Соединения и композиции в качестве модуляторов активности gpr119

























Формула / Реферат
1. Соединение формулы (I)

или его фармацевтически приемлемая соль, в котором
А может содержать вплоть до 2 кольцевых атомов углерода, замещенных оксогруппой;
n выбран из 0, 1 и 2;
t1 и t2, каждый независимо, выбраны из 0 и 1;
R1 выбран из цианогруппы, -S(O)2R6a, -N(R6a)S(O)2R6a, -S(O)2OR6a, -S(O)2C(O)R6a, -S(O)2C(O)OR6a и
-S(O)2NR6aR6b, где R6a выбран из C1-6алкила, где указанный алкил в R6a необязательно замещен 1-3 радикалами, независимо выбранными из гидроксигруппы и галогена;
каждый R2 независимо выбран из C1-6алкила, галогензамещенного C1-6алкила, гидроксизамещенного C1-6алкила;
R4 выбран из R9 и -C(O)OR9, где R9 выбран из C1-6алкила, C6-10арила и C1-10гетероарила, где указанный арил или гетероарил в R9 необязательно замещен 1-3 радикалами, независимо выбранными из галогена, цианогруппы, C1-6алкила, C3-12циклоалкила, C3-8гетероциклоалкила, галогензамещенного C1-6алкила, гидроксизамещенного C1-6алкила, C1-6алкоксигруппы, галогензамещенной C1-6алкоксигруппы и -C(O)OR17,
-C(O)R19 и -C(O)NR17R18, где R17 и R18 независимо выбраны из водорода и C1-6алкила или R17 и R18 вместе с атомом азота, к которому R17 и R18 присоединены, образуют С3-8гетероциклоалкил; R19 выбран из C1-6алкила и С3-8гетероциклоалкила; и указанные циклоалкильные или гетероциклоалкильные заместители в R9 необязательно далее замещены 1-3 C1-6алкильными радикалами;
каждый R6 независимо выбран из гидроксигруппы, нитрогруппы, цианогруппы, галогена, C1-6алкила, C2-6алкенила, галогензамещенного C1-6алкила, галогензамещенного C2-6алкенила, гидроксизамещенного C1-6алкила, C1-6алкоксигруппы, галогензамещенной C1-6алкоксигруппы, C6-10арила, C1-10гетероарила, C3-8гетероциклоалкила, С3-8циклоалкила и -X3OR20, -NR20X3OR21, -C(O)OR20, где Х3 выбран из связи, C1-4алкилена и C2-4алкенилена; R20 и R21 независимо выбраны из водорода и C1-6алкила;
W1 и W2 независимо выбраны из CR10 и N, где R10 выбран из водорода и C1-6алкила;
Y1 выбран из NR11 и О, где R11 выбран из водорода и C1-6алкила;
Y3 выбран из СН и N;
Y4 выбран из СН2 и ОСН2;
где C1-10гетероарил обозначает моноциклический или конденсированный бициклический ароматический кольцевой комплекс, содержащий от 1 до 10 атомов углерода и вплоть до 4 гетероатомов в качестве кольцевых атомов, где гетероатомы независимо выбраны из N и О;
С3-8гетероциклоалкил обозначает насыщенный или частично ненасыщенный, моноциклический, конденсированный бициклический или мостиковый полициклический кольцевой комплекс, содержащий от 3 до 8 атомов углерода и вплоть до 4 гетероатомов в качестве кольцевых атомов, где гетероатомы независимо выбраны из N и О.
2. Соединение по п.1 формулы (Ia)

в котором А может содержать кольцевой атом углерода, замещенный оксогруппой;
t1 и t2, каждый независимо, выбраны из 0 и 1;
W2 выбран из СН и N.
3. Соединение по п.2, в котором
R1 выбран из цианогруппы, -S(O)2R6a и -N(R6a)S(O)2R6a, где R6a выбран из водорода, C1-6алкила и C1-10гетероарила, необязательно замещенного C1-6алкилом.
4. Соединение по п.3, в котором
R4 выбран из R9 и -C(O)OR9, где R9 выбран из трет-бутила, пиридинила, пиримидинила, 1,2,4-оксадиазол-5-ила и тетразолила, где указанный пиридинил, пиримидинил, 1,2,4-оксадиазол-5-ил или тетразолил в R9 необязательно замещен радикалом, выбранным из галогена, цианогруппы, трифторметила, изопропила, метила, этила, метоксикарбонила, диметиламинокарбонила, аминокарбонила и морфолинокарбонила.
5. Соединение по п.4, в котором
R6 выбран из фтора, хлора, брома, трифторметоксигруппы, метила, метоксигруппы, метоксикарбонила, 3-метоксипроп-1-енила, метоксипропила, винила, фенила, пиразолила, 5-хлорпент-1-енила, гидроксипропила, метоксиэтиламиногруппы и морфолиногруппы;
Y1 представляет собой О;
Y3 представляет собой СН;
Y4 представляет собой CH2.
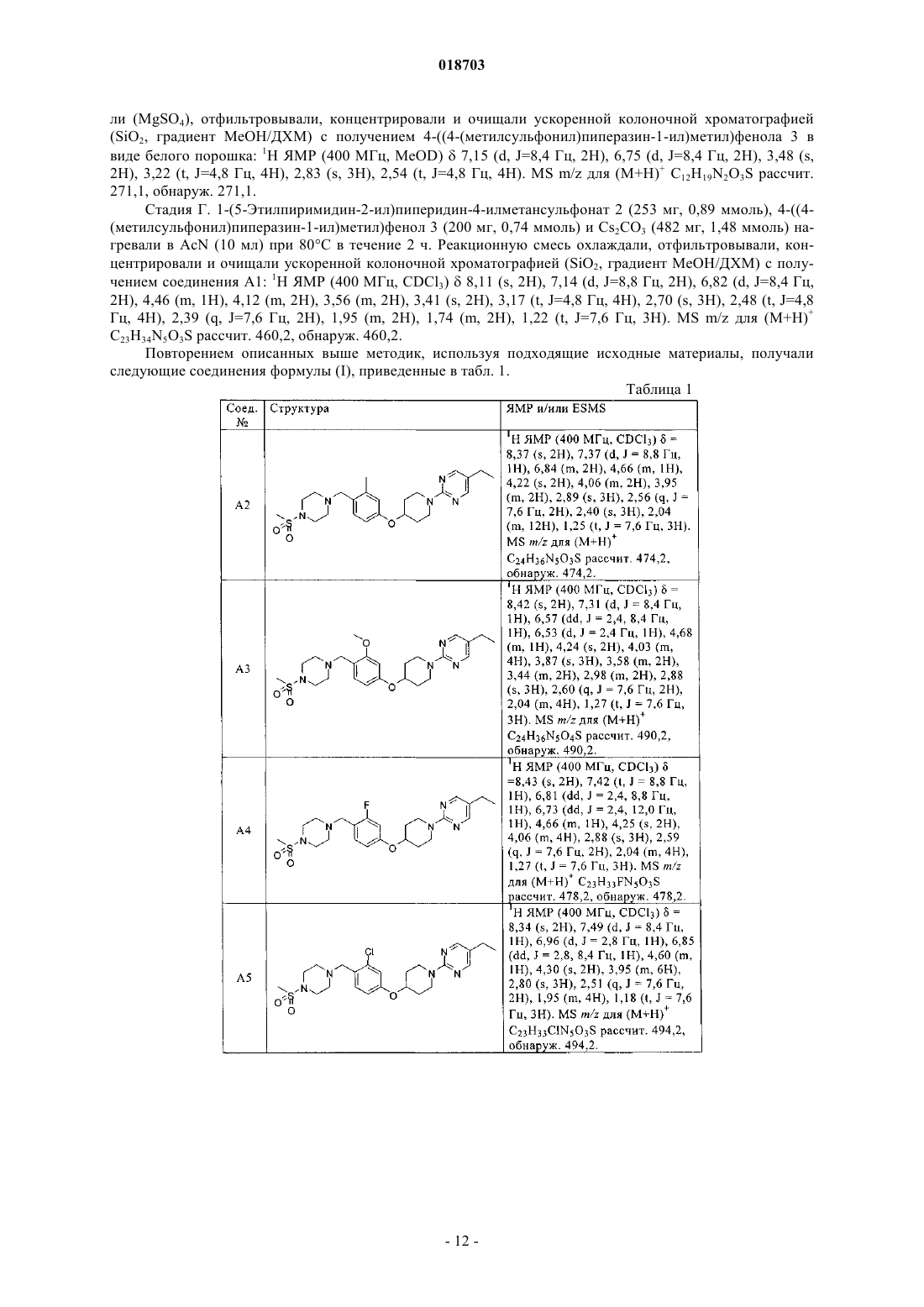
6. Соединение по п.1, выбранное из следующих соединений:
5-этил-2-(4-(4-((4-(метилсульфонил)пиперазин-1-ил)метил)фенокси)пиперидин-1-ил)пиримидин;
5-этил-2-(4-(3-метил-4-((4-(метилсульфонил)пиперазин-1-ил)метил)фенокси)пиперидин-1-ил)пиримидин;
5-этил-2-(4-(3-метокси-4-((4-(метилсульфонил)пиперазин-1-ил)метил)фенокси)пиперидин-1-ил)пиримидин;
5-этил-2-(4-(3-фтор-4-((4-(метилсульфонил)пиперазин-1-ил)метил)фенокси)пиперидин-1-ил)пиримидин;
2-(4-(3-хлор-4-((4-(метилсульфонил)пиперазин-1-ил)метил)фенокси)пиперидин-1-ил)-5-этилпиримидин;
5-этил-2-(4-(2-метил-4-((4-(метилсульфонил)пиперазин-1-ил)метил)фенокси)пиперидин-1-ил)пиримидин;
5-этил-2-(4-(2-фтор-4-((4-(метилсульфонил)пиперазин-1-ил)метил)фенокси)пиперидин-1-ил)пиримидин;
2-(4-(2-хлор-4-((4-(метилсульфонил)пиперазин-1-ил)метил)фенокси)пиперидин-1-ил)-5-этилпиримидин;
2-(4-(2-бром-4-((4-(метилсульфонил)пиперазин-1-ил)метил)фенокси)пиперидин-1-ил)-5-этилпиримидин;
5-этил-2-(4-(2-метокси-4-((4-(метилсульфонил)пиперазин-1-ил)метил)фенокси)пиперидин-1-ил)пиримидин;
5-этил-2-(4-(4-((4-(метилсульфонил)пиперазин-1-ил)метил)-2-(трифторметокси)фенокси)пиперидин-1-ил)пиримидин;
2-(4-(2,3-дифтор-4-((4-(метилсульфонил)пиперазин-1-ил)метил)фенокси)пиперидин-1-ил)-5-этилпиримидин;
2-(4-(2-хлор-6-фтор-4-((4-(метилсульфонил)пиперазин-1-ил)метил)фенокси)пиперидин-1-ил)-5-этилпиримидин;
5-этил-2-(4-(6-((4-(метилсульфонил)пиперазин-1-ил)метил)пиридин-3-илокси)пиперидин-1-ил)пиримидин;
трет-бутил 4-(4-((4-(метилсульфонил)пиперазин-1-ил)метил)фенокси)пиперидин-1-карбоксилат;
5-фтор-2-(4-(4-((4-(метилсульфонил)пиперазин-1-ил)метил)фенокси)пиперидин-1-ил)пиримидин;
1-(4-(1-(5-фторпиридин-2-ил)пиперидин-4-илокси)бензил)-4-(метилсульфонил)пиперазин;
1-(метилсульфонил)-4-(4-(1-(5-(трифторметил)пиридин-2-ил)пиперидин-4-илокси)бензил)пиперазин;
5-(4-(2-хлор-4-((4-(метилсульфонил)пиперазин-1-ил)метил)фенокси)пиперидин-1-ил)-3-изопропил-1,2,4-оксадиазол;
3-(4-(3-хлор-4-(1-(5-этилпиримидин-2-ил)пиперидин-4-илокси)бензил)пиперазин-1-илсульфонил)пропан-1-ол;
5-этил-2-(4-(4-((1-(метилсульфонил)пиперидин-4-ил)метил)фенокси)пиперидин-1-ил)пиримидин;
трет-бутил 4-(2-фтор-4-((4-(метилсульфонил)пиперазин-1-ил)метил)фениламино)пиперидин-1-карбоксилат;
1-(3-хлор-4-(1-(5-этилпиримидин-2-ил)пиперидин-4-илокси)бензил)-4-(метилсульфонил)пиперазин-2-он;
метил 2-(1-(5-этилпиримидин-2-ил)пиперидин-4-илокси)-5-((4-(метилсульфонил)пиперазин-1-ил)метил)бензоат;
метил 2-(4-(2-хлор-4-((4-(метилсульфонил)пиперазин-1-ил)метил)фенокси)пиперидин-1-ил)пиримидин-5-карбоксилат;
2-(4-(2-хлор-4-((4-(метилсульфонил)пиперазин-1-ил)метил)фенокси)пиперидин-1-ил)пиримидин;
5-бром-2-(4-(2-хлор-4-((4-(метилсульфонил)пиперазин-1-ил)метил)фенокси)пиперидин-1-ил)пиримидин;
5-хлор-2-(4-(2-хлор-4-((4-(метилсульфонил)пиперазин-1-ил)метил)фенокси)пиперидин-1-ил)пиримидин;
2-(4-(2-хлор-4-((4-(метилсульфонил)пиперазин-1-ил)метил)фенокси)пиперидин-1-ил)пиримидин-5-карбоксамид;
2-(4-(2-хлор-4-((4-(метилсульфонил)пиперазин-1-ил)метил)фенокси)пиперидин-1-ил)-N,N-диметилпиримидин-5-карбоксамид;
(2-(4-(2-хлор-4-((4-(метилсульфонил)пиперазин-1-ил)метил)фенокси)пиперидин-1-ил)пиримидин-5-ил)(морфолино)метанон;
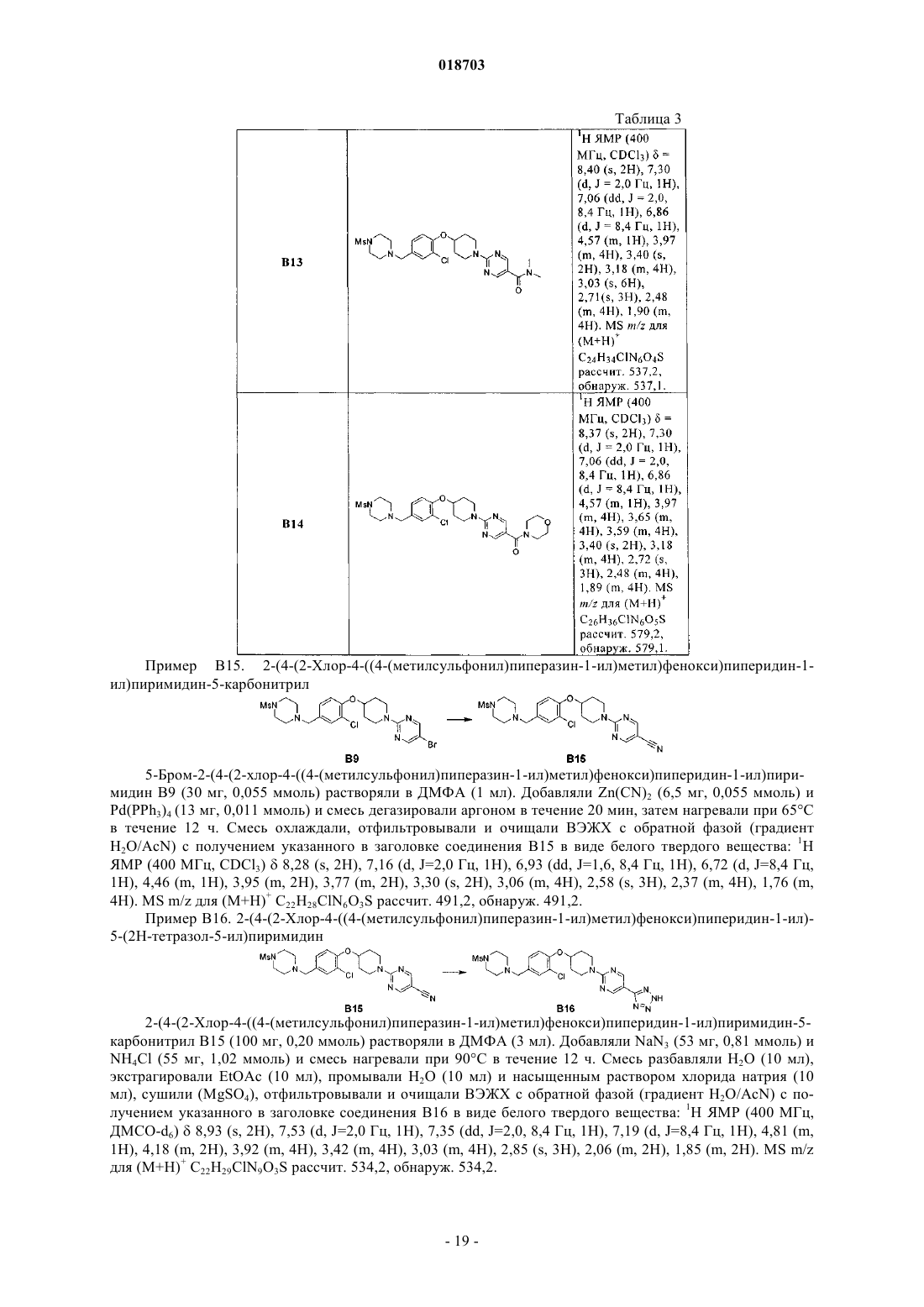
2-(4-(2-хлор-4-((4-(метилсульфонил)пиперазин-1-ил)метил)фенокси)пиперидин-1-ил)пиримидин-5-карбонитрил;
2-(4-(2-хлор-4-((4-(метилсульфонил)пиперазин-1-ил)метил)фенокси)пиперидин-1-ил)-5-(2Н-тетразол-5-ил)пиримидин;
2-(4-(2-хлор-4-((4-(метилсульфонил)пиперазин-1-ил)метил)фенокси)пиперидин-1-ил)-5-(2-метил-2Н-тетразол-5-ил)пиримидин;
(Е)-5-этил-2-(4-(2-(3-метоксипроп-1-енил)-4-((4-(метилсульфонил)пиперазин-1-ил)метил)фенокси)пиперидин-1-ил)пиримидин;
3-(2-(1-(5-этилпиримидин-2-ил)пиперидин-4-илокси)-5-((4-(метилсульфонил)пиперазин-1-ил)метил)фенил)пропан-1-ол;
5-этил-2-(4-(4-((4-(метилсульфонил)пиперазин-1-ил)метил)-2-винилфенокси)пиперидин-1-ил)пиримидин;
5-этил-2-(4-(5-((4-(метилсульфонил)пиперазин-1-ил)метил)бифенил-2-илокси)пиперидин-1-ил)пиримидин;
5-этил-2-(4-(4-((4-(метилсульфонил)пиперазин-1-ил)метил)-2-(1Н-пиразол-5-ил)фенокси)пиперидин-1-ил)пиримидин;
(Е)-2-(4-(2-(5-хлорпент-1-енил)-4-((4-(метилсульфонил)пиперазин-1-ил)метил)фенокси)пиперидин-1-ил)-5-этилпиримидин;
(Е)-4-(2-(1-(5-этилпиримидин-2-ил)пиперидин-4-илокси)-5-((4-(метилсульфонил)пиперазин-1-ил)метил)фенил)бут-3-ен-1-ол;
3-(2-(1-(5-этилпиримидин-2-ил)пиперидин-4-илокси)-5-((4-(метилсульфонил)пиперазин-1-ил)метил)фенил)пропан-1-ол;
2-(1-(5-этилпиримидин-2-ил)пиперидин-4-илокси)-N-(2-метоксиэтил)-5-((4-(метилсульфонил)пиперазин-1-ил)метил)анилин;
4-(2-(1-(5-этилпиримидин-2-ил)пиперидин-4-илокси)-5-((4-(метилсульфонил)пиперазин-1-ил)метил)фенил)морфолино;
2-(4-(2-хлор-4-((1-(метилсульфонил)пиперидин-4-ил)метил)фенокси)пиперидин-1-ил)-5-этилпиримидин;
3-(4-(3-хлор-4-(1-(5-этилпиримидин-2-ил)пиперидин-4-илокси)бензил)пиперидин-1-илсульфонил)пропан-1-ол;
2-(4-(2-хлор-4-((4-(метилсульфонил)пиперидин-1-ил)метил)фенокси)пиперидин-1-ил)-5-этилпиримидин;
2-(4-(2-хлор-4-((2-метил-4-(метилсульфонил)пиперазин-1-ил)метил)фенокси)пиперидин-1-ил)-5-этилпиримидин;
N-(3-хлор-4-(1-(5-этилпиримидин-2-ил)пиперидин-4-илокси)фенил)-N-метил-1-(метилсульфонил)пиперидин-4-амин;
4-(3-хлор-4-(1-(5-этилпиримидин-2-ил)пиперидин-4-илокси)бензил)пиперазин-1-карбонитрил;
1-(3-хлор-4-(1-(5-этилпиримидин-2-ил)пиперидин-4-илокси)бензил)пиперидин-4-карбонитрил;
2-(4-(2-хлор-4-((1-(метилсульфонил)азетидин-3-илокси)метил)фенокси)пиперидин-1-ил)-5-этилпиримидин;
2-(4-(2-хлор-4-((1-(метилсульфонил)азетидин-3-илокси)метил)фенокси)пиперидин-1-ил)-5-этилпиримидин и
N-(1-(3-хлор-4-(1-(5-этилпиримидин-2-ил)пиперидин-4-илокси)бензил)азетидин-3-ил)-N-метилметансульфонамид.
7. Фармацевтическая композиция, содержащая терапевтически эффективное количество соединения по п.1 в комбинации с фармацевтически приемлемым эксципиентом.
8. Применение соединения или его фармацевтически приемлемой соли по любому из пп.1-6 для лечения заболевания или состояния, в котором стимулирование активности GPR119 может предотвращать, ингибировать или облегчать патологию и/или симптоматику заболевания или состояния.
9. Применение по п.8, в котором указанное заболевание или состояние выбрано из ожирения, диабета I типа, сахарного диабета II типа, гиперлипидемии, идиопатического диабета I типа, латентного аутоиммунного диабета у взрослых, раннего диабета II типа, детского атипичного диабета, зрелого диабета у детей, связанного с недоеданием диабета и диабета у беременных.
10. Применение по п.8, в котором указанное заболевание или состояние выбрано из коронарной болезни, ишемического инсульта, рестеноза после ангиопластики, периферийного сосудистого заболевания, перемежающейся хромоты, инфаркта миокарда, дислипидемии, постпрандиальной липемии, состояний ослабленной толерантности к глюкозе, состояний пониженного уровня глюкозы в плазме натощак, метаболического ацидоза, кетоза, артрита, остеопороза, гипертензии, застойной сердечной недостаточности, гипертрофии левого желудочка, периферийного артериального заболевания, диабетической ретинопатии, дегенерации желтого пятна, катаракты, диабетической нефропатии, гломерулосклероза, хронической почечной недостаточности, диабетической невропатии, метаболического синдрома, синдрома X, предменструального синдрома, коронарной болезни, стенокардии, тромбоза, атеросклероза, инфаркта миокарда, преходящей ишемической атаки, инсульта, васкулярного рестеноза, гипергликемии, гиперинсулинемии, гиперлипидемии, гипертриглицеридемии, резистентности к инсулину, ослабленного метаболизма глюкозы, состояний ослабленной толерантности к глюкозе, состояний пониженного уровня глюкозы в плазме натощак, ожирения, эректильной дисфункции, нарушений кожи и соединительной ткани, язв ступней и язвенного колита, эндотелиальной дисфункции и слабости сосудов.
11. Применение фармацевтически приемлемой композиции по п.7 для лечения заболевания или состояния, в котором стимулирование активности GPR119 может предотвращать, ингибировать или облегчать патологию и/или симптоматику заболевания или состояния.
12. Применение по п.11, в котором указанное заболевание или состояние выбрано из ожирения, диабета I типа, сахарного диабета II типа, гиперлипидемии, идиопатического диабета I типа, латентного аутоиммунного диабета у взрослых, раннего диабета II типа, детского атипичного диабета, зрелого диабета у детей, связанного с недоеданием диабета и диабета у беременных.
13. Применение по п.11, в котором указанное заболевание или состояние выбрано из коронарной болезни, ишемического инсульта, рестеноза после ангиопластики, периферийного сосудистого заболевания, перемежающейся хромоты, инфаркта миокарда, дислипидемии, постпрандиальной липемии, состояний ослабленной толерантности к глюкозе, состояний пониженного уровня глюкозы в плазме натощак, метаболического ацидоза, кетоза, артрита, остеопороза, гипертензии, застойной сердечной недостаточности, гипертрофии левого желудочка, периферийного артериального заболевания, диабетической ретинопатии, дегенерации желтого пятна, катаракты, диабетической нефропатии, гломерулосклероза, хронической почечной недостаточности, диабетической невропатии, метаболического синдрома, синдрома X, предменструального синдрома, коронарной болезни, стенокардии, тромбоза, атеросклероза, инфаркта миокарда, преходящей ишемической атаки, инсульта, васкулярного рестеноза, гипергликемии, гиперинсулинемии, гиперлипидемии, гипертриглицеридемии, резистентности к инсулину, ослабленного метаболизма глюкозы, состояний ослабленной толерантности к глюкозе, состояний пониженного уровня глюкозы в плазме натощак, ожирения, эректильной дисфункции, нарушений кожи и соединительной ткани, язв ступней и язвенного колита, эндотелиальной дисфункции и слабости сосудов.
Текст
Изобретение относится к соединениям, фармацевтическим композициям, включающим такие соединения, и к способам применения таких соединений для лечения или профилактики заболеваний или нарушений, связанных с активностью GPR119, таких как, но не ограничиваясь ими, диабет, ожирение и связанные метаболические нарушения. Формула (I) представляет собой соединение, в котором А может содержать вплоть до 2 кольцевых атомов углерода, замещенных оксогруппой; W1 и W2 независимо выбраны из CR10 и N; где R10 выбран из водорода и C1-6 алкила;Y1 выбран из NR11 и О; где R11 выбран из водорода и C1-6 алкила; Y3 выбран из СН и N; Y4 выбран из СН 2 и ОСН 2, или его фармацевтически приемлемые соли. Ссылки на заявки-аналоги Данная заявка является продолжением заявки США 61/043100, поданной 07.04.2008. Полное описание данной заявки включено сюда полностью и во всех целях в качестве ссылки. Предпосылки для создания изобретения Область техники, к которой относится изобретение Настоящее изобретение относится к соединениям, фармацевтическим композициям, включающим такие соединения, и к способам применения таких соединений для лечения или профилактики заболеваний или нарушений, связанных с активностью GPR119. Уровень техникиGPR119 представляет собой рецептор, связанный с G-белком (GPCR), который экспрессируется главным образом в поджелудочной железе, малом кишечнике, толстой кишке и жировой ткани. Профиль экспрессии рецептора GPR119 человека показывает его потенциальную полезность в качестве мишени для лечения ожирения и диабета. Новые соединения настоящего изобретения модулируют активностьGPR119 и, следовательно, предполагается, что они являются полезными для лечения заболеваний или нарушений, связанных с GPR119, таких как, но не ограничиваясь ими, диабет, ожирение и связанные с ними метаболические нарушения. Сущность изобретения В одном варианте осуществления настоящее изобретение относится к соединению формулы (I) или его фармацевтически приемлемой соли, в котором А может содержать вплоть до 2 кольцевых атомов углерода, замещенных оксогруппой;-S(O)2NR6aR6b, где R6a выбран из С 1-6 алкила, где указанный алкил в R6a необязательно замещен 1-3 радикалами, независимо выбранными из гидроксигруппы и галогена; каждый R2 независимо выбран из C1-6 алкила, галогензамещенного C1-6 алкила, гидроксизамещенного C1-6 алкила;R4 выбран из R9 и -C(O)OR9, где R9 выбран из C1-6 алкила, C6-10 арила и C1-10 гетероарила, где указанный арил или гетероарил в R9 необязательно замещен 1-3 радикалами, независимо выбранными из галогена, цианогруппы, C1-6 алкила, С 3-12 циклоалкила, С 3-8 гетероциклоалкила, галогензамещенного C1-6 алкила,гидроксизамещенного C1-6 алкила, C1-6 алкоксигруппы, галогензамещенной C1-6 алкоксигруппы и-C(O)OR17, -C(O)R19 и -C(O)NR17R18, где R17 и R18 независимо выбраны из водорода и C1-6 алкила или R17 и R18 вместе с атомом азота, к которому R17 и R18 присоединены, образуют С 3-8 гетероциклоалкил; R19 выбран из C1-6 алкила и С 3-8 гетероциклоалкила; и указанные циклоалкильные или гетероциклоалкильные заместители в R9 необязательно далее замещены 1-3 C1-6 алкильными радикалами; каждый R6 независимо выбран из гидроксигруппы, нитрогруппы, цианогруппы, галогена,C1-6 алкила, C2-6 алкенила, галогензамещенного C1-6 алкила, галогензамещенного C2-6 алкенила, гидроксизамещенного C1-6 алкила, C1-6 алкоксигруппы, галогензамещенной C1-6 алкоксигруппы, С 6-10 арила,C1-10 гетероарила, С 3-8 гетероциклоалкила, С 3-8 циклоалкила и -X3OR20, -NR20X3OR21, -C(O)OR20, где Х 3 выбран из связи, C1-4 алкилена и С 2-4 алкенилена; R20 и R21 независимо выбраны из водорода и C1-6 алкила;W1 и W2 независимо выбраны из CR10 и N, где R10 выбран из водорода и C1-6 алкила;Y1 выбран из NR11 и О, где R11 выбран из водорода и C1-6 алкила;Y3 выбран из СН и N;Y4 выбран из СН 2 и ОСН 2; где C1-10 гетероарил обозначает моноциклический или конденсированный бициклический ароматический кольцевой комплекс, содержащий от 1 до 10 атомов углерода и вплоть до 4 гетероатомов в качестве кольцевых атомов, где гетероатомы независимо выбраны из N и О; С 3-8 гетероциклоалкил обозначает насыщенный или частично ненасыщенный, моноциклический,конденсированный бициклический или мостиковый полициклический кольцевой комплекс, содержащий от 3 до 8 атомов углерода и вплоть до 4 гетероатомов в качестве кольцевых атомов, где гетероатомы независимо выбраны из N и О. Во втором варианте осуществления настоящее изобретение относится к фармацевтической композиции, которая содержит соединение формулы (I) в комбинации с фармацевтически приемлемым эксципиентом. В третьем варианте осуществления настоящее изобретение относится к применению соединения формулы (I) или его фармацевтически приемлемой соли для лечения заболевания или состояния, в кото-1 018703 ром стимулирование активности GPR119 может предотвращать, ингибировать или облегчать патологию и/или симптоматику заболевания или состояния. В четвертом варианте осуществления настоящее изобретение относится к применению фармацевтически приемлемой композиции для лечения заболевания или состояния, в котором стимулирование активности GPR119 может предотвращать, ингибировать или облегчать патологию и/или симптоматику заболевания или состояния. Подробное описание изобретения Определения."Алкил" как группа и в качестве структурного элемента других групп, например галогензамещенного алкила и алкокси, может быть линейным или разветвленным. C1-4 Алкоксигруппа включает метоксигруппу, этоксигруппу и им подобные. Галогензамещенный алкил включает трифторметил, пентафторэтил и им подобные."Арил" обозначает моноциклический или конденсированный бициклический ароматический кольцевой комплекс, содержащий от 6 до 10 атомов углерода в кольце. Например, арил может представлять собой фенил или нафтил, предпочтительно фенил. "Арилен" обозначает двухвалентный радикал, полученный из арильной группы. "Гетероарил" имеет значение, определенное выше для арила, где один или несколько членов кольца являются гетероатомами. Например, гетероарил включает пиридил, индолил,индазолил, хиноксалинил, хинолинил, бензофуранил, бензопиранил, бензотиопиранил, бензо[1,3]диоксол, имидазолил, бензоимидазолил, пиримидинил, фуранил, оксазолил, изоксазолил, триазолил, тетразолил, пиразолил, тиенил, 1H-пиридин-2-онил, 6-оксо-1,6-дигидропиридин-3-ил и т.д."C6-10 арилС 0-4 алкил" обозначает арил, как описано выше, присоединенный через алкиленовую группу. Например, C6-10 арилС 0-4 алкил включает фенэтил, бензил и т.д. Гетероарил также включает N-оксидные производные, например пиридин N-оксидные производные следующей структуры:"Циклоалкил" обозначает насыщенный или частично ненасыщенный, моноциклический, конденсированный бициклический или мостиковый полициклический кольцевой комплекс, содержащий указанное количество атомов в кольце. Например, С 3-10 циклоалкил включает циклопропил, циклобутил, циклопентил, циклогексил и т.д. "Гетероциклоалкил" обозначает циклоалкил, как определено в настоящем описании, при условии, что один или несколько указанных атомов углерода в кольце замещены группой,выбранной из -О-, -N=, -NR-, -С(О)-, -S-, -S(O)- или -S(O)2-, где R представляет собой водород, C1-4 алкил или защитную группу для атома азота. Например, C3-8 гетероциклоалкил, как используется в настоящем описании для описания соединений по изобретению, включает морфолино, пирролидинил, пиперазинил,пиперидинил, пиперидинилон, 1,4-диокса-8-азаспиро[4,5]дец-8-ил, 2-оксопирролидин-1-ил, 2-оксопиперидин-1-ил и т.д.GPR119 обозначает рецептор 119, связанный с G-белком (GenBank Accession No. AAP72125), также в литературе известный как RUP3 и GPR116. Термин GPR119, как здесь используется, включает последовательности человека, помещенные в GeneBank с номером доступа AY288416, природные аллельные варианты, ортологи млекопитающих и рекомбинантные мутанты."Галоген" (или гало) предпочтительно представляет собой хлор или фтор, но также может быть бром или йод."Лечить", "излечение" и "лечение" относятся к способу облегчения или ослабления заболевания и/или сопровождающих его симптомов. Описание предпочтительных вариантов осуществления В одном варианте осуществления со ссылкой на соединения формулы (I) описаны соединения формулы (Ia) в которых А может содержать кольцевой атом углерода, замещенный оксогруппой;W2 выбран из СН и N. В другом варианте осуществления R1 выбран из цианогруппы, -S(O)2R6a и -N(R6a)S(O)2R6a, где R6a выбран из водорода, C1-6 алкила и C1-10 гетероарила, необязательно замещенного C1-6 алкилом. В другом варианте осуществления R4 выбран из R9 и -C(O)OR9, где R9 выбран из трет-бутила, пиридинила, пиримидинила, 1,2,4-оксадиазол-5-ила и тетразолила, где указанный пиридинил, пиримидинил,1,2,4-оксадиазол-5-ил или тетразолил в R9 необязательно замещен радикалом, выбранным из галогена,цианогруппы, трифторметила, изопропила, метила, этила, метоксикарбонила, диметиламинокарбонила,аминокарбонила и морфолинокарбонила. В другом варианте осуществления R6 выбран из фтора, хлора, брома, трифторметоксигруппы, ме-2 018703 тила, метоксигруппы, метоксикарбонила, 3-метоксипроп-1-енила, метоксипропила, винила, фенила, пиразолила, 5-хлорпент-1-енила, гидроксипропила, метоксиэтиламиногруппы и морфолиногруппы; Y1 представляет собой О; Y3 представляет собой СН и Y4 представляет собой СН 2. В другом варианте осуществления соединения выбраны из следующих соединений: 5-этил-2-(4-(44-(метилсульфонил)пиперазин-1-ил)метил)фенокси)пиперидин-1-ил)пиримидин; 5-этил-2-(4-(3-метил 4-4-(метилсульфонил)пиперазин-1-ил)метил)фенокси)пиперидин-1-ил)пиримидин; 5-этил-2-(4-(3 метокси-4-4-(метилсульфонил)пиперазин-1-ил)метил)фенокси)пиперидин-1-ил)пиримидин; 5-этил-2(4-(3-фтор-4-4-(метилсульфонил)пиперазин-1-ил)метил)фенокси)пиперидин-1-ил)пиримидин; 2-(4-(3 хлор-4-4-(метилсульфонил)пиперазин-1-ил)метил)фенокси)пиперидин-1-ил)-5-этилпиримидин; 5-этил 2-(4-(2-метил-4-4-(метилсульфонил)пиперазин-1-ил)метил)фенокси)пиперидин-1-ил)пиримидин; 5 этил-2-(4-(2-фтор-4-4-(метилсульфонил)пиперазин-1-ил)метил)фенокси)пиперидин-1-ил)пиримидин; 2(4-(2-хлор-4-4-(метилсульфонил)пиперазин-1-ил)метил)фенокси)пиперидин-1-ил)-5-этилпиримидин; 2(4-(2-бром-4-4-(метилсульфонил)пиперазин-1-ил)метил)фенокси)пиперидин-1-ил)-5-этилпиримидин; 5 этил-2-(4-(2-метокси-4-4-(метилсульфонил)пиперазин-1-ил)метил)фенокси)пиперидин-1-ил)пиримидин; 5 этил-2-(4-(4-4-(метилсульфонил)пиперазин-1-ил)метил)-2-(трифторметокси)фенокси)пиперидин-1 ил)пиримидин; 2-(4-(2,3-дифтор-4-4-(метилсульфонил)пиперазин-1-ил)метил)фенокси)пиперидин-1 ил)-5-этилпиримидин; 2-(4-(2-хлор-6-фтор-4-4-(метилсульфонил)пиперазин-1-ил)метил)фенокси)пиперидин-1-ил)-5-этилпиримидин; 5-этил-2-(4-(6-4-(метилсульфонил)пиперазин-1-ил)метил)пиридин-3 илокси)пиперидин-1-ил)пиримидин; трет-бутил 4-(4-4-(метилсульфонил)пиперазин-1-ил)метил)фенокси)пиперидин-1-карбоксилат; 5-фтор-2-(4-(4-4-(метилсульфонил)пиперазин-1-ил)метил)фенокси)пиперидин-1-ил)пиримидин; 1-(4-(1-(5-фторпиридин-2-ил)пиперидин-4-илокси)бензил)-4-(метилсульфонил)пиперазин; 1-(метилсульфонил)-4-(4-(1-(5-(трифторметил)пиридин-2-ил)пиперидин-4-илокси)бензил)пиперазин; 5-(4-(2-хлор-4-4-(метилсульфонил)пиперазин-1-ил)метил)фенокси)пиперидин-1-ил)-3 изопропил-1,2,4-оксадиазол; 3-(4-(3-хлор-4-(1-(5-этилпиримидин-2-ил)пиперидин-4-илокси)бензил)пиперазин-1-илсульфонил)пропан-1-ол; 5-этил-2-(4-(4-1-(метилсульфонил)пиперидин-4-ил)метил)фенокси)пиперидин-1-ил)пиримидин; трет-бутил 4-(2-фтор-4-4-(метилсульфонил)пиперазин-1 ил)метил)фениламино)пиперидин-1-карбоксилат; 1-(3-хлор-4-(1-(5-этилпиримидин-2-ил)пиперидин-4 илокси)бензил)-4-(метилсульфонил)пиперазин-2-он; метил 2-(1-(5-этилпиримидин-2-ил)пиперидин-4 илокси)-5-4-(метилсульфонил)пиперазин-1-ил)метил)бензоат; метил 2-(4-(2-хлор-4-4(метилсульфонил)пиперазин-1-ил)метил)фенокси)пиперидин-1-ил)пиримидин-5-карбоксилат; 2-(4-(2 хлор-4-4-(метилсульфонил)пиперазин-1-ил)метил)фенокси)пиперидин-1-ил)пиримидин; 5-бром-2-(4-(2 хлор-4-4-(метилсульфонил)пиперазин-1-ил)метил)фенокси)пиперидин-1-ил)пиримидин; 5-хлор-2-(4-(2 хлор-4-4-(метилсульфонил)пиперазин-1-ил)метил)фенокси)пиперидин-1-ил)пиримидин; 2-(4-(2-хлор-44-(метилсульфонил)пиперазин-1-ил)метил)фенокси)пиперидин-1-ил)пиримидин-5-карбоксамид; 2-(4(2-хлор-4-4-(метилсульфонил)пиперазин-1-ил)метил)фенокси)пиперидин-1-ил)-N,N-диметилпиримидин-5-карбоксамид; (2-(4-(2-хлор-4-4-(метилсульфонил)пиперазин-1-ил)метил)фенокси)пиперидин-1 ил)пиримидин-5-ил)(морфолино)метанон; 2-(4-(2-хлор-4-4-(метилсульфонил)пиперазин-1-ил)метил)фенокси)пиперидин-1-ил)пиримидин-5-карбонитрил; 2-(4-(2-хлор-4-4-(метилсульфонил)пиперазин-1 ил)метил)фенокси)пиперидин-1-ил)-5-(2 Н-тетразол-5-ил)пиримидин; 2-(4-(2-хлор-4-4-(метилсульфонил)пиперазин-1-ил)метил)фенокси)пиперидин-1-ил)-5-(2-метил-2 Н-тетразол-5-ил)пиримидин; Другие соединения по изобретению описаны далее в примерах и табл. I. Фармакология и полезность Соединения по изобретению модулируют активность GPR119 и как таковые полезны для лечения заболеваний или нарушений, в которых активность GPR119 участвует в патологии и/или симптоматике заболевания. Изобретение также относится к соединениям настоящего изобретения для применения для изготовления лекарственных средств для лечения заболеваний или нарушений, в которых активностьGPR119 участвует в патологии и/или симптоматике заболевания. Итоговыми патологиями диабета II типа являются ослабление передачи сигнала инсулина в его тканях-мишенях и нарушение продуцирования инсулина клетками поджелудочной железы в нужной степени секреции инсулина в ответ на гипергликемический сигнал. Используемые в настоящее время терапии для лечения последнего включают ингибиторы -клеточных АТФ-чувствительных калиевых каналов для запуска высвобождения запасов эндогенного инсулина, или введение экзогенного инсулина. Ни один из этих подходов не нормализует уровни глюкозы в крови, и оба несут риск, включающий гипогликемию. По этим причинам интенсивно разрабатываются фармацевтические средства, которые функционируют в глюкозозависимом действии, то есть усилители передачи сигнала глюкозы. Системы физиологической передачи сигнала, которые функционируют этим способом, хорошо охарактеризованы и включают пептиды кишечника GLP-1, GIP и РАСАР. Эти гормоны действуют через их родственный связанный с G-белком рецептор для стимулирования выработки цАМФ в панкреатических -клетках. Оказывается, повышенный уровень цАМФ не приводит к стимулированию высвобождения инсулина в процессе голодания или перед приемом пищи. Однако ряд биохимических мишеней передачи сигнала цАМФ, включая АТФ-чувствительный калиевый канал, чувствительные к напряжению калиевые каналы и экзоцитотический механизм, модифицируются таким способом, что отклик секреции инсулина на глюкозу после еды заметно повышается. Соответственно агонисты новых, аналогично функционирующих-клеточных GPCR, включая GPR119, также будут стимулировать высвобождение эндогенного инсулина и затем стимулировать нормогликемию при диабете II типа. Также установлено, что повышение цАМФ,например, в результате стимулирования GLP-1 промотирует пролиферацию -клеток, ингибирует гибель-клеток и таким образом улучшает массу островков поджелудочной железы. Предполагается, что это положительное действие на -клеточную массу благоприятно для диабета типа II, где присутствует недостаточная выработка инсулина, и диабета типа I, где -клетки разрушаются неподходящим аутоиммунным откликом. Некоторые -клеточные GPCR, включая GPR119, также присутствуют в гипоталамусе, где они модулируют чувство голода, насыщения, понижение потребления пищи, контроль или понижение веса и расход энергии. Следовательно, учитывая эту функцию с гипоталамической схемой, агонисты или обратные агонисты этих рецепторов уменьшают чувство голода, стимулируют насыщение и, следовательно, модулируют вес. Также установлено, что метаболические заболевания оказывают отрицательное влияние на другие физиологические системы. Так, часто присутствуют совместно развивающиеся многочисленные заболевания (например, диабет I типа, диабет II типа, нарушенная толерантность к глюкозе, резистентность к инсулину, гипергликемия, гиперлипидемия, гипертриглицеридемия, гиперхолестеролемия, дислипидемия, ожирение или сердечно-сосудистое заболевание при "синдроме X") или вторичные заболевания,которые часто встречаются при диабете (например, почечное заболевание, периферийная невропатия). Так, предполагается, что эффективное лечение диабетического состояния в свою очередь благоприятно скажется на таких взаимосвязанных заболеваниях. Соединения по изобретению могут использоваться в способе лечения метаболического заболевания и/или связанного с метаболическим нарушения у пациента, включающем введение пациенту, нуждающемуся в таком лечении, терапевтически эффективного количества соединения по изобретению или его фармацевтической композиции. Метаболические заболевания и связанные с метаболическими нарушения выбраны из следующих, но не ограничиваясь ими: гиперлипидемия, диабет I типа, сахарный диабетII типа, идиопатический диабет I типа (Ib типа), латентный аутоиммунный диабет у взрослых (LADA),ранний диабет II типа (EOD), детский атипичный диабет (YOAD), зрелый диабет у детей (MODY), связанный с недоеданием диабет, диабет у беременных, коронарная болезнь, ишемический инсульт, рестеноз после ангиопластики, периферийное сосудистое заболевание, перемежающаяся хромота, инфаркт миокарда (например, некроз и апоптоз), дислипидемия, постпрандиальная липемия, состояния ослабленной толерантности к глюкозе (IGT), состояния пониженного уровня глюкозы в плазме натощак, метаболический ацидоз, кетоз, артрит, ожирение, остеопороз, гипертензия, застойная сердечная недостаточность, гипертрофия левого желудочка, периферийное артериальное заболевание, диабетическая ретинопатия, дегенерация жлтого пятна, катаракта, диабетическая нефропатия, гломерулосклероз, хроническая почечная недостаточность, диабетическая невропатия, метаболический синдром, синдром X, предменструальный синдром, коронарная болезнь, стенокардия, тромбоз, атеросклероз, инфаркт миокарда, преходящая ишемическая атака, инсульт, васкулярный рестеноз, гипергликемия, гиперинсулинемия, гиперлипидемия, гипертриглицеридемия, резистентность к инсулину, ослабленный метаболизм глюкозы, со-4 018703 стояния ослабленной толерантности к глюкозе, состояния пониженного уровня глюкозы в плазме натощак, ожирение, эректильная дисфункция, нарушения кожи и соединительной ткани, язвы ступней и язвенный колит, эндотелиальная дисфункция и слабость сосудов. В варианте осуществления по изобретению описана терапевтическая польза модуляторов активности GPR119 от повышенных уровней GIP и панкреатического полипептида (PPY). Например, нейропротекция, обучение и память, приступы и периферийная невропатия. Показано, что GLP-1 и агонисты рецептора GLP-1 являются эффективными для лечения нейродегенеративных заболеваний и других неврологических нарушений. Показано, что GLP-1 и эксендин-4 стимулируют рост нейрита и повышают клеточную выживаемость после удаления фактора роста в клеткахPC12. На модели нейродегенерации у грызунов GLP-1 и эксендин-4 восстанавливают активность холинергических маркеров в базальной передней части мозга. Центральное введение GLP-1 и эксендина-4 также снижает уровни амилоид- пептида у мыши и понижает количество белка амилоидного предшественника в культивируемых клетках PC12. Было показано, что агонисты рецептора GLP-1 повышают обучаемость у крыс, и мыши с блокированным рецептором GLP-1 проявляют плохое поведение при обучении. Мыши с блокированным GLP-1 также проявляют повышенную чувствительность к каинатвызванным приступам, которые можно предотвратить введением агонистов рецептора GLP-1. Также было показано, что GLP-1 и эксендин-4 являются эффективными при лечении вызванной пиридоксином дегенерации периферийного нерва, экспериментальной модели периферийной сенсорной невропатии. Также было показано, что глюкозозависимый инсулинотропный полипептид (GIP) оказывает действия на пролиферацию гиппокампальных исходных клеток и на повышение сенсомоторной координации и реконструкции памяти. В варианте осуществления по изобретению описана терапевтическая польза модуляторов активности GPR119. Например, GLP-2 и краткосрочного кишечного синдрома (SBS). Несколько исследований на животных и клинические испытания показали, что GLP-2 является трофическим гормоном, который играет важную роль в кишечной адаптации. Его роль в клеточной пролиферации, апоптозе и абсорбции пищи хорошо описана. Краткосрочный кишечный синдром характеризуется мальабсорбцией пищи, воды и витаминов в результате заболевания или хирургического удаления части малого кишечника (например,болезнь Крона). Терапии, которые улучшают кишечную адаптацию, как полагают, будут успешны для лечения этого заболевания. Фактически, исследования II фазы на пациентах с SBS показали, что тедуглутид, аналог GLP-2, недостаточно повышает абсорбцию жидкости и пищи. В варианте осуществления по изобретению описана терапевтическая польза модуляторов активности GPR119, полученная при повышенных уровнях GIP и PPY. Например, GLP-1, GIP и остеопороз. Показано, что GLP-1 повышает секрецию кальцитонина и пептида гена, связанного с кальцитонином(CGRP), и экспрессию мышиных С-клеточных линий (СА-77). Кальцитонин ингибирует резорбцию кости остеокластами и промотирует минерализацию скелетной кости. Остеопороз является заболеванием,которое характеризуется пониженной минеральной плотностью кости, и тем самым вызванное GLP-1 повышение кальцитонина могло бы быть терапевтически выгодным. Описано, что GIP участвует в сверхрегулировании маркером образования новых костей в остеобластах, включая мРНК типа I коллагена, и в повышении минеральной плотности кости. Также показано,что GLP-1, GIP ингибируют резорбцию кости. В варианте осуществления по изобретению описана терапевтическая польза модуляторов активности GPR119, полученная при повышенных уровнях GIP и PPY. Например, PPY и опорожнение желудка.GPR119 расположен в клетках панкреатического полипептида (РР) островков поджелудочной железы и участвует в секреции PPY. Описано, что PPY оказывает сильные действия на различные физиологические процессы, включая модулирование опорожнения желудка и желудочно-кишечную моторику. Эти действия замедляют процесс пищеварения и усвоение пищи и тем самым предотвращают повышение уровня глюкозы в крови после еды. PPY может подавлять потребление пищи путем изменения экспрессии гипоталамических регулирующих прием пищи пептидов. Мыши со сверхэкспрессией РР проявляют слабый фенотип с пониженным потреблением пищи и скоростью опорожнения желудка. В соответствии с вышеизложенным соединения по изобретению также могут использоваться в способе профилактики или лечения любого из описанных выше заболеваний или нарушений у субъекта,нуждающегося в таком лечении, который включает введение указанному субъекту терапевтически эффективного количества (см. далее раздел "Введение и фармацевтические композиции") соединения формулы (I) или его фармацевтически приемлемой соли. Для любого из указанных выше применений необходимая дозировка будет зависеть от способа введения, конкретного излечиваемого состояния и желаемого эффекта. Введение и фармацевтические композиции Обычно соединения по изобретению вводят в терапевтически эффективных количествах любыми обычными и приемлемыми способами, известными из уровня техники, отдельно или в комбинации с одним или несколькими терапевтическими агентами. Терапевтически эффективное количество может широко варьироваться в зависимости от степени тяжести заболевания, возраста и относительного здоровья субъекта, активности используемого соединения и других факторов. Обычно удовлетворительные ре-5 018703 зультаты получают при систематических суточных дозировках от около 0,03 до 2,5 мг/кг веса тела. Показанная суточная дозировка для большого млекопитающего, например для людей, находится в диапазоне от около 0,5 до около 100 мг, которая обычно вводится, например, раздельными дозами вплоть до четырех раз в день или в замедленной форме. Подходящие единичные дозированные формы для перорального введения включают от около 1 до 50 мг активного ингредиента. Соединения по изобретению могут вводиться в форме фармацевтических композиций любым обычным способом, в частности энтерально,например перорально, например в форме таблеток или капсул, или парентерально, например в форме инъекционных растворов или суспензий, местно, например в форме лосьонов, гелей, мазей или кремов,или в назальной или суппозитарной форме. Фармацевтические композиции, включающие соединение настоящего изобретения в свободной форме или в форме фармацевтически приемлемой соли вместе по крайней мере с одним фармацевтически приемлемым носителем или разбавителем, могут быть изготовлены обычными способами смешения, гранулирования или покрытия. Например, пероральными композициями могут быть таблетки или желатиновые капсулы, содержащие активный ингредиент вместе с: а) разбавителями, например лактозой, декстрозой, сахарозой, маннитом, сорбитом, целлюлозой и/или глицином; б) лубрикантами, например оксидом кремния, тальком, стеариновой кислотой, ее солью магния или кальция и/или полиэтиленгликолем; для таблеток также в) связующими, например магнийалюмосиликатом, крахмальной пастой, желатином, трагакантом, метилцеллюлозой, натрийкарбоксиметилцеллюлозой и/или поливинилпирролидоном; при необходимости г) разрыхлителями, например крахмалами,агаром, альгиновой кислотой или ее натриевой солью, или смесями для сухого шипучего напитка; и/или д) абсорбентами, красителями, отдушками и подсластителями. Инъекционными композициями могут быть водные изотонические растворы или суспензии, и суппозитории могут быть получены из жирных эмульсий или суспензий. Композиции могут быть стерилизованы и/или содержать адъюванты, такие как консервирующие, стабилизирующие, увлажняющие или эмульгирующие агенты, промотеры раствора,соли для регулирования осмотического давления и/или буферы. Кроме того, они также могут содержать другие терапевтически активные вещества. Подходящие составы для трансдермальных применений включают эффективное количество соединения настоящего изобретения с носителем. Носитель может включать абсорбируемые фармакологически приемлемые растворители для обеспечения проникновения через кожу пациента. Например, трансдермальные устройства выполнены в форме бандажа, включающего подложку, емкость, содержащую соединение, необязательно с носителями, необязательно барьер для контроля скорости доставки соединения на кожу пациента с контролируемой и предварительно заданной скоростью в течение пролонгированного периода времени, и средства для удержания устройства на коже. Вязкие трансдермальные составы также могут использоваться. Подходящими составами для местного применения, например, на кожу и в глаза предпочтительно являются водные растворы, мази, кремы или гели, хорошо известные из предшествующего уровня техники. Они могут содержать солюбилизаторы, стабилизаторы, повышающие тоничность агенты, буферы и консерванты. Соединения по изобретению могут вводиться в терапевтически эффективных количествах в комбинации с одним или несколькими терапевтическими агентами (фармацевтические комбинации). Например, синергетические действия могут встречаться с другими агентами против ожирения, аноректическими агентами, веществами для снижения аппетита и аналогичными агентами. Диета и/или физические упражнения также могут проявлять синергетические действия. Агенты против ожирения включают, но не ограничиваются ими, ингибиторы белка секреции аполипопротеина-В/микросомального триглицеридного переноса (аро-В/МТР), агонисты MCR-4, агонисты холесцистокинина-А (ССК-А), ингибиторы перезахвата серотонина и норэпинефрина (например, сибутрамин), симпатомиметические агенты, агонисты 3 адренергического рецептора, агонисты допамина (например, бромкриптин), аналоги рецептора меланоцит-стимулирующего гормона, антагонисты каннабиноидного рецептора 1 (например,соединения, описанные в международной заявке на патент WO 2006/047516), антагонисты меланинконцентрирующего гормона, лептоны (белок ОВ), аналоги лептина, агонисты рецептора лептина, антагонисты галанина, ингибиторы липазы (такие как тетрагидролипстатин, например, орлистат), аноректические агенты (такие как агонист бомбезина), антагонисты нейропептида-Y, тиромиметические агенты, дегидроэпиандростерон или его аналог, агонисты или антагонисты глюкокортикоидного рецептора, антагонисты рецептора орексина, антагонисты урокортинсвязывающего белка, агонисты рецептора глюкагонподобного пептида-1, цилиарные нейротрофические факторы (такие как Axokine), агути-связанные белки человека (AGRP), антагонисты рецептора грелина, антагонисты или обратные агонисты рецептора гистамина 3, агонисты рецептора нейромедина U, норадренергические аноректические агенты (например,фентермин, мазиндол и им подобные) и вещества, подавляющие аппетит (например, бупропион). Когда соединения по изобретению вводят в сочетании с другими лекарственными препаратами, дозировки совместно вводимых соединений, конечно, будут зависеть от вида совместно вводимого лекарственного препарата, от конкретного используемого лекарственного препарата, от излечиваемого состояния и прочих условий. Комбинированный препарат или фармацевтическая композиция может включать соединение по изобретению, как определено выше, или его фармацевтически приемлемую соль и по крайней мере один активный ингредиент, выбранный из следующих агентов: а) антидиабетические агенты, такие как инсулин, производные и миметики инсулина; вещества,способствующие высвобождению инсулина, такие как сульфонилмочевины, например глипизид, глибурид и амарил; лиганды инсулинотропного рецептора сульфонилмочевины, такие как меглитиниды, например натеглинид и репаглинид; сенсибилизаторы инсулина, такие как ингибиторы протеинтирозинфосфатазы-1 В (РТР-1 В), такие как РТР-112; ингибиторы GSK3 (гликогенсинтазакиназа-3), такие как SB517955, SB-4195052, SB-216763, NN-57-05441 и NN-57-05445; лиганды RXR, такие как GW-0791 и AGN194204; ингибиторы натрийзависимого сотранспортера глюкозы, такие как Т-1095; ингибиторы гликогенфосфорилазы А, такие как BAY R3401; бигуаниды, такие как метформин; ингибиторы альфаглюкозидазы, такие как акарбоза; GLP-1 (глюкагоноподобный пептид-1), аналоги GLP-1, такие как эксендин-4 и миметики GLP-1; ингибиторы DPPIV (дипептидилпептидазы IV), такие как DPP728, LAF237(вильдаглиптин - пример 1 международной заявки на патент WO 00/34241), MK-0431, саксаглиптин,GSK23A; вещество, разрушающее AGE; производное тиазолидона (глитазон), такое как пиоглитазон,розиглитазон или (R)-1-4-[5-метил-2-(4-трифторметилфенил)оксазол-4-илметокси]бензолсульфонил 2,3-дигидро-1H-индол-2-карбоновая кислота, описанная в международной заявке на патент WO 03/043985, как соединение 19 примера 4, агонист неглитазонового типа PPAR гамма, например GI262570; ингибиторы диацилглицеринацетилтрансферазы (DGAT), такие как соединения, описанные в международных заявках на патент WO 2005044250, WO 2005013907, WO 2004094618 и WO 2004047755; б) гиполипидемические агенты, такие как ингибиторы 3-гидрокси-3-метилглутарила коэнзим A(HMG-CoA) редуктазы, например ловастатин и аналогичные соединения, такие как соединения, описанные в патенте US 4231938, питавастатин, симвастатин и аналогичные соединения, такие как соединения,описанные в патентах US 4448784 и 4450171, правастатин и аналогичные соединения, такие как соединения, описанные в патенте US 4346227, церивастатин, мевастатин и аналогичные соединения, такие как соединения, описанные в патенте US 3983140, велостатин, флувастатин, дальвастатин, аторвастатин, розувастатин и аналогичные статину соединения, описанные в патенте US 5753675, ривастатин, аналоги пиразола производных мевалонолактона, описанные в патенте US 4613610, аналоги индена производных мевалонолактона, описанные в международной заявке на патент WO 86/03488, 6-[2-(замещенныепиррол-1-ил)алкил]пиран-2-оны и его производные, описанные в патенте US 4647576, Searle's SC-45355(3-замещенное производное пентандионовой кислоты) дихлорацетат, аналоги имидазола мевалонолактона, описанные в международной заявке на патент WO 86/07054, производные 3-карбокси-2 гидроксипропанфосфоновой кислоты, описанные в патенте FR 2596393, 2,3-дизамещенные производные пиррола, фурана и тиофена, описанные в заявке на патент ЕР 0221025, нафтильные аналоги мевалонолактона, описанные в патенте US 4686237, октагидронафталены, такие как соединения, описанные в патенте US 4499289, кетоаналоги мевинолина (ловастатин), описанные в заявке на патент 0142146 А 2, и производные хинолила и пиридина, описанные в патенте US 5506219 и 5691322. Кроме того, подходящие для применения соединения фосфиновой кислоты, полезные для ингибирования HMG СоА редуктазы, описаны в патенте GB 2205837; ингибиторы скваленсинтазы; лиганды FXR (фарнезоидного X рецептора) и LXR (рецептор печени X); холестирамин; фибраты; никотиновая кислота и аспирин; в) агент против ожирения или агент, регулирующий аппетит, такой как модулятор активности СВ 1,агонисты рецептора меланокортина (MC4R), антагонисты рецептора меланин-концентрирующего гормона (MCHR), антагонисты рецептора вещества, высвобождающего гормон роста (GHSR), модуляторы рецептора галанина, антагонисты орексина, агонисты CCK, агонисты GLP-1 и другие Pre-проглюкагонполученные пептиды; антагонисты NPY1 или NPY5, модуляторы NPY2 и NPY4, агонисты кортикотропинвысвобождающего фактора, модуляторы гистаминового рецептора-3 (Н 3), ингибиторы аР 2, модуляторы PPAR гамма, модуляторы PPAR дельта, ингибиторы ацетил-СоА карбоксилазы (АСС), ингибиторы 11HSD-1, модуляторы рецептора адинопектина; агонисты бета 3 адренергического рецептора, такие как AJ9677 (Takeda/Dainippon), L750355 (Merck) или СР 331648 (Pfizer) или другие известные бета 3 агонисты, описанные в патентах US 5541204, 5770615, 5491134, 5776983 и 5488064, модуляторы тироидного рецептора бета, такие как лиганд тироидного рецептора, описанный в международных заявках на патентWO 97/21993 (U. Cal SF), WO 99/00353 (KaroBio) и патенте GB 98/284425 (KaroBio), ингибитор SCD-1,описанный в международной заявке на патент WO 2005011655, ингибитор липазы, такой как орлистат или ATL-962 (Alizyme), агонисты рецептора серотонина (например, BVT-933 (Biovitrum, ингибиторы перезахвата моноамина или высвобождающие его агенты, такие как фенфлурамин, дексфенфлурамин,флувоксамин, флуоксетин, пароксетин, сертралин, хлорфентермин, клофорекс, клортермин, пицилорекс,сибутрамин, дексамфетамин, фентермин, фенилпропаноламин или мазиндол, аноректические агенты,такие как топирамат (JohnsonJohnson), CNTF (цилиарный нейротрофический фактор)/Axokine (Regeneron), BDNF (нейротрофический фактор мозга), лептин и модуляторы рецептора лептина, фентермин,лептин, бромкриптин, дексамфетамин, амфетамин, фенфлурамин, дексфенфлурамин, сибутрамин, орлистат, дексфенфлурамин, мазиндол, фентермин, фендиметразин, диэтилпропион, флуоксетин, бупропион,топирамат, диэтилпропион, бензфетамин, фенилпропаноламин или экопипам, эфедрин, псевдоэфедрин; г) антигипертензивные агенты, например петлевые диуретики, такие как этакриновая кислота, фу-7 018703 росемид и торсемид; диуретики, такие как производные тиазида, хлортиазид, гидрохлортиазид, амилорид; ингибиторы ангиотензинпревращающего фермента (АСЕ), такие как беназеприл, каптоприл, эналаприл, фозиноприл, лизиноприл, моэксиприл, перинодоприл, хинаприл, рамиприл и трандолаприл; ингибиторы Na-K-АТФазы мембранного насоса, такие как дигоксин; ингибиторы нейтралэндопептидазы-адренергического рецептора, такие как ацебутолол, атенолол, бетаксолол, бисопролол, метопролол,надолол, пропранолол, соталол и тимолол; инотропные агенты, такие как дигоксин, добутамин и милринон; блокаторы кальциевых каналов, такие как амлодипин, бепридил, дилтиазем, фелодипин, никардипин, нимодипин, нифедипин, низолдипин и верапамил; антагонисты альдостеронового рецептора; ингибиторы альдостеронсинтазы и двойной антагонист ET/AII, такой как соединения, описанные в международной заявке на патент WO 00/01389. д) соединение, повышающее ЛВП; е) модулятор абсорбции холестерина, такой как Zetia и KT6-971; ж) аналоги и миметики Аро-А 1; з) ингибиторы тромбина, такие как ксимелагатран; и) ингибиторы альдостерона, такие как анастразол, фадразол, эплеренон; к) ингибиторы агрегации тромбоцитов, такие как аспирин, бисульфат клопидогреля; л) эстроген, тестостерон, селективный модулятор рецептора эстрогена, селективный модулятор рецептора андрогена; м) химиотерапевтический агент, такой как ингибиторы ароматазы, например фемара, антиэстрогены, ингибиторы топоизомеразы I, ингибиторы топоизомеразы II, микротубулярные активные агенты,алкилирующие агенты, антинеопластические антиметаболиты, платиновые соединения, соединения, понижающие активность протеинкиназы, такие как ингибитор рецептора тирозинкиназы PDGF, предпочтительно иматиниб (N-5-[4-(4-метилпиперазинометил)бензоиламидо]-2-метилфенил-4-(3-пиридил)-2 пиримидинамин), описанный в заявке на патент ЕР-А-0564409 как пример 21, или 4-метил-N-[3-(4 метилимидазол-1-ил)-5-трифторметилфенил]-3-(4-пиридин-3-илпиримидин-2-иламино)бензамид,описанный в международной заявке на патент WO 04/005281 как пример 92; и н) агент, взаимодействующий с рецептором 5-НТ 3 и/или агент, взаимодействующий с рецептором 5-НТ 4, такой как тегасерод, описанный в патенте US 5510353 как пример 13, гидромалеат тегасерода,цизаприд, цилансетрон; о) агент для лечения табакозависимости, например частичные агонисты никотинового рецептора,гипохлорид бупропиона (также известный под товарным знаком Zyban) и никотинзамещающие средства; п) агент для лечения эректильной дисфункции, например допаминергические агенты, такие как апоморфин, агенты ADD/ADHD (например, Ritalin, Strattera, Concerta и Adderall); р) агент для лечения алкоголизма, такой как опиоидные антагонисты (например, нальтрексон (также известный под товарным знаком ReVia) и нальмефен), дисульфирам (также известный под товарным знаком Antabuse) и акампросат (также известный под товарным знаком Campral. Кроме того,также могут совместно вводиться агенты для снижения симптомов алкогольной зависимости, такие как бензодиазепины, бета-блокаторы, клонидин, карбамазепин, прегабалин и габапентин (Neurontin); с) другие полезные агенты, включая противовоспалительные агенты (например, ингибиторы СОХ 2); антидепрессанты (например, гидрохлорид флуоксетина (Prozac; улучшающие когнитивное поведение агенты (например, гидрохлорид донепезила (Aircept) и другие ингибиторы ацетилхолинэстеразы); нейропротекторные агенты (например, мемантин); антипсихотические лекарственные средства (например, зипразидон (Geodon), рисперидон (Risperdal) и оланзапин (Zyprexa; или в каждом случае их фармацевтически приемлемые соли и необязательно фармацевтически приемлемый носитель. Соединения по изобретению также могут использоваться в фармацевтических комбинациях, например наборе, включающем а) первый агент, который представляет собой соединение по изобретению,как здесь описано, в свободной форме или в форме фармацевтически приемлемой соли, и б) по крайней мере один совместно вводимый агент. Набор может включать инструкции для его применения. Термины "совместное введение" или "объединенное введение" или им подобные используются здесь для обозначения введения выбранных терапевтических агентов отдельному пациенту и включают режимы лечения, в которых агенты необязательно вводятся одним и тем же путем введения или в одно и то же время. Термин "фармацевтическая комбинация", как здесь используется, обозначает продукт, который получают смешением или объединением более одного активного ингредиента, и включает фиксированные и нефиксированные комбинации активных ингредиентов. Термин "фиксированная комбинация" обозна-8 018703 чает, что активные ингредиенты, например соединение формулы (I) и совместно вводимый агент, оба,вводятся пациенту одновременно в форме одного целого или одной дозировки. Термин "нефиксированная комбинация" обозначает, что активные ингредиенты, например соединение формулы (I) и совместно вводимый агент, оба, вводятся пациенту раздельными частями совместно, одновременно или последовательно в неограниченном периоде времени, где такое введение обеспечивает терапевтически эффективные уровни двух соединений в организме пациента. Последнее также включает коктейльную терапию,например введение трех или более активных ингредиентов. Способы получения соединений по изобретению В описанных ниже реакциях может быть необходимо защитить реакционные функциональные группы, например гидроксигруппу, аминогруппу, иминогруппу, тиогруппу или карбоксигруппу, где они присутствуют в конечных продуктах для устранения их нежелательного участия в реакциях. Обычные защитные группы могут использоваться в соответствии со стандартной практикой, например см. книгуT.W. Greene и P.G.M. Wuts, "Protective Groups in Organic Chemistry", John Wiley and Sons, 1991. На следующих схемах некоторые способы получения соединений настоящего изобретения являются иллюстративными. Специалисту в данной области техники ясно, что эти способы являются показательными и не включают все способы получения соединений настоящего изобретения. Радикалы на схемах являются такими, как описано для формулы (I). Схема реакций I Соединение формулы (I) может быть получено реакцией соединения формулы 2 с соединением формулы 3, где Q представляет собой уходящую группу (например, Cl, Br, OCF3 и им подобные) в присутствии подходящего растворителя (например, диметилацетамида, диметилформамида и им подобных) и подходящего основания (например, карбоната калия, трет-бутоксида натрия и им подобных), необязательно в присутствии палладиевого катализатора (например, Pd2(dba)3 и им подобных) и подходящего лиганда (например, ксантфоса и им подобных). Реакцию проводят при температуре от около 0 до около 200 С, и может проводиться в течение 24 ч до окончания. Схема реакций II Соединение формулы (I), где Y1 представляет собой кислород, может быть получено реакцией соединения формулы 4 с соединением формулы 5, где Y представляет собой уходящую группу (например,OMs, Cl, Br, I и им подобные), в присутствии подходящего растворителя (например, диметилформамида,ацетонитрила и им подобных) и подходящего основания (например, K2CO3, Cs2CO3, триэтиламина и им подобных). Реакцию проводят при температуре от около 0 до около 120 С, и может проводиться в течение 24 ч до окончания. Схема реакций III Соединение формулы (I), где Y4 представляет собой CH2 и W2 представляет собой N, может быть получено реакцией соединения формулы 6 с соединением формулы 7 в присутствии подходящего растворителя (например, дихлорэтана, этанола и им подобных), затем добавлением подходящего восстанавливающего агента (например, цианоборгидрида натрия, триацетоксиборгидрида натрия и им подобных). Реакцию проводят при температуре от около 0 до около 120 С, и может проводиться в течение 24 ч до Соединение формулы (I), где Y4 представляет собой СН 2 и W2 представляет собой N, может быть получено реакцией соединения формулы 8 с соединением формулы 9, где Q1 представляет собой уходящую группу (например, OMs, Cl, Br, I и им подобные), в присутствии подходящего растворителя (например, диметилформамида, ацетонитрила и им подобных) и подходящего основания (например, K2CO3,Cs2CO3, триэтиламина и им подобных). Реакцию проводят при температуре от около 0 до около 120 С, и может проводиться в течение 24 ч до окончания. Подробные описания синтеза соединений по изобретению приведены далее в примерах. Дополнительные способы получения соединений по изобретению Соединение по изобретению может быть получено в виде фармацевтически приемлемой кислотной аддитивной соли реакцией формы свободного основания соединения с фармацевтически приемлемой неорганической или органической кислотой. Альтернативно, фармацевтически приемлемая основная аддитивная соль соединения по изобретению может быть получена реакцией формы свободной кислоты соединения с фармацевтически приемлемым неорганическим или органическим основанием. Альтернативно, формы соли соединений по изобретению могут быть получены с помощью солей исходных материалов или промежуточных соединений. Формы свободных кислот или свободных оснований соединений по изобретению могут быть получены из соответствующей формы основной аддитивной соли или кислотной аддитивной соли соответственно. Например, соединение по изобретению в форме кислотной аддитивной соли может быть превращено в соответствующее свободное основание обработкой подходящим основанием (например, раствором гидроксида аммония, гидроксида натрия и им подобными). Соединение по изобретению в форме основной аддитивной соли может быть превращено в соответствующую свободную кислоту обработкой подходящей кислотой (например, хлористо-водородной кислотой и т.д.). Соединения по изобретению в неокисленной форме могут быть получены из N-оксидов соединений по изобретению обработкой восстанавливающим агентом (например, серой, диоксидом серы, трифенилфосфином, боргидридом лития, боргидридом натрия, трихлоридом фосфора, трибромидом или им подобными) в подходящем инертном органическом растворителе (например, ацетонитриле, этаноле, водном диоксане или им подобных) при температуре от 0 до 80 С. Пролекарственные производные соединений по изобретению могут быть получены способами, известными специалисту в данной области техники (например, более подробно см. статью Saulnier и др.(1994) Bioorganic and Medicinal Chemistry Letters, т. 4, с. 1985). Например, подходящие пролекарства могут быть получены реакцией свободного соединения по изобретению с подходящим карбамилирующим агентом (например, 1,1-ацилоксиалкилкарбанохлоридатом, пара-нитрофенилкарбонатом или им подобными). Защищенные производные соединений по изобретению могут быть получены способами, известными специалисту в данной области техники. Подробное описание методик, используемых для создания защитных групп и их удаления, может быть обнаружено в книге Т.W. Greene, "Protecting Groups in Organic Chemistry", 3-е изд., John Wiley and Sons, Inc., 1999. Соединения настоящего изобретения могут обычно получаться или образовываться в процессе осуществления изобретения в виде сольватов (например, гидратов). Гидраты соединений настоящего изобретения могут обычно получаться перекристаллизацией из смеси водного/органического растворителя, используя органические растворители, такие как диоксин, тетрагидрофуран или метанол. Соединения по изобретению могут быть получены в виде их индивидуальных стереоизомеров реакцией рацемической смеси соединения с оптически активным расщепляющим агентом с образованием пары диастереоизомерных соединений, разделением диастереомеров и расщеплением оптически чистых энантиомеров. Хотя расщепление энантиомеров может осуществляться, используя ковалентные диастереомерные производные соединений по изобретению, диссоциирующие комплексы являются предпочтительными (например, кристаллические диастереомерные соли). Диастереомеры имеют различные физические свойства (например, точки плавления, точки кипения, растворимости, реакционноспособность и т.д.) и могут быть легко разделены, используя эти различия. Диастереомеры могут быть разделены хроматографией или предпочтительно методиками разделения/расщепления, основанными на различиях в растворимости. Оптически чистый энантиомер затем выделяют с расщепляющим агентом любыми практическими способами, которые не приводят к рацемизации. Более подробное описание методик, исполь- 10018703 зующихся для разделения стереоизомеров соединений из их рацемической смеси, могут быть обнаружены в книге Jean Jacques, Andre Collet, Samuel H. Wilen, "Enantiomers, Racemates and Resolutions", JohnWiley And Sons, Inc., 1981. Наконец, соединения формулы (I) могут быть получены способом, который включает:(а) реакции, приведенные на схемах I-IV;(б) необязательно превращение соединения по изобретению в фармацевтически приемлемую соль;(в) необязательно превращение формы соли соединения по изобретению в форму, отличную от формы соли;(г) необязательно превращение неокисленной формы соединения по изобретению в фармацевтически приемлемый N-оксид;(д) необязательно превращение N-оксидной формы соединения по изобретению в его неокисленную форму;(е) необязательно выделение индивидуального изомера соединения по изобретению из смеси изомеров;(ж) необязательно превращение свободного соединения по изобретению в его фармацевтически приемлемое пролекарственное производное;(з) необязательно превращение пролекарственного производного соединения по изобретению в его свободную форму. Несмотря на то что получение исходных материалов конкретно не описано, соединения являются известными или могут быть получены аналогично способам, известным из предшествующего уровня техники, или как описано далее в разделе "Примеры". Специалисту в данной области техники ясно, что описанные выше превращения только иллюстрируют способы получения соединений настоящего изобретения и что аналогично могут использоваться другие хорошо известные способы. Примеры Настоящее изобретение далее представлено, но не ограничено, следующими примерами, которые иллюстрируют получение соединений по изобретению. Пример А 1. 5-Этил-2-(4-(4-4-(метилсульфонил)пиперазин-1-ил)метил)фенокси)пиперидин-1 ил)пиримидин(14,5 г, 105 ммоль) растворяли в DMA (50 мл) и перемешивали при 150 С в течение 12 ч. Реакционную смесь охлаждали, разбавляли H2O (100 мл) и экстрагировали EtOAc (3100 мл). Органические слои объединяли, промывали H2O (100 мл) и насыщенным раствором хлорида натрия (100 мл), сушили (MgSO4),отфильтровывали и концентрировали с получением 1-(5-этилпиримидин-2-ил)пиперидин-4-ола 1, который использовали далее без очистки на стадии Б: MS m/z для (М+Н)+ C11H18N3O рассчит. 208,1, обнаруж. 208,2. Стадия Б. 1-(5-Этилпиримидин-2-ил)пиперидин-4-ол 1 растворяли в ДХМ (200 мл). Добавляли диизопропилэтиламин (25 мл, 140 ммоль), затем смесь охлаждали до 0 С. По каплям добавляли метансульфонилхлорид (6,5 мл, 84 ммоль) и смесь перемешивали в течение 1 ч при КТ. Смесь выливали в H2O (100 мл) и органический слой отделяли. Органический слой промывали насыщ. водн. раствором NaHCO3, сушили (MgSO4), отфильтровывали и концентрировали. Продукт перекристаллизовывали из смесиEtOAc/гексан с получением 1-(5-этилпиримидин-2-ил)пиперидин-4-илметансульфоната 2 в виде беловатого порошка: 1 Н ЯМР (400 МГц, CDCl3)8,20 (s, 2H), 4,99 (m, 1H), 4,21 (m, 2H), 3,61 (m, 2H), 3,07 (s,3H), 2,49 (q, J=7,6 Гц, 2H), 2,07 (m, 2H), 1,89 (m, 4H), 1,55 (m, 2H), 1,45 (d, J=6,8 Гц, 1H), 1,21 (t, J=7,6 Гц,3H). MS m/z для (М+Н)+ C12H20N3O3S рассчит. 286,1, обнаруж. 286,2. Стадия В. 4-Гидроксибензальдегид (500 мг, 4,09 ммоль) и 1-метансульфонилпиперазин (670 мг, 4,09 ммоль) растворяли в дихлорэтане (10 мл). Добавляли АсОН (50 мкл) и смесь нагревали при 80 С в течение 1 ч. Затем добавляли триацетоксиборгидрид натрия (1,7 г, 8,2 ммоль) и смесь нагревали при 80 С в течение 2 ч. Реакционную смесь охлаждали, разбавляли насыщ. водн. раствором NaHCO3 (50 мл) и экстрагировали ДХМ (30 мл). Органический слой промывали насыщенным раствором хлорида натрия, суши- 11018703 ли (MgSO4), отфильтровывали, концентрировали и очищали ускоренной колоночной хроматографией(SiO2, градиент МеОН/ДХМ) с получением 4-4-(метилсульфонил)пиперазин-1-ил)метил)фенола 3 в виде белого порошка: 1 Н ЯМР (400 МГц, MeOD)7,15 (d, J=8,4 Гц, 2 Н), 6,75 (d, J=8,4 Гц, 2 Н), 3,48 (s,2 Н), 3,22 (t, J=4,8 Гц, 4 Н), 2,83 (s, 3H), 2,54 (t, J=4,8 Гц, 4 Н). MS m/z для (М+Н)+ C12H19N2O3S рассчит. 271,1, обнаруж. 271,1. Стадия Г. 1-(5-Этилпиримидин-2-ил)пиперидин-4-илметансульфонат 2 (253 мг, 0,89 ммоль), 4-4(метилсульфонил)пиперазин-1-ил)метил)фенол 3 (200 мг, 0,74 ммоль) и Cs2CO3 (482 мг, 1,48 ммоль) нагревали в AcN (10 мл) при 80 С в течение 2 ч. Реакционную смесь охлаждали, отфильтровывали, концентрировали и очищали ускоренной колоночной хроматографией (SiO2, градиент МеОН/ДХМ) с получением соединения А 1: 1 Н ЯМР (400 МГц, CDCl3)8,11 (s, 2 Н), 7,14 (d, J=8,8 Гц, 2 Н), 6,82 (d, J=8,4 Гц,2 Н), 4,46 (m, 1H), 4,12 (m, 2H), 3,56 (m, 2H), 3,41 (s, 2H), 3,17 (t, J=4,8 Гц, 4 Н), 2,70 (s, 3H), 2,48 (t, J=4,8 Гц, 4H), 2,39 (q, J=7,6 Гц, 2H), 1,95 (m, 2H), 1,74 (m, 2H), 1,22 (t, J=7,6 Гц, 3H). MS m/z для (М+Н)+C23H34N5O3S рассчит. 460,2, обнаруж. 460,2. Повторением описанных выше методик, используя подходящие исходные материалы, получали следующие соединения формулы (I), приведенные в табл. 1. Таблица 1(20 мл). Добавляли диизопропилэтиламин (3,4 мл, 19,8 ммоль) и смесь охлаждали до 0 С, затем по каплям добавляли метансульфонилхлорид (0,92 мл, 11,93 ммоль) и смесь перемешивали в течение 1 ч при КТ. Смесь разбавляли H2O (20 мл) и разделяли. Органический слой промывали насыщ. водн. растворомNaHCO3, сушили (MgSO4), отфильтровывали, концентрировали и очищали ускоренной колоночной хроматографией(220 мл). Органические слои объединяли, промывали H2O (20 мл), затем насыщенным раствором хлорида натрия (10 мл), сушили (MgSO4), отфильтровывали, концентрировали и очищали ускоренной колоночной хроматографией (SiO2, градиент EtOAc/гексан) с получением N-трет-бутил 4-(4 формилфенокси)пиперидин-1-карбоксилата 5: 1 Н ЯМР (400 МГц, CDCl3)7,77 (d, J=8,8 Гц, 2 Н), 6,94 (d,J=8,8 Гц, 2 Н), 4,54 (m, 1 Н), 3,63 (m, 2 Н), 3,32 (m, 2 Н), 1,89 (m, 2 Н), 1,73 (m, 2 Н), 1,39 (s, 9H). MS m/z для(50 мкл) и смесь нагревали при 90 С в течение 1 ч, затем добавляли триацетоксиборгидрид натрия (725 мг, 3,42 ммоль) и смесь нагревали при 90 С в течение 2 ч. Реакционную смесь охлаждали, разбавляли насыщ. водн. раствором NaHCO3 (50 мл) и экстрагировали ДХМ (30 мл). Органический слой промывали насыщенным раствором хлорида натрия, сушили (MgSO4), отфильтровывали, концентрировали и очищали ускоренной колоночной хроматографией (SiO2, градиент МеОН/ДХМ) с получением указанного в заголовке соединения В 1 в виде белого порошка: 1 Н ЯМР (400 МГц, MeOD)7,22 (d, J=8,8 Гц, 2 Н), 6,88 Стадия А. N-трет-Бутил 4-(4-4-(метилсульфонил)пиперазин-1-ил)метил)фенокси)пиперидин-1 карбоксилат В 1 (340 мг, 0,75 ммоль) растворяли в ТФУК (20 мл) и перемешивали при КТ в течение 1 ч. Смесь концентрировали в вакууме и остаток растворяли в МеОН и концентрировали (2) с получением соли ТФУК 1-(метилсульфонил)-4-(4-(пиперидин-4-илокси)бензил)пиперазина 7 (563 мг), которую использовали непосредственно на стадии Б без дополнительной очистки. MS m/z для (М-С 4 Н 11+H+ фрагмент) C17H28N3O3S рассчит. 354,2, обнаруж. 354,2. Стадия Б. 1-(Метилсульфонил)-4-(4-(пиперидин-4-илокси)бензил)пиперазин 7 (36 мг, 0,06 ммоль) и 1-метилциклопропил 4-нитрофенилкарбонат (22 мг, 0,09 ммоль) растворяли в ДХМ (5 мл). Добавляли триэтиламин (60 мкл, 0,43 ммоль) и смесь перемешивали при КТ в течение 2 ч. Смесь концентрировали в вакууме и очищали ускоренной колоночной хроматографией (SiO2, градиент EtOAc/гексан) с получением указанного в заголовке соединения В 2: 1 Н ЯМР (400 МГц, CDCl3)7,13 (d, J=8,4 Гц, 2 Н), 6,78 (d,J=8,4 Гц, 2 Н), 4,38 (m, 1 Н), 3,60 (m, 2H), 3,40 (s, 2H), 3,29 (m, 2H), 3,16 (m, 4H), 2,70 (s, 3H), 2,47 (m, 4H),1,82 (m, 2H), 1,69 (m, 2H), 1,48 (s, 3H), 1,19 (m, 1H), 0,80 (m, 2H), 0,56 (m, 2H). MS m/z для (М+Н)+ Осуществлением тех же методик, как описано в примере В 1, но используя 3-хлор-4 гидроксибензальдегид в качестве фенола, получали указанное в заголовке соединение В 3: 1H ЯМР (400 МГц, CDCl3)7,42 (d, J=2,4 Гц, 1 Н), 7,32 (dd, J=2,4, 8,4 Гц, 1H), 6,98 (d, J=8,4 Гц, 1H), 4,63 (m, 1H), 4,15 1-(Метилсульфонил)-4-(4-(пиперидин-4-илокси)бензил) пиперазин 7 (30 мг, 0,05 ммоль), 2-хлор-5 фторпиримидин (7 мкл, 0,06 ммоль) и K2CO3 (24 мг, 0,17 ммоль) растворяли в DMA (1 мл) и подвергали микроволновому излучению (180 С, 5 мин). Реакционную смесь охлаждали, разбавляли H2O (10 мл) и экстрагировали EtOAc (10 мл). Органический слой промывали H2O (10 мл) и насыщенным раствором хлорида натрия (10 мл), затем сушили (MgSO4), отфильтровывали, концентрировали и очищали ВЭЖХ с обратной фазой (градиент H2O/AcN) с получением указанного в заголовке соединения В 4 в виде белого твердого вещества: 1 Н ЯМР (400 МГц, CDCl3)8,52 (s, 2H), 7,31 (d, J=8,8 Гц, 2 Н), 6,91 (d, J=8,4 Гц, 2 Н),4,52 (m, 1H), 4,08 (s, 2H), 4,03 (m, 2H), 3,79 (m, 2H) 3,63 (m, 2H), 3,48 (m, 4H), 2,80 (s, 3H), 1,95 (m, 2H),1,76 (m, 2H). MS m/z для (М+Н)+ C21H29FN5O3S рассчит. 450,2, обнаруж. 450,2. Повторением описанных выше методик, используя подходящие исходные материалы, получали следующие соединения формулы (I), приведенные в табл. 2. Таблица 2 Стадия А. Метил 2-(4-(2-хлор-4-4-(метилсульфонил)пиперазин-1-ил)метил)фенокси)пиперидин-1 ил)пиримидин-5-карбоксилат В 7 (216 мг, 0,41 ммоль) растворяли в ТГФ (5 мл) и добавляли LiOH (165 мкл 5 М раствора, 0,824 ммоль). Смесь нагревали при 80 С в течение 3 ч, затем охлаждали и концентрировали с получением сырой 2-(4-(2-хлор-4-4-(метилсульфонил)пиперазин-1 ил)метил)фенокси)пиперидин-1-ил)пиримидин-5-карбоновой кислоты, которую использовали на следующей стадии без дополнительной очистки. MS m/z для (МН+) C22H29ClN5O5S рассчит. 510,2, обнаруж. 510,1. Стадия Б. 2-(4-(2-Хлор-4-4-(метилсульфонил)пиперазин-1-ил)метил)фенокси)пиперидин-1 ил)пиримидин-5-карбоновую кислоту (21 мг, 0,41 ммоль) растворяли в смеси ДХМ (5 мл) и ТГФ (5 мл). Добавляли оксалилхлорид (40 мкл, 0,45 ммоль) и смесь перемешивали при КТ в течение 2 ч, затем добавляли гидроксид аммония (100 мкл 30% раствора в H2O) и смесь перемешивали при КТ в течение 12 ч. Смесь концентрировали и очищали ВЭЖХ с обратной фазой (градиент H2O/AcN) с получением указанного в заголовке соединения В 12 в виде белого твердого вещества: 1 Н ЯМР (400 МГц, CDCl3)8,65 (s,2 Н), 7,30 (d, J=2,0 Гц, 1 Н), 7,07 (dd, J=2,0, 8,4 Гц, 1 Н), 6,87 (d, J=8,4 Гц, 1H), 4,59 (m, 1 Н), 4,05 (m, 4 Н),3,41 (m, 2 Н), 3,19 (m, 4 Н), 2,72 (s, 3H), 2,50 (m, 4H), 1,90 (m, 4H). MS m/z для (М+Н)+ C22H30ClN6O4S рассчит. 509,2, обнаруж. 509,2. Повторением описанных выше методик, используя подходящие исходные материалы, получали следующие соединения формулы (I), представленные в табл. 3.Pd(PPh3)4 (13 мг, 0,011 ммоль) и смесь дегазировали аргоном в течение 20 мин, затем нагревали при 65 С в течение 12 ч. Смесь охлаждали, отфильтровывали и очищали ВЭЖХ с обратной фазой (градиентH2O/AcN) с получением указанного в заголовке соединения В 15 в виде белого твердого вещества: 1 Н ЯМР (400 МГц, CDCl3)8,28 (s, 2 Н), 7,16 (d, J=2,0 Гц, 1 Н), 6,93 (dd, J=1,6, 8,4 Гц, 1 Н), 6,72 (d, J=8,4 Гц,1H), 4,46 (m, 1H), 3,95 (m, 2H), 3,77 (m, 2H), 3,30 (s, 2H), 3,06 (m, 4H), 2,58 (s, 3H), 2,37 (m, 4H), 1,76 (m,4H). MS m/z для (М+Н)+ C22H28ClN6O3S рассчит. 491,2, обнаруж. 491,2. Пример В 16. 2-(4-(2-Хлор-4-4-(метилсульфонил)пиперазин-1-ил)метил)фенокси)пиперидин-1-ил)5-(2 Н-тетразол-5-ил)пиримидинNH4Cl (55 мг, 1,02 ммоль) и смесь нагревали при 90 С в течение 12 ч. Смесь разбавляли H2O (10 мл),экстрагировали EtOAc (10 мл), промывали H2O (10 мл) и насыщенным раствором хлорида натрия (10 мл), сушили (MgSO4), отфильтровывали и очищали ВЭЖХ с обратной фазой (градиент H2O/AcN) с получением указанного в заголовке соединения В 16 в виде белого твердого вещества: 1H ЯМР (400 МГц,ДМСО-d6)8,93 (s, 2 Н), 7,53 (d, J=2,0 Гц, 1 Н), 7,35 (dd, J=2,0, 8,4 Гц, 1 Н), 7,19 (d, J=8,4 Гц, 1 Н), 4,81 (m,1 Н), 4,18 (m, 2 Н), 3,92 (m, 4 Н), 3,42 (m, 4 Н), 3,03 (m, 4 Н), 2,85 (s, 3H), 2,06 (m, 2 Н), 1,85 (m, 2 Н). MS m/z для (М+Н)+ C22H29ClN9O3S рассчит. 534,2, обнаруж. 534,2. 2-(4-(2-Хлор-4-4-(метилсульфонил)пиперазин-1-ил)метил)фенокси)пиперидин-1-ил)-5-(2 Н-тетразол-5-ил)пиримидин В 16 (28 мг, 0,052 ммоль) растворяли в ДМФА (2 мл). Добавляли K2CO3 (40 мг, 0,29 ммоль) и MeI (4 мкл, 0,06 ммоль) и смесь перемешивали при КТ в течение 2 ч. Смесь разбавляли H2O (10 мл), экстрагировали EtOAc (10 мл), промывали H2O (10 мл) и насыщенным раствором хлорида натрия(10 мл), сушили (MgSO4), отфильтровывали и очищали ВЭЖХ с обратной фазой (градиент H2O/AcN) с получением указанного в заголовке соединения В 17 в виде белого твердого вещества: 1H ЯМР (400 МГц,ДМСО-d6)8,92 (s, 2 Н), 7,30 (d, J=2,0 Гц, 1 Н), 7,06 (dd, J=2,0, 8,4 Гц, 1 Н), 6,88 (d, J=8,8 Гц, 1 Н), 4,58 (m,1H), 4,32 (s, 3H), 4,04 (m, 2 Н), 3,96 (m, 2 Н), 3,40 (s, 2 Н), 3,18 (m, 4 Н), 2,72 (s, 3H), 2,48 (m, 4 Н), 1,91 (m,4 Н). MS m/z для (М+Н)+ C23H31ClN9O3S рассчит. 548,2, обнаруж. 548,2. Пример С 1. 2-(4-(2-Бром-4-4-(метилсульфонил)пиперазин-1-ил)метил)фенокси)пиперидин-1-ил)-5-этилпиримидин А 8 (60 мг, 0,11 ммоль), (Е)-2-(3-метокси-1-пропенил)-4,4,5,5-тетраметил-1,3,2-диоксаборолан (35 мкл, 0,16 ммоль), Na2CO3 (35 мг, 0,33 ммоль) и Pd(PPh3)4 (13 мг, 0,011 ммоль) растворяли в смеси диметоксиэтана (360 мкл), H2O (240 мкл) и EtOH (180 мкл) и подвергали микроволновому облучению (170 С,5 мин). Смесь охлаждали, разбавляли Н 2 О (10 мл) и экстрагировали EtOAc (20 мл). Органический слой сушили (MgSO4), отфильтровывали, концентрировали и очищали ВЭЖХ с обратной фазой (градиентH2O/AcN) с получением указанного в заголовке соединения С 1 в виде белого твердого вещества: 1 Н ЯМР(10 мл) и смесь перемешивали в H2 в аппарате Парра (55 psi) в течение 1 ч. Смесь отфильтровывали через целит, концентрировали и очищали ВЭЖХ с обратной фазой (градиент H2O/AcN) с получением указанного в заголовке соединения С 2 в виде белого твердого вещества: 1 Н ЯМР (400 МГц, CDCl3)8,12 (s,2 Н), 7,03 (d, J=2,0 Гц, 1 Н), 7,00 (dd, J=2,0, 8,4 Гц, 1 Н), 6,75 (d, J=8,4 Гц, 1 Н), 4,51 (m, 1 Н), 3,97 (m, 2 Н),3,71 (m, 2 Н), 3,40 (s, 2H), 3,31 (t, J=6,8 Гц, 2 Н), 3,25 (s, 3H), 3,17 (s, 3H), 2,71 (s, 3H), 2,60 (m, 1 Н), 2,48 (s,3H), 2,40 (q, J=7,6 Гц, 2 Н), 1,93 (m, 2 Н), 1,79 (m, 4 Н), 1,13 (t, J=7,6 Гц, 3H); MS m/z для (М+Н)+C27H42N5O4S рассчит. 532,3, обнаруж. 532,0. Повторением описанных выше методик, используя подходящие исходные материалы, получали следующие соединения формулы (I), представленные в табл. 4.Pd(PPh3)4 (25 мг, 0,022 ммоль) растворяли в триэтиламине (750 мкл), дегазировали в течение 20 мин аргоном, затем нагревали при 90 С в течение 3 ч. Смесь охлаждали, разбавляли H2O (10 мл) и экстрагировали EtOAc (20 мл). Органический слой сушили (MgSO4), отфильтровывали, концентрировали и очищали ускоренной колоночной хроматографией (SiO2, градиент EtOAc/гексан) с получением 4-(2-(1-(5 этилпиримидин-2-ил)пиперидин-4-илокси)-5-4-(метилсульфонил)пиперазин-1-ил)метил)фенил)бут-3 ин-1-ола в виде белого порошка: 1H ЯМР (400 МГц, CDCl3)8,11 (s, 2 Н), 7,27 (d, J=2,4 Гц, 1 Н), 7,10 (dd,J=2,4, 8,4 Гц, 1H), 6,81 (d, J=8,4 Гц, 1H), 4,54 (m, 1H), 4,07 (m, 2H), 3,66 (m, 4H), 3,38 (s, 2H), 3,17 (m, 4H),2,71 (s, 3H), 2,63 (t, J=6,0 Гц, 2 Н), 2,47 (m, 4H), 2,40 (q, J=7,6 Гц, 2H), 1,94 (m, 4H), 1,82 (m, 2H), 1,53 (m,2H), 1,12 (t, J=7,6 Гц, 3H); MS m/z для (М+Н)+ C27H38N5O4S рассчит. 528,3, обнаруж. 528,2. Стадия Б. 4-(2-(1-(5-Этилпиримидин-2-ил)пиперидин-4-илокси)-5-4-(метилсульфонил)пиперазин 1-ил)метил)фенил)бут-3-ин-1-ол (50 мг, 0,095 ммоль) и циклогексадиен (100 мкл) растворяли в EtOH (2 мл). Добавляли Pd/C (20 мг 10%, сырой) и смесь нагревали при 80 С в течение ночи, затем смесь охлаждали и отфильтровывали через фильтр с порами 0,2 мкм. Смесь концентрировали и очищали ВЭЖХ с обратной фазой (градиент H2O/AcN) с получением указанного в заголовке соединения С 7 в виде белого твердого вещества: 1 Н ЯМР (400 МГц, CDCl3)8,12 (s, 2 Н), 7,31 (d, J=2,4 Гц, 1 Н), 7,04 (dd, J=2,4, 8,4 Гц,1 Н), 6,80 (d, J=8,4 Гц, 1 Н), 6,75 (d, J=16 Гц, 1 Н), 6,15 (dt, J=7,2, 16 Гц, 1H), 4,49 (m, 1H), 4,06 (m, 2H), 3,64(1 мл) и дегазировали в течение 20 мин аргоном. Затем реакционный сосуд закрывали и нагревали при 120 С в течение 12 ч. Смесь охлаждали, отфильтровывали, концентрировали и очищали ВЭЖХ с обратной фазой (градиент H2O/AcN) с получением указанного в заголовке соединения С 9 в виде белого твердого вещества: 1H ЯМР (400 МГц, CDCl3)8,11 (s, 2H), 6,69 (d, J=8,0 Гц, 1H), 6,53 (d, J=1,6 Гц, 1 Н), 6,49 Той же методикой, описанной в примере С 9, но используя морфолин в качестве амина, получали указанное в заголовке соединение С 10: 1 Н ЯМР (400 МГц, CDCl3)8,12 (s, 2H), 6,82 (m, 3H), 4,55 (m,1H), 4,03 (m, 2H), 3,77 (m, 4H), 3,65 (m, 2H), 3,22 (m, 4H), 3,03 (m, 4H), 2,73 (m, 4H), 2,40 (q, J=7,6 Гц, 2 Н),1,95 (m, 3H), 1,78 (m, 3H), 1,12 (t, J=7,6 Гц, 3H); MS m/z для (М+Н)+ C27H41N6O4S рассчит. 545,3, обнаруж. 545,2. Пример D1. 3-(4-(3-Хлор-4-(1-(5-этилпиримидин-2-ил)пиперидин-4-илокси)бензил)пиперазин-1 илсульфонил)пропан-1-ол Стадия А. 3-Хлор-4-гидроксибензальдегид (500 мг, 3,19 ммоль) и N-трет-бутилпиперазин-1 карбоксилат (595 мг, 3,19 ммоль) растворяли в дихлорэтане (20 мл). Добавляли АсОН (50 мкл) и смесь нагревали при 80 С в течение 1 ч. Добавляли триацетоксиборгидрид натрия (1,35 г, 6,38 ммоль) и смесьNaHCO3 (50 мл) и экстрагировали ДХМ (30 мл). Органический слой промывали насыщенным раствором хлорида натрия, сушили (MgSO4), отфильтровывали, концентрировали и очищали ускоренной колоночной хроматографией (SiO2, градиент МеОН/ДХМ) с получением N-трет-бутил 4-(3-хлор-4 гидроксибензил)пиперазин-1-карбоксилата 8 в виде белого порошка: 1 Н ЯМР (400 МГц, CDCl3)7,29 (d,J=2,0, 1H), 7,11 (dd, J=2,0, 8,0 Гц, 1 Н), 6,95 (d, J=8,0 Гц, 1H), 3,44 (m, 6H), 2,39 (m, 4H), 1,47 (s, 9H). MSm/z для (М+Н)+ C16H24ClN2O3 рассчит. 327,1, обнаруж. 327,2. Стадия Б. 4-(3-Хлор-4-гидроксибензил)пиперазин-1-карбоксилат 8 (940 мг, 2,87 ммоль), 1-(5 этилпиримидин-2-ил)пиперидин-4-илметансульфонат 2 (1,23 г, 4,31 ммоль) и Cs2CO3 (1,8 г, 5,74 ммоль) нагревали в AcN (5 мл) при 60 С в течение 12 ч. Реакционную смесь охлаждали, отфильтровывали, концентрировали и очищали ускоренной колоночной хроматографией (SiO2, градиент EtOAc/гексан) с получением N-трет-бутил 4-(3-хлор-4-(1-(5-этилпиримидин-2-ил)пиперидин-4-илокси)бензил)пиперазин-1 карбоксилата 9: MS m/z для (М+Н)+ C27H39ClN5O3 рассчит. 516,3, обнаруж. 516,3. Стадия В.N-трет-Бутил 4-(3-хлор-4-(1-(5-этилпиримидин-2-ил)пиперидин-4 илокси)бензил)пиперазин-1-карбоксилат 9 (345 мг, 0,67 ммоль) растворяли в ТФУК (4 мл) и перемешивали при КТ в течение 1 ч. Смесь подщелачивали насыщ. водн. раствором NaHCO3 (20 мл) и экстрагировали ДХМ (20 мл). Органический слой сушили (MgSO4), отфильтровывали и концентрировали с получением 2-(4-(2-хлор-4-(пиперазин-1-илметил)фенокси)пиперидин-1-ил)-5-этилпиримидина 10, который использовали непосредственно на стадии Г без дополнительной очистки: MS m/z для (М+Н) C17H28N3O3S рассчит. 416,2, обнаруж. 416,2. Стадия Г. 2-(4-(2-Хлор-4-(пиперазин-1-илметил)фенокси)пиперидин-1-ил)-5-этилпиримидин 10(163 мг, 0,39 ммоль) растворяли в ДХМ (5 мл). Добавляли триэтиламин (60 мкл, 0,43 ммоль) и смесь охлаждали до 0 С. Добавляли 3-хлорпропансульфонилхлорид (52 мкл, 0,43 ммоль) и смесь перемешивали при КТ в течение 1 ч, затем разбавляли Н 2 О (10 мл) и экстрагировали ДХМ. Органический слой сушили(MgSO4), отфильтровывали и концентрировали с получением сырого 2-(4-(2-хлор-4-4-(3 хлорпропилсульфонил)пиперазин-1-ил)метил)фенокси)пиперидин-1-ил)-5-этилпиримидина 11, который использовали непосредственно на стадии Д без дополнительной очистки: MS m/z для (М+Н)+C25H36Cl2N5O3S рассчит. 556,2, обнаруж. 556,1. Стадия Д. 2-(4-(2-Хлор-4-4-(3-хлорпропилсульфонил)пиперазин-1-ил)метил)фенокси)пиперидин 1-ил)-5-этилпиримидин 11 (0,39 ммоль), NaI (60 мг, 0,395 ммоль) и NaOAc (96 мг, 1,17 ммоль) растворяли в ДМФА (2 мл) и нагревали при 120 С в течение 2 ч. Смесь охлаждали, разбавляли H2O (10 мл) и экстрагировали EtOAc (20 мл). Органический слой промывали H2O (10 мл) и насыщенным раствором хлорида натрия (10 мл), затем сушили (MgSO4), отфильтровывали и концентрировали с получением 3-(4-(3 хлор-4-(1-(5-этилпиримидин-2-ил)пиперидин-4-илокси)бензил)пиперазин-1-илсульфонил)пропил ацетата 12, который использовали непосредственно на стадии Е без дополнительной очистки: MS m/z для(М+Н)+ C27H39ClN5O5S рассчит. 580,2, обнаруж. 580,1. Стадия Е. 3-(4-(3-Хлор-4-(1-(5-этилпиримидин-2-ил)пиперидин-4-илокси)бензил)пиперазин-1 илсульфонил)пропилацетат 12 (0,39 ммоль) и LiOH-Н 2 О (50 мг, 1,19 ммоль) растворяли в ТГФ (3 мл) и Н 2 О (25 мкл) и нагревали при 60 С в течение 2 ч. Реакционную смесь концентрировали и очищали ускоренной колоночной хроматографией (SiO2, градиент EtOAc/гексан) с получением указанного в заголовке соединения D1 в виде белого порошка: 1 Н ЯМР (400 МГц, CDCl3)8,11 (s, 2 Н), 7,28 (d, J=2,0, 1 Н), 7,05(20 мл), затем добавляли диизопропилэтиламин (1,15 мл, 6,58 ммоль) и метансульфонилхлорид (0,34 мл,4,38 ммоль) и перемешивали при КТ в течение 2 ч. Смесь подщелачивали насыщ. водн. растворомNaHCO3 (20 мл) и разделяли. Органический слой промывали 1 N HCl (10 мл) и насыщенным раствором хлорида натрия (10 мл), сушили (MgSO4), отфильтровывали и концентрировали с получением 4-1(метилсульфонил)пиперидин-4-ил)метил)фенилметансульфоната 13, который использовали на стадии Б- 24018703 без дополнительной очистки: MS m/z для (М+Н)+ C14H22NO5S2 рассчит. 348,1, обнаруж. 348,2. Стадия Б. 4-1-(Метилсульфонил)пиперидин-4-ил)метил)фенилметансульфонат 13 (2,19 ммоль) растворяли в МеОН (3 мл), затем добавляли 10% NaOH (2 мл) и смесь нагревали при 80 С в течение 1 ч. Реакционную смесь охлаждали, гасили 1 N HCl (10 мл) и экстрагировали EtOAc (20 мл). Органический слой сушили (MgSO4), отфильтровывали, концентрировали и очищали ускоренной колоночной хроматографией (SiO2, градиент EtOAc/гексан) с получением 4-1-(метилсульфонил)пиперидин-4 ил)метил)фенола 14 в виде белого порошка: 1 Н ЯМР (400 МГц, CDCl3)6,92 (d, J=8,4 Гц, 2 Н), 6,69 (d,J=8,4 Гц, 2 Н), 3,70 (m, 2H), 2,68 (s, 3H), 2,51 (dt, J=2,4, 8,0 Гц, 2 Н), 2,43 (d, J=7,2 Гц, 2 Н), 1,67 (m, 2 Н),1,49 (m, 1 Н), 1,26 (m, 2 Н). MS m/z для (М+Н)+ C13H20NO3S рассчит. 270,1, обнаруж. 270,1. Стадия В. 4-1-(Метилсульфонил)пиперидин-4-ил)метил)фенол 14 (50 мг, 0,19 ммоль), 1-(5 этилпиримидин-2-ил)пиперидин-4-илметансульфонат 2 (79 мг, 0,28 ммоль) и Cs2CO3 (121 мг, 0,37 ммоль) нагревали в AcN (5 мл) при 60 С в течение 12 ч. Реакционную смесь охлаждали, отфильтровывали, концентрировали и очищали ускоренной колоночной хроматографией (SiO2, градиент EtOAc/гексан) с получением указанного в заголовке соединения Е 1 в виде белого порошка: 1 Н ЯМР (400 МГц, CDCl3)8,31 Стадия А. Гидрохлорид 4-(пиперидин-4-илметил)фенола (380 мг, 1,67 ммоль), Boc2O (400 мг, 1,83 ммоль) и бикарбонат натрия (1,4 г, 16,7 ммоль) растворяли в H2O (10 мл) и диоксане (10 мл) и перемешивали при КТ в течение 2 ч. Затем смесь экстрагировали EtOAc (20 мл), промывали насыщенным раствором хлорида натрия (10 мл), сушили (MgSO4), отфильтровывали и концентрировали с получением 4-1(метилсульфонил)пиперидин-4-ил)метил)фенилметансульфоната 15, который использовали на стадии Б без дополнительной очистки: MS m/z для (М-С 4 Н 9+Н)+ C13H18NO3 рассчит. 236,1, обнаруж. 236,1. Стадия Б. N-трет-Бутил 4-(4-гидроксибензил)пиперидин-1-карбоксилат 15 (236 мг, 0,811 ммоль) растворяли в ДХМ (3 мл), затем добавляли сульфурилхлорид (33 мкл, 0,40 ммоль) и смесь перемешивали при КТ в течение 1 ч. Реакционную смесь разбавляли водой (10 мл) и экстрагировали ДХМ (20 мл). Органический слой сушили (MgSO4), отфильтровывали, концентрировали и очищали ускоренной колоночной хроматографией (SiO2, градиент EtOAc/гексан) с получением N-трет-бутил 4-(3-хлор-4 гидроксибензил)пиперидин-1-карбоксилата 16 в виде белого порошка. MS m/z для (М-С 4 Н 9+Н)N-трет-Бутил 4-(3-хлор-4-(1-(5-этилпиримидин-2-ил)пиперидин-4 илокси)бензил)пиперидин-1-карбоксилат 17 (345 мг, 0,67 ммоль) растворяли в ТФУК (4 мл) и перемешивали при КТ в течение 1 ч. Смесь подщелачивали насыщ. водн. раствором NaHCO3 (20 мл) и экстрагировали ДХМ (20 мл). Органический слой сушили (MgSO4), отфильтровывали и концентрировали. Остаток растворяли в ДХМ (3 мл) и обрабатывали диизопропилэтиламином (34 мкл, 0,19 ммоль) и метансульфонилхлоридом (8 мкл, 0,10 ммоль). Смесь перемешивали при КТ в течение 1 ч, затем смесь концентрировали и очищали ВЭЖХ с обратной фазой (градиент H2O/AcN) с получением указанного в заголовке соединения Е 2 в виде белого твердого вещества: 1 Н ЯМР (400 МГц, CDCl3)8,20 (s, 2 Н), 7,18 (d, J=2,0 Гц,1 Н), 6,98 (dd, J=2,0, 8,0 Гц, 1 Н), 6,93 (d, J=8,4 Гц, 1 Н), 4,57 (m, 1 Н), 4,13 (m, 2 Н), 3,77 (m, 4 Н), 2,78 (s, 3H),2,62 (td, J=2,4, 12,0 Гц, 2 Н), 2,50 (m, 4 Н), 1,99 (m, 2 Н), 1,90 (m, 2 Н), 1,77 (m, 2 Н), 1,62 (m, 2 Н), 1,34 (m,2 Н), 1,12 (t, J=7,6 Гц, 3H); MS m/z для (М+Н)+ C24H34ClN4O3S рассчит. 493,2, обнаруж. 493,2. Стадия А. 3-Хлор-4-гидроксибензойную кислоту (1 г, 5,79 ммоль) растворяли в МеОН (5 мл), затем добавляли конц. H2SO4 (250 мкл) и смесь нагревали при кипении с обратным холодильником в течение 3 ч. Реакционную смесь охлаждали, подщелачивали насыщ. водн. раствором NaHCO3 (20 мл) и экстрагировали EtOAc (220 мл). Органические слои объединяли, сушили (MgSO4), отфильтровывали и концентрировали с получением метил 3-хлор-4-гидроксибензоата 18: MS m/z для (М+Н)+ C8H8ClO3 рассчит. 187,0, обнаруж. 187,1. Стадия Б. 1-(5-Этилпиримидин-2-ил)пиперидин-4-илметансульфонат 2 (458 мг, 1,61 ммоль), 3-хлор 4-гидроксибензоат 18 (200 мг, 1,07 ммоль) и Cs2CO3 (697 мг, 2,14 ммоль) растворяли в AcN (3 мл) и подвергали микроволновому излучению (180 С, 3 мин). Реакционную смесь охлаждали, отфильтровывали,концентрировали и очищали ускоренной колоночной хроматографией (SiO2, градиент EtOAc/гексан) с получением метил 3-хлор-4-(1-(5-этилпиримидин-2-ил)пиперидин-4-илокси)бензоата 19: 1H ЯМР (400 МГц, CDCl3)8,21 (s, 2H), 8,09 (d, J=2,0 Гц, 1 Н), 7,93 (dd, J=2,0, 8,8 Гц, 1 Н), 7,01 (d, J=8,8 Гц, 1 Н), 4,76(m, 1H), 4,06 (m, 2H), 3,92 (s, 3H), 3,87 (m, 2H), 2,48 (q, J=7,6 Гц, 2 Н), 2,04 (m, 2H), 1,94 (m, 2H), 1,22 (t,J=7,6 Гц, 3H). MS m/z для (М+Н)+ C19H23ClN3O3 рассчит. 376,1, обнаруж. 376,1. Стадия В. Метил 3-хлор-4-(1-(5-этилпиримидин-2-ил)пиперидин-4-илокси)бензоат 19 (536 мг, 1,53 ммоль) растворяли в сухом ТГФ (10 мл) и охлаждали до 0 С, затем добавляли 1 М раствор LiAlH4 и смесь перемешивали при 0 С в течение 10 мин. Реакцию гасили медленным добавлением по каплям H2O,затем экстрагировали EtOAc (30 мл). Органический слой промывали насыщ. водн. раствором NaHCO3(10 мл), H2O (10 мл) и насыщенным раствором хлорида натрия (10 мл), затем сушили (MgSO4), отфильтровывали и концентрировали с получением сырого (3-хлор-4-(1-(5-этилпиримидин-2-ил)пиперидин-4 илокси)фенил)метанола 20, который использовали на стадии Г без дополнительной очистки. MS m/z для(М+Н)+ C18H23ClN3O2 рассчит. 348,1, обнаруж. 348,1. Стадия Г. (3-Хлор-4-(1-(5-этилпиримидин-2-ил)пиперидин-4-илокси)фенил)метанол 20 (1,53 ммоль) растворяли в ДХМ (20 мл). Добавляли диизопропилэтиламин (535 мкл, 3,07 ммоль), затем метансульфонилхлорид (130 мкл, 1,68 ммоль) и смесь перемешивали при КТ в течение 2 ч. Смесь концентрировали и очищали ускоренной колоночной хроматографией (SiO2, градиент EtOAc/гексан) с получением 2-(4-(2-хлор-4-(хлорметил)фенокси)пиперидин-1-ил)-5-этилпиримидина 21: 1 Н ЯМР (400 МГц, CDCl3)8,11 (s, 2 Н), 7,36 (d, J=2,4 Гц, 1 Н), 7,16 (dd, J=2,4, 8,4 Гц, 1 Н), 6,89 (d, J=8,4 Гц, 1H), 4,55 (m, 1H), 4,45 (m,- 26018703m/z для (M+H)+ C18H22Cl2N3O рассчит. 366,1, обнаруж. 366,1. Стадия Д. N-Boc-оксопиперазин (40 мг, 0,20 ммоль) растворяли в ДМФА (5 мл). Добавляли гидрид натрия (12 мг, 0,3 ммоль) и смесь нагревали при 60 С в течение 1 ч, затем добавляли 2-(4-(2-хлор-4(хлорметил)фенокси)пиперидин-1-ил)-5-этилпиримидин 21 (76 мг, 0,20 ммоль) и смесь перемешивали при КТ в течение 12 ч. Смесь гасили H2O (20 мл) и экстрагировали EtOAc (20 мл). Органический слой промывали H2O (20 мл) и насыщенным раствором хлорида натрия (10 мл), затем сушили (MgSO4), отфильтровывали, концентрировали и очищали ускоренной колоночной хроматографией (SiO2, градиентEtOAc/гексан) с получением N-трет-бутил 4-(3-хлор-4-(1-(5-этилпиримидин-2-ил)пиперидин-4 илокси)бензил)-3-оксопиперазин-1-карбоксилата 22: MS m/z для (М+Н)+ C27H37ClN5O4 рассчит. 530,2,обнаруж. 366,1. Стадия Е. N-трет-Бутил 4-(3-хлор-4-(1-(5-этилпиримидин-2-ил)пиперидин-4-илокси)бензил)-3 оксопиперазин-1-карбоксилат 22 (79 мг, 0,15 ммоль) растворяли в смеси ДХМ (2 мл) и 4 N раствора HCl в диоксане (3 мл) и перемешивали при КТ в течение 1 ч. Смесь подщелачивали насыщ. водн. растворомNaHCO3 (40 мл) и экстрагировали ДХМ (20 мл). Органический слой сушили (MgSO4), отфильтровывали и концентрировали с получением 1-(3-хлор-4-(1-(5-этилпиримидин-2-ил)пиперидин-4 илокси)бензил)пиперазин-2-она 23, который использовали непосредственно на стадии Ж без дополнительной очистки: MS m/z для (М+Н)+ C22H29ClN5O2 рассчит. 430,2, обнаруж. 430,1. Стадия Ж. 1-(3-Хлор-4-(1-(5-этилпиримидин-2-ил)пиперидин-4-илокси)бензил)пиперазин-2-он 23(20 мг, 0,04 ммоль) растворяли в ДХМ (3 мл), затем добавляли диизопропилэтиламин (22 мкл, 0,13 ммоль) и метансульфонилхлорид (4 мкл, 0,05 ммоль) и перемешивали при КТ в течение 2 ч. Смесь подщелачивали насыщ. водн. раствором NaHCO3 (10 мл) и экстрагировали ДХМ (10 мл). Органический слой сушили (MgSO3), отфильтровывали, концентрировали и очищали ускоренной колоночной хроматографией (SiO2, градиент EtOAc/гексан) с получением указанного в заголовке соединения Е 4: 1H ЯМР (400 МГц, CDCl3)8,36 (s, 2H), 7,25 (d, J=2,4 Гц, 1H), 7,08 (dd, J=2,4, 8,4 Гц, 1 Н), 6,87 (d, J=8,4 Гц, 1H), 4,66 Стадия А. Той же методикой, как описано в примере Е 2 для стадии А, но используя гидрохлорид 4 метилтиопиперидина в качестве исходного материала, получали N-трет-бутил 4-(метилтио)пиперидин-1 карбоксилат. Стадия Б. N-трет-Бутил 4-(метилтио)пиперидин-1-карбоксилат (1,11 г, 4,8 ммоль) и периодат натрия (5 г, 24 ммоль) растворяли в AcN (20 мл) и H2O (8 мл) и нагревали при 100 С в течение 2 ч. Смесь охлаждали, разбавляли водой (20 мл) и экстрагировали EtOAc (20 мл). Органический слой промывали насыщенным раствором хлорида натрия (20 мл), сушили (MgSO4), отфильтровывали и концентрировали с получением N-трет-бутил 4-(метилсульфонил)пиперидин-1-карбоксилата: 1 Н ЯМР (400 МГц, CDCl3)4,32 (s, 2 Н), 2,99 (tt, J=3,6, 12,4 Гц, 1 Н), 2,86 (s, 3H), 2,77 (m, 2H), 2,14 (d, J=14,0 Гц, 2H), 1,73 (m, 2H),1,48 (s, 9H). MS m/z для (М-С 3 Н 9+Н)+ C7H14NO4S рассчит. 208,1, обнаруж. 208,1. Стадия В. N-трет-Бутил 4-(метилсульфонил)пиперидин-1-карбоксилат (0,77 г, 2,91 ммоль) растворяли в HCl (10 мл 4 N в диоксане) и перемешивали при КТ в течение 2 ч. Реакционную смесь концентрировали с получением гидрохлорида 4-(метилсульфонил)пиперидина 19, который использовали непосредственно на стадии Г без дополнительной очистки. MS m/z для (М+Н)+ C6H14NO2S рассчит. 164,1, обнаруж. 164,1. Стадия Г. Гидрохлорид 4-(метилсульфонил)пиперидина (19 мг, 0,09 ммоль), 2-(4-(2-хлор-4(хлорметил)фенокси)пиперидин-1-ил)-5-этилпиримидин 21 (23 мг, 0,06 ммоль) и Cs2CO3 нагревали при 60 С в AcN в течение 12 ч. Смесь охлаждали, отфильтровывали, концентрировали и очищали ВЭЖХ с обратной фазой (градиент H2O/AcN) с получением указанного в заголовке соединения Е 5 в виде белого твердого вещества: 1 Н ЯМР (400 МГц, CDCl3)8,11 (s, 2H), 7,27 (d, J=2,0 Гц, 1 Н), 7,10 (dd, J=2,0, 8,4 Гц,1H), 6,88 (d, J=8,4 Гц, 1H), 4,53 (m, 1H), 4,02 (m, 2H), 3,70 (m, 2H), 3,46 (m, 2H), 3,07 (m, 2H), 2,78 (m, 4H),2,39 (q, J=7,6 Гц, 2H), 1,94 (s, 3H), 1,93 (m, 2H), 1,84 (m, 2H), 1,12 (t, J=7,6 Гц, 3H); MS m/z для (М+Н)+C7H15N2O4S рассчит. 223,1, обнаруж. 223,1. Стадия Б. Той же методикой, как описано в примере Е 4 для стадии Е, но используя соединение 26 в качестве исходного материала, получали соединение 27. MS m/z для (М+Н)+ C6H15N2O2S рассчит. 179,1,обнаруж. 179,1. Стадия В. Той же методикой, как описано в примере Е 6 для стадии В, но используя соединение 27 в качестве исходного материала, получали указанное в заголовке соединение Е 7: 1 Н ЯМР (400 МГц,CDCl3)8,11 (s, 2H), 7,28 (d, J=2,0 Гц, 1 Н), 7,07 (dd, J=2,0, 8,4 Гц, 1H), 6,87 (d, J=8,4 Гц, 1H), 4,52 (m, 1H),4,02 (m, 3H), 3,70 (m, 2H), 3,37 (m, 4H), 2,79 (s, 3H), 2,39 (q, J=7,6 Гц, 2 Н), 1,92 (m, 2H), 1,93 (m, 2H), 1,31 ную смесь разбавляли насыщенным раствором NaHCO3 (10 мл) и экстрагировали EtOAc (20 мл). Органический слой сушили (MgSO4), отфильтровывали и концентрировали с получением 2-хлор-4-(1(метилсульфонил)пиперидин-4-иламино)фенола 26, который использовали на следующей стадии без дополнительной очистки. MS m/z для (М+Н)+ C12H18ClN2O3S рассчит. 305,1, обнаруж. 305,1. Стадия Б. Той же методикой, как описано в примере А 1 для стадии Г, но используя 2-хлор-4-(1(метилсульфонил)пиперидин-4-иламино)фенол 26 в качестве фенола, получали N-(3-хлор-4-(1-(5 этилпиримидин-2-ил)пиперидин-4-илокси)фенил)-1-(метилсульфонил)пиперидин-4-амин 27: 1 Н ЯМРAcOH/EtOH (5 мл) и нагревали при 80 С в течение 1 ч. Смесь охлаждали, затем добавляли NaBH3CN (60 мг, 0,95 ммоль) и смесь перемешивали в течение 2 ч при КТ. Смесь разбавляли насыщенным растворомNaHCO3 (10 мл) и экстрагировали EtOAc (20 мл). Органический слой промывали насыщенным раствором хлорида натрия (10 мл), сушили (MgSO4), отфильтровывали, концентрировали и очищали ВЭЖХ с обратной фазой (градиент H2O/AcN) с получением указанного в заголовке соединения Е 8 в виде белого твердого вещества: 1 Н ЯМР (400 МГц, CDCl3)8,44 (s, 2 Н), 7,19 (d, J=2,8 Гц, 1 Н), 7,10 (dd, J=2,8, 8,8 Гц,1 Н), 7,00 (d, J=8,8 Гц, 1 Н), 4,66 (m, 1 Н), 4,18 (m, 2 Н), 3,98 (m, 4 Н), 3,61 (m, 1 Н), 2,98 (s, 3H), 2,84 (s, 3H),2,80 (td, J=2,0, 12,0 Гц, 2 Н), 2,60 (q, J=7,6 Гц, 2 Н), 2,05 (m, 6 Н), 1,83 (m, 2 Н), 1,27 (t, J=7,6 Гц, 3H); MS m/z для (М+Н)+ C24H35ClN5O3S рассчит. 508,2, обнаруж. 508,1. Пример Е 9. 4-(3-Хлор-4-(1-(5-этилпиримидин-2-ил)пиперидин-4-илокси)бензил)пиперазин-1 карбонитрил(100 мг, 0,22 ммоль), цианобромид (26 мг, 0,24 ммоль) и K2CO3 (66 мг, 0,48 ммоль) растворяли в смеси 1:1 H2O и ДХМ (8 мл) и перемешивали при КТ в течение 12 ч. Смесь экстрагировали ДХМ (5 мл) и органический слой сушили (MgSO4), отфильтровывали, концентрировали и очищали ВЭЖХ с обратной фазой (градиент H2O/AcN) с получением указанного в заголовке соединения Е 9 в виде белого твердого вещества. MS m/z для (М+Н)+ C23H30ClN6O рассчит. 441,2, обнаруж. 441,1. Пример Е 10. 2-(4-(2-Хлор-4-4-(2-метил-2 Н-тетразол-5-ил)пиперазин-1-ил)метил)фенокси)пиперидин-1-ил)-5-этилпиримидин Стадия А. 4-(3-Хлор-4-(1-(5-этилпиримидин-2-ил)пиперидин-4-илокси)бензил)пиперазин-1 карбонитрил Е 9 (97 мг, 0,22 ммоль), азид натрия (38 мг, 0,59 ммоль) и NH4Cl (39 мг, 0,73 ммоль) растворяли в ДМФА (2 мл) и нагревали при 90 С в течение 12 ч. Смесь охлаждали, разбавляли H2O (10 мл) и экстрагировали EtOAc (20 мл). Органический слой промывали H2O (10 мл) и насыщенным раствором хлорида натрия (10 мл), сушили (MgSO4), отфильтровывали, концентрировали и очищали ВЭЖХ с обратной фазой (градиент H2O/AcN) с получением 2-(4-(4-4-(2 Н-тетразол-5-ил)пиперазин-1-ил)метил)-2 хлорфенокси)пиперидин-1-ил)-5-этилпиримидина в виде белого твердого вещества: MS m/z для (М+Н)+C23H31ClN9O рассчит. 484,2, обнаруж. 484,1. Стадия Б. 2-(4-(4-4-(2 Н-Тетразол-5-ил)пиперазин-1-ил)метил)-2-хлорфенокси)пиперидин-1-ил)-5 этилпиримидин (23 мг, 0,047 ммоль), K2CO3 (50 мг) и йодметан (3,5 мкл, 0,056 ммоль) растворяли в ацетоне (2 мл) и перемешивали при КТ в течение 3 ч. Смесь отфильтровывали, концентрировали и очищали ВЭЖХ с обратной фазой (градиент H2O/AcN) с получением указанного в заголовке соединения Е 10 в виде белого твердого вещества: 1 Н ЯМР (400 МГц, CDCl3)8,45 (s, 2 Н), 7,43 (d, J=2,0 Гц, 1 Н), 7,37 (dd,J=2,4, 8,4 Гц, 1H), 7,03 (d, J=8,4 Гц, 1H), 4,81 (m, 1H), 4,26 (m, 2H), 4,20 (m, 4H), 3,93 (m, 3H), 2,61 (q,J=7,6 Гц, 2 Н), 2,07 (m, 4H), 1,28 (t, J=7,6 Гц, 3H); MS m/z для (М+Н)+ C24H33ClN9O рассчит. 498,2, обнаруж. 498,1.
МПК / Метки
МПК: C07D 401/14, C07D 211/44, A61P 3/10, A61K 31/497, A61P 3/04, C07D 401/04
Метки: активности, качестве, соединения, модуляторов, композиции, gpr119
Код ссылки
<a href="https://eas.patents.su/30-18703-soedineniya-i-kompozicii-v-kachestve-modulyatorov-aktivnosti-gpr119.html" rel="bookmark" title="База патентов Евразийского Союза">Соединения и композиции в качестве модуляторов активности gpr119</a>
4-феноксиметилпиперидины в качестве модуляторов активности gpr119

Номер патента: 17260
Опубликовано: 30.11.2012
Авторы: Уэсткотт-Бейкер Лукас, Эппле Роберт, Леле Жеральд, Никулин Виктор
МПК: A61K 31/4523, C07D 211/30, A61P 3/10...
Метки: качестве, gpr119, 4-феноксиметилпиперидины, активности, модуляторов
Формула / Реферат:
1. Соединение формулы Iгде R1 выбран из фенила, пиридин-2-ила, пиридин-3-ила, пиридин-4-ила, пиримидин-5-ила, пиразол-4-ила, 1H-пиразол-4-ила, 6-оксо-1,6-дигидропиридин-3-ила, 2-оксо-1,2-дигидропиримидин-5-ила, 2-оксо-1,2-дигидропиридин-4-ила и 2-оксо-2,3-дигидрооксазоло[4,5-b]пиридин-6-ила; где указанный фенил, пиридин-2-ил, пиридин-3-ил, пиридин-4-ил, пиримидин-5-ил, пиразол-4-ил, 1H-пиразол-4-ил, 6-оксо-1,6-дигидропиридин-3-ил,...
Соединения и композиции в качестве модуляторов активности tlr

Номер патента: 18068
Опубликовано: 30.05.2013
Авторы: Ли Йонкай, Чжан Сяою, Мишра Пранаб, Зоу Йэфень, Валианте Николас, Сингх Манмохан, Кортес Алекс, Ву Том Яосянь, Скибински Дейвид
МПК: C07D 221/12, A61K 31/4375, A61P 37/04...
Метки: композиции, соединения, качестве, модуляторов, активности
Формула / Реферат:
1. Соединение формулы (I-A) или его фармацевтически приемлемая сольформула (I-A)где X3 обозначает N;X4 обозначает CR3;X5 обозначает -CR4=CR5-;R3 обозначает H;R4 обозначает H;R5 выбирают из галогена, -C(O)OR7, -N(R9)2, -NHN(R9)2, -(CH2)nR7, -LR10, C1-C6-алкила, C1-C6-галогеналкила, C2-C8-алкенила, C2-C8-алкинила и C1-C6-алкоксигруппы, причем указанный C1-C6-алкил, C2-C8-алкенил и C2-C8-алкинил в составе R5, каждый необязательно, замещен 1-3...
Соединения и композиции в качестве ингибиторов активности каннабиноидного рецептора 1

Номер патента: 17696
Опубликовано: 28.02.2013
Авторы: Ван Син, Чжу Сюэфэн, Ян Куньюн, Хе Сяосюй, Нгуэн Трук Нгок, Ло Томас, Филлипс Дин, Ву Баогэнь, Лю Хон, Сиэ Юнпин
МПК: C07D 233/16, C07D 207/40, C07D 233/12...
Метки: соединения, качестве, ингибиторов, активности, рецептора, каннабиноидного, композиции
Формула / Реферат:
1. Соединение формулы Iили его фармацевтически приемлемая соль, в которойY1 выбран из N и CR11;Y2 выбран из N и CR8;R1a выбран из циано, замещенного цианогруппой C1-6алкила, -X1R12, -X1NR13S(O)2R13, -X1OS(O)2R13, -X1NR13X1OR13, -X1OR13, -X1C(O)OR13, -X1S(O)2R12, -X1S(O)2NR13C(O)R13, -X1S(O)2R13, -X1C(O)R12, -X1R13R13, -X1S(O)2NR13R13, -X1OC(O)NR13R13, -X1C(O)NR12R13, -X1NR13X1C(O)R12, -X1NR13X1C(O)NR13R13, -X1C(O)NR13X1C(O)OR13,...
N-уреидоалкилпиперидины в качестве модуляторов активности рецепторов хемокинов

Номер патента: 3147
Опубликовано: 27.02.2003
Авторы: Ким Уи Тае, Сантелла Джозеф Б.III, Вэкер Дин А.К., Делукка Джордж В., Ко Соо С., Данкиа Джон В.
МПК: A61K 31/445, A61P 11/06, C07D 211/32...
Метки: рецепторов, качестве, n-уреидоалкилпиперидины, хемокинов, активности, модуляторов
Формула / Реферат:
1. Соединение формулы (I) или его стереоизомеры или фармацевтически приемлемые соли, где M отсутствует или выбран из CH2, CHR5, CHR13, CR13R13 и CR5R13; Q выбран из CH2, CHR5, CHR13, CR13R13 и CR5R13; J и K выбраны из CH2, CHR5, CHR6, CR6R6 и CR5R6; L представляет CHR5; при условии, что если M отсутствует, то J выбран из CH2, CHR5, CHR13 и CR5R13; Z выбран из O и S; E выбран из кольцо A представляет C3-6карбоциклический остаток, при условии,...
Соединения в качестве модуляторов пути hedgehog

Номер патента: 18302
Опубликовано: 30.07.2013
Авторы: Хань Дун, У Сюй, Гао Вэнци, Чэн Дай, Ван Юнцинь, Пан Шифенг, Цзян Цзицин
МПК: A61P 35/00, C07C 233/65, A61K 31/4433...
Метки: качестве, соединения, модуляторов, hedgehog, пути
Формула / Реферат:
1. Соединение формулы Iгде Y1 и Y2 независимо выбраны из N и СН;R1 выбран из циано, С1-6алкила, галогензамещенного С1-6алкила, С1-6алкокси, галогензамещенного С1-6алкокси, диметиламино, С1-6алкилсульфанила и пиперазинила, необязательно замещенного не более чем двумя C1-6алкильными радикалами;R2 и R5 независимо выбраны из водорода, циано, галогена, С1-6алкила, галогензамещенного С1-6алкила и С1-6алкокси;R3 и R4 независимо выбраны из водорода,...
Предыдущий патент: Аппарат биомеханический ротационно-корригирующий для нижних конечностей
Следующий патент: Новые формы метилового эфира 2-циано-3,12-диоксоолеан-1,9(11)-диен-28-овой кислоты (сddo-me)
Случайный патент: Малокалиберная деформационная пуля и способ ее изготовления