Дикетопиперазиновые и пиперидиновые производные в качестве противовирусных агентов
Номер патента: 14957
Опубликовано: 29.04.2011
Авторы: Жанг Жонгксинг, Кэдоу Джон Ф., Хан Йинг, Ванг Тао, Минвелл Николас А., Йин Живей, Карини Дэвид Дж., Регуэйро-Рен Алисия, Свидорски Джейкоб




















Формула / Реферат
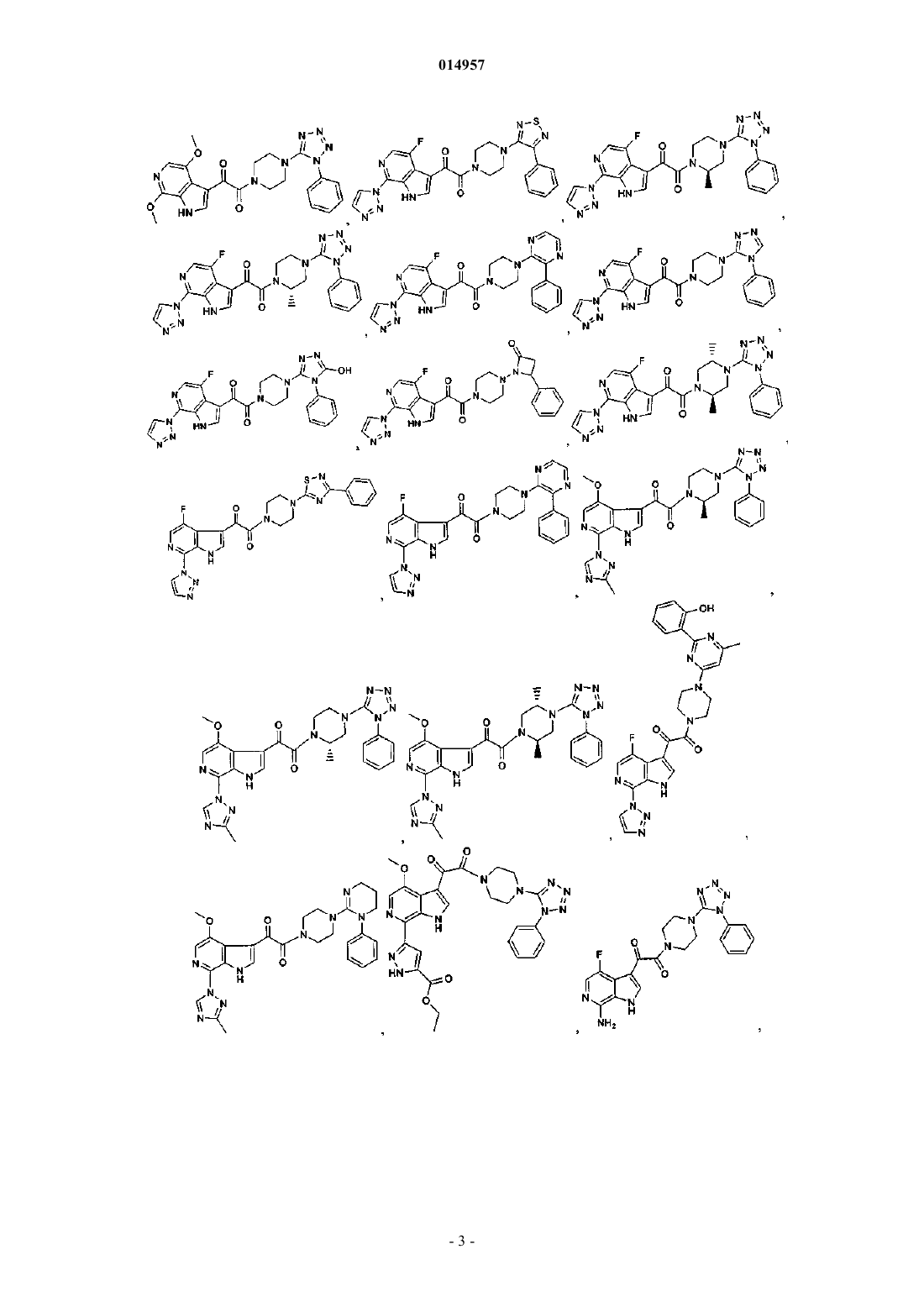
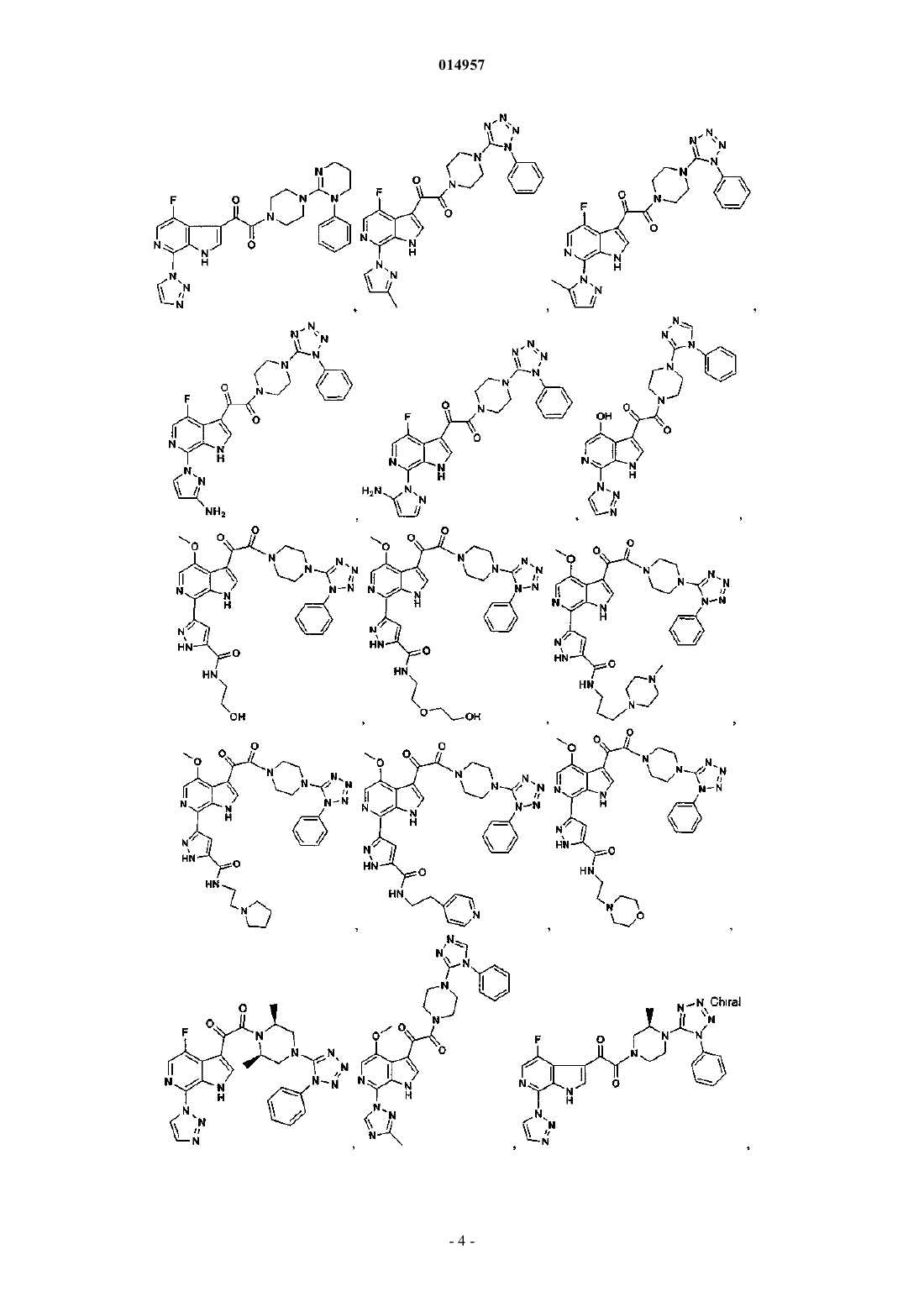
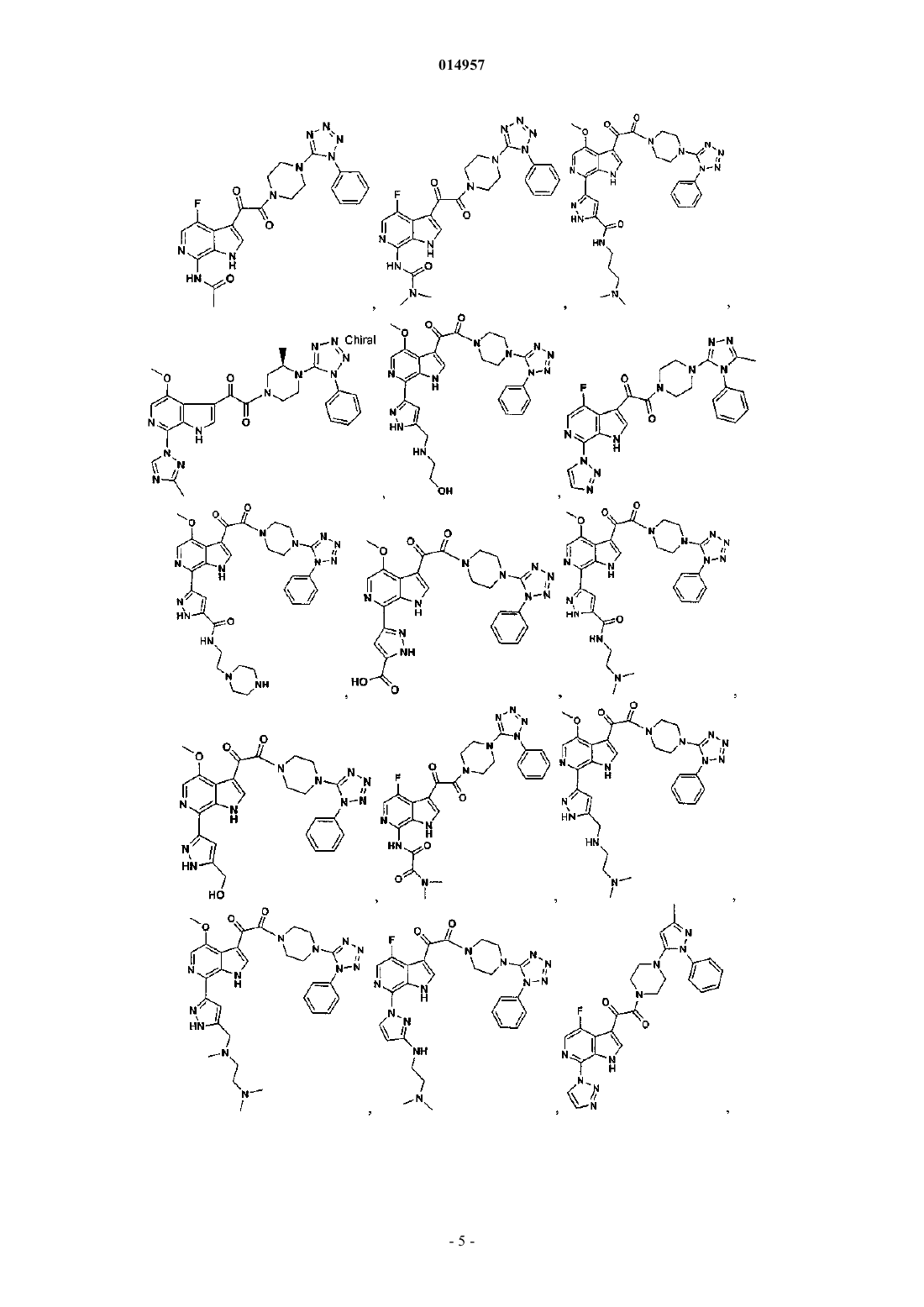
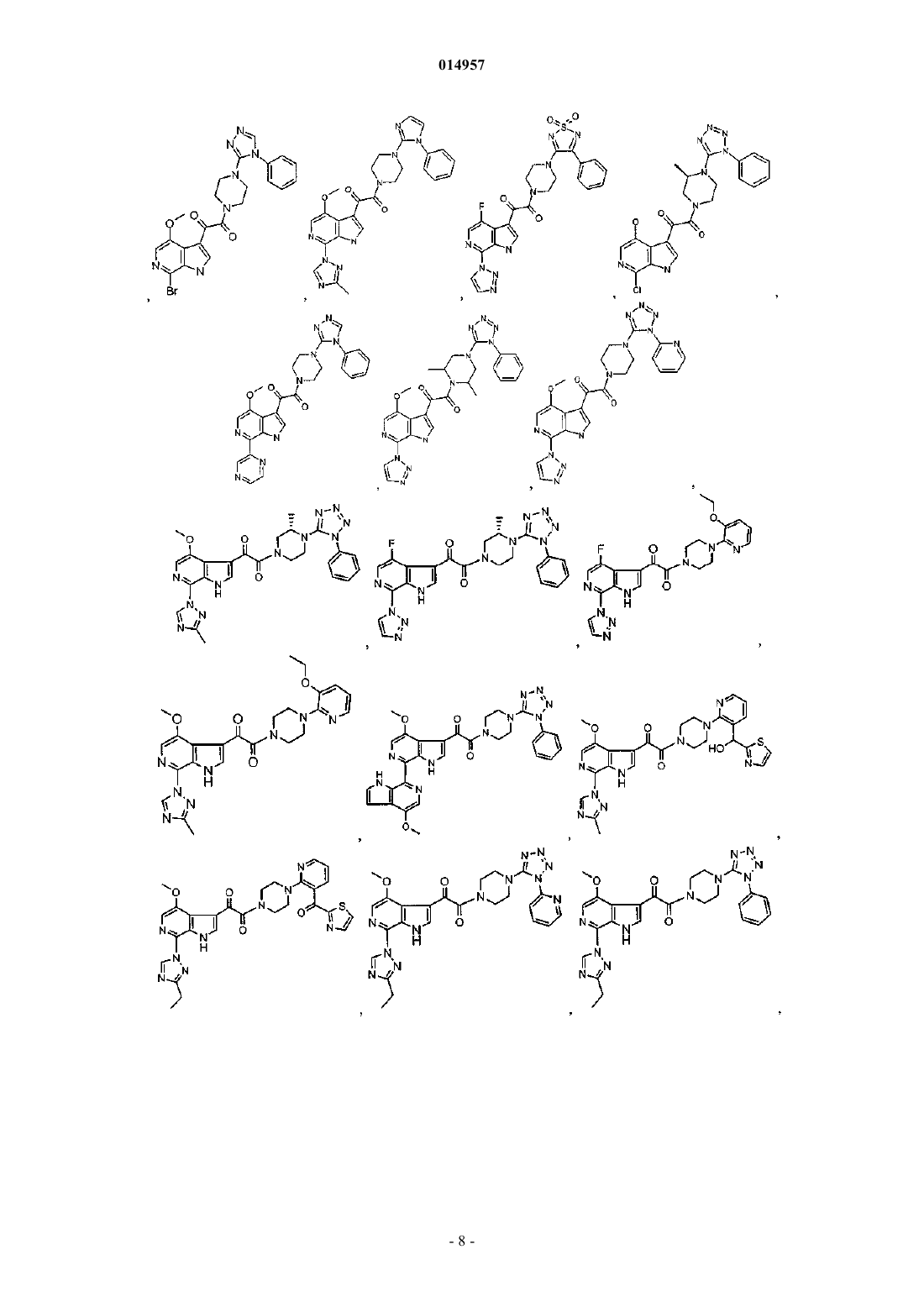
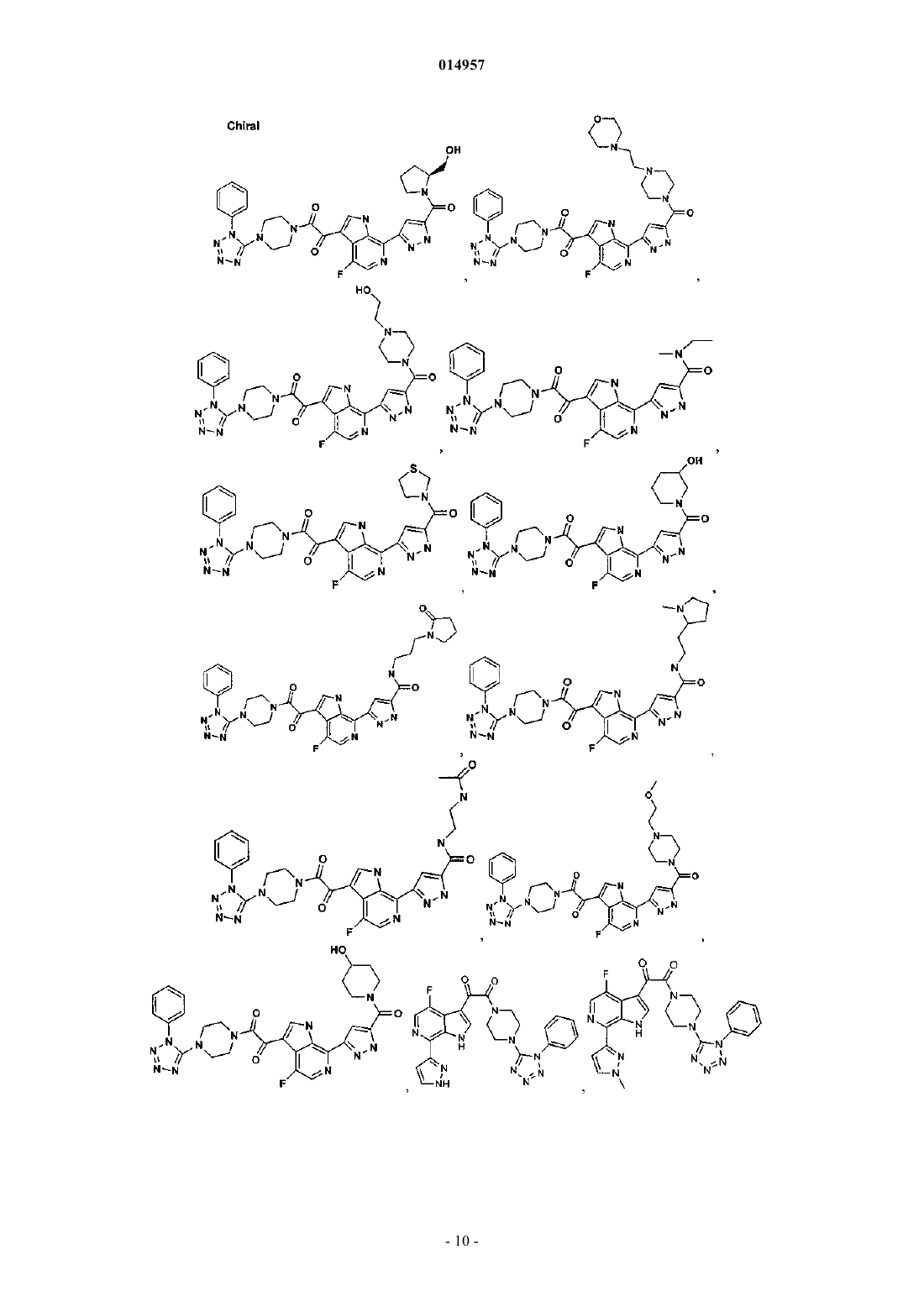
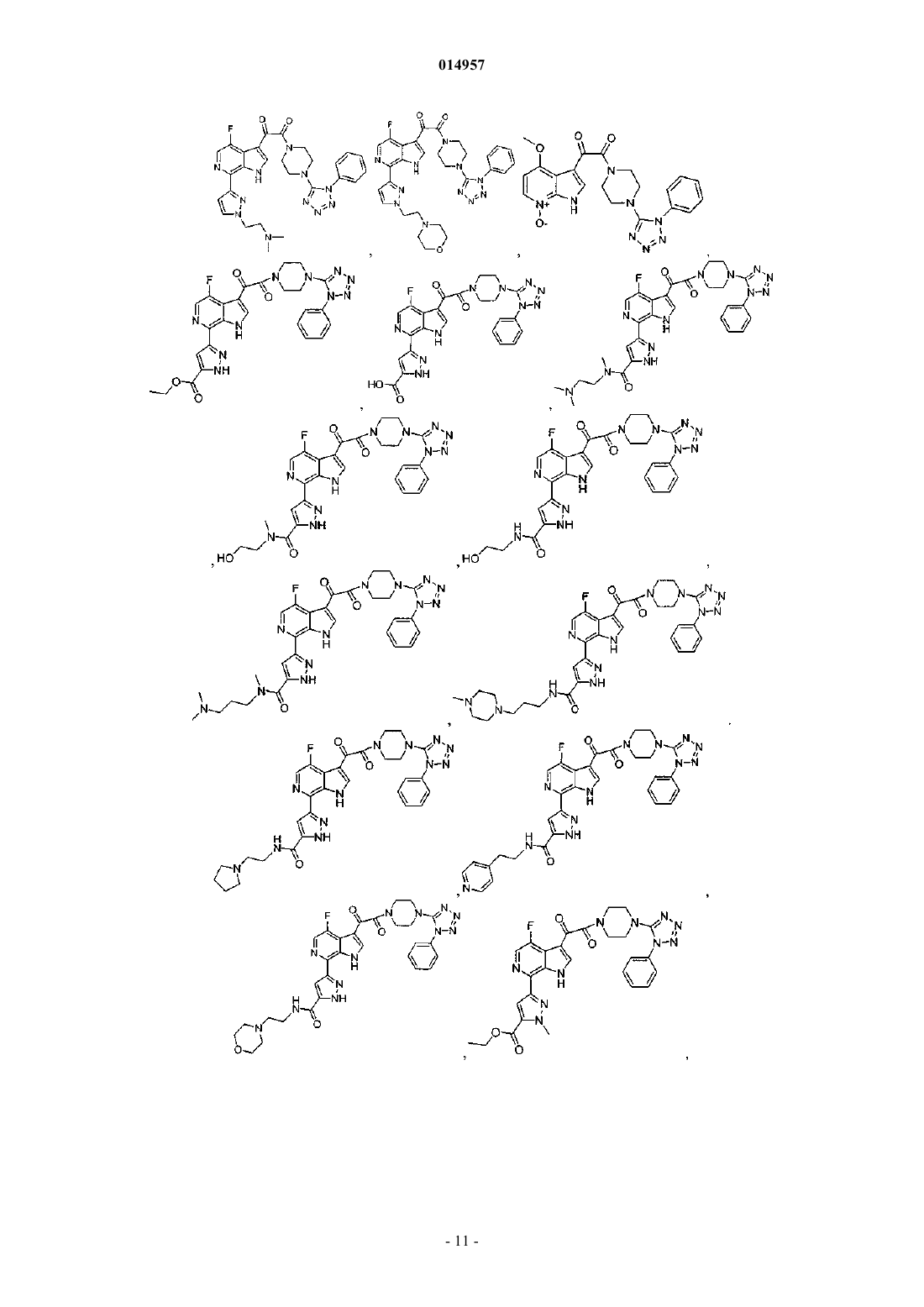
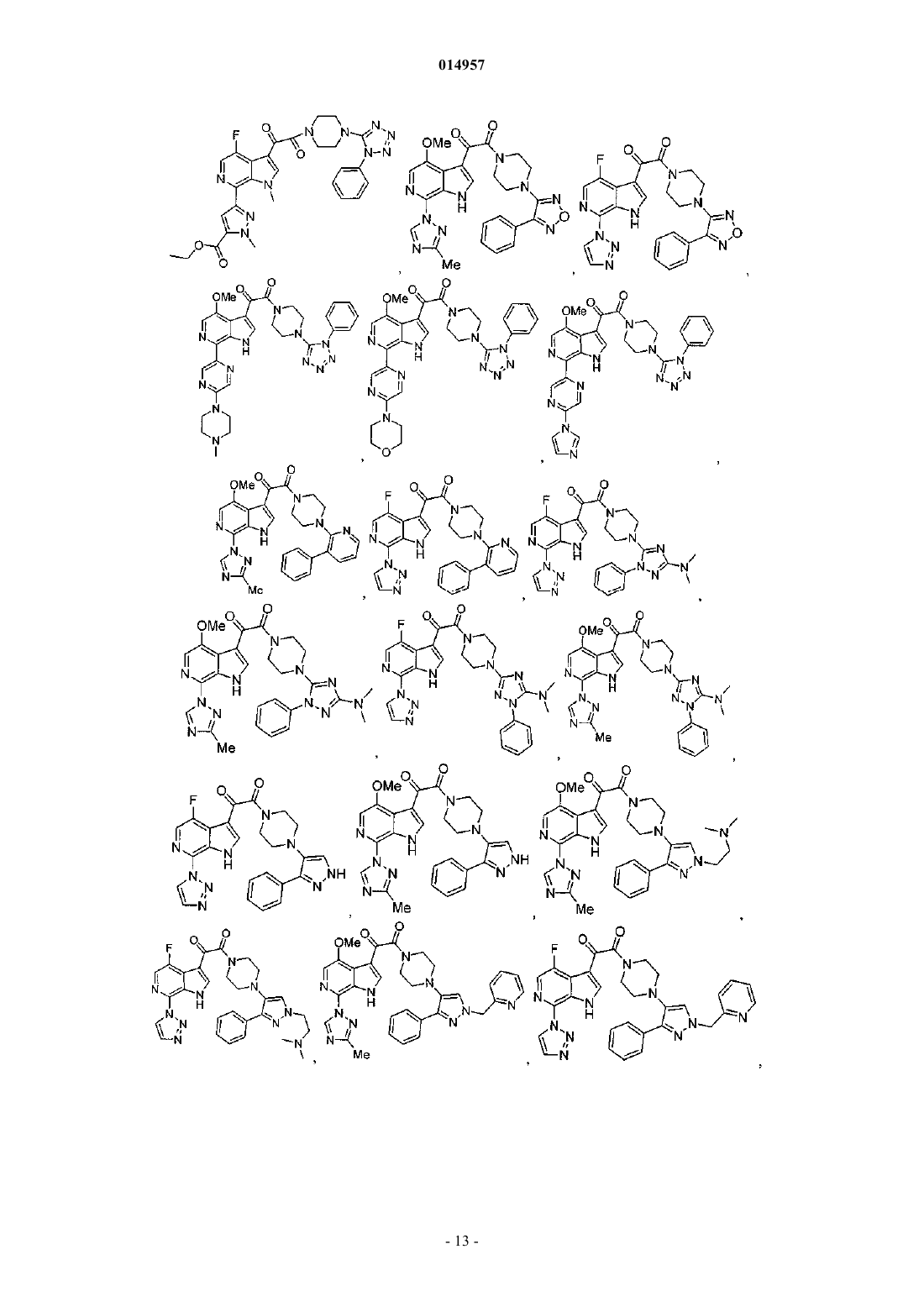
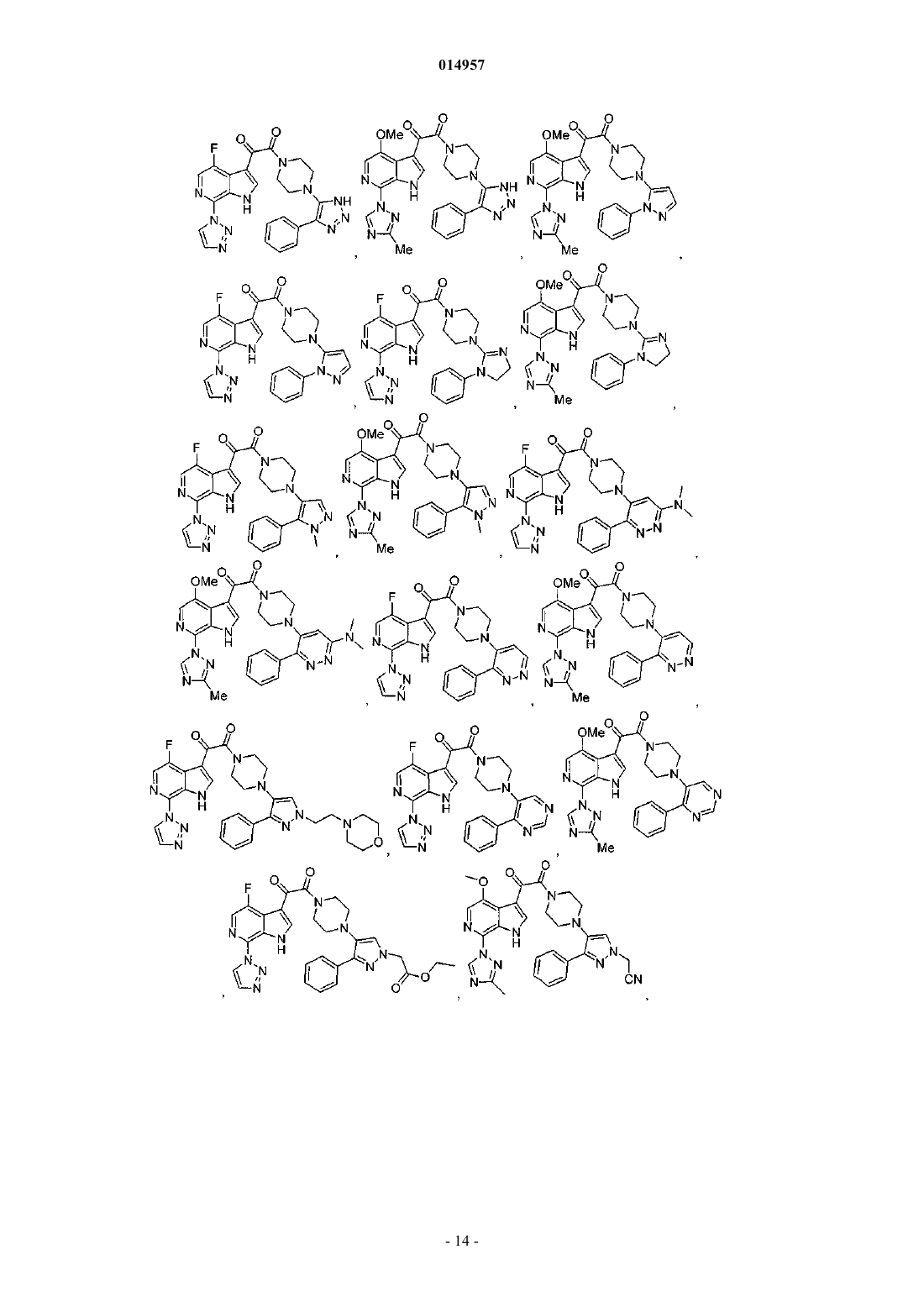
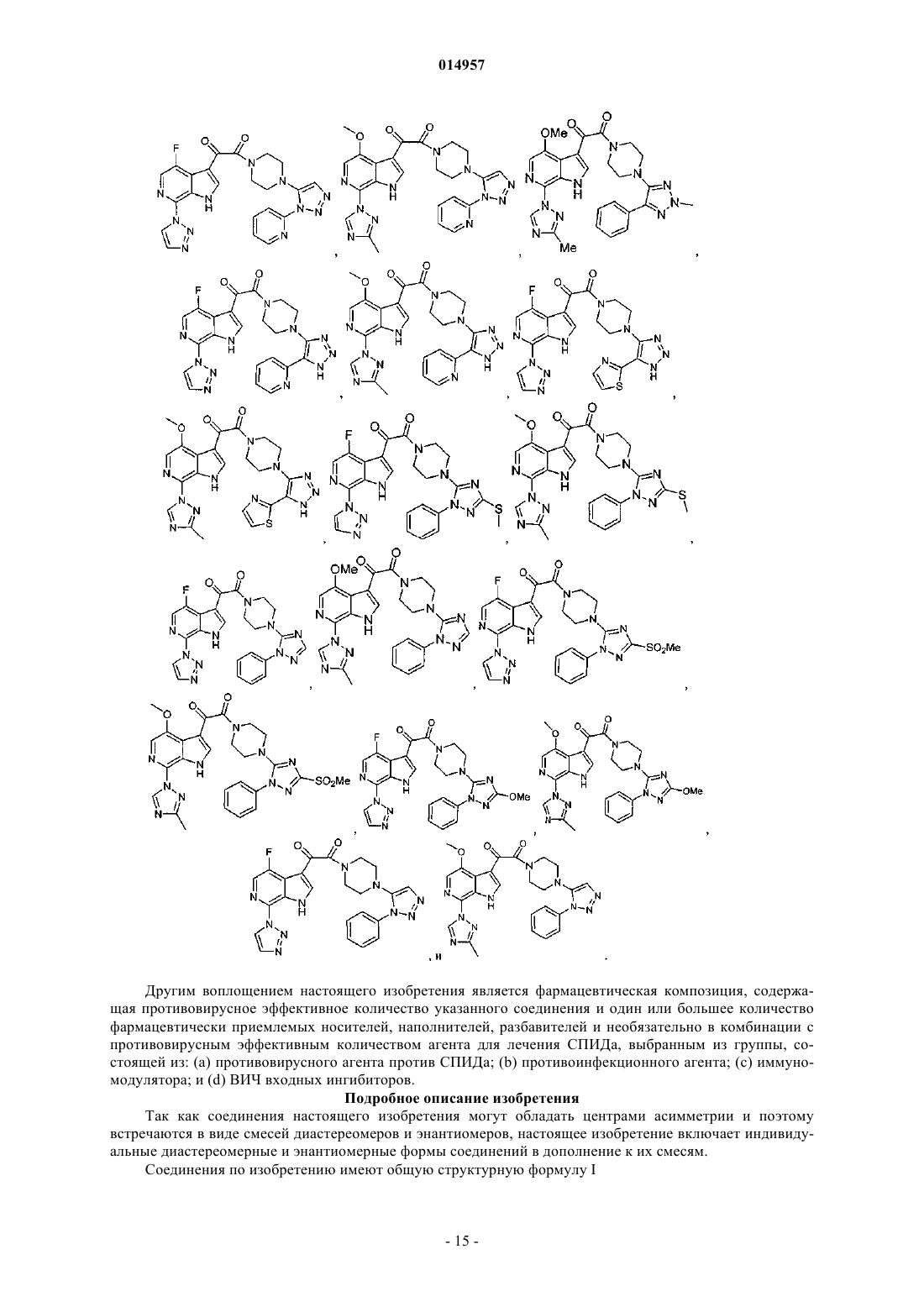
1. Соединение, выбранное из группы, состоящей из


















































2. Фармацевтическая композиция, содержащая противовирусное эффективное количество соединения по п.1 или его фармацевтически приемлемую соль и один или большее количество фармацевтически приемлемых носителей, наполнителей или разбавителей.
3. Композиция по п.2, дополнительно содержащая второе соединение, обладающее противо-ВИЧ активностью.
4. Фармацевтическая композиция по п.3, в которой соединение, обладающее противо-ВИЧ активностью, выбрано из группы, состоящей из:
(а) противовирусного агента против СПИДа;
(б) противоинфекционного агента;
(с) иммуномодулятора и
(д) ВИЧ входных ингибиторов.
Текст
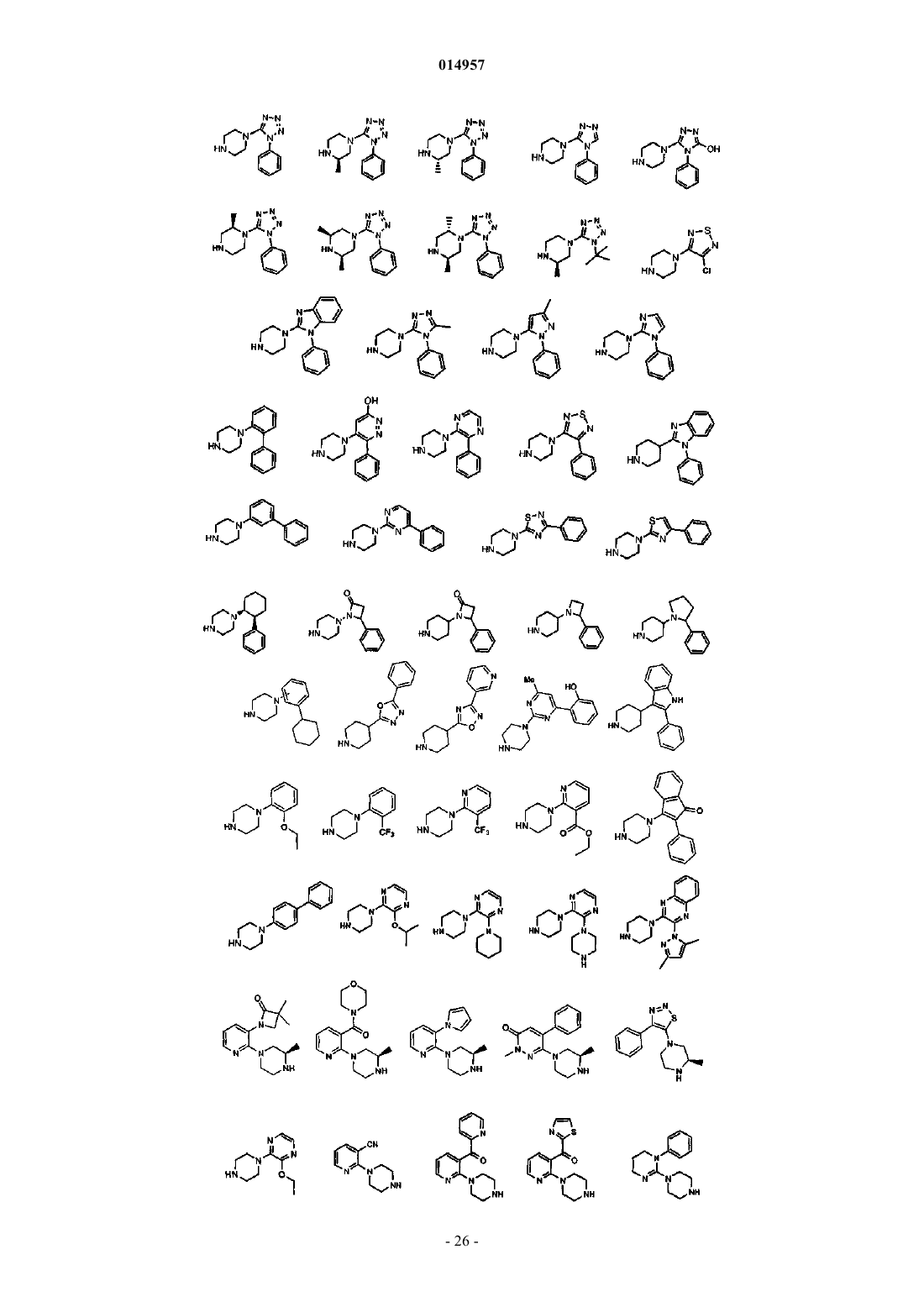
Изобретение предлагает соединения, обладающие фармакологическими и биологически активными свойствами, их фармацевтические композиции и способ их применения. В частности, изобретение относится к дикетопиперазиновым и пиперидиновым производным, которые обладают уникальной противовирусной активностью. В особенности, настоящее изобретение касается соединений, применимых для лечения ВИЧ-инфекции и СПИДа. 014957 Область техники Изобретение относится к соединениям, обладающим биологически активными и фармакологическими свойствами, их фармацевтическим композициям и способам их применения. В частности, изобретение относится к новым дикетопиперазиновым и пиперазиновым производным, которые обладают уникальной противовирусной активностью. В особенности, настоящее изобретение касается соединений, применимых для лечения ВИЧ-инфекции и СПИДа. Уровень техники ВИЧ-1 (вирус иммунодефицита человека 1-го типа) инфекция остается главной медицинской проблемой, касающейся приблизительно 40 миллионов людей, инфицированных во всем мире, по данным на конец 2005 г. Количество случаев заболевания ВИЧ и СПИДом (приобретенный синдром иммунодефицита) быстро увеличивается. В 2005 сообщалось приблизительно о 5,0 миллионах новых инфекций, 3,1 миллиона людей умерло от СПИДа. В настоящее время доступные препараты для лечения ВИЧ включают нуклеозидные ингибиторы обратной транскриптазы (RT) или улучшенные отдельные комбинации:(зидовудин или AZT или Retrovir), диданозин (или Videx), ставудин (или Zerit), ламивудин (или 3 ТС или Epivir), залцитабин (или DDC или Hivid), абакавира сукцинат (или Ziagen), соль тенофовира дизопроксила фумарата (или Viread), эмитрицитабин (или FTC), Combivir (содержит -3 ТС и AZT),Trizivir (содержит абакавир, ламивудин и зидовудин), Epzicom (содержит абакавир и ламивудин),Truvada (содержит Viread и эмтрицитабин); ненуклеозидные ингибиторы обратной транскриптазы: невирапин (или Viramune), делавирдин (или Rescriptor) и эфавиренц (или Sustiva) и пептидомиметические ингибиторы протеазы или улучшенные составы: саквинавир, индинавир, ритонавир, нелфинавир, ампренавир, лопинавир и Kaletra (лопинавир и Ритонавир). Каждое из этих лекарственных средств может лишь кратковременно сдерживать репликацию вируса при использовании их по отдельности. В то же время при использовании их в комбинации эти препараты оказывают сильное действие на виремию и прогрессирование заболевания. Фактически значительное снижение уровня смертности среди пациентов,заболевших СПИДом, в последнее время отмечается вследствие широкого применения комбинированной терапии. В то же время, несмотря на эти впечатляющие результаты у 30-50% пациентов, в конечном счете, комбинированное лечение оказывается безуспешным. Недостаточная эффективность препарата,несоблюдение пациентом режима терапии, ограниченное проникновение в ткани и лекарство-специфические ограничения для определенных типов клеток (например, большинство нуклеозидных аналогов не может быть фосфорилировано в покоящихся клетках) могут обуславливать неполную супрессию чувствительных вирусов. Более того, высокий уровень репликации и быстрый оборот ВИЧ-1, сочетающиеся с частым появлением мутаций, приводят к появлению резистентных форм и безуспешному лечению, когда применяются субоптимальные дозы препаратов. Поэтому новые анти-ВИЧ агенты, демонстрирующие особые модели резистентности и подходящую фармакокинетику, равно как и безопасные профили, требуются для обеспечения более широких терапевтических возможностей. Улучшенные ВИЧ ингибиторы слияния и ВИЧ входные корректирующие антагонисты представляют собой в настоящее время два примера новых классов агентов протв ВИЧ, изучаемых рядом исследователей. Свойства класса ВИЧ ингибиторов входа, именуемые ВИЧ ингибиторами прикрепления, были улучшены в попытке получить соединения с максимизируемой полезностью и эффективностью в качестве противовирусных агентов. Было выявлено раскрытие, описывающее индолы, структура которых, показанная ниже для BMS-705, является представительной [Противовирусные Индолоксоацетильные Пиперазиновые производные]. Два других соединения, названные в уровне техники как BMS-806 и BMS-043 описаны как в научной, так и в патентной литературе:-1 014957 Ряд описаний их свойств, выявленных в клинических испытаниях на человеке, раскрыты в уровне техники. Следует отметить, что во всех трех указанных структурах пиперазинамид (в этих трех структурах пиперазинфениламид) присутствует, и указанная группа непосредственно присоединена к оксоацетильному остатку. Оксоацетильная группа присоединена по 3-положению 4-фториндола в BMS-705 и по 3 положению замещенных азаиндолов в BMS-806 и BMS-043. В попытке получить улучшенные соединения против ВИЧ более поздние публикации описывают частично модифицированные структурные заместители на индолах и азаиндолах. Примеры таких попыток включают: (1) новые замещенные индолоксоуксусные пиперазиновые производные, (2) замещенные пиперазинилоксоацетилиндольные производные и (3) замещенные азаиндолоксоуксусные пиперазиновые производные. Замещение указанных групп с помощью других гетероароматических или замещенных гетероароаматических или бициклических углеводородов также представлено как реально выполнимое. Примеры включают: (1) индол, азаиндол и родственные гетероциклические амидопиперазиновые производные; (2) бицикло 4.4.0 противовирусные производные; и (3) диазаиндольные производные. Выбор нескольких замещений для пиперазинамидной части молекул также описан в уровне техники и среди указанных примеров представлены (1) некоторые пиперидиновые алкены; (2) некоторые пирролидиновые амиды; (3) некоторые N-арильные или гетероарильные пиперазины; (4) некоторые пиперазинильные мочевины; и (5) некоторые содержащие карболин соединения. Способ(ы) получения пролекарств для указанного класса соединений раскрыт в Prodrugs ofPiperazine and Substituted Piperidine Antiviral Agents (Ueda et al.), U.S. заявка No. 11/066,745, зарегистрированная 02/25/2005 или US 20050209246A1 или WO 2005090367Al). Опубликованная РСТ заявка WO 2003103607Al (Июнь 11, 2003) раскрывает анализ, полезный для определения некоторых ВИЧ ингибиторов. Ряд опубликованных заявок описывают комбинированное лечение с пиперазинбензамидными ингибиторами, например, US 20050215543 (WO 2005102328A1), US 20050215544 (WO 2005102391A1) и US 20050215545 (WO 2005102392 А 2). Опубликованы новые соединения этого класса ингибиторов прикрепления (Jinsong Wang et. al. Org.Biol. Chem. 2005, 3, 1781-1786.) и заявка на некоторые более удаленно связанные соединения WO 2005016344. Опубликованные заявки WO 2005016344 и WO 2005121094 также описывают пиперазиновые производные, которые являются ВИЧ ингибиторами. Соединения, описанные в указанных заявках, структурно отличны от соединений настоящего изобретения. Ничто в указанных ссылках не может быть рассмотрено как раскрытие или предложение новых соединений по настоящему изобретению и их использования для ингибирования ВИЧ-инфекции. Сущность изобретения Настоящее изобретение относится к соединениям, структурные формулы которых представлены ниже. Изобретение также отностится к фармацевтическим композициям, содержащим данные соединенияи или их фармацевтически приемлемые соли, которые обладают противовирусным действием. Другим воплощением настоящего изобретения является фармацевтическая композиция, содержащая противовирусное эффективное количество указанного соединения и один или большее количество фармацевтически приемлемых носителей, наполнителей, разбавителей и необязательно в комбинации с противовирусным эффективным количеством агента для лечения СПИДа, выбранным из группы, состоящей из: (а) противовирусного агента против СПИДа; (b) противоинфекционного агента; (с) иммуномодулятора; и (d) ВИЧ входных ингибиторов. Подробное описание изобретения Так как соединения настоящего изобретения могут обладать центрами асимметрии и поэтому встречаются в виде смесей диастереомеров и энантиомеров, настоящее изобретение включает индивидуальные диастереомерные и энантиомерные формы соединений в дополнение к их смесям. Соединения по изобретению имеют общую структурную формулу I где А может быть выбран из группы, состоящей изR4, R5, R6 и R7 каждый независимо выбран из группы, состоящей из водорода, гидрокси, галогена,циано, нитро, С 1-С 4 алкила, С 1-С 4 фторалкила, ORa, NRaRb, COORa и группы В;Y выбран из группы, состоящей из фенила, С 5-С 7 моноциклического гетероарила, С 9-С 10 бициклического арила, С 9-С 10 бициклического гетероарила, С 4-С 7 гетероалициклила и С 5-С 7 циклоалкила, где указанный гетероарил или гетероалициклил содержит от 1 до 4 гетероатомов, выбранных из О, N и S и при условии, что когда Y представляет собой бициклический гетероарил оба X и Y присоединены к общему кольцу, где указанный арил, гетероарил и гетероалициклил необязательно замещены от одного до трех атомами галогена или от одного до трех заместителями, выбранными из оксо, гидроксила, С 1-С 6 алкила,-NR55R56, -ОС 1-С 3 алкила, -S-R1, -S(O)2R1, CF3, CN; где указанный C1-С 6 алкил может быть необязательно замещен группой В;Z выбран из группы, состоящей из С 1-6 алкила, С 1-6 алкокси, С 3-7 циклоалкила, -COOR3, 4, 5 или 6 членного кольцевого циклического N-лактама, -C(O)NR42R43, C(O)R57, где R57 необязательно замещен CN или группой В; -NR55R56, арила и гетероарила; указанный арил или гетероарил необязательно замещен от одного до двух членами, выбранными из группы, состоящей из амино, нитро, циано, гидрокси, C1-6 алкокси, -C(O)NH2, C1-6 алкила, -NC(O)CH3, галогена, трифторметила и группы В; группа В выбрана из группы, состоящей из -C(O)NR40R41, арила, гетероарила, гетероалициклила,C(O)R3, C(=N-O-R1)R3, ацеталя, UR8a, (C1-6)алкин-NR40R41, (С 1-6)алкил-COOR8b; где указанный арил, гетероарил и гетероалициклил необязательно замещены от одного до трех атомами галогена или от одного до трех заместителями, выбранными из группы F; или группа В представляет собой (С 1-6)алкил и (С 2-6)алкенил; где указанный (С 1-6)алкил и (С 2-6)алкенил необязательно независимо замещены членом, выбранным из группы, состоящей из фенила, гетероарила или -C(O)NR55R56; или с помощью от одного до трех атомами галогена; Группа F выбрана из группы, состоящей из оксо, (С 1-6)алкила, (С 3-7)циклоалкила, арила, гетероарила, гетероалициклила, гидрокси, (С 1-6)алкокси, арилокси, (С 1-6)тиоалкокси, циано, галогена, нитро,-C(O)R57, бензила, -NR42C(O)-(С 1-6)алкила, -NR42C(O)-(C3-6)циклоалкила, -NR42C(O)-арила, -NR42C(O)гетероарила, -NR42C(O)-гетероалициклила, 4-, 5- или 6-членного кольцевого циклического N-лактама,NR42S(O)2-(C1-6)алкила,-NR42S(О)2-(С 3-6)циклоалкила,-NR42S(O)2 арила,-NR42S(O)2-гетероарила,42 42 43NR42R43, C(O)NR42R43, NHC(O)NR42R43, OC(O)NR42R43, NHC(O)OR54, (C1-6)алкил-NR42R43, COOR54 и (С 1-6) алкил-COOR54; где указанный (С 1-6)алкил, (С 3-7)циклоалкил, арил, гетероарил, гетероалициклил, (С 1-6) алкокси и арилокси необязательно замещены от одного до девяти атомами галогена или от одного до пяти заместителями, выбранными из группы G; Группа G выбрана из группы, состоящей из (С 1-6)алкила, (С 3-7)циклоалкила, арила, гетероарила, гетероалициклила, гидрокси, (С 1-6)алкокси, арилокси, циано, галогена, нитро, -C(O)R57, бензила, неароматического гетероциклила с 1-2 гетероатомами, -NR48C(O)-(C1-6)алкила, -NR48C(O)-(C3-6)циклоалкила,-NR48C(O)-арила, -NR48C(O)-гетероарила, -NR48C(O)-гетероалициклила, 4-, 5- или 6-членного кольцевого циклического N-лактама, -NR48S(O)2-(C1-6)алкила, -NR48S(O)2,-(C3-6)циклоалкила, -NR48S(O)2-арила,-NR48S(O)2-гетероарила, -NR48S(O)2-гетероалициклила, сульфинила, сульфонила, сульфонамида, NR48R49,(C1-6)алкил-C(O)NR48R49, C(O)NR48R49, NHC(O)NR48R49, OC(O)NR48R49, NHC(O)OR54', (С 1-6)алкилNR48R49, COOR54 и (С 1-6)алкил-COOR54;R3 выбран из группы, состоящей из С 1-С 4 алкила, арила, гетероарила и гетероалициклила; где указанный С 1-С 4 алкил, арил, гетероарил и гетероалициклил необязательно замещены от одного до трех атомами галогена или от одного до трех заместителями, выбранными из группы F;R8 выбран из группы, состоящей из водорода, (С 1-6)алкила, (С 3-7)циклоалкила, (С 2-6)алкенила, (С 3-7) циклоалкенила, (С 2-6)алкинила, арила, гетероарила и гетероалициклила; где указанный (С 1-6)алкил, (С 3-7) циклоалкил, (С 2-6)алкенил, (С 3-7)циклоалкенил, (С 2-6)алкинил, арил, гетероарил и гетероалициклил необя- 16014957 зательно замещены от одного до шести атомами галогена или от одного до пяти заместителями, выбранными из группы F;R8a представляет собой член, выбранный из группы, состоящей из арила, гетероарила и гетероалициклила; где каждый член необязательно независимо замещен от одного до шести атомами галогена или от одного до пяти заместителями, выбранными из группы F;R9, R10, R11, R12, R13, R14, R15, R16, каждый независимо, выбран из группы, состоящей из водорода и(С 1-6)алкила; где указанный (С 1-6)алкил необязательно замещен от одного до трех атомами галогена или одним гидрокси или одним О(С 1-6)алкилом или одним NR55R56; или один из R9, R10 или один из R11, R12 может образовывать соответственно с одним из R15, R16 или одним R13, R14 один, двух или трех атомный мостик, состоящий из алкила или атомов азота;X представляет собой N или СН, (когда X представляет собой СН, конфигурация на центре X может быть рацемической или чистой (R), или чистой (S) конфигурацией);R40 и R41 независимо выбраны из группы, состоящей из (а) водорода; (b) (С 1-6)алкила, замещенного от одного до трех атомами галогена; (с) (С 1-6)алкокси, арила, гетероарила или гетероалициклила; или R40 и R41 взятые вместе с атомом азота, к которому они присоединены, образуют член, выбранный из группы, состоящей из азиридина, азетидина, пирролидина, пиперазина, 4-NMe пиперазина, пиперидина, азепина и морфолина; и где указанный арил, гетероарил и гетероалициклил необязательно замещены от одного до двух заместителями, выбранными из C1-С 3 алкила, галогена, гидроксила, -OR55, -NR55R56;R42 и R43 независимо выбраны из группы, состоящей из водорода, (С 1-6)алкила, аллила, (С 1-6)алкокси, (С 3-7)циклоалкила, арила, гетероарила и гетероалициклила; или R42 и R43 взятые вместе с атомом азота, к которому они присоединены, образуют член, выбранный из группы, состоящей из азиридина, азетидина, пирролидина, пиперазина (необязательно замещенного группой В), 4-NMe пиперазина, пиперидина, азепина и морфолина; и где указанный (С 1-6)алкил, (С 1-6)алкокси, (С 3-7)циклоалкил, арил, гетероарил и гетероалициклил необязательно замещены от одного до трех атомами галогена или от одного до двух заместителями, выбранными из Группы G; где для R42 и R43;R48 и R49 независимо выбраны из группы, состоящей из водорода, (С 1-6)алкила и фенила;R50 выбран из группы, состоящей из Н, (С 1-6)алкила, (С 3-6)циклоалкила и бензила; где каждый из указанного (С 1-6)алкила, (С 3-7)ииклоалкила и бензила необязательно замещен от одного до трех атомами галогена, амино, ОН, CN или NO2;R54 выбран из группы, состоящей из водорода и (С 1-6)алкила;R55 и R56 независимо выбраны из группы, состоящей из водорода и (С 1-6)алкила; иR57 выбран из группы, состоящей из водорода, (С 1-6)алкила и фенила. Определения Физиологически приемлемые соли и пролекарства соединений, раскрытые в настоящем описании,входят в границы настоящего изобретения. Термин "фармацевтически приемлемая соль", как его используют в описании и в формуле изобретения, как предполагается, включает нетоксичные основноаддитивные соли. Пригодные соли включают производные от органических и неорганических кислот,таких как, без ограничения, соляная кислота, бромистоводородная кислота, фосфорная кислота, серная кислота, метансульфоновая кислота, уксусная кислота, винная кислота, молочная кислота, сульфиновая кислота, лимонная кислота, малеиновая кислота, фумаровая кислота, сорбиновая кислота, аконитовая кислота, салициловая кислота, фталевая кислота и им подобные. Термин "фармацевтически приемлемая соль", как его используют в настоящем описании, как предполагается, включает соли кислотных групп,таких как карбоксилат, с такими притивоионами как аммоний, соли щелочных металлов, в частности,натрия или калия, соли щелочно-земельных металлов, в частности, кальция или магния и соли с пригодными органическими основаниями, такими как низшие алкиламины (метиламин, этиламин, циклогексиламин и тому подобное) или замещенными низшими алкиламинами (например гидроксилзамещенные алкиламины, такие как диэтаноламин, триэтаноламин или трис(гидроксиметил)-аминометан) или с основаниями, такими как пиперидин или морфолин. В способе настоящего изобретения термин "противовирусное эффективное количество" означает общее количество каждого активного компонента в способе, которое достаточно, чтобы показать заметное улучшение при лечении больных, то есть достигнуть прекращения острого состояния, характеризуемого ингибированием ВИЧ-инфекции. В случае индивидуального активного ингредиента, вводимого одного, термин относится к одному ингредиенту. В случае комбинации, термин относится к объединенным количествам активных ингредиентов, приводящему к результату в виде терапевтического эффекта,при введении комбинации, последовательно или одновременно. Термины "обрабатывать, лечение, обработка", как их используют в настоящем изобретении и в формуле изобретения, означает предотвращение- 17014957 или улучшение состояния при заболеваниях, связанных с ВИЧ-инфекцией. Настоящее изобретение также направлено на комбинации соединений с одним или более агентами,пригодными для лечения СПИДа. Например, соединения настоящего изобретения могут эффективно вводиться либо в периоды предварительного выявления и/или последующего выявления, в комбинации с эффективным количеством противовирусных препаратов против СПИДа, иммуномодуляторов, противоинфекционных препаратов или вакцин, таких как приведены в настоящей таблице: Противовирусные препараты Кроме того, соединения по настоящему изобретению могут быть применены в комбинации с другим классом агентов для лечения СПИДа, которые называются ВИЧ ингибиторами внедрения. Примеры таких ВИЧ ингибиторов внедрения рассмотрены в DRUGS OF THE FUTURE 1999, 24(12), pp. 1355-1362;DiscoveryDevelopment (2003), 6(4), 451-461. В особенности соединения могут быть использованы в комбинации с другими присоединенными ингибиторами, ингибиторами конденсации, и антагонистами хемокинового рецептора, предназначенными или для CCR5 или для CXCR4 сорецептора. Подразумевается, что набор комбинаций соединений по настоящему изобретению с противовирусными препаратами против СПИДа, иммуномодуляторами, противоинфекционными препаратами, ВИЧ ингибиторами внедрения или вакцинами не ограничивается списком в вышеупомянутой таблице, и он может включать в принципе любую комбинацию с любой фармацевтической структурой, применимой для лечения СПИДа. Предпочтительными комбинациями являются одновременное или альтернативное введение соединения по настоящему изобретению и ингибитора ВИЧ-протеазы и/или ненуклеозидного ингибитора обратной ВИЧ транскриптазы. Необязательным четвертым компонентом в комбинации является нуклеозидный ингибитор обратной ВИЧ транскриптазы, такой как AZT, 3 ТС, ddC или ddl. Предпочтительным ингибитором ВИЧ-протеазы является Рейатаз (активный инградиент Атазанавир). Обычно вводимая однократная ежедневная доза составляет от 300 до 600 мг. Она может быть введена совместно с меньшей дозой Ритонавира (от 50 до 500 мг). Другим применимым ингибитором ВИЧ-протеазы является Kaletra. Другим применимым ингибитором ВИЧ-протеазы является индинавир, который представляет собой сульфатную соль этанолата N-(2(R)-гидрокси-1-(S)-инданил)-2(R)-фенилметил-4-(S)-гидрокси-5-(1-(4-(3 пиридинилметил)-2(S)-N'-(трет-бутилкарбоксамидо)пиперазинилпентанамида, который синтезируют согласно U.S. 5413999. Индинавир обычно вводят в дозе 800 мг три раза в день. Другие предпочтительные ингибиторы ВИЧ-протеазы представляют собой нелфинавир и ритонавир. Другой предпочтительный ингибитор ВИЧ-протеазы представляет собой саквинавир, который вводят в дозе 600 или 1200 мг в- 22014957 день. Предпочтительные ненуклеозидные ингибиторы ВИЧ - обратной транскриптазы включают эфавиренц. Получение ddC, ddl и AZT, также описано в ЕРО 0484071. Указанные комбинации могут оказывать неожиданное воздействие на снижение распространения и степени инфицирования ВИЧ-инфекцией. Предпочтительные комбинации включают те, которые являются следующими (1) индинавир с эфавиренцом и, необязательно, AZT и/или 3 ТС и/или ddl и/или ddC; (2) индинавир и любой из AZT и/или ddl и/или ddC и/или 3 ТС, в частности, индинавир и AZT и 3 ТС; (3) ставудин и 3 ТС и/или зидовудин; (4) зидовудин и ламивудин, и 141W94 и 1592U89; (5) зидовудин и ламивудин. В таких сочетаниях соединение по настоящему изобретению и другие активные агенты могут быть введены отдельно или все вместе. В дополнение к сказанному, введение одного элемента может быть осуществлено до, во время или последовательно с введением других(ого) агентов(а). Предпочтительными комбинациями являются одновременное или альтернативное введение соединения по настоящему изобретению и ингибитора ВИЧ-протеазы и/или ненуклеозидного ингибитора обратной ВИЧ транскриптазы. Необязательным четвертым компонентом в комбинации является нуклеозидный ингибитор обратной ВИЧ транскриптазы, такой как AZT, 3 ТС, ddC или ddl. Предпочтительным ингибитором ВИЧ-протеазы является индинавир, который представляет собой сульфатную соль этанолата N-(2(R)-гидрокси-1-(S)-инданил)-2(R)-фенилметил-4-(S)-гидрокси-5-(1-(4-(3-пиридилметил)-2(S)-N'(трет-бутилкарбоксамидо)пиперазинилпентанамида и синтезируют в соответствии с U.S. 5413999. Индинавир обычно вводят в дозе 800 мг три раза в день. Другие предпочтительные ингибиторы ВИЧпротеазы представляют собой нелфинавир и ритонавир. Другой предпочтительный ингибитор ВИЧпротеазы представляет собой саквинавир, который вводят в дозе 600 или 1200 мг в день. Предпочтительные ненуклеозидные ингибиторы ВИЧ-обратной транскриптазы включают эфавиренц. Получение ddC,ddl и AZT, также описано в ЕРО 0484071. Указанные комбинации могут оказывать неожиданное воздействие на снижение распространения и степени инфицирования ВИЧ-инфекцией. Предпочтительные комбинации включают те, которые являются следующими (1) индинавир с эфавиренцом и, необязательно,AZT и/или 3 ТС и/или ddl и/или ddC; (2) индинавир и любой из AZT, и/или ddl, и/или ddC, и/или 3 ТС, в частности, индинавир и AZT и 3 ТС; (3) ставудин и 3 ТС и/или зидовудин; (4) зидовудин и ламивудин, и 141W94 и 1592U89; (5) зидовудин и ламивудин. В таких сочетаниях соединение по настоящему изобретению и другие активные агенты могут быть введены отдельно или все вместе. В дополнение к сказанному, введение одного элемента может быть осуществлено до, во время или последовательно с введением других(ого) агентов(а). Сокращения Следующие сокращения, большинство из которых являются традиционными сокращениями, хорошо известны среднему специалисту в данной области, используют на всем протяжении описания изобретения и примеров. Используют некоторые из следующих сокращений:P-EDC = на полимерной подложке 1-(3-диметиламинопропил)-3-этилкарбодиимид;CBZ = бензилоксикарбонил; РСС = хлорхромат пиридиния. Соединения по настоящему изобретению могут вводиться перорально, парентерально (включая инъекции подкожную, внутривенную, внутримышечную, надчревную инъекцию или путем вливания),ингаляционным распылением или ректально, в виде состава одноразовой дозы, содержащей обычные нетоксичные фармацевтически приемлемые носители, адъюванты и разбавители. Таким образом, в соответствии с настоящим изобретением обеспечивается способ лечения и фармацевтическая композиция для лечения вирусных заболеваний, таких как ВИЧ-инфекция и СПИД. Лечение включает введение пациенту, нуждающемуся в такой терапии, фармацевтической композиции, состоящей из фармацевтического носителя и терапевтически эффективного количества соединения настоящего изобретения. Фармацевтическая композиция может быть в форме суспензий для перорального введения или таблеток; назального спрея, стерильного препарата для инъекций, например, стерильные водные или масляные суспензии для инъекций или суппозитории. При назначении внутрь в виде суспензий композиции могут быть приготовлены согласно методикам, хорошо известным специалистам в области фармацевтики и могут содержать микрокристаллическую целлюлозу для придания объема, альгиновую кислоту или натрия альгинат в качестве суспендирующего агента, метилцеллюлозу для усиления вязкости, а также подсластители или ароматизаторы,известные специалистам данной области. Такие композиции в виде таблеток с немедленным высвобождением препарата могут содержать микрокристаллическую целлюлозу, фосфат дикальция, крахмал, магния стеарат и лактозу и/или другие наполнители, связующие вещества, носители, дезинтегрирующие агенты, разбавители и смазывающие вещества, известные специалистам данной области. Растворы или суспензии для инъекций могут быть приготовлены согласно методикам, принятым в данной области, с использованием нетоксичных, подходящих для парентерального применения разбавителей или растворителей, таких как маннитол, 1,3-бутандиол, вода, раствор Рингера или изотонический раствор хлорида натрия или подходящих диспергирующих или смачивающих агентов, таких как стерильные мягкие нелетучие масла, включая синтетические моно- или диглицериды, а также жирные кислоты, включая олеиновую кислоту. Соединения по настоящему изобретению могут быть введены людям перорально в диапазоне дозирования от 1 до 100 мг/кг массы тела в разделенных дозах. Одним из предпочтительных диапазонов дозирования является от 1 до 10 мг/кг массы тела перорально в разделенных дозах. Другим предпочтительный диапазоном дозирования является от 1 до 20 мг/кг массы тела в разделенных дозах. Необходимо понимать, в то же время, что конкретные дозировки и частота введения для каждого конкретного пациента может быть изменена и будет зависеть от множества факторов, включая активность назначенного соединения, его метаболическую стабильность и длительность действия, возраст, массу тела, общее состояние здоровья, пол, диету, способ и время введения, скорость экскреции, лекарственные взаимодействия, степень тяжести заболевания и субъект, нуждающийся в лечении. Химия Настоящее изобретение включает соединения формулы I, их фармацевтические составы, и способы их применения на пациентах, страдающих от или восприимчивых к ВИЧ-инфекции. Соединениям формулы I включают их фармацевтически приемлемые соли. Общие методики для получения соединений формулы I и промежуточных соединений, пригодные для их синтеза, описаны на следующих схемах. Химические схемы получение соединений формулы I Получение по стандарту А-СО-СО-Cl и А-СО-СО-ОН описано подробно в US 6469006B1, US 6573262B2, US 6900323B2, US 20050090522A1, US 6825201, US 20050261296A1, US 20040186292A1, US- 24014957 20050267130A1, US 6900206B2, US 20040063746, WO-00076521, WO-00162255, WO-00204440, WO02062423, WO-02085301, WO-03068221 и US-2004/0063744. Средний специалист осведомлен о большинстве стандартных условий для взаимодействия амина с ацилгалоидом 1 (схема 1) и карбоновой кислотой 4 (схема 2), которые могут быть применены для превращения хлорангидрида или кислоты в желаемые амидные продукты. Некоторые общие ссылки указанных методологий и наставлений для применения содержатся в "Comprehensive Organic Transformation" by Схема 1 описывает общий способ образования амида из пиперазинамидина 2 и ацилхлорида 1. Подходящее основание (от каталитического до избыточного количества), выбранное из гидрида натрия,карбоната калия, триэтиламина, DBU, пиридина, DMAP или диизопропилэтиламина добавляют в раствор из агента 2 и ацилхлорида в подходящем растворителе, выбранном из дихлорметана, хлороформа, бензола, толуола, ТГФ, диэтилового эфира, диоксана, ацетона, N,N-диметилформамида или пиридина при комнатной температуре. Реакцию проводят или при комнатной температуре или экспериментально определенной оптимальной температуре вплоть до 150 С в течение определенного периода времени (от 30 минут до 16 ч), чтобы получить соединения 3, которые могут или быть соединениями формулы I или предшественниками. Ряд выбранных ссылок, касающихся таких реакций, включают а) Indian J. Chem.,Sect В 1990, 29, 1077; 2) Chem. Sci 1998, 53, 1216; 3) Chem. Pharm. Bull. 1992, 40, 1481; 4) Chem. Heterocycl. Compd. 2002,38, 539. Схема 2 Альтернативно, как показано на схеме 2, структура 2 может быть связана с кислотой 4, используя обычную амидную связь или пептидную связь, образованную реагентами связывания. Большинство реагентов связывания для амидной связи известны среднему специалисту в области органической химии и почти все из них являются пригодными для осуществления связывания амидных продуктов. Более часто применяют комбинацию из EDAC и триэтиламина в тетрагидрофуране или BOPCl и диизопропилэтиламина в хлороформе, но могут быть применены DEPBT или другие реагенты связывания, такие как РуВор. В другом пригодном условии связывания применяют HATU a) J.Chem.Soc. Chem Comm. 1994,201; (b) J. Am. Chem. Soc. 1994, 116,11580). Кроме того, DEPBT (3-(диэтоксифосфорилокси)-1,2,3 бензотриазин-4(3H)-он) и N,N-диизопропилэтиламин, как правило известное как основание Ханига,представляет собой другой эффективный способ образования амидной связи и обеспечения соединений Формулы I. DEPBT или приобретают от Adrich или получают в соответствии с методикой, описанной вOrganic Lett., 1999, J, 91. Обычно используют инертный растворитель, такой как ДМФА или ТГФ, но могут быть использованы другие апротонные растворители. Общий агент 2, использование которого представлено на схемах 1 и 2, является или коммерчески доступным или может быть получен с помощью общих способов, описанных в экспериментальной части. Ряд неограничивающих примеров из или коммерчески доступных или синтезируемых реагентов 2 перечислены ниже:- 26014957 Когда X представляет собой N в формуле I, он может быть также получен путем связывания ацилхлорида 1 или кислоты 4 с Boc-защищенным пиперазином в условиях, описанных на схемах 1 и 2, для получения соединения 5. Последующее хорошо разработанное снятие Boc защитной группы в кислотных условиях, должно обеспечить пиперазинамид 6. Используют TFA и водный раствор HCl, представляющие собой обычные кислоты для эффетивного удаления Boc группы, при этом обычно используют растворители представляющие собой либо эфир либо дихлорметан. TFA может быть применен в качестве как реагента так и растворителя в некоторых случаях. Для эффективного удаления ВОС могут также быть использованы другие кислотные агенты, такие как муравьиная кислота и растворители. Амид 6 может взаимодействовать с галоидом при SN2, SNAr и металл-опосредованным перекрестным связыванием для обеспечения соединения 3 (соединения формулы I или предшественники). Схема 3 Для SN2, SNAr реакции в апротонном (например, ТГФ, ДМФА, DMSO, бензол) или протонном растворителе (например, МеОН, EtOH, PrOH, BuOH), при температуре от комнатной до 150 С, в отсутствии или присутствии основания, такого как NaH, пиридин, Et3N, ди-Pr2NEt, Na2CO3, K2CO3, пиперазин, амид 6 может взаимодействовать с галоидом или гетероарилсульфоном 7, что дает соединение 3. Для металл-опосредованного перекрестного связывания температура может изменяться от комнатной до 150 С и растворители предпочтительно представляют собой апротонные растворители, такие как ТГФ, диоксан, DME, ДМФА и DMSO. Основания могут быть выбранное из NaH, KH, пиридина, Et3N,ди-Pr2NEt, Na2CO3, K2CO3, NaHMDS, LiHMDS, KHMDS, Na3PO4, Na2HPO4 и NaH2PO4, Pd, Rh, Ru, Ni илиPt агенты могут быть использованы как катализаторы. Примеры Нижеследующие примеры демонстрируют типичный синтез соединений формулы I как описаны вообщем выше. Указанные примеры являются только иллюстративными и не предназначены в любом случае для ограничения изобретения. Реагенты и исходные продукты являются легко доступными для среднего специалиста. Химия Типичные методики и характеристики выбранных примеров Если иное не установлено, растворители и реагенты используют непосредственно в виде тех, которые получены из коммерческих источников и реакции проводят в атмосфере азота. Флеш хроматографию проводят на Силикагеле 60 (0.040-0.063 размер частиц; ЕМ Science supply). 1 Н ЯМР спектр регистртруют на Bruker DRX-500f при 500 МГц (или Broker DPX-300B или Varian Gemini 300 при 300 МГц как указано). Химические сдвиги представляют в част. на млн. нашкале относительно TMS = 0. Следующие внутренние ссылки используют для остаточных протонов в следующих растворителях: CDCl3 (Н 7.26), CD3OD (Н 3.30) и DMSO-d6 (H 2.50). Обычные акронимы применяют для описания многочислен- 27014957 ных конфигураций: с (синглет), д (дуплет), т (триплет), кв (квартет), т (мультиплет), б (широкий), арр(видимый). Константа связывания (J) представлены в герцах. Все данные по жидкостной хроматографии(LC) зарегистрированы на жидкостном хроматографе Shimadzu LC-10AS, с использованием детектораSPD-10AV UV-Vis с определенными данными Масс-спектрометра (MS), с использованием электроаэрозольной методики Micromass Platform для LC. Все данные по жидкостной хроматографии (LC) зарегистрированы на жидкостном хроматографеShimadzu LC-10AS, с использованием детектора SPD-10AV UV-Vis с определенными данными массспектрометра (MS), с использованием электроаэрозольной методики Micromass Platform для LC. Получение из стандартов A-CO-CO-Cl и А-СО-СО-ОН, если иное не указано, описано подробно вUS 6469006 B1, US 6573262 B2, US 6900323 B2, US 20050090522 A1, US 6825201, US 20050261296 A1,US 20040186292 A1, US 20050267130 A1, US 6900206 B2, US 20040063746, WO-00076521, WO-00162255,WO-00204440, WO-02062423, WO-02085301, WO-03068221 или US-2004/0063744. В особенности 7-бром 4-фтор-1 Н-пирроло[2,3-с]пиридин, 2-(4-фтор-7-(1H-1,2,3-триазол-1-ил)-1 Н-пирроло[2,3-с]пиридин-3-ил)2-оксоуксусную кислоту, 2-(4-метокси-7-(1H-1,2,3-триазол-1-ил)-1H-пирроло[2,3-с]пиридин-3-ил)-2 оксоуксусную кислоту, 2-(7-хлор-4-метокси-1H-пирроло[2,3-с]пиридин-3-ил)-2-оксоуксусную кислоту и 2-(4-метокси-7-(3-метил-1 Н-1,2,4-триазол-1-ил)-1H-пирроло[2,3-с]пиридин-3-ил)-2-оксоуксусную кислоту получают как описано в US20050090522A1. Пример химической секции А Следующие общие способы применены для примера химической секции А:LC/MS Способы (то есть, идентификация соединения); колонка A: Xterra MS C18 5um 4.630 мм колонка; колонка В: Phenomenex 5u C18 4.630 мм колонка; колонка С: Xterra MS C18 4.630 мм колонка; колонка D: Phenomenex 4.650 мм С 18 5um колонка; колонка Е: Xterra 4.630 мм S5 колонка; колонка F: Phenomenex-Luna 4.650 мм S10 колонка; колонка G: Phenomenex 10u 3.050 мм колонка; колонка Н: Phenomenex-Luna 4.630 мм S5 колонка; колонка I: Phenomenex 4.630 мм 10n колонка; колонка J: Phenomenex C18 10u 3.050 мм колонка; колонка K: Phenomenex, Onyx Monolithic C18 504.6 мм колонка; градиент: 100% растворитель А/0% растворитель В до 0% растворитель А/100% растворитель В; градиент времени: Все LC-MS:, за исключением, которые иным образом определены, используют 2 мин градиента времени; время выдержки: 1 мин; скорость потока: а. 5 мл/мин;(все LC-MS, за исключением иным образом определенных, который используют скорость потока b,получают путем использования скорости потока а.); детекторная длина волны: 220 нм. Растворительная система I Растворитель А: 10% МеОН/90% Н 2 О /0.1% трифторуксусная кислота. Растворитель В: 10% Н 2 О/90% МеОН/0.1% трифторуксусная кислота. Растворительная система II Растворитель А: 5% MeCN/95% Н 2 О/10 мм ацетат аммония. Растворитель В: 95% MeCN/5% H2O/10 мм ацетат аммония.(Все LC-MS: в последующих разделах, за исключением иным образом определенных, которые используют систему растворителей II, получают путем использования системы растворителей I). Соединения, очищенные с помощью препаративной HPLC, разбавляют в метаноле (1.2 мл) и очищают, используя следующие способы на Shimadzu LC-10A автоматической препаративной HPLC системе. Препаративный HPLC способ (то есть, очистка соединения) Способ очистки: исходный градиент (40% В, 60% А) линейно увеличивают до конечного градиента- 28014957 Типичные методики и характеристика выбранных примеров Получение агента 2 Способ А Избыток пиперазина (5-10 экв.) добавляют к раствору электрофила в ТГФ, диоксане или ДМФА. Реакционную смесь перемешивают в течение 17 ч при комнатной температуре или при температуре 115 С, затем гасят насыщенным водным раствором NaHCO3. Водную фазу экстрагируют EtOAc. Объединенный органический слой промывают водой и сушат над MgSO4, фильтруют и фильтрат концентрируют до получения остатка, который используют в последующих реакциях без дополнительной очистки или очищают с помощью хроматографии на колонке с силикагелем или с помощью автоматической препаративной HPLC системы Шимадзу. Способ В Избыток основания (1-20 экв., такого как Et3N, iPr2NEt или NaH), добавляют к раствору N-Boc пиперазина (2-5 экв.) в ТГФ, диоксане или ДМФА, с последующим добавлением электрофила (1 экв.). Реакционную смесь перемешивают в течение 17 ч при комнатной температуре или при температуре 115 С,затем гасят насыщенным водным раствором NaHCO3. Водную фазу экстрагируют EtOAc. Объединенный органический слой сушат над MgSO4, фильтруют и фильтрат концентрируют до получения остатка, который используют в последующих реакциях без дополнительной очистки или очищают с помощью хроматографии на колонке с силикагелем или с помощью автоматической препаративной HPLC системы Шимадзу.N-Boc пиперазиновое производное растворяют в кислотном растворе TFA или HCl в CH2Cl2, эфире,диоксане или спирте. Через период времени от 0,5 до 17 ч раствор концентрируют в вакууме, что дает остаток соли, который распределяют между водным раствором NaHCO3 и EtOAc. Водную фазу экстрагируют EtOAc. Объединенный органический слой сушат над MgSO4, фильтруют и фильтрат концентрируют до получения остатка, который используют в последующих реакциях без дополнительной очистки или очищают с помощью хроматографии на колонке с силикагелем или с помощью автоматической препаративной HPLC системы Шимадзу. Способ С К перемешиваемому раствору пиперазина (2 экв.) в сухом ТГФ добавляют н-BuLi (от 2 до 4 экв.) в ТГФ при комнатной температуре. После перемешивания в течение 0.5 ч при комнатной температуре к раствору аниона добавляют арилгалоид (1 экв.) и полученную реакционную смесь перемешивают в течение еще от 1 до 17 ч в интервале от комнатной температуры до температуры 115 С. Реакционную смесь гасят МеОН и растворители упаривают. Остаток распределяют между EtOAc и насыщенным растворомNaHCO3. Водный слой экстрагируют EtOAc. Органический слой сушат над MgSO4 и концентрируют,чтобы получить сырой продукт, который очищают с помощью хроматографии на колонке с силикагелем или с помощью автоматической препаративной HPLC системы Шимадзу. Способ D К раствору метилтиопроизводного (1 экв.) в сухом CH2Cl2 или НОАс добавляют mCPBA (2-5 экв.) при комнатной температуре. После перемешивания в течение 17 ч при комнатной температуре реакционную смесь гасят NaHSO3 и растворители упаривают. Остаток распределяют между EtOAc и насыщенным раствором NaHCO3. Водный слой экстрагируют ЕtOAc. Органический слой сушат над MgSO4 и концентрируют, чтобы получить сырой продукт, который очищают с помощью хроматографии на колонке с силикагелем или с помощью автоматической препаративной HPLC системы Шимадзу. Избыток пиперазина (5-10 экв.) добавляют к раствору сульфонового или сульфоксидного производного в 1-бутаноле или 1-пентаноле. Реакционную смесь нагревают при температуре кипения с обрат- 29
МПК / Метки
МПК: C07D 519/00, A61P 31/18, A61K 31/4427, A61K 31/4436, C07D 417/14, C07D 403/12, C07D 471/04, A61K 31/437
Метки: пиперидиновые, производные, агентов, противовирусных, качестве, дикетопиперазиновые
Код ссылки
<a href="https://eas.patents.su/30-14957-diketopiperazinovye-i-piperidinovye-proizvodnye-v-kachestve-protivovirusnyh-agentov.html" rel="bookmark" title="База патентов Евразийского Союза">Дикетопиперазиновые и пиперидиновые производные в качестве противовирусных агентов</a>
Хинолинкарбоксамиды в качестве противовирусных агентов

Номер патента: 3945
Опубликовано: 30.10.2003
Авторы: Такер Джон Алан, Вэйлланкорт Валери А., Тернер Стивен Рональд, Таисривонгс Сувит, Стробач Джозеф Волтер, Шнуте Марк Э.
МПК: A61P 31/12, C07D 215/16, A61K 31/47...
Метки: хинолинкарбоксамиды, агентов, качестве, противовирусных
Формула / Реферат:
1. Соединение формулы I или его фармацевтически приемлемая соль, где X представляет a) O или b) S; W представляет a) R2, b) NR7R8, c) OR9 или d) SOiR9; R1 представляет a) Cl, b) F, c) Br, d) CN или e) NO2; R2 представляет a) (CH2CH2O)mR10, b) het, где указанный het связан через атом углерода, c) C1-7алкил, который может быть частично ненасыщенным и необязательно замещенным одним или несколькими заместителями, выбранными из группы,...
Новые замещенные 4-фенил-4-[1н-имидазол-2-ил] пиперидиновые производные и их применение в качестве селективных непептидных агонистов дельта-опиоидов

Номер патента: 6507
Опубликовано: 29.12.2005
Авторы: Мерт Тео Франс, Фернандес-Гадеа Франсиско Хавьер, Гомес-Санчес Антонио, Янссенс Франс Эдуард, Ленартс Йозеф Элизабет
МПК: A61P 25/04, C07D 401/04, A61K 31/445...
Метки: новые, селективных, пиперидиновые, дельта-опиоидов, применение, 4-фенил-4-[1н-имидазол-2-ил, качестве, производные, замещенные, агонистов, непептидных
Формула / Реферат:
1. Соединение формулы (I) где A=B представляет собой двухвалентный радикал с p-связью; X представляет собой ковалентную связь, -CH2- или CH2CH2-; R1 представляет собой водород, гидрокси, алкилокси, алкилкарбонилокси, Ar-окси, Het-окси, Ar-карбонилокси, Het-карбонилокси, Ar-алкилокси, Het-алкилокси, алкил, полигалогеналкил, алкилоксиалкил, Ar-алкил, Het-алкил, Ar, Het, тио, алкилтио, Ar-тио, Het-тио или NR9R10, где R9 и R10, каждый независимо,...
Производные индазоламида в качестве серотонинергических агентов

Номер патента: 2352
Опубликовано: 25.04.2002
Авторы: Бруфани Марио, Пинца Марио, Джаннанджели Марилена, Каццолла Никола, Алиси Алессандра
МПК: C07D 401/12, A61K 31/416
Метки: агентов, качестве, серотонинергических, производные, индазоламида
Формула / Реферат:
1. Индазоламидные соединения общей формулы в которой R6 выбран из группы, включающей фенил C1-3 алкил, С3-7-циклоалкил, 5- или 6-членное гетероциклическое кольцо, в котором от 1 до 2 членов являются одинаковыми или различными гетероатомами, выбранными из группы, состоящей из N и О, диметиламино C1-3 алкил, метокси C1-3 алкил, аминосульфонилметил, арил, замещенный гидроксигруппой; их соли присоединения к фармацевтически приемлемым органическим и...
Производные хинолина в качестве антибактериальных агентов

Номер патента: 14431
Опубликовано: 30.12.2010
Авторы: Коул Анил, Гийемон Жером Эмиль Жорж, Андрис Кунрад Йозеф Лодевейк Марсель, Лансуа Давид Франсис Ален, Мотт Магали Мадлен Симон
МПК: A61K 31/4353, C07D 215/227, A61P 31/04...
Метки: агентов, антибактериальных, хинолина, производные, качестве
Формула / Реферат:
1. Применение соединения для получения лекарственного средства для лечения бактериальной инфекции, причем указанное соединение является соединением формулы (Ia)его N-оксидом или его стереохимически изомерной формой, гдеА- обозначает фармацевтически приемлемый противоион;R1 обозначает водород или галоген;р является целым числом, равным 1, 2, 3 или 4;R2 обозначает алкилокси или алкилтио;R3 обозначает Ar, Ar-алкил или Het;q является целым числом,...
Производные хинолина в качестве антибактериальных агентов

Номер патента: 10601
Опубликовано: 30.10.2008
Авторы: Андрис Кунрад Йозеф Лодевейк Марсель, Коул Анил, Гийемон Жером Эмиль Жорж, Паскье Элизабет Тереза Жанна
МПК: C07D 215/26, A61K 31/47, A61K 31/4709...
Метки: хинолина, качестве, производные, антибактериальных, агентов
Формула / Реферат:
1. Применение соединения для изготовления лекарственного средства для лечения бактериальной инфекции, причем соединение является соединением формулы его фармацевтически приемлемой кислотно- или основно-аддитивной солью, его стереохимически изомерной или N-оксидной формой, где R1 представляет собой водород, галоген, полигалоC1-6алкил, C1-6алкил, Ar или Het; р представляет собой целое число, равное 1 или 2; R2 представляет собой C1-6алкилокси,...
Предыдущий патент: Гетероциклические соединения, применяемые для лечения воспалительных и аллергических нарушений
Следующий патент: Антагонисты рецептора il – 8
Случайный патент: Применения хлорида холина в агрохимических препаратах