить, что соединения по настоящему изобретению могут быть использованы в форме композиций с длительным высвобождением, контролируемым высвобождением и замедленным высвобождением, и эти формы также хорошо известны обычному специалисту в данной области. Соединения по данному изобретению также могут быть использованы в сочетании с другими фармацевтическими агентами для лечения заболеваний, состояний и/или расстройств, описанных в данном изобретении. Поэтому также предложены способы лечения, включающие введение соединений по настоящему изобретению в комбинации с другими фармацевтическими агентами. Подходящие фармацевтические агенты, которые могут быть использованы в комбинации с соединениями по настоящему изобретению, включают агенты против ожирения (включая средства для подавления аппетита), антидиабетические агенты, антигипергликемические агенты, агенты, снижающие уровень липидов, и антигипертензивные агенты. Подходящие агенты, снижающие уровень липидов, которые могут быть объединены с соединениями по настоящему изобретению, включают, например, агенты, описанные в WO 2011005611, с. 30, строка 20, до с. 31, строка 30. Агенты, снижающие уровень липидов, включают секвестранты желчных кислот, ингибиторы ГМГ-КоА-редуктазы (гидроксиметилглутарил-КоА-редуктазы), ингибиторы ГМГКоА-синтазы,ингибиторы абсорбции холестерина,ингибиторы ацил-кофермент А-9 022375 холестеринацилтрансферазы (АСАТ), ингибиторы СЕТР (белок-переносчик эфиров холестерина), ингибиторы скваленсинтетазы, агонисты PPAR(рецепторы, активируемые пероксисомными пролифераторами ), модуляторы рецептора FXR (фарнезоидный Х-рецептор), модуляторы рецептора LXR (печеночный Х-рецептор), ингибиторы синтеза липопротеинов, ингибиторы ренин-ангиотензиновой системы,частичные агонисты PPAR 5, ингибиторы реабсорбции желчных кислот, агонисты PPAR , ингибиторы синтеза триглицеридов, ингибиторы микросомального переноса триглицеридов, модуляторы транскрипции, ингибиторы скваленэпоксидазы, индукторы рецепторов липопротеинов низкой плотности, ингибиторы агрегации тромбоцитов, ингибиторы 5-LO (5-липоксигеназа) или FLAP (белок, активирующий 5 липоксигеназу), связанный ниацином хром и другие агенты, влияющие на липидный состав. Подходящие антигипертензивные агенты, которые могут быть объединены с соединениями по настоящему изобретению, включают, например, агенты, описанные в WO 2011005611, с. 31, строка 31, до с. 32, строка 18. Антигипертензивные агенты включают диуретики, бета-адренергические блокаторы, блокаторы кальциевых каналов, ингибиторы ангиотензин-превращающего фермента (АСЕ), ингибиторы нейтральных эндопептидаз, антагонисты эндотелина, вазодилататоры, антагонисты рецепторов ангиотензина II,/-адреноблокаторы, альфа-1-блокаторы, альфа-2-агонисты, ингибиторы альдостерона, ингибиторы минералокортикоидных рецепторов, ингибиторы ренина и ангиопоэтин-2-связывающие агенты. Подходящие антидиабетические агенты включают ингибитор ацетил-КоА-карбоксилазы (АСС), такой как описанные в WO 2009144554, WO 2003072197, WO 2009144555 и WO 2008065508, ингибитор диацилглицерин-О-ацилтрансферазы 1 (DGAT-1), такой как описанные в WO 09016462 или WO 2010086820, AZD7687 или LCQ908, ингибитор диацилглицерин-О-ацилтрансферазы 2 (DGAT-2), ингибиторы моноацилглицерин-О-ацилтрансферазы, ингибитор фосфодиэстеразы (PDE)-10, активатор АМРК(аденозинмонофосфат-активируемая протеинкиназа), сульфонилмочевину (например, ацетогексамид,хлорпропамид, диабинез, глибенкламид, глипизид, глибурид, глимепирид, гликлазид, глипентид, гликвидон, глизоламид, толазамид и толбутамид), меглитинид, ингибитор -амилазы (например, тендамистат, трестатин и AL-3688), ингибитор -глюкозидгидролазы (например, акарбоза), ингибитор глюкозидазы (например, адипозин, камиглибоза, эмиглитат, миглитол, воглибоза, прадимицин-Q и салбостатин), агонист PPAR (например, балаглитазон, циглитазон, дарглитазон, энглитазон, изаглитазон,пиоглитазон, розиглитазон и троглитазон), агонист PPAR / (например, CLX-0940, GW-1536, GW-1929,GW-2433, KRP-297, L-796449, LR-90, MK-0767 и SB-219994), бигуанид (например, метформин), модулятор глюкагоноподобного пептида 1 (GLP-1), такой как агонист (например, эксендин-3 и эксендин-4), лираглутид, албиглутид, эксенатид (Byetta), албиглутид, таспоглутид, ликсисенатид, дулаглутид, семаглутид, NN-9924, ТТР-054, ингибитор протеинтирозинфосфатазы-1 В (РТР-1 В) (например, тродусквемин,гиртиозаловый (hyrtiosal) экстракт и соединения, раскрытые в Zhang S., et al., Drug Discovery Today,12(9/10), 373-381 (2007, ингибитор SIRT-1 (сиртуина-1) (например, ресвератрол, GSK2245840 илиGSK184072), ингибитор дипептидилпептидазы IV (DPP-IV) (например, такой как в WO 2005116014, ситаглиптин, вилдаглиптин, алоглиптин, дутоглиптин, линаглиптин и саксаглиптин), усилитель секреции инсулина, ингибитор окисления жирных кислот, антагонист А 2, ингибитор c-jun-аминоконцевой киназы(JNK), активаторы глюкокиназы (GKa), такие как описанные в WO 2010103437, WO 2010103438, WO 2010013161, WO 2007122482, ТТР-399, ТТР-355, ТТР-547, AZD1656, ARRY403, МК-0599, TAK-329,AZD5658 или GKM-001, инсулин, миметик инсулина, ингибитор гликогенфосфорилазы (например,GSK1362885), агонист рецептора VPAC2, ингибиторы SGLT2 (натрий-зависимый переносчик глюкозы 2), такие как описанные в Е.С. Chao et al. Nature Reviews Drug Discovery 9, 551-559 (July 2010), включая дапаглифлозин, канаглифлозин, BI-10733, тофоглифлозин (CSG452), ASP-1941, THR1474, TS-071,ISIS388626 и LX4211, а также описанные в WO 2010023594, модулятор рецептора глюкагона, такой как описанные в Demong D.E. et al. Annual Reports in Medicinal Chemistry, 2008, 43, 119-137, модуляторыGPR119 (связанный с G-белком рецептор 119), в частности, агонисты, такие как описанные в WO 2010140092, WO 2010128425, WO 2010128414, WO 2010106457, Jones R.M. et al., Medicinal Chemistry,2009, 44, 149-170 (например, МВХ-2982, GSK1292263, APD597 и PSN821), производные или аналогиFGF21 (фактор роста фибробластов 21), такие как описанные в Kharitonenkov A. et al., Current Opinion inInvestigational Drugs, 2009, 10(4) 359-364, модуляторы рецептора TGR5 (также называемого GPBAR1), в частности, агонисты, такие как описанные в Zhong M., Current Topics in Medicinal Chemistry, 2010, 10(4),386-396, и INT777, агонисты GPR40, такие как описанные в Medina J.C., Annual Reports in MedicinalChemistry, 2008, 43, 75-85, включая, но не ограничиваясь этим, TAK-875, модуляторы GPR120, в частности агонисты, активаторы высокоаффинного рецептора никотиновой кислоты (НМ 74 А) и ингибиторыSGLT1, такие как GSK1614235. Дополнительный типичный перечень антидиабетических агентов, которые могут быть объединены с соединениями по настоящему изобретению, можно найти, например, в WO 2011005611, с. 28, строка 35, до с. 30, строка 19. Предпочтительными антидиабетическими агенты являются метформин и ингибиторы DPP-IV (например, ситаглиптин, вилдаглиптин, алоглиптин, дутоглиптин, линаглиптин и саксаглиптин). Другие антидиабетические агенты могут включать ингибиторы или модуляторы ферментов карнитинпальмитоилтрансфераз, ингибиторы фруктозо-1,6-дифосфатазы, инги- 10022375 биторы альдозоредуктазы, ингибиторы минералокортикоидных рецепторов, ингибиторы TORC2, ингибиторы CCR2 (С-С рецептор хемокина 2) и/или CCR5, ингибиторы изоформ PKC (протеинкиназа С) (например, PKC, PKC, PKC), ингибиторы синтетазы жирных кислот, ингибиторы серинпальмитоилтрансферазы, модуляторы GPR81, GPR39, GPR43, GPR41, GPR105, Kv1.3, ретинолсвязывающего белка 4, глюкокортикоидного рецептора, соматостатиновых рецепторов (например, SSTR1, SSTR2, SSTR3 иSSTR5), ингибиторы или модуляторы PDHK2 или PDHK4, ингибиторы МАР 4K4, модуляторы семействаIL1 (интерлейкинов 1), включая IL-1 бета, модуляторы RXR (ретиноидный Х-рецептор) альфа. Помимо это, подходящие антидиабетические агенты включают механизмы, перечисленные в Carpino P.A., Goodwin В. Expert Opin. Ther. Pat., 2010, 20(12), 1627-51. Подходящие агенты против ожирения (некоторые из которых также могут действовать в качестве антидиабетических агентов) включают ингибиторы 11-гидроксистероиддегидрогеназы-1 (11-HSD тип 1), ингибитор стеароил-КоА-десатуразы-1 (SCD-1), агонисты MCR-4, агонисты холецистокинина-А(ССК-А), ингибиторы обратного захвата моноаминов (такие как сибутрамин), симпатомиметические агенты, 3-адренергические агонисты, агонисты дофамина (такие как бромкриптин), аналоги меланоцитстимулирующего гормона, агонисты 5 НТ 2, антагонисты меланин-концентрирующего гормона, лептин(белок ОВ), аналоги лептина, агонисты лептина, антагонисты галанина, ингибиторы липаз (такие как тетрагидролипстатин, т.е. орлистат), аноректические агенты (такие как агонист бомбезина), антагонисты нейропептида-Y (например, антагонисты NPY Y5, такие как велнеперит), пептид PYY3-36 (включая его аналоги), модулятор BRS3, смешанные антагонисты подтипов опиоидных рецепторов, тиромиметические агенты, дегидроэпиандростерон или его аналог, агонисты или антагонисты глюкокортикоидов, антагонисты орексина, агонисты глюкагоноподобного пептида-1, цилиарные нейротрофические факторы(такие как Axokine, доступный от Regeneron Pharmaceuticals, Inc., Тарритаун, Нью-Йорк (NY) и ProcterGamble Company, Цинциннати, Огайо (ОН, ингибиторы агути-родственного белка (AGRP) человека,антагонисты или обратные агонисты гистамина 3, агонисты нейромедина U, ингибиторы МТР/АроВ (например, действующие избирательно в кишечнике ингибиторы МТР, такие как дирлотапид, JTT130, Usistapide, SLx4090), опиоидный антагонист, модуляторы мю-опиоидного рецептора, включая, но не ограничиваясь этим, GSK1521498, ингибиторы MetAp2, включая, но не ограничиваясь этим, ZGN-433, агенты со смешанной модуляторной активностью в отношении 2-х или более рецепторов глюкагона, GIP и GLP1 рецепторов, такие как MAR-701 или ZP2929, ингибиторы переносчика норэпинефрина, антагонист/обратные агонисты каннабиноидного рецептора 1, агонисты/антагонисты грелина, оксинтомодулин и аналоги, ингибиторы захвата моноаминов, такие как, но не ограничиваясь этим, тезофензин, антагонист орексина, комбинированные агенты (такие как бупропион плюс зонисамид, прамлинтид плюс метрелептин, бупропион плюс налтрексон, фентермин плюс топирамат) и т.п. Предпочтительные агенты против ожирения для применения в аспектах настоящего изобретения,относящихся к комбинациям, включают действующие избирательно в кишечнике ингибиторы МТР (например, дирлотапид, митратапид и имплитапид, R56918 (CAS403987) и CAS913541-47-6), агонисты CCKa (например, N-бензил-2-[4-(1H-индол-3-илметил)-5-оксо-1-фенил-4,5-дигидро-2,3,6,10bтетрааза-бензо[е]азулен-6-ил]-N-изопропилацетамид, описанный в публикации РСТWO 2005/116034 или публикации США 2005-0267100 А 1), агонисты 5 НТ 2 с (например, лоркасерин), агонист MCR4PYY3-36, например, описанные в публикации US 2006/0178501), опиоидные антагонисты (например, налтрексон), олеоилэстрон (CAS180003-17-2), обинепитид (ТМ 30338), прамлинтид (Symlin), тезофензин (NS2330), лептин, бромкриптин, орлистат, AOD-9604 (CAS221231-10-3) и сибутрамин. Предпочтительно, соединения по настоящему изобретению и комбинированные терапии вводят в сочетании с упражнениями и разумной диетой. Все цитированные патенты и публикации США (включая все технические бюллетени, на которые сделаны ссылки в примерах) включены в данную заявку посредством ссылки во всей своей полноте. Примеры, приведенные в данном изобретении ниже, предназначены лишь для иллюстративных целей. Композиции, способы и различные параметры, отраженные в данном изобретении, предназначены только для пояснения различных аспектов и воплощений изобретения и не предназначены для ограничения объема заявляемого изобретения каким-либо образом. Примеры Соединения и промежуточные соединения, описанные ниже, были названы с использованием способа присвоения названий, приведенного в Chemdraw Ultra, версия 11.0.1 (CambridgeSoft Corp., Кеймбридж, Массачусетс). Способ присвоения названий, приведенный в Chemdraw Ultra, версия 11.0.1, хорошо известен специалистам в данной области техники, и считается, что способ присвоения названий, приведенный в Chemdraw Ultra, версия 11.0.1, в целом согласуется с рекомендациями JUPAC (Международный союз теоретической и прикладной химии) по номенклатуре органической химии и правилами CASIndex. Если не указано иное, все реактивы получали из коммерческих источников. Все ссылки, приведенные в данном описании ниже, включены посредством ссылки.- 11022375 Флэш-хроматографию проводили согласно способу, описанному в Still et al., J. Org. Chem., 1978, 43,2923. Все очистки Biotage, обсуждаемые в данном изобретении, осуществляли с использованием колонок Biotage SNAP, содержащих диоксид кремния KP-SIL (40-63 мкм, 60 ангстрем) (Biotage AB; Уппсала, Швеция). Все очистки Combiflash, обсуждаемые в данном изобретении, осуществляли с использованием системы CombiFlash Companion (Teledyne Isco; Линкольн, Небраска), применяя упакованные колонки с диоксидом кремния RediSep. Масс-спектры регистрировали на спектрометре Waters (Waters Corp.; Милфорд, Массачусетс (MAMicromass Platform II. Если не указано иное, масс-спектры регистрировали на спектрометре Waters(Милфорд, MA) Micromass Platform II. Химические сдвиги протонного ЯМР приведены в миллионных долях в сторону слабого поля от тетраметилсилана, и их регистрировали на спектрометре Varian Unity 300, 400 или 500 МГц (мегагерц)(Varian Inc.; Пало-Альто, Калифорния (СА. Химические сдвиги ЯМР приведены в миллионных долях в сторону слабого поля от тетраметилсилана (для протона) или фтортрихлорметана (для фтора). Времена удерживания HPLC (высокоэффективная жидкостная хроматография) измеряли, используя следующие методы: метод А: колонка: Waters Atlantis dC18 (4,650 мм; 5 мкм); подвижная фаза А: 0,05%TFA (трифторуксусная кислота) в воде (об./об.); подвижная фаза В: 0,05% TFA в ацетонитриле (об./об.); градиент: линейный от 95% А/5% В до 5% А/95% В в течение 4,0 мин, выдерживание при 5% А/95% В до 5,0 мин; скорость потока: 2,0 мл/мин. Способы получения, описанные ниже, использовали в синтезе соединений, приведенных в следующих примерах. Следующие исходные вещества доступны из соответствующих источников: Получение промежуточных соединений и исходных веществ. Промежуточное соединение 1: 1-изопропил-4,6-дигидроспиро[индазол-5,4'-пиперидин]-7(1H)-он,показанный ниже, получали, как приведено далее Метилвинилкетон (146 мл; 1,78 моль) добавляли к раствору трет-бутил-4-формилпиперидин-1 карбоксилата (375 г; 1,76 моль) в тетрагидрофуране (18 л). Реакционную смесь охлаждали до -5 С и по каплям в течение 10 мин добавляли раствор гидроксида калия в этаноле (3 н., 0,243 л). Реакционную смесь оставляли нагреваться до комнатной температуры и перемешивали в течение 16 ч. Добавляли циклогексан (10 л) и раствор промывали насыщенным хлоридом натрия (310 л). Органический слой концентрировали до масла. Это масло растворяли в 2 л смеси 80:20 циклогексан/этилацетат и фильтровали через Celite для удаления нерастворенного вещества. Фильтрат очищали колоночной флэшхроматографией (30% этилацетат/гексаны), получая продукт в виде масла. Масло растирали в гексанах,получая указанное в заголовке соединение в виде бесцветного твердого вещества (131 г; 28%). Стадия 2: 1-изопропил-4,6-дигидроспиро[индазол-5,4'-пиперидин]-7(1H)-он. Раствор трет-бутил-9-оксо-3-азаспиро[5.5]ундец-7-ен-3-карбоксилата(250 г) и трис(диметиламинометана) (325 мл) в толуоле (1,9 л) нагревали при температуре дефлегмации в течение 4 ч. Смесь подвергали дистилляции и концентрировали до минимального перемешиваемого объема (110 С) и затем добавляли толуол (1,9 л). Реакционную смесь снова подвергали дистилляции до минимального перемешиваемого объема и охлаждали до комнатной температуры. Добавляли толуол (1,8 л) и гидрохлорид изопропилгидразина (135 г) и нагревали раствор до температуры дефлегмации в течение 5 ч. Реакционную смесь охлаждали до комнатной температуры и промывали лимонной кислотой (10%-ной водной,2150 мл) и водой (200 мл). Органический слой затем подвергали дистилляции до минимального перемешиваемого объема. Добавляли метанол (2 л) и подвергали дистилляции до минимального перемешиваемого объема. Это повторяли, используя метанол (2 л). Раствор снова переносили в метанол (2,5 л) и добавляли N-бромсукцинимид (176 г) в виде одной порции. Раствор перемешивали при 23 С в течение 2 ч. Добавляли водный раствор тиосульфата натрия (5 мас.%; 0,5 л) и смесь перемешивали в течение 15 мин. Реакционную смесь концентрировали посредством дистилляции (45 С; 210 мм рт. ст. (примерно 28 кПа до примерно 0,5 л и затем добавляли 2-метилтетрагидрофуран (2,5 л). После перемешивания в течение 15 мин водный слой отбрасывали. Органический слой концентрировали до примерно 0,2 л и добавляли тетрагидрофуран (0,5 л). К смеси добавляли раствор трет-бутилата калия в тетрагидрофуране(1,9 л; 1 М раствор). Раствор нагревали до 60 С и перемешивали в течение 1 ч. После охлаждения до комнатной температуры добавляли водную соляную кислоту (1 н.; 2,2 л) в течение 20 мин. Смесь перемешивали при комнатной температуре в течение 20 мин и далее давали возможность слоям разделиться. Водный слой удаляли и снова экстрагировали этилацетатом (1,75 л). Объединенные органические слои промывали водой (1 л) и концентрировали посредством дистилляции (удаляли 4 л растворителя). Добавляли этилацетат (1,8 л) и раствор концентрировали до минимального перемешиваемого объема. Добавляли этилацетат (3 л) и метанол (0,8 л) и раствор охлаждали до 0 С. По каплям в течение 20 мин добавляли ацетилхлорид (401 мл) и раствор перемешивали при 0 С в течение 4 ч. Осадок собирали фильтрацией в атмосфере азота. Фильтрат промывали этилацетатом (0,5 л) и сушили в вакуумном сушильном шкафу при 40 С, получая указанное в заголовке соединение в виде не совсем белого твердого вещества К раствору 6-бром-1H-индол-3-карбонитрила (328 мг; 1,48 ммоль) в этаноле (5 мл) в колбе Парра емкостью 500 мл добавляли ацетат натрия (370 мг; 4,47 ммоль) и [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладий(II), комплекс с дихлорметаном (242 мг; 0,297 ммоль). Реакционный сосуд продували азотом и вакуумировали трижды и затем заполняли монооксидом углерода до давления 30 ф/кв. дюйм (примерно 206,8 кПа. Реакционную смесь нагревали до 70 С, повышая давление внутри сосуда до 45 ф/кв. дюйм (примерно 310,3 кПа). Реакционную смесь перемешивали при 70 С в течение 24 ч. Реакционную смесь охлаждали до комнатной температуры. Смесь фильтровали через Celite, промывая этанолом. Фильтрат концентрировали при пониженном давлении и разбавляли дихлорметаном. Оставшиеся твердые вещества отфильтровывали и фильтрат концентрировали. Остаток очищали колоночной флэш-хроматографией (20-80% этилацетат/гептаны), получая этиловый эфир 3 циано-1 Н-индол-6-карбоновой кислоты (142 мг; 45%). -APCI (М-Н) 213,4. 1H ЯМР (400 МГц, DMSO-d6, ): 12.50 (br. s., 1H), 8.45 (s, 1 Н), 8.16 (s, 1H), 7.82 (dd, J=8,4, 1,4 Гц,1 Н), 7.73 (d, J=8,4 Гц, 1 Н), 4.32 (q, J=7,0 Гц, 2 Н), 1.33 (t, J=7,0 Гц, 3H). Стадия 2: 3-карбамоил-1H-индол-6-карбоновая кислота. Суспензию этилового эфира 3-циано-1H-индол-6-карбоновой кислоты (100 мг; 1,4 ммоль) в метаноле (1,12 мл) добавляли к раствору пероксида мочевины (453 мг; 4,67 ммоль) в 2,5 М гидроксиде натрия(1,12 мл; 2,80 ммоль) при 0 С. Суспензию оставляли нагреваться до комнатной температуры и перемешивали в течение ночи. Реакционную смесь концентрировали при пониженном давлении. Добавляли воду и раствор подкисляли 3 н. водной соляной кислотой до рН, равного 2. Образовывался осадок. Реакционную смесь перемешивали в течение 1 мин при комнатной температуре и затем фильтровали, получая оранжевое твердое вещество. Добавляли дополнительное количество пероксида мочевины (453 мг) в 2,5 М гидроксиде натрия и метаноле и смесь перемешивали в течение 2 ч. Реакционную смесь концентрировали, разбавляли водой, подкисляли до рН, равного 3, и фильтровали. Твердое вещество промывали водой и гептанами и сушили в вакуумном сушильном шкафу, получая 3-карбамоил-1H-индол-6 карбоновую кислоту в виде твердого вещества (66,5 мг; 70%). -ESI (М-1) 203,1. 1 Метил-3-йод-1H-индазол-5-карбоксилат (30,7 г; 102 ммоль), цианид цинка (20,3 г; 173 ммоль), цинковую пыль (4,05 г; 61,9 ммоль), [1,1'-бис-(дифенилфосфино)ферроцен]дихлорпалладий(II), комплекс с дихлорметаном (12 г; 15 ммоль), и йодид меди(I) (19,7 г; 103 ммоль) объединяли в круглодонной колбе емкостью 1 л. Добавляли N,N-диметилацетамид (500 мл) и реакционную смесь продували азотом в течение 10 мин. Реакционную смесь нагревали до 120 С в течение 1 ч. Реакционную смесь охлаждали до комнатной температуры, разбавляли этилацетатом (1 л) и оставляли перемешиваться в течение 20 мин. Реакционную смесь фильтровали через пробку Celite, промывая 500 мл этилацетата. Фильтрат добавляли к раствору насыщенного хлорида аммония и концентрированного гидроксида аммония (2 л) (полученного путем добавления гидроксида аммония к насыщенному водному раствору хлорида аммония до получения рН, равного 8) и двухфазный раствор энергично перемешивали в течение 1 ч. Полученную эмульсию фильтровали через небольшую набивку Celite. Слои разделяли и водный слой дополнительно экстрагировали два раза этилацетатом (1,1 л), каждый раз фильтруя полученную эмульсию через Celite. Объединенные органические слои промывали водой (2900 мл) и рассолом (900 мл), сушили над сульфатом натрия, фильтровали и концентрировали. К неочищенному веществу добавляли метанол (100 мл) и смесь перемешивали в течение 20 мин. Полученный осадок отфильтровывали и промывали метанолом (10 мл). Фильтрат концентрировали, получая указанное в заголовке соединение (13,2 г; 65%) в виде твердого вещества. -ESI (M-H) 200,0. 1H ЯМР (400 МГц, DMSO-d6, ): 8.43-8.45 (m, 1H), 8.05 (dd, J=8,8, 1,6 Гц, 1H), 7.85 (dd, J=8,9, 0,9 Гц,1H), 3.88 (s, 3H). Стадия 2: 3-карбамоил-1H-индазол-5-карбоновая кислота Суспензию метил-3-циано-1H-индазол-5-карбоксилата (50,0 г; 249 ммоль) в метаноле (1 л) охлаждали до 10 С. По каплям добавляли раствор пероксида мочевины (241 г; 2,49 моль) в гидроксиде натрия (1 л; 2,5 н.) и воду (100 мл), поддерживая внутреннюю температуру ниже 25 С. По завершении добавления ледяную баню удаляли и реакционную смесь оставляли перемешиваться при комнатной температуре в течение 16 ч. Используя HPLC, наблюдали небольшое количество непрореагировавшего исходного вещества. Реакционную смесь охлаждали до 15 С и порциями добавляли дополнительное количество пероксида мочевины (50 г). Было отмечено интенсивное образование пузырьков газа. Реакционную смесь оставляли перемешиваться в течение еще 2 ч. Неочищенную реакционную смесь фильтровали для удаления присутствующих в ней твердых веществ и фильтрат концентрировали для удаления метанола. Оставшийся раствор охлаждали в ледяной бане и по каплям добавляли 6 н. соляную кислоту (420 мл) для доведения рН до 4. Раствор перемешивали в течение 20 мин и полученное рыжевато-коричневое твердое вещество собирали фильтрацией и сушили, получая 57,2 г неочищенного продукта. К неочищенному продукту добавляли ацетонитрил (700 мл) и дихлорметан (700 мл) и смесь перемешивали при комнатной температуре в течение 1 ч. Твердое вещество собирали фильтрацией, промывали смесью 1:1 ацетонитрил:дихлорметан (400 мл) и сушили, получая указанное в заголовке соединение (39,5 г; 77%) в виде рыжевато-коричневого твердого вещества. +ESI (М+Н) 206,1. 1 К раствору 1 Н-пирроло[3,2-b]пиридин-6-карбоновой кислоты (1,37 г; 8,45 ммоль) в N,Nдиметилформамиде (55 мл) добавляли карбонат цезия (2,79 г; 8,56 ммоль) и бензилбромид (1,05 мл; 8,64 ммоль). Реакционную смесь оставляли перемешиваться при комнатной температуре в течение 17 ч. Добавляли дополнительные количества карбоната цезия (500 мг; 1,54 ммоль) и бензилбромида (0,186 мл; 1,53 ммоль) и реакционную смесь перемешивали в течение еще 4 ч. Затем реакционную смесь гасили водой и разбавляли этилацетатом. Слои разделяли и водный слой трижды экстрагировали этилацетатом. Объединенные органические слои промывали водой и рассолом, сушили над сульфатом натрия, фильтровали и концентрировали. После очистки колоночной флэш-хроматографией (0-100% этилацетат/гептаны) получали указанное в заголовке соединение (1,42 г; 67%). +ESI (М+Н) 253,3. 1 К раствору бензил-1H-пирроло[3,2-b]пиридин-6-карбоксилата (830 мг; 3,29 ммоль) в N,Nдиметилформамиде (19 мл), имеющему температуру 0 С, добавляли N-бромсукцинимид (609 мг; 3,42 ммоль). Реакционную смесь оставляли постепенно нагреваться до комнатной температуры и перемешивали в течение уикэнда. Реакционную смесь разбавляли этилацетатом и промывали последовательно насыщенным водным тиосульфатом натрия, насыщенным водным бикарбонатом натрия, водой и рассолом. Органические слои сушили над сульфатом натрия, фильтровали и концентрировали, получая указанное в заголовке соединение (1,08 г; количественный выход). +ESI (М+1+Н) 333,0. 1(d, J=2,9 Гц, 1H), 7.52 (m, 2H), 7.41 (m, 3H), 5.41 (s, 2H). Стадия 3: 3-карбамоил-1H-пирроло[3,2-b]пиридин-6-карбоновая кислота. Указанное в заголовке соединение получали способом, аналогичным описанному на стадиях 3-4 для промежуточного соединения 6, используя бензил-3-бром-1H-пирроло[3,2-b]пиридин-6-карбоксилат. К раствору 5-(метоксикарбонил)-1H-индол-2-карбоновой кислоты (2,50 г; 11,4 ммоль) в тетрагидрофуране (20 мл) добавляли 1,1-карбонилдиимидазол (3,70 г; 22,8 ммоль). Желтую суспензию перемешивали в течение 2 ч. Затем добавляли концентрированный гидроксид аммония (20 мл) и смесь перемешивали при комнатной температуре в течение 5 ч. Бледно-зеленую суспензию фильтровали, промывали водой и 5 мл метанола и сушили на воздухе, получая метил-2-карбамоил-1H-индол-5-карбоксилат (2,04 г; 82%) в виде бесцветного твердого вещества.(4,5 мл) и этиленгликоле (4,5 мл) добавляли гидроксид калия (3,16 г; 56,4 ммоль). Смесь нагревали до температуры дефлегмации и перемешивали в течение 2 ч. Реакционную смесь охлаждали до комнатной температуры и разбавляли водой. Тетрагидрофуран удаляли при пониженном давлении. Твердые вещества отфильтровывали и фильтрат подкисляли до рН 4 концентрированной соляной кислотой. Полученный осадок собирали фильтрацией и сушили под вакуумом, получая 2-карбамоил-1H-индол-5 карбоновую кислоту (230 мг, 82%). 1 К имеющей температуру -78 С смеси 2-хлор-6-метоксихинолин-3-карбальдегида (4,64 г; 20,9 ммоль) в дихлорметане (130 мл) добавляли трибромид бора (4,0 мл; 42 ммоль). Смесь оставляли нагреваться до комнатной температуры и перемешиваться в течение 4 ч. Реакционную смесь нейтрализовали путем осторожного добавления насыщенного водного бикарбоната натрия. Затем смесь экстрагировали 2-метилтетрагидрофураном (3). Объединенные органические экстракты фильтровали и фильтрат промывали насыщенным водным бикарбонатом натрия (3) и один раз рассолом. Органические экстракты сушили над сульфатом натрия, фильтровали и концентрировали до желтого твердого вещества. Твердое вещество частично растворяли в 2-метилтетрагидрофуране и фильтровали. Фильтрат концентрировали до получения твердого вещества и вновь частично растворяли в 2-метилтетрагидрофуране, фильтровали и концентрировали. После очистки колоночной флэш-хроматографией (10-100% этилацетат/гептаны) получали указанное в заголовке соединение (2,65 г; 61%) в виде бледно-желтого твердого вещества. +ESI(3,13 г; 22,6 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 1,5 ч. Реакционную смесь разбавляли водой (100 мл) и оставляли перемешиваться в течение 1 ч. Полученное твердое вещество собирали фильтрацией, промывали водой и сушили под вакуумом, получая указанное в заголовке соединение (3,62 г; 99%) в виде не совсем белого твердого вещества. +ESI (М+Н) 322,1. 1 К суспензии трет-бутил-2-(2-хлор-3-формилхинолин-6-илокси)ацетата (3,6 г; 11 ммоль) в третбутаноле (150 мл) добавляли 2-метил-2-бутен (14,9 мл; 133 ммоль) и раствор хлорита натрия (8,27 г; 73,1- 17022375 ммоль) и дигидрофосфат натрия (8,10 г; 58,7 ммоль) в воде (50 мл). Реакционную смесь оставляли перемешиваться при комнатной температуре в течение 1 ч. Проводили удаление трет-бутанола в вакууме. Смесь разбавляли водой (60 мл) и подкисляли до рН, равного 4, 1 н. водной соляной кислотой. Полученный осадок отфильтровывали, промывали водой и сушили под вакуумом, получая указанное в заголовке соединение (3,7 г; 100%) в виде белого твердого вещества. +ESI (М+Н) 338,1. 1(d, J=2,93 Гц, 1H), 4.76 (s, 2H), 1.49 (s, 9H). Стадия 4: 6-(2-трет-бутокси-2-оксоэтокси)хинолин-3-карбоновая кислота Метанол (100 мл) и триэтиламин (4,5 мл; 33 ммоль) добавляли к 6-(2-трет-бутокси-2-оксоэтокси)-2 хлорхинолин-3-карбоновой кислоте (1,27 г; 3,76 ммоль). Добавляли 10% палладий на угле (350 мг) и в реакционном сосуде создавали давление водорода до 17 ф/кв. дюйм (примерно 117,2 кПа). Реакционную смесь перемешивали при комнатной температуре в течение 3 ч. Реакционную смесь разбавляли метанолом и фильтровали через Celite. Фильтрат концентрировали до получения желтого твердого вещества. Твердое вещество растворяли в воде и смесь подкисляли до рН, равного 4, 1 н. водной соляной кислотой. Твердое вещество собирали фильтрацией, промывали водой и сушили под вакуумом, получая указанное в заголовке соединение (960 мг, 84%) в виде бледно-желтого твердого вещества. +ESI (М+Н) 304,2. 1 К раствору 5-бром-2-нитробензальдегида (2 г, 9 ммоль) в этаноле (46 мл) добавляли дигидрат хлорида олова(II) (7,95 г; 35,2 ммоль) и этиловый эфир 3,3-диэтоксипропионовой кислоты (4,2 мл; 22 ммоль). Реакционную смесь нагревали до 90 С в течение 16 ч. Затем реакционную смесь оставляли охлаждаться до комнатной температуры и перемешиваться в течение ночи. Реакционную смесь концентрировали и остаток растворяли в этилацетате. Смесь выливали в насыщенный водный бикарбонат натрия. Полученную эмульсию фильтровали через Celite, промывая этилацетатом. Проводили разделение слоев и водный слой экстрагировали этилацетатом. Объединенные органические слои промывали рассолом, сушили над сульфатом магния, фильтровали и концентрировали. После очистки колоночной флэшхроматографией (0-50% этилацетат/гептаны) получали указанное в заголовке соединение (1,41 г; 60%) в виде твердого вещества. Стадия 2: 6-карбамоилхинолин-3-карбоновая кислота Указанное в заголовке соединение получали способом, аналогичным описанному на стадиях 3-4 для промежуточного соединения 6, используя этил-6-бромхинолин-3-карбоксилат. +ESI (М+Н) 217,0. 1- 18022375 добавляли N-бромсукцинимид (8,52 г; 47,9 ммоль). Реакционную смесь нагревали до 100 С в течение 1,5 ч. Реакционную смесь охлаждали до комнатной температуры и разбавляли насыщенным водным тиосульфатом натрия. Смесь экстрагировали этилацетатом (3). Объединенные органические экстракты промывали водой, насыщенным тиосульфатом натрия и рассолом, сушили над сульфатом натрия, фильтровали и концентрировали. Остаток очищали колоночной флэш-хроматографией (0-30% этилацетат/гептаны), получая указанное в заголовке соединение (2,6 г; чистота примерно 43%) в виде бледножелтого твердого вещества. 1 Неочищенный 1-бром-2-метокси-4-метил-5-нитробензол (2,6 г; 11 ммоль) растворяли в N,Nдиметилформамиде (15 мл) и добавляли диметилацеталь N,N-диметилформамида (5 мл; 38 ммоль). Реакционную смесь нагревали до 120 С в течение 18 ч. Реакционную смесь охлаждали до комнатной температуры и сразу же добавляли к имеющей температуру 0 С суспензии перйодата натрия (12,2 г; 57,2 ммоль) в воде (20 мл) и N,N-диметилформамиде (5 мл). Реакционную смесь перемешивали при 0 С в течение 2 ч, затем оставляли нагреваться до комнатной температуры и перемешиваться в течение еще 6 ч. Реакционную смесь фильтровали, промывая этилацетатом, толуолом и водой. Фильтрат экстрагировали этилацетатом (3). Объединенные органические экстракты промывали водой и рассолом, сушили над сульфатом натрия, фильтровали и концентрировали. После очистки колоночной флэш-хроматографией(0-50% этилацетат/гептаны) получали указанное в заголовке соединение (550 мг, 19%) в виде бледножелтого твердого вещества. 1 Н ЯМР (400 МГц, CDCl3, ): 10.47 (s, 1H), 8.41 (s, 1H), 7.35 (s, 1H), 4.05 (s, 3H). Стадия 3: этил-7-бром-6-метоксихинолин-3-карбоксилат К суспензии 4-бром-5-метокси-2-нитробензальдегида (475 мг; 1,83 ммоль) в этаноле (15 мл) добавляли дигидрат хлорида олова(II) (1,65 г; 7,31 ммоль) и этил-3,3-диэтоксипропионат (1,0 мл; 5,1 ммоль). Реакционную смесь нагревали до 90 С в течение 3 ч. Данные LCMS (жидкостная хроматография/массспектрометрия) показали, что реакция прошла не до конца. Добавляли дополнительные количества дигидрата хлорида олова(II) (600 мг; 2,7 ммоль) и этил-3,3-диэтоксипропионата (0,35 мл; 1,8 ммоль) и реакционную смесь оставляли нагреваться в течение ночи. Данные LCMS показали, что реакция прошла не до конца. Добавляли дигидрат хлорида олова(II) (200 мг; 0,89 ммоль) и этил-3,3-диэтоксипропионат (0,10 мл; 0,51 ммоль) и реакционную смесь нагревали при 90 С в течение еще 3 ч. Реакционную смесь концентрировали и остаток распределяли между этилацетатом и насыщенным водным бикарбонатом натрия. Водный слой фильтровали и экстрагировали еще раз этилацетатом. Объединенные органические экстракты промывали рассолом, сушили над сульфатом натрия, фильтровали и концентрировали. Остаток очищали колоночной флэш-хроматографией (0-100% этилацетат/гептаны), получая желтое твердое вещество (410 мг), которое содержало желаемый продукт и примеси. Это вещество переносили в тетрагидрофуран (5 мл) и добавляли этил-3,3-диэтоксипропионат (0,17 мл; 0,87 ммоль) и моногидрат nтолуолсульфоновой кислоты (12,0 мг; 0,63 ммоль). Смесь нагревали до 75 С и перемешивали в течение 3 ч. Реакционную смесь концентрировали и остаток распределяли между этилацетатом и насыщенным водным бикарбонатом натрия. Слои разделяли и водный слой экстрагировали этилацетатом (2). Объединенные органические экстракты промывали рассолом, сушили над сульфатом натрия, фильтровали и концентрировали. После очистки колоночной флэш-хроматографией (0-25% этилацетат/гептаны) получали указанное в заголовке соединение (256 мг, 45%) в виде желтого твердого вещества. +ESI (М+Н+1) 313,3-Стадия 4: 7-бром-6-метоксихинолин-3-карбоновая кислота К раствору этил-7-бром-6-метоксихинолин-3-карбоксилата (250 мг; 0,80 ммоль) в тетрагидрофуране(5 мл) добавляли 1 н. водный гидроксид лития (1,6 мл; 1,6 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение ночи. Реакционную смесь концентрировали и остаток переносили в воду и 1 н. водный гидроксид лития (0,4 мл). Раствор дважды экстрагировали этилацетатом. Затем водный слой подкисляли до рН, равного 4, 1 н. водной соляной кислотой. Полученный осадок отфильтровывали, промывали водой и сушили под вакуумом, получая указанное в заголовке соединение (165 мг, 73%) в виде не совсем белого твердого вещества. +ESI (М+Н+1) 284,1. К раствору этил-хинолин-7-карбоксилата (1,02 г; 5,05 ммоль) в дихлорметане (20 мл) добавляли перуксусную кислоту (2,13 мл; 10,1 ммоль; 32 мас.%, в уксусной кислоте). Реакционную смесь перемешивали при комнатной температуре в течение ночи. Реакционную смесь распределяли между водой и дихлорметаном. Слои разделяли и водный слой экстрагировали дихлорметаном (4). Объединенные органические экстракты промывали водой и рассолом, сушили над сульфатом натрия, фильтровали и концентрировали. Твердое вещество несколько раз концентрировали из гептанов и этилацетата, затем сушили под вакуумом, получая указанное в заголовке соединение (1,01 г; 92%) в виде желтого твердого вещества. +ESI (М+Н) 218,2. 1(d, J=8,39 Гц, 1H), 7.82 (d, J=8,58 Гц, 1H), 7.42 (dd, J=8,49, 6,15 Гц, 1H), 4.47 (q, J=7,02 Гц, 2 Н), 1.45 (t,J=7,1 Гц, 3H). Стадия 2: 2-(трет-бутиламино)хинолин-7-карбоновая кислота. К имеющему температуру 0 С раствору 7-(этоксикарбонил)хинолин-1-оксида (500 мг; 2,3 ммоль) и трет-бутиламина (1,46 мл; 13,8 ммоль) в трифторметилбензоле (25 мл) порциями добавляли nтолуолсульфоновый ангидрид (1,96 г; 5,76 ммоль), поддерживая температуру внутри реакционного сосуда ниже 5 С. Реакционную смесь перемешивали в течение 1 ч. Добавляли насыщенный водный бикарбонат натрия и реакционную смесь оставляли перемешиваться в течение 15 мин. Далее фазы разделяли и водную фазу экстрагировали дихлорметаном (2). Объединенные органические экстракты промывали рассолом, сушили над сульфатом магния, фильтровали и концентрировали. После очистки колоночной флэш-хроматографией получали 499 мг прозрачного масла, которое отвердевало при стоянии. Это вещество переносили в тетрагидрофуран (5 мл) и добавляли 1 н. водный гидроксид лития (3,6 мл; 3,6 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение ночи. Реакционную смесь концентрировали и остаток разбавляли водой и подкисляли до рН, равного 4, 1 н. водной соляной кислотой. Полученный осадок отфильтровывали и сушили под вакуумом, получая указанное в заголовке соединение (330 мг, 75%) в виде бледно-желтого порошка. -ESI (М-Н) 243,1. 1 К имеющему температуру -70 С раствору 7-(этоксикарбонил)хинолин-1-оксида (1,67 г; 7,70 ммоль) в дихлорметане (80 мл) по каплям в атмосфере азота добавляли трифторметансульфоновый ангидрид(1,43 мл; 8,47 ммоль). Смесь перемешивали при -70 С в течение 5 мин. Затем по каплям при -70 С добавляли раствор метиламина в тетрагидрофуране (21 мл; 42 ммоль; 2,0 М). Смесь перемешивали в течение 5 мин и затем реакцию гасили водой (20 мл). Слои разделяли и водный слой экстрагировали дихлорметаном (330 мл). Объединенные органические экстракты промывали рассолом, сушили над сульфатом- 20022375 натрия, фильтровали и концентрировали. После очистки колоночной флэш-хроматографией получали указанное в заголовке соединение (720 мг, 41%) в виде желтого твердого вещества. 1(br. s., 1H), 4.44-4.38 (m, 2 Н), 3.12-3.11 (m, 3H), 1.44-1.40 (m, 3H). Стадия 2: 2-(метиламино)хинолин-7-карбоновая кислота. Указанное в заголовке соединение получали способом, аналогичным описанному на стадии 2 для промежуточного соединения 13, используя этил-2-(метиламино)хинолин-7-карбоксилат. 1 Смесь 3-карбамоил-1H-индол-6-карбоновой кислоты (25 мг; 0,12 ммоль), 1-изопропил-4,6 дигидроспиро[индазол-5,4'-пиперидин]-7(1H)-она (38 мг; 0,13 ммоль), гексафторфосфата (1 Н-7 азабензотриазол-1-ил)-1,1,3,3-тетраметилурония (46 мг; 0,12 ммоль) и диизопропилэтиламина (85 мкл; 0,49 ммоль) в 0,5 мл диметилформамида перемешивали при комнатной температуре в течение ночи. Реакционную смесь разбавляли этилацетатом и промывали 0,1 н. соляной кислотой. Органический слой сушили над сульфатом магния, фильтровали и концентрировали в вакууме. Остаток очищали обращенно-фазовой HPLC, получая указанное в заголовке соединение (14,6 мг; 28%). Аналитическая LCMS: +ESI(М+Н) 434,1; время удерживания 2,24 мин (колонка Waters Atlantis C18 4,650 мм, 5 мкм; линейный градиент от 95% вода/ацетонитрил до 5% вода/ацетонитрил в течение 4,0 мин, выдерживание при 5% вода/ацетонитрил до 5,0 мин; модификатор 0,05%-ная трифторуксусная кислота; скорость потока 2,0 мл/минута) (метод А). Типичный пример 2: 5-[(1-изопропил-7-оксо-1,4,6,7-тетрагидро-1H-спиро[индазол-5,4'-пиперидин]1'-ил)карбонил]-1H-индазол-3-карбоксамид К суспензии 1-изопропил-4,6-дигидроспиро[индазол-5,4'-пиперидин]-7(1H)-она (16,8 г; 53,6 ммоль) и 3-карбамоил-1H-индазол-5-карбоновой кислоты (10,0 г; 48,7 ммоль) в N,N-диметилформамиде (120 мл) добавляли триэтиламин (40 мл; 290 ммоль). Смесь перемешивали при комнатной температуре в течение 5 мин и затем охлаждали до 5 С. По каплям в течение 20 мин добавляли циклический ангидрид 1 пропанфосфоновой кислоты (60 мл; 100 ммоль; 50%-ный мас. раствор в этилацетате), поддерживая внутреннюю температуру от 5 до 10 С. Реакционную смесь оставляли перемешиваться в течение ночи, постепенно нагревая до комнатной температуры. Реакционную смесь медленно выливали в 700 мл воды при 5 С. Полученный осадок отфильтровывали и сушили. К фильтрату добавляли смесь 10% метанол/этилацетат (1 л) и полученный осадок отфильтровывали и сушили. Собранные твердые вещества объединяли (8,9 г) и растворяли в N,N-диметилформамиде (34 мл) при 130 С. Раствор охлаждали до 100 С и добавляли метанол (65 мл). Раствор оставляли медленно охлаждаться до комнатной температуры. Полученный осадок собирали фильтрацией и сушили, получая указанное в заголовке соединение (6,0 г; 28%). +ESI (М+Н) 435,1. 1 К раствору гидрохлорида 1-(3-диметиламинопропил)-3-этилкарбодиимида (252 мг; 1,31 ммоль) и гидрата 1-гидроксибензотриазола (199 мг; 1,30 ммоль) в дихлорметане (5 мл) добавляли 3-карбамоил-1Hпирроло[3,2-b]пиридин-6-карбоновую кислоту (212 мг; 1,04 ммоль). Смесь перемешивали при комнатной температуре в течение 40 мин. Затем добавляли 1-изопропил-4,6-дигидроспиро[индазол-5,4'-пиперидин]7(1H)-он (312 мг; 1,26 ммоль) и триэтиламин (0,434 мл; 3,11 ммоль) и реакционную смесь оставляли перемешиваться при комнатной температуре в течение 16 ч. Реакционную смесь разбавляли насыщенным водным хлоридом аммония и экстрагировали дважды дихлорметаном. Объединенные органические слои промывали последовательно насыщенным бикарбонатом натрия, водой и рассолом. Органические слои сушили над сульфатом натрия, фильтровали и концентрировали. После очистки с использованием колоночной флэш-хроматографии (0-100% раствора 20% метанол/дихлорметан) получали указанное в заголовке соединение (254 мг, 57%). +ESI (М+Н) 435,5. 1H ЯМР (400 МГц, CD3OD, ): 8.57 (d, J=1,8 Гц, 1H), 8.26 (s, 1H), 7.98 (d, J=1,0 Гц, 1H), 7.43 (s, 1H),5.38 (m, 1H), 3.63 (m, 4 Н), 2.90 (s, 2H), 2.66 (s, 2H), 1.66 (m, 4 Н), 1.42 (d, J=6,6 Гц, 6 Н). Соединения, перечисленные ниже в табл. 1, получали, используя методики, аналогичные описанным выше для синтеза соединений типичных примеров 1-3, с применением соответствующих исходных веществ, имеющихся в продаже, полученных с использованием способов, хорошо известных специалистам в данной области, или полученных путем, описанным выше. Перечисленные ниже соединения сначала были выделены в виде свободного основания, и для тестирования их можно превратить в фармацевтически приемлемую соль. Таблица 1 Указанное в заголовке соединение получали способом, аналогичным описанному для примера 3,используя 2-(трет-бутиламино)хинолин-7-карбоновую кислоту. +APCI (М+Н) 474,6. 1H ЯМР (400 МГц, CDCl3, ): 7.72 (d, J=8,8 Гц, 1H), 7.64 (s, 1H), 7.55 (d, J=8,2 Гц, 1H), 7.36 (s, 1H),7.16 (dd, J=8,1, 1,3 Гц, 1H), 6.59 (d, J=9,2 Гц, 1H), 5.36 (quin, J=6,6 Гц, 1H), 3.31-3.96 (m, 4H), 2.79 (s, 2H),2.58 (s, 2H), 1.55-1.75 (m, 4 Н), 1.52 (s, 9 Н), 1.44 (d, J=6,4 Гц, 6 Н). Стадия 2: трифторацетатная соль 1'-[(2-аминохинолин-7-ил)карбонил]-1-изопропил-1,4 дигидроспиро[индазол-5,4'-пиперидин]-7(6 Н)-она. Трифторуксусную кислоту (0,90 мл; 12 ммоль) добавляли к 1'-(2-(трет-бутиламино)хинолин-7 карбонил)-1-изопропил-4,6-дигидроспиро[индазол-5,4'-пиперидин]-7(1H)-ону (50 мг; 0,11 ммоль). Реакционную смесь нагревали до 70 С в течение 3 ч, затем охлаждали до комнатной температуры и оставляли перемешиваться в течение ночи. Реакционную смесь концентрировали до сухого состояния и после очистки обращенно-фазовой HPLC получали указанное в заголовке соединение (41 мг, 93%). +ESI(М+Н) 418,2; время удерживания при HPLC 2,11 мин (метод А). 1(br. s., 6H). Фармакологические данные Биологические протоколы. Полезность соединений по настоящему изобретению в лечении заболеваний (таких как подробно описанные в данном изобретении) у животных, в частности млекопитающих (например, людей), можно продемонстрировать по их активности в традиционных анализах, известных обычному специалисту в данной области, включая анализы in vitro и in vivo, описанные ниже. Такие анализы также предоставляют средство, с помощью которого можно провести сравнение активностей соединения по настоящему изобретению с активностями других известных соединений. Прямое ингибирование активностей АСС 1 и АСС 2 АСС-ингибирующую активность соединения по настоящему изобретению демонстрировали способами, основанными на стандартных методиках. Например, прямое ингибирование активности АСС для соединения по настоящему изобретению определяли, используя препараты рекомбинантной АСС 1 человека (rhACC1) и рекомбинантной АСС 2 человека (rhACC2). Типичные последовательности рекомбинантных АСС 1 и АСС 2 человека, которые могут быть использованы в анализе, приведены в данном изобретении как SEQ ID NO: 1 и SEQ ID NO: 2 соответственно.[1] Получение rhACC1. 2 л клеток SF9, инфицированных рекомбинантным бакуловирусом, содержащим полноразмерную кДНК АСС 1 человека, суспендировали в охлажденном на льду лизирующем буфере (25 мМ Трис, рН 7,5; 150 мМ NaCl; 10% глицерин; 5 мМ имидазол (EMD Bioscience; Гиббстаун, NJ); 2 мМ ТСЕР (трис-(2 карбоксиэтил)фосфин) (BioVectra; Шарлоттаун, Канада); нуклеаза Benzonase (10000 ед./100 г клеточной пасты; Novagen; Мадисон, WI); не содержащая EDTA (этилендиаминтетрауксусная кислота) смесь ингибиторов протеаз (1 таб./50 мл; Roche Diagnostics; Мангейм, Германия). Клетки лизировали при помощи 3 циклов замораживания-оттаивания, и центрифугировали при 40000g в течение 40 мин (4 С). Супернатант наносили непосредственно на колонку HisTrap FF crude (HisTrap FF для необработанных образцов)(GE Healthcare; Пискатавэй, NJ) и элюировали градиентом имидазола вплоть до 0,5 М, используя 20 объемов колонки (CV). Объединяли АСС 1-содержащие фракции, разбавляли 1:5, используя 25 мМ Трис, рН 7,5; 2 мМ ТСЕР, 10% глицерин, наносили непосредственно на колонку CaptoQ (GE Healthcare) и элюировали градиентом NaCl вплоть до 1 М, используя 20 CV. Удаление фосфатных групп из очищенной АСС 1 проводили посредством инкубации с лямбда-фосфатазой (100 ед./10 мкМ целевого белка; New EnglandBiolabs; Беверли, MA) в течение 14 ч при 4 С; для ингибирования фосфатазы добавляли окадаиковую кислоту (конечная концентрация 1 мкМ; Roche Diagnostics). Выполняли замену буфера в очищенной АСС 1 на 25 мМ Трис, рН 7,5; 2 мМ ТСЕР, 10% глицерин, 0,5 М NaCl, используя диализ при 4 С в течение 6 ч. Готовили аликвоты и замораживали при -80 С.[2] Измерение ингибирования rhACC1. Анализ с использованием hACC1 проводили в 384-луночном планшете Costar3676 (Costar,Кеймбридж, MA) с применением набора для детекции АДФ (аденозиндифосфат) посредством анализа FP(поляризация флуоресценции) Transcreener (Bellbrook Labs, Мадисон, Висконсин), следуя рекомендованным производителем условиям для реакции с 50 мкМ АТФ (аденозинтрифосфат). Для анализа использовали следующие конечные условия: 50 мМ HEPES (4-(2-гидроксиэтил)-1-пиперазинэтансульфоновая кислота), рН 7,2; 10 мМ MgCl2, 7,5 мМ трикалия цитрат, 2 мМ DTT (дитиотреитол); 0,1 мг/мл BSA (бычий сывороточный альбумин); 30 мкМ ацетил-КоА; 50 мкМ АТФ и 10 мМ KHCO3. Обычно реакцию в объеме 10 мкл проводили в течение 120 мин при 25 С, добавляли 10 мкл буфера Transcreener Stop andDetect buffer (буфер для остановки реакции и детекции) и смесь инкубировали при комнатной температуре в течение еще 1 ч. Данные получали с использованием флуоресцентного ридера Envision (PerkinElmer), применяя общее двойное зеркало Су 5 FP возбуждения 620, фильтр Су 5 FP возбуждения 620,фильтр эмиссии 688 (S) и эмиссии 688 (Р).[3] Получение rhACC2. Ингибирование АСС 2 человека измеряли, используя очищенную рекомбинантную АСС 2 человека(hrACC2). Кратко, полноразмерную клонированную АСС 2 (Cytomax) приобретали у Cambridge Bioscience Limited, секвенировали и субклонировали в PCDNA5 FRT TO-TOPO (Invitrogen, Карлсбад, СА). АСС 2 экспрессировали в клетках СНО (яичники китайского хомячка) путем индукции тетрациклином и собирали в 5 литрах DMEM/F12 (модифицированная Дульбекко среда Игла с питательной смесью ХэмаF12) с глутамином, биотином, гигромицином и бластицидином с 1 мкг/мл тетрациклина (Invitrogen, Карлсбад, СА). Кондиционированную среду, содержащую АСС 2, затем наносили на колонку Softlink SoftRelease Avidin (колонка с авидином с мягким высвобождением) (Promega, Мадисон, Висконсин) и элюировали 5 мМ биотином. Элюировали 4 мг АСС 2 в концентрации 0,05 мг/мл (определенной по A280) с предполагаемой чистотой 95% (определенной по A280)- Очищенную АСС 2 диализовали против 50 мМ Трис, 200 мМ NaCl, 4 мМ DTT, 2 мМ EDTA и 5% глицерина. Собранный белок замораживали и хранили при -80 С без потери активности при оттаивании. Для измерения активности АСС 2 и оценки ингибирования АСС 2 тестируемые соединения растворяли в DMSO (диметилсульфоксид) и добавляли к ферментуrhACC2 в виде 5-кратного концентрированного раствора с получением конечной концентрации DMSO 1%.[4] Измерение ингибирования АСС 2 человека. Анализ с использованием hACC2 проводили в 384-луночном планшете Costar3676 (Costar,Кеймбридж, MA) с применением набора для детекции АДФ посредством анализа FP Transcreener (Bellbrook Labs, Мадисон, Висконсин), следуя рекомендованным производителем условиям для реакции с 50 мкМ АТФ. Для данного анализа использовали следующие конечные условия: 50 мМ HEPES, рН 7,2; 5 мМ MgCl2, 5 мМ трикалия цитрат, 2 мМ DTT; 0,1 мг/мл BSA; 30 мкМ ацетил-КоА; 50 мкМ АТФ и 8 мМKHCO3. Обычно реакцию в объеме 10 мкл проводили в течение 50 мин при 25 С, добавляли 10 мкл буфера Transcreener Stop and Detect buffer и смесь инкубировали при комнатной температуре в течение еще 1 ч. Данные получали с использованием флуоресцентного ридера Envision (PerkinElmer), применяя общее двойное зеркало Су 5 FP возбуждения 620, фильтр Су 5 FP возбуждения 620, фильтр эмиссии 688 (S) и эмиссии 688 (Р). Результаты анализов рекомбинантной hACC1 и рекомбинантной hACC2 с применением Transcreener суммированы ниже в таблице для соединений по настоящему изобретению, приведенных выше в примерах.- 24022375 Согласно SEQ ID NO: 1 предложена последовательность рекомбинантной АСС 1 человека (SEQ IDNO: 1), которая может быть использована в анализе in vitro с применением Transcreener. Последовательность hACC1 Согласно SEQ ID NO: 2 предложена последовательность рекомбинантной АСС 2 человека (SEQ IDNO: 2), которая может быть использована в анализе in vitro с применением Transcreener. Оценка ингибирования АСС in vivo в остром опыте на экспериментальных животных. АСС-ингибирующая активность соединений по настоящему изобретению может быть подтверждена in vivo посредством оценки их способности снижать уровни малонил-КоА в печени и мышечной тка- 26022375 ни обработанных животных. Измерение ингибирования образования малонил-КоА у экспериментальных животных может быть проведено с использованием приведенной далее методологии. При применении этого метода самцов крыс Sprague-Dawley (225-275 г), содержащихся на стандартном корме и с доступом к воде без ограничения, случайным образом распределяли по группам перед исследованием. Одних животных кормили, другим не давали корма в течение 18 ч перед началом эксперимента. Через 2 ч после начала светового цикла животным перорально вводили дозу 0,5%-ной метилцеллюлозы (разбавителя) или соответствующего соединения (приготовленного в разбавителе) в объеме 5 мл/кг. Контрольные крысы, получавшие корм и разбавитель, были включены для определения фоновых уровней малонил-КоА в тканях, в то время как животные, не получавшие корма, были включены для определения того, как влияло голодание на уровни малонил-КоА. Через один час после введения соединений у животных вызывали асфиксию, используя СО 2, и ткани извлекали. Конкретно, кровь отбирали путем сердечной пункции и помещали в пробирки BD Microtainer, содержащие EDTA (BD Biosciences,NJ), перемешивали и помещали на лед. Для определения воздействия лекарственного средства использовали плазму крови. Печень и четырехглавую мышцу извлекали, незамедлительно фиксировали замораживанием, заворачивали в фольгу и хранили в жидком азоте. Ткани измельчали под жидким N2 для обеспечения однородности при приготовлении образцов. Из ткани (150-200 мг) экстрагировали малонил-КоА, используя 5 объемов 10%-ной трикарбоновой кислоты в Lysing Matrix А (лизирующей матрице А) (МР Biomedicals, PN 6910) на приборе FastPrep FP120 (Thermo Scientific, скорость равна 5,5; в течение 45 секунд). Супернатант, содержащий малонил-КоА, отделяли от клеточного дебриса после центрифугирования при 15000g в течение 30 мин (Eppendorf Centrifuge 5402). Образцы надежно замораживали при -80 С до завершения анализа. Оценка уровней малонил-КоА в печени и мышечной ткани может быть проведена в анализах с использованием приведенной далее методологии. В методе были использованы следующие вещества: тетралитиевая соль малонил-КоА и трилитиевая соль малонил- С 3-КоА, которые были приобретены у Isotec (Майамисберг, ОН, США), перхлорат натрия (Sigma, номер по каталогу 410241), трихлоруксусная кислота (ACROS, номер по каталогу 42145),фосфорная кислота (J.T. Baker, номер по каталогу 0260-01), формиат аммония (Fluka, номер по каталогу 17843), метанол (квалификация "для HPLC", J.T. Baker, номер по каталогу 9093-33) и вода (квалификация "для HPLC", J.T. Baker, 4218-03), которые были использованы для приготовления необходимых подвижных фаз. Колонки для твердофазной экстракции в режиме он-лайн Strata-X 25 мкм; 20 мм 2,0 мм в.д.(внутренний диаметр) (номер по каталогу 00M-S033-B0-CB) были получены от Phenomenex (Торранс,СА, США). Колонки для обращенно-фазовой хроматографии SunFire C18 3,5 мкм; 100 мм 3,0 мм в.д.(номер по каталогу 186002543) были приобретены у Waters Corporation (Милфорд, MA, США). Этот метод можно осуществлять, примененяя следующее оборудование. Для двумерной хроматографии использовали бинарный насос Agilent 1100, насос для четырехкомпонентных смесей Agilent 1100 и два 6-канальных двухпозиционных клапана Valco Cheminert. Образцы вводили, применяя автоматический дозатор НТС PAL от LEAP с охлаждаемым посредством использования эффекта Пельтье штативом,поддерживаемым при 10 С, и 20 мкл петлей для образцов. Для промывки иглы автоматического дозатора использовали следующие растворы: 10%-ную трихлоруксусную кислоту в воде (масс/об.) для промывки 1 и смесь 90:10 метанол:вода для промывки 2. Температуру в аналитической колонке (Sunfire) поддерживали при 35 С, используя термостат для микроколонок для LC (жидкостная хроматография) (Micro-LCColumn Oven) от MicroTech Scientific. Элюент анализировали на тройном квадрупольном массспектрометре API3000 от ABI Sciex с ионизатором Turbo Ion Spray. Двумерную хроматографию осуществляли параллельно, используя различные условия градиентного элюирования для он-лайн твердофазной экстракции и обращенно-фазовой хроматографии. Общая схема метода состояла в том, что первое измерение использовали для очистки образца и захвата представляющего интерес аналита, с последующим кратковременным соединением обеих измерений для элюирования с первого измерения на второе измерение. Затем измерения разъединяли, чтобы осуществить градиентное элюирование аналита со второго измерения для количественного определения, одновременно подготавливая первое измерение для следующего образца в последовательности. В момент кратковременного соединения обеих измерений друг с другом, движение потока подвижной фазы в первом измерении меняли на противоположное для элюирования аналита во втором измерении, что позволяло получить оптимальные ширину пика, форму пика и время элюирования. В первом измерении в HPLC-системе использовали колонку для твердофазной экстракции в режиме он-лайн Strata-X от Phenomenex и подвижную фазу, составленную из смеси 100 мМ перхлорат натрия/0,1% (об./об.) фосфорной кислоты для растворителя А и метанола для растворителя В. Во втором измерении в HPLC-системе использовали колонку для обращенно-фазовой хроматографии SunFire C18 от Waters и подвижную фазу, составленную из 100 мМ формиата аммония для растворителя А и метанола для растворителя В. Начальное состояние градиента выдерживали в течение 2 мин,и в течение этого времени происходил перенос аналита в аналитическую колонку. Было важно, чтобы- 27022375 элюирующая сила в начальном состоянии была достаточной для осуществления элюирования аналита с колонки для он-лайн SPE (твердофазной экстракции) с одновременным удержанием его на аналитической. Потом градиент линейно увеличивали до 74,5% А в течение 4,5 мин, после чего выполняли стадию промывки и повторного уравновешивания. Масс-спектрометрия в сочетании с HPLC может представлять собой высокоселективный и чувствительный метод количественного определения аналитов в матрицах сложного состава, однако она попрежнему подвержена влиянию интерференции и подавления. В результате сочетания двумерной HPLC и масс-спектрометрии эти интерференции были значительно снижены. Кроме того, благодаря использованию в тройном квадрупольном масс-спектрометре режима мониторинга множественных реакций(MRM) было значительно улучшено соотношение сигнала к шуму. Что касается этого анализа, то работу на масс-спектрометре осуществляли в режиме положительных ионов с напряжением на ионизаторе TurbolonSpray, составляющем 2250 В. Распыляющий газ нагревали до 450 С. Потенциал декластеризации (DP), фокусирующий потенциал (FP) и энергию соударений(СЕ) устанавливали на 60, 340 и 42 В соответственно. Для квадруполя 1 (Q1) устанавливали единичное разрешение, для квадруполя 3 (Q3) устанавливали низкое разрешение. Заданное значение в отношении газа для CAD (столкновительно активируемая диссоциация) составляло 8. Регистрировали следующиеMRM-переходы: для малонил-КоА 854,1347,0 m/z (L. Gao et al. (2007) J. Chromatogr. В 853, 303-313); и для малонил- 13 С 3-СоА 857,1350,0 m/z с временами измерения 200 мс. Элюент направляли в массспектрометр в момент, близкий к ожидаемому времени элюирования аналита, в остальное время его направляли в слив для сохранения источника и улучшения устойчивости оборудования. Интегрирование полученных хроматограмм выполняли, используя программное обеспечение Analyst (AppliedBiosystems). Концентрации малонил-КоА в тканях рассчитывали, используя калибровочную кривую,полученную с применением 10%-ного раствора трихлоруксусной кислоты в воде. Образцы для построения калибровочной кривой с целью количественного определения малонилКоА в экстрактах тканей готовили в 10%-ной (мас./об.) трихлоруксусной кислоте (ТСА) и в диапазоне от 0,01 до 1 пмоль/мкл. В качестве внутреннего стандарта в каждый компонент для калибровочной кривой и образец добавляли малонил-13 С 3-КоА (в конечной концентрации 0,4 пмоль/мкл). Готовили шесть внутрианалитических качественных контролей: три из объединенного экстракта,полученного от животных, не получавших корма, и три из объединенного экстракта, полученного от животных, получавших корм. Их анализировали в качестве независимых образцов, дополненных 12 Смалонил-КоА в концентрации 0, 0,1 или 0,3 пмоль/мкл, а также малонил-13 С 3-КоА (0,4 пмоль/мкл). Каждый внутрианалитический качественный контроль содержал 85% водного тканевого экстракта, а остальную часть составлял внутренний стандарт (0,4 пмоль/мкл) и 12 С-малонил-КоА. При проведении каждого измерения включали межаналитические контроли; они состоят из объединенных образцов четырехглавых мышц, по одному образцу от животных, не получавших корма и получавших корм, и/или из объединенных образцов печени, по одному образцу от животных, не получавших корма и получавших корм. Все подобные контроли дополнены малонил-13 С 3-КоА (0,4 пмоль/мкл). Все публикации, включая, но не ограничиваясь этим, выданные патенты, заявки на патент и журнальные статьи, цитированные в этом изобретении, включены по отдельности в настоящее описание посредством ссылки во всей своей полноте. Хотя данное изобретение изложено выше со ссылкой на описанные воплощения, специалистам в данной области техники будет несложно понять, что подробно описанные конкретные эксперименты являются только иллюстрациями изобретения. Следует понимать, что могут быть выполнены различные модификации без отступления от сущности изобретения. Соответственно, данное изобретение ограничено только приведенной далее формулой изобретения. ФОРМУЛА ИЗОБРЕТЕНИЯ 1. Соединение структуры или его фармацевтически приемлемая соль. 2. Соединение структуры или его фармацевтически приемлемая соль. 3. Соединение структуры или его фармацевтически приемлемая соль. 4. Соединение структуры или его фармацевтически приемлемая соль. 5. Фармацевтическая композиция для лечения заболеваний, опосредованных активностью ацетилКоА-карбоксилазы, содержащая соединение по любому из пп.1-4 или его фармацевтически приемлемую соль и фармацевтически приемлемый эксципиент, разбавитель или носитель. 6. Применение соединения по любому из пп.1-4 в изготовлении лекарственного средства для лечения заболевания, состояния или расстройства, модулируемого посредством ингибирования фермента(ов),представляющего(их) собой ацетил-КоА-карбоксилазу(ы). 7. Применение по п.6, где заболевание, состояние или расстройство представляет собой диабет 2 типа, связанные с диабетом расстройства, неалкогольную жировую болезнь печени или печеночную инсулинорезистентность. 8. Применение по п.6, где заболевание, состояние или расстройство представляют собой диабет 2 типа.
МПК / Метки
МПК: A61P 3/10, C07D 519/00, C07D 471/10, A61P 3/04, A61K 31/438
Метки: ацетил-коа-карбоксилаз, применения, качестве, производные, ингибиторов, пиразолоспирокетонов
Код ссылки
<a href="https://eas.patents.su/30-22375-proizvodnye-pirazolospiroketonov-dlya-primeneniya-v-kachestve-ingibitorov-acetil-koa-karboksilaz.html" rel="bookmark" title="База патентов Евразийского Союза">Производные пиразолоспирокетонов для применения в качестве ингибиторов ацетил-коа-карбоксилаз</a>
Производные хинолина для применения в качестве микобактериальных ингибиторов

Номер патента: 11572
Опубликовано: 28.04.2009
Авторы: Ланкуа Давид Франсис Ален, Паскье Элизабет Терез Жанн, Гийемон Жером Эмиль Жорж
МПК: C07D 215/18, A61K 31/47, A61P 31/06...
Метки: производные, микобактериальных, качестве, применения, хинолина, ингибиторов
Формула / Реферат:
1. Соединение основной формулы (I) его фармацевтически приемлемые кислотно- или основно-аддитивные соли, его четвертичные амины, его стереохимически изомерные формы, его таутомерные и N-оксидные формы, где R1 представляет собой галоген или алкилокси; р представляет собой целое число, равное 1; s представляет собой целое число, равное 0 или 1; R2 представляет собой водород; галоген или алкил; R3 представляет собой Ar; q представляет собой целое...
Производные 1,2,3-триазола для применения в качестве ингибиторов стеароил-соа десатуразы

Номер патента: 16621
Опубликовано: 30.06.2012
Авторы: Тротте Лионель, Буйо Анн Мари Жанн, Лароз Ален
МПК: A61P 3/06, A61K 31/4192, C07D 249/04...
Метки: качестве, применения, производные, 1,2,3-триазола, ингибиторов, десатуразы, стеароил-соа
Формула / Реферат:
1. Соединение формулы (I)где X является -CONH-, -NHCO- или -CH2NH-;R1 является -С6-10арилом, необязательно замещенным одной, двумя или тремя группами, независимо выбранными из:(a) -С1-6алкила, -ОСН3, -С1-6галогеналкила, -ОС1-6галогеналкила, -С3-6циклоалкила, -ОС3-6циклоалкила или галогена;(b) фенила, необязательно замещенного одной, двумя или тремя группами, независимо выбранными из галогена;R2 является -С1-6алкилом;R3 является -С6-10арилом,...
Производные n5-(2-этоксиэтил)-n3-(2-пиридинил)-3,5-пиперидиндикарбоксамида, предназначенные для применения в качестве ингибиторов ренина

Номер патента: 16446
Опубликовано: 30.05.2012
Авторы: Хитоми Юко, Йококава Фумиаки, Эхара Такеру, Каваками Шимпей, Тояо Атсуши, Сузуки Масаки, Ирие Осаму
МПК: A61K 31/4545, C07D 401/12, A61P 9/12...
Метки: n5-(2-этоксиэтил)-n3-(2-пиридинил)-3,5-пиперидиндикарбоксамида, качестве, ренина, применения, ингибиторов, предназначенные, производные
Формула / Реферат:
1. Соединение формулы (I)в которой R1 обозначает С1-С7-алкил, который необязательно содержит 1, 2 или 3 заместителя, выбранных из группы, включающей гидроксигруппу, галоген и C1-С7-алкоксигруппу;R2 обозначает водород, С1-С7-алкил, С1-С7-алкокси, галоген-С1-С7-алкил, галоген-С1-С7-алкоксигруппу или С1-С7-алкокси-С1-С7-алкоксигруппу;R3 обозначает С1-С7-алкил или С3-С8-циклоалкил;R4 обозначает водород или гидроксигруппу иR5 обозначает С1-С7-алкил...
Производные пиримидина, применимые в качестве ингибиторов pi3 киназы, и способы их применения

Номер патента: 18083
Опубликовано: 30.05.2013
Авторы: Иванович Эдуин, Хендриксон Томас, Уагман Аллан, Волива Чарлз, Луи Алисия, Смит Аарон, Аталлах Гордана, Синь Сяхуа, Визманн Марион, Пик Тереса, Верхаген Джоелл, Фрейзьер Келли, Берджер Маттью, Бартулис Сара, Пун Даниэль, Барсанти Пол, Пекки Сабина, Ни Чжицзи, Чжан Яньчэнь, Кнапп Марк, Фантл Уэнди, Нг Саймон, Меритт Ханне, Пфистер Кейт
МПК: A61P 35/00, C07D 401/04, A61K 31/506...
Метки: применения, пиримидина, применимые, качестве, ингибиторов, производные, способы, киназы
Формула / Реферат:
1. Соединение формулы Iили его стереоизомер, таутомер или фармацевтически приемлемая соль, в которойW выбран из СН и N;R1 выбран из группы, включающей:(1) замещенный и незамещенный гетероциклил, представляющий собой 3- или 4-членное кольцо, содержащее гетероатом, выбранный из азота, кислорода и серы, или 5- или 6-членное кольцо, содержащее от 1 до 3 гетероатомов, выбранных из группы, состоящей из азота, кислорода или серы, где любое из указанных...
Конформационно ограниченные бифенильные производные для применения в качестве ингибиторов вируса гепатита с

Номер патента: 20511
Опубликовано: 28.11.2014
Авторы: Минвелл Николас А., Лэнгли Дэвид Р., Гудрих Джейсон, Дэон Даниэль Х., Ванг Гэн, Джеймс Клинт А., Рюдигер Эдвард Х., Ст.Лоран Денис Р., Бачанд Кэрол, Гуд Эндрю К., Мартел Алан, Лавуа Рико, Нгуен Ван Н., Ромин Джеффри Ли, Снайдер Лоуренс Б., Белема Маконен, Хаманн Лоуренс Г., Янг Фуканг, Лопез Омар Д.
МПК: A61P 31/14, C07D 403/14, A61K 31/4178...
Метки: гепатита, бифенильные, ингибиторов, качестве, вируса, ограниченные, применения, конформационно, производные
Формула / Реферат:
1. Соединение формулы (I)или его фармацевтически приемлемая соль, гдеD и Е представляют собой имидазолил;R2 и R2' вместе с атомами углерода, к которым они присоединены, образуют пяти-семизвенное ненасыщенное кольцо, которое может содержать один гетероатом, независимо выбранный из азота и кислорода, необязательно замещенное С1-С6алкоксиС1-С6алкилом, С1-С6алкилом, гидрокси, -NRaRb, оксогруппой или С2-6алкиленовым дирадикалом, образующим...
Предыдущий патент: Гетероарильные соединения в качестве лигандов 5-ht4 рецептора
Следующий патент: Замещенные производные бензамида
Случайный патент: Применение композиции на основе простагландина для лечения расстройства сексуального возбуждения у женщин