Сульфоксиминзамещенные анилинопиримидиновые производные в качестве cdk ингибиторов, их получение и применение в качестве лекарственных средств
Номер патента: 19230
Опубликовано: 28.02.2014
Авторы: Зимайстер Герхард, Яутелат Рольф, Шульце Юлия, Люккинг Ульрих, Линау Филип







Формула / Реферат
1. Соединения общей формулы (I)
в которых X представляет собой -O- или -NH-; и
R1 представляет собой метильную, этильную, пропильную или изопропильную группу; и
R2 и R3, независимо друг от друга, представляют собой водород, метильную или этильную группу; и
R4 представляет собой C1-C6-алкильную группу или C3-C7-циклоалкильное кольцо,
и их соли, диастереомеры и энантиомеры.
2. Соединения, как заявлено в п.1, которые отличаются тем, что X представляет собой -O-, и их соли, диастереомеры и энантиомеры.
3. Соединения, как заявлено в любом из пп.1 или 2, которые отличаются тем, что R1 представляет собой метильную группу, и их соли, диастереомеры и энантиомеры.
4. Соединения, как заявлено в любом из пп.1-3, которые отличаются тем, что R2 представляет собой метильную группу, и их соли, диастереомеры и энантиомеры.
5. Соединения, как заявлено в любом из пп.1-4, которые отличаются тем, что R3 представляет собой водород или метильную группу, и их соли, диастереомеры и энантиомеры.
6. Соединения, как заявлено в любом из пп.1-5, которые отличаются тем, что R4 представляет собой метильную или этильную группу или представляет собой циклопропильное кольцо, и их соли, диастереомеры и энантиомеры.
7. Соединения общей формулы (I), как заявлено в п.1, в которых
X представляет собой -O- или -NH-; и
R1 представляет собой метильную группу; и
R2 представляет собой метильную группу; и
R3 представляет собой водород или метильную группу; и
R4 представляет собой метильную или этильную группу или представляет собой циклопропильное кольцо,
и их соли, диастереомеры и энантиомеры.
8. (RS)-S-Циклопропил-S-(4-{[4-{[(1R,2R)-2-гидрокси-1-метилпропил]окси}-5-(трифторметил)пиримидин-2-ил]амино}фенил)сульфоксимид и его соли, диастереомеры и энантиомеры.
9. (R)-S-Циклопропил-S-(4-{[4-{[(1R,2R)-2-гидрокси-1-метилпропил]окси}-5-(трифторметил)пиримидин-2-ил]амино}фенил)сульфоксимид и его соли, диастереомеры и энантиомеры.
10. (S)-S-Циклопропил-S-(4-{[4-{[(1R,2R)-2-гидрокси-1-метилпропил]окси}-5-(трифторметил)пиримидин-2-ил]амино}фенил)сульфоксимид и его соли, диастереомеры и энантиомеры.
11. Способ получения соединений общей формулы (Ia), которые являются соединениями формулы (I), где X представляет собой -O-, включающий следующие стадии a)-h):
а) окисление соединения формулы (IVd), получая сульфоксид формулы (IVc)
b1) прямое иминирование сульфоксида формулы (IVc), получая защищенный сульфоксимин формулы (IVa)
или
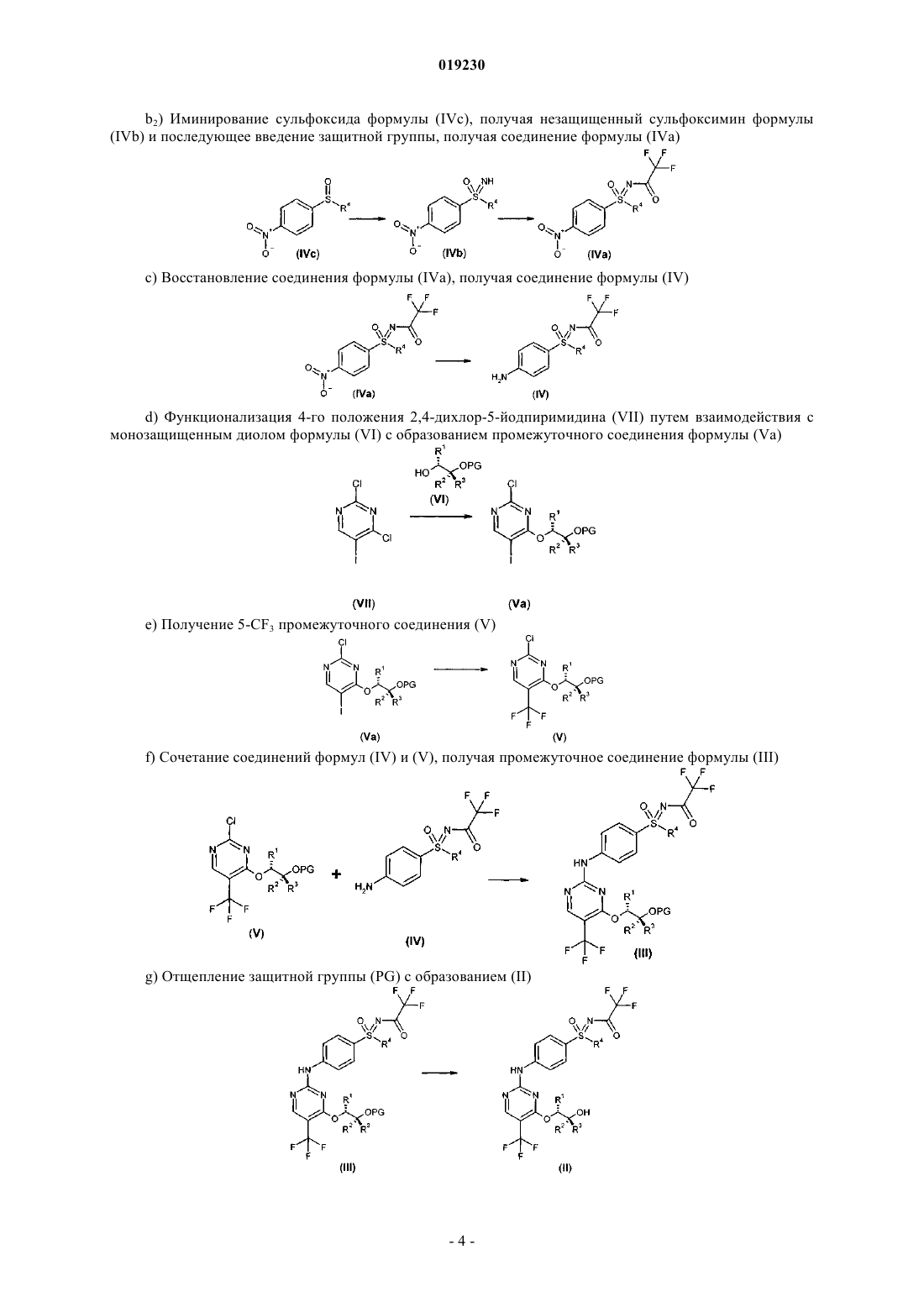
b2) иминирование сульфоксида формулы (IVc), получая незащищенный сульфоксимин формулы (IVb), и последующее введение защитной группы, получая соединение формулы (IVa)
с) восстановление соединения формулы (IVa), получая соединение формулы (IV)
d) функционализация 4-го положения 2,4-дихлор-5-йодпиримидина (VII) путем взаимодействия с монозащищенным диолом формулы (VI) с образованием промежуточного соединения формулы (Va)
е) получение 5-CF3 промежуточного соединения (V)
f) сочетание соединений формулы (IV) и (V) с получением промежуточного соединения формулы (III)
g) отщепление защитной группы PG с образованием (II)
h) отщепление защитной группы на сульфоксимине с образованием (Ia)
которые отличаются тем, что заместители R1, R2, R3 и R4 имеют значения, приведенные в общей формуле (I) пп.1-7.
12. Способ получения соединений общей формулы (Ib), которые являются соединениями формулы (I), где X представляет собой -NH-, включающий следующие стадии a)-f):
а) окисление соединения формулы (IVd), получая сульфоксид формулы (IVc)
b1) прямое иминирование сульфоксида формулы (IVc) с получением защищенного сульфоксимина формулы (IVa)
или
b2) иминирование сульфоксида формулы (IVc) с получением незащищенного сульфоксимина формулы (IVb) и последующее введение защитной группы, получая соединение формулы (IVa)
с) восстановление соединения формулы (IVa), получая соединение формулы (IV)
d) функционализация 4-го положения 2,4-дихлор-5-трифторметилпиримидина (VIIb) путем взаимодействия с амином формулы (VIa) с образованием промежуточного соединения формулы (Vb)
е) сочетание соединений формулы (Vb) и (IV) с получением промежуточного соединения формулы (IIb)
f) отщепление защитной группы на сульфоксимине с образованием формулы (Ib)
которые отличаются тем, что заместители R1, R2, R3 и R4 имеют значения, приведенные в общей формуле (I) пп.1-7.
13. Применение соединения, как заявлено в любом из пп.1-10, в качестве лекарственных средств.
14. Применение соединения, как заявлено в любом из пп.1-10, для получения лекарственного средства для лечения злокачественного новообразования.
15. Применение соединения, как заявлено в любом из пп.1-10, в качестве лекарственных средств против злокачественного новообразования.
16. Фармацевтический состав, содержащий соединение, как заявлено в любом из пп.1-10.
Текст
СУЛЬФОКСИМИНЗАМЕЩЕННЫЕ АНИЛИНОПИРИМИДИНОВЫЕ ПРОИЗВОДНЫЕ В КАЧЕСТВЕ CDK ИНГИБИТОРОВ, ИХ ПОЛУЧЕНИЕ И ПРИМЕНЕНИЕ В КАЧЕСТВЕ ЛЕКАРСТВЕННЫХ СРЕДСТВ Изобретение относится к сульфоксиминзамещенным анилинопиримидиновым производным формулы (I) способам их получения и их применению в качестве лекарственного средства для лечения различных заболеваний.(71)(73) Заявитель и патентовладелец: БАЙЕР ИНТЕЛЛЕКЧУАЛ ПРОПЕРТИ ГМБХ (DE) Настоящее изобретение относится к выбранным сульфоксиминзамещенным анилинопиримидиновым производным, способам их получения и их применению в качестве лекарственного средства для лечения различных заболеваний. Циклинзависимые киназы (CDK) представляют собой семейство ферментов, которые играют важную роль в регуляции клеточного цикла и, таким образом, представляют собой чрезвычайно интересную мишень для развития низкомолекулярных ингибиторных молекул. Селективные ингибиторы CDK могут использоваться для лечения злокачественного новообразования или других заболеваний, которые вызваны расстройствами клеточной пролиферации. Пиримидины и аналоги уже были описаны в качестве активных веществ, например 2-анилинопиримидины в качестве фунгицидов (DE 4029650) или замещенные пиримидиновые производные для лечения неврологических или нейродегенеративных заболеваний (WO 99/19305). Очень разные пиримидиновые производные описаны в качестве CDK ингибиторов, например 2-амино-4-замещенные пиримидины (WO 01/14375), пурины (WO 99/02162), 5-цианопиримидины (WO 02/04429), анилинопиримидины (WO 00/12486) и 2-гидрокси-3-N,N-диметиламинопропоксипиримидины (WO 00/39101). В частности, производные пиримидина описаны в WO 02/096888 и WO 03/076437, которые обладают ингибирующим действием по отношению к CDK. Соединения, которые содержат фенилсульфонамидную группу, являются известными ингибиторами карбоангидразы человека (в особенности, карбоангидразы-2) и применяются в качестве диуретиков, в частности, для лечения глаукомы. Атом азота и атомы кислорода сульфонамида связываются посредством водородных мостиков с ионом цинка 2+ и аминокислотой Thr 199 в активном центре карбоангидразы-2 и, таким образом, блокируют его ферментативную функцию (A. Casini, F. Abbate, A. Scozzafava, C.T. Supuran, Bioorganic. Med. Chem. Lett. 2003, 1,2759). Клиническое применение CDK ингибиторов, содержащих фенилсульфонамидную группу, может быть ограничено вследствие возможности ингибирования карбоангидраз и получающегося в результате этого спектра побочных действий. Примерами сульфоксиминовых активных веществ являются сульфонимидоилмодифицированные триазолы в качестве фунгицидов (Н. Kawanishi, H. Morimoto, Т. Nakano, T. Watanabe, K. Oda,K. Tsujihara, Heterocycles, 1998, 49, 181) или аралкилсульфоксимины в качестве гербицидов и пестицидов(Shell International Research, Ger. P. 2, 129-678). В WO 2005/037800 описаны незамкнутые сульфоксиминзамещенные анилинопиримидиновые производные в качестве ингибиторов циклинзависимых киназ. Представленные примеры представляют собой структуры, которые либо незамещены, либо замещены галогеном, в частности бромом в 5-м положении пиримидина. Никакие из конкретно описанных структур не имели 5-трифторметильного заместителя. С учетом раскрытого известного уровня техники задачей, лежащей в основе настоящего изобретения, является обеспечение соединений, которые являются не только эффективными CDK ингибиторами,но также могут эффективно ингибировать рост опухоли. В действительности, эффективное CDK ингибирование является необходимым, но не достаточным требованием для эффективного ингибирования опухоли. Для структур также необходимы другие свойства, например свойства проникновения в опухолевую клетку. Сейчас было обнаружено, что соединения общей формулы (I)R1 представляет собой метильную, этильную, пропильную или изопропильную группу; иR2 и R3 представляют собой, независимо друг от друга, водород, метильную или этильную группу;R4 представляет собой C1-C6-алкильную группу или C3-C7-циклоалкильное кольцо,и их соли, диастереомеры и энантиомеры не только эффективно ингибируют CDK, но также чрезвычайно эффективно ингибируют рост опухоли. Описание основывается на следующих определениях.C1-C6-Алкильная группа понимается в каждом случае как линейный или разветвленный алкильный остаток, например метильный, этильный, пропильный, изопропильный, бутильный, изобутильный, вторбутильный, трет-бутильный, пентильный, изопентильный или гексильный остаток.C3-C7-Циклоалкильное кольцо понимается как циклопропильное, циклобутильное, циклопентильное, циклогексильное или циклогептильное кольцо. В общей формуле (I) X может представлять собой -O- или -NH-. Предпочтительно X представляет собой -O-. В общей формуле (I) R1 может представлять собой метильную, этильную, пропильную или изопропильную группу. Предпочтительно R1 представляет собой метильную группу. В общей формуле (I) R2 и R3, независимо друг от друга, могут представлять собой водород, метильную или этильную группу. Предпочтительно R2 и R3 представляют собой, независимо друг от друга, водород или метильную группу. Особенно предпочтительно R2 представляет собой метильную группу и R3 представляет собой водород или метильную группу. В общей формуле (I) R4 может представлять собой C1-C6-алкильный остаток илиC3-C7-циклоалкильное кольцо. Предпочтительно R4 представляет собой метильную или этильную группу или представляет собой циклопропильное кольцо. Предпочтительная подгруппа соединений в соответствии с общей формулой (I) включает соединения в соответствии с общей формулой (I), в которыхR1 представляет собой метильную группу; иR2 представляет собой метильную группу; иR3 представляет собой водород или метильную группу; иR4 представляет собой метильную или этильную группу или представляет собой циклопропильное кольцо,и их соли, диастереомеры и энантиомеры. Соединения в соответствии с изобретением пригодны для лечения злокачественного новообразования, такого как солидные опухоли, опухолевые метастазы, и гематологические опухоли, в частности опухоли головы и шеи; легочные и бронхиальные опухоли; опухоли желудочно-кишечного тракта, например рак желудка, колоректальная карцинома, карцинома поджелудочной железы, печеночно-клеточный рак; эндокин-активные опухоли; рак молочной железы и гинекологические опухоли; опухоли мочеполовой системы, например карцинома почки, карцинома мочевого пузыря, рак предстательной железы; опухоли кожи; саркомы; лейкозы и лимфомы; вирусных заболеваний и сердечно-сосудистых заболеваний, таких как стенозы, артериосклерозы и рестенозы, рестенозы,индуцированные стентом. Приготовление лекарственных форм соединений в соответствии с изобретением в виде фармацевтических препаратов осуществляют с помощью способа, известного per se, путем превращения активного вещества или веществ с обычными наполнителями, используемыми для галеновых препаратов с получением желательной дозированной формы. В качестве наполнителей можно использовать, например, носители, заполнители, агенты, вызывающие дезинтеграцию, связующие, гигроскопические вещества, вещества, способствующие скольжению, абсорбенты и адсорбенты, разбавители, растворители, сорастворители, эмульсификаторы, солюбилизаторы, корригенты, красители, консерванты, стабилизаторы, смачивающие вещества, соли для модификации осмотического давления или буферы. В качестве ссылки можно привести Remington's Pharmaceutical Science, 15-e изд. Mack PublishingCompany, East Pennsylvania (1980). Лекарственные средства могут быть представлены в твердой форме, например в виде таблеток, таблеток с сахарным покрытием, пилюль, суппозиториев, капсул, трансдермальных систем; или в полутвердой форме, например в виде мазей, кремов, гелей, суппозиториев, эмульсий; или в жидкой форме, например в виде растворов, настоек, суспензий или эмульсий. Наполнители в контексте изобретения могут представлять собой, например, соли, сахариды (моно-,ди-, три-, олиго- и/или полисахариды), белки, аминокислоты, пептиды, жиры, воски, масла, углеводороды и их производные, где наполнители могут быть природного происхождения или могут быть получены синтетически или частично синтетически. Таблетки, таблетки с сахарным покрытием, капсулы, пилюли, порошки, гранулы, пастилки, суспензии, эмульсии или растворы могут, в частности, рассматриваться для орального или перорального применения. Суспензии, эмульсии и, главным образом, растворы, могут, в частности, рассматриваться для парентерального применения. Получение соединений в соответствии с изобретением. Сульфоксимины, как правило, обладают высокой стабильностью в отношении структуры и конфигурации (С. Bolm, J.P. Hildebrand, J. Org. Chem. 2000, 65, 169). Эти свойства функциональной группы часто представляют возможность даже сильнодействующих условий реакции и позволяют осуществить простую дериватизацию сульфоксиминов на иминном азоте и-углероде. Энатиомерно чистые сульфоксимины также используются в качестве вспомогательных веществ при диастереоселективном синтезе a) S.G. Pyne, Sulfur Reports. 1992, 12, 57; (b) C.R. Johnson,Aldrichimica Acta. 1985, 18, 3). Описано получение энантиомерно чистых сульфоксиминов, например, путем разделения рацемата с энантиомерно чистой камфор-10-сульфоновой кислотой a) C.R. Johnson, C.W. Schroeck, J. Am. Chem.Soc. 1973, 95, 7418; (b) C.S. Shiner, A.H. Berks, J. Org. Chem. 1988, 53, 5542). Другой способ получения оптически активных сульфоксиминов представляет собой стереоселективное иминирование оптически активных сульфоксидов а) С. Bolm, P. Mller, K. Harms, ActChem. Scand. 1996, 50, 305; (b) Y. Tamura, J.Minamikawa, K. Sumoto, S. Fujii, M. Ikeda, J. Org. Chem. 1973, 38, 1239; (c) H. Okamura, С. Bolm, Org. Lett. 2004, 6, 1305). Для обзора статей относительно сульфоксиминов, см., например: (а) М. Regglin, С. Zur, Synthesis. 2000, 1; (b) C.R. Johnson, Aldrichimica Acta. 1985, 18, 3). Следующие примеры поясняют приготовление соединений в соответствии с изобретением, не ограничивая объем заявленных соединений этими примерами. Получение соединений формулы (Ia) (4-O производные). Соединения в соответствии с изобретением могут быть получены с помощью способа, который характеризуется следующими стадиями. а) Окисление соединения формулы (IVd), получая сульфоксид формулы (IVc)b1) Прямое иминирование сульфоксида формулы (IVc), получая защищенный сульфоксимин формулы (IVa)b2) Иминирование сульфоксида формулы (IVc), получая незащищенный сульфоксимин формулы(IVb) и последующее введение защитной группы, получая соединение формулы (IVa) с) Восстановление соединения формулы (IVa), получая соединение формулы (IV)d) Функционализация 4-го положения 2,4-дихлор-5-йодпиримидина (VII) путем взаимодействия с монозащищенным диолом формулы (VI) с образованием промежуточного соединения формулы (Va)f) Сочетание соединений формул (IV) и (V), получая промежуточное соединение формулы (III)g) Отщепление защитной группы (PG) с образованием (II)h) Отщепление защитной группы на сульфоксимине с образованием (Ia) где заместители R1, R2, R3 и R4 имеют значения, указанные в общей формуле (I). Стадия а). Соединение формулы (IVd) окисляют, получая сульфоксид формулы (IVc). Различные методы,включая стереоселективные методы, доступны для превращения простого тиоэфира, получая сульфоксид(см., например: (а) М.Н. Ali и др., Synthesis 1997, 764; b) M.C. Carreno, Chem. Rev. 1995, 95, 1717; с) I. Patel и др., Org. Proc. Res. Dev. 2002, 6, 225; с) N. Khiar и др., Chem. Rev. 2003, 103, 3651). Описанное применение перйодной кислоты/хлорида железа (III) чрезвычайно пригодно для получения соединений формулы (IVc). Стадия b1). Взаимодействие сульфоксида формулы (IVc) с трифторацетамидом в комбинации с диацетатом йодбензола, оксидом магния и каталитическими количествами димера ацетата родия (II) предоставляет возможность получения защищенного сульфоксимина формулы (IVa). Эта реакция является стереоспецифической и осуществляется с сохранением конфигурации в стереоцентре (см.: (а) Н. Okamura, С. Bolm,Org. Lett. 2004, 6, 1305). Стадия b2). Сначала осуществляют реакцию сульфоксида формулы (IVc), получая незащищенный сульфоксимин формулы (IVb). Подходящими методами являются, например:(a) C.R. Johnson, R.A. Kirchhoff, H.G. Corkins, J. Org. Chem. 1974, 39, 2458). Последующее введение защитной группы с образованием соединений формулы (IVa) можно осуществлять, например, как описано, путем взаимодействия с трифторуксусным ангидридом в щелочных условиях. Стадия с). Для последующего восстановления ароматической нитрогруппы с получением соединения формулы (IV) доступны, в принципе, различные условия реакций (см., например: R.C. Larock, ComprehensiveOrganic Transformations, VCH, New York, 1989, 411). Чрезвычайно пригодным является описанное применение хлорида титана (III) или гидрирование, используя палладий на угле. Стадия d). Взаимодействие 2,4-дихлор-5-йодпиримидина (VII) со спиртом формулы (VI) предоставляет возможность получения промежуточного соединения формулы (Va) (см., например: U. Lcking и др.,WO 2007/071455). Стадия е). По существу, доступны различные методы для замены галогена на трифторметильную группу в азотсодержащем гетероароматическом соединении (см., например: (a) G.E. Carr, R.D. Chambers,T.F. Holmes, J. Chem. Soc. Perkin Trans. 1, 1988, 921; (b) F. Cottet, M. Schlosser, Eur. J. Org. Chem. 2002,327; (c) F.G. Njoroge и др., J. Med. Chem. 1997, 40, 4290). В частности, описанное применение йодида меди(I), фторида калия и (трифторметил)триметилсилана в N-метил-2-пирролидиноне и ТГФ пригодно для введения пиримидина (Va) в 5-е положение с образованием промежуточного соединения (V). Стадия f). 2-Хлорпиримидин формулы (V) можно подвергать реакции с анилином формулы (IV), получая промежуточное соединение формулы (III) (см., например: (a) J. Bryant и др., WO 2004/048343). Стадия g). Отщепление защитной группы (PG) от промежуточного соединения (III) приводит к получению промежуточного соединения (II) (см., например: P.J. Kocienski, Protecting Groups, Georg Thieme VerlagStuttgart, New York, 1994). Описанное гидрирование чрезвычайно пригодно для описанного отщепления бензильной группы. Отщепление ТГП группы можно, при необходимости, уже осуществлять в условиях стадии f). Стадия h). Отщепление трифторацето группы на сульфоксимине (II) приводит к получению соединения формулы (Ia). Описанная техника, с использованием карбоната калия в метаноле при комнатной температуре, чрезвычайно пригодна для этого (см., например: (а) Н. Okamura, С. Bolm, Org. Lett. 2004, 6, 1305). Общая информация. Все реакции с соединениями, чувствительными к окислению или чувствительными к гидролизу,осуществляли в атмосфере аргона, с безводными растворителями. За исключением сульфоксиминовых производных, вещества называли с помощью программыAutonom 2000 Name, которая реализована в MDL ISIS Draw. Autonom 2000 Name не принимает любых сульфоксиминов, следовательно, сульфоксимины называли согласно правилам ИЮПАК (IUPAC,Nomenclature of Organic Chemistry, 1979 издание, С-6.3. Sulfoxides and Sulfones, Rule C-633, 633.1 1,78 г (44,6 ммоль) гидрида натрия (60%) порциями добавляли к раствору 3,00 г (40,5 ммоль) циклопропан тиола (получение в соответствии с Е. Block и др., J. Am. Chem. Soc. 1992, 114, 3492) в 100 мл ТГФ/100 мл простого диэтилового эфира и перемешивали в течение 30 мин при комнатной температуре. Затем порциями добавляли 6,00 г (38,7 ммоль) 1-фтор-4-нитробензола. Смесь перемешивали в течение 2 ч при 40 С. После ее охлаждения смесь вводили в воду и экстрагировали бензолом (3). Объединенные органические фазы концентрировали путем упаривания и остаток очищали хроматографически (гексан/этилацетат 95:5). Получали 4,6 г (23,6 ммоль; выход: 61%) продукта. 1 Н-ЯМР (400 МГц, ДМСО):= 8,12 (m, 2 Н), 7,54 (m, 2 Н), 2,35 (m, 1 Н), 1,16 (m, 2 Н), 0,61 (m, 2 Н). Соединение 1.2. 179 мг (1,11 ммоль) хлорида железа (III) добавляли к смеси 7,2 г (36,88 ммоль) 1-циклопропилсульфанил-4-нитробензола в 140 мл ацетонитрила и ее перемешивали в течение 15 мин при комнатной температуре. Затем порциями добавляли 9,25 г (40,57 ммоль) перйодной кислоты при 25 С. Смесь перемешивали в течение 30 мин и после этого добавляли, при перемешивании, к охлажденному насыщенному раствору тиосульфата натрия. После этого ее экстрагировали этилацетатом (2). Объединенные органические фазы высушивали (Na2SO4), фильтровали и концентрировали путем упаривания. Полученный остаток очищали хроматографически (гексан/этилацетат 1:1). Получали 5,93 г 1,58 г (3,58 ммоль) димера ацетата родия (II) добавляли, в атмосфере аргона, к суспензии 15,1 г(71,53 ммоль) (RS)-1-циклопропан сульфинил-4-нитробензола, 17,8 г (157,37 ммоль) трифторацетамида,38,0 г (118,02 ммоль) диацетата йодбензола и 12,7 г (314,73 ммоль) оксида магния в 801 мл ДХМ и перемешивали в течение ночи при комнатной температуре. Смесь фильтровали через целит с отсасыванием и концентрировали путем упаривания. Остаток, который остался, очищали хроматографически (гексан/этилацетат 2:1). Получали 18,0 г (55,97 ммоль; выход: 78%) продукта. 1 Н-ЯМР (400 МГц, ДМСО):= 8,49 (m, 2 Н), 8,25 (m, 2 Н), 3,56 (m, 1 Н), 1,51 (m, 1 Н), 1,41 (m, 1 Н),1,18 (m, 2 Н). Соединение 1.4. 1,4 г палладия на угле (10%/50% влажность) добавляли к раствору 6,9 г (21,44 ммоль)(RS)-S-циклопропил-S-(4-нитрофенил)-N-(трифторацетил)сульфоксимида в 214 мл этанола и 39 мл ТГФ и гидрировали в течение 1 ч при атмосферном давлении при 25 С. Дополнительно добавляли 1,4 г палладия на угле и его гидрировали дополнительно в течение 4,5 ч при атмосферном давлении. Смесь фильтровали, к фильтрату снова добавляли 1,4 г палладия на угле и в завершение гидрировали в течение 45 мин. Смесь фильтровали и концентрировали путем упаривания. Получали 5,8 г (19,91 ммоль; выход: 93%) продукта. 5,00 г (44,6 ммоль) трет-бутилата калия добавляли к раствору 4,00 г (44,4 ммоль) (2R,3R)бутан-2,3 диола в 300 мл ТГФ при комнатной температуре и смесь нагревали в колбе с обратным холодильником в течение 15 мин. Смесь охлаждали приблизительно до 50 С и добавляли 5,3 мл (44,6 ммоль) бензил бромида. Ее нагревали в колбе с обратным холодильником в течение 3 ч и после этого ее перемешивали в течение ночи при комнатной температуре. Смесь разводили этилацетатом и раствором хлорида натрия и после этого промывали с помощью 1 н. раствора соляной кислоты (1) и раствора хлорида натрия (2). Органическую фазу высушивали (Na2SO4), фильтровали и концентрировали путем упаривания. Полученный остаток очищали хроматографически (гексан/этилацетат 1:1). Получали 3,41 г (18,9 ммоль; выход: 43%) продукта. 1 Н-ЯМР (400 МГц, ДМСО):= 7,35 (m, 4 Н), 7,28 (m, 1 Н), 4,52 (m, 3 Н), 3,67 (m, 1 Н), 3,37 (m, 1 Н),1,05 (d, 3 Н), 1,01 (d, 3 Н). Соединение 1.6. 4-1R,2R)-2-Бензилокси-1-метилпропокси)-2-хлор-5-йодпиримидин 2,07 г гидрида натрия (55%) порциями добавляли к 8,55 г (47,4 ммоль) (2R,3R)-3-бензилоксибутан 2-ола в 56 мл простого диэтилового эфира при 0 С при перемешивании. Через 10 мин ледяную баню удаляли и смесь перемешивали дополнительно в течение 3 мин при комнатной температуре. Образованную суспензию добавляли при 0 С к раствору 6,52 г (23,7 ммоль) 2,4-дихлор-5-йодпиримидина. Смесь перемешивали в течение 4 ч при 40 С и после этого добавляли разведенный раствор хлорида натрия. После этого ее экстрагировали этилацетатом (2). Объединенные органические фазы высушивали (Na2SO4),фильтровали и концентрировали путем упаривания. Полученный остаток подвергали хроматографии(трифторметил)триметилсилана добавляли при перемешивании к раствору 4,66 г (11,1 ммоль) 4-1R,2R)2-бензилокси-1-метилпропокси)-2-хлор-5-йодпиримидина в 15,8 мл NMP и 15,8 мл ТГФ при комнатной температуре. Смесь перемешивали в течение 5,5 ч при 80 С. После охлаждения смесь добавляли к разведенному раствору хлорида натрия и экстрагировали этилацетатом (2). Объединенные органические фазы высушивали (Na2SO4), фильтровали и концентрировали путем упаривания. Полученный остаток подвергали хроматографии (гексан/этилацетат 4:1). Получали 2,17 г (6,0 ммоль; выход: 54%) продукта. 1 Н-ЯМР (400 МГц, ДМСО):= 8,81 (s, 1 Н), 7,21 (m, 5 Н), 5,40 (m, 1 Н), 4,57 (d, 1 Н), 4,42 (d, 1 Н), 3,70 0,96 мл 4 н. раствора хлористого водорода в диоксане добавляли к 1,39 г (3,85 ммоль) 4-1R,2R)-2 бензилокси-1-метилпропокси)-2-хлор-5-трифторметилпиримидина и 1,35 г(RS)-S-(4-аминофенил)-S-циклопропил-N-(трифторацетил)сульфоксимида в 18,8 мл ацетонитрила и перемешивали в течение 5 ч при 80 С. После охлаждения смесь разводили этилацетатом и промывали насыщенным раствором гидрокарбоната натрия и насыщенным раствором хлорида натрия, высушивали(Na2SO4), фильтровали и концентрировали путем упаривания. Полученный остаток очищали хроматографически (гексан/этилацетат 4:1). Получали 1,32 г (2,14 ммоль, выход: 56%) продукта. 1 Н-ЯМР (400 МГц, ДМСО):= 10,71 (s, 1H), 8,84 (s, 1H), 8,08 (m, 2H), 7,93 (m, 2 Н), 7,26 (m, 5 Н),5,52 (m, 1 Н), 4,62 (d, 1 Н), 4,51 (d, 1 Н), 3,78 (m, 1 Н), 3,35 (m, 1 Н), 1,37 (m, 5 Н), 1,16 (m, 5 Н). Соединение 1.9. 1,64 г палладия на угле (10%) добавляли к раствору 1,31 г (2,12 ммоль) (RS)-S-(4-[4-[(1R,2R)-2(бензилокси)-1-метилпропил]окси-5-(трифторметил)пиримидин-2-ил]аминофенил)-S-циклопропил-N(трифторацетил)сульфоксимида в 66 мл этанола и гидрировали при атмосферном давлении при комнатной температуре. Смесь фильтровали и концентрировали путем упаривания. Получали 0,88 г(1,67 ммоль; выход: 79%) продукта. 1 Н-ЯМР (400 МГц, ДМСО):= 10,65 (s, 1H), 8,58 (s, 1H), 8,04 (m, 2H), 7,89 (m, 2 Н), 5,28 (m, 1 Н),4,86 (d, 1 Н), 3,82 (m, 1 Н), 3,35 (m, 1 Н), 1,45 (m, 5 Н), 1,15 (m, 5 Н). 1b) Получение конечного продукта. 1130 мг (8,20 ммоль) карбоната калия добавляли к 863 мг (1,64 ммоль) (RS)-S-циклопропил-S-(4[4-[(1R,2R)-2-гидрокси-1-метилпропил]окси-5-(трифторметил)пиримидин-2-ил]аминофенил)-N(трифторацетил)сульфоксимида в 35 мл метанола и перемешивали в течение 1,5 ч при комнатной температуре. Смесь разводили с помощью насыщенного раствора хлорида натрия и экстрагировали этилацетатом (3). Объединенные органические фазы высушивали (Na2SO4), фильтровали и концентрировали путем упаривания. Получали 709 мг (1,64 ммоль) неочищенного продукта. 1 Н-ЯМР (400 МГц, ДМСО):= 10,50 (s, 1H), 8,59 (s, 1H), 7,94 (m, 2H), 7,84 (m, 2 Н), 5,32 (m, 1 Н),4,91 (d, 1 Н), 4,07 (s, 1 Н), 3,86 (m, 1 Н), 2,63 (m, 1 Н), 1,30 (d, 3 Н), 1,11 (m, 4 Н), 0,91 (m, 3 Н). МС: 431 (ES+). Смесь диастереомеров разделяли на чистые стереоизомеры путем препаративной ВЭЖХ: 0,70 г (10,76 ммоль) азида натрия добавляли к 1,56 г (8,42 ммоль) 1-(метилсульфинил)-4 нитробензола в 20 мл ДХМ. 2,3 мл концентрированной серной кислоты медленно добавляли к смеси при 0 С и затем ее нагревали до 45 С. Через 16 ч смесь охлаждали до комнатной температуры и после добавления воды ее экстрагировали с помощью ДХМ. Водную фазу доводили до рН 11 с помощью 15% раствора гидроксида натрия и экстрагировали с помощью ДХМ (2). Объединенные органические фазы высушивали (Na2SO4), фильтровали и концентрировали путем упаривания. Получали 1,08 г (5,39 ммоль; выход: 63%) продукта. 1 Н-ЯМР (400 МГц, ДМСО):= 8,43 (m, 2 Н), 8,17 (m, 2 Н), 4,62 (s, 1H), 3,18 (s, 3 Н). Соединение 2.2. 1,00 мл (7,08 ммоль) трифторуксусного ангидрида добавляли по каплям, при охлаждении с помощью льда, к раствору 1000 мг (4,99 ммоль) (RS)-S-(4-нитрофенил)-S-метилсульфоксимида, 55 мг(0,45 ммоль) DMAP и 0,76 мл (5,49 ммоль) триэтиламина в 32 мл ДХМ. Смесь перемешивали дополнительно в течение 2 ч на ледяной бане. Ее разводили с помощью ДХМ и промывали с помощью полуконцентрированного раствора хлорида натрия. Органическую фазу высушивали (Na2SO4), фильтровали и концентрировали путем упаривания. Полученный остаток очищали хроматографически (гексан/этилацетат 60:40). Полученный продукт в завершение перемешивали с простым диизопропиловым эфиром. Твердое вещество отфильтровали с отсасыванием и высушивали. Получали 1444 мг (4,87 ммоль; выход: 98%) продукта. 1 Н-ЯМР (400 МГц, ДМСО):= 8,50 (m, 2 Н), 8,24 (m, 2 Н), 3,87 (s, 3 Н). Соединение 2.3. 292 мг палладия на угле (10%/50% влажность) добавляли к раствору 1,34 г (4,52 ммоль)(RS)-S-метил-S-(4-нитрофенил)-N-(трифторацетил)сульфоксимида в 45 мл этанола и 8 мл ТГФ и гидрировали в течение 45 мин при атмосферном давлении при 24 С. Смесь фильтровали и концентрировали путем упаривания. Полученный остаток перемешивали с простым диизопропиловым эфиром. Твердое вещество отфильтровали с отсасыванием и высушивали. Получали 1,07 г (4,03 ммоль; выход: 89%) продукта. 1 Н-ЯМР (400 МГц, ДМСО):= 7,54 (m, 2 Н), 6,67 (m, 2 Н), 6,35 (s, 2H), 3,55 (s, 3 Н). 0,97 мл 4 н. раствора хлористого водорода в диоксане добавляли к 1,40 г (3,88 ммоль) 4-1R,2R)-2 бензилокси-1-метилпропокси)-2-хлор-5-трифторметилпиримидина и 1,20 г(RS)-S-(4-аминофенил)-S-метил-N-(трифторацетил)сульфоксимида в 19,0 мл ацетонитрила и после этого смесь перемешивали в течение 6 ч при 80 С. После охлаждения смесь разводили этилацетатом и промывали насыщенным раствором гидрокарбоната натрия и насыщенным раствором хлорида натрия, высушивали (Na2SO4), фильтровали и концентрировали путем упаривания. Полученный остаток очищали хроматографически (гексан/этилацетат 1:1). Получали 1,76 г (2,98 ммоль, выход: 77%) продукта. 1 Н-ЯМР (400 МГц, ДМСО):= 10,66 (s, 1H), 8,60 (s, 1H), 8,02 (m, 2H), 7,93 (m, 2 Н), 7,21 (m, 5 Н),5,46 (m, 1 Н), 4,57 (d, 1 Н), 4,46 (d, 1 Н), 3,72 (m, 1 Н), 3,68 (s, 3 Н), 1,31 (d, 3H), 1,16 (d, 3H). Соединение 2.5. 0,18 г палладия на угле (10%) добавляли к раствору 1,75 г (2,96 ммоль) (RS)-S-(4-[4-[(1R,2R)-2(бензилокси)-1-метилпропил]окси-5-(трифторметил)пиримидин-2-ил]аминофенил)-S-метил-N(трифторацетил)сульфоксимида в 35 мл этанола и его гидрировали при атмосферном давлении при комнатной температуре. Смесь фильтровали и концентрировали путем упаривания. Получали 1,40 г неочищенного продукта. 1 Н-ЯМР (400 МГц, ДМСО):= 10,65 (s, 1H), 8,58 (s, 1H), 8,04 (m, 2H), 7,93 (m, 2 Н), 5,28 (m, 1 Н),4,86 (d, 1 Н), 3,83 (m, 1 Н), 3,70 (s, 3 Н), 1,26 (d, 3 Н), 1,07 (d, 3 Н). 2b) Получение конечного продукта. 1,92 г (13,89 ммоль) карбоната калия добавляли к 1,39 г (2,78 ммоль) (RS)-S-(4-[4-[(1R,2R)-2 гидрокси-1-метилпропил]окси-5-(трифторметил)пиримидин-2-ил]аминофенил)-S-метил-N(трифторацетил)сульфоксимида в 60 мл метанола и перемешивали в течение 1,5 ч при комнатной температуре. Смесь разводили с помощью насыщенного раствора хлорида натрия и экстрагировали с помощью этилацетата (2). Объединенные органические фазы высушивали (Na2SO4), фильтровали и концентрировали путем упаривания. Остаток очищали хроматографически (ДХМ/EtOH 9:1). Получали 862 мг Смесь диастереомеров разделяли на чистые стереоизомеры путем препаративной ВЭЖХ: Раствор 10,0 г (96,1 ммоль) (R)-(+)-метил лактата в 20 мл ТГФ медленно добавляли по каплям к 160 мл (480,0 ммоль) охлажденному на льду 3 н. раствору хлорида метилмагния в ТГФ. Смесь сначала медленно нагревали до комнатной температуры и после этого нагревали с обратным холодильником в течение 30 мин. После охлаждения смесь добавляли к насыщенному раствору хлорида аммония и экстрагировали этилацетатом (3). Объединенные органические фазы фильтровали через фильтр Ватмана и концентрировали путем упаривания. Получали 4,5 г (43,1 ммоль) неочищенного продукта и использовали без дополнительной очистки. 1 Н-ЯМР (400 МГц, ДМСО):= 4,21 (d, 1H), 3,93 (s, 1H), 3,29 (m, 1H), 0,97 (m, 9 Н). Соединение 3.2. 1,84 г (42,3 ммоль) гидрида натрия (55%) порциями добавляли, при перемешивании при 0C, к раствору 4,40 г (42,3 ммоль) (R)-2-метилбутан-2,3-диола в 83 мл простого диэтилового эфира и перемешивали в течение 10 мин. Ее перемешивали дополнительно в течение 3 мин при комнатной температуре и после этого смесь добавляли к охлажденному на льду раствору 9,68 г (35,2 ммоль) 2,4-дихлор-5 йодпиримидина в 97 мл ацетонитрила. Смесь перемешивали в течение 4 ч при 40C и, после охлаждения,добавляли лед и насыщенный раствор NaCl. После этого ее экстрагировали этилацетатом (3). Объединенные органические фазы высушивали (Na2SO4), фильтровали и концентрировали путем упаривания. Полученный остаток очищали хроматографически (гексан/этилацетат 4:1). Получали 4,96 г (14,5 ммоль; выход: 41%) продукта. 1 Н-ЯМР (400 МГц, ДМСО):= 8,73 (s, 1H), 4,96 (q, 1H), 4,62 (s, 1H), 1,21 (d, 3 Н), 1,13 (s, 6H). 2,64 мл (29,0 ммоль) дигидропирана и 0,36 г (1,5 ммоль) тозилата пиридиния добавляли к раствору 4,96 г (14,5 ммоль) (R)-3-(2-хлор-5-йодпиримидин-4-илокси)-2-метилбутан-2-ола в 30 мл ДХМ и перемешивали в течение 22 ч при комнатной температуре. Смесь разводили с помощью ДХМ и промывали насыщенным раствором гидрокарбоната натрия. Органическую фазу высушивали (Na2SO4), фильтровали и концентрировали путем упаривания. Полученный остаток очищали хроматографически (гексан/этилацетат 4:1). Получали 5,50 г (12,9 ммоль; выход: 89%) смеси диастереомеров. 1 Н-ЯМР (400 МГц, ДМСО):= 8,75 (s, 1 Н), 8,74 (s, 1 Н), 5,15 (m, 2 Н), 4,91 (m, 2 Н), 3,70 (m, 2 Н), 3,30(трифторметил)триметилсилана добавляли при комнатной температуре к раствору 1,00 г (2,34 ммоль) 2-хлор-4-[(R)-1,2-диметил-2-(тетрагидропиран-2-илокси)пропокси]-5-йодпиримидина в 3,3 мл NMP и 3,3 мл ТГФ. Смесь перемешивали в течение 2 ч при 90 С. После охлаждения смесь добавляли к разведенному раствору хлорида натрия и экстрагировали этилацетатом (3). Объединенные органические фазы высушивали (Na2SO4), фильтровали и концентрировали путем упаривания. Полученный остаток очищали хроматографически (гексан/этилацетат 4:1). Получали 0,53 г (1,43 ммоль; выход: 61%) продукта. 1 Н-ЯМР (400 МГц, ДМСО):= 8,84 (s, 1 Н), 5,32 (m, 1 Н), 4,85 (m, 1 Н), 3,68 (m, 1 Н), 3,30 (m, 1 Н),1,31 (m, 15 Н). 3b) Получение конечного продукта. 200 мг (0,54 ммоль) 2-хлор-4-[(R)-1,2-диметил-2-(тетрагидропиран-2-илокси)пропокси]-5 трифторметилпиримидина и 87 мг(RS)-S-(4-аминофенил)-S-метил-N(трифторацетил)сульфоксимида в 5 мл этанола перемешивали в течение 6 ч при 70 С. Смесь упаривали насухо на роторном испарителе и остаток ресуспендировали в 11,6 мл. 373 мг (2,70 ммоль) карбоната калия добавляли к раствору и его перемешивали в течение 1,5 ч при комнатной температуре. Смесь разводили с помощью насыщенного раствора хлорида натрия и экстрагировали этилацетатом (2). Объединенные органические фазы высушивали (Na2SO4), фильтровали и концентрировали путем упаривания. Остаток очищали путем ВЭЖХ. Получали 31 мг (0,07 ммоль; выход: 14%) продукта. Н-ЯМР (400 МГц, ДМСО):= 10,48 (s, 1H), 8,56 (s, 1H), 7,90 (m, 2H), 7,83 (m, 2 Н), 5,13 (q, 1 Н),4,67 (s, 1 Н), 4,06 (s, 1 Н), 3,01 (s, 3 Н), 1,28 (d, 3 Н), 1,12 (m, 6 Н). Получение соединений общей формулы (Ib) (4-N производные). Соединения в соответствии с изобретением могут быть получены с помощью способа, который характеризуется следующими стадиями. а) Окисление соединения формулы (IVd), получая сульфоксид формулы (IVc)b1) Прямое иминирование сульфоксида формулы (IVc), получая защищенный сульфоксимин формулы (IVa)b2) Иминирование сульфоксида формулы (IVc), получая незащищенный сульфоксимин формулы(IVb), и последующее введение защитной группы, получая соединение формулы (IVa) с) Восстановление соединения формулы (IVa), получая соединение формулы (IV)d) Функционализация 4-го положения 2,4-дихлор-5-трифторметилпиримидина (VIIb) путем взаимодействия с амином формулы (VIa) с образованием промежуточного соединения формулы (Vb) е) Сочетание соединений формулы (Vb) и (IV), получая промежуточное соединение формулы (IIb)f) Отщепление защитной группы на сульфоксимине с образованием (Ib) где заместители R1, R2, R3 и R4 имеют значения, приведенные в общей формуле (I). Стадии а)-с). Эти стадии идентичны стадиям a)-d) для получения соединений в соответствии с общей формулой(Ia). Стадия d). Взаимодействие 2,4-дихлор-5-трифторметилпиримидина (VIIb) с амином формулы (VIa) обеспечивает смесь продуктов (Vb) и (Vc). Желательный продукт (Vb) может быть выделен, например, хроматографически (см., например: (a) J. Bryant и др., WO 2004/048343). Стадия е). 2-Хлорпиримидин формулы (Vb) можно подвергать реакции с анилином формулы (IV), получая промежуточное соединение формулы (IIb) (см., например: (a) J. Bryant и др., WO 2004/048343). Стадия f). Отщепление трифторацето группы на сульфоксимине (IIb) обеспечивает получение соединения формулы (Ib). Описанная методика с использованием карбоната калия в метаноле при комнатной температуре чрезвычайно пригодна для этого. Пример 4. 72,2 мл (520,71 ммоль) триэтиламина добавляли по каплям при 0 С к 56,5 г (260,35 ммоль) 2,4-дихлор-5-трифторметилпиримидина и 32,7 г (260,35 ммоль) гидрохлорида (2R,3R)-3-аминобутан-2 ола в 1035 мл ацетонитриле. Смесь медленно нагревали в течение ночи до комнатной температуры. Смесь добавляли к полуконцентрированному раствору хлорида натрия и экстрагировали этилацетатом(2). Объединенные органические фазы высушивали (Na2SO4), фильтровали и концентрировали путем упаривания. Остаток, который остался, очищали хроматографически (гексан/этилацетат 0-100%). Получали 18,6 г (68,97 ммоль; выход: 27%) продукта. 1 Н-ЯМР (400 МГц, ДМСО):= 8,38 (s, 1H), 6,71 (d, 1H), 5,00 (d, 1H), 4,08 (m, 1H), 3,71 (m, 1 Н), 1,12(d, 3 Н), 1,01 (d, 3 Н). Получение (RS)-S-(4-аминофенил)-S-циклопропил-N-(трифторацетил)сульфоксимида осуществляли, как описано для соединения 1.4. 4b) Получение конечного продукта. 0,21 мл 4 н. раствора хлористого водорода в диоксане добавляли к 250 мг (0,86 ммоль)(2R,3R)-3-(2-хлор-5-трифторметилпиримидин-4-иламино)бутан-2-ола в 3,75 мл ацетонитрила и затем перемешивали в течение 3,5 ч при 60 С. Смесь упаривали насухо. 18,4 мл метанола и 590 мг (4,28 ммоль) карбоната калия добавляли и ее перемешивали в течение 1 ч при комнатной температуре. Смесь разводили с помощью насыщенного раствора хлорида натрия и экстрагировали этилацетатом (2). Объединенные органические фазы высушивали (Na2SO4), фильтровали и концентрировали путем упаривания. Остаток, который остался, очищали хроматографически (ДХМ/МеОН 4:1). Получали 242 мг (0,56 ммоль; выход: 65%) продукта. 1 Н-ЯМР (400 МГц, ДМСО):= 10,04 (s, 1H), 8,25 (s, 1H), 7,91 (m, 2H), 7,74 (m, 2 Н), 6,05 (d, 1 Н),5,07 (d, 1 Н), 4,14 (m, 1 Н), 3,97 (s, 1 Н), 3,77 (m, 1 Н), 2,56 (m, 1 Н), 1,19 (d, 3 Н), 1,05 (m, 4 Н), 0,86 (m, 3 Н). МС: 430 (ESI+). Смесь диастереомеров разделяли на чистые стереоизомеры путем препаративной ВЭЖХ: 3,6 г (35,03 ммоль) (R)-3-амино-2-метилбутан-2-ола добавляли по каплям к раствору 7,6 г(70,1 ммоль) триэтиламина по каплям при 0 С и смесь медленно нагревали в течение ночи до комнатной температуры. Ее перемешивали дополнительно в течение 2 дней при комнатной температуре. Смесь добавляли к полуконцентрированному раствору хлорида натрия и экстрагировали этилацетатом (2). Объединенные органические фазы высушивали (Na2SO4), фильтровали и концентрировали путем упаривания. Остаток, который остался, очищали путем препаративной ВЭЖХ. Получали 3,0 г (10,65 ммоль; выход: 30%) продукта. Н-ЯМР (400 МГц, ДМСО):= 8,42 (s, 1H), 6,52 (d, 1H), 5,01 (s, 1H), 4,10 (m, 1H), 1,11 (m, 9 Н). Получение (RS)-S-(4-аминофенил)-S-циклопропил-N-(трифторацетил)сульфоксимида осуществляли, как описано для соединения 1.4. 5b) Получение конечного продукта. 0,34 мл 4 н. раствора хлористого водорода в диоксане добавляли к 400 мг (1,37 ммоль)(R)-3-(2-хлор-5-трифторметилпиримидин-4-иламино)-2-метилбутан-2-ола в 6,0 мл ацетонитрила и перемешивали в течение 3,5 ч при 60 С. Смесь упаривали насухо. 29,4 мл метанола и 950 мг (6,84 ммоль) карбоната калия добавляли и ее перемешивали в течение 1 ч при комнатной температуре. Смесь разводили с помощью насыщенного раствора хлорида натрия и экстрагировали этилацетатом (2). Объединенные органические фазы высушивали (Na2SO4), фильтровали и концентрировали путем упаривания. Получали 600 мг (1,35 ммоль) неочищенного продукта. 1 Н-ЯМР (400 МГц, ДМСО):= 10,08 (s, 1H), 8,30 (s, 1H), 7,94 (m, 2H), 7,80 (m, 2 Н), 6,07 (d, 1 Н),4,95 (s, 1 Н), 4,16 (m, 1 Н), 4,02 (s, 1 Н), 2,62 (m, 1 Н), 1,20 (m, 6 Н), 1,10 (m, 4 Н), 0,89 (m, 3 Н). МС: 444 (ESI+). Смесь диастереомеров разделяли на чистые стереоизомеры путем препаративной ВЭЖХ: 16,56 г (106,72 ммоль) 4-нитротиофенола добавляли, при охлаждении с помощью воды, к раствору 4,27 г (106,76 ммоль) гидроксида натрия в 320 мл этанола и его перемешивали в течение 15 мин при комнатной температуре. Затем, при охлаждении с помощью воды, добавляли 8,63 мл (106,79 ммоль) этил йодида и смесь перемешивали в течение ночи при комнатной температуре. Смесь добавляли к насыщенному раствору хлорида натрия и экстрагировали этилацетатом (2). Объединенные органические фазы высушивали (Na2SO4), фильтровали и концентрировали путем упаривания. Затем их растворяли в ДХМ и снова фильтровали и упаривали насухо. Получали 16,86 г (92,02 ммоль) неочищенного продукта. 1 Н-ЯМР (400 МГц, ДМСО):= 8,14 (m, 2 Н), 7,49 (m, 2 Н), 3,14 (q, 2H), 1,31 (tr, 3 Н). Соединение 6.2. 428 мг (2,64 ммоль) хлорида железа (III) добавляли к смеси 16,86 г (92,02 ммоль) 1-этилсульфанил 4-нитробензола в 75 мл ацетонитрила и ее перемешивали в течение 10 мин при комнатной температуре. Затем порциями добавляли 22,44 г (98,44 ммоль) перйодной кислоты таким образом, что температура не превышала 30 С. Смесь перемешивали в течение 50 мин и после этого добавляли, при перемешивании, к смеси 170 мл ДХМ, 500 мл смеси воды со льдом и 100 г пентагидрата тиосульфата натрия. Ее экстрагировали с помощью ДХМ (2). Объединенные органические фазы высушивали (Na2SO4), фильтровали и концентрировали путем упаривания. Полученный остаток перекристаллизовывали из этилацетата/гексана. Получали 12,49 г (62,69 ммоль; выход: 68%) продукта. 1 Н-ЯМР (400 МГц, ДМСО):= 8,35 (m, 2 Н), 7,88 (m, 2 Н), 3,12 (m, 1 Н), 2,84 (m, 1 Н), 0,99 (tr, 3 Н). Соединение 6.3. 30,5 мл дымящей серной кислоты (20% SO3) добавляли осторожно, на ледяной бане, к 6,00 г(30,12 ммоль) (RS)-1-этилсульфинил-4-нитробензола. Затем, в атмосфере аргона, осторожно добавляли 2,35 г (36,14 ммоль) азида натрия, порциями и при перемешивании, и смесь затем нагревали до 45 С. Через 6 ч смесь охлаждали до комнатной температуры и осторожно вливали в лед. Смесь подщелачивали с помощью гидрокарбоната натрия и экстрагировали этилацетатом (2). Объединенные органические фазы высушивали (Na2SO4), фильтровали и концентрировали путем упаривания. Получали 5,74 г 4,53 мл (32,04 ммоль) трифторуксусного ангидрида добавляли по каплям, при охлаждении на льду,к раствору 5,72 г (26,70 ммоль) (RS)-S-этил-S-(4-нитрофенил)сульфоксимида и 4,07 мл (29,37 ммоль) триэтиламина в 175 мл ДХМ. Смесь перемешивали дополнительно в течение 3 ч на ледяной бане, где температура повышалась приблизительно до 10 С. Ее разводили с помощью ДХМ и промывали с помощью полуконцентрированного раствора хлорида натрия. Органическую фазу высушивали (Na2SO4),фильтровали и концентрировали путем упаривания. Получали 8,17 г (26,33 ммоль) продукта. 1 Н-ЯМР (400 МГц, ДМСО):= 8,52 (m, 2 Н), 8,22 (m, 2 Н), 3,99 (m, 2 Н), 1,16 (tr, 3 Н). Соединение 6.5. 88,5 мл 15% раствора хлорида титана (III) приблизительно в 10% соляной кислоте медленно добавляли, при охлаждении на льду, к раствору 4,05 г (13,05 ммоль) (RS)-S-этил-S-(4-нитрофенил)-N(трифторацетил)сульфоксимида в 191 мл ТГФ. Смесь перемешивали в течение 3,5 ч при комнатной температуре, разводили с помощью этилацетата и после этого промывали полуконцентрированным раствором хлорида натрия (3). Органическую фазу высушивали (Na2SO4), фильтровали и концентрировали путем упаривания. Получали 3,17 г (11,31 ммоль) продукта. 1 Н-ЯМР (400 МГц, ДМСО):= 7,48 (m, 2 Н), 6,68 (m, 2 Н), 3,64 (m, 2 Н), 1,06 (tr, 3 Н). Получение (2R,3R)-3-(2-хлор-5-трифторметилпиримидин-4-иламино)бутан-2-ола осуществляли, как описано для соединения 4.1. 6b) Получение конечного продукта. 0,36 мл 4 н. раствора хлористого водорода в диоксане добавляли к 400 мг (1,43 ммоль) (RS)-S-(4 аминофенил)-S-этил-N-(трифторацетил)сульфоксимида и 385 мг (1,43 ммоль) (2R,3R)-3-(2-хлор-5 трифторметилпиримидин-4-иламино)бутан-2-ола в 7,0 мл ацетонитрила и перемешивали в течение 4,5 ч при 60 С. Смесь упаривали насухо. Добавляли 19,0 мл метанола и 608 мг (4,40 ммоль) карбоната калия и ее перемешивали в течение 1 ч при комнатной температуре. Смесь разводили с помощью насыщенного раствора хлорида натрия и экстрагировали этилацетатом (2). Объединенные органические фазы высушивали (Na2SO4), фильтровали и концентрировали путем упаривания. Получали 590 мг (1,41 ммоль) неочищенного продукта. 1 Н-ЯМР (400 МГц, ДМСО):= 10,10 (s, 1H), 8,30 (s, 1H), 7,96 (m, 2H), 7,78 (m, 2 Н), 6,10 (d, 1 Н),5,11 (d, 1 Н), 4,19 (m, 1 Н), 4,00 (s, 1 Н), 3,82 (m, 1 Н), 3,07 (q, 2H), 1,24 (d,3H), 1,06 (m,6 Н).MC: 418 (ESI+). Смесь диастереомеров разделяли на чистые стереоизомеры путем препаративной ВЭЖХ: 7 а) Получение промежуточных соединений. Получение (RS)-S-(4-аминофенил)-S-этил-N-(трифторацетил)сульфоксимида осуществляли, как описано для соединения 6.5. Получение (R)-3-(2-хлор-5-трифторметилпиримидин-4-иламино)-2-метилбутан-2-ола осуществляли, как описано для соединения 5.1. 7b) Получение конечного продукта. 0,36 мл 4 н. раствора хлористого водорода в диоксане добавляли к 400 мг (1,43 ммоль)(5,70 ммоль) карбоната калия и ее перемешивали в течение 1 ч при комнатной температуре. Смесь разводили с помощью насыщенного раствора хлорида натрия и экстрагировали этилацетатом (2). Объединенные органические фазы высушивали (Na2SO4), фильтровали и концентрировали путем упаривания. Получали 620 мг (1,43 ммоль) неочищенного продукта. 1 Н-ЯМР (400 МГц, ДМСО):=10,06 (s, 1H), 8,28 (s, 1H), 7,92 (m, 2H), 7,74 (m, 2 Н), 6,03 (d, 1 Н), 4,90(s, 1 Н), 4,12 (m, 1 Н), 3,96 (s, 1 Н), 3,03 (q, 2 Н), 1,16 (m, 6 Н), 1,08 (m, 3 Н), 1,02 (tr, 3 Н). МС: 432 (ESI+). Смесь диастереомеров разделяли на чистые стереоизомеры путем препаративной ВЭЖХ: 8 а) Получение промежуточных соединений. Получение (RS)-S-(4-аминофенил)-S-метил-N-(трифторацетил)сульфоксимида осуществляли, как описано для соединения 2.3. Получение (2R,3R)-3-(2-хлор-5-трифторметилпиримидин-4-иламино)бутан-2-ола осуществляли, как описано для соединения 4.1. 8b) Получение конечного продукта. 0,38 мл 4 н. раствора хлористого водорода в диоксане добавляли к 399 мг (1,50 ммоль)(2R,3R)-3-(2-хлор-5-трифторметилпиримидин-4-иламино)бутан-2-ола в 7,3 мл ацетонитрила и перемешивали в течение 9 ч при 60 С. Смесь упаривали насухо. 32,2 мл метанола и 1040 мг (7,50 ммоль) карбоната калия добавляли и ее перемешивали в течение 1,5 ч при комнатной температуре. Смесь разводили с помощью насыщенного раствора хлорида натрия и экстрагировали этилацетатом (3). Объединенные органические фазы высушивали (Na2SO4), фильтровали и концентрировали путем упаривания. Получали 565 мг (1,40 ммоль) неочищенного продукта. 1 Н-ЯМР (400 МГц, ДМСО): 10,09 (s, 1 Н), 8,30 (s, 1 Н), 7,96 (m, 2 Н), 7,83 (m, 2 Н), 6,10 (d, 1 Н), 5,11 (d,1 Н), 4,18 (m, 1 Н), 4,03 (s, 1 Н), 3,82 (m, 1 Н), 3,03 (s, 3H), 1,25 (d, 3 Н), 1,10 (d, 3 Н). Смесь диастереомеров разделяли на чистые стереоизомеры путем препаративной ВЭЖХ: 9 а) Получение промежуточных соединений. Получение (RS)-S-(4-аминофенил)-S-метил-N-(трифторацетил)сульфоксимида осуществляли, как описано для соединения 2.3. Получение (R)-3-(2-хлор-5-трифторметилпиримидин-4-иламино)-2-метилбутан-2-ола осуществляли, как описано для соединения 5.1. 9b) Получение конечного продукта. 0,38 мл 4 н. раствора хлористого водорода в диоксане добавляли к 399 мг (1,50 ммоль)(7,50 ммоль) карбоната калия и ее перемешивали в течение 1,5 ч при комнатной температуре. Смесь разводили с помощью насыщенного раствора хлорида натрия и экстрагировали этилацетатом (2). Объединенные органические фазы высушивали (Na2SO4), фильтровали и концентрировали путем упаривания. Получали 600 мг (1,44 ммоль) неочищенного продукта. 1 Н-ЯМР (400 МГц, ДМСО):= 10,05 (s, 1H), 8,26 (s, 1H), 7,91 (m, 2H), 7,79 (m, 2 Н), 6,03 (d, 1 Н),4,91 (s, 1 Н), 4,11 (m, 1 Н), 3,99 (s, 1 Н), 2,99 (s, 3 Н), 1,16 (m, 6 Н), 1,10 (m, 3 Н).MC: 418 (ESI+). Смесь диастереомеров разделяли на чистые стереоизомеры путем препаративной ВЭЖХ: Получение сравнительных веществ. Соединения в соответствии с изобретением, которые характеризуются, в частности, 5-CF3 заместителем в 5-м положении пиримидина, сравнивали, относительно их эффективности в условиях in vitro и invivo, с их 5-Br аналогами, которые либо раскрыты подробно в WO 2005/037800, либо альтернативно охватываются ее общим раскрытием. Сравнительное вещество в примере 11 представляет собой более активный стереоизомер смеси диастереомеров (RS)-S-[4-(5-бром-4-[(R)-(2-гидрокси-1,2-диметилпропил)амино]пиримидин-2-иламино)фенил]-S-циклопропилсульфоксимид, который описан в виде примера 1.6 в заявке WO 2005/037800 Для сравнения в условиях in vivo диастереомер (R)-S-[4-(5-бром-4-[(R)-(2-гидрокси-1,2 диметилпропил)амино]пиримидин-2-иламино)фенил]-S-циклопропилсульфоксимид, который имеет более высокую эффективность в условиях in vitro по сравнению с (S)-S-[4-(5-бром-4-[(R)-(2-гидрокси 1,2-диметилпропил)амино]пиримидин-2-иламино)фенил]-S-циклопропилсульфоксимидом, использовали в примере 11. Для этой цели V11 получали с помощью следующего способа. 0,07 мл 4 н. раствора хлористого водорода в диоксане добавляли к 988 мг (3,35 ммоль)U. Lcking и др., WO 2007/071455, с. 112, пример 4) в 16,50 мл бутанола и 1,65 мл метанола и перемешивали в течение 3 дней при 60 С. После охлаждения смесь упаривали насухо и очищали хроматографически (ДХМ/EtOH 9:1). Получали 319 мг (0,61 ммоль, выход: 22%) продукта. 1 Н-ЯМР (400 МГц, ДМСО):= 9,96 (s, 1H), 8,13 (s, 1H), 7,92 (m, 2H), 7,73 (m, 2 Н), 6,28 (d, 1 Н), 4,05V11b) Получение конечного продукта. 2,0 мл свежеприготовленного 1,5 М раствора этанолята натрия добавляли к 319 мг (0,61 ммоль) промежуточного соединения в 4,3 мл этанола и перемешивали в течение 18 ч при 60 С. После охлаждения смесь добавляли к насыщенному раствору хлорида натрия и экстрагировали этилацетатом (3). Объединенные органические фазы высушивали (Na2SO4), фильтровали и концентрировали путем упаривания. После конечной перекристаллизации (ДХМ/этилацетат) получали 215 мг (0,47 ммоль; выход: 78%) продукта. 1 Н-ЯМР (400 МГц, ДМСО):= 9,68 (s, 1H), 8,08 (s, 1H), 7,86 (m, 2H), 7,70 (m, 2 Н), 6,07 (d, 1 Н), 4,82WO 2005/037800. Пример 1.52 обладает более высокой эффективностью в условиях in vitro по сравнению с примером 1.53.WO 2005/037800. Пример 3.13 обладает более высокой эффективностью в условиях in vitro по сравнению с примером 3.12. 1,5 мл 4 н. раствора хлористого водорода в диоксане добавляли к 845 мг (3,00 ммоль)WO 2005/037800, с. 93) и 877 мг (3,00 ммоль) (RS)-S-(4-аминофенил)-S-циклопропил-N(трифторацетил)сульфоксимида в 13,1 мл ацетонитрила и перемешивали в течение 5 ч при 80 С. К смеси дополнительно добавляли 422 мг (1,50 ммоль) (2R,3R)-3-(5-бром-2-хлорпиримидин-4-илокси)бутан-2 ола и ее перемешивали дополнительно при 80 С. Через 3 ч снова добавляли 0,75 мл 4 н. раствора хлористого водорода в диоксане и перемешивали дополнительно при 80 С. Через 33 ч снова добавляли 175 мг(0,60 ммоль) (RS)-S-(4-аминофенил)-S-циклопропил-N-(трифторацетил)сульфоксимида и смесь перемешивали в заключение в течение 18 ч при 80 С. После охлаждения смесь концентрировали путем упаривания и остаток, который остался, очищали хроматографически (ДХМ/EtOH 9:1). Получали 740 мгV14b) Получение конечного продукта. 950 мг (6,84 ммоль) карбоната калия добавляли к 735 мг (1,37 ммоль) промежуточного соединения в 29 мл метанола и перемешивали в течение 1,5 ч при комнатной температуре. Смесь разводили с помощью насыщенного раствора хлорида натрия и экстрагировали этилацетатом (3). Объединенные органические фазы высушивали (Na2SO4), фильтровали и концентрировали путем упаривания. Получали 607 мг(1,37 ммоль) неочищенного продукта. 1 Н-ЯМР (400 МГц, ДМСО):= 10,08 (s, 1H), 8,40 (s, 1H), 7,86 (m, 2H), 7,75 (m, 2 Н), 5,19 (m, 1 Н),4,87 (d, 1 Н), 3,97 (s, 1 Н), 3,82 (m, 1 Н), 2,56 (m, 1 Н), 1,25 (d, 3 Н), 1,09 (d, 3 Н), 1,05 (m, 1 Н), 0,86 (m, 3 Н). Смесь диастереомеров разделяли на чистые стереоизомеры путем препаративной ВЭЖХ: Стереоизомер 2 проявлял более высокую эффективность в условиях in vitro по сравнению со стереоизомером 1 и, следовательно, его использовали в качестве сравнительного вещества в примере 14. Пример 10. 10.1. Исследование 1. Исследование киназы CDK1/CycB. Рекомбинантные CDK1 и CycB-GST слитые белки, очищенные из клеток насекомых, инфицированных бакуловирусами (Sf9), получали от ProQinase GmbH, Freiburg. Гистон IIIS, который использовали в качестве субстрата киназы, коммерчески доступный от компании Sigma.CDK1/CycB (200 нг/точку измерения) инкубировали в течение 10 мин при 22 С в присутствии различных концентраций тестируемых веществ (0 мкМ и в диапазоне 0,01-100 мкМ) в буфере для исследования [50 мМ Трис/HCl pH 8,0, 10 мМ MgCl2, 0,1 мМ отро-ванадат Na, 1,0 мМ дитиотреитол, 0,5 мкМ аденозин трифосфат (АТР), 10 мкг/точку измерения гистон IIIS, 0,2 мкКи/точку измерения 33 Р-гамма АТР, 0,05% NP40, 1,25% диметилсульфоксид]. Реакцию останавливали путем добавления раствораEDTA (250 мМ, pH 8,0, 15 мкл/точку измерения). Из каждой реакционной смеси 15 мкл наносили на Р 30 фильтрующие полосы (от Wallac) и инкорпорированный 33 Р-АТР удаляли путем промывания фильтрующих полос три раза, в течение 10 мин каждый раз, в 0,5% фосфорной кислоте. После высушивания фильтрующих полос в течение 1 ч при 70 С фильтрующие полосы покрывали сцинтилляционными полосами (MeltiLex А, от Wallac) и обжигали в течение 1 ч при 90 С. Количество инкорпорированного 33 Р (субстратное фосфорилирование) определяли путем измерения сцинтилляции в приборе для измерения гамма-излучения (Wallac). Измеренные данные стандартизировали относительно 0% ингибирования (ферментативная реакция без ингибитора) и 100% ингибирования (все анализируемые компоненты, за исключением фермента). IC50 значения определяли с помощью 4-параметрической подгонки, используя собственное программное обеспечение компании. 10.2. Исследование 2. Исследование киназы CDK2/CycE. Рекомбинантные CDK2 и CycE-GST слитые белки, очищенные из клеток насекомых, инфицированных бакуловирусами (Sf9), получали от ProQinase GmbH, Freiburg. Гистон IIIS, который использовали в качестве субстрата киназы, получали от компании Sigma.CDK2/CycE (50 нг/точку измерения) инкубировали в течение 10 мин при 22 С в присутствии различных концентраций тестируемых веществ (0 мкМ и в диапазоне 0,01-100 мкМ) в буфере для исследования [50 мМ Трис/HCl pH 8,0, 10 мМ MgCl2, 0,1 мМ отро-ванадат Na, 1,0 мМ дитиотреитол, 0,5 мкМ аденозин трифосфат (АТР), 10 мкг/точку измерения гистон IIIS, 0,2 мкКи/точку измерения 33 Р-гамма АТР, 0,05% NP40, 1,25% диметилсульфоксид]. Реакцию останавливали путем добавления раствораEDTA (250 мМ, pH 8,0, 15 мкл/точку измерения). Из каждой реакционной смеси 15 мкл наносили на Р 30 фильтрующие полосы (от Wallac) и инкорпорированный 33 Р-АТР удаляли путем промывания фильтрующих полос три раза, в течение 10 мин каждый раз, в 0,5% фосфорной кислоте. После высушивания фильтрующих полос в течение 1 ч при 70 С фильтрующие полосы покрывали сцинтилляционными полосами (MeltiLex А, от Wallac) и обжигали в течение 1 ч при 90 С. Количество инкорпорированного 33 Р (субстратное фосфорилирование) определяли путем измерения сцинтилляции в приборе для измерения гамма-излучения (Wallac). Измеренные данные стандартизировали относительно 0% ингибирования (ферментативная реакция без ингибитора) и 100% ингибирования (все анализируемые компоненты, за исключением фермента). IC50 значения определяли с помощью 4-параметрической подгонки, используя собственное программное обеспечение компании. 10.3. Исследование 3. Исследование VEGF рецепторной-2 киназы. Рекомбинантную VEGF рецепторную тирозинкиназу-2 очищали в виде GST слитого белка из клеток насекомых, инфицированных бакуловирусами (Sf9). Poly-(Glu4Tyr), который использовали в качестве субстрата киназы, получали от компании Sigma.VEGF рецепторную тирозинкиназу (90 нг/точку измерения) инкубировали в течение 10 мин при 22 С в присутствии различных концентраций тестируемых веществ (0 мкМ и в диапазоне 0,001-30 мкМ) в 30 мкл буфера для анализа [40 мМ Трис/HCl рН 5,5, 10 мМ MgCl2, 1 мМ MnCl2, 3 мкМ отро-ванадатNa, 1,0 мМ дитиотреитол, 8 мкМ аденозин трифосфат (АТР), 0,96 мкг/точку измерения poly-(Glu4Tyr),0,2 мкКи/точку измерения 33 Р-гамма АТР, 1,4% диметилсульфоксид]. Реакцию останавливали путем добавления раствора EDTA (250 мМ, pH 8,0, 15 мкл/точку измерения). Из каждой реакционной смеси 15 мкл наносили на Р 30 фильтрующие полосы (от Wallac) и инкорпорированный 33 Р-АТР удаляли путем промывания фильтрующих полос три раза, в течение 10 мин каждый раз, в 0,5% фосфорной кислоте. После высушивания фильтрующих полос в течение 1 ч при 70 С фильтрующие полосы покрывали сцинтилляционными полосами (MeltiLex А, от Wallac) и обжигали в течение 1 ч при 90 С. Количество инкорпорированного 33 Р (субстратное фосфорилирование) определяли путем измерения сцинтилляции в приборе для измерения гамма-излучения (Wallac). Измеренные данные стандартизировали относительно 0% ингибирования (ферментативная реакция без ингибитора) и 100% ингибирования (все анализируемые компоненты, за исключением фермента). IC50 значения определяли с помощью 4-параметрической подгонки, используя собственное программное обеспечение компании. 10.4. Исследование 4. Исследование пролиферации. Культивированные HeLa-MaTu клетки рака шейки матки человека (полученные от EPO-GmbH,Berlin) высевали при плотности 3000 клеток/точку измерения в многоярусном планшете на 96 лунок в 200 мкл ростовой среды (DMEM/HAMS F12, 2 мМ L-глутамин, 10% фетальная телячья сыворотка). Через 24 ч клетки одного планшета (планшет нулевой точки) окрашивали с помощью кристаллического фиолетового (см. ниже), тогда как среду других планшетов заменяли свежей культуральной средой(200 мкл), к которой добавляли тестируемые вещества в различных концентрациях (0 мкМ и в интервале 0,01-30 мкМ; конечная концентрация растворителя диметилсульфоксида составляла 0,5%). Клетки инкубировали в течение 4 дней в присутствии тестируемых веществ. Клеточную пролиферацию определяли путем окрашивания клеток с помощью кристаллического фиолетового: клетки фиксировали путем добавления 20 мкл/точку измерения 11% раствора глутаральдегида в течение 15 мин при комнатной температуре. После промывания фиксированных клеток водой три раза планшеты высушивали при комнатной температуре. Клетки окрашивали путем добавления 100 мкл/точку измерения 0,1% раствора кристаллического фиолетового (рН доводили до рН 3 путем добавления уксусной кислоты). После промывания окрашенных клеток водой три раза планшеты высушивали при комнатной температуре. Краситель растворяли путем добавления 100 мкл/точку измерения 10% раствора уксусной кислоты. Экстинкцию определяли фотометрически при длине волны 595 нм. Относительное изменение роста клеток, выраженное в процентах, рассчитывали путем стандартизации измеренных значений к значениям экстинкции планшета нулевой точки (=0%) и экстинкции необработанных (0 мкМ) клеток (=100%). Измеренные данные стандартизировали относительно 0% ингибирования (пролиферация клеток без ингибитора) и 100% ингибирования (планшет нулевой точки). IC50 значения определяли с помощью 4-параметрической подгонки,используя собственное программное обеспечение компании. 10.5. Результаты исследований фермента и пролиферации. Результаты исследований фермента и пролиферации не показывают любого системного преимущества соединений в соответствии с изобретением относительно соединений из уровня техники. Сравнение в условиях in vivo. Эффективность в условиях in vivo примера 5-SI-2 исследовали в примере 11 с V11 для сравнения. Эффективность в условиях in vivo примера 6-SI-2 исследовали в примере 12 с примером 1.52 изHeLa-MaTu (EPO-GmbH, Berlin) клетки рака шейки матки человека, которые росли в клеточной культуре, имплантировали подкожно в бок самкам NMRI бестимусных мышей. Лечение начинали, как только опухоли вырастали до объема приблизительно 20 мм 2. Исследование останавливали, как только опухоли в одной из групп достигали размера приблизительно 150 мм 2. Использовали следующие тестируемые группы. Группа 1: Контроль, обработка солюбилизатором (40% PEG 400/60% вода). Группа 2: Стереоизомер 2, приготовленный в соответствии с примером 5 (= пример 5-SI-2) (5 мг/кг,орально, 2 сутки в дни 4, 5, 11, 12). Исследование создавали для определения начальной реакции модели ксенотрансплантата рака шейки матки человека на лечения с помощью примера 5-SI-2. Рост-ингибирующее действие соединений тестировали в HeLa-MaTu модели опухоли шейки матки в качестве ксенотрансплантата у NMRI бестимусных мышей. Пример 5-SI-2 растворяли полностью в солюбилизаторе 40% полиэтиленгликоль (PEG) 400/60% вода до конечной концентрации 0,5 мг/мл. Обработку сформировавшихся опухолей начинали в день 4 после инокуляции опухолей. Исследование останавливали в день 17 после того, как размер опухоли у животных в группе 1 (контроль) превышал размер приблизительно 150 мм 2. Результаты исследований (фиг. 1) свидетельствуют о том, что пример 5-SI-2 в выбранной дозировке 5 мг/кг орально в циклической схеме лечения (на протяжении 2 последующих дней 2 сутки лечения с последующими 5 днями без лечения) значительно ингибирует рост опухоли (группа 3, уменьшение веса опухоли при окончании исследования на 7% относительно контрольной группы, р 0,05). 5-Br сравнительное вещество (= V11).HeLa-MaTu (EPO-GmbH, Berlin) клетки рака шейки матки человека, которые росли в клеточной культуре, имплантировали подкожно в бок самкам NMRI бестимусных мышей. Лечение начинали, как только опухоли вырастали до объема приблизительно 20 мм 2. Исследование останавливали, как только- 24019230 опухоли в одной из групп достигали размера приблизительно 150 мм 2. Использовали следующие тестируемые группы: Группа 1: Контроль, обработка солюбилизатором (30% HPCD/70% воды). Группа 2: Соединение в соответствии с WO 2005/037800, полученное в соответствии с приготовлением V11, описанным выше (8 мг/кг, орально, 2 сутки в дни 4, 5, 11, 12) Исследование создавали для определения начальной реакции модели ксенотрансплантата рака шейки матки человека на лечения с помощью 5-Br сравнительного вещества V11. Рост-ингибирующее действие соединений тестировали в HeLa-MaTu модели опухоли шейки матки в качестве ксенотрансплантата у NMRI бестимусных мышей. V11 растворяли полностью в солюбилизаторе 30%-гидроксипропилциклодекстрин (HPCD)/70% вода до конечной концентрации 0,8 мг/мл. Обработку сформировавшихся опухолей начинали в день 4 после инокуляции опухолей. Исследование останавливали в день 17 после того, как размер опухоли у животных в группе 1 (контроль) превышал размер приблизительно 150 мм 2. Результаты исследований (фиг. 2) свидетельствуют о том, что V11, в выбранной дозировке 8 мг/кг орально 2 сутки в течение 2 последовательных дней с последующими 5 днями без лечения, ингибирует рост опухоли на модели ксенотрансплантата HeLa-MaTu (группа 2, уменьшение веса опухоли при окончании исследования на 39% относительно контрольной группы, р 0,05). Вывод. Стереоизомер 2 из примера 5 (пример 5-SI-2 (5-CF3 обеспечивает в циклической схеме лечения,состоящей из оральных применений два раза в сутки в дозе 5 мг/кг в течение двух последовательных дней с последующими 5 днями без лечения, полное ингибирование роста опухоли на модели ксенотрансплантата HeLa-MaTu (соотношение лечение/контроль Л/К=0,07). В соответствующей хорошо переносимой циклической схеме лечения в дозе 8 мг/кг, 5-Br сравнительное вещество (= V11) обеспечивает только замедление роста опухоли на модели ксенотрансплантата HeLa-MaTu (соотношение лечение/контроль Л/К=0,39). Неожиданно, по сравнению с V11 (5-Br), пример 5-SI-2 (5-CF3) проявляет более высокую эффективность (5 мг/кг доза для примера 5-SI-2 по сравнению с 8 мг/кг для V11) и значительно лучшую противоопухолевую эффективность (полное ингибирование роста опухоли с Л/К=0,07 для примера 5-SI-2 по сравнению с замедлением роста опухоли с Л/К=0,39 для V11). Пример 12.HeLa-MaTu (EPO-GmbH, Berlin) клетки рака шейки матки человека, которые росли в клеточной культуре, имплантировали подкожно в бок самкам NMRI бестимусных мышей. Лечение начинали, как только опухоли вырастали до объема приблизительно 20 мм 2. Исследование останавливали, как только опухоли в одной из групп достигали размера приблизительно 150 мм 2. Использовали следующие тестируемые группы: Группа 1: Контроль, обработка солюбилизатором (40% PEG 400/60% вода). Группа 2: Стереоизомер 2, приготовленный в соответствии с примером 6 (= пример 6-SI-2) (3 мг/кг,орально, 2 сутки в дни 4, 5, 11, 12, 17, 18). Группа 3: Стереоизомер 2, приготовленный в соответствии с примером 6 (= пример 6-SI-2) (4 мг/кг,орально, 2 сутки в дни 4, 5, 11, 12, 17, 18). Группа 4: Стереоизомер 2, приготовленный в соответствии с примером 6 (= пример 6-SI-2) (5 мг/кг,орально, 2 сутки в дни 4, 5, 11, 12, 17, 18). Исследование создавали для определения начальной реакции модели ксенотрансплантата рака шейки матки человека на лечения с помощью примера 6-SI-2. Рост-ингибирующее действие соединений тестировали в HeLa-MaTu модели опухоли шейки матки в качестве ксенотрансплантата у NMRI бестимусных мышей. Пример 6-SI-2 растворяли полностью в солюбилизаторе 40% полиэтиленгликоль 400(PEG 400)/60% вода до конечной концентрации 0,3 мг/мл (группа 2), 0,4 мг/мл (группа 3) или 0,5 мг/мл(группа 4). Обработку сформировавшихся опухолей начинали в день 4 после инокуляции опухолей. Исследование останавливали в день 20 после того, как размер опухоли у животных в группе 1 (контроль) превышал размер приблизительно 150 мм 2. Результаты исследований (фиг. 3) свидетельствуют о том, что в схеме лечения, состоящей из оральных применений два раза в сутки в течение 2 последовательных дней с последующими 5 днями без лечения, пример 6-SI-2 обеспечивает дозозависимое ингибирование роста опухоли на модели ксенотрансплантата HeLa-MaTu. В группе с наивысшей дозой (группа 4) рост опухоли ингибировался почти полностью и обеспечивалось уменьшение веса опухоли при окончании исследования на 8% относительно контрольной группы (Л/К=0,08, р 0,05). 5-Br сравнительное вещество (= V12).HeLa-MaTu (EPO-GmbH, Berlin) клетки рака шейки матки человека, которые росли в клеточной культуре, имплантировали подкожно в бок самкам NMRI бестимусных мышей. Лечение начинали, как только опухоли вырастали до объема приблизительно 20 мм 2. Исследование останавливали, как только опухоли в одной из групп достигали размера приблизительно 150 мм 2. Использовали следующие тестируемые группы: Группа 1: Контроль, обработка солюбилизатором (40% PEG 400/60% вода). Группа 2: Соединение в соответствии с примером 1.52 из WO 2005/037800 (= V12) (7 мг/кг, орально, 2 сутки в дни 4, 5, 11, 12, 17, 18, 23, 24). Группа 3: Соединение в соответствии с примером 1.52 из WO 2005/037800 (= V12) (8,5 мг/кг, орально, 2 сутки в дни 4, 5, 11, 12, 17, 18, 23, 24). Группа 4: Соединение в соответствии с примером 1.52 из WO 2005/037800 (= V12) (10 мг/кг, орально, 2 сутки в дни 4, 5, 11, 12, 17, 18, 23, 24). Исследование создавали для определения начальной реакции модели ксенотрансплантата рака шейки матки человека на лечения с помощью 5-Br сравнительного вещества V12. Рост-ингибирующее действие соединений тестировали в HeLa-MaTu модели опухоли шейки матки в качестве ксенотрансплантата у NMRI бестимусных мышей. V12 растворяли полностью в солюбилизаторе 40% полиэтиленгликоль 400 (PEG 400)/60% вода до конечной концентрации 0,7 мг/мл (группа 2), 0,85 мг/мл (группа 3) или 1,0 мг/мл (группа 4). Обработку сформировавшихся опухолей начинали в день 4 после инокуляции опухолей. Исследование останавливали в день 28 после того, как размер опухоли у животных в группе 1(контроль) превышал размер приблизительно 150 мм 2 Результаты исследований (фиг. 4) свидетельствуют о том, что в схеме лечения, состоящей из оральных применений два раза в сутки в течение 2 последовательных дней с последующими 5 днями без лечения, V12 продуцирует дозозависимое слабое ингибирование роста опухоли на модели ксенотрансплантата HeLa-MaTu. В группе с наивысшей дозой (группа 4) рост опухоли ингибировался приблизительно наполовину по сравнению с контрольной группой (Л/К=0,51). Вывод. В циклической схеме лечения, состоящей из оральных применений два раза в сутки в дозе 5 мг/кг в течение двух последовательных дней с последующими 5 днями без лечения, стереоизомер 2 из примера 6(пример 6-SI-2 (5-CF3 обеспечивает почти полное ингибирование роста опухоли на модели ксенотрансплантата HeLa-MaTu (соотношение лечение/контроль Л/К=0,08). В соответствующей хорошо переносимой циклической схеме лечения в дозе 10 мг/кг соединение в соответствии с примером 1.52 изWO 2005/037800 (= V12 (5-Br обеспечивает только замедление роста опухоли на модели ксенотрансплантата HeLa-MaTu (соотношение лечение/контроль Л/К=0,51). Неожиданно, по сравнению с V12(5-Br) пример 6-SI-2 (5-CF3) проявляет более высокую эффективность (5 мг/кг доза для примера 6-SI-2 по сравнению с 10 мг/кг для V12) и значительно лучшую противоопухолевую эффективность (полное ингибирование роста опухоли с Л/К=0,08 для примера 6-SI-2 по сравнению с замедлением роста опухоли с Л/К=0,51 для V12). Пример 13.HeLa-MaTu (EPO-GmbH, Berlin) клетки рака шейки матки человека, которые росли в клеточной культуре, имплантировали подкожно в бок самкам NMRI бестимусных мышей. Лечение начинали, как только опухоли вырастали до объема приблизительно 20 мм 2. Исследование останавливали, как только опухоли в одной из групп достигали размера приблизительно 160 мм 2. Использовали следующие тестируемые группы: Группа 1: Контроль, обработка солюбилизатором (40% PEG 400/60% вода). Группа 2: Стереоизомер 2, приготовленный в соответствии с примером 2 (= пример 2-SI-2)(2,5 мг/кг, орально, 2 сутки в дни 5, 6, 12, 13, 19, 20). Исследование создавали для определения начальной реакции модели ксенотрансплантата рака шейки матки человека на лечения с помощью примера 2-SI-2. Рост-ингибирующее действие соединений тестировали в HeLa-MaTu модели опухоли шейки матки в качестве ксенотрансплантата у NMRI бестимусных мышей. Пример 2-SI-2 растворяли полностью в солюбилизаторе 40% полиэтиленгликоль 400(PEG 400)/60% вода до конечной концентрации 0,15 мг/мл (группа 2), 0,2 мг/мл (группа 3) или 0,25 мг/мл(группа 4). Обработку сформировавшихся опухолей начинали в день 5 после инокуляции опухолей. Исследование останавливали в день 20 после того, как размер опухоли у животных в группе 1 (контроль) превышал размер приблизительно 160 мм 2. Результаты исследований (фиг. 5) свидетельствуют о том, что в схеме лечения, состоящей из оральных применений два раза в сутки в течение 2 последовательных дней с последующими 5 днями без лечения, пример 2-SI-2 продуцирует дозозависимое ингибирование роста опухоли на модели ксенотрансплантата HeLa-MaTu. В группе с наивысшей дозой (2,5 мг/кг, группа 4) рост опухоли ингибировался почти полностью и наблюдалось уменьшение веса опухоли при окончании исследования на 18% относительно контрольной группы (Л/К=0,18, р 0,05).HeLa-MaTu (EPO-GmbH, Berlin) клетки рака шейки матки человека, которые росли в клеточной культуре, имплантировали подкожно в бок самкам NMRI бестимусных мышей. Лечение начинали, как только опухоли вырастали до объема приблизительно 20 мм 2. Исследование останавливали, как только опухоли в одной из групп достигали размера приблизительно 160 мм 2. Использовали следующие тестируемые группы: Группа 1: Контроль, обработка солюбилизатором (40% PEG 400/60% вода). Группа 2: Соединение в соответствии с примером 3.13 из WO 2005/037800 (= V13) (6 мг/кг, орально, 2 сутки в дни 5, 6, 12, 13, 19, 20). Группа 3: Соединение в соответствии с примером 3.13 из WO 2005/037800 (= V13) (8 мг/кг, орально, 2 сутки в дни 5, 6, 12, 13, 19, 20). Группа 4: Соединение в соответствии с примером 3.13 из WO 2005/037800 (= V13) (10 мг/кг, орально, 2 сутки в дни 5, 6, 12, 13, 19, 20). Исследование создавали для определения начальной реакции модели ксенотрансплантата рака шейки матки человека на лечения с помощью 5-Br сравнительного вещества V13. Рост-ингибирующее действие соединений тестировали в HeLa-MaTu модели опухоли шейки матки в качестве ксенотрансплантата у NMRI бестимусных мышей. V13 растворяли полностью в солюбилизаторе 40% полиэтиленгликоль 400(PEG 400)/60% вода до конечной концентрации 0,6 мг/мл (группа 2), 0,8 мг/мл (группа 3) или 1,0 мг/мл(группа 4). Обработку сформировавшихся опухолей начинали в день 5 после инокуляции опухолей. Исследование останавливали в день 20 после того, как размер опухоли у животных в группе 1 (контроль) превышал размер приблизительно 160 мм 2. Результаты исследований (фиг. 6) свидетельствуют о том, что в схеме лечения, состоящей из оральных применений два раза в сутки в течение 2 последовательных дней с последующими 5 днями без лечения, V13 продуцирует дозозависимое ингибирование роста опухоли на модели ксенотрансплантата HeLa-MaTu. В группе с наивысшей дозой (10 мг/кг, группа 4), рост опухоли ингибировался очень заметно и обеспечивалось уменьшение веса опухоли при окончании исследования на 23% относительно контрольной группы (Л/К=0,23, р 0,05). Вывод. В циклической схеме лечения, состоящей из оральных применений два раза в сутки в дозе 2,5 мг/кг в течение двух последовательных дней с последующими 5 днями без лечения, стереоизомер 2 из примера 2 (пример 2-SI-2 (5-CF3 обеспечивалось почти полное ингибирование роста опухоли на модели ксенотрансплантата HeLa-MaTu (соотношение лечение/контроль Л/К=0,18). В соответствующей хорошо переносимой циклической схеме лечения в дозе 10 мг/кг соединение в соответствии с примером 3.13 изWO 2005/037800 (= V13 (5-Br обеспечивало чуть меньшее замедление роста опухоли на модели ксенотрансплантата HeLa-MaTu (соотношение лечение/контроль Л/К=0,23). Неожиданно, по сравнению сV13 (5-Br) пример 2-SI-2 (5-CF3) обеспечивал значительно большую эффективность (2,5 мг/кг доза для примера 2-SI-2 по сравнению с 10 мг/кг для V13) и слегка лучшую противоопухолевую эффективность(ингибирование роста опухоли с Л/К=0,18 для примера 2-SI-2 по сравнению с ингибированием роста опухоли с Л/К=0,23 для V13). Пример 14.HeLa-MaTu (EPO-GmbH, Berlin) клетки рака шейки матки человека, которые росли в клеточной культуре, имплантировали подкожно в бок самкам NMRI бестимусных мышей. Лечение начинали, как только опухоли вырастали до объема приблизительно 20 мм 2. Исследование останавливали, как только опухоли в одной из групп достигали размера приблизительно 160 мм 2. Использовали следующие тестируемые группы: Группа 1: Контроль, обработка солюбилизатором (40% PEG 400/60% вода). Группа 2: Стереоизомер 2, приготовленный в соответствии с примером 1 (= пример 1-SI-2)(2,5 мг/кг, орально, 2 сутки в дни 5, 6, 12, 13, 19, 20). Исследование создавали для определения начальной реакции модели ксенотрансплантата рака шейки матки человека на лечения с помощью примера 1-SI-2. Рост-ингибирующее действие соединений тестировали в HeLa-MaTu модели опухоли шейки матки в качестве ксенотрансплантата у NMRI бестимусных мышей. Пример 1-SI-2 растворяли полностью в солюбилизаторе 40% полиэтиленгликоль 400(PEG 400)/60% вода до конечной концентрации 0,15 мг/мл (группа 2), 0,2 мг/мл (группа 3) или 0,25 мг/мл(группа 4). Обработку сформировавшихся опухолей начинали в день 5 после инокуляции опухолей. Исследование останавливали в день 20 после того, как размер опухоли у животных в группе 1 (контроль) превышал размер приблизительно 160 мм 2. Результаты исследований (фиг. 7) свидетельствуют о том, что в схеме лечения, состоящей из оральных применений два раза в сутки в течение 2 последовательных дней с последующими 5 днями без лече- 27019230 ния, пример 1-SI-2 продуцирует дозозависимое ингибирование роста опухоли на модели ксенотрансплантата HeLa-MaTu. В группе с наивысшей дозой (2,5 мг/кг, группа 4) рост опухоли ингибировался почти полностью и обеспечивалось уменьшение веса опухоли при окончании исследования на 19% относительно контрольной группы (Л/К=0,19, р 0,05). 5-Br сравнительное вещество (= V14).HeLa-MaTu (EPO-GmbH, Berlin) клетки рака шейки матки человека, которые росли в клеточной культуре, имплантировали подкожно в бок самкам NMRI бестимусных мышей. Лечение начинали, как только опухоли вырастали до объема приблизительно 20 мм 2. Исследование останавливали, как только опухоли в одной из групп достигали размера приблизительно 160 мм 2. Использовали следующие тестируемые группы: Группа 1: Контроль, обработка солюбилизатором (40% PEG 400/60% вода). Группа 2: Соединение в соответствии с WO 2005/037800, полученное в соответствии с приготовлением V14, описанным выше (6 мг/кг, орально, 2 сутки в дни 5, 6, 12, 13, 19, 20). Группа 3: Соединение в соответствии с WO 2005/037800, полученное в соответствии с приготовлением V14, описанным выше (8 мг/кг, орально, 2 сутки в дни 5, 6, 12, 13, 19, 20). Группа 4: Соединение в соответствии с WO 2005/037800, полученное в соответствии с приготовлением V14, описанным выше (10 мг/кг, орально, 2 сутки в дни 5, 6, 12, 13, 19, 20). Исследование создавали для определения начальной реакции модели ксенотрансплантата рака шейки матки человека на лечения с помощью 5-Br сравнительного вещества V14. Рост-ингибирующее действие соединений тестировали в HeLa-MaTu модели опухоли шейки матки в качестве ксенотрансплантата у NMRI бестимусных мышей. V14 растворяли полностью в солюбилизаторе 40% полиэтиленгликоль 400(PEG 400)/60% вода до конечной концентрации 0,6 мг/мл (группа 2), 0,8 мг/мл (группа 3) или 1,0 мг/мл(группа 4). Обработку сформировавшихся опухолей начинали в день 5 после инокуляции опухолей. Исследование останавливали в день 20 после того, как размер опухоли у животных в группе 1 (контроль) превышал размер приблизительно 160 мм 2. Результаты исследований (фиг. 8) свидетельствуют о том, что в схеме лечения, состоящей из оральных применений два раза в сутки в течение 2 последовательных дней с последующими 5 днями без лечения, V14 продуцирует дозозависимое ингибирование роста опухоли на модели ксенотрансплантата HeLa-MaTu. В группе с наивысшей дозой (10 мг/кг, группа 4) рост опухоли слабо ингибировался и обеспечивалось уменьшение веса опухоли при окончании исследования на 44% относительно контрольной группы (Л/К=0,44, статистическую достоверность не наблюдали). Вывод. В циклической схеме лечения, состоящей из оральных применений два раза в сутки в дозе 2,5 мг/кг в течение двух последовательных дней с последующими 5 днями без лечения, стереоизомер 2 из примера 1 (пример 1-SI-2 (5-CF3 обеспечивалось почти полное ингибирование роста опухоли на модели ксенотрансплантата HeLa-MaTu (соотношение лечение/контроль Л/К=0,19). В соответствующей хорошо переносимой циклической схеме лечения в дозе 10 мг/кг 5-Br соединение в соответствии сWO 2005/037800 (= V14 (5-Br обеспечивало слабое ингибирование роста опухоли на модели ксенотрансплантата HeLa-MaTu (соотношение лечение/контроль Л/К=0,44). Неожиданно, по сравнению сV14 (5-Br), пример 1-SI-2 (5-CF3) обеспечивал значительно большую эффективность (2,5 мг/кг доза для примера 1-SI-2 по сравнению с 10 мг/кг для V14) и значительно лучшую противоопухолевую эффективность (ингибирование роста опухоли с Л/К=0,19 для примера 1-SI-2 по сравнению с ингибированием роста опухоли с Л/К=0,44 для V14). Описание фигур На фиг. 1 показано ингибирование роста опухоли на модели ксенотрансплантированной опухоли шейки матки HeLa-MaTu человека при осуществлении лечения с помощью примера 5-SI-2. HeLa-MaTu клетки имплантировали подкожно NMRI бестимусным мышам в день 0. Лечение начинали в день 4 после того, как опухоли достигали размера приблизительно 20 мм 2. Лечения осуществляли со следующими дозировками и схемами применения: Группа 1: (Контрольная группа) - солюбилизатор (40% PEG 400/60% вода). Группа 2: Пример 5-SI-2, циклическая схема лечения, 2 сутки, орально, в дни 4, 5, 11, 12, дозировка 5 мг/кг.A) рост ксенотрансплантированных опухолей HeLa-MaTu в зависимости от времени;B) вес опухолей HeLa-MaTu в день 17 и соотношение среднего веса опухоли в леченной группе к среднему весу опухоли в контрольной группе (Л/К). На фиг. 2 показано ингибирование роста опухоли на модели ксенотрансплантированной опухоли шейки матки HeLa-MaTu человека при осуществлении лечения с помощью V11. HeLa-MaTu клетки имплантировали подкожно NMRI бестимусным мышам в день 0. Лечение начинали в день 4 после того, как опухоли достигали размера приблизительно 20 мм 2. Лечения осуществляли со следующими дозировками и схемами применения:A) рост ксенотрансплантированных опухолей HeLa-MaTu в зависимости от времени;B) вес опухолей HeLa-MaTu в день 17 и соотношение среднего веса опухоли в соответствующих леченных группах к среднему весу опухоли в контрольной группе (Л/К). На фиг. 3 показано ингибирование роста опухоли на модели ксенотрансплантированной опухоли шейки матки HeLa-MaTu человека при осуществлении лечения с помощью примера 6-SI-2. HeLa-MaTu клетки имплантировали подкожно NMRI бестимусным мышам в день 0. Лечение начинали в день 4 после того, как опухоли достигали размера приблизительно 20 мм 2. Лечения осуществляли в следующих дозировках и в циклической схеме лечения, состоящей из оральных применений два раза в сутки в дни 4,5, 11, 12, 17 и 18: Группа 1: (Контрольная группа) - солюбилизатор (40% PEG 400/60% вода). Группа 2: Пример 6-SI-2, дозировка 3 мг/кг. Группа 3: Пример 6-SI-2, дозировка 4 мг/кг. Группа 4: Пример 6-SI-2, дозировка 5 мг/кг.A) рост ксенотрансплантированных опухолей HeLa-MaTu в зависимости от времени;B) вес опухолей HeLa-MaTu в день 20 и соотношение среднего веса опухоли в соответствующих леченных группах к среднему весу опухоли в контрольной группе (Л/К). На фиг. 4 показано ингибирование роста опухоли на модели ксенотрансплантированной опухоли шейки матки HeLa-MaTu человека при осуществлении лечения с помощью V12. HeLa-MaTu клетки имплантировали подкожно NMRI бестимусным мышам в день 0. Лечение начинали в день 4 после того, как опухоли достигали размера приблизительно 20 мм 2. Лечения осуществляли в следующих дозировках и в циклической схеме лечения, состоящей из оральных применений два раза в сутки в дни 4, 5, 11, 12, 17,18, 23 и 24: Группа 1: (Контрольная группа) - солюбилизатор (40% PEG 400/60% вода). Группа 2: V12, дозировка 7 мг/кг. Группа 3: V12, дозировка 8,5 мг/кг. Группа 4: V12, дозировка 10 мг/кг. А) рост ксенотрансплантированных опухолей HeLa-MaTu в зависимости от времени; В) вес опухолей HeLa-MaTu в день 28, и соотношение среднего веса опухоли в соответствующих леченных группах к среднему весу опухоли в контрольной группе (Л/К). На фиг. 5 показано ингибирование роста опухоли на модели ксенотрансплантированной опухоли шейки матки HeLa-MaTu человека при осуществлении лечения с помощью примера 2-SI-2. HeLa-MaTu клетки имплантировали подкожно NMRI бестимусным мышам в день 0. Лечение начинали в день 5 после того, как опухоли достигали размера приблизительно 20 мм 2. Лечения осуществляли в следующих дозировках и в циклической схеме лечения, состоящей из оральных применений два раза в сутки в дни 5,6, 12, 13, 19 и 20: Группа 1: (Контрольная группа) - солюбилизатор (40% PEG 400/60% вода). Группа 2: Пример 2-SI-2, дозировка 1,5 мг/кг. Группа 3: Пример 2-SI-2, дозировка 2 мг/кг. Группа 4: Пример 2-SI-2, дозировка 2,5 мг/кг.A) рост ксенотрансплантированных опухолей HeLa-MaTu в зависимости от времени;B) вес опухолей HeLa-MaTu в день 20 и соотношение среднего веса опухоли в соответствующих леченных группах к среднему весу опухоли в контрольной группе (Л/К). На фиг. 6 показано ингибирование роста опухоли на модели ксенотрансплантированной опухоли шейки матки HeLa-MaTu человека при осуществлении лечения с помощью V13. HeLa-MaTu клетки имплантировали подкожно NMRI бестимусным мышам в день 0. Лечение начинали в день 5 после того, как опухоли достигали размера приблизительно 20 мм 2. Лечения осуществляли в следующих дозировках и в циклической схеме лечения, состоящей из оральных применений два раза в сутки в дни 5, 6, 12, 13, 19 и 20: Группа 1: (Контрольная группа) - солюбилизатор (40% PEG 400/60% вода). Группа 2: V13, дозировка 6 мг/кг. Группа 3: V13, дозировка 8 мг/кг. Группа 4: V13, дозировка 10 мг/кг.A) рост ксенотрансплантированных опухолей HeLa-MaTu в зависимости от времени;B) вес опухолей HeLa-MaTu в день 20 и соотношение среднего веса опухоли в соответствующих леченных группах к среднему весу опухоли в контрольной группе (Л/К). На фиг. 7 показано ингибирование роста опухоли на модели ксенотрансплантированной опухоли шейки матки HeLa-MaTu человека при осуществлении лечения с помощью примера 1-SI-2. HeLa-MaTu клетки имплантировали подкожно NMRI бестимусным мышам в день 0. Лечение начинали в день 5 по- 29
МПК / Метки
МПК: C07D 239/47, A61P 35/00, A61K 31/505
Метки: ингибиторов, сульфоксиминзамещенные, лекарственных, средств, качестве, применение, производные, получение, анилинопиримидиновые
Код ссылки
<a href="https://eas.patents.su/30-19230-sulfoksiminzameshhennye-anilinopirimidinovye-proizvodnye-v-kachestve-cdk-ingibitorov-ih-poluchenie-i-primenenie-v-kachestve-lekarstvennyh-sredstv.html" rel="bookmark" title="База патентов Евразийского Союза">Сульфоксиминзамещенные анилинопиримидиновые производные в качестве cdk ингибиторов, их получение и применение в качестве лекарственных средств</a>
Замещённые в положении 6 индолиноновые производные, их получение и их применение в качестве лекарственных средств

Номер патента: 8623
Опубликовано: 29.06.2007
Авторы: Ван-Мэл Якобус, Клей Йорг, Тонч-Грунт Ульрике, Хильберг Франк, Леманн-Линтц Торстен, Хеккель Армин, Рот Геральд Юрген
МПК: A61K 31/404, A61P 35/00, A61K 31/4045...
Метки: индолиноновые, производные, получение, качестве, средств, положении, замещённые, применение, лекарственных
Формула / Реферат:
1. Соединения общей формулы в которой X обозначает атом кислорода, R1 обозначает атом водорода, R2 обозначает атом фтора, хлора либо брома или цианогруппу, R3 обозначает фенильную группу, замещенную С1-С2алкилкарбониламиногруппой, карбокси-С1-С3алкилом, карбокси-С1-С4алкоксигруппой, С1-С4алкоксикарбонил-С1-С3алкилом, С1-С4алкоксикарбонил-С1-С3алкоксигруппой, аминокарбонил-С1-С3алкилом, (С1-С2алкиламино)карбонил-С1-С3алкилом,...
Новые производные бензимидазолов и бензотиазолов, способ их получения, их применение в качестве лекарственных средств, фармацевтические композиции и новое применение, в частности, в качестве ингибиторов cmet

Номер патента: 14315
Опубликовано: 29.10.2010
Авторы: Клерк Франсуа, Немесек Консепсьон
МПК: C07D 235/30, C07D 235/32, C07D 277/82...
Метки: ингибиторов, получения, бензотиазолов, новое, способ, лекарственных, средств, производные, бензимидазолов, качестве, применение, частности, новые, фармацевтические, композиции
Формула / Реферат:
1. Соединения формулы (I)в которой А обозначает NH или S;R1 и R2, одинаковые или разные, выбирают из атомов водорода, радикала NH2, атомов галогена и алкильных радикалов, необязательно замещенных одним или несколькими атомами галогена,и R3 обозначает атом водорода или выбран из значений R1 и R2, причем по меньшей мере один из R1, R2 и R3 не является водородом,R обозначает циклоалкил или алкил, необязательно замещенный фенилом, гетероарилом,...
Новые производные имидазолонов в качестве лекарственных средств, способ их получения, фармацевтические композиции и применение в качестве ингибиторов протеинкиназ, в частности cdc7

Номер патента: 18496
Опубликовано: 30.08.2013
Авторы: Консейер Эмманюэль, Ронан Батист, Штайнметц Анке, Леруа Венсан, Бак Эрик, Леталлек Жан-Филипп
МПК: A61K 31/437, C07D 471/04, A61P 35/00...
Метки: способ, частности, новые, фармацевтические, применение, композиции, протеинкиназ, имидазолонов, производные, ингибиторов, качестве, средств, лекарственных, получения
Формула / Реферат:
1. Соединения формулы (I)в которой X-Y обозначает NH-C(S), N=C-NR7R8, N=C-SR, N=C-R или N=C-OR;R1 обозначает атом водорода, радикал циклоалкил или радикал алкил, гетероциклоалкил, арил или гетероарил, причем все эти радикалы необязательно замещены;R, идентичный или отличающийся от R1, выбран из значений R1;R2 обозначает атом водорода, атом галогена или радикал алкил;R3 обозначает атом водорода, атом галогена, радикал гидроксил или радикал алкил...
Замещенные индолиноны, их получение и их применение в качестве лекарственных средств

Номер патента: 3514
Опубликовано: 26.06.2003
Авторы: Хеккель Армин, Редеманн Норберт, Грелль Вольфганг, Вальтер Райнер, Ван Меель Якобус С.А.
МПК: C07D 209/34, A61K 31/4045, A61P 35/00...
Метки: средств, применение, качестве, получение, лекарственных, замещенные, индолиноны
Формула / Реферат:
1. Замещенные индолиноны общей формулы в которой X обозначает атом кислорода или серы, R1 обозначает атом водорода, C1-C4алкоксикарбонильную или C2-C4алканоильную группу, R2 обозначает карбокси- или C1-C4алкоксикарбонильную группу либо необязательно замещенную одной или двумя C1-C3алкильными группами аминокарбонильную группу, при этом заместители могут быть идентичными или разными, R3 обозначает атом водорода или C1-C6алкильную группу,...
Бензимидазолы, их получение и их применение в качестве лекарственных средств

Номер патента: 3947
Опубликовано: 30.10.2003
Авторы: Кауффманн Ириз, Нар Герберт, Припке Хеннинг, Винен Вольфганг, Риз Уве, Хауэль Норберт, Штассен Еан Марие
МПК: C07D 235/14, A61K 31/415
Метки: применение, получение, качестве, бензимидазолы, средств, лекарственных
Формула / Реферат:
1. Бензимидазолы общей формулы I в которой Ar обозначает необязательно замещенную атомом фтора, хлора либо брома, трифторметильной, C1-C3алкильной либо C1-C3алкоксигруппой фениленовую или нафтиленовую группу, или обозначает необязательно замещенную в углеродном скелете C1-C3алкильной группой тиениленовую, тиазолиленовую, пиридиниленовую, пиримидиниленовую, пиразиниленовую или пиридазиниленовую группу, A обозначает C1-C3алкиленовую группу, B...
Предыдущий патент: Гербицидный композиционный состав "горчак"
Следующий патент: Синхронный явнополюсный генератор (варианты)
Случайный патент: Способ получения удобрения-мочевины со свойствами низкого влагопоглощения