4′-азидо,3′-фторзамещенные производные нуклеозидов в качестве ингибиторов репликации рнк вируса гепатита с
Номер патента: 24847
Опубликовано: 31.10.2016
Авторы: Чжан Цзин, Таламас Франсиско Ксавьер, Чжан Чжумин, Смит Марк


























Формула / Реферат
1. Соединение формулы I

в котором
R1 представляет собой Н, фенил или нафтил;
R2a представляет собой Н;
R2b представляет собой C1-7-алкил;
R3 представляет собой С1-7-алкил или фенил-С1-7-алкил;
R4 представляет собой Н;
R5 представляет собой Н, C(=O)R1c, где R1c представляет собой C1-7-алкил;
R6 представляет собой

или его фармацевтически приемлемая соль.
2. Соединение по п.1, в котором R1 представляет собой нафтил или фенил.
3. Соединение по п.2, в котором R2a представляет собой Н.
4. Соединение по п.3, в котором R2b представляет собой метил.
5. Соединение по п.4, в котором R3 представляет собой изопропил, этил или бензил.
6. Соединение по п.5, в котором R5 представляет собой Н.
7. Соединение по п.5, в котором R5 представляет собой C(=O)R1c.
8. Соединение по п.7, в котором R1c представляет собой этил.
9. Соединение по п.6, в котором R6 представляет собой

10. Соединение по п.6, в котором R6 представляет собой

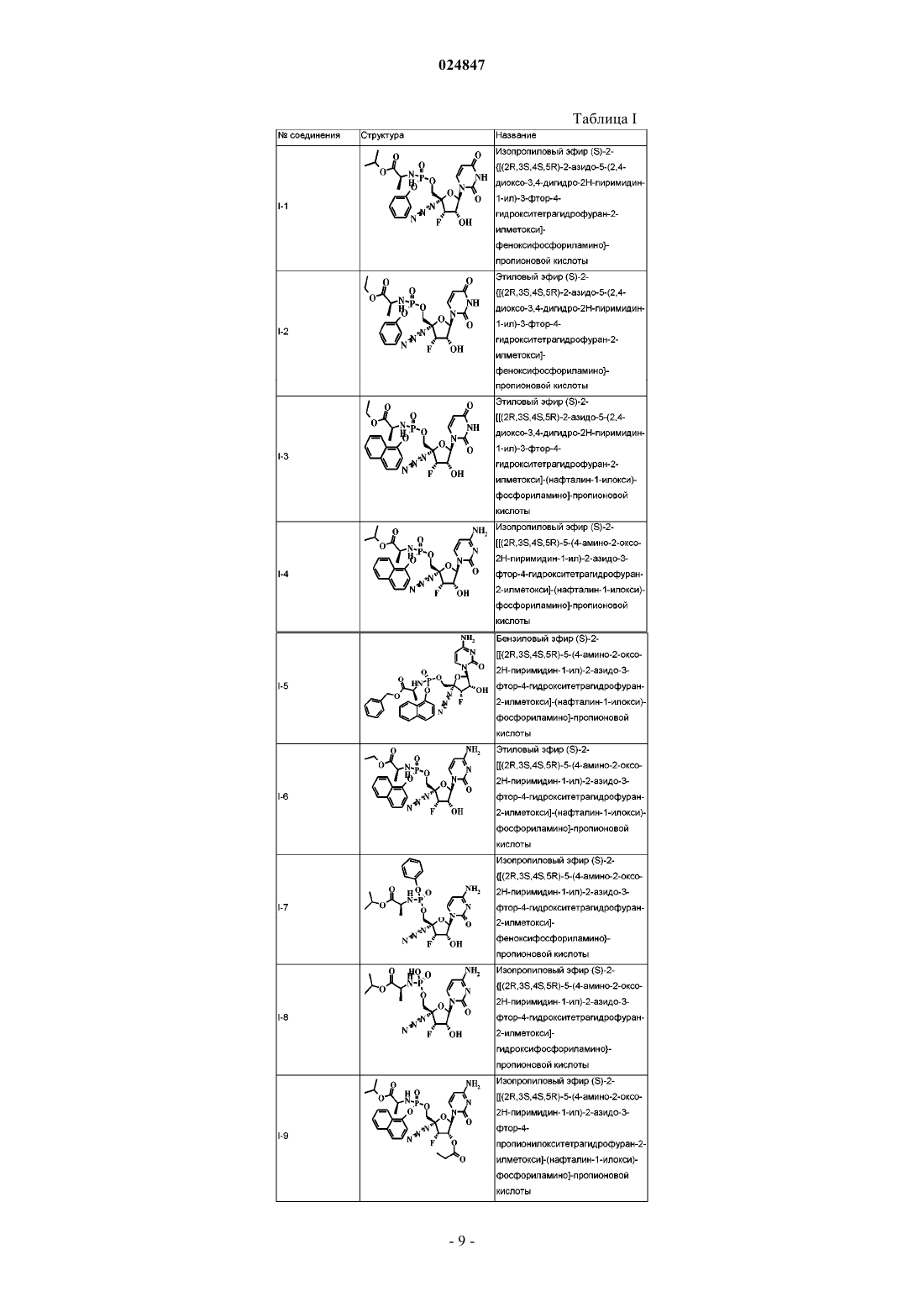
11. Соединение, выбранное из группы, состоящей из следующих соединений:
изопропиловый эфир (S)-2-{[(2R,3S,4S,5R)-2-азидо-5-(2,4-диоксо-3,4-дигидро-2H-пиримидин-1-ил)-3-фтор-4-гидрокситетрагидрофуран-2-илметокси]феноксифосфориламино}пропионовой кислоты;
этиловый эфир (S)-2-{[(2R,3S,4S,5R)-2-азидо-5-(2,4-диоксо-3,4-дигидро-2H-пиримидин-1-ил)-3-фтор-4-гидрокситетрагидрофуран-2-илметокси]феноксифосфориламино}пропионовой кислоты;
этиловый эфир (S)-2-[[(2R,3S,4S,5R)-2-азидо-5-(2,4-диоксо-3,4-дигидро-2H-пиримидин-1-ил)-3-фтор-4-гидрокситетрагидрофуран-2-илметокси]-(нафталин-1-илокси)фосфориламино]пропионовой кислоты;
изопропиловый эфир (S)-2-[[(2R,3S,4S,5R)-5-(4-амино-2-оксо-2H-пиримидин-1-ил)-2-азидо-3-фтор-4-гидрокситетрагидрофуран-2-илметокси]-(нафталин-1-илокси)фосфориламино]пропионовой кислоты;
бензиловый эфир (S)-2-[[(2R,3S,4S,5R)-5-(4-амино-2-оксо-2H-пиримидин-1-ил)-2-азидо-3-фтор-4-гидрокситетрагидрофуран-2-илметокси]-(нафталин-1-илокси)фосфориламино]пропионовой кислоты;
этиловый эфир (S)-2-[[(2R,3S,4S,5R)-5-(4-амино-2-оксо-2H-пиримидин-1-ил)-2-азидо-3-фтор-4-гидрокситетрагидрофуран-2-илметокси]-(нафталин-1-илокси)фосфориламино]пропионовой кислоты;
изопропиловый эфир (S)-2-{[(2R,3S,4S,5R)-5-(4-амино-2-оксо-2H-пиримидин-1-ил)-2-азидо-3-фтор-4-гидрокситетрагидрофуран-2-илметокси]феноксифосфориламино}пропионовой кислоты;
изопропиловый эфир (S)-2-{[(2R,3S,4S,5R)-5-(4-амино-2-оксо-2H-пиримидин-1-ил)-2-азидо-3-фтор-4-гидрокситетрагидрофуран-2-илметокси]гидроксифосфориламино}пропионовой кислоты и
изопропиловый эфир (S)-2-[[(2R,3S,4S,5R)-5-(4-амино-2-оксо-2H-пиримидин-1-ил)-2-азидо-3-фтор-4-пропионилокситетрагидрофуран-2-илметокси]-(нафталин-1-илокси)фосфориламино]пропионовой кислоты.
12. Применение соединения по любому из пп.1-11 для лечения или профилактики инфекции вируса гепатита С (ВГС).
13. Применение соединения по любому из пп.1-11 для изготовления лекарственного средства для лечения или профилактики инфекции вируса гепатита С (ВГС).
14. Способ лечения инфекции вируса гепатита С (ВГС), включающий введение пациенту, который в этом нуждается, терапевтически эффективного количества соединения по любому из пп.1-11.
15. Фармацевтическая композиция, включающая соединение по любому из пп.1-11 и терапевтически инертные носители.
Текст
4'-АЗИДО,3'-ФТОРЗАМЕЩЕННЫЕ ПРОИЗВОДНЫЕ НУКЛЕОЗИДОВ В КАЧЕСТВЕ ИНГИБИТОРОВ РЕПЛИКАЦИИ РНК ВИРУСА ГЕПАТИТА С Изобретение относится к применению нуклеозидных производных формулы I где символы имеют значения, определенные в описании, и их фармацевтически приемлемых солей,а также к фармацевтическим композициям, содержащим такие соединения. Область техники Настоящее изобретение относится к нуклеозидным производным в качестве ингибиторов репликации РНК репликона ВГС. В частности, настоящее изобретение касается применения пуриновых и пиримидиновых нуклеозидных производных в качестве ингибиторов репликации субгеномной РНК вируса гепатита С (ВГС) и фармацевтических композиций, содержащих такие соединения. Вирус гепатита С является основной причиной хронических заболеваний печени в мире. Пациенты, инфицированные ВГС, имеют риск развития цирроза печени и последующего печеночно-клеточного рака,и, следовательно, ВГС является главным показанием к пересадке печени. В настоящее время доступны только два одобренных терапевтических способа для лечения ВГС-инфекции (R.G. Gish, Sem. Liver. Dis.,1999, 19, 35). Это монотерапия с помощью интерферона- и, с недавнего времени, комбинированная терапия с помощью нуклеозидного аналога, рибавирина (Virazole), и интерферона-. Многие лекарственные вещества, одобренные для лечения вирусных инфекций, представляют собой нуклеозиды или аналоги нуклеозидов, и большинство таких лекарственных веществ - нуклеозидных аналогов ингибируют репликацию вируса после их превращения в соответствующие трифосфаты путем ингибирования ферментов вирусной полимеразы. Это превращение в трифосфат обычно опосредовано клеточными киназами, и поэтому прямую оценку нуклеозидов в качестве ингибиторов репликации ВГС удобно проводить только с помощью клеточного анализа. Для ВГС отсутствует истинный клеточный анализ репликации вируса или животная модель инфицирования. Вирус гепатита С принадлежит к семейству Flaviridae. Он представляет собой РНК-вирус, при этом РНК кодирует большой полипротеин, который после процессирования генерирует необходимый механизм репликации, чтобы обеспечить синтез дочерней РНК. Полагают, что большинство неструктурных белков, кодируемых геномом РНК ВГС, вовлечено в репликацию РНК. Lohmann и соавт. [V. Lohmann etal., Science, 1999, 285, 110-113] описали конструкцию клеточной линии человеческой гепатомы (Huh7), в которую вводили молекулы субгеномной РНК ВГС, и было показано, что она реплицируется с высокой эффективностью. Полагают, что механизм репликации РНК в этих линиях клеток идентичен репликации полноразмерного генома РНК ВГС в инфицированных гепатоцитах. Клоны субгеномной кДНК ВГС,которые были использованы для выделения этих клеточных линий, послужили основой для разработки клеточного анализа для идентификации ингибиторов репликации ВГС на основе нуклеозидных аналогов. Сущность изобретения Соединения формулы I полезны для лечения заболеваний, опосредованных вирусом гепатита С(ВГС), и для изготовления фармацевтических композиций, включающих такие соединения. В настоящей заявке предложено соединение формулы I или его фармацевтически приемлемая соль. В настоящей заявке предложен способ лечения инфекции вируса гепатита С (ВГС), включающий введение пациенту, который в этом нуждается, терапевтически эффективного количества соединения формулы I. В настоящей заявке предложена фармацевтическая композиция, включающая соединение формулыI и терапевтически инертные носители. Подробное описание изобретения Показали, что соединения формулы I являются ингибиторами субгеномной репликации вирус гепатита С в линии клеток гепатомы. Эти соединения потенциально эффективны в качестве противовирусных лекарственных средств для лечения ВГС-инфекций у человека. Термин "алкил", используемый в настоящем описании, обозначает углеводородный остаток с линейной или разветвленной цепью, содержащий от 1 до 7 атомов углерода. Наиболее предпочтительны метил, этил, пропил, изопропил, н-бутил, изобутил, трет-бутил или пентил. Алкил может быть незамещенным или замещенным. Заместители выбирают из одной или нескольких следующих групп: циклоалкил, нитрогруппа, аминогруппа, алкиламиногруппа, диалкиламиногруппа, алкилкарбонил и циклоалкилкарбонил. Термин "циклоалкил", используемый в настоящем описании, обозначает возможно замещенную циклоалкильную группу, содержащую от 3 до 7 атомов углерода, например циклопропил, циклобутил,циклопентил, циклогексил или циклогептил. Термин "алкокси", используемый в настоящем описании, обозначает возможно замещенную алкилоксигруппу с линейной или разветвленной цепью, в которой алкильный фрагмент раскрыт выше, такую как метокси-, этокси-, н-пропилокси-, изопропилокси-, н-бутилокси-, изобутилокси-, трет-бутилокси-,пентилокси-, гексилокси-, гептилоксигруппа, включая их изомеры. Термин "алкоксиалкил", используемый в настоящем описании, обозначает алкоксигруппу, раскрытую выше, которая присоединена к алкильной группе, раскрытой выше. Примерами являются метоксиметил, метоксиэтил, метоксипропил, этоксиметил, этоксиэтил, этоксипропил, пропилоксипропил, метоксибутил, этоксибутил, пропилоксибутил, бутилоксибутил, трет-бутилоксибутил, метоксипентил, этоксипентил, пропилоксипентил, включая их изомеры. Термин "алкенил", используемый в настоящем описании, обозначает радикал с незамещенной или замещенной углеводородной цепью, включающий от 2 до 7 атомов углерода, предпочтительно от 2 до 4 атомов углерода, и содержащий одну или две олефиновые двойные связи, предпочтительно одну олефиновую двойную связь. Примерами являются винил, 1-пропенил, 2-пропенил (аллил) или 2-бутенил (кротил). Термин "алкинил", используемый в настоящем описании, обозначает радикал с незамещенной или замещенной углеводородной цепью, содержащий от 2 до 7 атомов углерода, предпочтительно от 2 до 4 атомов углерода, и включающий одну или, если возможно, две тройные связи, предпочтительно одну тройную связь. Примерами являются этинил, 1-пропинил, 2-пропинил, 1-бутинил, 2-бутинил или 3 бутинил. Термин "гидроксиалкил", используемый в настоящем описании, обозначает алкильную группу с линейной или разветвленной цепью, раскрытую выше, в которой 1, 2, 3 или более атомов водорода замещены гидроксигруппой. Примерами являются гидроксиметил, 1-гидроксиэтил, 2-гидроксиэтил, 1 гидроксипропил, 2-гидроксипропил, 3-гидроксипропил, гидроксиизопропил, гидроксибутил и т. п. Термин "галогеналкил", используемый в настоящем описании, обозначает алкильную группу с линейной или разветвленной цепью, раскрытую выше, в которой 1, 2, 3 или более атомов водорода замещены атомами галогена. Примерами являются 1-фторметил, 1-хлорметил, 1-бромметил, 1-иодметил,трифторметил, трихлорметил, трибромметил, трииодметил, 1-фторэтил, 1-хлорэтил, 1-бромэтил, 1 иодэтил, 2-фторэтил, 2-хлорэтил, 2-бромэтил, 2-иодэтил, 2,2-дихлорэтил, 3-бромпропил или 2,2,2 трифторэтил и т. п. Термин "алкилтио", используемый в настоящем описании, обозначает (алкил)S-группу с линейной или разветвленной цепью, в которой алкильный фрагмент раскрыт выше. Примерами являются метилтио-, этилтио-, н-пропилтио-, изопропилтио-, н-бутилтио-, изобутилтио- или трет-бутилтиогруппа. Термин "арил", используемый в настоящем описании, обозначает возможно замещенный фенил и нафтил (например 1-нафтил, 2-нафтил или 3-нафтил). Подходящие заместители для арила можно выбирать из тех, которые указаны для алкила, и к этому перечню для выбора дополнительно можно добавить следующие заместители: галоген, гидроксигруппа и возможно замещенный алкил, галогеналкил, алкенил, алкинил и арилоксигруппа. Термин "гетероциклил", используемый в настоящем описании, обозначает возможно замещенные насыщенные, частично ненасыщенные или ароматические моноциклические, бициклические или трициклические гетероциклические системы, содержащие один или более гетероатомов, выбранных из азота, кислорода и серы, которые могут быть также конденсированы с возможно замещенным насыщенным,частично ненасыщенным или ароматическим моноциклическим карбоциклом или гетероциклом. Примерами подходящих гетероциклов являются оксазолил, изоксазолил, фурил, тетрагидрофурил,1,3-диоксоланил, дигидропиранил, 2-тиенил, 3-тиенил, пиразинил, изотиазолил, дигидрооксазолил, пиримидинил, тетразолил, 1-пирролидинил, 2-пирролидинил, 3-пирролидинил, пирролидинонил, (Nоксид)-пиридинил, 1-пирролил, 2-пирролил, триазолил, например 1,2,3-триазолил или 1,2,4-триазолил, 1 пиразолил, 2-пиразолил, 4-пиразолил, пиперидинил, морфолинил (например, 4-морфолинил), тиоморфолинил (например, 4-тиоморфолинил), тиазолил, пиридинил, дигидротиазолил, имидазолидинил, пиразолинил, пиперазинил, 1-имидазолил, 2-имидазолил, 4-имидазолил, тиадиазолил, например 1,2,3 тиадиазолил, 4-метилпиперазинил, 4-гидроксипиперидин-1-ил. Подходящие заместители для гетероциклила можно выбирать из тех, которые были указаны для алкила, и к этому перечню для выбора дополнительно можно добавить следующие заместители: возможно замещенный алкил, алкенил, алкинил, оксогруппа (=O) или аминосульфонил. Термин "ацил" ("алкилкарбонил"), используемый в настоящем описании, обозначает группу формулы C(=O)R, в которой R представляет собой водород, незамещенный или замещенный углеводородный остаток с линейной или разветвленной цепью, содержащий от 1 до 7 атомов углерода, или фенильную группу. Наиболее предпочтительно ацильные группы представляют собой те, в которых R представляет собой водород, незамещенный углеводородный остаток с линейной или разветвленной цепью, содержащий от 1 до 4 атомов углерода, или фенильную группу. Термин галоген обозначает фтор, хлор, бром или иод, предпочтительно фтор, хлор, бром. В графическом представлении соединений, приведенных в настоящей заявке, утолщенная заостренная линия обозначает заместитель, который находится выше плоскости кольца, к которому принадлежит ассиметрический атом углерода, а точечная линия обозначает заместитель, который расположен ниже плоскости кольца, к которому принадлежит ассиметрический атом углерода. Соединения формулы I проявляют стереоизомерию. Эти соединения могут представлять собой любой изомер соединения формулы I или смеси таких изомеров. Соединения и промежуточные продукты по настоящему изобретению, содержащие один или более ассиметрических атомов углерода, могут быть получены в виде рацемических смесей стереоизомеров, которые можно разделять. Соединения формулы I проявляют таутомерию, и это означает, что соединения по настоящему изобретению могут существовать в виде двух или более химических соединений, которые способны легко превращаться друг в друга. Во многих случаях это просто означает обмен атомом водорода между двумя другими атомами, с каждым из которых он образует ковалентную связь. Таутомерные соединения существуют в подвижном равновесии друг с другом, и попытки получить отдельные соединения обычно приводят к образованию смеси со всеми химическими и физическими свойствами, которые ожидались, исходя из структуры ее компонентов. Наиболее распространенный тип таутомерии включает карбонильные, или кето-соединения, и ненасыщенные гидроксисоединения или енолы. Структурное изменение заключается в перемещении атома водорода между атомами углерода и кислорода с перегруппировкой связей. Например, во многих алифатических альдегидах и кетонах, таких как ацетальдегид, преобладает кето-форма; в фенолах основным компонентом является енольная форма. Соединения формулы I, которые являются основными, могут образовывать фармацевтически приемлемые соли с неорганическими кислотами, такими как галогеноводородные кислоты (например, хлороводородная кислота и бромоводородная кислота), серная кислота, азотная кислота и фосфорная кислота и т. п., а также с органическими кислотами (например, с уксусной кислотой, винной кислотой, янтарной кислотой, фумаровой кислотой, малеиновой кислотой, яблочной кислотой, салициловой кислотой,лимонной кислотой, метансульфоновой кислотой и п-толуолсульфоновой кислотой и т. п.). Получение и выделение таких солей можно проводить способами, известными в данной области техники. Ингибиторы ВГС. В настоящей заявке предложено соединение формулы I или его фармацевтически приемлемая соль. В настоящей заявке предложено соединение формулы I, в котором R4 представляет собой Н. В настоящей заявке предложено соединение формулы I, в котором R6 представляет собой В настоящей заявке предложено соединение формулы I, в котором R6 представляет собой В настоящей заявке предложено соединение формулы I, в котором R6 представляет собойВ настоящей заявке предложено соединение формулы I, в котором R4 представляет собой Н, a R6 представляет собой В настоящей заявке предложено соединение формулы I, в котором R4 представляет собой Н, a R6 представляет собой В настоящей заявке предложено соединение формулы I, в котором R4 представляет собой Н, a R6 представляет собой В настоящей заявке предложено соединение формулы I, в котором R1 представляет собой нафтил или фенил. В настоящей заявке предложено соединение формулы I, в котором R1 представляет собой нафтил. В настоящей заявке предложено соединение формулы I, в котором R1 представляет собой фенил. В настоящей заявке предложено соединение формулы I, в котором R1 представляет собой фенил, a 4R представляет собой Н. В настоящей заявке предложено соединение формулы I, в котором R1 представляет собой фенил, R4 представляет собой Н, a R6 представляет собой В настоящей заявке предложено соединение формулы I, в котором R1 представляет собой фенил, R4 представляет собой Н, a R6 представляет собой В настоящей заявке предложено соединение формулы I, в котором R1 представляет собой нафтил, aR представляет собой Н. В настоящей заявке предложено соединение формулы I, в котором R1 представляет собой нафтил,4 В настоящей заявке предложено соединение формулы I, в котором R1 представляет собой нафтил,-4 024847 В настоящей заявке предложено соединение формулы I, в котором R2a представляет собой Н. В настоящей заявке предложено соединение формулы I, в котором R2b представляет собой метил. В настоящей заявке предложено соединение формулы I, в котором R2a представляет собой Н, a R2b представляет собой метил. В настоящей заявке предложено соединение формулы I, в котором R1 представляет собой нафтил,4 В настоящей заявке предложено соединение формулы I, в котором R1 представляет собой нафтил,R представляет собой Н, R2b представляет собой метил, a R6 представляет собой 4 В настоящей заявке предложено соединение формулы I, в котором R1 представляет собой нафтил,R представляет собой Н, R2a представляет собой Н, R2b представляет собой метил, a R6 представляет собой 4 В настоящей заявке предложено соединение формулы I, в котором R3 представляет собой изопропил. В настоящей заявке предложено соединение формулы I, в котором R3 представляет собой этил. В настоящей заявке предложено соединение формулы I, в котором R3 представляет собой бензил. В настоящей заявке предложено соединение формулы I, в котором R1 представляет собой нафтил, a 3R представляет собой изопропил. В настоящей заявке предложено соединение формулы I, в котором R1 представляет собой нафтил,4R представляет собой Н, a R3 представляет собой изопропил. В настоящей заявке предложено соединение формулы I, в котором R1 представляет собой нафтил,4 В настоящей заявке предложено соединение формулы I, в котором R1 представляет собой нафтил,R представляет собой Н, R3 представляет собой этил, a R6 представляет собой 4 В настоящей заявке предложено соединение формулы I, в котором R1 представляет собой нафтил,R представляет собой Н, R3 представляет собой бензил, a R6 представляет собой 4 В настоящей заявке предложено соединение формулы I, в котором R1 представляет собой нафтил,R представляет собой Н, R3 представляет собой изопропил, a R6 представляет собой 4 В настоящей заявке предложено соединение формулы I, в котором R1 представляет собой нафтил,-5 024847 В настоящей заявке предложено соединение формулы I, в котором R1 представляет собой нафтил,R представляет собой Н, R3 представляет собой бензил, a R6 представляет собой 4 В настоящей заявке предложено соединение формулы I, в котором R1 представляет собой нафтил,R представляет собой Н, R2a представляет собой Н, R3 представляет собой изопропил, a R6 представляет собой 4 В настоящей заявке предложено соединение формулы I, в котором R1 представляет собой нафтил,R представляет собой Н, R2a представляет собой Н, R2b представляет собой метил, R3 представляет собой изопропил, a R6 представляет собой 4 В настоящей заявке предложено соединение формулы I, в котором R1 представляет собой нафтил,R представляет собой Н, R2a представляет собой Н, R3 представляет собой изопропил, a R6 представляет собой 4 В настоящей заявке предложено соединение формулы I, в котором R1 представляет собой нафтил,R представляет собой Н, R2a представляет собой Н, R2b представляет собой метил, R3 представляет собой изопропил, a R6 представляет собой 4 В настоящей заявке предложено соединение формулы I, в котором R5 представляет собой Н. В настоящей заявке предложено соединение формулы I, в котором R1 представляет собой нафтил, a 5R представляет собой Н. В настоящей заявке предложено соединение формулы I, в котором R1 представляет собой нафтил,4R представляет собой Н, a R5 представляет собой Н. В настоящей заявке предложено соединение формулы I, в котором R1 представляет собой нафтил,4 В настоящей заявке предложено соединение формулы I, в котором R1 представляет собой нафтил,R представляет собой Н, R2a представляет собой Н, R5 представляет собой Н, a R6 представляет собой 4 В настоящей заявке предложено соединение формулы I, в котором R1 представляет собой нафтил,-6 024847 В настоящей заявке предложено соединение формулы I, в котором R1 представляет собой нафтил,R представляет собой Н, R2a представляет собой Н, R2b представляет собой метил, R3 представляет собой изопропил, R5 представляет собой Н, a R6 представляет собой 4 В настоящей заявке предложено соединение формулы I, в котором R1 представляет собой нафтил,R представляет собой Н, R2a представляет собой Н, R2b представляет собой метил, R3 представляет собой этил, R5 представляет собой Н, a R6 представляет собой 4 В настоящей заявке предложено соединение формулы I, в котором R1 представляет собой нафтил,R представляет собой Н, R2a представляет собой Н, R2b представляет собой метил, R3 представляет собой бензил, R5 представляет собой Н, a R6 представляет собой 4 В настоящей заявке предложено соединение формулы I, в котором R1 представляет собой нафтил,R представляет собой Н, R2a представляет собой Н, R2b представляет собой метил, R3 представляет собой изопропил, R5 представляет собой Н, a R6 представляет собой 4 В настоящей заявке предложено соединение формулы I, в котором R1 представляет собой нафтил,R представляет собой Н, R2a представляет собой Н, R2b представляет собой метил, R3 представляет собой этил, R5 представляет собой Н, a R6 представляет собой 4 В настоящей заявке предложено соединение формулы I, в котором R1 представляет собой нафтил,R представляет собой Н, R2a представляет собой Н, R2b представляет собой метил, R3 представляет собой бензил, R5 представляет собой Н, a R6 представляет собой 4 В настоящей заявке предложено соединение формулы I, в котором R5 представляет собой C(=O)R1c. В настоящей заявке предложено соединение формулы I, в котором R1 представляет собой нафтил,4 В настоящей заявке предложено соединение формулы I, в котором R1c представляет собой этил. В настоящей заявке предложено соединение формулы I, в котором R1 представляет собой нафтил,4 В настоящей заявке предложено соединение, выбранное из группы, состоящей из следующих соединений: изопропиловый эфир (S)-2-[(2R,3S,4S,5R)-2-азидо-5-(2,4-диоксо-3,4-дигидро-2H-пиримидин-1-ил)3-фтор-4-гидрокситетрагидрофуран-2-илметокси]феноксифосфориламинопропионовой кислоты; этиловый эфир (S)-2-[(2R,3S,4S,5R)-2-азидо-5-(2,4-диоксо-3,4-дигидро-2H-пиримидин-1-ил)-3 фтор-4-гидрокситетрагидрофуран-2-илметокси]феноксифосфориламинопропионовой кислоты; этиловый эфир (S)-2-(2R,3S,4S,5R)-2-азидо-5-(2,4-диоксо-3,4-дигидро-2H-пиримидин-1-ил)-3 фтор-4-гидрокситетрагидрофуран-2-илметокси]-(нафталин-1-илокси)фосфориламино]пропионовой кислоты; изопропиловый эфир (S)-2-(2R,3S,4S,5R)-5-(4-амино-2-оксо-2H-пиримидин-1-ил)-2-азидо-3-фтор 4-гидрокситетрагидрофуран-2-илметокси]-(нафталин-1-илокси)фосфориламино]пропионовой кислоты; бензиловый эфир (S)-2-(2R,3S,4S,5R)-5-(4-амино-2-оксо-2H-пиримидин-1-ил)-2-азидо-3-фтор-4 гидрокситетрагидрофуран-2-илметокси]-(нафталин-1-илокси)фосфориламино]пропионовой кислоты; этиловый эфир (S)-2-(2R,3S,4S,5R)-5-(4-амино-2-оксо-2H-пиримидин-1-ил)-2-азидо-3-фтор-4 гидрокситетрагидрофуран-2-илметокси]-(нафталин-1-илокси)фосфориламино]пропионовой кислоты; изопропиловый эфир (S)-2-[(2R,3S,4S,5R)-5-(4-амино-2-оксо-2H-пиримидин-1-ил)-2-азидо-3-фтор 4-гидрокситетрагидрофуран-2-илметокси]феноксифосфориламинопропионовой кислоты; изопропиловый эфир (S)-2-[(2R,3S,4S,5R)-5-(4-амино-2-оксо-2H-пиримидин-1-ил)-2-азидо-3-фтор 4-гидрокситетрагидрофуран-2-илметокси]гидроксифосфориламинопропионовой кислоты; и изопропиловый эфир (S)-2-(2R,3S,4S,5R)-5-(4-амино-2-оксо-2H-пиримидин-1-ил)-2-азидо-3-фтор 4-пропионилокситетрагидрофуран-2-илметокси]-(нафталин-1-илокси)фосфориламино]пропионовой кислоты. В настоящей заявке предложен способ лечения инфекции вируса гепатита С (ВГС), включающий введение пациенту, который в этом нуждается, терапевтически эффективного количества соединения формулы I. В настоящей заявке предложен указанный выше способ, дополнительно включающий введение модулятора иммунной системы или противовирусного агента, который ингибирует репликацию ВГС, или их комбинацию. В настоящей заявке предложен указанный выше способ, в котором модулятор иммунной системы представляет собой интерферон или химически дериватизированный интерферон. В настоящей заявке предложены указанные выше способы, в которых противовирусный агент выбирают из группы, состоящей из ингибитора протеазы ВГС, ингибитора полимеразы ВГС, ингибитора геликазы ВГС, ингибитора праймазы ВГС, ингибитора слияния ВГС и их комбинации. В настоящей заявке предложен способ ингибирования репликации ВГС в клетке, включающий введение соединения формулы I. В настоящей заявке предложена фармацевтическая композиция, включающая соединение формулыI и терапевтически инертные носители. В настоящей заявке предложено применение соединения формулы I для изготовления лекарственного средства для лечения или профилактики инфекции ВГС. В настоящей заявке предложены соединение, фармацевтическая композиция и способ, раскрытые в настоящем описании. Соединения. Выборочные примеры соединений, охватываемых настоящим изобретением и входящих в объем настоящего изобретения, представлены в нижеследующей таблице. Эти примеры и способы получения,которые следуют далее, приведены, для того чтобы помочь специалисту в данной области техники более ясно понять и осуществить на практике настоящее изобретение. Их следует рассматривать не как ограничение объема настоящего изобретения, а лишь как его иллюстрацию и наглядный пример. В общем случае номенклатура, использованная в настоящей заявке, основана на AUTONOMv.4.0, компьютеризированной системе Института Бельштейна для генерации систематической номенклатуры IUPAC. При несоответствии между изображенной структурой и ее названием преимущество имеет изображенная структура. Кроме того, если стереохимия структуры или ее части не отражена, например,посредством жирной или пунктирной линий, следует понимать, что такая структура или ее часть охватывает все ее стереоизомеры. В табл. I изображены примеры соединений общей формулы I. Синтез. Настоящая заявка связана со следующими публикациями: Smith, David В.; Kalayanov Genadiy; SundIsabel; Benkestock Kurt; et al. Journal of Medicinal Chemistry (2009), 52(9), 2971-2978. Общие схемы. Способы, обсуждавшиеся выше, описаны ниже более детально. Имеющийся в продаже нуклеозид 3'-фтор-3'-дезоксиуридин (1) можно получить также способами,описанными в литературе: Gosselin G. et al., Collect. Czech. Chem. Commun. (2006), Vol. 71, No. 7, 9911010. Иодирование с последующим отщеплением иода в основных условиях может приводить к промежуточному продукту 3. Введение азидогруппы по положению 4' в промежуточном продукте 3 с последующим окислительным замещением 5'-иодида с помощью м-хлорпербензойной кислоты в промежуточном продукте 4 с получением соединения 5 можно осуществить способами, описанными в литературе:Smith D.В. et al., J. Med. Chem. (2009), 52(9), 2971-2978. Снятие защитной 5'-м-хлорбензойной группы в промежуточном продукте 5 дает промежуточный уридин 6 (схема 1). Схема 1 Получение промежуточного цитидина 10 было раскрыто в литературе Smith D.В. et al., J. Med.Chem. (2009), 52(9), 2971-2978. Как вариант, соединение 10 можно эффективно получить синтетическим путем, который показан на схеме 2 Схема 2 Фосфорамидаты по настоящему изобретению можно получить путем конденсации нуклеозида 6 или 10 с подходящим образом замещенным фосфохлоридатом 13 в присутствии сильного основания(схема 4). Конденсацию можно проводить по незамещенному нуклеозиду 6 или 10. Конденсированный продукт 16 в формуле I изначально получают в виде смеси двух диастереомеров при реакции конденсации, и их можно разделить на соответствующие оптические изомеры на хиральной колонке путем хиральной ВЭЖХ или хиральной сверхкритической флюидной хроматографии. Схема 4 Реакцию конденсации можно также проводить по защищенному нуклеозиду 6 или 10. Например,нуклеозид 6 можно защищать по положению 2' с образованием промежуточного продукта 17. Реакция конденсации с соединением 17 может приводить к соединению 18 в формуле I с улучшенным выходом. В случае если R5 представляет собой триэтилсилильную группу, в соединении 18 можно селективно снимать защиту с удалением 2'-триэтилсилильной группы путем обработки уксусной кислотой или муравьиной кислотой при комнатной температуре с получением соединения 16, в котором R6 представляет собой цитозин (схема 5) Схема 5 Дозировка и введение. Как показано в приведенной ниже табл. II, соединения формулы I потенциально эффективны в качестве противовирусных лекарственных средств для лечения ВГС-инфекций у человека или метаболизируют до соединения, которое обладает такой активностью. В другом воплощении настоящего изобретения действующее вещество, или его производное, или его соль можно вводить в комбинации с другим противовирусным агентом, таким как агент против гепатита, включая соединения формулы I. Если действующее вещество, или его производное, или его соль вводят в комбинации с другим противовирусным агентом, его активность может возрастать по сравнению с исходным соединением. Это можно легко оценить путем получения производного и тестирования его активности против ВГС способом, раскрытым в настоящем описании. Способ введения действующего вещества может варьироваться от непрерывного (внутривенное вливание) до нескольких пероральных введений в сутки (например, четыре раза в сутки) и может включать пероральное, местное, парентеральное, внутримышечное, внутривенное, подкожное, трансдермальное (возможно, с агентом, усиливающим проницаемость), трансбуккальное введение и введение с помощью суппозиториев и другие пути введения. 4'-Замещенные нуклеозидные производные, а также их фармацевтически применимые соли можно применять в качестве лекарственных средств в форме любой фармацевтической композиции. Фармацевтическую композицию можно вводить энтерально, как перорально, например в форме таблеток, таблеток, покрытых оболочкой, драже, твердых и мягких желатиновых капсул, растворов, эмульсий, сиропов или суспензий, так и ректально, например в форме суппозиториев. Их можно также вводить парентерально (внутримышечно, внутривенно, подкожно или путем внутригрудинной инъекции или с помощью инфузии), например в форме растворов для инъекции; интраназально, например в форме назальных спреев или спреев для ингаляции; местно и т. д. Для изготовления фармацевтических препаратов 4'-замещенные нуклеозидные производные, а также их фармацевтически применимые соли можно включать в лекарственную форму совместно с терапевтически инертным неорганическим или органическим эксципиентом для изготовления таблеток, таблеток, покрытых оболочкой, драже, твердых и мягких желатиновых капсул, растворов, эмульсий или суспензий. Соединения формулы I можно включать в лекарственную форму в смеси с фармацевтически приемлемым носителем. Например, соединения по настоящему изобретению можно вводить перорально в виде фармацевтически приемлемых солей. Поскольку соединения по настоящему изобретению по большей части растворимы в воде, их можно вводить внутривенно в физиологическом солевом растворе (например, забуференном примерно до рН 7,2-7,5). В этих целях можно применять стандартные буферы,например фосфаты, бикарбонаты или цитраты. Разумеется, средний специалист в данной области техники может модифицировать композиции, не выходя за рамки идеи настоящего описания, с получением многочисленных композиций для конкретных способов введения, не делая композиции по настоящему изобретению нестабильными и не ограничивая их терапевтическую активность. В частности, модификацию предложенных соединений с целью улучшения их растворимости в воде или другом растворителе можно легко осуществить, например, за счет небольших модификаций (композиция в виде соли, этерификация и т. п.), что соответствует уровню навыков среднего специалиста в данной области техники. Кроме того, средний специалист в данной области техники сможетмодифицировать способ введения и схему дозирования конкретного соединения, с тем чтобы привести фармакокинетические свойства предложенных соединений к максимальному благоприятному эффекту у пациентов. Для парентеральных композиций носитель обычно включает стерильную воду или водный раствор хлорида натрия, хотя могут быть включены и другие ингредиенты, в том числе способствующие диспергированию. Разумеется, в случае, если необходимо использовать стерильную воду и поддерживать стерильность, такие композиции и носители также нужно стерилизовать. Можно изготавливать также инъе- 11024847 цируемые суспензии, при этом можно использовать подходящие жидкие носители, суспендирующие агенты и т. п. Подходящими эксципиентами для таблеток, таблеток, покрытых оболочкой, драже и твердых желатиновых капсул являются, например, лактоза, кукурузный крахмал и его производные, тальк и стеариновая кислота или ее соли. По желанию таблетки или капсулы могут быть покрыты кишечно-растворимой оболочкой или изготовлены для пролонгированного высвобождения стандартными способами. Подходящими эксципиентами для мягких желатиновых капсул являются, например, растительные масла, воски, жиры, полутвердые и жидкие полиолы. Подходящими эксципиентами для растворов для инъекций являются, например, вода, солевой раствор, спирты, полиолы, глицерин или растительные масла. Подходящими эксципиентами для суппозиториев являются, например, природные и отвержденные масла, воски, жиры, полужидкие и жидкие полиолы. Подходящими эксципиентами для растворов и сиропов для энтерального применения являются, например, вода, полиолы, сахароза, инвертированный сахар и глюкоза. Фармацевтические препараты по настоящему изобретению могут быть также представлены в виде композиций с пролонгированным высвобождением или других подходящих композиций. Фармацевтические препараты могут также содержать консерванты, солюбилизаторы, стабилизаторы, увлажняющие агенты, эмульгаторы, подсластители, красители, ароматизаторы, соли для регуляции осмотического давления, буферы, маскирующие агенты или антиоксиданты. Фармацевтические препараты могут также содержать другие терапевтически активные агенты, известные в данной области техники. Дозировка может варьироваться в широких пределах и ее, разумеется, следует подбирать в соответствии с индивидуальными требованиями в каждом конкретном случае. В случае перорального введения подходит суточная доза примерно в интервале от 0,01 до 100 мг/кг массы тела в сутки в монотерапии и/или в комбинированной терапии. Предпочтительная суточная дозировка находится в интервале примерно от 0,1 и 500 мг/кг массы тела, более предпочтительно от 0,1 до 100 мг/кг массы тела и наиболее предпочтительно от 1,0 до 100 мг/кг массы тела в сутки. Стандартный препарат содержит примерно от 5 до 95% действующего вещества (вес/вес). Суточную дозу можно вводить в виде единственной дозы или разделенными дозами, как правило, от 1 до 5 доз в сутки. В некоторых лекарственных формах предпочтительна пролекарственная форма соединения, в частности, включая ацилированные (ацетилированные и другие) производные, пиридиновые эфиры и различные солевые формы предложенных соединений. Среднему специалисту в данной области техники известно, каким образом можно легко модифицировать предложенные соединения до их пролекарственной формы, чтобы способствовать доставке действующего вещества к целевому сайту в организме хозяина или пациента. Средний специалист в данной области техники сможет также использовать преимущества подходящих фармакокинетических параметров пролекарственных форм, где это применимо, в доставке предложенных соединений к целевому сайту в организме хозяина или пациента, чтобы сделать максимальным предполагаемый эффект соединения. Показания к применению и способ лечения. Соединения по настоящему изобретению, их изомерные формы и их фармацевтически приемлемые соли полезны для лечения и профилактики инфекции ВГС. В настоящей заявке предложен способ лечения инфекции вируса гепатита С (ВГС), включающий введение пациенту, который в этом нуждается, терапевтически эффективного количества соединения формулы I. В настоящей заявке предложен способ ингибирования репликации ВГС в клетке, включающий введение соединения формулы I. Комбинированная терапия. Соединения по настоящему изобретению, их изомерные формы и их фармацевтически приемлемые соли полезны для лечения и профилактики инфекции ВГС в отдельности или при использовании в комбинации с другими соединениями, направленными на вирусные или клеточные элементы либо функции,вовлеченные в жизненный цикл ВГС. Классы соединений, полезных в настоящем изобретении, включают, без ограничения, все классы противовирусных средств против ВГС. Для комбинированной терапии механистические классы агентов, которые могут быть полезны при их комбинации с соединениями по настоящему изобретению, включают, например, нуклеозидные и ненуклеозидные ингибиторы полимеразы ВГС, ингибиторы протеазы, ингибиторы геликазы, ингибиторыNS4B и медицинские агенты, которые функционально ингибируют внутренний сайт связывания рибосомы (IRES), а также другие лекарственные средства, которые ингибируют присоединение ВГС к клетке или вхождение вируса, трансляцию РНК ВГС, транскрипцию РНК ВГС, репликацию или созревание ВГС, сборку или высвобождение вируса. Конкретные соединения этих классов, полезные в целях настоящего изобретения, включают, без ограничения, макроциклические, гетероциклические и линейные ингибиторы протеазы ВГС, такие как телапревир (VX-950), боцепревир (SCH-503034), нарлапревир(SCH-9005 18), ITMN-191 (R-227), ТМС-435350 (известный также как ТМС-435), МК-7009, BI-201335,BI-2061 (цилупревир), BMS-650032, АСН-1625, АСН-1095 (ингибитор кофактора протеазы ВГС NS4A),VX-500, VX-8 13, PHX-1766, PHX-2054, IDX-136, IDX-316, ABT-450 EP-0 13420 (и родственные соединения) и VBY-376; нуклеозидные ингибиторы полимеразы ВГС (репликазы), полезные в целях настоящего изобретения, включают, без ограничения, R7128, PSI-785 1, IDX-184, IDX-102, R1479, UNX-08189,PSI-6130, PSI-938 и PSI-879, а также различные другие нуклеозидные и нуклеотидные аналоги и ингибиторы ВГС, включая (без ограничения) те, которые получают как 2'-С-метил-модифицированные нуклеоз(т)иды, 4'-аза-модифицированные нуклеоз(т)иды и 7'-деаза-модифицированные нуклеоз(т)иды. Ненуклеозидные ингибиторы полимеразы ВГС (репликазы), полезные в целях настоящего изобретения,включают, без ограничения, HCV-796, HCV-371, VCH-759, VCH-916, VCH- 222, ANA-598, MK-3281,АВТ-333, АВТ-072, PF-00868554, BI-207127, GS-9190, А-837093, JKT-109, GL-59728 и GL-60667. Кроме того, соединения по настоящему изобретению можно применять в комбинации с антагонистами циклофиллина и иммунофиллина (например, без ограничения, соединения DEBIO, NM-811, а также циклоспорин и его производные), ингибиторы киназ, ингибиторы белков теплового шока (например,HSP90 и HSP70), другие иммуномодулирующие агенты, которые могут включать, без ограничения, интерфероны (-альфа, -бета, -омега, -гамма, -лямбда или синтетические), такие как Intron A, Roferon-A,Canferon-А 300, Advaferon, Infergen, Humoferon, Sumiferon MP, Alfaferone, IFN-bbb, Feron и т. п.; полиэтиленгликоль-дериватизированные (пегилированные) соединения интерферона, такие как ПЭГинтерферон 2 а (Pegasys), ПЭГ-интерферон 2b (PEGIntron), пегилированный IFNcon1 и т. п.; композиции длительного действия и производные соединения интерферона, такие как слитый с альбумином интерферон, Albuferon, Locteron, и т.п.; интерфероны с системами контролируемой доставкой различных типов (например, ITCA-638, омега-интерферон, доставляемый подкожной системой доставки DUROS); соединения, которые стимулируют синтез интерферона в клетках, такие как резиквимод и т. п.; интерлейкины; соединения, которые повышают развитие Т-клеточного ответа Т-хелперов 1 типа, такие какSCV-07 и т. п.; агонисты TOLL-подобного рецептора, такие как CpG-10101 (актилон), изоторабин,ANA773 и т. п.; тимозин -1; ANA-245 и ANA-246; гистамин дигидрохлорид; пропагерманий; тетрахлордекаоксид; амплиген; IMP-321; KRN-7000; антитела, такие как цивацир, XTL-6865 и т. п. и профилактические и терапевтические вакцины, такие как InnoVac С, HCV E1E2/MF59 и т. п. Кроме того, любой из выше описанных способов, включающий введение ингибитора NS5A, агониста рецептора интерферона I типа (например, IFN-) и агониста рецептора интерферона II типа (например, IFN-), можно усилить введением эффективного количества антагониста TNF-. Не ограничивающие примеры антагонистов TNF-, которые подходят для применения в такой комбинированной терапии, включают ENBREL, REMICADE и HUMIRA. Кроме того, соединения по настоящему изобретению можно применять в комбинации со средствами против простейших и другими противовирусными средствами, которые считаются эффективными в лечении инфекции ВГС, такими как, без ограничения, пролекарство нитазоксанид. Нитазоксанид можно применять в качестве агента в комбинации с соединениями, раскрытыми в настоящем изобретении, а также в комбинации с другими агентами, полезными в лечении инфекции ВГС, такими как пегилированный интерферон -2 а и рибавирин. Соединения по настоящему изобретению можно также применять с другими формами интерферонов и пегилированных интерферонов, рибавирина или его аналогов (например, тарабаварин, левовирон),соединениями микроРНК, малой интерферирующей РНК (например, SIRPLEX-140-N и т. п.), аналогами нуклеотидов или нуклеозидов, иммуноглобулинами, гепатопротекторами, противовоспалительными агентами и другие ингибиторами NS5A. Ингибиторы других мишеней жизненного цикла ВГС включают ингибиторы NS3 геликазы; ингибиторы кофактора NS4A; ингибиторы антисмыслового олигонуклеотида,такие как ISIS-14803, AVI-4065 и т. п.; кодируемая вектором короткая шпилечная РНК (кшРНК); ВГСспецифичные рибозимы, такие как гептазим, RPI, 13919 и т. п.; ингибиторы вхождения, такие как НереХС, HuMax-НерС и т. п.; ингибиторы альфа-глюкозидазы, такие как целгозивир, UT-231B и т. п.; КРЕ 02003002 и BIVN 401 и ингибиторы IMPDH. Другие наглядные примеры соединений-ингибиторов ВГС включают те, что раскрыты в следующих публикациях: патенты США 5807876; 6498178; 6344465; и 6054472; опубликованные патентные заявки РСТWO 97/40028; WO 98/40381; WO 00/56331, WO 02/04425; WO 03/007945; WO 03/010141; WO 03/000254; WO 01/32153; WO 00/06529; WO 00/18231; WO 00/10573; WO 00/13708; WO 01/85172; WO 03/037893; WO 03/037894; WO 03/037895; WO 02/100851; WO 02/100846; WO 99/01582; WO 00/09543; WO 02/18369; WO 98/17679, WO 00/056331; WO 98/22496; WO 99/07734; WO 05/073216, WO 05/073195 и WO 08/021927. Кроме того, комбинации, например, рибавирина и интерферона, можно вводить в рамках множественной комбинированной терапии по меньшей мере с одним соединением по настоящему изобретению. Настоящее изобретение не ограничено упомянутыми выше классами соединений и подразумевает известные и новые соединения, а также комбинации биологически активных агентов. Следует понимать,что комбинированная терапия по настоящему изобретению включают любую химически совместимую комбинацию соединений этой изобретательской группы с другими соединениями изобретательской группы или другими соединениями вне изобретательской группы, и, кроме того, такая комбинация не исключает противовирусную активность соединений данной изобретательской группы или противовирусную активность фармацевтической композиции как таковой. Комбинированная терапия может быть последовательной, что представляет собой терапию сначала первым агентом, а затем вторым агентом (например, каждая терапия включает различное соединение по настоящему изобретению, или одна терапия включает соединение по настоящему изобретению, а другая терапия включает один или более биологически активных агентов), или представляет собой терапию двумя агентами одновременно (параллельно). Последовательная терапия может включать достаточный период времени после завершения первой терапии до начала второй терапии. Лечение двумя агентами одновременно может быть в одной и той же суточной дозе или в отдельных дозах. Комбинированная терапия не обязательно ограничена двумя агентами и может включать три или более агентов. Дозировки как для параллельной, так и для последовательной комбинированной терапии зависят от абсорбции, распределения, метаболизма и скорости экскреции компонентов комбинированной терапии, а также от других факторов, известных специалисту в данной области техники. Величина дозы также будет варьироваться в зависимости от тяжести состояния, которое необходимо улучшить. Кроме того, следует понимать, что для каждого конкретного субъекта можно подбирать особые режимы дозирования и схемы во времени в соответствии с индивидуальными потребностями и мнением специалиста в данной области техники, который осуществляет введение лекарственных средств и наблюдение за ходом комбинированной терапии. В настоящей заявке предложен способ лечения инфекции вируса гепатита С (ВГС), включающий введение пациенту, который в этом нуждается, терапевтически эффективного количества соединения любой формулы I. В настоящей заявке предложен указанный выше способ, дополнительно включающий введение модулятора иммунной системы или противовирусного агента, который ингибирует репликацию ВГС, или их комбинацию. В настоящей заявке предложен указанный выше способ, в котором модулятор иммунной системы представляет собой интерферон или химически дериватизированный интерферон. В настоящей заявке предложены указанные выше способы, отличающиеся тем, что противовирусный агент выбирают из группы, состоящей из ингибитора протеазы ВГС, ингибитора полимеразы ВГС,ингибитора геликазы ВГС, ингибитора праймазы ВГС, ингибитора слияния ВГС и их комбинации. Примеры Общие условия. Соединения по настоящему изобретению можно получать различными способами, представленными в наглядных реакциях синтеза, описанных ниже в разделе примеров. Исходные вещества и реагенты, используемые при получении этих соединений, как правило, либо можно приобрести у фирм-поставщиков, таких как Aldrich Chemical Co., либо можно получить способами, известными специалисту в данной области техники, руководствуясь процедурами, представленными в литературе, например: Fieser and Fieser's Reagents for Organic Synthesis; WileySons: New York, 1991,т. 1-15; Rodd's Chemistry of Carbon Compounds, Elsevier Science Publishers, 1989, т. 1-5 с приложениями;Organic Reactions, WileySons: New York, 1991, т. 1-40. Следует понимать, что схемы реакций синтеза,приведенные в разделе примеров, являются лишь иллюстрацией способов, которыми соединения по настоящему изобретению можно синтезировать, при этом возможны различные модификации этих схем реакций синтеза, которые можно посоветовать осуществить специалисту в данной области техники, обратившемуся к содержанию настоящей заявки. Исходные вещества и промежуточные продукты в схемах реакций синтеза можно по желанию выделять и очищать обычными способами, включая, без ограничения, фильтрацию, перегонку, кристаллизацию, хроматографию и т. п. Можно получать характеристики этих веществ обычными средствами,включая физические константы и данные спектров. Если не указано иное, описанные здесь реакции обычно проводят в инертной атмосфере при атмосферном давлении, в диапазоне температур реакции примерно от -78 до 150 С, часто примерно от 0 до 125 С и наиболее часто и в целях удобства примерно при комнатной температуре, например около 20 С. Различные заместители в соединениях по настоящему изобретению могут присутствовать в исходных соединениях, быть введены в любой из промежуточных продуктов или быть введены после образований конечных продуктов известными способами, с помощью реакций замещения или перегруппировки. Если заместители сами по себе реакционноспособны, то заместители можно сами по себе защищать способами, известными в данной области техники. В данной области техники известно большое количество защитных групп, которые можно применять. Примеры многих из возможных групп можно найти в следующем издании: "Protective Groups in Organic Synthesis", Green et al., John Wiley and Sons, 1999. Например, нитрогруппы можно вводить нитрованием, и нитрогруппу можно превращать в другие группы,такие как аминогруппа (путем восстановления) и галоген (путем диазотирования аминогруппы и заменой диазогруппы на галоген). Ацильные группы можно вводить ацилированием по Фриделю-Крафтсу. Ацильные группы можно затем превращать в соответствующие алкильные группы различными способа- 14024847 ми, включая восстановление по Кижнеру-Вольфу и восстановление по Клеменсону. Аминогруппы можно алкилировать с образованием моно- и диалкиламиногрупп; меркапто- и гидроксигруппы можно алкилировать с образованием соответствующих эфиров. Первичные спирты можно окислять с помощью окислителей, известных в данной области техники с образованием карбоновых кислот или альдегидов, а вторичные спирты можно окислять с образованием кетонов. Таким образом, можно использовать реакции замещения или перегруппировки с получением различных заместителей в молекулах исходного вещества, промежуточных продуктов или конечного продукта, включая выделение продуктов. Аббревиатура. Используемая в настоящей заявке аббревиатура включает следующее: ацетил (Ас), уксусная кислота (НОАс), азо-бис-изобутирилнитрил (AIBN), 1-N-гидроксибензотриазол (HOBt), атмосферы (Атм), высокоэффективная жидкостная хроматография (ВЭЖХ), 9-борабицикло[3.3.1]нонан (9-BBN или BBN),метил (Me), трет-бутоксикарбонил (Вос), ацетонитрил (MeCN), ди-трет-бутил пирокарбонат или bocангибрид (ВОС 2 О), 1-(3-диметиламинопропил)-3-этилкарбодиимид гидрохлорид (EDCl), бензоил (Bz),бензил (Bn), м-хлорпербензйная кислота (МСРВА), м-хлорбензойная кислота (МСВА), бутил (Bu), метанол (МеОН), бензилоксикарбонил (cbz или Z), точка плавления (mp), карбонил диимидазол (CDI),MeSO2- (мезил или Ms), 1,4-диазабицикло[2.2.2]октан (DABCO), масс-спектр (ms), трифторид диэтиламиносеры (DAST), метил-трет-бутиловый эфир (МТВЕ), дибензилиденацетон (Dba), Nкарбоксиангидрид (NCA), 1,5-диазабицикло[4.3.0]нон-5-ен (DBN), N-бромсукцинимид (NBS), 1,8 диазабицикло[5.4.0]ундец-7-ен (DBU), N-метилморфолин (NMM), N-метилпирролидон (NMP), 1,2 дихлорэтан (DCE), пиридиний хлорхромат (РСС), N,N'-дициклогексилкарбодиимид (DCC), пиридиний дихромат (PDC), дихлорметан (DCM), пропил (Pr), диэтил азодикарбоксилат (DEAD), фенил (Ph), диизопропилазодикарбоксилат, DIAD, фунты на дюйм квадратный (psi), ди-изопропилэтиламин (DIPEA),пиридин (pyr), ди-изо-бутилалюминийгидрид, DIBAL-H, комнатная температура, кт или КТ, N,N(TBDMS),4-N,Nдиметилацетамид(TMHD),1,1'-бис(дифенилфосфино)ферроцен (dppf), тонкослойная хроматография (ТСХ), этилацетат (EtOAc, EA), тетрагидрофуран (THF), диэтиловый эфир (Et2O), триметилсилил или Me3Si (TMS), этил (Et), птолуолсульфоновая кислота моногидрат (TsOH или pTsOH), литий гексаметилдисилазан (LiHMDS), 4Me-C6H4SO2- или тозил (Ts), изопропил (i-Pr), N-уретан-N-карбоксиангидрид (UNCA), этанол (EtOH),петролейный эфир (РЕ). Общепринятая номенклатура, включая префиксы нормальный (н-), изо-, вторичный (втор-), третичный (трет-) и нео- употребляются в своем обычном значении, если они используются с алкильной группой (J. Rigaudy and D.P. Klesney, Nomenclature in Organic Chemistry, IUPAC 1979Pergamon Press, Oxford). Примеры получения. Способ получения 1. Получение промежуточного хирального 1-2R,3S,4S,5S)-4-фтор-3-гидрокси 5-иодметилтетрагидрофуран-2-ил)-1H-пиримидин-2,4-диона Хиральный 3'-дезокси-3'-фторуридин (Green ChemPharma) (5,2 г; 21 ммоль) и PPh3 (7,7 г; 29 ммоль) растворяли в смеси CH3CN/пиридин (95:5, 250 мл). Добавляли иод (7,0 г; 27,5 ммоль), и реакционную смесь перемешивали при комнатной температуре в атмосфере азота в течение 12 ч. Добавляли воду (80 мл), и растворитель выпаривали досуха при пониженном давлении. Осуществляли азеотропную перегонку с CH3CN и затем с CHCl3 для удаления остаточной воды. Полученный остаток очищали хроматографией на колонке с силикагелем (2-4% EtOH в CH2Cl2) с получением указанного в заголовке продукта (5 г). 1H ЯМР (300 МГц, DMSO-d6): 11,45 (s, 1H), 7,72-7,69 (d, J=8,1Hz, 1H), 5,87-5,84 (d, J=8,1 Гц, 1H),5,74-5,71 (m, 2 Н), 5,00-4,80 (dd, J=54,3 Гц, 4,2 Гц, 1 Н), 4,53-4,41 (m, 1H), 4,32-4,19 (m, 1H), 3,56-3,40 (m,2H). Способ получения 2. Получение промежуточного хирального 1-2R,3S,4S)-4-фтор-3-гидрокси-5 метилентетрагидрофуран-2-ил)-1H-пиримидин-2,4-диона К хиральному 1-2R,3S,4S,5S)-4-фтор-3-гидрокси-5-иодметил-тетрагидро-фуран-2-ил)-1Hпиримидин-2,4-диону (10,7 г) в метаноле (650 мл) добавляли NaOMe (16,2 г; 30 ммоль). Реакционную смесь нагревали при температуре флегмы в течение 2 ч. Эту смесь охлаждали до комнатной температуры, добавляли порциями катионообменную смолу (Н+, промытую водой) при 0 С до достижения рН 67. Смолу удаляли фильтрацией и промывали метанолом, и фильтрат выпаривали досуха при пониженном давлении. Полученный остаток очищали хроматографией на колонке с силикагелем (использовали этилацетат в качестве элюента) с получением указанного в заголовке соединения в виде желтого твердого вещества (2,3 г). 1H ЯМР (300 МГц, DMSO-d6): 11,53 (s, 1 Н), 7,79-7,76 (d, J=8,1 Гц, 1 Н), 6,14-6,12 (d, J=6,3 Гц, 1 Н),6,08-6,06 (d, J=7,5 Гц, 1H), 5,74-5,71 (d, J=8,1 Гц, 1 Н), 5,40-5,19 (dd, J=55,8 Гц, 4,5 Гц, 1 Н), 4,71-4,54 (m,3H). MS [M+H]+=229,2 Способ получения 3. Получение промежуточного хирального 1-2R,3S,4S,5S)-5-азидо-4-фтор-3 гидрокси-5-иодметилтетрагидрофуран-2-ил)-1H-пиримидин-2,4-диона[Bn(Et)3N]Cl (17 г; 75 ммоль) и NaN3 (4,5 г; 69 ммоль) суспендировали в безводном CH3CN (200 мл) и перемешивали при комнатной температуре в течение ночи. Полученную тонкую суспензию фильтровали в раствор хирального 1-2R,3S,4S)-4-фтор-3-гидрокси-5-метилентетрагидрофуран-2-ил)-1Hпиримидин-2,4-диона (2 г; 8,8 ммоль) в сухом THF (30 мл). Добавляли 4-метилморфолин (0,3 мл, 2,6 ммоль), полученный раствор охлаждали на водно-ледяной бане и добавляли по каплям раствор иода (8 г; 31 ммоль) в безводном THF (30 мл) в течение 60 мин. Реакционную смесь перемешивали при 0-9 С в течение 16 ч. Добавляли N-ацетил-L-цистеин, и этот раствор перемешивали до уменьшения образования пузырьков. Растворитель концентрировали при пониженном давлении до половины объема, затем добавляли 0,1 М раствор Na2S2O3 и насыщенный водный раствор NaHCO3. Эту смесь экстрагировали с помощью 10% EtOH в CH2Cl2 и промывали солевым раствором. Органические слои высушивали над Na2SO4,фильтровали и выпаривали досуха при пониженном давлении. Смесь очищали хроматографией на колонке с силикагелем [этилацетат:CH2Cl2:EtOH; 200:100:3] с получением неочищенного желаемого продукта. Неочищенный продукт очищали хроматографией на силикагеле (03% EtOH в CH2Cl2) дважды с получением указанного в заголовке соединения в виде белого твердого вещества (0,73 г; 21%). 1 Н ЯМР (300 М Гц, CDCl3): 9,09 (s, 1 Н), 7,38-7,35 (d, J=8,1 Гц, 1 Н), 5,83-5,80 (d, J=8,1 Гц, 1 Н), 5,765,75 (d, J=4,2 Гц, 1 Н), 5,45-5,26 (dd, J=52,2 Гц, 5,7 Гц, 1 Н), 4,84-4,77 (m, 1 Н), 3,60-3,48 (m, 1H). Способ получения 4. Получение промежуточного хирального (2R,3S,4S,5R)-2-азидо-5-(2,4-диоксо 3,4-дигидро-2 Н-пиримидин-1-ил)-3-фтор-4-гидрокситетрагидрофуран-2-илметилового эфира 3 хлорбензойной кислоты Раствор хирального 1-2R,3S,4S,5S)-5-азидо-4-фтор-3-гидрокси-5-иодметилтетрагидрофуран-2-ил)1H-пиримидин-2,4-диона (0,76 г; 1,9 ммоль) в CH2Cl2 (120 мл) объединяли со смесью (Bu)4NHSO4 (796 мг; 2,35 ммоль) и м-хлорбензойной кислотой (500 мг; 3,2 ммоль) в K2HPO4 (1,75 М, 40 мл). Полученную двухфазную систему интенсивно перемешивали при комнатной температуре, и добавляли одну порцию м-хлорпербензойной кислоты (3,6 г) [55% в равновесии с 3-хлорбензойной кислотой (10%) и водой(35%)]. Через 1 ч вносили 3 раза по 1,2 г этой смеси реагентов с интервалом 1 ч. После последнего внесения полученную смесь интенсивно перемешивали при комнатной температуре в течение 18 ч. Добавляли раствор Na2S3O3 (0,1 М) и насыщенный водный раствор NaHCO3 (pH 78). Эту смесь интенсивно перемешивали при комнатной температуре в течение 15 мин. Органический слой отделяли, и водный слой экстрагировали с помощью CH2Cl2. Объединенный органический экстракт промывали насыщенным водным раствором NaHCO3. Органический слой отделяли и концентрировали при пониженном давлении. Полученный остаток очищали хроматографией на колонке с силикагелем (03% EtOH в CH2Cl2) с получением указанного в заголовке соединения в виде белого твердого вещества (0,35 г; 43%). 1 Н ЯМР (300 МГц, CDCl3): 9,00 (s, 1H), 8,09-7,91 (m, 2H), 7,59-7,56 (m, 1H), 7,44-7,39 (m, 1 Н),7,32-7,29 (d, J=8,1 Гц, 1 Н), 5,79-5,73 (m, 2H), 5,61-5,42 (dd, J=51,9 Гц, 5,7 Гц, 1 Н), 4,81-4,78 (m, 1H). Способ получения 5. Получение промежуточного хирального 1-2R,3S,4S,5R)-5-азидо-4-фтор-3 гидрокси-5-гидроксиметилтетрагидрофуран-2-ил)-1H-пиримидин-2,4-диона Раствор NH3 в МеОН (7 н; 10 мл) добавляли к хиральному (2R,3S,4S,5R)-2-азидо-5-(2,4-диоксо-3,4 дигидро-2 Н-пиримидин-1-ил)-3-фтор-4-гидрокситетрагидрофуран-2-илметиловому эфиру 3-хлорбензойной кислоты (100 мг; 0,24 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 2 ч. Растворитель выпаривали и полученный остаток очищали с помощью препаративной ВЭЖХ с получением указанного в заголовке соединения в виде белого твердого вещества (34,5 мг; 51 %).MS [М+Н]+=288,0; 1 Н ЯМР (300 МГц, DMSO-d6): 11,52 (s, 1H), 7,84-7,82 (d, J=8,1 Гц, 1 Н), 6,186,15 (m, 2H), 5,88-5,77 (m, 2H), 5,23-5,03 (dd, J=53,7 Гц, 4,5 Гц, 1 Н), 4,60-4,40 (m, 1H), 3,54-3,53 (d, J=5,1 Гц,2 Н). Способ получения 6. Получение промежуточного хирального (2R,3S,4S,5S)-5-азидо-2-(2,4-диоксо 3,4-дигидро-2 Н-пиримидин-1-ил)-4-фтор-5-иодметилтетрагидрофуран-3-илового эфира бензойной кислоты К раствору хирального 1-2R,3S,4S,5S)-5-азидо-4-фтор-3-гидрокси-5-иодметилтетрагидрофуран-2 ил)-1 Н-пиримидин-2,4-диона, полученного по способу получения 3 (1,5 г; 3,78 ммоль), и DMAP (0,87 г; 7,56 ммоль) в сухом THF (20 мл) в атмосфере азота при 0 С добавляли по каплям BzCl (0,67 мл, 5,67 ммоль). Реакционную смесь перемешивали при 0 С в течение 5 мин. Эту смесь затем разбавляли с помощью ЕА, промывали солевым раствором и водным раствором HCl (0,1 М). Органический слой отделяли, высушивали над Na2SO4 и концентрировали. Полученный остаток очищали хроматографией на колонке с силикагелем (РЕ:ЕА=от 5:1 до 2:1) с получением указанного в заголовке соединения в виде белого твердого вещества (1,78 г; 94%).MS [M+H]+=502 Способ получения 7. Получение промежуточного хирального (2R,3S,4S,5S)-5-азидо-2-(2,4-диоксо 3,4-дигидро-2 Н-пиримидин-1-ил)-4-фтор-5-бензоилоксиметилтетрагидрофуран-3-илового эфира бензойной кислоты К смеси хирального (2R,3S,4S,5S)-5-азидо-2-(2,4-диоксо-3,4-дигидро-2 Н-пиримидин-1-ил)-4-фтор 5-иодметилтетрагидрофуран-3-илового эфира бензойной кислоты (1,78 г; 3,55 ммоль) в DMSO добавляли бензоат натрия (2,56 г; 17,76 ммоль) и 18-краун-6 (0,187 г; 0,71 ммоль). Реакционную смесь нагревали в атмосфере азота при 100 С в течение 18 ч. Эту смесь охлаждали до комнатной температуры, разбавляли ЕА, затем промывали солевым раствором и водой. Органический слой отделяли и концентрировали при пониженном давлении. Полученный остаток очищали хроматографией на колонке с силикагелем(РЕ:ЕА=5:1) с получением указанного в заголовке соединения в виде белого твердого вещества (1,30 г; 74%).MS [M+H]+=496 Способ получения 8. Получение промежуточного хирального 4-амино-1-2R,3S,4S,5R)-5-азидо-4 фтор-3-гидрокси-5-гидроксиметилтетрагидрофуран-2-ил)-1H-пиримидин-2-она К раствору хирального (2R,3S,4S,5S)-5-азидо-2-(2,4-диоксо-3,4-дигидро-2 Н-пиримидин-1-ил)-4 фтор-5-бензоилоксиметилтетрагидрофуран-3-илового эфира бензойной кислоты (0,2 г; 2,6 ммоль) и 1Hтетразола (0,283 г; 26 ммоль) в сухом пиридине (5 мл) в атмосфере азота при 0 С добавляли 4 хлорфенилфосфородихлоридат (0,297 г; 7,9 ммоль). Реакционную смесь перемешивали при 0-5 С в течение 5 мин, затем доводили до комнатной температуры и перемешивали в течение 5 ч. Эту смесь концентрировали при пониженном давлении. Полученный остаток делили между DCM и насыщенным раствором NaHCO3. Органический слой отделяли, промывали солевым раствором, высушивали над Na2SO4 и концентрировали с получением неочищенного 1 Н-тетразолового промежуточного продукта 9 на схеме 2 и использовали непосредственно без дополнительной очистки на следующей стадии. 1 Н-тетразоловый продукт 9 (0,2 г) растворяли в диоксане (70 мл) при комнатной температуре, добавляли NH3H2O (10 мл). Реакционную смесь перемешивали при комнатной температуре в течение 0,5 ч. Анализ с помощью ТСХ показал, что исходное вещество было полностью израсходовано. Растворитель удаляли при пониженном давлении, и полученный остаток растворяли в метанольном растворе NH3 (7 н, 10 мл). Реакционную смесь перемешивали при комнатной температуре в течение 16 ч. Эту смесь концентрировали и полученный остаток очищали с помощью препаративной ВЭЖХ с получением указанного в заголовке соединения в виде белого твердого вещества (50 мг; 50%).(dd, 1H, J=4,5, J=53,4), 4,548-4,436 (m, 1H), 3,539-3,462 (m, 2H). Пример 1. Получение изопропилового эфира (S)-2-[(2R,3S,4S,5R)-2-азидо-5-(2,4-диоксо-3,4 дигидро-2 Н-пиримидин-1-ил)-3-фтор-4-гидрокситетрагидрофуран-2-илметокси]феноксифосфориламинопропионовой кислоты(S)-изопропил-2-аминопропионат гидрохлорид (Oakwood, 300 мг; 1,95 ммоль) и фенилфосфородихлоридат (Aldrich, 397 мг; 280 мкл; 1,79 ммоль) суспендировали в безводном DCM (10 мл). Реакционную смесь охлаждали до -78 С. Добавляли по каплям триэтиламин (362 мг; 498 мкл; 3,58 ммоль). Реакционную смесь перемешивали при -78 С в течение 1 ч, затем нагревали до комнатной температуры и перемешивали в течение 5 ч. Растворитель удаляли, полученный остаток промывали сухим эфиром. Фильтрат концентрировали с получением неочищенного(2S)-изопропил-2-(хлор(фенокси)фосфориламино)пропионата в виде светло-желтого масла (0,5 г; 91%), который использовали без дополнительной очистки. Стадия В. К раствору хирального 1-2R,3S,4S,5R)-5-азидо-4-фтор-3-гидрокси-5-гидроксиметилтетрагидрофуран-2-ил)-1 Н-пиримидин-2,4-диона, полученного по способу получения 5 (54 мг; 188 мкмоль), в безводном THF (3,75 мл) добавляли по каплям раствор трет-бутилмагнийхлорида (470 мкл; 470 мкмоль) в THF (Aldrich, 1 М). Эту смесь перемешивали при комнатной температуре в течение 15 мин, затем добавляли по каплям раствор (0,5 М) неочищенного (2S)-изопропил-2-(хлор(фенокси)фосфориламино)пропионата (940 мкл; 470 мкмоль) в THF. Реакционную смесь перемешивали при комнатной температуре в течение 3 ч. Затем добавляли MeOH (2 мл). Растворитель удаляли. Полученный остаток очищали флэшхроматографией (силикагель, 40 г; 0-15% МеОН в DCM) с получением указанного в заголовке соединения в виде грязно-белого твердого вещества (22 мг; 21%).LC-MS (M+H)+=557,0 Пример 2. Получение этилового эфира (S)-2-[(2R,3S,4S,5R)-2-азидо-5-(2,4-диоксо-3,4-дигидро-2 Нпиримидин-1-ил)-3-фтор-4-гидрокситетрагидрофуран-2-илметокси]феноксифосфориламинопропионовой кислоты(Aldrich, 434 мг; 306 мкл; 1,95 ммоль) суспендировали в безводном DCM (20 мл). Реакционную смесь охлаждали до -78 С. Добавляли по каплям триэтиламин (395 мг; 544 мкл; 3,91 ммоль). Реакционную смесь перемешивали при -78 С в течение 1 ч, затем нагревали до комнатной температуры и перемешивали в течение 5 ч. Растворитель удаляли, полученный остаток промывали сухим эфиром. Фильтрат концентрировали с получением неочищенного (2S)-этил-2-(хлор(фенокси)фосфориламино)пропионата в виде светло-желтого масла (0,5 г; 88%) и использовали без дополнительной очистки. Стадия В. К раствору хирального 1-2R,3S,4S,5R)-5-азидо-4-фтор-3-гидрокси-5-гидроксиметилтетрагидрофуран-2-ил)-1 Н-пиримидин-2,4-диона, полученного по способу получения 5 (50 мг; 174 мкмоль) в безводном THF (5 мл) добавляли по каплям раствор трет-бутилмагнийхлорида (435 мкл; 435 мкмоль) вTHF (Aldrich, 1 М). Эту смесь перемешивали при комнатной температуре в течение 15 мин, затем добавляли по каплям раствор неочищенного (2S)-этил-2-(хлор(фенокси)фосфориламино)пропионата (870 мкл; 435 мкмоль) в THF (0,5 М). Реакционную смесь перемешивали при комнатной температуре в течение ночи. Затем добавляли МеОН (2 мл). Растворитель удаляли. Полученный остаток очищали флэш-хроматографией (силикагель,40 г; 0-15% МеОН в DCM) с получением указанного в заголовке соединения в виде грязно-белого твердого вещества (7 мг; 7,4%).LC-MS (M+H)+=543,0 Пример 3. Получение этилового эфира (S)-2-(2R,3S,4S,5R)-2-азидо-5-(2,4-диоксо-3,4-дигидро-2 Нпиримидин-1-ил)-3-фтор-4-гидрокситетрагидрофуран-2-илметокси]-(нафталин-1-илокси)фосфориламино]пропионовой кислоты Стадия А. Нафталин-1-ол (Aldrich, 0,72 г; 4,99 ммоль) и оксихлорид фосфора (V) (Aldrich, 767 мг; 466 мкл; 5,00 ммоль) суспендировали в безводном эфире (20 мл), и температуру понижали до -78 С. Добавляли по каплям триэтиламин (505 мг; 695 мкл; 4,99 ммоль), и реакционную смесь перемешивали при -78 С в течение 0,5 ч. Реакционную смесь нагревали до комнатной температуры и перемешивали в течение ночи. Эту смесь фильтровали, и фильтрат концентрировали с получением неочищенного нафталин-1-илфосфородихлоридата в виде светло-желтого масла (1,3 г; 100%), который использовали в следующей стадии без дополнительной очистки. Стадия В.(S)-Этил-2-аминопропионат гидрохлорид (Aldrich, 300 мг; 1,95 ммоль) и нафталин-1-ил фосфородихлоридат (510 мг; 1,95 ммоль) суспендировали в безводном DCM (30 мл). Реакционную смесь охлаждали до -78 С. Добавляли по каплям триэтиламин (395 мг; 544 мкл; 3,91 ммоль). Реакционную смесь перемешивали при -78 С в течение 1 ч, затем нагревали до комнатной температуры и перемешивали в течение 5 ч. Растворитель удаляли, и полученный остаток промывали сухим эфиром. Фильтрат концентрировали с получением неочищенного (2S)-этил-2-(хлор(нафталин-1-илокси)фосфориламино)пропионата в виде светло-желтого масла (0,6 г; 90%), которое использовали без дополнительной очистки. Стадия С. К раствору хирального 1-2R,3S,4S,5R)-5-азидо-4-фтор-3-гидрокси-5-гидроксиметилтетрагидрофуран-2-ил)-1 Н-пиримидин-2,4-диона, полученного по способу получения 5 (90 мг; 313 мкмоль), в безводном THF (6,25 мл) добавляли по каплям раствор трет-бутилмагнийхлорида (783 мкл; 783 мкмоль) вTHF (Aldrich, 1 М). Эту смесь перемешивали при комнатной температуре в течение 15 мин, затем добавляли по каплям раствор в THF (0,5 М) неочищенного (2S)-этил-2-(хлор(нафталин-1 илокси)фосфориламино)пропионата (1,57 мл, 783 мкмоль). Реакционную смесь перемешивали при комнатной температуре в течение 3 ч. Затем добавляли МеОН (2 мл). Растворитель удаляли. Полученный остаток очищали флэш-хроматографией (силикагель, 40 г; 0-15% МеОН в DCM) с получением указанного в заголовке соединения в виде светло-коричневого твердого вещества (70 мг; 38%).LC-MS (M+H)+=593,0 Способ получения 11. Получение промежуточного хирального 4-амино-1-2R,3S,4S,5R)-5-азидо-4 фтор-5-гидроксиметил-3-триэтилсиланилокситетрагидрофуран-2-ил)-1H-пиримидин-2-она К раствору хирального 4-амино-1-2R,3S,4S,5R)-5-азидо-4-фтор-3-гидрокси-5 гидроксиметилтетрагидрофуран-2-ил)-1H-пиримидин-2-она, полученного по способу получения 8 (300 мг; 1,05 ммоль), в пиридине (24,5 мл) при -5 С добавляли по каплям хлортриэтилсилан (Fluka, 440 мг; 2,92 ммоль) в течение 15 мин. Реакционную смесь перемешивали при 0 С в течение 2 ч, затем гасили добавлением метанола (5 мл). Эту смесь очищали непосредственно флэш-хроматографией (5-15% МеОН в DCM) с получением указанного в заголовке соединения в виде белого твердого вещества (0,29 г; 69%). Способ получения 12. Получение промежуточного изопропилового эфира (S)-2-(2R,3S,4S,5R)-5(4-амино-2-оксо-2 Н-пиримидин-1-ил)-2-азидо-3-фтор-4-триэтилсиланилокси-тетрагидрофуран-2 илметокси]-(нафталин-1-илокси)фосфориламино]пропионовой кислотыDCM (25 мл). Реакционную смесь охлаждали до -78 С. Добавляли по каплям триэтиламин (852 мг; 1,17 мл, 8,42 ммоль). Реакционную смесь перемешивали при -78 С в течение 1 ч, затем нагревали до комнатной температуры и перемешивали в течение 5 ч. Растворитель удаляли, и полученный остаток промывали с сухим этиловым эфиром и фильтровали. Фильтрат концентрировали с получением неочищенного(1,3 г; 87%), который использовали без дополнительной очистки. Стадия В. К раствору хирального 4-амино-1-2R,3S,4S,5R)-5-азидо-4-фтор-5-гидроксиметил-3 триэтилсиланилокситетрагидрофуран-2-ил)-1H-пиримидин-2-она, полученного по способу получения 11(0,29 г; 724 мкмоль), в безводном THF (42 мл) добавляли по каплям раствор трет-бутилмагнийхлорида(1,81 мл, 1,81 ммоль) в THF (Aldrich, 1 М). Эту смесь перемешивали при комнатной температуре в течение 15 мин, затем добавляли по каплям раствор в THF (0,5 М) неочищенного (2S)-изопропил-2(хлор(нафталин-1-илокси)фосфориламино)пропионата (3,62 мл, 1,81 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 1 ч, а затем последовательно вносили раствор третбутилмагнийхлорида (0,9 мл, 0,9 ммоль) в THF (Aldrich, 1 М) и раствор неочищенного (2S)-изопропил-2(хлор(нафталин-1-илокси)фосфориламино)пропионата (1,81 мл, 0,9 ммоль) в THF (0,5 М). Реакционную смесь перемешивали при комнатной температуре еще в течение 2 ч. Добавляли МеОН (5 мл). Растворитель удаляли. Полученный остаток очищали флэш-хроматографией (силикагель, 40 г; 2-18% МеОН вLC-MS (M+H)+=720,3 Пример 4. Получение изопропилового эфира (S)-2-(2R,3S,4S,5R)-5-(4-амино-2-оксо-2 Нпиримидин-1-ил)-2-азидо-3-фтор-4-гидрокситетрагидрофуран-2-илметокси]-(нафталин-1-илокси)фосфориламино]пропионовой кислоты 4-триэтилсиланилокситетрагидрофуран-2-илметокси]-(нафталин-1-илокси)фосфориламино]пропионовой кислоты, полученный по способу получения 12 (0,43 г; 597 мкмоль), растворяли в уксусной кислоте(80%, 28 мл). Реакционную смесь перемешивали при комнатной температуре в течение 5 ч. Растворитель выпаривали при пониженном давлении и остатки уксусной кислоты удаляли азеотропным концентрированием с МеОН три раза. Полученный остаток очищали флэш-хроматографией (силикагель, 2-18% МеОН в DCM) с получением указанного в заголовке соединения в виде белого твердого вещества (0,2 г; 55%).LC-MS (M+H)+=606,1 Пример 5. Получение бензилового эфира (S)-2-(2R,3S,4S,5R)-5-(4-амино-2-оксо-2H-пиримидин-1 ил)-2-азидо-3-фтор-4-гидрокситетрагидрофуран-2-илметокси]-(нафталин-1-илокси)фосфориламино]пропионовой кислотыDCM (15 мл). Реакционную смесь охлаждали до -78 С. Добавляли по каплям триэтиламин (619 мг; 852 мкл; 6,12 ммоль). Реакционную смесь перемешивали при -78 С в течение 1 ч, затем нагревали до комнатной температуры и перемешивали в течение 5 ч. Растворитель удаляли, и полученный остаток промывали с сухим этиловым эфиром и фильтровали. Фильтрат концентрировали с получением неочищенного (2S)-бензил-2-(хлор(нафталин-1-илокси)фосфориламино)пропионата в виде светло-желтого масла(1 г; 81%), который использовали без дополнительной очистки. Стадия В. К раствору хирального 4-амино-1-2R,3S,4S,5R)-5-азидо-4-фтор-3-гидрокси-5-гидроксиметилтетрагидрофуран-2-ил)-1H-пиримидин-2-она, полученного по способу получения 8 (85 мг; 292 мкмоль), в безводном THF (8,5 мл) добавляли по каплям раствор трет-бутилмагнийхлорида (742 мкл; 742 мкмоль) вTHF (Aldrich, 1 М). Эту смесь перемешивали при комнатной температуре в течение 1 ч, затем добавляли по каплям раствор неочищенного (2S)-бензил-2-(хлор(нафталин-1-илокси)фосфориламино)пропионата(1,48 мл, 742 мкмоль) в THF (0,5 М). Реакционную смесь перемешивали при комнатной температуре в течение 1 ч, а затем последовательно вносили раствор трет-бутилмагнийхлорида (371 мкл; 371 мкмоль) вTHF (Aldrich, 1 М) и раствор неочищенного (2S)-бензил-2-(хлор(нафталин-1-илокси)фосфориламино)пропионата (0,74 мл, 371 мкмоль) в THF (0,5 М). Реакционную смесь перемешивали при комнатной температуре еще в течение 2 ч. Добавляли МеОН (2 мл). Растворитель удаляли. Полученный остаток очищали флэш-хроматографией (силикагель, 0-20% МеОН в DCM) с получением указанного в заголовке соединения в виде светло-желтого твердого вещества (10 мг; 5%).LC-MS(M+H)+=654,1 Пример 6. Получение этилового эфира (S)-2-(2R,3S,4S,5R)-5-(4-амино-2-оксо-2H-пиримидин-1 ил)-2-азидо-3-фтор-4-гидрокситетрагидрофуран-2-илметокси]-(нафталин-1-илокси)фосфориламино]пропионовой кислоты К раствору хирального 4-амино-1-2R,3S,4S,5R)-5-азидо-4-фтор-3-гидрокси-5 гидроксиметилтетрагидрофуран-2-ил)-1H-пиримидин-2-она, полученного по способу получения 8 (50 мг; 175 мкмоль), в безводном THF (5 мл) добавляли по каплям раствор трет-бутилмагнийхлорида (437 мкл; 437 мкмоль) в THF (Aldrich, 1 М). Эту смесь перемешивали при комнатной температуре в течение 15 мин,затем добавляли по каплям раствор неочищенного(2S)-этил-2-(хлор(нафталин-1 илокси)фосфориламино)пропионата в THF (0,5 М), полученного в примере 3, стадия В (873 мкл; 437 мкмоль). Реакционную смесь перемешивали при комнатной температуре в течение 1 ч, а затем последовательно вносили раствор трет-бутилмагнийхлорида (219 мкл; 219 мкмоль) в THF (Aldrich, 1 М) и раствор неочищенного (2S)-бензил-2-(хлор(нафталин-1-илокси)фосфориламино)пропионата (437 мкл; 219 мкмоль) в THF (0,5 М). Реакционную смесь перемешивали при комнатной температуре еще в течение 2 ч. Добавляли МеОН (2 мл). Растворитель удаляли. Полученный остаток очищали флэш-хроматографией(силикагель, 0-16% МеОН в DCM) с получением указанного в заголовке соединения в виде белого твердого вещества (5 мг; 5%).LC-MS (M+H)+=592,2 Пример 7. Получение изопропилового эфира (S)-2-[(2R,3S,4S,5R)-5-(4-амино-2-оксо-2 Нпиримидин-1-ил)-2-азидо-3-фтор-4-гидрокситетрагидрофуран-2-илметокси]феноксифосфориламинопропионовой кислоты К раствору хирального 4-амино-1-2R,3S,4S,5R)-5-азидо-4-фтор-3-гидрокси-5 гидроксиметилтетрагидрофуран-2-ил)-1H-пиримидин-2-она, полученного в примере 8 (43 мг; 150 мкмоль), в безводном THF (8 мл) добавляли по каплям раствор трет-бутилмагнийхлорида (376 мкл; 376 мкмоль) в THF (Aldrich, 1 М). Эту смесь перемешивали при комнатной температуре в течение 15 мин,затем добавляли по каплям раствор неочищенного(2S)-изопропил-2(хлор(фенокси)фосфориламино)пропионата в THF (0,5 М), полученного в примере 1 стадия А (751 мкл; 376 мкмоль). Реакционную смесь перемешивали при комнатной температуре в течение 1 ч, а затем последовательно вносили раствор трет-бутилмагнийхлорида (188 мкл; 188 мкмоль) в THF (Aldrich, 1 М) и раствор неочищенного (2S)-бензил-2-(хлор(нафталин-1-илокси)фосфориламино)пропионата (376 мкл; 188 мкмоль) в THF (0,5 М). Реакционную смесь перемешивали при комнатной температуре в течение ночи. Добавляли МеОН (2 мл). Растворитель удаляли. Полученный остаток очищали флэш-хроматографией(силикагель, 0-18% МеОН в DCM) с получением указанного в заголовке соединения в виде белого твердого вещества (3 мг; 4%).LC-MS (M+H)+=556,0 Способ получения 13. Получение промежуточного хирального 4-амино-1-[(2R,3S,4S,5R)-5-азидо-3(трет-бутил-дифенил-силанилокси)-4-фтор-5-гидроксиметилтетрагидрофуран-2-ил]-1 Н-пиримидин-2-она К раствору хирального 4-амино-1-2R,3S,4S,5R)-5-аазидо-4-фтор-3-гидрокси-5 гидроксиметилтетрагидрофуран-2-ил)-1 Н-пиримидин-2-она, полученного в примере 8 (50 мг; 175 мкмоль), и имидазола (119 мг; 1,75 ммоль) в безводном DMF (2,62 мл) добавляли третбутилхлордифенилсилан (Aldrich, 480 мг; 1,75 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 20 мин. Эту смесь разбавляли этилацетатом, промывали водой несколько раз и солевым раствором. Органический слой отделяли, высушивали над MgSO4 и концентрировали. Полученный остаток очищали флэш-хроматографией (0-18% МеОН в DCM) с получением указанного в заголовке соединения в виде белого твердого вещества (36 мг; 39%). Способ получения 14. Получение промежуточного изопропилового эфира (S)-2-(2R,3S,4S,5R)-5(4-амино-2-оксо-2 Н-пиримидин-1-ил)-2-азидо-4-(трет-бутилдифенилсиланилокси)-3-фтортетрагидрофуран-2-илметокси]-(нафталин-1-илокси)фосфориламино]пропионовой кислотыTHF (4 мл) добавляли по каплям раствор трет-бутилмагнийхлорида (95,3 мкл; 95,3 мкмоль) в THF (Aldrich, 1 М). Эту смесь перемешивали при комнатной температуре в течение 15 мин, затем добавляли по каплям раствор неочищенного (2S)-изопропил-2-(хлор(нафталин-1-илокси)фосфориламино)пропионата вTHF (0,5 М), полученного по способу получения 12, стадия А (191 мкл; 95,3 мкмоль). Реакционную смесь перемешивали при комнатной температуре в течение 1 ч, а затем последовательно вносили раствор трет- 22024847 бутилмагнийхлорида (48 мкл; 48 мкмоль) в THF (Aldrich, 1 М) и раствор неочищенного (2S)-изопропил-2(хлор(нафталин-1-илокси)фосфориламино)пропионата в THF (0,5 М), полученного по способу получения 12, стадия А (96 мкл; 48 мкмоль). Реакционную смесь перемешивали при комнатной температуре в течение ночи. Добавляли МеОН (2 мл). Растворитель удаляли. Полученный остаток очищали флэшхроматографией (силикагель, 40 г; 2-18% МеОН в DCM) с получением указанного в заголовке соединения в виде белого твердого вещества (22 мг; 68%).LC-MS (M+H)+=844,2 Пример 8. Получение хирального изопропилового эфира (S)-2-[(2R,3S,4S,5R)-5-(4-амино-2-оксо 2 Н-пиримидин-1-ил)-2-азидо-3-фтор-4-гидрокситетрагидрофуран-2-илметокси]гидроксифосфориламинопропионовой кислоты К раствору изопропилового эфира (S)-2-(2R,3S,4S,5R)-5-(4-амино-2-оксо-2 Н-пиримидин-1-ил)-2 азидо-4-(трет-бутилдифенилсиланилокси)-3-фтортетрагидрофуран-2-илметокси]-(нафталин-1-илокси)фосфориламино]пропионовой кислоты (18 мг; 21,3 мкмоль) в THF (2,88 мл) добавляли раствор TBAF(21,3 мкл; 21,3 мкмоль) в THF (1 М). Реакционную смесь перемешивали при комнатной температуре в течение 30 мин. Растворитель удаляли и полученный остаток очищали с помощью препаративной ВЭЖХ с получением указанного в заголовке соединения в виде белого твердого вещества (6 мг; 59%).LC-MS (M+H)+=479,9 Пример 9. Получение изопропилового эфира (S)-2-(2R,3S,4S,5R)-5-(4-амино-2-оксо-2 Нпиримидин-1-ил)-2-азидо-3-фтор-4-пропионилокситетрагидрофуран-2-илметокси]-(нафталин-1-илокси)фосфориламино]пропионовой кислоты К раствору хирального (2R,3S,4S,5R)-2-(4-амино-2-оксо-2H-пиримидин-1-ил)-5-азидо-4-фтор-5 гидроксиметилтетрагидрофуран-3-илового эфира пропионовой кислоты (способ получения будет описан отдельно, 25 мг; 73,0 мкмоль) в безводном THF (5 мл) добавляли по каплям раствор третбутилмагнийхлорида (183 мкл; 183 мкмоль) в THF (Aldrich, 1 М). Эту смесь перемешивали при комнатной температуре в течение 15 мин, затем добавляли по каплям раствор неочищенного (2S)-изопропил-2(хлор(нафталин-1-илокси)фосфориламино)пропионата в THF (0,5 М), полученного по способу получения 12, стадия А (365 мкл; 183 мкмоль). Реакционную смесь перемешивали при комнатной температуре в течение 1 ч, а затем последовательно вносили раствор трет-бутилмагнийхлорида (92 мкл; 92 мкмоль) в(2S)-изопропил-2-(хлор(нафталин-1 илокси)фосфориламино)пропионата в THF (0,5 М), полученного по способу получения 12, стадия А (183 мкл; 92 мкмоль). Реакционную смесь перемешивали при комнатной температуре в течение 2 ч. Добавляли МеОН (2 мл). Растворитель удаляли. Полученный остаток очищали флэш-хроматографией (силикагель, 40 г; 2-18% MeOH в DCM), затем проводили препаративную ВЭЖХ с получением указанного в заголовке соединения в виде белого твердого вещества (28 мг; 58%).LC-MS (M+H)+=662,2 Биологические примеры. Анализ репликона ВГС. С помощью этого анализа можно оценить способность соединений формулы I ингибировать репликацию РНК ВГС и соответственно их потенциальную пользу для лечения инфекций ВГС. В данном анализе используется ген-репортер в формате простого считывания для определения внутриклеточного уровня репликона РНК ВГС. Ген люциферазы Renilla вводили в первую открытую рамку считывания в конструкт репликона NK5.1 генотипа 1b (N. Krieger et al., J. Virol. 2001 75(10):4614), сразу за последовательностью внутреннего сайта связывания рибосомы (IRES), и осуществляли слияние с геном неомицинфосфотрансферазы (NPTII) через саморасщепляющийся пептид 2 А из вируса ящура (M.D. RyanJ.Drew, EMBO 1994 13(4):928-933). После in vitro транскрипции эту РНК электропорировали в клетки гепатомы человека Huh7, и колонии, устойчивые к G418, выделяли и наращивали. Стабильно селектированная клеточная линия 2209-23 содержит репликативную субгеномную РНК ВГС, и активность люциферазы Renilla, экспрессируемой репликоном, отражает уровень ее РНК в клетках. Исследование прово- 23024847 дили на дублирующих планшетах, один анализ на матово-белом, а другой - на прозрачном планшете, с тем чтобы параллельно измерить противовирусную активность и цитотоксичность химического соединения, для гарантии того, что наблюдаемая активность не обусловлена сниженной пролиферацией клеток или их гибелью. Клетки с репликоном ВГС (2209-23), которые экспрессируют ген-репортер люциферазы Renilla,культивировали в среде Игла, модифицированной Дюльбекко (Invitrogen, кат.10569-010), содержащей 5%-ную фетальную сыворотку коровы (FBS, Invitrogen, кат.10082-147), высевали на 96-луночный планшет в количестве 5000 клеток в лунку и инкубировали в течение ночи. Через 24 ч к клеткам добавляли различные разведения химических соединений в питательной среде, а затем инкубировали далее при 37 С в течение трех суток. По окончании инкубации клетки в белых планшетах собирали и измеряли активность люциферазы с помощью набора R. luciferase Assay system (Promega, кат.E2820). Все реагенты, описанные в ниже следующем абзаце, входят в этот набор, и приготовление реагентов осуществляли по инструкции изготовителя. Клетки однократно промывали фосфатно-солевым буфером (рН 7,0) (PBS), по 100 мкл в лунку, и лизировали с помощью 20 мкл однократного аналитического лизирующего буфера R. Luciferase, после чего инкубировали при комнатной температуре в течение 20 мин. Затем планшет помещали в люминометр для микропланшетов Centra LB 960 (Berthold Technologies), вносили по 100 мкл аналитического буфера R. luciferase в каждую лунку и измеряли сигнал с использованием 2-секундной измерительной программы с 2-секундной задержкой. Величину IC50, представляющую собой концентрацию лекарственного вещества, которая требуется для снижения уровня репликона на 50%, по сравнению с контрольным значением для интактных клеток, можно рассчитать из графика процентного снижения активности люциферазы в зависимости от концентрации лекарственного вещества, как описано выше. Для анализа цитотоксичности использовали реагент WST-1 производства Roche Diagnostic (кат.1644807). В каждую лунку на прозрачном планшете вносили по 10 мкл реагента WST-1, включая лунки,содержащие только среду, используемые в качестве холостой пробы. После этого клетки инкубировали в течение 2 ч при 37 С и измеряли величину оптической плотности на спетрофотометре для считывания микропланшетов MRX Revelation (Lab System) при 450 нм (фильтр сравнения при 650 нм). Аналогичным образом, величину CC50, представляющую собой концентрацию лекарственного вещества, которая требуется для снижения пролиферации клеток на 50%, по сравнению с контрольной величиной для интактных клеток, можно рассчитать по графику процентного снижения величины WST-1 в зависимости от концентрации лекарственного вещества, как описано выше. Выборочные биологические данные приведены ниже в табл. II. Таблица II Следует понимать, что в настоящем описании упоминание терапии охватывает профилактику, а также лечение существующих состояний, и что терапия животных включает терапию человека, а также других млекопитающих. Кроме того, терапия инфекции вируса гепатита С (ВГС), используемая в настоящем описании, также подразумевает лечение или профилактику заболевания или состояния, связанного с или опосредованного инфекцией вируса гепатита С (ВГС), или его клинических симптомов. Признаки, раскрытые в приведенном выше описании, или следующей далее формуле изобретения,или сопровождающих иллюстрациях, представленные в их конкретной форме либо с помощью средств для осуществления раскрытой функции, способа или процесса для достижения раскрытого результата,соответствующим образом могут быть использованы по отдельности или в любой комбинации таких признаков для осуществления настоящего изобретения в его различных формах. Представленное выше изобретение детально описано посредством иллюстраций и примеров в целях большей ясности и лучшего понимания. Специалисту в данной области техники очевидно, что можно внести изменения и модификации в пределах объема прилагаемой формулы изобретения. Таким образом, подразумевается, что приведенное выше описание носит иллюстративный, а не ограничивающий характер. Соответственно объем настоящего изобретения следует определять исходя не из приведенного выше описания, а исходя из прилагаемой далее формулы изобретения наряду с полным объемом эквивалентов, на которые дают право пункты формулы. Все патенты, патентные заявки и публикации, на которые даны ссылки в настоящей заявке, вклю- 24024847 чены полностью в ее содержание посредством ссылок во всех отношениях и в той же мере, как если бы каждый из индивидуальных патентов, патентных заявок или публикаций были указаны отдельно. ФОРМУЛА ИЗОБРЕТЕНИЯ 1. Соединение формулы I или его фармацевтически приемлемая соль. 2. Соединение по п.1, в котором R1 представляет собой нафтил или фенил. 3. Соединение по п.2, в котором R2a представляет собой Н. 4. Соединение по п.3, в котором R2b представляет собой метил. 5. Соединение по п.4, в котором R3 представляет собой изопропил, этил или бензил. 6. Соединение по п.5, в котором R5 представляет собой Н. 7. Соединение по п.5, в котором R5 представляет собой C(=O)R1c. 8. Соединение по п.7, в котором R1c представляет собой этил. 9. Соединение по п.6, в котором R6 представляет собой 11. Соединение, выбранное из группы, состоящей из следующих соединений: изопропиловый эфир (S)-2-[(2R,3S,4S,5R)-2-азидо-5-(2,4-диоксо-3,4-дигидро-2H-пиримидин-1-ил)3-фтор-4-гидрокситетрагидрофуран-2-илметокси]феноксифосфориламинопропионовой кислоты; этиловый эфир (S)-2-[(2R,3S,4S,5R)-2-азидо-5-(2,4-диоксо-3,4-дигидро-2H-пиримидин-1-ил)-3 фтор-4-гидрокситетрагидрофуран-2-илметокси]феноксифосфориламинопропионовой кислоты; этиловый эфир (S)-2-(2R,3S,4S,5R)-2-азидо-5-(2,4-диоксо-3,4-дигидро-2H-пиримидин-1-ил)-3 фтор-4-гидрокситетрагидрофуран-2-илметокси]-(нафталин-1-илокси)фосфориламино]пропионовой кислоты; изопропиловый эфир (S)-2-(2R,3S,4S,5R)-5-(4-амино-2-оксо-2H-пиримидин-1-ил)-2-азидо-3-фтор 4-гидрокситетрагидрофуран-2-илметокси]-(нафталин-1-илокси)фосфориламино]пропионовой кислоты; бензиловый эфир (S)-2-(2R,3S,4S,5R)-5-(4-амино-2-оксо-2H-пиримидин-1-ил)-2-азидо-3-фтор-4 гидрокситетрагидрофуран-2-илметокси]-(нафталин-1-илокси)фосфориламино]пропионовой кислоты; этиловый эфир (S)-2-(2R,3S,4S,5R)-5-(4-амино-2-оксо-2H-пиримидин-1-ил)-2-азидо-3-фтор-4 гидрокситетрагидрофуран-2-илметокси]-(нафталин-1-илокси)фосфориламино]пропионовой кислоты; изопропиловый эфир (S)-2-[(2R,3S,4S,5R)-5-(4-амино-2-оксо-2H-пиримидин-1-ил)-2-азидо-3-фтор 4-гидрокситетрагидрофуран-2-илметокси]феноксифосфориламинопропионовой кислоты; изопропиловый эфир (S)-2-[(2R,3S,4S,5R)-5-(4-амино-2-оксо-2H-пиримидин-1-ил)-2-азидо-3-фтор 4-гидрокситетрагидрофуран-2-илметокси]гидроксифосфориламинопропионовой кислоты и изопропиловый эфир (S)-2-(2R,3S,4S,5R)-5-(4-амино-2-оксо-2H-пиримидин-1-ил)-2-азидо-3-фтор- 25024847 4-пропионилокситетрагидрофуран-2-илметокси]-(нафталин-1-илокси)фосфориламино]пропионовой кислоты. 12. Применение соединения по любому из пп.1-11 для лечения или профилактики инфекции вируса гепатита С (ВГС). 13. Применение соединения по любому из пп.1-11 для изготовления лекарственного средства для лечения или профилактики инфекции вируса гепатита С (ВГС). 14. Способ лечения инфекции вируса гепатита С (ВГС), включающий введение пациенту, который в этом нуждается, терапевтически эффективного количества соединения по любому из пп.1-11. 15. Фармацевтическая композиция, включающая соединение по любому из пп.1-11 и терапевтически инертные носители.
МПК / Метки
МПК: C07H 19/06, A61K 31/708, A61P 31/14, C07H 19/16, A61K 31/7072
Метки: репликации, производные, качестве, нуклеозидов, вируса, гепатита, ингибиторов, 4'-азидо,3'-фторзамещенные, рнк
Код ссылки
<a href="https://eas.patents.su/27-24847-4-azido3-ftorzameshhennye-proizvodnye-nukleozidov-v-kachestve-ingibitorov-replikacii-rnk-virusa-gepatita-s.html" rel="bookmark" title="База патентов Евразийского Союза">4′-азидо,3′-фторзамещенные производные нуклеозидов в качестве ингибиторов репликации рнк вируса гепатита с</a>
2′,4′-дифтор-2′-метилзамещенные нуклеозидные производные в качестве ингибиторов репликации рнк вируса гепатита с

Номер патента: 24297
Опубликовано: 30.09.2016
Авторы: Чжан Чжумин, Чжан Цзин
МПК: A61P 31/14, C07H 19/06, A61K 31/7072...
Метки: нуклеозидные, гепатита, репликации, вируса, ингибиторов, 2',4'-дифтор-2'-метилзамещенные, рнк, качестве, производные
Формула / Реферат:
1. Соединение формулы Iгде R1 представляет собой С6-10арил;каждый R1c представляет собой C1-7алкил;R2a и R2b представляют собой (i) независимо H, C1-7алкил;R3 представляет собой C1-7алкил;R4 представляет собой H;R5 представляет собой H, C(=O)R1c или P(=O)(OR1)(NR4R7);R6 представляет собой H или гало;R7 представляет собой C(R2aR2b)COOR3,или его фармацевтически приемлемые соли.2. Соединение по п.1, где R6 представляет собой H или Br.3. Соединение...
Макроциклические карбоновые кислоты и ацилсульфонамиды в качестве ингибиторов репликации вируса гепатита с

Номер патента: 11857
Опубликовано: 30.06.2009
Авторы: Блэтт Лоренс М., Джоси Джон Энтони, Маддуру Мачендер Р., Джианг Ютонг, Догерти Джордж Эндрю, Кондроски Кевин Рональд, Венгловски Стивен Марк, Стенджел Питер Джон, Вуддард Бенджамин Т., Кеннеди Эйприл Лайн, Эндрьюс Стивен Вейд
МПК: A61K 38/00, C08H 1/00
Метки: гепатита, ингибиторов, макроциклические, ацилсульфонамиды, карбоновые, вируса, качестве, репликации, кислоты
Формула / Реферат:
1. Макроциклическая карбоновая кислота или ацилсульфонамид, имеющие формулу I, VIII или IX где (a) R1 и R2, каждый независимо, представляют собой H, галоген, циано, гидрокси, С1-6алкил, С1-6алкокси, C1-6алкил, возможно замещенный не более чем тремя атомами фтора, тиазолил, C(O)NR6R7, NR6R7, C(O)OR8, NHC(O)R8, OCHnNR6R7 или OCHnR9a; где R9a представляет собой имидазолил или пиразолил; указанный тиазолил в определении R1 и R2 является...
Производные пиримидина в качестве ингибиторов репликации вируса иммунодефицита человека

Номер патента: 2973
Опубликовано: 26.12.2002
Авторы: Андрис Кунрад Йозеф Лодевейк Марсель, Янссен Марсель Огюст Констант, Де Корт Барт, Янссен Поль Адриан Жан, Де Жонж Марк Рене, Кукла Майкл Джозеф, Хэрес Ян, Хо Чих Юнг, Ван Акен Кун Жанн Альфонс, Лудовики Дональд Уилльям, Койманс Люсьен Мария Хенрикус
МПК: A61P 31/18, A61K 31/505, C07D 239/50...
Метки: репликации, ингибиторов, человека, иммунодефицита, качестве, пиримидина, производные, вируса
Формула / Реферат:
1. Применение соединения формулы его N-оксида, фармацевтически приемлемой соли присоединения или стереохимически изомерной формы, где А является СН или N; n равен 0, 1, 2, 3 или 4; Q является водородом или -NR1R2; R1 и R2, каждый независимо, выбран из водорода, гидрокси, C1-12aлкила, С1-12алкилокси, С1-12алкилкарбонила, С1-12алкилоксикарбонила, где каждая из указанных С1-12алкильных групп необязательно и каждая независимо может быть замещена...
Производные бензимидазола и имидазопиридина в качестве ингибиторов репликации респираторно-синцитиального вируса

Номер патента: 4746
Опубликовано: 26.08.2004
Авторы: Лякрамп Жан Фернан Арман, Гийемон Жером Эмиль Жорж, Вене Марк Гастон, Янссенс Франс Эдуард, Андрис Кунрад Йозеф Лодевейк Марсель
МПК: A61P 11/00, A61K 31/4709, C07D 401/14...
Метки: имидазопиридина, вируса, бензимидазола, репликации, респираторно-синцитиального, качестве, производные, ингибиторов
Формула / Реферат:
1. Соединение формулы его аддитивная соль или стереохимически изомерная форма, где в указанной формуле -a1=a2-a3=a4- представляет двухвалентный радикал формулы -CH=CH-CH=CH- (a-1); -N=CH-CH=CH- (a-2); -CH=N-CH=CH- (a-3); -CH=CH-N=CH- (a-4) или -CH=CH-CH=N- (a-5); где каждый атом водорода в...
Макроциклические производные индола, пригодные в качестве ингибиторов вируса гепатита с

Номер патента: 19008
Опубликовано: 30.12.2013
Авторы: Амссомс Кати Ингрид Эдуард, Линь Тсе-И, Тахри Абделлах, Вендевилль Сандрин Мари Элен, Рабуассон Пьер Жан-Мари Бернар
МПК: C07D 513/18, A61K 31/55, A61P 31/14...
Метки: гепатита, ингибиторов, индола, вируса, производные, пригодные, макроциклические, качестве
Формула / Реферат:
1. Соединение формулы (I)включая его стереохимически изомерные формы, и N-оксиды, соли, гидраты и сольваты, гдеR1 означает двухвалентную цепь, выбранную изикаждая группа R3 независимо выбрана из группы, состоящей из водорода, C1-4алкила и C3-5циклоалкила;а равно 3, 4, 5 или 6;каждый b независимо равен 1 или 2;с равно 1 или 2;макроцикл А содержит от 14 до 18 атомов в цикле, в частности макроцикл А имеет 17 или 18 атомов в цикле;каждая группа R2...
Предыдущий патент: Бициклическое гетероциклическое соединение
Следующий патент: Катализатор окислительной конверсии углеводородных газов с получением оксида углерода и водорода
Случайный патент: Литая концевая крышка для сетчатых фильтров для очистки бурового раствора