Производные пирролидина в качестве антагонистов окситоцина
Номер патента: 8479
Опубликовано: 29.06.2007
Авторы: Дорбре Жером, Жоран-Лебрюн Катрин, Каттропани Анна, Валонье Дельфина, Шварц Маттиас



























Формула / Реферат
1. Производное пирролидина формулы I

его геометрические изомеры, его оптически активные формы, такие как энантиомеры, диастереомеры, их смеси и формы рацематов, а также его соли, в которых
R1 выбран из группы, включающей или состоящей из Н и C1-С6-алкила;
R2 выбран из группы, включающей или состоящей из водорода, C1-С6-алкила, C1-С6-алкиларила, гетероарила, C1-С6-алкилгетероарила, C2-С6-алкенила, C2-С6-алкениларила, C2-С6-алкенилгетероарила, C2-С6-алкинила, C2-С6-алкиниларила, C2-С6-алкинилгетероарила, C3-С8-циклоалкила, гетероциклоалкила, C1-С6-алкилциклоалкила, C1-С6-алкилгетероциклоалкила, C1-С6-алкилкарбокси, ацила, C1-С6-алкилацила, C1-С6-алкилацилокси, C1-С6-алкилалкокси, алкоксикарбонила, C1-С6-алкилалкоксикарбонила, аминокарбонила, C1-С6-алкиламинокарбонила, C1-С6-алкилациламино, C1-С6-алкилмочевины, амино, C1-С6-алкиламино, сульфонилокси, C1-С6-алкилсульфонилокси, сульфонила, C1-С6-алкилсульфонила, сульфинила, C1-С6-алкилсульфинила, C1-С6-алкилсульфанила, C1-С6-алкилсульфониламино;
R3 выбран из группы, включающей или состоящей из арила и гетероарила;
X выбран из группы, состоящей из О или NR4;
R4 выбран из группы, включающей или состоящей из Н, C1-С6-алкила, C1-С6-алкиларила, C1-С6-алкилгетероарила, арила, гетероарила; или
R2 и R4 могут вместе с атомом N, с которым они связаны, образовывать 5-8-членное насыщенное или ненасыщенное гетероциклоалкильное кольцо; и
n имеет значение от 1 до 3.
2. Производное пирролидина по п.1, в котором R1 представляет собой метил.
3. Производное пирролидина по п.1 или 2, в котором R3 представляет собой фенильную, бифенильную или 2-метилбифенильную группу.
4. Производное пирролидина по любому из предшествующих пунктов, в котором n имеет значение 1 или 2.
5. Производное пирролидина по любому из предшествующих пунктов, в котором R2 и R4 вместе с атомом N, в которым они связаны, образует 5 или 6-членное циклоалкильное или гетероциклоалкильное кольцо.
6. Производное пирролидина по пп.1-4, в котором X представляет собой О или NH.
7. Производное пирролидина по любому из предшествующих пунктов, выбранное из следующей группы:
О-метилоксим (3ЕZ,5S)-5-(гидроксиметил)-1-[(2'-метил-1,1'-бифенил-4-ил)карбонил]пирролидин-3-он;
О-метилоксим (3EZ,5S)-1-(1,1'-бифенил-4-илкарбонил)-5-(гидроксиметил)пирролидин-3-он;
О-метилоксим (3E,5S)-1-(1,1'-бифенил-4-илкарбонил)-5-(гидроксиметил)пирролидин-3-он;
О-метилоксим (3Z,5S)-1-(1,1'-бифенил-4-илкарбонил)-5-[(4-метилпиперазин-1-ил)метил]пирролидин-3-он;
трет-бутил {[(2S,4EZ)-1-(1,1'-бифенил-4-илкарбонил)-4-(метоксиимино)пирролидин-2-ил]метокси} ацетат;
{[(2S,4EZ)-1-(1,1'-бифенил-4-илкарбонил)-4-(метоксиимино)пирролидин-2-ил]метокси}уксусная кислота;
2-{[(2S,4EZ)-1-(1,1'-бифенил-4-илкарбонил)-4-(метоксиимино)пирролидин-2-ил]метокси}-N-(2-пирролидин-1-илэтил)ацетамид;
О-метилоксим (3EZ,5S)-1-(1,1'-бифенил-4-илкарбонил)-5-(метоксиметил)пирролидин-3-он;
О-метилоксим (3EZ,5S)-1-(1,1'-бифенил-4-илкарбонил)-5-[(4-метилпиперазин-1-ил)метил]пирролидин-3-он;
О-метилоксим (3EZ,5S)-1-(1,1'-бифенил-4-илкарбонил)-5-{[(4-метоксифенил)амино]метил}пирролидин-3-он;
О-метилоксим (3EZ,5S)-1-(1,1'-бифенил-4-илкарбонил)-5-({[2-(1Н-пиразол-1-ил)этил]амино}метил) пирролидин-3-он;
2-{[(2S,4EZ)-1-(1,1'-бифенил-4-илкарбонил)-4-(метоксиимино)пирролидин-2-ил]метил}-1Н-изоиндол-1,3(2Н)-дион;
О-метилоксим (3EZ,5S)-5-(аминометил)-1-(1,1'-бифенил-4-илкарбонил)пирролидин-3-он;
N-{[(2S,4EZ)-1-(1,1'-бифенил-4-илкарбонил)-4-(метоксиимино)пирролидин-2-ил]метил}ацетамид;
О-метилоксим (3EZ,5S)-1-(1,1'-бифенил-4-илкарбонил)-5-(пиперидин-1-илметил)пирролидин-3-он;
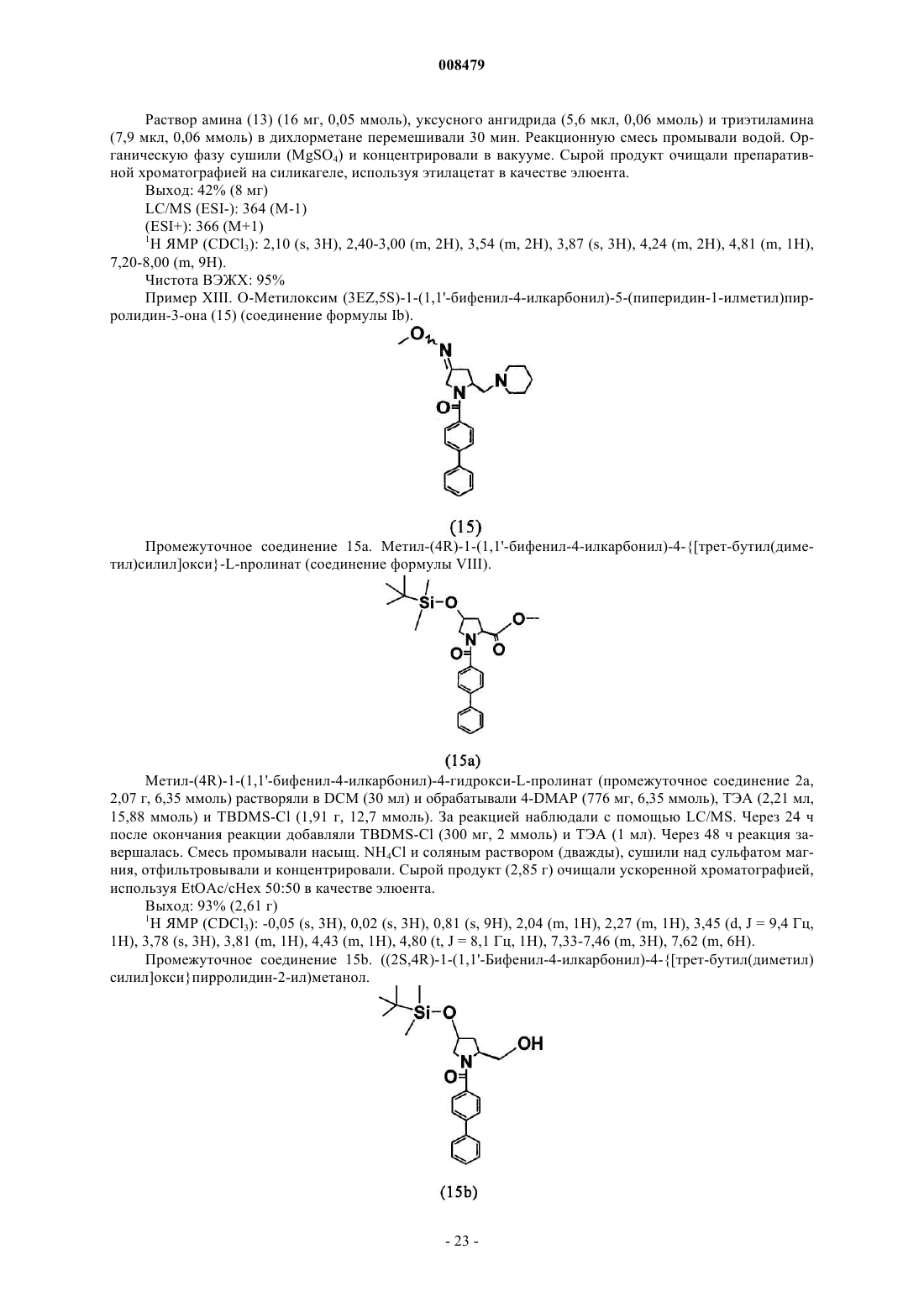
О-метилоксим (3ЕZ,5S)-1-(1,1'-бифенил-4-илкарбонил)-5-(2-гидроксиэтил)пирролидин-3-он.
8. Применение производного пирролидина по любому из пп.1-7, а также его изомеров, оптически активных форм, таких как энантиомеры, диастереомеры и их смеси, а также его солей для получения лекарственного средства для предотвращения и/или лечения выкидыша, преждевременных родов или дисменореи.
9. Применение производного пирролидина по пп.1-7 для получения лекарственного средства для лечения заболеваний, требующих модулирование рецептора окситоцина.
10. Применение по п.9 для лечения или предотвращения заболеваний, связанных с активностью рецептора окситоцина.
11. Применение по п.9 или 10, в котором указанное модулирование заключается в блокировании рецептора окситоцин или антагонистическом действии в отношении связывания окситоцина с его рецептором.
12. Фармацевтическая композиция, содержащая производное пирролидина по любому из пп.1-7 и фармацевтически приемлемый носитель, разбавитель или эксципиент.
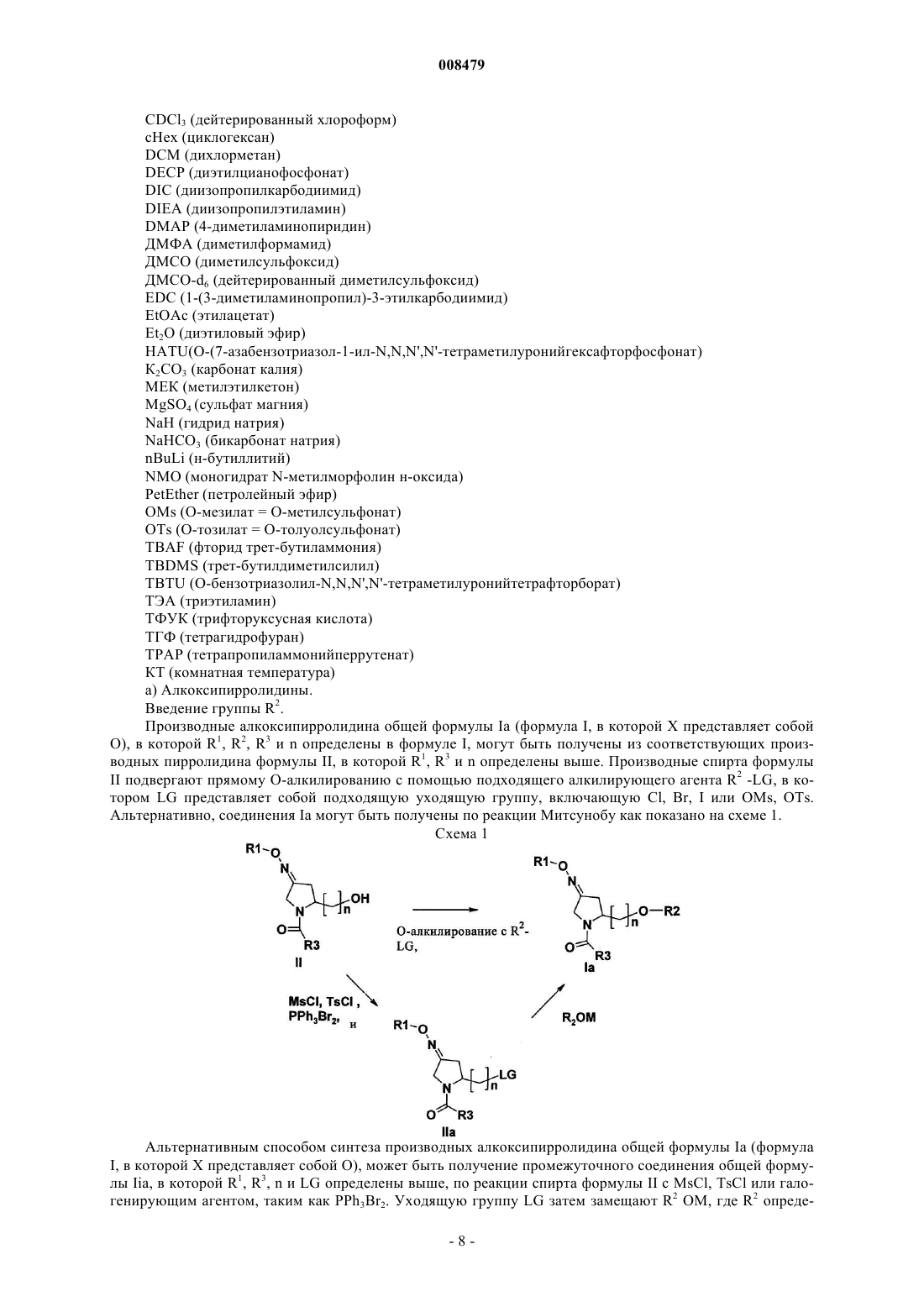
13. Способ получения производного пирролидина по любому из пп.1-7, в котором X представляет собой О, включающий стадию О-алкилирования производных спирта формулы (II) алкилирующим агентом R2-LG, в котором LG представляет собой уходящую группу, где R1, R2, R3 и n определены выше.

14. Способ получения производного пирролидина по любому из пп.1-7, в котором X представляет собой NR4, включающий стадию восстановительного аминирования производного альдегида формулы (XI) с амином HNR2R4, где R1, R2, R3, R4 и n определены выше.

Текст
008479 Область изобретения Настоящее изобретение относится к новым производным пирролидина, в частности к их применению в качестве лекарственных средств, а также к фармацевтическим составам, содержащим такие производные пирролидина. Указанные производные пирролидина являются полезными для лечения и/или предотвращения выкидыша, преждевременных родов, дисменореи. Предпочтительно производные пирролидина проявляют модулирующую, особенно антагонистическую активность в отношении рецептора окситоцина. Более предпочтительно указанные соединения полезны для лечения и/или предотвращения болезненных состояний, опосредованных окситоцином, включая выкидыш, преждевременные роды и дисменорею. Предпосылки изобретения Окситоцин (ОТ) представляет собой циклический нонапептид, действие которого опосредовано активацией специфических рецепторов, связанных с G-белком, в настоящее время классифицируемых в ОТ рецепторы (OT-R) (1). Окситоцин (ОТ) вызывает сокращение матки у млекопитающих в процессе родов. Соответствующий рецептор окситоцина принадлежит к семейству рецепторов, связанных с G-белком, и подобен рецепторам вазопрессина V1 и V2. Количество рецепторов ОТ сильно увеличивается в процессе беременности. Показано, что концентрация рецепторов ОТ соотносится с непосредственной активностью матки(2-3). Вызванные ОТ сокращения матки в процессе родов приводят к расширению шейки матки и в конечном счете к продвижению плода через вагинальный канал. В некоторых случаях эти сокращения происходят до того, как плод становится полностью жизнеспособным, что приводит к преждевременным родам. Преждевременные роды и выкидыш являются нежелательными, поскольку они являются главными причинами перинатальной заболеваемости. Следовательно, контроль над выкидышем представляет собой существенную проблему в области акушерства. Недавно было показано, что гормон окситоцин играет главную роль в начале родов у млекопитающих, особенно у людей. Таким образом, предполагается, что окситоцин проявляет указанный эффект прямым, а также косвенным путем, сжимая миометрий матки и увеличивая синтез и высвобождение сжимающих простагландинов из эндометрии/отпадающей оболочки матки. Эти простагландины, кроме того, могут играть роль в процессе созревания цервикальной шейки. Это "сверхрегулирование" рецепторов окситоцина и повышенная чувствительность матки, как оказывается, обусловлены трофическими действиями повышающихся со временем уровней эстрогена в плазме. При подрегулировании окситоцина ожидается, что прямое (сжимание) и косвенное (увеличение синтеза простагландина) действия окситоцина в матке могли бы быть блокированы. Модулятор окситоцина, например блокатор или антагонист,был бы вероятно эффективен для лечения выкидыша. Другим состоянием, связанным с окситоцином, является дисменорея, которая характеризуется болью или дискомфортом, связанными с менструациями. Боль, как полагают, является причиной сокращений матки и ишемии, вероятно опосредованных действием простагландинов, производимых в секретирующем эндометрии. Блокируя косвенное и прямое действие окситоцина в матке, антагонист окситоцина был бы полезен для лечения дисменореи. Некоторые агенты, противодействующие окситоцину, в настоящее время проходят клинические испытания (4). Такие токолитические агенты (то есть агенты, расслабляющие матку) включают бета-2 адренергические агонисты, сульфат магния и этанол. Ведущим бета-2-адренергическим агонистом является ритодрин, который проявляет множество сердечно-сосудистых и метаболических побочных эффектов, включая тахикардию, увеличение секреции ренина, гиперглицемию и реактивную гипоглицемию у младенца. Далее, бета-2-адренергические агонисты, включая тербуталин и альбутерол, имеют побочные эффекты, подобные побочным эффектам ритодрина. Сульфат магния при концентрациях в плазме выше терапевтического диапазона от 4 до 8 мг/дл может вызывать ингибирование сердечной проводимости и нейромышечной передачи, дыхательную депрессию и остановку сердца, таким образом, этот агент не подходит при повреждении функции почек. Этанол так же эффективен как ритодрин для предотвращения преждевременных родов, но он не вызывает соответствующего снижения действия эмбрионального дыхательного синдрома, который вызывает введение ритодрина. Атозибан, пептидный ОТ антагонист, имеет проблему большинства пептидов: низкая оральная биодоступность из-за разрушения в кишечнике. Такие соединения должны вводиться парентерально. Развитие непептидных лигандов для пептидных рецепторов гормонов, как ожидается, решит эту проблему. Merck были описаны селективные антагонисты окситоцина небольшой массы. В дополнение к циклическим гексапептидам, Merck предложил инданилпиперидины и толилпиперазины в качестве орально доставляемых антагонистов ОТ (5). В WO 96/22775 и US 5756497 Merck описаны бензоксазинилпиперидины или бензоксазиноны в качестве антагонистов рецептора ОТ. Сообщалось, что некоторые сульфонамиды антагонизируют окситоцин на рецепторе окситоцина.Elf Sanofi ЕР-А-0469984 и ЕР-А-0526348 описывают н-сульфонилиндолины в качестве антагонистов рецепторов вазопрессина и окситоцина.American Cyanamid US 5889001 описывает производные пиразолбензодиазепина в качестве антагонистов вазопрессина и окситоцина.-1 008479 Недавно были описаны производные пирролидина, такие как пирролидинамиды и пирролидины, замещенные конденсированным гетероарилом, в качестве антагонистов рецептора окситоцина (WO 01/72705). Сущность изобретения В первом варианте осуществления изобретение относится к новым производным пирролидина формулы IR3 в формуле (I) выбран из группы, состоящей из арила и гетероарила.X в формуле (I) выбран из группы, состоящей из О или NR4. R4 выбран из группы, состоящей из Н,C1-С 6-алкила, арила, гетероарила, C1-С 6-алкиларила, C1-С 6-алкилгетероарила. Предпочтительно R4 представляет собой Н или С 1-С 6-алкил, такой как метильная или этильная группа. Альтернативно, R2 и R4 в формуле (I) могут образовывать вместе с атомом N, с которым они связаны, 5-8-членное насыщенное или ненасыщенное гетероциклоалкильное кольцо, например пиперидинильную или пиперазинильную группу, которая необязательно может быть конденсирована с арильным,гетероарильным, циклоалкильным или гетероциклоалкильным кольцом,n в формуле (I) имеет значение от 1 до 3, более предпочтительно имеет значение 1 или 2. Во втором варианте осуществления настоящее изобретение относится к новым производным пирролидина формулы I для применения в качестве лекарственного средства. В третьем варианте осуществления изобретение относится к фармацевтической композиции, включающей соединение формулы I, вместе с фармацевтически приемлемым эксципиентом или носителем. В четвертом варианте осуществления изобретение относится к соединению формулы I для получения фармацевтической композиции, полезной для лечения и/или предотвращения выкидыша, преждевременных родов, дисменореи. В пятом варианте осуществления изобретение относится к соединению формулы I для модулирования функции рецептора ОТ. В шестом варианте осуществления изобретение относится к применению соединения формулы I для лечения заболевания, связанного с рецептором ОТ, такого как выкидыш, преждевременные роды,дисменорея. В седьмом варианте осуществления изобретение относится к способу лечения заболевания, связанного с рецептором ОТ, такого как выкидыш, преждевременные роды, дисменорея, включающему введение нуждающемуся в этом пациенту эффективного количества соединения формулы I. В восьмом варианте осуществления изобретение относится к способу получения соединения формулы I. Подробное описание изобретения Целью настоящего изобретения является получение веществ, которые являются полезными для лечения и/или предотвращения выкидыша, преждевременных родов и дисменореи. Очевидно, целью настоящего изобретения является обеспечение химических соединений, которые способны подрегулировать, включая антагонизм, функцию ОТ при заболевании у млекопитающих, особенно у людей. Также целью настоящего изобретения является обеспечение химических соединений небольшого размера для модулирования, предпочтительно для подрегулирования или антагонизма рецептора окситоцина. Кроме того, целью настоящего изобретения является обеспечение способов получения указанных химических соединений небольшого размера. Также целью настоящего изобретения является обеспечение новых категорий фармацевтических составов для лечения выкидыша и дисменореи и/или заболева-2 008479 ний, опосредованных рецептором окситоцина. Наконец, целью настоящего изобретения является обеспечение способа лечения и/или предотвращения заболеваний, опосредованных рецептором окситоцина, таких как выкидыш, с помощью антагонистов окситоцина, являющихся, например, антагонистами связывания окситоцина с его рецептором. Следующие абзацы раскрывают определения различных химических групп, которые входят в соединения настоящего изобретения и предназначены для раскрытия терминов во всем описании и формуле изобретения, если явно изложенное определение не имеет более широкого значения."С 1-С 6-алкил" обозначает моновалентные алкильные группы, имеющие от 1 до 6 атомов углерода. Этот термин представлен такими группами, как метил, этил, н-пропил, изопропил, н-бутил, изобутил,трет-бутил, н-гексил и т.д."Арил" обозначает ненасыщенную ароматическую карбоциклическую группу, имеющую от 6 до 14 атомов углерода в одном кольце (например, фенил) или в нескольких конденсированных кольцах (например, нафтил). Предпочтительно арил включает фенил, нафтил, фенантренил и т.д."Гетероарил" обозначает моноциклическую гетероароматическую или бициклическую или трициклическую конденсированную гетероароматическую группу. Конкретные примеры гетероароматических групп включают необязательно замещенный пиридил, пирролил, фурил, тиенил, имидазолил, оксазолил,изоксазолил, тиазолил, изотиазолил, пиразолил, 1,2,3-триазолил, 1,2,4-триазолил, 1,2,3-оксадиазолил,1,2,4-оксадиазолил, 1,2,5-оксадиазолил, 1,3,4-оксадиазолил, 1,3,4-триазинил, 1,2,3-триазинил, бензофурил, [2,3-дигидро]бензофурил, изобензофурил, бензотиенил, бензотриазолил, изобензотиенил, индолил,изоиндолил, 3 Н-индолил, бензимидазолил, имидазо[1,2-а]пиридил, бензотиазолил, бензоксазолил, хинолизинил, хиназолинил, фталазинил, хиноксалинил, циннолинил, нафтиридинил, пиридо[3,4-b]пиридил,пиридо[3,2-b]пиридил, пиридо[4,3-b]пиридил, хинолил, изохинолил, тетразолил, 5,6,7,8-тетрагидрохинолил, 5,6,7,8-тетрагидроизохинолил, пуринил, птеридинил, карбозолил, ксантенил или бензохинолил."С 2-С 6-алкенил" обозначает алкенильные группы, предпочтительно имеющие от 2 до 6 атомов углерода и имеющие по крайней мере 1 или 2 алкенильных ненасыщенных участка. Предпочтительные алкенильные группы включают этенил (-СН=СН 2), н-2-пропенил (аллил, -СН 2 СН=СН 2) и т.д."С 2-С 6-алкинил" обозначает алкинильные группы, предпочтительно имеющие от 2 до 6 атомов углерода и имеющие по крайней мере 1-2 алкинильных ненасыщенных участка, предпочтительно алкинильные группы включают этинил (-ССН), пропаргил (-СН 2 ССН), и т.д."С 3-С 8-циклоалкил" обозначает насыщенную карбоциклическую группу, имеющую от 3 до 8 атомов углерода, представленную одним кольцом (например, циклогексил) или несколькими конденсированными кольцами (например, норборнил). Предпочтительно циклоалкил включает циклопентил, циклогексил,норборнил и т.д."Гетероциклоалкил" обозначает С 3-С 8-циклоалкильную группу, определенную выше, в которой до 3 атомов углерода замещены гетероатомами, выбранными из группы, состоящей из О, S, NR, R представляет собой водород или метил. Предпочтительно гетероциклоалкил включает пирролидин, пиперидин,пиперазин, 1-метилпиперазин, морфолин и т.д."Аминокарбонил" обозначает группу -C(O)NRR', где каждый R, R' независимо представляет собой водород или С 1-С 6-алкил или арил или гетероарил или "С 1-С 6-алкиларил" или "С 1-С 6-алкилгетероарил"."Ациламино" обозначает группу -NRC(O)R' где каждый R, R' независимо представляет собой водород или "C1-С 6-алкил" или "арил" или "гетероарил" или "С 1-С 6-алкиларил" или "С 1-С 6-алкилгетероарил"."Мочевина" обозначает группу -NRC(O)NR'R", где каждый R, R', R" независимо представляет собой водород, "С 1-С 6-алкил", "С 2-С 6-алкенил", "С 2-С 6-алкинил", "С 3-С 8-циклоалкил", "гетероциклоалкил","арил", "гетероарил", "C1-С 6-алкиларил" или "C1-С 6-алкилгетероарил", "С 2-С 6-алкениларил", "С 2-С 6 алкенилгетероарил", "С 2-С 6-алкиниларил", "С 2-С 6-алкинилгетероарил", "С 1-С 6-алкилциклоалкил", "С 1-С 6 алкилгетероциклоалкил", и где R' и R" вместе с атомом азота, к которому они присоединены, могут необязательно образовывать 3-8-членное гетероциклоалкильное кольцо."Карбамат" обозначает группу -NRC(O)OR', где каждый R, R' независимо представляет собой водород, "С 1-С 6-алкил", "С 2-С 6-алкенил", "С 2-С 6-алкинил", "С 3-С 8-циклоалкил", "гетероциклоалкил", "арил","гетероарил", "С 1-С 6-алкиларил" или "С 1-С 6-алкилгетероарил", "С 2-С 6-алкениларил", "С 2-С 6-алкенилгетероарил", "С 2-С 6-алкиниларил", "С 2-С 6-алкинилгетероарил", "C1-С 6-алкилциклоалкил", "С 1-С 6-алкилгетероциклоалкил"."Амино" обозначает группу -NRR', где каждый R, R' независимо представляет собой водород или"циклоалкил" или "гетероциклоалкил", и где R и R', вместе с атомом азота, к которому они присоединены, могут необязательно образовывать 3-8-членное гетероциклоалкильное кольцо."Аммоний" обозначает положительно заряженную группу -N+ RR'R", где каждый R, R', R" независимо представляет собой "С 1-С 6-алкил" или "С 1-С 6-алкиларил" или "С 1-С 6-алкилгетероарил" или "циклоалкил" или "гетероциклоалкил", и где R и R', вместе с атомом азота, к которому они присоединены, могут необязательно образовывать 3-8-членное гетероциклоалкильное кольцо."Сульфанил" обозначает группы -S-R, где R включает "С 1-С 6-алкил" или "арил" или "гетероарил" или "С 1-С 6-алкиларил" или "С 1-С 6-алкилгетероарил". Предпочтительно сульфанильные группы включают метилсульфанил, этилсульфанил и т.д."Сульфониламино" обозначает группу -NRSO2-R', где каждый R, R' независимо представляет собой водород или "С 1-С 6-алкил" или "арил" или "гетероарил" или "С 1-С 6-алкиларил" или "С 1-С 6-алкилгетероарил"."Замещенный или незамещенный": если не подразумевается иное для определения индивидуального заместителя, указанные выше группы, такие как "алкил", "алкенил", "алкинил", "арил" и "гетероарил" и другие группы могут быть необязательно замещены 1-5 заместителями, выбранными из группы, состоящей из "С 1-С 6-алкила", "С 2-С 6-алкенила", "С 2-С 6-алкинила", "циклоалкила", "гетероциклоалкила","С 1-С 6-алкиларила", "С 1-С 6-алкилгетероарила", "С 1-С 6-алкилциклоалкила", "С 1-С 6-алкилгетероциклоалкила", "амино", "аммония", "ацила", "ацилокси", "ациламино", "аминокарбонила", "алкоксикарбонила","мочевины", "карбамата," "арила", "гетероарила", "сульфинила", "сульфонила", "алкокси", "сульфанила","галогена", "карбокси", тригалогенметила, циано, гидрокси, меркапто, нитро и т.д. Альтернативно, указанный заместитель может также включать случаи, когда соседние заместители замыкаются в кольцо,особенно когда включены вицинальные функциональные заместители, образуя, например, лактамы, лактоны, циклические ангидриды, а также ацетали, тиоацетали, аминали, образованные закрытием кольца,например, при получении защитной группы."Фармацевтически приемлемые соли или комплексы" обозначают соли или комплексы указанных соединений формулы (I). Примеры таких солей включают, но не ограничиваются ими, основные аддитивные соли, полученные реакцией соединений формулы (I) с органическими или неорганическими основаниями, такими как гидроксид, карбонат или бикарбонат катиона металла, такого как металл, выбранный из группы, состоящей из щелочных металлов (натрий, калий или литий), щелочно-земельных металлов (например, кальций или магний), или с органическими первичным, вторичным или третичным алкиламином. Соли с аминами, полученными из метиламина, диметиламина, триметиламина, этиламина,диэтиламина, триэтиламина, морфолина, H-Me-D-глюкамина, N,N'-бис(фенилметил)-1,2-этандиамина,трометаамина, этаноламина, диэтаноламина, этилендиамина, н-метилморфолина, прокаина, пиперидина,пиперазина и т.д., входят в объем настоящего изобретения. Также включаются кислотные аддитивные соли, которые получены с неорганическими кислотами(например, соляная кислота, бромо-водородная кислота, серная кислота, фосфорная кислота, азотная кислота и т.д.), а также соли, полученные с органическими кислотами, такими как уксусная кислота, оксалиновая кислота, винная кислота, янтарная кислота, яблочная кислота, фумаровая кислота, малеиновая кислота, аскорбиновая кислота, бензойная кислота, дубильная кислота, памовая кислота, альгиновая кислота, полиглутаминовая кислота, нафталенсульфоновая кислота, нафталендисульфоновая кислота и полигалактуроновая кислота."Фармацевтически активное производное" обозначает любое соединение, которое при введении пациенту прямо или косвенно способно обеспечить описанную здесь активность."Энантиомерный избыток" (ее) обозначает продукты, которые получают асимметричным синтезом,т.е. синтезом, включающим нерацемическое исходное сырье и/или реагенты, или синтезом, включающим по крайней мере одну энантиоселективную стадию, "ее" представляет собой процентное соотношение избытка основного энантиомера по отношению к минорному энантиомеру [%ее=%главного%минорного]. При отсутствии асимметричного синтеза рацемические продукты обычно получают таким образом, что они также имеют активность антагонистов OT-R. Термин "выкидыш" или термин "преждевременные роды" обозначает отторжение из матки эмбриона ранее нормального окончания беременности или, более конкретно, начало родов с изменением и расширением шейки матки до 37-й недели беременности. Это может сопровождаться вагинальным кровотечением или разрывом мембран. Термин "дисменорея" обозначает боли при менструации. Термин "кесарево сечение" обозначает разрез стенок брюшины и матки для извлечения плода. Настоящее изобретение также включает геометрические изомеры, оптически активные формы, энантиомеры, диастереомеры соединений формулы I, их смеси, рацематы, а также фармацевтически приемлемые соли. Соединениями настоящего изобретения являются также соединения формулы IR3 в формуле (I) выбран из группы, состоящей из арила и гетероарила.X в формуле (I) выбран из группы, состоящей из О или NR4 , в которой R4 выбран из группы, состоящей из Н, С 1-С 6-алкила, арила, гетероарила, C1-С 6-алкиларила и С 1-С 6-алкилгетероарила. Предпочтительно, R4 представляет собой Н или С 1-С 6-алкил, такой как метил или этил. Альтернативно, R2 и R4 в формуле (I) вместе с атомом N, с которым они связаны, могут образовывать 5-8-членное насыщенное или ненасыщенное гетероциклоалкильное кольцо, например пиперидинильную или пиперазинильную группу, которая необязательно может быть конденсирована с арильным,гетероарильным, циклоалкильным или гетероциклоалкильным кольцом.n в формуле (I) имеет значение от 1 до 3, более предпочтительно имеет значение 1 или 2. Предпочтительно R2 в соединениях формулы I выбран из группы, состоящей из Н, ацила, предпочтительно ацетильной группы, арила, необязательно замещенного замещенным или незамещенным С 1-С 6 алкокси, например, метилоксифенильной группы, С 1-С 3-алкила, такого как метил или этил, необязательно замещенного замещенной или незамещенной ацильной группой или эфирной группой, предпочтительно трет-бутилового эфира муравьиной кислоты или уксусной кислоты, н-(2-пирролидин-1-илэтил)ацетамида, и необязательно замещенного замещенным или незамещенным гетероарилом, предпочтительно н-пиразолом. Предпочтительно R3 в соединениях формулы I выбран из группы, состоящей из арильной группы,необязательно замещенной или незамещенной арильной группой. Особенно предпочтительным R3 является бифенильная или 2-метилбифенильная группа. Особенно предпочтительным вариантом осуществления настоящего изобретения является производное пирролидина формулы I, в котором X представляет собой О или NH и n имеет значение 1 или 2. Другим предпочтительным вариантом осуществления настоящего изобретения является производное пирролидина формулы I, в котором X представляет собой NR4 и в котором R4 и R2 образуют насыщенное или ненасыщенное, замещенное или незамещенное, конденсированное или неконденсированное,гетероциклическое кольцо с атомом N, с которым они связаны, предпочтительно 5 или 6-членное кольцо,более предпочтительно пиперидин, метилпиперазин или изоиндол-1,3-дион. Соединения формулы I могут использоваться для лечения заболевания. Конкретно, соединения формулы I полезны для применения для лечения заболеваний, таких как выкидыш, преждевременные роды, дисменорея и для остановки родов до кесарева сечения. Соединения настоящего изобретения особенно полезны для лечения выкидыша, преждевременных родов и дисменореи. Предпочтительно соединения формулы I отдельно или в форме фармацевтической композиции подходят для модулирования функции(й) окситоцина, таким образом позволяя лечение и/или предотвращение заболеваний, которые являются опосредованными рецептором окситоцина. Такое модулирование предпочтительно включает ингибирование OT-R функции(й), особенно антагонизируя рецептор окситоцина у млекопитающих и особенно у людей. Ненормальная активность или гиперактивность рецептора окситоцина часто включена в различные нарушения, включая вышеупомянутые перечисленные нарушения и заболевания. Следовательно, соединения по изобретению могут использоваться для лечения заболеваний, модулируя функцию OT-R или его путь. Модулирование функции OT-R или его пути может включать подрегулирование и/или ингибирование рецептора окситоцина. Соединения по изобретению могут использоваться отдельно или в комбинации с другими фармацевтическими агентами, например с другим модулятором OT-R. При использовании в качестве фармацевтических средств, производные пирролидина настоящего изобретения обычно вводят в форме фармацевтической композиции. Следовательно, фармацевтические композиции, включающие соединение формулы I и фармацевтически приемлемый носитель, разбавитель или эксципиент, также включены в объем настоящего изобретения. Среднему специалисту в данной области известны различные такие носители, разбавители или эксципиенты, подходящие для составления фармацевтической композиции. Соединения по изобретению вместе с обычно применяемым адьювантом, носителем, разбавителем или эксципиентом могут быть составлены в фармацевтические композиции и единичные дозировки и в такой форме могут использоваться в виде твердых веществ, таких как таблетки или наполненные капсулы, или жидких веществ, таких как растворы, суспензии, эмульсии, эликсиры, или в виде капсул, напол-6 008479 ненных этими веществами, для орального применения, или в форме стерильных инъекционных растворов для парентерального (включая подкожное) применения. Такие фармацевтические композиции и формы единичных дозировок могут включать ингредиенты в обычных пропорциях, с дополнительными активными соединениями или компонентами или без них, и такие формы единичных дозировок могут содержать любое подходящее эффективное количество активного ингредиента, соразмерное с предназначенным применяемым ежедневным диапазоном дозировки. При использовании в качестве фармацевтических веществ, производные пирролидина данного изобретения обычно вводят в форме фармацевтической композиции. Такие композиции могут быть получены известным в фармацевтике способом и включают по крайней мере одно активное соединение. Вообще, соединения данного изобретения вводятся в фармацевтически эффективном количестве. Фактически вводимое количество соединения обычно определяется врачом по различным факторам, включая состояние пациента, выбранный способ введения, фактически вводимое соединение, возраст, вес и реакция конкретного пациента, серьезность симптомов пациента и т.д. Фармацевтические композиции по изобретению могут вводиться различными способами, включая оральное, ректальное, чрезкожное, подкожное, внутривенное, внутримышечное и интраназальное введение. В зависимости от предназначенного способа введения, соединения предпочтительно составляются в виде инъекционных или оральных композиций. Композиции для орального введения могут иметь форму объемных жидких растворов, или суспензий, или объемных порошков. Обычно, однако, композиции находятся в формах единичных дозировок для облегчения точного приема. Термин "формы единичных дозировок" обозначает физически раздельные единицы, подходящие для отдельных дозировок для человека и других млекопитающих, где каждая единица, содержащая определенное количество активного материала, оказывает желательное терапевтическое действие, вместе с подходящим фармацевтическим эксципиентом. Обычные формы единичных дозировок включают предварительно наполненные, предварительно взвешенные ампулы или шприцы с жидкими композициями, или пилюли, таблетки, капсулы или т.д. в случае твердых композиций. В таких композициях соединение пирролидина обычно является минорным компонентом (приблизительно от 0,1 до приблизительно 50 вес.% или предпочтительно приблизительно от 1 до приблизительно 40 вес.%) с остатком, являющимся различным переносчиком или носителем и обеспечивающим получение желаемой дозированной формы. Жидкие формы, подходящие для орального введения, могут включать подходящий водный или неводный носитель с буферами, суспендирующими и диспергирующими агентами, красителями, отдушками и т.д. Твердые формы могут включать, например, любой из следующих ингредиентов или соединений подобной природы: связующее вещество, такое как микрокристаллическая целлюлоза, смола трагаканта или желатин; эксципиент, такой как крахмал или лактоза, разрыхлитель, такой как альгиновая кислота,Primogel или кукурузный крахмал; лубрикант, такой как стеарат магния; глидант, такой как коллоидный диоксид кремния; подсластитель, такой как сахароза или сахарин; или отдушка, такая как мята, метилсалицилат или апельсиновая отдушка. Инъекционные композиции обычно основаны на инъекционном стерильном соляном растворе или фосфатно-буферном соляном растворе или других инъекционных носителях, известных из уровня техники. Как упомянуто выше, производные пирролидина Формулы I в таких композициях обычно являются минорным компонентом, часто составляющим от 0,05 до 10 вес.% с остатком, являющимся инъекционным носителем и т.д. Вышеупомянутые описанные компоненты для оральных вводимых или инъекционных композиций являются просто примерными. Другие материалы, а также способы обработки и т.д. изложены в части 8(6). Соединения настоящего изобретения могут также вводиться в форме длительного высвобождения или в направленных системах длительного высвобождения лекарственного средства. Описание примерных материалов для длительного высвобождения могут также быть обнаружены в (6). Другим объектом настоящего изобретения является способ получения производных пирролидина формулы I. Производные пирролидина, представленные в настоящем изобретении, могут быть получены из легко доступных или ранее описанных исходных реагентов, используя следующие общие способы и процедуры. Следует понимать, что где приведены обычные или предпочтительные условия эксперимента (т.е. температуры реакций, время, мольные количества реактивов, растворители и т.д.), могут также использоваться другие условия эксперимента, если не указано иное. Оптимальные условия реакций могут изменяться для конкретных реагентов или используемых растворителей, но такие условия могут быть определены средним специалистом в данной области в соответствии с обычными процедурами оптимизации. Синтез соединений по изобретению Примеры схем синтеза соединений формулы I описаны ниже. Следующие сокращения соответственно относятся к приведенным ниже обозначениям:TBTU (О-бензотриазолил-N,N,N',N'-тетраметилуронийтетрафторборат) ТЭА (триэтиламин) ТФУК (трифторуксусная кислота) ТГФ (тетрагидрофуран) ТРАР (тетрапропиламмонийперрутенат) КТ (комнатная температура) а) Алкоксипирролидины. Введение группы R2. Производные алкоксипирролидина общей формулы Iа (формула I, в которой X представляет собой О), в которой R1, R2, R3 и n определены в формуле I, могут быть получены из соответствующих производных пирролидина формулы II, в которой R1, R3 и n определены выше. Производные спирта формулыII подвергают прямому О-алкилированию с помощью подходящего алкилирующего агента R2 -LG, в котором LG представляет собой подходящую уходящую группу, включающую Сl, Вr, I или OMs, OTs. Альтернативно, соединения Iа могут быть получены по реакции Митсунобу как показано на схеме 1. Схема 1 Альтернативным способом синтеза производных алкоксипирролидина общей формулы Iа (формулаI, в которой X представляет собой О), может быть получение промежуточного соединения общей формулы Iiа, в которой R1, R3, n и LG определены выше, по реакции спирта формулы II с MsCl, TsCl или галогенирующим агентом, таким как РРh3Br2. Уходящую группу LG затем замещают R2 ОМ, где R2 опреде-8 008479 лен выше и М представляет собой Н или металл, такой как Na, с получением соединения формулы Iа. Введение группы оксима. Соединение общей формулы II, где R1, R3 и n определены выше, могут быть получены из соединений общей формулы IV, в которой R3 и n определены выше и где PG1 представляет собой подходящую защитную группу для спирта, предпочтительно TBDMS. Кетон общей формулы IV подвергают реакции с производным гидроксиламина общей формулы V, в которой R1 определен выше. PG1 удаляют стадией снятия защиты стандартными синтетическими методиками, как показано на схеме 2. Схема 2 Гидроксиламин V, если не является коммерчески доступным может, например, синтезироваться по реакции н-Вос-дидроксиламина с соответствующим алкилирующим агентом формулы VI, где R1 определен выше и Ха = С 1, Вr, I, в стандартных условиях, как показано на схеме 3. Схема 3 Получение кетопирролидинов. Кетопирролидины общей формулы IV, в которой R3, n и PG1 определены выше, могут быть получены из соответствующих производных гидроксипирролидина формулы VII, в которой R3, n и PG1 определены выше, обработкой подходящим окислительным агентом, например ДМСO/(СOСl)2/ТЭА (условия Шверна) или ТРАР, в присутствии NMO, как показано на схеме 4. Схема 4 Стадия восстановления. Производные гидроксипирролидина общей формулы VII, в которой R3 и n определены выше и PG1 представляет собой защитную группу, могут быть получены восстановлением соответствующих пирролидинкарбоновых производных формулы VIII, в которой R3 и n определены выше, R7 представляет собой Н или алкильную группу и PG2 представляет собой подходящую защитную группу, после подходящей стадии защиты/снятия защиты, как показано на схеме 5. Предпочтительным восстанавливающим агентом является LiBH4, когда R7 представляет собой алкильную группу, или LAH, или ВН 3.DMS, когда R7 представляет собой Н. Схема 5 Стадия конденсации. Защищенные пирролидинкарбоновые производные общей формулы VIII, в которой R3, n, R7 и PG2 определены выше, получают реакцией соединения общей формулы IX, в которой n, R7 и PG2 определены выше и PG3 представляет собой Н или подходящую N-защитную группу, предпочтительно Вос, с ацилирующим агентом общей формулы R3-CO-Y, в которой R3 определен выше и Y представляет собой любую подходящую уходящую группу, как показано на схеме 6. Предпочтительно ацилирующими агентами являются хлориды кислот (Y = Сl) или карбоновые кислоты (Y = ОН), используемые вместе с подходящим пептидным конденсирующим агентом, таким как,например, DIC, EDC, HATU, DECP или др. Обычно, исходными реагентами являются соединения формулы IX, которые могут быть получены из коммерческих источников (например, защищенный 3-гидроксипролин, гомо-3-гидроксипролин, 3 гидроксипирролидин 5-пропановая кислота). Другие исходные реагенты (такие как соединения формулы XV, XVI, XIX и XX) могут быть получены из коммерчески доступных соединений формулы IX через промежуточные соединения формулыXII. В этом случае первые карбоновые производные общей формулы IX могут быть восстановлены до производных общей формулы XII, где PG2, PG3, R7 определены выше и n=2 или 3, как описано на схеме 7. Схема 7 Затем соединения формулы XII подвергают классическим процедурам защиты/снятия защиты и превращения функциональных групп, особенно процедурам гомологизации одного или двух атомов углерода, хорошо известным специалисту в данной области (7, 8). Один предпочтительный способ заключается в гомологизации одного атома углерода соединений общей формулы XII, в которой PG2 и PG3 определены выше и n имеет значение 2 или 3, замещением уходящей группы цианидом с последующим гидролизом с получением карбоновых кислот общей формулы XV, где PG2, PG3 и n определены выше, или восстановлением с получением аминосоединений общей формулы XVI, где PG2, PG3 и n определены выше, как описано на схеме 8. Схема 8 В случае гомологизации двух атомов углерода одна процедура предпочтительно включает реакцию альдегида общей формулы XVIII, полученного окислением соединения общей формулы XII, где PG2, PG3 и n определены выше, с реагентом Виттига-Хорнера, как описано на схеме 9. Полученное таким образом соединение затем восстанавливают до соединений общей формулы XIX, где PG2, PG3, n и R7 определены выше. Четыре описанные выше основные схемы химических превращений, т.е. стадия конденсации, стадия восстановления, получение оксима и введение группы R2, могут осуществляться в различном порядке. Наиболее подходящий выбор последовательности синтеза зависит от природы заместителей R1 -R4, n,X и других параметров, которые могут быть ясны специалисту в данной области техники. б) Аминопирролидины. Введение группы R2. Производные аминоалкилпирролидина общей формулы Ib (формула I, в которой X представляет собойNR4), где R1-R4 и n определены в формуле I, могут быть получены из соответствующих производных пирролидина Iia, полученных в а), из соединений формулы II в соответствии со схемой 1), в которой R1, R3, n иLG определены выше, замещением группы LG соответствующим амином HNR2R4, как показано на схеме 10. Схема 10 Альтернативный способ получения производных аминоалкилпирролидина общей формулы Ib(формула I, в которой X представляет собой NR4), в которой R1-R4 и n определены выше, описан на схеме 11. В соответствии с этой процедурой, гидроксигруппу производного пирролидина общей формулы II(которое может быть получено из соединений формулы IV; см. схему 2), в которой R1, R3 и n определены выше, окисляют до соответствующего альдегида в условиях, известных для такого превращения, например, ДМСО/(СОСl)2, ТЭА (условия Шверна) или реагент Десса-Мартина. Альдегид затем подвергают реакции с аминами HNR2R4, где R2 и R4 определены выше, в условиях восстановления. Схема 11 В случае производных аминоалкилпирролидина общей формулы Ib, в которой R4 представляет собой Н и R1, R2, R3 и n определены выше, может применяться альтернативный синтетический подход. Производные аминоалкилпирролидина общей формулы Ib (формула I, в которой X представляет собойNH), затем могут быть получены из соответствующих производных аминоалкилпирролидина формулыIII, в которой R1, R3 и n определены выше, прямым алкилированием с R2-LG, где R2 и LG определены выше, или восстановительным алкилированием альдегидом формулы R2 СНО, в которой R2 определен выше, и используя подходящий восстановительный агент, как показано на схеме 12. Схема 12 Производные аминоалкила формулы III, в которой R1, R3 и n определены выше, могут быть получены из гидроксиалкилпирролидина формулы II (который может быть получен из соединений формулы IV;- 11008479 см. схему 2), в которой R1, R3 и n определены выше, или производных общей формулы IIа (которые могут быть получены из соединений формулы II в соответствии со схемой 1), в которых R1, R3, n и LG определены выше, хорошо известными процедурами для таких превращений функциональных групп. Два примера таких превращений представлены на схеме 13. Схема 13 Для соединений формулы Ib (формула I, в которой X представляет собой NR4), в которой R2 представляет собой COR5, SO2R5, COOR5, CONR5R6, SO2NR5R6 , где R5 и R6 представляют собой замещенную или незамещенную алкильную или арильную группу и R4 представляет собой Н или замещенную или незамещенную алкильную или арильную группу, описанные выше на схемах 10, 11 и 12 способы являются неприменимыми. В этом случае соединения формулы Ib затем могут быть получены обработкой соединения общей формулы Iс (соединение формулы Ib, в которой R2 представляет собой Н, полученное способами, представленными на схемах 10, 11 или 12, с подходящими ацилирующими агентами, включая ацилхлорид или карбоновую кислоту в сочетании с пептидным конденсирующим агентом, например, DIC или EDC) сульфонирующим агентом и другими, как показано на схеме 14. Схема 14 Другие стадии, а именно, введение группы оксима, получение кетопирролидинов, стадия восстановления и стадия конденсации, описаны выше в разделе а). Однако как и для синтеза алкоксипирролидинов описанные выше четыре схемы основных химических превращений могут применяться в различном порядке. Наиболее подходящий выбор последовательности синтеза будет зависеть от природы заместителей R1-R4, n, X и других параметров, которые могут быть ясны специалисту в данной области техники. В качестве примера соединения общей формулы I, где R1-R4, n и X определены выше, могут быть получены из кетопирролидина общей формулы X, где R2, R3, n и X определены выше, реакцией с гидроксиламином V, где R1 определен выше, как описано на схеме 15, которая приводит к введению группы оксима. Схема 15 Кетопирролидин формулы X может быть получен окислением спирта общей формулы XI, где R2,R , n и X определены выше и PG2 представляет собой Н или подходящую О-защитную группу, в аналогичных условиях, как и для превращения соединений общей формулы VII в соединения общей формулы 3IV, как описано на схеме 16. Схема 16 Соединения общей формулы XI, в которой R2, R3, n, PG2 и X определены выше, могут быть получены из соединений общей формулы VII, в которой R3, n и PG1 определены выше, введением группы R2 с помощью одной из процедур, описанных на схемах 1, 10, 11, 12, 13 или 14. Выбор способа будет зависеть от природы R3, R2, n и X и ясен специалисту в данной области, так же как и выбор и последовательность подходящих стадий защиты/снятия защиты, как описано на схеме 17. Схема 17 Получение соединений формулы VII стадией восстановления уже описано выше. Последовательности реакций, показанные на приведенных выше схемах, приводят к получению энантиомерно чистых соединений формулы I, если использовались энантиомерно чистые исходные реагенты. (R)-, а также (S)-энантиомеры могут быть получены в зависимости от того, (R)- или (S)-формы коммерчески доступных соединений формул IX использовались в качестве исходных реагентов. Последовательности реакций, показанные на приведенных выше схемах реакций, обычно приводят к получению смесей (Е)- и (Z)-изомеров относительно заместителей на экзоциклической двойной связи пирролидинового кольца. (E)/(Z)-изомеры могут быть разделены стандартными хроматографическими методами, хорошо известными специалисту в данной области, такими как высокоэффективной жидкостной хроматографией с обратной фазой (ВЭЖХ) или ускоренной хроматографией на силикагеле (FC). Альтернативно, любой из (E)/(Z)-изомеров может быть успешно получен селективной кристаллизацией из подходящих растворителей или смеси растворителей. Значение абсолютной конфигурации экзоциклической двойной связи подтверждается с помощью ЯМР-технологий, описанных в уровне техники и хорошо известных специалисту в данной области (для конфигурации, например, групп оксима (9. Для повышения общего выхода одного изомера (обычно (Z)-изомер), другой изомер (обычно (Е)-изомер) может быть рециклизован преднамеренной переизомеризацией в органических растворителях, содержащих следовые количества кислоты, такой как НСl, с последующим повторным (E)/(Z)-разделением хроматографически и/или кристаллизацией. В соответствии с другим общим способом (схема 18), соединения формулы I могут быть преобразованы в альтернативные соединения формулы I с помощью подходящих методик внутреннего превращения, таких как описаны далее в примерах. Схема 18 Если указанные выше общие способы синтеза не применимы для получения соединений формулы I и/или необходимых промежуточных соединений для синтеза соединений формулы I, могут использоваться подходящие способы получения, известные специалисту в данной области. Вообще, путь синтеза индивидуального соединения формулы I будет зависеть от конкретных заместителей каждой молекулы и от легкодоступности необходимых промежуточных соединений; и опять же эти факторы ясны специалисту в данной области. Все способы защиты, снятия защиты см. (7, 10).- 13008479 Примеры Изобретение представлено следующими примерами, которые не служат для ограничения объема настоящего изобретения. Соединения настоящего изобретения могут синтезироваться различными способами синтеза, приведенными выше. Следующие примеры иллюстрируют предпочтительные способы получения соединений формулы I и определение их биологической активности. Пример I. О-метилоксим (3 ЕZ,5S)-5-(гидроксиметил)-1-[(2'-метил-1,1'-бифенил-4-ил)карбонил]пирролидин-3-она (1) (соединение формулы II или формулы Iа, в которой R2 представляет собой Н) Получение кетопирролидина. Коммерчески доступную (2S,4R)-1-(трет-бутоксикарбонил)-4-гидрокси-2-пирролидинкарбоновую кислоту (30 г, 0,13 моль) растворяли в ацетоне (1500 мл). В колбу помещали механическую мешалку и раствор энергично перемешивали. Свежеприготовленный раствор 8N хромовой кислоты получали растворением триоксида хрома (66,7 г, 0,667 моль) в воде (40 мл), добавляя концентрированную серную кислоту (53,3 мл) и добавляя достаточное количество воды до объема раствора 115 мл. 8N раствор хромовой кислоты (115 мл) затем по каплям добавляли в течение 30 мин, продолжая энергичное перемешивание, экзотермическую реакцию поддерживали при оптимальной температуре 25 С, применяя ледяную баню. После окончания добавления хромовой кислоты реакционную смесь перемешивали в течение 15 мин, поддерживая оптимальную температуру 25 С. Реакционную смесь затем гасили добавлением метанола (20 мл). Экзотермическую реакцию контролировали с помощью ледяной бани и, при необходимости, прямым добавлением небольшого количества раскрошенного льда непосредственно к реакционной смеси. Реакционную смесь отфильтровывали через целит pad и затем концентрировали в вакууме. Полученный кислотный раствор затем экстрагировали этилацетатом (3300 мл) и объединенные органические слои промывали соляным раствором (2100 мл), затем сушили сульфатом магния и концентрировали в вакууме. Сырой продукт перекристаллизовывали из этилацетата с получением белого кристаллического продукта, (2S)-1-(трет-бутоксикарбонил)-4-оксо-2-пирролидинкарбоновой кислоты (22,55 г, 76%) (1a). 1 Введение группы оксима. Готовили раствор, содержащий (2S)-1-(трет-бутоксикарбонил)-4-оксо-2-пирролидинкарбоновую кислоту (промежуточное соединение 1 а, 5,0 г, 21 ммоль) и О-гидрохлорид метилгидроксиламина (2,7 г,32,8 ммоль) в хлороформе (100 мл), содержащем триэтиламин (5,5 г, 55 ммоль). Реакционную смесь затем перемешивали при комнатной температуре в течение ночи до удаления растворителя. Полученную сырую реакционную смесь растворяли в этилацетате (150 мл) и промывали 1N HCl (40 мл). Кислотный слой затем экстрагировали этилацетатом (320 мл) и объединенные органические слои промывали соля- 14008479 ным раствором перед высушиванием над сульфатом магния, отфильтровыванием и удалением растворителя в вакууме. Полученный продукт (1b) (5,3 г, 94%) выделяли в виде светло-желтого масла. 1 Н ЯМР (400 МГц, CDCl3): 1,45 (m, 9H), 2,8-3,2 (m, 2H), 3,9 (s, 3 Н), 4,2 (m, 2H), 4,5-4,7 (m, 1H). Промежуточное соединение 1 с. 1-трет-Бутил-2-метил-(2S,4EZ)-4-(метоксиимино)пирролидин-1,2 дикарбоксилат. Получали раствор оксимэфира (2S,4EZ)-1-(трет-бутоксикарбонил)-4-(метоксиимино)-2-пирролидинкарбоновой кислоты (промежуточного соединения 1b, 0,648 г, 2,5 ммоль), в 1:1 смеси метанола и толуола (35 мл). Затем по каплям добавляли триметилсилилдиазометан (3,8 мл 2 М раствора в гексане, 7,5 ммоль) к перемешиваемому раствору при комнатной температуре под азотом. После окончания выделения азота полученный желтый раствор упаривали в вакууме и остаток отфильтровывали через фильтр с силикагелем, промывая этилацетатом. Удаление растворителя из фильтрата приводило к получению метилэфирного (1 с) продукта в виде желтого масла (0,646 г, выход 95%). Промежуточное соединение 1d. Метил-(2S,4EZ)-4-(метоксиимино)-1-[(2'-метил[1,1'-бифенил]-4-ил) карбонил]-2-пирролидинкарбоксилат. Конденсация группы R3. Готовили раствор, содержащий 1-трет-бутил-2-метил-(2S,4EZ)-4-(метоксиимино)-1,2-пирролидиндикарбоксилат (промежуточное соединение 1 с, 0,892 г, 3,28 ммоль) в безводном DCM (28 мл). По каплям добавляли ТФУК (20%, 7 мл). Смесь перемешивали при комнатной температуре в течение 20 мин. Растворители упаривали и желаемый продукт (0,564 г, количеств.) выделяли в виде желтого масла. Его растворяли в смеси 7:3 DCM и ДМФА (30 мл) и обрабатывали 2'-метил[1,1'-бифенил]-4-карбоновой кислотой (0,765 г, 3,60 ммоль) и 4-диметиламинопиридином (0,880 г, 7,21 ммоль). Медленно при 0 С добавляли EDC (0,691 мг, 3,60 ммоль) и реакционную смесь перемешивали в течение ночи при комнатной температуре. Ее промывали водой (дважды по 20 мл), сушили над MgSO4, отфильтровывали и упаривали в вакууме. Промежуточное соединение 1 е. (2S,4EZ)-4-(Метоксиимино)-1-[(2'-метил[1,1'-бифенил]-4-ил)карбонил]-2-пирролидинкарбоновая кислота. Введение группы R2. Метил(2S,4EZ)-4-(метоксиимино)-1-[(2'-метил[1,1'-бифенил]-4-ил)карбонил]-2-пирролидинкарбок- 15008479 силат (промежуточное соединение 1d, 391 мг, 1,06 ммоль) перемешивали при комнатной температуре в течение 4 ч в растворе, содержащем диоксан (9 мл), воду (3 мл) и NaOH (1,13 мл, 1,6N раствор). Диоксан удаляли в вакууме и раствор подкисляли обработкой 0,1N НСl. Его экстрагировали EtOAc, промывали соляным раствором, сушили над сульфатом магния, отфильтровывали и концентрировали с получением желаемого продукта (1 е) (342 мг, выход=91%). 1 Н ЯМР (300 МГц, CDCl3): 2,23 (s, 1,5H), 2,25 (s, 1,5 Н), 3,10 (m, 2 Н), 3,83 (s, 1,5H), 3,85 (s, 1,5H),4,10 (m, 2H), 5,18 (m, 1H), 7,18 (m, 4H), 7,37 (m, 2H), 7,57 (m, 2H).MS (APCI+): 353 (M+1) (APCI-): 351 (M+1). Пример I. (2S,4EZ)-4-(Метоксиимино)-1-[(2'-метил[1,1'-бифенил]-4-ил)карбонил]-2-пирролидинкарбоновую кислоту (промежуточное соединение 1 е, 50 мг, 0,14 ммоль) растворяли в ТГФ (1 мл) и обрабатывали этилхлороформатом (163 мкл, 0,17 ммоль) и ТЭА (29 мкл, 0,76 ммоль) при -15 С. Реакционную смесь перемешивали при этой температуре и под атмосферой азота в течение 30 мин перед добавлением боргидрида натрия (13,4 мг в 0,65 мл воды, 0,35 ммоль). Затем нагревали до комнатной температуры. Через 3 ч реакцию гасили 2,5 мл 1N раствора НСl и экстрагировали ЕtOАс трижды. Объединенные органические слои промывали 0,1N раствором НСl (трижды), водой (трижды), сушили над сульфатом магния, отфильтровывали и концентрировали с получением соединения (1). Выход: 12% (6 мг) Вид: желтое масло(16,9 г, 88,3 ммоль), HOBt (11,9 г, 88,3 ммоль) и DIEA (27,9 мл, 183,9 ммоль). Смесь затем перемешивали при комнатной температуре в течение 10 мин перед добавлением гидрохлорида транс-гидрокси-Lпролинметилэфира (10,7 г, 73,6 ммоль) и оставляли на 48 ч при комнатной температуре под атмосферой азота. Его затем концентрировали под высоким вакуумом и растворяли в этилацетате, промывали водой,1N раствором соляной кислоты, насыщенным раствором гидрокарбоната натрия и соляным раствором. Наконец его сушили над сульфатом магния, отфильтровывали и концентрировали. Сырой продукт очищали ускоренной хроматографией с циклогексан/этилацетатом 90:10 (соединение 2 а). Выход: 53% (12,6 г) Вид: коричневое твердое вещество 1 Н ЯМР (CDCl3): 2,11 (m, 1H), 2,36 (m, 1H), 3,58 (d, J=11,5 Гц, 1 Н), 3,77 (s, 3H), 3,86 (dd, J=3,4 и 11,1 Гц, 1H), 4,51 (s, 1H), 4,86 (t, J=8,3 Гц, 1 Н), 7,33-7,62 (m, 9H).- 16008479 Боргидрид лития (600 мг, 25,8 ммоль) медленно добавляли к раствору метилового эфира (2 а) (5,6 г,17,2 ммоль) в ТГФ (80 мл) при 0 С под атмосферой азота. Смесь перемешивали при комнатной температуре в течение 2 ч и боргидрид нейтрализовали водой. Белый осадок, содержащий соединение (2b), отфильтровывали и промывали эфиром. Выход: 82% (4,2 г) Вид: белое твердое вещество 1 Н ЯМР (ДМСО): 1,90-2,02 (m, 2 Н), 3,24-3,30 (m, 2 Н), 3,57 (m, 2 Н), 3,67 (m, 1 Н), 4,18 (m, 1 Н), 4,28DCM (150 мл) и обрабатывали DBU (421 мкл, 2,81 ммоль) и ТЭА (1,96 мл, 14,1 ммоль). Реакционную смесь затем перемешивали в течение 16 ч при комнатной температуре под атмосферой азота. После разбавления этилацетатом органическую фазу промывали водой. Водную фазу экстрагировали опять этилацетатом и объединенные органические фазы промывали насыщенным раствором хлорида аммония и три раза соляным раствором перед сушкой над сульфатом магния, отфильтровывали и концентрировали. Сырой продукт очищали ускоренной хроматографией с DCM/MeOH 95:5 (соединение 2 с). Выход: 75% (4,39 г) Вид: белый порошок 1 Н ЯМР (ДМСО): 0,03 (s, 6 Н), 0,88 (s, 9 Н), 1,92-2,04 (m, 2 Н), 3,30 (m, 1 Н), 3,54 (m, 1 Н), 3,72 (brd,J=9,0 Гц, 1 Н), 3,92 (m, 1 Н), 4,20 (m, 1 Н), 4,30 (m, 1 Н), 4,83 (m, 1 Н), 7,37-7,56 (m, 5 Н), 7,77 (m, 4 Н). Раствор сухого ДМСО (2,04 мл, 28,8 ммоль) в DCM (15 мл) медленно добавляли к раствору оксалилхлорида (1,34 мл, 15,7 ммоль) в DCM (5 мл) при -78 С под атмосферой азота. Смесь перемешивали в течение 30 мин перед медленным добавлением спирта (2 с) (5,38 г, 13,1 ммоль) в DCM (50 мл). Реакционную смесь перемешивали в течение 3 ч при -78 С, обрабатывали по каплям ТЭА (9,06 мл, 65,3 ммоль) и нагревали до комнатной температуры. Ее затем промывали соляным раствором, 1N раствором НСl, опять соляным раствором, сушили над сульфатом магния, отфильтровывали и концентрировали с получением соединения 2d. Выход: 91% (4,88 г) Вид: коричневое масло 1 Смесь кетона 2d (4,78 г, 11,7 ммоль), гидрохлорида метилгидроксиламина (2,44 г, 29,2 ммоль) и ТЭА (4,05 мл, 29,2 ммоль) в хлороформе (80 мл) нагревали при 65 С в течение 16 ч. Смесь затем промывали соляным раствором, 1N раствором НСl, опять соляным раствором и сушили над сульфатом магния,отфильтровывали и концентрировали с получением соединения 2 е. Выход: 86% (4,41 г) Вид: коричневое масло 1 Н ЯМР (CDCl3): 0,06 (s, 6H), 0,88 (s, 9H), 2,68-2,90 (m, 2H), 3,42 (m, 1H), 3,78 (s, 1,5H), 3,83 (s,1,5H), 4,1 (m, 2H), 4,31 (m, 1H), 4,83 (m, 1H), 7,34-7,64 (m, 9H). Раствор TBAF (14,1 мл раствора 1M B ТГФ, 14,1 ммоль) добавляли к раствору оксима (2 е) (4,13 г,9,41 ммоль) в ТГФ (100 мл). Реакционную смесь перемешивали при комнатной температуре в течение ночи. Затем концентрировали и разбавляли этилацетатом. Органическую фазу промывали водой, 1 Н раствором НСl и соляным раствором перед сушкой над сульфатом магния, отфильтровывали и концентрировали. Выход: количественный EZ смеси (2) Вид: белая пенаLC/MS: ESI (+): 325 (М+1) Два изомера Е и Z разделяли ускоренной хроматографией с помощью этилацетата/циклогексана 80:20 в качестве элюента. Менее полярная фракция: О-метилоксим (3 Е,5S)-1-(1,1'-бифенил-4-илкарбонил)-5-(гидроксиметил)пирролидин-3-она (3).Rf: 0,22 (AcOEt/циклогексан 80:20) Выход: 33% (1013 мг) Вид: белый порошок Температура плавления: 189 С 1 Н ЯМР (ДМСО): 2,64-2,82 (m, 2 Н), 3,20-3,57 (m, 3 Н), 3,70-3,80 (m, 3 Н), 3,98-4,60 (m, 2 Н), 5,0 (t,J=8,0 Гц, 1 Н), 7,37-7,76 (m, 9 Н). К перемешиваемому раствору спирта (2) (смесь EZ, 58 мг, 0,18 ммоль) и трет-бутилбромацетата(530 мкл, 3,6 ммоль) в дихлорметане (0,2 мл) добавляли 50% водный NaOH (0,8 мл) и тетрабутиламмонийхлорид (50 мг, 0,18 ммоль) при комнатной температуре и всю реакционную смесь перемешивали в течение 1 ч. После разбавления водой смесь экстрагировали этилацетатом, органическую фазу промывали соляным раствором, высушивали (MgSO4) и концентрировали. Продукт (соединение 5) очищали колоночной хроматографией на силикагеле, используя DCM:MeOH, 95:5 в качестве элюента. Выход: 99% (85 мг) К раствору трет-бутилового эфира (5) (45 мг, 0,1 ммоль) в дихлорметане (0,5 мл) добавляли трифторуксусную кислоту (0,1 мл) при комнатной температуре. После окончания реакции смесь концентрировали в вакууме. Сырой продукт растворяли в дихлорметане и промывали 1 М НСl. Органический слой сушили (MgSO4) и концентрировали. Выход: 40% (20 мг) Раствор кислоты (6) (15 мг, 0,04 ммоль), 1-(2-аминоэтил)пирролидина (6 мкл, 0,05 ммоль), DIC (7,2 мкл,0,05 ммоль) и DMАР (1 мг, 0,01 ммоль) в дихлорметане (1 мл) перемешивали под аргоном при комнатной температуре в течение 18 ч. Смесь концентрировали в вакууме и очищали препаративной хроматографией на силикагеле, используя DCM:MeOH, 50:50 в качестве элюента. Выход: 80% (17 мг) 1 Н ЯМР (CDCl3): 1,71 (s, 4H), 2,30-4,00 (m, 8H), 3,37 (m, 2H), 3,50-4,40 (m, 9 Н), 4,96 (m, 1 Н), 6,87 (m,1H), 7,30-7,70 (m, 9H). К раствору спирта (2) (смесь EZ, 20 мг, 0,06 ммоль) и гидрида натрия (3 мг, 0,12 ммоль) в тетрагидрофуране (1 мл) под аргоном добавляли метилйодид (7,7 мкл, 0,12 ммоль). Реакционную смесь перемешивали в течение ночи и гасили водой. Смесь разбавляли этилацетатом и промывали соляным раствором, сушили (MgSO4) и концентрировали в вакууме. Сырой продукт очищали препаративной хроматографией на силикагеле, используя DCM:MeOH 100:0, затем 95:5. Выход: 94% (21 мг) 1 Н ЯМР (CDCl3): 2,80 (m, 2H), 3,35 (m, 3 Н), 3,46 (m, 1H), 3,67 (m, 1H), 3,84 (s, 3H), 4,27 (m, 2H), 4,91DCM (8 мл), охлаждали до 0 С и оставляли под атмосферой азота. Реакционную смесь затем нагревали до комнатной температуры и наблюдали с помощью ТСХ. Реакция завершалась через 1 ч 30 мин. Органическую фазу промывали насыщенным раствором хлорида аммония и соляным раствором, сушили над сульфатом магния, отфильтровывали и концентрировали. Выход: количеств. (115 мг) Чистота ВЭЖХ: 87% Пример VII. Мезилат (9 а) (60 мг, 0,15 ммоль) растворяли в MEK/ACN (1:1, 10 мл) и обрабатывали бромидом лития (16 мг, 0,18 ммоль). Реакционную смесь нагревали до 85 С перед добавлением н-метилпиперазина (22 мг, 0,22 ммоль) и ТЭА (31 мкл, 0,22 ммоль) и перемешивали при этой температуре в течение ночи. Затем концентрировали, повторно растворяли в этилацетате и промывали насыщенным раствором NaHCO3,соляным раствором, сушили над сульфатом магния, отфильтровывали и концентрировали. Сырой продукт(48 мг) в конце очищали ускоренной хроматографией с DCM/MeOH/NH4OH 92:8:1 с получением соединения 9 а.(22 мкл, 0,16 ммоль) в метилэтилкетоне/ацетонитриле (2 мл, 1:1) перемешивали в течение 2 дней. Реакционную смесь разбавляли этилацетатом и промывали насыщ. NH4Cl. Органическую фазу сушили(MgSO4) и концентрировали. Сырой продукт очищали ВЭЖХ, используя систему PARALLEX FLEX. Выход: 21% (10 мг) Чистота ВЭЖХ: 72%(41 мг, 0,30 ммоль) и йодида натрия (225 мг, 1,50 ммоль) в тетрагидрофуране (5 мл) перемешивали в течение 2 дней. Реакционную смесь разбавляли этилацетатом и промывали 1N HCl, затем соляным раствором. Органическую фазу сушили (MgSO4) и концентрировали. Сырой продукт очищали, используя картридж С 8 SPE. Выход: 5% (3 мг) Раствор спирта (2) (смесь EZ, 51 мг, 0,16 ммоль), фталимида (70 мг, 0,48 ммоль), трифенилфосфина, связанного с полимером (158 мг, 0,48 ммоль) и диэтилазодикарбоксилата (40% в толуоле, 205 мкл, 0,48 ммоль) в тетрагидрофуране (5 мл) перемешивали в течение 2 дней. Смолу отфильтровывали и реакционную смесь концентрировали в вакууме. Сырой продукт очищали препаративной хроматографией на силикагеле, используя DCM в качестве элюента. Выход: 59% (50 мг) 1 Н ЯМР (CDCl3): 2,76 (m, 2 Н), 3,60-4,50 (m, 7 Н), 5,32 (m, 1H), 7,20-8,00 (m, 13H). Раствор фталимида (12) (42 мг, 0,09 ммоль), гидразина моногидрата (45 мкл, 0,93 ммоль) в этанолететрагидрофуране (1:1, 1 мл) перемешивали в течение ночи. Белый осадок отфильтровывали и фильтрат концентрировали в вакууме с получением желаемого амина. Выход: 76% (26 мг)(7,9 мкл, 0,06 ммоль) в дихлорметане перемешивали 30 мин. Реакционную смесь промывали водой. Органическую фазу сушили (MgSO4) и концентрировали в вакууме. Сырой продукт очищали препаративной хроматографией на силикагеле, используя этилацетат в качестве элюента. Выход: 42% (8 мг) Метил-(4R)-1-(1,1'-бифенил-4-илкарбонил)-4-гидрокси-L-пролинат (промежуточное соединение 2 а,2,07 г, 6,35 ммоль) растворяли в DCM (30 мл) и обрабатывали 4-DMAP (776 мг, 6,35 ммоль), ТЭА (2,21 мл,15,88 ммоль) и TBDMS-Cl (1,91 г, 12,7 ммоль). За реакцией наблюдали с помощью LC/MS. Через 24 ч после окончания реакции добавляли TBDMS-Cl (300 мг, 2 ммоль) и ТЭА (1 мл). Через 48 ч реакция завершалась. Смесь промывали насыщ. NH4Cl и соляным раствором (дважды), сушили над сульфатом магния, отфильтровывали и концентрировали. Сырой продукт (2,85 г) очищали ускоренной хроматографией,используя EtOAc/cHex 50:50 в качестве элюента. Выход: 93% (2,61 г) 1 Н ЯМР (CDCl3): -0,05 (s, 3 Н), 0,02 (s, 3 Н), 0,81 (s, 9H), 2,04 (m, 1H), 2,27 (m, 1H), 3,45 (d, J = 9,4 Гц,1H), 3,78 (s, 3H), 3,81 (m, 1H), 4,43 (m, 1H), 4,80 (t, J = 8,1 Гц, 1H), 7,33-7,46 (m, 3H), 7,62 (m, 6H). Промежуточное соединение 15b. 2S,4R)-1-(1,1'-Бифенил-4-илкарбонил)-4-[трет-бутил(диметил) силил]оксипирролидин-2-ил)метанол.- 23008479 Раствор метил-(4R)-1-(1,1'-бифенил-4-илкарбонил)-4-[трет-бутил(диметил)силил]окси-L-пролината (промежуточного соединения 15 а, 2,61 г, 5,94 ммоль) в ТГФ (60 мл) охлаждали до 0 С и обрабатывали боргидридом лития (95%, 206 мг, 8,9 ммоль). Реакционную смесь перемешивали в течение 3 ч и гасили медленно водой. ТГФ удаляли при пониженном давлении, сырой продукт повторно растворяли вAcOEt, промывали насыщ. NH4Cl, соляным раствором, сушили над сульфатом магния, отфильтровывали и концентрировали. Выход: 92% (2,268 г) К раствору спирта (15b) (200 мг, 0,49 ммоль) в дихлорметане (5 мл) под аргоном добавляли реагент Десса-Мартина (227 мг, 053 ммоль). Реакционную смесь перемешивали в течение 24 ч, затем разбавляли дихлорметаном и промывали насыщ. NaHCO3. Водный слой экстрагировали дихлорметаном. Органические фазы промывали водой, сушили (MgSO4) и концентрировали. Полученный альдегид непосредственно использовали на следующей стадии. К раствору альдегида (184 мг, 0,45 ммоль) в 1,2-дихлорэтане добавляли пиперидин (49 мкл, 0,50 ммоль), уксусную кислоту (28 мкл, 0,50 ммоль) и затем триацетоксиборгидрид натрия (143 мг, 0,68 ммоль). Реакцию перемешивали в течение ночи и затем разбавляли этилацетатом. Органическую фазу промывали насыщ. NaHCO3, затем соляным раствором. Органическую фазу сушили (MgSO4) и концентрировали с получением желаемого третичного амина. Выход: 95% (230 мг) Раствор защищенного спирта (15 с) (200 мг, 0,42 ммоль) и TBAF (0,63 мл, 1M B ТГФ) в тетрагидрофуране перемешивали при комнатной температуре в течение 1 ч. Реакционную смесь концентрировали и затем разбавляли ацетон-этилацетатом (1-2) и промывали насыщенным раствором NaHCO3. Органическую фазу сушили (MgSO4) и концентрировали с получением желаемого спирта. Выход: 51% (90 мг) Раствор ДМСО (46,8 мкл, 0,66 ммоль) в дихлорметане (1 мл) по каплям добавляли к раствору оксалилхлорида (28,2 мкл, 0,33 ммоль) в дихлорметане (2 мл) при -78 С под аргоном. Через 15 мин при -78 С по каплям добавляли раствор спирта (15d) (80 мг, 0,22 ммоль) в дихлорметане (1 мл). Реакционную смесь перемешивали при -78 С в течение 1 ч, обрабатывали триэтиламином (0,152 мл, 1,1 ммоль) и оставляли нагреваться до комнатной температуры. Реакционную смесь разбавляли этилацетатом, промывали водой, затем соляным раствором. Органическую фазу сушили (MgSO4) и концентрировали с получением желаемого кетона. Выход: 84% (78 мг)(ESI+): 363 (М+1) Чистота ВЭЖХ: 86% Пример XIII. Раствор кетона (15 е) (70 мг, 0,19 ммоль), гидрохлорида гидроксиламинметилового эфира (48 мг, 0,58 ммоль) и триэтиламина (80 мкл, 0,58 ммоль) в хлороформе (3 мл) перемешивали при 70 С в течение 2 дней. Реакционную смесь разбавляли дихлорметаном и промывали 1N HCl. Органическую фазу сушили (MgSO4) и концентрировали с получением эфира оксима. Выход: 90% (73 мг) К раствору коммерчески доступного гидрохлорида L-бета-гомогидроксипролина (245 мг, 1,35 ммоль),триэтиламина (1,13 мл, 8,09 ммоль) в воде (0,8 мл) и тетрагидрофуране (2 мл) при 0 С под аргоном по каплям добавляли раствор 4-фенилбензоилхлорида (438 мг, 2,02 ммоль) в тетрагидрофуране (1 мл). Реакционную смесь нагревали до комнатной температуры и перемешивали в течение 18 ч. Затем разбавляли смесью ацетона с этилацетатом (1-2) и промывали 1N HCl. Органическую фазу сушили (MgSO4) и концентрировали с получением смеси желаемого продукта и 4-фенилбензойной кислоты. Небольшое количество кислоты может быть получено осаждением этилацетатом. 1 Н ЯМР (ДМСО): 1,82 (m, 1 Н), 2,11 (m, 1 Н), 2,5 (m, 1 Н), 2,81 (dd, J=15,6 Гц, J=3,2, 1 Н), 3,3 (m, 1 Н),3,51 (dd, J=11,7 Гц, J=2,6 Гц, 1H), 4,16 (m, 1H), 4,40 (m, 1H), 7,30-8,00 (m, 9H). К раствору ранее полученной кислотной смеси (промежуточное соединение 16 а) в смеси толуол-метанол(10 мл, 1-1) добавляли диазометилтриметилсилан (2,76 мл, 2 М в гексане). Через 3 ч реакционную смесь концентрировали и очищали колоночной хроматографией на силикагеле, используя этилацетат в качестве элюента. Выход: 40% (с двух стадий, 256 мг) 1 Н ЯМР (ДМСО): 1,82 (т, 1 Н), 2,11 (m, 1 Н), 2,6 (dd, J=15,4 Гц, J= 8,3 Гц, 1 Н), 2,97 (dd, J=15,3 Гц,J=3,4 Гц, 1 Н), 3,25 (d, J=ll,4 Гц, 1 Н), 3,62 (s, 3H), 3,67 (dd, J=11,4 Гц, J=3,4 Гц, 1 Н), 4,16 (m, 1 Н), 4,44 (m,1 Н), 4,86 (d, J= 3,4 Гц, ОН) 7,30-8,00 (m, 9 Н). К раствору метилового эфира (16b) (310 мг, 0,91 ммоль) в тетрагидрофуране при 0 С под аргоном добавляли боргидрид лития (30 мг, 1,37 ммоль). Реакционную смесь нагревали до комнатной температуры и перемешивали в течение 12 ч. LiBH4 гасили водой и тетрагидрофуран упаривали в вакууме. Добавляли ацетонитрил и белый осадок отфильтровывали, промывали ацетонитрилом, затем эфиром и сушили. Выход: 97% (280 мг) 1 Н ЯМР (ДМСО): 1,69 (m, 1 Н), 1,87 (m, 1H), 2,15 (m, 2 Н), 3,35 (m, 1 Н), 3,57 (m, 2 Н), 3,72 (d, J=11,3 Гц,1 Н), 4,25 (m, 1H), 4,40 (m, 1 Н), 4,56 (m, ОН), 4,87 (m, ОН) 7,30-8,00 (m, 9 Н). К раствору диола (16 с) (270 мг, 0,87 ммоль) в диметилформамиде (10 мл) по каплям добавляли раствор трет-бутилдиметилсилилхлорида (131 мг, 0,87 ммоль) и триэтиламина (120 мкл, 0,87 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 2 дней. Добавляли этилацетат и реакционную смесь промывали водой. Водную фазу экстрагировали этилацетатом. Органические фазы сушили (MgSO4) и концентрировали в вакууме. Сырой продукт очищали препаративной хроматографией на силикагеле, используя смесь этилацетатциклогексан, 50:50 в качестве элюента. Раствор ДМСО (31,4 мкл, 0,44 ммоль) в дихлорметане (1 мл) по каплям добавляли к раствору оксалилхлорида (19 мкл, 0,22 ммоль) в дихлорметане (2 мл) при -78 С под аргоном. Через 15 мин при -78 С по каплям добавляли раствор спирта (16d) (63 мг, 0,15 ммоль) в дихлорметане (1 мл). Реакционную смесь перемешивали при -78 С в течение 1 ч и добавляли триэтиламин (0,102 мл, 0,74 ммоль) и нагревали до комнатной температуры. Реакционную смесь разбавляли этилацетатом, промывали водой, затем соляным раствором. Органическую фазу сушили (MgSO4) и концентрировали с получением желаемого кетона. Выход: 100% (64 мг) Раствор кетона (16 е) (64 мг, 0,15 ммоль), гидрохлорида гидроксиламинметилового эфира (38 мг,0,45 ммоль) и триэтиламина (62 мкл, 0,45 ммоль) в хлороформе (4 мл) перемешивали при 70 С в течение 5 дней. Реакционную смесь разбавляли дихлорметаном и промывали 1N HCl. Органическую фазу сушили (MgSO4) и концентрировали с получением желаемого эфира оксима. Выход: 96% (68 мг) 1LC/MS (ESI+): 453 (M+1) Пример XIV. Раствор защищенного спирта (16f) (68 мг, 0,15 ммоль) и TBAF (0,225 мл, 1M B ТГФ) в тетрагидрофуране перемешивали при комнатной температуре в течение 1 ч. Реакционную смесь концентрировали и затем разбавляли этилацетатом и промывали водой. Органическую фазу сушили (MgSO4) и концентрировали. Выход: 85% 1LC/MS (ESI+): 339 (M+1) Чистота ВЭЖХ: 96% ПримерХV. Получение фармацевтических составов. Следующие примеры составов иллюстрируют примерные фармацевтические композиции в соответствии с настоящим изобретением. Состав 1. Таблетки. Соединение пирролидина формулы I смешивали в виде сухого порошка с сухим желатиновым связующим веществом в приблизительном весовом соотношении 1:2. Добавляли минорное количество стеарата магния в качестве лубриканта. Смесь формовали в таблетки по 240-270 мг (80-90 мг активного соединения пирролидина на таблетку) в прессе для таблеток.- 27008479 Состав 2. Капсулы. Соединение пирролидина формулы I смешивали в виде сухого порошка с крахмальным разбавителем в приблизительном весовом соотношении 1:1. Смесь наполняли в 250 мг капсулы (125 мг активного соединения пирролидина на капсулу). Состав 3. Жидкость. Соединение пирролидина формулы I (1250 мг), сахарозу (1,75 г) и ксантановую смолу (4 мг) смешивали, просеивали через сито 10 US и затем смешивали с предварительно полученным раствором микрокристаллической целлюлозы и карбоксиметилцеллюлозы натрия (11:89, 50 мг) в воде. Бензоат натрия (10 мг), отдушку и краситель разбавляли водой и добавляли при перемешивании. Затем добавляли достаточное количество воды до общего объема 5 мл. Состав 4. Таблетки. Соединение пирролидина формулы I смешивали в виде сухого порошка с сухим желатиновым связующим веществом в приблизительном весовом соотношении 1:2. Добавляли минорное количество стеарата магния в качестве лубриканта. Смесь формовали в таблетки по 450-900 мг (150-300 мг активного соединения пирролидина) в прессе для таблеток. Состав 5. Инъекция. Соединение пирролидина формулы I растворяли в буферной стерильной соляной инъекционной водной среде до концентрации приблизительно 5 мг/мл. Пример XVI. Биологические испытания. Соединения формулы I могут быть подвергнуты следующим испытаниям.a) In vitro анализ конкурентного связывания с hOT рецептором с помощью Scintillation Proximity Assay (11). Этот анализ позволяет определять сродство тестируемого соединения относительно рецептора окситоцина человека (hOТ). Мембраны HEK293EBNA (клетки, экспрессирующие рецептор hOT) суспендировали в буфере, содержащем 50 мМ Трис-HCl, рН 7,4, 5 мМ MgCl2 и 0,1% БСА (вес/об.). Мембраны(2-4 мкг) смешивали с 0,1 мг шариками SPA, покрытыми аглютинином зародыша пшеницы (WGA-PVTполиэтилениминовые шарики от Amersham) и 0,2 нМ радиоактивно меченого [125I]-OVTA (OVTA орнитин вазоактивный, аналог ОТ для экспериментов конкурентного связывания). Неспецифическое связывание определяли в присутствии 1 мМ окситоцина. Общий объем испытания составлял 100 мкл. Пластины (Corning NBS пластины) инкубировали при комнатной температуре в течение 30 мин и подсчитывали на стинтилляционном счетчике Mibrobeta. Конкурентное связывание проводили в присутствии соединения формулы (I) при следующих концентрациях: 30 мкМ, 10 мкМ, 1 мкМ, 300 нМ, 100 нМ, 10 нМ,1 нМ, 100 пМ, 10 пМ. Данные конкурентного связывания анализировали с помощью итерационной, нелинейной программы построения кривых "Prism" (GraphPad Software, Inc.). Способность производных пирролидина формулы (I) ингибировать связывание 1-OVTA с ОТ-рецептором оценивали с помощью описанного выше биологического испытания in vitro. Полученные значения для некоторых примерных соединений приведены в табл. I, где сродство связывания тестируемых соединений в вышеупомянутых примерах выражено константой ингибирования (Ki; нМ). Из этих значений может быть видно, что указанные тестируемые соединения формулы I проявляют существенное связывание с рецептором окситоцина. Таблица IFLIPR (Fluorimetric Imaging Plate Reader). Действие ОТ на ОТ-рецептор вызывает сложный каскад в ячейке, который приводит к увеличению концентрации Са 2+ в цитоплазме. Это увеличение концентрации Са 2+ обусловлено высвобождением кальция из саркоплазмического ретикулума (хранилище кальция) в цитоплазму и притоком кальция из внеклеточного пространства через Са 2+ каналы. Эта мобилизация Са 2+ в цитоплазме вызывает машинальное сокращение миометрических клеток, которое приводит к сокращениям матки (1 и 3). Это испытание позволяет измерить ингибирование OT/OT-R медиированной мобилизации кальция тестируемых соединений формулы (I).FLIPR является флуориметрическим отображающим устройством, использующим лазер (лазер иона аргона) для одновременного освещения и считки (охлаждающая CCD камера) каждой ячейки 96 ячеечной подложки, таким образом позволяя быстрые измерения на большом количестве образцов. Подготовка пластин: FLIPR-пластины предварительно покрывали PLL (поли-L-лизин) 10 мкг/мл + 0,1% желатина для присоединения HEK293EBNA клеток (эмбриональные клетки почки человека, экспрессирующие рецептор hOT) и инкубировали в течение 30 мин 2 дня при 37 С. Клетки помещали в 96 ячеечные пластины (60000 клеток/ячейка). Мечение fluo-4: 50 мкг fluo-4 (Са 2+ чувствительный флуоресцентный краситель) растворяли в 20 мкл плурониевой кислоты (20% в ДМСО). Растворенный fluo-4 затем разбавляли 10 мл DMEM (минимальная среда для существования Dubecco)-F12 культуральной среды. Пластины промывали один раз средойDMEM-F12. 100 мкл fluo-4 содержащей-DМЕМ-F12 среды добавляли к НЕК-клеткам, которые инкубировали в течение 1,5-2 ч в этой флуоресцентной среде. Fluo-4 переносили в цитоплазму клеток. Буфер: 145 мМ NaCl, 5 мМ KСl, 1 мМ MgCl2, 10 мМ Hepes, 10 мМ глюкозы, EGTA (этилен-бисоксиэтиленнитрилотетрауксусная кислота). рН доводили до 7,4. Осуществление испытания: получали минимальное количество 80 мкл/ячейка соединений формулы (I)(5) в вышеупомянутом буфере (1) (96-ячеечные пластины). Соединения формулы (I) добавляли к 96-ячеечным пластинами в различных концентрациях (30 мкМ, 10 мкМ, 1 мкМ, 300 нМ, 100 нМ, 10 нМ, 1 нМ,100 пМ, 10 пМ). ОТ добавляли в концентрации 40 нМ. Затем измеряли относительную флюоресценцию Fluo-4 (ех=488 нм, еm=590 нм), с помощью FLIPR в присутствии или отсутствии соединений формулы (I). Флюоресценция маркера является чувствительной к количеству Са 2+, поэтому движения Са 2+ могут быть обнаружены. Затем можно определить способность соединений формулы (I) антагонизировать вызванной окситоцином внутриклеточной мобилизации Са 2+, опосредованной рецептором окситоцина. Активности производных пирролидина формулы I оценивали с помощью описанного выше биологического испытания in vitro. Полученные значения для некоторых примерных соединений представлены в табл. II. Значения относятся к концентрации тестируемых соединений формулы I, необходимой для антагонизма 50% OT/OTR внутриклеточной мобилизации Са 2+. Из этих значений видно, что указанные примерные соединения формулы I проявляют существенную активность в качестве антагонистов рецептора окситоцина. Таблица II в) Функциональный анализ 2. Ингибирование IP3 (инозитолтрифосфата)-Синтез в HEK/EBNAOTR клетках. Взаимодействие ОТ на ОТ-рецепторе приводит к синтезу IP3, вторичному мессенджеру для высвобождения Са 2+ из саркоплазмического ретикулума, включенного в процесс сокращения матки (3). Это испытание может использоваться для иллюстрации ингибирования OT/OT-R медиируемого синтеза IP3 с помощью тестируемых соединений формулы (I). Стимулирование клеток: клетки HEK/EBNA OTR (крысы или человека) помещали в 12-ячеечные(без FCS, инозитол), 20 мМ Hepes (4-(2-гидроксиэтил)-1-пиперазинэтансульфоновая кислота), 1 мг/мл БСА, содержащую 10 мМ LiCl (свежеприготовленную), и инкубировали в течение 10-15 мин при 37 С. В определенное (15-45 мин) время могут быть добавлены агонист (т.е. окситоцин, используемый при концентрации 10 нМ) и антагонисты (т.е. тестируемые соединения формулы (I), которые могут использоваться в концентрации 10 мкМ, 1 мкМ, 300 нМ, 100 нМ, 10 нМ, 1 нМ, 100 пМ, 10 пМ, 3 пМ), следом за введением среды. В присутствии ОТ радиоактивно меченый инозитол преобразуется в радиоактивно меченый IP3. Антагонистическое действие ОТ на ОТ-рецепторе ингибирует получение IP3. Количество радиоактивно меченого IP3 может быть определено следующим образом. Реакцию останавливали 1 мл STOP-раствора (т.е. 0,4 М перхлорной кислотой) и оставляли в течение 5-10 мин при комнатной температуре. Затем 0,8 мл переносили в пробирки, содержащие 0,4 мл нейтрализующего раствора (0,72 М KОН/0,6 М KНСО 3), и пробирки укупоривали и хранили в холоде по крайней мере в течение 2 ч. Разделение IP: образцы центрифугировали в настольной центрифуге при 3000-4000 об./мин в течение 15 мин. 1 мл супернатанта переносили в новые пробирки, содержащие 2,5 мл H2O. Упакованную смолу (Dowex AG1X8) уравновешивали с 20 мл Н 2 О и цельные образцы выливали на хроматографические колонки, разделяя таким образом смесь. Для удаления свободного инозитола осуществляли две промывки 10 мл Н 2 О. Элюирование общего IP. Элюирование осуществляли, используя 3 мл 1 М формата аммония/0,1 М муравьиную кислоту. Элюента собирали в пробирки стинцилляционного счетчика, затем добавляли 7 мл стинтилляционной жидкости. Количество [3 Н]-IР 3 определяли стинцилляционным счетчиком. Способность соединений формулы (I) эффективно антагонизировать окситоцин-вызванный синтезIP3, опосредованный рецептором окситоцина, может быть оценена с помощью описанного выше биологического испытания in vitro. г) In vivo модель для ингибирования сокращений матки. Испытание позволяет оценить биологическое действие тестируемых соединений in vivo в модели выкидыша, преждевременных родов. Небеременных Charles River CD (SD) BR самок крыс (9-10 недельного возраста, 200-250 г) обрабатывали за 18 и 24 ч перед экспериментом 250 мкг/кг внутрибрюшинно диэтилстилбестролом (DES). Для испытания животное анестезировали уретаном (1,75 г/кг, внутрибрюшинно) и помещали на гомеотермический операционный стол. Вырезали трахею и канюлировали подходящей полиэтиленовой (РЕ) трубкой. Делали средний разрез на уровне гипогастрия и один маточный роговой отросток выставляли, его цефалическое окончание канюлировали РЕ 240 трубкой и после наполнения внутренней полости 0,2 мл стерильного физиологического соляного раствора соединяли с усиливающей/записывающей системой"Gemini" через преобразователь давления P23ID Gould Statham. Одну яремную вену выделяли, канюлировали РЕ 60 трубкой и соединяли с иглой в виде бабочки для обеспечения внутривенного способа введения тестируемых соединений через распределительный шприц. В случае интрадуоденального введения тестируемых соединений, двенадцатиперстная кишка может быть выделена и подобным образом канюлирована через маленький разрез в его стенке. Одну сонную артерию также выделяли и канюлировали РЕ 60 катетером и соединяли с подходящим шприцом для забора образцов крови. После периода стабилизации и в процессе эксперимента, ту же дозу окситоцина неоднократно вводили внутривенно с интервалами 30 мин. Когда получали восстанавливаемые сокращения матки на тот же ОТ стимул (выбранная доза окситоцина), вводили дозу тестируемого соединения или ссылки (транспортное средство). Дальнейшие циклы введения той же дозы окситоцина продолжали (введение ОТ с интервалами 30 мин) в течение подходящего времени после лечения для оценки ингибирующих действий и обратимости этих действий. Сокращение матки в ответ на окситоцин определяли количественно, измеряя внутриматочное давление и число сокращений. Действие ссылки и тестируемых соединений оценивали, сравнивая значения давления перед и после лечения. Кроме того, сокращение матки измеряли через 5, 40, 75, 110, 145 и 180 мин после введения тестируемого соединения. Действия производных пирролидина формулы I могут быть оценены с помощью описанного выше биологического испытания in vivo. Полученные значения для одного примерного соединения приведены в табл. III. Значения относятся к способности примерного соединения формулы I эффективно антагонизировать окситоцинвызванные сокращения матки у крыс при введении внутривенным или оральным путем через 40 мин после введения тестируемого соединения. Из значений, показанных в табл. III, видно,что указанное примерное тестируемое соединение формулы I проявляет существенную активность в качестве токолитического, т.е. расслабляющего матку агента.
МПК / Метки
МПК: A61P 5/10, A61K 31/4025, C07D 521/00, C07D 403/06, C07D 207/22
Метки: качестве, производные, окситоцина, пирролидина, антагонистов
Код ссылки
<a href="https://eas.patents.su/30-8479-proizvodnye-pirrolidina-v-kachestve-antagonistov-oksitocina.html" rel="bookmark" title="База патентов Евразийского Союза">Производные пирролидина в качестве антагонистов окситоцина</a>
Производные индолин-2-она, их получение и их применение в качестве лигандов рецепторов окситоцина

Номер патента: 5093
Опубликовано: 28.10.2004
Авторы: Серрадейль-Ле Галь Клодин, Гарсия Жорж, Валетт Жерар, Фулон Луак
МПК: A61P 5/10, A61K 31/404, C07D 209/34...
Метки: получение, индолин-2-она, рецепторов, окситоцина, лигандов, качестве, применение, производные
Формула / Реферат:
1. Соединения в форме чистого энантиомера или смеси энантиомеров формулы в которой R0 представляет собой группу, выбранную из 1) в которой Z1 представляет собой атом хлора, брома, йода или фтора или (C1-C4)алкильную, (C1-C4)алкокси или трифторметильную группу; Z2 представляет собой атом водорода, хлора, брома, йода или фтора или (C1-C4)алкильную, (C3-C5)циклоалкильную, (C1-C4)алкокси, (C3-C5)циклоалкокси или полифтор(C1-C4)алкильную...
Производные пирролидина в качестве ингибиторов фосфодиэстеразы, специфичной к циклическому амф

Номер патента: 5686
Опубликовано: 28.04.2005
Авторы: Ньюхаус Брэдли Дж., Оливер Эйми, Мартинс Тимоти Дж., Кесицки Эдвард А., Бэрджесс Лоренс И., Фаулер Керри У., Шлахтер Стивен, Одинго Джошуа, Джоунс Закари С., Годино Джон Дж.
МПК: A61K 31/40, C07D 295/20
Метки: качестве, амф, пирролидина, ингибиторов, циклическому, производные, специфичной, фосфодиэстеразы
Формула / Реферат:
1. Соединение, имеющее формулу где R1 выбран из группы, включающей водород, C1-6алкил, мостиковый алкил, выбранный из группы, включающей норборнил, адамантил, бицикло[2.2.2]октил, бицикло[3.2.1]гептил, бицикло[3.2.1]октил, бицикло[4.1.0]гептил, бицикло[3.1.0]гексил и декагидронафтил, замещенный или незамещенный арил, выбранный из группы включающей фенил, нафтил, бифенил, тетрагидронафтил, инданил, 2-хлорфенил, 3-хлорфенил, 4-хлорфенил,...
N-замещённые производные пирролидина в качестве ингибиторов дипептидилпептидазы

Номер патента: 7968
Опубликовано: 27.02.2007
Авторы: Бёрингер Маркус, Лёффлер Бернд Михаэль, Хунцикер Даниель, Кюне Хольгер, Вессель Ханс Петер, Сарабу Рамакант
МПК: A61K 31/4025, A61K 31/401, A61K 31/4155...
Метки: дипептидилпептидазы, производные, n-замещённые, пирролидина, ингибиторов, качестве
Формула / Реферат:
1. Соединения формулы (I) в которой R1 обозначает Н или CN, R2 обозначает -C(R3,R4)-(CH2)n-R5, -C(R3,R4)-CH2-NH-R6, -C(R3,R4)-CH2-O-R7, тетралинил, тетрагидрохинолинил или тетрагидроизохинолинил, причем эта тетралинильная, тетрагидрохинолинильная или тетрагидроизохинолинильная группа может быть необязательно замещенной 1-3 заместителями, независимо выбранными из ряда, включающего (низш.)алкил, (низш.)алкокси, атом галогена, CN и CF3, R3...
Производные пиридилкарбамоилиндолинов в качестве антагонистов 5-нт2с-рецепторов

Номер патента: 1780
Опубликовано: 27.08.2001
Авторы: Бромидж Стивен Марк, Форбес Ян Томсон
МПК: C07D 401/14, A61P 25/28, A61K 31/4439...
Метки: 5-нт2с-рецепторов, производные, антагонистов, пиридилкарбамоилиндолинов, качестве
Формула / Реферат:
1. Соединение формулы (I) или его соль где Х обозначает СН или N; R1 обозначает водород или C1-6-алкил; R2 и R3 обозначают независимо C1-6-алкил или трифторметил. 2. Соединение по п.1, в котором Х обозначает СН. 3. Соединение по п.1 или 2, в котором R1 обозначает метил. 4. Соединение по любому из пп.1-3, в котором R2 обозначает СF3. 5. Соединение по любому из пп.1-4, в котором R3 обозначает C1-6-алкил. 6. Соединение по любому из пп.1-5, в...
Тиеноазепиновые производные в качестве антагонистов вазопрессина.

Номер патента: 1427
Опубликовано: 26.02.2001
Авторы: Делос Сантос Эфрен Гильермо, Олбрайт Джей Дональд
МПК: C07D 471/04, A61P 9/00, A61K 31/55...
Метки: антагонистов, вазопрессина, качестве, производные, тиеноазепиновые
Формула / Реферат:
1. Тиеноазепиновые производные общей формулы I где E-Y выбраны из радикалов -СН=СН-, -СН2-СН2- и R1 выбран из водорода и галогена; R3 представляет собой -СОАr, где Аr представляет собой радикал, выбранный из группы, включающей R5 выбран из водорода, (C1-С3)алкила, (C1-С3)алкокси и галогена; R6 выбран из (а) радикалов формул: где J представляет Ar' выбран из радикалов формул где W' выбран из О или S; (b) радикала формулы -M-Rd , ...