Способ получения промежуточных соединений, применимых для получения противораковых соединений
Номер патента: 5561
Опубликовано: 28.04.2005
Авторы: Сантафьянос Динос Пол, Лехнер Ричард Шелтон, Норрис Тимоти




Формула / Реферат
1. Способ получения соединения формулы 3

где R1 и R2, каждый независимо, выбраны из C1-C10алкила и C1-C10алкокси, причем указанные алкил и алкокси необязательно имеют до 2 заместителей, независимо выбранных из гидрокси и C1-C6алкокси;
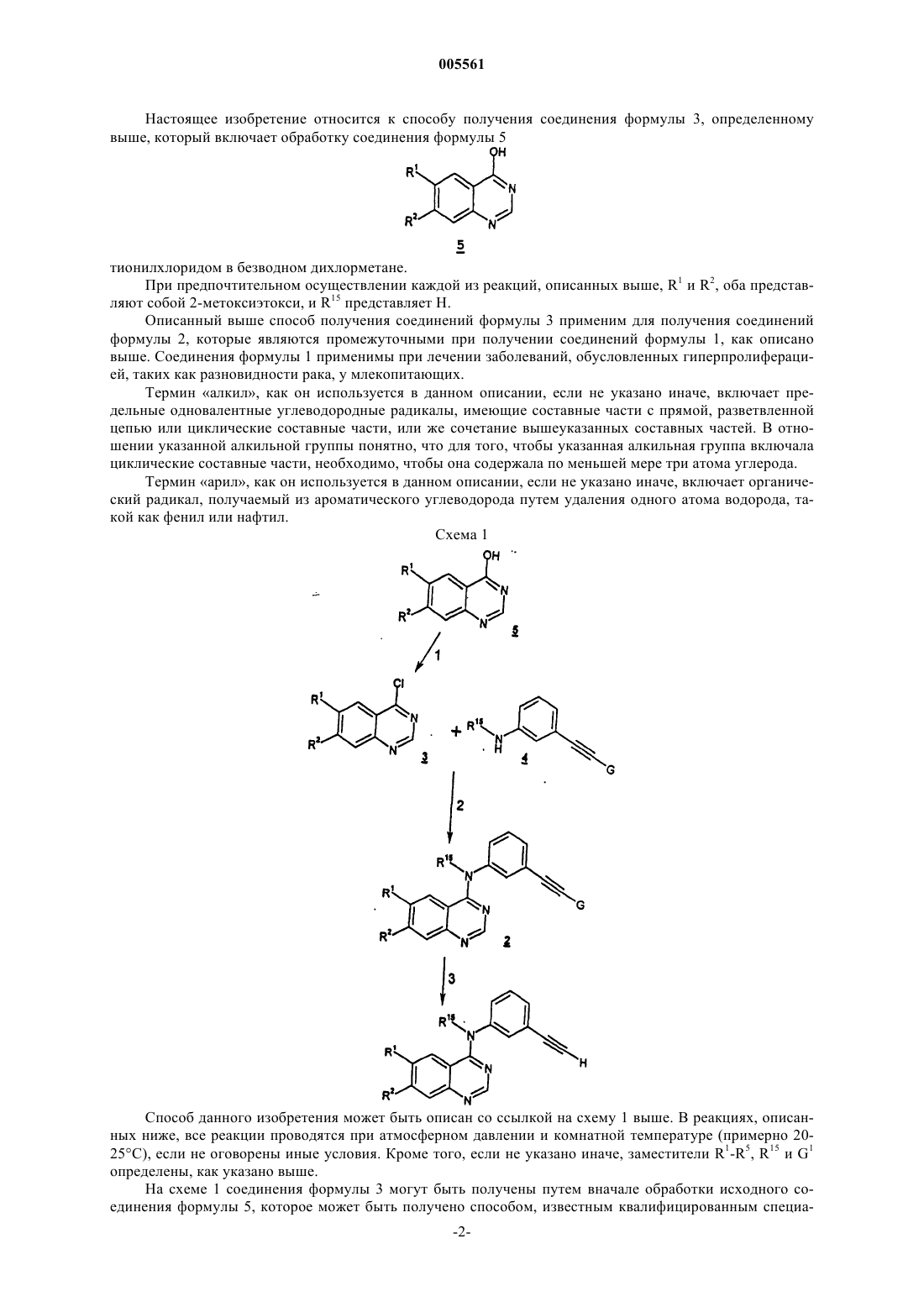
где соединение формулы 3 получают обработкой соединения формулы 5

где R1 и R2 определены как указано выше,
тионилхлоридом в безводном дихлорметане.
2. Способ по п.1, где как R1, так и R2 являются 2-метоксиэтокси.
Текст
005561 Настоящее изобретение относится к способам получения промежуточного вещества, предназначенного для конечного получения соединений, которые применимы для лечения заболеваний, обусловленных гиперпролиферацией, таких как различные виды рака, у млекопитающих. Патент Соединенных Штатов 5747498, который опубликован 5 мая 1998 г. и упомянут здесь для сведения во всей его полноте, относится к новым сериям хиназолиновых производных, включая [6,7 бис(2-метоксиэтокси)хиназолин-4-ил]-(3-этинилфенил)амина гидрохлорид, которые являются ингибиторами семейства erbB онкогенных и протоонкогенных белковых тирозинкиназ, таких как рецепторы эпидермального фактора роста (РЭФР), и поэтому применимы для лечения заболеваний, обусловленных пролиферацией, таких как разновидности рака у людей. Заявка на предварительный патент США на изобретение, озаглавленная Безводный N-(3-этинилфениламино)-6,7-бис(2-метоксиэтокси)-4-хиназолинаминмезилат и его моногидрат, зарегистрированная 29 апреля 1998 г., авторами которой названы Т.Norris, D. Santafianos, D.J.M. Alien, R.M. Shanker, and J.W. Raggon, досье поверенного PC 10074, которая упомянута здесь для сведения во всей полноте, относится к N-(3-этинилфениламино)-6,7-бис(2 метоксиэтокси)-4-хиназолинаминмезилату, безводному и его гидратным формам, которые обладают таким же противораковым действием, что и соответствующая гидрохлоридная соль, упомянутая выше. Данное изобретение относится к способу получения промежуточного соединения, предназначенного для использования при получении противораковых соединений, о которых говорится в патенте Соединенных Штатов и в патентной заявке, упомянутых выше. В евразийском патенте 004654 раскрыт способ получения соединений формулы и фармацевтически приемлемых солей и сольватов указанных соединений, у которыхR1 и R2, каждый независимо, выбираются из C1-С 10 алкила и C1-С 10 алкокси, причем указанные алкил и алкокси необязательно имеют до 2 заместителей, независимо выбранных из гидрокси и C1-С 6 алкокси;R15 представляет собой Н, C1-С 10 алкил или -(СН 2)q(С 6-С 10 арил), где q является целым числом от 0 до 4; который включает обработку соединения формулы 2R3, R4 и R5, каждый независимо, представляют собой C1-С 6 алкил; либо (а) гидроксидом щелочного или щелочно-земельного металла в растворителе, содержащем замещенный гидроксилом C1-С 10 алкил, когда G является -C(OH)R3R4, либо (b) тетра-(C1-С 6 алкил) аммонийфторидным соединением в апротонном растворителе, когда G представляет собой -SiR3R4R5. В названном патенте ЕА 004654 описывается также способ получения соединения формулы 2, описанного выше, который включает обработку соединения формулы 3 где G и R15 определены, как указано для соединения формулы 2.-1 005561 Настоящее изобретение относится к способу получения соединения формулы 3, определенному выше, который включает обработку соединения формулы 5 тионилхлоридом в безводном дихлорметане. При предпочтительном осуществлении каждой из реакций, описанных выше, R1 и R2, оба представляют собой 2-метоксиэтокси, и R15 представляет Н. Описанный выше способ получения соединений формулы 3 применим для получения соединений формулы 2, которые являются промежуточными при получении соединений формулы 1, как описано выше. Соединения формулы 1 применимы при лечении заболеваний, обусловленных гиперпролиферацией, таких как разновидности рака, у млекопитающих. Термин алкил, как он используется в данном описании, если не указано иначе, включает предельные одновалентные углеводородные радикалы, имеющие составные части с прямой, разветвленной цепью или циклические составные части, или же сочетание вышеуказанных составных частей. В отношении указанной алкильной группы понятно, что для того, чтобы указанная алкильная группа включала циклические составные части, необходимо, чтобы она содержала по меньшей мере три атома углерода. Термин арил, как он используется в данном описании, если не указано иначе, включает органический радикал, получаемый из ароматического углеводорода путем удаления одного атома водорода, такой как фенил или нафтил. Схема 1 Способ данного изобретения может быть описан со ссылкой на схему 1 выше. В реакциях, описанных ниже, все реакции проводятся при атмосферном давлении и комнатной температуре (примерно 2025 С), если не оговорены иные условия. Кроме того, если не указано иначе, заместители R1-R5, R15 и G1 определены, как указано выше. На схеме 1 соединения формулы 3 могут быть получены путем вначале обработки исходного соединения формулы 5, которое может быть получено способом, известным квалифицированным специа-2 005561 листам, тионилхлоридом в безводном дихлорметане при температуре дефлегмации (примерно 38-42 С при атмосферном давлении) с получением соединения формулы 3. В дальнейшем может быть получено соединение формулы 2 путем обработки соединения формулы 3 соединением формулы 4 в органическом растворителе, таком как ДМФА, ДМСО, ТГФ, MeCN или смеси двух или более вышеназванных растворителей, предпочтительно MeCN, при температуре в интервале от 50 С до температуры дефлегмации, предпочтительно при температуре дефлегмации. Далее может быть получено соединение формулы 1 путем взаимодействия соединения формулы 2 с гидроксидом щелочного металла или щелочно-земельного металла в растворителе, содержащем C1-С 10 алкил, замещенный по меньшей мере одной гидроксильной группой, когда G представляет собой -C(OH)R3R4, или с тетра-(C1-С 6 алкил)аммонийфторидным соединением в апротонном растворителе, когда G представляет собой -SiR3R4R5. Представленные ниже иллюстрирующие примеры дополнительно иллюстрируют применение соединения 3, полученного способом данного изобретения. Иллюстрирующие примеры 1 и 3 показывают получение соединений формулы 2, в которых G является блокирующей группой -SiR3R4R5 или -C(OH)R3R4 соответственно, из соединений формулы 3, получаемых способом согласно настоящему изобретению. Иллюстрирующие примеры 2 и 4 показывают дальнейшее использование соединений формулы 2, полученных в примерах 1 и 3 соответственно, для дальнейшего получения из них конечных целевых соединений формулы 1, применимых при лечении заболеваний, обусловленных гиперпролиферацией. Иллюстрирующий пример 1. Получение 6,7-бис(2-метоксиэтокси)-N-[3-[(триметилсилил)этинил] фенил]-4-хиназолинамина моногидрохлорида. 4-Хлор-6,7-бис(2-метоксиэтокси)хиназолин (942 мг, 3,01 ммоль) обрабатывали раствором 3[(триметилсилил)этинил]анилина, полученного как описано в патенте ЕА 004654 (645 мг, 3,41 ммоль), в 2-пропаноле (14 мл) и нагревали при температуре дефлегмации в течение 2,5 ч. Смеси давали остыть до комнатной температуры и перемешивали в течение 1 ч. Твердое вещество собирали фильтрованием,промывали 2-пропанолом (5 мл) и сушили в вакууме в течение ночи с получением продукта, указанного в заголовке (1,33 г, 88%) в виде белого твердого вещества. Н (400 МГц; CDCl3) 0,21 (9 Н, с), 3,38 (3 Н, с), 3,41 (3 Н, с), 3,72 (2 Н, м), 3,77 (2 Н, м), 4,10 (2 Н, с),4,53 (2 Н, с), 7,20 (1 Н, т, J=7,8 Гц), 7,23-7,28 (2 Н, м), 7,75 (1 Н, д, J=7,8 Гц), 7,88 (1 Н, с), 8,20 (1 Н, с), 8,42m/е 466(М+Н)+. Иллюстрирующий пример 2. Получение N-(3-этинилфенил)-6,7-бис(2-метоксиэтокси)-4-хиназолинамин, моногидрохлорид. Густую суспензию силильного соединения, а именно 6,7-бис(2-метоксиэтокси)-N-[3[(триметилсилил)этинил]фенил]-4-хиназолинамина моногидрохлорида, полученного выше (1,22 г, 2,43 ммоль) в тетрагидрофуране (6,1 мл), обрабатывали 1 М раствором тетра-н-бутиламмонийфторида в тетрагидрофуране (2,6 мл, 2,55 ммоль) и перемешивали при комнатной температуре в течение 1 ч. Раствор обрабатывали 2-пропанолом (12,2 мл) и концентрировали выпариванием. Масло в 2-пропаноле (20 мл) обрабатывали концентрированной соляной кислотой (0,2 мл), получая осадок. Смесь перемешивали при комнатной температуре в течение 1 ч. Твердое вещество собирали фильтрованием, промывали 2 пропанолом (2 мл) и сушили в вакууме с получением соединения, указанного в заголовке (747 мг, 72%) в виде не совсем белого твердого вещества (т.пл. 226-229 С). Н (300 МГц; d6-ДMCO) 3,36 (6 Н, с), 3,77-3,80 (4 Н, м), 4,30 (1 Н, с), 7,39 (1H, с), 7,41 (1H, д, J=7,8 Гц), 7,50 (1H, т, J=7,9 Гц), 7,79 (1H, д, J=8,1 Гц), 7,88 (1H, с), 8,40 (1H, с), 8,86 (1H, с), 11,49 (1H, ушир.с);m/e 394 (М+Н)+. Иллюстрирующий пример 3. Получение 4-[3-6,7-бис(2-метоксиэтокси)-4-хиназолинил]амино] фенил]-2-метил-3-бутин-2-ола, моногидрохлорида. 4-Хлор-6,7-бис(2-метоксиэтокси)хиназолин (15 г, 48 ммоль), 4-(3-аминофенил)-2-метил-3-бутин-2 ол (9,2 г, 52,8 ммоль) и ацетонитрил (225 мл) нагревали с обратным холодильником в течение 5 ч. Смесь охлаждали до 5-10 С и перемешивали в течение 1 ч. Твердое вещество собирали фильтрованием, промывали ацетонитрилом (15 мл) и сушили в вакууме в течение ночи с получением продукта, указанного в заголовке (23,4 г, 100%) в виде белого твердого вещества.m/e 45,2 (М+Н)+. Иллюстрирующий пример 4. Получение N-(3-этинилфенил)-6,7-бис(2-метоксиэтокси)-4-хиназолинамина, моногидрохлорида. 4-[3-6,7-Бис(2-метоксиэтокси)-4-хиназолинил]амино]фенил]-2-метил-3-бутин-2-ола моногидрохлорид, полученный, как описано выше (32,34 г, 66,3 ммоль), воду (300 мл) и бутан-1-ол (600 мл) пере-3 005561 мешивали вместе при комнатной температуре до образования смеси. Показатель рН смеси доводили до рН 10-12 с помощью 50% водного раствора гидроксида натрия с получением двух прозрачных слоев. Органический слой отделяли от водного слоя и упаривали при атмосферном давлении так, что вода удалялась из раствора в бутан-1-оле в виде азеотропной смеси. Конечный объем бутан-1-ольного раствора составлял 300 мл. К осушенному азеотропной перегонкой бутан-1-ольному раствору добавляли безводный твердый гидроксид натрия (0,13 г, 3,3 ммоль), и полученную смесь нагревали с обратным холодильником при 115-120 С в течение 24 ч. Бутан-1-ол (150 мл) удаляли путем отгонки и концентрированную реакционную смесь охлаждали до 15-25 С. К охлажденному концентрату добавляли концентрированную соляную кислоту (6,1 мл) и бутан-1-ол (60 мл) и смесь гранулировали в течение ночи при 20-25 С до наступления кристаллизации. Кристаллы продукта, указанного в заголовке, выделяли фильтрованием и сушили под пониженным давлением при 45-50 С для удаления бутан-1-ола. Выход (21,0 г, 73,7%). Чистота по ВЭЖХ 96,5%. ФОРМУЛА ИЗОБРЕТЕНИЯ 1. Способ получения соединения формулы 3 где R1 и R2, каждый независимо, выбраны из C1-С 10 алкила и C1-C10 алкокси, причем указанные алкил и алкокси необязательно имеют до 2 заместителей, независимо выбранных из гидрокси и C1-С 6 алкокси; где соединение формулы 3 получают обработкой соединения формулы 5 где R1 и R2 определены как указано выше,тионилхлоридом в безводном дихлорметане. 2. Способ по п.1, где как R1, так и R2 являются 2-метоксиэтокси.
МПК / Метки
МПК: C07D 239/94
Метки: соединений, промежуточных, противораковых, получения, применимых, способ
Код ссылки
<a href="https://eas.patents.su/5-5561-sposob-polucheniya-promezhutochnyh-soedinenijj-primenimyh-dlya-polucheniya-protivorakovyh-soedinenijj.html" rel="bookmark" title="База патентов Евразийского Союза">Способ получения промежуточных соединений, применимых для получения противораковых соединений</a>
Способ получения пиразоло[4,3-d]пиримидин-7-он-3- пиридилсульфонильных соединений и их промежуточных соединений

Номер патента: 3145
Опубликовано: 27.02.2003
Авторы: Леветт Филип Чарльз, Вуд Альберт Шо, Девриз Кейт Майкл, Негри Джоанна Тереза
МПК: C07D 401/14, C07D 401/12, C07D 487/04...
Метки: способ, получения, промежуточных, пиридилсульфонильных, пиразоло[4,3-d]пиримидин-7-он-3, соединений
Формула / Реферат:
1. Способ получения соединения формулы (I) где R представляет собой C1-C6алкил, необязательно замещенный одним или двумя заместителями, выбранными из C3-C5циклоалкила, OH, C1-C4алкокси, бензилокси, NR5R6, фенила, фуранила и пиридинила; C3-C6циклоалкил; 1-(C1-C4алкил)пиперидинил; тетрагидрофуранил или тетрагидропиранил, и где указанные C1-C6алкильные или C1-C4алкоксильные группы необязательно замещены галогеналкилом; R1 (который может быть...
Способы получения и промежуточные соединения для получения противораковых соединений

Номер патента: 4654
Опубликовано: 24.06.2004
Авторы: Норрис Тимоти, Лехнер Ричард Шелтон, Сантафьянос Динос Пол
МПК: C07D 239/94
Метки: соединения, противораковых, соединений, получения, способы, промежуточные
Формула / Реферат:
1. Способ получения соединения формулы 1 или фармацевтически приемлемых солей и сольватов указанного соединения, где R1 и R2, каждый независимо, выбраны из C1-C10алкила и C1-C10алкокси, причем указанные алкил и алкокси необязательно имеют до 2 заместителей, независимо выбранных из гидрокси и C1-C6алкокси; R15 представляет собой H или C1-C10алкил; который включает обработку соединения формулы 2 где R15, R1 и R2 определены как указано выше, а G...
Способ получения циклопропанкарбоновых кислот и их промежуточных соединений

Номер патента: 683
Опубликовано: 28.02.2000
Авторы: Винкельманн Иб, Колинн-Андерсен Ханс, Клемменсен Пер Дауселль
МПК: C07D 307/93, C07C 61/35
Метки: получения, циклопропанкарбоновых, промежуточных, способ, соединений, кислот
Формула / Реферат:
1. Способ получения соединений общей формулы I где R' представляет Н, а два атома водорода циклопропанового кольца находятся в цис-положении по отношению друг к другу, включающий взаимодействие между соединением общей формулы II и соединением СF3-CClХ2, где Х представляет атом галогена, в частности атом хлора или брома, в инертной среде в присутствии Zn и при подходящей температуре от 0 до 150шС, предпочтительно от 20 до 100шС, в...
Способы получения промежуточных соединений для пестицидов.

Номер патента: 955
Опубликовано: 28.08.2000
Авторы: Вилкинсон Джон Херри, Клавель Жан-Луи, Хоукинс Девид Вилльям, Робертс Девид Ален
МПК: C07D 231/38, C07C 253/00
Метки: получения, пестицидов, способы, промежуточных, соединений
Формула / Реферат:
1. Способ получения соединения формулы (I) где R представляет собой алкил с нормальной или разветвленной цепью, содержащий от 1 до 18 атомов углерода, или его соли, который включает реакцию цианоацетата формулы (II) RO2C-CH2CN (II) где R определен выше, с солью цианисто-водородной кислоты и формальдегидом или источником последнего. 2. Способ по п.1, отличающийся тем, что соль цианисто-водородной кислоты представляет собой соль...
Способ синтеза промежуточных соединений хлорпурина

Номер патента: 3183
Опубликовано: 27.02.2003
Авторы: Джоунз Мартин Фрэнсис, Уоллис Кристофер Джон
МПК: C07D 473/00
Метки: хлорпурина, синтеза, промежуточных, соединений, способ
Формула / Реферат:
1. Способ получения соединения формулы (I) возможно в форме его соли или комплекса, включающий гидролиз в присутствии кислоты соединения формулы (IV) где Р - защитная группа, конденсацию образующегося продукта формулы (V) in situ в полярном растворителе в присутствии основания с соединением формулы (VI) где R представляет собой CHO или H, с последующим in situ замыканием кольца образующегося промежуточного продукта формулы (VII) где R...
Предыдущий патент: Подключение широкополосных модемов к линиям электропитания
Следующий патент: Способ повышения селективности гербицида на основе 1,3 – циклогександиона
Случайный патент: Металлургическая печь