Производные карболина, полезные в ингибировании ангиогенеза
Номер патента: 16575
Опубликовано: 30.06.2012
Авторы: Хванг Сеонгвоо, Моон Йоунг-Чоон, Леннокс Уилльям Джозеф, Цао Лянсянь, Корсон Дональд Томас, Ци Хунянь, Тамиларасу Надараджан, Чой Соонгиу







Формула / Реферат
1. Соединение формулы (IV)

или фармацевтически приемлемая соль, рацемат или стереоизомер указанного соединения,
в которой X обозначает водород; С1-С6 алкил, необязательно замещенный одним или более галогенами; гидроксильную группу; галоген; C1-C5 алкокси, необязательно замещенный фенилом;
R0 обозначает галоген; циано; нитро; сульфонил, причем сульфонил может быть замещен C1-C6 алкилом или морфолинилом; аминогруппу, причем аминогруппа может быть замещена C1-С6 алкилом,
-C(O)-Rb, -C(O)O-Rb, алкилсульфонилом, морфолинилом или тетрагидропиранилом; C1-С6 алкил, причем алкил может быть замещен по меньшей мере одним независимо выбранным гидроксилом, галогеном или амино, группой -C(O)-Rn или группой -ORa;
Ra обозначает водород; С2-С8 алкенил; группу -C(O)O-Rb; -C(O)-NH-Rb; C1-C8 алкил, причем
алкил может быть замещен по меньшей мере одним независимо выбранным гидроксилом, галогеном,
C1-C4 алкокси, C1-C4 алкокси-С1-С4 алкокси, амино, алкиламино, диалкиламино, ацетамидо, -С(О)-Rb,
-C(O)O-Rb, арилом, морфолинилом; тиоморфолинил, пирролидинил, пиперидинил, пиперазинил,
1,3-диоксолан-2-он, оксиранил, тетрагидрофуранил, тетрагидропиранил, 1,2,3-триазол, 1,2,4-триазол, фуран, имидазол, изоксазол, изотиазол, оксазол, пиразол, тиазол, тиофен или тетразол;
где аминогруппа может быть замещена С1-С4 алкоксикарбонилом, имидазолом, изотиазолом, пиразолом, пиридином, пиразином, пиримидином, пирролом, тиазолом, где пиридин и тиазол могут быть необязательно замещены С1-С4 алкилом;
причем алкиламино и диалкиламино могут быть по алкилу замещены гидроксилом, С1-С4 алкокси, имидазолом, пиразолом, пирролом или тетразолом и
причем морфолинил, тиоморфолинил, пирролидинил, пиперидинил, пиперазинил и оксиранил могут быть замещены группами -C(O)-Rn, -C(O)O-Rn или С1-С4 алкил, причем С1-С4 алкил может быть замещен гидроксилом;
Rb обозначает гидроксил; амино; алкиламино, причем алкиламино может быть по алкилу замещен гидроксилом, амино, алкиламино или С1-С4 алкокси; C1-C4 алкокси; С2-С8 алкенил; С2-C8 алкинил; арил, причем арил может быть замещен по меньшей мере одним заместителем, независимо выбранным из галогена и C1-С4 алкокси; фуран или C1-C8 алкил, причем алкил может быть замещен по меньшей мере одним независимо выбранным С1-С4 алкокси, арилом, амино; морфолинил, пиперидинил, пиперазинил;
Rd обозначает фенил; причем фенил может быть замещен по меньшей мере одним независимо выбранным галогеном, нитрогруппой, C1-С6 алкилом, -C(O)O-Re или -ORe;
Re обозначает водород; C1-С6 алкил, причем алкил может быть замещен по меньшей мере одним независимо выбранным галогеном или алкоксигруппой; или фенил, причем фенил может быть замещен по меньшей мере одним независимо выбранным галогеном или алкоксигруппой; и
Rn обозначает гидроксил, C1-C4 алкокси, амино или C1-C6 алкил.
2. Соединение по п.1, в котором
X обозначает C1-С6 алкил, необязательно замещенный одним или более галогенами; галоген;
C1-C5 алкокси, необязательно замещенный фенилом;
R0 обозначает галоген; циано; нитро; сульфонил, замещенный C1-C6 алкилом или морфолинилом; аминогруппу, причем аминогруппа может быть замещена C1-С6 алкилом, -C(O)-Rb, -C(O)O-Rb, алкилсульфонилом и тетрагидропиранилом; C1-C6 алкил, причем алкил может быть замещен по меньшей мере одним независимо выбранным галогеном; группой -C(O)-Rn или группой -ORa;
Ra обозначает водород; С2-С8 алкенил; группу -C(O)O-Rb; C1-C8 алкил, причем алкил может быть замещен по меньшей мере одним независимо выбранным гидроксилом, галогеном, C1-C4 алкокси,
C1-C4 алкокси-С1-С4 алкокси, амино, алкиламино, диалкиламино, ацетамидо, -C(O)-Rb, -C(O)O-Rb, арилом, морфолинилом, тиоморфолинилом, пирролидинилом, пиперидинилом, пиперазинилом,
1,3-диоксолан-2-оном, оксиранилом, 1,2,3-триазолом, 1,2,4-триазолом, имидазолом или пиразолом;
где аминогруппа может быть замещена С1-С4 алкоксикарбонилом, пиридином и тиазолом, необязательно замещеными С1-С4 алкилом;
причем алкиламино и диалкиламино могут быть по алкилу замещены гидроксилом, C1-C4 алкокси или имидазолом и
причем морфолинил, тиоморфолинил, пирролидинил, пиперидинил, пиперазинил и оксиранил могут быть замещены группами -C(O)-Rn, -C(O)O-Rn или С1-С4 алкил, причем С1-С4 алкил может быть замещен гидроксилом;
Rb обозначает гидроксил; С1-С4 алкокси; С2-С8 алкенил; фенил, причем фенил может быть замещен по меньшей мере одним заместителем, независимо выбранным из галогена; фуран или C1-С8 алкил, причем алкил может быть замещен по меньшей мере одним независимо выбранным C1-C4 алкокси, фенилом, амино или морфолинилом; и
Rd обозначает фенил; причем фенил может быть замещен по меньшей мере одним независимо выбранным галогеном, нитрогруппой, С1-С6 алкилом или -ORe; где все остальные заместители определены выше.
3. Соединение по п.2, в котором
X обозначает C1-C6 алкил; галоген; C1-C5 алкокси, необязательно замещенный фенилом;
R0 обозначает галоген; циано; нитро; сульфонил, замещенный C1-С6 алкилом или морфолинилом; аминогруппу, причем аминогруппа может быть замещена -C(O)-Rb, -C(O)O-Rb, алкилсульфонилом и тетрагидропиранилом; C1-C6 алкил, причем алкил может быть замещен по меньшей мере одним галогеном; группой -C(O)-Rn или группой -ORa;
Ra обозначает водород; С2-С8 алкенил; группу -C(O)O-Rb; C1-C8 алкил, причем алкил может быть замещен по меньшей мере одним независимо выбранным гидроксилом, галогеном, С1-С4 алкокси,
С1-С4 алкокси-С1-С4 алкокси, амино, алкиламино, диалкиламино, ацетамидо, -C(O)O-Rb, морфолинилом, тиоморфолинилом, пирролидинилом, пиперидинилом, пиперазинилом, 1,3-диоксолан-2-оном, оксиранилом, 1,2,3-триазолом, 1,2,4-триазолом, имидазолом или пиразолом; где аминогруппа может быть замещена C1-C4 алкоксикарбонилом, пиридином и тиазолом, где пиридин и тиазол, каждый, необязательно могут быть замещены С1-С4 алкилом;
причем алкиламино и диалкиламино могут быть по алкилу замещены гидроксилом, С1-С4 алкокси или имидазолом и
причем морфолинил, тиоморфолинил, пирролидинил, пиперидинил, пиперазинил и оксиранил могут быть замещены группами -C(O)-Rn, -C(O)O-Rn или С1-С4 алкил, причем С1-С4 алкил может быть замещен гидроксилом;
Rb обозначает гидроксил; С1-С4 алкокси; С2-С8 алкенил; фенил, причем фенил может быть замещен по меньшей мере одним заместителем, независимо выбранным из галогена; фуран или С1-С8 алкил, причем алкил может быть замещен по меньшей мере одним независимо выбранным С1-С4 алкокси или морфолинилом;
и где все остальные заместители определены выше.
4. Соединение по любому из пп.1-3, в котором указанное соединение или его фармацевтически приемлемая соль имеет хиральный атом углерода в месте присоединения фенила, замещенного R0, в соединении формулы (IV) и указанное соединение представляет собой (S) изомер по указанному хиральному атому аглерода.
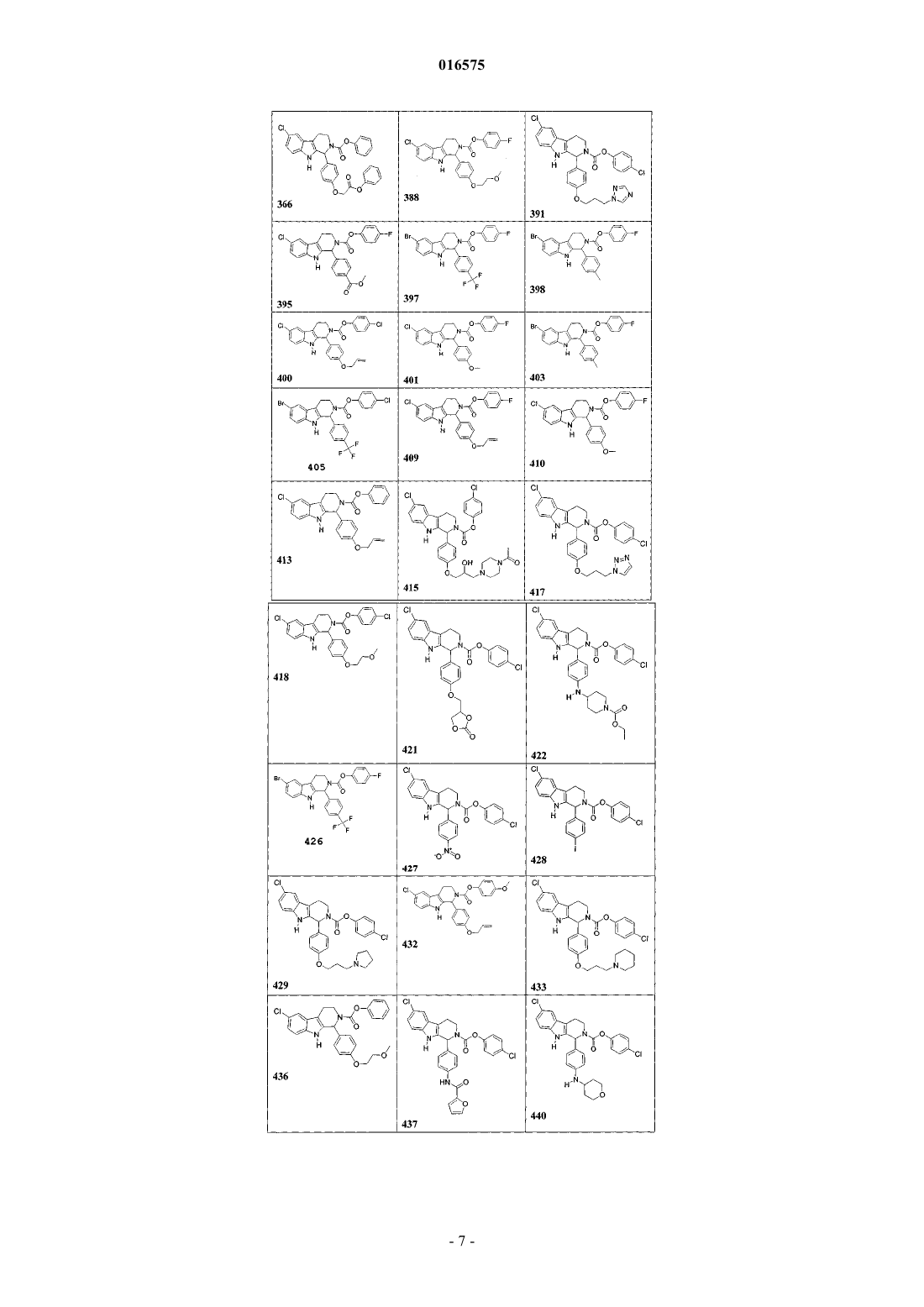
5. Соединение, причем указанное соединение представлено следующей структурой, выбранной из группы состоящей из

















или фармацевтически приемлемая соль, рацемат или стереоизомер указанного соединения.
6. Соединение по п.5, в котором указанное соединение или его фармацетически приемлемая соль имеет структурообразующую молекулу карболина и хиральный атом углерода в месте присоединения необязательно замещенного фенильного кольца к указанной структурообразующей молекуле и где указанное соединение представляет собой (S) изомер по указанному хиральному атому углерода.
7. Соединение по п.6, в котором указанное соединение выбирают из группы состоящей из

или его фармацевтически приемлемая соль.
8. Соединение по п.7, в котором указанное соединение представляет собой

или его фармацевтически приемлемую соль.
9. Соединение по п.7, в котором указанное соединение представляет собой

или его фармацевтически приемлемую соль.
10. Соединение по п.7, в котором указанное соединение представляет собой

или его фармацевтически приемлемую соль.
11. Соединение по п.7, в котором указанное соединение представляет собой

или его фармацевтически приемлемую соль.
12. Соединение по п.7, в котором указанное соединение представляет собой

или его фармацевтически приемлемую соль.
13. Соединение по п.7, в котором указанное соединение представляет собой

или его фармацевтически приемлемую соль.
14. Соединение по п.7, в котором указанное соединение представляет собой

или его фармацевтически приемлемую соль.
15. Фармацевтическая композиция, содержащая соединение по любому из пп.1-6 или его фармацевтически приемлемую соль, рацемат или стереоизомер и фармацевтически приемлемый эксципиент.
16. Фармацевтическая композиция, содержащая соединение по любому из пп.7-14 или его фармацевтически приемлемую соль, рацемат и фармацевтически приемлемый эксципиент.
Текст
В соответствии с настоящим изобретением были идентифицированы соединения общей формулыIV, которые посттранскрипционно ингибируют экспрессию VEGF. Изобретение относится к соединениям, которые являются полезными в ингибировании продуцирования VEGF, в ингибировании ангиогенеза и/или в лечении рака, диабетической ретинопатии или экссудативной дегенерации желтого пятна. 016575 Перекрестная ссылка на родственные заявки По настоящей заявке испрашивается приоритет согласно предварительной заявке на патент США 60/552725, поданной 15 марта 2004 г., которая включена в настоящую заявку путем ссылки. Область изобретения Настоящее изобретение относится к соединениям для ингибирования ангиогенеза. Уровень техники Аберрантный ангиогенез играет критическую роль в патогенезе многочисленных заболеваний,включая злокачественные, ишемические, воспалительные и иммунные нарушения (Carmeliet, Nat. Med.,9 (6): 653-60 (2003), Ferrara, Semin. Oncol., 29 (6 Suppl 16): 10-4 (2002. Самые известные из этих нарушений - рак, экссудативная дегенерация желтого пятна и диабетическая ретинопатия (DR), последние два из которых являются ведущей причиной слепоты в Соединенных Штатах (Witmer et al., Prog. RetinEye Res., 22(1): 1-29 (2003), Clark et al., Nat. Rev. Drug Discovery, 2: 448-459 (2003. В течение прошлого десятилетия понимание молекулярной базы ангиогенеза значительно выросло. Были идентифицированы многочисленные цитокины и факторы роста, которые стимулируют ангиогенез, такие как VEGF, FGF-2,PDGF, IGF-1, TGF, TNF-, G-CSF (Ferrara et al., Nat. Med., 5 (12): 1359-64 (1999), Kerbel et al., Nat. Rev.Cancer, 2 (10): 727-39 (2002), Rofstad et al., Cancer Res., 60 (17): 4932-8 (2000. Среди этих факторов роста сосудистый эндотелиальный фактор роста (VEGF) играет центральную роль в ангиогенезе (Ferrara,Semin., Oncol., 29 (6 Suppl 16): 10-4 (2002.VEGF, также известный как VEGF-A, был первоначально идентифицирован в отношении способности индуцировать проницаемость сосудов и вызывать пролиферацию клеток эндотелия сосудов (LeungChem., 264:20017-20024 (1989. VEGF кодируется отдельным геном, который дает начало четырем изоформам путем альтернативного сплайсинга (Tischer et al., J. Biol. Chem., 266: 11947-11954 (1991. Все четыре изоформы используют одну и ту же необычно длинную и богатую парами GC 5'-UTR, а также 3'UTR, которая включает многократные детерминанты стабильности РНК. Рецепторы VEGFR-2 (также известный как KDR или Flk-1) и VEGFR-1 (ранее известный как Fltl) распознают димерную форму VEGF(Ortega et al., Front. Biosci., 4:D141-52 (1999), Sato et al., Annals of New York Academy of Science, 902:201207, (2000. Высокоспецифичный рецептор VEGFR-2 экспрессируется на эндотелиальных клетках.VEGF, связывающийся с рецептором VEGFR-2, активирует активность тирозинкиназы рецептора, приводя к пролиферации клеток эндотелия, дифференцировке и примитивному формированию сосуда(Shalaby et al., Nature, 376: 62-66 (1995. VEGFR-1 ингибирует рост клеток эндотелия, либо действуя как приманка, либо подавляя сигнальные пути через VEGFR-2 (Fong et al., Nature, 376: 66-70 (1995. Более 30 лет назад было сделано предположение, что ингибирование ангиогенеза опухоли могло бы быть эффективным подходом для лечения рака (Folkman, N. Engl. J. Med., 285 (21): 1182-6 (1971. Было показано, что VEGF и его рецептор играют центральную роль в ангиогенезе опухоли, особенно в ранних стадиях роста опухоли (Hanahan et al., Cell, 86:353-364, 1996. Действительно, увеличенные уровни экспрессии VEGF коррелировали с плотностью микрососудов в первичных тканях опухоли (Gasparini et al.,J. Natl. Cancer Inst., 89:139-147 (1997. Кроме того, увеличенные уровни транскрипта VEGF обнаружены фактически во всех обычных солидных опухолях (Ferrara et al., Endocr. Rev., 18:4-25, 1997. В общем,онкологические пациенты имеют более высокое уровни VEGF по сравнению с их уровнями у людей без опухоли, и высокие уровни VEGF в сыворотке/плазме были связаны с неблагоприятным прогнозомVEGF-отрицательные зародышевые стволовые клетки показали резко сниженную способность образовывать опухоли у голых мышей (Carmeliet et al., Nature, 380:435-439 (1996. Прямое свидетельство участия VEGF в туморогенезе было продемонстрировано при использовании антител, специфичных кVEGF, в человеческих ксенотрансплантатах, внедренных голым мышам (Kim et al., Nature, 362:841-844(1993), Hichlin et al., Drug Discovery Today, 6:517-528 (2001. В этих исследованиях ингибирование роста опухоли положительно коррелировало со сниженным формированием сосудов в обработанных антителами опухолях. Последующие эксперименты, в которых использовали растворимые рецепторы, доказали важность активности VEGF в росте опухоли (Lin et al., Cell Growth Differ., 9(1): 49-58 (1998 и продемонстрировали, что инактивация VEGF обработкой специфичными антителами непосредственно привела к почти полной супрессии связанной с опухолью неоваскуляризации (Borgstrom et al., Prostate, 35:1-10(1998), Yuan et al., Proc. Natl. Acad. Sci. USA, 93:14765-14770 (1996. В отношении экссудативной дегенерации желтого пятна и диабетической ретинопатии, доклинические эксперименты и клинические испытания продемонстрировали, что сверхпродуцирование VEGF является критической для аберрантной ретинальной или хориоидальной неоваскуляризации (рассматривается в Witmer et al., Prog Retin Eye Res., 22(1):1-29 (2003. Было с очевидностью показано, что внутриглазные уровни VEGF сильно коррелируют с активной ретинальной/хориоидальной неоваскуляризацией(CNV) у больных с такими заболеваниями, как диабетическая ретинопатия и влажная дегенерация желтого пятна (Funatsu et al., Am. J. Ophthalmol., 133 (4):537-43 (2002), Lip et al., Ophthalmology, 108(4): 70510 (2001. Кроме того, исследования с использованием трансгенных мышей продемонстрировали, что суперэкспрессия VEGF в ретинальных пигментных эпителиоцитах или клетках зрительного рецептора-1 016575 приводит к хориоидальной или ретинальной неоваскуляризации (Schwesinger et al., Am. J. Pathol., 158(3): 1161-72 (2001), Ohno-Matsui et al., Am. J. Pathol., 160(2): 711-9 (2002. В недавних исследованиях нейтрализирующие антитела, растворимый рецептор, антагонисты рецептора или siRNA оказались эффективными в ослаблении VEGF-опосредованного формирования кровеносных сосудов в моделях животных и в клинике (Eyetech Study Group, 22(2): 143-52 (2002), Krzystolik et al., Arch. Ophthalmol., 120(3): 338-46al., J. Cell. Physiol., 195(2): 241-8 (2003. Экспрессия VEGF регулируется множеством факторов и средств, включая цитокины, факторы роста, стероидные гормоны и химические вещества, и мутации, которые модулируют активность онкогенов,таких как ras или ген супрессора опухоли VHL (Maxwell et al., Nature, 399: 271-275 (1999), Rak et al., Cancer Res., 60:490-498 (2000. Однако гипоксия представляет собой самый значительный физиологический сигнал для регуляции экспрессии VEGF. Гипоксия приводит к увеличенной экспрессии VEGF, увеличивая как уровень транскрипции, так и стабильность транскрипта VEGF (Ikeda et al., J. Biol. Chem. 270: 19761-19766 (1995), Stein et al., Mol. Cell. Biol. 18:3112-3119 (1998), Levy et al., J. Biol. Chem. 271:27462753 (1996. Индуцируемый гипоксией фактор 1 (HIF-1) представляет собой транскрипционный фактор, который усиливает экспрессию гена VEGF в клетках, испытывающих гипоксию, путем связывания с элементами ответа на гипоксию (HRE), расположенными в промоторе VEGF (Liu et al., Circ. Res., 77:638643 (1995), Semenza, Annu. Rev. Cell. Dev. Biol., 5:551-578 (1999. Стабильность мРНК VEGF также значительно увеличивается вследствие связывания факторов с элементами в 3'-UTR (Goldberg et al., J. Biol.Cell. J. Biol. Chem., 277 (16):13635-40 (2002. Кроме того, инициация трансляции транскрипта VEGF уникально регулируется. В условиях гипоксии трансляция большинства клеточных транскриптов, опосредуемая сар-зависимым процессом инициации трансляции в значительной степени ухудшается (Kraggerud et al., Anticancer Res., 15:683-686 (1995. Тем не менее, инициация трансляции мРНК VEGF является в условиях гипоксии уникальной в том, что она опосредуется через внутренний сайт посадки рибосомы (IRES) в составе 5'UTR VEGF (Stein et al., Mol. Cell. Biol. 18:3112-3119 (1998), Levy et al., J. Biol.Chem. 271:2746-2753 (1996), Huez et al., Mol. Cell. Biol., 18:6178-6190 (1998), Akiri et al., Oncogene,17:227-236 (1998. Многочисленные эксперименты наглядно показали, что рост опухоли может ингибироваться путем предупреждения неоваскуляризации (Lin et al., Cell Growth Differ., 9(1):49-58 (1998), Zhu et a., Invest. NewDrugs, 17:195-212 (1999. Сосуды опухоли обычно являются незрелыми, подвергаются постоянной перестройке (Carmeliet, Nat. Med., 9(6):653-60 (2003), Carmeliet et al., Nature, 407:249-257 (2000. Активный и аберрантный ангиогенез является результатом нарушения в нормальном балансе проангиогенных и антиангиогенных факторов, включая различные цитокины, факторы роста и стероидные гормоны. Несмотря на сложность регулирования ангиогенеза опухоли, накопленные свидетельства указывают на то, что нацеливание единственного проангиогенного фактора могло бы быть достаточно, чтобы ингибировать ангиогенез опухоли и подавить рост опухоли (Kim et al., Nature, 362:841-844 (1993), Millauer et al., Nature,367:576-579 (1994), Fong et al., Cancer Res., 59:99-106 (1999. Среди многих мишеней ангиогенеза, VEGF и его рецептор являются самыми привлекательными (Carmeliet, Nat. Med., 9(6): 653-60 (2003), Ortega etal., Front. Biosci., 4: D141-52 (1999. Как отмечено выше, лечение с использованием моноклонального антитела, специфически нацеливающего VEGF, ингибирует рост опухолей в человеческих ксенотрансплантатах, введенных голым мышам. Впоследствии различные подходы, имевшие целью инактивировать передачу сигналов VEGF, были проверены в моделях опухоли и оказались очень эффективными в широком диапазоне линий опухолевых клеток, включая различные виды рака, саркомы и глиомы (Ferrara et(1994), Fong et al., Cancer Res., 59: 99-106 (1999), Geng et al., Cancer Res., 61: 2413-2419 (2001. Кроме того, ингибирование VEGF анти-VEGF антителом не приводило к значительным побочным эффектам у полностью развившихся грызунов или приматов (Ryan et al, Toxicol. Pathol., 27: 78-86 (1999), Ferrara etal., Nat. Med., 4: 336-340 (1998. Вместе эти результаты указывают, что VEGF представляет собой действительную цель для разработки терапии опухоли. Действительно, во множестве клинических испытаний в стадии осуществления используют ингибиторы VEGF (Matter, Drug Discovery Today, 6: 1005-1024(2001), Hichlin et al., Drug Discovery Today, 6: 517-528 (2001. Хотя несколько проангиогенных факторов участвуют в патологии экссудативной возрастной дегенерации желтого пятна, VEGF представляется наиболее значимым в патогенезе и развитии этого заболевания (Witmer et al., Prog. Retin Eye Res., 22(1): 1-29 (2003), Holash et al., Science, 284: 1994-1998 (1999. Данные от доклинических экспериментов и клинических испытаний продемонстрировали, что блокада одного только VEGF достаточна, чтобы облегчить или стабилизировать развитие заболевания (EyetechStudy Group, 22(2): 143-52 (2002), Krzystolik et al., Arch. Ophthalmol., 120 (3): 338-46 (2002), Shen et al.,Lab Invest., 82(2): 167-82 (2002), Honda et al., Gene Ther., 7(11): 978-85 (2000), Saishin et al., Cell. Physiol.,195(2): 241-8 (2003. Например, ингибирование передачи сигналов VEGFR специфическим ингибитором тирозинкиназы достаточно, чтобы полностью предотвратить ретинальную неоваскуляризацию в мышиной модели преждевременной ретинопатии (Ozaki H., Seo MC, Ozaki et al., Am. J. Pathol., 156(2): 697-707(2000. Кроме того, недавно было продемонстрировано, что короткие интерферирующие РНК (siRNA),-2 016575 направленные к мышиному VEGF, значительно ингибировали глазную неоваскуляризацию после лазерной фотокоагуляции на модели мыши (Reich et al., Mol. Vis. 30; 9:210-6 (2003. Эти результаты показывают, что селективное ингибирование экспрессии VEGF может быть достигнуто и подтверждает приемлемость этого подхода для лечения заболеваний, связанных с глазной неоваскуляризацией, таких как экссудативная дегенерация желтого пятна и диабетическая ретинопатия. Три подхода использовались для ингибирования активности VEGF, включая (1) нейтрализацию активности VEGF при использовании специфического антитела, растворимого рецептора VEGF или аптамера олиго против взаимодействия VEGF/VEGFR (Kim et al., Nature, 362:841-844 (1993), Lin et al., Cell(1999), Brekken et al., Cancer Res., 60(18): 5117-24 (2000; (2) ингибирование VEGFR-опосредованной трансдукции сигнала специфическими малыми молекулами ингибиторов тирозинкиназы (Fong et al.,Cancer Res., 59:99-106 (1999), Wedge et al., Cancer Res., 60(4): 970-5 (2000), Laird et al., Cancer Res., 60(15): 4152-60 (2000; и (3) ингибирование экспрессии VEGF/VEGFR при использовании антисмысловой последовательности, siRNA или рибозима (Reich et al., Mol. Vis. 30; 9:210-6 (2003), Parry et al., Nucleic AcidsRes., 27: 2569-2577 (1999), Ellis et al., Surgery, 120: 871-878 (1996), Filleur et al., Cancer Res., 63 (14):391922 (2003. Хотя все эти подходы показывают значительное ингибирование ангиогенеза in vivo, они связаны со значительными ограничениями. Например, терапевтические белки (антитело и растворимые рецепторы) или олигомеры (антисмысловой, siRNA и рибозим) представляют собой большие молекулы со слабой проницаемостью, которые обычно требуют парентерального введения и являются дорогостоящими в получении. Для лечения хронической глазной неоваскуляризации, многократные инъекции могут быть непрактичными вследствие потенциальных осложнений, таких как отслоение сетчатки и связанные с процедурой инфекции. Кроме того, ингибиторы тирозинкиназы имеют потенциально ограниченную специфичность. VEGF конститутивно экспрессируется в небольших количествах в нормальных глазных и других тканях, и таким образом может быть вредно полностью подавить функцию VEGF путем системного введения антитела или ингибиторов тирозинкиназы, особенно в случае пациентов с AMD и RD,многие из которых также являются гипертониками (Giles et al., Cancer, 97(8): 1920-8 (2003), Sugimoto etClinical Oncology 36h Annual Meeting, 20-23 May, 2000, New Orleans, LA, USA, Abstract 1896). Таким образом, сохраняется потребностью в разработке, характеризации и оптимизации молекул для развития новых препаратов против ангиогенеза. Соответственно, задачей настоящего изобретения является получение таких соединений. Все документы, упомянутые здесь, включены в настоящую заявку путем ссылки, как если бы они были полностью раскрыты здесь. Сущность изобретения В соответствии с настоящим изобретением были идентифицированы соединения, которые ингибируют экспрессию VEGF на пост-транскрипционном уровне. В одном аспекте изобретение относится к соединениям формулы (IV), которые являются полезными в ингибировании продуцирования VEGF, в ингибировании ангиогенеза и/или в лечении рака, диабетической ретинопатии или экссудативной дегенерации желтого пятна. В другом аспекте изобретение относится к ингибированию ангиогенеза и/или лечения рака, диабетической ретинопатии, ревматоидного артрита, псориаза, атеросклероза, хронического воспаления, других хронических связанных с воспалением заболеваний и нарушений, ожирения или экссудативной дегенерации желтого пятна, с использованием соединений, описанных здесь. Эти и другие аспекты изобретения будут более понятны в отношении следующих предпочтительных вариантов осуществления и подробного описания. Краткое описание фигур Фиг. 1 иллюстрирует ингибирование экспрессии VEGF определенным соединением изобретения; фиг. 2 показывает, что соединение изобретения не действует на активность фосфодиэстеразы 5(PDE-5). Подробное описание изобретения Аберрантная повышающая регуляция сосудистого эндотелиального фактора роста (VEGF), ключевого фактора для ангиогенеза, является важным компонентом в патогенезе болезненных состояний, таких как рак, диабетическая ретинопатия, ревматоидный артрит, псориаз, атеросклероз, хроническое воспаление, другие хронические связанные с воспалением заболевания и нарушения, ожирение или экссудативная дегенерация желтого пятна. В соответствии с настоящим изобретением были идентифицированы соединения, которые ингибируют экспрессию VEGF на посттранскрипционном уровне, и разработаны способы их применения. Соединения изобретения обладают от наномолярной до субнаномолярной активностью в отношении ингибирования экспрессии VEGF. А. Соединения данного изобретения. В одном аспекте изобретение относится к соединениям, которые являются полезными в ингибиро-3 016575 вании продуцирования VEGF, в ингибировании ангиогенеза и/или в лечении рака, диабетической ретинопатии или экссудативной дегенерации желтого пятна. В некоторых вариантах осуществления соединения изобретения специфически ингибируют продуцирование VEGF, в то время как в других вариантах осуществления соединения изобретения ингибируют экспрессию VEGF, а также экспрессию других факторов ангиогенеза, таких как FGF-2. В этом отношении пан-ангиогенный ингибитор может быть предпочтительным в способах ингибирования роста опухоли, в то время как специфические ингибиторыVEGF могут быть предпочтительными для лечения нарушения, связанного с глазной неоваскуляризацией (Eyetech Study Group, 22(2):143-52 (2002. Соединения изобретения обычно включают один или более хиральных центров и также могут существовать как рацемические смеси (R/S) или как энантиомерно чистые композиции. Соединения могут существовать как (R) или (S) изомеры (когда присутствует один хиральный центр) в энантиомерно чистых композициях. В предпочтительном варианте осуществления соединения изобретения представляют собой (S) изомеры и могут существовать как энантиомерно чистые композиции, включающие только (S) изомер. Специалисту понятно, что когда присутствует более одного хирального центра, соединения изобретения могут существовать как (R,R), (R,S), (S,R), (S,S) и т.д. изомеры. Предпочтительные соединения включают (S,S) и (S,R) изомеры. В рамках изобретения "энантиомерно чистый" относится к композициям, состоящим в основном из индивидуального изомера, предпочтительно состоящим на 90, 92, 95, 98, 99 или 100% из индивидуального изомера. Предпочтительные соединения согласно настоящему изобретению, полезные в ингибировании продуцирования VEGF, включают соединения формулы (I) как показано ниже.X обозначает водород; С 1-С 6 алкил, необязательно замещенный одним или более галогенами; гидроксильную группу; галоген; С 1-С 5 алкокси, необязательно замещенный фенилом; А обозначает N; В обозначает С;R0 обозначает галоген; циано; нитро; сульфонил, причем сульфонил может быть замещен C1-C6 алкилом или морфолинилом; аминогруппу, причем аминогруппа может быть замещена C1-C6 алкилом,-C(O)-Rb, -C(O)O-Rb, алкилсульфонилом, морфолинилом или тетрагидропиранилом; C1-С 6 алкил, причем алкил может быть замещен по меньшей мере одним независимо выбранным гидроксилом, галогеном или амино; группой -C(O)-Rn; или группой -ORa;Ra обозначает водород; С 2-С 8 алкенил; группу -C(O)O-Rb; -C(O)-NH-Rb; С 1-С 8 алкил, причем алкил может быть замещен по меньшей мере одним независимо выбранным гидроксилом, галогеном, C1-C4 алкокси, C1-C4 алкокси-С 1-С 4 алкокси, амино, алкиламино, диалкиламино, ацетамидо, -C(O)-Rb, -C(O)ORb, арилом, морфолинилом, тиоморфолинил, пирролидинил, пиперидинил, пиперазинил, 1,3-диоксолан 2-он, оксиранил, тетрагидрофуранил, тетрагидропиранил, 1,2,3-триазол, 1,2,4-триазол, фуран, имидазол,изоксазол, изотиазол, оксазол, пиразол, тиазол, тиофен или тетразол; где аминогруппа может быть замещена С 1-С 4 алкоксикарбонилом, имидазолом, изотиазолом, пиразолом, пиридином, пиразином, пиримидином, пирролом, тиазолом, где пиридин и тиазол могут быть необязательно замещены С 1-С 4 алкилом; причем алкиламино и диалкиламино могут быть по алкилу замещены гидроксилом, C1-C4 алкокси,имидазолом, пиразолом, пирролом или тетразолом и причем морфолинил, тиоморфолинил, пирролидинил, пиперидинил, пиперазинил и оксиранил могут быть замещены группами -C(O)-Rn, -C(O)O-Rn или С 1-С 4 алкил, причем С 1-С 4 алкил может быть замещен гидроксилом;Rb обозначает гидроксил; амино; алкиламино, причем алкиламино может быть по алкилу замещен гидроксилом, амино, алкиламино или C1-C4 алкокси; C1-C4 алкокси; С 2-С 8 алкенил; С 2-С 8 алкинил; арил,причем арил может быть замещен по меньшей мере одним заместителем, независимо выбранным из галогена и С 1-С 4 алкокси; фуран; или C1-C8 алкил, причем алкил может быть замещен по меньшей мере одним независимо выбранным С 1-С 4 алкокси, арилом, амино, морфолинил, пиперидинил, пиперазинил;Rd фенил; причем фенил может быть замещен по меньшей мере одним независимо выбранным галогеном, нитрогруппой, C1-C6 алкилом, -C(O)O-Re, или -ORe;Re обозначает водород; C1-C6 алкил, причем алкил может быть замещен по меньшей мере одним независимо выбранным галогеном или алкоксигруппой; или фенил, причем фенил может быть замещен по меньшей мере одним независимо выбранным галогеном или алкоксигруппой; иRn обозначает гидроксил, С 1-С 4 алкокси, амино или С 1-С 6 алкил. Как очевидно для специалиста, соединения формулы (I) включают по меньшей мере один стереогенный центр (например, в заместителе R1) и могут существовать как рацемическая смесь или как энантиомерно чистая композиция. В предпочтительном варианте осуществления соединения формулы (I) представляют собой (S) изомер в энантиомерно чистой композиции. В рамках изобретения термин "алкил" в целом относится к насыщенным углеводородным радикалам прямой, разветвленной или циклической конфигурации, включающим метил, этил, н-пропил, изопропил, н-бутил, изобутил, втор-бутил, трет-бутил, н-пентил, н-гексил, циклогексил, н-гептил, октил, ноктил и т.п. В некоторых вариантах осуществления алкильные заместители могут включать С 1-С 8, C1-С 6 или C1-C4 алкильные группы. Алкил может быть замещен одним или более галогенами или алкоксигруппами. Например, алкил может представлять собой галогеналкил, дигалогеналкил или тригалогеналкил. В рамках настоящего изобретения "алкенил" в целом относится к прямым, разветвленным или циклическим алкеновым радикалам, имеющим одну или более углерод-углеродных двойных связей, таким как С 2-С 8 и С 2-С 6 алкенильные группы, включая 3-пропенил. В рамках изобретения "алкинил" в целом относится к прямым, разветвленным или циклическим алкиновым радикалам, имеющим одну или более углерод-углеродных тройных связей, таким как С 2-С 8 и С 2-С 6 алкинильные группы, включая гекс-3-ин. В рамках изобретения "арил" относится к карбоциклической ароматической кольцевой структуре. К арильным группам относятся ароматические кольца, содержащие от пяти до двадцати атомов углерода. Кольцевые арильные структуры включают соединения, имеющие одну или более кольцевых структур,такие как моно-, би- или трициклические соединения. Примеры арильных групп, которые включают фенил, толил, антраценил, флуоренил, инденил, азуленил, фенантренил (то есть фенантрен) и нафтил (то есть нафталин) кольцевые структуры. В определенных вариантах осуществления арильная группа может быть замещена. В рамках изобретения "гетероарил" относится к циклическим ароматическим кольцевым структурам, в которых один или более атомов в кольце, гетероатом(ы) являются элементом, отличным от углерода. Гетероатомы обычно представляют собой атомы О, S или N. К гетероарилам относятся и независимо выбираются О-, N- и S-гетероарильные кольцевые структуры. Кольцевая структура может включать соединения, имеющие одну или более кольцевых структур, таких как моно-, би- или трициклические соединения. В некоторых вариантах осуществления гетероарильные группы могут быть выбраны из гетероарильных групп, которые содержат один или более гетероатом, два или более гетероатома, три или более гетероатома или четыре или более гетероатома. Кольцевые гетероарильные структуры могут быть выбраны из тех, которые содержат пять или более атомов, шесть или более атомов или восемь или более атомов. Примеры кольцевых структур гетероарила включают акридин, бензимидазол, бензоксазол, бензодиоксол, бензофуран, дигидрохромен-4-онил, 1,3-диазин, 1,2-диазин, 1,2-диазол, 1,4-диазанафталин,фуран, фуразан, имидазол, индол, изоксазол, изохинолин, изотиазол, изоиндолил, оксазол, пурин, пиридазин, пиразол, пиридин, пиразин, пиримидин, пиррол, хинолин, хиноксалин, тиазол, тиофен, 1,3,5 триазин, 1,2,4-триазин, 1,2,3-триазин, тетразол и хиназолин. В определенных вариантах осуществления гетероарил может быть замещен. В рамках изобретения "гетероцикл" относится к циклическим кольцевым структурам, в которых один или более атомов в кольце, гетероатом(ы) являются элементом, отличным от углерода. Гетероатомы обычно представляют собой атомы О, S или N. К гетероциклам относятся и независимо выбираются О-, N- и S-гетероциклические кольцевые структуры. Кольцевая структура может включать соединения,имеющие одну или более кольцевых структур, таких как моно-, би-, или трициклические соединения. В некоторых вариантах осуществления гетероциклические группы могут быть выбраны из гетероциклических групп, которые содержат один или более гетероатом, два или более гетероатома, три или более гетероатома или четыре или более гетероатома. Примеры гетероциклических групп включают морфолинил, пирролидинонил, пирролидинил, пиперидинил, пиперазинил, гидантоинил, валеролактамил, оксиранил, оксетанил, тетрагидрофуранил, тетрагидропиранил, тетрагидропиридинил, тетрагидропиримидинил, тетрагидротиофенил или тетрагидротиопиранил и т.п. В определенных вариантах осуществления гетероцикл может быть замещен. В рамках изобретения "алканоил" в целом относится к группе со структурой -C(O)-R. В определенных вариантах осуществления R может являться водородом, алкилом, 4-морфолинилом или тиазоламиногруппой. В рамках изобретения "алкокси" в целом относится к группе со структурой -O-R. В определенных вариантах осуществления R может представлять собой алкил, такой как C1-C5 алкил. В рамках настоящего изобретения галогеновые заместители могут быть независимо выбраны из таких галогенов, как фтор, хлор, бром, йод и астат. В некоторых предпочтительных вариантах осуществления X обозначает водород; C1-C6 алкил, не-5 016575 обязательно замещенный одним или более галогенами; гидроксильную группу; галоген; C1-С 5 алкокси,необязательно замещенный фенилом.Rd фенил; причем фенил может быть замещен по меньшей мере одним независимо выбранным галогеном, нитрогруппой, C1-C6 алкилом, -C(O)O-Re или -ORe. В рамках настоящпего изобретения в случае, если одна или более функциональных групп, охватывающих X, R1, R2, R0, Ra, Rb, Rc Rd и Re, включены в молекулу формулы (IV), каждая из этих функциональных групп в любом положении может быть независимо выбрана и, необязательно, независимо замещена. Далее, где указано родовое название заместителя для любого положения в молекулах согласно настоящему изобретению, следует понимать, что этот родовой заместитель может быть заменен более конкретными заместителями, и полученные молекулы находятся в рамках молекул настоящего изобретения. Предпочтительные соединения изобретения включают следующие. В определенных вариантах осуществления предпочтительные соединения включают соединения с ЕС 50 в тесте ELISA VEGF, описанном в примере 2, менее чем приблизительно 2 мкМ, более предпочтительно от приблизительно 2 до приблизительно 0,04 мкМ (от 200 до 40 нМ); более предпочтительно от приблизительно 0,04 до приблизительно 0,008 мкМ (от 40 до 8 нМ) и более предпочтительно менее чем приблизительно 0,008 мкМ (8 нМ). Особенно предпочтительными соединениями являются соединения 2, 4, 5, 7, 8, 10, 11, 12, 17, 23, 25, 81, 102, 112, 140, 328, 329, 330, 331, 332, 355, 816, 817, 818, 823,824, 825, 830, 831, 832, 837, 838, 841, 842, 843 и их региоизомеры. В одном варианте осуществления предпочтительные соединения изобретения образуют рацемическую смесь, и в другом варианте осуществления соединения изобретения являются изомерами (R), (S), (R,R), (S,S), (R,S), (S,R) в энантиомерно чистой композиции. Более предпочтительно соединения изобретения представляют собой (S)-изомеры в энантиомерно чистой композиции. Указанные выше соединения перечислены только в качестве примеров, которые могут использоваться в способах данного изобретения. На основании настоящего раскрытия для специалиста будет очевидно, какие другие соединения, включенные в рамки заявленного изобретения, будут пригодны для использования в описанных здесь способах.- 14016575 В. Получение соединений данного изобретения. Соединения изобретения могут быть получены любым способом, известным из уровня техники. Например, соединения изобретения могут быть получены согласно следующим общим схемам. Более конкретно схема I может быть использована для получения соединения формулы I. Схема Ia может быть использована в сочетании со схемой I, когда R2 обозначает -СН 2-фуранил. Альтернативно для асимметричного синтеза, когда R2 обозначает водород или гидроксил, может быть использована схема Ib. Схема I Схема II может быть использована для получения соединений формулы I-h. Схема II Схемы IIIa или IIIb могут быть использованы для получения соединений формулы I-i. Схема IIIaRef: Magid Abou-Gharbia et al., J. Med. Chem. 1987, 30, 1818. В предпочтительном варианте осуществления соединения изобретения могут быть переведены в энантиомерно чистые композиции с использованием любого способа, известного из уровня техники. Например, соединения изобретения могут быть получены прямой кристаллизацией энантиомерных смесей,получением диастереомерной соли энантиомеров, получением диастереомеров и разделением или ферментативным получением. В предпочтительном варианте осуществления соединения изобретения могут быть получены путем кристаллизации с использованием, например, N-ацетил-L-фенилаланина с получением (S) изомера или В определенных вариантах осуществления примеры способов согласно схеме I для получения предпочтительных соединений формулы I включают образование продуктов реакции ПиктеШпенглера/промежуточных продуктов в форме свободных аминов, как описано ниже в методике I. Методика I В одном варианте осуществления методика I может включать добавление желаемого альдегида(II) к суспензии 5-замещенного триптаминаHCl (I) в 0,1 н. серной кислоте. Раствор может затем быть перемешан при температуре около 110-120 С в закрытом реакторе в течение времени, достаточного для завершения реакции, например в течение от приблизительно 15 мин до приблизительно 20 ч. После завершения реакции реакционная смесь может быть охлаждена до комнатной температуры, и осажденная соль может быть отфильтрована. Отфильтрованный остаток может затем быть промыт эфиром, EtOAc или смесью DCM и DMF и высушен с получением продукта (III) в виде кислой соли. Альтернативно, желаемый альдегид(II) может быть добавлен к суспензии 5-замещенного триптаминаHCl (I) в уксусной кислоте, и смесь может быть нагрета с обратным холодильником в течение времени, достаточного для завершения реакции, например в течение от приблизительно 15 мин до приблизительно 20 ч. После завершения реакции реакционная смесь может быть охлаждена до комнатной температуры, и кислая соль может быть отфильтрована. Отфильтрованный остаток может быть промыт уксусной кислотой, затем DCM и высушен с получением продукта (III) в виде кислой соли. Свободный амин (III) может быть получен экстракцией с EtOAc и промыванием водным раствором гидроксида аммония или 1M водным раствором гидроксида натрия. Свободный амин или его соль могут быть использованы для получения других предпочтительных соединений формулы I, таких как карбаматные аналоги (формула 1-c, методика-II), амидные аналоги,включая N-ацетильные аналоги (формула 1-c, методика-IIIa и методика-IIIb), аналоги мочевины и аналоги тиомочевины (формула I-е и 1-f, методика-IV и методика-V соответственно), аналоги сульфоксида(формула 1-g, методика-VI) и аналоги пиримидина (методика-VII). Более конкретно методика-II может быть использована для синтеза карбаматных аналогов свободных аминов(III) или их солей. В соответствии с методикой-II диизопропилэтиламин (DIEA) может быть добавлен к свободному амину (III) или его кислой соли в дихлорметане (DCM) с последующим медленным добавлением замещенного хлороформиата. Реакционная смесь может перемешиваться при комнатной температуре в течение приблизительно 1-20 ч. Растворитель может затем быть выпарен и сырой продукт может быть очищен либо ВЭЖХ, либо хроматографией на колонке с силикагелем. Методика-IIIa может быть использована для синтеза амидных аналогов свободного амина(III) или их солей. Методика-IIIa В соответствии с методикой-IIIa предварительно перемешивают в течение 15 мин смесь R2-кислоты и диизопропилкарбодиимида (DIC), и эта смесь может быть добавлена к свободному амину(III), или его кислой соли в DCM и DIEA. Реакционная смесь может перемешиваться в течение приблизительно 1 ч. Растворители могут затем быть выпарены и сырой продукт может быть очищен ВЭЖХ. Альтернативно, методика-IIIb может быть использована для синтеза N-ацетильных аналогов свободных аминов (III) или их солей. Методика-IIIb В соответствии с методикой-IIIb пиридин может быть добавлен к свободному амину(III) или его кислой соли в DCM с последующим добавлением уксусного ангидрида. Реакционная смесь может перемешиваться при комнатной температуре в течение приблизительно 8-20 ч. Растворители могут затем быть выпарены, и сырой продукт может быть очищен ВЭЖХ. Методика-IV может быть использована для синтеза аналогов мочевины свободных аминов(III) или их солей. Методика-IV В соответствии с методикой-IV, DIEA и R2-изоцианат могут быть добавлены к свободному амину(III) или его кислой соли в DCM. Реакционная смесь может быть нагрета с обратным холодильником приблизительно в течение 1,5 ч. Растворители могут затем быть выпарены и сырой продукт может быть очищен ВЭЖХ. Методика-V может быть использована для синтеза аналогов тиомочевины свободных аминов(III) или их солей. Методика-V В соответствии с методикой-V DIEA и R2-изотиоцианат могут быть добавлены к свободному амину(III) или его кислой соли в DCM. Реакционная смесь может быть нагрета с обратным холодильником в течение приблизительно 12 ч. Растворители могут затем быть выпарены и сырой продукт может быть очищен ВЭЖХ. Методика-VI может быть использована для синтеза сульфонильных аналогов свободных аминов В соответствии с методикой-VI DIEA и R2-сульфонилхлорид могут быть добавлены к свободному амину (III) или его кислой соли в DCM. Реакционная смесь может перемешиваться при комнатной температуре в течение приблизительно 12 ч. Растворители могут затем быть выпарены и сырой продукт может быть очищен ВЭЖХ. Методика-VII может быть использована для синтеза пиримидиновых аналогов свободных аминов В соответствии с методикой-VII триэтиламин и 2-бромпиримидин в N,N-диметилформамиде (DMF) могут быть добавлены к свободному амину (III) или его кислой соли в DCM. Реакционная смесь может быть нагрета приблизительно при 120 С в течение приблизительно 12 ч. Растворители могут затем быть выпарены и сырой продукт может быть очищен ВЭЖХ. Эти и другие методологии реакции могут быть пригодными в получении соединений изобретения,как понятно специалисту. Различные модификации вышеупомянутых схем и методик будут очевидны для специалиста, и изобретение не ограничено конкретными способами получения соединений изобретения. С. Способы данного изобретения. В другом аспекте изобретение относится к способам ингибирования продуцирования VEGF, ингибирования ангиогенеза и/или лечения рака, диабетической ретинопатии, ревматоидного артрита, псориаза, атеросклероза, хронического воспаления, других хронических связанных с воспалением заболеваний и нарушений, ожирения или экссудативной дегенерации желтого пятна с использованием описанных здесь соединений. В одном варианте осуществления изобретение относится к способам ингибирования продуцирования VEGF, включающим введение пациенту ингибирующего VEGF-экспрессию количества по меньшей мере одного соединения изобретения. В другом варианте осуществления изобретение относится к способам ингибирования ангиогенеза,включающим введение пациенту антиангиогенного количества по меньшей мере одного соединения изобретения. В еще одном варианте осуществления изобретение относится к способам лечения рака, диабетической ретинопатии, ревматоидного артрита, псориаза, атеросклероза, хронического воспаления, других хронических связанных с воспалением заболеваний и нарушений, ожирения или экссудативной дегенерации желтого пятна, включающим введение пациенту терапевтически эффективного количества по меньшей мере одного соединения изобретения. Вне связи с какой-либо теорией считается, что способы согласно настоящему изобретению действуют через комбинацию механизмов, которые модулируют активность VEGF. В предпочтительных вариантах осуществления способы изобретения включают введение терапевтически эффективного количества по меньшей мере одного соединения изобретения, причем соединение представляет собой (S) изомер. Согласно способам изобретения соединение(я) может вводиться пациенту любым путем доставки лекарственного средства, известным из уровня техники. Частные примеры пути введения включают пероральный, глазной, ректальный, буккальный, топический, назальный, офтальмический, подкожный,внутримышечный, внутривенный (болюс и инфузия), внутрицеребральный, чрескожный и легочный. Термины "VEGF-ингибирующее количество", "антиангиогенное количество" и "терапевтически эффективное количество" в рамках изобретения относятся к количеству фармацевтического средства для лечения, улучшения состояния или профилактики идентифицируемого заболевания или состояния или для проявления поддающегося регистрации терапевтического или ингибирующего эффекта. Эффект мо- 18016575 жет быть обнаружен, например, с помощью тестов, раскрытых в следующих примерах. Точное эффективное количество для пациента будет зависеть от массы тела, размера и здоровья пациента; природы и степени состояния и терапевтических средств или комбинации терапевтических средств, выбранных для введения. Терапевтически эффективные количества для данной ситуации могут быть определены обычным экспериментированием, которое находится в пределах квалификации и общих знаний клинического врача. Для любого соединения терапевтически эффективное количество может быть оценено первоначально либо с помощью тестов на культуре клеток, например опухолевых клеток, либо на моделях животных, обычно крыс, мышей, кроликов, собак или свиней. Модель животных может также использоваться для определения адекватного диапазона концентраций и пути введения. Такая информация может быть использована для определения полезных доз и путей введения человеку. Терапевтическая/профилактическая эффективность и токсичность могут быть определены в соответствии со стандартными фармацевтическими методиками в культурах клеток или на подопытных животных, например,ED50 (доза, терапевтически эффективная для 50% популяции) и LD50 (доза, летальная для 50% популяции). Отношение дозы между терапевтическими и токсическими эффектами составляет терапевтический индекс, и он может быть выражен как отношение ED50/LD50. Фармацевтические композиции, которые показывают большие терапевтические индексы, являются предпочтительными. Данные, полученные в тестах на культуре клеток и исследованиях на животных, могут использоваться при составлении диапазона доз для использования человеком. Доза, содержавшаяся в таких композициях, предпочтительно находится в пределах диапазона циркулирующих концентраций, которые включают ED50 с низкой токсичностью или ее отсутствием. Доза может варьировать в пределах этого диапазона в зависимости от используемой лекарственной формы,чувствительности пациента и пути введения. Более конкретно, соотношения концентрации и биологического действия, наблюдаемые относительно соединения(й) согласно настоящему изобретению, указывают начальную целевую концентрацию в плазме от приблизительно 0,1 до приблизительно 100 мкг/мл, предпочтительно от приблизительно 5 до приблизительно 50 мкг/мл, более предпочтительно от приблизительно 5 до приблизительно 10 мкг/мл. Для достижения таких концентраций в плазме соединения изобретения могут вводиться в дозах, которые составляют от 0,1 мкг до 100000 мг, в зависимости от пути введения. Руководство относительно конкретных доз и способов доставки содержится в литературе и в целом доступно для практикующих специалистов в данной области техники. В целом доза будет в диапазоне от приблизительно 1 мг/сутки до приблизительно 10 г/сутки, или от приблизительно 0,1 до приблизительно 3 г/сутки, или от приблизительно 0,3 до приблизительно 3 г/сутки, или от приблизительно 0,5 до приблизительно 2 г/сутки в виде разового, разделенного или непрерывного введения пациенту с массой тела от приблизительно 40 до приблизительно 100 кг (причем доза может быть уточнена для пациентов с массой тела выше или ниже этого диапазона, в частности, для детей с массой тела менее 40 кг). Точная доза должна быть определена практиком в свете факторов, относящихся к пациенту. Дозу и путь введения выбирают таким образом, чтобы обеспечить достаточные уровни активного агента(агентов) или поддержать желаемый эффект. Факторы, которые могут быть приняты во внимание,включают серьезность состояния, общее здоровье пациента, возраст, массу тела и пол пациента, диету,время и частоту введения, комбинацию(комбинации) лекарственного средства, чувствительность реакции и переносимость/реакцию на терапию. Фармацевтические композиции длительного действия могут вводиться каждые 3-4 дня, каждую неделю или один раз в две недели в зависимости от периода полураспада и скорости выведения конкретного состава.D. Метаболиты соединений данного изобретения. Также в рамки настоящего изобретения попадают продукты метаболизма in vivo описанных здесь соединений. Такие продукты могут образовываться в результате, например, окисления, восстановления,гидролиза, амидирования, этерификации и т.п. введенного соединения, прежде всего, вследствие ферментативных процессов. Соответственно, изобретение включает соединения, произведенные процессом,включающим контактирование соединения изобретения с тканью млекопитающих или с млекопитающим в течение времени, достаточного для получения продукта его метаболизма. Такие продукты обычно идентифицируют, получая меченное радиоактивным изотопом (например, С 14 или Н 3) соединение изобретения, вводя его в поддающейся определению дозе (например, больше чем приблизительно 0,5 мг/кг) млекопитающему, такому как крыса, мышь, гвинейская свинка, обезьяна, или человеку, оставляя достаточное время для метаболизма (обычно от приблизительно 30 с до 30 ч) и выделяя его продукты превращения из мочи, крови или других биологических проб. Эти продукты легко выделяются, так как они являются мечеными (другие выделяют при помощи антител, способных к связыванию с эпитопами, сохраняющимися в метаболите). Структуры метаболита определяют обычным способом, например анализом ЯМР или МС. В целом, анализ метаболитов может быть осуществлен таким же образом, как и изучение метаболизма обычных лекарственных средств, обычное для специалиста. Продукты превращения при условии, если для них in vivo не показано иное, являются пригодными в диагностических тестах для терапевтического дозирования соединений изобретения, даже если они не обладают никакой собственной- 19016575 биологической активностью. Е. Фармацевтические композиции данного изобретения. Хотя соединения согласно настоящему изобретению могут вводиться индивидуально, может быть предпочтительно составить соединения в форме фармацевтических композиций. Также в еще одном аспекте изобретение относится к фармацевтическим композициям, пригодным для использования в способах изобретения. Фармацевтические композиции изобретения могут быть составлены с фармацевтически приемлемыми эксципиентами, такими как носители, растворители, стабилизаторы, адъюванты, разбавители и т.д., в зависимости от конкретного способа введения и лекарственной формы. Фармацевтические композиции в целом составляют таким образом, чтобы достигнуть физиологически совместимого рН, и рН может составлять от приблизительно 3 до приблизительно 11, предпочтительно от приблизительно 3 до приблизительно 7 в зависимости от состава и пути введения. В альтернативных вариантах осуществления может быть предпочтительным установить рН в диапазоне от приблизительно 5,0 до приблизительно 8,0. Более конкретно фармацевтические композиции изобретения включают терапевтически или профилактически эффективное количество по меньшей мере одного соединения согласно настоящему изобретению вместе с одним или более фармацевтически приемлемыми эксципиентами. Необязательно,фармацевтические композиции изобретения могут включать комбинацию соединений согласно настоящему изобретению или могут включать второй активный ингредиент, полезный в лечении рака, диабетической ретинопатии или экссудативной дегенерации желтого пятна. Составы согласно настоящему изобретению, например, для парентерального или перорального введения являются чаще всего твердыми веществами, жидкими растворами, эмульсиями или суспензиями, в то время как ингаляционные составы для легочного введения обычно представляют собой жидкости или порошки, причем порошковые составы обычно являются предпочтительными. Предпочтительная фармацевтическая композиция изобретения может также быть составлена в форме лиофилизованного твердого вещества, которое перед введением ресуспендируют в физиологически совместимом растворителе. Альтернативные фармацевтические композиции изобретения могут быть составлены в форме сиропов, кремов, мазей, таблеток и т.п. Термин "фармацевтически приемлемый эксципиент" относится к эксципиенту для введения фармацевтического агента, такого как соединения согласно настоящему изобретению. Термин относится к любому фармацевтическому эксципиенту, который может быть введен без проявления нежелательной токсичности. Фармацевтически приемлемые эксципиенты определяются отчасти конкретной вводимой композицией, а также конкретным способом, используемым для введения композиции. Соответственно, существует широкая разновидность подходящих составов фармацевтических композиций согласно настоящему изобретению (см., например, Remington's Pharmaceutical Sciences). Подходящие эксципиенты могут быть молекулами носителя, которые включают большие, медленно метаболизирующиеся макромолекулы, такие как белки, полисахариды, полимолочные кислоты, полигликолевые кислоты, полимерные аминокислоты, сополимеры аминокислоты, и неактивные вирусные частицы. Другие примеры эксципиентов включают антиоксиданты, такие как аскорбиновая кислота; хелатирующие агенты, такие как EDTA; углеводы, такие как декстрин, гидроксиалкилцеллюлозу, гидроксиалкилметилцеллюлозу, стеариновую кислоту; жидкости, такие как масла, вода, солевой раствор, глицерин и этанол; смачивающие агенты или эмульгаторы; буферы рН и т.п. Липосомы также входят в ряд фармацевтически приемлемых эксципиентов. Фармацевтические композиции изобретения могут быть составлены в любой форме, подходящей для намеченного способа введения. Когда они предназначены для перорального использования, могут быть получены, например, таблетки, лепешки, пастилки, водные или масляные суспензии, неводные растворы, диспергируемые порошки или гранулы (включая микронизированные частицы или наночастицы),эмульсии, твердые или мягкие капсулы, сиропы или эликсиры. Композиции, предназначенные для перорального использования, могут быть получены согласно любому способу, известному из уровня техники как пригодный для получения фармацевтических композиций, и такие композиции могут содержать одно или более средств, включая подсластители, ароматизаторы, красители и консерванты, чтобы обеспечить приемлемый препарат. Фармацевтически приемлемые эксципиенты, особенно подходящие для использования в таблетках,включают, например, инертные разбавители, такие как целлюлоза, карбонат кальция или натрия, лактозу, фосфат кальция или натрия; дезинтеграторы, такие как кроскармеллоза натрия, поперечно-сшитый повидон, кукурузный крахмал или альгиновая кислота; связующие вещества, такие как повидон, крахмал, желатин или гуммиарабик; и лубриканты, такие как стеарат магния, стеариновая кислота или тальк. Таблетки могут не иметь покрытия или покрытие может быть на них нанесено известными способами,включая микрокапсуляцию, чтобы задержать измельчение и адсорбцию в желудочно-кишечном тракте и таким образом обеспечить пролонгированное действие за более длительный период. Например, может использоваться материал для замедленного высвобождения, такой как глицерилмоностеарат или глицерилдистеарат, индивидуально или вместе с воском. Составы для перорального использования могут быть также представлены как твердые желатино- 20016575 вые капсулы, где активный ингредиент смешан с инертным твердым разбавителем, таким как, например,целлюлоза, лактоза, фосфат кальция или каолин, или как мягкие желатиновые капсулы, где активный ингредиент смешан с неводной или масляной средой, такой как глицерин, пропиленгликоль, полиэтиленгликоль, арахисовое масло, вазелиновое масло или оливковое масло. В другом варианте осуществления фармацевтические композиции изобретения могут быть составлены как суспензии, включающие соединение согласно настоящему изобретению в смеси по меньшей мере с одним фармацевтически приемлемым эксципиентом, подходящим для получения суспензии. В еще одном варианте осуществления фармацевтические композиции изобретения могут быть составлены в форме диспергируемых порошков и гранул, подходящих для получения суспензии путем добавления подходящих эксципиентов. Эксципиенты, подходящие для использования в суспензиях, включают суспендирующие агенты,такие как натрийкарбоксиметилцеллюлозу, метилцеллюлозу, гидроксипропилметилцеллюлозу, альгинат натрия, поливинилпирролидон, трагакант, аравийскую камедь, диспергирующие или смачивающие вещества, такие как природный фосфатид (например, лецитин), продукт конденсации эпоксида с жирной кислотой (например, стеарат полиэтиленоксида), продукт конденсации этиленоксида с длинноцепочечным алифатическим спиртом (например, гептадекаэтиленоксицетанол), продукт конденсации этиленоксида с неполным эфиром многоатомного спирта, полученного из жирной кислоты и ангидрида гексита(например, сорбитан моноолеат полиоксиэтилена); и загустители, такие как карбомер, пчелиный воск,твердый парафин или цетиловый спирт. Суспензии могут также содержать одно или более консервантов,таких как уксусная кислота, метил и/или н-пропил-п-гидроксибензоат; один или более красителей; один или более ароматизаторов и один или более подслащивающих агентов, таких как сахароза или сахарин. Фармацевтические композиции изобретения могут также быть в форме эмульсий "масло-в-воде". Масляная фаза может быть растительным маслом, таким как оливковое масло или арахисовое масло,минеральным маслом, таким как вазелиновое масло, или их смесью. Подходящие эмульгаторы включают природные смолы, такие как аравийская камедь и трагакант; природные фосфатиды, такие как лецитин сои, сложные эфиры или неполные эфиры многоатомного спирта, полученного из жирных кислот; ангидриды гексита, такие как сорбитан моноолеат; и продукты конденсации этих неполных эфиров многоатомного спирта с этиленоксидом, такие как сорбитан моноолеат полиоксиэтилена. Эмульсия может также содержать подсластители и ароматизаторы. Сиропы и эликсиры могут быть составлены с подсластителями, такими как глицерин, сорбит или сахароза. Такие составы могут также содержать средство,уменьшающее раздражение, консервант, ароматизатор или краситель. Дополнительно фармацевтические композиции изобретения могут быть в форме стерильного инъецируемого препарата, такого как стерильная инъецируемая водная эмульсия или маслянистая суспензия. Эта эмульсия или суспензия могут быть составлены согласно известному уровню техники с использованием подходящих диспергаторов или смачивающих веществ и суспендирующих агентов, которые были упомянуты выше. Стерильный инъецируемый препарат может также быть стерильным инъецируемым раствором или суспензией в нетоксичном парентерально приемлемом разбавителе или растворителе, таком как раствор в 1,2-пропандиоле. Стерильный инъецируемый препарат может также быть получен в форме лиофилизованного порошка. Среди приемлемых носителей и растворителей, которые могут использоваться, можно назвать воду, раствор Рингера и изотонический раствор хлорида натрия. Кроме того, стерильные нелетучие масла могут использоваться как растворитель или суспендирующая среда. С этой целью может использоваться любое мягкое нелетучее масло, включая синтетические моно- или диглицериды. Кроме того, жирные кислоты, такие как олеиновая кислота, могут аналогичным образом использоваться в получении инъецируемых препаратов. Вообще, соединения согласно настоящему изобретению, пригодные для использования в способах согласно настоящему изобретению, в основном не растворимы в воде и умеренно растворимы в большинстве фармацевтически приемлемых протонных растворителей и в растительных маслах. Однако соединения вообще растворимы в жирных кислотах со средней длиной цепи (например, каприловых и каприновых кислотах) или триглицеридах и имеют высокую растворимость в сложных эфирах пропиленгликоля жирных кислот со средней длиной цепи. Также в изобретении рассматриваются соединения, которые были модифицированы путем замещений или присоединений химических или биохимических звеньев, которые делают их более подходящими для доставки (например, за счет увеличения растворимости, биоактивности, приятного вкуса, уменьшения побочных реакций и т.д.), например, этерификацией, гликозилированием, ПЭГилированием и т.д. В предпочтительном варианте осуществления соединения согласно настоящему изобретению могут быть составлены для перорального введения в составе на липидной основе, подходящем для соединений с низкой растворимостью. Составы на липидной основе могут вообще усиливать пероральную биодоступность таких соединений. Также предпочтительная фармацевтическая композиция изобретения включает терапевтически или профилактически эффективное количество соединения согласно настоящему изобретению вместе по меньшей мере с одним фармацевтически приемлемым эксципиентом, выбранным из группы, состоящей из жирных кислот со средней длиной цепи или их эфиров с пропиленгликолем(например, сложные эфиры пропиленгликоля и пищевых жирных кислот, таких как каприловые и капри- 21016575 новые жирные кислоты) и фармацевтически приемлемых поверхностно-активных веществ, таких как полиоксил 40 гидрированное касторовое масло. В альтернативном предпочтительном варианте осуществления циклодекстрины могут быть добавлены в качестве усилителей растворимости в воде. Предпочтительные циклодекстрины включают гидроксипропиловые, гидроксиэтиловые, глюкозиловые, мальтозиловые и мальтотриозиловые производные , и -циклодекстрина. Особенно предпочтительным циклодекстриновым усилителем растворимости является гидроксипропилциклодекстрин (НРВС), который может быть добавлен к любой из вышеописанных композиций, чтобы дополнительно улучшить характеристики растворимости в воде соединений согласно настоящему изобретению. В одном варианте осуществления композиция включает от 0,1 до 20% гидроксипропилциклодекстрина, более предпочтительно от 1 до 15% гидроксипропил-циклодекстрина и еще более предпочтительно от 2,5 до 10% гидроксипропилциклодекстрина. Количество используемого усилителя растворимости будет зависеть от количества соединения согласно настоящему изобретению в композиции.I. Комбинированная терапия. Также возможно комбинировать любое соединение согласно настоящему изобретению с одним или более другими активными ингредиентами, полезными в лечении рака, включая соединения в форме разовой дозы, или в отдельных лекарственных формах, предназначенных для одновременного или последовательного введения пациенту. При последовательном введении комбинация может вводиться в виде двух или более введений. В альтернативном варианте осуществления возможно вводить одно или более соединений согласно настоящему изобретению и одно или более дополнительных активных ингредиентов разными путями. Для специалиста понятно, что различные активные ингредиенты могут вводиться в комбинации с соединениями согласно настоящему изобретению, которые могут увеличивать или синергически усиливать VEGF-ингибирующую и/или антиангиогенезную активность соединений изобретения. В соответствии со способами изобретения комбинация активных ингредиентов может быть (1) совместно составленной и вводимой или доставляемой одновременно в комбинированном составе; (2) доставляемой при чередовании или параллельно в форме отдельных составов или (3) любым другим комбинированным режимом терапии, известным в уровне техники. При введении с чередованием способы изобретения могут включать введение или доставку активных ингредиентов последовательно, например, в отдельном растворе, эмульсии, суспензии, таблетках, пилюлях или капсулах, или различными инъекциями в отдельных шприцах. В целом, в ходе терапии с чередованием эффективная доза каждого активного ингредиента вводится последовательно, то есть сериями, тогда как при одновременной терапии эффективные дозы двух или более активных ингредиентов вводятся вместе. Различные последовательности чередующейся комбинированной терапии также могут использоваться. Для лучшего понимания настоящего изобретения включены следующие примеры. Эксперименты,касающиеся настоящего изобретения, не должны, конечно, рассматриваться как ограничивающие изобретение и изменения изобретения, уже известные или которые будут открыты позднее, которые находятся в пределах общих знаний специалиста, входят в рамки изобретения, как оно описано здесь и далее в формуле изобретения. Примеры Настоящее изобретение описывается более подробно со ссылкой на следующие не имеющие ограничительного характера примеры, которые предлагаются, чтобы более полно проиллюстрировать изобретение, но не должны рассматриваться как ограничение его объема. Примеры иллюстрируют получение конкретных соединений изобретения, и испытания этих соединений in vitro и/или in vivo. Для специалиста очевидно, что методики, описанные в этих примерах, представляют собой методики, описанные авторами как хорошо подходящими в практике осуществления изобретения, и также составляют предпочтительные способы для его осуществления на практике. Однако следует понимать, что для специалиста в свете настоящего раскрытия очевидно, что в конкретные описанные способы могут быть внесены изменения с получением подобного или схожего результата и без отступления от духа и объема изобретения. Пример 1. Получение соединений изобретения. Используя схемы и методики, описанные выше в разделе В, можно получить некоторые соединения изобретения следующим образом. Другие предпочтительные соединения изобретения, такие как представленные ниже в табл. 5, могут быть получены аналогично. Пример 1 А. Соединения формулы I, схема I. Некоторые соединения формулы I могут быть получены согласно схеме I с использованием продуктов/промежуточных продуктов в форме свободного амина или их солей, полученных в соответствии с методикой I. Например, некоторые свободные амины (III) или их соли получают, используя методику I. Следующая таблица иллюстрирует некоторые свободные амины (III) или их соли, промежуточные продукты-1-11. Промежуточный продукт-1. Этот промежуточный продукт получают, используя методику-1 с 5-хлортриптаминомHCl (5,8 г, 25 ммоль), п-анизальдегид (6,13 мл, 50 ммоль) и 0,1 н. серную кислоту (60 мл), чтобы получить целевое соединение в форме кислой соли (6,1 г, 59%). ES-MC: 313 (М+Н)+. Альтернативно этот промежуточный продукт получают, используя методику-IB с 5-хлортриптаминомHCl (20 г, 86,5 ммоль), п-анизальдегид(15,9 мл, 130 ммоль) и уксусную кислоту (250 мл), с получением целевого соединения в форме кислой соли (25,8 г, 79%). ES-MC: 313 (М+Н)+. Промежуточный продукт-2. Этот промежуточный продукт получают, используя методику-1 с 5-хлортриптаминомHCl (116 мг,0,5 ммоль), 2,3-дифторбензальдегид (109 мкл, 1 ммоль) и 0,1 н. серную кислоту (2 мл), с получением целевого соединения в форме кислой соли (158 мг, 75%). ES-MC: 319 (М+Н)+. Промежуточный продукт-3. Этот промежуточный продукт получают, используя методику-I с 5-хлортриптаминомHCl (462 мг, 2 ммоль), 4-хлорбензальдегид (562 мг, 4 ммоль) и 0,1 н. серную кислоту (8 мл), с получением целевого соединения в форме кислой соли (825 мг, 99%). ES-MC: 317 (М+Н)+. Промежуточный продукт-4. Этот промежуточный продукт получают, используя методику-1 с 5-хлортриптаминомHCl (462 мг,2 ммоль), 4-цианобензальдегид (525 мг, 4 ммоль) и 0,1 н. серную кислоту (8 мл), с получением целевого соединения в форме кислой соли (810 мг, 100%). ES-MC: 308 (M+H)+. Промежуточный продукт-5. Этот промежуточный продукт получают, используя методику-1 с 5-хлортриптаминомHCl (374 мг,1,5 ммоль), 4-фторбензальдегид (322 мкл, 3 ммоль) и 0,1 н. серную кислоту (4 мл), с получением целевого соединения в форме кислой соли (250 мг, 42%). ES-MC: 301 (М+Н)+. Промежуточный продукт-6. Этот промежуточный продукт получают, используя методику-1 с 5-хлортриптаминомHCl (1,15 г, 5 ммоль), 4-изопропилбензальдегид (1,516 мл, 10 ммоль) и 0,1 н. серную кислоту (12 мл), с получением целевого соединения в форме кислой соли (628 мг, 30%). ES-MC: 325 (М+Н)+. Промежуточный продукт-7. Этот промежуточный продукт получают, используя методику-1 с 5-бромтриптаминомHCl (551 мг,2 ммоль), 4-хлорбензальдегид (562 мг, 4 ммоль) и 0,1 н. серную кислоту (8 мл), с получением целевого соединения в форме кислой соли (330 мг, 36%). ES-MC: 363 (М+Н)+. Промежуточный продукт-8. Этот промежуточный продукт получают, используя методику-1 с 5-бромтриптаминомНС 1 (551 мг,2 ммоль), п-толуолальдегид (471 мкл, 4 ммоль) и 0,1 н. серную кислоту (8 мл), с получением целевого соединения в форме гидросульфата (257 мг, 29%). ES-MC: 341 (М+Н)+. Альтернативно, этот промежуточный продукт получают, используя методику-IB с 5-бромтриптаминомHCl (10 г, 36,3 ммоль), птолуолальдегид (6,41 мл, 54,5 ммоль) и уксусную кислоту (120 мл), с получением целевого соединения в форме ацетата (14,5 г, 100%). ES-MC: 341 (М+Н)+. Промежуточный продукт-9 (соединение 112). Этот продукт/промежуточный продукт получают, используя методику-I с 5-бромтриптаминомHCl(551 мг, 2 ммоль), 4-изопропилбензальдегид (606 мкл, 4 ммоль) и 0,1 н. серную кислоту (8 мл), с получе- 23016575 нием целевого соединения в форме гидросульфата (329 мг, 35%). ES-MC: 369 (М+Н)+. Альтернативно, этот промежуточный продукт получают, используя методику-IB с 5 бромтриптаминомHCl (10 г, 36,3 ммоль), бензальдегид с 4 изопропилами (8,24 мл, 54,5 ммоль) и уксусную кислоту (120 мл), с получением целевого соединения в форме ацетата (13 г, 77%). ES-MC: 369(М+Н)+. Промежуточный продукт-10. Этот промежуточный продукт получают, используя методику-I с 5-бромтриптаминомHCl (551 мг,2 ммоль), 3-хлорбензальдегид (453 мкл, 4 ммоль) и 0,1 н. серную кислоту (8 мл), с получением целевого соединения в форме кислой соли (662 мг, 72%). ES-MC: 361 (М+Н)+. Промежуточный продукт-11. Этот промежуточный продукт получают, используя методику-I с 5-бромтриптаминомHCl (551 мг,2 ммоль), п-анизальдегид (491 мкл, 4 ммоль) и 0,1 н. серную кислоту (8 мл), с получением целевого соединения в форме кислой соли (611 мг, 67%). ES-MC: 357 (М+Н)+. Промежуточный продукт-12. 4-(2-Морфолин-4-ил-этокси)бензальдегид, промежуточный продукт реакции, получают, комбинируя 4-гидроксибензальдегид (1,2 г, 10,0 ммоль), 4-(2-хлорэтил)морфолин гидрохлорид (2,0 г, 11,0 ммоль),карбонат калия (4,1 г, 30,0 ммоль) и йодид калия (170 мг, 1 ммоль) в 100 мл ацетона и нагревая с обратным холодильником при перемешивании. После полного израсходования 4-гидроксибензальдегида (48 ч ЖХ/МС) твердые вещества фильтруют и растворитель удаляют в вакууме. Выход составляет 4,1 г. Затем получают промежуточный продукт 12 в соответствии с методикой-IB. Так, 5-хлортриптамин гидрохлорид (231 мг, 1,0 ммоль) комбинируют с 4-(2-морфолин-4-ил-этокси)бензальдегидом (565 мг,1,2 ммоль) в 3 мл ледяной уксусной кислоты. Суспензию нагревают при приблизительно 120 С в течение 10 мин с постоянным охлаждением и максимальной мощностью 300W с использованием СЕМ Explorer microwave system. Ацетонитрил (2 мл) добавляют к охлажденной реакционной смеси и твердое вещество отфильтровывают и промывают 1 мл ацетонитрила, чтобы получить соль уксусной кислоты промежуточного соединения 12 (6-хлор-1-[4-(2-морфолин-4-ил-этокси)фенил]-2,3,4,9-тетрагидро-1 Н-карболин) (179 мг, 34%). Промежуточные продукты 1-12 могут затем использоваться для получения соединений изобретения согласно методикам II-VII следующим образом. Соединение 2. Этот продукт получают в соответствии с методикой-II, используя промежуточный продукт-1 (3 г,9,6 ммоль), этилхлорформиат (1,37 мл, 14,4 ммоль) и DIEA (2,5 мл, 14,4 ммоль) в дихлорметане (70 мл), с получением целевого соединения в форме порошка белого цвета (1,56 г, 42%). ES-MC: 385 (М+Н)+. Соединение 4. Этот продукт получают в соответствии с методикой-II, используя промежуточный продукт-7 (72 мг,0,2 ммоль), этилхлорформиат (29 мкл, 0,3 ммоль) и DIEA (52 мкл, 0,3 ммоль) в дихлорметане (2 мл), с получением целевого соединения в форме порошка белого цвета (37 мг, 43%). ES-MC: 435 (М+Н)+. Соединение 5. Этот продукт получают в соответствии с методикой-II, используя промежуточный продукт-2 (50 мг,0,16 ммоль), этилхлорформиат (23 мкл, 0,24 ммоль) и DIEA (42 мкл, 0,24 ммоль) в дихлорметане (2 мл), с получением целевого соединения в форме порошка белого цвета (25 мг, 41%). ES-MC: 391 (М+Н)+. Соединение 7. Этот продукт получают в соответствии с методикой-II, используя промежуточный продукт-9 (74 мг,0,2 ммоль), этилхлорформиат (29 мкл, 0,3 ммоль) и DIEA (52 мкл, 0,3 ммоль) в дихлорметане (2 мл), с получением целевого соединения в форме порошка белого цвета (34 мг, 38%). ES-MC: 441 (М+Н)+. Соединение 8. Этот продукт получают в соответствии с методикой-II, используя промежуточный продукт-8 (72 мг,0,2 ммоль), этилхлорформиат (2 9 мкл, 0,3 ммоль) и DIEA (52 мкл, 0,3 ммоль) в дихлорметане (2 мл), с получением целевого соединения в форме порошка белого цвета (39 мг, 47%). ES-MC: 413 (М+Н)+. Соединение 10. Этот продукт получают в соответствии с методикой-II, используя ацетат промежуточного продукта 1 (10,5 г, 2 8,2 ммоль), 4-хлорфенилхлорформиат (4,74 мл, 33,8 ммоль) и DIEA (9,8 мл, 56,4 ммоль) в дихлорметане (300 мл), с получением целевого соединения в форме порошка белого цвета (10,2 г, 78%). ESMC: 467 (М+Н)+. Соединение 11. Этот продукт получают в соответствии с методикой-II, используя промежуточный продукт-3 (63 мг,0,2 ммоль), этилхлорформиат (29 мкл, 0,3 ммоль) и DIEA (52 мкл, 0,3 ммоль) в дихлорметане (2 мл), с получением целевого соединения в форме порошка белого цвета (31 мг, 40%). ES-MC: 389 (М+Н)+. Соединение 12. Этот продукт получают в соответствии с методикой-II, используя промежуточный продукт-4 (31 мг,0,1 ммоль), 2-хлорэтилхлорформиат (16 мкл, 0,15 ммоль) и DIEA (26 мкл, 0,15 ммоль) в дихлорметане (2- 24016575 мл), с получением целевого соединения в форме порошка белого цвета (22 мг, 53%). ES-MC: 414 (М+Н)+. Соединение 17. Этот продукт получают в соответствии с методикой-II, используя промежуточный продукт-1 (47 мг,0,15 ммоль), 4-метилфенилхлорформиат (33 мкл, 0,2 3 ммоль) и DIEA (39 мкл, 0,23 ммоль) в дихлорметане (2 мл), с получением целевого соединения в форме порошка белого цвета (34 мг, 51%). ES-MC: 447(М+Н)+. Соединение 23. Этот продукт получают в соответствии с методикой-II, используя промежуточный продукт-5 (30 мг,0,1 ммоль), этилхлорформиат (14 мкл, 0,15 ммоль) и DIEA (26 мкл, 0,15 ммоль) в дихлорметане (2 мл), с получением целевого соединения в форме порошка белого цвета (21 мг, 56%). ES-MC: 373 (М+Н)+. Соединение 25. Этот продукт получают в соответствии с методикой-VII, используя промежуточный продукт-9 (74 мг, 0,2 ммоль), 2-бромпиримидин (48 мг, 0,3 ммоль) и триэтиламин (42 мкл, 0,3 ммоль) в DMF (2 мл), с получением целевого соединения (42 мг, 47%). ES-MC: 447 (М+Н)+. Соединение 102. Этот продукт получают в соответствии с методикой-IIIb, используя промежуточный продукт-9 (74 мг, 0,2 ммоль), уксусный ангидрид (47 мкл, 0,5 ммоль) и пиридин (41 мкл, 0,5 ммоль) в дихлорметане (2 мл), с получением целевого соединения в форме порошка белого цвета (31 мг, 38%). ES-MC: 411 (М+Н)+. Соединение 140. Этот продукт получают в соответствии с методикой IV, используя промежуточный продукт-10 (72 мг, 0,2 ммоль), циклогексилизоцианат (26 мкл, 0,2 ммоль) и DIEA (37 мкл, 0,21 ммоль) в дихлорметане (2 мл), с получением целевого соединения в форме порошка белого цвета (51 мг, 53%). ES-MC: 486 (М+Н)+. Соединение 166. Этот продукт получают в соответствии с методикой-IIIa, используя промежуточный продукт в форме свободного амина (141 мг, 0,5 ммоль), Boc-L-аланин (105 мг, 0,6 ммоль) , DIC (94 мкл, 0,6 ммоль),DIEA (105 мкл, 0,6 ммоль) и дихлорметан (4 мл), с получением целевого соединения (105 мг, 46%). ESMC: 420 (М+Н)+. Соединение 225. Этот продукт получают в соответствии с методикой-VI, используя промежуточный продукт в форме свободного амина (78 мг, 0,2 ммоль), метилсульфонилхлорид (16 мкл, 0,2 ммоль) и DIEA (37 мкл, 0,21 ммоль) и дихлорметан (2 мл), с получением целевого соединения (32 мг, 34%). ES-MC: 461 (М+Н)+. Соединение 242. Этот продукт получают в соответствии с методикой-V, используя промежуточный продукт в форме свободного амина (59 мг, 0,2 ммоль), циклогексилизотиоцианат (29 мкл, 0,2 ммоль), DIEA (35 мкл, 0,2 ммоль) и дихлорметан (4 мл), с получением целевого соединения (52 мг, 60%). ES-MC: 438 (М+Н)+. Соединение 279. Этот продукт получают, производя промежуточный продукт-12 (6-хлор-1-[4-(2-морфолин-4-илэтокси)фенил]-2,3,4,9-тетрагидро-1 Нкарболин) с использованием методики-I. Промежуточный продукт-12 затем используют для получения соединения 279 (этиловый эфир 6-хлор-1-[4-(2-морфолин-4-илэтокси)фенил]-1,3,4,9-тетрагидрокарболин-2-карбоновой кислоты) с использованием методики-II. В соответствии с методикой-II промежуточный продукт-12 (82 мг, 0,20 ммоль), этилхлорформиат(2 мл) и перемешивают при комнатной температуре в течение 15 мин, чтобы получить соединение 279. Растворитель удаляют под потоком азота. Сырую смесь очищают препаративной ВЭЖХ с обратной фазой на колонке С-18, используя градиент ацетонитрила в воде, буферизованной 0,2% трифторуксусной кислотой (ТФК). Соль ТФК соединения 279 (3,7 мг, 3%) выделяют в форме твердого вещества желтого цвета. Та же самая методика может использоваться для других реакций с образованием карбамата в соответствии с методикой-II. Соединение 320. Этот продукт/промежуточный продукт получают,используя методику-I с 5 бензилокситриптаминомHCl (100 мг, 0,33 ммоль), пиридин-3-карбоксальдегид (62 мкл, 0,66 ммоль) и 0,1 н. серную кислоту (2 мл), с получением целевого соединения в форме гидросульфата (64 мг, 55%). ESМС: 356 (М+Н)+. Соединение 329. Этот продукт получают в соответствии с методикой-VII, используя промежуточный продукт-11 (71 мг, 0,2 ммоль), 2-бромпиримидин (48 мг, 0,3 ммоль) и триэтиламин (42 мкл, 0,3 ммоль) в DMF (2 мл), с получением целевого соединения (41 мг, 49%). ES-MC: 434 (М+Н)+. Соединение 330. Этот продукт получают в соответствии с методикой-II, используя промежуточный продукт-6 (65 мг,0,2 ммоль), 2-фторэтилхлорформиат (38 мкл, 0,3 ммоль) и DIEA (70 мкл, 0,4 ммоль) в дихлорметане (2 мл), с получением целевого соединения в форме порошка белого цвета (34 мг, 41%). ES-MC: 415 (М+Н)+.- 25016575 Соединение 332. Этот продукт получают в соответствии с методикой-II, используя промежуточный продукт-7 (36 мг,0,1 ммоль), 4-метоксифенилхлорформиат (22 мкл, 0,15 ммоль) и DIEA (26 мкл, 0,15 ммоль) в дихлорметане (2 мл), с получением целевого соединения в форме порошка белого цвета (41 мг, 81%). ES-MC: 511(М+Н)+. Пример 1 В. Некоторые исходные вещества, схема Ia. Схема Ia может использоваться в сочетании со схемой I (выше) для получения исходных веществ,когда R2 обозначает -СН 2-фуранил, следующим образом. Фурфурол (0,05 мл, 1,1 экв.) добавляют к раствору 5-хлортриптамина (114 мг, 0,586 ммоль) в 2 мл МеОН. Реакционную смесь перемешивают при комнатной температуре в течение приблизительно 1 ч. Медленно добавляют NaBH4 (110 мг, 5 экв.). Реакционную смесь перемешивают при комнатной температуре приблизительно в течение 30 мин. МеОН выпаривают и остаток распределяют между водой и метиленхлоридом. Органический слой отделяют и высушивают над K2CO3. Собранный органический слой концентрируют , получая 134,9 мг цистамина (84%). Пример 1 С. Соединения формулы I, схема Ib. Альтернативно, некоторые соединения формулы I могут быть получены согласно схеме Ib следующим образом. Суспензию материала реакции А (8,05 г, 35,9 ммоль) и CH3COONH4 (4,15 г, 1,5 экв.) в 60 млCH3NO2 нагревают с обратным холодильником в масляной бане приблизительно при 110 С. Приблизительно после 30 мин реакционную смесь охлаждают на ледяной бане. Осажденное твердое вещество отфильтровывают и промывают водой (3100 мл), затем гексаном (250 мл), получая сырой продукт В в форме индола. Собранное твердое вещество высушивают под вакуумом при приблизительно 40 С в течение приблизительно 30 мин, получая 6,97 г твердого вещества коричневого цвета (73%). Раствор индолового продукта В (12,32 г, 46,1 ммоль) в ТГФ (130 мл) медленно обрабатывают раствором тетрабутиламмонийборогидрида (11,9 г, 1 экв.) в 75 мл ТГФ в течение приблизительно 60 мин при приблизительно -5 С. Реакционную смесь перемешивают при комнатной температуре в течение приблизительно 1 ч и разбавляют дихлорметаном (200 мл). Органический слой промывают водой дважды и раствором соли. Объединенные органические слои высушивают и выпаривают под вакуумом. Остаток очищают на силикагеле, получая 10,28 г твердого вещества С (83%). Хлорид аммония (9,9 мл водного раствора (100 мг/мл), 2 экв.) и Zn (725 мг, 1,2 экв.) добавляют к раствору индолового продукта С (2,49 г, 9,24 ммоль) в 161 мл ТГФ. Реакционную смесь перемешивают при комнатной температуре в течение приблизительно 10 мин и добавляют Zn (725 мг, 1,2 экв.). Приблизительно после 30 мин добавляют дополнительный Zn (967 мг, 1,6 экв.) и перемешивают в течение приблизительно 2 ч, после чего снова добавляют Zn (845 мг, 1,4 экв.). После перемешивания при комнатной температуре в течение приблизительно 15 мин Zn отфильтровывают и остаток концентрируют и растворяют в ТГФ. Полученный раствор обрабатывают п-хлорбензальдегидом (0,7 экв.) и перемешивают при комнатной температуре в течение приблизительно 15 ч. Реакционную смесь концентрируют под вакуумом и очищают на силикагеле, получая 953,5 мг желаемого нитронового продукта D.(+)-DIP-Cl (6,93 мл, 2 экв., 85,8 мг/мл в CH2Cl2) добавляют к раствору нитронового продукта D (350 мг, 0,93 ммоль) в 60 мл дихлорметана. Реакционную смесь перемешивают при приблизительно -78 С в течение приблизительно 10 дней и реакцию останавливают смесью 10% NaHCO3 (7 мл) и 10 мл воды. Водный слой экстрагируют трижды дихлорметаном. Объединенные органические слои концентрируют и очищают на силикагеле, получая желаемый продукта гидроксиламина Е (98% ее). Воду (11,5 мл), NH4Cl (2,5 мл, 5 экв.) и Zn (908 мг, 15 экв.) добавляют к раствору продукта гидроксиламина Е (0,927 ммоль) в ТГФ (28 мл). Реакционную смесь перемешивают при комнатной температуре в течение приблизительно 1 дня. Добавляют дополнительный ТГФ (10 мл), NH4Cl (5 мл, 10 экв.) и Zn(1,8 г, 30 экв.) и перемешивают в течение приблизительно еще 21 ч. Снова добавляют ТГФ (10 мл),NH4Cl (5 мл, 10 экв.) и Zn (1,8 г, 30 экв.) и перемешивают в течение приблизительно еще 20 ч. Реакционную смесь фильтруют через целит и промывают МС. Собранный слой дихлорметана промывают водой и раствором соли. Органический слой высушивают и концентрируют, получая бороновый комплекс карболина. Этот продукт растворяют в 20 мл ТГФ. Этот раствор загружают на готовую катионобменную смолу (предварительно обработанную МеОН и ТГФ) и промывают ТГФ. Объединенный раствор ТГФ концентрируют, получая 390 мг свободного амина. Твердое вещество последовательно промывают эфиром и гексаном, чтобы получить 130 мг энантиомерно чистого соединения F. Пример 1D. Соединения формулы I, схема II. Соединения формулы I-h могут быть получены согласно схеме II следующим образом. п-Анизальдегид (2,16 г, 15,9 ммоль, 1,93 мл) добавляют к суспензии 5-бромтрилтофана А (3 г, 10,6 ммоль) в 100 мл уксусной кислоты при комнатной температуре. Реакционную смесь нагревают с обратным холодильником при приблизительно 125 С в ванне силиконового масла и поддерживают при этой температуре в течение приблизительно 3 ч 20 мин. Полученный раствор концентрируют под вакуумом. Остаток растирают с дихлорметаном, диэтиловым эфиром и гексаном, чтобы получить порошкообразное твердое вещество коричневого цвета. Ацетатные соли промежуточного продукта В собирают и трижды промывают гексаном. Промежуточный продукт В суспендируют (70 мг, 0,174 ммоль) в 2 мл дихлорметана и к суспензии добавляют триэтиламин (52,8 мг, 0,522 ммоль), 5-метил-2-аминотиазол (37,6 мг, 0,2 6 ммоль) и РуВОР(135,8 мг, 0,2 6 ммоль). Реакционную смесь перемешивают при комнатной температуре в течение приблизительно 6 ч и реакцию останавливают насыщенным раствором NaHCO3. Водный слой экстрагируют дихлорметаном. Объединенные органические слои высушивают над K2CO3 и концентрируют. Очистка на силикагеле с 40%-ным этилацетатом в гексане приводит к 8,1 мг желаемого амида С. ЖХ-МС [МН+] 498,Rt = 2,54. Пример 1E. Соединения формулы I, схема III. Соединения формулы I-i могут быть получены согласно схеме III следующим образом. Триптофан А (1,0 г, 5,0 ммоль) и 3-метоксибензальдегид (670 мкл, 5,5 ммоль) суспендируют/растворяют в ацетонитриле (100 мл) и добавляют концентрированную серную кислоту (100 мкл). Ре- 27016575 акционную смесь нагревают с обратным холодильником до полного израсходования альдегида (в течение ночи). Растворитель удаляют в вакууме и остаток растворяют в 5 мл этанола. Продукт осаждают диэтиловым эфиром, фильтруют и промывают 10 мл простого эфира. Желаемый -карболиновый продукт/промежуточный проукт В (1-(3-метоксифенил)-2,3,4,9-тетрагидро-1 Нкарболин-3-карбоновая кислота) выделяют в форме твердого вещества бежевого цвета (1,2 г, 76%). LC/MC RT = 2,33 мин. M/Z+ 323, 100%.-карболиновый продукт/промежуточный продукт В (200 мг, 0,62 ммоль) растворяют в 5 мл сухого ТГФ и охлаждают до приблизительно 0 С. Раствор алюмогидрида лития (LAH) (1,2 мл, 1,0 М в диэтиловом эфире, 1,2 ммоль) добавляют к охлажденной реакционной смеси в атмосфере азота. После завершения добавления (приблизительно 10 мин) реакционной смеси дают нагреться до комнатной температуры в течение приблизительно 4 ч. Реакционную смесь затем снова охлаждают до 0 С и добавляют насыщенный раствор (750 мкл) сульфата натрия и смесь перемешивают в течение приблизительно 5 мин при 0 С. Реакционную смесь фильтруют и промывают ТГФ (100 мл). Растворитель удаляют в вакууме и сырой продукт очищают препаративной ВЭЖХ. Продукт С ([1-(3-метоксифенил)-2,3,4,9-тетрагидро-1 Н-карболин-3-ил]метанол) выделяют в форме твердого вещества белого цвета (106 мг, 55%). LC/MC RT = 2,25 мин. M/Z+ 309, 100%. Пример 1F. Химическое разделение соединений данного изобретения. Соединения изобретения могут быть необязательно химически выделены в форме энантиомерно чистых композиций, предпочтительно энантиомерно чистых композиций (S) изомера следующим образом. Рацемический амин А (18,21 г, 58,2 ммоль) смешивают с N-ацетил-L-фенилаланином (12,05 г, 58,2 ммоль) в EtOH (1,28 л) и нагревают с обратным холодильником, чтобы получить прозрачный раствор. Раствору дают охладиться до комнатной температуры. По истечении ночи осажденное твердое вещество отфильтровывают и промывают EtOH (200 мл), получая соль В (16,4 г). Соль В помещают в EtOAc (500 мл) и промывают водным 1 н. NaOH (300 мл 2) или NH4OH (200 мл 2), высушивают и выпаривают,получая S-изомер свободного амина С (7,4 г). R-изомер получают в соответствии с подобной методикой,используя N-ацетил-D-фенилаланин. Пример 1G. Примеры других соединений изобретения. В качестве других неограничивающих примеров следующие соединения могут быть получены в соответствии с методикой, подобной описанной выше, как будет очевидно для специалиста.
МПК / Метки
МПК: C07D 471/04, A61K 31/541, A61K 31/5355, A61K 31/497, A61P 35/00, A61K 31/551, A61K 31/506, A61K 31/437
Метки: полезные, производные, карболина, ингибировании, ангиогенеза
Код ссылки
<a href="https://eas.patents.su/30-16575-proizvodnye-karbolina-poleznye-v-ingibirovanii-angiogeneza.html" rel="bookmark" title="База патентов Евразийского Союза">Производные карболина, полезные в ингибировании ангиогенеза</a>
Производные азобициклооктанов и нонанов обладающие активностью в ингибировании dpp – iv

Номер патента: 8166
Опубликовано: 27.04.2007
Авторы: Шушан Эдит, Сабо Тибор, Араньи Петер, Балаж Ласло, Боронкаи Эва, Варга Мартон, Бата Имре, Батори Шандор, Урбан-Сабо Каталин, Капуи Зольтан, Т.Надь Лайош
МПК: C07D 207/06, A61K 31/46, A61P 3/10...
Метки: обладающие, ингибировании, азобициклооктанов, производные, нонанов, активностью
Формула / Реферат:
1. Соединения общей формулы (I) в которой R означает азотсодержащий ароматический фрагмент с одним или двумя циклами, состоящий из одного или двух ароматических циклов, предпочтительно пиридила, пиридазинила, пиримидинила, пиразинила, имидазолила, пиразолила, тиазолила, изотиазолила, оксазолила, изоксазолила, оксадиазолила, хинолинила, изохинолинила, циннолинила, фталазинила, хиназолинила, хиноксалинила, бензимидазолила, индазолила,...
Производные 6-фенилпиридил-2-амина, полезные в качестве ингибиторов nos

Номер патента: 2907
Опубликовано: 31.10.2002
Автор: Лоу Джон Адамс III
МПК: C07D 213/73, A61K 31/4418, A61P 11/06...
Метки: ингибиторов, полезные, производные, 6-фенилпиридил-2-амина, качестве
Формула / Реферат:
1. Соединение формулы где R1 и R2 независимо выбраны из водорода, гидрокси, метила и метокси; и G является группой формулы где n равно нулю или единице; Y представляет собой NR3R4, (С1-С6)алкил или аралкил, где арильная группировка указанного аралкила является фенилом или нафтилом, а алкильная группировка является нормальной или разветвленной и содержит от 1 до 6 атомов углерода и где указанный (С1-С6)алкил и арильная группировка указанного...
Производные тропана, полезные для терапии

Номер патента: 5382
Опубликовано: 24.02.2005
Авторы: Прайс Дэвид Энтони, Вуд Энтони, Перрос Маноуссос, Стаммен Бланда Люциа Криста
МПК: A61P 29/00, A61K 31/46, C07D 451/04...
Метки: производные, полезные, терапии, тропана
Формула / Реферат:
1. Соединение формулы где R1 означает C3-6-циклоалкил, необязательно замещенный одним или более атомами фтора, или C1-6-алкил, необязательно замещенный одним или более атомами фтора, или C3-6-циклоалкилметил, необязательно замещенный по кольцу одним или более атомами фтора, и R2 означает фенил, необязательно замещенный одним или более атомами фтора; или его фармацевтически приемлемая соль или сольват. 2. Соединение по п.1 формулы где R1...
Производные тиенопиримидина и тиенопиридина, полезные в качестве противораковых агентов

Номер патента: 5889
Опубликовано: 30.06.2005
Авторы: Соболов-Джейнз Сьюзен Бет, Манчхоф Майкл Джон
МПК: A61K 31/505, C07D 495/04
Метки: тиенопиридина, производные, агентов, противораковых, качестве, тиенопиримидина, полезные
Формула / Реферат:
1. Соединение формулы 1 или 2 или его фармацевтически приемлемая соль или гидрат, где X1 представляет собой CH; R1 представляет собой H, C1-C6алкил или -C(O)(C1-C6алкил); R2 представляет собой C6-C10арил или 5-13-членный гетероцикл, причем указанные группы R2 возможно замещены заместителями R5 в количестве от 1 до 5; каждый R5 независимо выбран из галогено, циано, нитро, трифторметокси, трифторметила, азидо, -C(O)R8, -C(O)OR8, -OC(O)R8,...
Новые производные бензимидазола, полезные в качестве антипролиферативных агентов

Номер патента: 8448
Опубликовано: 29.06.2007
Авторы: Уонг Хьюйфен Фэйе, Кат Джон Чарльз, Лиссикэйтос Джозеф Питер
МПК: A61K 31/415, C07D 215/00, A61P 35/00...
Метки: агентов, производные, антипролиферативных, качестве, бензимидазола, новые, полезные
Формула / Реферат:
1. Соединение, выбранное из группы, состоящей из (+)-1-{2-[5-(тетрагидрофуран-3-илокси)бензимидазол-1-ил]хинолин-8-ил}пиперидин-4-иламина; (+)-1-{2-[5-(тетрагидрофуран-3-илокси)бензимидазол-1-ил]хинолин-8-ил}пиперидин-4-иламина; (-)-1-{2-[5-(тетрагидрофуран-3-илокси)бензимидазол-1-ил]хинолин-8-ил}пиперидин-4-иламина; 1-{2-[5-(3-метилоксетан-3-илметокси)бензимидазол-1-ил]хинолин-8-ил}пиперидин-4-иламина;...
Предыдущий патент: Летательный аппарат
Следующий патент: Применение сложных эфиров гидроксибензойной кислоты для получения лекарственного средства для предупреждения и лечения папилломавирусной инфекции человека
Случайный патент: Псевдоожижение популяции каталитических частиц, характеризующейся низким содержанием каталитической мелочи