Производные 2-амино-3-(имидазол-2-ил)пиридин-4-она и их применение в качестве ингибиторов киназы рецептора vegf
Номер патента: 23580
Опубликовано: 30.06.2016
Авторы: Мартен Валери, Брон Ален, Лассалль Жильбер, Дюкло Оливье, Лорж Франц, Стрюб Орели, Ритцелер Олаф























Формула / Реферат
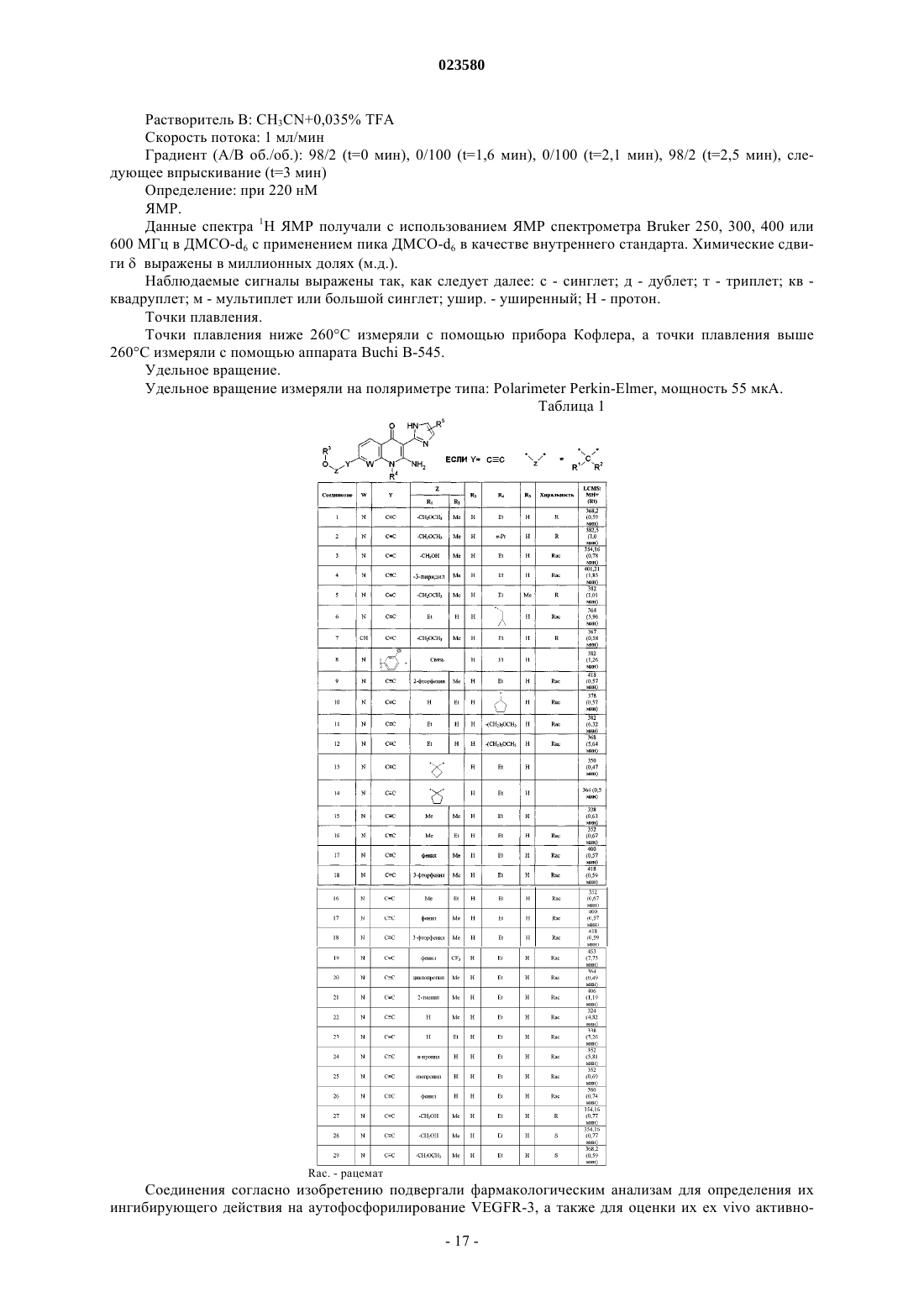
1. Соединение, соответствующее формуле (I)

в которой W представляет собой атом азота или группу CH;
Y представляет собой С2-С3-алкинилен, 1,4-фенилен, необязательно замещенный R7, где R7 представляет собой один или более атом(ов) галогена;
Z представляет собой связь или группу CR1R2;
R1 и R2 независимо друг от друга представляют собой группу, выбранную из атома водорода, С1-С6-алкильной группы, трифторметильной группы, группы (CH2)nOR6, С3-С7-циклоалкила, гетероарила или арила, где гетероарил или арил необязательно замещен одним или более атомом(ами) галогена; арил представляет собой моноциклическую или бициклическую ароматическую группу, содержащую от 6 до 10 атомов углерода, и гетероарил представляет собой 5-12-членную моноциклическую или бициклическую ароматическую группу, включающую от 1 до 5 гетероатомов, выбранных из О, S или N; или
R1 и R2 образуют вместе с атомом углерода, к которому они присоединены, С3-С7-циклоалкил;
R3 представляет собой атом водорода;
R4 представляет собой группу, выбранную из C1-C6-алкильной группы, группы (CH2)nOR6, С3-С7-циклоалкила или C1-C6-алкила, необязательно замещенного С3-С7-циклоалкилом;
R5 представляет собой группу, выбранную из атома водорода или C1-C6-алкильной группы;
R6 представляет собой группу, выбранную из атома водорода или C1-C6-алкильной группы;
n равен 1, 2 или 3;
в форме основания, или аддитивной соли с фармацевтически приемлемыми кислотами, или энантиомера, или диастереоизомера, или их смеси.
2. Соединение формулы (I) по п.1, отличающееся тем, что W представляет собой атом азота или группу CH в форме основания, или аддитивной соли с фармацевтически приемлемыми кислотами, или энантиомера, или диастереоизомера, или их смеси.
3. Соединение формулы (I) по п.1 или 2, отличающееся тем, что W представляет атом азота в форме основания, или аддитивной соли с фармацевтически приемлемыми кислотами, или энантиомера, или диастереоизомера, или их смеси.
4. Соединение формулы (I) по любому из пп.1-3, отличающееся тем, что Y представляет собой С2-С3-алкинилен в форме основания, или аддитивной соли с фармацевтически приемлемыми кислотами, или энантиомера, или диастереоизомера, или их смеси.
5. Соединение формулы (I) по п.4, отличающееся тем, что Y представляет собой этинилен в форме основания, или аддитивной соли с фармацевтически приемлемыми кислотами, или энантиомера, или диастереоизомера, или их смеси.
6. Соединение формулы (I) по любому из пп.1-4, отличающееся тем, что
Z представляет собой связь, группу CR1R2;
R1 представляет собой группу, выбранную из атома водорода, С1-С6-алкильной группы, группы (CH2)nOR6, C3-C7-циклоалкила, арила или 5- или 6-членного гетероарила;
R2 представляет собой группу, выбранную из атома водорода, C1-C6-алкильной группы или трифторметила;
R6 представляет собой группу, выбранную из атома водорода или C1-С6-алкильной группы;
n равен 1, 2 или 3;
в форме основания, или аддитивной соли с фармацевтически приемлемыми кислотами, или энантиомера, или диастереоизомера, или их смеси.
7. Соединение формулы (I) по любому из пп.1-6, отличающееся тем, что
Z представляет собой группу CR1R2;
R1 представляет собой группу, выбранную из атома водорода, C1-C6-алкильной группы, группы (CH2)nOR6, C3-C7-циклоалкила, арила или 5- или 6-членного гетероарила;
R2 представляет собой группу, выбранную из атома водорода, C1-C6-алкильной группы или трифторметила;
R6 представляет собой группу, выбранную из атома водорода или C1-C6-алкильной группы; и
n равен 1, 2 или 3;
в форме основания, или аддитивной соли с фармацевтически приемлемыми кислотами, или энантиомера, или диастереоизомера, или их смеси.
8. Соединение формулы (I) по любому из пп.1-7, отличающееся тем, что R4 представляет собой группу, выбранную из С1-С6-алкильной группы, группы (CH2)nOR6, С3-С7-циклоалкила или С1-С6-алкила, необязательно замещенного С3-С7-циклоалкилом, в форме основания, или аддитивной соли с фармацевтически приемлемыми кислотами, или энантиомера, или диастереоизомера, или их смеси.
9. Соединение формулы (I) по любому из пп.1-7, отличающееся тем, что R4 представляет собой C1-C6-алкил в форме основания, или аддитивной соли с фармацевтически приемлемыми кислотами, или энантиомера, или диастереоизомера, или их смеси.
10. Соединение формулы (I) по п.9, отличающееся тем, что R4 представляет собой этил в форме основания, или аддитивной соли с фармацевтически приемлемыми кислотами, или энантиомера, или диастереоизомера, или их смеси.
11. Соединение формулы (I) по любому из пп.1-10, отличающееся тем, что R5 представляет собой группу, выбранную из атома водорода или C1-C6-алкильной группы, в форме основания, или аддитивной соли с фармацевтически приемлемыми кислотами, или энантиомера, или диастереоизомера, или их смеси.
12. Соединение формулы (I) по любому из пп.1-11, отличающееся тем, что R5 представляет собой атом водорода в форме основания, или аддитивной соли с фармацевтически приемлемыми кислотами, или энантиомера, или диастереоизомера, или их смеси.
13. Соединение формулы (I) по п.1, отличающееся тем, что
W представляет собой атом азота или группу CH;
Y представляет собой С2-С3-алкинилен, 1,4-фенилен, необязательно замещенный R7, который представляет собой атом галогена;
Z представляет собой связь или группу CR1R2;
R1 представляет собой группу, выбранную из атома водорода, C1-C6-алкильной группы, группы (CH2)nOR6, С3-С7-циклоалкила, арила или 5- или 6-членного гетероарила, причем арил или гетероарил необязательно замещен атомом галогена;
R2 представляет собой группу, выбранную из атома водорода, C1-C6-алкильной группы или трифторметила;
R3 представляет собой атом водорода;
R4 представляет собой группу, выбранную из C1-C6-алкильной группы, группы (CH2)nOR6, С3-С7-циклоалкила или C1-C6-алкила, необязательно замещенного С3-С7-циклоалкилом;
R5 представляет собой группу, выбранную из атома водорода или C1-C6-алкильной группы;
R6 представляет собой группу, выбранную из атома водорода или C1-C6-алкильной группы;
n равен 1, 2 или 3;
в форме основания, или аддитивной соли с фармацевтически приемлемыми кислотами, или энантиомера, или диастереоизомера, или их смеси.
14. Соединение формулы (I) по любому из пп.1-13, отличающееся тем, что R1 и R2 вместе с атомом углерода, к которому они присоединены, образуют С3-С7-циклоалкил в форме основания, или аддитивной соли с фармацевтически приемлемыми кислотами, или энантиомера, или диастереоизомера, или их смеси.
15. Соединение формулы (I) по п.1, выбранное из
2-амино-1-этил-7-((3R)-3-гидрокси-4-метокси-3-метилбут-1-инил)-3-(1Н-имидазол-2-ил)-1Н-[1,8]нафтиридин-4-она;
2-амино-1-пропил-7-((3R)-3-гидрокси-4-метокси-3-метилбут-1-инил)-3-(1Н-имидазол-2-ил)-1Н-[1,8]нафтиридин-4-она;
2-амино-7-(3,4-дигидрокси-3-метилбут-1-инил)-1-этил-3-(1Н-имидазол-2-ил)-1Н-[1,8]нафтиридин-4-она;
2-амино-1-этил-7-(3-гидрокси-3-пиридин-2-илбут-1-инил)-3-(1Н-имидазол-2-ил)-1Н-[1,8]нафтиридин-4-она;
2-амино-1-этил-7-[(3R)-3-гидрокси-4-метокси-3-метилбут-1-инил]-3-(4-метил-1Н-имидазол-2-ил)-1Н-[1,8]нафтиридин-4-она;
2-амино-1-(циклопропилметил)-7-(3-гидроксипент-1-инил)-3-(1Н-имидазол-2-ил)-1Н-[1,8]нафтиридин-4-она;
2-амино-1-этил-7-[(3R)-3-гидрокси-4-метокси-3-метилбут-1-инил]-3-(1Н-имидазол-2-ил)-1Н-хинолин-4-она;
2-амино-7-(3-хлор-4-гидроксифенил)-1-этил-3-(1Н-имидазол-2-ил)-1Н-[1,8]нафтиридин-4-она;
2-амино-1-этил-7-[3-(2-фторфенил)-3-гидроксибут-1-инил]-3-(1Н-имидазол-2-ил)-1Н-[1,8]нафтиридин-4-она;
2-амино-1-циклопентил-7-(3-гидроксипент-1-инил)-3-(1Н-имидазол-2-ил)-1Н-[1,8]нафтиридин-4-она;
2-амино-7-(3-гидроксипент-1-инил)-3-(1Н-имидазол-2-ил)-1-(3-метоксипропил)-1Н-[1,8]нафтиридин-4-она;
2-амино-7-(3-гидроксипент-1-инил)-3-(1Н-имидазол-2-ил)-1-(2-метоксиэтил)-1Н-[1,8]нафтиридин-4-она;
2-амино-1-этил-7-[(1-гидроксициклобутил)этинил]-3-(1Н-имидазол-2-ил)-1Н-[1,8]нафтиридин-4-она;
2-амино-1-этил-7-[(1-гидроксициклопентил)этинил]-3-(1Н-имидазол-2-ил)-1Н-[1,8]нафтиридин-4-она;
2-амино-1-этил-7-(3-гидрокси-3-метилбут-1-инил)-3-(1Н-имидазол-2-ил)-1Н-[1,8]нафтиридин-4-она;
2-амино-1-этил-7-(3-гидрокси-3-метилпент-1-инил)-3-(1Н-имидазол-2-ил)-1Н-[1,8]нафтиридин-4-она;
2-амино-1-этил-7-(3-гидрокси-3-фенилбут-1-инил)-3-(1Н-имидазол-2-ил)-1Н-[1,8]нафтиридин-4-она;
2-амино-1-этил-7-[3-(3-фторфенил)-3-гидроксибут-1-инил]-3-(1Н-имидазол-2-ил)-1Н-[1,8]нафтиридин-4-она;
2-амино-1-этил-3-(1Н-имидазол-2-ил)-7-(4,4,4-трифтор-3-гидрокси-3-фенилбут-1-инил)-1Н-[1,8]нафтиридин-4-она;
2-амино-7-(3-циклопропил-3-гидроксибут-1-инил)-1-этил-3-(1Н-имидазол-2-ил)-1Н-[1,8]нафтиридин-4-она;
2-амино-1-этил-7-[3-гидрокси-3-(тиофен-2-ил)бут-1-инил]-3-(1Н-имидазол-2-ил)-1Н-[1,8]нафтиридин-4-она;
2-амино-1-этил-7-(3-гидроксибут-1-инил)-3-(1Н-имидазол-2-ил)-1Н-[1,8]нафтиридин-4-она;
2-амино-1-этил-7-(3-гидроксипент-1-инил)-3-(1Н-имидазол-2-ил)-1Н-[1,8]нафтиридин-4-она;
2-амино-1-этил-7-(3-гидроксигекс-1-инил)-3-(1Н-имидазол-2-ил)-1Н-[1,8]нафтиридин-4-она;
2-амино-1-этил-7- (3-гидрокси-4-метилпент-1-инил)-3-(1Н-имидазол-2-ил)-1Н-[1,8]нафтиридин-4-она;
2-амино-1-этил-7-(3-гидрокси-3-фенилпрол-1-инил)-3-(1Н-имидазол-2-ил)-1Н-[1,8]нафтиридин-4-она;
2-амино-7-((3R)-3,4-дигидрокси-3-метилбут-1-инил)-1-этил-3-(1Н-имидазол-2-ил)-1,8-нафтиридин-4(1Н)-она;
2-амино-7-((3S)-3,4-дигидрокси-3-метилбут-1-инил)-1-этил-3-(1Н-имидазол-2-ил)-1,8-нафтиридин-4(1Н)-она;
2-амино-1-этил-7-((3S)-3-гидрокси-4-метокси-3-метилбут-1-инил)-3-(1Н-имидазол-2-ил)-1,8-нафтиридин-4(1Н)-она,
или их фармацевтически приемлемых солей, или энантиомеров, или диастереоизомеров, или их смеси.
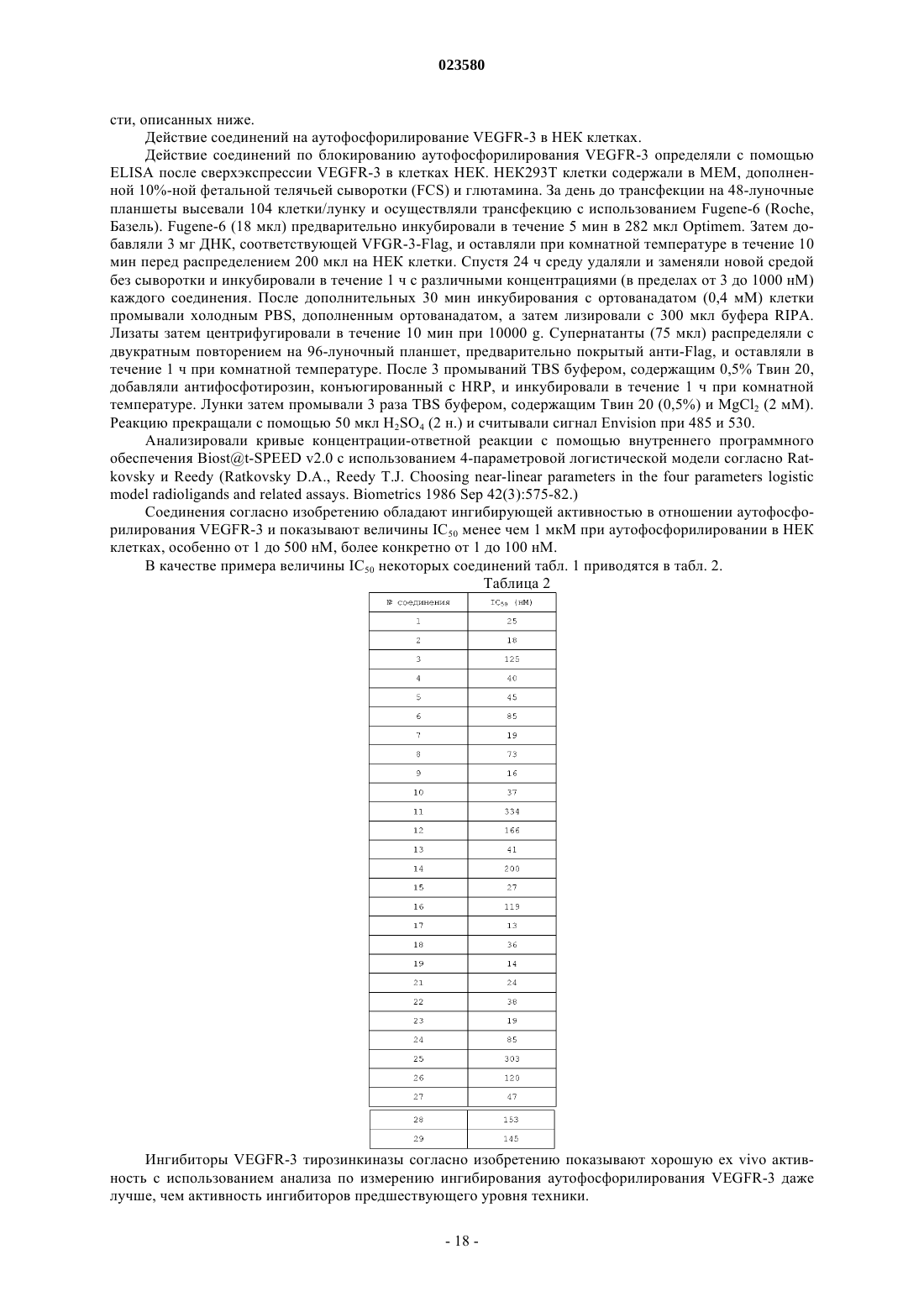
16. Способ получения соединения формулы (I), где радикал W представляет собой атом азота, по любому из пп.1-13, или его фармацевтически приемлемой соли, или энантиомера, или диастереоизомера, или их смеси, характеризующийся тем, что соединение формулы (VII)

в которой X представляет собой хлор или бром, PG представляет собой защитную группу, a R4 и R5 имеют значения, как определено в общей формуле (I), по любому из пп.1-13, подвергают реакции с соединением общей формулы (XVa)

в которой R1, R2 и R3 имеют значения, как определено в общей формуле (I), по любому из пп.1-13, или подвергают реакции с соединением общей формулы (XVb)

в которой R3 и R7 имеют значения, как определено в общей формуле (I), по любому из пп.1-13,
стадию снятия защиты проводят до или после реакции соединения формулы (VII) с соединением общей формулы (XVa) или с соединением общей формулы (XVb) в присутствии HCl в диоксане или трифторуксусной кислоты в этаноле или дихлорметане при температуре между -5 и 60°C с получением соединения формулы (I).
17. Лекарственное средство для предотвращения и/или лечения заболеваний, в которые вовлечен рецептор фактора роста эндотелия сосудов VEGFR-3, включающее соединение формулы (I) по любому из пп.1-15, или его фармацевтически приемлемую соль, или энантиомер, или диастереоизомер, или их смесь.
18. Фармацевтическая композиция для предотвращения и/или лечения заболеваний, в которые вовлечен рецептор VEGFR-3, включающая соединение по любому из пп.1-15, или его фармацевтически приемлемую соль, или энантиомер, или диастереоизомер, или их смесь, а также по меньшей мере один фармацевтически приемлемый эксципиент.
19. Применение соединения формулы (I) по любому из пп.1-15 для получения лекарственного средства для предотвращения и/или лечения заболеваний, в которые вовлечен рецептор VEGFR-3.
20. Применение соединения формулы (I) по любому из пп.1-15 для получения лекарственного средства для предотвращения и/или лечения рака и метастазов.
21. Применение соединения формулы (I) по любому из пп.1-15 для получения лекарственного средства для предотвращения и/или лечения заболеваний, выбранных из глиабластомы, множественной миеломы, миелодиспластического синдрома, саркомы Капоши, кожной ангиосаркомы, солидных опухолей, лимфомы, меланомы, рака груди, колоректального рака, рака легкого, рака поджелудочной железы, рака простаты, рака почек, рака головы и шеи, рака печени, рака яичников, рака дыхательного тракта и грудной клетки.
22. Применение соединения формулы (I) по любому из пп.1-15 для получения лекарственного средства для предотвращения и/или лечения неонкологических пролиферативных заболеваний и патологического ангиогенеза, связанного с VEGFR-3.
23. Применение соединения формулы (I) по любому из пп.1-15 для получения лекарственного средства для предотвращения и/или лечения заболеваний, выбранных из группы, состоящей из артроза, рестеноза, псориаза, гемангимы, лимфангиомы, глаукомы, гломерунефрита, диабетической нефропатии, нефросклероза, тромботического микроангиопатического синдрома, цирроза печени, атеросклероза, отторжения трансплантируемого органа, заболеваний глаз, включающих процесс ангиогенеза или лимфоангиогенеза.
24. Применение соединения формулы (I) по любому из пп.1-15 для получения лекарственного средства для предотвращения и/или лечения хронического или нехронического воспаления, инфицирования микроорганизмами и аутоиммунных заболеваний.
25. Применение по п.24, где заболеванием является ревматоидный артрит.
26. Применение соединения формулы (I) по любому из пп.1-15 для получения лекарственного средства для предотвращения и/или лечения редких заболеваний, выбранных из лимфангиолейомиоматоза или болезни Горхема.
Текст
ПРОИЗВОДНЫЕ 2-АМИНО-3-(ИМИДАЗОЛ-2-ИЛ)ПИРИДИН-4-ОНА И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ ИНГИБИТОРОВ КИНАЗЫ РЕЦЕПТОРА VEGF Изобретение относится к соединениям общей формулы (I); способу получения и терапевтическому применению 023580 Данное изобретение относится к производным 7-фенол или 7-алкинил-3-(имидазол-2-ил)-1,8 нафтиридин-4-она и их возможным хинолиноновым аналогам, которые являются ингибиторами киназной активности рецептора VEGF, к их получению и их терапевтическому применению. Семейство белков VEGF (фактор роста эндотелия сосудов) связывается с тремя структурно родственными рецепторными тирозинкиназами, известными как VEGF-R1 (Flt-1), VEGF-R2 (KDR), VEGF-R3(Flt-4). Все три рецептора являются жизненно важными для развития сосудистой системы во время эмбриогенеза и во время индуцируемого опухолью ангиогенеза. В дополнение VEGFR-3 играет важную роль в развитии лимфатической системы и в индуцируемом опухолью лимфоангиогенезе. В частности, WO 2009/007535 описывает замещенные производные 7-алкинил-4-оксо-1,8 нафтиридин-3-карбоксамидов, которые являются ингибиторами киназной активности рецептора VEGF. Соединения данного изобретения отличаются от этих соединений предшествующего уровня техники, по меньшей мере,присутствием имидазольного кольца в 3 положении бицикла. Критериями, которые следует принять во внимание при разработке лекарственного средства, являются воздействие соединения на ткани и его эффективность. Данные критерии могли бы быть повышены путем улучшения по меньшей мере одного из следующих показателей среди эффективности, абсорбции,распределения, метаболизма, выведения из организма и токсичности. Остается необходимость в устранении ингибиторов киназной активности рецептора VEGF с повышенной активностью, и это успешно достигается с новыми соединениями согласно изобретению. Первый объект изобретения относится к соединениям, соответствующим общей формуле (I), представленной далее. Другой объект изобретения относится к способам получения соединений общей формулы (I). Другой объект изобретения относится к применению соединений общей формулы (I), особенно в лекарственных средствах и фармацевтических композициях. Соединения по изобретению соответствуют общей формуле (I) в которой W представляет собой атом азота или группу CH;Y представляет собой С 2-С 3-алкинилен, 1,4-фенилен, необязательно замещенный R7, который представляет собой один или более атом(-ов) галогена;Z представляет собой связь или группу CR1R2;R1 и R2 независимо друг от друга представляют собой группу, выбранную из атома водорода, C1-C6 алкильной группы, трифторметильной группы, группы (CH2)nOR6, C3-C7-циклоалкила, гетероарила или арила, необязательно замещенного одним или более атомом(-ами) галогена;R1 и R2 могут образовывать вместе с атомом углерода, к которому они присоединены, C3-C7 циклоалкил;R3 представляет собой атом водорода;R5 представляет собой группу, выбранную из атома водорода или C1-C6-алкильной группы;R6 представляет собой группу, выбранную из атома водорода или C1-C6-алкильной группы;n равен 1, 2 или 3. Соединения формулы (I) могут включать один или более асимметричных атомов углерода. Они могут, таким образом, существовать в виде энантиомеров или диастереомеров. Эти энантиомеры и диастереомеры, а также их смеси, включая рацемические смеси, составляют часть изобретения. Соединения формулы (I) могут существовать в виде оснований или кислотно-аддитивных солей. Такие аддитивные соли составляют часть изобретения. Эти соли могут быть получены с фармацевтически приемлемыми кислотами, но соли других кислот, которые также являются полезными, например, для очистки или выделения соединений формулы(I), также составляют часть изобретения. В контексте данного изобретения применяют следующие обозначения: атом галогена: атом фтора, хлора, брома или йода;Ct-Cz - цепь на основе углерода, возможно содержащая от t до z атомов углерода, в которой t и z могут иметь значения от 1 до 7; например C1-C3: цепь на основе углерода, возможно содержащая от 1 до 3 атомов углерода; алкил - линейная или разветвленная насыщенная алифатическая группа. Примеры, которые могут быть упомянуты, включают метил, этил, н-пропил, изопропил, бутил, изобутил, трет-бутил, пентил и т.д.; алкилен - бивалентный радикал, полученный из алкана путем удаления атома водорода от каждого из двух концевых атомов углерода цепи, необязательно замещенного алкильной группой; например C1-1 023580C3-алкилен представляет собой линейную или разветвленную двухвалентную цепь на основе углерода из 1-3 атомов углерода, более конкретно метилен, этилен, метилэтилен или пропилен; алкенилен - бивалентный радикал, полученный из алкена путем удаления атома водорода от каждого из двух концевых атомов углерода цепи, необязательно замещенного алкильной или алкенильной группой; например C2-C3-алкенилен представляет собой линейную или разветвленную двухвалентную цепь на основе углерода из 2-3 атомов углерода, более конкретно этенилен или пропенилен; алкинилен - бивалентный радикал, полученный из алкина путем удаления атома водорода от каждого из двух концевых атомов углерода цепи, необязательно замещенного алкильной, алкенильной или алкинильной группой; например C2-C3-алкинилен представляет собой линейную или разветвленную двухвалентную цепь на основе углерода из 2-3 атомов углерода, более конкретно этинилен или пропинилен; циклоалкил - насыщенная или частично ненасыщенная циклическая алкильная группа. Примеры,которые могут быть упомянуты, включают циклопропильную, циклобутильную, циклопентильную, циклогексильную, циклопропенильную, циклобутенильную, циклопентенильную, циклогексенильную группы и т.д.; циклоалкилокси - радикал -O-циклоалкил, в котором циклоалкильная группа является такой, как определено выше; фторалкил - алкильная группа, у которой один или более атомов водорода заменены атомом фтора; алкокси: радикал -O-алкил, в котором алкильная группа является такой, как определено выше; фторалкокси - алкокси группа, у которой один или более атомов водорода заменены атомом фтора; тиоалкил или алкилтио - радикал -S-алкил, в котором алкильная группа является такой, как определено выше; арил - моноциклическая или бициклическая ароматическая группа, содержащая от 6 до 10 атомов углерода. Примеры арильных групп, которые могут быть упомянуты, включают фенильную и нафтильную группы; арилен - бивалентная группа, полученная из арила путем удаления атома водорода от двух кольцевых атомов углерода. Примеры ариленовой группы, которые могут быть упомянуты, включают фениленовую группу; гетероцикл - насыщенная или частично ненасыщенная 5-7-членная моноциклическая группа, включающая от 1 до 3 гетероатомов, выбранных из О, S и N. Примеры гетероциклов, которые могут быть упомянуты, включают азетидинил, пиперидинил, азепинил, морфолинил, тиоморфолинил, пиперазинил,гомопиперазинил, дигидрооксазолил, дигидротиазолил, дигидроимидазолил, дигидропирролил или тетрагидропиридил, [1,3]диоксолил, [1,3]диоксинил, дигидро[1,4]диоксинил, дигидро[1,2]оксазинил, дигидро[1,3]оксазинил,дигидрооксазолил,дигидроизоксазолил,дигидро[1,4]оксазинил,тетрагидро[1,3]оксазепинил,тетрагидро[1,4]оксазепинил,тетрагидро[1,3]диазепинил и тетрагидро[1,4]диазепинил; гетероарил - 5-12-членная моноциклическая или бициклическая ароматическая группа, содержащая от 1 до 5 гетероатомов, выбранных из О, S и N. Примеры моноциклических гетероарилов, которые могут быть упомянуты, включают имидазолил, пиразолил, тиазолил, оксазолил, изотиазолил, изоксазолил, фурил, тиенил, оксадиазолил, тиадиазолил, триазолил, тетразолил, пиридил, пиразинил, пиримидинил, пиридазинил и триазинил. Примеры бициклических гетероарилов, которые могут быть упомянуты, включают индолил, изоиндолил, бензофурил, бензотиофенил, бензоксазолил, бензимидазолил, индазолил,бензотиенил, изобензофурил, изобензотиазолил, пирроло[2,3-c]пиридил, пирроло[2,3-b]пиридил, пирроло[3,2-b]пиридил, пирроло[3,2-c]пиридил, пирроло[1,2-а]пиридил, хинолил, изохинолил, циннолинил,хиназолинил,хиноксалинил,пирроло[1,2-а]имидазолил,имидазо[1,2-а]пиридил,имидазо[1,2 а]пиридазинил, имидазо[1,2-c]пиримидинил, имидазо[1,2-а]пиримидинил, имидазо[1,2-а]пиразинил,имидазо[4,5-b]пиразинил, имидазо[4,5-b]пиридил, имидазо[4,5-c]пиридил, пиразоло[2,3-а]пиридил, пиразоло[2,3-а]пиримидинил, пиразоло[2,3-а]пиразинил, тиазоло[5,4-b]пиридил, тиазоло[5,4-с]пиридил, тиазоло[4,5-c]пиридил, тиазоло[4,5-b]пиридил, оксазоло[5,4-b]пиридил, оксазоло[5,4-c]пиридил, оксазоло[4,5-с]пиридил, оксазоло[4,5-b]пиридил, изотиазоло[5,4-b]пиридил, изотиазоло[5,4-c]пиридил, изотиазоло[4,5-c]пиридил, изотиазоло[4,5-b]пиридил, изоксазоло[5,4-b]пиридил, изоксазоло[5,4-c]пиридил,изоксазоло[4,5-c]пиридил и изоксазоло[4,5-b]пиридил."тио" означает "-S-". В контексте данного изобретения используются следующие сокращения и эмпирические формулы:HCl - соляная кислота ИК - инфракрасныйNa2SO4 - сульфат натрия ЯМР - ядерно-магнитный резонансXphos - 2-дициклогексилфосфино-2',4',6'-триизопропилбифенил Среди соединений общей формулы (I), которые являются объектами изобретения, первую подгруппу соединений составляют соединения, для которых W представляет собой атом азота или группу CH. Среди соединений общей формулы (I), которые являются объектами изобретения, вторую подгруппу соединений составляют соединения, для которых W представляет собой атом азота. Среди соединений общей формулы (I), которые являются объектами изобретения, третью подгруппу соединений составляют соединения, для которых Y представляет собой C2-C3-алкинилен, более конкретно этинилен. Среди соединений общей формулы (I), которые являются объектами изобретения, четвертую подгруппу соединений составляют соединения, для которыхR1 представляет собой группу, выбранную из атома водорода, C1-C6-алкильной группы, группы(CH2)nOR6, С 3-С 7-циклоалкила, арила или 5- или 6-членного гетероарила, необязательно замещенного одним или более атомом(-ами) галогена;R2 представляет собой группу, выбранную из атома водорода, C1-C6-алкильной группы или трифторметила;R6 представляет собой группу, выбранную из атома водорода или C1-C6-алкильной группы;n равен 1, 2 или 3. Среди соединений общей формулы (I), которые являются объектами изобретения, пятую подгруппу соединений составляют соединения, для которыхZ представляет собой группу CR1R2;R1 представляет собой группу, выбранную из атома водорода, C1-C6-алкильной группы, группыR2 представляет собой группу, выбранную из атома водорода, C1-C6-алкильной группы или трифторметила;R6 представляет собой группу, выбранную из атома водорода или C1-C6-алкильной группы; иn равен 1, 2 или 3. Когда Y представляет собой C2-C3-алкинилен, более конкретно этинилен, тогда Z представляет собой группу CR1R2, причем R1 и R2 имеют значения, определенные выше. Среди соединений общей формулы (I), которые являются объектами изобретения, шестую подгруппу соединений составляют соединения, для которых R4 представляет собой группу, выбранную из C1-C6 алкильной группы, группы (CH2)nOR6, C3-C7-циклоалкила или C1-C6-алкила, необязательно замещенногоC3-C7-циклоалкилом. Среди соединений общей формулы (I), которые являются объектами изобретения, седьмую подгруппу соединений составляют соединения, для которых R4 представляет собой C1-C6-алкил, более конкретно этил. Среди соединений общей формулы (I), которые являются объектами изобретения, восьмую подгруппу соединений составляют соединения, для которых R5 представляет собой группу, выбранную из-3 023580 атома водорода или C1-C6-алкильной группы. Среди соединений общей формулы (I), которые являются объектами изобретения, девятую подгруппу соединений составляют соединения, для которых R5 представляет собой атом водорода. Среди соединений общей формулы (I), которые являются объектами изобретения, десятую подгруппу соединений составляют соединения, для которых объединены определения W, E, Z, R3, R4 и R5,данные выше. Среди соединений общей формулы (I), которые являются объектами изобретения, одиннадцатую подгруппу соединений составляют соединения, для которыхW представляет собой атом азота или группу CH;Y представляет собой C2-C3-алкинилен, 1,4-фенилен, необязательно замещенный атомом галогена;R1 представляет собой группу, выбранную из атома водорода, C1-C6-алкильной группы, группы(CH2)nOR6, С 3-С 7-циклоалкила, арила или 5- или 6-членного гетероарила, необязательно замещенного атомом галогена;R2 представляет собой группу, выбранную из атома водорода, C1-C6-алкильной группы или трифторметила;R3 представляет собой атом водорода;R5 представляет собой группу, выбранную из атома водорода или C1-C6-алкильной группы;R6 представляет собой группу, выбранную из атома водорода или С 1-С 6-алкильной группы;n равен 1, 2 или 3. Среди соединений общей формулы (I), которые являются объектами изобретения, двенадцатую подгруппу соединений составляют соединения, для которыхW представляет собой атом азота или группу CH;Y представляет собой группу С 2-С 3-алкинилен;Z представляет собой группу CR1R2;R1 представляет собой группу, выбранную из атома водорода, C1-C6-алкильной группы, группы(CH2)nOR6, С 3-С 7-циклоалкила, арила или 5- или 6-членного гетероарила, необязательно замещенного атомом галогена;R2 представляет собой группу, выбранную из атома водорода, C1-C6-алкильной группы или трифторметила;R3 представляет собой атом водорода;R5 представляет собой группу, выбранную из атома водорода или C1-C6-алкильной группы;R6 представляет собой группу, выбранную из атома водорода или C1-C6-алкильной группы;n равен 1, 2 или 3. Среди соединений общей формулы (I), которые являются объектами изобретения, тринадцатую подгруппу соединений составляют соединения, для которых R1 и R2 образуют вместе с атомом углерода,к которому они присоединены, С 3-С 7-циклоалкил. Само собой разумеется, каждая из этих подгрупп, упомянутых выше, может быть объединена с одной или более другими подгруппами, и соответствующие соединения также являются объектами изобретения. Среди соединений общей формулы (I), которые являются объектами изобретения, особенно могут быть упомянуты следующие соединения: Далее в тексте термин "уходящая группа" означает группу, которая может быть легко отщеплена от молекулы путем разрыва гетеролитической связи с потерей электронной пары. Данная группа, таким образом, может быть легко заменена другой группой, например, во время реакции замещения. Такими уходящими группами являются, например, галогены или активированная гидроксильная группа, такая как метансульфонат, бензолсульфонат, п-толуолсульфонат, трифлат, ацетат и др. Примеры уходящих групп и ссылки на их получение даны в "Advances in Organic Chemistry", J. March, 5-е издание, Wiley Interscience, 2001.-5 023580 Далее в тексте термин "защитная группа" (PG) означает группу, которая может быть немедленно включена в химическую структуру для цели временной инактивации части молекулы во время реакции и которая может быть легко удалена на следующей стадии синтеза. Примеры защитных групп и ссылки в отношении их свойств даны в "Protective Groups in Organic Synthesis", T.W. Green, P.G.M. Wutz, 3-е издание, Wiley Interscience 1999. В соответствии с изобретением соединения общей формулы (I) могут быть получены согласно способу, представленному общими схемами 1, 2 и 3 Схема 1 Согласно схеме 1 на стадии (i) 2,6-дигалогенникотиновую кислоту формулы (II) подвергают монозамещению во 2 положении амином формулы R4-NH2 (где R4 имеет значение, как определено ранее в отношении соединения формулы (I, при комнатной температуре или при температуре от 50 до 100C с обычным нагревом или микроволновым нагреванием и в протонном растворителе, таком как спирт, например, этанол, н-бутанол, трет-бутанол или вода. Кислоту (III), полученную на стадии (i), затем активируют до производного формулы (IV), на следующей стадии (ii), либо в виде фторангидрида путем активации цианурилфторида при комнатной температуре в присутствии основания, такого как триэтиламин или пиридин, и в апротонном растворителе, таком как дихлорметан или THF, как описано авторами G.Olah и другими в Synthesis (1973), 487, либо в виде имидазола путем активации карбонилдиимидазола в полярном апротонном растворителе, таком как DMF или THF, либо любыми другими методами, известными специалистам в данной области, такими, как описаны авторами Mukaiyama и Tanaka в Chem. Lett.(1976), 303 или авторами Ishikawa и Sasaki в Chem. Lett. (1976), 1407. Цианометилимидазолы формулы (V) получают путем проведения двух стадий из имидазол-2 карбоксальдегида, незамещенного или замещенного в положении (4,5) имидазола. На стадии (iii) свободный азот имидазол-2-карбоксальдегида защищают защитной группой, обозначенной PG на схеме 1, например, такой как SEM, Boc или тритильная группа, в обычных рабочих условиях, известных специалистам в данной области, как описано, например, в "Protective Groups in Organic Synthesis", Green et al., 3 издание (John WileySons, Inc. New York). Если применимо, два изомера Tau и Pi защищенного имидазола получают и используют без разграничения в последующих реакциях. Защищенный имидазол-2-6 023580 карбоксальдегид затем преобразуют на стадии (iv) в цианометилимидазол формулы (V) путем взаимодействия альдегидной функциональной группы с анионом TOSMIC, образованным путем добавления трет-бутилата калия к безводному DME раствору TOSMIC при низкой температуре (-50C) с последующим раскрытием кольца образованного анионного производного, 4-тозил-2-оксазолина, затем реакционную смесь нагревают при температуре кипения с обратным холодильником в присутствии метанола,чтобы допустить образование ацетилнитрильной функциональной группы, следуя методике, описаннойVan Leusen A. et al. (Synthetic Comm, 10(5) 1980, 399-403). Фторангидрид или имидазол формулы (IV), полученный в конце стадии (ii), очень реакционноспособный, но стабильный, подвергают затем реакции на стадии (v) с цианометилимидазолом формулы (V),незамещенным или замещенным в положении (4,5), в присутствии одного эквивалента основания, такого как гидрид натрия или трет-бутоксид калия, в полярном апротонном растворителе, таком как THF илиDMF, при температуре от -5C до комнатной температуры, затем добавляют второй эквивалент примененного основания, и соединение формулы (VI), которое при этом образуется, подвергают циклизации insitu, при комнатной температуре, давая пиримидино-пиридиноновое соединение формулы (VII), на следующей стадии (vi). Согласно схеме 2 на стадии (vii) 2,4-дигалогентолуол формулы (VIII) окисляют до соответствующего производного кислоты (IX) с использованием сильного окислителя, такого как перманганат калия, при комнатной температуре или при температуре от 50 до 100C с обычным нагревом или микроволновым нагреванием и в протонном растворителе, таком как вода, и в присутствии основания, такого как пиридин, или любыми другими методами, известными специалистам в данной области, такими, как описаны в следующем патенте: US 6187950. Кислоту (IX), полученную на стадии (vii), подвергают монозамещению в положении 2 амином формулы R4-NH2 (где R4 имеет значение, как определено ранее в отношении соединений формулы (I, при комнатной температуре или при температуре от 50 до 100C с обычным нагревом или микроволновым нагреванием и в протонном растворителе, таком как спирт, например, этанол, н-бутанол, трет-бутанол или вода. Кислоту (X), полученную на стадии (viii), затем циклизуют в бензо-1,3-оксазин-2,4-дион (XI) под воздействием карбонилдиимидазола или трифосгена при комнатной температуре или при температуре от 50 до 120C с обычным нагревом или микроволновым нагреванием и в апротонном растворителе, таком как DMF, толуол, THF, диоксан. Бензо-1,3-оксазин-2,4-дион (XI) затем обрабатывают малононитрилом при комнатной температуре или при температуре от 50 до 120C с обычным нагревом или микроволновым нагреванием и в апротонном растворителе, таком как DMF, толуол, диоксан, и в присутствии основания, такого как триэтиламин или пиридин, или любыми другими методами, известными специалистам в данной области, такими, как описано авторами Iminov et al. в Synthesis (2008), 1535, с получением нитрила (XII). Данный нитрил (XII), полученный на стадии (X), взаимодействует с аминоацетальдегид диэтилацеталем в присутствии медного катализатора, такого как CuCl,при комнатной температуре или при температуре от 50 до 120C с обычным нагревом или микроволновым нагреванием и в апротонном растворителе, таком как DME, DMF, толуол, диоксан. Ацеталь (XIII),полученный на стадии (xi), подвергали затем циклизации в имидазол (XIV) с применением условий сильной кислоты, такой как HCl (12 н.), при комнатной температуре или при температуре от 50 до 120C,с обычным нагревом или микроволновым нагреванием и в протонном растворителе, таком как спирт,например этанол, н-бутанол, трет-бутанол или вода. Для получения соединений формулы (I) согласно данному изобретению могут применяться два способа, описанные на схеме 3, начиная с галогенированных промежуточных соединений формулы (VII) или (XIV). Следуя первому способу, ведущему к соединению формулы (I), которое является предметом данного изобретения, на стадии (xiii) применяют галогенированное промежуточное соединение формулы (VII) или (XIV), или в реакции сочетания Соногашира с подходящим производным пропаргилового спиртаR1R2CH (OR3)CCH формулы (XVa), где R1, R2 и R3 имеют значения, как определено ранее, или в реакции сочетания Сузуки с подходящей арилбороновой кислотой формулы (XVb). Реакцию Соногашира(xiii) проводят в присутствии палладиевого комплекса (в степени окисления (0) или (II), например, такого как Pd(PPh3)4, PdCl2(PPh3)2, в присутствии йодида меди, триэтиламина, в апротонном полярном растворителе, таком как THF или DMF, с обычным нагреванием от 80 до 120C или микроволновым нагреванием. Реакцию Сузуки (xiii) проводят в присутствии палладиевого комплекса (в степени окисления (0) или (II, например, такого как Pd(PPh3)4, PdCl2(PPh3)2, Pd2dba3, Xphos или PdCl2(dppf), в протонном или апротонном полярном растворителе, таком как DME, этанол, DMF, диоксан или смеси данных растворителей, в присутствии основания, такого как карбонат цезия, водный раствор бикарбоната натрия илиK3PO4, с обычным нагреванием между 80 и 120C или микроволновым нагреванием между 130 и 170C. В заключение, продукт реакции Соногашира (XVIa) или Сузуки (XVIb) подвергают реакции снятия защиты согласно общепринятой стадии снятия защиты (xiv), например, в присутствии кислоты, такой как HCl (4 н.) в диоксане или трифторуксусная кислота в растворителе, таком как этанол или дихлорметан, при температуре между -5 и 60C, получая соединение формулы (I). Следуя второму способу получения соединения формулы (I), которое является объектом данного-7 023580 изобретения, галогенированное промежуточное соединение формулы (VII) или (XIV) сначала подвергают реакции снятия защиты (xv), согласно той же общепринятой процедуре, как для стадии (xiv). И полученное незащищенное соединение (XVII) применяют или в реакции сочетания Соногашира с подходящим производным пропаргилового спирта R1R2CH(OR3)CCH формулы (XVa), где R1, R2 и R3 имеют значения, как определено ранее; или в реакции сочетания Сузуки с подходящей арилбороновой кислотой формулы (XVb), согласно тем же условиям, которые описаны ранее для стадии (xiii). Обе реакции сочетания ведут непосредственно к соединению (I). Если необходимо, во время стадий реакции, представленных на схеме 1, гидроксильная группа или некоторые реакционноспособные функции, расположенные на группах R1, R2 и R3, могут быть временно защищены защитными группами, известными специалистам в данной области, как описано в "ProtectiveGroups in Organic Synthesis", Green et al., 2 издание (John WilleySons, Inc., New York). Согласно другому аспекту изобретения объектом изобретения являются также соединения формулы(VII), как представлено на схеме 1. Данные соединения могут быть использованы как промежуточные соединения синтеза для соединений формулы (I). Согласно другому аспекту изобретения объектом изобретения является также способ получения соединения формулы (I), характеризующийся тем, что соединение формулы (VII) в которой X представляет собой хлор или бром, и R4 и R5 имеют значения, как определено в общей формуле (I), подвергают реакции с соединением общей формулы (XVa) в которой R1, R2 и R3 имеют значения, как определено в общей формуле (I), или подвергают реакции с соединением общей формулы (XVb) в которой R3 и R7 имеют значения, как определено в общей формуле (I), общепринятая стадия снятия защиты проводится до или после реакции соединения формулы (VII) с соединением общей формулы(XVa) или с соединением общей формулы (XVb). Примеры, которые следуют далее, описывают получение некоторых соединений в соответствии с изобретением. Эти примеры не являются ограничивающими и служат только для иллюстрации данного изобретения. Представленные номера соединений относятся к номерам в табл. 1. Элементный микроанализ, анализ LC/MS и ИК или ЯМР спектр подтверждают структуры полученных соединений. Пример 1 (соединение 1). 2-Амино-1-этил-7-3R)-3-гидрокси-4-метокси-3-метилбут-1-инил)-3(1 Н-имидазол-2-ил)-1 Н-[1,8]нафтиридин-4-он. 1.1. 6-Хлор-2-этиламиноникотиновая кислота. Раствор 18,0 г (84,4 ммоль) 2,6-дихлорникотиновой кислоты в 180 мл раствора этиламина (70% в воде) перемешивали при температуре окружающей среды в течение 72 ч. Избыток амина затем выпаривали при пониженном давлении и добавляли 10%-ный водный раствор уксусной кислоты до тех пор, пока продукт не осаждался. Бежевое твердое вещество сушили на центробежном фильтре, промывали холодной водой и сушили в сушильном шкафу. Получалось 10,5 г ожидаемого продукта. Точка плавления=158-160C. Выход=62% 1.2. 6-Хлор-2-этиламиноникотиноилфторид. 2 мл (24,8 ммоль) пиридина и 4,2 мл (49,8 ммоль) 2,4,6-трифтортриазина добавляли к суспензии 5,0 г (24,8 ммоль) 6-хлор-2-этиламиноникотиновой кислоты в 125 мл дихлорметана. Смесь перемешивали в течение 3 ч при температуре окружающей среды и затем фильтровали. Твердое вещество промывали 50 мл дихлорметана, и фильтрат дважды промывали 60 мл охлажденной льдом воды. Органическую фазу сушили над Na2SO4, и растворитель выпаривали при пониженном давлении. Получали 5,01 г продукта в виде оранжевого масла, которое использовали без дальнейшей очистки. Выход=99% 1.3. 1-(2-Триметилсиланилэтоксиметил)-1 Н-имидазол-2-карбальдегид. Маслянистую суспензию 2 0,8 г гидрида натрия в минеральном масле (50%, 0,52 моль) промывали от минерального масла при перемешивании с гексаном три раза и суспендировали в 4 00 мл DMF. При перемешивании при температуре окружающей среды к суспензии добавляли 50,0 г (0,52 0 моль) имидазол-2-карбальдегида. Через 1,5 ч добавляли 101,0 мл(триметилсиланил)этоксиметилхлорида, и реакционную смесь перемешивали еще 1 ч. К суспензии с избытком добавляли воду, и реакционную смесь экстрагировали три раза этилацетатом. Органическую фазу сушили над Na2SO4, и растворитель выпаривали при пониженном давлении. Сырой материал затем очищали с помощью колоночной хроматографии (DCM), получая 85,0 г (0,376 моль) SEM-защищенного имидазол-2-карбальдегида. Выход=72% МН+=227,1(C10H18N2O2Si, Mr=226,35) 1(т, 2H); 0,02 (с, 9H). 1.4. [1-(2-Триметилсиланилэтоксиметил)-1 Н-имидазол-2-ил]ацетонитрил. 1,73 г (8,84 ммоль) тозилметилизоцианида растворяли в 10 мл DME и охлаждали до -60C. При данной температуре сначала добавляли 1,98 г трет-бутоксида калия, затем медленно добавляли раствор 2,00 г (8,84 ммоль) 1-(2-триметилсиланилэтоксиметил)-1 Н-имидизол-2-карбальдегида в 5 мл DME. Через 2 ч перемешивания при -60C реакционной смеси давали нагреться до 0C и к раствору добавляли 5 мл метанола (123,60 ммоль). Реакционную смесь перемешивали в течение следующих 24 ч при температуре окружающей среды и в течение 2 ч при 40C. С избытком добавляли воду, и раствор экстрагировали три раза дихлорметаном. Органическую фазу сушили над Na2SO4, после выпаривания растворителя при пониженном давлении сырой материал очищали с помощью колоночной хроматографии с обращенной фазой (вода 0,1% TFA/ацетонитрил=80/20), получая 0,87 г (0,367 моль) SEM-защищенного имидазолацетонитрила. Выход=41% МН+=238,1(C11H19N3OSi, Mr=237,38) 1(т, 2H); 0,02 (с, 9H). 1.5. 3-(6-Хлор-2-этиламинопиридин-3-ил)-3-гидрокси-2-[1-(2-триметилсиланилэтоксиметил)-1 Нимидазол-2-ил]акрилонитрил. 0,283 г (2,53 ммоль) трет-бутилата калия в небольших количествах добавляли к охлажденному до 0C раствору 0,600 г (2,53 ммоль) [1-(2-триметилсиланилэтоксиметил)-1 Н-имидазол-2-ил]ацетонитрила в 10 мл безводного THF. Смесь перемешивали в течение 45 мин при температуре окружающей среды и затем снова охлаждали до 0C. Затем добавляли раствор 0,512 г (2,53 ммоль) 6-хлор-2 этиламиноникотиноилфторида в 10 мл THF, и смесь перемешивали при температуре окружающей среды в течение ночи, снова охлаждали до 0C и добавляли второй эквивалент трет-бутилата калия (0,283 г,2,53 ммоль). Через 2 ч перемешивания при температуре окружающей среды добавляли 50 мл насыщенного водного раствора хлорида аммония, рН доводили до 7 с помощью 2 н. HCl, затем экстрагировали три раза этилацетатом. Объединенные органические фазы сушили над MgSO4, и растворители выпаривали при пониженном давлении. Сырой материал затем очищали с помощью колоночной хроматографии(DCM/Метанол=90:10), получая 418 мг (выход=38%) указанного в заголовке соединения в виде промежуточного вещества, которое затем использовали на следующей стадии. МН+=421(C19H26ClN5O2Si, Mr=419,99) 1(д, 1H); 5,59 (с, 2H); 3,58 (т, 2H); 3,34 (д.кв, 2H); 1,13 (т, 3H); -0,03 (3 с, 9H). 1.6. 2-Амино-7-хлор-1-этил-3-[1-(2-триметилсиланилэтоксиметил)-1 Н-имидазол-2-ил]-1 Н-[1,8]нафтиридин-4-он. 0,112 г (1 ммоль) трет-бутилата калия в небольших количествах добавляли к охлажденному до 0C раствору 418 мг (1 ммоль) промежуточного соединения, полученному в 1.5, 3-(6-хлор-2-этиламинопиридин-3-ил)-3-гидрокси-2-[1-(2-триметилсиланил-этоксиметил)-1 Н-имидазол-2-ил]акрилонитрила в 5 мл безводного THF. Смесь перемешивали в течение 48 ч при температуре окружающей среды, после чего добавляли 50 мл насыщенного водного раствора хлорида аммония, рН доводили до 7 с помощью 2 н.HCl, и реакционную смесь экстрагировали три раза этилацетатом. Объединенные органические фазы сушили над MgSO4, и растворители выпаривали при пониженном давлении, получая 400 мг указанного в заголовке соединения. Выход=38% МН+=421 (C19H26ClN5O2Si, Mr=419,99) 1(с, 2H); 4,58 (кв, 2H); 3,57 (т, 2H); 1,42 (т, 3H); 0,85 (т, 2H); -0,03 (3 с, 9H). 1.7. -2-Метилбут-3-ин-1,2-диол. Коммерчески доступный 0,5 М раствор этинилмагний хлорида в тетрагидрофуране разбавляли 200 мл тетрагидрофурана и охлаждали до 0C. Затем добавляли раствор гидроксиацетона в 200 мл тетрагидрофурана, и смесь перемешивали при температуре окружающей среды в течение 3 ч. Реакционную смесь охлаждали и добавляли водный раствор NH4Cl. Смесь три раза экстрагировали этилацетатом, и органические фазы объединяли, сушили над сульфатом натрия, фильтровали и концентрировали в вакууме(примерно 200 мбар). В итоге получали 20 г ожидаемого продукта в виде коричневого масла, которое использовали без последующей очистки (количественный сырой выход) в рацемической форме или которое могло быть разделено на чистые энантиомеры с помощью препаративной ВЭЖХ на хиральных ВЭЖХ колонках. Для того чтобы получить оптически чистые энантиомеры, соответствующую рацемическую смесь подвергали препаративной хроматографии на хиральной стационарной фазе (Chiralpak ADH колонка, 25021 мм, 5 мм) с применением в качестве подвижной фазы: или CO2/2-пропанола (70/30%) со скоростью потока 60 мл/мин при давлении 100 бар или смеси изогексан/этанол (70/30%) с 0,3% TFA и скоростью потока 120 мл/мин. После разбавления и выпаривания каждый энантиомер отделяли, и химическую чистоту и энантиомерную чистоту каждого определяли с помощью аналитических методов, известных специалистам в данной области. 1.8. 2-Амино-1-этил-7-3R)-3-гидрокси-4-метокси-3-метилбут-1-инил)-3-[1-(2-триметилсиланилэтоксиметил)-1 Н-имидазол-2-ил]-1 Н-[1,8]нафтиридин-4-он. В заполненную аргоном микроволновую реакционную емкость помещали 500 мг (1,2 ммоль) 2 амино-7-хлор-1-этил-3-[1-(2-триметилсиланилэтоксиметил)-1 Н-имидазол-2-ил]-1 Н-[1,8]нафтиридин-4 она, 204 мг (1,8 ммоль) (3R)-1-метокси-2-метилбут-3-ин-2-ола, 84 мг (0,120 ммоль) дихлорида бис(трифенилфосфин)палладия (II), 30 мг (0,16 ммоль) йодида меди (I), 2 мл DMF (дегазированного), 2 мл триэтиламина (дегазированного) и подвергали облучению микроволнами таким образом, чтобы реакционная смесь выдерживалась при 120C в течение 24 ч. Растворители выпаривали, и твердое вещество повторно суспендировали в 30 мл DMF и фильтровали. Фильтрат затем очищали с помощью ВЭЖХ, получая 430 мг (0,702 ммоль) TFA соли соединения, указанного в заголовке. Выход=59% МН+=498,2(C25H35N5O4Si, Mr=497,67) 1DCM. Раствор выдерживали при 3-5C в течение ночи до тех пор, пока аналитическая ВЭЖХ не показывала завершение снятия защиты с нафтиридинона. Раствор нейтрализовали путем добавления избытка водного раствора NaHCO3. Смесь затем экстрагировали три раза этилацетатом. Объединенные органические фазы сушили над MgSO4, и растворители выпаривали при пониженном давлении. Полученный таким образом сырой материал очищали на силикагеле (DCM:MeOH=4:1), получая 143 мг (количественный выход) незащищенного соединения, указанного в заголовке. МН+=368,2 (C19H21N5O3, Mr=367,41) 1H ЯМР (ДМСО-d6, 500 МГц):13,15 (с, 1H); 11,55 (ушир.с, 1H); 8,59 (д, 1H); 8,10 (ушир.с, 1H); 7,47 (д, 1H); 7,25 (с, 1H); 7,02 (с, 1H); 5,85 (с, 1H); 4,58 (уширенный сигнал, 2H); 3,51-3,37 0 (уширенный сигнал, пик воды+4H); 1,4 8 (с, 3H); 1,25 (т, 3H). Время удерживания (аналитическая ВЭЖХ): 4,806 мин. Пример 2 (соединение 2). 2-Амино-1-пропил-7-3R)-3-гидрокси-4-метокси-3-метилбут-1-инил)3-(1 Н-имидазол-2-ил)-1 Н-[1,8]нафтиридин-4-он. Применяя процедуру, описанную до стадии 1.6, 2-амино-7-хлор-1-пропил-3-[1-(2-триметилсиланилэтоксиметил)-1 Н-имидазол-2-ил]-1 Н-[1,8]нафтиридин-4-он синтезировали с использованием нпропиламина вместо этиламина на стадии 1.1. Присоединяя данное промежуточное соединение к (3R)-1 метокси-2-метилбут-3-ин-2-олу, следуя аналогичным образом подробной процедуре, изложенной для примера 1, получали соединение, указанное в заголовке. МН+=382,48(C20H23N5O3, Mr=381,44) 1 Н ЯМР (ДМСО-d6, 500 МГц):ушир.с. 8,45 (м, 1 Н); 7,4 (м, 3H); 5,85 (с, 1 Н); 4,58 (м, 3H); 3,513,370 (пик воды+4H); 1,70 (м, 2H); отчетливые сигналы: 1,48 (с, 3H); 0, 95 (т, 3H). Время удерживания (аналитическая ВЭЖХ): 4,98 мин. Пример 3 (соединения 3, 27 и 28). 2-Амино-7-(3,4-дигидрокси-3-метилбут-1-инил)-1-этил-3-(1 Н-имидазол-2-ил)-1,8-нафтиридин 4(1 Н)-он. 2-Амино-7-3R)3,4-дигидрокси-3-метилбут-1-инил)-1-этил-3-(1 Н-имидазол-2-ил)-1,8-нафтиридин 4(1 Н)-он. 2-Амино-7-3S)3,4-дигидрокси-3-метилбут-1-инил)-1-этил-3-(1 Н-имидазол-2-ил)-1,8-нафтиридин 4(1 Н)-он. 3.1. 2-Амино-1-этил-7-(3,4-дигидрокси-3-метилбут-1-инил)-1-этил-3-[1-(2-триметилсиланилэтоксиметил)-1 Н-имидазол-2-ил]-1,8-нафтиридин-4(1 Н)-он Получали, следуя подробной процедуре, изложенной для стадии 1.8, с использованием промежу- 10023580 точного соединения, описанного как 1.6 (2-амино-7-хлор-1-этил-3-[1-(2-триметилсиланил-этоксиметил)1 Н-имидазол-2-ил]-1,8-нафтиридин-4(1 Н)-он) и -2-метилбут-3-ин-1,2-диола, полученного ранее, как описано в процедуре на стадии 1.7. МН+=354,16 (C18H19N5O3, Mr=353,38) Время удерживания (аналитическая ВЭЖХ): 4,48 мин. 3.2. 2-Амино-1-этил-7-3R)3,4-дигидрокси-3-метилбут-1-инил)-1-этил-3-[1-(2-триметилсиланилэтоксиметил)-1 Н-имидазол-2-ил]-1,8-нафтиридин-4(1 Н)-он. 2-Амино-1-этил-7-3S)3,4-дигидрокси-3-метилбут-1-инил)-1-этил-3-[1-(2-триметилсиланилэтоксиметил)-1 Н-имидазол-2-ил]-1,8-нафтиридин-4(1 Н)-он. Рацемическое соединение, полученное на стадии 3.1, подвергалось очистке посредством препаративной хиральной SFC (сверхкритическая флюидная хроматография), с применением методов, Bergerprep SFC, UV детекция при 230 нм, стационарная фаза Chiralpak IC 20250 нм 5 мкм, подвижная фаза 65/35% CO2/(MeOH+0,5% изопропиламин), 50 мл/мин, 100 бар), приводя к разделению двух энантиомеров. Хиральную чистоту для двух энантиомеров определяли с использованием методов хиральной SFC,Berger SFC, UV детекция при 210 нм, стационарная фаза Chiralpak AD-H (250 нм 4,6) 5 мкм, подвижная фаза б 5/35% CO2/ (изопропанол+0,5% изопропиламин), 2,4 мл/мин, 100 бар.S энантиомер (время удерживания=5,9 мин, энатиомерная чистота=96,8%) 3.3. 2-Амино-7-(3,4-дигидрокси-3-метилбут-1-инил)-1-этил-3-(1 Н-имидазол-2-ил)-1,8-нафтиридин 4(1 Н)-он 2-Амино-7-3R)-3,4-дигидрокси-3-метилбут-1-инил)-1-этил-3-(1 Н-имидазол-2-ил)-1,8-нафтиридин 4(1 Н)-он 2-Амино-7-3S)-3,4-дигидрокси-3-метилбут-1-инил)-1-этил-3-(1 Н-имидазол-2-ил)-1,8-нафтиридин 4(1 Н)-он После стадии снятия защиты согласно стадии 1.9 выделяли соединения 3, 27 и 28 в виде желтого порошка. МН+=354,16 (C18H19N5O3, Mr=353,38) Время удерживания=0,77 мин. 1(ушир.с, 1H); 7,47 (д, 1 Н, J=6,4 Гц); 7,15 (с, 1H); 7,02 (с, 1H); 5,6 (с, 1H); 5,1 (т, 1 Н, J=6,4 Гц) 4,53 (ушир.д,2H); 3,49 (дд, 1 Н, J=6,4; 10,4 Гц); 3,41 (дд, 1 Н, J=6,4; 10,4 Гц); 1,48 (с, 3H); 1,27 (т, 3 Н, J=7,2 Гц). Хиральную чистоту для двух энантиомеров определяли с использованием методов хиральной SFC,Berger SFC, UV детекция при 230 нм, стационарная фаза Chiralpak AD-H (250 нм 4,6) 5 мкм, подвижная фаза 60/40% CO2/(изопропанол+0,5% изопропиламин), 2,4 мл/мин, 100 бар.S энантиомер (время удерживания=7,29 мин, энатиомерная чистота=98,5%) Пример 4 (соединение 4). 2-Амино-1-этил-7-(3-гидрокси-3-пиридин-2-илбут-1-инил)-3-(1 Нимидазол-2-ил)-1 Н-[1,8]нафтиридин-4-он. Применяя промежуточное соединение, описанное на стадии 1.6 (2-амино-7-хлор-1-этил-3-[1-(2 триметилсиланилэтоксиметил)-1 Н-имидазол-2-ил]-1 Н-[1,8]нафтиридин-4-он), и присоединяя его к -2 придин-2-илбут-3-ин-2-олу, следуя подробной процедуре, изложенной для примера 1 аналогичным образом, получали соединение, указанное в заголовке. МН+=401,21 (C22H20N6O2, Mr=400,44) Время удерживания (аналитическая ВЭЖХ): 4,49 мин. Пример 5 (соединение 5). 2-Амино-1-этил-7-[(3R)-3-гидрокси-4-метокси-3-метилбут-1-ин-1-ил]3-(4-метил-1 Н-имидазол-2-ил)-1 Н-[1,8]нафтиридин-4-он. 5.1. 4-Метил-1-(2-триметилсиланилэтоксиметил)-1 Н-имидазол-2-карбальдегид. Применяя ту же процедуру, что и для стадии 1.3 примера 1, начиная с 4 г (36,3 ммоль) 4 (5)-метил 1 Н-имидазол-2-карбальдегида, 1,5 г (38 ммоль) гидрида натрия и 6,7 г (40 ммоль) 2(триметилсиланил)этоксиметилхлорида в 73 мл DMF, получали 8,7 г указанного в заголовке соединения в виде коричневого масла (количественный выход). МН+=241(C11H20N2O2Si, 240,377) 5.2. [4-Метил-1-(2-триметилсиланил-этоксиметил)-1 Н-имидазол-2-ил]ацетонитрил. По той же процедуре, которая описана на стадии 1.4 примера 1, начиная с 8,7 г (32,7 ммоль) 4 метил-1-(2-триметилсиланилэтоксиметил)-1 Н-имидазол-2-карбальдегида, 6,7 г (34,3 ммоль) TOSMIC и 7,3 г (65 ммоль) трет-бутилата калия в безводном растворе DME (54 мл), получали 6,6 г соединения в виде желтого масла в виде 70/30 смеси региоизомеров tau и Pi. Выход=80% МН+=252(C12H21N3OSi, 251,404) Время удерживания=6,38 и 6,55 мин.- 11023580 5.3. 2-Амино-7-хлор-1-этил-3-[4-метил-1-(2-триметилсиланилэтоксиметил)-1 Н-имидазол-2-ил]-1 Н[1,8]нафтиридин-4-он. По той же процедуре, которая описана на стадии (1.5-1.6) примера 1, начиная с 6,3 г (25 ммоль) соединения, полученного в конце стадии 6.2, 5 г (25 ммоль) соединения, полученного в конце стадии 1.2, и 7,2 г (62 ммоль) трет-бутилата калия в безводном растворе THF (83 мл), получали 4 г продукта в виде бежевого порошка в виде смеси 80/20 2 изомеров Tau и Pi, используемых в виде смеси на следующей стадии. Выход=38% Точка плавления=120C МН+=435(C20H28ClN5O2Si, 434,013) Время удерживания=10,5 и 10,6 мин. 1 Н ЯМР обоих изомеров (500 МГц, ДМСО-d6):(м.д.) 8,45 (д, J=8,07 Гц, 2H), 7,70 (ушир.с, 2H),7,56 (ушир.с, 2H), 7,43 (д, J=8,07 Гц, 1H), 7,42 (д, J=8,07 Гц, 1H), 6,99 (м, 1H), 6,84 (м, 1H), 5,18 (с, 2H),5,17 (с, 2H), 4,43 (кв, J=6,85 Гц, 4H), 3,19 (м, 2 Н) , 3,12 (м, 2 Н) , 2,25 (д, J=0,98 Гц, 3H) , 2,16 (д, J=0,98 Гц,3H), 1,24-1,29 (м, 6H), 0,57-0,64 (м, 4H), -0,22 (с, 9H), -0,22 (с, 9H). 5.4. 2-Амино-1-этил-7-[(3R)-3-гидрокси-4-метокси-3-метилбут-1-ин-1-ил]-3-(4-метил-1-(2-триметилсиланилэтоксиметил)-1 Н-имидазол-2-ил)-1 Н-[1,8]нафтиридин-4-он. В заполненной аргоном микроволновой реакционной емкости 0,5 г (1,15 ммоль) соединения, полученного в конце стадии 5.3, 0,26 г (2,3 ммоль) (R)-1-метокси-2-метилбут-3-ин-2-ола, 40 мг (0,06 ммоль) дихлорида бис(трифенилфосфин)палладия (II), 22 мг (0,12 ммоль) йодида меди (I), 3 мл DMF (дегазированного), 3 мл триэтиламина (дегазированного) нагревали при 80C в течение 3 ч. Растворители выпаривали; твердое вещество растворяли в этилацетате и промывали последовательно водным насыщенным раствором NaHCO3 и HCl (1 н.). Органическую фазу затем сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении. Остаток затем очищали с помощью флеш-хроматографии на силикагеле (DCM/THF 95/5: MeOH (1% NH4OH) от 0 до 10%), получая 0,19 г указанного в заголовке соединения. Выход=32% МН+=512 (C26H37N5O4Si, 511,695) 1H ЯМР (400 М Гц, ДМСО-d6):(м.д.) 8,43 (д, J=7,91 Гц, 2H), 7,69 (ушир.с, 2H), 7,55 (ушир.с, 2H),7,41 (д, J=7,79 Гц, 2H), 6,99 (с, 1H), 6,84 (с, 1H), 5,81 (с, 2H), 5,18 (с, 4H), 4,40-4,58 (м, 4H), 3,47 (д, J=9,54 Гц, 2H), 3,37-3,43 (м, 8H), 3,19 (т, J=8,02 Гц, 2H), 3,12 (т, J=8,08 Гц, 2H), 2,26 (с, 3H), 2,17 (с, 3H), 1,47 (с,6H), 1,26 (т, J=6,57 Гц, 6H), 0,54-0,67 (м, 4H), -0,23 (с, 18H). 5.5. 2-Амино-1-этил-7-[(3R)-3-гидрокси-4-метокси-3-метилбут-1-ин-1-ил]-3-(4-метил-1 Н-имидазол 2-ил)-1 Н-[1,8]нафтиридин-4-он. 0,18 г (0,36 ммоль) соединения, полученного в конце стадии 5.4, растворяли при 0C в 1,7 мл TFA и 1,7 мл DCM. Раствор выдерживали при 3-5C в течение ночи. Раствор нейтрализовали путем добавления с избытком водного раствора NaHCO3. Смесь затем экстрагировали три раза этилацетатом. Объединенные органические фазы сушили над Na2SO4 и растворители выпаривали при пониженном давлении. Сырой материал очищали с помощью кристаллизации в DCM, получая 62 мг (выход 46%) указанного в заголовке незащищенного соединения. МН+=382 (C20H23N5O3, 381,434) 1 Н ЯМР (ДМСО-d6, 500 МГц): (2 топоизомера на имидазоле определяются как соотношение 60/40)=12,9-12,8 (2 с, 1H), 11,6 (ушир.с, 1H), 8,52 (д, J=7,9 Гц, 1H), 8,0 (ушир.с, 1H), 7,42 (д, J=7,9 Гц, 1H), 6,846,70 (2 с, 1H), 5,8 (с, 1H), 4,56 (м, 2H), 3,46 (д, J=9,5 Гц, 1H), 3,3 (с, 3H), 3,4 (д, J=9,5 Гц, 1H), 2,28-2,2 (2 с,3H), 1,47 (с, 3H) 1,28 (т, J=6,6 Гц, 3H). Пример 6 (соединение 6). 2-Амино-1-циклопропилметил-7-(3-гидроксипент-1-инил)-3-(1 Нимидазол-2-ил)-1 Н-[1,8]-нафтиридин-4-он. 6.1. 6-Хлор-2-(циклопропилметиламино)никотиновая кислота. В герметичной колбе 3 г (42 ммоль) циклопропилметиламина добавляли к 3 г (14 ммоль) 2,6 дихлорникотиновой кислоты в растворе трет-бутанола (14 мл), колбу закупоривали и нагревали при 170C в течение 30 мин в микроволновой печи Biotage Initiator. Реакционную смесь охлаждали до комнатной температуры, разбавляли дихлорметаном (100 мл) и промывали 10%-ным водным раствором уксусной кислоты (12 мл). Органическую фазу сушили над Na2SO4, фильтровали, концентрировали и сушили в вакууме. Получали 3,4 г продукта в виде оранжевого масла. Выход является количественным. МН+=227 Время удерживания=4,54 мин. 6.2. 6-Хлор-2-(циклопропилметиламино)никотиноилфторид. Та же процедура, которая описана на стадии 1.2 примера 1, начиная с 0,334 г (1,4 ммоль) соединения, полученного в конце стадии 7.1, в растворе с 4 мл дихлорметана, 0,38 г (2,8 ммоль) фторангидрида циануровой кислоты, и 0,28 г (2,8 ммоль) триэтиламина. Продукт, полученный в виде зеленого масла,- 12023580 использовали на следующей стадии без очистки. 6.3. 2-Амино-7-хлор-1(циклопропилметил)-3-[1-(2-триметилсиланилэтоксиметил)-1 Н-имидазол-2 ил]-1 Н-[1,8]нафтиридин-4-он. Та же процедура, которая описана на стадии (1.5-1.6) примера 1, начиная с неочищенного соединения, полученного в конце стадии 6.2, 0,32 г (1,4 ммоль) соединения, полученного в конце стадии 1.3, в растворе из 5 мл безводного THF и 0,4 г (0,35 ммоль) трет-бутилата калия. Получали 0,56 г продукта в виде коричневого порошка. Выход=90% Точка плавления=70C МН+=447(C21H28ClN5O2Si). Время удерживания=6,68 мин. 1H ЯМР (400 М Гц, ДМСО-d6):(м.д.) 8,46 (д, J=8,05 Гц, 1H), 7,67 (ушир.с, 2H), 7,43 (д, J=8,05 Гц,1H), 7,35 (д, J=1,37 Гц, 1H), 7,12 (д, J=1,19 Гц, 1H), 5,27 (с, 2H), 4,38 (д, J=7,04 Гц, 2H), 3,19-3,25 (м, 2H),1,21-1,32 (м, 1H), 0,59-0,68 (м, 2H), 0,45-0,57 (м, 4H), -0,21 (с, 9H). 6.4. 2-Амино-1-(циклопропилметил)-7-(3-гидроксипент-1-инил]-3-[1-(2-триметилсиланилэтоксиметил)-1 Н-имидазол-2-ил]-1 Н-[1,8]нафтиридин-4-он. Та же процедура, которая описана на стадии 5.4 примера 5, начиная с 0,5 г (1,2 ммоль) соединения,полученного в конце стадии 6.3, 0,22 г (2,5 ммоль) пент-4-ин-3-ола, 43 мг (0,06 ммоль) дихлорида бис(трифенилфосфин)палладия (II), 23 мг (0,12 ммоль) йодида меди (I), 3 мл DMF (дегазированного), 3 мл триэтиламина (дегазированного). Получали 0,19 г указанного в заголовке соединения. Выход=30% МН+=494 (C26H35N5O3Si 493,68) 1H ЯМР (400 М Гц, ДМСО-d6):(м.д.) 8,44 (д, J=7,87 Гц, 1H), 7,65 (ушир.с, 2H), 7,42 (д, J=7,87 Гц,1H), 7,33 (с, 1H), 7,10 (с, 1H), 5,63 (д, J=5,58 Гц, 1H), 5,28 (с, 2H), 4,38-4,52 (м, 3H), 3,17-3,25 (м, 2H), 1,671,76 (м, 2H), 1,22-1,31 (м, 1H), 1,01 (т, J=7,36 Гц, 3H), 0,59-0,66 (м, 2H), 0,44-0,57 (м, 4H), -0,24-0,20 (м,9H). 6.5. 2-Амино-1-циклопропилметил-7-(3-гидроксипент-1-инил]-3-1 Н-имидазол-2-ил)-1 Н-[1,8]нафтиридин-4-он. По той же процедуре, которая описана на стадии 5.5 примера 5, начиная с 0,18 г (0,36 ммоль) соединения, полученного в конце стадии 7.4, в 1,7 мл TFA и 1,7 мл DCM, получали 19 мг указанного в заголовке соединения. Выход=15% Точка плавления=252C МН+=364 (C20H21N5O2, 363,419) 1(м, 1H), 1,05 (т, J=7,36 Гц, 3H), 0,5 (м, 2H), 0,48 (м, 2H). Пример 7 (соединение 7). 2-Амино-1-этил-7-R)-3-гидрокси-4-метокси-3-метилбут-1-инил)-3(1 Н-имидазол-2-ил)-1 Н-хинолин-4-он. 7.1. 2-Фтор-4-йодбензойная кислота. 13,39 г (84,74 ммоль) перманганата калия добавляли к суспензии из 5 мг (21,18 ммоль) 2-фтор-4 йодтолуола и 25,13 г (317,77 ммоль) пиридина в воде. Смесь нагревали и перемешивали при 70C в течение 18 ч. Пока реакция не заканчивалась, к реакционной смеси при комнатной температуре добавляли 3,34 г (21,18 ммоль) перманганата калия, и смесь перемешивали еще 6 ч при 70C. Затем реакционную смесь фильтровали через подушку из целита, которую затем промывали водой и этилацетатом. После декантирования, водную фазу подкисляли 6 н. водным раствором HCl до рН 1. Белое твердое вещество сначала фильтровали, и водную фазу экстрагировали три раза этилацетатом. Объединенные органические фазы сушили над MgSO4 и растворители выпаривали при пониженном давлении. Отфильтрованное белое твердое вещество и твердое вещество, экстрагированное этилацетатом,объединяли, получая 4,1 г. Выход=73% МН+=266, 9 (C7H4FIO2) 1H ЯМР (ДМСО-d6, 400 МГц):13,49 (ушир.с, 1H); 7,88 (д, 1H); 7,78 (д, 1H); 7,65 (д, 1H). 7.2. 2-Этиламино-4-йодбензойная кислота. В герметичной колбе 3,5 г (13,16 ммоль) 2-фтор-4-йодбензойной кислоты смешивали с 16,11 мл раствора этиламина (70% в воде). Реакционную емкость нагревали, и смесь перемешивали при 125C в течение 24 ч. Через реакционную смесь барботировали азот для удаления избытка этиламина. Затем реакционную смесь вливали в ледяной водный раствор, и смесь подкисляли до рН 3-4 уксусной кислотой. Полученное белое твердое вещество затем отфильтровывали, промывали водой и сушили, получая 2,2 гH ЯМР (ДМСО-d6, 400 МГц):12,5 (ушир.с, 1H); 7,55 (д, 1H); 7,11 (с, 1H); 6,9 (д, 1H). 7.3. 1-Этил-7-йод-1 Н-бензо[d][1,3]оксазин-2,4-дион. При комнатной температуре к 2,2 г (7,55 ммоль) 2-этиламино-4-йодбензойной кислоты в 30 мл диоксана добавляли 0,785 г (2,65 ммоль) трифосгена. Затем реакционную смесь нагревали и перемешивали при 110C в течение 2 ч. Раствор выпаривали досуха и снова дважды выпаривали после двух добавлений по 20 мл толуола, получая 2,39 г (7,5 ммоль) твердого вещества. Выход=100% МН+=317,7 (C10H8INO3) 1H ЯМР (ДМСО-d6, 400 МГц):7,87 (с, 1H); 7,68 (с, 2H); 4,04 (м, 2H); 1,19 (т, 3H). 7.4. 2-Амино-1-этил-7-йод-4-оксо-1,4-дигидрохинолин-3-карбонитрил. 0,32 г (6,31 ммоль) малононитрила и 1,45 г (14,35 ммоль) триэтиламина добавляли к 2 г (6,31 ммоль) 1-этил-7-йод-1 Н-бензо[d][1,3]оксазин-2,4-диона, растворенного в 25 мл DMF. Раствор перемешивали в течение 2 ч при 120C, и после добавления 0,73 г (7,17 ммоль) триэтиламина перемешивали еще 1 ч при 110C. DMF затем выпаривали при пониженном давлении, и остаток добавляли в смесь воды и дихлорметана. Фильтрование данной смеси давало первую фракцию ожидаемого соединения: 0,55 г (1,62 ммоль). Выход=26% МН+=339,7(C10H8INO3) 1DME. Раствор перемешивали и облучали микроволнами в течение 0,5 ч при 100C. Раствор затем отфильтровывали и выпаривали досуха. Сырой материал затем очищали с помощью колоночной хроматографии (DCM/MeOH: 9/1), получая 0,57 г (1,2 ммоль) твердого вещества. Выход=78% МН+=473(C18H25IN4O3) 7.6. 2-Амино-1-этил-3-(1 Н-имидазол-2-ил)-7-йод-1 Н-хинолин-4-он. 0,37 мл 12 н. раствора HCl добавляли при 0C к суспензии из 0,13 г (0,28 ммоль) 2-амино-N-(2,2 диэтоксиэтил)-1-этил-7-йод-4-оксо-1,4-дигидрохинолин-3-карбоксамидина. Реакционную смесь перемешивали при комнатной температуре в течение 16 ч. Затем реакционную смесь разбавляли 0,55 мл воды,подщелачивали с помощью 0,32 мл 1 н. раствора NaOH и 0,134 мл раствора NH4OH. Смесь затем фильтровали, и полученное твердое вещество промывали водой, ацетонитрилом и пентаном, получая 0,08 г(0,21 ммоль) коричневого твердого вещества. Выход=76% МН+=381(C14H13IN4O) 7.7. 2-Амино-1-этил-7-R)-3-гидрокси-4-метокси-3-метилбут-1-инил)-3-(1 Н-имидазол-2-ил)-1 Нхинолин-4-он. 0,43 мг (1,15 ммоль) 2-амино-1-этил-3-(1 Н-имидазол-2-ил)-7-йод-1 Н-хинолин-4-она, 0,262 (23 ммоль) (R)-3-гидрокси-4-метокси-3-метилбут-1-ина и 0,397 г (3,45 ммоль) перемешивали в 15 мл DMF. Через данный раствор в течение 10 мин продували аргон. После добавления 0,126 мг (0,17 ммоль) дихлорида 1,1'-бис(дифенилфосфино)ферроценпалладия и 0,033 мг (0,17 ммоль) йодида меди реакционную смесь перемешивали при 80C в течение 6 ч. Реакционную смесь затем выпаривали досуха, и остаток вливали в смесь DCM и воды. Черное нерастворимое твердое вещество отфильтровывали (100 мг). Водную фазу три раза экстрагировали дихлорметаном. Объединенные органические фазы сушили надMgSO4, и растворители выпаривали при пониженном давлении. Остаток затем очищали с помощью колоночной хроматографии (DCM/MeOH/NH4OH водн.: 9/1/0,1), получая 0,80 г (1,2 ммоль) желтого твердого вещества. Данное твердое вещество перекристаллизовывали в дихлорметане, получая 0,02 г бежевого твердого вещества. Выход=4,5% МН+=367(C20H22N4O3, 366,419) 1- 14023580 гидроксифенилбороновой кислоты, 0,098 г (0,11 ммоль) трис(дибензилиденацетон)дипалладия (0), 0,030 мг (0,011 ммоль) трициклогексилфосфина, 0,303 г трехосновного фосфата калия и 8 мл смеси диоксан/вода (50/50) (дегазированной) перемешивали и нагревали при 85C в течение 8 ч. Растворители выпаривали, и остаток очищали с помощью колоночной хроматографии (DCM/MeOH: 9/1), получая 0,4 г коричневого твердого вещества. Данное твердое вещество использовали на следующей стадии без дальнейшей очистки. 8.2. 2-Амино-7-(3-хлор-4-гидроксифенил)-1-этил-3-(1 Н-имидазол-2-ил)-1 Н-[1,8]нафтиридин-4-он. 400 мг (0,59 ммоль) неочищенного 2-амино-7-(3-хлор-4-гидроксифенил)-1-этил-3-[1-(2 триметилсиланилэтоксиметил)-1 Н-имидазол-2-ил]-1 Н-[1,8]нафтиридин-4-она растворяли в 20 мл DCM. При 0C добавляли 3,39 г TFA, и смесь перемешивали при комнатной температуре в течение 24 ч. Раствор нейтрализовали путем добавления избытка водного раствора NaHCO3. Смесь затем три раза экстрагировали DCM. Объединенные органические фазы сушили над MgSO4, и растворители выпаривали при пониженном давлении. Полученный таким образом неочищенный материал очищали на силикагеле(DCM/MeOH/NH4OH=4/10,1), получая 0,035 г указанного в заголовке незащищенного соединения. Выход за две стадии=13% МН+=382(C19H16ClN5O2, 381,821). 1(с, 1H); 7,09 (д, 1H); 7,3 (с, 1H); 4,72-4,66 (м, 2H); 1,37 (т, 3H). Пример 9 (соединение 9). 2-Амино-1-этил-7-[3-(2-фторфенил)-3-гидроксибут-1-инил]-3-(1 Нимидазол-2-ил)-1 Н-[1,8]-нафтиридин-4-он. 9.1. 2-Амино-1-этил-7-хлор-3-(1 Н-имидазол-2-ил)-1 Н-[1,8]нафтиридин-4-он. 2-Амино-7-хлор-1-этил-3-[1-(2-триметилсиланилэтоксиметил)-1 Н-имидазол-2-ил]-1 Н-[1,8]нафтиридин-4-он (1,0 г, 2,38 ммоль) растворяли в 30 мл дихлорметана. Добавляли 30 мл трифторуксусной кислоты, и реакционную смесь перемешивали при комнатной температуре в течение 3 ч до тех пор, пока ВЭЖХ не показывала полное израсходование исходного материала. Растворители удаляли при пониженном давлении, и к остатку добавляли этилацетат. Раствор промывали насыщенным водным раствором бикарбоната натрия. Образованный осадок собирали с помощью фильтрования, затем подвергали сушке в течение ночи при 40C в вакууме, получая 574 мг бежевого порошка. Выход=83% 1H ЯМР (ДМСО-d6, 600 МГц):(м.д.) 13,09 (с, 1H); 8,57 (д, 1H); 7,47 (д, 1H); 7,15 (с, 1H); 7,03 (с,1H); 4,51 (ушир.кв, 2H); 1,29 (т, 3H). 9.2 2-Амино-1-этил-7-[3-(2-фторфенил)-3-гидроксибут-1-инил]-3-(1 Н-имидазол-2-ил)-1 Н-[1,8]нафтиридин-4-он. Йодид меди (I) (2,37 мг 0,12 ммоль), N-этилморфолин (130 мкл, 1,04 ммоль) и 2-(2-фторфенил)бут 3-ин-2-ол (0,76 мкл, 0,52 ммоль) добавляли к 2,5 мл DMF. Смесь дегазировали аргоном. Добавляли комплекс хлорида [1'1-бис(дифенилфосфино)ферроцен]палладия (II) с дихлорметаном (5,6 мг, 0,01 ммоль) 1:1 и промежуточное соединение 20.1 (100 мг, 0,35 ммоль). Смесь перемешивали при 80C в атмосфере аргона в течение 2 ч до тех пор, пока не подтверждалось данными LCMS, что не осталось никакого исходного материала. Добавляли этилацетат. Органический слой последовательно промывали водой, 1 н. водным раствором гидроксида натрия, насыщенным водным раствором хлорида натрия, затем сушили над сульфатом магния. Растворитель выпаривали при пониженном давлении. Остаток затем очищали с помощью колоночной хроматографии (DCM/MeOH/NH4OH водный: 100/0/095/5/0,5), получая 30 мг не совсем белого порошка. Выход=21% МН+=418 Время удерживания=0,57 мин (C23H20FN5O2, 417,442). 1H ЯМР (ДМСО-d6, 600 МГц):13,10 (ушир.с, 1H); 8,54 (д, 1H); 7,72 (дт, 1H); 7,44 (д, 1H); 7,38 (м,1H); 7,25-7,19 (м, 2H); 7,13 (д, 1H); 7,02 (д, 1H); 6, 62 (с, 1H); 4,54 (ушир.кв, 2H); 1,84 (с, 3H); 1,27 (т, 3H). Пример 10 (соединение 12). 2-Амино-7-(3-гидроксипент-1-инил)-3-(1 Н-имидазол-2-ил)-1-(2 метоксиэтил)-1 Н-[1,8]нафтиридин-4-он. По аналогичной процедуре, которая описана на стадии 1 и 2 примера 6, начиная стадию 1 с 2 метоксиэтиламина вместо циклопропилметиламина и затем следуя той же процедуре, которая описана на стадии 2 примера 20, начиная с пент-1-ин-3-ола вместо 2-(2-фторфенил)бут-3-ин-2-ола, получали 15 мг продукта в виде порошка. МН+=368 Время удерживания=0,46 мин (C19H21N5O3 367,407). Пример 11 (соединение 19). 2-Амино-1-этил-3-(1 Н-имидазол-2-ил)-7-(4,4,4-трифтор-3-гидрокси 3-фенилбут-1-инил)-1 Н-[1,8]нафтиридин-4-он. 11.1. 2-Амино-1-этил-7-(4,4,4-трифтор-3-гидрокси-3-фенилбут-1-инил)-3-[1-(2-триметилсиланилэтоксиметил)-1 Н-имидазол-2-ил]-1 Н-[1,8]нафтиридин-4-он. По той же процедуре, которая описана на стадии 5.4 примера 5, начиная с 0,4 г (0,95 ммоль) 2- 15023580 амино-7-хлор-1-этил-3-[1-(2-триметилсиланилэтоксиметил)-1 Н-имидазол-2-ил]-1 Н-[1,8]нафтиридин-4 она, 0,4 г (1,9 ммоль) 1,1,1-трифтор-2-фенилбут-3-ин-2-ола, 33 мг (0,05 ммоль) дихлорида бис(трифенилфосфино)палладия (II), 18 мг (0,1 ммоль) йодида меди (I), 3 мл DMF (дегазированного), 3 мл триэтиламина (дегазированного) получали 0,15 г указанного в заголовке соединения. Выход=30% МН+=584 (C29H32N5O3Si) 1(с, 2H), 4,44-4,57 (м, 2H), 3,16-3,27 (м, 2H), 1,28 (т, J=6,91 Гц, 3H), 0,57-0,69 (м, 2H), -0,22 (с, 9H). 11.2. 2-Амино-1-этил-3-(1 Н-имидазол-2-ил)-7-(4,4,4-трифтор-3-гидрокси-3-фенилбут-1-инил)-1 Н[1,8]-нафтиридин-4-он. По той же процедуре, которая описана на стадии 5.5 примера 5, начиная с 0,145 г (0,25 ммоль) соединения, полученного в конце стадии 21.1, в 1,2 мл TFA и 1,2 мл DCM получали 97 мг указанного в заголовке соединения. Выход=86% Точка плавления=260C МН+=454(C23H18F3N5O2) Время удерживания=7,29 мин. 1(ушир.с, 2H), 4,57 (кв, J=6,59 Гц, 2H), 1,30 (т, J=7,00 Гц, 3H). Соединения 10 и 11 получали по процедуре, аналогичной той, которая описана в примере 6. Соединения 13, 14, 15, 16, 17, 18, 20, 21, 25 получали по процедуре, аналогичной той, которая описана в примере 10. Соединения 22, 23 и 24 получали по процедуре, аналогичной той, которая описана в примере 11. Соединение 29 получали по процедуре, аналогичной той, которая описана в примере 1. Оборудование, используемое для примеров 5 и 6. Микроволновое устройство: Biotage, инициатор Аналитический метод LC/UV/MS, определение времени удерживания (Rt). Аналитический метод LC/UV/MS, используемый для анализа соединений (Rt) 1, 2, 3 и 4: Колонка: Merck Chromolith, характеристика RP18e, 1004,6 мм, 3,5 мкм Растворитель A: H2O/TFA (99,9/0,1) Растворитель В: ACN/TFA (99,9/0,1) Скорость потока: 2 мл/мин Градиент (А/В): 98/2 (0 мин) до 0/100 (8 мин) до 98/2 (10 мин) Определение: 254,16 нМ Аналитический метод LC/UV/MS, используемый для анализа соединений (Rt) 9, 12, 13, 14, 11, 18 и 20:UPLC SQD Электрораспылительная ионизация, положительный режим (30 В) Колонка: Ascentis Express 502,1 мм, 2,7 мкм, Т=55C Растворитель А: Н 2 О+0,02% TFA Растворитель В: CH3CN+0,014% TFA Скорость потока: 1 мл/мин Градиент (А/В об./об.): 98/2 (t=0 мин), 2/98 (t=1 мин), 2/98 (t=1,3 мин), 98/2 (t=1,33 мин), следующее впрыскивание (t=1,5 мин) Определение: 220 нМ Аналитический метод, используемый для анализа соединений 5, 6, 10, 11, 19, 22, 23 и 24: ВЭЖХ цепь: Series 1100, масс-спектрометр MCD SL (Agilent) Программное обеспечение: Chemstation версия В.01.03 от Agilent Вид ионизации: электрораспыление, положительный режим ESI+ Пределы массы: 90-150 0 мкмайт (1 майт=3,24 мг) Колонка: Symmetry C18 3,5 мкм (2,150 мм) (Waters) T=25C, рН 3 Элюенты: А: Н 2 О+0,005% TFA/B: CH3CN+0,005% TFA Поток: 0,4 мл/мин Градиент: 0-10 мин 0-100% В и от 10 до 15 мин 100% В Определение: 220 нМ Аналитический метод LC/UV/MS, используемый для соединений (Rt) 7, 8, 15, 16, 21, 25, 26:- 16023580 Растворитель В: CH3CN+0,035% TFA Скорость потока: 1 мл/мин Градиент (А/В об./об.): 98/2 (t=0 мин), 0/100 (t=1,6 мин), 0/100 (t=2,1 мин), 98/2 (t=2,5 мин), следующее впрыскивание (t=3 мин) Определение: при 220 нМ ЯМР. Данные спектра 1 Н ЯМР получали с использованием ЯМР спектрометра Bruker 250, 300, 400 или 600 МГц в ДМСО-d6 с применением пика ДМСО-d6 в качестве внутреннего стандарта. Химические сдвигивыражены в миллионных долях (м.д.). Наблюдаемые сигналы выражены так, как следует далее: с - синглет; д - дублет; т - триплет; кв квадруплет; м - мультиплет или большой синглет; ушир. - уширенный; Н - протон. Точки плавления. Точки плавления ниже 260C измеряли с помощью прибора Кофлера, а точки плавления выше 260C измеряли с помощью аппарата Buchi B-545. Удельное вращение. Удельное вращение измеряли на поляриметре типа: Polarimeter Perkin-Elmer, мощность 55 мкА. Таблица 1 Соединения согласно изобретению подвергали фармакологическим анализам для определения их ингибирующего действия на аутофосфорилирование VEGFR-3, а также для оценки их ex vivo активно- 17023580 сти, описанных ниже. Действие соединений на аутофосфорилирование VEGFR-3 в НЕК клетках. Действие соединений по блокированию аутофосфорилирования VEGFR-3 определяли с помощьюELISA после сверхэкспрессии VEGFR-3 в клетках НЕК. НЕК 293 Т клетки содержали в MEM, дополненной 10%-ной фетальной телячьей сыворотки (FCS) и глютамина. За день до трансфекции на 48-луночные планшеты высевали 104 клетки/лунку и осуществляли трансфекцию с использованием Fugene-6 (Roche,Базель). Fugene-6 (18 мкл) предварительно инкубировали в течение 5 мин в 282 мкл Optimem. Затем добавляли 3 мг ДНК, соответствующей VFGR-3-Flag, и оставляли при комнатной температуре в течение 10 мин перед распределением 200 мкл на НЕК клетки. Спустя 24 ч среду удаляли и заменяли новой средой без сыворотки и инкубировали в течение 1 ч с различными концентрациями (в пределах от 3 до 1000 нМ) каждого соединения. После дополнительных 30 мин инкубирования с ортованадатом (0,4 мМ) клетки промывали холодным PBS, дополненным ортованадатом, а затем лизировали с 300 мкл буфера RIPA. Лизаты затем центрифугировали в течение 10 мин при 10000 g. Супернатанты (75 мкл) распределяли с двукратным повторением на 96-луночный планшет, предварительно покрытый анти-Flag, и оставляли в течение 1 ч при комнатной температуре. После 3 промываний TBS буфером, содержащим 0,5% Твин 20,добавляли антифосфотирозин, конъюгированный с HRP, и инкубировали в течение 1 ч при комнатной температуре. Лунки затем промывали 3 раза TBS буфером, содержащим Твин 20 (0,5%) и MgCl2 (2 мМ). Реакцию прекращали с помощью 50 мкл H2SO4 (2 н.) и считывали сигнал Envision при 485 и 530. Анализировали кривые концентрации-ответной реакции с помощью внутреннего программного обеспечения Biostt-SPEED v2.0 с использованием 4-параметровой логистической модели согласно Ratkovsky и Reedy (Ratkovsky D.A., Reedy T.J. Choosing near-linear parameters in the four parameters logisticmodel radioligands and related assays. Biometrics 1986 Sep 42(3):575-82.) Соединения согласно изобретению обладают ингибирующей активностью в отношении аутофосфорилирования VEGFR-3 и показывают величины IC50 менее чем 1 мкМ при аутофосфорилировании в НЕК клетках, особенно от 1 до 500 нМ, более конкретно от 1 до 100 нМ. В качестве примера величины IC50 некоторых соединений табл. 1 приводятся в табл. 2. Таблица 2 Ингибиторы VEGFR-3 тирозинкиназы согласно изобретению показывают хорошую ex vivo активность с использованием анализа по измерению ингибирования аутофосфорилирования VEGFR-3 даже лучше, чем активность ингибиторов предшествующего уровня техники.Ex vivo анализ. Протокол введения продуктов мышам. Продукты получают в ступке с 0,5% Твин 80 и 0,6% метилцеллюлозы с равновесным набуханием в конечном объеме. Суспензии вводят с помощью желудочного зонда (10 мл/кг) самцам мышей Balb/c в возрасте 8-15 недель. Через 3 или 6 ч после одноразовых оральных введений 30 мг/кг животных анестезировали пентобарбиталом, брали пробы крови (400 мкл) из полой вены и переносили в стеклянные пробирки, содержащие литийгепарин. После центрифугирования (1550-2000 g в течение 10 мин) образцы плазмы замораживали при температуре, близкой к -20C до проведения анализа. Для того чтобы определить ex vivo активность продуктов в плазме, использовали анализ аутофосфорилирования в НЕК клетках, описанный ранее. Для этой цели трансфицированные клетки инкубировали с плазмой (10%) вместо соединений. Результаты выражают как процент ингибирования аутофосфорилирования VEGFR-3 в сравнении с необработанными клетками (максимум аутофосфорилирования) и с нетрансфицированными клетками (основа). Ex vivo активность соединений изобретения приведена в следующей табл. 3. Результаты выражены в виде процентного ингибирования VEGFR-3 аутофосфорилирования НЕК клеток в присутствии образца плазмы, взятого (за время) после введения через рот (р.о.) соединений. Для того чтобы оценить увеличение данной активности в отношении соединений по изобретению, сравнение с соединениями предшествующего уровня техники (WO 2009/007535) и подвергнутыми тому же измерению, приводят в табл. 3. Таблица 3 Сравнение ex vivo активности между соединениями данного изобретения и соответствующего соединения известного уровня техники после введения р.о. самцам мышей Balb/c Следует, очевидно, что соединения согласно изобретению имеют ингибирующую активность в отношении аутофосфорилирования VEGFR-3, следовательно, они могут быть использованы в получении лекарственных средств, в частности лекарственных средств, которые ингибируют VEGFR-3. Увеличение проявления соединением, в частности, биодоступности является одним из критериев увеличения ингибирования ex vivo аутофосфорилирования соединения по изобретению. Ингибиторы VEGFR-3 тирозинкиназы согласно изобретению показывают хорошую биодоступность, даже лучшую, чем биодоступность ингибиторов предшествующего уровня техники. Биодоступность относится к степени и скорости, при которых активный фрагмент (лекарственное средство или метаболит), входящий в большой круг кровообращения, достигает тем самым участка действия. Биодоступность лекарственного средства в значительной степени определяется свойствами дозированной формы (которые зависят частично от ее дизайна и производства) скорее, чем физикохимическими свойствами лекарственного средства, которые определяют абсорбционный потенциал. Различия в биодоступности среди рецептурных форм заданного лекарственного средства могут иметь клиническое значение; зная, таким образом, является ли эквивалент рецептурных форм лекарственного средства существенным.- 19023580 Биодоступность используется для описания фракции вводимой дозы неизменного лекарственного средства, которая достигает большого круга кровообращения и которая является одним из главных фармакокинетических свойств лекарственных средств. По определению, когда лекарственное средство вводится внутривенно, его биодоступность составляет 100%. Однако, когда лекарственное средство вводится иными путями (такими как пероральный), его биодоступность снижается (вследствие неполного поглощения и пресистемного метаболизма) или может варьировать от пациента к пациенту (вследствие взаимоиндивидуального варьирования). Биодоступность является одним из существенных инструментов в фармакокинетике, так как биодоступность должна учитываться при расчете дозировок для невнутривенных способов введения. Таким образом, согласно одному из аспектов объектом изобретения являются лекарственные средства, которые включают соединение формулы (I) или его аддитивную соль с фармакологически приемлемой кислотой и основанием, а также энантиомером или диастереоизомером, включая их смесь, соединения формулы (I). Еще один аспект изобретения включает комбинацию по меньшей мере одного соединения согласно изобретению и по меньшей мере одного терапевтического агента. В частности, соединения настоящего изобретения могут быть использованы отдельно или в смеси по меньшей мере с одним терапевтическим агентом, который может быть выбран из: алкилирующих агентов,интеркалирующих агентов,антимикротубулиновых агентов,антимитотических агентов,антиметаболитов,антипролиферативных агентов,антибиотиков,иммуномодулирующих веществ,противовоспалительных агентов,ингибиторов киназы,антиангиогенных агентов,антиваскулярных агентов,эстрогенных и андрогенных гормонов. Можно также комбинировать соединения согласно изобретению с лучевой терапией. Комбинации соединений по изобретению с терапевтическими агентами, приведенными выше, и/или с радиационным облучением составляют еще один объект настоящего изобретения. Терапевтические агенты, упомянутые выше, и/или радиационное облучение могут применяться одновременно, раздельно или последовательно. Лечение обычно регулируется практикующим врачом в соответствии с подвергаемым лечению пациентом. Данные лекарственные средства используются в терапии, в частности, при лечении и/или предотвращении: рака и его метастазов, таких как глиабластома, множественная миелома, миелодиспластический синдром, саркома Капоши, кожная ангиосаркома, солидные опухоли, лимфома, меланома, рак груди,колоректальный рак, рак легкого, включая немелкоклеточный рак, рак поджелудочной железы, рак простаты, рак почек, рак головы и шеи, рак печени, рак яичников, рак дыхательного тракта и грудной клетки, другие опухоли, экспрессирующие VEGFR-3 или вовлекающие процесс ангиогенеза или лимфоангиогенеза,неонкологических пролиферативных заболеваний и патологического ангиогенеза, связанного сVEGFR-3, таких как артроз, рестеноз, псориаз, гемангиома, лимфангиома, глаукома, гломерунефрит,диабетическая нефропатия, нефросклероз,тромботический микроангиопатический синдром, цирроз печени, атеросклероз, отторжение трансплантируемого органа, заболевания глаз, включая процесс ангиогенеза или лимфоангиогенеза, такие как диабетическая ретинопатия или дегенерация желтого пятна,или еще при лечении и профилактике воспаления (хронического или нехронического), инфицирования микроорганизмами и аутоиммунных заболеваний, таких как ревматоидный артрит,или еще в лечении редких заболеваний, таких как лимфангиолейомиоматоз. Согласно еще одному из аспектов изобретение относится к фармацевтическим композициям, содержащим в качестве активного ингредиента соединение согласно изобретению. Данные фармацевтические композиции содержат эффективную дозу по меньшей мере одного соединения согласно изобретению или его фармацевтически приемлемой соли, гидрата или сольвата, а также по меньшей мере одного фармацевтически приемлемого эксципиента. Указанные эксципиенты выбирают в соответствии с фармацевтической формой и способом желаемого введения из обычных эксципиентов, которые известны специалистам в данной области. В фармацевтических композициях по изобретению для перорального, подъязычного, подкожного,внутримышечного, внутривенного, топического или местного, внутритрахейного, внутриназального,- 20023580 трансдермального или ректального введения активный ингредиент формулы (I), указанной выше, или его возможная соль, сольват или гидрат могут быть введены в единичной форме в виде смеси с общепринятыми фармацевтическими эксципиентами животным или человеку для лечения или профилактики указанных выше расстройств или заболеваний. Соответствующие единичные формы для введения включают формы для перорального введения,такие как таблетки, мягкие или твердые гелевые капсулы, порошки, гранулы и пероральные растворы или суспензии, формы для подъязычного, защечного, внутритрахейного, внутриглазного и внутриназального введения, формы для введения с помощью ингаляции, формы для введения топически, трансдермально, подкожно, внутримышечно или внутривенно, формы для введения ректально и имплантанты. Для топического введения соединения согласно изобретению могут использоваться в виде кремов, гелей,мазей или лосьонов. Например, единичная форма для введения соединения согласно изобретению в виде таблетки может включать следующие компоненты: Согласно другим аспектам настоящее изобретение относится также к способу лечения патологических состояний, указанных выше, включающему введение пациенту эффективной дозы соединения согласно изобретению или его фармацевтически приемлемой соли. ФОРМУЛА ИЗОБРЕТЕНИЯ 1. Соединение, соответствующее формуле (I) в которой W представляет собой атом азота или группу CH;Y представляет собой С 2-С 3-алкинилен, 1,4-фенилен, необязательно замещенный R7, где R7 представляет собой один или более атом(ов) галогена;Z представляет собой связь или группу CR1R2;R1 и R2 независимо друг от друга представляют собой группу, выбранную из атома водорода, С 1-С 6 алкильной группы, трифторметильной группы, группы (CH2)nOR6, С 3-С 7-циклоалкила, гетероарила или арила, где гетероарил или арил необязательно замещен одним или более атомом(ами) галогена; арил представляет собой моноциклическую или бициклическую ароматическую группу, содержащую от 6 до 10 атомов углерода, и гетероарил представляет собой 5-12-членную моноциклическую или бициклическую ароматическую группу, включающую от 1 до 5 гетероатомов, выбранных из О, S или N; илиR3 представляет собой атом водорода;R5 представляет собой группу, выбранную из атома водорода или C1-C6-алкильной группы;R6 представляет собой группу, выбранную из атома водорода или C1-C6-алкильной группы;n равен 1, 2 или 3; в форме основания, или аддитивной соли с фармацевтически приемлемыми кислотами, или энантиомера, или диастереоизомера, или их смеси. 2. Соединение формулы (I) по п.1, отличающееся тем, что W представляет собой атом азота или группу CH в форме основания, или аддитивной соли с фармацевтически приемлемыми кислотами, или энантиомера, или диастереоизомера, или их смеси. 3. Соединение формулы (I) по п.1 или 2, отличающееся тем, что W представляет атом азота в форме основания, или аддитивной соли с фармацевтически приемлемыми кислотами, или энантиомера, или диастереоизомера, или их смеси. 4. Соединение формулы (I) по любому из пп.1-3, отличающееся тем, что Y представляет собой С 2 С 3-алкинилен в форме основания, или аддитивной соли с фармацевтически приемлемыми кислотами,или энантиомера, или диастереоизомера, или их смеси. 5. Соединение формулы (I) по п.4, отличающееся тем, что Y представляет собой этинилен в форме основания, или аддитивной соли с фармацевтически приемлемыми кислотами, или энантиомера, или диастереоизомера, или их смеси.- 21023580 6. Соединение формулы (I) по любому из пп.1-4, отличающееся тем, чтоR1 представляет собой группу, выбранную из атома водорода, С 1-С 6-алкильной группы, группыR2 представляет собой группу, выбранную из атома водорода, C1-C6-алкильной группы или трифторметила;R6 представляет собой группу, выбранную из атома водорода или C1-С 6-алкильной группы;n равен 1, 2 или 3; в форме основания, или аддитивной соли с фармацевтически приемлемыми кислотами, или энантиомера, или диастереоизомера, или их смеси. 7. Соединение формулы (I) по любому из пп.1-6, отличающееся тем, чтоZ представляет собой группу CR1R2;R1 представляет собой группу, выбранную из атома водорода, C1-C6-алкильной группы, группыR2 представляет собой группу, выбранную из атома водорода, C1-C6-алкильной группы или трифторметила;R6 представляет собой группу, выбранную из атома водорода или C1-C6-алкильной группы; иn равен 1, 2 или 3; в форме основания, или аддитивной соли с фармацевтически приемлемыми кислотами, или энантиомера, или диастереоизомера, или их смеси. 8. Соединение формулы (I) по любому из пп.1-7, отличающееся тем, что R4 представляет собой группу, выбранную из С 1-С 6-алкильной группы, группы (CH2)nOR6, С 3-С 7-циклоалкила или С 1-С 6-алкила,необязательно замещенного С 3-С 7-циклоалкилом, в форме основания, или аддитивной соли с фармацевтически приемлемыми кислотами, или энантиомера, или диастереоизомера, или их смеси. 9. Соединение формулы (I) по любому из пп.1-7, отличающееся тем, что R4 представляет собой C1C6-алкил в форме основания, или аддитивной соли с фармацевтически приемлемыми кислотами, или энантиомера, или диастереоизомера, или их смеси. 10. Соединение формулы (I) по п.9, отличающееся тем, что R4 представляет собой этил в форме основания, или аддитивной соли с фармацевтически приемлемыми кислотами, или энантиомера, или диастереоизомера, или их смеси. 11. Соединение формулы (I) по любому из пп.1-10, отличающееся тем, что R5 представляет собой группу, выбранную из атома водорода или C1-C6-алкильной группы, в форме основания, или аддитивной соли с фармацевтически приемлемыми кислотами, или энантиомера, или диастереоизомера, или их смеси. 12. Соединение формулы (I) по любому из пп.1-11, отличающееся тем, что R5 представляет собой атом водорода в форме основания, или аддитивной соли с фармацевтически приемлемыми кислотами,или энантиомера, или диастереоизомера, или их смеси. 13. Соединение формулы (I) по п.1, отличающееся тем, чтоW представляет собой атом азота или группу CH;Y представляет собой С 2-С 3-алкинилен, 1,4-фенилен, необязательно замещенный R7, который представляет собой атом галогена;Z представляет собой связь или группу CR1R2;R1 представляет собой группу, выбранную из атома водорода, C1-C6-алкильной группы, группы(CH2)nOR6, С 3-С 7-циклоалкила, арила или 5- или 6-членного гетероарила, причем арил или гетероарил необязательно замещен атомом галогена;R2 представляет собой группу, выбранную из атома водорода, C1-C6-алкильной группы или трифторметила;R3 представляет собой атом водорода;R5 представляет собой группу, выбранную из атома водорода или C1-C6-алкильной группы;R6 представляет собой группу, выбранную из атома водорода или C1-C6-алкильной группы;n равен 1, 2 или 3; в форме основания, или аддитивной соли с фармацевтически приемлемыми кислотами, или энантиомера, или диастереоизомера, или их смеси. 14. Соединение формулы (I) по любому из пп.1-13, отличающееся тем, что R1 и R2 вместе с атомом углерода, к которому они присоединены, образуют С 3-С 7-циклоалкил в форме основания, или аддитивной соли с фармацевтически приемлемыми кислотами, или энантиомера, или диастереоизомера, или их смеси. 15. Соединение формулы (I) по п.1, выбранное из 2-амино-1-этил-7-3R)-3-гидрокси-4-метокси-3-метилбут-1-инил)-3-(1 Н-имидазол-2-ил)-1 Н[1,8]нафтиридин-4-она;- 22023580 2-амино-1-пропил-7-3R)-3-гидрокси-4-метокси-3-метилбут-1-инил)-3-(1 Н-имидазол-2-ил)-1 Н[1,8]нафтиридин-4-она; 2-амино-7-(3,4-дигидрокси-3-метилбут-1-инил)-1-этил-3-(1 Н-имидазол-2-ил)-1 Н-[1,8]нафтиридин 4-она; 2-амино-1-этил-7-(3-гидрокси-3-пиридин-2-илбут-1-инил)-3-(1 Н-имидазол-2-ил)-1 Н-[1,8]нафтиридин-4-она; 2-амино-1-этил-7-[(3R)-3-гидрокси-4-метокси-3-метилбут-1-инил]-3-(4-метил-1 Н-имидазол-2-ил)1 Н-[1,8]нафтиридин-4-она; 2-амино-1-(циклопропилметил)-7-(3-гидроксипент-1-инил)-3-(1 Н-имидазол-2-ил)-1 Н-[1,8]нафтиридин-4-она; 2-амино-1-этил-7-[(3R)-3-гидрокси-4-метокси-3-метилбут-1-инил]-3-(1 Н-имидазол-2-ил)-1 Н-хинолин-4-она; 2-амино-7-(3-хлор-4-гидроксифенил)-1-этил-3-(1 Н-имидазол-2-ил)-1 Н-[1,8]нафтиридин-4-она; 2-амино-1-этил-7-[3-(2-фторфенил)-3-гидроксибут-1-инил]-3-(1 Н-имидазол-2-ил)-1 Н-[1,8]нафтиридин-4-она; 2-амино-1-циклопентил-7-(3-гидроксипент-1-инил)-3-(1 Н-имидазол-2-ил)-1 Н-[1,8]нафтиридин-4 она; 2-амино-7-(3-гидроксипент-1-инил)-3-(1 Н-имидазол-2-ил)-1-(3-метоксипропил)-1 Н-[1,8]нафтиридин-4-она; 2-амино-7-(3-гидроксипент-1-инил)-3-(1 Н-имидазол-2-ил)-1-(2-метоксиэтил)-1 Н-[1,8]нафтиридин 4-она; 2-амино-1-этил-7-[(1-гидроксициклобутил)этинил]-3-(1 Н-имидазол-2-ил)-1 Н-[1,8]нафтиридин-4 она; 2-амино-1-этил-7-[(1-гидроксициклопентил)этинил]-3-(1 Н-имидазол-2-ил)-1 Н-[1,8]нафтиридин-4 она; 2-амино-1-этил-7-(3-гидрокси-3-метилбут-1-инил)-3-(1 Н-имидазол-2-ил)-1 Н-[1,8]нафтиридин-4 она; 2-амино-1-этил-7-(3-гидрокси-3-метилпент-1-инил)-3-(1 Н-имидазол-2-ил)-1 Н-[1,8]нафтиридин-4 она; 2-амино-1-этил-7-(3-гидрокси-3-фенилбут-1-инил)-3-(1 Н-имидазол-2-ил)-1 Н-[1,8]нафтиридин-4 она; 2-амино-1-этил-7-[3-(3-фторфенил)-3-гидроксибут-1-инил]-3-(1 Н-имидазол-2-ил)-1 Н-[1,8]нафтиридин-4-она; 2-амино-1-этил-3-(1 Н-имидазол-2-ил)-7-(4,4,4-трифтор-3-гидрокси-3-фенилбут-1-инил)-1 Н-[1,8]нафтиридин-4-она; 2-амино-7-(3-циклопропил-3-гидроксибут-1-инил)-1-этил-3-(1 Н-имидазол-2-ил)-1 Н-[1,8]нафтиридин-4-она; 2-амино-1-этил-7-[3-гидрокси-3-(тиофен-2-ил)бут-1-инил]-3-(1 Н-имидазол-2-ил)-1 Н-[1,8]нафтиридин-4-она; 2-амино-1-этил-7-(3-гидроксибут-1-инил)-3-(1 Н-имидазол-2-ил)-1 Н-[1,8]нафтиридин-4-она; 2-амино-1-этил-7-(3-гидроксипент-1-инил)-3-(1 Н-имидазол-2-ил)-1 Н-[1,8]нафтиридин-4-она; 2-амино-1-этил-7-(3-гидроксигекс-1-инил)-3-(1 Н-имидазол-2-ил)-1 Н-[1,8]нафтиридин-4-она; 2-амино-1-этил-7-(3-гидрокси-4-метилпент-1-инил)-3-(1 Н-имидазол-2-ил)-1 Н-[1,8]нафтиридин-4 она; 2-амино-1-этил-7-(3-гидрокси-3-фенилпрол-1-инил)-3-(1 Н-имидазол-2-ил)-1 Н-[1,8]нафтиридин-4 она; 2-амино-7-3R)-3,4-дигидрокси-3-метилбут-1-инил)-1-этил-3-(1 Н-имидазол-2-ил)-1,8-нафтиридин 4(1 Н)-она; 2-амино-7-3S)-3,4-дигидрокси-3-метилбут-1-инил)-1-этил-3-(1 Н-имидазол-2-ил)-1,8-нафтиридин 4(1 Н)-она; 2-амино-1-этил-7-3S)-3-гидрокси-4-метокси-3-метилбут-1-инил)-3-(1 Н-имидазол-2-ил)-1,8-нафтиридин-4(1 Н)-она,или их фармацевтически приемлемых солей, или энантиомеров, или диастереоизомеров, или их смеси. 16. Способ получения соединения формулы (I), где радикал W представляет собой атом азота, по любому из пп.1-13, или его фармацевтически приемлемой соли, или энантиомера, или диастереоизомера,или их смеси, характеризующийся тем, что соединение формулы (VII)- 23023580 в которой X представляет собой хлор или бром, PG представляет собой защитную группу, a R4 и R5 имеют значения, как определено в общей формуле (I), по любому из пп.1-13, подвергают реакции с соединением общей формулы (XVa) в которой R1, R2 и R3 имеют значения, как определено в общей формуле (I), по любому из пп.1-13,или подвергают реакции с соединением общей формулы (XVb) в которой R3 и R7 имеют значения, как определено в общей формуле (I), по любому из пп.1-13,стадию снятия защиты проводят до или после реакции соединения формулы (VII) с соединением общей формулы (XVa) или с соединением общей формулы (XVb) в присутствии HCl в диоксане или трифторуксусной кислоты в этаноле или дихлорметане при температуре между -5 и 60C с получением соединения формулы (I). 17. Лекарственное средство для предотвращения и/или лечения заболеваний, в которые вовлечен рецептор фактора роста эндотелия сосудов VEGFR-3, включающее соединение формулы (I) по любому из пп.1-15, или его фармацевтически приемлемую соль, или энантиомер, или диастереоизомер, или их смесь. 18. Фармацевтическая композиция для предотвращения и/или лечения заболеваний, в которые вовлечен рецептор VEGFR-3, включающая соединение по любому из пп.1-15, или его фармацевтически приемлемую соль, или энантиомер, или диастереоизомер, или их смесь, а также по меньшей мере один фармацевтически приемлемый эксципиент. 19. Применение соединения формулы (I) по любому из пп.1-15 для получения лекарственного средства для предотвращения и/или лечения заболеваний, в которые вовлечен рецептор VEGFR-3. 20. Применение соединения формулы (I) по любому из пп.1-15 для получения лекарственного средства для предотвращения и/или лечения рака и метастазов. 21. Применение соединения формулы (I) по любому из пп.1-15 для получения лекарственного средства для предотвращения и/или лечения заболеваний, выбранных из глиабластомы, множественной миеломы, миелодиспластического синдрома, саркомы Капоши, кожной ангиосаркомы, солидных опухолей,лимфомы, меланомы, рака груди, колоректального рака, рака легкого, рака поджелудочной железы, рака простаты, рака почек, рака головы и шеи, рака печени, рака яичников, рака дыхательного тракта и грудной клетки. 22. Применение соединения формулы (I) по любому из пп.1-15 для получения лекарственного средства для предотвращения и/или лечения неонкологических пролиферативных заболеваний и патологического ангиогенеза, связанного с VEGFR-3. 23. Применение соединения формулы (I) по любому из пп.1-15 для получения лекарственного средства для предотвращения и/или лечения заболеваний, выбранных из группы, состоящей из артроза, рестеноза, псориаза, гемангимы, лимфангиомы, глаукомы, гломерунефрита, диабетической нефропатии,нефросклероза, тромботического микроангиопатического синдрома, цирроза печени, атеросклероза, отторжения трансплантируемого органа, заболеваний глаз, включающих процесс ангиогенеза или лимфоангиогенеза. 24. Применение соединения формулы (I) по любому из пп.1-15 для получения лекарственного средства для предотвращения и/или лечения хронического или нехронического воспаления, инфицирования микроорганизмами и аутоиммунных заболеваний. 25. Применение по п.24, где заболеванием является ревматоидный артрит. 26. Применение соединения формулы (I) по любому из пп.1-15 для получения лекарственного средства для предотвращения и/или лечения редких заболеваний, выбранных из лимфангиолейомиоматоза или болезни Горхема.
МПК / Метки
МПК: A61K 31/4375, A61K 31/4709, A61P 35/00, C07D 471/04, C07D 401/04
Метки: киназы, качестве, 2-амино-3-(имидазол-2-ил)пиридин-4-она, рецептора, производные, применение, ингибиторов
Код ссылки
<a href="https://eas.patents.su/25-23580-proizvodnye-2-amino-3-imidazol-2-ilpiridin-4-ona-i-ih-primenenie-v-kachestve-ingibitorov-kinazy-receptora-vegf.html" rel="bookmark" title="База патентов Евразийского Союза">Производные 2-амино-3-(имидазол-2-ил)пиридин-4-она и их применение в качестве ингибиторов киназы рецептора vegf</a>
Производные пиримидина и их применение в терапии в качестве ингибиторов киназы csbp/rk/p38

Номер патента: 12875
Опубликовано: 30.12.2009
Авторы: Ливия Стефано, Нортон Бет А., Купер Энтони Уилльям Джеймс, Каллахан Джеймс Фрэнсис, Невинс Нейса, Вань Цзехун, Боэм Джеффри С.
МПК: A61K 31/5365, A61P 29/00, A61K 31/519...
Метки: качестве, терапии, киназы, применение, ингибиторов, пиримидина, производные
Формула / Реферат:
1. Соединение формул в которых G3 представляет CH2; G4 представляет CH; R1 представляет C(Z)N(R10')(CR10R20)vRb, C(Z)O(CR10R20)vRb, N(R10')C(Z)(CR10R20)vRb, N(R10')C(Z)N(R10')(CR10R20)vRb или N(R10')OC(Z)(CR10R20)vRb; R1' независимо выбран в каждом случае из атома галогена, C1-4алкила, замещенного атомом галогена C1-4алкила, циано, нитро, (CR10R20)v'NRdRd', (CR10R20)v'C(О)R12, SR5, S(O)R5, S(O)2R5 или (CR10R20)v'OR13; Rb представляет атом...
Замещенные пиразолохиназолиновые производные, способ их получения и их применение в качестве ингибиторов киназы

Номер патента: 17769
Опубликовано: 29.03.2013
Авторы: Карузо Микеле, Постери Элена, Вальсазина Барбара, Браска Мария Габриелла, Берия Итало, Фергюсон Рон
МПК: A61K 31/519, A61P 35/00, C07D 295/135...
Метки: пиразолохиназолиновые, получения, способ, качестве, применение, ингибиторов, производные, киназы, замещенные
Формула / Реферат:
1. Соединение формулы (I)где R1 представляет собой ортозамещенный ариламино формулыгде R'4 и R''4 независимо выбраны из группы, включающей галоген, нитро, циано, C1-C6-алкил, полифторированный алкил, полифторированный алкокси, алкенил, алкинил, гидроксиалкил, арил, арилалкил, гетероциклил, С3-С6-циклоалкил, гидрокси, алкокси, арилокси, гетероциклилокси, алкилкарбонилокси, арилкарбонилокси, гетероциклилкарбонилокси, карбокси, алкоксикарбонил,...
Новые аминоциклические производные мочевины, их получение и фармацевтическое применение в качестве ингибиторов киназы

Номер патента: 11088
Опубликовано: 30.12.2008
Авторы: Бенар Дидье, Руф Свен, Эль Амад Юссеф, Бушар Эрве, Лебрэн Анн, Штробель Хартмут, Гюссреген Штефан, Лесюисс Доминик, Иттэнжер Огюстэн, Риттер Курт, Немесек Консепсьон
МПК: A61K 31/4166, C07D 401/14, C07D 401/06...
Метки: качестве, фармацевтическое, мочевины, новые, применение, киназы, получение, производные, аминоциклические, ингибиторов
Формула / Реферат:
1. Соединение формулы (I) в которой р представляет собой целые числа 0, 1 и 2; A представляет собой арильный, гетероарильный или моноциклический или бициклический конденсированный карбоциклический или гетероциклический 5-11-членный радикал, все указанные радикалы необязательно замещены одним или несколькими заместителями, которые могут быть одинаковыми или различными, выбранными из значений R3; X представляет собой прямую связь или следующие...
Производные хиназолина в качестве ингибиторов киназы

Номер патента: 5809
Опубликовано: 30.06.2005
Авторы: Цукуда Еидзи, Скарборо Роберт М., Ода Содзи, Мацуно Кендзи, Иде Синити, Итимура Митио, Номото Юдзи, Панди Анджали, Ирие Дзунко
МПК: C07D 403/12
Метки: ингибиторов, качестве, киназы, хиназолина, производные
Формула / Реферат:
1. Азотсодержащее гетероциклическое соединение формулы в которой R1 обозначает радикал, выбранный из группы, в которую входят -CN, -O-метил, -O-этил, -O-пропил, -O-изопропил, -O-бутил, -O-трет-бутил, -O-изоамил, 1-нафтилокси, 2-нафтилокси, 4-индолилокси, 5-индолилокси, 5-изохинолилокси и их пространственные изомеры и гомологи; R2 и R4 каждый представляют собой -O-CH3 или -O(-CH2)n-R3; причем, когда R2 представляет собой -O-CH3, R4 представляет...
Производные аминоизоксазола в качестве ингибиторов киназы

Номер патента: 6769
Опубликовано: 28.04.2006
Авторы: Салом Барбара, Вульпетти Анна, Кавиккьоли Марчелло, Певарелло Паоло
МПК: A61K 31/4439, A61K 31/422, A61K 31/427...
Метки: ингибиторов, производные, киназы, аминоизоксазола, качестве
Формула / Реферат:
1. Способ лечения заболеваний, вызванных и/или ассоциированных с изменением активности протеинкиназы, который включает введение млекопитающему, нуждающемуся в этом, эффективного количества производного аминоизоксазола, представленного формулой (I) где R представляет 5- или 6-членную гетероарильную группу, содержащую 1-3 гетероатома, выбранных из азота, кислорода или серы, необязательно дополнительно конденсированную с 5-7-членным ароматическим...
Предыдущий патент: Замещенные производные индазола, активные в качестве ингибиторов киназы
Следующий патент: Производные [1,2,3]триазоло[4,5-d]пиримидина в качестве агонистов каннабиноидного рецептора 2
Случайный патент: Способ профилактики и лечения диабетической периферической невропатии депротеинизированным гемодериватом