Производные пиразола, используемые в качестве антагонистов рецептора ccr4
Номер патента: 21740
Опубликовано: 31.08.2015
Авторы: Прокопиоу Панайиотис Александроу, Парр Найджел Джеймс, Нидхэм Дебора, Ритчи Тимоти Джон, Ходжсон Саймон Тинби, Лакруа Янник Морис, Хоббс Хизер, Вудроу Майкл Дэвид




























Формула / Реферат
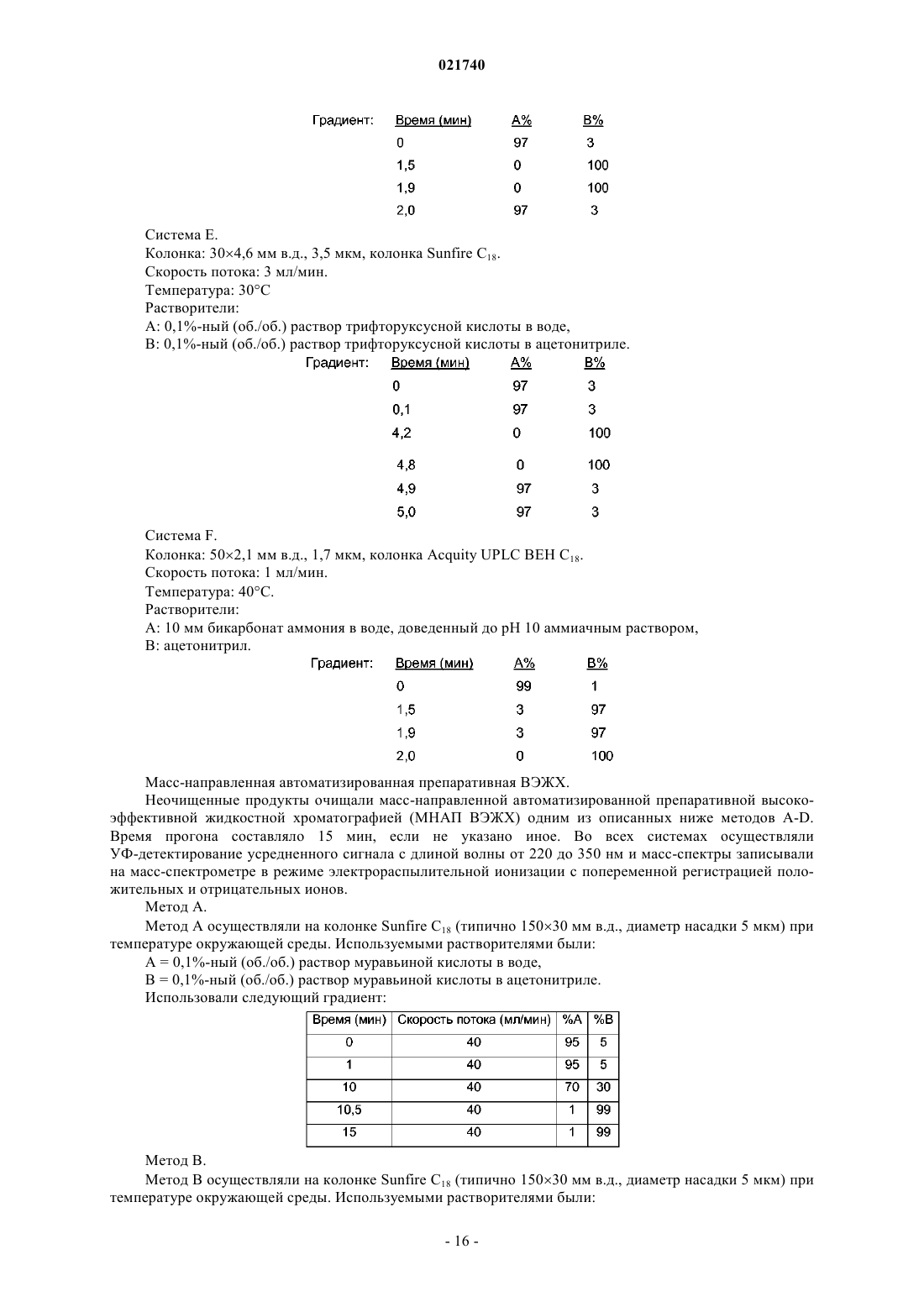
1. Соединение формулы (I) или его соль

где R1 выбран из группы, состоящей из следующих групп:
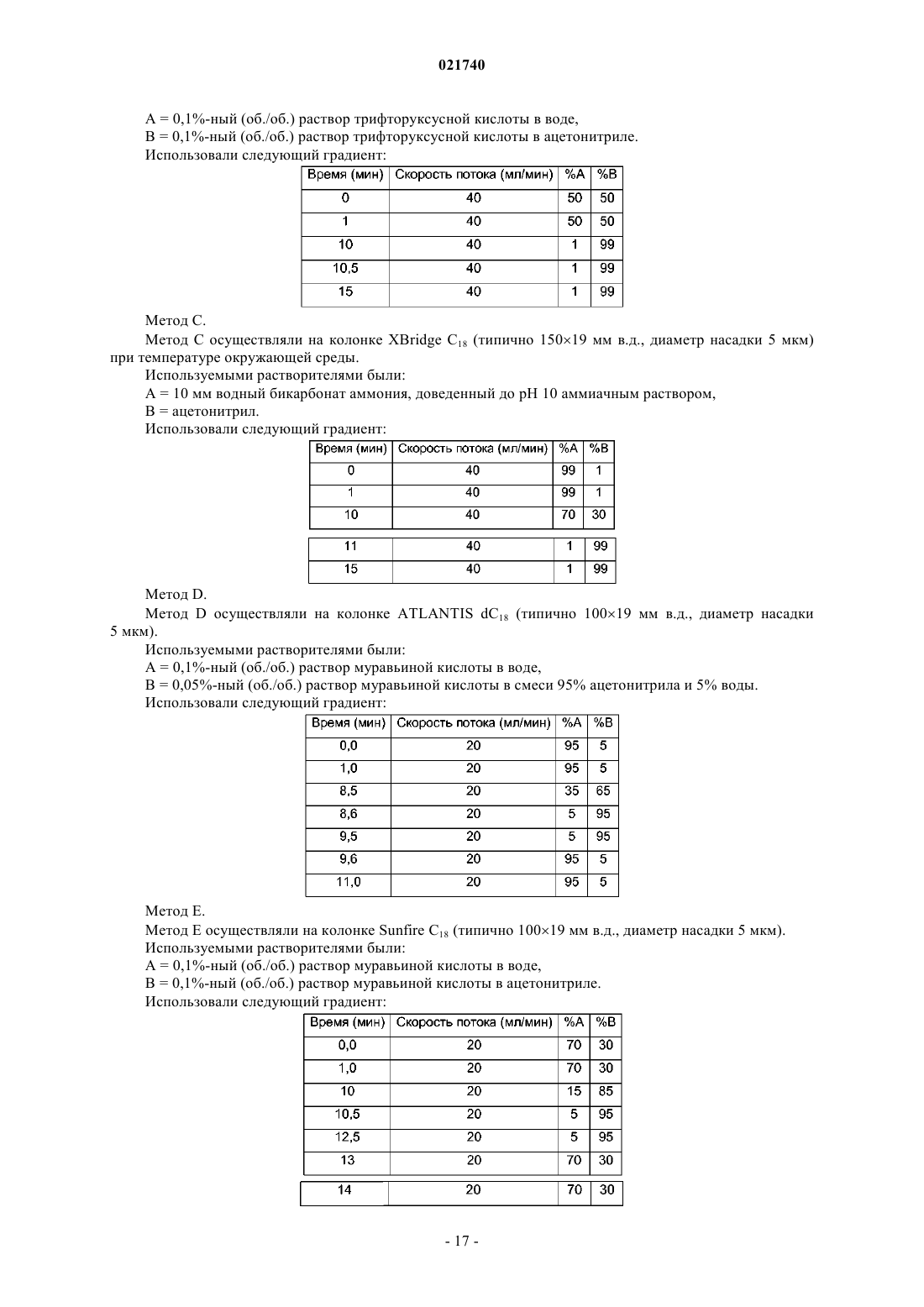
(1) группа формулы (а)

где A выбран из группы, состоящей из:
(i) водорода;
(ii) C1-6алкила, возможно замещенного одним или двумя заместителями, выбранными из группы, состоящей из -NRaRb, -ORc, -C(O)NRaRb, -C(O)ORc, насыщенного 4-7-членного моноциклического кольца, содержащего один или два гетероатома, выбранных из азота, кислорода или серы; фенила или тиенила, фурила, пирролила, триазолила, имидазолила, оксазолила, тиазолила, оксадиазолила, изотиазолила, изоксазолила, тиадиазолила, пиразолила, пиримидила, пиридазинила, пиразинила, пиридила, хинолинила, изохинолинила, индолила, бензофурила, бензотиенила, бензимидазолила, бензоксазолила;
(iii) С3-7циклоалкила, возможно замещенного группой -C(O)ORc или -NRaRb;
(iv) насыщенного 4-7-членного моноциклического кольца, содержащего один или два гетероатома, выбранных из азота, кислорода или серы, возможно замещенного одним или более С1-6алкилом;
(v) тиенила, фурила, пирролила, триазолила, имидазолила, оксазолила, тиазолила, оксадиазолила, изотиазолила, изоксазолила, тиадиазолила, пиразолила, пиримидила, пиридазинила, пиразинила и пиридила, хинолинила, изохинолинила, индолила, бензофурила, бензотиенила, бензимидазолила, бензоксазолила;
(vi) -NRaRb;
(vii) фенила, замещенного группой -(CH2)pC(O)OH, где р означает 0, 1, 2 или 3; или
(viii) -(CH2)7C(O)OH;
(2) группа формулы (b)

где В представляет собой С1-6алкил;
(3) -C(O)NRaRb или -CH2C(O)NRaRb;
(4) -S(O)2NRaRb;
(5) С1-6алкокси, возможно замещенный группой NRaRb, где Ra, Rb и Rc независимо представляют собой водород или С1-6алкил;
R2 представляет собой галоген, C1-6алкил, CF3, гидрокси или С1-6алкокси;
R3 представляет собой галоген, CF3, гидрокси, C1-6алкокси, CRdReOH или CHF2, где Rd и Re независимо представляют собой водород или метил;
R4 представляет собой водород, галоген, C1-6алкил или CF3;
R5 и R6 независимо представляют собой водород, галоген или С1-6алкил;
n означает 0 или 1.
2. Соединение по п.1 или его соль, где R1 выбран из группы, состоящей из следующих групп:
(1) группа формулы (а)

где A выбран из группы, состоящей из:
(i) водорода;
(ii) C1-6алкила, возможно замещенного одним или двумя заместителями, выбранными из группы, состоящей из -NRaRb, -ORc, -C(O)NRaRb, -C(O)ORc, насыщенного 4-7-членного моноциклического кольца, содержащего один или два гетероатома, выбранных из азота, кислорода или серы; фенила или тиенила, фурила, пирролила, триазолила, имидазолила, оксазолила, тиазолила, оксадиазолила, изотиазолила, изоксазолила, тиадиазолила, пиразолила, пиримидила, пиридазинила, пиразинила, пиридила, хинолинила, изохинолинила, индолила, бензофурила, бензотиенила, бензимидазолила, бензоксазолила;
(iii) С3-7циклоалкила, возможно замещенного группой -NRaRb;
(iv) насыщенного 4-7-членного моноциклического кольца, содержащего один или два гетероатома, выбранных из азота, кислорода или серы, возможно замещенного одним или более чем одним С1-6алкилом;
(v) тиенила, фурила, пирролила, триазолила, имидазолила, оксазолила, тиазолила, оксадиазолила, изотиазолила, изоксазолила, тиадиазолила, пиразолила, пиримидила, пиридазинила, пиразинила, пиридила, хинолинила, изохинолинила, индолила, бензофурила, бензотиенила, бензимидазолила, бензоксазолила;
(vi) -NRaRb;
(2) группа формулы (b)

где В представляет собой C1-6алкил;
(3) -C(O)NRaRb;
(4) -S(O)2NRaRb;
(5) C1-6алкокси, возможно замещенного группой NRaRb, где Ra, Rb и Rc независимо представляют собой водород или С1-6алкил;
R2 представляет собой галоген, C1-6алкил, CF3, гидрокси или С1-6алкокси;
R3 представляет собой галоген, CF3, гидрокси, C1-6алкокси, CH2OH или CHF2;
R4 представляет собой водород, галоген, С1-6алкил или CF3;
R5 и R6 независимо представляют собой водород, галоген или С1-6алкил;
n означает 0 или 1.
3. Соединение по п.1 или 2, которое представляет собой соединение формулы (Ia), или его соль

где R1 и R3 такие, как определено в п.1 или 2.
4. Соединение по любому из пп.1-3 или его соль, где R1 представляет собой группу формулы (а)

5. Соединение по п.4 или его соль, где A представляет собой С1-6алкил.
6. Соединение по п.4 или его соль, где A представляет собой С1-6алкил, замещенный одним или двумя заместителями, выбранными из группы, состоящей из -NRaRb, -ORc, -C(O)NRaRb, -C(O)ORc, пирролидинила, фенила или имидазолила.
7. Соединение по п.4 или его соль, где A представляет собой пирролидинил, пиперидинил или морфолинил.
8. Соединение по п.4 или его соль, где A представляет собой фурил, имидазолил, пиразолил или оксазолил.
9. Соединение по любому из пп.1-8 или его соль, где R3 представляет собой метокси.
10. Соединение, представляющее собой любое из следующих соединений:
N-[(3-{[3-{[(5-хлор-2-тиенил)сульфонил]амино}-4-(метилокси)-1H-индазол-1-ил]метил}фенил)метил]ацетамид,
N-[(3-{[3-{[(5-хлор-2-тиенил)сульфонил]амино}-4-(метилокси)-1Н-индазол-1-ил]метил}фенил)метил]пропанамид,
N-[(3-{[3-{[(5-хлор-2-тиенил)сульфонил]амино}-4-(метилокси)-1Н-индазол-1-ил]метил}фенил)метил]-2-метилпропанамид,
N-[(3-{[3-{[(5-хлор-2-тиенил)сульфонил]амино}-4-(метилокси)-1Н-индазол-1-ил]метил}фенил)метил]-2,2-диметилпропанамид,
5-хлор-N-[1-({3-[(формиламино)метил]фенил}метил)-4-(метилокси)-1Н-индазол-3-ил]-2-тиофенсульфонамид,
трифторацетат N-[(3-{[3-{[(5-хлор-2-тиенил)сульфонил]амино}-4-(метилокси)-1Н-индазол-1-ил]метил}фенил)метил]-3-морфолинкарбоксамида,
гидрохлорид (3R)-N-[(3-{[3-{[(5-хлор-2-тиенил)сульфонил]амино}-4-(метилокси)-1Н-индазол-1-ил]метил}фенил)метил]-3-морфолинкарбоксамида,
гидрохлорид (3S)-N-[(3-{[3-{[(5-хлор-2-тиенил)сульфонил]амино}-4-(метилокси)-1Н-индазол-1-ил]метил}фенил)метил]-3-морфолинкарбоксамида,
соль трифторацетат (2R)-N-[(3-{[3-{[(5-хлор-2-тиенил)сульфонил]амино}-4-(метилокси)-1Н-индазол-1-ил]метил}фенил)метил]-2-пиперидинкарбоксамида,
гидрохлорид N1-[(3-{[3-{[(5-хлор-2-тиенил)сульфонил]амино}-4-(метилокси)-1Н-индазол-1-ил]метил}фенил)метил]-N2-метил-D-аланинамида,
гидрохлорид 3-амино-N-[(3-{[3-{[(5-хлор-2-тиенил)сульфонил]амино}-4-(метилокси)-1Н-индазол-1-ил]метил}фенил)метил]-2,2-диметилпропанамида,
гидрохлорид N1-[(3-{[3-{[(5-хлор-2-тиенил)сульфонил]амино}-4-(метилокси)-1Н-индазол-1-ил]метил}фенил)метил]-N2-метилглицинамида,
N-[(3-{[3-{[(5-хлор-2-тиенил)сульфонил]амино}-4-(метилокси)-1Н-индазол-1-ил]метил}фенил)метил]-2-метил-D-пролинамид,
N-[(3-{[3-{[(5-хлор-2-тиенил)сульфонил]амино}-4-(метилокси)-1H-индазол-1-ил]метил}фенил)метил]-2-метил-2-пиперидинкарбоксамид,
гидрохлорид N-[(3-{[3-{[(5-хлор-2-тиенил)сульфонил]амино}-4-(метилокси)-1Н-индазол-1-ил]метил}фенил)метил]-1-(метиламино)циклопропанкарбоксамида,
гидрохлорид N-[(3-{[3-{[(5-хлор-2-тиенил)сульфонил]амино}-4-(метилокси)-1Н-индазол-1-ил]метил}фенил)метил]-2-пиперазинкарбоксамида,
трифторацетат N1-[(3-{[3-{[(5-хлор-2-тиенил)сульфонил]амино}-4-(метилокси)-1H-индазол-1-ил]метил}фенил)метил]-N2-метил-1-аланинамида,
трифторацетат N1-[(3-{[3-{[(5-хлор-2-тиенил)сульфонил]амино}-4-(метилокси)-1Н-индазол-1-ил]метил}фенил)метил]-L-аланинамида,
трифторацетат N1-[(3-{[3-{[(5-хлор-2-тиенил)сульфонил]амино}-4-(метилокси)-1Н-индазол-1-ил]метил}фенил)метил]-D-аланинамида,
трифторацетат N1-[(3-{[3-{[(5-хлор-2-тиенил)сульфонил]амино}-4-(метилокси)-1Н-индазол-1-ил]метил}фенил)метил]глицинамида,
N-[(3-{[3-{[(5-хлор-2-тиенил)сульфонил]амино}-4-(метилокси)-1Н-индазол-1-ил]метил}фенил)метил]пропандиамид,
трифторацетат N4-[(3-{[3-{[(5-хлор-2-тиенил)сульфонил]амино}-4-(метилокси)-1Н-индазол-1-ил]метил}фенил)метил]-N1-метил-L-аспартамида,
трифторацетат метил-N4-[(3-{[3-{[(5-хлор-2-тиенил)сульфонил]амино}-4-(метилокси)-1Н-индазол-1-ил]метил}фенил)метил]аспарагината,
трифторацетат N-[(3-{[3-{[(5-хлор-2-тиенил)сульфонил]амино}-4-(метилокси)-1Н-индазол-1-ил]метил}фенил)метил]-2-(2-пирролидинил)ацетамида,
трифторацетат 3-амино-N-[(3-{[3-{[(5-хлор-2-тиенил)сульфонил]амино}-4-(метилокси)-1Н-индазол-1-ил]метил}фенил)метил]бутанамида,
трифторацетат 3-амино-N-[(3-{[3-{[(5-хлор-2-тиенил)сульфонил]амино}-4-(метилокси)-1Н-индазол-1-ил]метил}фенил)метил]-2-метилпропанамида,
трифторацетат N-[(3-{[3-{[(5-хлор-2-тиенил)сульфонил]амино}-4-(метилокси)-1Н-индазол-1-ил]метил}фенил)метил]-2-метил-L-пролинамида,
трифторацетат (4S)-N-[(3-{[3-{[(5-хлор-2-тиенил)сульфонил]амино}-4-(метилокси)-1Н-индазол-1-ил]метил}фенил)метил]-4-фтор-L-пролинамида,
трифторацетат N1-[(3-{[3-{[(5-хлор-2-тиенил)сульфонил]амино}-4-(метилокси)-1Н-индазол-1-ил]метил}фенил)метил]-N3-метил-β-аланинамида,
N-[(3-{[3-{[(5-хлор-2-тиенил)сульфонил]амино}-4-(метилокси)-1Н-индазол-1-ил]метил}фенил)метил]-2-(метилокси)ацетамид,
трифторацетат N1-[(3-{[3-{[(5-хлор-2-тиенил)сульфонил]амино}-4-(метилокси)-1Н-индазол-1-ил]метил}фенил)метил]-N2,N2-диметилаланинамида,
N-[(3-{[3-{[(5-хлор-2-тиенил)сульфонил]амино}-4-(метилокси)-1Н-индазол-1-ил]метил}фенил)метил]-2-гидроксиацетамид,
N-[(3-{[3-{[(5-хлор-2-тиенил)сульфонил]амино}-4-(метилокси)-1Н-индазол-1-ил]метил}фенил)метил]-1Н-пиразол-4-карбоксамид,
трифторацетат N1-[(3-{[3-{[(5-хлор-2-тиенил)сульфонил]амино}-4-(метилокси)-1Н-индазол-1-ил]метил}фенил)метил]-N2,N2,2-триметилаланинамида,
N-[(3-{[3-{[(5-хлор-2-тиенил)сульфонил]амино}-4-(метилокси)-1Н-индазол-1-ил]метил}фенил)метил]тетрагидро-3-фуранкарбоксамид,
трифторацетат N1-[(3-{[3-{[(5-хлор-2-тиенил)сульфонил]амино}-4-(метилокси)-1Н-индазол-1-ил]метил}фенил)метил]-N2-метил-N2-[2-(метилокси)этил]глицинамида,
N-[(3-{[3-{[(5-хлор-2-тиенил)сульфонил]амино}-4-(метилокси)-1Н-индазол-1-ил]метил}фенил)метил]-2-гидроксипропанамид,
N-[(3-{[3-{[(5-хлор-2-тиенил)сульфонил]амино}-4-(метилокси)-1Н-индазол-1-ил]метил}фенил)метил]-1,3-оксазол-5-карбоксамид,
трифторацетат N-[(3-{[3-{[(5-хлор-2-тиенил)сульфонил]амино}-4-(метилокси)-1Н-индазол-1-ил]метил}фенил)метил]-1-метил-L-пролинамида,
трифторацетат N-[(3-{[3-{[(5-хлор-2-тиенил)сульфонил]амино}-4-(метилокси)-1Н-индазол-1-ил]метил}фенил)метил]-1-метил-D-пролинамида,
N-[(3-{[3-{[(5-хлор-2-тиенил)сульфонил]амино}-4-(метилокси)-1Н-индазол-1-ил]метил}фенил)метил]-3-фуранкарбоксамид,
трифторацетат N-[(3-{[3-{[(5-хлор-2-тиенил)сульфонил]амино}-4-(метилокси)-1Н-индазол-1-ил]метил}фенил)метил]-1-метил-2-пиперидинкарбоксамида,
N-[(3-{[3-{[(5-хлор-2-тиенил)сульфонил]амино}-4-(метилокси)-1Н-индазол-1-ил]метил}фенил)метил]тетрагидро-2Н-пиран-4-карбоксамид,
N-[(3-{[3-{[(5-хлор-2-тиенил)сульфонил]амино}-4-(метилокси)-1Н-индазол-1-ил]метил}фенил)метил]-L-гистидинамид,
N1-[(3-{[3-{[(5-хлор-2-тиенил)сульфонил]амино}-4-(метилокси)-1Н-индазол-1-ил]метил}фенил)метил]-D-лейцинамид,
N1-[(3-{[3-{[(5-хлор-2-тиенил)сульфонил]амино}-4-(метилокси)-1H-индазол-1-ил]метил}фенил)метил]-D-аллоизолейцинамид,
N-[(3-{[3-{[(5-хлор-2-тиенил)сульфонил]амино}-4-(метилокси)-1H-индазол-1-ил]метил}фенил)метил]-L-фенилаланинамид,
N1-[(3-{[3-{[(5-хлор-2-тиенил)сульфонил]амино}-4-(метилокси)-1Н-индазол-1-ил]метил}фенил)метил]-D-валинамид,
N1-[(3-{[3-{[(5-хлор-2-тиенил)сульфонил]амино}-4-(метилокси)-1H-индазол-1-ил]метил}фенил)метил]-L-лизинамид,
трифторацетат (2R)-N-[(3-{[3-{[(5-хлор-2-тиенил)сульфонил]амино}-4-(метилокси)-1Н-индазол-1-ил]метил}фенил)метил]-2-азетидинкарбоксамида,
трифторацетат (2S)-N-[(3-{[3-{[(5-хлор-2-тиенил)сульфонил]амино}-4-(метилокси)-1Н-индазол-1-ил]метил}фенил)метил]-2-пиперидинкарбоксамида,
трифторацетат (4R)-N-[(3-{[3-{[(5-хлор-2-тиенил)сульфонил]амино}-4-(метилокси)-1Н-индазол-1-ил]метил}фенил)метил]-4-фтор-1-пролинамида,
N-[1-{[3,4-бис(метилокси)фенил]метил}-4-(метилокси)-1Н-индазол-3-ил]-5-хлор-2-тиофенсульфонамид,
трифторацетат N1-[(3-{[3-{[(5-хлор-2-тиенил)сульфонил]амино}-4-(метилокси)-1Н-индазол-1-ил]метил}фенил)метил]-О-метилсеринамида,
трифторацетат N-[(3-{[3-{[(5-хлор-2-тиенил)сульфонил]амино}-4-(метилокси)-1Н-индазол-1-ил]метил}фенил)метил]-4,4-дифтор-D-пролинамида,
формиат N1-[(3-{[3-{[(5-хлор-2-тиенил)сульфонил]амино}-4-(метилокси)-1Н-индазол-1-ил]метил}фенил)метил]-N2,2-диметилаланинамида,
N-[(3-{[3-{[(5-хлор-2-тиенил)сульфонил]амино}-4-(метилокси)-1Н-индазол-1-ил]метил}фенил)метил]-2-гидрокси-2-метилпропанамид,
5-хлор-N-{4-(метилокси)-1-[(3-метилсульфонил)амино]метил}фенил)метил]-1H-индазол-3-ил}-2-тиофенсульфонамид,
N-[1-[(3-{[(аминокарбонил)амино]метил}фенил)метил]-4-(метилокси)-1Н-индазол-3-ил]-5-хлор-2-тиофенсульфонамид,
5-хлор-N-[1-({3-[({[(1,1-диметилэтил)амино]карбонил}амино)метил]фенил}метил)-4-(метилокси)-1Н-индазол-3-ил]-2-тиофенсульфонамид,
5-хлор-N-[1-({3-[({[(1-метилэтил)амино]карбонил}амино)метил]фенил}метил)-4-(метилокси)-1Н-индазол-3-ил]-2-тиофенсульфонамид,
N-[1-{[4-(аминосульфонил)фенил]метил}-4-(метилокси)-1Н-индазол-3-ил]-5-хлор-2-тиофенсульфонамид,
2-(3-{[3-{[(5-хлор-2-тиенил)сульфонил]амино}-4-(метилокси)-1Н-индазол-1-ил]метил}фенил)-N-метилацетамид,
3-{[3-{[(5-хлор-2-тиенил)сульфонил]амино}-4-(метилокси)-1H-индазол-1-ил]метил}бензамид,
3-{[3-{[(5-хлор-2-тиенил)сульфонил]амино}-4-(метилокси)-1Н-индазол-1-ил]метил}-N-[2-(диметиламино)этил]бензамид,
3-[(4-(метилокси)-3-{[(5-метил-2-тиенил)сульфонил]амино}-1Н-индазол-1-ил)метил]бензамид,
3-{[3-{[(5-бром-2-тиенил)сульфонил]амино}-4-(метилокси)-1Н-индазол-1-ил]метил}бензамид,
3-{[3-{[(5-хлор-2-тиенил)сульфонил]амино}-7-фтор-4-(метилокси)-1Н-индазол-1-ил]метил}бензамид,
3-[(3-{[(5-хлор-2-тиенил)сульфонил]амино}-7-фтор-4-гидрокси-1Н-индазол-1-ил)метил]бензамид,
3-[(3-{[(5-хлор-2-тиенил)сульфонил]амино}-4-гидрокси-1Н-индазол-1-ил)метил]бензамид,
соль N1-({3-[(3-{[(5-хлор-2-тиенил)сульфонил]амино}-4-гидрокси-1Н-индазол-1-ил)метил]фенил}метил)-N2-метил-D-аланинамида с муравьиной кислотой,
N-({3-[(3-{[(5-хлор-2-тиенил)сульфонил]амино}-4-гидрокси-1H-индазол-1-ил)метил]фенил}метил)ацетамид,
формиатная соль N-({3-[(3-{[(5-хлор-2-тиенил)сульфонил]амино}-4-гидрокси-1Н-индазол-1-ил)метил]фенил}метил)-3-морфолинкарбоксамида,
5-хлор-N-[1-[(3-{[2-(диметиламино)этил]окси}фенил)метил]-4-(метилокси)-1Н-индазол-3-ил]-2-тиофенсульфонамид,
5-хлор-N-[1-[(4-{[2-(диметиламино)этил]окси}фенил)метил]-4-(метилокси)-1Н-индазол-3-ил]-2-тиофенсульфонамид,
5-хлор-N-[1-{[4-{[2-(диметиламино)этил]окси}-3-(метилокси)фенил]метил}-4-(метилокси)-1Н-индазол-3-ил]-2-тиофенсульфонамид,
N-[1-{[3-(аминосульфонил)фенил]метил}-4-(метилокси)-1H-индазол-3-ил]-5-хлор-2-тиофенсульфонамид,
трифторацетат N1-[(3-{[3-{[(5-хлор-2-тиенил)сульфонил]амино}-4-(метилокси)-1Н-индазол-1-ил]метил}фенил)метил]-β-аланинамида,
соль формиат N1-[(3-{[3-{[(5-хлор-2-тиенил)сульфонил]амино}-4-(метилокси)-1Н-индазол-1-ил]метил}фенил)метил]-L-глутамамида,
трифторацетат N4-[(3-{[3-{[(5-хлор-2-тиенил)сульфонил]амино}-4-(метилокси)-1Н-индазол-1-ил]метил}фенил)метил]-L-аспарагина,
трифторацетат 1,1-диметилэтил-N4-[(3-{[3-{[(5-хлор-2-тиенил)сульфонил]амино}-4-(метилокси)-1Н-индазол-1-ил]метил}фенил)метил]-L-аспарагината,
трифторацетат N-[(3-{[3-{[(5-хлор-2-тиенил)сульфонил]амино}-4-(метилокси)-1Н-индазол-1-ил]метил}фенил)метил]-D-пролинамида,
трифторацетат 1-амино-N-[(3-{[3-{[(5-хлор-2-тиенил)сульфонил]амино}-4-(метилокси)-1Н-индазол-1-ил]метил}фенил)метил]циклобутанкарбоксамида,
трифторацетат 1-амино-N-[(3-{[3-{[(5-хлор-2-тиенил)сульфонил]амино}-4-(метилокси)-1Н-индазол-1-ил]метил}фенил)метил]циклопропанкарбоксамида,
трифторацетат N1-[(3-{[3-{[(5-хлор-2-тиенил)сульфонил]амино}-4-(метилокси)-1Н-индазол-1-ил]метил}фенил)метил]-N2-метилглицинамида,
трифторацетат N1-[(3-{[3-{[(5-хлор-2-тиенил)сульфонил]амино}-4-(метилокси)-1Н-индазол-1-ил]метил}фенил)метил]-N2,N2-диметилглицинамида,
N-[(3-{[3-{[(5-хлор-2-тиенил)сульфонил]амино}-4-(метилокси)-1Н-индазол-1-ил]метил}фенил)метил]-3-(метилокси)пропанамид,
N-[(3-{[3-{[(5-хлор-2-тиенил)сульфонил]амино}-4-(метилокси)-1Н-индазол-1-ил]метил}фенил)метил]-1H-пиррол-3-карбоксамид,
N-[(3-{[3-{[(5-хлор-2-тиенил)сульфонил]амино}-4-(метилокси)-1H-индазол-1-ил]метил}фенил)метил]тетрагидро-2-фуранкарбоксамид,
N-[(3-{[3-{[(5-хлор-2-тиенил)сульфонил]амино}-4-(метилокси)-1Н-индазол-1-ил]метил}фенил)метил]-1,3-оксазол-4-карбоксамид,
N-[(3-{[3-{[(5-хлор-2-тиенил)сульфонил]амино}-4-(метилокси)-1Н-индазол-1-ил]метил}фенил)метил]-1Н-пиразол-3-карбоксамид,
N-[(3-{[3-{[(5-хлор-2-тиенил)сульфонил]амино}-4-(метилокси)-1Н-индазол-1-ил]метил}фенил)метил]-1Н-пиррол-2-карбоксамид,
N-[(3-{[3-{[(5-хлор-2-тиенил)сульфонил]амино}-4-(метилокси)-1Н-индазол-1-ил]метил}фенил)метил]-1Н-имидазол-2-карбоксамид,
N-[(3-{[3-{[(5-хлор-2-тиенил)сульфонил]амино}-4-(метилокси)-1H-индазол-1-ил]метил}фенил)метил]-1Н-имидазол-4-карбоксамид,
N-[(3-{[3-{[(5-хлор-2-тиенил)сульфонил]амино}-4-(метилокси)-1H-индазол-1-ил]метил}фенил)метил]-2-фуранкарбоксамид,
N-[(3-{[3-{[(5-хлор-2-тиенил)сульфонил]амино}-4-(метилокси)-1Н-индазол-1-ил]метил}фенил)метил]-4-метил-3-морфолинкарбоксамид,
N-[(4-{[3-{[(5-хлор-2-тиенил)сульфонил]амино}-4-(метилокси)-1H-индазол-1-ил]метил}фенил)метил]ацетамид,
трифторацетат N-[(4-{[3-{[(5-хлор-2-тиенил)сульфонил]амино}-4-(метилокси)-1Н-индазол-1-ил]метил}фенил)метил]-3-морфолинкарбоксамида,
трифторацетат N1-[(4-{[3-{[(5-хлор-2-тиенил)сульфонил]амино}-4-(метилокси)-1Н-индазол-1-ил]метил}фенил)метил]-N2-метил-D-аланинамида,
3-{[3-{[(5-хлор-2-тиенил)сульфонил]амино}-4-(метилокси)-1Н-индазол-1-ил]метил}-N-метилбензамид,
3-{[3-{[(5-хлор-2-тиенил)сульфонил]амино}-4-(метилокси)-1Н-индазол-1-ил]метил}-N,N-диметилбензамид,
N1-[(3-{[3-{[(5-хлор-2-тиенил)сульфонил]амино}-4-(метилокси)-1Н-индазол-1-ил]метил}фенил)метил]-L-серинамид,
4-{[3-{[(5-хлор-2-тиенил)сульфонил]амино}-4-(метилокси)-1Н-индазол-1-ил]метил}бензамид,
5-хлор-N-(4-(метилокси)-1-{[4-(метилокси)фенил]метил}-1H-индазол-3-ил)-2-тиофенсульфонамид,
5-хлор-N-(4-(метилокси)-1-{[2-(метилокси)фенил]метил}-1Н-индазол-3-ил)-2-тиофенсульфонамид,
5-хлор-N-[1-({3-[(метиламино)сульфонил]фенил}метил)-4-(метилокси)-1Н-индазол-3-ил]-2-тиофенсульфонамид,
5-хлор-N-[1-({3-[(диметиламино)сульфонил]фенил}метил)-4-(метилокси)-1Н-индазол-3-ил]-2-тиофенсульфонамид,
3-({4-(метилокси)-3-[(2-тиенилсульфонил)амино]-1Н-индазол-1-ил}метил)бензамид,
3-{[3-{[(4,5-дихлор-2-тиенил)сульфонил]амино}-4-(метилокси)-1Н-индазол-1-ил]метил}бензамид,
5-хлор-N-[1-{[3-{[2-(диметиламино)этил]окси}-4-(метилокси)фенил]метил}-4-(метилокси)-1Н-индазол-3-ил]-2-тиофенсульфонамид,
формиатная соль (3R)-N-({3-[(3-{[(5-хлор-2-тиенил)сульфонил]амино}-4-гидрокси-1Н-индазол-1-ил)метил]фенил}метил)-3-морфолинкарбоксамида,
формиатная соль N1-({3-[(3-{[(5-хлор-2-тиенил)сульфонил]амино}-4-гидрокси-1H-индазол-1-ил)метил]фенил}метил)-N2-метилглицинамида,
5-хлор-N-[1-{[3-({[(этиламино)карбонил]амино}метил)фенил]метил}-4-(метилокси)-1Н-индазол-3-ил]-2-тиофенсульфонамид,
N-[(3-{[3-{[(5-хлор-2-тиенил)сульфонил]амино}-4-(метилокси)-1Н-индазол-1-ил]метил}фенил)метил]-1-триптофанамид,
формиатная соль 1-[(3-{[3-{[(5-хлор-2-тиенил)сульфонил]амино}-4-(метилокси)-1Н-индазол-1-ил]метил}фенил)метил]-D-аллотреонинамида,
N-{[3-({4-(метилокси)-3-[(2-тиенилсульфонил)амино]-1Н-индазол-1-ил}метил)фенил]метил}ацетамид,
4-[(4-(метилокси)-3-{[(5-метил-2-тиенил)сульфонил]амино}-1Н-индазол-1-ил)метил]бензамид,
N-[1-{[3,4-бис-(метилокси)фенил]метил}-4-(метилокси)-1H-индазол-3-ил]-5-метил-2-тиофенсульфонамид,
N-({3-[(4-(метилокси)-3-{[(5-метил-2-тиенил)сульфонил]амино}-1Н-индазол-1-ил)метил]фенил}метил)ацетамид,
трифторацетат (3R)-N-({4-[(4-(метилокси)-3-{[(5-метил-2-тиенил)сульфонил]амино}-1Н-индазол-1-ил)метил]фенил}метил)-3-морфолинкарбоксамида,
N-({3-[(3-{[(5-хлор-2-тиенил)сульфонил]амино}-4-фтор-1Н-индазол-1-ил)метил]фенил}метил)ацетамид,
N-({3-[(4-хлор-3-{[(5-хлор-2-тиенил)сульфонил]амино}-1Н-индазол-1-ил)метил]фенил}метил)ацетамид,
N-[(3-{[3-{[(5-хлор-2-тиенил)сульфонил]амино}-4-(метилокси)-1Н-индазол-1-ил]метил}фенил)метил]-4-метил-3-морфолинкарбоксамид,
N-[(3-{[3-{[(5-хлор-2-тиенил)сульфонил]амино}-4-(метилокси)-1Н-индазол-1-ил]метил}фенил)метил]-4-метил-3-морфолинкарбоксамид, энантиомер 1,
N-[(3-{[3-{[(5-хлор-2-тиенил)сульфонил]амино}-4-(метилокси)-1Н-индазол-1-ил]метил}фенил)метил]-4-метил-3-морфолинкарбоксамид, энантиомер 2,
(2S)-N-[(3-{[3-{[(5-хлор-2-тиенил)сульфонил]амино}-4-(метилокси)-1Н-индазол-1-ил]метил}фенил)метил]-2-гидроксипропанамид, S-энантиомер,
N1-[(3-{[3-{[(5-хлор-2-тиенил)сульфонил]амино}-4-(метилокси)-1H-индазол-1-ил]метил}фенил)метил]-2-метилаланинамид,
N-[(3-{[3-{[(5-хлор-2-тиенил)сульфонил]амино}-7-фтор-4-(метилокси)-1Н-индазол-1-ил]метил}фенил)метил]ацетамид,
N-[(3-{[3-{[(5-хлор-2-тиенил)сульфонил]амино}-6-фтор-4-(метилокси)-1Н-индазол-1-ил]метил}фенил)метил]ацетамид,
3-{[3-{[(5-хлор-2-тиенил)сульфонил]амино}-6-фтор-4-(метилокси)-1Н-индазол-1-ил]метил}бензамид,
(3R)-N-[(3-{[3-{[(5-хлор-2-тиенил)сульфонил]амино}-6-фтор-4-(метилокси)-1Н-индазол-1-ил]метил}фенил)метил]-3-морфолинкарбоксамид,
N-[(3-{[3-{[(5-хлор-2-тиенил)сульфонил]амино}-6-фтор-4-(метилокси)-1Н-индазол-1-ил]метил}фенил)метил]-2-гидрокси-2-метилпропанамид,
N-[(3-{[3-{[(5-хлор-2-тиенил)сульфонил]амино}-6-фтор-4-(метилокси)-1Н-индазол-1-ил]метил}фенил)метил]-4-метил-3-морфолинкарбоксамид,
N-[(3-{[3-{[(5-хлор-2-тиенил)сульфонил]амино}-5-фтор-4-(метилокси)-1H-индазол-1-ил]метил}фенил)метил]ацетамид,
(3S)-N-[(3-{[3-{[(5-хлор-2-тиенил)сульфонил]амино}-5-фтор-4-(метилокси)-1Н-индазол-1-ил]метил}фенил)метил]-3-морфолинкарбоксамид,
(3R)-N-[(3-{[3-{[(5-хлор-2-тиенил)сульфонил]амино}-5-фтор-4-(метилокси)-1Н-индазол-1-ил]метил}фенил)метил]-3-морфолинкарбоксамид,
N-[(3-{[3-{[(5-хлор-2-тиенил)сульфонил]амино}-5-фтор-4-(метилокси)-1Н-индазол-1-ил]метил}фенил)метил]-2-гидрокси-2-метилпропанамид,
4-{[(3-{[3-{[(5-хлор-2-тиенил)сульфонил]амино}-4-(метилокси)-1Н-индазол-1-ил]метил}фенил)метил]амино}-4-оксобутановая кислота,
5-{[(3-{[3-{[(5-хлор-2-тиенил)сульфонил]амино}-4-(метилокси)-1Н-индазол-1 -ил]метил}фенил)метил]амино}-5-оксопентановая кислота,
6-{[(3-{[3-{[(5-хлор-2-тиенил)сульфонил]амино}-4-(метилокси)-1Н-индазол-1-ил]метил}фенил)метил]амино}-6-оксогексановая кислота,
3-({[(3-{[3-{[(5-хлор-2-тиенил)сульфонил]амино}-4-(метилокси)-1Н-индазол-1-ил]метил}фенил)метил]амино}карбонил)бензойная кислота,
4-({[(3-{[3-{[(5-хлор-2-тиенил)сульфонил]амино}-4-(метилокси)-1H-индазол-1-ил]метил}фенил)метил]амино}карбонил)бензойная кислота,
транс-4-({[(3-{[3-{[(5-хлор-2-тиенил)сульфонил]амино}-4-(метилокси)-1H-индазол-1-ил]метил}фенил)метил]амино}карбонил)циклогексанкарбоновая кислота,
8-{[(3-{[3-{[(5-хлор-2-тиенил)сульфонил]амино}-4-(метилокси)-1Н-индазол-1-ил]метил}фенил)метил]амино}-8-оксооктановая кислота,
9-{[(3-{[3-{[(5-хлор-2-тиенил)сульфонил]амино}-4-(метилокси)-1Н-индазол-1-ил]метил}фенил)метил]амино}-9-оксононановая кислота,
2-({[(3-{[3-{[(5-хлор-2-тиенил)сульфонил]амино}-4-(метилокси)-1Н-индазол-1-ил]метил}фенил)метил]амино}карбонил)бензойная кислота,
N-[(3-{[3-{[(5-хлор-2-тиенил)сульфонил]амино}-4-(дифторметил)-1Н-индазол-1-ил]метил}фенил)метил]ацетамид,
N-[(3-{[3-{[(5-хлор-2-тиенил)сульфонил]амино}-4-(гидроксиметил)-1Н-индазол-1-ил]метил}фенил)метил]ацетамид,
N-[(3-{[3-{[(5-хлор-2-тиенил)сульфонил]амино}-4-(1-гидроксиэтил)-1H-индазол-1-ил]метил}фенил)метил]ацетамид,
N-[(3-{[3-{[(5-хлор-2-тиенил)сульфонил]амино}-4-(1-гидрокси-1-метилэтил)-1Н-индазол-1-ил]метил}фенил)метил],
N-[(3-{[3-{[(5-хлор-2-тиенил)сульфонил]амино}-4-(1-гидроксиэтил)-1Н-индазол-1-ил]метил}фенил)метил]-2-гидрокси-2-метилпропанамид,
формиатная соль (3R)-N-[(3-{[3-{[(5-хлор-2-тиенил)сульфонил]амино}-4-(1-гидроксиэтил)-1Н-индазол-1-ил]метил}фенил)метил]-3-морфолинкарбоксамида,
формиатная соль (3S)-N-[(3-{[3-{[(5-хлор-2-тиенил)сульфонил]амино}-4-(1-гидроксиэтил)-1Н-индазол-1-ил]метил}фенил)метил]-3-морфолинкарбоксамида,
N-[(3-{[3-{[(5-хлор-2-тиенил)сульфонил]амино}-4-(1-гидрокси-1-метилэтил)-1Н-индазол-1-ил]метил}фенил)метил]-2-гидрокси-2-метилпропанамид,
формиатная соль (3R)-N-[(3-{[3-{[(5-хлор-2-тиенил)сульфонил]амино}-4-(1-гидрокси-1-метилэтил)-1Н-индазол-1-ил]метил}фенил)метил]-3-морфолинкарбоксамида,
(3S)-N-[(3-{[3-{[(5-хлор-2-тиенил)сульфонил]амино}-4-(1-гидрокси-1-метилэтил)-1Н-индазол-1-ил]метил}фенил)метил]-3-морфолинкарбоксамид,
трифторацетат 1-[(3-{[3-{[(5-хлор-2-тиенил)сульфонил]амино}-4-(метилокси)-1Н-индазол-1-ил]метил}фенил)метил]-D-серинамида,
трифторацетат N-[(3-{[3-{[(5-хлор-2-тиенил)сульфонил]амино}-4-(метилокси)-1Н-индазол-1-ил]метил}фенил)метил]-L-пролинамида,
трифторацетат (2S)-N-[(3-{[3-{[(5-хлор-2-тиенил)сульфонил]амино}-4-(метилокси)-1Н-индазол-1-ил]метил}фенил)метил]-2-азетидинкарбоксамида,
(3R)-N-({3-[(4-(метилокси)-3-{[(5-метил-2-тиенил)сульфонил]амино}-1Н-индазол-1-ил)метил]фенил}метил)-3-морфолинкарбоксамид,
N-[(3-{[3-{[(5-хлор-2-тиенил)сульфонил]амино}-5-фтор-4-(метилокси)-1Н-индазол-1-ил]метил}фенил)метил]-4-метил-3-морфолинкарбоксамид, энантиомер 1,
N-[(3-{[3-{[(5-хлор-2-тиенил)сульфонил]амино}-5-фтор-4-(метилокси)-1Н-индазол-1-ил]метил}фенил)метил]-4-метил-3-морфолинкарбоксамид, энантиомер 2,
4-{[3-{[(5-хлор-2-тиенил)сульфонил]амино}-4-(метилокси)-1Н-индазол-1-ил]метил}-N,N-диэтилбензамид,
5-хлор-N-(4-(метилокси)-1-{[3-(метилокси)фенил]метил}-1Н-индазол-3-ил)-2-тиофенсульфонамид,
N-[1-{[3-фтор-4-(метилокси)фенил]метил}-4-(метилокси)-1Н-индазол-3-ил]-5-метил-2-тиофенсульфонамид,
формиатная соль (3S)-N-({3-[(3-{[(5-хлор-2-тиенил)сульфонил]амино}-4-гидрокси-1H-индазол-1-ил)метил]фенил}метил)-3-морфолинкарбоксамида,
или его соль.
11. Соединение, представляющее собой N-[(3-{[3-{[(5-хлор-2-тиенил)сульфонил]амино}-4-(метилокси)-1Н-индазол-1-ил]метил}фенил)метил]-2-гидрокси-2-метилпропанамид

12. Фармацевтическая композиция, содержащая соединение по любому из пп.1-11 или его фармацевтически приемлемую соль и один или более фармацевтически приемлемых носителей, разбавителей и эксципиентов.
13. Применение соединения по любому из пп.1-11 или его фармацевтически приемлемой соли для лечения астмы.
14. Применение соединения по любому из пп.1-11 или его фармацевтически приемлемой соли в лечении заболевания или состояния, при котором показан антагонист рецептора CCR4.
15. Применение соединения по любому из пп.1-11 или его фармацевтически приемлемой соли в изготовлении лекарственного средства для лечения заболевания или состояния, при котором показан антагонист рецептора CCR4.
16. Способ лечения заболевания или состояния, при котором показан антагонист рецептора CCR4, у субъекта, нуждающегося в таком лечении, включающий введение терапевтически эффективного количества соединения по любому из пп.1-11 или его фармацевтически приемлемой соли.
Текст
ПРОИЗВОДНЫЕ ПИРАЗОЛА, ИСПОЛЬЗУЕМЫЕ В КАЧЕСТВЕ АНТАГОНИСТОВ РЕЦЕПТОРА CCR4 Индазольные соединения, способы их получения, промежуточные соединения, используемые в этих способах, фармацевтические композиции, содержащим такие соединения, и их применение в терапии в качестве антагонистов рецептора CCR4.(71)(73) Заявитель и патентовладелец: ГЛАКСО ГРУП ЛИМИТЕД (GB) Область изобретения Настоящее изобретение относится к индазольным соединениям, к способам их получения, к промежуточным соединениям, используемым в этих способах, к фармацевтическим композициям, содержащим такие соединения, и к их применению в терапии. Предшествующий уровень техники Считается, что хемокины играют важную роль в иммунных и воспалительных ответах при ряде заболеваний или состояний. СС-хемокиновый рецептор 4 (далее CCR4) первоначально был клонирован из базофильной клеточной линии (Power et al., J. Biol. Chem. 270:19495, 1995). Антагонисты рецептораCCR4, имеющие небольшую молекулу, известны в данной области, и примеры таких антагонистов приведены в Andrews et al., Mol. Pharmacol. 73:855, 2008. Краткое изложение сущности изобретения В первом аспекте настоящего изобретения предложено соединение формулы (I) или его соль, более конкретно - соединение формулы (I) или его фармацевтически приемлемая соль: Во втором аспекте настоящего изобретения предложена фармацевтическая композиция, содержащая соединение формулы (I) или его фармацевтически приемлемую соль и один или более фармацевтически приемлемых носителей, разбавителей и эксципиентов. В третьем аспекте настоящего изобретения предложено соединение формулы (I) или его фармацевтически приемлемая соль для применения в терапии, в частности в лечении заболевания или состояния,при котором показан антагонист рецептора CCR4. В четвертом аспекте настоящего изобретения предложен способ лечения заболевания или состояния, при котором показан антагонист рецептора CCR4, у субъекта, нуждающегося в таком лечении,включающий введение терапевтически эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли. В пятом аспекте настоящего изобретения предложено применение соединения формулы (I) или его фармацевтически приемлемой соли в изготовлении лекарственного средства для лечения заболевания или состояния, при котором показан антагонист рецептора CCR4. Подробное описание изобретения Настоящее изобретение относится к соединению формулы (I) или его соли в которой R1 выбран из группы, состоящей из следующих групп:(ii) C1-6 алкила, возможно замещенного одной или более группами -NRaRb, -ORc, -C(O)NRaRb,-C(O)ORc, гетероциклила, фенила или гетероарила;(iii) С 3-7 циклоалкила, возможно замещенного группой -C(O)ORc или -NRaRb;(iv) гетероциклила, возможно замещенного одним или более чем одним C1-6 алкилом;(v) гетероарила, возможно замещенного одним или более чем одним галогеном или C1-6 алкилом;(5) C1-6 алкокси, возможно замещенный группой NRaRb, где Ra, Rb и Rc независимо представляют собой водород или C1-6 алкил;R3 представляет собой галоген, CF3, гидрокси, C1-6 алкокси, CRdReOH или CHF2, где Rd и Re независимо представляют собой водород или метил;R5 и R6 независимо представляют собой водород, галоген или C1-6 алкил;n означает 0 или 1. В одном из воплощений изобретение относится к соединению формулы (I) или его соли где R1 выбран из группы, состоящей из следующих групп:(ii) C1-6 алкила, возможно замещенного одной или более группами -NRaRb, -ORc, -C(O)NRaRb,-C(O)ORc, гетероциклила, фенила или гетероарила;(iii) С 3-7 циклоалкила, возможно замещенного группой -NRaRb;(iv) гетероциклила, возможно замещенного одним или более C1-6 алкилом;(v) гетероарила, возможно замещенного одним или более галогеном или C1-6 алкилом;(5) C1-6 алкокси, возможно замещенный группой NRaRb, гдеRa, Rb и Rc независимо представляют собой водород или C1-6 алкил;R5 и R6 независимо представляют собой водород, галоген или C1-6 алкил;-2 021740 В одном из воплощений группа R1 замещена в мета-положении (положение 3). В одном из воплощений R1 представляет собой группу формулы (а), где A представляет собой С 1-6 алкил, такой как метил, этил, изопропил или трет-бутил. В другом воплощении R1 представляет собой группу формулы (а), где A замещен одной или более группами -NRaRb (например, NH2, NHMe или NMe2), -ORc (например, OH или OMe), -C(O)NRaRb (например, C(O)NH2), -C(O)ORc (например, С(О)OH или С(О)OMe), пирролидинил, фенил или имидазолил. В еще одном воплощении R1 представляет собой группу формулы (а), где A представляет собой гетероциклил, выбранный из группы, состоящей из пирролидинила, пиперидинила и морфолинила. В еще одном воплощении R1 представляет собой группу формулы (а), где A представляет собой гетероарил, выбранный из группы, состоящей из фурила, имидазолила, пиразолила и оксазолила. В одном из воплощений, когда n означает 1, тогда R2 выбран из группы, состоящей из галогена (такого как фтор или хлор), C1-6 алкила (например, метила) или C1-6 алкокси (например, метокси). В одном из воплощений R3 представляет собой галоген (например, фтор), гидрокси или С 1-4 алкокси(например, метокси). В еще одном воплощении R3 представляет собой метокси. В одном из воплощений R4 представляет собой водород или фтор. В одном из воплощений R5 и R6 независимо представляют собой водород, галоген (например, хлор) или C1-6 алкил (например, метил). В еще одном воплощении R5 представляет собой хлор и R6 представляет собой водород. В одном из воплощений Ra, Rb и Rc независимо представляют собой водород или метил. В конкретном воплощении настоящего изобретения предложены соединения формулы (Ia) или их соли где R1 и R3 такие, как определено выше. Для каждого воплощения переменные в общем перечислены выше, для каждой переменной по отдельности данное изобретение охватывает те соединения, в которых несколько воплощений или каждое воплощение в формуле (I) выбраны из каждого из воплощений, перечисленных выше. Таким образом,данное изобретение охватывает все комбинации воплощений для каждой переменной, указанной выше,включая их соли. Конкретные соединения по изобретению включают соединения примеров 1-166, приведенных в данном описании, или их соли. Конкретными соединениями по изобретению являются: или их соли. Если не указано иное, то по всему описанию настоящего изобретения: термин "галоген" использован для описания группы, выбранной из фтора, хлора или брома; термин "C1-6 алкил" использован для описания группы или части группы, содержащей линейную или разветвленную алкильную группу, содержащую от 1 до 6 атомов углерода соответственно; подходящие примеры таких групп включают метил, этил, пропил, изопропил, н-бутил, изобутил, трет-бутил,пентил и гексил; термин "C1-6 алкокси" использован для описания группы или части группы, содержащей линейную или разветвленную алкильную группу, содержащую от 1 до 6 атомов углерода; подходящие примеры включают метокси, этокси, пропокси, изопропокси, н-бутокси, изобутокси, трет-бутокси, пентокси или гексокси; термин "гетероциклил" или "гетероциклильное кольцо" использован для описания насыщенного 4-7-членного моноциклического кольца, содержащего один или два гетероатома, выбранные из азота,кислорода или серы; подходящие примеры включают азетидинил, пирролидинил, пиперидинил, пиперазинил, морфолинил, тетрагидрофуранил и тетрагидропиранил; термин "гетероарил" использован для описания ароматического или бензоконденсированного ароматического кольца, содержащего от 1 до 3 гетероатомов, выбранных из кислорода, азота и серы; подходящие примеры таких ароматических колец включают тиенил, фурил, пирролил, триазолил, имидазолил,оксазолил, тиазолил, оксадиазолил, изотиазолил, изоксазолил, тиадиазолил, пиразолил, пиримидил, пиридазинил, пиразинил и пиридил; подходящие примеры таких бензоконденсированных ароматических колец включают хинолинил, изохинолинил, индолил, бензофурил, бензотиенил, бензимидазалолил, бензоксазолил; термин "С 3-7 циклоалкил" использован для описания неароматического карбоциклического кольца,содержащего по меньшей мере три или максимум семь атомов углерода; примеры С 3-7 циклоалкила включают циклопропил, циклобутил, циклопентил, циклогексил или циклогептил. Следует иметь в виду, что настоящее изобретение охватывает соединения формулы (I) в форме свободного основания и в форме их солей, например в форме их фармацевтически приемлемых солей. В одном из воплощений изобретение относится к соединениям формулы (I) или их фармацевтически приемлемым солям. В связи с их потенциальным применением в медицине соли соединений формулы (I) представляют собой желательно фармацевтически приемлемые соли. Подходящие фармацевтически приемлемые соли могут включать соли присоединения кислоты или основания (см. обзор подходящих солей в Berge et al.,J. Pharm. Sci., 66:1-19 (1977. Типичная фармацевтически приемлемая соль без труда может быть получена с использованием желаемой кислоты или желаемого основания, где как подходит. Полученная соль может быть осаждена из раствора и собрана фильтрованием или может быть выделена выпариванием растворителя. Фармацевтически приемлемая соль присоединения основания может быть образована путем взаимодействия соединения формулы (I) с подходящим неорганическим или органическим основанием (например, триэтиламином, этаноламином, триэтаноламином, холином, аргинином, лизином или гистидином), возможно в подходящем растворителе, с образованием соли присоединения основания, которую обычно выделяют, например, кристаллизацией и фильтрованием. Фармацевтически приемлемые соли присоединения основания включают аммониевые соли, соли щелочных металлов, такие как натриевые и калиевые соли, соли щелочно-земельных металлов, такие как кальциевые и магниевые соли, и соли с органическими основаниями, включая соли с первичными, вторичными и третичными аминами, такими как изопропиламин,диэтиламин,этаноламин,триметиламин,дициклогексиламин иN-метил-D-глюкамин. Фармацевтически приемлемая соль присоединения кислоты может быть образована путем взаимодействия соединения формулы (I) с подходящей неорганической или органической кислотой (такой как бромоводородная, соляная, серная, азотная, фосфорная, янтарная, малеиновая, уксусная, пропионовая,фумаровая, лимонная, винная, молочная, бензойная, салициловая, глутаминовая, аспарагиновая,п-толуолсульфоновая, бензолсульфоновая, метансульфоновая, этансульфоновая, нафталинсульфоновая,например 2-нафталинсульфоновая, или капроновая кислота), возможно в подходящем растворителе, таком как органический растворитель, с получением соли, которую обычно выделяют, например, кристаллизацией и фильтрованием. Фармацевтически приемлемая соль присоединения кислоты соединения формулы (I) может содержать или представлять собой, например, соль гидробромид, гидрохлорид, сульфат, нитрат, фосфат, сукцинат, малеат, ацетат, пропионат, фумарат, цитрат, тартрат, лактат, бензоат, салицилат, глутамат, аспартат, п-толуолсульфонат, бензолсульфонат, метансульфонат, этансульфонат,нафталинсульфонат (например, 2-нафталинсульфонат) или гексаноат. Другие нефармацевтически приемлемые соли, например формиаты, оксалаты или трифторацетаты,могут быть использованы, например, в процессе выделения соединений формулы (I), и они входят в объем данного изобретения. Данное изобретение в своем объеме охватывает все возможные стехиометрические и нестехиометрические формы солей соединений формулы (I) Очевидно, что многие органические соединения могут образовывать комплексы с растворителями,в которых их подвергают взаимодействию или из которых их осаждают или кристаллизуют. Эти комплексы известны как "сольваты". Например, комплекс с водой известен как "гидрат". Растворители с высокими точками кипения и/или способные образовывать водородные связи, такие как вода, ксилол,N-метилпирролидинон, метанол и этанол, могут быть использованы для образования сольватов. Способы идентификации сольватов включают, без ограничения, ЯМР (ядерный магнитный резонанс) и микроанализы. Сольваты соединений формулы (I) входят в объем данного изобретения. Данное изобретение охватывает в своем объеме все возможные стехиометрические и нестехиометрические формы сольватов соединений формулы (I). Соединения формулы (I) могут быть в кристаллической или аморфной форме. Кроме того, некоторые кристаллические формы соединений формулы (I) могут существовать в виде полиморфов, которые входят в объем настоящего изобретения. Полиморфные формы соединений формулы (I) могут быть охарактеризованы и дифференцированы с использованием целого ряда общепринятых аналитических методов, включая, без ограничения, картины дифракции рентгеновских лучей на порошке (ДРЛП), инфракрасные (ИК) спектры, спектры комбинационного рассеяния, дифференциальную сканирующую калориметрию (ДСК), термогравиметрический анализ (ТГА) и твердотельный ядерный магнитный резонанс(ТТЯМР). Некоторые соединения, описанные в данном изобретении, могут содержать один или более хиральных атомов, так что могут быть образованы оптические изомеры, например энантиомеры или диастереоизомеры. Соответственно, настоящее изобретение охватывает все изомеры соединений формулы (I), либо выделенные индивидуальные изомеры, такие как изомеры, по существу, не содержащие другого изомера (т.е. чистые), либо в виде смесей (т.е. рацематы и рацемические смеси). Индивидуальный изомер,выделенный таким образом, что он, по существу, не содержит другой изомер (т.е. чистый), может быть выделен таким образом, что он содержит менее 10%, в частности, менее примерно 1%, например, примерно менее 0,1% другого изомера. Разделение изомеров может быть осуществлено общепринятыми методами, известными специалистам в данной области техники, например фракционной кристаллизацией, хроматографией или ВЭЖХ(высокоэффективная жидкостная хроматография). Некоторые соединения формулы (I) могут существовать в одной из нескольких таутомерных форм. Следует иметь в виду, что настоящее изобретение охватывает все таутомеры соединений формулы (I),будь то индивидуальные таутомеры или их смеси. Из вышесказанного ясно, что в объем изобретения входят сольваты, гидраты, комплексы, изомеры и полиморфные формы соединений формулы (I) и их солей. Соединения по изобретению могут быть получены различными способами, включая стандартные химические способы. Любая определенная выше переменная далее будет иметь определенное выше значение, если не указано иное. Иллюстративные общие способы синтеза изложены ниже, и получение конкретных соединений по изобретению описано в рабочих примерах. Согласно настоящему изобретению предложен также способ получения соединения формулы (I) или его соли, включающий способ, выбранный из (а), (b), (с) или (d), где:(а) включает взаимодействие соединения формулы (II) или его соли где R2-R6 и n такие, как определено в п.1,с соединением формулы (IIIa), (IIIb), (IIIc) или (IIId) или его защищенным производным где A такой, как определено здесь выше; и возможно после этого снятие защиты с полученного продукта;(b) включает взаимодействие соединения формулы (IV) или его защищенного производногоHal представляет собой галоген; и возможно после этого снятие защиты с полученного продукта;(с) включает взаимодействие соединения формулы (VI)(d) включает превращение соединения формулы (I) или его соли в другое соединение формулы (I) или его соль. Способ (а). Соединения формулы (II) и карбоновую кислоту формулы (IIIa) подвергают взаимодействию в условиях образования амида, которые известны специалистам в данной области техники. Такие реакции могут быть проведены в подходящем органическом растворителе (например, в DMF или ацетонитриле) с основанием (например, DIPEA или триэтиламином) в присутствии подходящей активирующей группы(например, HATU или TBTU). Соединения формулы (II) и карбоновую кислоту формулы (IIIa) можно также подвергать взаимодействию в присутствии активирующего реагента, такого как 1-хлор-N,N,2-триметил-1-пропен-1-амин, в подходящем органическом растворителе (например, в THF или дихлорметане) с подходящим основанием(например, DIPEA или триэтиламином). Такая технология описана в Schmidt et al., Synthesis, 1988, 475. Подходящими примерами соединений формулы (IIIb) являются хлорангидриды кислот или ангидриды кислот (т.е. соединения, в которых уходящая группа LG1 представляет собой Cl или OC(O)R). Взаимодействие между соединениями формулы (II) и соединениями формулы (IIIb) обычно проводят в инертном органическом растворителе (таком как тетрагидрофуран, DMF, хлороформ или дихлорметан) при температуре окружающей среды или при более низкой температуре, возможно в присутствии подходящего основания, например органического основания (такого как триэтиламин или диизопропилэтиламин), карбоната щелочного металла (такого как карбонат калия) или гидрокарбоната щелочного металла (такого как гидрокарбонат натрия). Подходящими примерами соединений формулы (IIIc) являются соединения, в которых уходящая группа LG2 представляет собой хлоро. Соединения формулы (II) и соединения формулы (IIIc) обычно подвергают взаимодействию в подходящем растворителе (таком как тетрагидрофуран или дихлорметан) в присутствии подходящего основания (такого как триэтиламин или пиридин). Соединения формулы (II) и соединения формулы (IIId) обычно подвергают взаимодействию в подходящем органическом растворителе (например, в дихлорметане) в присутствии подходящего амина (например, триэтиламина). Очевидно, что для взаимодействия соединения формулы (II) с соединением формулы (III) может быть предпочтительным защитить одну или более функциональных групп соединения формулы (III). Примеры защитных групп и способы их удаления можно найти в Т.W. Greene "Protective Groups inOrganic Synthesis" (3rd edition, J. WileySons, 1999). Подходящие защитные группы для аминогруппы включают ацил (например, ацетил), карбамат (например, 2',2',2'-трихлорэтоксикарбонил, бензилоксикарбонил или трет-бутоксикарбонил) и арилалкил (например, бензил), которые могут быть удалены гидро-6 021740 лизом (например, с использованием кислоты, например соляной кислоты в диоксане или трифторуксусной кислоты в дихлорметане) или восстановлением (например, гидрогенолизом бензильной или бензилоксикарбонильной группы или восстановительным удалением 2',2',2'-трихлорэтоксикарбонильной группы с использованием цинка в уксусной кислоте), где как подходит. Другие подходящие защитные группы для аминогруппы включают трифторацетил (-COCF3), который может быть удален гидролизом,катализируемым основанием. Соединения формулы (II) могут быть получены описанными в данном документе способами. В качестве иллюстрации репрезентативное соединение формулы (II) (т.е. соединение, в котором n означает О, R4 и R6 представляют собой водород, R5 представляет собой хлоро и R3 представляет собой метокси) может быть получено способами, представленными на схеме 1. Схема 1d) 1 М раствор LiAlH4 в диэтиловом эфире, THF, 2 M HCl, MeOH, 77%. Соединения формулы (III) (a)-(d) могут быть приобретены коммерческим путем. Способ (b). Для соединений формулы (Va) подходящей группой Hal является хлор, бром, йод, в частности хлор. Обычно соединение формулы (IV) находится в форме его защищенного производного (например, с использованием силилэфирной группы, такой как -(триметилсилил)этокси)метил (SEM), в качестве защитной группы). Реакция алкилирования между защищенными производными соединений формулы (IV) и соединениями формулы (Va) может быть проведена под действием микроволнового излучения в инертном органическом растворителе (таком как DMF) при температуре окружающей среды или при повышенной температуре, возможно в присутствии подходящего основания, такого как карбонат калия или цезия. Силилэфирная защитная группа может быть удалена из образованного таким образом продукта стандартными способами, такими как реакция с тетра-н-бутиламмонийфторидом (TBAF) в подходящем растворителе, таком как THF. Реакция между соединениями формулы (IV) и (Vb) может быть проведена в условиях реакции Мицунобу, которые известны специалистам в данной области. Соединения формулы (IV) обычно находятся в форме их защищенных производных (например, с использованием сульфонамидной защитной группы). Обычно реакцию проводят с использованием трифенилфосфина с азодикарбоксилатным соединением(таким как TBAD, DIAD или DEAD) в подходящем органическом растворителе (таком как THF илиDMF). Сульфонамидная защитная группа может быть удалена из образованного таким образом продукта путем обработки гидроксидом натрия в метаноле. Соединения формулы (IV) или их защищенные производные могут быть получены способами, описанными в данном изобретении. Соединения формулы (Va) или (Vb) либо коммерчески доступны, либо могут быть получены описанными в данном документе способами. Способ (с). Взаимодействие соединения формулы (VI) с соединением формулы (VII) обычно проводят в подходящем органическом растворителе (таком как пиридин). Соединения формулы (VI) могут быть получены описанными в данном документе способами. В качестве иллюстрации репрезентативное соединение формулы (VI) (т.е. соединение, в котором n означает 0, R4 представляет собой водород, R3 представляет собой метокси и R1 представляет собой C(O)NH2) может быть получено способами, представленными на схеме 2. Схема 2 Подходящие соединения формулы (VII), например 5-хлор-2-тиофенсульфонилхлорид,5-метил-2-тиофенсульфонилхлорид и 5-бром-2-тиофенсульфонил хлорид, коммерчески доступны. Способ (d). Очевидно, что некоторые соединения формулы (I) могут быть подвергнуты реакции с образованием другого соединения формулы (I). Например, соединения, в которых R3 представляет собой C1-6 алкокси(например, метокси), может быть превращено в соответствующее соединение, в котором R3 представляет собой гидрокси, путем взаимодействия с деметилирующим агентом (таким как трибромид бора) в подходящем органическом растворителе (таком как DCM). Очевидно, что в любом из путей синтеза (a)-(d), описанных выше, точный порядок стадий синтеза,в результате которого различные группы и группировки вводят в молекулу, может быть изменен. Квалифицированный специалист-практик в данной области сможет обеспечить, чтобы группы или группировки, введенные на одной стадии способа, не подвергались воздействию последующих превращений и реакций, и выбрать соответствующий порядок стадий синтеза. В еще одном аспекте настоящего изобретения предложено соединение формулы (II) или его соль. Некоторые соединения формул (IV) и (VI) также являются новыми и поэтому составляют еще один аспект данного изобретения. Соединения формулы (I) и их соли являются ингибиторами активности СС-хемокиновых рецепторов, в частности активности рецептора CCR4, и, следовательно, полезны в лечении заболеваний или состояний, при которых показано применение ингибитора CCR4. Таким образом, согласно настоящему изобретению предложено соединение формулы (I) или его фармацевтически приемлемая соль для применения в терапии. Соединение формулы (I) или его фармацевтически приемлемую соль можно применять в лечении заболевания или состояния, при котором показан антагонист рецептора CCR4. Таким образом, согласно настоящему изобретению предложено соединение формулы (I) или его фармацевтически приемлемая соль для применения в лечении заболевания или состояния, при котором показан антагонист рецептора CCR4. Предложено также применение соединения формулы (I) или его фармацевтически приемлемой соли в изготовлении лекарственного средства для лечения заболевания или состояния, при котором показан антагонист рецептора CCR4. Предложен также способ лечения заболеваний или состояний, при которых показан антагонист рецептора CCR4, у субъекта, нуждающегося в таком лечении, включающий введение терапевтически эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли. Соответственно, субъектом, нуждающимся в таком лечении, является млекопитающее, в частности человек. Используемый в данном описании термин "эффективное количество" означает такое количество лекарственного средства или фармацевтического агента, вызывающего биологическую или медицинскую ответную реакцию ткани, системы, животного или человека, которое определяет, например, исследователь или клиницист. Кроме того, термин "терапевтически эффективное количество" означает любое количество, которое, по сравнению с соответствующим субъектом, не получившим такое количество, приводит к улучшению лечения, излечению, предупреждению, или к уменьшению симптомов заболевания,расстройства, или к уменьшению побочных эффектов, или к снижению скорости прогрессирования заболевания или расстройства. Этот термин также охватывает количества, эффективные для усиления нормальной физиологической функции. Считается, что антагонисты CCR4 полезны в лечении различных заболеваний или состояний, таких как иммунорегуляторные, воспалительные и/или аллергические заболевания. Примеры включают астму,хроническое обструктивное заболевание легких (COPD), в том числе хронический бронхит и эмфизему,идиопатический легочный фиброз, атопический или контактный дерматит, крапивницу, аллергический ринит (сезонный или круглогодичный), вазомоторный ринит, назальные полипы, аллергический конъюнктивит, весенний конъюнктивит, профессиональный конъюнктивит, инфекционный конъюнктивит,эозинофильные синдромы, эозинофильную гранулему, псориаз, ревматоидный артрит, язвенный колит,болезнь Крона, тромбоз, реперфузионное повреждение миокарда и головного мозга, хронический гломерулонефрит, сепсис, респираторный дистресс-синдром взрослых, рассеянный склероз, ухудшение памяти(включая болезнь Альцгеймера), боль и рак. Считается также, что антагонисты CCR4 полезны в лечении таких заболеваний или состояний, как аллергический бронхолегочный аспергиллез, аллергический грибковый синусит, тяжелая астма с грибковой сенситизацией и заболевания, в которые вовлечена патогенная роль грибков, включая инвазию или колонизацию (например, инвазивный аспергиллез, аспергиллома или кандидоз). Термин "заболевание или состояние, при котором показан ингибитор CCR4" охватывает любое или все вышеуказанные болезненные состояния. В одном из воплощений заболевание или состояние, при котором показан ингибитор CCR4, выбрано из астмы, COPD, ринита, идиопатического легочного фиброза, псориаза и контактного дерматита. В конкретном воплощении заболевание или состояние, при котором показан ингибитор CCR4, представляет собой астму. Несмотря на то что для применения в терапии соединение формулы (I), а также его фармацевтически приемлемые соли можно вводить в виде химического вещества, обычно оно присутствует в качестве активного ингредиента в фармацевтической композиции. Таким образом, в следующем аспекте настоящего изобретения предложена фармацевтическая композиция, содержащая соединение формулы (I) или его фармацевтически приемлемую соль и один или более фармацевтически приемлемых носителей, разбавителей и/или эксципиентов. Соединения формулы(I) и фармацевтически приемлемые соли описаны выше. Носитель(и), разбавитель(и) или эксципиент(ы) должны быть приемлемыми с точки зрения совместимости с другими ингредиентами композиции и не должны быть опасными для реципиента. В соответствии с другим аспектом изобретения предложен также способ приготовления фармацевтической композиции, включающий смешивание соединения формулы (I) или его фармацевтически приемлемой соли с одним или более фармацевтически приемлемыми носителями, разбавителями или эксципиентами. Фармацевтическая композиция может быть предназначена для применения в лечении любого из состояний, описанных в данном документе. Предложена также фармацевтическая композиция для лечения заболеваний или состояний, при которых показан ингибитор CCR4, содержащая соединение формулы (I) или его фармацевтически приемлемую соль. Предложена также фармацевтическая композиция, содержащая от 0,05 до 1000 мг соединения формулы (I) или его фармацевтически приемлемой соли и от 0,1 до 2 г одного или более фармацевтически приемлемых носителей, разбавителей или эксципиентов. Поскольку соединения формулы (I) предназначены для использования в фармацевтических композициях, ясно, что каждое из них предпочтительно предоставляется, по существу, в чистой форме, например имеет по меньшей мере 60%-ную чистоту, лучше по меньшей мере 75%-ную чистоту и предпочтительно по меньшей мере 85%-ную чистоту, особенно по меньшей мере 98%-ную чистоту (в мас.%). Фармацевтические композиции могут быть представлены в стандартных лекарственных формах,содержащих предопределенное количество активного ингредиента на стандартную дозу. Предпочтительными стандартными лекарственными композициями являются композиции, содержащие суточную дозу или субдозу активного ингредиента или соответствующие их доли. Такие стандартные дозы можно,следовательно, вводить более чем один раз в сутки. Предпочтительными стандартными лекарственными композициями являются композиции, содержащие суточную дозу или субдозу активного ингредиента(для введения более чем один раз в сутки), как указано здесь выше, или соответствующие их доли. Фармацевтические композиции могут быть адаптированы для введения подходящим путем, например пероральным (включая трансбуккальный или сублингвальный), ректальным, ингаляционным, интраназальным, местным (включая трансбуккальный, сублингвальный или трансдермальный), вагинальным или парентеральным (включая подкожный, внутримышечный, внутривенный или интрадермальный) путем. Такие композиции могут быть приготовлены любым способом, известным в области фармации, например путем объединения активного ингредиента с носителем(ями) или эксципиентом(ами). В одном из воплощений фармацевтическая композиция адаптирована для перорального введения. Фармацевтические композиции, адаптированные для перорального введения, могут быть представлены в виде дискретных единиц, таких как капсулы или таблетки; порошки или гранулы; растворы или суспензии в водных или неводных жидкостях; съедобные пены или муссы либо эмульсии типа масло-вводе или эмульсии типа вода-в-масле. Например, для перорального введения в форме таблетки или капсулы активный лекарственный компонент может быть объединен с пероральным, нетоксичным фармацевтически приемлемым инерт-9 021740 ным носителем, таким как этанол, глицерин, вода и т.п. Порошки, подходящие для введения в таблетки или капсулы, могут быть приготовлены путем измельчения соединения до подходящего тонкого размера(например, микронизацией) и смешивания с аналогичным образом приготовленного фармацевтического носителя, такого как пищевой углевод, например крахмал или маннит. Корригент, консервант, диспергирующий агент и краситель также могут присутствовать. Капсулы могут быть изготовлены путем приготовления порошковой смеси, как описано выше, и заполнения этой смесью сформованных желатиновых капсул. Глиданты и смазывающие вещества, такие как коллоидный диоксид кремния, тальк, стеарат магния, стеарат кальция или твердый полиэтиленгликоль, могут быть добавлены в порошковую смесь перед операцией заполнения. Разрыхляющий или солюбилизирующий агент, такой как агар-агар, карбонат кальция или карбонат натрия, также могут быть добавлены для улучшения доступности лекарственного средства при проглатывании капсулы. Кроме того, при желании или необходимости, в смесь могут быть введены подходящие связывающие вещества, глиданты, смазывающие вещества, подсластители, корригенты, разрыхляющие агенты и красители. Подходящие связывающие вещества включают крахмал, желатин, природные сахара, такие как глюкоза или бета-лактоза, зерновые подсластители, природные и синтетические камеди, такие как аравийская камедь, трагакант или альгинат натрия, карбоксиметилцеллюлозу, полиэтиленгликоль, воски и т.п. Смазывающие вещества, используемые в этих лекарственных формах, включают олеат натрия, стеарат натрия, стеарат магния, бензоат натрия, ацетат натрия, хлорид натрия и т.п. Разрыхлители включают, без ограничения, крахмал, метилцеллюлозу, агар, бентонит, ксантановую камедь и т.п. Таблетки изготавливают, например, путем приготовления порошковой смеси, гранулирования или агрегирования,добавления смазывающего вещества и разрыхлителя и прессования в таблетки. Порошковую смесь готовят путем смешивания соединения, подходящим образом измельченного, с разбавителем или основанием, как описано выше, и возможно со связывающим веществом, таким как карбоксиметилцеллюлоза,альгинат, желатин или поливинилпирролидон, замедлителем растворения, таким как парафин, ускорителем десорбции, таким как четвертичная соль, и/или агентом адсорбции, таким как бентонит, каолин или дикальцийфосфат. Порошковая смесь может быть гранулирована путем увлажнения связывающим веществом, таким как сироп, крахмальная паста, акадийская слизь или растворы целлюлозных или полимерных веществ, и протирания через сито. В качестве альтернативы гранулированию порошковая смесь может быть пропущена через таблеточную машину, и в результате можно получить неидеально сформованные агломераты, разломанные в гранулы. Гранулы могут быть смазаны для предотвращения прилипания к матрицам для формования таблеток путем добавления стеариновой кислоты, стеаратной соли,талька или вазелинового масла. Смазанную смесь затем прессуют в таблетки. Соединения по настоящему изобретению могут быть также объединены с сыпучим инертным носителем и непосредственно спрессованы в таблетки без выполнения стадий гранулирования или агломерации. Таблетки могут быть покрыты прозрачной или матовой защитной оболочкой, состоящей из шеллакового покрытия, покрытия из сахара или полимерного материала и полировочного покрытия из воска. В эти покрытия могут быть добавлены красители, чтобы различать разные стандартные дозировки. Пероральные жидкости, такие как растворы, сиропы и эликсиры, могут быть приготовлены в стандартной лекарственной форме таким образом, чтобы заданное количество содержало предопределенное количество соединения. Сиропы могут быть приготовлены путем растворения соединения в соответственно корригированном водном растворе, а эликсиры могут быть приготовлены путем использования нетоксичного спиртового разбавителя. Суспензии могут быть приготовлены путем диспергирования соединения в нетоксичном разбавителе. Могут быть добавлены также солюбилизаторы и эмульгаторы, такие как этоксилированные изостеариловые спирты и простые эфиры полиоксиэтиленсорбита, консерванты, корригент, такой как масло мяты перечной, или натуральные подсластители, или сахарин, или другие искусственные подсластители и т.п. В тех случаях, когда это подходит, композиции в стандартной лекарственной форме для перорального введения могут быть микроинкапсулированными. Препарат композиции может быть также приготовлен для пролонгированного или длительного высвобождения, например путем нанесения оболочки или заключения дисперсного материала в полимеры, воск или т.п. Соединения по изобретению можно также вводить в форме липосомной системы доставки, такой как небольшие однослойные везикулы,большие однослойные везикулы и многослойные везикулы. Липосомы могут быть образованы из различных фосфолипидов, таких как холестерин, стеариламин или фосфатидилхолины. Фармацевтические композиции, адаптированные для трансдермального введения, могут быть представлены в виде отдельных пластырей, предназначенных для оставления в тесном контакте с эпидермисом реципиента в течение длительного периода времени. Фармацевтические композиции, адаптированные для местного введения, могут быть изготовлены в виде мазей, кремов, суспензий, лосьонов, порошков, растворов, паст, гелей, спреев, аэрозолей или масел. Для лечения глаз или других наружных тканей, например ротовой полости и кожи, композиции предпочтительно наносят в виде местной мази или крема. При приготовлении мази активный ингредиент может быть использован либо с парафиновой, либо с водосмешиваемой мазевой основой. Альтернативно, активный ингредиент может быть введен в состав крема с основой для кремов типа масло-в-воде или вода-в-масле. Фармацевтические композиции, адаптированные для местного введения в глаз, включают глазные капли, где активный ингредиент растворен или суспендирован в подходящем носителе, в частности водном растворителе. Фармацевтические композиции, адаптированные для местного введения в ротовую полость, включают пластинки, пастилки и полоскания. Фармацевтические композиции, адаптированные для ректального введения, могут быть представлены в виде суппозиториев или в виде клизм. Лекарственные формы для назального введения или ингаляционного введения могут быть для удобства изготовлены в виде аэрозолей, растворов, суспензий, гелей или сухих порошков. Для композиций, подходящих и/или адаптированных для ингаляционного введения, предпочтительно, чтобы соединение по изобретению было в форме измельченных частиц, более предпочтительно форму измельченных частиц получают путем микронизации. Предпочтительный размер частиц измельченного (например, микронизированного) соединения или соли определяется значением D50 примерно от 0,5 до примерно 10 мкм (измеряют, например, методом лазерной дифракции). Аэрозольные препараты, например, для ингаляционного введения могут содержать раствор или тонкую суспензию активного вещества в фармацевтически приемлемом водном или неводном растворителе. Аэрозольные препараты могут быть представлены в одно- или многодозовых количествах в стерильном виде в герметично закрытом контейнере, который может принимать форму картриджа или блока для использования с распылительным устройством или ингалятором. Альтернативно, герметично закрытый контейнер может представлять собой одноразовое дозирующее устройство, которое выбрасывают сразу после использования его содержимого, такое как однодозовый назальный ингалятор или дозатор аэрозоля, снабженный отмеривающим дозу клапаном (ингалятор отмеренной дозы). Если дозировочная форма содержит дозатор аэрозоля, то он предпочтительно содержит подходящий пропеллент под давлением, такой как сжатый воздух, диоксид углерода, или органический пропеллент, такой как гидрофторуглерод (HFC). Подходящие HFC пропелленты включают 1,1,1,2,3,3,3 гептафторпропан и 1,1,1,2-тетрафторэтан. Формы дозатора аэрозоля могут также представлять собой нагнетательный распылитель. Аэрозоль под давлением может содержать раствор или суспензию активного соединения. Для этого может потребоваться включение в состав дополнительных эксципиентов,например сорастворителей и/или поверхностно-активных веществ, для улучшения дисперсионных характеристик и гомогенности суспензионных композиций. Композиции раствора также могут требовать добавления сорастворителей, таких как этанол. Другие эксципиенты-модификаторы также могут быть включены в состав для улучшения, например, стабильности, и/или вкуса, и/или характеристик массы тонкодисперсных частиц (количество и/или профиль) композиции. Для фармацевтических композиций, подходящих и/или адаптированных для ингаляционного введения, фармацевтическая композиция может представлять собой порошковую ингаляционную композицию. Такая композиция может содержать порошковую основу, такую как лактоза, глюкоза, трегалоза,маннит или крахмал, соединение формулы (I) или его соль (предпочтительно в измельченной форме, например в микронизированной форме) и возможно модификатор характеристик, такой как L-лейцин или другая аминокислота и/или соли металлов стеариновой кислоты, такие как стеарат магния или кальция. Предпочтительно сухая порошковая ингаляционная композиция содержит сухую порошковую смесь лактозы и соединения формулы (I) или его соли. Лактоза представляет собой предпочтительно гидрат лактозы, например моногидрат лактозы, и/или представляет собой предпочтительно лактозу сорта для ингаляций и/или лактозу высшего сорта. Предпочтительно размер частиц лактозы составляет 90% или более (по массе или по объему) частиц лактозы диаметром менее 1000 мкм (например, 10-1000 мкм, 301000 мкм) и/или 50% или более частиц лактозы диаметром менее 500 мкм (например, 10-500 мкм). Более предпочтительно размер частиц лактозы составляет 90% или более частиц лактозы диаметром менее 300 мкм (например, 10-300 мкм, 50-300 мкм) и/или 50% или более частиц лактозы диаметром менее 100 мкм. Возможно, размер частиц лактозы составляет 90% или более частиц лактозы диаметром менее 100-200 мкм и/или 50% или более частиц лактозы диаметром менее 40-70 мкм. Наиболее важно предпочтительно, чтобы от примерно 3 до примерно 30% (например, примерно 10%) (по массе или по объему) частиц имели диаметр менее 50 или менее 20 мкм. Например, без ограничения, подходящей лактозой сорта для ингаляций является лактоза Е 9334 (10% тонких фракций) (Borculo Domo Ingredients, Hanzeplein 25, 8017 JD Zwolle, Netherlands). Возможно, в частности, для сухих порошковых ингаляционных композиций фармацевтическая композиция для введения ингаляцией может быть заключена во множество герметично закрытых дозовых контейнеров (например, содержащих сухую порошковую композицию), размещенных в продольном направлении на полоске или ленте внутри подходящего ингаляционного устройства. Контейнер разрывается или открывается путем снятия предохранительной пленки при срабатывании, и доза сухой порошковой композиции может быть введена ингаляцией с помощью устройства, такого как устройствоDISKUS, продаваемое фирмой GlaxoSmithKline. Ингаляционное устройство DISKUS описано, например, в GB 2242134 А, и в таком устройстве по меньшей мере один контейнер для фармацевтической композиции в форме порошка (контейнер или контейнеры, предпочтительно представляющие собой множество герметично закрытых дозовых контейнеров, размещенных в продольном направлении на полоске или ленте) зафиксирован между двумя элементами, соединенными друг с другом с возможностью отсоединения друг от друга, и это устройство содержит средство определения местоположения открытия указанного(ых) контейнера или контейнеров; средство для отсоединения друг от друга указанных элементов в открывающем положении для открытия контейнера и выпускное отверстие, соединяющееся с открытым контейнером, через которое пользователь может вдыхать фармацевтическую композицию в порошковой форме из открытого контейнера. Таким образом, соединения по изобретению могут быть приготовлены в виде жидкой композиции для доставки из жидкостного дозатора, например жидкостного дозатора, имеющего дозирующую насадку или дозирующее отверстие, через которую(ое) отмеренная доза жидкой композиции дозируется при нажатии пользователем на нагнетательный механизм жидкостного дозатора. Такие жидкостные дозаторы обычно обеспечены резервуаром с множеством отмериваемых доз жидкой композиции, причем дозы дозируются при последовательных срабатываниях насоса. Дозирующее сопло или насадка могут иметь конфигурацию, подходящую для введения в ноздри пользователя для распылительного дозирования жидкой композиции в носовую полость. Жидкостной дозатор вышеупомянутого типа описан и проиллюстрирован в WO-А-2005/044354, полное содержание которого включено в данное описание посредством ссылки. Дозатор имеет корпус, в котором размещено выпускающее жидкость устройство с нагнетательным насосом, установленным на контейнере, в котором находится жидкая композиция. Корпус имеет по меньшей мере один приводимый в действие пальцами боковой рычаг, который двигается внутрь относительно корпуса, перемещая контейнер вверх в корпусе посредством эксцентрика, заставляя насос сжимать и нагнетать отмеренную дозу композиции из ствола насоса через назальное сопло корпуса. Особенно предпочтительным жидкостным дозатором является дозатор общего типа, проиллюстрированный на фиг. 30-40, приведенных в WO-A-2005/044354. Фармацевтические композиции, адаптированные для вагинального введения, могут быть представлены в виде пессариев, тампонов, кремов, гелей, паст, пен или спреев. Фармацевтические композиции, адаптированные для парентерального введения, включают водные и неводные стерильные инъекционные растворы, которые могут содержать антиоксиданты, буферные агенты, бактериостатические факторы и растворенные вещества, которые делают композицию изотоничной с кровью реципиента; и водные и неводные стерильные суспензии, которые могут включать суспендирующие агенты и загустители. Композиции могут быть представлены в контейнерах для стандартной дозы или в многодозовых контейнерах, например запаянных ампулах и флаконах, и могут храниться в высушенном сублимацией (лиофилизированном) состоянии, требующем только добавления стерильного жидкого носителя, например воды для инъекций, непосредственно перед использованием. Приготавливаемые для немедленного введения инъекционные растворы и суспензии могут быть получены из стерильных порошков, гранул и таблеток. Терапевтически эффективное количество соединения по изобретению будет зависеть от ряда факторов, включая, например, возраст и массу субъекта, точное состояние, требующее лечения, и его тяжесть, природу композиции и путь введения, и, в конечном счете, будет определяться лечащим врачом или ветеринаром. В фармацевтической композиции каждая дозировочная единица для перорального или парентерального введения предпочтительно содержит от 0,01 до 3000 мг, более предпочтительно от 0,5 до 1000 мг соединения по изобретению в расчете на свободное основание. Каждая дозировочная единица для назального или ингаляционного введения предпочтительно содержит от 0,001 до 50 мг, более предпочтительно от 0,01 до 5 мг соединения формулы (I) или его фармацевтически приемлемой соли в расчете на свободное основание. Фармацевтически приемлемые соединения по изобретению можно вводить в суточной дозе (для взрослого пациента), например пероральной или парентеральной дозе от 0,01 до 3000 мг/сутки или от 0,5 до 1000 мг/сутки или назальной или ингаляционной дозы от 0,001 до 50 мг/сутки или от 0,01 до5 мг в сутки соединения формулы (I) или его фармацевтически приемлемой соли в расчете на свободное основание. Это количество можно вводить однократной дозой один раз в сутки или чаще с несколькими (например, двумя, тремя, четырьмя, пятью или шестью) субдозами в сутки, но так, чтобы суммарная суточная доза была той же. Эффективное количество соли может быть определено как пропорциональное эффективному количеству самого соединения формулы (I). Соединения по изобретению можно использовать одни или в комбинации с другими терапевтическими агентами. Комбинированные терапии согласно настоящему изобретению могут включать введение по меньшей мере одного соединения формулы (I) или его фармацевтически приемлемой соли и применение по меньшей мере одного другого фармацевтически активного агента. Предпочтительно комбинированные терапии согласно настоящему изобретению включают введение по меньшей мере одного соединения формулы (I) или его фармацевтически приемлемой соли и по меньшей мере одного другого фармацевтически активного агента. Соединения формулы (I) и их фармацевтически приемлемые соли и другой(ие) фармацевтически активный(ые) агент(ы) можно вводить вместе в единой фармацевтической композиции или по отдельности, и при введении по отдельности введение можно проводить одновременно или последовательно в любом порядке. Количества соединения(й) по изобретению и другого(их) фармацевтически активного(ых) агента(ов) и относительные моменты времени введения выбирают так,чтобы достигался желаемый комбинированный терапевтический эффект. Таким образом, в еще одном аспекте предложена комбинация, содержащая соединение по изобретению и по меньшей мере один другой фармацевтически активный агент. Таким образом, в одном из аспектов соединение и фармацевтические композиции по изобретению могут быть использованы в комбинации с одним или более другими терапевтическими агентами или могут включать в себя один или более других терапевтических агентов, например, выбранных из противовоспалительных агентов (включая стероидные), антихолинергических агентов (в частности, антагонистов рецепторов М 1/М 2/М 3), агонистов 2-адренорецепторов, противоаллергических агентов, противоинфекционных агентов (таких как антибиотики или противовирусные средства) или антигистаминных средств. Согласно изобретению предложен в еще одном аспекте комбинированный фармацевтический продукт, содержащий соединение формулы (I) или его фармацевтически приемлемую соль вместе с одним или более другими терапевтически активными агентами, например, выбранными из противовоспалительного агента, такого как кортикостероид или NSAID (нестероидное противовоспалительное лекарственное средство), антихолинергического агента, агониста 2-адренорецепторов, противоаллергического агента, противоинфекционного агента или антигистаминного средства. Понятно, что если соединение по изобретению вводят в комбинации с другими терапевтическими агентами, которые обычно вводят ингаляционным, внутривенным, пероральным или интраназальным путем, то фармацевтическую композицию можно вводить таким же путем. Альтернативно, индивидуальные компоненты композиции можно вводить разными путями. Одно из воплощений изобретения охватывает комбинации, содержащие один или два других терапевтических агента. Подходящие противовоспалительные агенты включают кортикостероиды. Противовоспалительные кортикостероиды общеизвестны в данной области. Репрезентативные примеры включают флутиказона пропионат, беклометазона 17-пропионат, беклометазона 17,21-дипропионат, дексаметазон или его эфир,мометазон или его эфир (например, мометазона фуроат), циклесонид, будесонид, флунисолид, метилпреднизолон, преднизолон и дексаметазон. Дополнительные примеры противовоспалительных кортикостероидов описаны в WO 02/12266 А 1 (Glaxo Group Ltd), в частности соединение примера 1S-фторметиловый эфир) или их фармацевтически приемлемые соли. Примеры агонистов 2-адренорецепторов включают сальметерол (например, в виде рацемата или единственного энантиомера, например R-энантиомер), сальбутамол, формотерол, сальмефамол, фенотерол или тербуталин и их соли, например ксинафоатная соль сальметерола, сульфатная соль или свободное основание сальбутамола или фумаратная соль формотерола. В одном из воплощений агонисты 2-адренорецепторов являются длительно действующими агонистами 2-адренорецепторов, например,оказывающими терапевтический эффект в течение периода времени 24 ч, например сальметерол или формотерол. Дополнительным примером агониста 2-адренорецепторов является соединение 4-(1R)-2-[(6-2-[(2,6-дихлорфенил)метилокси]этоксигексил)амино]-1-гидроксиэтил-2-(гидроксиэтил)фенола трифенилацетат (вилантерола трифенатат). Примеры антихолинергических соединений, которые можно использовать в комбинации с соединением формулы (I) или его фармацевтически приемлемой солью, описаны в WO 03/011274 А 2 иWO 02/069945 A2/US 2002/0193393 А 1 и US 2002/052312 А 1. Например, антихолинергические агенты включают антагонисты мускариновых рецепторов, в частности соединения, которые являются антагонистами рецепторов M1 или М 3, дуальные антагонисты рецепторов M1/М 3 или М 2/М 3 или пан-антагонисты рецепторов М 1/М 2/М 3, такие как ипратропия бромид, окситропия бромид или тиотропия бромид. Антигистаминное средство, используемое в комбинации с соединением по изобретению, может представлять собой, например, метапирилен или антагонисты Н 1. Примеры антагонистов Н 1 включают,без ограничения, амелексанокс, астемизол, азатанид, азеластин, акривастин, бромфенирамин, цетиризин,левоцетиризин, эфлетиризин, хлорфенирамин, клемастин, циклизин, каребастин, ципрогептадин, карбиноксамин, дезкарбоэтоксилоратидин, доксиламин, диметинден, эбастин, эпинастин, эфлетиризин, фексофенадин, гидроксизин, кетотифен, лоратидин, левокабастин, мизоластин, меквитазин, миансерин, ноберастин, меклизин, норастемизол, олопатадин, пикумаст, пириламин, прометазин, терфенадин, трипеленнамин, темеластин, тримепразин и трипролидин, в частности цетиризин, левоцетиризин, эфлетиризин и фексофенадин. В другом воплощении изобретения предложена комбинация, содержащая соединение по изобретению вместе с антагонистом Н 3 (и/или обратным агонистом). Примеры антагонистов Н 3 включают, например, соединения, описанные в WO 2004/035556 и в WO 2006/045416. Другие подходящие комбинации включают, например, комбинации, содержащие соединение по изобретению вместе с другими противовоспалительными агентами, такими как противовоспалительный кортикостероид, или нестероидное противовоспалительное лекарственное средство (NSAID), такое как антагонист лейкотриенов (например, монтелукаст), ингибитор iNOS (индуцибельная синтаза оксида азота), ингибитор триптазы, ингибиторы IKK2, ингибитор р 38, ингибиторы Syk, ингибитор эластазы, антагонист бета-2 интегрина, агонист аденозина 2 а, антагонист хемокинов, такой как антагонист CCR3, ингибитор высвобождения медиаторов, такой как натрия кромогликат, ингибитор 5-липоксигеназы, антагонист DP1, антагонист DP2, ингибитор CTTh2, ингибитор pI3K дельта, ингибитор ITK, ингибитор LP(липофосфатидный) и ингибитор FLAP (белок, активирующий пять-липоксигеназу). Другие подходящие комбинации включают соединение по изобретению вместе с противоинфекционным агентом (например, антибиотиком или противовирусным средством), антигипертензивным агентом, антитромботическим агентом, статином или ингибитором холинэстеразы. Комбинации, упомянутые выше, могут для удобства быть представлены для применения в форме фармацевтической композиции, и поэтому фармацевтические композиции, содержащие комбинацию, как она определена выше, вместе с фармацевтически приемлемым разбавителем или носителем составляют еще один аспект изобретения. В одном из аспектов настоящего изобретения предложено также так называемое "тройное комбинированное" терапевтическое средство, содержащее соединение по изобретению вместе с агонистом 2-адренорецепторов и противовоспалительным кортикостероидом. Предпочтительно эта комбинация предназначена для лечения и/или профилактики астмы, COPD или аллергического ринита. Агонист 2-адренорецепторов и/или противовоспалительный кортикостероид могут быть такими, как описано выше и/или как описано в WO 03/030939 А 1. Репрезентативная "тройная" комбинация содержит соединение по изобретению, сальметерол или его фармацевтически приемлемую соль (например, сальметерола ксинафоат) и флутиказона пропионат. Еще одним примером "тройной" комбинации является комбинация,содержащая соединение по изобретению,4-(1R)-2-[(6-2-[(2,6-дихлорфенил)метилокси]этоксигексил)амино]-1-гидроксиэтил-2-(гидроксиэтил)фенола трифенилацетат (вилантерола трифенатат) и 6,9-дифтор-17-[(2-фуранилкарбонил)окси]-11-гидрокси-16-метил-3 оксоандроста-1,4-диен-17-карботиокислоты S-фторметиловый эфир (флутиказона фуроат). Ревматоидный артрит (RA) является еще одним воспалительным заболеванием, при котором предусматривается комбинированная терапия. Таким образом, в еще одном аспекте настоящего изобретения предложено соединение по изобретению в комбинации с дополнительным терапевтическим агентом,полезным в лечении ревматоидного артрита, причем указанная комбинация полезна для лечения ревматоидного артрита. Соединение и фармацевтические композиции по изобретению могут быть использованы в комбинации с или включают один или более других терапевтических агентов, например, выбранных из NSAID,кортикостероидов, ингибиторов СОХ-2, ингибиторов цитокинов, анти-TNF агентов, ингибиторов онкостатина М, противомалярийных средств, иммунодепрессантов и цитостатиков. Анти-TNF агенты включают инфликсимаб (Remicade), этанерцепт (Enbrel) и адалимум (Humira). Другие "биологические" терапевтические средства включают анакинра (Kineret), ритуксимаб, лимфостат-В, ингибиторы BAFF/APRIL и CTLA-4-lg или его миметики. Другие ингибиторы цитокинов включают лефлюономид (Arava). Дополнительные лекарственные средства второй линии включают препараты золота (ауранофин (таблетки Ridaura) или ауротиомалат (инъекция Myocrisin, лекарственные средства, применяемые против малярии: (гидроксихлороквин (Plaquenil, лекарственные средства, подавляющие иммунную систему (азатиоприн (Imuran, Thioprine), метотрексат (Methoblastin, Ledertrexate,Emethexate), циклоспорин (Sandimmun, Neoral, циклофосфамид (Cycloblastin), цитоксан, эндоксан, Dпеницилламин (D-Penamine), сульфасалазин (Salazopyrin), нестероидные противовоспалительные лекарственные средства (включая аспирин и ибупрофен). Специалисту в данной области будет ясно, что в тех случаях, когда это подходит, другой(ие) терапевтический(ие) ингредиент(ы) можно применять в форме соли, например соли щелочного металла, или аминной соли, или соли присоединения кислоты, или в форме пролекарств, или в форме эфиров, например низших алкиловых эфиров, или в форме сольватов, например гидратов, для оптимизации активности, и/или стабильности, и/или физических характеристик, таких как растворимость, терапевтического ингредиента. Должно быть ясно, что в тех случаях, когда это подходит, терапевтические ингредиенты можно применять в оптически чистой форме. Комбинации, упомянутые выше, могут быть для удобства представлены для применения в форме фармацевтической композиции, и поэтому фармацевтические композиции, содержащие комбинацию, как определено выше, вместе с фармацевтически приемлемым разбавителем или носителем составляют еще один аспект изобретения. Индивидуальные соединения таких комбинаций можно вводить либо последовательно, либо одновременно в отдельных или в объединенных фармацевтических композициях. Предпочтительно индивидуальные соединения следует вводить одновременно в объединенной фармацевтической композиции. Специалисты в данной области без труда определят целесообразные дозы известных терапевтических агентов. Общие подробности экспериментов. Аналитическая ЖХ/МС. Аналитическую ЖХ/МС (жидкостная хроматография/масс-спектрометрия) проводили на одной из следующих систем A-F. Во всех системах осуществляли УФ-детектирование усредненного сигнала с длиной волны от 220 до 350 нм, и масс-спектры записывали на масс-спектрометре в режиме электрораспылительной ионизации с поочередной регистрацией положительных и отрицательных ионов. Экспериментальные подробности ЖХ/МС систем A-F, упомянутых здесь выше, следующие. Система А. Колонка: 502,1 мм в.д. (внутренний диаметр), 1,7 мкм, Acquity для ультраэффективной жидкостной хроматографии (UPLC) ВЕН C18. Скорость потока: 1 мл/мин. Температура: 40 С. Растворители: А: 0,1%-ный (об./об.) раствор муравьиной кислоты в воде,В: 0,1%-ный (об./об.) раствор муравьиной кислоты в ацетонитриле. Система В. Колонка: 304,6 мм в.д., 3,5 мкм, колонка Sunfire C18. Скорость потока: 3 мл/мин. Температура: 30 С. Растворители: А: 0,1%-ный (об./об.) раствор муравьиной кислоты в воде,В: 0,1%-ный (об./об.) раствор муравьиной кислоты в ацетонитриле. Система С. Колонка: 504,6 мм в.д., 3,5 мкм, колонка XBridge C18. Скорость потока: 3 мл/мин. Температура: 30 С. Растворители: А: 10 мМ бикарбонат аммония в воде, доведенный до рН 10 аммиачным раствором,В: ацетонитрил. Система D. Колонка: 502,1 мм в.д., 1,7 мкм, Acquity UPLC ВЕН C18. Скорость потока: 1 мл/мин. Температура: 40 С. Растворители: А: 0,1%-ный (об./об.) раствор трифторуксусной кислоты в воде,В: 0,1%-ный (об./об.) раствор трифторуксусной кислоты в ацетонитриле. Система Е. Колонка: 304,6 мм в.д., 3,5 мкм, колонка Sunfire C18. Скорость потока: 3 мл/мин. Температура: 30 С Растворители: А: 0,1%-ный (об./об.) раствор трифторуксусной кислоты в воде,В: 0,1%-ный (об./об.) раствор трифторуксусной кислоты в ацетонитриле. Система F. Колонка: 502,1 мм в.д., 1,7 мкм, колонка Acquity UPLC ВЕН C18. Скорость потока: 1 мл/мин. Температура: 40 С. Растворители: А: 10 мм бикарбонат аммония в воде, доведенный до рН 10 аммиачным раствором,В: ацетонитрил. Масс-направленная автоматизированная препаративная ВЭЖХ. Неочищенные продукты очищали масс-направленной автоматизированной препаративной высокоэффективной жидкостной хроматографией (МНАП ВЭЖХ) одним из описанных ниже методов A-D. Время прогона составляло 15 мин, если не указано иное. Во всех системах осуществляли УФ-детектирование усредненного сигнала с длиной волны от 220 до 350 нм и масс-спектры записывали на масс-спектрометре в режиме электрораспылительной ионизации с попеременной регистрацией положительных и отрицательных ионов. Метод А. Метод A осуществляли на колонке Sunfire C18 (типично 15030 мм в.д., диаметр насадки 5 мкм) при температуре окружающей среды. Используемыми растворителями были: А = 0,1%-ный (об./об.) раствор муравьиной кислоты в воде,В = 0,1%-ный (об./об.) раствор муравьиной кислоты в ацетонитриле. Использовали следующий градиент: Метод В. Метод В осуществляли на колонке Sunfire C18 (типично 15030 мм в.д., диаметр насадки 5 мкм) при температуре окружающей среды. Используемыми растворителями были: А = 0,1%-ный (об./об.) раствор трифторуксусной кислоты в воде,В = 0,1%-ный (об./об.) раствор трифторуксусной кислоты в ацетонитриле. Использовали следующий градиент: Метод С. Метод С осуществляли на колонке XBridge C18 (типично 15019 мм в.д., диаметр насадки 5 мкм) при температуре окружающей среды. Используемыми растворителями были: А = 10 мм водный бикарбонат аммония, доведенный до рН 10 аммиачным раствором,В = ацетонитрил. Использовали следующий градиент: Метод D. Метод D осуществляли на колонке ATLANTIS dC18 (типично 10019 мм в.д., диаметр насадки 5 мкм). Используемыми растворителями были: А = 0,1%-ный (об./об.) раствор муравьиной кислоты в воде,В = 0,05%-ный (об./об.) раствор муравьиной кислоты в смеси 95% ацетонитрила и 5% воды. Использовали следующий градиент: Метод Е. Метод Е осуществляли на колонке Sunfire C18 (типично 10019 мм в.д., диаметр насадки 5 мкм). Используемыми растворителями были:A = 0,1%-ный (об./об.) раствор муравьиной кислоты в воде,В = 0,1%-ный (об./об.) раствор муравьиной кислоты в ацетонитриле. Использовали следующий градиент: Сокращения. Приведенный ниже список дает определения некоторым сокращениям, использованным в данном описании. Понятно, что этот список не является исчерпывающим, но значение тех сокращений, которые не определены ниже, очевидны специалистам в данной области. Ас - ацетил;TMS - триметилсилил. Все ссылки на эфир относятся к диэтиловому эфиру, и рассол относится к насыщенному водному раствору NaCl. Промежуточное соединение 1. 4-(Метилокси)-1H-индазол-3-амин(9,63 мл, 198 ммоль) в н-бутаноле (100 мл) нагревали при температуре дефлегмации в атмосфере азота в течение 18 ч. Реакционной смеси давали возможность охладиться, добавляли воду (300 мл) и органическую фазу удаляли. Твердое вещество в водной фазе собирали фильтрованием и сушили в вакууме при 40 С с получением белого твердого вещества (0,6 г). Бутанольную фазу упаривали в вакууме и остаток и водные маточные жидкости объединяли и экстрагировали с использованием этилацетата (2200 мл). Объединенные этилацетатные экстракты сушили над сульфатом магния и упаривали в вакууме. Остаток растворяли в DCM и наносили на картридж с диоксидом кремния (100 г). Его элюировали циклогексаном(500 мл), смесью циклогексан-этилацетат (1:1 об./об., 500 мл) и этилацетатом (500 мл). Нужные фракции объединяли и упаривали в вакууме с получением указанного в заголовке соединения (9,92 г, 92%) в виде не совсем белого твердого вещества. ЖХ/МС (система A) RT (время удерживания) = 0,5 мин, ЭРИ+ (электрораспылительная ионизация,регистрация положительных ионов) m/z 164 (М+Н)+. К раствору измельченного гидроксида калия (6,75 г, 120 ммоль) в DMSO (300 мл) при комнатной температуре в атмосфере азота добавляли 4-(метилокси)-1 Н-индазол-3-амин (промежуточное соединение 1) (7,85 г, 48,1 ммоль), и это привело к образованию темно-красного раствора. Через 5 мин одной порцией добавляли 3-(хлорметил)бензонитрил (8,84 г, 58,3 ммоль). Реакционную смесь перемешивали в течение 20 мин и затем вливали в воду (500 мл) с образованием эмульсии. Эту эмульсию экстрагировали с использованием хлороформа (3500 мл). Объединенные органические растворы промывали водой(400 мл) и пропускали через гидрофобную фритту. Растворитель удаляли в вакууме, остаток наносили на картридж с диоксидом кремния (340 г) и элюировали с градиентом 0-100% этилацетата в циклогексане при пропускании 8 КО. Это привело к образованию оранжевого твердого вещества, которое обрабатывали этилацетатом (10 мл) и циклогексаном (90 мл). Твердое вещество собирали фильтрованием и промывали циклогексаном (50 мл). Твердое вещество сушили в вакууме с получением указанного в заголовке соединения (7,89 г, 59%) в виде бледно-оранжевого твердого вещества. ЖХ/МС (система A) RT = 0,93 мин, ЭРИ+ m/z 279 (М+Н)+. Промежуточное соединение 3. 5-Хлор-N-[1-[(3-цианофенил)метил]-4-(метилокси)-1 Н-индазол-3-ил]-2-тиофенсульфонамид(7,89 г, 28,3 ммоль) добавляли раствор 5-хлор-2-тиофенсульфонилхлорида (Aldrich) (6,15 г, 28,3 ммоль) в пиридине (9,17 мл, 113 ммоль) в атмосфере азота при комнатной температуре. Реакция была экзотермической, и реакционная смесь становилась темно-красной. Через 40 мин реакционную смесь распределяли между этилацетатом (500 мл) и 2 н. соляной кислотой (500 мл). Водную фазу промывали этилацетатом(400 мл). Объединенные органические растворы сушили над сульфатом магния и упаривали в вакууме. Темно-красный остаток растворяли в DCM и наносили на картридж с диоксидом кремния (340 г). Картридж элюировали с градиентом 0-10% этилацетата в дихлорметане при пропускании 8 КО. Упаривание соответствующих фракций привело к получению указанного в заголовке соединения (11,1 г, 85%) в виде не совсем белого твердого вещества. ЖХ/МС (система A) RT = 1,15 мин, ЭРИ+ m/z 459/461 (М+Н)+. Промежуточное соединение 4. Гидрохлорид К охлажденному до 0 С раствору 5-хлор-N-[1-[(3-цианофенил)метил]-4-(метилокси)-1 Н-индазол-3 ил]-2-тиофенсульфонамида (промежуточное соединение 3) (11,1 г, 24,2 ммоль) в THF (150 мл) по каплям добавляли, поддерживая температуру ниже 10 С, 1,0 М раствор алюмогидрида лития в эфире (60,5 мл,60,5 ммоль), при этом происходило газовыделение и эту суспензию перемешивали при комнатной температуре в течение 1 ч. Реакцию гасили добавлением воды (7 мл) с последующим добавлением 2,0 М раствора гидроксида натрия (42,5 мл). После перемешивания в течение 30 мин твердое вещество удаляли фильтрованием и промывали THF. Объединенные фильтрат и промывочные жидкости обрабатывали 140 г SCX диоксида кремния. Его отфильтровывали и промывали метанолом (1 л), а затем 10% раствором 2 н. соляной кислоты в метаноле (2 л). Нужные фракции объединяли и концентрировали. Полученное твердое вещество собирали фильтрованием и промывали водой. Твердое вещество сушили в вакууме с получением указанного в заголовке соединения (N7916-76-1) (9,3 г, 77%) в виде белого твердого вещества. ЖХ/МС (система A) RT = 0,85 мин, ЭРИ+ m/z 463/465 (М+Н)+. Промежуточное соединение 5. 1,1-Диметилэтил-3-амино-4-(метилокси)-1 Н-индазол-1-карбоксилат К раствору 4-(метилокси)-1 Н-индазол-3-амина (5,56 г, 34 ммоль), DMAP (2,08 г, 17 ммоль) и триэтиламина (4,75 мл, 34 ммоль) в DCM (30 мл) добавляли по каплям при 0 С раствор ди-третбутилдикарбоната (7,44 г, 34 ммоль) в DCM (20 мл). Реакционную смесь перемешивали при комнатной температуре в течение 1 ч, затем добавляли 50 мл рассола. Органический слой отделяли и водный слой дополнительно экстрагировали 50 мл DCM. Органические растворы объединяли, промывали рассолом(100 мл), сушили над гидрофобной фриттой и концентрировали при пониженном давлении. Неочищенное твердое вещество растирали с 50 мл Et2O, фильтрат концентрировали при пониженном давлении и снова растирали с 20 мл Et2O. Твердые вещества объединяли с получением указанного в заголовке соединения (8,97 г, 67%), которое использовали без дополнительной очистки. ЖХ/МС (система В) RT = 2,9 мин, ЭРИ+ m/z 264 (М+Н)+. Промежуточное соединение 6. 1,1-Диметилэтил-3-[(5-хлор-2-тиенил)сульфонил]амино-4-(метилокси)-1 Н-индазол-1-карбоксилат(30 мл) добавляли по каплям при 0 С раствор пиридина (3,7 мл, 46 ммоль) в DCM. Реакционную смесь перемешивали, давая возможность нагреться с 0 С до комнатной температуры в течение 2 ч, затем перемешивали при комнатной температуре в течение выходных дней. Добавляли пиридин (50 мл) и полученную смесь перемешивали при 50 С в течение еще 24 ч. Реакционную смесь затем гасили добавлением 50 мл насыщенного раствора NaHCO3. Органический слой отделяли и водный слой дополнительно экстрагировали 50 мл DCM. Органические растворы объединяли, промывали водой (50 мл), затем рассолом(50 мл) и сушили с использованием гидрофобной фритты. Неочищенный продукт очищали хроматографией на двух Si-картриджах (100 г) на Flashmaster, элюируя с градиентом от 0 до 25% EtOAc в DCM за 60 мин, с получением целевого продукта в виде бледно-оранжевого твердого вещества: (2,21 г, 22%). ЖХ/МС (система A) RT = 1,35 мин, ЭРИ+ m/z 444 (М+Н)+. Промежуточное соединение 7. 1,1-Диметилэтил-3-(5-хлор-2-тиенил)сульфонил]([2-(триметилсилил)этил]оксиметил)амино]-4(метилокси)-1 Н-индазол-1-карбоксилат К раствору 1,1-диметилэтил-3-[(5-хлор-2-тиенил)сульфонил]амино-4-(метилокси)-1 Н-индазол-1 карбоксилата (промежуточное соединение 6) (2,21 г, 4,98 ммоль) в DCM (20 мл) добавляли при 0 С 2-(триметилсилил)этоксиметилхлорид (1,23 мл, 6,97 ммоль). Затем по каплям добавляли при 0 С диизопропиламин (1,419 мл, 9,96 ммоль) и эту реакционную смесь перемешивали при комнатной температуре. Реакцию гасили водой (50 мл), органический слой отделяли и водный слой дополнительно экстрагировали 30 мл DCM. Органические растворы объединяли, сушили с использованием гидрофобной фритты и концентрировали при пониженном давлении с получением желтого масла, которое использовали без дополнительной очистки (3,12 г). ЖХ/МС (система A) RT = 1,64 мин, ЭРИ+ m/z 574/576 (М+Н)+. Промежуточное соединение 8. 5-Хлор-N-[4-(метилокси)-1H-индазол-3-ил]-N-([2-(триметилсилил)этил]оксиметил)-2 тиофенсульфонамид К раствору 1,1-диметилэтил-3-(5-хлор-2-тиенил)сульфонил]([2-(триметилсилил)этил]оксиметил)амино]-4-(метилокси)-1 Н-индазол-1-карбоксилата (промежуточное соединение 7) (3,12 г,5,43 ммоль) в DMF (20 мл) добавляли раствор карбоната натрия (0,864 г, 8,15 ммоль) в воде (20 мл) при комнатной температуре. Получали белую суспензию и отмечалось нагревание среды. Полученную суспензию перемешивали в течение ночи при 80 С, реакционную смесь затем охлаждали до комнатной температуры и добавляли карбонат натрия (300 мг). Полученную смесь затем перемешивали при 85 С. После перемешивания в течение 3 ч в реакционную смесь добавляли 50 мл воды и 50 мл DCM. Органический слой отделяли и водный слой дополнительно экстрагировали 30 мл DCM. Органические растворы объединяли, сушили с использованием гидрофобной фритты и концентрировали при пониженном давлении. Неочищенное масло очищали хроматографией на двух Si-картриджах (100 г) на Flashmaster, элюируя градиентом от 0 до 25% EtOAc в циклогексане за 25 мин. Чистые фракции объединяли с получением указанного в заголовке продукта в виде оранжевой смолы (1,62 г, 63%). ЖХ/МС (система A) RT = 1,41 мин, ЭРИ+ m/z 474/476 (М+Н)+. Фракции, содержащие примеси, объединяли и повторно очищали хроматографией на картридже с диоксидом кремния (70 г) на Flashmaster, элюируя с градиентом от 0 до 25% EtOAc в циклогексане за 40 мин. Фракции, содержащие чистый продукт, объединяли с получением дополнительного количества целевого продукта (277 мг, 11%). Промежуточное соединение 9. 1,1-Диметилэтил-3-бис-[(5-хлор-2-тиенил)сульфонил]амино-4-(метилокси)-1 Н-индазол-1 карбоксилат(50 мл) добавляли пиридин (9,95 мл, 123 ммоль). Реакционную смесь перемешивали при 45 С в атмосфере азота в течение 3 суток. Реакционную смесь гасили, используя насыщенный раствор бикарбоната натрия (100 мл), и разбавляли DCM (100 мл). Фазы разделяли и водную фазу экстрагировали, используяDCM (100 мл). Объединенные органические растворы промывали рассолом и фильтровали через гидрофобную фритту. Растворитель удаляли в вакууме и остаток растворяли в DCM (30 мл). Раствор наносили на картридж с диоксидом кремния (330 г) и элюировали с градиентом 0-25% этилацетата в DCM при пропускании 10 КО. Нужные фракции упаривали в вакууме с получением указанного в заголовке соединения (4,17 г, 54%) в виде белого твердого вещества, ЖХ/МС (система В) RT = 4,12 мин, ЭРИ+ m/z 624/626/628, и 1,1-диметилэтил-3-[(5-хлор-2-тиенил)сульфонил]амино-4-(метилокси)-1 Н-индазол-1- 21021740(4,17 г, 6,68 ммоль) в DCM (20 мл) добавляли TFA (10,29 мл, 134 ммоль) и этот раствор перемешивали при комнатной температуре в течение 4 ч. Реакционную смесь разбавляли DCM (100 мл) и водой (50 мл). Органическую фазу пропускали через гидрофобную фритту и растворитель удаляли в вакууме с получением продукта (3,78 г) в виде белого твердого вещества. Его сушили в вакууме при 45 С в течение 2 суток с получением указанного в заголовке соединения (3,55 г, 100%) в виде белого твердого вещества. ЖХ/МС (система A) RT = 1,33 мин, ЭРИ+ m/z 524/526/528 (М+Н)+. Промежуточное соединение 11. 2-[4-(Метилокси)-1 Н-индазол-3-ил]-1 Н-изоиндол-1,3(2H)-дион 4-(Метилокси)-1 Н-индазол-3-амин (промежуточное соединение 1) (500 мг, 3 ммоль) и фталевый ангидрид (680 мг, 4,6 ммоль) (доступен от Aldrich) растворяли в 1,4-диоксане (5 мл) при комнатной температуре. Реакционную смесь перемешивали при 110 С в течение ночи, а затем ее охлаждали и концентрировали при пониженном давлении. Остаток растирали и обрабатывали ультразвуком с 50 мл эфира. Твердое вещество собирали фильтрованием и сушили в вакуумном шкафу с получением коричневого твердого вещества (711 мг, 81%). ЖХ/МС (система В) RT = 2,82 мин, ЭРИ+ m/z 294 (М+Н)+. Промежуточное соединение 12. 3-[3-Амино-4-(метилокси)-1H-индазол-1-ил]метилбензамид К раствору 2-[4-(метилокси)-1 Н-индазол-3-ил]-1H-изоиндол-1,3(2 Н)-диона (промежуточное соединение 11) (1 г, 3,4 ммоль) в DMF (3 мл) добавляли при температуре окружающей среды карбонат калия(0,707 г, 5,11 ммоль) и 3-(хлорметил)бензамид (Maybridge) (0,752 г, 4,43 ммоль) и полученную смесь перемешивали при 80 С в течение ночи. Растворитель выпаривали в потоке азота в продувочной системе. Остаток растворяли в 20 мл DCM и добавляли 20 мл воды. Образовавшийся осадок собирали фильтрованием и непосредственно использовали на следующей стадии. ЖХ/МС (система A) RT = 0,92 мин, ЭРИ+ m/z 459 (М+Н)+. Твердое вещество растворяли в этаноле (3 мл) и по каплям при температуре окружающей среды добавляли к гидразин гидрату (0,497 мл, 10,2 ммоль). Полученную суспензию перемешивали при температуре окружающей среды в течение 1 ч, затем добавляли ацетон (3 мл) и полученную суспензию перемешивали в течение ночи. Реакционную смесь разбавляли DCM (30 мл) и водой (30 мл). Образовался осадок, который собирали фильтрованием, однако ЖХ/МС показала, что реакция не прошла до конца. Твердое вещество снова суспендировали в этаноле (3 мл) и обрабатывали гидразингидратом (0,497 мл,10,2 ммоль) и перемешивали при температуре окружающей среды в течение ночи. Реакционную смесь обрабатывали ацетоном (3 мл) и полученную суспензию перемешивали в течение ночи. Твердое вещество собирали фильтрованием и промывали DCM (10 мл) с получением указанного в заголовке соединения(785 мг, 78%) в виде белого твердого вещества. ЖХ/МС RT = 0,81 мин, ЭРИ+ m/z 297 (M+H)+. Промежуточное соединение 13. 2-[7-Фтор-4-(метилокси)-1 Н-индазол-3-ил]-1 Н-изоиндол-1,3(2 Н)-дион 2,3-Дифтор-6-метоксибензонитрил (600 мг, 3,5 ммоль) (доступен от JRD Fluorochemicals) растворяли в сухом N-метилпирролидиноне (3 мл), затем обрабатывали гидразин гидратом (4 экв., 0,65 мл) (доступен от Aldrich) и полученную смесь нагревали в микроволновом реакторе Biotage при высоком уровне поглощения в течение 15 мин при 150 С. Добавляли ацетон (3 мл), раствор перемешивали в течение 15 мин и оставляли стоять в течение ночи. Раствор затем наносили на картридж SCX-2 (50 г). Колонку элюировали метанолом и затем 2 М раствором аммиака в этаноле. Основные фракции объединяли и упаривали при пониженном давлении. Полученное неочищенное масло обрабатывали фталевым ангидридом(789 мг, 5,3 ммоль) (доступен от Aldrich) и сухим 1,4-диоксаном (10 мл). Реакционную смесь перемешивали в течение 48 ч при 110 С. Диоксан удаляли при пониженном давлении и остаток распределяли между дихлорметаном (50 мл) и водой (50 мл). Органическую фазу отделяли и водную фазу дополнительно экстрагировали 50 мл дихлорметана. Объединенные органические растворы сушили через гидрофобную фритту и концентрировали при пониженном давлении. Остаток очищали хроматографией на диоксиде кремния (картридж 100 г) на Flashmaster, элюируя с градиентом 0-100% этилацетата в циклогексане за 80 мин, с получением указанного в заголовке соединения (510 мг, 47%). ЖХ/МС (система A) RT = 0,98 мин, ЭРИ+ m/z 312 (М+Н)+. Промежуточное соединение 14. 3-[3-Амино-7-фтор-4-(метилокси)-1 Н-индазол-1-ил]метилбензамид Раствор 2-[7-фтор-4-(метилокси)-1 Н-индазол-3-ил]-1 Н-изоиндол-1,3(2H)-диона (промежуточное соединение 13) (62,0 мг, 0,2 ммоль) в безводном DMF (2,5 мл) обрабатывали карбонатом калия (55,1 мг,0,4 ммоль), затем 3-(хлорметил)бензамидом (40,5 мг, 0,24 ммоль) и перемешивали при 80 С в течение 17 ч. Охлажденную реакционную смесь распределяли между насыщенным водным раствором гидрокарбоната натрия и этилацетатом. Органический слой отделяли, промывали смесью рассол-вода (1:1), пропускали через гидрофобную фритту и упаривали в вакууме с получением бледно-коричневого твердого вещества, которое использовали на следующей стадии без дополнительной очистки. ЖХ/МС (система В) RT = 2,45 мин, ЭРИ+ m/z 445 (М+Н)+. Твердое вещество суспендировали в безводном этаноле (3 мл) и обрабатывали при комнатной температуре моногидратом гидразина (0,048 мл, 1 ммоль). Полученную суспензию перемешивали при комнатной температуре в течение 1 ч. Реакционную смесь обрабатывали ацетоном (3 мл) и перемешивали при комнатной температуре в течение 30 мин. Смесь упаривали в вакууме с получением оранжевого твердого вещества, которое распределяли между насыщенным водным раствором гидрокарбоната натрия и этилацетатом. Органический слой отделяли, пропускали через гидрофобную фритту и упаривали в вакууме с получением оранжевого твердого вещества. Остаток растворяли в смеси MeOH-DMSO (1 мл) и очищали методом МНАП на колонке Sunfire C18, элюируя растворителями А/В (А: 0,1%-ный (об./об.) раствор муравьиной кислоты в воде, В: 0,1%-ный (об./об.) раствор муравьиной кислоты в ацетонитриле) за 25 мин. Соответствующую фракцию распределяли между насыщенным водным раствором гидрокарбоната натрия и дихлорметаном. Органический слой пропускали через гидрофобную фритту и упаривали в вакууме с получением белого твердого вещества (22 мг, 35%). ЖХ/МС (система В) RT = 1,96 мин, ЭРИ+ m/z 315 (М+Н)+.(7,5 мл) в атмосфере азота обрабатывали по каплям раствором трибромида бора в DCM (1 M, 0,533 мл,0,533 ммоль) при комнатной температуре и реакционную смесь перемешивали при комнатной температуре в течение 1 ч (реакционная смесь представляла собой не раствор, а липкую оранжевую смолу). Добавляли дополнительную порцию трибромида бора (1 М, 0,533 мл) и реакционную смесь перемешивали при комнатной температуре в течение 1 ч (реакционная смесь все еще не представляла собой раствор). Безводный метанол добавляли по каплям до тех пор, пока реакционная смесь не становилась раствором. Реакционную смесь затем перемешивали при комнатной температуре в течение последующих 30 мин. Осторожно по каплям добавляли насыщенный раствор бикарбоната натрия (10 мл), чтобы погасить реакцию, при этом образовались как белый осадок (суспендированный в водном слое), так и темно-зеленая липкая смола (прилипшая к боковой стенке колбы). Водный и органический слои наносили на гидрофобную фритту и органический слой собирали и отставляли. Оставшуюся в колбе зеленую смолу растворяли в смеси 1:1 метанол-дихлорметан, пропускали через гидрофобную фритту, объединяли с отставленным органическим слоем и упаривали в вакууме с получением темно-зеленого липкого твердого вещества (265 мг). Его растворяли в DMSO (31 мл) и очищали методом МНАП ВЭЖХ на колонке SunfireC18, элюируя растворителями А/В (А: 0,1%-ный (об./об.) раствор муравьиной кислоты в воде, В: 0,1%ный (об./об.) раствор муравьиной кислоты в ацетонитриле) (прогон 25 мин). Соответствующие фракции объединяли и упаривали в вакууме с получением указанного в заголовке соединения в виде бесцветного масла (86 мг, 33%). ЖХ/МС (система В) RT = 1,63 мин, ЭРИ+ m/z 449/451 (М+Н)+. Промежуточное соединение 16. Метил-3-[2-(диметиламино)-2-оксоэтил]оксибензоат(10 мл) перемешивали в течение 1 ч. Добавляли 2-хлор-N,N-диметилацетамид (Merck) (1,03 мл,10 ммоль), смесь перемешивали в течение 1 ч при комнатной температуре и затем выдерживали при комнатной температуре в течение 6 суток. Смесь затем подкисляли 2 М HCl и экстрагировали EtOAc. Водный слой дважды экстрагировали EtOAc и объединенные органические растворы промывали 2 МHCl (3), насыщенным водным раствором NaHCO3, рассолом, сушили (MgSO4) и упаривали при пониженном давлении с получением указанного в заголовке соединения (2,22 г, 94%) в виде белого твердого вещества. ЖХ/МС (система A) RT = 0,80 мин, 91%, ЭРИ+ m/z 238 (М+Н)+. Промежуточное соединение 17. Метил-3-[2-(диметиламино)-2-оксоэтил]оксибензоат (промежуточное соединение 16) (2,22 г,9,36 ммоль) растворяли в THF (15 мл) и охлаждали во льду. Раствор затем обрабатывали по каплям эфирным раствором LiAlH4 (1 M, 10 мл) в атмосфере азота и смесь перемешивали в ледяной бане в течение 1,5 ч. ЖХ/МС показала почти завершение реакции. Смесь охлаждали во льду и в нее добавляли насыщенный водный раствор сульфата натрия до образования белого осадка. Смесь перемешивали в течение 2 ч и затем оставляли стоять в течение выходных дней при комнатной температуре. Смесь затем фильтровали, твердое вещество промывали этилацетатом и фильтрат и промывочные жидкости концентрировали при пониженном давлении. Полученное бесцветное масло растворяли в метаноле и наносили на предварительно промытый картридж SCX-2. Картридж промывали метанолом и затем элюировали 10%-ным водным раствором аммиака в метаноле. Аммиачные фракции объединяли и упаривали при по- 24021740(150 мг,0,28 ммоль),(3-[2-(диметиламино)этил]оксифенил)метанола (73 мг, 0,37 ммоль), трифенилфосфина (113 мг,0,43 ммоль) и ди-трет-бутилазодикарбоксилата (132 мг, 0,57 ммоль) в THF (15 мл) нагревали до 65 С в течение ночи. Смесь упаривали при пониженном давлении, остаток растворяли в MeOH, а затем наносили на ионообменный картридж SCX-2 (20 г) и элюировали последовательно метанолом, 2 М аммиаком в метаноле. Соответствующие фракции объединяли и упаривали в вакууме с получением неочищенного продукта. Его растворяли в DCM и очищали хроматографией на диоксиде кремния (картридж 20 г) наFlashmaster, используя смесь 0-15% метанола (содержащего 1% Et3N)-дихлорметан за период времени 20 мин. Соответствующую фракцию упаривали в вакууме с получением указанного в заголовке продукта(10 мл) обрабатывали 2-хлор-N,N-диметилацетамидом (Merck) (1,03 мл, 10 ммоль) и смесь перемешивали при комнатной температуре в течение ночи. Реакционную смесь распределяли между этилацетатом и 2 М соляной кислотой, промывали HCl, рассолом, сушили (MgSO4) и упаривали досуха. Остаток растворяли в DMF (5 мл) и обрабатывали карбонатом калия (1,38 г, 10 ммоль) и 2-хлор-N,N-диметилацетамидом (Merck) (1,03 мл, 10 ммоль). Смесь перемешивали при комнатной температуре в течение выходных дней и распределяли между этилацетатом и 2 М HCl и органический раствор промывали HCl (2), рассолом (3), сушили (MgSO4) и упаривали с получением указанного в заголовке соединения (1,62 г, 68%) в виде белого твердого вещества. ЖХ/МС (система A) RT = 0,73 мин, ЭРИ+ m/z 238 (М+Н)+. Промежуточное соединение 20. Метил-4-[2-(диметиламино)-2-оксоэтил]оксибензоат (промежуточное соединение 19) (1,61 г,6,79 ммоль) в THF (40 мл) охлаждали во льду и затем обрабатывали 1 М раствором LiAlH4 в диэтиловом эфире (6,8 мл) и смесь перемешивали в атмосфере азота в течение 4 ч. Реакционную смесь осторожно обрабатывали насыщенным водным раствором сульфата натрия до прекращения газовыделения. Смесь затем перемешивали в атмосфере азота в течение 1 ч и фильтровали. Твердое вещество промывали диэтиловым эфиром и объединенный фильтрат и промывочные жидкости упаривали при пониженном давлении с получением указанного в заголовке соединения (1,32 г, 100%). ЖХ/МС (система В) RT = 1,70 мин, ЭРИ+ m/z 196 (М+Н)+.(4-[2-(диметиламино)этил]оксифенил)метанола (98 мг, 0,5 ммоль), трифенилфосфина (197 мг,0,75 ммоль) и ди-трет-бутилазодикарбоксилата (230 мг, 1 ммоль) в THF (30 мл) нагревали до 65 С в течение 1 ч. ЖХ/МС показала, что реакция завершена. Смесь концентрировали и остаток растворяли в метаноле, наносили на ионообменный картридж SCX-2 (50 г) и элюировали метанолом, затем 10%-ным водным раствором аммиака в метаноле. Аммиачные фракции объединяли и упаривали в вакууме с получением неочищенного продукта (400 мг) в виде желтой смолы. Смесь наносили в дихлорметане на картридж с диоксидом кремния (20 г) и очищали хроматографией на Flashmaster, используя градиент 0-15% метанола (содержащего 1% Et3N)-дихлорметан за период времени 40 мин. Соответствующие фракции объединяли и упаривали в вакууме с получением продукта, загрязненного примесями, (167 мг) в виде бесцветной смолы. Образец растворяли в смеси 1:1 MeOH-DMSO (2 мл) и очищали методом МНАП ВЭЖХ на колонке Xbridge, используя систему растворителей ацетонитрил:вода с модификатором карбонатом аммония. Соответствующие фракции объединяли и растворитель выпаривали в вакууме с получением указанного в заголовке соединения (91 мг, 26%). ЖХ/МС (система С) (прогон 5 мин) RT = 3,76 мин, ЭРИ+ m/z 701/703 (М+Н)+. Промежуточное соединение 22. Метил-4-[2-(диметиламино)-2-оксоэтил]окси-3-(метилокси)бензоат Метил-4-гидрокси-3-(метилокси)бензоат (метилваниллат, Aldrich) (3,85 г, 21,1 ммоль) и карбонат калия (2,92 г, 21,1 ммоль) в DMF (10 мл) обрабатывали при комнатной температуре 2-хлор-N,N-диметилацетамидом (Merck) (2,18 мл, 21,1 ммоль) и смесь перемешивали в течение выходных дней. Смесь распределяли между этилацетатом и 2 М HCl. Органическую фазу промывали 2 М HCl,насыщенным раствором бикарбоната натрия (становится слегка окрашенной), кислотой (теряет окраску),рассолом, сушили (MgSO4) и упаривали при пониженном давлении с получением продукта, содержащего примесь исходного вещества (5,0 г). Смесь затем растворяли в DMF (5 мл) и обрабатывали карбонатом калия (1,38 г, 10 ммоль) и 2-хлор-N,N-диметилацетамидом (1 мл, 10 ммоль) в течение следующих суток. Смесь затем распределяли между этилацетатом и 2 М HCl и обрабатывали так, как указано выше. Фильтрат упаривали с получением твердого вещества (2,614 г), которое загружали в хлороформе в картридж с диоксидом кремния (70 г) и очищали хроматографией на Flashmaster, используя градиент 0-100% этилацетата-циклогексан в течение 30 мин. Соответствующие фракции объединяли и упаривали в вакууме с получением указанного в заголовке соединения (1,4 г, 25%) в виде белого твердого вещества. ЖХ/МС (система A) RT = 0,69 мин, ЭРИ+ m/z 268 (М+Н)+. Промежуточное соединение 23. Метил-4-[2-(диметиламино)-2-оксоэтил]окси-3-(метилокси)бензоат (промежуточное соединение 22) (1,4 г, 5,2 ммоль) растворяли в THF (50 мл) и охлаждали в бане лед-вода в атмосфере азота. Смесь затем осторожно обрабатывали 1 М эфирным раствором алюмогидрида лития (5,5 мл) и смесь перемешивали при 5 С, давая возможность нагреться до комнатной температуры в течение ночи. Реакционную смесь гасили осторожным добавлением насыщенного водного раствора NaHCO3 (1 мл) с последующим добавлением MgSO4 (2 г) и перемешиванием в течение 30 мин. Твердые вещества удаляли фильтрованием, промывали этилацетатом и фильтрат упаривали с получением указанного в заголовке соединения К раствору 3-(аминосульфонил)бензойной кислоты (Fluorochem) (150 мг, 0,746 ммоль) в THF (1 мл) добавляли по каплям при 0 С комплекс боран-THF (3,73 мл, 3,73 ммоль). Полученный раствор перемешивали 10 мин при 0 С и затем при комнатной температуре в течение ночи. Реакционную смесь осторожно гасили 10 мл воды при 0 С. Реакционную смесь разбавляли 30 мл EtOAc и 20 мл воды. Водный слой затем экстрагировали 30 мл EtOAc. Объединенные органические растворы промывали рассолом(30 мл), сушили над гидрофобной фриттой и концентрировали при пониженном давлении. Остаток наносили в дихлорметане на картридж с диоксидом кремния (20 г) и очищали хроматографией на Flashmaster,используя градиент 0-100% этилацетата-дихлорметан, в течение 40 мин. Соответствующие фракции объединяли и упаривали в вакууме с получением указанного в заголовке соединения (86,4 мг, 62%) в виде смолы. ЖХ/МС (система A) RT = 0,41 мин, ЭРИ+ m/z 188 (М+Н)+. Промежуточное соединение 25. 1-[3,4-бис-(Метилокси)фенил]метил-4-(метилокси)-1H-индазол-3-амин Порошкообразный KOH (0,494 г, 8,80 ммоль) растворяли в DMSO (20 мл) в атмосфере азота и добавляли 4-(метилокси)-1 Н-индазол-3-амин (промежуточное соединение 1) (0,653 г, 4 ммоль). Смесь перемешивали в течение 15 мин с получением темно-красного раствора. Добавляли одной порцией 4-(хлорметил)-1,2-бис-(метилокси)бензол (0,896 г, 4,80 ммоль) и смесь перемешивали в течение 2 ч. Смесь добавляли к воде (150 мл) и экстрагировали дихлорметаном (350 мл). Высушенный (Na2SO4) экстракт упаривали и остаток растирали с диэтиловым эфиром (33 мл) с получением указанного в заголовке соединения (1,05 г, 84%) в виде оранжевого твердого вещества. ЖХ/МС (система В) RT = 1,42 мин, ЭРИ+ m/z 314 (М+Н)+. Промежуточное соединение 26. 5-Хлор-N-[4-(метилокси)-1H-индазол-3-ил]-2-тиофенсульфонамид Раствор 1,1-диметилэтил-3-[(5-хлор-2-тиенил)сульфонил]амино-4-(метилокси)-1 Н-индазол-1 карбоксилата (промежуточное соединение 6) (155 мг, 0,35 ммоль) растворяли в DCM (0,2 мл) и добавляли TFA (0,4 мл). Раствор выдерживали при комнатной температуре в течение 30 мин и затем продували досуха в потоке азота в продувочной системе Radley с получением указанного в заголовке соединения Перемешиваемую суспензию N-[1-[3-(аминометил)фенил]метил-4-(метилокси)-1 Н-индазол-3-ил]5-хлор-2-тиофенсульфонамида (40,5 г, 87 ммоль) в дихлорметане (1000 мл) и триэтиламине (36,4 мл,262 ммоль) охлаждали и затем по каплям добавляли 2-хлор-1,1-диметил-2-оксоэтилацетат (12,52 мл,87 ммоль). Реакционную смесь перемешивали при комнатной температуре в атмосфере азота в течение 1 ч и 20 мин. Затем ее промывали 2 М HCl (300 мл), раствором NaHCO3 (300 мл), рассолом, сушили над сульфатом магния и фильтровали. Фильтрат упаривали в вакууме с получением белого твердого вещества. Этот остаток растворяли в DCM и наносили на колонку с диоксидом кремния (1500 г) и элюировали с градиентом 40-100% этилацетата в циклогексане при пропускании 8 КО. Нужные фракции объединяли и упаривали в вакууме с получением указанного в заголовке соединения двумя порциями (37,87 г, 73%) и(3,39 г) в виде белой пены. ВЭЖХ показала, что последняя порция не является чистой, поэтому ее снова очищали на колонке с диоксидом кремния (120 г), используя градиент 40-75% этилацетата в циклогексане при пропускании 8 КО. Нужные фракции объединяли и упаривали в вакууме с получением указанного в заголовке соединения (2,16 г, 4%) в виде белой пены. ЖХ/МС (система A) RT = 1,10 мин, ЭРИ+ m/z 591/593 (М+Н)+. Промежуточное соединение 28. 1,1-Диметилэтил-[(4-[3-бис-[(5-хлор-2-тиенил)сульфонил]амино-4-(метилокси)-1 Н-индазол-1 ил]метилфенил)метил]карбамат К раствору 5-хлор-N-[(5-хлор-2-тиенил)сульфонил]-N-[4-(метилокси)-1 Н-индазол-3-ил]-2 тиофенсульфонамида (промежуточное соединение 10) (400 мг, 0,763 ммоль), трифенилфосфина (400 мг,1,52 ммоль) и 1,1-диметилэтил-[4-(гидроксиметил)фенил]метилкарбамата (Maybridge) (362 мг,1,52 ммоль) в THF (3 мл) при комнатной температуре добавляли DIAD (0,300 мл, 1,52 ммоль). Полученную смесь перемешивали при 65 С в течение 3 ч. Реакционную смесь распределяли между DCM (3 мл) и водой (3 мл). Органическую фазу отделяли и водный слой дополнительно экстрагировали DCM (3 мл). Объединенные органические растворы сушили над гидрофобной фриттой и упаривали в потоке азота в продувочной системе. Остаток наносили в дихлорметане на картридж с диоксидом кремния (50 г) и очищали хроматографией на Flashmaster II, используя градиент 0-100% дихлорметана-циклогексан за период времени 40 мин. Соответствующие фракции объединяли и упаривали в вакууме с получением указанного в заголовке соединения (498 мг, 88%) в виде смолы. ЖХ/МС (система A) RT = 1,48 мин, ЭРИ+ m/z 760/762 (M+NH4)+. Промежуточное соединение 29. 1,1-Диметилэтил-[(4-[3-[(5-хлор-2-тиенил)сульфонил]амино-4-(метилокси)-1 Н-индазол-1 ил]метилфенил)метил]карбамат К раствору 1,1-диметилэтил-[(4-[3-бис-[(5-хлор-2-тиенил)сульфонил]амино-4-(метилокси)-1 Ниндазол-1-ил]метилфенил)метил]карбамата (промежуточное соединение 28) (497 мг, 0,67 ммоль) в метаноле (3 мл) добавляли гидроксид натрия (2 М, 3,34 мл) и эту смесь перемешивали при 60 С в течение 3 ч. Растворители выпаривали в потоке азота в продувочной системе. Остаток распределяли между DCM(5 мл) и водой (5 мл) и добавляли разбавленный раствор HCl до рН водного слоя примерно 1. Органическую фазу отделяли и водный слой дополнительно экстрагировали DCM (5 мл). Объединенные органи- 28021740 ческие растворы промывали рассолом (2 мл), сушили над гидрофобной фриттой и концентрировали в потоке азота в продувочной системе. Остаток растворяли в смеси 1:1 MeOH-DMSO (1 мл) и очищали в продувочной системе Radley (колонка Supelcosil ABZ+Plus), элюируя растворителями А/В (А: вода + 0,1% муравьиной кислоты, В: MeCN:вода 95:5 + 0,05% муравьиной кислоты). Растворитель удаляли в потоке азота в продувочной системе Radley с получением указанного в заголовке соединения (112 мг,30%) в виде белого твердого вещества. ЖХ/МС (система A) RT = 1,26 мин, ЭРИ+ m/z 563/565 (М+Н)+. Промежуточное соединение 30. К раствору 1,1-диметилэтил-[(4-[3-[(5-хлор-2-тиенил)сульфонил]амино-4-(метилокси)-1 Ниндазол-1-ил]метилфенил)метил]карбамата (промежуточное соединение 29) (92 мг, 0,16 ммоль) в дихлорметане (3 мл) по каплям добавляли хлористый водород (0,041 мл, 0,163 ммоль). Полученную смесь перемешивали при комнатной температуре в течение 4 ч, а затем по каплям добавляли дополнительную порцию хлористого водорода (0,041 мл, 0,163 ммоль) и эту смесь перемешивали при комнатной температуре в течение ночи. Растворители затем упаривали в потоке азота в продувочной системе. Остаток растворяли в смеси 1:1 MeOH-DMSO (1 мл) и очищали масс-направленной автоматизированной хроматографией (колонка supelcosil ABZ+Plus), элюируя растворителями А/В (А: вода + 0,1% муравьиной кислоты, В: MeCN:вода 95:5 + 0,05% муравьиной кислоты). Растворитель сушили в потоке азота в продувочной системе Radley с получением указанного в заголовке соединения (69 мг, 83%). ЖХ/МС (система Е) RT = 2,10 мин, ЭРИ+ m/z 463/465 (М+Н)+. Промежуточное соединение 31. Метил-3-[3-(5-хлор-2-тиенил)сульфонил]([2-(триметилсилил)этил]оксиметил)амино]-4(метилокси)-1 Н-индазол-1-ил]метилбензоат Раствор 5-хлор-N-[4-(метилокси)-1 Н-индазол-3-ил]-N-([2-(триметилсилил)этил]оксиметил)-2 тиофенсульфонамида (промежуточное соединение 8) (1 г, 2,11 ммоль) в DMF (10 мл) обрабатывали при температуре окружающей среды карбонатом калия (0,583 г, 4,22 ммоль) и метил-3(бромметил)бензоатом (Alfa Aesar) (0,580 г, 2,53 ммоль). Полученную смесь перемешивали при 50 С в течение 2 ч, и ЖХ/МС затем показала продукт как основной пик. Смесь разбавляли DCM (20 мл) и водой(20 мл). Органическую фазу отделяли и водный слой дополнительно экстрагировали DCM (20 мл). Объединенные органические растворы промывали рассолом (30 мл), сушили над гидрофобной фриттой и концентрировали при пониженном давлении. Остаток наносили в дихлорметане на картридж с диоксидом кремния (100 г) и очищали хроматографией на Flashmaster II, используя градиент 0-100% В в А,причем A представлял собой чистый DCM, а В представлял собой 5%-ный раствор EtOAc в DCM, за период времени 40 мин, и затем дополнительно очищали на втором картридже с диоксидом кремния (70 г),используя градиент 0-100% В в А, причем A представлял собой чистый DCM, а В представлял собой 5%ный раствор EtOAc в DCM, за период времени 40 мин. Соответствующие фракции объединяли и упаривали в вакууме с получением указанного в заголовке соединения (571 мг, 44%) в виде смолы. ЖХ/МС (система A) RT = 1,60 мин, ЭРИ+ m/z 639/641 (M+NH4)+.
МПК / Метки
МПК: C07D 409/14, C07D 413/14, A61P 11/08, A61P 29/00, C07D 409/12, A61K 31/5377, A61P 37/08, A61K 31/416, A61P 11/06
Метки: используемые, качестве, производные, рецептора, пиразола, антагонистов
Код ссылки
<a href="https://eas.patents.su/30-21740-proizvodnye-pirazola-ispolzuemye-v-kachestve-antagonistov-receptora-ccr4.html" rel="bookmark" title="База патентов Евразийского Союза">Производные пиразола, используемые в качестве антагонистов рецептора ccr4</a>
Серосодержащие производные пиразола в качестве избирательных антагонистов рецептора каннабиноидов св1

Номер патента: 15107
Опубликовано: 30.06.2011
Авторы: Ланге Йозефус Х.М., Ван Влиет Бернард Й., Крюсе Корнелис Г.
МПК: C07D 231/18, A61P 25/30, A61K 31/4155...
Метки: рецептора, избирательных, антагонистов, серосодержащие, пиразола, св1, качестве, производные, каннабиноидов
Формула / Реферат:
1. Соединения общей формулы (I)где R1означает Н, Cl или Br;R2 означает Cl или Br;X означает атом серы, сульфоксидную (S=O) или сульфоновую (SO2) группу;Y означает метильную или этильную группу;n может иметь значения 1, 2 или 3,таутомеры, стереоизомеры указанных соединений и изотопно меченые соединения формулы (I), а также фармакологически приемлемые соли, гидраты и сольваты указанных соединений формулы (I) и их таутомеры, стереоизомеры или...
Трициклические производные пиразола в качестве модуляторов каннабиноидного рецептора

Номер патента: 11307
Опубликовано: 27.02.2009
Авторы: Лохрай Видья Бхушан, Лохрай Брадж Бхушан, Сривастава Бриджеш
МПК: C07D 231/54, A61K 31/416, A61K 31/4162...
Метки: производные, каннабиноидного, модуляторов, качестве, пиразола, трициклические, рецептора
Формула / Реферат:
1. Соединения общей формулы (I) их фармацевтически приемлемые соли, где Ar представляет собой фенильную группу, замещенную 1-2 атомами галогена; А представляет собой замещенную фенильную группу, Х выбирают из атома кислорода или атома серы, m и n представляют собой целые числа, так что m+n=2; или А представляет собой замещенную дигидротиазолильную группу; Х выбирают из -CH2- или атома серы, m и n представляют собой целые числа, так что 1_m+n_2;...
Производные триазола в качестве антагонистов рецептора тахикинина

Номер патента: 7720
Опубликовано: 29.12.2006
Авторы: Хонг Дзиан Эрик, Юнгхайм Луи Николас, Робертсон Майкл Алан, Эмбре Эрик Джеймс, Ремик Дэвид Майкл, Мюль Брайан Стефен, Амегадзие Альберт Кудзови, Савин Кеннет Аллен, Гардинье Кевин Мэттью
МПК: C07D 401/04, A61K 31/4192, C07D 249/06...
Метки: рецептора, антагонистов, тахикинина, производные, триазола, качестве
Формула / Реферат:
1. Соединение формулы (I) где D означает (C1-C3)алкандиил; R1 означает фенил, который необязательно замещен от одного до трех заместителями, независимо выбираемыми из группы, состоящей из атома галогена, (C1-C4)алкила, (C1-C4)алкоксила, цианогруппы, дифторметила, трифторметила и трифторметоксигруппы; R4 означает радикал, выбираемый из группы, состоящей из где -А1-А2-А3-А4- вместе с атомами, с которыми они связаны, образуют ароматический...
Производные циклопропила в качестве антагонистов рецептора nk3

Номер патента: 9477
Опубликовано: 28.02.2008
Авторы: Поульсен Андерс, Келер Ян, Нергор Мортен Банг, Бьернхольм Берит, Нильсен Серен Меллер, Хансен Туре, Руланд Томас
МПК: A61P 25/00, A61K 31/435, A61K 31/495...
Метки: рецептора, качестве, производные, циклопропила, антагонистов
Формула / Реферат:
1. Соединение формулы (I) или его фармацевтически приемлемая соль; где R1-R5 независимо выбирают из водорода и галогена; R6 выбирают из водорода и C1-6алкила; R7 является группой фенил-CR8R9-, R8 и R9 независимо выбирают из водорода и C1-6алкила; n равно 0, 1 или 2; Q выбирают из (ii), (iii), стрелки указывают на место присоединения: где R11 выбирают из фенила или бензила; R12 является фенилом; R13 является водородом, или гидрокси, или одной...
Производные циклогексиламида и их применение в качестве антагонистов рецептора crf-1

Номер патента: 20921
Опубликовано: 27.02.2015
Авторы: Станли Эмили, Битти Дейвид, Колсон Анни-Одиль, Калшо Эндрью Джеймс, Руни Лайза, Свириденко Лиля
МПК: A61K 31/427, A61K 31/44, A61K 31/166...
Метки: циклогексиламида, рецептора, применение, качестве, производные, crf-1, антагонистов
Формула / Реферат:
1. Соединение формулы XIв которой R7 обозначает водород или C1-C6-алкил;R8 обозначает фенил или гетероарил, который представляет собой 5-14-членную моноциклическую или бициклическую или полициклическую ароматическую кольцевую систему, содержащую от 1 до 8 гетероатомов, выбранных из N, О или S, каждый из которых необязательно может содержать один или более заместителей, выбранных из группы, включающей C1-C6-алкил, галоген-C1-C6-алкил, галоген,...
Предыдущий патент: Фрикционный болт
Следующий патент: Мультивалентная адъювантная конструкция
Случайный патент: Устройство для измерения деформаций грунта