2′-фтор-2′-дезокситетрагидроуридины в качестве ингибиторов цитидиндеаминазы
Номер патента: 18757
Опубликовано: 30.10.2013
Авторы: Дюваль Бриджит, Феррарис Дана В., Цукамото Такаси, Лапидус Рена, Гамильтон Грегори С.

















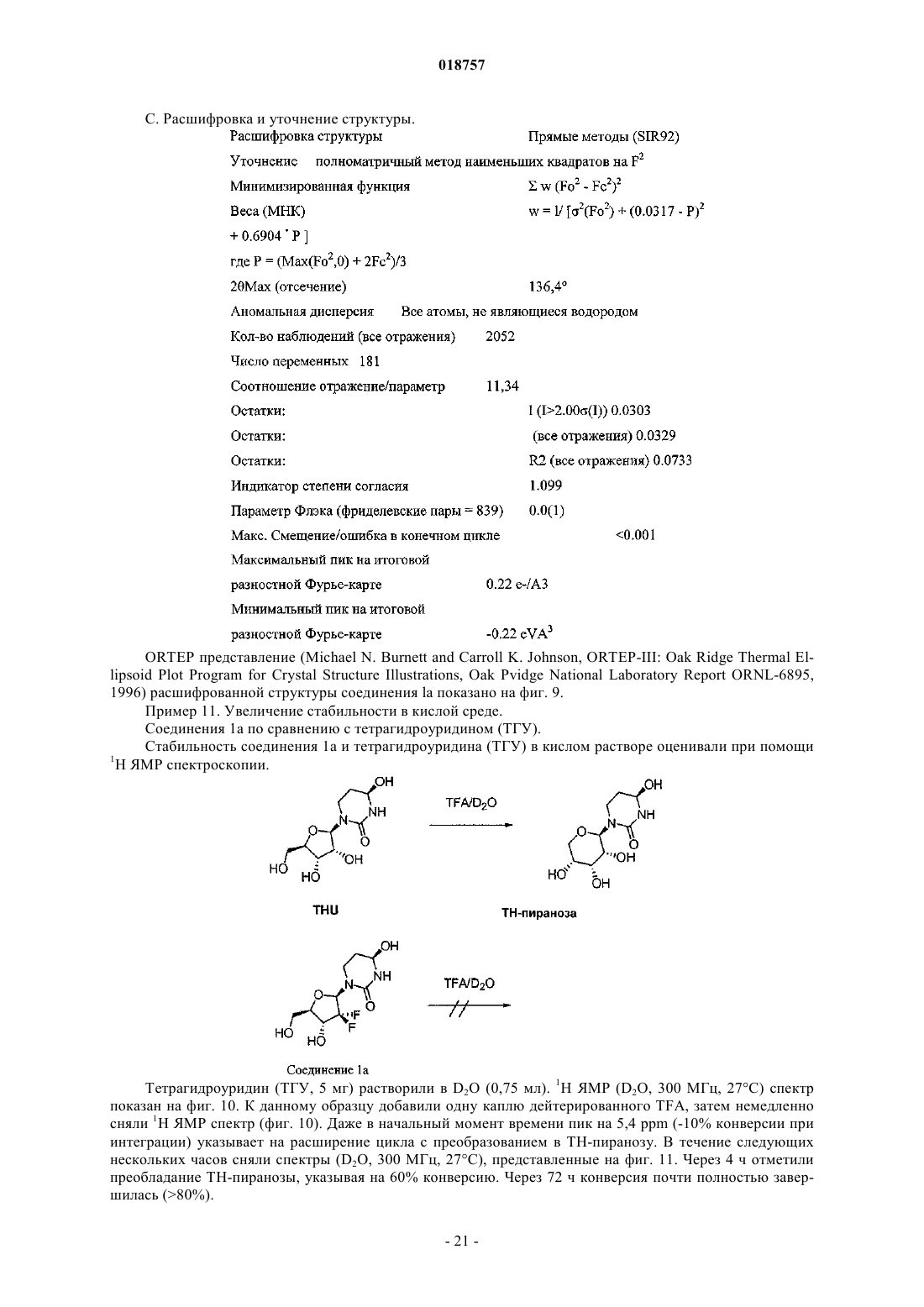




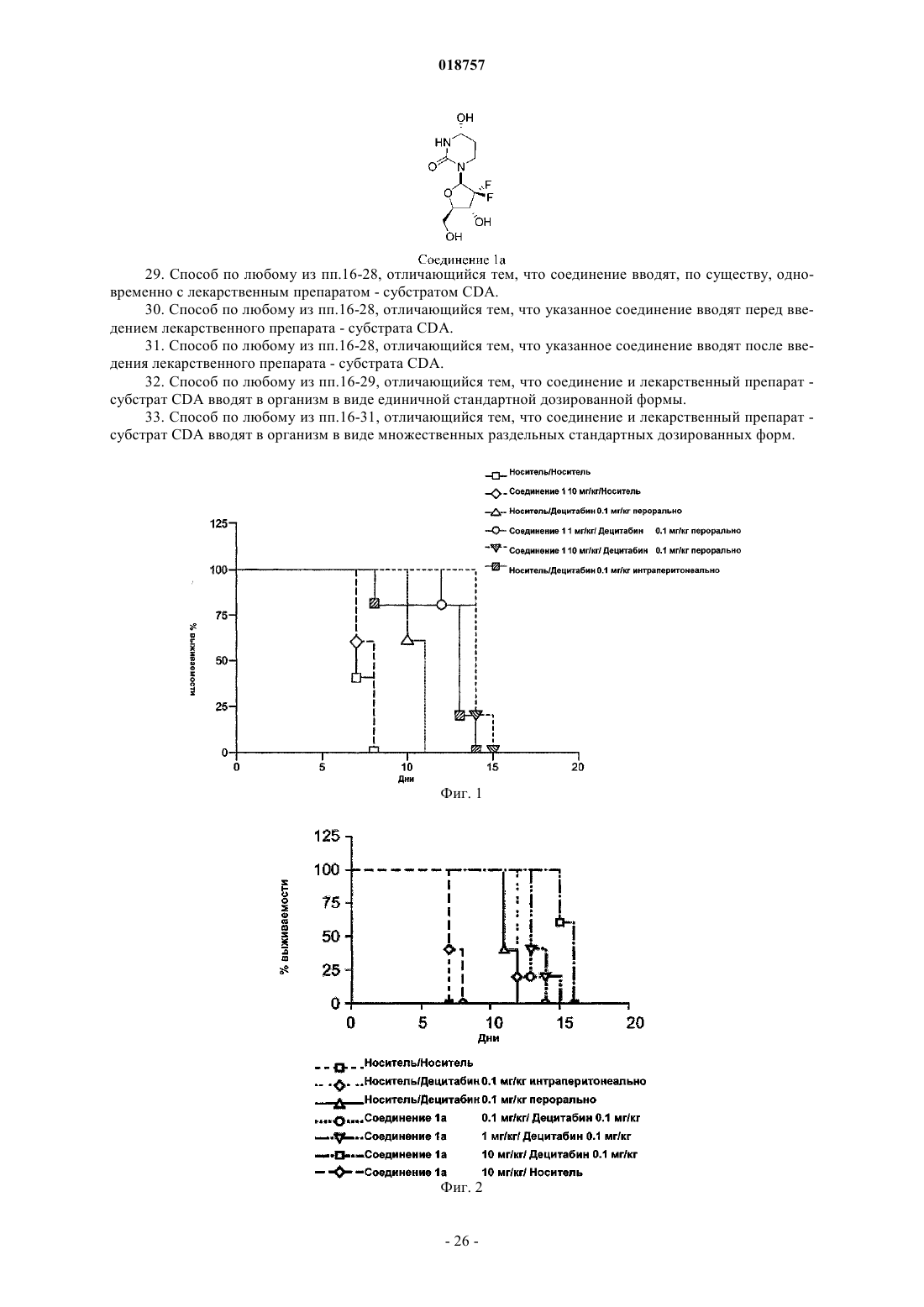
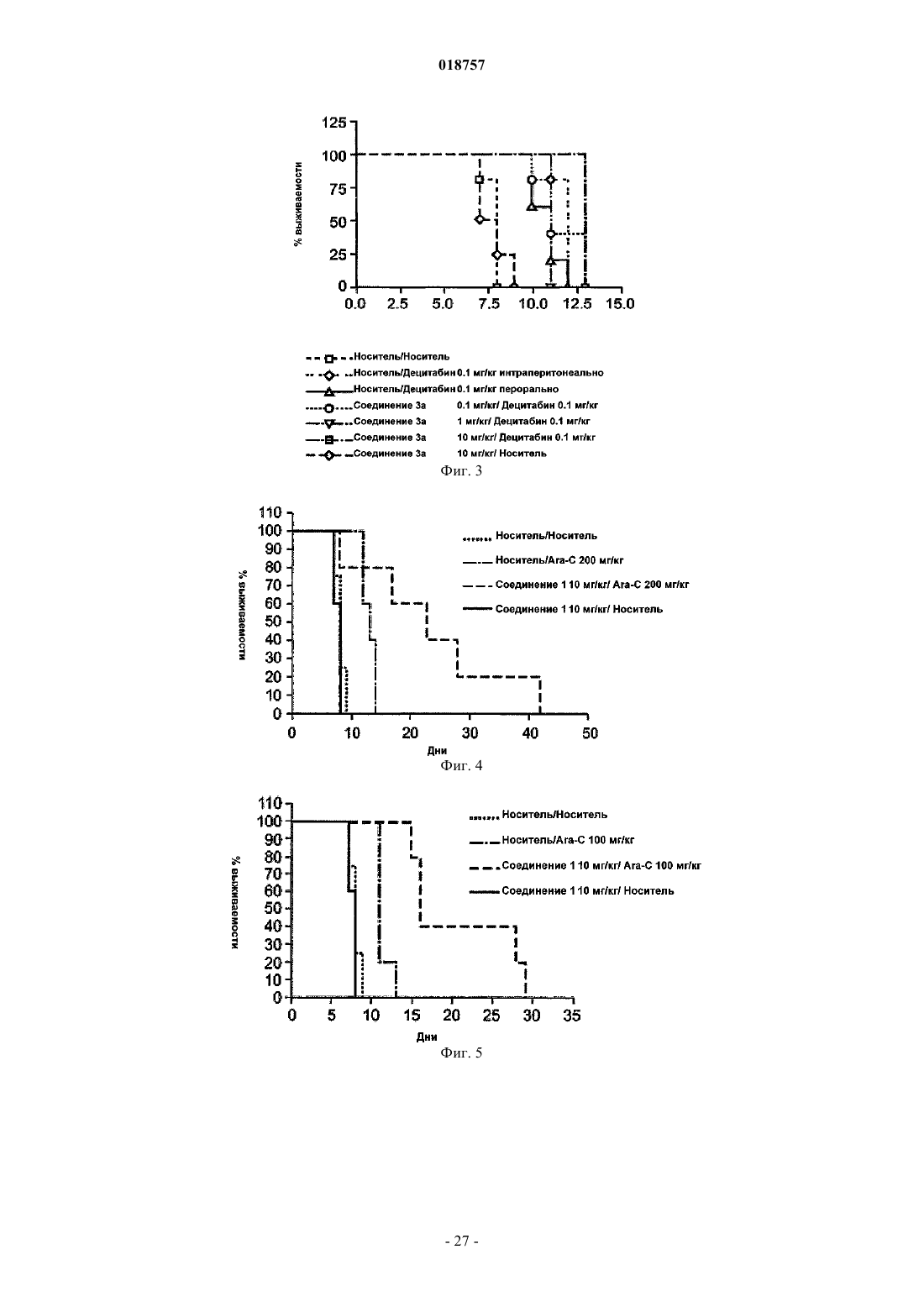
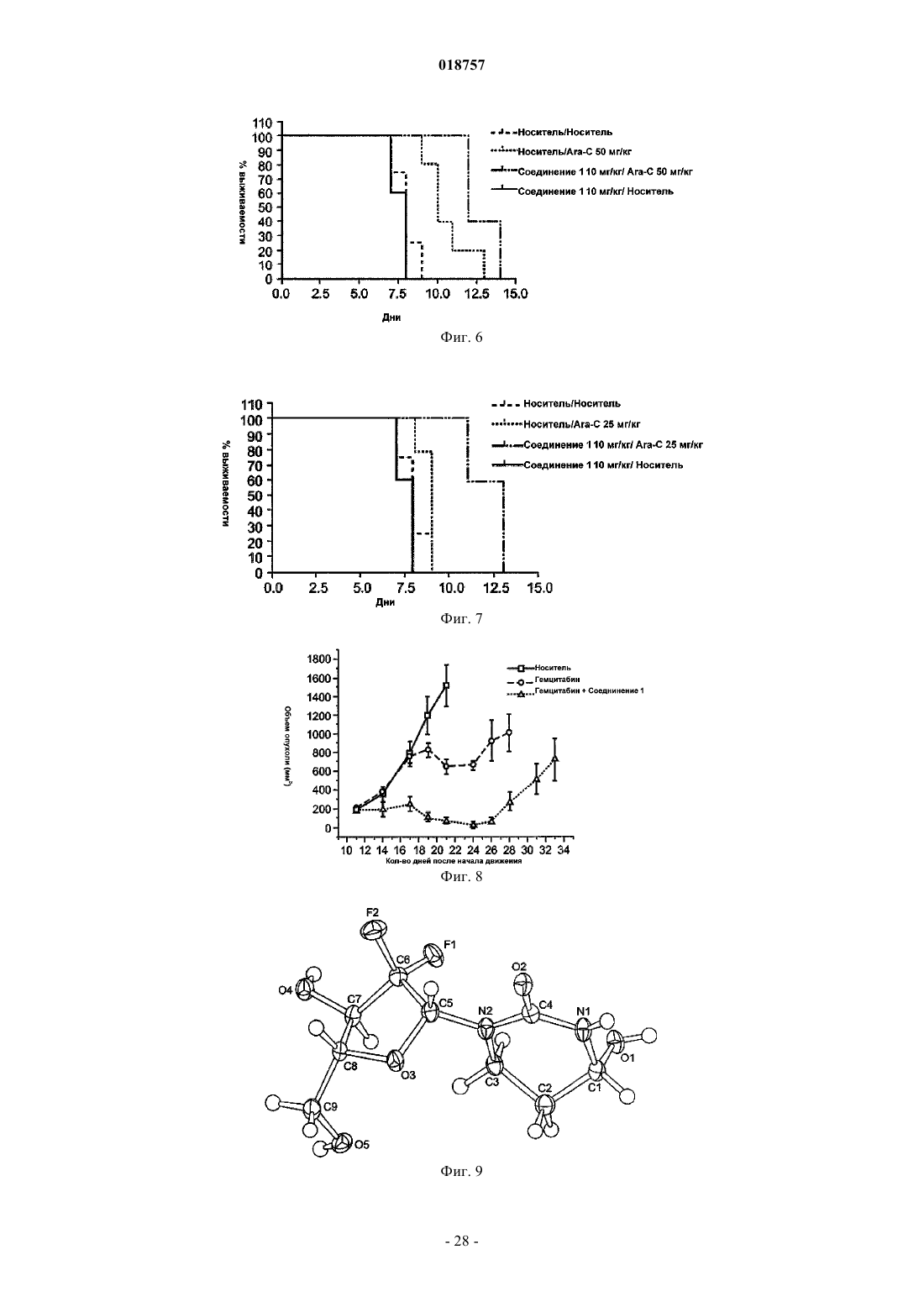

Формула / Реферат
1. Соединение формулы I или его фармацевтически приемлемая соль

где атом углерода, отмеченный символом *, может находиться в (R)- или (S)-конфигурации и где R1 и R2 представляют собой фтор.
2. Соединение по п.1, где указанное соединение представляет собой соединение 1a или его фармацевтически приемлемую соль

3. Соединение по п.1, где указанное соединение представляет собой соединение 1а

4. Фармацевтическая композиция для ингибирования ферментной активности CDA, по существу, состоящая из соединения формулы I или его фармацевтически приемлемой соли

где атом углерода, отмеченный символом *, может находиться в (R)- или (S)-конфигурации и где R1 и R2 представляют собой фтор и фармацевтически приемлемого эксципиента.
5. Фармацевтическая композиция по п.4, отличающаяся тем, что указанное соединение представляет собой соединение 1а или его фармацевтически приемлемую соль
6. Фармацевтическая композиция по п.4, отличающаяся тем, что указанное соединение представляет собой соединение 1а
7. Фармацевтическая композиция, содержащая:
(i) соединение формулы I или его фармацевтически приемлемую соль

где атом углерода, отмеченный символом *, может находиться в (R)- или (S)-конфигурации и где R1 и R2 представляют собой фтор; и
(ii) лекарственный препарат - субстрат CDA, выбранный из группы, состоящей из 5-азацитидина, гемцитабина, ара-С, тезацитабина, 5-фтор-2'-дезоксицитидина и цитохлора.
8. Фармацевтическая композиция, содержащая:
(i) соединение формулы I или его фармацевтически приемлемую соль

где атом углерода, отмеченный символом *, может находиться в (R)- или (S)-конфигурации и где R1 и R2 представляют собой фтор; и
(ii) лекарственный препарат - субстрат CDA, полезный для лечения рака;
при этом лекарственный препарат - субстрат CDA не является децитабином.
9. Фармацевтическая композиция, содержащая:
(i) соединение формулы I или его фармацевтически приемлемую соль

где атом углерода, отмеченный символом *, может находиться в (R)- или (S)-конфигурации и где R1 и R2 представляют собой фтор; и
(ii) лекарственный препарат - субстрат CDA;
при этом лекарственный препарат - субстрат CDA представляет собой децитабин.
10. Фармацевтическая композиция по любому из пп.7 или 8, отличающаяся тем, что лекарственный препарат - субстрат CDA представляет собой гемцитабин.
11. Фармацевтическая композиция по любому из пп.7 или 8, отличающаяся тем, что лекарственный препарат - субстрат CDA представляет собой ара-С.
12. Фармацевтическая композиция по любому из пп.7 или 8, отличающаяся тем, что лекарственный препарат - субстрат CDA представляет собой 5-азацитидин.
13. Фармацевтическая композиция по любому из пп.7-12, отличающаяся тем, что указанное соединение представляет собой соединение 1а или его фармацевтически приемлемую соль
14. Фармацевтическая композиция по любому из пп.7-12, отличающаяся тем, что указанное соединение представляет собой соединение 1а
15. Фармацевтическая композиция, содержащая соединение по любому из пп.1-3 и фармацевтически приемлемый эксципиент.
16. Способ лечения рака, чувствительного к субстрату CDA, включающий:
(i) введение в организм млекопитающего, нуждающегося в лечении, соединения формулы I или его фармацевтически приемлемой соли

где атом углерода, отмеченный символом *, может находиться в (R)- или (S)-конфигурации и где R1 и R2 представляют собой фтор; и
(ii) введение в организм млекопитающего, нуждающегося в лечении, лекарственного препарата - субстрата CDA, выбранного из группы, состоящей из 5-азацитидина, гемцитабина, ара-С, тезацитабина, 5-фтор-2'-дезоксицитидина и цитохлора.
17. Способ лечения рака, чувствительного к субстрату CDA, включающий:
(i) введение в организм млекопитающего, нуждающегося в лечении, соединения формулы I или его фармацевтически приемлемой соли

где атом углерода, отмеченный символом *, может находиться в (R)- или (S)-конфигурации и где R1 и R2 представляют собой фтор; и
(ii) введение в организм млекопитающего, нуждающегося в лечении, лекарственного препарата - субстрата CDA, полезного для лечения рака;
при этом лекарственный препарат - субстрат CDA не является децитабином.
18. Способ лечения рака, чувствительного к субстрату CDA, включающий:
(i) введение в организм млекопитающего, нуждающегося в лечении, соединения формулы I или его фармацевтически приемлемой соли

где атом углерода, отмеченный символом *, может находиться в (R)- или (S)-конфигурации и где R1 и R2 представляют собой фтор; и
(ii) введение в организм млекопитающего, нуждающегося в лечении, лекарственного препарата - субстрата CDA;
при этом лекарственный препарат - субстрат CDA представляет собой децитабин.
19. Способ по любому из пп.16, 17, отличающийся тем, что лекарственный препарат - субстрат CDA представляет собой гемцитабин.
20. Способ по любому из пп.16, 17, отличающийся тем, что лекарственный препарат - субстрат CDA представляет собой ара-С.
21. Способ по любому из пп.16, 17, отличающийся тем, что лекарственный препарат - субстрат CDA представляет собой 5-азацитидин.
22. Способ по любому из пп.16-21, отличающийся тем, что раковое заболевание выбрано из группы, состоящей из злокачественных гематологических заболеваний и солидных форм рака.
23. Способ по п.22, отличающийся тем, что рак представляет собой злокачественное гематологическое заболевание, выбранное из группы, состоящей из миелодиспластического синдрома и лейкоза.
24. Способ по п.23, отличающийся тем, что злокачественное гематологическое заболевание представляет собой миелодиспластический синдром.
25. Способ по п.23, отличающийся тем, что злокачественное гематологическое заболевание представляет собой острый миелоидный лейкоз или хронический миелоидный лейкоз.
26. Способ по п.22, отличающийся тем, что рак представляет собой солидный рак, выбранный из группы, состоящей из рака поджелудочной железы, рака яичников, перитонеального рака, немелкоклеточного рака легкого и метастатического рака молочной железы.
27. Способ по любому из пп.16-26, отличающийся тем, что указанное соединение представляет собой соединение 1а или его фармацевтически приемлемую соль
28. Способ по любому из пп.16-26, отличающийся тем, что указанное соединение представляет собой соединение 1а
29. Способ по любому из пп.16-28, отличающийся тем, что соединение вводят, по существу, одновременно с лекарственным препаратом - субстратом CDA.
30. Способ по любому из пп.16-28, отличающийся тем, что указанное соединение вводят перед введением лекарственного препарата - субстрата CDA.
31. Способ по любому из пп.16-28, отличающийся тем, что указанное соединение вводят после введения лекарственного препарата - субстрата CDA.
32. Способ по любому из пп.16-29, отличающийся тем, что соединение и лекарственный препарат - субстрат CDA вводят в организм в виде единичной стандартной дозированной формы.
33. Способ по любому из пп.16-31, отличающийся тем, что соединение и лекарственный препарат - субстрат CDA вводят в организм в виде множественных раздельных стандартных дозированных форм.
Текст
Настоящее изобретение относится к соединениям формулы I или их фармацевтически приемлемым солям Область техники, к которой относится изобретения Настоящее изобретение относится к соединениям - определенным производным тетрагидроуридина, которые являются ингибиторами цитидиндеаминазы, фармацевтическим композициям и наборам,включающим указанные соединения, а также способам получения и применению указанных соединений. Сведения о предшествующем уровне техники Действие ферментов аденозин деаминазы (ADA, EC 3.5.4.4) и цитидиндеаминазы (CDA, ЕС 3.5.4.5) направлено на деаминирование природных аминопуриновых и аминопиридиновых нуклеозидов, соответственно, в организме человека и в других организмах. Кроме того, они могут конвертировать активные лекарства на основе нуклеозидов в неактивные метаболиты. Например, при деаминировании содержащего пуриновый нуклеозид лекарства арабинозиладенин (флударабин, ара-А) под действием ферментаADA образуется соединение, в котором исходная аминогруппа замещена гидроксилом; данное соединение не является активным противоопухолевым агентом по сравнению с исходным соединением. Аналогично, применяемое для лечения лейкоза лекарство арабинозилцитозин (цитарабин, ара-С) при метаболической деградации под действием CDA преобразуется в неактивный арабинозилурацил.CDA является компонентом, участвующим в реутилизации пиримидина. Она конвертирует цитидин и дезоксицитидин в уридин и дезоксиуридин, соответственно, путем гидролитического деаминирования(Arch. Biochem. Biophys. 1991, 290, 285-292; Methods Enzymol. 1978, 51, 401-407; Biochem. J. 1967, 104,7P). Кроме того, CDA деаминирует ряд синтетических аналогов цитозина, являющихся клинически полезными лекарствами, такими как вышеуказанное лекарство ара-С (Cancer Chemother. Pharmacol. 1998,42, 373- 378; Cancer Res. 1989, 49, 3015-3019; Antiviral Chem. Chemother. 1990, 1, 255-262). Конверсия соединений цитозина в производные уридина обычно приводит к потере терапевтической активности или появлению дополнительных побочных эффектов. Кроме того, показано, что в раковых клетках, проявляющих резистентность к лекарствам - аналогам цитозина, зачастую наблюдается сверхэкспрессияCDA (Leuk. Res. 1990, 14, 751-754). Лейкемические клетки с высоким уровнем экспрессии CDA могут проявлять резистентность к антиметаболитам цитозина и таким образом ограничивать антинеопластическую активность подобной терапии (Biochem. Pharmacol. 1993, 45, 1857-1861). Таким образом, ингибиторы CDA могут оказаться полезными адъювантами при комбинированной химиотерапии. В течение уже нескольких лет известно, что тетрагидроуридин (ТГУ) является ингибитором цитидиндеаминазы. В различных публикациях высказаны предположения о том, что совместное с ТГУ введение повышает эффективность и активность лекарств на основе цитидина при пероральном введении. Например,показано, что ТГУ увеличивает активность антилейкемического агента 5-азацитидина у мышей L1210 с лейкозом (Cancer Chemotherapy Reports 1975, 59, 459-465). Комбинацию ТГУ и 5-азацитидина также исследовали на модели серповидноклеточной анемии у бабуинов (Am. J. Hematol. 1985, 18, 283-288), а также у пациентов-людей с серповидноклеточной анемией в комбинации с 5-азацитодином, вводимым перорально (Blood 1985, 66, 527-532). Показано также, что ТГУ увеличивает эффективность ара-С при пероральном введении у мышейL1210 с лейкозом (Cancer Research 1970, 30, 2166; Cancer Invest 1987, 5, (4), 293-9) и у мышей с опухолями (Cancer Treat. Rep. 1977, 61, 1355-1364). Комбинацию вводимого внутривенно ара-С с вводимым внутривенно ТГУ исследовали в ходе нескольких клинических испытаниях на людях (Cancer Treat. Rep. 1977, 61, 1347-1353; Cancer Treat. Rep. 1979, 63, 1245-1249; Cancer Res. 1988, 48, 1337-1342). В частности,провели исследования комбинаций среди пациентов с острым миелолейкозом (AML) и хроническим миелолейкозом (CML) (Leukemia 1991, 5, 991-998; Cancer Chemother. Pharmacol. 1993, 31, 481-484). 5-Аза-2'-дезоксицитидин (децитабин) является антинеопластическим агентом, применяемым для лечения миелодиспластического синдрома (MDS), с потенциальной возможностью применения для лечения AML и CML. Как и для других лекарств на основе цитидина, его пероральная биодоступность ограничена из-за деактивации под действием CDA. Показано, что ТГУ усиливает эффективность децитабина в модели заболевания серповидных клеток у бабуинов (Am. J. Hematol. 1985, 18, 283-288). Кроме того, показано, что другой ингибитор CDA, зебуларин, усиливает эффективность децитабина в модели лейкоза у мышей L1210 (Anticancer Drugs 2005, 16, 301-308). Другое антинеопластический препарат,гемцитабин, также исследовали в сочетании с ингибиторами CDA (Biochem. Pharmacol. 1993, 45, 18571861). Показано, что совместное введение с ТГУ изменяет фармакокинетику и биодоступность гемцитабина у мышей (Abstr. 1556, 2007 AACR Annual Meeting, April 14-18, 2007, Los Angeles, CA; Clin. CancerRes. 2008, 14, 3529-3535). 5-Фтор-2'-дезоксицитидин (фторцитидин, FdCyd) представляет собой другой еще один противораковый препарат на основе цитидина, являющийся ингибитором ДНК метилтрансферазы. Исследовали изменения его метаболизма и фармакокинетики под действием ТГУ в мышиной модели (Clin Cancer Res., 2006, 12, 7483-7491; Cancer Chemother. Pharm. 2008, 62, 363-368). Результаты вышеуказанных исследований подтверждают терапевтическую пользу при введении ингибиторов CDA вместе с лекарствами на основе цитокина, такими как ара-С, децитабин, 5-азацитидин и другие. Тем не менее, ингибиторы CDA на начальной стадии, такие как ТГУ, обладают определенными недостатками, включая нестабильность в кислой среде (J. Med. Chem. 1986, 29, 2351) и низкую биодоступность (J. Clin. Pharmacol. 1978, 18, 259). Вследствие этого существует насущная необходимость в разработке новых, эффективных и терапевтически полезных ингибиторов CDA. Сущность изобретения Настоящее изобретение относится к соединениям, определенным производным тетрагидроуридина,фармацевтическим композициям и наборам, включающим указанные соединения, а также способам получения и применению указанных соединений. Указанные соединения, композиции, наборы и способы согласно изобретению могут обладать определенными преимуществами. Например, соединения и композиции согласно изобретению могут ингибировать ферментную активность CDA и/или увеличить период полувыведения, биодоступность и/или эффективность лекарств, представляющих собой субстраты CDA. Кроме того, соединения, композиции, наборы и способы согласно изобретению могут иметь улучшенные характеристики, такие как растворимость в воде, химическая стабильность, уровни абсорбции лекарственных средств, уровни токсичности, срок годности, воспроизводимость при производстве и изготовлении составов, а также терапевтическая эффективность. В одном варианте реализации согласно настоящему изобретению предложено соединение формулы I или фармацевтически приемлемая соль указанного соединения, гдеR1 и R2 представляют собой фтор. Согласно некоторым вариантам реализации соединение представлено формулой где атом углерода, отмеченный символом , может находиться в (R)- или (S)-конфигурации. Согласно некоторым вариантам реализации фармацевтическая композиция или способ согласно настоящему изобретению возможно включает соединение (R)-конфигурации или (S)-конфигурации или смесь (R)- и (S)-2 018757 конфигураций. Согласно следующим вариантам реализации стереохимия соединения формулы I представлена структурами Ia или Ib Согласно следующим вариантам реализации соединение формулы I выбирают из группы, состоящей из соединений Ia и Ib и их фармацевтически приемлемых солей. Поскольку соединения согласно изобретению содержат по меньшей мере один хиральный центр,они могут существовать в форме энантиомеров, диастереомеров, рацемических смесей или других стереомеров. Настоящее изобретение охватывает все возможные изомеры подобного вида, а также геометрические изомеры и таутомеры. Согласно другому аспекту настоящее изобретение относится к фармацевтической композиции, содержащей:(i) эффективное количество описанного соединения согласно изобретению, включая все конкретные варианты реализации без ограничения; и(ii) фармацевтически приемлемый эксципиент. Согласно следующим вариантам реализации фармацевтическая композиция включает дополнительное эффективное количество по меньшей мере одного дополнительно терапевтического агента, такого как лекарственный препарат - субстрат CDA или химиотерапевтический агент. Эффективное количество соединения согласно изобретению может составлять от 0,1 до приблизительно 100 вес.%. Согласно некоторым вариантам реализации эффективное количество соединения составляет от 0,1 до 20% вес./вес. Согласно другим вариантам реализации эффективное количество составляет от 1 до 10% вес./вес. Согласно другим вариантам реализации эффективное количество составляет от 2 до 5% вес./вес. Фармацевтические композиции согласно изобретению можно приготовить в виде жидких и твердых лекарственных форм, включая формы, адаптированные для (1) перорального введения, например жидкие дозированные формы (например, водные и неводные растворы или суспензии); таблетки (например,предназначенные для буккального, сублингвального или системного поглощения); капсулы, болюсы,порошки, гранулы, пасты для нанесения на язык, твердые желатиновые капсулы, мягкие желатиновые капсулы, аэрозоли для орошения полости рта, лепешки, леденцы, пилюли, сиропы, суспензии, эликсиры,жидкости, эмульсии и микроэмульсии; (2) парентерального введения, например, путем подкожной, внутримышечной, внутривенной или эпидуральной инъекции, например, стерильного раствора или суспензии; (3) местного применения, например, в виде крема, мази, пластыря, мягкой прокладки или спрея, наносимого на кожу; (4) вагинального или ректального введения, например, в виде пессария, крема или пены; (5) сублингвального введения; (6) окулярного введения; (7) трансдермального введения или (8) интраназального введения. Можно изготовить фармацевтические композиции с немедленным, замедленным или контролируемым высвобождением. Согласно некоторым вариантам реализации фармацевтические композиции изготавливают для перорального введения. Согласно следующим вариантам реализации фармацевтические композиции изго-3 018757 тавливают для перорального введения в виде твердой лекарственной формы. Другой вариант реализации настоящего изобретения относится к способу ингибирования цитидиндеаминазы, включающему введение в организм субъекта, нуждающегося в лечении, эффективного количества описанной фармацевтической композиции согласно изобретению, включая все конкретные варианты реализации без ограничения. Согласно некоторым вариантам реализации субъект является млекопитающим. Согласно дальнейшим вариантам реализации субъект является человеком. Другой вариант реализации настоящего изобретения относится к способу лечения рака, включающему введение в организм субъекта, нуждающегося в лечении:(i) эффективного количества описанного соединения или фармацевтической композиции согласно изобретению, включая все конкретные варианты реализации без ограничения; и(ii) лекарственного препарата - субстрата CDA, включая все описанные здесь конкретные варианты реализации без ограничения. Согласно некоторым вариантам реализации субъект является млекопитающим. Согласно дальнейшим вариантам реализации субъект является человеком. Согласно некоторым вариантам реализации рак выбирают из злокачественных гематологических заболеваний и солидных форм рака. Согласно следующим вариантам реализации гематологический рак выбирают из МДС (миелодиспластического синдрома) и лейкемии (лейкоза). Согласно следующим вариантам реализации солидный рак выбирают из рака поджелудочной железы, перитонеального рака, немелкоклеточного рака легкого и рака молочной железы. Согласно другим вариантам реализации лейкоз представляет собой острый миелолейкоз (ACL) или хронический миелолейкоз (CML). Другой вариант реализации настоящего изобретения относится к способу ингибирования деградации лекарственного препарата - субстрата CDA под действием цитидиндеаминазы, включающий введение эффективного количества соединения или фармацевтической композиции согласно изобретению,включая все частные варианты реализации без ограничения, субъекту, получающему лечение лекарственным препаратом - субстратом CDA. "Лекарственный препарат - субстрат CDA" включает все описанные здесь частные варианты реализации без ограничения. Согласно отдельным вариантам реализации субъект является млекопитающим. Согласно другим вариантам реализации субъект является человеком. Другой вариант реализации настоящего изобретения относится к набору, содержащему по меньшей мере одну стандартную дозированную форму, причем стандартная дозированная форма включает соединение или фармацевтическую композицию согласно изобретению. Кроме того, набор может содержать контейнер и/или упаковку, приемлемую для коммерческой продажи. Контейнер, изготовленный из фармацевтически приемлемого материала, может иметь любую традиционную форму или вид, известный специалистам в данной области, такой как бумажная или картонная коробка, стеклянная или пластиковая бутылка или банка, повторно герметизируемый пакет или блистер-упаковка с индивидуальными дозами, извлекаемыми из упаковки для приема согласно терапевтическому расписанию. Одна упаковка может содержать более одного контейнера одновременно. Например, таблетки могут находиться в блистер-упаковке, которая в свою очередь может находиться в коробке. Перечень фигур На фиг. 1 представлен график, демонстрирующий влияние соединения 1 на выживаемость мышей,получавших децитабин, в модели лимфомы L 1210 на мышах; на фиг. 2 - график, демонстрирующий влияние соединения 1 а на выживаемость мышей, получавших децитабин, в модели лимфомы L 1210 на мышах; на фиг. 3 - график, демонстрирующий влияние соединения 3 а на выживаемость мышей, получавших децитабин, в модели лимфомы L 1210 на мышах; на фиг. 4 - график, демонстрирующий влияние соединения 1 а на выживаемость мышей, получавших Ара-С (200 мг/кг), в модели L 1210 на мышах; на фиг. 5 - график, демонстрирующий влияние соединения 1 а на выживаемость мышей, получавших Ара-С (100 мг/кг), в модели L 1210 на мышах; на фиг. 6 - график, демонстрирующий влияние соединения 1 а на выживаемость мышей, получавших Ара-С (50 мг/кг), в модели L 1210 на мышах; на фиг. 7 - график, демонстрирующий влияние соединения 1 а на выживаемость мышей, получавших Ара-С (25 мг/кг), в модели L 1210; на фиг. 8 - график, демонстрирующий влияние соединения 1 на уменьшение размеров опухоли, вызванное введением гемцитабина, в мышиной модели ксенотранстплантата раковых клеток яичника человека линии А 2780; на фиг. 9 - изображение в программе ORTEP кристаллической структуры соединения 1 а; на фиг. 10 - 1H ЯМР спектр ТГУ (тетрагидроуридина) в D2O; на фиг. 11 - 1 Н ЯМР спектр ТГУ в D2O в присутствии трифторуксусной кислоты, в разные моменты времени; на фиг. 12 - 1 Н ЯМР спектр соединения 1 а в D2O; на фиг. 13 - 1H ЯМР спектр соединения 1 а в D2O в присутствии трифторуксусной кислоты, в разные моменты времени. Сведения, подтверждающие возможность осуществления изобретения Настоящее изобретение относится к соединениям - определенным производным тетрагидроуридина, фармацевтическим композициям и наборам, содержащим указанные соединения, а также способам получения и применению указанных соединений. Соединения, композиции, наборы и способы согласно изобретению могут обладать определенными преимуществами. Например, соединения и композиции согласно изобретению способны ингибировать ферментную активность CDA и/или увеличить период полувыведения, биодоступность и/или эффективность лекарственных препаратов, представляющих собой субстраты CDA. Кроме того, соединения, композиции, наборы и способы согласно изобретению могут иметь улучшенные характеристики, такие как растворимость в воде, химическая стабильность, уровни абсорбции лекарственных средств, уровни токсичности, срок годности, воспроизводимость при производстве и изготовлении составов, а также терапевтическая эффективность. Определения В тексте заявки и формуле изобретения используют следующие определения. В тексте заявки и формуле изобретения слова (термины) в единственном числе включают и множественное число, если в тексте не указано иного. Так, например, ссылка на фармацевтическую композицию, содержащую "соединение" может относиться к двум и более соединениям. Термин "алкил" относится к насыщенному углеводородному радикалу с прямой или разветвленной углеродной цепью. Примеры включают без ограничения метил, этил, пропил, изопропил, бутил, изобутил, трет-бутил, n-пентил и n-гексил. Согласно некоторым вариантам реализации алкильная цепь представляет собой C1-C6 углеродную цепь, прямую или разветвленную. Согласно некоторым вариантам реализации алкильная цепь представляет собой C2-C5 углеродную цепь, прямую или разветвленную. Согласно некоторым вариантам реализации алкильная цепь представляет собой С 1-С 4 углеродную цепь, прямую или разветвленную. Согласно некоторым вариантам реализации алкильная цепь представляет собой С 2-С 4 углеродную цепь, прямую или разветвленную. Согласно некоторым вариантам реализации алкильная цепь представляет собой С 3 С 5 углеродную цепь, прямую или разветвленную. Согласно некоторым вариантам реализации алкильная цепь представляет собой C1-С 2 углеродную цепь, прямую или разветвленную. Согласно некоторым вариантам реализации, алкильная цепь представляет собой С 2-С 3 углеродную цепь, прямую или разветвленную. Термин "алкенил" относится к углеводородному радикалу с прямой или разветвленной углеводородной цепью, включающему по меньшей мере одну углерод-углеродную двойную связь. Примеры включают без ограничения этенил, пропенил, изопропенил, бутенил, изобутенил, трет-бутенил, nпентенил и n-гексенил. Согласно некоторым вариантам реализации алкенильная цепь представляет собой С 2-С 6 углеродную цепь, прямую или разветвленную. Согласно некоторым вариантам реализации алкенильная цепь представляет собой С 2-С 5 углеродную цепь, прямую или разветвленную. Согласно некоторым вариантам реализации алкенильная цепь представляет собой С 2-С 4 углеродную цепь, прямую или разветвленную. Согласно некоторым вариантам реализации алкенильная цепь представляет собой С 3-С 5 углеродную цепь, прямую или разветвленную. Термин "алкокси" относится к алкильной группе, связанной через атом кислорода. Термин "алкенокси" относится к алкенильной группе, связанной через атом кислорода. Термин "циклоалкил" относится к неароматическому циклическому алкильному радикалу. Термин "циклоалкенил" относится к неароматическому циклическому алкенильному радикалу. Термин "галоген" относится к фтор-, хлор-, бром- или йод-радикалам. Термин "замещенный" означает, что по меньшей мере один водород указанной группы замещен другим радикалом при условии, что нормальная валентность указанной группы не превышена. По отношению ко всем группам, содержащим один или несколько заместителей, замещение подобными группами не должно приводить к образованию соединения, которое стерически невыгодно, не осуществимо с точки зрения синтеза и/или не обладает естественной стабильностью. Термин "лекарственный препарат - субстрат CDA" относится к лекарству, которое можно деаминировать воздействием CDA. Примеры субстратов CDA включают без ограничения аналоги цитидина, такие как децитабин, 5-азацитидин, гемцитабин, ара-С, троксацитабин, тезацитабин, 5'-фтор-2'дезоксицитидин и цитохлор (cytochlor). Термин "эффективное количество" относится к количеству, необходимому для достижения желательного эффекта (например, увеличения периода пслувыведения, биодоступности или эффективности лекарственного препарата - субстрата CDA, лечения субъекта, больного раком, ингибирования цитидиндеаминазы в организме субъекта или ингибирования деградации субстрата CDA при воздействии цитидиндеаминазы). Термин "период полувыведения" относится к периоду времени, необходимого для снижения кон-5 018757 центрации или содержания соединения в организме субъекта ровно наполовину относительно заданной концентрации или содержания. Термин "фармацевтически приемлемый" относится к характеристикам и/или веществам, приемлемым для пациента с фармакологической и токсикологической точки зрения, и/или для химикафармацевта, изготавливающего лекарство, с физической и/или химической точки зрения, включая композицию, состав, стабильность, переносимость пациентом, биодоступность и совместимость с другими ингредиентами. Термин "фармацевтически приемлемый эксципиент" относится ко всем веществам, не являющимся терапевтическими агентами, применяемым в качестве наполнителя, разбавителя, адъюванта, связующего агента и/или носителя, доставляющего терапевтический агент в организм субъекта; или же к веществам,добавляемым к фармацевтической композиции для улучшения характеристик, связанных с обработкой и хранением, или для обеспечения возможности или облегчения приготовления единичной дозированной формы для приема соединения или композиции. Фармацевтически приемлемые эксципиенты хорошо известны специалистам в области фармацевтики, они описаны, например, в Remington's PharmaceuticalSciences, Mack Publishing Co., Easton, Pa (e.g., 20th Ed., 2000) и Handbook of Pharmaceutical Excipients,American Pharmaceutical Association, Washington, D.C., (например, 1st, 2nd и 3rd Eds., 1986, 1994 и 2000 соответственно). Как известно специалистам в данной области, эксципиенты могут выполнять различные функции; их называют смачивающими агентами, буферными агентами, суспендирующими агентами,смазывающими агентами, эмульгаторами, разрыхлителями, консервантами, сурфактантами, колорантами, ароматизаторами и подсластителями. Примеры фармацевтически приемлемых эксципиентов включают без ограничения (1) сахара, такие как лактоза, глюкоза и сахароза; (2) крахмалы, такие как кукурузный и картофельный крахмал; (3) целлюлозу и ее производные, такие как натрийкарбоксиметилцеллюлоза, этилцеллюлоза, ацетат целлюлозы, гидроксипропилметилцеллюлоза и гидроксипропилцеллюлоза; (4) порошкообразный тракагант; (5) солод; (6) желатин; (7) тальк; (8) наполнители, такие как масло какао или суппозиторные воски; (9) масла, такие как арахисовое масло, хлопковое масло, сафлоровое масло,кунжутное масло, оливковое масло, кукурузное масло или соевое масло; (10) гликоли, такие как пропиленгликоль; (11) полиолы, такие как глицерин, сорбитол, маннитол и полиэтиленгликоль; (12) сложные эфиры, такие как этил олеат и этил лаурат; (13) агар; (14) буферные агенты, такие как гидроксид магния и гидроксид алюминия; (15) альгиновая кислота; (16) апирогенная вода; (17) изотонический солевой раствор; (18) раствор Рингера; (19) этиловый спирт; (20) рН буферные растворы; (21) полиэфиры, поликарбонаты и/или полиангидриды; и (22) другие нетоксичные совместимые вещества, применяемые в фармацевтических составах. Термин "фармацевтически приемлемая соль" относится к соли кислоты или основания соединения согласно настоящему изобретению, обладающей желательной фармацевтической активностью и не являющейся неблагоприятной ни в биологическом, ни в любом другом смысле. Соли, образованные кислотами, включают без ограничения ацетат, адипат, альгинат, аспартат, бензоат, бензосульфонат, бисульфат,бутират, цитрат, камфорат, камфоросульфонат, циклопентанпропионат, диглюконат, додецилсульфат,этансульфонат, фумарат, глюкогептаноат, глицерофосфат, гемисульфат, гептаноат, гексаноат, гидрохлорид, гидробромид, гидроиодид, 2-гидроксиэтан-сульфонат, лактат, малеат, метансульфонат, 2 нафталинсульфонат, никотинат, оксалат, тиоцианат, тозилат и ундеканоат. Примеры солей оснований включают без ограничения соли аммония, соли щелочных металлов, такие как соли натрия и калия, соли щелочно-земельных металлов, такие как соли кальция и магния, соли органических оснований, такие как соли дициклогексиламина, N-метил-D-глюкамина, а также соли аминокислот, таких как аргинин и лизин. Согласно некоторым вариантам реализации основные азотсодержащие группы, могут быть кватернизированы с помощью различных агентов, включая низшие алкилгалиды, такие как метил, этил, пропил и бутил хлориды, бромиды и иодиды; диалкил сульфаты, такие как диметил, диэтил, дибутил и диамил сульфаты; галиды с длинными углеродными цепями, такие как децил, лаурил, миристил и стеарил хлориды, бромиды и иодиды; а также аралкилгалиды, такие как фенэтилбромиды. Термин "единичная дозированная форма" относится к физически дискретным единицам, пригодным для приема в качестве единичной дозы человеком или другим субъектом. Каждая единичная дозированная форма содержит определенное рассчитанное количество активного вещества (например, соединения или композиции согласно изобретению, лекарственного препарата - субстрата CDA и/или другого терапевтического агента), вызывающее желательный эффект. Термин изомеры относится к соединениям, содержащим одинаковые атомы в одинаковом количестве, следовательно, имеющим одинаковый молекулярный вес, но отличающиеся по расположению или конфигурации атомов. Термин "стереоизомеры" относится к изомерам, отличающимся только расположением атомов в пространстве. Термин "диастереомеры" относится к стереоизомерам, не являющимся зеркальным отражением друг друга. Термин "энантиомеры" относится к стереоизомерам, представляющим собой неналагающиеся зеркальные отображения друг друга. Энантиомеры включают "энантиометрически чистые" изомеры, вклю-6 018757 чающие, в основном, один изомер, например, в количестве, превышающем 90, 92, 95, 98 или 99, или на 100%, состоящие из одного изомера. Термин "эпимеры" относится к стереоизомерам соединения, различающимся конфигурацией одного из нескольких стереогенных центров. Термин "рацемическая" относится к смеси, содержащей индивидуальные энантиомеры в равных частях. Термин "нерацемическая" относится к смеси, содержащей индивидуальные энантиомеры в неравных частях. Нерацемическую смесь можно обогатить энантиомером R- или S-конфигурации, включая без ограничения соотношения, составляющие приблизительно 50/50, приблизительно 60/40 и приблизительно 70/30 R- к S-энантиомеру или S- к R- энантиомеру в смесях. Термин "необязательно" или "необязательный" означает, что описанное далее событие или обстоятельство может произойти или не произойти и что описание включает ситуации, где событие происходит, и ситуации, где событие не происходит. Например, термин "алкил, необязательно замещенный" охватывает как алкил замещенный, так и алкил незамещенный. Термин "субъект" относится к клетке или ткани, in vitro или in vivo, к человеку или животному. Субъект, представляющий собой человека или животное, может быть назван "пациент"."Животное" относится к живому организму, обладающему ощущениями и способного к произвольному движению, для поддержания жизни которого необходим кислород и органическая пища. Термин "млекопитающее" относится к теплокровным позвоночным животным с мехом или волосами. Примеры включают без ограничения человека, а также лошадей, свиней, крупный рогатый скот,мышевидных грызунов, собак или кошек. Понятие "лечение" по отношению к болезни, расстройству или состоянию относится к (i) ингибированию болезни, расстройства или состояния, например прекращению развития; и/или (ii) облегчению тяжести заболевания, расстройства или состояния, например индуцированию регрессии клинических симптомов. Понятие "профилактика" по отношению к болезни, расстройству или состоянию относится к предотвращению болезни, расстройства или состояния, например к прекращению развития клинических симптомов заболевания, расстройства или состояния. Термин "рак" относится к аномальному росту клеток с тенденцией к пролиферации неконтролируемым образом и в некоторых случаях к распространению с образованием метастазов. Специфические типы рака включают без ограничения раковые заболевания, упомянутые в публикацииUS 2006/0014949 и следующие: кардиальные: саркома (например, такие как ангиосаркома, фибросаркома, рабдомиосаркома, липосаркома и т.п.), миксома, рабдомиома, фиброма, липома и тератома; легочные: бронхогенный рак (например, такой как плоскоклеточный рак, недифференцированный мелкоклеточный рак, недифференцированный крупноклеточный рак, аденокарцинома и т.п.), альвеолярная (например, бронхиолярная) карцинома, аденома бронха, саркома, лимфома, хондроматозная гамартома, мезотиелома; желудочно-кишечные: пищевода (например, плоскоклеточная карцинома, аденокарцинома, леймиосаркома, лимфома и т.п.), желудка (например, карцинома, лимфома, леймиосаркома и т.п.), поджелудочной железы (например, протоковая аденокарцинома, инсулинома, глюкагонома, гастринома, карциноидные опухоли, випома и т.п.), тонкого кишечника (например, аденокарцинома, лимфома, карциноидные опухоли, саркома Капоши, лейомиома, гемангиома, липома, нейрофиброма, фиброма и т.п.), толстого кишечника (например, аденокарцинома, тубулярная аденома, ворсинчатая аденома, гамартома, лейомиома и т.п.); урогенитальных путей: почки (напримр, аденокарцинома, опухоль Уилма, нефробластома, лимфома, лейкемия и т.п.), мочевого пузыря и уретры (например, плоскоклеточная карцинома, переходноклеточный рак, аденокарцинома и т.п.), предстательной железы (например, аденокарцинома, саркома),яичка (например, семинома, тератома, эмбриональная карцинома, хориокарцинома, саркома, интерстициально-клеточная карцинома, фиброма, фиброаденома, аденоматоидные опухоли, липома и т.п.); печени: гепатома (например, гепатоцеллюлярная карцинома и т.п.), холангиокарцинома, гепатобластома, ангиосаркома, гепатоцеллюлярная аденома, гемангиома; кости: остеогенная саркома (например, остеосаркома и т.п.), фибросаркома, злокачественная фиброзная гистиома, хондросаркома, саркома Эвинга, злокачественная лимфома (например, ретикулоклеточная саркома), множественная миелома, злокачественная гигантоклеточная опухоль, хордома, osteochronfroma (например, костно-хрящевой экзостоз), доброкачественная хондрома, хондробластома, хондромиксофиброма, остеоидная остеома и гигантоклеточные опухоли; нервной системы: черепа (например, остеома, гемангиома, гранулема, ксантома, остит деформирующий и т.п.), мягких мозговых оболочек (например, менингиома, менингиосаркома, глиоматоз и т.п.),мозга (например, астроцитома, медуллобластома, глиома, эпендимома, герминома [пинеалома], глиобластома мультиформная, олигодендроглиома, шваннома, ретинобластома, врожденные опухоли и т.п.),спинного мозга (например, нейрофиброма, менингиома, глиома, саркома и т.п.); гинекологические: матки (например, эндометриальная карцинома и т.п.), шейки матки (например,цервикальная карцинома, предопухолевая цервикальная дисплазия и т.п.), яичника (например, карцинома яичника [серозная цистаденокарцинома, муцинозная цистаденокарцинома, неклассифицированный рак],гранулезо-текальноклеточные опухоли, опухоль из клеток Сертоли-Лейдига, дисгерминома, злокачественная тератома и т.п.), вульвы (например, плоскоклеточная карцинома, интраэпителиальная карцинома,аденокарцинома, фибросаркома, меланома и т.п.), влагалища (например, светлоклеточный рак, плоскоклеточный рак, кистевидная саркома (эмбриональная рабдомиосаркома)], фаллопиевых труб (карцинома и т.п.); гематологические: крови (например, миелоидный лейкоз [острый и хронический], острый лимфобластический лейкоз, хронический лимфолейкоз, миелопролиферативные заболевания, множественная миелома, миелодиспластический синдром и т.п.), болезнь Ходжкина, лимфома неходжкинская; кожи: злокачественная меланома, базальноклеточный рак, плоскоклеточный рак, саркома Капоши,бородавки, диспластический невус, липома, ангиома, дерматофиброма, келоиды, псориаз и т.п. и надпочечников: нейробластома. Соединения Один аспект настоящего изобретения относится к соединению формулы I или фармацевтически приемлемой соли этого соединения, где R1 и R2 представляют собой фтор. Согласно некоторым вариантам реализации предложено соединение формулы где возможна (R)- или (S)-конфигурация атома углерода, отмеченного символом . Согласно некоторым вариантам реализации описанная фармацевтическая композиция или способ ее применения может содержать соединение (R)-конфигурации или (S)-конфигурации или смесь соединений (R)- и (S)конфигурации. Согласно дальнейшим вариантам реализации стереохимию соединения формулы I описывает структура Ia или Ib Согласно следующим вариантам реализации соединение формулы I выбирают из группы, состоящей из соединений Ia и Ib и их фармацевтически приемлемых солей. Поскольку соединения согласно изобретению могут включать по меньшей мере один хиральный центр, они могут существовать в форме энантиомеров, диастереомеров, рацемических смесей, не рацемических смесей или других стереомеров. Настоящее изобретение охватывает все возможные подобные изомеры, а также геометрические изомеры и таутомеры. Стереоизомеры можно приготовить или выделить известными способами. Например, диастереомеры можно разделить физическими методами разделения, такими как дробная кристаллизация и хроматографические методы; энантиомеры можно разделить методом селективной кристаллизации солейдиастереомеров с оптически активными кислотами или основаниями или при помощи хиральной хроматографии. Кроме того, чистые стереомеры можно приготовить путем синтеза из соответствующих стереохимически чистых исходных материалов или путем стереоселективных реакций. Фармацевтические композиции Другой аспект настоящего изобретения относится к фармацевтической композиции, содержащей:(i) эффективное количество описанного соединения согласно изобретению, включая все отдельные варианты реализации без ограничения; и(ii) фармацевтически приемлемый эксципиент. Согласно следующим вариантам реализации фармацевтическая композиция включает сверх описанного по меньшей мере, один дополнительный терапевтический агент, такой как субстрат CDA или химиотерапевтический агент. Лекарственным препаратом - субстратом CDA может быть любое лекарство, способное к деаминированию под действием CDA. Примеры субстратов CDA включают без ограничения аналоги цитозина и цитидина, такие как децитабин, 5-азацитидин, гемцитабин, ара-С, троксацитабин, тезацитабин, 5'-фтор 2'-дезоксицитидин, цитохлор и соединения, описанные в публикации США 2006/0014949. Согласно некоторым вариантам реализации лекарственным препаратом - субстратом CDA является децитабин. Согласно другим вариантам реализации лекарственным препаратом - субстратом CDA является 5 азацитидин. Согласно другим вариантам реализации лекарственным препаратом - субстратом CDA является гемцитабин. Согласно следующим вариантам реализации лекарственным препаратом - субстратомCDA является ара-С. Примеры химиотерапевтических агентов включают без ограничения: алкилирующие агенты (например, возможно включающие доксорубицин, циклофосфамид, эстрамустин, кармустин, митомицин,блеомицин и т.п.); антиметаболиты (например, возможно включающие 5-фторурацил, капецитабин, гемцитабин, неларабин, флударабин, метотрексат и т.п.); платинирующие агенты (например, возможно включающие цисплатин, оксалиплатин, карбоплатин и т.п.); ингибиторы топоизомеразы (например, возможно включающие топотекан, иринотекан, этопозид и т.п.); тубулиновые агенты (например, возможно включающие паклитаксел, доцетаксел, винорелбин, винбластин, винкристин, другие таксаны, эпотилоны и т.п.); сигнальные ингибиторы (например, ингибиторы киназы, антитела, ингибиторы фарнесилтрансферазы и т.п.) и другие химиотерапевтические агенты(например, тамоксифен, антимитотические агенты,такие как ингибиторы поло-подобных киназ или ингибиторы аврора-киназ, и т.п.)."Эффективное количество" соединения согласно изобретению может варьировать приблизительно от 0,1 до 100 вес.%. Согласно некоторым вариантам реализации эффективное количество соединения составляет от 0,1 до 20% вес./вес. Согласно другим вариантам реализации эффективное количество составляет 1-10% вес./вес. Согласно некоторым другим вариантам реализации эффективное количество составляет 2-5% вес./вес. Фармацевтические композиции согласно изобретению можно приготовить в виде жидких и твердых лекарственных форм, включая формы, адаптированные для (1) перорального введения, например жидкие дозированные формы (например, водные и неводные растворы или суспензии); таблетки (например,предназначенные для буккального, сублингвального или системного поглощения); капсулы, болюсы,порошки, гранулы, пасты для нанесения на язык, твердые желатиновые капсулы, мягкие желатиновые капсулы, аэрозоли для орошения полости рта, лепешки, леденцы, пилюли, сиропы, суспензии, эликсиры,жидкости, эмульсии и микроэмульсии; (2) парентерального введения, например, путем подкожной, внутримышечной, внутривенной или эпидуральной инъекции, например, стерильного раствора или суспен-9 018757 зии; (3) местного применения, например, в виде крема, мази, пластыря, мягкой прокладки или спрея, наносимого на кожу; (4) вагинального или ректального введения, например, в виде пессария, крема или пены; (5) сублингвального введения; (6) окулярного введения; (7) трансдермального введения или (8) интраназального введения. Можно изготовить фармацевтические композиции с немедленным, замедленным или контролируемым высвобождением. Согласно некоторым вариантам реализации, фармацевтические композиции предназначены для перорального введения. Согласно следующим вариантам реализации, фармацевтические композиции для перорального введения изготавливают в виде твердой лекарственной формы. Фармацевтические композиции согласно изобретению можно изготовить при помощи известных материалов и способов, включающих без ограничения смешивание и/или измельчение соединения согласно изобретению с фармацевтически приемлемым эксципиентом и с необязательным дополнительным терапевтическим агентом(агентами). Способы Другой аспект настоящего изобретения относится к способу ингибирования цитидиндеаминазы,включающему введение субъекту, нуждающемуся в лечении, эффективного количества соединения или фармацевтической композиции согласно изобретению, включая все отдельные примеры без ограничения. Согласно некоторым вариантам реализации субъект является млекопитающим. Согласно следующим вариантам реализации субъект является человеком. Другой аспект настоящего изобретения относится к способу лечения рака, включающему введение субъекту, нуждающемуся в лечении:(i) эффективного количества соединения или фармацевтической композиции согласно изобретению,включая все отдельные варианты реализации без ограничения; и(ii) лекарственного препарата - субстрата CDA, включая все описанные здесь отдельные варианты реализации без ограничения. Согласно некоторым вариантам реализации, субъект является млекопитающим. Согласно другим вариантам реализации, субъект является человеком. Согласно некоторым вариантам реализации раковое заболевание выбрано из гематологических раковых заболеваний и солидных форм рака. Согласно другим вариантам реализации, гематологический рак выбран из МДС и лейкемии (лейкоза). Согласно другим вариантам реализации солидная форма рака выбрана из рака поджелудочной железы, рака яичника, перитонеального рака, немелкоклеточного рака легкого и рака молочной железы. Согласно следующим вариантам реализации лейкоз представляет собой острый миелоидный лейкоз(AML) или хронический миелоидный лейкоз (CML). Другой аспект настоящего изобретения относится к способу ингибирования деградации лекарственного препарата - субстрата CDA под действием цитидиндеаминазы, включающему введение эффективного количества соединения или фармацевтической композиции согласно изобретению, включая все описанные здесь отдельные варианты реализации без ограничения, субъекту, получающему лечение лекарственным препаратом - субстратом CDA. "Лекарственный препарат - субстрат CDA" включает без ограничения все описанные здесь отдельные варианты реализации без ограничения. Согласно некоторым вариантам реализации субъект является млекопитающим. Согласно другим вариантам реализации субъект является человеком. Возможно введение соединения или композиции согласно изобретению любым приемлемым путем,известным специалистам в данной области, например пероральным, парентеральным путем, ингаляцией распыленного вещества, ректальным, назальным, буккальным, вагинальным, интраокулярным, интрапульмонарным путем, через местное применение или имплантированный резервуар. Термин "парентеральный путь" включает без ограничения введение путем подкожной, внутривенной, внутримышечной,внутрибрюшинной, интратекальной, интравентрикулярной, интрастернальной, интракраниальной, эндостальной инъекции или путем инфузии. Любой режим введения, известный специалистам в данной области техники, для регулирования времени и последовательности доставки лекарства может быть использован и повторен столько раз, сколько будет необходимо для достижения технического результата в способах согласно настоящему изобретению. Например, соединение или композицию согласно изобретению можно вводить 1, 2, 3 или 4 раза в сутки в виде единичной дозы, множественных дискретных доз или путем непрерывной инфузии. Соединение или композицию согласно изобретению можно вводить перед, после или одновременно с приемом лекарственного препарата - субстрата CDA. Режим введения возможно включает предварительный прием и/или совместное введение по меньшей мере с одним дополнительным терапевтическим агентом. В данном случае соединение или композицию согласно изобретению, лекарственный препарат - субстрат CDA и по меньшей мере один дополнительный терапевтический агент можно вводить одновременно, отдельно, последовательно. Примеры режимов введения включают без ограничения введение каждого соединения, композиции,лекарственного препарата - субстрата CDA и/или терапевтического агента последовательно; и совместное введение каждого соединения, композиции, лекарственного препарата - субстрата CDA и/или терапевтического агента, в основном, одновременно (например, в виде единичной дозированной формы) или в виде множественных, отдельных стандартных дозированных форм для каждого соединения, композиции, лекарственного препарата - субстрата CDA и/или терапевтического агента. Для специалистов в данной области очевидно, что "эффективное количество" или "уровень дозы" зависит от многих факторов,таких как путь введения, режим введения, выбор соединения или композиции, а также конкретного заболевания и состояния пациента, получающего лечение. Например, назначенный уровень дозы может варьироваться в зависимости от активности, скорости экскреции и возможной токсичности конкретного применяемого соединения или композиции; возраста, веса, общего здоровья, пола и диеты пациента, получающего лечения; частоты введения доз; другого терапевтического агента (агентов), совместно вводимого; а также типа и степени тяжести заболевания. Согласно настоящему изобретению уровни доз составляют приблизительно от 0,001 до 10,000 мг/кг/сутки. Согласно некоторым вариантам реализации уровень дозы составляет приблизительно от 0,1 до 1,000 мг/кг/сутки. Согласно другим вариантам реализации уровень дозы составляет приблизительно от 1 до 100 мг/кг/сутки. Согласно другим вариантам реализации уровень дозы составляет приблизительно от 1 до 50 мг/кг/сутки. Согласно другим вариантам реализации уровень дозы составляет приблизительно от 1 до 25 мг/кг/сутки. Соответствующие уровни дозирозания, способ введения и режим введения может уточнить специалист в данной области, используя известные методики. Наборы Другой аспект настоящего изобретения относится к набору, содержащему по меньшей мере одну единичную дозированную форму; данная единичная дозированная форма включает соединение или фармацевтическую композицию согласно изобретению. Кроме того, набор может содержать контейнер и/или упаковку, приемлемую для коммерческой продажи. Контейнер, изготовленный из фармацевтически приемлемого материала, может иметь любую традиционную форму или вид, известный специалистам в данной области, такой как бумажная или картонная коробка, стеклянная или пластиковая бутылка или банка, повторно герметизируемый пакет или блистер-упаковка с индивидуальными дозами, извлекаемыми из упаковки для приема согласно терапевтическому режиму. Одна упаковка можем содержать более одного контейнера одновременно. Например, таблетки в блистер-упаковке могут находиться в коробке. Кроме того, набор может содержать информацию. Информация может быть представлена на текстоносителе. Текстоноситель, возможно, включает этикетку. Информация может быть обращена к врачу,фармацевту или пациенту. Информация может содержать указание на то, что единичная дозированная форма способна вызвать один или несколько неблагоприятных побочных эффектов. Информация может содержать инструкции по введению единичной дозированной формы, например, описанный здесь способ(режим) введения. Эти инструкции можно представлять в различных формах. Например, информация может включать таблицу, где указаны веса или весовые интервалы и дозы, соответствующие определенному весу или весовому интервалу. Информация может прилагаться к контейнеру, например содержаться на этикетке (например, на сигнатуре или на отдельной этикетке), приклеенной к контейнеру; находиться внутри контейнера в виде вложения с текстом; быть нанесенной непосредственно на контейнер,например в виде текста, напечатанного на стенке коробки или упаковки-блистера; или содержаться в тексте, привязанном или прикрепленном, например на инструкции, прикрепленной к горлышку бутылки при помощи веревки, лески или другого типа шнура, или при помощи приспособления типа клипсы или веревки с петлей. Для специалиста в данной области очевидно, что частные варианты реализации настоящего изобретения возможно относятся к одному, нескольким или ко всем вышеупомянутым аспектам; точно так же другие аспекты могут охватывать один, несколько или все варианты реализации, вышеупомянутые и нижеследующие, наряду с другими вариантами реализации. Отличные от упомянутых в рабочих примерах"приблизительно". Соответственно, если не указано иное, эти числа являются приближенными значениями и могут варьировать в зависимости от желательных характеристик в соответствии с целями настоящего изобретения. И, наконец, без намерения ограничить доктрины эквивалентов рамками изобретения, каждый численный параметр следует рассматривать с точки зрения количества значимых цифр и обычных методик округления. В то время как числовые интервалы и параметры, устанавливающие рамки настоящего изобретения, являются приблизительными, численные значения, представленные в рабочих примерах, даны с максимальной точностью. Любое численное значение, тем не менее, по сути содержит определенные ошибки, возникающие вследствие стандартных отклонений соответствующих измерительных приборов. Примеры Следующие примеры являются иллюстративными и не ограничивают настоящего изобретения. Синтез соединений. Соединение согласно настоящему изобретению можно приготовить описанным здесь способом и/или с применением или адаптацией известных методик. Для специалиста в данной области очевидно,- 11018757 что один или несколько реагентов, этапов и/или условий, указанных в реакционных схемах, возможно,потребуется изменить для того, чтобы ввести другие заместители при R1 и R2. Пример 1. Схема 1 Синтез производных дифтортетрагидроуридина (соединения 1 а и 1b) 2'2'-Дифтордигидроуридин (DFDHU, 25). Гемцитабин 24 (3,0 г, 11,4 ммоль) растворили в Н 2 О (50 мл). К раствору добавили родий на окиси алюминия (900 мг) и гидрогенизировали смесь в течение ночи при 40 psi. На следующий день смесь профильтровали, отогнали воду под вакуумом и вновь растворили образовавшееся липкое твердое вещество в Н 2 О. К раствору добавили родий на окиси алюминия (900 мг) и гидрогенировали смесь в течение ночи при 40 psi. Родий отфильтровали, фильтрат сконцентрировали, в результате получили неочищенную смесь дифтордигидроуридина (5, DFDHU) и 10% дифтортетрагидроуридина Ia и Ib (DFTHU). Полученную смесь очистили обратнофазной ВЭЖХ (HPLC) (обращенная фаза С 185% CH3CN/H2O), в результате получили 1,84 г (61%, 14,5 мин) DFDHU 25 и 175 мг (17%, Ia, 9,5 мин и Ib, 13,9 мин) эпимеров ДФТГУ. Абсолютную конфигурацию С-4 в соединении Ia определили при помощи рентгеновской дифракции монокристалла; данные согласуются с описанными в литературе прецедентами - кристаллическими структурами цитидиндеаминазы в комплексе с одним эпимером тетрагидроуридина. 1DFDHU 25 (1,2 г, 4,9 ммоль) растворили в 30 мл МеОН и охладили до 0 С. К раствору порциями добавили борогидрид натрия (540 мг, 14,6 ммоль) и медленно нагревали реакционную смесь до комнатной температуры. После перемешивания при комнатной температуре в течение 4 ч удалили МеОН под вакуумом, а остаток растворили в 15 мл Н 2 О. Раствор нейтрализовали добавлением 2,0N HCl до рН 7. Затем раствор очистили при помощи препаративной HPLC (обращенная фаза C185% CH3CN/H2O). Элюирование солей происходит через 5,2 мин. Один пик появляется через 7,5 мин (12%). Один эпимер ДФТГУ Ia элюирует через 9,5 мин (350, 29%). Другой эпимер Ib элюирует через 14,3 мин (370 мг, 31%). Дезоксигенированный продукт 26 элюирует через 17 мин (200 мг, 17%). 2'(R)-фтор-2'дезоксидигидроуридин [(R)-FDHU, 28]. 2'(R)-фтор-2'-дезоксиуридин 27 (1,2 г, 4,9 ммоль) растворили в Н 2 О (30 мл) с несколькими каплями концентрированного гидроксида аммония (5 капель). К раствору добавили родий на окиси алюминия(300 мг) и гидрогенизировали смесь в течение ночи при 40 psi. На следующий день смесь профильтровали, фильтрат сконцентрировали и очистили при помощи препаративной (обращенная фаза C185%CH3CN/H2O). Основной продукт 28, (R)-FDHU элюирует через 9,2 мин (780 мг, 64%). Остаток исходного материала 7 а (5,5 мин, 95 мг, 8%) и малое количество ФТГУ 2 а и 2b (7,2 мин, 50 мг, 4% и 8,6 мин, 45 мг,4%) отделили. 1The (R)-FDHU (600 мг, 2,4 ммоль) растворили в 20 мл MeOH и охладили до 0 С. К раствору порциями добавили борогидрид натрия (355 мг, 9,6 ммоль) и медленно нагревали реакционную смесь до комнатной температуры в течение ночи. Удалили МеОН под вакуумом, а остаток растворили в 10 мл Н 2 О. Раствор нейтрализовали 2.0N HCl до рН 7. Затем раствор очистили при помощи препаративнойHPLC (обращенная фаза C185% CH3CN/H2O). Желательный продукт 2 а элюирует через 7,2 мин (275 мг, 46%), затем элюирует другой эпимер 2b через 8,6 мин (125 мг, 21%), и через 9,2 мин - остаточный исходный материал и полностью восстановленное вещество 29 (50 мг, 9%) - через 14,9 мин. Стереохимическая структура у С-4 приписана 2 а и 2b на основании описанных в литературе прецедентов кристаллических структур цитидиндеаминазы в комплексе с одним эпимером тетрагидроуридина. 2 а (7,2 мин): 1HNMR (DMSO-J6): 7.21 (d, 1H), 5.93 (dd, 1H), 5.59 (d, 1H), 5.39 (d, 1H), 4.99-4.75 (m,3H), 3.95 (m, 1H), 3.62-3.21 (m, 5H), 1.69 (m, 2H); 13 2'(S)-фтор-2'дезоксидигидроуридин [(S)-FDHU, 31]. Соединение 30 (1,2 г, 4,0 ммоль) растворили в Н 2 О (40 мл). К раствору добавили родий на окиси алюминия (200 мг) и гидрогенизировали смесь в течение ночи при 50 psi. На следующий день смесь профильтровали через слой целлитов и сконцентрировали под вакуумом. Желательный продукт 31 получили с количественным выходом (1,0 г). 1HNMR (D2O): 6.08 (dd, 1H), 5.09 (dt, 1H), 4.28 (m, 1H), 3.85-3.80 (m, 2H), 3.72 (m, 2H), 3.51 (m, 1H),2.65 (t, J = 9 Гц, 2H). 2'(S)-фтор-2'дезокситетрагидроуридин [(S)-ФТГУ, 3 а и 3b]. Соединение 31 (1.12 мг, 4.55 ммоль) растворили в 28 мл МеОН и охладили до 0 С. К раствору порциями добавили борогидрид натрия (475 мг, 12,55 ммоль) и оставили реакционную смесь для продолжения реакции на 1 ч и 15 мин. МеОН удалили под вакуумом и растворили остаток в 15 мл 5% CH3CN/H2O. Раствор нейтрализовали 2,0N HCl до рН 7 (3 мл). Затем раствор очистили при помощи препаративнойHPLC (обращенная фаза C18 Phenomenex Luna с 5% CH3CN/H2O (изократический элюент и рефрактометрический детектор). Желательный продукт 3 а элюирует через 9,3 мин (163 мг, 14%), затем были детектированы другой эпимер 3b через 13,4 мин (236 мг, 21%), остаточный исходный материал (не определен количественно), а также полностью восстановленный продукт 32 (не определен количественно). Стереохимическая структура у С-4 приписана 3 а и 3b на основании описанных в литературе прецедентов - кристаллических структур цитидиндеаминазы в комплексе с одним эпимером тетрагидроуридина. 3 а (9,3 мин): 1HNMR (D2O)6.12 (dd, 1H), 5.04 (dt, 1H), 5.03 (m, 1H), 4.27 (m, 1H), 3.83-3.59 (m, 4H),3.34 (m, 1H), 1.86 (m, 2H). 13C, 41.96; H, 6.19; N, 10.87. Найдено: С, 41.99; H, 6.15; N, 10.91. Пример 4. Ферментативная активность CDA. Способность соединений согласно изобретению ингибировать ферментативную активность CDA можно продемонстрировать при помощи исследования, проведенного следующим исследованием. Методика определения ферментативной активности CDA основана на опубликованных методологических подходах (например, Cacciamani, T. et al., Arch. Biochem. Biophys. 1991, 290, 285-92; Cohen R. etal., J. Biol. Chem., 1971, 246, 7566-8; Vincenzetti S. et al, Protein Expr. Purif. 1996, 8, 247-53). В ходе исследования отслеживали изменения поглощения на 286 нм при катализированном CDA деаминировании цитидина с образованием уридина. Реакцию проводят в калий-фосфатном буфере (рН 7.4, 20 мМ, содержащем 1 мМ DTT) в общем объеме 200 мкл на 96-луночном планшете. Конечная реакционная смесь содержит цитидин (50 мкМ) и очищенную рекомбинантную CDA человека. Очищенный фермент разбавляют, чтобы обеспечить изменение величины поглощения приблизительно на 2 миллиоптических единицы/мин. Периодически выполняют фоновые измерения до добавления CDA для того, чтобы убедиться в отсутствии изменений величин поглощения в отсутствии CDA. После добавления CDA проводят мониторинг изменений величины поглощения в течение 20-30 мин. В присутствии потенциальных ингибито- 14018757 ров исследуют по восемь концентраций каждого образца в интервале 0,1-1 мМ для расчета величин IC50. Скорость изменения величины поглощения во времени для образцов, содержащих как цитидин, так иCDA, но не содержащих ингибитор (суммарная), принимают за 100%. Ферментативную активность CDA в присутствии соединения, выраженную в процентах от суммарной активности, вычитают из 100%, чтобы получить величину ингибирования (в процентах) при различных концентрациях соединения. По результатам вышеописанного исследования рассчитали ингибирующий эффект соединений 1 и 2. Величины IC50 представлены в табл. 1. "Ia" и "Ib" обозначают отдельные стереоизомеры; "1" обозначает смесь эпимеров. Таблица 1 Ингибирующий эффект тестируемых соединений Увеличение эффективности лекарственных препаратов - субстратов CDA. Способность соединений согласно изобретению к увеличению эффективности лекарственных препаратов - субстратов CDA можно продемонстрировать на модели лимфомы L1210 у мышей. Пример 5. Влияние ингибитора CDA, соединения 1, на выживаемость мышей, получавших децитабин (0,1 мг/кг), в модели лимфомы L1210. Методы. Самок мышей 30 CD2F1 в возрасте 6-7 недель случайным образом разделили на 6 групп:L1210 внутривенная (i.v.) инъекция. Перед экспериментом клетки L1210 перевивали по меньшей мере 3 раза мышам-самкам CD2F1. За неделю до умерщвления при помощи СО 2 мыши получали интраперитонеально (i.p.) инъекцию L1210 асцитической жидкости. Каждую мышь фиксировали в положении брюшком вверх, протирали брюшко тампоном, смоченным в спирте, и делали небольшой разрез для проникновения в брюшную полость. В полость ввели 2 мл охлажденного на ледяной бане 2,1% бычьего сывороточного альбумина (BSA) в солевом растворе. Жидкость удалили из полости, при помощи 18G 3 сс шприца перенесли в чистую стерильную пробирку и хранят на льду. Жидкость разбавили 1:10 в 2,1%BSA в солевом растворе, добавили к 2 мл разбавленной асцитической жидкости и добавили одну каплюZap-o-globin'a. Разбавленную асцитическую жидкость (вновь разбавленную 1:10) поместили в гемоцитометр и рассчитали количество клеток/мл. Исходный раствор асцитической жидкости в BSA развели до 1104 клеток/0,1 мл. Мыши получили инъекцию 0,1 мл клеточного раствора через иглу 27G. Приготовление раствора дозирования: подготовленные мыши получали дозу носителя или соединения 1 р.о. за 30 мин до введения децитабина. Приготовили основной раствор децитабина концентрации 1 мг/мл в PBS и разбавили соответственно для получения раствора дозирования концентрации 0,01 и 0,02 мг/мл. Схема дозирования: свежий раствор децитабина готовили дважды в день. Все растворы дозирования во время дозирования держали на льду. Мыши получали дозы i.p. или перорально (р.о.) дважды в день (с интервалом в 8 ч) в течение 4 последовательных дней. Итоговая схема дозирования и суммарные дозы децитабина и соединения 1 представлены в табл. 2. Выживаемость и аутопсия: выживаемость мышей проверяли ежедневно и проводили взвешивание(с понедельника по пятницу) на протяжении исследования (30 дней). Проводили аутопсию умерших мышей и отмечали наличие опухолей в органах. Смерть связывали с наличием опухоли, если вес печени превышал 1,6 г и вес селезенки превышал 150 мг (согласно Covey et al., Eur. J. Cancer Oncol. 1985). Мыши, получавшие децитабин и децитабин + соединение 1, жили дольше, чем мыши контрольной группы, получавшие только носитель или только соединение 1 (фиг. 1 и табл. 3; р 0,05). Тенденцию к отклику на дозу наблюдали при введении соединения 1 вместе с децитабином. Децитабин (0,1 мг/кг) р.о. менее эффективен, чем 0,1 мг/кг децитабина i.p., однако разница не существенна (табл. 3; фиг. 1,р=0,052). Совместное введение 10 мкг/кг соединения 1 с 0,1 мг/кг децитабина р.о. значительно повышает выживаемость по сравнению с 0,1 мг/кг р.о. только децитабина (р=0,0031) и 0,1 мг/кг децитабина i.p.(p=0,016; табл. 3, фиг. 1). Совместное введение 1 мкг/кг соединения 1 с 0,1 мг/кг децитабина р.с. значительно повышает выживаемость по сравнению с 0,1 мг/кг р.о. только децитабина (р=0,0031), но не по сравнению с 0,1 мг/кг децитабина i.p. (p=0,13; табл. 3, фиг. 1). В табл. 3 представлена средняя выживаемость в каждой группе, получавшей лекарства и процентILS (увеличенной продолжительности жизни), по сравнению с группой, получавшей носитель. Во всех группах, получавших лекарство, продолжительность жизни была значительно больше, чем в контрольной группе, получавшей носитель, и в группе CDA (р 0,05). В табл. 3 представлен вес печени и селезенки, взятых у мышей при аутопсии. Причиной смерти всех мышей, кроме одной - получавшей децитабин (0,1 мг/кг) i.p., явилась опухоль, на что указывает вес печени, превышающий 1 г, и вес селезенки, превышающий 80 мг (Covey et al., supra). Вес печени и селезенки у 3 контрольных мышей составили 0,970,08 г и 0,080,02 г. Описаны общие наблюдения относительно состояния перитонеальных и торакальных полостей. Таблица 3 Влияние децитабина и соединения 1 на выживаемость и вес печени и селезенки в модели выживаемости L1210 IV% ILS = средняя выживаемость экспериментальной группы (дни) - средняя выживаемость контрольной группы (дни)100 Средняя выживаемость контрольной группы (дни). На фиг. 1 представлен график, демонстрирующий влияние соединения 1 на выживаемость мышей,получавших децитабин, в модели лимфомы L1210 у мышей. Пример 6. Влияние ингибитора CDA, соединения 1 а на выживаемость мышей, получавших децитабин (0,1 мг/кг), в модели лимфомы L1210 у мышей. В модели L1210 на мыши исследовали соединение 1 а по методике, изложенной в примере 5. Мыши,получавшие децитабин (DAC), и DAC + соединение 1a, жили дольше, чем мыши, получавшие носитель(контроль) и только ингибитор CDA (фиг. 2; р 0,05). В комбинации с DAC доза соединения 1 а 10 мг/кг более эффективно влияла на выживаемость по сравнению с меньшими дозами. Пример 7. Влияние ингибитора CDA, соединения 3 а на выживаемость мышей, получавших децитабин (0,1 мг/кг), в L1210 модели выживаемости. В модели L1210 на мыши исследовали соединение 3 а по методике, изложенной в примере 5. Мыши,- 16018757(контроль) и только ингибитор CDA (фиг. 3; р 0,05). Пример 8. Влияние ингибитора CDA, соединения 1 на выживаемость мышей, получавших цитарабин (ара-С), в L1210 модели выживаемости. Самок мышей 50 CD2F1 в возрасте 6-7 недель случайным образом разделяли на 10 групп. Инъекция L1210: за неделю до умерщвления (СО 2) мыши получали интраперитонеально (i.p.) инъекцию L1210 асцитической жидкости. Перед экспериментом клетки L1210 перевивали по меньшей мере 3 раза in vivo. Мышь фиксировали в положении брюшком вверх, протирали брюшко тампоном, смоченным в спирте, и делали небольшой разрез для проникновения в брюшную полость. В полость ввели 2 мл охлажденного в ледяной бане 2,1% бычьего сывороточного альбумина (BSA) в солевом растворе. Жидкость удалили из полости, при помощи 18G 3 cc шприца перенесли в чистую стерильную пробирку и держали на льду. Жидкость разбавили 1:10 в 2,1% BSA в солевом растворе, добавили к 2 мл разбавленной асцитической жидкости и добавили одну каплю Zap-o-globin'a. Разбавленную асцитическую жидкость (вновь разбавленную 1:10) поместили в гемоцитометр и рассчитали количество клеток/мл. Исходный раствор асцитической жидкости в BSA развели до 1104 клеток/0,1 мл. Мыши получали инъекцию 0,1 мл клеточного раствора через иглу 27G. Суммарное время проведения i.v. инъекций составило около 50 мин. Приготовление раствора дозирования: подготовленные мыши получали дозу носителя или соединения 1 р.о. за 30 мин до введения Ара-С. Приготовили основной раствор соединения 1 концентрации 1 мг/мл в PBS; приготовили основной раствор Ара-С концентрации 20 мг/мл в PBS, затем разбавили соответственно для получения раствора меньшей концентрации. Схема дозирования: соединение 1 готовили в начале исследования и хранили при 4 С. Свежий раствор Ара-С готовили дважды в день. Все растворы дозирования во время дозирования держали на льду. Мыши получали дозы перорально (р.о.) дважды в день (с интервалом в 8 ч) в течение 4 последовательных дней. Итоговая схема дозирования и суммарные дозы Ара-С и соединения 1 представлены в табл. 4. Таблица 4 Схема дозирования Выживаемость и аутопсия: выживаемость мышей проверяли ежедневно и проводили взвешивание(с понедельника по пятницу) на продолжении исследования (45 дней). Проводили аутопсию умерших мышей и отмечали наличие опухолей в органах. Смерть связывали с наличием опухоли, если вес печени превышал 1,6 г и вес селезенки превышал 150 мг (согласно Covey et al., Eur. J. Cancer Oncol. 1985). Мыши, получавшие только Ара-С 50 мг/кг, 100 мг/кг и 200 мг/кг) и Ара-С + соединение 1, жили дольше, чем мыши, получавшие носитель (контроль) и только ингибитор CDA (фиг. 4-7; р 0,05). Одно лишь соединение 1 не влияло на выживаемость по сравнению с контрольным носителем (фиг. 4). Введение 25 мг/кг Ара-С не влияло на увеличение выживаемости по сравнению с выживаемостью мышей, получавших носитель и 10 мг/кг соединения 1. 50, 100 и 200 мг/кг Ара-С значительно увеличивает выживаемость (в днях) с зависимостью эффекта от дозы, по сравнению с выживаемостью мышей контрольной группы. Совместное введение 10 мг/кг соединения 1 и Ара-С р.о. значительно повышает выживаемость по сравнению со временем выживаемости мышей, получавших те же дозы одного лишь Ара-С (фиг. 4-7). Пример 9. Воздействие соединения 1 на уменьшение объема опухоли, вызванное приемом гемцитабина, в модели на мыши ксенотрансплантанта А 2780 рака яичника человека. Исследовали эффективность гемцитобина при пероральном приеме в комбинации с соединением 1 в модели ксенотрансплантанта А 2780 рака яичника человека. Самкам мышей NCr nu/nu в возрасте 5-6 недель подкожно имплантировали фрагменты опухоли 30-60 мг. На 11 день, когда объем опухоли достигал приблизительно 200 мм 3, начинается лечение, описанное в табл. 5. Таблица 5 Схема дозирования дозу соединения 1 вводят приблизительно за 30 мин до введения Гемцитабина. Объем опухоли отслеживали на протяжении всего эксперимента. Объем опухоли измеряли три раза в неделю. Опухолевую массу (мг = мм 3) рассчитывают по результатам кавернометрических измерений по формуле объема вытянутого эллипсоида (LW2)/2, где L и W представляют собой соответственно ортогональные длину и ширину (мм). Основными критериями при оценке эффективности в модели А 2780 явились полная и частичная регрессия опухоли, замедление роста опухоли и количество выживших особей, не имевших опухолей, к концу исследования. Полный ответ (CR) определяли как уменьшение размера опухоли до недетектируемого размера (50 мм 3). Частичный ответ (PR) определяли как 50% уменьшение опухолевой массы по сравнению с начальным размером опухоли. Если опухоль, состояние которой оценивали как CR или PR в конце исследования, начинала расти вновь, ее состояние, тем не менее, оценивали как CR или PR. Выжившие особи, не имевшие опухолей (TFS) в конце исследования, не имели детектируемых (50 мм 3) опухолей на момент окончания исследования (день 74). Замедление роста опухоли (TGD) в данном эксперименте определяли как среднее число дней, за которые опухоль достигает размера 750 мм 3, для группы, получавшей лекарство, по сравнению с контрольной группой. Пероральное введение 10 мг/кг РО q3d4 гемцитабина оказалось не достаточно эффективным в данной модели, где замедление роста опухоли составило 6,1 дней (статистически не значимо), при отсутствии CR, PR или выживших особей, не имевших опухолей, на момент окончания исследования (день 74). При введении дозы 10 мг/кг Гемцитабина в комбинации с соединением 1, отмечали значительное замедление роста опухоли по сравнению с замедлением при введении такой же дозы одного гемцитабина(19,2 дней; р 0,05 по сравнению с одним Гемцитабином); при этом 75% опухолей проявили CR, однако к концу эксперимента не осталось TFS. На фиг. 8 (объем опухоли - время (дни) после начала лечения) видно, что одно соединение 1 не оказывает воздействия на рост опухоли, в то время как комбинированное лечение соединением 1 и гемцитабином более эффективно, чем лечение одним гемцитабином. Эффект перорального введения гемцитабина 30 мг/кг РО q3d4 (MTD) проявился в статистически значимом замедлении роста опухоли на 22 дня при 63% CR, однако при отсутствии TGF. При комбинированной химиотерапии гемцитабином (30 мг/кг РО) + соединение 1 отметили статистически значимое замедление роста опухоли (57 дней) и 100% CR и 50% случаев TFS на момент окончания исследования(день 74). Таблица 6 Воздействие комбинированного лечения Гемцитабином и соединением 1 на замедление роста опухоли в модели А 2780 рака яичника Пример 10. Характеристика соединения 1 а в твердом состоянии. Сбор данных. Бесцветный кристалл соединения 1a (F2O5N2C9H14) в форме призмы с приблизительными размерами 0,480,430,32 мм устанавливали в держателе. Все измерения проводят на оборудовании Rigaku RAXISSPIDER детекторе, с излучением Cu-K через графитовый монохроматор. Индексацию проводили по 5 осцилляциям, время экспозиции - 60 с. Расстояние кристалл-детектор составляет 127,40 мм. Постоянные ячейки и матрица ориентации кристалла для сбора данных соответствуют элементарной тригональной ячейке (класс Лауэ: - 3 мл) с размерами а =9.78961(18) А с = 20.4588(7) АV = 1698.02(7) А 3 При Z = 6 и F.W. (полный вес) = 268.22, рассчитанная плотность составила 1,574 г/см 3. На основании систематического отсутствия 0001:13n и успешной расшифровки и уточнения структуры, определили, что пространственная группа является Р 321 (152). Данные получили при температуре -1231 С при максимальном значении 2 136,4. Всего получено 111 осцилляционных картин. Сбор данных осуществляют присканировании от 20,0 до 200,0 при шаге сканирования 5,0; при =0,0 и= 180,0. Второй сбор данных:сканирование от 20,0 до 200,0 при шаге сканирования 5,0; от =54,0 и = 180,0. Третий сбор данных:сканирование от 20,0 до 185,0 при шаге сканирования 5,0, при= 54,0 и= 90,.0, и окончательный сбор данных:сканирование от 20,0 до 50,0 при шаге сканирования 5,0, при= 0,0 о и= 0,0. Экспозиционная доза -12,0[sec./0]. Расстояние кристалл-детектор - 127,40 мм. Считывание в режиме 0,100 мм пиксель. Предварительная обработка данных. Из 11772 собранных изображений 2052 являются уникальными (Rint = 0,038); эквивалентные изображения объединяли. Коэффициент линейного поглощениядля Cu-K-излучения составил 13,035 см-1. После эмпирической коррекции величин поглощения получили интервал величин коэффициентов пропускания 0,5400,659. Данные скорректировали с учетом эффекта Лоренца и поляризационного эффекта. Провели коррекцию на вторичную экстинкцию (Larson, А.С., Crystallographic Computing 1970, 291-294; уравнение 22,V заменили на объем ячейки) (коэффициент = 0,005900). Расшифровка и уточнение структуры. Структуру расшифровали прямыми методами (SIR92: Larson, A.C., J. Appl. Cryst, 1994, 27, 435) и уточнили при помощи метода Фурье (DIRDIF99: Beurskens, P.T. et al., The DIRD-99 Program System.Technical Report of the Crystallography Laboratory, 1999, University of Nijmegen, The Netherlands). Уточнение для неводородных атомов проводили в анизотропном приближении. Уточнения для некоторых атомов водорода провели в изотропном приближении, а остальные включены в уточнение в модели "наездника". Последний цикл полноматричного уточнения по методу наименьших квадратов (среднеквадратичные веса = w(Fo2-Fc2)2) на F2 основан на 2052 наблюдавшихся отражениях и 181 переменном параметре и сходится (максимальный сдвиг параметра составляет менее 0,01 от величины стандартного отклонения с невзвешенными и взвешенными факторами согласия Стандартное отклонение по наблюдаемым единичным массам (стандартное отклонение = [w(F02No = количество наблюдений, Nv = количество переменных) равняется 1.10. Максимальные и минимальные пики итоговой разностной Фурье-карты соответствуют 0,22 и -0,22 е'/А 3 соответственно. Абсолютную структуру определили на основании параметра Флэка, 0,0(1), уточнили по 839 фриделевским парам (Flack, H.D., Acta Cryst. 1983, A 39, 876-881). Множители рассеяния нейтральных атомов взяты из Cromer, D.Т. et al., International Tables for X-rayCrystallography 1974, IV, Table 2.2 А. Аномальные дисперсионные эффекты включены в Fcalc (Ibers, J.A.Crystallography 1992, C, Table 4.2.6.8, 219-222. Значения массовых коэффициентов ослабления взяты из производили при помощи пакета кристаллографических программ Crystal Structure 3.8, за исключением уточнения, которое проводили при помощи SHELXL-97. Подробности эксперимента.ORTEP представление (Michael N. Burnett and Carroll K. Johnson, ORTEP-III: Oak Ridge Thermal Ellipsoid Plot Program for Crystal Structure Illustrations, Oak Pvidge National Laboratory Report ORNL-6895,1996) расшифрованной структуры соединения la показано на фиг. 9. Пример 11. Увеличение стабильности в кислой среде. Соединения 1 а по сравнению с тетрагидроуридином (ТГУ). Стабильность соединения 1 а и тетрагидроуридина (ТГУ) в кислом растворе оценивали при помощи 1 Н ЯМР спектроскопии. Тетрагидроуридин (ТГУ, 5 мг) растворили в D2O (0,75 мл). 1 Н ЯМР (D2O, 300 МГц, 27C) спектр показан на фиг. 10. К данному образцу добавили одну каплю дейтерированного TFA, затем немедленно сняли 1 Н ЯМР спектр (фиг. 10). Даже в начальный момент времени пик на 5,4 ppm (-10% конверсии при интеграции) указывает на расширение цикла с преобразованием в ТН-пиранозу. В течение следующих нескольких часов сняли спектры (D2O, 300 МГц, 27C), представленные на фиг. 11. Через 4 ч отметили преобладание ТН-пиранозы, указывая на 60% конверсию. Через 72 ч конверсия почти полностью завершилась (80%). Характерные изменения в интервале 4,0-4,5 также указывают на разложение ТГУ и образование ТН-пиранозы. Соединение 1 а (5 мг) растворили в D2O (0,75 мл). 1 Н ЯМР спектр (D2O, 300 МГц, 27 С) представлен на фиг. 12. К данному образцу добавили одну каплю дейтэрированного TFA, затем немедленно сняли 1 Н ЯМР спектр (фиг. 12). После первой временной точки единственным существенным изменением была эпимеризация аминала, на которую указывают дополнительные пики на 5,92 и 5,93. Через 4 и 72 ч (фиг. 13) других заметных изменений в спектре не было (D2O, 300 МГц, 27C). Данные результаты обозначают, что образования пиранозы из соединения 1 а при данных условиях не происходит. Настоящее изобретение описано через частные варианты реализации; тем не менее, для специалиста в данной области очевидно, что в рамках настоящего изобретения возможно внесение изменений и эквивалентные замены. Кроме того, при адаптации к конкретной ситуации в рамках настоящего изобретения возможны многочисленные модификации материалов, композиций веществ, процесса, этапа или этапов процесса. Все подобные модификации существуют в рамках настоящего изобретения. Все вышеуказанные патенты и публикации включены в данную заявку посредством ссылки. ФОРМУЛА ИЗОБРЕТЕНИЯ 1. Соединение формулы I или его фармацевтически приемлемая соль где атом углерода, отмеченный символом , может находиться в (R)- или (S)-конфигурации и где R1 и R2 представляют собой фтор. 2. Соединение по п.1, где указанное соединение представляет собой соединение 1a или его фармацевтически приемлемую соль 3. Соединение по п.1, где указанное соединение представляет собой соединение 1 а 4. Фармацевтическая композиция для ингибирования ферментной активности CDA, по существу,состоящая из соединения формулы I или его фармацевтически приемлемой соли представляют собой фтор; и фармацевтически приемлемого эксципиента. 5. Фармацевтическая композиция по п.4, отличающаяся тем, что указанное соединение представляет собой соединение 1 а или его фармацевтически приемлемую соль 6. Фармацевтическая композиция по п.4, отличающаяся тем, что указанное соединение представляет собой соединение 1 а(i) соединение формулы I или его фармацевтически приемлемую соль(i) соединение формулы I или его фармацевтически приемлемую соль(ii) лекарственный препарат - субстрат CDA, полезный для лечения рака; при этом лекарственный препарат - субстрат CDA не является децитабином. 9. Фармацевтическая композиция, содержащая:(i) соединение формулы I или его фармацевтически приемлемую соль(ii) лекарственный препарат - субстрат CDA; при этом лекарственный препарат - субстрат CDA представляет собой децитабин. 10. Фармацевтическая композиция по любому из пп.7 или 8, отличающаяся тем, что лекарственный препарат - субстрат CDA представляет собой гемцитабин. 11. Фармацевтическая композиция по любому из пп.7 или 8, отличающаяся тем, что лекарственный препарат - субстрат CDA представляет собой ара-С. 12. Фармацевтическая композиция по любому из пп.7 или 8, отличающаяся тем, что лекарственный препарат - субстрат CDA представляет собой 5-азацитидин. 13. Фармацевтическая композиция по любому из пп.7-12, отличающаяся тем, что указанное соединение представляет собой соединение 1 а или его фармацевтически приемлемую соль 14. Фармацевтическая композиция по любому из пп.7-12, отличающаяся тем, что указанное соединение представляет собой соединение 1 а 15. Фармацевтическая композиция, содержащая соединение по любому из пп.1-3 и фармацевтически приемлемый эксципиент. 16. Способ лечения рака, чувствительного к субстрату CDA, включающий:(i) введение в организм млекопитающего, нуждающегося в лечении, соединения формулы I или его фармацевтически приемлемой соли(ii) введение в организм млекопитающего, нуждающегося в лечении, лекарственного препарата субстрата CDA, выбранного из группы, состоящей из 5-азацитидина, гемцитабина, ара-С, тезацитабина,5-фтор-2'-дезоксицитидина и цитохлора. 17. Способ лечения рака, чувствительного к субстрату CDA, включающий:(i) введение в организм млекопитающего, нуждающегося в лечении, соединения формулы I или его фармацевтически приемлемой соли(ii) введение в организм млекопитающего, нуждающегося в лечении, лекарственного препарата субстрата CDA, полезного для лечения рака; при этом лекарственный препарат - субстрат CDA не является децитабином. 18. Способ лечения рака, чувствительного к субстрату CDA, включающий:(i) введение в организм млекопитающего, нуждающегося в лечении, соединения формулы I или его фармацевтически приемлемой соли(ii) введение в организм млекопитающего, нуждающегося в лечении, лекарственного препарата субстрата CDA; при этом лекарственный препарат - субстрат CDA представляет собой децитабин. 19. Способ по любому из пп.16, 17, отличающийся тем, что лекарственный препарат - субстратCDA представляет собой гемцитабин. 20. Способ по любому из пп.16, 17, отличающийся тем, что лекарственный препарат - субстратCDA представляет собой ара-С. 21. Способ по любому из пп.16, 17, отличающийся тем, что лекарственный препарат - субстратCDA представляет собой 5-азацитидин. 22. Способ по любому из пп.16-21, отличающийся тем, что раковое заболевание выбрано из группы,состоящей из злокачественных гематологических заболеваний и солидных форм рака. 23. Способ по п.22, отличающийся тем, что рак представляет собой злокачественное гематологическое заболевание, выбранное из группы, состоящей из миелодиспластического синдрома и лейкоза. 24. Способ по п.23, отличающийся тем, что злокачественное гематологическое заболевание представляет собой миелодиспластический синдром. 25. Способ по п.23, отличающийся тем, что злокачественное гематологическое заболевание представляет собой острый миелоидный лейкоз или хронический миелоидный лейкоз. 26. Способ по п.22, отличающийся тем, что рак представляет собой солидный рак, выбранный из группы, состоящей из рака поджелудочной железы, рака яичников, перитонеального рака, немелкоклеточного рака легкого и метастатического рака молочной железы. 27. Способ по любому из пп.16-26, отличающийся тем, что указанное соединение представляет собой соединение 1 а или его фармацевтически приемлемую соль 28. Способ по любому из пп.16-26, отличающийся тем, что указанное соединение представляет собой соединение 1 а 29. Способ по любому из пп.16-28, отличающийся тем, что соединение вводят, по существу, одновременно с лекарственным препаратом - субстратом CDA. 30. Способ по любому из пп.16-28, отличающийся тем, что указанное соединение вводят перед введением лекарственного препарата - субстрата CDA. 31. Способ по любому из пп.16-28, отличающийся тем, что указанное соединение вводят после введения лекарственного препарата - субстрата CDA. 32. Способ по любому из пп.16-29, отличающийся тем, что соединение и лекарственный препарат субстрат CDA вводят в организм в виде единичной стандартной дозированной формы. 33. Способ по любому из пп.16-31, отличающийся тем, что соединение и лекарственный препарат субстрат CDA вводят в организм в виде множественных раздельных стандартных дозированных форм.
МПК / Метки
МПК: A61K 31/706, C07H 19/04, A61P 35/00
Метки: 2'-фтор-2'-дезокситетрагидроуридины, ингибиторов, качестве, цитидиндеаминазы
Код ссылки
<a href="https://eas.patents.su/30-18757-2-ftor-2-dezoksitetragidrouridiny-v-kachestve-ingibitorov-citidindeaminazy.html" rel="bookmark" title="База патентов Евразийского Союза">2′-фтор-2′-дезокситетрагидроуридины в качестве ингибиторов цитидиндеаминазы</a>
Гетероариламины в качестве ингибиторов гликогенсинтаза-киназы 3-бета (ингибиторов gsk3)

Номер патента: 7298
Опубликовано: 25.08.2006
Авторы: Эмбрехтс Вернер Констант Йохан, Хэрес Ян, Бейнстерс Петер Якобус Йоханнес Антониус, Виллемс Марк, Леви Паулус Йоаннес, Лав Кристофер Джон, Де Жонж Марк Рене, Фрейн Эдди Жан Эдгар, Дильс Гастон Станислас Марселла, Винкерс Хендрик Мартен, Ван Акен Кун Жанн Альфонс, Жанссен Поль Адриан Ян, Койманс Люсьен Мария Хенрикус
МПК: A61K 31/50, A61K 31/505, A61K 31/435...
Метки: 3-бета, gsk3, качестве, гетероариламины, ингибиторов, гликогенсинтаза-киназы
Формула / Реферат:
1. Соединение формулы его N-оксид, фармацевтически приемлемая аддитивная соль, четвертичный амин и стереохимически изомерная форма, где R1 обозначает водород; X обозначает -O-; -О-С1-4алкил- или прямую связь; Z обозначает прямую связь, -NH-, -С1-4алкил-NH- или -С(=O)-; R2 обозначает водород или фенил; R3 обозначает водород, С1-4алкил, полигалогенC1-6алкил или циано; R4 обозначает моноциклический или бициклический насыщенный гетероцикл;...
Гетероариламины-производные пиримидина и пиридазина в качестве ингибиторов гликогенсинтаза-киназы 3-бета (ингибиторов gskз)

Номер патента: 10859
Опубликовано: 30.12.2008
Авторы: Леви Паулус Йоаннес, Фрейн Эдди Жан Эдгар, Ван Акен Кун Жанн Альфонс, Бейнстерс Петер Якобус Йоханнес Антониус, Де Жонж Марк Рене, Койманс Люсьен Мария Хенрикус, Хэрес Ян, Лав Кристофер Джон, Виллемс Марк, Жанссен Поль Адриан Ян, Винкерс Хендрик Мартен, Дильс Гастон Станислас Марселла, Эмбрехтс Вернер Констант Йохан
МПК: A61K 31/505, A61K 31/50, A61K 31/435...
Метки: гликогенсинтаза-киназы, качестве, ингибиторов, гетероариламины-производные, пиридазина, пиримидина, gskз, 3-бета
Формула / Реферат:
1. Соединение формулы его N-оксид, фармацевтически приемлемая аддитивная соль, четвертичный амин и стереохимически изомерная форма, где кольцо А является R1 обозначает водород; X обозначает -O-; -O-C1-6алкил- или прямую связь; Z обозначает прямую связь, -С(=O)- или -NR1-C1-6алкил-; R обозначает водород или R20; R3 обозначает водород; галоген; циано; полигалогенС1-6алкил; R4 обозначает моноциклический, насыщенный или частично насыщенный или...
Сульфонамидные производные в качестве ингибиторов рассасывания костной ткани и ингибиторов адгезии клеток,способ их получения, применение и фармацевтическая композиция

Номер патента: 3102
Опубликовано: 26.12.2002
Авторы: Вилл Дэвид Вильям, Гурвест Жан-Франсуа, Шойнеманн Карлхайнц, Гадек Томас, Макдауэлл Роберт, Карниато Дени, Бодари Сара Кэтрин, Катбертсон Роберт Эндрю, Кнолле Йохен, Пейман Ануширван
МПК: A61P 19/10, A61K 31/505, C07D 239/42...
Метки: рассасывания, получения, клеток,способ, фармацевтическая, костной, адгезии, ткани, производные, применение, композиция, ингибиторов, сульфонамидные, качестве
Формула / Реферат:
1. Сульфонамидные производные общей формулы I где R1 и R2 вместе образуют двухвалентный (С2-С3)алкиленовый радикал; R4 является Н или (С1-С6)алкилом; R5 является (С1-С10)алкилом, (С6-С14)арилом, (C5-C14)гетероарилом или (С6-С14)арил(С1-С10)алкильной группой, где арил, гетероарил или алкил возможно замещены R3, или 2-оксобицикло[2.2.1]гепт-1-илметильной группой; R3 является (C1-C4)алкилом, (C1-C4)алкилокси, галогеном, трифторметилом, циано,...
Триамид-замещённые индолы, бензофураны и бензотиофены в качестве ингибиторов микросомального белка, переносящего триглицериды, (мтр) и/или ингибиторов секреции аполипопротеина в (аро в)

Номер патента: 7008
Опубликовано: 30.06.2006
Авторы: Ли Дзин, Бронк Брайан Скотт, Блайз Алан Элвуд, Ченг Хенгмиао, Мейсон Клайв Филип, Бертинато Питер, Хуатан Хип
МПК: A61K 31/343, A61K 31/404, A61K 31/4439...
Метки: переносящего, ингибиторов, индолы, белка, секреции, триглицериды, бензофураны, качестве, аро, аполипопротеина, бензотиофены, микросомального, мтр, триамид-замещённые
Формула / Реферат:
1. Соединение формулы 1b или его фармацевтически приемлемая соль, где R1 представляет собой заместитель в 5 или 6 положении формулы 1b и имеет структуру или, когда R7 представляет собой фенил, пиридил, фенил-Z1-или пиридил-Z1-, необязательно замещенные от одного до пяти, независимо, выбранными R12, то R1 представляет собой (C1-С6)алкил, (С3-С8)циклоалкил, (С5-С10)бициклоалкил, -(CRaRb)tO(C1-C6 алкил), -(СRaRb)tS(C1-C6 алкил), -(CRaRb)rC(O)R15,...
2-аминопиридины, содержащие в качестве заместителей конденсированные кольца, применяемые в качестве ингибиторов nos (no синтазы)

Номер патента: 3839
Опубликовано: 30.10.2003
Автор: Лоуэ Джон Адамс III
МПК: C07D 401/10, A61K 31/495
Метки: применяемые, 2-аминопиридины, качестве, содержащие, кольца, конденсированные, синтазы, заместителей, ингибиторов
Формула / Реферат:
1. Соединение формулы где кольцо A является 5-7-членным насыщенным или ненасыщенным конденсированным кольцом, X является кислородом или связью; n представляет собой целое число от 2 до 6 и R1 и R2 независимо выбирают из водорода (C1-C6)алкила, арила, тетрагидронафталина и аралкила, где указанный арил и арильная часть указанного аралкила представляет собой фенил или нафтил, а алкильная часть является линейной или разветвленной и содержит от 1 до...
Предыдущий патент: Антитела к склеростину, композиции, их содержащие, и их применение
Следующий патент: Котел водогрейный
Случайный патент: Устройство для подачи рабочей жидкости противовыбросового превентора