Производные бициклических гетероароматических соединений
Номер патента: 8410
Опубликовано: 27.04.2007
Авторы: Брукингз Дэниел Кристофер, Лэнгхэм Бэрри Джон, Дейвис Джереми Мартин







Формула / Реферат
1. Соединение общей формулы (1)

где пунктирная линия, соединяющая А и C(Ra), представляет собой связь и А означает группу -C(Rb);
Ra и Rb, каждый независимо, обозначает атом водорода или С1-4алкильную группу;
R обозначает атом водорода или линейную или разветвлённую C1-6алкильную группу;
X обозначает -S- атом;
Y обозначает -СН= или -C(R10);
R10 обозначает -CN или -CONH2;
n равно 0 или 1;
Alk1 обозначает C1-6алкиленовую цепь;
L1 обозначает ковалентную связь;
Су1 обозначает атом водорода или возможно замещённый С3-7циклоалкил, фенил, фурил, тиенил, пирролил, оксазолил, тиазолил, пиридил, пиримидинил, триазинил или индолил;
Аr обозначает возможно замещённые фенил, фурил, тиенил, пирролил, оксазолил, тиазолил, пиридил, пиримидинил или триазинил, причем возможные заместители Су1 и Аr выбраны из галогена, C1-6алкила, галоC1-6алкила, C1-6алкокси, галоC1-6алкокси, циано(-CN), -СО2СН3, -СО2С(СН3)3, нитро(-NO2), амино(-NH2), -NHCH3, -N(CH3)2, -C(O)CH3 и -NHCOCH3;
и его соли, сольваты, гидраты и N-окиси.
2. Соединение по п.1, отличающееся тем, что R обозначает атом водорода.
3. Соединение по п.1 или 2, отличающееся тем, что Ra обозначает атом водорода.
4. Соединение по любому из пп.1-3, отличающееся тем, что Rb обозначает атом водорода.
5. Соединение по любому из пп.1-4, отличающееся тем, что n равен 1 и Аlk1 обозначает -СН2- или
-СН2СН2-.
6. Соединение по любому из пп.1-4, отличающееся тем, что n равен 0.
7. Соединение по любому из пп.1-6, отличающееся тем, что Су1 обозначает возможно замещённую С3-7циклоалкильную группу.
8. Соединение по п.7, отличающееся тем, что Су1 обозначает циклопропильную группу.
9. Соединение по любому из пп.1-6, отличающееся тем, что Су1 обозначает возможно замещённую фенильную группу.
10. Соединение по п.9, отличающееся тем, что Су1 обозначает фенильную группу.
11. Соединение по любому из пп.1-6, отличающееся тем, что Су1 обозначает возможно замещённый фурил, тиенил, пирролил, оксазолил, тиазолил, пиридил, пиримидинил, триазолил или индолил.
12. Соединение по п.11, отличающееся тем, что Су1 обозначает тиенил, пиридил или индолил.
13. Соединение по любому из предшествующих пунктов, отличающееся тем, что Аr обозначает возможно замещённый фенил.
14. Соединение по любому из пп.1-13, отличающееся тем, что Y обозначает -C(R10), где R10 обозначает -CONH2.
15. Соединение по п.13, отличающееся тем, что Аr обозначает 3-метилфенил.
16. Фармацевтическая композиция, содержащая соединение по любому из пп.1-13 вместе с одним или несколькими фармацевтически приемлемыми носителями, эксципиентами или разбавителями.
17. 3-[(3-Метилфенил)амино]-6-оксо-7-фенил-6,7-дигидротиено[2,3-b]пиридин-2-карбоксамид или его фармацевтически приемлемая соль.
18. Фармацевтическая композиция, содержащая соединение по п.17 или его фармацевтически приемлемую соль вместе с одним или несколькими фармацевтически приемлемыми носителями, эксципиентами или разбавителями.
19. Применение соединения по п.17 для изготовления средств для лечения ревматоидных артритов.
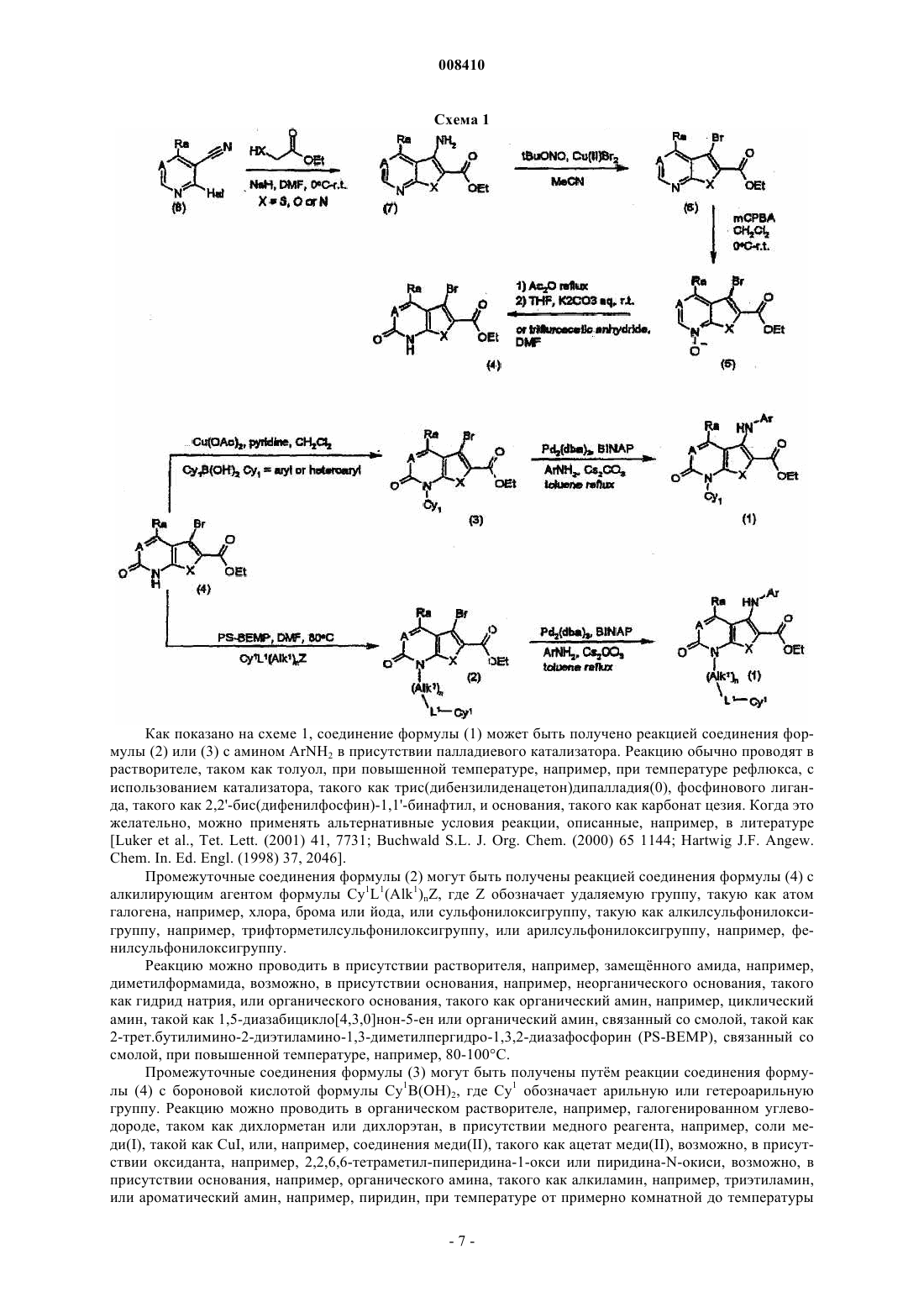
Текст
008410 Данное изобретение относится к группе 5-6 сконденсированных бициклических гетероароматических соединений, к композициям, содержащим их, к способам их получения и их применению в медицине. Иммунные и воспалительные реакции вовлекают многообразные типы клеток с регулированием и координацией различных видов взаимодействий, возникающих при контактах клетка-клетка (например,взаимодействие интегринов с их рецепторами) и внутриклеточных сигнальных молекул. Большое количество различных сигнальных молекул участвуют в этих процессах, включая цитокины, лимфоциты, хемокины и факторы роста. Клетки отвечают на такие внутриклеточные сигнальные молекулы посредством внутриклеточных сигнальных механизмов, которые включают протеинкиназы, фосфатазы и фосфолипазы. Существуют пять классов протеинкиназ, основными из которых являются тирозинкиназы и серин/треонинкиназы[Hunter Т., Methods in Enzymology (Protein Kinase Classification) p. 3, Hunter T. and Sefton B.M.; eds. vol. 200, Academic Press; San Diego, 1991]. Один подкласс серин/треонинкиназ представляет собой активирующие митоген протеинкиназы(MAP), среди которых существуют по меньшей мере три семейства, которые отличаются последовательностью и размером петли активации [Adams, J.L. et al., Progress in Medicinal Chemistry p. 1-60, King, F.D.and Oxford, A.W.; eds. vol. 38, Elsevier Science, 2001]: (i) внеклеточные регулируемые киназы (ERKS), (ii)c-Jun киназы с концевыми NH2-группами или киназы стрессов (JNKS или SAP киназы) и (iii) p38 киназы,которые содержат треонин-глицин-тирозин (TGY) фрагмент активации. И JNKS, и р 38 МАР-киназы в основном активируются стрессовыми раздражителями, включая, но не ограничиваясь этим, провоспалительные цитокины, например, фактор некроза опухоли (TNF) и интерлейкин-1 (IL-1), ультрафиолетовый свет, эндотоксин и химический или осмотический шок. Описаны четыре изоформы р 38 (р 38///). Фермент р 38 человека вначале был идентифицирован как мишень подавляющих цитокин противовоспалительных лекарств (CSAIDS) и два обнаруженных изофермента вначале обозначали как связывающий CSAID белок-1 (CSBP-1) и CSBP-2 [Lee J.C. et al.,Nature (London) 1994, 372, 739-46]. Теперь CSBP-2 повсеместно называют р 38, и он отличается отCSBP-1 внутренней последовательностью 25 аминокислот в результате дифференциального сплайсинга двух экзонов, которые сохраняются как у мыши, так и у человека [McDonnell P.С. et al., Genomics 1995,29, 301-2]. CSBP-1 и р 38 экспрессируются повсеместно и между двумя изоформами нет различия в отношении распределения в тканях, профиля активации, предпочтительности субстрата или связывания СSAID. Второй изоформой является р 38, которая идентична р 38 на 70%. Вторая форма р 38, называемая р 382, также известна и полагают, что из двух форм она является предпочтительной. р 38 и р 382 экспрессируются во многих различных тканях. Однако в моноцитах и макрофагах р 38 является более активной киназой [Lee J.C., ibid; Jing Y. et al., J. Biol. Chem. 1996, 271, 10531-34; Hale K.K. et al., J.Immun. 1999, 162, 4246-52]. p38 и р 38 (также называемые SAP-киназа-3 и SАР-киназа-4, соответственно) гомологичны р 38 на 63% и 61%, соответственно. р 38 преимущественно экспрессируется в мышцах, в то время как р 38 обнаруживается в яичках, поджелудочной железе, простате, тонкой кишке и в некоторых эндокринных тканях. Все гомологи р 38 и варианты сплайсинга содержат петлю активации из 12 аминокислот, которая включает фрагмент Thr-Grly-Tyr. Двойное фосфорилирование как Thr-180 и Thr-182 во фрагменте TGY при помощи киназы с двойной специфичностью против хода транскрипции является существенным для активации р 38 и приводит к более чем тысячекратному увеличению специфической активности этих ферментов [Doza Y.N. et al., FEBS Lett., 1995, 364, 7095-8012]. Это двойное фосфорилирование осуществляется при помощи МКК 6 и в определнных условиях при помощи фермента МКК 3 [Enslen H. et al., J.Biol. Chem., 1998, 273, 1741-48]. МКК 3 и МКК 6 принадлежат к семейству ферментов, называемых МАРКК (киназы, активируемые митогенами протеинкиназ), которые в свою очередь активируются МАРККК (киназами киназы МАР-киназы), иначе называемыми МАР 3 К. Некоторые MAP3KS были идентифицированы как активируемые самыми различными раздражителями, включая окружающую среду, воспалительные цитокины и другие факторы. МЕКК 4/МТК 1 (MAP или ERK киназа киназы/MAP трикиназы-1), ASK1 (стимулируемая апоптозом киназа) и ТАК 1 (активируемая TGF- киназа) представляют собой некоторые ферменты, идентифицируемые как активаторы против хода транскрипции для MAPKKS. Полагают, что МЕКК 4/МТК 1 активируется несколькими генами, подобными GADD-45, которые индуцируются в ответ на раздражители окружающей среды и которые в конечном счте приводят к активации р 38 [Takekawa M. and Saito H. Cell, 1998, 95, 521-30]. Было показано, что ТАК 1 активирует МКК 6 в ответ на трансформирующий фактор- роста (TGF-). Считается, что стимулируемая TNF активация р 38 опосредована рекрутированием TRAF2 (фактор, ассоциируемый с рецептором TNF) и адаптерным белком Fas, Daxx, который приводит к активации ASK1 и затем р 38. Были идентифицированы несколько субстратов р 38, включая другие киназы [например, протеинкиназа 2/3/5, активируемая МАРК (МАРКАР 2/3/5), протеинкиназа, регулируемая/активируемая р 38(PRAK), киназа 1/2, взаимодействующая с МАР-киназой (MNK1/2), протеинкиназа 1, активируемая ми-1 008410 тогеном и стрессом (MSK1/RLPK) и рибосомальная S6-киназа-В (RSK-B)]; факторы транскрипции [например, активирующий фактор транскрипции 2/6 (ATF2/6), энхансер моноцитов фактор-2 А/С(MEF2A/C), С/ЕВР гомологичный белок (CHOP), Elk1 и Sap-lal]; и другие субстраты (например, cPLA2,p47phox). МАРКАР К 2 активируется р 38 в ответ на стресс окружающей среды. Мыши, полученные без МАРКАР К 2, не продуцируют TNF в ответ на действие липополисахарида (LPS). Продуцирование нескольких других цитокинов, таких как IL-1, IL-6, IFN-g и IL-10, также частично ингибируется [Kotlyarov A. et al.,Natural Cell Biol. 1999, 1, 94-7]. Далее, МАРКАР К 2 из эмбриональных стволовых клеток из р 38 голых мышей не активируется стрессом, и эти клетки не продуцируют IL-6 в ответ на IL-1 [Allen М. et al., J.Exp. Med. 2000, 191, 859-69]. Эти результаты показывают, что МАРКАР К 2 является существенным не только для продуцирования TNF и IL-1, но также и для сигнализации, индуцированной цитокинами. Кроме того, МАРКАР К 2/3 фосфорилируют и тем самым регулируют протеины теплового шока HSP 25 и HSP 27, которые вовлечены в реорганизацию цитоскелета. Были описаны несколько ингибиторов р 38, являющихся малыми молекулами, которые ингибируют синтез IL-1 и TNF в моноцитах человека в концентрациях в области низких значений мкМ [Lee J.C. et al.,Int. J. Immunopharm, 1988, 10, 835] и проявляют активность в животных моделях, которые рефракторны на ингибиторы циклооксигеназы [Lee J.C. et al., Annals N.Y. Acad. Sci. 1993, 696, 149]. В добавление эти ингибиторы с малыми молекулами, как известно, снижают образование большого числа провоспалительных белков, включая IL-6, IL-8, гранулоцито-макрофагный колониестимулирующий фактор (GMCSF) и циклооксигеназу-2 (СОХ-2). Индуцированное TNF фосфорилирование и активация цитозольногоPLA2, индуцированная TNF экспрессия VCAM-1 на эндотелиальных клетках и стимулируемый IL-1 синтез коллагеназы и стромелизина также ингибируются малыми молекулами р 38 [Cohen P. Trends Cell.Biol. 1997, 7, 353-61]. Многие клетки, включая моноциты и макрофаги, продуцируют TNF и IL-1. Избыточное или нерегулируемое продуцирование TNF влечт за собой ряд болезненных состояний, включая болезнь Крона,язвенный колит, изжогу, ревматоидный артрит, ревматоидный спондилит, остеоартрит, подагрический артрит и другие артриты, синдром токсического шока, эндотоксический шок, сепсис, септический шок,грам-отрицательный сепсис, резорбцию костей, повреждения, вызываемые реперфузией, реакцию организма хозяина на чужеродное тело, отторжение аллографта, синдром респираторного дистресса у взрослых, хроническое воспаление лгких, силикоз, лгочный саркоидоз, церебральную малярию, образование шрамов, образование келоидных рубцов, лихорадку и миалгии, вызванные инфекцией, такой как грипп,кахексию, вызванную синдромом приобретнного иммунодефицита (AIDS), кахексию, вызванную инфекцией или злокачественными образованьями, СПИД или осложнения, связанные со СПИДом. Избыточное или нерегулируемое продуцирование IL-1 влечт за собой ревматоидный артрит, остеоартрит, травматический артрит, артрит, вызванный вирусом Rubella, острый синовит, псориазный артрит, кахексию, синдром Рейтера, эндотоксемию, синдром токсического шока, туберкулз, атеросклероз, дегенерацию мышц и другие острые или хронические воспалительные заболевания, такие как воспалительная реакция, вызванная эндотоксином, или воспаление кишечника. Кроме того, IL-1 связан с диабетом и -клетками поджелудочной железы [Dinarello С.A. J. Clinical Immunology, 1985, 5, 287-97].IL-8 является хемотактическим фактором, продуцируемым различными типами клеток, включая эндотелиальные клетки, моноядерные клетки, фибробласты и кератиноциты. IL-1, TNF и LPS - все индуцируют образование IL-8 эндотелиальными клетками. Было показано, что in vitro IL-8 имеет ряд функций, включая функцию хемоаттрактанта для нейтрофилов, Т-лимфоцитов и базофилов. IL-8 также увеличивает поверхностную экспрессию Мас-1 (CD11b/CD18) на нейтрофилах без de novo синтеза белков,который может способствовать повышенной адгезии нейтрофилов к сосудистым эндотелиальным клеткам. Многие заболевания характеризуются массовой инфильтрацией нейтрофилов. Высвобождение гистамина из базофилов (атопических и нормальных) индуцируется IL-8 как высвобождение лизосомальных ферментов и респираторый взрыв в нейтрофилах. Центральная роль IL-1 и TNF вместе с другими цитокинами из лейкоцитов как важных и определяющих медиаторов воспалительных реакций хорошо известна. Ингибирование этих цитокинов было описано и ожидается, что это будет благоприятным для регулирования, ослабления или уменьшения многих из этих болезненных состояний. Центральное положение, которое р 38 занимает в каскаде сигнальных молекул, опосредующих внеклеточные сигналы во внутриклеточные, и его влияние не только на продуцирование IL-1, TNF и IL-8, но также и на синтез и/или действие других провоспалительных белков (например, IL-6, GM-CSF,COX-2,коллагеназу и стромелизин) делает его привлекательной мишенью для ингибирования малыми молекулами с ожиданием того, что такое ингибирование будет высокоэффективным механизмом регулирования избыточной и деструктивной активации иммунной системы. Такое ожидание поддерживается сильной и разнообразной противовоспалительной активностью, описанной для ингибиторов р 38 киназы [Adams,ibid; Badger et al., J. Pharm. Exp. Ther. 1996, 279, 1453-61; Griswold et al., Pharmacol. Comm. 1996, 7, 32329].-2 008410 В соответствии с настоящим изобретением найдена группа соединений, которые являются эффективными и селективными ингибиторами киназы р 38 (р 38 , ,и ) и е изоформы и варианты сплайсинга, особенно, р 38, р 38 и р 382. Эти соединения могут применяться в медицине, например, для профилактики и лечения иммунных и воспалительных заболеваний, как описано в данном изобретении. Согласно одному аспекту изобретения мы получили соединение общей формулы (1) где пунктирная линия, соединяющая А и C(Ra), представляет собой связь и А означает группу -C(Rb);Ra и Rb каждый независимо обозначает атом водорода или С 1-4 алкильную группу;R обозначает атом водорода или линейную или разветвлнную С 1-6 алкильную группу;L1 обозначает ковалентную связь; Су 1 обозначает атом водорода или возможно замещнный С 3-7 циклоалкил, фенил, фурил, тиенил,пирролил, оксазолил, тиазолил, пиридил, пиримидинил, триазинил или индолил; Аr обозначает возможно замещнные фенил, фурил, тиенил, пирролил, оксазолил, тиазолил, пиридил, пиримидинил или триазинил, причем возможные заместители Су 1 и Аr выбраны из галогена,C1-6 алкила, галоC1-6 алкила, C1-6 алкокси, галоC1-6 алкокси, циано(-CN), -СО 2 СН 3, -О 2 С(СН 3)3, нитро(-NO2),амино(-NH2), -NHCH3, -N(CH3)2, -C(O)CH3 и -NHCOCH3; и его соли, сольваты, гидраты и N-окиси. Особенно пригодные соединения по изобретению включают каждое из соединений, описанных ниже в примерах, и их соли, сольваты, гидраты и N-окиси. Наиболее предпочтительным является соединение 3-[(3-метилфенил)амино]-6-оксо-7-фенил-6,7 дигидротиено[2,3-b]пиридин-2-карбоксамид или его фармацевтически приемлемая соль. Соединения согласно изобретению являются эффективными и селективными ингибиторами киназ р 38, включая все изоформы и варианты сплайсинга. Более конкретно, соединения по изобретению являются ингибиторами р 38, р 38 и р 382. Способность соединений действовать таким образом можно определить просто с применением исследований, описанных ниже в примерах. Соединения формулы (1) применяются при модулировании активности киназ р 38 и, в частности,они полезны для профилактики и лечения любых заболеваний и расстройств, опосредованных любой киназой р 38, у человека или другого млекопитающего. Изобретение охватывает такое применение и применение соединений для изготовления лекарства для лечения таких заболеваний или расстройств. Далее, изобретение охватывает введение человеку эффективного количества ингибитора р 38 для лечения любого такого заболевания или расстройства. Данное изобретение относится также к профилактике и лечению любого заболевания или расстройства, в которых играет роль киназа р 38, включая условия, вызванные избыточным или нерегулированным продуцированием противовоспалительного цитокина, включая, например, избыточное или нерегулируемое образование TNF, IL-1, IL-6 и IL-8 у человека или другого млекопитающего. Изобретение охватывает такое применение и применение соединений для изготовления лекарства для лечения таких болезней или расстройств, опосредованных цитокинами. Далее, изобретение относится к введению человеку эффективного количества ингибитора р 38 для лечения любого такого заболевания или расстройства. Заболевания или расстройства, в которых киназа р 38 играет роль или непосредственно, или через провоспалительные цитокины, включая цитокины TNF, IL-1, IL-6 и IL-8, включают, без ограничения,аутоиммунные заболевания, воспалительные заболевания, разрушение костной ткани, пролиферативные заболевания, нейродегенеративные расстройства, вирусные заболевания, аллергию, инфекционные болезни, сердечные приступы ангиогенные расстройства, ишемию при реперфузии в результате инсульта,сосудистую гиперплазию, гипоксию органов, сердечную гипертрофию, агрегацию тромбоцитов, вызванную тромбином, и состояния, ассоциированные простагландином-эндопероксидазой-синтетазой-2 (СОХ 2). Аутоиммунные заболевания, которые можно предотвратить или подвергнуть лечению, включают,-3 008410 но не ограничиваются этим, ревматоидный артрит, воспаление кишечника, язвенный колит, болезнь Крона, рассеянный склероз, диабет, гломерулонефрит, системную атрофическую волчанку, склеродермию, хронический тиреоидит, болезнь Грейва, гемолитическую анемию, аутоиммунный гастрит, аутоиммунную нейтропению, тромбоцитопению, хронический активный гепатит, миастению gravis, атопический дерматит, реакцию трансплантат против хозяина или псориаз. Настоящее изобретение, далее, относится к конкретному аутоиммунному заболеванию - ревматоидному артриту. Воспалительные болезни, которые можно предотвратить или подвергнуть лечению, включают, но не ограничиваются этим, астму, аллергию, респираторный дистресс-синдром или острый или хронический панкреатит. Деструктивные заболевания костей, которые можно предотвратить или подвергнуть лечению,включают, но не ограничиваются этим, остеопороз, остеоартрит и связанное с множественными миеломами разрушение костной ткани. Пролиферативные заболевания, которые можно предотвратить или подвергнуть лечению, включают, но не ограничиваются этим, острую или хроническую миелогенную лейкемию, саркому Капоши,метастатическую меланому и множественную миелому. Нейродегенеративные заболевания, которые можно предотвратить или подвергнуть лечению,включают, но не ограничиваются этим, болезнь Паркинсона, болезнь Альцгеймера, церебральную ишемию или нейродегенеративное заболевание, вызванное травмой. Вирусные болезни, которые можно предотвратить или подвергнуть лечению, включают, но не ограничиваются этим, острый гепатит (включая гепатит А, гепатит В и гепатит С), ВИЧ-инфекцию и CMV ретинит. Инфекционные болезни, которые можно предотвратить или подвергнуть лечению, включают, но не ограничиваются этим, септический шок, сепсис и шигеллз. Кроме того, ингибиторы р 38 по изобретению также ингибируют экспрессию индуцируемых провоспалительных белков, таких как простагландин-эндопероксидаза-синтетаза-2, известная как циклооксигеназа-2 (СОХ-2) и поэтому их можно применять в терапии. Провоспалительные медиаторы циклооксигеназного пути на основе архидоновой кислоты продуцируются индуцируемым ферментом СОХ-2. Регуляция СОХ-2 будет регулировать эти провоспалительные медиаторы, такие как простагландины,которые влияют на разнообразные клетки и являются важными и критическими воспалительными медиаторами большого количества заболеваний и состояний. В частности, эти воспалительные медиаторы вовлечены в проявление боли, такое как сенсибилизация болевых рецепторов или отк. Соответственно,дополнительные состояния, опосредованные р 38, которые можно предотвратить или подвергнуть лечению, - отк, аналгезию, лихорадку и боль, например нервно-мышечную боль, головную боль, зубную боль, артритную боль и боль, вызванную раком. В результате их ингибирующей р 38 активности соединения по изобретению полезны при профилактике и лечении болезней, связанных с образованием цитокинов, включая, но не ограничиваясь этим,болезни, ассоциируемые с образованием TNF, IL-1, IL-6 и IL-8. Так, опосредованные TNF болезни или состояния включают, например, ревматоидный артрит, ревматоидный спондилит, остеоартрит, подагрический артрит и другие артриты, сепсис, синдром септического шока, респираторный дистресс-синдром у взрослых, церебральную малярию, хроническое воспаление лгких, силикоз, лгочный саркоидоз, резорбция костной ткани, повреждение при реперфузии,реакцию имплантата на хозяина, отторжение аллографта, лихорадку и миалгию вследствие инфекции,кахексию, вызванную инфекцией, СПИД, АРС или злокачественные образования, келоидные рубцы, образование шрамов, болезнь Крона, язвенный колит, изжогу, вирусные инфекции, такие как ВИЧ, CMV,грипп и герпес; и ветеринарные вирусные инфекции, такие как инфекции, вызванные лентивирусом,включая, но не ограничиваясь этим, вирус оспы, вирус козьего артрита, вирус висны или маеди; или ретровирусные инфекции, включая вирус иммунодефицита кошки, вирус иммунодефицита коровы или вирус иммунодефицита собаки. Соединения по изобретению можно использовать для лечения вирусных инфекций, когда такие вирусы способствуют in vivo образованию TNF или чувствительны к нерегуляции при помощи TNF. Такие вирусы включают те, которые продуцируют TNF в результате инфекции, и те, которые чувствительны к ингибированию, например, в результате пониженной репликации, непосредственно или косвенно, соединениями по изобретению, ингибирующими TNF. Такие вирусы включают, но не ограничиваются этим,ВИЧ-1, ВИЧ-2 и ВИЧ-3, цитомегаловирус (CMV), вирус гриппа, аденовирус и группу вирусов герпеса,такого как герпес Зостера и герпес Simplex. Опосредованные IL-1 заболевания или состояния включают, например, ревматоидный артрит, остеоартрит, псориазный артрит, травматический артрит, артрит rubella, воспаление кишечника, удар, наличие в крови эндотоксинов и/или синдром токсического шока, воспалительную реакцию, вызванную эндотоксином, диабет, заболевание клеток поджелудочной железы, болезнь Альцгеймера, туберкулз,атеросклероз, мышечную дегенерацию и кахексию. Опосредованные IL-8 заболевания или состояния включают, например, такие, которые характери-4 008410 зуются массивной инфильтрацией нейтрофилов, такие как псориаз, воспаление кишечника, астма, заболевания сердца, повреждения при реперфузии мозга и почек, респираторный дистресс-синдром у взрослых, тромбоз и гломерулонефрит. Повышенное образование IL-8, связанное с каждым из этих заболеваний, ответственно за хемотаксис нейтрофилов в воспалительные сайты. Это обусловлено уникальным свойством IL-8 (по сравнению с TNF, IL-1 и IL-6) промотировать хемотаксис и активацию нейтрофилов. Следовательно, ингибирование образования IL-8 приведт к прямому уменьшению инфильтрации нейтрофилов. Известно также, что и IL-6, и IL-8 продуцируются риновирусом (HRV) и вносят вклад в патогенез обычной простуды и обострение астмы, связанные с HRV инфекцией [Turner et al., Clin. Infec. Dis., 1997,26, 840; Grunberg et al., Am. J. Crit. Care Med. 1997, 155, 1362; Zhu et al., J. Clin. Invest. 1996, 97, 421]. Было также показано in vitro, что инфекция лгочных эпителиальных клеток (которые являются первичным сайтом инфекции HRV), вызванная HRV, приводит к продуцированию IL-6 и IL-8 [Sabauste et al., J. Clin.Invest. 1995, 96, 549]. Следовательно, ингибиторы р 38 по изобретению могут быть использованы для лечения или профилактики обычной простуды или респираторной вирусной инфекции, вызванной риновирусом человека (HRV), другими энтеровирусами, коронавирусом, вирусом гриппа, вирусом парагриппа,респираторным синциальным вирусом или аденовирусом. Для профилактики или лечения болезней, опосредованных р 38 или провоспалительными цитокинами, соединения по изобретению могут вводиться человеку или другому млекопитающему в виде фармацевтических композиций, и согласно дальнейшему аспекту изобретения предусмотрена фармацевтическая композиция, которая включает соединения формулы (1) вместе с одним или более фармацевтически приемлемыми носителями, эксципиентами или разбавителями. Фармацевтические композиции по изобретению могут быть в форме, пригодной для орального,буккального, парентерального, назального, топического, офтальмического или ректального введения или в форме, пригодной для введения путм ингаляции или инсуффляции. Для орального введения фармацевтические композиции могут быть в форме, например, таблеток,леденцов или капсул, полученных обычными средствами с фармацевтически приемлемыми эксципиентами, такими как связующие агенты (например, желатинизированный маисовый крахмал, поливинилпирролидон или гидроксипропилметилцеллюлоза); наполнителями (например, лактоза, микрокристаллическая целлюлоза или кислый фосфат кальция); смазочными агентами (например, стеарат магния, тальк или двуокись кремния); дезинтегрантами (например, картофельный крахмал или гликолят натрия); или смачивающими агентами (например, лаурилсульфат натрия). Таблетки могут содержать покрытие, нанеснное способами, хорошо известными из уровня техники. Жидкие препараты для орального введения могут быть в виде, например, растворов, сиропов или суспензий, или они могу быть в виде сухого продукта, воссоздаваемого водой или другим подходящим носителем перед употреблением. Такие жидкие препараты могут быть получены обычными методами с фармацевтически приемлемыми добавками, такими как суспендирующие агенты, эмульгирующие агенты, неводные носители и стабилизаторы. Препараты могут также содержать буферные соли, ароматизаторы, красители и подсластители. Препараты для орального введения могут быть получены в виде форм с регулируемым высвобождением активного соединения. Для буккального введения композиции могут быть в виде таблеток или леденцов, полученных обычными методами. Соединения формулы (1) могут входить в состав препарата дли парентерального введения путм инъекции, например, инъекции или инфузии болюса. Составы для инъекции могут быть в виде единичных дозированных форм, например в стеклянной ампуле, или многодозированных контейнерах, например в стеклянных пузырьках. Композиции для инъекции могут быть в виде суспензий, растворов или эмульсий в масляных или водных носителях и могут содержать целевые добавки, например, суспендирующие, стабилизирующие агенты, консерванты и/или диспергаторы. Альтернативно, активный ингредиент может быть в виде порошка для разбавления подходящим носителем, например стерильной апирогенной водой, перед использованием. Кроме препаратов, описанных выше, соединения формулы (1) могут быть в виде препаратов-депо. Такие составы, действующие длительное время, могут вводиться путм имплантации или внутримышечной инъекции. Для назального введения или введения путм ингаляции соединения по изобретению обычно поставляются в виде аэрозолей для ингалятора или распылителя с использованием подходящего распыляющего газа, например, дихлордифторметана, трихлорфторметана, дихлортетрафторэтана, двуокиси углерода или другого подходящего газа или смеси газов. Если желательно, композиции могут быть представлены в упаковке или в системе, которая может содержать одну или несколько единичных дозированных форм, содержащих активный ингредиент. Упаковка или система для доставки могут быть снабжены инструкциями по применению. Для топического введения соединения по изобретению могут входить в состав подходящей мази,содержащей активный компонент, суспендированный или растворнный в одном или более фармацевтически приемлемых носителей. Конкретные носители включают, например, минеральное масло, жидкую нефть, пропиленгликоль, полиоксиэтилен, полиоксипропилен, эмульгирующий воск и воду. Кроме того,-5 008410 соединения по изобретению могут входить в состав подходящего лосьона, содержащего активный компонент, суспендированный или растворнный в одном или нескольких фармацевтически приемлемых носителях. Конкретные носители включают, например, минеральное масло, моностеарат сорбитана, полисорбат 60, воскоподобные цетиловые эфиры, цетариловый спирт, бензиловый спирт, 2-октилдодеканол и воду. Для офтальмического введения соединения по изобретению могут входить в состав микронизированных суспензий в изотоническом стерильном физиологическом растворе с определнным рН или с консервантом, таким как бактерицидный или фунгицидный агент, например нитрат фенилртути, хлорид бензалконил или ацетат хлоргексидина, или без консерванта. Кроме того, для офтальмического введения можно применять мазь, содержащую соединения по изобретению и петролатум. Для ректального введения соединения по изобретению могут входить в состав суппозиториев. Они могут быть приготовлены смешением активного компонента с подходящим, не раздражающим эксципиентом, который является тврдым при комнатной температуре и жидким при ректальной температуре и будет плавиться в заднем проходе с высвобождением активного компонента. Такие вещества включают,например, масло какао, пчелиный воск и полиэтиленгликоли. Количество соединения по изобретению, требующееся для профилактики или лечения конкретного состояния, будет колебаться в зависимости от выбранного соединения и состояния пациента, подвергающегося лечению. В общем, однако, дневные дозы могут колебаться от примерно 100 нг/кг до 100 мг/кг, например, от примерно 0,01 до 40 мг/кг веса пациента для орального или буккального введения, от примерно 10 до 50 мг/кг веса пациента для парентерального введения и от примерно 0,05 до 1000 мг,например, от примерно 0,5 до примерно 1000 мг для назального введения или введения путм ингаляции или инсуффляции. Соединения по изобретению могут быть получены несколькими способами, описанными в общем ниже и более конкретно в примерах. При описании способов символы Аr, Су 1, Alk1, n, L1, R, Ra, Rb, Rc, A,X и Y, используемые в формулах на схемах, представляют группы, описанные выше для формул (1 а) и(1b), если не указано иное. В реакциях, описанных ниже, может быть необходимым защитить реакционноспособные функциональные группы, например, гидроксил, амино, тио или карбоксильную группы,когда они желательны в конечном продукте, чтобы избежать их нежелательного участия в реакциях. В соответствии с обычной практикой можно использовать обычные защитные группы [см., например,Green, T.W. in Protective Groups in Organic Synthesis, John Wiley and Sons, 1999]. В некоторых случаях снятие защиты может быть конечной стадией синтеза соединения формулы (1), и способы по изобретению, описанные ниже, распространяются и на удаление защитных групп. Таким образом, согласно дальнейшему аспекту изобретения соединение формулы (1), где А обозначает -C(Rb)=, X обозначает -О- или -S- или группу -NH- и Y обозначает насыщенный атом углерода, у которого заместителем является этерифицированная карбоксильная группа, например, -CO2Alk6, может быть получено согласно реакциям, показанным на схеме 1 ниже. На схеме показано получение конкретного этилового эфира, но следует иметь в виду, что другие эфиры могут быть получены путм применения другого исходного вещества и, если нужно, изменением условий реакции. Как показано на схеме 1, соединение формулы (1) может быть получено реакцией соединения формулы (2) или (3) с амином ArNH2 в присутствии палладиевого катализатора. Реакцию обычно проводят в растворителе, таком как толуол, при повышенной температуре, например, при температуре рефлюкса, с использованием катализатора, такого как трис(дибензилиденацетон)дипалладия(0), фосфинового лиганда, такого как 2,2'-бис(дифенилфосфин)-1,1'-бинафтил, и основания, такого как карбонат цезия. Когда это желательно, можно применять альтернативные условия реакции, описанные, например, в литературеChem. In. Ed. Engl. (1998) 37, 2046]. Промежуточные соединения формулы (2) могут быть получены реакцией соединения формулы (4) с алкилирующим агентом формулы Су 1L1(Alk1)nZ, где Z обозначает удаляемую группу, такую как атом галогена, например, хлора, брома или йода, или сульфонилоксигруппу, такую как алкилсульфонилоксигруппу, например, трифторметилсульфонилоксигруппу, или арилсульфонилоксигруппу, например, фенилсульфонилоксигруппу. Реакцию можно проводить в присутствии растворителя, например, замещнного амида, например,диметилформамида, возможно, в присутствии основания, например, неорганического основания, такого как гидрид натрия, или органического основания, такого как органический амин, например, циклический амин, такой как 1,5-диазабицикло[4,3,0]нон-5-ен или органический амин, связанный со смолой, такой как 2-трет.бутилимино-2-диэтиламино-1,3-диметилпергидро-1,3,2-диазафосфорин (PS-BEMP), связанный со смолой, при повышенной температуре, например, 80-100 С. Промежуточные соединения формулы (3) могут быть получены путм реакции соединения формулы (4) с бороновой кислотой формулы Су 1 В(ОН)2, где Су 1 обозначает арильную или гетероарильную группу. Реакцию можно проводить в органическом растворителе, например, галогенированном углеводороде, таком как дихлорметан или дихлорэтан, в присутствии медного реагента, например, соли меди(I), такой как CuI, или, например, соединения меди(II), такого как ацетат меди(II), возможно, в присутствии оксиданта, например, 2,2,6,6-тетраметил-пиперидина-1-окси или пиридина-N-окиси, возможно, в присутствии основания, например, органического амина, такого как алкиламин, например, триэтиламин,или ароматический амин, например, пиридин, при температуре от примерно комнатной до температурыLetters, 2001, 3415]. Промежуточные соединения формулы (3), где Су 1 обозначает арильную или гетероарильную группу, могут быть также получены путм нуклеофильного ароматического замещения подходящим образом активированного арил- или гетероарилгалоидных соединений с соединением формулы(4). Реакцию можно осуществить в диалкиламидном растворителе, таком как диметилформамид, в присутствии основания, такого как гидрид металла, например, гидрид натрия, при температуре от примерно комнатной до 100 С. Подходящим образом активированные арил- или гетероарилгалоидные соединения - это такие галоидангидриды, которые содержат заместитель, оттягивающий электроны, такой как нитро-, циано или сложноэфирная группа, например, хлор- или фторнитробензол или 2-хлор-5 нитропиридин. Альтернативно, азотсодержащее гетероарилгалоидное соединение может быть активировано с получением нуклеофильного замещения путм N-окисления, например, галоидпиридин-N-оксид, такой как хлорпиридина N-оксид, например, 2-хлорпиридина N-оксид. Следует отметить, что, если желательно, только что описанные реакции могут проводиться в обратном порядке, так что аминирование с применением ArNH2 осуществляется вначале с получением промежуточного продукта формулы (4) с последующим алкилированием/арилированием с получением соединения формулы (1). Может быть необходимым защитить азотную функцию соединений формулы (4) во время осуществления этих реакций. Такая защита может быть достигнута О-алкилированием алкилгалоидом, например, циклопропилметил-бромидом или арилалкилбромидом, например бензилбромидом,как показано на схеме 1 а. Схема 1 а Реакцию О-алкилирования можно проводить в органическом растворителе, таком как диметилформамид, в присутствии основания, например, неорганического основания, такого как Cs2CO3, или органического основания, такого как амин, например циклический амин, такой как 1,5-диазабицикло[4,3,0]нон 5-ен, при повышенной температуре, например, при 80-100 С, с образованием соединения формулы (13). Реакцию защищенного соединения (13) с ArNH2 в присутствии палладиевых катализаторов можно проводить, как описано ранее, с образованием соединения (14). Снятие защиты можно осуществить путм обработки раствора этого соединения в спирте, например метаноле, минеральной кислотой, такой как концентрированная НСl, при повышенной температуре, например, при температуре рефлюкса с получением соединения формулы (15). Альтернативно, когда используется бензильная защита, эта группа может быть удалена путм восстановления при обработке раствора соединения (14) в растворителе, например воде или органическом растворителе, таком как этанол, с использованием палладиевого или платинового катализатора, например, палладия на угле или РtO2, при повышенном давлении водорода при температуре от примерно комнатной до 60 С. Соединения формулы (15) могут затем подвергаться реакциям алкилирования/арилирования, как описано выше, с получением соединений формулы (1). Промежуточные пиридиноны формулы (4) могут быть получены из пиридина N-окиси формулы (5) путм последовательной реакции с ангидридом, например уксусным ангидридом, при повышенной температуре,например, при температуре рефлюкса, с последующей реакцией с неорганическим основанием, например карбонатом, таким как водный карбонат калия, в растворителе, таком как простой эфир, например циклический эфир, такой как тетрагидрофуран, при примерно комнатной температуре. Альтернативно, реакцию можно проводить с применением трифторуксусного ангидрида в диметилформамиде при температуре от 0 С до комнатной температуры [см., например, Коnnо et al., Heterocycles (1985) 24, 2169].N-оксиды пиридина формулы (5) могут быть получены путм окисления пиридинов формулы (6) с применением окисляющего агента, такого как перекись водорода, в присутствии кислоты, такой как уксусная кислота, при повышенной температуре, например при примерно 70-80 С, или же путм реакции перкислотой, такой как перуксусная кислота или м-хлорпероксибензойная кислота, в среде растворителя,-8 008410 такого как галогенированный углеводород, например, дихлорметан или спирт, например, трет.бутанол,при температуре от комнатной температуры до температуры рефлюкса. Промежуточные пиридины формулы (6) на схеме 1 могут быть получены стандартными методами, например, по реакции Сандмейера(Sandmeyer). Так, например, бромид формулы (6) может быть получен обработкой ариламина формулы(7) алкилнитритом, например трет.бутилнитритом, и солью меди, например, бромидом меди(II) в присутствии растворителя, например, нитрила, такого как ацетонитрил, при температуре от примерно 0 С до примерно 65 С. Амины формулы (7) могут быть получены из 2-галопиридин-3-карбонитрилов формулы (8) путм реакции с реагентом формулы HXCH2CO2Et (где Et обозначает этильную группу и X обозначает атом -Оили -S- или группу -NH-). Реакцию можно осуществлять в присутствии растворителя, такого как замещнный амид, например диметилформамид, или простой эфир, например циклический эфир, такой как тетрагидрофуран, или спирт, такой как этанол, в присутствии основания, например неорганического основания, такого как карбонат натрия, или гидрид, например гидрид натрия, или органического основания, такого как 1,5-диазабицикло[4,3,0]нон-5-ен, или триалкиламин, например триэтиламин, при температуре между примерно 0 и 100 С. Исходные карбонитрилы являются легко доступными или могут быть получены из известных соединений по стандартным методикам. Согласно другому способу по изобретению соединение формулы (1), где А обозначает -C(Rb)=, X обозначает атом -О- или -S- или группу -NH-, и Y обозначает -C(CN)=, может быть получено с применением реакций, представленных на схеме 2 ниже. Схема 2 Так, на схеме 2 показано, что 2-циан-промежуточное соединение формулы (9) можно аминировать и алкилировать или арилировать на конечной стадии с получением соединения по изобретению с использованием реакций и реагентов, описанных выше в отношении аминирования, алкилирования и арилирования промежуточных формулы (4). Нитрилы формулы (9) могут быть получены дегидратацией соответствующего амида формулы (10) с использованием дегидратирующего агента, такого как трифторуксусный ангидрид, в присутствии основания, такого как пиридин, в растворителе, например в галогенированном углеводороде, таком как дихлорметан, при примерно комнатной температуре. Альтернативно,можно использовать цианурхлорид в среде растворителя, такого как диметилформамид, при температуре от примерно 0 до 110 С. Амиды формулы (10) могут быть получены из соответствующих кислот формулы (11) с использованием обычных процедур, например, путм реакции с 1,1'-карбонилдиимидазолом и водным аммиаком в растворителе, таком как диметилформамид, при комнатной температуре. Промежуточные кислоты формулы (11) можно получить гидролизом сложных эфиров формулы (4), используя основание, такое как гидроокись лития, в воде или растворителе, таком как тетрагидрофуран. Согласно другому способу по изобретению соединение формулы (1), где А обозначает -C(Rb)=, X обозначает атом -О- или -S-, Y обозначает группу -C(CN)=, Аr обозначает гетероароматическую группу иAlk1, L, n и Су 1 указаны ранее, может быть получено путм нуклеофильного ароматического замещения надлежащим образом активированного гетероарилгалоидного соединения соединением формулы (16). Эту реакцию можно осуществить в среде органического растворителя, такого как простой эфир, например тетрагидрофуран, или диалкиламид, например диметилформамид, или растворителя, такого как диметилсульфоксид, в присутствии неорганического основания, такого как гидрид натрия, при повышенной температуре 50-80 С. Активированные гетероарил-галоидные соединения включают галопиридины,например хлорпиридин, такой как 2,6-дихлорпиридин, или галопиридина N-окиси, например хлорпири-9 008410 дина N-окиси, такие как 2-хлор-6-метилпиридина N-окись. Амиды формулы (1 а) можно получить из кислот формулы (17), используя обычные процедуры, показанные на схеме 4. Схема 4 Так, амиды могут быть получены по реакции кислоты (17) с 1,1'-карбонилдиимидазолом и амином формулы HNR15R15 (где каждый R15 независимо обозначает Н или R11, описанные ранее) или водного аммиака в среде растворителя, такого как амидный растворитель, например диметилформамид, при температуре от примерно комнатной до 60 С. Или же кислота может реагировать с активирующим агентом,таким как карбодиимид, например 1-(3-диметиламинопропил)-3-этилкарбодиимид или N,N'дициклогексилкарбодиимид, и амином HNR15R15, возможно, в присутствии основания, такого как амин,например триэтиламин или N-метилморфолин, в среде растворителя, такого как диметилформамид или простой эфир, например тетрагидрофуран, и галогенированный углеводород, например дихлорметан,при температуре от примерно комнатной до температуры рефлюкса. Согласно другому способу реакция кислоты формулы (17) с 1-(3-диметиламинопропил)-3-этилкарбодиимидом и пентафторфенолом в среде растворителя, такого как амид, например, ДМФ, при примерно комнатной температуре позволяет получить пентафторфениловый эфир формулы (18). Амиды формулы (1 а) могут быть получены затем по реакции этого эфира с аминами формулы HNR15R15 или с водным аммиаком в среде растворителя, такого как галогенированный углеводород, например дихлорметан, при примерно комнатной температуре. Промежуточные кислоты формулы (17) могут быть получены гидролизом эфиров формулы (1), что показано на схеме 1, с использованием основания, такого как гидроокись щелочного металла, например гидроокись натрия или гидроокись лития, в среде воды или растворителя, такого как тетрагидрофуран или спирт, такой как этанол, при температуре от примерно комнатной до температуры рефлюкса. Амиды формулы (1 а) могут быть также получены непосредственно из эфиров формулы (1), показанных на схеме 1, путм нагревания их с амином формулы HNRI5R15 при нагревании до температуры рефлюкса этого амина или при атмосферном давлении, или под давлением в герметичной трубке. Карбоксамиды формулы (1b), где X обозначает атом -О- или -S- и Y обозначает группу -С(СONH2)=, и где А,Аr, Alk1, L, n и Су 1 указаны выше, также могут быть получены по реакции эфира формулы (1) с жидким аммиаком в среде растворителя, такого как 2-этоксиэтанол, при повышенном давлении (200-400 ф/дюйм 2, что соответствует 1379-2758 кПа) в автоклаве Parr и при температуре 60-120 С. Согласно другому способу карбоксамиды формулы (1b), описанные выше, могут быть также получены гидролизом нитрила формулы (1 с) с основанием, таким как гидроокись щелочного металла, например гидроокись натрия или гидроокись калия, в среде воды и растворителя, такого как спирт, например этанол, при температуре от примерно комнатной до температуры рефлюкса. Согласно другому способу соединения формулы (1), где А обозначает -С(Н)=, Ra обозначает Н, X обозначает атом -S-, Y обозначает группу -C(CR10a)=, где R10a обозначает -CN, -NO2, -CONH2, -CONHR11,-CON(R11)2, -COR11 или -CO2Alk6, L обозначает ковалентную связь, n=0 и Су 1, R11, Alk6 и Аr указаны выше, могут быть получены, как показано на схеме 5. Схема 5 Так, на стадии (А) реакционной схемы соединение формул (19) или (20), где Rx обозначает возможно замещнную алкильную группу, например метил, и W обозначает атом водорода, ион металла или соль амина, реагирует с тиоамидом формулы (21). Реакцию можно проводить в присутствии основания. Соответствующие основания могут включать, но не ограничиваются этим, основания лития, такие как нбутиллитий или трет.бутиллитий, или диизопропиламид лития (LDA), или силазаны, например гексаметилдисилазан лития (LiHMDS) или гексаметилдисилазан натрия (NaHMDS), или карбонат, например карбонат калия, алкоксид, например этоксид натрия, метоксид натрия, трет.бутоксид калия, гидроокись,например NaOH, или гидрид, например гидрид натрия, или органический амин, например триэтиламин или N,N-диизопропилэтиламин, или циклический амин, такой как N-метилморфолин или пиридин. Реакцию можно проводить в среде органического растворителя, такого как амид, например замещнный амид, такой как диметилформамид, простой эфир, например циклический эфир, такой как тетрагидрофуран или диоксан, или спирт, например метанол, этанол или пропанол, или ацетонитрил, при температуре от комнатной до температуры рефлюкса. Согласно одному конкретному аспекту способа реакция проходит с использованием основания алкоксида, особенно этоксида натрия или метоксида натрия, в среде спиртового растворителя, особенно этанола, при температуре рефлюкса. Промежуточные продукты формулы (19), если они не являются коммерчески доступными, могут быть получены по стандартным методикам (см., например, Mir Hedayatullah, J. Heterocyclic Chem., 18,339 (1981. Точно также, промежуточные продукты формулы (20), не являющиеся коммерчески доступными, могут быть получены по стандартным методикам. Например, они могут быть получены in situ взаимодействием ацетата, например этилацетата, с основанием, таким как метоксид натрия, с последующим добавлением формиата, например метилформиата. Подробным образом промежуточные продукты формулы (21), не являющиеся коммерчески доступными, могут быть получены известными методами (см., например, Adhikari et al., Aust. J. Chem., 52, 6367, (1999. Например, изотиоцианат формулы Cy1NCS может реагировать с ацетонитрилом в присутствии основания, например, NaHMDS, в среде подходящего растворителя, например тетрагидрофурана,возможно, при низкой температуре, например, примерно -78 С. Согласно природе группы Су 1 промежуточное формулы (21) может быть получено in situ, например, с применением описанных здесь методов с последующим добавлением соединения формулы (19) или (20). В ходе процесса может образоваться промежуточное формулы (22). Если желательно, это промежуточное может быть выделено в конце стадии (А) и затем реагировать с промежуточным соединением (23) с получением желательного амина (24). Промежуточное соединение формулы (22) и реакция (В) могут осуществляться непосредственно с реакционной смесью со стадии (А). Если на второй стадии процесса используют другой растворитель, может быть необходимым выпа- 11008410 ривание растворителя в вакууме после первой стадии процесса перед второй стадией. После выпаривания сырой продукт со стадии (А) можно использовать на следующей стадии, или он может быть очищен,например, кристаллизацией, с получением выделенного промежуточного продукта, такого как соединение формулы (22). Во время стадии (В) промежуточное соединение формулы (23) может быть добавлено в реакционную смесь или к сырому продукту, или к очищенному продукту со стадии (А) в подходящем растворителе. Подходящие растворители включают, но не ограничиваются этим, амиды, например замещнный амид, такой как диметилформамид, спирты, например этанол, метанол или изопропиловый спирт, простые эфиры, например циклический эфир, такой как тетрагидрофуран или диоксан, или ацетонитрил. Реакцию можно проводить при температуре от комнатной до температуры рефлюкса. Во время стадии (В) может получаться промежуточное соединение формулы (26) Оно даже может быть выделено в зависимости от природы группы R. Промежуточное соединение формулы (26) может быть превращено в соединение формулы (24) с использованием методов, описанных выше. В этой ситуации может быть необходимым добавление основания, чтобы реакция полностью завершилась. Соответствующие основания включают карбонаты, например карбонат цезия или калия,или алкоксиды, например трет.бутоксид калия, или гидриды, например гидрид натрия, или органические амины, например триэтиламин или N,N-диизопропилэтиламин или циклические амины, такие как Nметилморфолин или пиридин. Амины формулы (24) могут быть превращены в бромиды формулы (25) стандартными методами,такими как реакция Сандмейера, описанная ранее для соединений формулы (7). Затем из этих бромидов могут быть получены соединения формулы (1) по реакции аминирования, катализируемой палладием,которая описана выше. Следует отметить, что промежуточные соединения формулы (23), не являющиеся коммерчески доступными, могут быть получены стандартными методами, известными специалистам. Например, спиртовые группы могут быть превращены в удаляемые, такие как атомы галогена или сульфонилоксигруппы,при условиях, известных специалистам. Например, спирт может реагировать с тионилхлоридом в среде галогенированного углеводорода, например, дихлорметана, с получением соответствующегохлорида. В реакции может быть также использовано основание, например триэтиламин. Следует отметить, что промежуточные соединения, такие как соединения (19), (20), (21) или (23), не являющиеся коммерчески доступными, могут быть также получены известными способами, описанными в таких источниках, какSons, 1992). Согласно дальнейшему аспекту изобретения соединение формулы (1), где X обозначает атом -S-, Y обозначает группу -CS(O2)NR16R16= (где каждый R16 независимо обозначает Н или R11, указанный ранее),может быть получено способом, показанным на схеме 6. Схема 6 Так, соединение формулы (28) может быть получено по реакции соединения (27) с амидом металла,таким как бис(триметилсилил)амид натрия, в среде растворителя, такого как простой эфир, например циклический эфир, такой как тетрагидрофуран, при температуре примерно 0 С с последующим добавлением ди-трет.бутилдикарбоната в среде растворителя, такого как тетрагидрофуран, при перемешивании при комнатной температуре. Затем может быть получено соединение формулы (1) по следующей схеме.- 12008410 Соединение формулы (28) обрабатывают основанием, таким как литийалкил, например литий-н.бутил, в среде растворителя, такого как эфир, например циклический эфир, такой как тетрагидрофуран, при температуре примерно -78 С. Газообразный серный ангидрид пропускают через реакционную смесь до того,как дать температуре реакции подняться до комнатной. Растворители удаляют в вакууме и сырой продукт растворяют в растворителе, таком как галогенированный углеводород, например дихлорметан, и смесь обрабатывают хлорирующим реагентом, таким как N-хлорсукцинимид, при примерно комнатной температуре. Затем к реакционной смеси для получения соединения формулы (29), где R обозначает трет.бутоксикарбонил, можно добавить амин формулы HNR16R16. Сульфонамид формулы (1) может быть затем получен обработкой соединения формулы (29) кислотой, например минеральной, такой как НСl,или органической кислотой, такой как трифторуксусная кислота, в среде растворителя, такого как галогенированный углеводород, например дихлорметан. Промежуточные соединения формулы (27) могут быть получены декарбоксилированием соединений формулы (17) при помощи кислоты, такой как минеральная кислота, например НСl, в среде растворителя, такого как простой эфир, например циклический эфир, такой как тетрагидрофуран или 1,4-диоксан, при температуре от 50 С до температуры рефлюкса. Соединения формулы (1), где А обозначает атом -N=, может быть получено с использованием методов синтеза, показанных на схемах (1) и (2), из исходных пиримидинов формулы (12) где Hal обозначает атом галогена и LG обозначает галоген, такой как хлор или такую группу, как метилсульфанил или метилсульфонил. Соединение (30) и его аналоги, где LG обозначает метилсульфанильную группу, известны [Santilli et al., J. Heterocycl. Chem. 8, 445-453 (1971)]. Соединение формулы(30) может подвергаться реакции, такой как реакция Sandmeyer, описанная для соединений формулы (7) с получением бромида формулы (31). На последующих стадиях метилсульфанильная группа или другая уходящая группа LG, содержащиеся в соединениях формулы (31), могут быть гидролизованы с использованием основания, такого как гидроокись натрия или калия, в среде растворителя, такого как спирт,например метанол или этанол, при повышенной температуре, например, при температуре рефлюкса. Альтернативно, группа LG вначале может быть превращена в простую эфирную группу по реакции с алкоксидом, таким как метоксид натрия или фенилметанолят натрия, в среде растворителя, такого как спирт, например, метанола или этанола, при температуре от 0 С до температуры рефлюкса, полученный эфир затем расщепляют, используя стандартные процедуры, например, восстановление газообразным водородом в присутствии катализатора, такого как палладиевый катализатор, например палладий на древесном угле, или когда эфир представляет собой алкиловый эфир, путм реакции с триалкилсилилгалоидным соединением, таким как триметилсилилхлорид, в присутствии неорганического галоидсодержащего соединения, такого как иодид натрия, в среде растворителя, такого как галогенированный углеводород, например дихлорметан, или нитрил, такой как ацетонитрил. Это позволит открыть пиримидиноновые функциональные группы, которые могут быть или алкилированы, или арилированы, как описано для соединений формулы (4), и могут быть превращены в соединения формулы (1), как описано ранее. Соединения по изобретению или их промежуточные, где А обозначает -N(Rb)- или -C(Rb)(Rc)-, могут быть получены из соответствующих соединений по изобретению и промежуточных соединений, где А обозначает -N= или -C(Rb)=, путм восстановления, например, каталитическим гидрированием, с использованием катализатора на основе металла, такого как палладий на древесном угле, в присутствии газообразного водорода при повышенном давлении в среде растворителя, такого как спирт, например этанол, возможно, при повышенной температуре, например, при 40-60 С. Если в описанных выше общих способах промежуточные соединения, такие как алкилирующие агенты формулы Cy1L1(Alk1)nZ, реагенты формулы HXCH2CO2Et и любые другие промежуточные соединения, требующиеся для синтеза соединений по изобретению, не являются коммерчески доступными или известными из литературы, они могут быть легко получены из более простых известных соединений одним или несколькими стандартными методами, с использованием реакций замещения, окисления, восстановления или расщепления. Реакции замещения включают обычное алкилирование, арилирование,гетероарилирование, ацитилирование, тиоацитилирование, галогенирование, сульфонилирование, нитрование, формилирование и сочетание. Следует иметь в виду, что эти методы могут быть также использованы для получения или модификации других промежуточных соединений и, в частности, соединений формулы (1), когда соответствующие функциональные группы содержатся в этих соединениях. Конкретные примеры этих методов приведены в нижеследующих примерах. Так, например, ароматические галогенсодержащие заместители в соединениях могут быть подвергнуты обмену галоген-металл при помощи основания, например, литиевого основания, такого как нбутил- или трет.бутиллитий, возможно, при низкой температуре, например, около -78 С, в среде растворителя, такого как тетрагидрофуран, затем реакцию обрывают электрофильным реагентом для введения- 13008410 желательного заместителя. Таким образом, например, формильная группа может быть введена с применением в качестве электрофильного реагента диметилформамида, тиометильную группу можно ввести,используя диметилдисульфид в качестве электрофила, спиртовую группу можно ввести при использовании альдегида в качестве электрофильного реагента и кислотную группу можно ввести, используя в качестве электрофильного реагента двуокись углерода. Ароматические кислоты формулы АrСО 2 Н также могут быть получены при действии двуокиси углерода на реактивы Гриньяра формулы ArMgHal. Ароматические кислоты формулы АrСO2 Н, полученные этим способом, и соединения, содержащие кислотные группы, вообще могут быть превращены в активированные производные, например, галоидангидриды кислоты, путм взаимодействия с галогенирующим агентом, таким как трихлорид фосфора или пентагалогенид фосфора, такой как пентахлорид фосфора, возможно в среде инертного растворителя, такого как ароматический углеводород, например толуол, или хлорированный углеводород, например дихлорметан, при температуре от примерно 0 С до температуры рефлюкса, или могут быть превращены в амиды Вайнреба (Weinreb) формулы ArC(O)N(OMe)Me путм конверсии в галоидангидрид кислоты,как только что описано, и последующей реакции с амином формулы HN(OMe)Me или его солью, возможно, в присутствии основания, такого как органический амин, например триэтиламин, в среде инертного растворителя, такого как ароматический углеводород, например толуол, или хлорированный углеводород, например дихлорметан, при температуре от примерно 0 С до комнатной температуры. Соединения по изобретению и промежуточные соединения для них могут быть получены алкилированием, арилированием или гетероарилированием. Например, соединения, содержащие группу -L1H(где L1 - атом линкера или линкерная группа), могут быть обработаны алкилирующим агентом Cy1Z2, гдеZ2 - удаляемый атом или группа, такие как атом галогена, например, фтора, хлора, брома или йода, или сульфонилоксигруппа, такая как алкилсульфонилоксигруппа, например трифторметилсульфонилоксигруппа, или арилсульфонилоксигруппа, например, п-толуолсульфонилоксигруппа. Реакцию можно осуществлять в присутствии основания, такого как карбонат, например карбонат цезия или калия, алкоксида, например трет.бутоксида калия, или гидрида, например гидрида натрия, в диполярном апротонном растворителе, таком как амид, например замещнный амид, такой как диметилформамид, или простой эфир, например циклический эфир, такой как тетрагидрофуран. Согласно другому варианту соединения, содержащие -L2H группы, указанные выше, можно функционализировать путм ацилирования или тиоацилирования, например, при реакции с алкилирующими агентами, описанными выше, но в которых Z замещн -C(O)Z3, C(S)Z3, -N(R2)COZ3 или -N(R2)C(S)Z3 группами, в которых Z3 обозначает удаляемую группу или атом, описанные для Z2. Реакцию можно проводить в присутствии основания, такого как гидрид, например гидрид натрия, или амин, например триэтиламин или N-метилморфолин, в среде растворителя, такого как галогенированный углеводород, например дихлорметан или четырххлористый углерод, или амид, например диметилформамид, например,при комнатной температуре. Альтернативно, ацилирование можно проводить в тех же условиях при помощи кислоты (например, одного из алкилирующих агентов, описанных выше, в которых Z2 замещн-СО 2 Н группой), в присутствии конденсирующего агента, например, диимида, такого как 1-(3-диметиламинопропил)-3-этилкарбодиимид или N,N'-дициклогексилкарбодиимид, или бензотриазола, такого как [О-(7-азабензотриазол-1-ил)-1,1,3,3-тетраметилуроний]гексафторфосфат, преимущественно, в присутствии катализатора, такого как N-гидроксисоединение, например, N-гидрокситриазол, такой как 1 гидроксибензотриазол. Альтернативно, кислота может взаимодействовать с хлорформиатом, например этилхлорформиатом, перед реакцией ацилирования. Далее, соединения по изобретению могут быть получены сульфонилированием соединения, содержащим группу -ОН, по реакции с одним из вышеуказанных алкилирующих агентов, но в котором Z2 замещн -S(O)Hal или -SO2Hal (где Hal обозначает атом галогена, такой как атом хлора), в присутствии основания, например, неорганического основания, такого как гидрид натрия, в среде растворителя, такого как амид, например замещнный амид, такой как диметилформамид, например, при комнатной температуре. В другом примере соединения, содержащие -L2H-группу, описанную выше, могут вступать в реакцию сочетания с одним из алкилирующих агентов, описанных только что выше, но в котором Z2 замещн группой -ОН, в среде растворителя, такого как тетрагидрофуран, в присутствии фосфина, например трифенилфосфина, и активатора, такого как диэтил-, диизопропил- или диметилазодикарбоксилат. Сложноэфирные группы, такие как -CO2Alk6 и -CO2R4 в соединении формулы (1) и в промежуточных соединениях могут быть превращены в соответствующую кислоту (-СО 2 Н) путм гидролиза, катализируемого кислотой или основанием, в зависимости от природы группы Alk6 или R4. Гидролиз, катализируемый кислотой или основанием, может быть осуществлн, например, путм обработки органической или неорганической кислотой, например трифторуксусной кислотой, в среде органического растворителя, например дихлорметана, или минеральной кислоты, такой как соляная кислота, в растворителе, таком как диоксан, или гидроокиси щелочного металла, например гидроокиси лития в водном спирте, например водном метаноле. Ещ в одном примере группа -OR6 (где R6 обозначает алкильную группу, такую как метил) в соединениях формулы (1) и в промежуточных соединениях может быть превращена в соответствующую спир- 14008410 товую группу -ОН по реакции с трхбромистым бором в среде растворителя, такого как галогенированный углеводород, например дихлорметан, при низкой температуре, например, около -78 С. Спиртовые группы -ОН могут быть также получены гидрированием соответствующей -OCH2R31 группы (где R31 обозначает арил) с использованием катализатора на основе металла, например палладия на носителе, таком как уголь, в среде растворителя, такого как этанол, в присутствии формиата аммония,циклогексадиена или водорода при температуре от примерно комнатной до температуры рефлюкса. Согласно другому примеру группы -ОН могут получаться из соответствующего эфира (например, -СО 2Alk6) или альдегида (-СНО) путм восстановления с применением, например, комплекса гидрида металла, такого как литийалюминийгидрид или боргидрид натрия, в среде растворителя, такого как метанол. В другом примере спиртовые группы -ОН в соединениях могут быть превращены в соответствующую группу -OR6 путм сочетания с реагентом R6OH в среде растворителя, такого как тетрагидрофуран,в присутствии фосфина, например трифенилфосфина, и активатора, такого как диэтил-, диизопропилили диметилазодикарбоксилат. Аминосульфониламиногруппы [-NHSO2NH2] в соединениях могут быть получены реакцией соответствующего амина (-NH2) с сульфамидом в присутствии органического основания, такого как пиридин,при повышенной температуре, например, при температуре рефлюкса. Согласно другому примеру соединения, содержащие -NHCSR7 или -CSNHR7-группы, могут быть получены путм обработки соответствующего соединения, содержащего группу -NHCOR7 или -CONHR7,реагентом, содержащим серу, таким как реагент Лавессона или P2S5, в безводном растворителе, например, циклическом эфире, таком как тетрагидрофуран, при повышенной температуре, например, при температуре рефлюкса. Согласно ещ одному примеру группы (-NH2) могут быть алкилированы с применением восстановительного алкилирования с использованием альдегида и восстановителя. Подходящие восстановители включают боргидриды, например триацетоксиборгидрид натрия или цианборгидрид натрия. Восстановление можно проводить в растворителе, таком как галогенированный углеводород, например дихлорметан, кетон, такой как ацетон, или спирт, например этанол, где это необходимо, в присутствии кислоты,такой как уксусная кислота, примерно при комнатной температуре. Альтернативно, амин и альдегид могут вначале реагировать в среде растворителя, такого как ароматический углеводород, например, толуол,а затем смесь подвергают гидрированию в присутствии катализатора на основе металла, например, палладия на носителе, таком как углерод, в растворителе, таком как спирт, например этанол. Согласно ещ одному примеру аминогруппы (-NH2) в соединении формулы (1) и в промежуточных соединениях могут быть получены гидролизом соответствующего имида по реакции с гидразином в среде растворителя, такого как спирт, например этанол, при комнатной температуре. Согласно другому примеру нитрогруппа (-NO2) может быть восстановлена до аминогруппы (-NH2),например, каталитическим гидрированием с применением, например, водорода в присутствии катализатора на основе металла, например, палладия на носителе, таком как уголь, в среде растворителя, таком как простой эфир, например тетрагидрофуран, или спирт, например метанол, или путм химического восстановления с применением, например, металла, например олова или железа, в присутствии кислоты,такой как соляная кислота. Согласно ещ одному примеру группы (-CH2NH2) в соединениях формулы (1) и в промежуточных соединениях могут быть получены восстановлением нитрилов (-CN), например, путм каталитического гидрирования с применением, например, водорода в присутствии катализатора на основе металла, например, палладия на носителе, таком как уголь, или никеля Ренея, в среде растворителя, такого как простой эфир, например циклический эфир, такой как тетрагидрофуран, или спирт, например метанол или этанол, возможно, в присутствии раствора аммиака при температуре от комнатной до температуры рефлюкса, или путм химического восстановления с применением, например, гидрида металла, например, литийалюминийгидрида, в среде растворителя, такого как простой эфир, например циклический эфир, такой как тетрагидрофуран, при температуре от 0 С до температуры рефлюкса. В другом примере атомы серы в соединениях, например, когда они содержатся в группе L1 или L2,могут быть окислены с получением соответствующего сульфоксида или сульфона с применением окисляющего агента, такого как пероксикислота, например, 3-хлорпероксибензойная кислота, в среде инертного растворителя, такого как галогенированный углеводород, например дихлорметан, примерно при комнатной температуре. Ещ в одном примере N-окиси соединений формулы (1) в общем могут быть получены, например,путм окисления соответствующего азотсодержащего основания, как описано выше при получении промежуточных соединений формулы (5). Соли соединений формулы (1) могут быть получены по реакции соединений формулы (1) с соответствующим основанием в подходящем растворителе или смеси растворителей, например, в органическом растворителе, таком как простой эфир, например диэтиловый эфир, или спирт, например этанол, по обычным методикам. Если желательно получить конкретный энантиомер соединения формулы (1), это можно сделать из соответствующей смеси энантиомеров с использованием любой подходящей известной процедуры для- 15008410 разрешения энантиомеров. Так, например, диастереомерные производные, например соли, могут быть получены путм взаимодействия смеси энантиомеров формулы (1), например, рацемата и соответствующего хирального соединения, например хирального основания. Диастереомеры могут быть затем разделены любым обычным методом, например кристаллизацией, и выделением желаемого энантиомера, например, обработкой кислотой в случае, когда диастереомер представляет собой соль. Согласно другому способу разрешения рацемат формулы (1) может быть разделн с применением хиральной жидкостной хроматографии высокого разрешения. Или же, если желательно, конкретный энантиомер может быть получен с использованием соответствующего хирального промежуточного соединения в одном из вышеописанных способов. Кроме того, конкретный энантиомер может быть получен путм осуществления специфической ферментативной биотрансформации энантиомера, например,гидролизом сложного эфира с применением эстеразы, и последующей очисткой только энантиомерно чистой гидролизованной кислоты от не прореагировавшего сложноэфирного антипода. Когда желательно получить конкретный геометрический изомер по изобретению, для промежуточных соединений или конечных продуктов можно также использовать методы хроматографии, перекристаллизации и другие обычные методы разделения. Нижеследующие примеры иллюстрируют данное изобретение. Все температуры указаны в С. Используются следующие сокращения:p.s.i. - фунт на квадратный дюйм. Все ЯМР были получены при 300 или 400 МГц. Соединения были названы с помощью Beilstein Autonom, supplied by MDL Information SystemsGmbH, Theodor-Heuss-Allee 108, D-60486, Frankfurt, Germany или ACD Labs Name (v.5.0 или v.6.0), supplied by Advanced Chemical Development, Toronto, Canada. Указанные величины времени удерживания(RT) LCMS были получены на Hewlett Packard 1100 LC/MS с применением следующего метода: колонка%В нач. 5 2,00 95 3,00 95 5 конец Где указано, использовались альтернативные условия LCMS (условия В): время удерживания (RT) измерялось в системе Hewlett Packard 1100/TermoFinnigan LCQ Duo LC/MS с применением ионизации при электрораспылении и следующего метода ЖХ: колонка Phenomenex Luna C18 (2) 5 мкм 100 мм х 4,6 мм; подвижная фаза А=0,08% муравьиная кислота в воде; подвижная фаза В=0,08% муравьиная кислота в MeCN; скорость истечения 3,0 млмин-1; температура колонки 35 С.EtOH (1,2 л) нагревали до кипения с обратным холодильником в течение 4,5 ч. Затем смесь охлаждали до комнатной температуры, добавляли к воде (10 л) и промывали водой (5 л). Полученную суспензию перемешивали в течение 30 мин и затем фильтровали. Осадок на фильтре промывали двумя порциями воды(2 х 2,5 л) и сушили. Затем полученное тврдое вещество сушили до постоянного веса под вакуумом при 45 С с получением указанного в заголовке соединения в виде коричневого тврдого вещества (493,1 г,93,2%).(CDCl3) 8,68 (1H, dd, J 4,7, 1,2 Гц), 7,93 (1H, dd, J 8,5, 1,2 Гц), 7,29 (1H, dd, J 8,5, 4,7 Гц), 5,90(2 Н, b), 4,38 (2 Н, q, J 7,0 Гц), 1,40 (3 Н, t, J 7,0 Гц). LCMS RT 2,9 мин, 223 (М+Н)+. Промежуточное соединение 2. Этил-3-бромтиено[2,3-b]пиридин-2-карбоксилат. Промежуточное соединение 1 (363,6 г) добавляли порциями в течение 2 ч к смеси бромида меди (II)(403,3 г), трет.бутилнитрита (220,6 г) и ацетонитрила (3,6 л) перемешиваемой при температуре 20-25 С. Смесь перемешивали при 20 С в течение 2 ч перед медленным добавлением к 2 М HСl (водн.) (4,2 л). Суспензию реакционной смеси фильтровали и тврдое вещество промывали водой (500 мл). Объединнный фильтрат экстрагировали этилацетатом (8 л), этот раствор в этилацетате промывали 2 М НСl (водн.)(2,2 л). Тврдый продукт растворяли в этилацетате (6 л), этот раствор промывали дважды 2 М НСl (водн.)(4,4 л и 2,2 л). Два этилацетатных раствора затем соединяли и промывали 2 М НСl (водн.) (2,2 л) и дважды водой (2 х 2,2 л). Раствор в этилацетате затем сушили (MgSO4), фильтровали и концентрировали в вакууме при 40 мбар и 60 С с получением тврдого осадка. Осадок измельчали и сушили до постоянного веса под вакуумом при 45 С с получением указанного в заголовке соединения в виде коричневого тврдого продукта (458,5 г, 97,6%). H (DMSO-d6) 8,89 (1H, d, J 4,7 Гц), 8,47 (1H, d, J 8,6 Гц), 7,71 (1 Н, dd, J 8,6, 4,7 Гц), 4,46 (2 Н, q, J 7,2 Гц), 1,40 (3 Н, t, J 7,2 Гц), LCMS RT 3,8 мин, 288 (М+Н)+. Промежуточное соединение 3. Этил-3-бромтиено[2,3-b]пиридин-2-карбоксилата N-окись. К суспензии промежуточного соединения 2 (214 г, 0,747 Мол) в DCM (2140 мл) в атмосфере азота добавляли МСРВА (240 г 70% = 168 г, 0,97 Мол) порциями в течение 0,5 ч. Реакционную смесь перемешивали при комнатной температуре в течение 18 ч. Реакционную смесь обрывали водой (800 мл), доводили рН до 8,5 при помощи 10% вес./об. раствора карбоната натрия (1250 мл). Водный слой основания концентрировали в вакууме и сырой продукт, указанный в заголовке, был выделен в виде коричневого тврдого вещества. Сырой продукт очищали путм получения суспензии в трет.бутилметиловом эфире(3 Н, t, J 7,1 Гц). LCMS (ES+) RT 2,61 мин, 302 (М+Н)+. Промежуточное соединение 4. Этил-3-бром-6-оксо-6,7-дигидротиено[2,3-b]пиридин-2-карбоксилат. Смесь промежуточного соединения 3 (500 мг, 1,66 ммол) и DMF (10 мл) перемешивали при 0 С в атмосфере азота. К этой реакционной смеси добавляли трифторуксусный ангидрид (3,49 г, 2,36 мл, 16,6 ммол) одной порцией при помощи шприца. После перемешивания в течение 16 ч удаляли летучие в вакууме и осадок обрабатывали толуолом (2x20 мл). Сырой продукт затем экстрагировали ЕtOАс (2x100 мл). Экстракты в EtOAc сушили (MgSO4) и концентрировали в вакууме. Сырой продукт очищали получением новой суспензии в толуоле (10 мл) с получением указанного в заголовке соединения в виде тврдого вещества бежевого цвета (260 мг, 52%). Н (DMSO-d6) 12,20 (1H, br s), 7,75 (1 Н, d, J 9,0 Гц), 6,50 (1 Н, d, J 9,0 Гц), 4,15 (2 Н, q, J 7,1 Гц), 1,12 (3 Н, t, J 7,1 Гц). LCMS (ES+) RT 2,86 мин, 302 М+Н)+, 100%). МР=261,7-268,1 С. Промежуточное соединение 5. Этил-3-бром-6-оксо-7-фенил-6,7-дигидротиено[2,3-b]пиридин-2-карбоксилат. В двухгорлую круглодонную колбу последовательно добавляли промежуточное соединение 4 (302 мг, 1,00 ммол), ацетат меди(II) (278 мг, 1,50 ммол), фенилбороновую кислоту (488 мг, 4,00 ммол), DCM(5 мл) и пиридин (158 мг, 2,00 ммол). Смесь перемешивали при комнатной температуре в течение 18 ч в отсутствие влаги. Затем смесь разбавляли DCM (50 мл), промывали 2 М НСl (водн.) (50 мл), водный слой экстрагировали DCM (50 мл), сушили (MgSO4) и концентрировали в вакууме. Сырой продукт очищали суспендированием в метаноле (12 мл) с получением соединения, указанного в заголовке, в виде тврдого вещества бежевого цвета (270 мг, 72%). Н (CDCl3) 7,82 (1H, d, J 8,5 Гц), 7,70-7,62 (3 Н, m), 7,54-7,42 (2 Н,- 17008410MP=201,6-206,0C. Промежуточное соединение 6. 3-Бром-6-оксо-6,7-дигидротиено[2,3-b]пиридин-2-карбоновая кислота. Моногидрат гидроксида лития (1,39 г, 33,1 ммол) добавляли к суспензии промежуточного соединения 4 (5,0 г, 16,55 ммол) в воде (100 мл) и перемешивали 5 мин. Добавляли THF (10 мл) и реакционную смесь перемешивали при r.t. в течение 18 ч. Добавляли 2 М НСl (водн.) для получения рН 1-2 и полученный осадок собирали фильтрованием, промывали EtOH и сушили в вакууме с получением соединения,указанного в заголовке, в виде тврдого вещества белого цвета (4,5 г). Н (DMSO-d6) 7,9 (1H, d, J 9,2 Гц),6,67 (1 Н, d, J 9,2 Гц), пиридиновые фрагменты и протоны карбоновой кислоты не наблюдались. Промежуточное соединение 7. 3-Бром-6-оксо-6,7-дигидротиено[2,3-b]пиридин-2-карбоксамид. 1,1'-Карбонилдиимидазол (3,18 г, 19,6 ммол) добавляли к суспензии промежуточного соединения 6(4,30 г, 15,7 ммол) в безводном DMF (50 мл) и перемешивали смесь при r.t. в атмосфере азота до получения раствора (30 мин). Добавляли гидроокись аммония (50 мл 28% NH3 в воде) и перемешивали смесь 15 мин перед удалением растворителей в вакууме. Остаток суспендировали в воде (75 мл) и обрабатывали 2 М НСl (водн.) (20 мл). Полученный тврдый продукт собирали фильтрованием, промывали водой и сушили в вакуумной печке с получением указанного в заголовке соединения в виде тврдого вещества светло-коричневого цвета (3,70 г). Н (DMSO-d6) 7,69 (1H, d, J 9,2 Гц), 6,49 (1H, d, J 9,1 Гц), 7,30 (1H, bs).LCMS (ES+) 273 (М+Н)+. Промежуточное соединение 8. 3-Бром-6-оксо-6,7-дигидротиено[2,3-b]пиридин-2-карбонитрил. К суспензии промежуточного соединения 7 (3,70 г, 13,55 ммол) в DCM (200 мл) добавляли пиридин(2,70 мл, 34 ммол) и затем трифторуксусный ангидрид (2,40 мл, 17 ммол). Реакционную смесь перемешивали ещ 8 ч и затем концентрировали в вакууме. Остаток суспендировали в воде, подкисляли до рН 2 водным 2 М раствором НСl и полученное тврдое вещество собирали фильтрованием, промывали водой и сушили в вакууме с получением соединения, указанного в заголовке, в виде тврдого вещества бледножлтого цвета (3,20 г).(DMSO-d6) 7,92 (1H, d, J 8,8 Гц), 6,73 (1H, d, J 8,8 Гц). LCMS (ES+) 255 (М+Н)+. Промежуточное соединение 9. 3-Бром-6-оксо-7-фенил-6,7-дигидротиено[2,3-b]пиридин-2-карбонитрил. В высушенную в печке колбу добавляли промежуточное соединение 8 (2,0 г, 7,84 ммол), фенилбороновую кислоту (1,19 г, 15,7 ммол), ацетат меди (II) (1,42 г, 7,84 ммол), безводный пиридин (1,3 мл, 16 ммол) и безводный DCM (50 мл). Реакционную смесь перемешивали при r.t. в отсутствие влаги в течение 48 ч. Смесь разбавляли DCM (50 мл), промывали 2 М HCl (водн.) (100 мл), насыщенным NаНСО 3 (водн.)(50 мл), сушили (Na2SO4) и концентрировали в вакууме. Остаток очищали методом хроматографии на двуокиси кремния (5-10% ЕtОАс в DCM) с получением соединения, указанного в заголовке, в виде белого вещества (1,24 г). Н (DMSO-d6) 7,67 (1H, d, J 9,6 Гц), 7,58-7,50 (3 Н, m), 7,32-7,29 (2 Н, m), 6,70 (1 Н, d,J 9,6 Гц). Промежуточное соединение 10. 3-Циан-6-оксо-1-фенил-1,6-дигидропиридин-2-тиолят натрия. Раствор метоксида натрия в МеОН (30 вес.%, 202,2 г) добавляли к абсолютному этанолу (360 мл) с последующим добавлением 1,3-диметилурацила (75 г) и 2-циан-N-фенилтиоацетамида (Adhikari et al.,Australian J. Chem., 1999, 52, 63-67) (90 г). Полученную смесь нагревали при кипении с обратным холодильником 8 ч и давали ей охладиться до комнатной температуры, оставив на ночь. Затем реакционную смесь охлаждали до +5 и выдерживали при этой температуре по меньшей мере 1 ч, полученный продукт выделяли фильтрованием. Остаток на фильтре промывали холодным (+5) абсолютным этанолом (450 мл) и сушили до постоянного веса в вакууме при 45 с получением соединения, указанного в заголовке, в виде продукта розового цвета (130,0 г). Полученный таким образом продукт содержит остаточные EtOH и МеОН, в количестве 12,2 вес.%, выход 114,1 г.(DMSO-d6) 7,32 (2H, m), 7,27-7,18 (1H, m), 7,16 (1 Н,d, J 9,1 Гц), 6,92 (2 Н, m), 5,63 (1H, d, J 9,1 Гц). LCMS (Условия В) (ES+) RT 2,43 мин, 229 (М+Н)+. Промежуточное соединение 11. 3-Амино-6-оксо-7-фенил-6,7-дигидротиено[2,3-b]пиридин-2-карбонитрил. Смесь промежуточного продукта 10 (100 г при 100%) и хлорацетонитрила (30,4 мл) в ацетонитриле(500 мл) нагревали при температуре рефлюкса 2 ч. Смесь охлаждали, вначале до 40, когда добавляли воду (300 мл) и затем до +10. Смесь выдерживали при (+10) по меньшей мере 1 ч, полученный продукт выделяли фильтрованием. Осадок на фильтре промывали холодной (+10) водой (500 мл) и затем холодной (+10) смесью ацетонитрила и воды (1:1, 300 мл). Продукт сушили под вакуумом при 50 до постоянного веса с получением соединения, указанного в заголовке, в виде не совсем белого тврдого вещества (100,9 г).(DMSO-d6) 7,90 (1H, d, J 9,6 Гц), 7,46-7,33 (3 Н, m), 7,25 (2 Н, m), 6,95 (2 Н, br s), 6,35 (1H,d, J 9,6 Гц). LCMS (Условия В) (ES+) RT 2,69 мин, 268 (М+Н)+. Промежуточное соединение 12. 3-Амино-6-оксо-7-фенил-6,7-дигидротиено[2,3-b]пиридин-2-карбоновой кислоты этиловый эфир. Смесь промежуточного соединения 10 (0,34 г при 100%) и этилбромацетата (0,197 мл) в этаноле (6 мл) перемешивали при комнатной температуре 1 ч. Затем добавляли воду (10 мл). Отфильтровывали- 18008410 тврдое вещество и промывали водой (2 мл). Продукт сушили под вакуумом при 40 до постоянного веса с получением соединения, указанного в заголовке, в виде тврдого продукта розового цвета (0,35 г). Н(альтернативный способ). 3-Бром-6-оксо-7-фенил-6,7 дигидротиено[2,3-b]пиридин-2-карбонитрил. К смеси безводного бромида меди (II) (23,4 г) и трет.бутилнитрита (14,8 мл) в ацетонитриле (600 мл) при комнатной температуре добавляли порционно промежуточный продукт 12 (20 г) с такой скоростью, чтобы температура была ниже 25 С. Добавление происходило в течение почти 1 ч. ЖХВР показала почти полную конверсию исходного продукта через 30 мин при перемешивании. Затем реакционную смесь выливали в 500 мл 1 М HCl (внимание: выделяются коричневые пары). Затем осуществляли экстракцию дихлорметаном (2 х 400 мл). Соединнные органические экстракты промывали затем 1 М HCl (3 х 300 мл), сушили над MgSO4 и выпаривали досуха. Полученный сырой продукт затем перекристаллизовывали из метилизобутилкетона (700 мл). Продукт сушили в вакууме при 50 до постоянного веса с получением соединения, указанного в заголовке, в виде продукта светло-коричневого цвета (15,14 г).-78 С добавляли ацетонитрил (10 мл) с получением толстого белого осадка. Добавляли 2 хлорфенилизотиоцианат (7,72 г, 45,45 ммол) с получением коричневого раствора. Смеси давали нагреться до r.t. в течение 1 ч, затем разбавляли EtOH (50 мл). Добавляли N,N-диметилурацил (6,4 г, 45 ммол) и нагревали смесь при температуре рефлюкса в течение 24 ч. Летучие удаляли в вакууме и растворяли осадок в ацетонитриле (100 мл). Добавляли хлорацетонитрил (2,85 мл, 45 ммол) и нагревали смесь при 50 С в течение 1 ч, снова добавляли хлорацетонитрил (2,85 мл, 45 ммол) и продолжали нагревание в течение 1,5 ч. Часть ацетонитрила (50 мл) удаляли в вакууме и добавляли воду для осаждения продукта. Отфильтровывали коричневое тврдое вещество, промывали водой (50 мл) и Еt2 О (50 мл) и сушили с получением соединения, указанного в заголовке, в виде продукта коричневого цвета (14,3 г, колич.). Н(DMSO-d6) 8,10 (1H, d, J 9,7 Гц), 7,75-7,73 (1H, m), 7,65-7,54 (3 Н, m), 7,14 (2 Н, br s, NH2), 6,54 (1H, d, J 9,7 Гц). LCMS (ES+) RT 2,97 мин, 302 (M+H)+. Промежуточное соединение 14. 3-Бром-7-(2-хлорфенил)-6-оксо-6,7-дигидротиено[2,3-b]пиридин-2 карбонитрил. Промежуточное соединение 13 (1,17 г, 3,88 ммол) суспендировали в ацетонитриле (20 мл). Добавляли бромид меди (II) (953 мг, 4,27 ммол) и затем трет.бутилнитрил (0,64 мл, 5,43 ммол). Смесь перемешивали при комнатной температуре в течение 3 ч, затем распределяли между 2 М HCl (водн.) (100 мл) иEtOAc (100 мл). Органический слой промывали 2 М HCl (водн.) (50 мл), 2 М NaOH (водн.) (50 мл) и водой(25 мл), сушили (Na2SO4) и концентрировали в вакууме. Очистка методом хроматографии на колонке(двуокись кремния, 0-5% ЕtOАс в DCM) привела к получению соединения, указанного в заголовке, в виде продукта светло-коричневого цвета (980 мг, 67%). Н (CDCl3) 7,70 (1H, d, J 9,7 Гц), 7,61 (1 Н, dd, J 1,7, 7,7 Гц), 7,52-7,44 (2 Н, m), 7,34 (1H, dd, J 1,7, 7,7 Гц), 6,70 (1 Н, d, J 9,7 Гц). LCMS (ES+) RT 3,56 мин,365 (М+Н)+. Промежуточное соединение 15. 3-Циан-1-(4-метилфенил)-6-оксо-1,6-дигидропиридин-2-тиолят натрия. Бис(триметилсилил)амид натрия (36,8 мл, 1,0 М в THF, 36,8 ммол) медленно добавляли к раствору 4-толилизотиоцианата (2,5 г, 16,75 ммол) в THF (30 мл) и ацетонитриле (5 мл) при -78 С. Смесь нагревали до r.t. в течение 1 ч. Добавляли N,N-диметилурацил (2,35 г, 16,75 ммол) и EtOH (20 мл) и нагревали смесь при температуре рефлюкса в течение 4 ч. Летучие удаляли в вакууме и остаток растворяли в EtOH(6 мл). Добавляли медленно Et2O (60 мл) с получением мелкодисперсного продукта не совсем белого цвета. Суспензию охлаждали до 0 С и отфильтровывали тврдый продукт, промывали его Еt2O и сушили с получением соединения, указанного в заголовке, в виде тврдого продукта не совсем белого цвета (1,7 г, 39% ). Н (DMSO-d6) 7,15-7,12 (3 Н, m), 6,80-6,77 (2 Н, m), 5,60 (1H, d, J 9,1 Гц), 2,30 (3 Н, s). Промежуточное соединение 16. 3-Амино-7-(4-метилфенил)-6-оксо-6,7-дигидротиено[2,3-b]пиридин-2-карбонитрил. К суспензии промежуточного соединения 15 (1,7 г, 6,44 ммол) в ацетонитриле (40 мл) добавляли хлорацетонитрил (0,41 мл, 6,4 ммол). Смесь нагревали при 45 С в течение 2 ч. Растворитель удаляли в вакууме и остаток суспендировали в воде (30 мл). Тврдый продукт отфильтровывали, промывали водой(3 х 10 мл) и простым эфиром (5 мл) и сушили, получали соединение, указанное в заголовке, в виде продукта не совсем белого цвета (1,22 г, 67%). Н (DMSO-d6) 8,01 (1H, d, J 9,7 Гц), 7,34-7,32 (2 Н, m), 7,277,25 (2 Н, m), 7,00 (2 Н, br s), 6,45 (1H, d, J 9,7 Гц), 2,34 (3 Н, s). LCMS (ES+) RT 3,03 мин, 282,0 (M+H)+. Промежуточное соединение 17. 3-Бром-7-(4-метилфенил)-6-оксо-6,7-дигидротиено[2,3-b]пиридин- 19008410 2-карбонитрил. Промежуточное соединение 16 (562 мг, 2,0 ммол) суспендировали в ацетонитриле (15 мл). Бромид меди (II) (419 мг, 2,2 ммол) и затем трет.бутилнитрил (0,33 мл, 2,8 ммол) добавляли в смесь. Смесь перемешивали при r.t. в течение 3 ч, затем разбавляли DCM (100 мл), промывали 2 М HCl (водн.) (50 мл) и 1 МNaOH (водн.) (50 мл), сушили (Na2SO4) и концентрировали в вакууме. После очистки методом хроматографии на колонке (двуокись кремния, 0-20% ЕtOАс в DCM) получали соединение, указанное в заголовке, в виде жлтого продукта (450 мг, 65%). Н (CDCl3) 7,59 (1H, d, J 9,7 Гц), 7,29-7,27 (2 Н, m), 7,13-7,10(2 Н, m), 6,62 (1H, d, J 8,7 Гц), 2,33 (3 Н, s). LCMS (ES+) RT 3,64 мин, 345,0/347,0 (79Br/81Br)(M+H)+. Промежуточное соединение 18. 3-[(4-Фтор-3-метилфенил)амино]-7-(4-метилфенил)-6-оксо-6,7-дигидротиено[2,3-b]пиридин-2-карбонитрил. Смесь промежуточного соединения 17 (225 мг, 0,65 ммол), 4-фтор-3-метиланилина (98 мг, 0,78 ммол), карбоната цезия (297 мг, 0,91 ммол), BINAP (41 мг, 0,065 ммол, 10 мол.%) и трис(дибензилиденацетон)дипалладия(0) (30 мг, 0,0325 ммол, 5 мол.%) в толуоле (5 мл) нагревали при 100 С в атмосфере азота в течение 18 ч. Тврдый продукт отфильтровывали, промывали водой (2 х 15 мл) и эфиром (3 х 10 мл) и получали соединение, указанное в заголовке, в виде коричневого продукта. Этот продукт использовали на следующей стадии без дальнейшей очистки. LCMS (ES ) RT 3,66 мин,390,0 (М+Н)+. Промежуточное соединение 19. 3-Циан-1-циклопропил-6-оксо-1,6-дигидропиридин-2-тиолят натрия. Раствор бис(триметилсилил)амида натрия (122 мл 1,0 М раствора в THF, 122 ммол) добавляли к раствору циклопропилизотиоцианата (4,85 г, 48,9 ммол) и ацетонитрила (25,5 мл, 10 экв.) в THF (50 мл) при-78 С. Смеси давали нагреться до r.t. в течение 2 ч. Добавляли N,N-диметилурацил (59 г, 49 ммол) иEtOH (60 мл) и смесь нагревали при температуре рефлюкса в течение 3 ч, перемешивали при r.t. в течение ночи. Летучие удаляли в вакууме. Остаток обрабатывали смесью EtOH и ЕtOАс, затем добавлялиEt2O. Липкий продукт отфильтровывали и сушили, получали соединение, указанное в заголовке (11 г,техн. продукт), который далее использовали без дальнейшей очистки. Промежуточное соединение 20. 3-Амино-7-циклопропил-6-оксо-6,7-дигидротиено[2,3-b]пиридин-2 карбонитрил. Смесь сырого промежуточного продукта 19 (9 г, примерно 42 ммол) и хлорацетонитрила (2,7 мл, 42 ммол) в ацетонитриле (100 мл) нагревали при температуре рефлюкса в течение 3 ч. Летучие удаляли в вакууме. К остатку добавляли воду (100 мл), тврдый полученный продукт отфильтровывали и сушили. Сырой продукт распределяли между водой и ЕtOАс и экстрагировали водную фазу ЕtOАс. Соединнные органические фазы концентрировали в вакууме. Остаток растворяли в ЕtOН и обрабатывали раствор Еt2 О, получая тврдый продукт, который отфильтровывали и высушивали, получали соединение, указанное в заголовке, в виде коричневого продукта (2,5 г). Н (CDCl3) 7,42 (1H, d, J 9,6 Гц), 6,52 (1H, d, J 9,6 Гц), 4,6 (2 Н, br s), 3,08-3,00 (1H, m), 1,2-1,1 (2 Н, m), 1,08-1,0 (2 Н, m). LCMS (ES+) RT 2,532 мин, 232(М+Н)+. Промежуточное соединение 21. 3-Бром-7-циклопропил-6-оксо-6,7-дигидротиено[2,3-b]пиридин-2 карбонитрил. Бромид меди (II) (0,53 г, 2,37 ммол) и трет.бутилнитрит (0,40 мл, 3,02 ммол) добавляли к раствору промежуточного соединения 20 (0,5 г, 2,16 ммол) в ацетонитриле (15 мл). Реакционную смесь перемешивали при r.t. в течение 4 ч. Добавляли DCM (100 мл) и промывали смесь 2 М HCl (водн.) и 2 М NaOH-78 С. Смеси давали нагреться до r.t. в течение 3 ч. Добавляли N,N-диметилурацил (4,62 г, 33 ммол) и ЕtOН (75 мл), смесь нагревали 3 ч при температуре рефлюкса и затем перемешивали при r.t. Летучие удаляли в вакууме. Остаток использовали без очистки на следующей стадии. Промежуточное соединение 23. 3-Амино-7-(2-метилфенил)-6-оксо-6,7-дигидротиено[2,3-b]пиридин-2-карбонитрил. Смесь сырого промежуточного соединения 22 (половину полученного продукта) и хлорацетонитрила (1,94 мл) в ацетонитриле (25 мл) нагревали при температуре рефлюкса в течение 5 ч. Летучие удаляли в вакууме. Остаток обрабатывали водой и получали соединение, указанное в заголовке (3,0 г). НRT 2,932 мин, 281,9 (М+Н)+. Промежуточное соединение 24. 3-Бром-7-(2-метилфенил)-6-оксо-6,7-дигидротиено[2,3-b]пиридин- 20008410 2-карбонитрил. Получали из промежуточного соединения 23, как промежуточное соединение 14.(CDCl3) 7,8(1H, d, J 9,6 Гц), 7,55-7,4 (4 Н, m), 6,8 (1H, d, J 9,6 Гц), 2,12 (3 Н, s). LCMS (ES+) RT 4,10 мин, масс-ионы не наблюдались. Промежуточное соединение 25. 7-(2-Метилфенил)-3-[(3-метилфенил)амино]-6-оксо-6,7-дигидротиено[2,3-b]пиридин-2-карбонитрил. Получали из промежуточного соединения 24 и м-толуидина методом, описанным в примере 1.LCMS (ES+) RT 3,64 мин, 372,0 (М+Н)+. Промежуточное соединение 26. Этил-3-бром-7-(1 Н-индол-5-ил)-6-оксо-6,7-дигидротиено[2,3b]пиридин-2-карбонитрил. Смесь 5-индолилбороновой кислоты (644 мг, 4 ммол), промежуточного соединения 4 (604 мг, 2 ммол) и ацетата меди (I) (363 мг, 2 ммол) в пиридине (2 мл) перемешивали при r.t. в течение ночи. Смесь распределяли между DCM и 2 М НСl (водн.). Органическую фазу сушили (Na2SO4) и концентрировали в вакууме. После очистки хроматографией на колонке (двуокись кремния, 10% ЕtOАс в DCM) получали соединение, указанное в заголовке, в виде бледно-жлтого продукта (650 мг, 78%). Н (DMSO-d6) 11,61(1H, br s), 8,01 (1H, d, J 9,3 Гц), 7,80 (1H, d, J 2,0 Гц), 7,74 (1H, d, J 8,6 Гц), 7,65 (1H, m), 7,23 (1 Н, dd, J 8,6,2,0 Гц), 6,81 (1H, d, J 9,6 Гц), 6,68 (1H, m), 4,33 (2 Н, q, J 7,1 Гц), 1,32 (3 Н, t, J 7,1 Гц). LCMS (ES+) RT 3,53 мин, 419/420 (79Br/81Br) (M+H)+. Промежуточное соединение 27. Этил-3-бром-7-[1-(метилсульфонил)-1 Н-индол-5-ил]-6-оксо-6,7 дигидротиено[2,3-b]пиридин-2-карбоксилат. Гидрид натрия (60% в минеральном масле) (58 мг, 1,44 ммол) добавляли к раствору промежуточного соединения 26 (500 мг, 1,2 ммол) в THF (30 мл) при 0 С. Смесь перемешивлаи 5 мин при 0 С и 15 мин при r.t., затем добавляли метансульфонилхлорид (0,124 мл, 1,6 ммол) и продолжали перемешивание при комнатной температуре в течение ночи. Удаляли растворитель в вакууме и остаток растворяли в EtOAc. Раствор в EtOAc промывали рассолом (х 2), сушили (MgSO4) и концентрировали в вакууме. После очистки хроматографией на колонке (двуокись кремния, 10% EtOAc в DCM) получали соединение, указанное в заголовке (240 мг, 40%).(CDCl3) 8,20 (1H, d, J 8,8 Гц), 7,82 (1 Н, d, J 9,7 Гц), 7,70 (1H, d, J 2,0 Гц),7,55 (1H, d, J 3,7 Гц), 7,37 (1H, dd, J 8,8, 2,0 Гц), 6,80 (1 Н, d, J 3,7 Гц), 6,75 (1H, d, J 15 9,7 Гц), 4,30 (2 Н, q,J 7,1 Гц), 3,21 (3 Н, s), 1,31 (3 Н, t, J 7,1 Гц). Промежуточное соединение 28. Этил-3-бром-7-(метил-1 Н-индол-5-ил)-6-оксо-6,7-дигидротиено[2,3-b]пиридин-2-карбоксилат. Из промежуточного соединения 26 и метилиодида получают соединение 28 способом, описанным для соединения 27. Н (CDCl3) 7,77 (1 Н, d, J 9,7 Гц), 7,56 (1 Н, s), 7,45 (1H, d, J 8,6 Гц), 7,11-7,07 (2 Н, m),6,68 (1H, d, J 9,7 Гц), 6,51 (1H, s), 4,21 (2 Н, q, J 7,1 Гц), 3,80 (3 Н, s), 1,22 (3 Н, t, J 7,1 Гц). LCMS (ES+) RT 3,84 мин, 4,32 (М+Н)+. Промежуточное соединение 29. Этил-3-бром-7-(циклопропилметил)-6-оксо-6,7-дигидротиено[2,3b]пиридин-2-карбоксилат. Гидрид натрия (60% в минеральном масле) (3,27 г, 81,4 ммол) добавляли порциями к раствору промежуточного соединения 4 (22,3 г, 74 ммол) в DMF (300 мл) при 0 С. Смесь перемешивали при r.t. в течение 30 мин, затем медленно добавляли циклопропилметилбромид (10 г, 74 ммол) и смесь нагревали в течение ночи при 60 С. DMF удаляли в вакууме, а остаток распределяли между EtOAc и рассолом. Органическую фазу сушили (MgSO4) и концентрировали в вакууме. После очистки хроматографией на колонке (двуокись кремния, 0-10% EtOAc в DCM) получали соединение, указанное в заголовке, в виде продукта жлтого цвета (12,5 г, 47%). Н (CDCl3) 7,57 (1H, d, J 9,5 Гц), 6,47 (1 Н, d, J 9,5 Гц), 4,22 (2H, q, J 7,0 Гц), 3,87 (2H, d, J 7,1 Гц), 1,26-1,19 (4H, m), 0,43-0,37 (4H, m). LCMS (ES+) RT 3,80 мин, 357 (M+H)+. Промежуточное соединение 30. Этил-3-бром-7-(циклопропилметил)-6-оксо-6,7-дигидротиено[2,3b]пиридин-2-карбонитрил. Это соединение получали из промежуточного соединения 8 способом, описанным для соединения 29. Продукт не совсем белого цвета. H (CDCl3) 7,67 (1H, d, J 9,6 Гц), 6,68 (1H, d, J 9,6 Гц), 4,02 (2 Н, d, J 7,1 Гц), 1,36-1,23 (1H, m), 0,75-0,51 (4 Н, m). LCMS (ES+) RT 3,45 мин, 309,0 (М+Н)+. Промежуточное соединение 31. 3-Анилин-7-(циклопропилметил)-6-оксо-6,7-дигидротиено[2,3-b] пиридин-2-карбоксилат аммония. Раствор из примера 44 (1,38 г, 3,75 ммол) и NaOH (160 мг, 4,0 ммол) в ЕtOН (50 мл) и воду (20 мл) нагревали при температуре рефлюкса в течение 2 ч. ЕtOН удаляли в вакууме и добавляли воду (30 мл). Раствор обрабатывали 10% NH4Cl водн. (100 мл) и охлаждали. Осадок отфильровывали и сушили, получали соединение, указанное в заголовке, в виде продукта не совсем белого цвета (1,15 г, 90%). Н(DMSO-d6) 7,03-6,98 (2 Н, m), 6,91 (1H, d, J 9,5 Гц), 6,72-6,68 (3 Н, m), 6,02 (1H, d, J 9,5 Гц), 3,70 (2 Н, d, J 7,0 Гц), 1,16-1,03 (1H, m), 0,32-0,24 (4 Н, m). Промежуточное соединение 32. Этил-3-бром-6-(циклопропилметокси)тиено[2,3-b]пиридин-2 карбоксилат. Смесь промежуточного соединения 4 (5,0 г, 16,5 ммол), карбоната цезия (5,39 г, 16,5 ммол) и цик- 21008410 лопропилметилбромида (1,58 мл, 16,5 ммол) в DMF (150 мл) нагревали при 90 С в течение 3 дней. Добавляли EtOAc и промывали раствор рассолом, сушили (MgSO4) и концентрировали в вакууме. Очистка хроматографией на колонке (двуокись кремния, 20% EtOAc в гексане) позволила получить соединение,указанное в заголовке (2,64 г, 45%). Н (CDCl3) 7,95 (1H, d, J 8,85 Гц), 6,81 (1H, d, J 8,88 Гц), 4,34 (2 Н, q, J 7,2 Гц), 4,18 (2 Н, d, J 8,8 Гц), 1,35 (3 Н, t, J 7,1 Гц), 1,26-1,22 (1H, m), 0,58 (2 Н, dd, J 4,8, 5,4 Гц), 0,31-0,27(2 Н, m). LCMS (ES+) RT 5,43 мин, 357 (М+Н)+. Промежуточное соединение 33. Этил-3-анилин-6-(циклопропилметокси)тиено[2,3-b]пиридин-2 карбоксилат. Смесь промежуточного соединения 32 (2,0 г, 5,6 ммол), анилина (0,61 мл, 6,72 ммол), карбоната цезия (2,55 г, 7,8 ммол), BINAP (690 мг, 20 мол.%) и трис-(дибензилиденацетон)дипалладия(0) (510 мг, 10 мол.%) в толуоле (50 мл) нагревали при температуре рефлюкса в течение 3 дней. Добавляли рассол и смесь обрабатывали EtOAc. Органический экстракт сушили (MgSO4) и концентрировали в вакууме. После очистки хроматографией на колонке (двуокись кремния, 5% EtOAc в гексане) получали соединение,указанное в заголовке (1,8 г, 86%). Н (CDCl3) 8,72 (1 Н br s), 7,29-7,26 (2 Н, m), 7,18-7,13 (2 Н, m), 6,937,06 (3 Н, m), 6,39 (1H, d, J 9,01 Гц), 4,22 (2 Н, q, J 7,1 Гц), 4,05 (2 Н, d, J 7,2 Гц), 1,26 (3 Н, t, J 7,1 Гц), 0,480,46 (2 Н, m), 0,25-0,23 (2 Н, m). Промежуточное соединение 34. Этил-3-анилин-6-оксо-6,7-дигидротиено[2,3-b]пиридин-2-карбоксилат. Промежуточное соединение 33 (1,8 г, 4,9 ммол) растворяли в МеОН (100 мл), добавляли конц. HCl водн. (10 мл) и смесь нагревали при температуре рефлюкса в течение 6 ч. Реакционную смесь экстрагировали СНСl3 и органическую фазу промывали рассолом, сушили (MgSO4) и концентрировали в вакууме,получали соединение, указанное в заголовке, в виде продукта жлтого цвета (1,3 г, 84%). Н (CDCl3) 8,75(1H, br s), 7,34-7,20 (4 Н, m), 7,18-7,02 (2 Н, m), 6,26 (1H, d, J 9,63 Гц), 4,25 (2H, q, J 7,1 Гц), 1,29 (3 Н, t, J 7,1 Гц). LCMS (ES+) RT 3,296 мин, 314,9 (M+H)+. Промежуточное соединение 35. Этил-3-бром-6-оксо-7-(3-тиенил)-6,7-дигидротиено[2,3-b]пиридин 2-карбоксилат. Смесь промежуточного соединения 4 (2,36 г, 7,5 ммол), 2-тиофенбороновой кислоты (2,0 г, 15 ммол) и ацетат меди (II) (1,41 г, 7,5 ммол) в пиридине (3,7 мл) перемешивали при r.t. в течение 3 дней. Смесь разбавляли DCM, промывали 2 М НСl водн., сушили (MgSO4) и концентрировали в вакууме. После очистки хроматографией на колонке (двуокись кремния, 50% EtOAc в гексане) получили соединение,указанное в заголовке (270 мг). Н (CDCl3) 7,83 (1H, d, J 9,6 Гц), 7,60-7,58 (2 Н, m), 7,18-7,16 (1 Н, m), 6,68(1H, d, J 9,6 Гц), 4,31 (2 Н, q, J 7,1 Гц), 1,32 (3 Н, t, J 7,1 Гц). LCMS (ES+) 386 (М+Н)+. Промежуточное соединение 36. Этил-3-[(4-фтор-3-метилфенил)амино]-6-(циклопропилметокси) тиено[2,3-b]пиридин-2-карбоксилат. Способом, описанным для соединения 33, получали указанное соединение из промежуточного соединения 32 и 4-фтор-3-метиланилина в виде вещества белого цвета. Н (CDCl3) 8,64 (1H, br s), 7,24-7,13[2,3-b]пиридин-2-карбоксилат. Это соединение получали из соединения 36 способом, описанным для соединения 34. Н (CDCl3) 7,18-7,13 (3 Н, m), 6,95-6,84 (3 Н, m), 6,39 (1H, d, J 9,6 Гц), 4,26 (2 Н, q, J 7,2 Гц), 2,19 (3 Н, s), 1,30 (3 Н, t, J 7,2 Гц). LCMS (ES+) RT 3,585 мин, 346 (М+Н)+. Промежуточное соединение 38. 3-[(6-Метил-1-оксидопиридин-2-ил)амино]-6-оксо-7-фенил-6,7 дигидротиено[2,3-b]пиридин-2-карбонитрил. Гидрид натрия (330 мг, 60% в минеральном масле, 8,25 ммол) добавляли к суспензии промежуточного соединения 11 (2 г, 7,5 ммол) в THF (150 мл) и затем нагревали смесь до 70 С в течение 5 мин. Приr.t. добавляли 6-хлорпиколина N-оксид (1,13 г, 8,25 ммол) и смесь нагревали при 70 С в течение 24 ч. Растворитель удаляли в вакууме. После очистки на хроматографической колонке (двуокись кремния,50% THF в DCM - 40% МеОН в DCM) получили соединение, указанное в заголовке (1,54 г, 55%). LCMS(ES+) RT 2,79 мин, 375 (М+Н)+. Промежуточное соединение 39. Этил-3-бром-7-[(4-метилтио)фенил]-6-оксо-6,7-дигидротиено[2,3b]пиридин-2-карбоксилат. Это соединение получали способом, описанным для соединения 5, из промежуточного соединения 4 и 4-тиометилбороновой кислоты. Н (CDCl3) 7,76 (1 Н, d, J 9,7 Гц), 7,36 (2 Н, d, J 8,7 Гц), 7,23 (2 Н, d, J 8,7 Гц), 6,65 (1H, d, J 9,7 Гц), 4,26 (2 Н, q, J 7,1 Гц), 2,48 (3 Н, s), 1,27 (3 Н, t, J 7,1 Гц). LCMS (ES+) RT 3,91 мин, 426 (M+H)+. Промежуточное соединение 40. Этил-3-бром-7-(4-формилфенил)-6-оксо-6,7-дигидротиено[2,3b]пиридин-2-карбоксилат. Это соединение получают способом, описанным для соединения 5, из промежуточного соединения 4 и 4-формилбороновой кислоты. Н (CDCl3) 10,03 (1H, s), 8,12-8,0 (2 Н, m), 7,85-7,78 (1H, m), 7,54 (2 Н, d,- 22008410(М+Н)+. Промежуточное соединение 41. Этил-3-бром-6-оксо-7-[(4-пирролидин-1-илметил)фенил]-6,7 дигидротиено[2,3-b]пиридин-2-карбоксилат. Смесь промежуточного соединения 40 (150 мг, 0,36 ммол), пирролидина (0,03 мл, 0,36 ммол) и триацетоксиборгидрида натрия (117 мг, 0,55 ммол) в 1,2-дихлорэтане (5 мл) нагревали при температуре рефлюкса в течение ночи. Смесь промывали насыщенным водным раствором NaHCO3. Органическую фазу сушили (MgSO4) и концентрировали в вакууме. После очистки на хроматографической колонке(1H, t, J 5,8 Гц), 3,19 (5 Н, br s), 1,35 (3 Н, t, J 7,1 Гц). LCMS (ES+) RT 2,367 мин, 463 (M+H)+. Промежуточное соединение 42. Этил-3-бром-7-(4-[трет.бутил(диметил)силил]оксифенил)-6-оксо 6,7-дигидротиено[2,3-b]пиридин-2-карбоксилат. Это соединение получали способом, описанным для соединения 5, из промежуточного соединения 4 и (4-[трет.бутил(диметил)силил]оксифенил)бороновой кислоты. Н (CDCl3) 7,75 (1H, d, J 9,6 Гц),7,17 (2 Н, d, J 8,8 Гц), 6,96 (2 Н, d, J 8,8 Гц), 6,64 (1H, d, J 9,6 Гц), 4,26 (2 Н, q, J 7,1 Гц), 1,27 (3 Н, t, J 7,1 Гц),0,94 (9 Н, S), 0,20 (6 Н, s). LCMS (ES+) RT 3,405 мин, 508/510 (79Br/81Br) (M+H)+. Промежуточное соединение 43. Этил-7-(4-[трет.бутил(диметил)силил]оксифенил)-3-[(2,4 дифторфенил)амино]-6-оксо-6,7-дигидротиено[2,3-b]пиридин-2-карбоксилат. Получают способом, описанным в примере 1 из промежуточного соединения 42 и 2,4 дифторанилина. Н (CDCl3) 8,45 (1 Н, br s), 7,20-7,10 (3 Н, m), 7,00-6,77 (5 Н, m), 6,29 (1H, d, J 9,8 Гц), 4,20(2 Н, q, J 7,1 Гц), 1,23 (3 Н, t, J 7,1 Гц), 0,94 (9 Н, s), 0,20 (6 Н, s). LCMS (ES+) RT 3,890 мин, 557 (М+Н)+. Промежуточное соединение 44. Этил-3-[(2,4-дифторфенил)амино]-7-(4-дигидроксифенил)-6-оксо 6,7-дигидротиено[2,3-b]пиридин-2-карбоксилат. Тетрабутиламмоний фторид (483 мг, 1,85 ммол) добавляли к раствору промежуточного соединения 43 (1,0 г, 1,80 ммол) в THF (20 мл). Через 15 мин реакционную смесь распределяли между ЕtOАс (50 мл) и водой (50 мл). Органическую фазу промывали рассолом, сушили (MgSO4) и концентрировали в вакууме, получая соединение, указанное в заголовке (745 мг, 95%). H (DMSO-d6) 8,65 (1H, br s), 7,50-7,27 (4 Н,m), 7,20-7,10 (2 Н, m), 6,99 (2 Н, d, J 8,8 Гц), 6,40 (1 Н, d, J 9,8 Гц), 4,19 (2H, q, J 7,1 Гц), 1,21 (3 Н, t, J 7,1 Гц). LCMS (ES+) RT 3,590 мин, 443 (M+H)+. Промежуточное соединение 45. Этил-7-[4-(2-бромэтокси)фенил]-3-[(2,4-дифторфенил)амино]-6 оксо-6,7-дигидротиено[2,3-b]пиридин-2-карбоксилат. Диизопропилазодикарбоксилат (0,334 мл, 1,70 ммол) добавляли к раствору промежуточного соединения 44 (500 мг, 1,13 ммол) и трифенилфосфина (445 мг, 1,70 ммол) в смеси DCM (10 мл) и THF (5 мл). Добавляли 2-бромэтанол (0,125 мл, 1,70 ммол) и смесь перемешивали в течение 3 дней. Добавляли воду(30 мл), органическую фазу сушили (MgSO4) и концентрировали в вакууме. После очистки хроматографией на колонке (двуокись кремния, 5% ЕtOАс в DCM) получали соединение, указанное в заголовке (375 мг, 60%). Н (CDCl3) 8,40 (1H, br s), 7,18-7,11 (2 Н, m), 7,03-6,87 (4 Н, m), 6,81-6,68 (2 Н, m), 6,19 (1 Н, d, J 9,8 Гц), 4,19 (2 Н, t, J 6,2 Гц), 4,09 (2 Н, q, J 7,1 Гц), 3,50 (2 Н, t, J 6,2 Гц), 1,12 (3 Н, t, J 7,1 Гц). LCMS (ES+)RT 4,206 мин, 549/551 (79Br/81Br) (M+H)+. Промежуточное соединение 46. Этил-3-бром-6-оксо-7-(4-винилфенил)-6,7-дигидротиено[2,3b]пиридин-2-карбоксилат. Получали способом, описанным для соединения 5, из промежуточного соединения 4 и (4 винилфенил)бороновой кислоты. Н (CDCl3) 7,95 (1 Н, d, J 9,7 Гц), 7,76-7,73 (2 Н, m), 7,47-7,44 (2 Н, m),6,90 (1H, dd, J 17,6, 11,1 Гц), 6,83 (1H, d, J 9,7 Гц), 5,96 (1H, dd, J 17,6, 0,4 Гц), 5,51 (1H, d, J 11,1 Гц), 4,43(2 Н, q, J 7,1 Гц), 1,44 (3 Н, t, J 7,1 Гц). LCMS (ES+) RT 3,947 мин, 404/406 (79Br/81Br) (M+H)+. Промежуточное соединение 47. 3-Бром-7-(4-[трет.бутил(диметил)силил]оксифенил)-6-оксо-6,7 дигидротиено[2,3-b]пиридин-2-карбонитрил. Это соединение получали способом, описанным для соединения 5, из промежуточного соединения 8 и (4-[трет.бутил(диметил)силил]оксифенил)бороновой кислоты. Н (CDCl3) 7,52 (1H, d, J 9,6 Гц),7,05-6,99 (2 Н, m), 6,84-6,79 (2 Н, m), 6,54 (1H, d, J 9,6 Гц), 0,80 (9 Н, s),0,05 (6 Н, s). LCMS (ES+) RT 4,65 мин, 461/463 (79 Вr/81 Вr) (М+Н)+. Промежуточное соединение 48. 7-(4-[трет.Бутил(диметил)силил]оксифенил)-3-[(2,4-дифторфенил)амино]-6-оксо-6,7-дигидротиено[2,3-b]диридин-2-карбонитрил. Получают способом, описанным в примере 1, из промежуточного соединения 47 и 2,4 дифторанилина. Н (CDCl3) 7,14 (1 Н, d, J 9,7 Гц), 7,04-7,00 (2 Н, m), 6,95-6,87 (1H, m), 6,80-6,63 (4 Н, m),6,32 (1H, d, J 9,7 Гц), 5,93 (1H, br s), 0,78 (9 Н, s), 0,04 (6H,s). LCMS (ES+) RT 4,81 мин, 510 (M+H)+. Промежуточное соединение 49. 3-[(2,4-Дифторфенил)амино]-7-(4-гидроксифенил)-6-оксо-6,7 дигидротиено[2,3-b]пиридин-2-карбонитрил. Получают указанное соединение способом, описанным для соединения 44, из промежуточного соединения 48. Н (d3-MeOD) 8,14 (1H, d, J 9,6 Гц), 7,54-7,32 (3 Н, m), 7,22-7,06 (4 Н, m), 6,74 (1 Н, d, J 9,6- 23008410 Гц). LCMS (ES+) RT 3,113 мин, 396 (M+H)+. Промежуточное соединение 50. 3-[(2,4-Дифторфенил)амино]-7-4-[(2,2-диметил-1,3-диоксолан-4 ил)метокси]фенил-6-оксо-6,7-дигидротиено[2,3-b]пиридин-2-карбонитрил. Смесь промежуточного соединения 49 (370 мг, 0,94 ммол), (R)-(2,2-диметил-1,3-диоксолан-4 ил)метил-4-метилбензолсульфоната (538 мг, 1,88 ммол) и карбоната цезия (365 мг, 1,1 ммол) в DMF (2 мл) нагревали при 80 С в течение 3 дней. Смесь распределяли между EtOAc и рассолом. Органическую фазу сушили (MgSO4) и концентрировали в вакууме. После очистки на хроматографической колонке(силикагель, 20% ЕtOАс в DCM) получали соединение, указанное в заголовке (54 мг). Н (CDCl3) 7,327,20 (3 Н, m), 7,12-7,02 (3 Н, m), 6,93-6,78 (2 Н, m), 6,49 (1H, d, J 9,7 Гц), 6,12 (1 Н, s), 4,49-4,41 (1H, m),4,15-3,84 (4 Н, m), 1,41 (3 Н, s ), 1,35 (3 Н, s ). LCMS (ES+) RT 3,516 мин, 510 (М+Н)+. Промежуточное соединение 51. Этил-3-бром-7-(2-нитрофенил)-6-оксо-6,7-дигидротиено[2,3b]пиридин-2-карбоксилат. Гидрид натрия (440 мг, 60% в минеральном масле, 11 ммол) добавляли порциями в суспензию промежуточного соединения 4 (3,02 г. 10 ммол) в DMF (40 мл) при r.t. Через 15 мин добавляли 2 фторнитробензол (2,11 мл, 20 ммол) и нагревали смесь при 90 С в течение 4 дней. DMF удаляли в вакууме и остаток очищали на хроматографической колонке (двуокись кремния, 0-3% THF в DCM). Соединение, указанное в заголовке, получали в виде жлтого тврдого вещества (1,36 г, 32%). Н (DMSOd6) 8,37 (1H, dd, J 1,4, 8,1 Гц), 8,10-8,06 (1H, m), 8,01-7,92 (3 Н, m), 6,71 (1H, d, J 9,7 Гц), 4,27 (2 Н, q, J 7,1 Гц), 1,24 (3 Н, t, J 7,1 Гц). LCMS (ES+) RT 3,644 мин, 424,9 (M+H)+. Промежуточное соединение 52. 3-[(2,4-Дифторфенил)амино]-7-(2-нитрофенил)-6-оксо-6,7 дигидротиено[2,3-b]пиридин-2-карбоксилат натрия. Смесь примера 95 (405 мг, 0,86 ммол) и гидроокись натрия (38 мг, 0,946 ммол) в воде (5 мл) и EtOH(10 мл) нагревали 90 мин при температуре рефлюкса. Растворитель удаляли в вакууме, получая вещество коричневого цвета. Этот сырой продукт является соединением, указанным в заголовке, и использовался на следующей стадии без очистки. LCMS (ES+) RT 3,262 мин, 443,8 (М+Н)+. Промежуточное соединение 53. 2-Амино-4-метил-6-оксо-1-фенил-1,6-дигидропиридин-3-карбонитрил. Получали, как описано Habashi et al., Liebigs Ann. Chem. 1986, 1632-1638.(DS-d 6) 7,63-7,45 (3, m), 7,25 (2, m), 6,70 (2, br s), 5,68 (1, s), 2,18 (3, s).LCMS (Условия В) (ES+) RT 1,97 мин, 226 (М+H)+. Промежуточное соединение 54. 2,5-Дибром-4-метил-6-оксо-1-фенил-1,6-дигидропиридин-3 карбонитрил. К перемешиваемой смеси бромида меди (II) (10,35 г) и трет.бутилнитрила (6,6 мл) в среде ацетонитрила (75 мл) добавляли порциями в течение 15 мин промежуточное соединение 53 (7,45 г). Полученную смесь перемешивали при контатной температуре ещ 2 ч, затем выливали в 2 М HCl (водн.) (100 мл). Добавляли этилацетат (150 мл) и смесь перемешивали около 1 ч. Затем слои разделяли и водную фазу обрабатывали для экстракции этилацетатом (50 мл). Соединнные органические экстракты промывали 2 М НСl (водн.) (100 мл). Все нерастворимые вещества удаляли фильтрованием и полученный фильтрат промывали рассолом и концентрировали в вакууме. Остаток очищали на колонке с силикагелем, используя DCM в качестве элюента. Получали указанное в заголовке соединение в виде бледно-жлтого продукта (5,63 г). Н (CDCl3) 7,55 (3 Н, m), 7,17 (2 Н, m), 2,65 (3 Н, s). LCMS (Условия В) (ES+) RT 3,59 мин,369 (М+Н)+. Промежуточное соединение 55. Этил-3-амино-5-бром-4-метил-6-оксо-7-фенил-6,7-дигидротиено[2,3-b]пиридин-2-карбоксилат. Смесь промежуточного соединения 54 (5,6 г), этил-2-меркаптоацетата, (2,1 мл) и карбоната натрия(1,70 г) в абсолютном этаноле (56 мл) нагревали 2,5 ч при температуре рефлюкса. Затем реакционную смесь охлаждали до комнатной температуры, добавляли воду и полученную смесь перемешивали ещ 1 ч. Продукт выделяли фильтрованием, промывали водой (56 мл) и сушили в вакууме, получая указанное в заголовке соединение в виде продукта не совсем белого цвета (5,83 г). Н (CDCl3) 7,55 (3 Н, m), 7,35 (2 Н,m), 6,15 (2 Н, br s), 4,25 (2 Н, q, J 7,0 Гц), 2,86 (3 Н, s), 1,28 (3 Н, t, J 7,0 Гц). LCMS (Условия В) (ES+) RT 3,77 мин, 407 (М+Н)+. Промежуточное соединение 56. Этил-3-амино-4-метил-6-оксо-7-фенил-6,7-дигидротиено[2,3b]пиридин-2-карбоксилат. К расвору промежуточного соединения 55 (5,83 г) в DCM (650 мл), содержащему триэтиламин (4 мл), добавляли 5% палладия на угле (Johnson Matthey type 38H paste; 0,58 г) и полученную смесь перемешивали в атмосфере водорода до тех пор, пока не прекратилось потребление водорода. Катализатор удаляли фильтрованием и органическую фазу промывали водой (дважды) и затем выпаривали в вакууме,получая соединение, указанное в заголовке, в виде белого продукта (4,73 г). Н (CDCl3) 7,57 (3 Н, m), 7,35- 24008410 пиридин-2-карбоксилат. К перемешиваемой смеси бромида меди (II) (2,9 г) и трет.бутилнитрита (1,84 мл) в ацетонитриле(90 мл) добавляли промежуточное соединение 56 (3,05 г) порциями в течение 15 мин. Полученную смесь перемешивали при комнатной температуре ещ в течение 0,5 ч и добавляли 2 М НСl (водн.) (150 мл). Реакционную смесь обрабатывали этилацетатом (дважды) и соединнные органические экстракты промывали водой (75 мл), рассолом (100 мл) и затем сушили (Na2SO4) и выпаривали в вакууме. Остаток очищали хроматографией на силикагеле с применением элюента DCM, получая соединение, указанное в заголовке, в виде вещества не совсем белого цвета (1,23 г). Н (CDCl3) 7,63 (3 Н, m), 7,35 (2 Н, m), 6,53 (1H, s),4,30 (2 Н, q, J 7,0 Гц), 2,79 (3 Н, s), 1,32 (3 Н, t, J 7,0 Гц). LCMS (Условия В) (ES+) RT 3,98 мин, 392 (М+Н)+. Промежуточное соединение 58. 3-[(2,4-Дифторфенил)амино]-7-фенилтиено[2,3-b]пиридин-6(7 Н)он. Получали из промежуточного соединения 67 способом по примеру 15. Н (CDCl3) (DMSO-d6) 7,94(1 Н, d, J 9,4 Гц), 7,64-7,53 (3 Н, m), 7,46 (2 Н, m), 7,45 (1H, m), 7,20 (1H, m), 6,99 (1H, m), 6,48 (1H, d, J 9,4 Гц), 5,74 (1 Н, s). LCMS (ES+) RT 3,54 мин, 354,9 (M+H)+. Промежуточное соединение 59. трет.Бутил-(2,4-дифторфенил)(6-оксо-7-фенил-6,7-дигидротиено[2,3-b]пиридин-3-ил)карбамат. бис(Триметилсилил)амид натрия (6,0 мл, 1,0 М в THF, 6 ммол) добавляли к раствору промежуточного соединения 58 (2,0 г, 5,65 ммол) в THF (50 мл) при 0 С. Через 30 мин добавляли дитрет.бутилдикарбонат (1,36 г, 6,22 ммол) и смесь перемешивали при r.t. в течение 1 ч. Реакционную смесь распределяли между EtOAc и рассолом. Водную фазу обрабатывали EtOAc (х 3), соединнные органические экстракты промывали рассолом, сушили (MgSO4) и концентрировали в вакууме. После очистки на хроматографической колонке (силикагель, 10% ЕtOАс в DCM) получали соединение, указанное в заголовке (1,2 г, 47%). Н (CDCl3) 7,45 (1H, d, J 9,5 Гц), 7,43-7,30 (3 Н, m),7,22-7,19 (2H, m), 7,08-7,03 (1H, m),6,76-6,65 (2 Н, m), 6,49- 6,45 (2 Н, m), 1,26 (9 Н, s). LCMS (ES+) RT 3,79 мин, 455 (М+Н)+. Промежуточное соединение 60. трет.Бутил-[2-(аминосульфонил)-6-оксо-7-фенил-6,7 дигидротиено[2,3-b]пиридин-3-ил] (2,4-дифторфенил)карбамат. н-Бутиллитий (0,18 мл 2,5 М раствора в гексанах, 0,44 ммол) добавляли к раствору промежуточного соединения 59 (200 мг, 0,44 ммол) в THF (10 мл) при -78 С. Через 3 мин через раствор пропускали газообразный серный ангидрид в течение 2 мин. Реакционной смеси давали нагреться до r.t. и удаляли растворитель в вакууме. Остаток растворяли в DCM (15 мл) и добавляли N-хлорсукцинимид (60 мг, 0,44 ммол). После выдержки с течение 20 мин при r.t. добавляли водный аммиак (конц.) (5 мл) и смесь перемешивали ещ 10 мин. Смесь разбавляли DCM и промывали рассолом. Органическую фазу сушили(MgSO4) и концентрировали в вакууме. После очистки на хроматографической колонке (силикагель, 10% ЕtOАс в DCM) получали соединение, указанное в заголовке (50 мг, 21%). Н (CDCl3) 7,42-7,36 (3 Н, m),7,23-7,16 (4 Н, m), 6,81-6,77 (1H, m), 6,75-6,63 (1H, m), 6,39 (1H, d, J 10,3 Гц), 5,43 (2 Н, br s), 1,27 (9 Н, s).[2,3-b]пиридин-2-карбоксилат. Гидрид натрия (60% в минеральном масле, 240 мг, 6 ммол) добавляли к суспензии промежуточного соединения 4 (1,50 г, 4,97 ммол) в DMF (12 мл). Через 10 мин при r.t добавляли бромметоксиэтоксиэтан(1,0 г, 5,5 ммол) и смесь нагревали при 60 С в течение 4 ч. Затем смесь распределяли между рассолом(200 мл) и DCM (200 мл). Ограническую фазу промывали рассолом (2 х 100 мл), сушили (Na2SO4) и концентрировали в вакууме. После очистки на хроматографической колонке (силикагель, 5-15% THF вDCM) получали соединение, указанное в заголовке, в виде белого продукта (420 мг, 21%). Н (CDCl3) 7,78 (1 Н, d, J 9,6 Гц), 6,65 (1H, d, J 9,6 Гц), 4,42 (2 Н, q, J 7,1 Гц), 7,38-7,34 (2 Н, m), 3,96-3,07 (2 Н, m), 3,683,64 (2 Н, m), 3,48-3,45 (2 Н, m), 3,32 (3 Н, s), 1,41 (3 Н, t, J 7,1 Гц). Промежуточное соединение 62. Этил-3-бром-6-оксо-7-(тетрагидро-2 Н-пиран-2-илметил)-6,7 дигидротиено[2,3-b]пиридин-2-карбоксилат. Гидрид натрия (60% в минеральном масле, 320 мг, 8 ммол) добавляли к суспензии промежуточного соединения 4 (2,0 г, 6,67 ммол) в DMF (15 мл). Через 10 мин при r.t. добавляли 2-(бромметил)тетрагидро-2 Н-пиран (0,895 мл, 7 ммол) и смесь нагревали при 80 С в течение 18 ч. Смесь распределяли между рассолом (200 мл) и ЕtOАс (120 мл). Органическую фазу промывали рассолом (3 х 75 мл), сушили(Na2SO4) и концентрировали в вакууме. После очистки на хроматографической колонке (силикагель, 010% ЕtOАс в DCM) получили соединение, указанное в заголовке, в виде белого тврдого продукта (642 мг, 24%). Н (CDCl3) 7,76 (1 Н, d, J 9,6 Гц), 6,63 (1H, d, J 9,6 Гц), 4,45-4,30 (3 Н, m), 3,93-3,78 (3 Н, m), 3,323,26 (1 Н, m), 1,90-1,86 (1H, m), 1,77-1,74 (1H, m), 1,66-1,38 (4 Н, m), 1,42 (3 Н, t, J 7,1 Гц). LCMS (ES+) RT 3,90 мин, 440,0/402,0 (79Br/81Br) (M+H)+. Промежуточное соединение 63. Этил-3-бром-7-бензил-6-оксо-6,7-дигидротиено[2,3-b]пиридин-2 карбоксилат. Получали способом, описанным для промежуточного соединения 61, из промежуточного соединения 4 и бензилбромида. Н (CDCl3) 7,80 (1 Н, d, J 9,6 Гц), 7,41-7,32 (5 Н, m), 6,74 (1H, d, J 9,6 Гц), 5,37 (2 Н,- 25008410s), 4,38 (2 Н, q, J 7,1 Гц), 1,40 (3 Н, t, J 7,1 Гц). LCMS (ES+) RT 3,89 мин, 392,0/394,0 (79Br/81Br) (M+H)+. Промежуточное соединение 64. 3-Анилин-7-бензил-6-оксо-6,7-дигидротиено[2,3-b]пиридин-2 карбоксилат аммония. Получали из соединений по примеру 102 способом, описанным для. промежуточного соединения 31. Н (DMSO-d6) 7,47-7,25 (9 Н, m), 7,19-7,02 (3 Н, m), 6,43 (1 Н, d, J 9,6 Гц), 5,35 (2 Н, s). Промежуточное соединение 65. Этил-3-бром-7-[4-(диметиламино)фенил]-6-оксо-6,7-дигидротиено[2,3-b]пиридин-2-карбоксилат. Получали из промежуточного соединения 4 и 4-(диметиламино)фенилбороновой кислоты способом, описанным для промежуточного соединения 5, в виде светло-коричневого продукта. Н (CDCl3) 7,68 (1H, d, J 9,7 Гц), 7,08 (2H, d, J 9,0 Гц), 6,71 (2H, d, J 9,0 Гц), 6,59 (1H, d, J 9,7 Гц), 4,20 (2H, q, J 7,1 Гц), 2.93 (6H, s): 1,21 (3H, t, J 7, 1 Гц). Промежуточное соединение 66. 3 Анилин-7-[4-(диметиламино)фенил]-6-оксо-6,7-дигидротиено[2,3b]пиридин-2-карбоксилат аммония. Получали из соединений по примеру 105 способом, описанным в примере 31. Н (DMSO-d6) 7,267,17 (6 Н, m), 6,93-6,89 (2 Н, m), 6,83 (2 Н, d, J 9,0 Гц), 6,26 (1H, d, J 9,6 Гц), 2.97 (6H, s). LCMS (ES+) RT 3,30 мин, 406 (M+H)+. Промежуточное соединение 67. 3-[(2,4-Дифторфенил)амино]-6-оксо-6,7-дигидротиено[2,3b]пиридин-2-карбоксилат аммония. Получали из соединения по примеру 12 способом, описанным для промежуточного соединения 31. Н (DMSO-d6) 9,75 (1 Н, br s), 7,75-7,50 (3 Н, m), 7,49-7,37 (2 Н, m), 7,31-7,27 (2 Н, m), 7,00-6,80 (2 Н, br m),6,35-6,31 (1H, br m). LCMS (ES+) RT 3,25 мин, (М+Н)+ не наблюдалось. Промежуточное соединение 68. Этил-3-бром-7-(4-фторфенил)-6-оксо-6,7-дигидротиено[2,3b]пиридин-2-карбоксилат. Получали способом, описанным для промежуточного соединения 5, из промежуточного соединения 4 и 4-фторфенилбороновой кислоты. Н (CDCl3) 7,84 (1 Н, d, J 9,7 Гц), 7,41-7,37 (2 Н, m), 7,32-7,25 (2 Н,m), 6,72 (1 Н, d, J 9,7 Гц), 4,33 (2H, q, J 7,1 Гц), 1,34 (3H, t, 77,1 Гц). LCMS (ES+) RT 3,729 мин, 397,8(M+H)+. Промежуточное соединение 69. Этил-3-бром-7-(4-хлорфенил)-6-оксо-6,7-дигидротиено[2,3-b]пиридин-2-карбоксилат. Получали из промежуточного соединения 4 и 4-хлорфенилбороновой кислоты способом, описанным для промежуточного соединения 5. Н (CDCl3) 7,86 (1H, d, J 9,6 Гц), 7,60 (2 Н, d, J 8,5 Гц), 7,37 (2H,d, J 8,5 Гц), 6,74 (1H, d, J 9,6 Гц), 4,35 (2H, q, J 7,1 Гц), 1,36 (3H, t, J 7,1 Гц). LCMS (ES+) RT 3,937 мин,413 (M+H)+. Промежуточное соединение 70. Этил-3-бром-7-(3-метилфенил)-6-оксо-6,7-дигидротиено[2,3-b] пиридин-2-карбоксилат. Получали из промежуточного соединения 4 и 3-метилфенилбороновой кислоты способом, описанным для промежуточного соединения 5. H (CDCl3) 7,85 (1H, d, J 9,6 Гц), 7,51-7,48 (1H, m), 7,38- 7,27LCMS (ES+) RT 3,865 мин, 393 (M+H)+. Промежуточное соединение 71. 3-[(3-(Трифторметил)фенил)амино]-6-оксо-7-фенил-6,7 дигидротиено[2,3-b]пиридин-2-карбоксилат. Получали из промежуточного соединения 9 и 3-(трифторметил)анилина способом по примеру 1.LCMS (ES+) RT 3,63 мин, 412 (М+Н)+. Промежуточное соединение 72. 3-Бром-7-(4-фторфенил)-6-оксо-6,7-дигидротиено[2,3-b]пиридин-2 карбонитрил. Получали из промежуточного соединения 8 и 4-фторфенилбороновой кислоты способом, описанным для промежуточного соединения 5. Н (DMSO-d6) 8,1-8,0 (1H, m), 7,8-7,7 (2 Н, m), 7,65-7,6 (2 Н, m),6,9-6,85 (1H, m). LCMS (ES+) RT 3,460 мин, 350,8 (М+Н)+. Промежуточное соединение 73. 3-Бром-7-(4-метоксифенил)-6-оксо-6,7-дигидротиено[2,3-b]пиридин-2-карбонитрил. Получали из промежуточного соединения 8 и 4-метоксифенилбороновой кислоты способом, описанным для промежуточного соединения 5. Н (CDCl3) 7,53 (1H, d, J 9,7 Гц), 7,10-7,05 (2 Н, m), 6,92-6,87(2 Н, m), 6,56 (1H, d, J 9,7 Гц), 3,69 (3H, s). LCMS (ES+) RT 3,452 мин, 361/363 (79Br/81Br) (M+H)+. Промежуточное соединение 74. 7-(4-Ацетилфенил)-3-бром-6-оксо-6,7-дигидротиено[2,3-b]пиридин 2-карбонитрил. Получали из промежуточного соединения 8 и 4-ацетилфенилбороновой кислоты способом, описанным для промежуточного соединения 5. Н (CDCl3) 7,98 (2 Н, d, J 8,7 Гц), 7,55 (1H, d; J 9,7 Гц), 7,29 (2H,d, J 8,7 Гц), 6,56 (1H, d, J 9,7 Гц), 2,46 (3H, s). LCMS (ES+) 375 (M+Na)+. Промежуточное соединение 75. 3-[(3-Хлор-4-фторфенил)амино]-6-оксо-7-фенил-6,7-дигидротиено[2,3-b]пиридин-2-карбоксилат аммония. Получали из соединений по примеру 129 способом, описанным для промежуточного соединения 31.- 26008410 Н (DMSO-d6) 7,65-7,59 (3 Н, m), 7,52-7,50 (2 Н, m), 7,34-7,29 (2 Н, m), 7,10-7,21 (3 Н, br m), 6,99-6,95 (1H,m), 6,40 (1H, d, J 9,6 Гц). LCMS (ES+) RT 3,363 мин. Промежуточное соединение 76. 3-[(4-Фторфенил)амино]-6-оксо-7-фенил-6,7-дигидротиено[2,3b]пиридин-2-карбоксилат аммония. Получали из соединений по примеру 127 способом, описанным для промежуточного соединения 31. Промежуточное соединение 77. 3-[(3-Хлорфенил)амино]-6-оксо-7-фенил-6,7-дигидротиено[2,3-b] пиридин-2-карбоксилат аммония. Получали из соединений по примеру 3 способом, описанным для промежуточного соединения 31. Н (DMSO-d6) 7,65-7,57 (3 Н, m), 7,51-7,49 (2 Н, m), 7,34 (1H, d, J 9,6 Гц), 7,28-7,22 (1H, m), 7,00-6,99 (1 Н,m), 6,96-6,89 (2 Н, m), 6,39 (1H, d, J 9,6 Гц). LCMS (ES+) RT 3,372 мин, 396,8 (M+H)+. Промежуточное соединение 78. трет.Бутил(имино-3-[(3-метилфенил)амино]-6-оксо-7-фенил-6,7 дигидротиено[2,3-b]пиридин-2-илметил)-2-карбамат. Раствор этилбромида магния (1 М, 2,8 мл, 2,8 ммол) добавляли к трет.бутилкарбамату (328 мг, 2,8 ммол) в среде простого эфира при r.t. Через 10 мин добавляли соединение по примеру 139 (200 мг, 0,56 ммол) и нагревали смесь при температуре рефлюкса в течение 1 ч. Добавляли THF (7 мл) и нагревали смесь при 60 С в течение 18 ч. Добавляли воду и экстрагировали смесь DCM. Органическую фазу высушивали (Na2SO4) и концентрировали в вакууме. После очистки на хроматографической колонке (силикагель, Et2O) получали соединение, указанное в заголовке, в виде вещества жлтого цвета (46 мг). Н(CDCl3) 9,53 (1 Н, s), 7,58-7,45 (3 Н, m), 7,34 (2 Н, d, J 7,7 Гц), 7,19-7,12 (2 Н, m), 6,88-6,80 (3 Н, m), 6,29 (1 Н,d, J 9,7 Гц), 2,27 (3H, s), 1,41 (9H, s). LCMS (ES+) RT 3,343 мин, 475 (M+H)+. Промежуточное соединение 79. Пентафторфенил-3-[(2,4-дифторфенил)амино]-6-оксо-7-фенил-6,7 дигидротиено[2,3-b]пиридин-2-карбоксилат. К раствору промежуточного соединения 67 (2,5 г, 6,18 ммол) в DMF (100 мл) добавляли CDI (1,42 г,7,42 ммол). Через 20 мин добавляли пентафторфенол (1,71 г, 9,27 ммол) и перемешивали смесь при r.t. в течение ночи. Растворитель удаляли в вакууме, остаток распределяли между DCM и водой и водную фазу обрабатывали DCM. Соединнные органические слои сушили (MgSO4) и концентрировали в вакууме. После очистки на хроматографической колонке (силикагель, 20-100% ЕtOАс в гексане) получали соединение, указанное в заголовке, в виде белого продукта (666 мг, 19%). Н (CDCl3) 8,66 (1 Н, br s), 7,76 (3 Н,m), 7,58 (2 Н, m), 7,47 (1H, m), 7,14 (3 Н, m), 6,54 (1H, d, J 9,9 Гц). LCMS (ES+) RT 4,57 мин, 564,9 (M+H)+. Промежуточное соединение 80. 2-[4-(5,5-Диметил-1,3,2-диоксаборинан-2-ил)фенил]пропан-2-ол. Смесь 2-(4-бромфенил)пропан-2-ола (3,50 г, 16,3 ммол), бис(неопентилгликолят)дибора (4,05 г,17,93 ммол), КОАс (2,40 г, 24,45 ммол) и [(1,1'-бисдифенилфосфин)ферроцен]дихлорпалладия (II) (665 мг, 0,815 ммол) в DMF (25 мл) нагревали при 60 С в течение 18 ч. DMF удаляли в вакууме и получали азеотроп остатка с толуолом (х 3). Остаток растворяли в Et2O, фильтровали и фильтрат концентрировали в вакууме, получая соединение, указанное в заголовке, в виде коричневого продукта (3,08 г). Н (CDCl3) 7,70 (2 Н, d, J 9,8 Гц), 7,41 (2 Н, d, J 9,8 Гц), 3,69 (4H, s), 1,51 (6H, s), 0,95 (6H, s). Промежуточное соединение 81. Этил-3-бром-7-[4-(1-гидрокси-1-метилэтил)фенил]-6-оксо-6,7 дигидротиено[2,3-b]пиридин-2-карбоксилат. Получали из промежуточного соединения 4 и промежуточного соединения 80 способом, описанным для промежуточного соединения 5, в виде белого продукта. Н (CDCl3) 7,77 (1H, d, J 9,7 Гц), 7,66 (2 Н, d,J 8,5 Гц), 7,28 (2 Н, d, J 8,5 Гц), 6,66 (1H, d, J 9,6 Гц), 4,26 (2H, q, J 7,1 Гц), 1,75 (1H, br 5), 1,58 (6H, s), 1,27(3H, t, J 7,1 Гц). LCMS (ES+) RT 3,41 мин, 438 (M+H)+. Промежуточное соединение 82. Этил-3-бром-7-4-[(трет.бутоксикарбонил)амино]фенил-6-оксо 6,7-дигидротиено[2,3-b]пиридин-2-карбоксилат. Получали из промежуточного соединения 4 и 4-[(трет.бутоксикарбонил)амино]фенилбороновой кислоты способом, описанным для промежуточного соединения 5. Н (CDCl3) 8,85 (1H, d, J 8,8 Гц), 7,61(2 Н, d, J 8,8 Гц), 7,32 (2H, d, J 8,8 Гц), 6,75 (1H, s), 6,73 (1H, d, J 9,6 Гц), 4,34 (2H, q, J 7,1 Гц), 1,56 (9H, s),1,36 (3H, t, J 7,1 Гц). Промежуточное соединение 83. Этил-7-4-[(трет.бутоксикарбонил)амино]фенил-3-[(2,4 дифторфенил)амино]-6-оксо-6,7-дигидротиено[2,3-b]пиридин-2-карбоксилат. Получали из промежуточного соединения 82 и 2,4-дифторанилина способом по примеру 1. НLCMS (ES+) RT 4,288 мин, 542 (M+H)+. Промежуточное соединение 84. Этил-7-(4-аминофенил)-3-[(2,4-дифторфенил)амино]-6-оксо-6,7 дигидротиено[2,3-b]пиридин-2-карбоксилат. Трифторуксусную кислоту (0,5 мл) добавляли к раствору промежуточного соединения 83 в DCM (2 мл). Через 1 ч при r.t. удаляли в вакууме растворитель и получали азеотроп остатка с гептаном. Остаток растворяли в DCM и раствор промывали водным раствором NаНСО 3. Органическую фазу сушили(Na2SO4) и концентрировали в вакууме. После очистки на хроматографической колонке (силикагель,ЕtOАс) получали соединение, указанное в заголовке (72 мг, 82%). Н (CDCl3) 8,30 (1 Н, s), 7,05-6,97 (3 Н,- 27008410m), 6,89 (1H, d, J 9,7 Гц), 6,83-6,68 (3 Н, m), 6,66 (1H, d, J 8,6 Гц), 6,20 (1 Н, d, J 9,8 Гц), 4,11 (2H, q, J 7,1 Гц), 3,80 (2H, br s), 1,14 (3H, t, J 7,1 Гц). Промежуточное соединение 85. Этил-7-4-[бис(метилсульфонил)амино]фенил-3-[(2,4-дифторфенил)амино]-6-оксо-6,7-дигидротиено[2,3-b]пиридин-2-карбоксилат. Смесь промежуточного соединения 84 (70 мг, 0,16 ммол), метансульфонилхлорида (18 мг, 0,16 ммол) и триэтиламина (0,023 мл, 0,16 ммол) в DCM (2 мл) перемешивали при r.t. в течение ночи. Летучие удаляли в вакууме и остаток растворяли в DCM. Раствор промывали водным раствором NaHCO3, сушили(Na2SO4) и концентрировали в вакууме. После очистки на хроматографической колонке (силикагель,EtOAc) получали соединение, указанное в заголовке (50 мг, 53%). Н (CDCl3) 8,50 (1 Н, s), 7,51 (2 Н, d, J 6,5 Гц), 7,45 (2 Н, d, J 6,5 Гц), 7,11 (1H, m), 7,01 (1H, d, J 9,8 Гц), 6,93-6,72 (2H, m), 6,30 (1 H, d, J 9,8 Гц),4,22 (2H, q, J 7,1 Гц), 3,39 (6H, s), 1,25 (3H, t, J 7,1 Гц). LCMS (ES+) RT 3,704 мин, 598 (M+H)+. Промежуточное соединение 86. Этил-3-[(4-фтор-3-метилфенил)амино]-6-оксо-6,7-дигидротиено[2,3-b]пиридин-2-карбоксилат. Получали из промежуточного соединения 5 и 4-фтор-3-метиланилина способом, описанным в примере 1, в виде белого тврдого вещества. Н (DMSO-d6) 8,72 (1 Н, s), 7,67-7,60 (3 Н, m), 7,51-7,49 (2 Н, m),7,18-7,10 (3 Н, m), 7,09-6,99 (1H, m), 6,39 (1H, d, J 9,7 Гц), 4,15 (2 Н, q, J 7,07 Гц), 2.22 (3 Н, s), 1,72 (3 Н, t, J 7,08 Гц). LCMS (ES+) 423 (M+H)+. Промежуточное соединение 87. 3-[(4-Фтор-3-метилфенил)амино]-6-оксо-7-фенил-6,7-дигидротиено[2,3-b]пиридин-2-карбоксилат аммония. Получали в виде продукта бежевого цвета из промежуточного соединения 86 способом, описанным для промежуточного соединения 31. Н (DMSO-d6) 7,81-7,75 (3 Н, m), 7,64-7,62 (2 Н, m), 7,41-7,38 (1H, d, J 9,55 Гц), 7,20-7,15 (1H, t, J 9,01 Гц), 7,04-7,03 (1H, br m), 6,93-6,90 (1H, br m), 6,48-6,46 (1H, d, J 9,54 Гц),2.35 (3H, s). LCMS (ES+) 395 (M+H)+. Промежуточное соединение 88. Пентафторфенил-3-[(4-фтор-3-метилфенил)амино]-6-оксо-7-фенил 6,7-дигидротиено[2,3-b]пиридин-2-карбоксилат. К раствору промежуточного соединения 87 (0,284 г, 0,710 ммол) в DMF (10 мл) добавляли EDC(0,163 г, 0,852 ммол) и смесь перемешивали при r.t. в течение 30 мин. Добавляли пентафторфенол (0,196 г, 1,065 ммол) и смесь перемешивали при r.t. в течение 24 ч. Растворитель удаляли в вакууме и остаток растворяли в DCM, затем промывали водой, сушили (MgSO4) и концентрировали в вакууме. После очистки на хроматографической колонке (силикагель, 50% гексан/EtOAc) получали соединение, указанное в заголовке, в виде продукта белого цвета (0,226 г). Н (DMSO-d6) 8,96 (1H, s), 7,07-6,95 (5 Н, br m), 7,557,39 (4 Н, br s), 6,29 (1H, d, J 9,86 Гц), 2,08 (3 Н, s). LCMS (ES+) 561 (M+H)+. Промежуточное соединение 89. 2-Ацетил-3-амино-7-фенилтиено[2,3-b]пиридин-6(7 Н)-он. Смесь промежуточного соединения 10 (2,5 г, 10 ммол) и хлорацетона (0,88 мл, 11 ммол) в ацетонитриле (45 мл) нагревали при 50 С в течение 2 ч. Растворитель удаляли в вакууме и после очистки на хроматографической колонке (силикагель, 2,5-3% МеОН в DCM) получали соединение, указанное в заголовке, в виде жлтого продукта (2,32 г, 82%). Н (DMSO-d6) 8,27 (1H, d, J 9,6 Гц), 7,92 (2 Н, br s), 7,727,63 (3 Н, m), 7,59-7,50 (2 Н, m), 6,61 (1H, d, J 9,6 Гц), 2,18 (3 Н, s). LCMS (ES+) RT 2.883 мин, 284,9(M+H)+. Промежуточное соединение 90. 2-Ацетил-3-бром-7-фенилтиено[2,3-b]пиридин-6(7 Н)-он. Получали в виде светло-коричневого продукта из промежуточного соединения 89 способом, описанным для промежуточного соединения 14. Н (DMSO-d6) 7,98 (1H, d, J 9,7 Гц), 7,74-7,65 (3 Н, m), 7,597,56 (2 Н, m), 6,77 (1H, d, J 9,7 Гц), 2,68 (3 Н, s). LCMS (ES+) RT 3,388 мин, 349,8 (M+H)+. Промежуточное соединение 91. 3-Амино-6-оксо-7-пиридин-3-ил-6,7-дигидротиено[2,3-b]пиридин 2-карбонитрил. К раствору бис(триметилсилил)амида натрия (16,1 мл 1 М раствора в THF, 16,1 ммол) в THF (50 мл) при -78 С добавляли ацетонитрил (5 мл). Добавляли пиридилизотиоцианат (0,820 мл, 7,34 ммол) и смеси давали нагреться до r.t. в течение 2 ч. Добавляли ЕtOН (30 мл) и N,N-диметилурацил (1,03 г, 7,34 ммол) и смесь нагревали в течение 18 ч при температуре рефлюкса. Летучие удаляли в вакууме. Остаток растворяли в ацетонитриле (20 мл), добавляли хлорацетонитрил (0,470 мл, 7,34 ммол) и нагревали смесь при 50 С в течение 3 ч. Растворитель удаляли в вакууме, добавляли воду (50 мл) и смесь охлаждали до 0 С. Осадок отфильтровывали, промывали водой и эфиром, сушили, получая соединение, указанное в заголовке, в виде коричневого продукта (1,47 г, 75%). Н (DMSO-d6) 8,80-8,64 (2 Н, m), 8,13-7,95 (2 Н, m),7,60-7,56 (1H, m), 7,09 (2 Н, s), 6,48 (1H, d, 79,5 Гц). LCMS (ES+) RT 2,493 мин, 268,9 (M+H)+. Промежуточное соединение 92. 3-Бром-6-оксо-7-пиридин-3-ил-6,7-дигидротиено[2,3-b]пиридин-2 карбонитрил. Получали из промежуточного соединения 91 способом, описанным для промежуточного соединения 14. LCMS (ES+) RT 2,954 мин, 331,8 (М+Н)+. Промежуточное соединение 93. Бензил-3-[(3-[(2,4-дифторфенил)амино]-6-оксо-7-фенил-6,7 дигидротиено[2,3-b]пиридин-2-илкарбонил)амино]пирролидин-1-карбоксилат. К раствору промежуточного соединения 79 (250 мг, 0,44 ммол) в DCM (5 мл) добавляли бензил-3- 28008410 аминопирролидин-1-карбоксилат (350 мг, 1,6 ммол) и смесь перемешивали при r.t в течение 18 ч. Добавляли дополнительный эквивалент аминопирролидина (96 мг, 0,44 ммол) и смесь перемешивали ещ 18 ч. Растворитель удаляли в вакууме и сырой продукт очищали на хроматографической колонке (силикагель,60% EtOAc в изогексане) с получением соединения, указанного в заголовке, в виде жлтого масла (141 мг, 53%). LCMS (ES+) RT 3,63 мин, 601 (М+Н)+. Пример 1. Этил-3-(фениламино)-6-оксо-7-фенил-6,7-дигидротиено[2,3-b]пиридин-2-карбоксилат. Трис(дибензилиденацетон)дипалладий(0) (12 мг, 0,0133 ммол, 5 мол.%) добавляли к смеси промежуточного соединения 5 (100 мг, 0,265 ммол), карбоната цезия (120 мг, 0,37 ммол), анилина (0,030 мл,0,32 ммол) и BINAP (17 мг, 0,027 ммол, 10 мол.%) в безводном толуоле (2 мл) и нагревали смесь при температуре рефлюкса в атмосфере азота в течение 18 ч. Растворитель удаляли в вакууме и сырой продукт очищали на хроматографической колонке на силикагеле (0-20% EtOAc в DCM) с получением соединения, указанного в заголовке, в виде белого продукта (80 мг). Н (CDCl3) 8,70 (1H, bs), 7,57-7,47 (3 Н,m), 7,33-7,25 (4 Н, m), 7,20-7,10 (4 Н, m), 6,27 (1H, d, J 9,7 Гц), 4,19 (2 Н, q, J 7,1 Гц), 1,22 (3 Н, t, J 7,1 Гц).LCMS (ES+) RT 4,10 мин, 391 (M+H)+. Общая методика получения этил-3-анилино-6-оксо-7-фенил-6,7-дигидротиено[2,3-b]пиридин-2 карбоксилатов. Соединения по примерам 2-13 получали методом параллельного синтеза, используя установку для реакции Radleys Carousel (Radleys Ltd., Saffron Walden, U.K.), процесс осуществляли аналогично способу,описанному в примере 1. В каждую трубку Carousel, высушенную в печке, вводили магнитную мешалку,замещнный анилин (0,64 ммол), безводный толуол (3 мл), промежуточное соединение 5 (200 мг, 0,53 ммол), карбонат цезия (240 мг, 0,74 ммол) и трис(бензилиденацетон)дипалладий(0) (48 мг, 0,053 ммол, 10 мол.%) и BINAP (66 мг, 0,106 ммол, 20 мол.%). Реакционную смесь нагревали при температуре рефлюкса в атмосфере азота при перемешивании магнитной мешалкой в течение 48 ч. Затем каждую полученную смесь разбавляли DCM (10 мл), промывали водой (10 мл), сушили (MgSO4) и концентрировали в вакууме. Сырой продукт очищали на силикагеле, используя в качестве элюента 0-20% EtOAc в DCM,получая соединения, указанные в заголовках, в виде тврдых веществ. Пример 2. Этил-3-[(2-хлорфенил)амино]-6-оксо-7-фенил-6,7-дигидротиено[2,3-b]пиридин-2-карбоксилат. Соединение, указанное в заголовке, получали из 2-хлоранилина (92 мг). Н (CDCl3) 8,60 (1H, bs),7,56-7,48 (3 Н, m), 7,40-7,38 (1H, m), 7,36-7,32 (2 Н, m), 7,20-7,15 (2 Н, m), 7,14-7,05 (1H, m), 7,05-6,98 (1H,m), 6,35 (1H, d, J 9,8 Гц), 4,21 (2 Н, q, J 7,1 Гц), 1,23 (3 Н, t, J 7,1 Гц). LCMS (ES+) RT 4,38 мин, 425 (M+H)+. Пример 3. Этил-3-[(3-хлорфенил)амино]-6-оксо-7-фенил-6,7-дигидротиено[2,3-b]пиридин-2-карбоксилат. Соединение, указанное в заголовке, получали из 3-хлоранилина (65 мг). Н (CDCl3) 8,60 (1H, bs),7,57-7,50 (3 Н, m), 7,36-7,30 (2 Н, m), 7,20-7,18 (1H, m), 7,18 (1H, d, J 9,7 Гц), 7,05 (1H, d, J 1,5 Гц), 7,057,04 (1H, m), 6,96-6,92 (1H, m), 6,35 (1H, d, J 9,8 Гц), 4,19 (2 Н, q, J 7,1 Гц), 1,22 (3 Н, t, J 7,1 Гц). LCMS(M+H)+. Пример 12. Этил-3-[(2,4-дифторфенил)амино]-6-оксо-7-фенил-6,7-дигидротиено[2,3-b]пиридин-2 карбоксилат. Соединение, указанное в заголовке, получали из 2,4-дифторфениланилина (99 мг). Н (CDCl3) 8,49 30 (1H, bs), 7,58-7,40 (3 Н, m), 7,32-7,25 (2 Н, m), 7,13-7,04 (1H, m), 7,01 (1H, d, J 9,8 Гц), 6,93-6,86 (1H, m),6,82-6,75 (1H, m), 6,31 (1H, d, J, 9,8 Гц), 4,20 (2H, q, J 7,1 Гц), 1,23 (3 Н, J 7,1 Гц). LCMS (ES+) RT 4,06 мин;427 (М+Н)+. Пример 13. Этил-6-оксо-7-фенил-3-[(3-толил)амино]-6,7-дигидротиено[2,3-b]пиридин-2-карбоксилат. Соединение, указанное в заголовке, получали из 3-толуидина (95 мг). Н (CDCl3) 8,66 (1H, bs), 7,597,41 (3 Н, m), 7,36-7,27 (2 Н, m), 7,22-7,13 (1H, m), 7,11 (1H, d, J 9,8 Гц), 6,95-6,84 (3 Н, m), 6,27 (1H, d, J 9,8 Гц), 4,18 (2 Н, q, J 7,1 Гц), 2,28 (3 Н, s), 1,22 (3 Н, t, J 7,1 Гц). LCMS (ES+) RT 4,36 мин, 405 (М+Н)+. Пример 14. Аммоний-6-оксо-3-(фениламино)-7-фенил-6,7-дигидротиено[2,3-b]пиридин-2-карбоксилат. Моногидрат гидроокиси лития (302 мг, 7,2 ммол) добавляли к суспензии соединения по примеру 1(1,49 г, 3,6 ммол) в THF (20 мл) и воде (20 мл), смесь нагревали при 60 С в течение 18 ч. Реакционную смесь охлаждали до r.t. и в вакууме удаляли большую часть THF. Оставшийся концентрат разбавляли насыщенным водным раствором хлорида аммония (50 мл), осадок фильтровали и промывали водой (2x20 мл), Et2O (2x20 мл) и сушили в вакууме с получением соединения, указанного в заголовке, с количественным выходом в виде белого продукта. LCMS (ES+) RT 3,24 мин, 363 (М+Н)+. Пример 15. 3-(Фениламино)-7-фенилтиено[2,3-b]пиридин-6(7 Н)-он. К раствору соединения по примеру 14 (200 мг) в 1,4-диоксане (10 мл) добавляли 2 М НСl (водн.) (0,5 мл) и смесь нагревали при 70 С в течение 1 ч. Реакционную смесь разбавляли водой (30 мл), обрабатывали EtOAc (3x20 мл) и экстракты сушили (MgSO4) и концентрировали в вакууме. После очистки сырого продукта хроматографией на силикагеле (0-5% ЕtOАс в DCM), получали соединение, указанное в заголовке, в виде белого продукта (90 мг). Н (DMSO-d6) 8,21 (1H, bs), (1 Н, bs), 7,96 (1 Н, d, J 9,6 Гц), 7,637,47 (3 Н, m), 7,43-7,36 (2 Н, m), 7,25-7,11 (2 Н, m), 7,10-7,03 (2 Н, m), 6,82-6,71 (1 Н, m), 6,46 (1H, d, J 9,6 Гц), 6,44 (1H, s). LCMS (ES+) RT 3,54 мин, 319 (M+H)+. Пример 16. 6-оксо-3-(Фениламино)-7-фенил-6,7-дигидротиено[2,3-b]пиридин-2-карбоксамид. К суспензии по примеру 14 (370 мг, 1,02 ммол) в безводном DMF (5 мл) добавляли 1,1'карбонилдиимидазол (182 мг, 1,12 ммол) и перемешивали смесь при r.t. в атмосфере азота в течение 20 мин. Добавляли гидроокись аммония (2 мл 28% NH3 в воде) и перемешивали смесь 72 ч. Растворители удаляли в вакууме и сырой остаток очищали хроматографией на колонке на силикагеле, (0-15% THF вDCM) с получением соединения, указанного в заголовке, в виде продукта белого цвета (123 мг). Н(DMSO-d6) 8,74 (1H, s), 7,67-7,34 (3 Н, m), 7,33-7,27 (2 Н, m), 7,22-7,00 (5 Н,m), 6,82-6,71 (3 Н, m), 6,21 (1H,d, J 9,7 Гц). LCMS (ES+) RT 3,04 мин, 362 (М+Н)+. Пример 16 (альтернативный способ). 6-оксо-7-Фенил-3-фениламино-6,7-дигидротиено[2,3-b]пиридин-2-карбоксамид. В круглодонную колбу объмом 100 мл, снабжнную вводом и выводом для азота, добавляли соединение примера 18 (1,45 г) и 13,3 мл раствора 0,382 г гидроокиси натрия в воде (20 мл) и 30 мл абсолютного этанола. Затем смесь нагрели до температуры рефлюкса. Примерно через 1 ч выдержки при температуре рефлюкса реакция завершилась. Смесь охлаждали до комнатной температуры и выливали в
МПК / Метки
МПК: A61P 29/00, A61K 31/44, C07D 495/04, A61P 37/00
Метки: соединений, бициклических, гетероароматических, производные
Код ссылки
<a href="https://eas.patents.su/30-8410-proizvodnye-biciklicheskih-geteroaromaticheskih-soedinenijj.html" rel="bookmark" title="База патентов Евразийского Союза">Производные бициклических гетероароматических соединений</a>
Способ получения пиразоло[4,3-d]пиримидин-7-он-3- пиридилсульфонильных соединений и их промежуточных соединений

Номер патента: 3145
Опубликовано: 27.02.2003
Авторы: Негри Джоанна Тереза, Леветт Филип Чарльз, Девриз Кейт Майкл, Вуд Альберт Шо
МПК: C07D 401/14, C07D 401/12, C07D 487/04...
Метки: пиразоло[4,3-d]пиримидин-7-он-3, пиридилсульфонильных, соединений, способ, получения, промежуточных
Формула / Реферат:
1. Способ получения соединения формулы (I) где R представляет собой C1-C6алкил, необязательно замещенный одним или двумя заместителями, выбранными из C3-C5циклоалкила, OH, C1-C4алкокси, бензилокси, NR5R6, фенила, фуранила и пиридинила; C3-C6циклоалкил; 1-(C1-C4алкил)пиперидинил; тетрагидрофуранил или тетрагидропиранил, и где указанные C1-C6алкильные или C1-C4алкоксильные группы необязательно замещены галогеналкилом; R1 (который может быть...
Производные октагидро-6, 10-диоксо-6н-пиридазино [1,2-а] [1,2] диазепин-1-карбоновой кислоты, способ их получения и их применение для получения терапевтически активных соединений

Номер патента: 3280
Опубликовано: 24.04.2003
Авторы: Крок Вероник, Ларкин Джон Патрик, Руссель Патрик, Колладан Колетт
МПК: C07D 487/04
Метки: 1,2, кислоты, получения, производные, 1,2-а, терапевтически, способ, активных, 10-диоксо-6н-пиридазино, применение, соединений, диазепин-1-карбоновой, октагидро-6
Формула / Реферат:
1. Соединения общей формулы (I) имеющие конфигурацию SR или находящиеся в виде смеси SR+SS, в формуле которых R представляет собой атом водорода, радикал алкил, содержащий от 1 до 4 атомов углерода, a R1 и R2 означают H или защитную группу аминофункции. 2. Соединения по п.1, в которых аминогруппа защищена в форме фталимида, формулы (IA1) имеющие конфигурацию SR или находящиеся в виде смеси SR+SS. 3. Соединения формулы (I) согласно любому из...
Конъюгаты специфичных к иммунным клеткам макролидных соединений и противовоспалительных соединений с улучшенной способностью к целевому поступлению в клетку при противовоспалительной терапии

Номер патента: 7061
Опубликовано: 30.06.2006
Авторы: Комач Марияна, Маркович Стрибор, Томаскович Линда, Хрвачич Бошка, Мерчеп Младен, Месич Милан
МПК: A61K 31/58, C07J 43/00, A61P 5/44...
Метки: специфичных, противовоспалительной, клетку, макролидных, соединений, улучшенной, конъюгаты, способностью, иммунным, терапии, поступлению, клеткам, целевому, противовоспалительных
Формула / Реферат:
1. Соединение формулы I где М обозначает макролидную субъединицу, обладающую способностью накапливаться в клетках воспаления, выбранную из группы, включающей где R1 обозначает водород или метильную группу, R2 и R3 оба обозначают водород или вместе образуют связь или R2 обозначает аминогруппу, описываемую субструктурой -NR'R", где R' и R" могут быть независимо друг от друга водородом или любой алкильной или циклоалкильной группой, содержащей...
Производные хиноксалин-2,3-диона и их фармацевтически приемлемые соли, фармацевтические композиции, обладающие антагонистической активностью в отношении рецептора глютамата и противосудорожной активностью, способы лечения пациентов, страдающих от удара или заболеваний, при помощи этих соединений.

Номер патента: 762
Опубликовано: 24.04.2000
Авторы: Корнберг Брайэн Эдвард, Рафферти Майкл Фрэнсис, Никам Шам
МПК: C07D 241/44
Метки: противосудорожной, рецептора, лечения, композиции, приемлемые, соли, хиноксалин-2,3-диона, производные, активностью, заболеваний, страдающих, способы, помощи, обладающие, фармацевтические, отношении, пациентов, фармацевтически, соединений, глютамата, этих, антагонистической, удара
Формула / Реферат:
1. Производные хиноксалин-2,3-диона общей формулы I где R - группа - NR4R5, где R4 и R5 независимо друг от друга означают водород, низший алкил, незамещенный или замещенный низшим алкилом, низший циклоалкил, который может содержать 1 или 2 атома кислорода в качестве гетероатома, R1 и R2 независимо друг от друга означают водород или нитрогруппу, R3 - низший алкил, при этом радикалы R3 и R-CH2- могут каждый находиться в положениях 5 или...
Способ получения промежуточных соединений, применимых для получения противораковых соединений

Номер патента: 5561
Опубликовано: 28.04.2005
Авторы: Норрис Тимоти, Лехнер Ричард Шелтон, Сантафьянос Динос Пол
МПК: C07D 239/94
Метки: способ, противораковых, получения, применимых, промежуточных, соединений
Формула / Реферат:
1. Способ получения соединения формулы 3 где R1 и R2, каждый независимо, выбраны из C1-C10алкила и C1-C10алкокси, причем указанные алкил и алкокси необязательно имеют до 2 заместителей, независимо выбранных из гидрокси и C1-C6алкокси; где соединение формулы 3 получают обработкой соединения формулы 5 где R1 и R2 определены как указано выше, тионилхлоридом в безводном дихлорметане. 2. Способ по п.1, где как R1, так и R2 являются...
Предыдущий патент: Фармацевтический состав для эффективного введения апоморфина, 6аr-(-)-n-пропил-норапоморфина, их производных и их пролекарств
Следующий патент: Способ синтеза высокочистого оксима d-(17α)-13-этил-17-гидрокси-18,19-динорпрегн-4-ен-20-ин-3-она
Случайный патент: Добыча in situ из нагретых остаточным теплом участков в пласте, содержащем углеводороды