Производные индазоламида в качестве серотонинергических агентов
Номер патента: 2352
Опубликовано: 25.04.2002
Авторы: Алиси Алессандра, Бруфани Марио, Пинца Марио, Каццолла Никола, Джаннанджели Марилена






Формула / Реферат
1. Индазоламидные соединения общей формулы

в которой R6 выбран из группы, включающей фенил C1-3 алкил, С3-7-циклоалкил, 5- или 6-членное гетероциклическое кольцо, в котором от 1 до 2 членов являются одинаковыми или различными гетероатомами, выбранными из группы, состоящей из N и О, диметиламино C1-3 алкил, метокси C1-3 алкил, аминосульфонилметил, арил, замещенный гидроксигруппой;
их соли присоединения к фармацевтически приемлемым органическим и неорганическим кислотам и их фармацевтически приемлемые четвертичные соли.
2. Соединение по п.1, характеризующееся тем, что гетероциклическими кольцами являются фуранил, пиранил, пирролил, имидазолил, пиразолил, изоксазолил, пиридинил, пиразинил, пиримидинил, пиридазинил, фуразанил, пирролинил, имидазолинил, пиразолидинил, пиразолинил, пиперидинил, пиперазинил, морфолинил.
3. Соединение по п.1, характеризующееся тем, что R6 выбран из группы, включающей циклопропил, циклогексил, пиридинил, морфолинил, метоксиметил, метоксипропил, гидроксифенил, диметиламинометил и аминосульфонилметил.
4. Соединение по п.1, характеризующееся тем, что R6 представляет циклогексил.
5. Соединение по п.1, характеризующееся тем, что R6 представляет собой пиридинил.
6. Соединение по п.1, характеризующееся тем, что R6 представляет диметиламинометил.
7. Соединение по п.1, характеризующееся тем, что R6 представляет морфолинил.
8. Соединение по п.1, характеризующееся тем, что R6 представляет аминосульфонилметил.
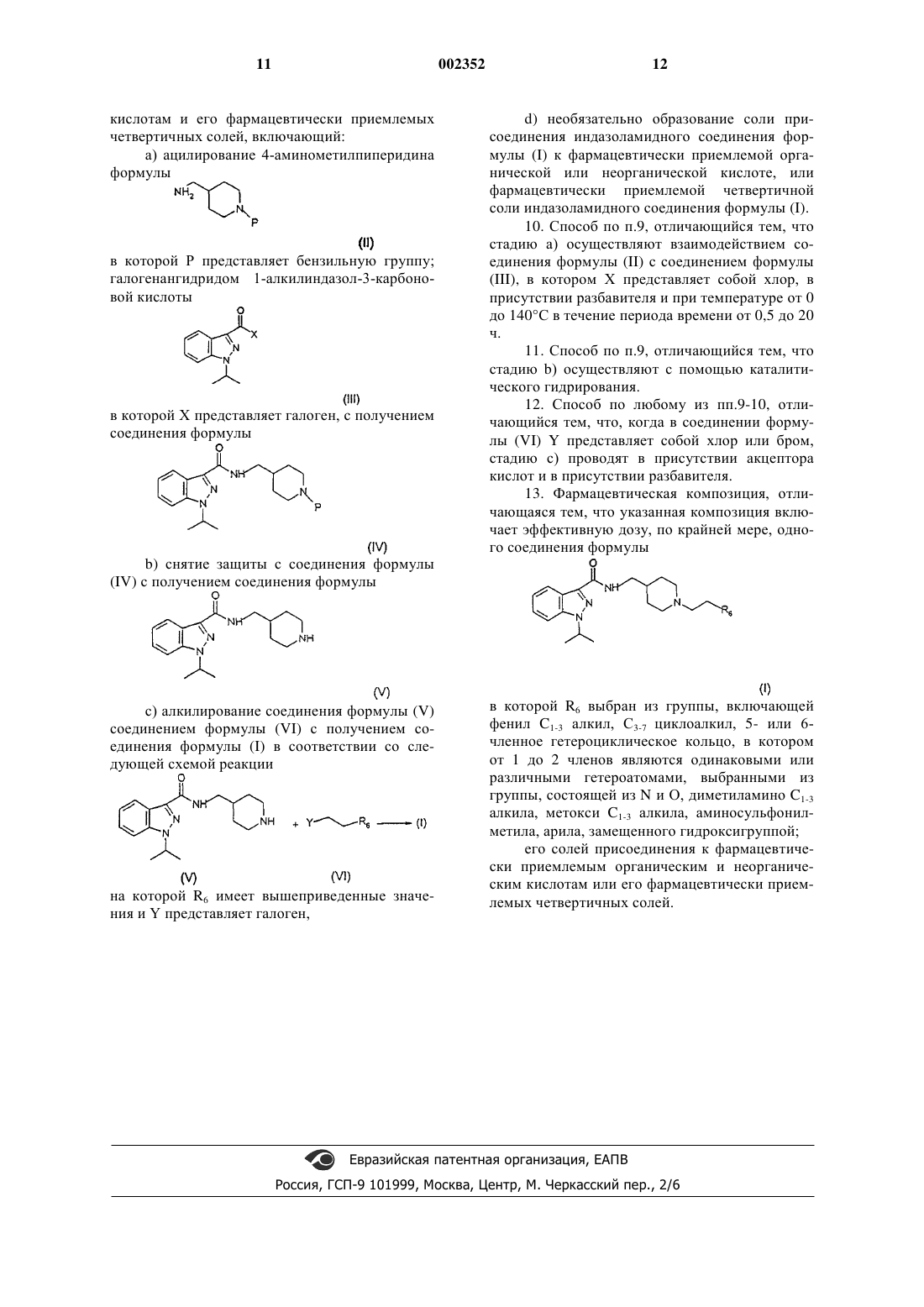
9. Способ получения соединения формулы (I), его солей присоединения к фармацевтически приемлемым органическим и неорганическим кислотам и его фармацевтически приемлемых четвертичных солей, включающий:
а) ацилирование 4-аминометилпиперидина формулы

в которой Р представляет бензильную группу; галогенангидридом 1-алкилиндазол-3-карбоновой кислоты

в которой Х представляет галоген, с получением соединения формулы

b) снятие защиты с соединения формулы (IV) с получением соединения формулы

с) алкилирование соединения формулы (V) соединением формулы (VI) с получением соединения формулы (I) в соответствии со следующей схемой реакции

на которой R6 имеет вышеприведенные значения и Y представляет галоген,
d) необязательно образование соли присоединения индазоламидного соединения формулы (I) к фармацевтически приемлемой органической или неорганической кислотe, или фармацевтически приемлемой четвертичной соли индазоламидного соединения формулы (I).
10. Способ по п.9, отличающийся тем, что стадию а) осуществляют взаимодействием соединения формулы (II) с соединением формулы (III), в котором Х представляет собой хлор, в присутствии разбавителя и при температуре от 0 до 140шС в течение периода времени от 0,5 до 20 ч.
11. Способ по п.9, отличающийся тем, что стадию b) осуществляют с помощью каталитического гидрирования.
12. Способ по любому из пп.9-10, отличающийся тем, что, когда в соединении формулы (VI) Y представляет собой хлор или бром, стадию с) проводят в присутствии акцептора кислот и в присутствии разбавителя.
13. Фармацевтическая композиция, отличающаяся тем, что указанная композиция включает эффективную дозу, по крайней мере, одного соединения формулы

в которой R6 выбран из группы, включающей фенил C1-3 алкил, С3-7 циклоалкил, 5- или 6-членное гетероциклическое кольцо, в котором от 1 до 2 членов являются одинаковыми или различными гетероатомами, выбранными из группы, состоящей из N и О, диметиламино C1-3 алкила, метокси C1-3 алкила, аминосульфонилметила, арила, замещенного гидроксигруппой;
его солей присоединения к фармацевтически приемлемым органическим и неорганическим кислотам или его фармацевтически приемлемых четвертичных солей.
Текст
1 Настоящее изобретение относится к соединению индазоламида, обладающему серотонинергическим действием, к способу его получения и к содержащим его фармацевтическим композициям. Среди многочисленных известных семейств рецепторов серотонина рецепторы 5 НТ 4 были лишь недавно идентифицированы в мочевом пузыре, гладкой и сердечной мышце и в особых областях центральной нервной системы. Соединения, обладающие агонистическим, частично агонистическим и антагонистическим действием по отношению к таким рецепторам представляют потенциальный интерес для фармакологического лечения расстройств желудочно-кишечной подвижности (перистальтики),расстройств центральной нервной системы, недержания мочи и сердечной аритмии. Действие таких соединений в действительности происходит путем подражания или антагонизма способности серотонина стимулировать кишечную подвижность активацией кишечных нейронов для модулирования важных мозговых процессов, таких как обучение, память и беспокойство и для индуцирования релаксации мочевого пузыря и увеличения частоты сердечных сжатий. В настоящее время было найдено семейство соединений индазоламида, которые обладают сродством к рецепторам 5 НТ 4 и действуют как антагонисты серотонина. Поэтому первым объектом настоящего изобретения является соединение индазоламида,имеющее общую формулу в которой R6 выбран из группы, включающей фенил C1-3 алкил, С 3-7 циклоалкил, гетероциклическое кольцо, имеющее 5 или 6 членов, в котором от 1 до 2 членов являются одинаковыми или различными гетероатомами, выбранными из группы, состоящей из N и О, диметиламино C1-3 алкил, метокси C1-3 алкил, аминосульфонилметил, арил, замещенный группой гидрокси; его соли присоединения кислот (или кислотно-аддитивные соли) с фармацевтически приемлемыми органическими и неорганическими кислотами и его фармацевтически приемлемые четвертичные соли. Предпочтительными примерами арила являются фенил, нафтил и бифенил. Предпочтительными примерами гетероциклического кольца являются тиенил, фуранил, пиранил, пирролил, имидазолил, пиразолил, изоксазолил, пиридинил, пиразинил, пиримидинил, пиридазинил, фуразанил, пирролинил,имидазолинил, пиразолидинил, пиразолинил,пиперидинил, пиперазинил, морфолинил. Типичными примерами R6 являются циклопропил, 002352 2 циклогексил, пиридинил, морфолинил, метоксиметил, метоксипропил, гидроксифенил, диметиламинометил и аминосульфонилметил. Вторым объектом настоящего изобретения является способ получения соединения формулы (I), его солей присоединения кислот с фармацевтически приемлемыми органическими и неорганическими кислотами и его фармацевтически приемлемых четвертичных солей, включающий: а) ацилирование 4-аминометилпиперидина формулы с) алкилирование соединения формулы (V) соединением формулы (VI) с получением соединения формулы (I) по следующей схеме реакции где R6 имеет вышеприведенные значения, и Y представляет галоген,d) необязательно образование соли присоединения кислоты соединения индазоламида формулы (I) с фармацевтически приемлемой органической или неорганической кислотой или фармацевтически приемлемой четвертичной соли соединения индазоламида формулы (I). Типичными примерами защитных групп 3 Стадию а) предпочтительно проводят с помощью взаимодействия соединения формулы(II) с соединением формулы (III), в котором Х представляет хлор, в присутствии подходящего разбавителя и при температуре от 0 до 140 С в течение периода времени от 0,5 до 20 ч. Предпочтительно разбавитель является апротонным, полярным или неполярным. Еще более предпочтительным является апротонный неполярный разбавитель. Примерами подходящих апротонных неполярных разбавителей являются ароматические углеводороды, такие как,например, бензол, толуол и ксилолы. Примерами подходящих апротонных полярных разбавителей являются диметилформамид и диметилсульфоксид. Еще более предпочтительно реакцию проводят при температуре от 15 до 40 С в течение периода времени от 1 до 14 ч. В свою очередь стадию b) проводят в соответствии с методиками, известными специалистам в области защитных групп (Theodora W.Sons, Inc., N.Y., 1991). В случае бензила или бензилоксикарбонила снятие защитной группы предпочтительно проводят путем каталитического гидрирования. Примером подходящего катализатора является палладий на активированном угле. Предпочтительно снятие защиты проводят гидрированием в присутствии подходящего разбавителя, такого как, например, низший алифатический спирт, низшая алифатическая кислота и их смеси. Примером предпочтительного разбавителя является смесь этиловый спирт/уксусная кислота. Стадию с) предпочтительно осуществляют с соединением формулы (VI), в котором Y представляет хлор или бром, в присутствии подходящего акцептора кислоты, такого, например,как щелочные карбонаты или бикарбонаты,низшие триалкиламины, и подходящего разбавителя, такого как, например, ароматические углеводороды, диметилформамид и диметилсульфоксид и низшие алифатические спирты. Типичными примерами предпочтительных органических и неорганических кислот для образования солей присоединения кислоты по настоящему изобретению (стадия d) являются щавелевая, малеиновая, винная, метансульфоновая,серная, фосфорная кислоты, бромистый водород и хлористый водород. Метилйодид является типичным примером предпочтительного соединения, образующего фармацевтически приемлемую четвертичную соль по изобретению. Получение вышеупомянутых солей включает присоединение (стадия d) фармацевтически приемлемой органической или неорганической кислоты или метилйодида к соединению инда 002352 4 золамида формулы (I), полученному на стадии с). Промежуточные соединения формул (IV) и(V) являются новыми. Поэтому они являются дополнительным объектом настоящего изобретения. Альтернативно соединение индазоламида формулы (I) может быть получено путем ацилирования подходящего 4-аминометилпиперидина соединением формулы (III). Типичными примерами патологических состояний, для которых может быть полезным лечение фармацевтической композицией согласно изобретению, являются все патологические состояния, которые дают ответную реакцию на лечение антагонистами рецептора 5-НТ 4,такие как, например, желудочно-кишечные расстройства, связанные с высокой кишечной подвижностью, такие как IBS (синдром раздраженной толстой кишки или слизистый колит), недержание мочи, и сердечные аритмии, такие как фибрилляция предсердий или мерцательная аритмия. Предпочтительно фармацевтические композиции по настоящему изобретению приготавливаются в виде подходящих дозировочных форм, включающих эффективную дозу, по меньшей мере, одного соединения формулы (I) или его фармацевтически приемлемой аддитивной соли или его четвертичной соли и, по меньшей мере, один фармацевтически приемлемый инертный ингредиент. Примерами подходящих дозировочных форм являются таблетки, капсулы, таблетки с покрытием, гранулы, растворы и сиропы для перорального введения; кремы, мази и лечебные полоски липкого пластыря для местного применения; свечи для ректального введения и стерильные растворы для инъекций, аэрозольного или офтальмогического введения. Дозировочные формы могут также содержать другие общепринятые ингредиенты, такие как стабилизаторы, консерванты, поверхностноактивные вещества, буферы, соли для регулирования осмотического давления, эмульгаторы,подсластители, красящие агенты, ароматизирующие или вкусовые агенты, и аналогичные. Если требуется для конкретного лечения,фармацевтическая композиция по настоящему изобретению может содержать другие фармакологически активные ингредиенты, сопутствующее введение которых является терапевтически полезным. Количество соединения формулы (I) или его фармацевтически приемлемой соли может варьироваться в широких пределах в зависимости от известных факторов, таких как, например, тип заболевания, которое подвергается лечению, тяжести заболевания, веса тела пациента, дозировочной формы, выбранного способа введения, количества дозировочных форм, вводимого в течение суток, и эффективности вы 5 бранного соединения формулы (I). Однако оптимальное количество может легко и обычно определяться специалистом в данной области. Обычно количество соединения формулы(I) или его соли в фармацевтической композиции по изобретению должно быть таким, чтобы обеспечить уровень введенной дозы от 0,001 до 50 мг/кг/сутки. Дозировочные формы фармацевтической композиции по изобретению могут быть приготовлены с помощью способов, которые известны фармацевтическим химикам и включают смешение, гранулирование, прессование, растворение, стерилизацию и т.п. Следующие примеры предназначены для иллюстрации настоящего изобретения, никоим образом не ограничивая его. Пример 1. Получение 1-изопропил-1 Н-3 индазолкарбонилхлорида (III:Х=Сl). а) 2-Метилпропил-1-изопропил-1 Н-3 индазолкарбоксилат. К раствору 2-метилпропил-1 Н-3-индазолкарбоксилата (50 г; 0,24 моль) в 1,2 диметоксиэтане (300 мл) добавляют раствор изопропилбромида (27,5 мл; 0,29 моль) в 1,2 диметоксиэтане (100 мл) и КОН (13,5 г; 0,24 моль), и смесь нагревают в условиях дефлегмации в течение 8 ч. После удаления растворителя остаток растворяют в толуоле (300 мл), полученный таким образом раствор промывают 1NNaOH (100 мл), H20 (2 х 100 мл), и затем сушат и концентрируют в вакууме. Остаток очищают от изомера 2-метилпропил-2-изопропил-2 Н-3-индазолкарбоксилата с помощью флэш-хроматографии (элюент : гексан/этилацетат = 95:5), получая указанное в заголовке соединение (23 г) в виде масла. 1b) 1-изопропил-1 Н-3-индазолкарбоновая кислота. Суспензию соединения примера 1 а) (10 г; 0,04 моль) в 0,75N NaOH (100 мл) нагревают в условиях дефлегмации в течение 12 ч. Затем раствор охлаждают, подкисляют 6N НСl (40 мл), твердый осадок отфильтровывают и перекристаллизуют из смеси гексан/этилацетат 1:1,получая указанное в заголовке соединение (5,5 г), т.пл. 162-3 С (Harada H. et al., "Chem. Pharm.c) 1-Изопропил-1 Н-3-индазолкарбонилхлорид. Тионилхлорид (4 мл, 0,054 моль) добавляют к перемешиваемому раствору соединения примера 1b), и смесь перемешивают при кипя 002352 6 чении в условиях дефлегмации в течение 2 ч. После удаления растворителя под вакуумом остаток перекристаллизуют из гексана, получая 3,5 г указанного в заголовке соединения, т.пл. 63-4 С. Элементный анализ дляH ЯМР (CDCl3, ); 1,69 (д, J=7 Гц, 6H); 5,00 (гепт., J=7 Гц, 1H); 7,20-7,70 (м, 3 Н); 8,038,33 (м,1H). Пример 2. Получение N3-[1-(фенилметил)-4-пиперидинил]метил-1-изопропил-1 Н-3 индазолкарбоксамида (IV: Р=-CH2C6H5). К перемешиваемому раствору 1 изопропил-1 Н-3-индазолкарбонилхлорида (52 г; 0,234 моль) в толуоле (300 мл) добавляют по каплям раствор [1-(фенилметил)-4-пиперидинил]метиламина, полученного, как описано вWO 94/10174, (47,7 г; 0,234 моль) в толуоле (200 мл). Спустя 5 ч растворитель удаляют выпариванием при пониженном давлении. Реакционную смесь обрабатывают 2N NaOH, экстрагируют дихлорметаном и концентрируют в вакууме. Твердый остаток (95 г) перекристаллизуют из смеси гексан/этилацетат 7:3, получая указанное в заголовке соединение в виде белого твердого вещества (45 г), т.пл. 72-74 С. 1H ЯМР (CDCl3, ); 1,59 (д, J=7 Гц, 6H); 1,10-2,25 (м, 7 Н); 2,80-3,15 (м, 2 Н); 3,27-3,60 (м,4 Н); 4,86 (гепт., J=7 Гц, 1 Н); 7,00-7,60 (м, 9 Н); 8,27-8,52 (м, 1H). ИК (KBr): Vco 1641 см-1. Пример 3. Получение гидрохлорида N3-(4 пиперидинилметил)-1-изопропил-1 Н-3-индазолкарбоксамида (V). Суспензию продукта примера 2 (28 г; 0,076 моль) в этиловом спирте (1500 мл) и ледяной уксусной кислоте (66 мл) гидрируют над 10%Pd-C (13,4 г) при 35 фунт/кв.дюйм (2,5 ати) в течение 24 ч. Смесь фильтруют и фильтрат концентрируют в вакууме. Остаток растворяют в воде, обрабатывают 5N NaOH и перемешивают в течение 2 ч при комнатной температуре. Полученное твердое вещество отфильтровывают К перемешиваемой суспензии продукта примера 3 в виде свободного основания (5,27 г; 15,6 ммоль) в этиловом спирте (20 мл) добавляют К 2 СО 3 (6,5 г; 50 ммоль) и 4-фенилбромбутан("Braun", B-44, 2872, 1911) (3,6 г; 17,1 ммоль). Реакционную смесь перемешивают при кипячении в условиях дефлегмации в течение 10 ч. После удаления растворителя остаток распределяют между этилацетатом и 1N НСl. Водную фазу подщелачивают 2N NaOH, экстрагируют этилацетатом и концентрируют в вакууме. Твердое вещество превращают в соответствующую оксалатную соль (2 г), т.пл. 154-155 С. Элементный анализ для С 29 Н 38N4O51/2 Н 2O найдено, %: вычислено, %:(AFR604) (I: R6=С 6 Н 11). По методике примера 4 N3-(4-пиперидинилметил)-1-изопропил-1 Н-3-индазолкарбоксамид (4,42 г) и (2-бромэтил)циклогексан(AFR606) (I: R6=-СН 2NС 2 Н 6). Согласно методике примера 4 N3-(4 пиперидинилметил)-1-изопропил-1 Н-3-индазолкарбоксамид (3 г) и гидрохлорид N-(3 хлорпропил)-N,N-диметиламин (580 мг) дают указанное в заголовке соединение (950 мг), т.пл. 155-156 С. 1 Элементный анализ для С 30 Н 43 Н 5O91/2 Н 2O найдено, %: вычислено, %: 8 Согласно методике примера 4 N3-(4 пиперидинилметил)-1-изопропил-1 Н-3 индазолкарбоксамид (3 г) и 4-(2-хлорэтил) морфолинил (3,42 г) дают указанное в заголовке соединение (3,2 г), т.пл. 266-267 С (разл.) Элементный анализ дляVco 1652 см-1. Пример 8. Получение гидрохлорида N3[(1-2-[(метилсульфонил)амино]этил-4-пиперидинил)метил]-1-изопропил-1 Н-3-индазолкарбоксамида (AFR703) (I: R6=CH3SO2NH-). Согласно методике примера 4 N3-(4 пиперидинилметил)-1-изопропил-1 Н-3-индазолкарбоксамид (5 г) и N-(2-бромэтил) метансульфонамид (WO 93/18036) (3 г) дают указанное в заголовке соединение (1,5 г), т.пл. 186-187 С Элементный анализ для C20H32ClN5O3S найдено, %: вычислено, %:(AFR605) (I: R6=C5H4N). К перемешиваемой суспензии продукта примера 4 в виде свободного основания (10 г; 33,3 ммоль) добавляют 2-винилпиридин (3,6 г; 34 ммоль), ледяную уксусную кислоту (2 мл) и воду (2,5 мл). После 16 ч при 95 С, реакционную смесь подщелачивают 2N NaOH, экстрагируют этилацетатом и концентрируют под вакуумом. Остаток очищают флэш-хроматографией на силикагеле со смесью СНСl3:МеОН=97:3 в качестве элюента, получая твердое вещество,которое превращают в гидрохлоридную соль (5 г), т.пл. 122-123 С (разл.) 1 Элементный анализ для С 24 Н 32 СlN5OН 2O найдено, %: вычислено, %:H ЯМР (ДМСО, ): 1,54 (д., J=9 Гц, 6 Н); 1,8-1,9 (м., 5 Н); 2,28 (м., 3 Н); 2,90-3,65 (м., 13 Н); 5,09 (гепт., J=7 Гц, 1 Н); 7,10 (д., J=9 Гц, 2 Н); 7,25 (т., J=8 Гц, 1 Н); 7.4-7,5 (м., 3 Н); 7,80 (д., J=9 Гц, 1 Н); 8,17 (д., J=9 Гц, 1 Н); 8,32 (т. шир., 1 Н); 9,0 (с. шир., 1 Н) Пример 12. Получение гидрата дималеата 1-(1-метилэтил)-N-(1-[2-(1-метил-2-пирролидинил)этил]-4-пиперидинилметил)-1 Н-индазол-3 карбоксамида (AFR615) (R6=С 4 Н 7NСН 3). В соответствии с процедурой примера 4(т. шир., 1H). Тест 1. Антагонистическое действие на рецептор 5-НТ 4. Антагонистическое действие соединений формулы (I) оценивали путем проверки влияния испытуемого соединения на индуцируемую серотонином релаксацию оболочки пищевода крыс, предварительно сжатой карбахолом по методике, описанной J.D. Gale et al. в "Br. J. 10 Все испытанные соединения по изобретению показали рА 28. Конкретные значения дляAFR 306 9,36 0,38 ФОРМУЛА ИЗОБРЕТЕНИЯ 1. Индазоламидные соединения общей формулы в которой R6 выбран из группы, включающей фенил C1-3 алкил, С 3-7-циклоалкил, 5- или 6 членное гетероциклическое кольцо, в котором от 1 до 2 членов являются одинаковыми или различными гетероатомами, выбранными из группы, состоящей из N и О, диметиламино C1-3 алкил, метокси C1-3 алкил, аминосульфонилметил, арил, замещенный гидроксигруппой; их соли присоединения к фармацевтически приемлемым органическим и неорганическим кислотам и их фармацевтически приемлемые четвертичные соли. 2. Соединение по п.1, характеризующееся тем, что гетероциклическими кольцами являются фуранил, пиранил, пирролил, имидазолил,пиразолил, изоксазолил, пиридинил, пиразинил,пиримидинил, пиридазинил, фуразанил, пирролинил, имидазолинил, пиразолидинил, пиразолинил, пиперидинил, пиперазинил, морфолинил. 3. Соединение по п.1, характеризующееся тем, что R6 выбран из группы, включающей циклопропил, циклогексил, пиридинил, морфолинил, метоксиметил, метоксипропил, гидроксифенил, диметиламинометил и аминосульфонилметил. 4. Соединение по п.1, характеризующееся тем, что R6 представляет циклогексил. 5. Соединение по п.1, характеризующееся тем, что R6 представляет собой пиридинил. 6. Соединение по п.1, характеризующееся тем, что R6 представляет диметиламинометил. 7. Соединение по п.1, характеризующееся тем, что R6 представляет морфолинил. 8. Соединение по п.1, характеризующееся тем, что R6 представляет аминосульфонилметил. 9. Способ получения соединения формулы(I), его солей присоединения к фармацевтически приемлемым органическим и неорганическим кислотам и его фармацевтически приемлемых четвертичных солей, включающий: а) ацилирование 4-аминометилпиперидина формулы с) алкилирование соединения формулы (V) соединением формулы (VI) с получением соединения формулы (I) в соответствии со следующей схемой реакции на которой R6 имеет вышеприведенные значения и Y представляет галоген, 12d) необязательно образование соли присоединения индазоламидного соединения формулы (I) к фармацевтически приемлемой органической или неорганической кислотe, или фармацевтически приемлемой четвертичной соли индазоламидного соединения формулы (I). 10. Способ по п.9, отличающийся тем, что стадию а) осуществляют взаимодействием соединения формулы (II) с соединением формулы(III), в котором Х представляет собой хлор, в присутствии разбавителя и при температуре от 0 до 140 С в течение периода времени от 0,5 до 20 ч. 11. Способ по п.9, отличающийся тем, что стадию b) осуществляют с помощью каталитического гидрирования. 12. Способ по любому из пп.9-10, отличающийся тем, что, когда в соединении формулы (VI) Y представляет собой хлор или бром,стадию с) проводят в присутствии акцептора кислот и в присутствии разбавителя. 13. Фармацевтическая композиция, отличающаяся тем, что указанная композиция включает эффективную дозу, по крайней мере, одного соединения формулы в которой R6 выбран из группы, включающей фенил C1-3 алкил, С 3-7 циклоалкил, 5- или 6 членное гетероциклическое кольцо, в котором от 1 до 2 членов являются одинаковыми или различными гетероатомами, выбранными из группы, состоящей из N и О, диметиламино C1-3 алкила, метокси C1-3 алкила, аминосульфонилметила, арила, замещенного гидроксигруппой; его солей присоединения к фармацевтически приемлемым органическим и неорганическим кислотам или его фармацевтически приемлемых четвертичных солей.
МПК / Метки
МПК: A61K 31/416, C07D 401/12
Метки: качестве, серотонинергических, индазоламида, агентов, производные
Код ссылки
<a href="https://eas.patents.su/7-2352-proizvodnye-indazolamida-v-kachestve-serotoninergicheskih-agentov.html" rel="bookmark" title="База патентов Евразийского Союза">Производные индазоламида в качестве серотонинергических агентов</a>
Производные индола в качестве эстрогенных агентов

Номер патента: 815
Опубликовано: 24.04.2000
Авторы: Миллер Крис П., Коллини Майкл Д., Тран Бэч Д.
МПК: A61K 31/405, C07D 209/08
Метки: индола, качестве, производные, агентов, эстрогенных
Формула / Реферат:
1. Соединение, имеющее структуру где R1 выбран из Н, ОН, -ОС(=O)(С1-С4алкил), -О(С1-С4алкил) или галогена; R2, R3, R4, R5 и R6 независимо выбраны из Н, ОН, -ОС(=O)(C1-С4алкил), -О(С1-C4алкил), галогена или C1-С6алкила, при условии, что в случае, когда R1 представляет собой Н, R2 не является группой ОН; Х выбран из Н, C1-С6алкила, нитро или галогена; Z выбран из где n равно 1, 2 или 3; Y выбран из: а) группы где R7 и R8...
Производные спиропиперидина и их использование в качестве терапевтических агентов

Номер патента: 1574
Опубликовано: 25.06.2001
Авторы: Свейн Кристофер Джон, Харрисон Тимоти, Сьюард Эйлин Мэри, Вилльямс Брайан Джон, Кертис Нил Рой, Эллиотт Джейсон Мэттью, Кулаговский Януш Йозеф, Джексон Филип Стефен, Бейкер Рэймонд, Холлингворт Грегори Джон
МПК: A61P 1/08, A61K 31/4355, C07D 491/10...
Метки: качестве, использование, терапевтических, спиропиперидина, производные, агентов
Формула / Реферат:
1. Соединение формулы (I) где R1 означает водород, гидрокси, C1-6-алкил, С2-6-алкенил, С3-7-циклоалкил, С3-7-циклоалкил-С1-4-алкил, C1-6-алкокси, фтор-С1-6-алкокси, C1-6-алкокси-С1-4-алкил, С1-б-алкокси-С1-4-алкокси, фтор-С1-6-алкокси-С1-4-алкил, С2-6-алкенилокси, С3-7-циклоалкокси, С3-7-циклоалкил-С1-4-алкокси, фенокси, бензилокси, циано, галоген, NRaRb, SRa, SORa, SO2Ra, OSO2Ra, NRaCOR14, CORa, CO2Ra или CONRaRb, где Ra и Rb независимо друг...
Производные дистамицина, способ их получения и применение в качестве противоопухолевых агентов

Номер патента: 2273
Опубликовано: 28.02.2002
Авторы: Коцци Паоло, Берия Итало, Джерони Мария Кристина, Кальдарелли Марина, Песенти Энрико
МПК: A61K 31/40, A61P 35/00, C07D 207/34...
Метки: получения, применение, производные, качестве, способ, дистамицина, противоопухолевых, агентов
Формула / Реферат:
1. Соединение, которое представляет собой производное дистамицина формулы (I) где n равен 2, 3 или 4; R0 представляет С1-С4-алкил или C1-С3-галогеналкил; R1 и R2, которые могут быть одинаковыми или различными, выбирают, каждый, из водорода, C1-C4-алкила, необязательно замещенного одним или несколькими атомами фтора, и C1-C4-алкокси; Х представляет атом галогена; В выбирают из групп следующих формул: где R3, R4, R5, R6, R7, R8 и R9, которые...
Производные 2-фенил-1- [4-(2-аминоэтокси)бензил] индола в качестве эстрогенных агентов

Номер патента: 1448
Опубликовано: 23.04.2001
Авторы: Миллер Крис Пол, Трэн Бэч Динх, Коллини Мишель Дэвид, Сантилли Артур Аттилио
МПК: A61P 9/00, C07D 209/22, A61K 31/404...
Метки: производные, 2-фенил-1, эстрогенных, 4-(2-аминоэтокси)бензил, качестве, индола, агентов
Формула / Реферат:
1. Производные 2-фенил-1-[4-(2-аминоэтокси)бензил] индола общей структурной формулы I или II где R1 выбран из Н, ОН, -О-(С=O)-(C1-C12)алкила (где алкил представляет собой линейную цепь или разветвленную), -О-(C1-C12)алкила (где алкил представляет собой линейную цепь, разветвленную или циклическую цепь), галогена; или -O-(C1-C4)-галогенированного алкила (включая трифторметокси и трихлорметокси); R2, R3, R4, R5 и R6 независимо выбраны из Н, ОН,...
Комплексы лимфотоксина альфа/бета и антител против рецептора лимфотоксина-бета в качестве противоопухолевых агентов

Номер патента: 96
Опубликовано: 27.08.1998
Авторы: Мейер Вернер, Броунинг Джеффри Л., Бенджамин Кристофер
МПК: A61K 38/19, G01N 33/53
Метки: качестве, агентов, рецептора, лимфотоксина, антител, комплексы, лимфотоксина-бета, против, противоопухолевых
Формула / Реферат:
1. Способ лечения или уменьшения прогрессирования, тяжести или эффектов неоплазии, включающий введение терапевтически эффективного количества гетеромерного комплекса лимфотоксина a/b в присутствии терапевтически эффективного количества антитела к рецептору лимфотоксина b и/или g-интерферона. 2. Способ лечения или уменьшения прогрессирования, тяжести или эффектов неоплазии, включающий введение терапевтически эффективного количества антитела к...
Предыдущий патент: Производные сульфонамида, способ их получения и их использование в качестве лекарственных средств.
Следующий патент: Лептин как ингибитор пролиферации опухолевых клеток
Случайный патент: Каландр