Бензотиепины, обладающие активностью ингибиторов перемещения желчных кислот в подвздошной кишке и поглощения таурохолата
Номер патента: 4650
Опубликовано: 24.06.2004
Авторы: Ли Джинглин Дж., Миллер Раймонд Е., Рейтц Дейвид Б., Ли Лен Ф., Тремонт Сэмюэль Дж., Ханг Хонг-Чи, Банерджи Шиамал С.






Формула / Реферат
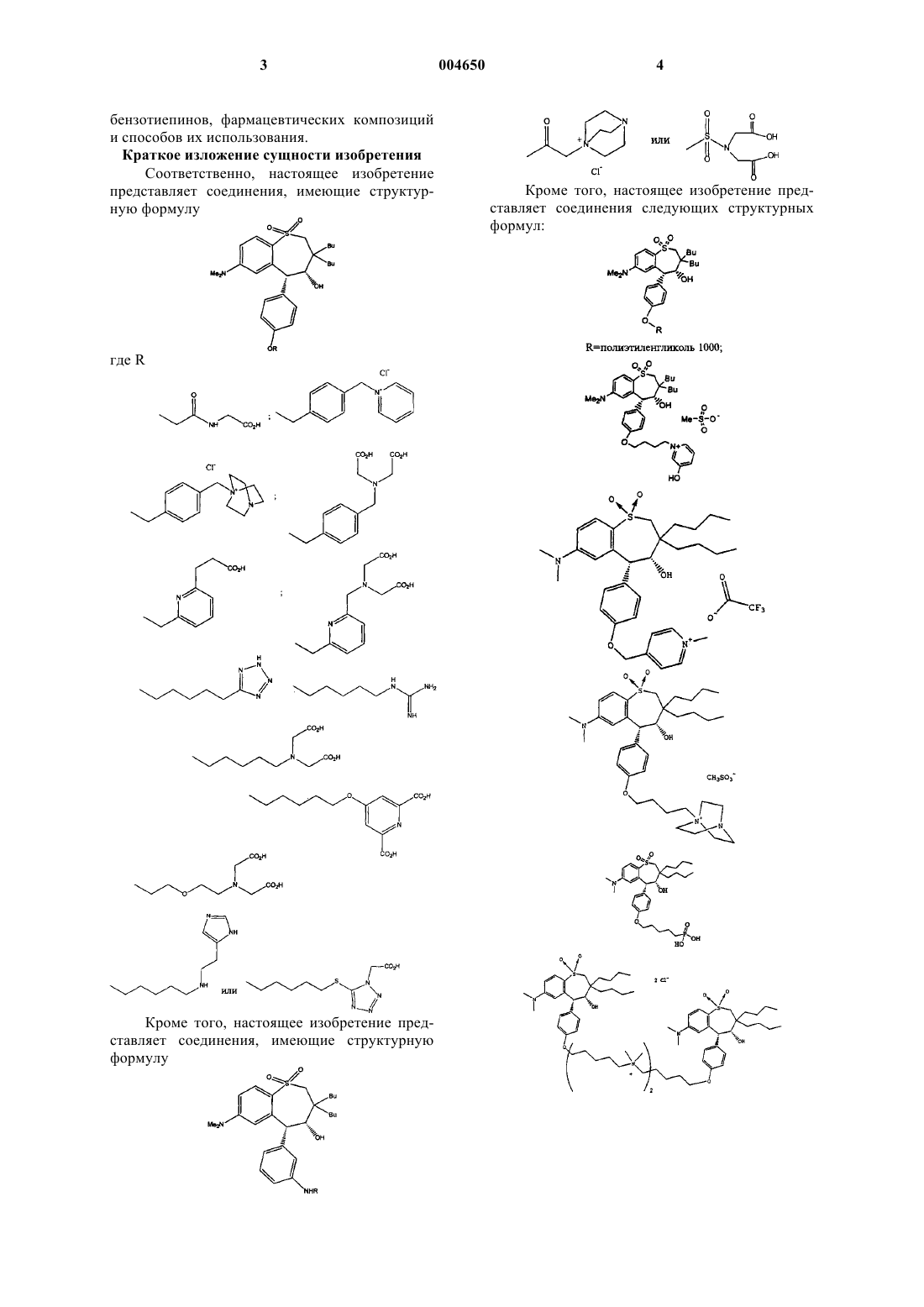
1. Соединение, имеющее структурную формулу

где R


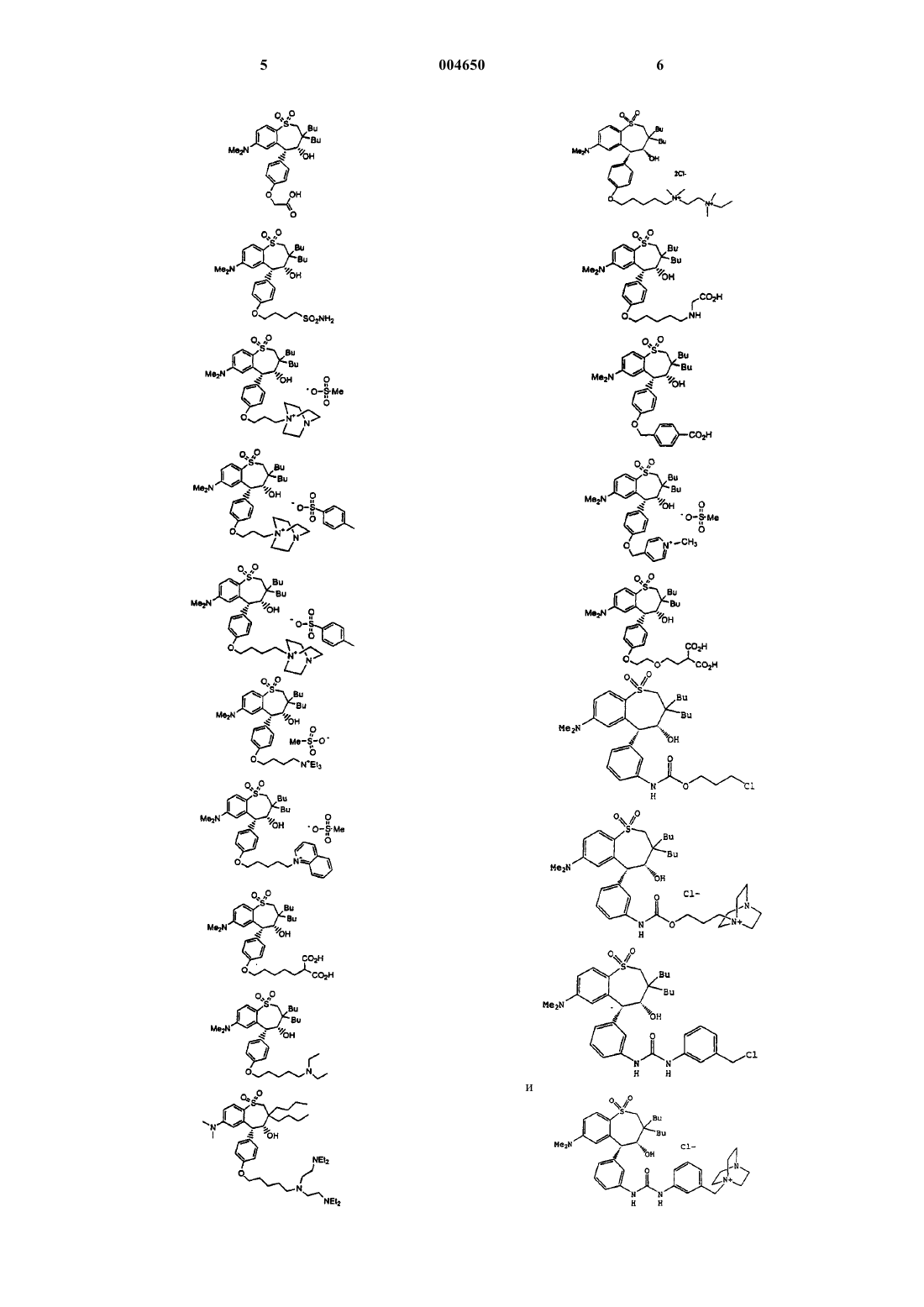
2. Соединение по п.1, имеющее структурную формулу

3. Соединение по п.1, имеющее структурную формулу

4. Соединение по п.1, имеющее структурную формулу

5. Соединение по п.1, имеющее структурную формулу

6. Соединение по п.1, имеющее структурную формулу

7. Соединение по п.1, имеющее структурную формулу

8. Соединение по п.1, имеющее структурную формулу

9. Соединение по п.1, имеющее структурную формулу

10. Соединение по п.1, имеющее структурную формулу

11. Соединение по п.1, имеющее структурную формулу

12. Соединение по п.1, имеющее структурную формулу

13. Соединение по п.1, имеющее структурную формулу

14. Соединение по п.1, имеющее структурную формулу

15. Соединение, имеющее структурную формулу

где R

16. Соединение по п.15, имеющее структурную формулу

17. Соединение по п.15, имеющее структурную формулу

18. Соединение, выбранное из группы, включающей







и

19. Фармацевтическая композиция, содержащая соединение по любому из пп.1-18 в антигиперлипидемически эффективном количестве и фармацевтически приемлемый носитель.
20. Фармацевтическая композиция, содержащая соединение по любому из пп.1-18 в антиатеросклеротически эффективном количестве и фармацевтически приемлемый носитель.
21. Фармацевтическая композиция, содержащая соединение по любому из пп.1-18 в антигиперхолестеринемически эффективном количестве и фармацевтически приемлемый носитель.
22. Способ профилактики или лечения гиперлипидемического состояния, включающий введение нуждающемуся пациенту композиции по п.19 в виде дозированной лекарственной формы.
23. Способ профилактики или лечения атеросклеротического состояния, включающий введение нуждающемуся пациенту композиции по п.20 в виде дозированной лекарственной формы.
24. Способ профилактики или лечения гиперхолестеринемии, включающий введение нуждающемуся пациенту композиции по п.21 в виде дозированной лекарственной формы.
25. Применение соединения по любому из пп.1-18 для изготовления лекарственного препарата, применяемого для профилактики или лечения гиперлипидемического состояния.
26. Применение соединения по любому из пп.1-18 для изготовления лекарственного препарата, применяемого для профилактики или лечения атеросклеротического состояния.
27. Применение соединения по любому из пп.1-18 для изготовления лекарственного препарата, применяемого для профилактики или лечения гиперхолестеринемического состояния.
Текст
1 Предпосылки создания изобретения Область, к которой относится изобретение Настоящее изобретение относится к новым бензотиепинам, их производным и аналогам,фармацевтическим композициям, в состав которых они входят, и их применению в медицине, в частности, для профилактики и лечения гиперлипидемических состояний, например, связанных с атеросклерозом либо гиперхолестеринемией, у млекопитающих. Описание известного уровня техники Достоверно установлено, что гиперлипидемические состояния, связанные с повышенными концентрациями общего содержания холестерина и холестерина липопротеидов низкой плотности, являются основными факторами риска для ишемической болезни сердца, и в частности атеросклероза. Установлено, что нарушение циркуляции желчных кислот в просвете кишечника находится в причинной взаимосвязи со снижением уровней содержания холестерина в сыворотке. Накопленные эпидемиологические данные свидетельствуют о том, что подобное снижение ведет к улучшению при болезненном атеросклеротическом состоянии. Стедронскиcholesterol with nonsystemic agents having hypocholesterolemic properties", Biochimica et Biophysica Acta, 1210 (1994), 255-287, обсуждает биохимию, физиологию и известные активные вещества, в том числе желчные кислоты и холестерин. Хьюби Дж.Е. (Heubi J.E.) и др. показано,что патофизиологические изменения у людей согласуются с нарушением энтерогепатической циркуляции желчных кислот. См. работу "PrimaryActive Bile Acid Transport", Gastroenterology,1982:83:804-11. Фактически холестирамин связывает желчные кислоты в кишечнике, нарушая тем самым их нормальную энтерогепатическую циркуляцию (Райнер Е. (Reihner E.) и др., в работе "Regulation of hepatic cholesterol metabolismhamster with cholestyramine treatment", Atherosclerosis, 89 (1991), 183-190). Следствием этого является интенсификация синтеза желчных кислот печенью с использованием холестерина, а также активация рецепторов липопротеидов низкой плотности в печени, что стимулирует выведение холестерина и снижает уровни содержания холестерина липопротеидов низкой плотности в сыворотке. В ином варианте подхода к снижению рециркуляции желчных кислот, система переноса желчных кислот в подвздошной кишке представляет собой предполагаемую фармацевтическую 2 цель для лечения гиперхолестеринемии, основываясь на нарушении энтерогепатической циркуляции с помощью специфических ингибиторов переноса (Крамер (Kramer) и др., "Intestinal Bile AcidAbsorption", The Journal of Biological Chemistry,т. 268,24 от 25 августа, 18035-18046, 1993). В ряде заявок на патенты, например в заявках на патенты Канады 2025294; 2078588; 2085782 и 2085830 и в заявках на европатент 0379161; 0549967; 0559064 и 0563731, компания Hoechst Aktiengesellschaft раскрывает полимеры различных природных составляющих энтерогепатической системы циркуляции и их производные, в том числе желчные кислоты, которые ингибируют физиологический перенос желчных кислот с целью снижения уровня содержания холестерина липопротеидов низкой плотности в степени, достаточной для того, чтобы быть эффективными фармацевтическими препаратами, и в частности для использования их в качестве гипохолестеринемических агентов. В международной заявке WO 93/16055"Hypolipidemic Benzothiazepine Compounds" компания The Wellcome Foundation Limited раскрывает ингибирование переноса желчных кислот in vitro для демонстрации гиполипидемической активности. В международной заявке WO 93/321146 раскрываются некоторыe бензотиепины, пригодные для различных вариантов использования, включая метаболизм жирных кислот и коронарную болезнь сердца. Известны другие избранные бензотиепины, пригодные для использования в качестве гиполипидемических и гипохолестеринемических агентов, в частности, для лечения либо профилактики атеросклероза, как раскрывается в заявках ЕР 508425, FR 2661676 и WO 92/18462, каждая из которых ограничивается амидом, связанным с атомом углерода, примыкающим к фенильному кольцу слитого бициклобензотиепинового кольца. В вышеупомянутых источниках показаны непрерывные усилия, направленные на обнаружение безопасных, эффективных агентов для профилактики и лечения гиперлипидемических заболеваний и возможности их использования в качестве гипохолестеринемических агентов. Дополнительно некоторые бензотиепины выявлены пригодными для применения при различных болезненных состояниях, которые не входят в область применения настоящего изобретения. Это приведено в заявке ЕР 568898 АAbstr.65860 Т-В) и заявке WO 92/18462. Настоящее изобретение дополняет подобные усилия посредством предоставления новых 3 бензотиепинов, фармацевтических композиций и способов их использования. Краткое изложение сущности изобретения Соответственно, настоящее изобретение представляет соединения, имеющие структурную формулу Кроме того, настоящее изобретение представляет соединения, имеющие структурную формулу Кроме того, настоящее изобретение представляет соединения следующих структурных формул: 7 Настоящим изобретением предлагается также фармацевтическая композиция, содержащая любое из раскрытых выше соединений в антигиперлипидемически эффективном количестве и фармацевтически приемлемый носитель,а также способ профилактики или лечения гиперлипидемического состояния, включающий введение нуждающемуся пациенту такой композиции в виде дозированной лекарственной формы. Кроме того, настоящим изобретением предлагается фармацевтическая композиция,содержащая любое из раскрытых выше соединений в антиатеросклеротически эффективном количестве и фармацевтически приемлемый носитель, а также способ профилактики или лечения атеросклеротического состояния,включающий введение нуждающемуся пациенту такой композиции в виде дозированной лекарственной формы. Кроме того, настоящим изобретением предлагается фармацевтическая композиция,содержащая любое из раскрытых выше соединений в антигиперхолестеринемически эффективном количестве и фармацевтически приемлемый носитель, а также способ профилактики или лечения гиперхолестеринемии, включающий введение нуждающемуся пациенту такой композиции в виде дозированной лекарственной формы. Настоящим изобретением предлагается также применение любого из раскрытых выше соединений для изготовления лекарственного препарата, применяемого для профилактики или лечения гиперлипидемического состояния, атеросклеротического состояния или гиперхолестеринемического состояния. Настоящее изобретения станет очевидным из приведенного ниже подробного описания. Следует, однако, понимать, что приведенные ниже подробное описание и примеры, которые показывают предпочтительные примеры осуществления настоящего изобретения, приведены лишь с иллюстративными целями, поскольку специалистам в данной отрасли из приведенного подробного описания станут очевидны многочисленные изменения и модификации настоящего изобретения, не выводящие за его пределы. Подробное описание изобретения Последующее подробное описание приведено в помощь специалистам в данной отрасли при практическом осуществлении настоящего изобретения. Даже в этом случае приведенное подробное описание не должно использоваться для неправомерного ограничения настоящего изобретения, поскольку модификации и изменения в вариантах осуществления, обсуждение которых предоставлено в настоящем описании,могут вноситься средними специалистами в данной отрасли без отступления от духа и объема настоящего изобретения. 8 Содержание каждого источника, который упоминался в данном описании в качестве ссылки, включая содержание источников, которые упоминались в указанных источниках,включено в полном объеме в данное описание в качестве ссылки. Определения Представленные далее определения приведены для оказания помощи читателю в понимании последующего подробного описания. Каждый из упомянутых терминов "алкил","алкенил" и "алкинил", в случае отсутствия иных определений, обозначает в настоящем изобретении углеводород с неразветвленной либо разветвленной цепью, состоящей из 1-20 атомов углерода в случае алкила либо 2-20 атомов углерода в случае алкенила и алкинила и,таким образом, обозначает, например, метил,этил, пропил, бутил, пентил либо гексил и этенил, пропенил, бутенил, пентенил либо гексенил и этинил, пропинил, бутинил, пентинил либо гексинил, соответственно, и их изомеры. Упомянутый термин "арил" обозначает полностью ненасыщенный одно-либо многоядерный углеродный цикл, включая, однако, не ограничиваясь, замещенный либо незамещенный фенил, нафтил либо антраценил. Упомянутый термин "гетероцикл" обозначает насыщенный либо ненасыщенный однолибо многоядерный углеродный цикл, где один либо несколько атомов углерода могут быть заменены N, S, Р либо О. Данное определение включает, например, следующие структуры: где Z, Z', Z" или Z представляют собой С,S, Р, О или N, при условии, что один из Z, Z', Z" или Z не является углеродом, но и не О или S,в случае, когда он соединен с другим атомом Z с помощью двойной связи, либо в случае, когда он соединен с другим атомом О или S. В дополнение к этому, подразумевается, что факультативные заместители присоединяются к Z, Z', Z" либо Z лишь в том случае, когда каждый из них представляет собой С. Упомянутый термин "гетероарил" обозначает полностью ненасыщенный гетероцикл. У "гетероцикла" либо "гетероарила" точка присоединения к молекуле, представляющей интерес, может находиться на гетероатоме либо в любом месте в пределах кольца. Упомянутый термин "четвертичный гетероцикл" обозначает гетероцикл, в котором один либо несколько гетероатомов, например, О, N, S либо Р имеют такое количество связей, что он оказывается положительно заряженным. Точка присоединения упомянутого четвертичного гетероцикла к молекуле, представляющей интерес, может находиться на гетероатоме либо в любом ином месте. 9 Упомянутый термин "четвертичный гетероарил" обозначает гетероарил, у которого один либо несколько гетероатомов, например, О, N, S либо Р имеют такое количество связей, что он оказывается положительно заряженным. Точка присоединения упомянутого четвертичного гетероарила к молекуле, представляющей интерес,может находиться на гетероатоме либо в любом ином месте. Упомянутый термин "галоген" обозначает фтор, хлор, бром либо йод. Упомянутый термин "галогеналкил" обозначает алкил, замещенный одним либо несколькими галогенами. Упомянутый термин "циклоалкил" обозначает одно- либо многоядерный углеродный цикл, где в состав каждого кольца входит от 3 до 10 атомов углерода и где любое кольцо может иметь одну либо несколько двойных либо тройных связей. К числу примеров относятся такие радикалы, как циклопропил, циклобутил,циклопентил, циклогексил, циклоалкенил и циклогептил. Упомянутый термин "циклоалкил" дополнительно обозначает спиросистемы, где упомянутое циклоалкильное кольцо имеет общий атом углерода с семичленным гетероциклическим кольцом бензотиепина. Упомянутый термин "диил" обозначает двухвалентную радикальную составляющую,где упомянутая составляющая имеет две точки присоединения к молекулам, представляющим интерес. Упомянутый термин "оксо" обозначает атом кислорода с двойной связью. Упомянутый термин "полиалкил" обозначает разветвленную либо неразветвленную углеводородную цепь, имеющую молекулярную массу приблизительно 20000, в более предпочтительном варианте приблизительно 10000 и в наиболее предпочтительном варианте приблизительно 5000. Упомянутый термин "полиэфир" обозначает полиалкил, где один либо несколько атомов углерода заменены атомом кислорода, где упомянутый полиэфир имеет молекулярную массу приблизительно 20000, в более предпочтительном варианте приблизительно 10000 и в наиболее предпочтительном варианте приблизительно 5000. Упомянутый термин "полиалкокси" обозначает полимер алкиленоксидов, где упомянутый полиалкокси имеет молекулярную массу приблизительно 20000, в более предпочтительном варианте приблизительно 10000 и в наиболее предпочтительном варианте приблизительно 5000. Упомянутый термин "циклоалкилиден" обозначает одно- либо многоядерный углеродный цикл, где атом углерода в пределах кольцевой структуры имеет двойную связь с атомом,который находится за пределами упомянутой кольцевой структуры. 10 Упомянутый термин "углеводород" обозначает моно-, ди-, три- либо полисахарид, где упомянутый полисахарид может иметь молекулярную массу приблизительно до 20000, например гидроксипропилметилцеллюлоза либо хитозан. Упомянутый термин "пептид" обозначает полиаминокислоту, в состав которой входит до приблизительно 100 аминокислотных единиц. Упомянутый термин "полипептид" обозначает полиаминокислоту, в состав которой входит от приблизительно 100 аминокислотных единиц до приблизительно 1000 аминокислотных единиц, в более предпочтительном варианте от приблизительно 100 аминокислотных единиц до приблизительно 750 аминокислотных единиц, в наиболее предпочтительном варианте от приблизительно 100 аминокислотных единиц до приблизительно 500 аминокислотных единиц. Упомянутый термин "алкиламмонийалкил" обозначает NН 2-группу либо моно-, диили тризамещенную аминогруппу, любая из которых связана с алкилом, где упомянутый алкил связан с упомянутой молекулой, представляющей интерес. Упомянутый термин "триазолил" включает все изомеры положения. У всех других гетероциклов и гетероарилов, которые содержат более одного кольцевого гетероатома и для которых возможно существование изомеров, такие изомеры включаются в определение упомянутых гетероциклов и гетероарилов. Упомянутый термин "сульфо" обозначает сульфогруппу, -SО 3 Н, либо ее соли. Упомянутый термин "сульфоалкил" обозначает алкильную группу, с которой связана сульфонатная группа, причем упомянутая алкильная группа связана с молекулой, представляющей интерес. Упомянутый термин "арилалкил" обозначает арилзамещенный алкильный радикал, такой как бензил. Упомянутый термин "алкиларилалкил" обозначает арилалкильный радикал,замещенный на упомянутой арильной группе одной либо несколькими алкильными группами. Упомянутый термин "гетероциклилалкил" обозначает алкильный радикал, замещенный одной либо несколькими гетероциклическими группами. Предпочтительными гетероциклилалкильными радикалами являются "низшие гетероциклилалкильные" радикалы, имеющие одну либо несколько гетероциклических групп,присоединенных к алкильному радикалу,имеющему от 1 до 10 атомов углерода. Упомянутый термин "гетероарилалкил" обозначает алкильный радикал, замещенный одной либо несколькими гетероарильными группами. Предпочтительными гетероарилалкильными радикалами являются "низшие гетероарилалкильные" радикалы, имеющие одну либо несколько гетероарильных групп, присое 11 диненных к алкильному радикалу, имеющему от 1 до 10 атомов углерода. Упомянутый термин "четвертичный гетероциклилалкил" обозначает алкильный радикал,замещенный одной либо несколькими четвертичными гетероциклическими группами. Предпочтительными четвертичными гетероциклилалкильными радикалами являются "низшие четвертичные гетероциклилалкильные" радикалы, имеющие одну либо несколько четвертичных гетероциклических групп, присоединенных к алкильному радикалу, имеющему от 1 до 10 атомов углерода. Упомянутый термин "четвертичный гетероарилалкил" обозначает алкильный радикал,замещенный одной либо несколькими четвертичными гетероарильными группами. Предпочтительными четвертичными гетероарилалкильными радикалами являются "низшие четвертичные гетероарилалкильные" радикалы, имеющие одну либо несколько четвертичных гетероарильных групп, присоединенных к алкильному радикалу, имеющему от 1 до 10 атомов углерода. Упомянутый термин "алкилгетероарилалкил" обозначает гетероарилалкильный радикал,замещенный одной либо несколькими алкильными группами. Предпочтительными алкилгетероарилалкильными радикалами являются"низшие алкилгетероарилалкильные" радикалы с алкильными участками, имеющими от 1 до 10 атомов углерода. Упомянутый термин "алкокси" обозначает алкильный радикал, присоединенный к остатку молекулы атомом кислорода, например метоксильный радикал. Более предпочтительными алкоксильными радикалами являются "низшие алкоксильные" радикалы, имеющие от 1 до 6 атомов углерода. Примерами таких радикалов являются метокси, этокси, пропокси, изопропокси, бутокси и трет-бутокси. Упомянутый термин "карбокси" обозначает карбоксильную группу, -СО 2 Н, либо ее соли. Упомянутый термин "карбоксиалкил" обозначает алкильный радикал, замещенный одной либо несколькими карбоксильными группами. Предпочтительными карбоксиалкильными радикалами являются "низшие карбоксиалкильные" радикалы, имеющие одну либо несколько карбоксильных групп, присоединенных к алкильному радикалу, имеющему от 1 до 6 атомов углерода. Упомянутый термин "карбоксигетероцикл" обозначает гетероциклический радикал,замещенный одной либо несколькими карбоксильными группами. Упомянутый термин "карбоксигетероарил" обозначает гетероарильный радикал, замещенный одной либо несколькими карбоксильными группами. Упомянутый термин "карбоалкоксиалкил" обозначает алкильный радикал, замещенный 12 одной либо несколькими алкоксикарбонильными группами. Предпочтительными карбоалкоксиалкильными радикалами являются "низшие карбоалкоксиалкильные" радикалы, имеющие одну либо несколько алкоксикарбонильных групп, присоединенных к алкильному радикалу,имеющему от 1 до 6 атомов углерода. Упомянутый термин "карбоксиалкиламино" обозначает аминорадикал, моно- либо дизамещенный карбоксиалкилом. В предпочтительном варианте упомянутым карбоксиалкильным заместителем является "низший карбоксиалкильный" радикал, карбоксильная группа которого связана с алкильным радикалом, имеющим от 1 до 6 атомов углерода. Упомянутый термин "активное соединение" обозначает соединение, соответствующее настоящему изобретению, которое угнетает перенос желчных кислот. В случае использования в сочетаниях, например, "алкиларил" либо "арилалкил", отдельные вышеперечисленные термины имеют указанное выше значение. Упомянутый термин "ингибитор переноса желчных кислот" обозначает соединение, способное угнетать абсорбирование желчных кислот из кишечника в систему кровообращения млекопитающего, например человека. Это включает повышение выделения желчных кислот с фекальными массами, а также снижение концентраций холестерина и эфира холестерина в сыворотке либо плазме крови и, более специфично, снижение холестерина липопротеидов низкой и очень низкой плотности. К числу состояний либо заболеваний, на которые благотворно сказывается профилактика либо лечение посредством угнетения переноса желчных кислот, относятся, например, гиперлипидемические состояния, такие как атеросклероз. Соединения Соединения, соответствующие настоящему изобретению, могут иметь как минимум 2 асимметричных атома углерода и, следовательно,включать рацематы и стереоизомеры, например диастереоизомеры и энантиомеры как в чистом виде, так и в виде смесей. Такие стереоизомеры могут быть получены с помощью традиционных методов, либо посредством реагирования энантиомерных исходных материалов, либо посредством разделения изомеров соединений, соответствующих настоящему изобретению. Изомеры могут включать геометрические изомеры, например цис-изомеры либо трансизомеры через двойную связь. Все такие изомеры предусматриваются в числе соединений, соответствующих настоящему изобретению. Упомянутые соединения, соответствующие настоящему изобретению, включают также таутомеры. Упомянутые соединения, соответствующие настоящему изобретению, как обсуждается далее,включат также соли, сольваты и пролекарства. 13 Синтез соединений Исходные материалы для использования при получении упомянутых соединений, соответствующих настоящему изобретению, известны либо могут быть получены посредством традиционных методов, известных специалисту,либо способом, аналогичным способам, описанным в данной отрасли. Вообще, упомянутые соединения, соответствующие настоящему изобретению, могут быть получены способами, описание которых приведено далее. Например, как показано на cхеме 1, в результате реакции альдегида II с формальдегидом и гидроксидом натрия может быть получен гидроксиальдегид III, который превращают в мезилат IV с помощью метансульфонилхлорида и триэтиламина способом, подобным описанному в Сhеm. Веr, 98, 728-734 (1965). Посредством реакции мезилата IV с тиофенолом V, полученным способом, описание которого приведено вWO 93/16055, в присутствии триэтиламина получают кетоальдегид VI, который может быть циклизован с помощью реактива, полученного из цинка и трихлорида титана в этиленгликольдиметиловом эфире (DME) при нагревании в колбе с обратным холодильником при температуре перегонки, с получением смеси 2,3-дигидробензотиепина VII и двух рацемических стереоизомеров бензотиепин-(5 Н)-4-она VIII в случае неэквивалентности R1 и R2. Посредством оксидирования VII тремя эквивалентами mхлорпербензойной кислоты (МСРВА) получают изомерные сульфоэпоксиды IX, которые, в случае гидрогенизации с использованием палладия на углероде в качестве катализатора, дают смесь четырех рацемических стереоизомеров 4-гидрокси 2,3,4,5-тетрагидробензотиепин-1,1-диоксидов Х и двух рацемических стереоизомеров 2,3,4,5 тетрагидробензотиепин-1,1-диоксидов ХI в случае неэквивалентности R1 и R2. Оптически активные соединения, соответствующие настоящему изобретению, могут быть получены посредством использования оптически активного исходного материала III либо посредством разделения соединений Х с помощью оптически разделяющих веществ, хорошо известных в данной отрасли, описание чего приведено в J. Org. Chem., 39, 3904 (1974), там же, 42, 2781 (1977), и там же, 44, 4891 (1979). Схема 1 В соответствии с альтернативным вариантом кетоальдегид VI, где R2 - Н, может быть получен посредством реагирования тиофенола Бензотиепин-(5 Н)-4-он VIII может быть оксидирован m-хлорпербензойной кислотой(МСРВА) с получением бензотиепин-(5 Н)-4-он 1,1-диоксида XII, который может быть восстановлен борогидридом натрия с получением четырех рацемических стереоизомеров X. Два стереоизомера X, Ха и Хb, имеющие ОН группу и R5 на противоположных сторонах бензотиепинового кольца, могут быть превращены в два других изомера X, Хс и Xd, имеющие ОН группу и R5 на той же самой стороне упомянутого бензотиепинового кольца, посредством реакции в метиленхлориде с 40-50% гидроксида натрия в присутствии межфазного катализатора (РТС). Упомянутое преобразование может также быть осуществлено с помощью t-бутоксида калия в тетрагидрофуране (THF). 15 МСРВА=m-хлорпербензойная кислота РТС=межфазный катализатор когда R1=бутил, R2=этил, R5=фенил, Х=Н, q=4 6 а=Ха 6b=Хb 6 с=Хс 6d=Xd 3 Упомянутые соединения, соответствующие настоящему изобретению, где R5 - OR,NRR' и S(O)nR, a R4 - гидрокси, могут быть получены посредством реагирования эпоксида IX,где R5 - Н, с тиолом, спиртом и амином в присутствии основания. Другой способ получения Хс и Xd, соответствующих настоящему изобретению, показан на cхеме 2. Соединение VI оксидируется до соединения XIII с помощью двух эквивалентов mхлорпербензойной кислоты. Посредством гидрогенолиза соединения XIII с использованием палладия на углероде получают соединениеXIV, которое может быть циклизовано tбутоксидом калия либо гидроксидом натрия в условиях фазового переноса с получением смеси Хс и Xd. Разделение Хс и Xd может осуществляться посредством высокоэффективной жидкостной хроматографии либо фракционированной кристаллизации. Упомянутые тиофенолы XVIII и V, используемые в настоящем изобретении, могут также быть получены по cхеме 3. Алкилирование фенола XV арилметилхлоридом в неполярном растворителе по способу, описание которого приведено в J. Chem. Soc., 2431-2432 (1958), обеспечивает получение ортозамещенного фенола XVI. Упомянутый фенол XVI может быть превращен в упомянутый тиофенол XVIII через тиокарбаматXVII по способу, описание которого приведено вJ. Org. Chem., 31, 3980 (1966). Вначале упомянутый фенол XVI реагирует с диметилтиокарбамоилхлоридом и триэтиламином с образованием тиокарбамата XVII, который подвергается тепловой перегруппировке при температуре 200-300 С, и полученный перегруппированный продукт гидролизуется гидроксидом натрия с получением упомянутого тиофенола XVIII. Подобным же образом, тиофенол V может также быть получен из 2-ацилфенола XIX через промежуточный тиокарбамат XX. На cхеме 4 показан другой способ получения бензотиепин-1,1-диоксидов Хс и Xd, начиная с тиофенола XVIII. Соединение XVIII может реагировать с мезилатом IV с образованием альдегида сульфокислоты XXI. Посредством оксидирования XXI двумя эквивалентами МСРВА получают альдегид сульфокислотыXIV, который может быть циклизован с помощью t-бутоксида калия до получения смеси Хс иXd. Циклизация альдегида сульфокислоты tбутоксидом калия также дает смесь бензотиепина XXII. Схема 4 Примеры амин- и гидроксиламинсодержащих соединений, соответствующих настоящему изобретению, могут быть получены, как показано на cхеме 5 и cхеме 6. 2-Xлор-4-нитробензофенон восстанавливают триэтилсиланом и трифторметансульфокислотой до 2-хлор-4-нитродифенилметана 32. Реакция 32 с сульфидом лития,с последующим реагированием полученного сульфида с мезилатом IV, дает альдегид сульфокислоты XXIII. Оксидирование XXIII двумя эквивалентами МСРВА дает альдегид сульфокислоты XXIV, который может быть восстановлен гидрогенизацией до гидроксиламина XXV. Введение в гидроксиламин XXV защитной группы с помощью ди-t-бутилдикарбоната обеспечивает получение производного N,O-ди-(tбутоксикарбонил)гидроксиламина XXVI. Цик 17 лизация XXVI t-бутоксидом калия и отщепление t-бутоксикарбонильной защитной группы дает смесь производных гидроксиламинаXXVIIc и XXVIId. Упомянутые производные первичного амина XXXIIIc и XXXIIId могут быть также получены посредством дополнительной гидрогенизации XXIV или XXVIIc и Схема 7 описывает один из способов введения заместителя в арильное кольцо в 5 положении бензотиепина. Йодирование 5 фенильного производного XXX йодом, катализированное трифлатом ртути, обеспечивает получение производного йода XXXI, которое, в результате катализированного палладием карбонилирования в спирте, обеспечивает получение карбоксилата XXXII. Гидролиз полученного карбоксилата и дериватизация полученной кислоты до кислотных производных хорошо известны в данной отрасли. Схема 7 На схеме 6 восстановление альдегида сульфокислоты XXV водородом с последующим гидроалкилированием полученного аминопроизводного водородом и альдегидом с использованием палладия на углероде в качестве катализатора в том же самом реакционном сосуде обеспечивает получение замещенного аминопроизводного XXVIII. Циклизация XXVIII tбутоксидом калия обеспечивает получение смеси замещенных аминопроизводных XXIXc иAliquart 336 - метилтрикаприламмонийхлорид МСРВА - m-хлорпербензойная кислота Целит - вспомогательное фильтровальное вещество (разновидность диатомовой земли)DME - этиленгликольдиметиловый эфир ВОС - t-бутоксикарбонильная группаEt2O - диэтиловый эфир СН 2 Сl2 - метиленхлоридNaOH - натрия гидроксид СН 3 ОН - метанол НСl - хлористо-водородная кислотаNMR - спектроскопия ядерного магнитного резонансаMPLC - жидкостная хроматография среднего давленияHPLC - жидкостная хроматография высокого давленияRPHPLC - жидкостная хроматография высокого давления с обращенной фазойmin - минута (минуты) Термин "энантиомерно обогащенный" (е.е.) означает, что один энантиомер либо набор диастереоизомеров преобладает над дополнительным энантиомером либо набором диастереоизомеров. Энантиомерное обогащение смеси энантиомеров вычисляется посредством деления концентрации преобладающего энантиомера на концентрацию другого энантиомера, умножением делимого на 100 и выражением результата в виде процента. Энантиомерное обогащение может составлять от приблизительно 1% до приблизительно 100%, в предпочтительном варианте от приблизительно 10% до приблизительно 100% и в более предпочтительном варианте от приблизительно 20 до 100%.R1 и R2 могут выбираться из числа замещенных и незамещенных C1-С 10 алкилов, где упомянутый заместитель (заместители) может выбираться из группы, включающей алкилкарбонил, алкокси, гидрокси и азотсодержащих гетероциклов, связанных с упомянутыми C1-С 10 алкилами через посредство эфирной связи. К числу заместителей атома углерода в 3 положении могут быть отнесены этил, n-пропил, n-бутил, nпентил, изобутил, изопропил, -СН 2 С(=O)С 2 Н 5, 004650-СН 2 ОС 2 Н 5 и -СН 2O-(4-пиколин). Предпочтение отдается этилу, n-пропилу, n-бутилу и изобутилу. В некоторых особо предпочтительных соединениях, соответствующих настоящему изобретению, заместители R1 и R2 идентичны, например, n-бутил/n-бутил, благодаря чему упомянутое соединение является ахиральным на атоме углерода в 3 положении. Ликвидация оптической изомерии на атоме углерода в 3 положении упрощает отбор, синтез, разделение и контроль качества соединения, используемого в качестве ингибитора перемещения желчных кислот в подвздошной кишке. У обоих соединений, имеющих хиральный атом углерода в 3 положении, и у соединений,имеющих ахиральный атом углерода в 3 положении, в число заместителей (Rx) на бензольном кольце могут входить водород, арил, алкил,гидрокси, галоген, алкокси, алкилтио, алкилсульфинил, алкилсульфонил, галогеналкил, галогеналкокси, (N)-гидроксикарбонилалкиламин,галогеналкилтио, галогеналкилсульфинил, галогеналкилсульфонил, амино, N-алкиламино, N,Nдиалкиламино, (N)-алкоксикарбамоил, (N)-арилоксикарбамоил, (N)-аралкилоксикарбамоил, триалкиламмоний (в частности, с противоионом галогенида), (N)-амидо, (N)-алкиламидо, -N-алкиламидо, -N,N-диалкиламидо, (N)-галогеналкиламидо, (N)-сульфонамидо, (N)-алкилсульфонамидо, (N)-галогеналкилсульфонамидо, карбоксиалкиламино, соль триалкиламмония, (N)карбаминовая кислота, алкиловый либо бензиловый эфир, N-ациламин, гидроксиламин, галогенациламин, углеводород, тиофен, соль триалкиламмония, имеющая карбоновую кислоту либо гидроксильный заместитель на одном либо нескольких алкильных заместителях, алкиленовая мостиковая связь, имеющая в качестве заместителя соль четвертичного аммония,-[O(CH2)w]x-X, где х - 2-12, w - 2 либо 3 и X представляет собой галоген либо соль четвертичного аммония, и (N)-азотсодержащий гетероцикл, где упомянутый азот упомянутого гетероцикла факультативно кватернизован. К числу предпочтительных разновидностей, которые могут образовывать Rx, относятся метил, этил,изопропил, t-бутил, гидрокси, метокси, этокси,изопропокси, метилтио, йод, бром, фтор, метилсульфинил, метилсульфонил, этилтио, амино,гидроксиламин, N-метиламино, N,N-диметиламино, N,N-диэтилaминo, (N)-бензилоксикарбамоил, триметиламмоний, A-, -NHC(=O)CH3,-NHC(=O)C5H11, -NHC(=O)C6H13, карбоксиэтиламино, (N)-мopфoлинил, (N)-aзeтидинил, (N)-Nмeтилазетидиний А-, (N)-пирролидинил, пирролил, (N)-N-метилпиридиний А-, (N)-N-метилморфолиний А- и N-N'-мeтилпиперазинил, (N)бpoммeтилaмидo, (N)-N-гексиламино, тиофен,-N+(CH3)2CO2H I-, -NCH3CH2CO2H, -(N)-N'-диметилпиперазиний I-, (N)-t-бутилоксикарбамоил,(N)-метилсульфонамидо, (N)N'-мeтилпиppoлидиний и -(ОСН 2 СН 2)3I, где А- представляет со 21 бой фармацевтически приемлемый анион. Упомянутое бензольное кольцо может быть монозамещеннным в 6, 7 либо 8 положении или же дизамещенным в 7 либо 8 положениях. Сюда же входят 6,7,8-триалкоксильные соединения, например 6,7,8-триметоксильные соединения. В 6,7, 8 и/или 9 положениях упомянутого бензольного кольца могут с таким же успехом присутствовать различные другие заместители, в том числе, например, гуанидинил, циклоалкил, углеводород (например, моносахарид с 5 либо 6 атомами углерода), пептид и соли четвертичного аммония, связанные с упомянутым кольцом посредством поли(оксиалкиленовых) связей,например -(OCH2CH2)x-N+R13R14R15A-, где х - от 2 до 10. В дополнительных соединениях, соответствующих настоящему изобретению, R5 и R6 независимо выбирают из группы, включающей арил, тиофен, пиридин, пиррол, тиазол, имидазол, пиразол, пиримидин, морфолин, Nалкилпиридиний,N-алкилпиперазиний,Nалкилморфолиний либо фуран с замещенными либо незамещенными атомами водорода либо углерода кольца, заместитель(и) которых выбираются из группы, включающей галоген, гидроксил, тригалогеналкил, алкокси, амино, Nалкиламино, N,N-диалкиламино, соли четвертичного аммония, C1-C4 алкиленовую мостиковую связь, имеющую в качестве заместителя соль четвертичного аммония, алкоксикарбонил,арилоксикарбонил, алкилкарбонилокси и арилкарбонилокси, (O,O)-диоксиалкилен, -[O(CH2)w]xX,где х - от 2 до 12, w - 2 либо 3 и X включает галоген либо соль четвертичного аммония, тиофен, пиридин, пиррол, тиазол, имидазол, пиразол либо фуран. Упомянутой арильной группойR5 или R6 в предпочтительном варианте является фенил, фенилен либо бензолтриил, т.е. они могут быть незамещенными, монозамещенными либо дизамещенными. К числу разновидностей,которые могут выступать в качестве заместителей на упомянутом арильном кольце R5 и R6,относятся фтор, хлор, бром, метокси, этокси,изопропокси, триметиламмоний (в предпочтительном варианте с противоионом йодида либо хлорида), метоксикарбонил, этоксикарбонил,формил, ацетил, пропаноил, (N)-гeкcилдимeтилaммoний, гексилентриметиламмоний, три(оксиэтилен)йодид и тетра(оксиэтилен)триметиламмонийодид, каждый из которых может замещаться на упомянутом кольце в пара-, металибо обоих положениях. К числу других заместителей, которые могут присутствовать на фениленовом, бензолтриильном либо другом ароматическом кольце, относятся 3,4-диоксиметилен (5-членное кольцо) и 3,4-диоксиэтилен (6 членное кольцо). К числу соединений, у которых уже были установлены либо могут быть продемонстрированы необходимые свойства ингибитора перемещения желчных кислот в подвздошной кишке, относятся соединения, у 22 которых R5 и R6 выбирают из группы, включающей фенил, p-фторфенил, m-фторфенил, pгидроксифенил, m-гидроксифенил, p-метоксифенил, m-метоксифенил, p-N,N-диметиламинофенил, m-N,N-диметиламинофенил, I- m-(СН 3)3N+-фенил, I- m-(СН 3)3-N+-фенил, I- m-(СН 3)3-N+CH2CH2-(OCH2CH2)2-O-фeнил, I- p-(CH3)3-N+CH2CH2-(OCH2CH2)2-O-фенил, I- m-(N,N-диметилпиперазиний)-(N')-СН 2-(ОСН 2 СН 2)2-O-фенил,3-метокси-4-фторфенил, тиенил-2-ил, 5-хлортиенил-2-ил, 3,4-дифторфенил, I- p-(N,N-димeтилпипepaзиний)-(N')-CH2-(OCH2CH2)2-O-фeнил,3-фтор-4-метоксифенил, 4-пиридинил, 2-пиридинил, 3-пиридинил, N-метил-4-пиридиний, IN-метил-3-пиридиний, 3,4-диоксиметиленфенил,3,4-диоксиэтиленфенил и p-метоксикарбонилфенил. К числу предпочтительных соединений относятся 3-этил-3-бутил и 3-бутил-3-бутил,каждое из которых имеет вышеприведенные предпочтительные заместители R5 в сочетании с заместителями Rx, представленными в табл. 1. Особое предпочтение отдается тому, чтобы один, но не оба R5 и R6, был водородом. Особое предпочтение отдается тому, чтобы R4 и R6 были водородом, чтобы R3 и R5 не были водородом и чтобы R3 и R5 были ориентированы в одинаковом направлении относительно плоскости молекулы, т.е. оба в - либо оба в-конфигурации. Дополнительное предпочтение отдается тому, чтобы в случае, когда R2 - бутил,а R1 - этил, R1 имел такую же ориентацию относительно плоскости молекулы, что и R3 и R5. Далее, в табл. 1 А, приведен перечень иллюстративных разновидностей R1/R2, R5/R6 и Rx. Таблица 1 А Альтернативные R группы Еще одни предпочтительные соединения,соответствующие настоящему изобретению,включают остовную структуру, имеющую две либо несколько фармацевтически активных бензотиепиновых структур, как описывалось ранее,ковалентно связанных с упомянутой остовной составляющей через посредство функциональных связей. Такие активные бензотиепиновые структуры в предпочтительном варианте включают где R1, R2, R3, R4, R5, R6, R7, R8, X, q и n соответствуют определению, приведенному ранее, а R55 ковалентная связь либо арилен. Упомянутая остовная составляющая может включать алкандиил, алкендиил, алкиндиил,полиалкандиил, алкоксидиил, полиэфирдиил,полиалкоксидиил, углеводород, аминокислоту и пептид, полипептид, где алкандиил, алкендиил,алкиндиил, полиалкандиил, алкоксидиил, полиэфирдиил, полиалкоксидиил, углеводород, аминокислота и пептид, полипептид могут факультативно иметь 1 либо несколько атомов углерода, замещенных О, NR7, N+R7R8, S, SO, SO2,S+R7R8, PR7, P+R7R8, фениленом, гетероциклом,четвертичным гетероциклом, четвертичным гетероарилом либо арилом,где алкандиил, алкендиил, алкиндиил, полиалкандиил, алкоксидиил, полиэфирдиил, полиалкоксидиил, углеводород, аминокислота и пептид, полипептид могут быть замещены одной либо несколькими замещающими группами,независимо выбранными из группы, включающей алкил, алкенил, алкинил, полиалкил, полиэфир, арил, галогеналкил, циклоалкил, гетероцикл, арилалкил, галоген, оксогруппу, OR13,NR13R14, SR13, S(O)R13, SO2R13, SO3R13,NR13OR14, NR13NR14R15, NO2, CO2R13, CN, OM,SO2OM, SO2NR13R14, C(O)NR13R14, C(O)OM,COR13, P(O)R13R14, P+R13R14R15A-, P(OR13)OR14,S+R13R14A- и N+R9R11R12A-; где упомянутые алкил, алкенил, алкинил,полиалкил, полиэфир, арил, галогеналкил, циклоалкил и гетероцикл могут быть дополнительно замещены одной либо несколькими замещающими группами, выбранными из группы,включающей OR7, NR7R8, SR7, S(O)R7, SO2R7,SO3R7, СО 2R7, CN, оксогруппу, CONR7R8,N+R7R8R9A-, алкил, алкенил, алкинил, арил,циклоалкил, гетероцикл, арилалкил, четвертичный гетероцикл, четвертичный гетероарил,P(O)R7R8, P+R7R8A- и P(O)(OR7)OR8, и где упомянутые алкил, алкенил, алкинил,полиалкил, полиэфир, арил, галогеналкил, циклоалкил и гетероцикл могут факультативно иметь 1 либо несколько атомов углерода, заме 25 щенных О, NR7, N+R7R8A-, S, SO, SO2, S+R7A-,PR7, P(O)R7, P+R7R8A- или фениленом. Примерами остовных составляющих являютсяR26 и R27 независимо выбирают из группы,включающей где R26, R29, R30 и R31 независимо выбирают из группы, включающей алкил, алкенил, алкинил,алкиларил, арил, арилалкил, циклоалкил, гетероцикл и гетероциклоалкил,А- представляет собой фармацевтически приемлемый анион и k=oт 1 до 10. В соединениях формулы DIV, R20, R21, R22 в формулах DII и DIII и R23 в формуле DIII могут быть связаны в любом из своих 6-, 7-, 8- либо 9 положениях с R19. В соединениях формулыDIVA предпочтение отдается тому, чтобы R55 включал фениленовую составляющую, связанную в m- либо p-положении с R19. В другом варианте осуществления основа остовной составляющей, R19, как обсуждалось в случае формул DII и DIII, может многократно замещаться более чем четырьмя боковыми активными бензотиепиновыми единицами, например R20, R21, R22 и R23, как обсуждалось ра 004650 26 нее, через многочисленные функциональные группы в пределах основы остовной составляющей. Основная единица остовной составляющий, R19, может включать однокомпонентную единицу остовной составляющей, ее мультимеры и мультимерные смеси различных единиц остовной составляющей, которые обсуждались в этом описании, например, самостоятельно либо в комбинациях. Количество отдельных единиц основы остовной составляющей может колебаться в пределах от приблизительно 1 до приблизительно 100, в предпочтительном варианте от приблизительно 1 до приблизительно 80, в более предпочтительном варианте от приблизительно 1 до приблизительно 50 и в еще более предпочтительном варианте от приблизительно 1 до приблизительно 25. Количество точек присоединения одинаковых либо различных боковых активных бензотиепиновых единиц в пределах одной основной единицы остовной составляющей может колебаться в пределах от приблизительно 1 до приблизительно 100, в предпочтительном варианте от приблизительно 1 до приблизительно 80, в более предпочтительном варианте от приблизительно 1 до приблизительно 50 и в еще более предпочтительном варианте от приблизительно 1 до приблизительно 25. Такие точки присоединения могут включать связи с С, S, О, N либо Р в любой из групп, охватываемых определением R19. Более предпочтительные бензотиепиновые составляющие, включающие R20, R21, R22 и/илиR23, соответствуют упомянутым предпочтительным структурам, описание которых было представлено ранее для формулы I. Атом углерода в 3 положении на каждой бензотиепиновой составляющей может быть ахиральным, и упомянутые заместители R1, R2, R3, R4, R5 и Rх могут выбираться из предпочтительных групп и комбинаций заместителей, как обсуждалось ранее. Упомянутые остовные структуры могут включать,например, поли(эксиалкилен) либо олиго(оксиалкилен), в частности поли- либо олиго(оксиэтилен) либо поли- или олиго(оксипропилен). Дозировки, лекарственные формы и пути введения Соединения-ингибиторы перемещения желчных кислот в подвздошной кишке, соответствующие настоящему изобретению, могут вводиться для профилактики и лечения гиперлилидемических заболеваний либо состояний любыми путями, в предпочтительном варианте перорально, которые обеспечивают контактирование упомянутых соединений с местами их действия в организме, например в подвздошной кишке млекопитающего, например человека. Для профилактики либо лечения вышеупомянутых состояний соединения, соответствующие настоящему изобретению, могут использоваться в виде соединения per se. Фармацевтически приемлемые соли особо пригодны для медицинского применения благо 27 даря более высокой растворимости в воде по сравнению с исходным соединением. Вполне очевидно, что подобные соли должны иметь фармацевтически приемлемый анион либо катион. Подходящие фармацевтически приемлемые соли соединений, соответствующих настоящему изобретению, получаемые путем добавления кислот, по возможности, включают соли, полученные с помощью неорганических кислот, например хлористо-водородной, бромисто-водородной, фосфорной, метафосфорной,азотной, сульфоновой и серной кислот, и органических кислот, например уксусной, бензолсульфоновой, бензойной, лимонной, этансульфоновой, фумаровой, глюконовой, гликолевой,изотионовой, молочной, лактобионовой, малеиновой, яблочной, метансульфоновой, янтарной,толуолсульфоновой, винной и трифторуксусной кислот. Хлорид является особо предпочтительным для медицинских целей. К числу подходящих фармацевтически приемлемых солей, получаемых посредством добавления основания,относятся соли аммония, соли щелочных металлов, например соли натрия и калия, и соли щелочно-земельных металлов, например соли магния и кальция. Анионы, соответствующие определению А- в описании настоящего изобретения, естественно, также должны быть фармацевтически приемлемыми и также выбираться из вышеприведенного перечня. Упомянутые соединения, соответствующие настоящему изобретению, могут представляться с приемлемым носителем в форме фармацевтической композиции. Упомянутый носитель, конечно, должен быть приемлемым с точки зрения совместимости с другими ингредиентами упомянутой композиции и не должен причинять вреда реципиенту. Носитель может быть как твердым, так и жидким либо комбинированным и в предпочтительном варианте вводиться в состав лекарственной формы с упомянутым соединением с получением стандартной композиции, например таблетки, в состав которой может входить от 0,05 до 95 мас.% упомянутого активного соединения. Присутствовать могут также другие фармакологически активные вещества, включая другие соединения, соответствующие настоящему изобретению. Фармацевтические композиции, соответствующие настоящему изобретению, могут готовиться с помощью любых хорошо известных фармацевтических способов, заключающихся существенным образом в смешивании компонентов. Эти соединения могут вводиться посредством любых традиционных средств, доступных для использования в сочетании с фармацевтическими препаратами в виде отдельных терапевтических соединений либо в виде сочетания терапевтических соединений. Количество соединения, необходимое для достижения необходимого биологического эф 004650 28 фекта, будет, естественно, зависеть от ряда факторов, например конкретного выбранного соединения, применения, для которого оно предназначено, способа введения и клинического состояния реципиента. В общем, суточная доза может находиться в пределах от приблизительно 0,3 мг до приблизительно 100 мг/кг массы тела/день, в предпочтительном варианте от приблизительно 1 мг до приблизительно 50 мг/кг массы тела/день, в более предпочтительном варианте от приблизительно 3 мг до приблизительно 10 мг/кг массы тела/день. Эта общая суточная доза может вводиться пациенту в виде разовой дозы либо в виде небольших доз, повторяемых через определенные промежутки времени. Небольшие дозы могут вводиться от 2 до 6 раз в течение дня. Дозы могут быть в форме, предназначенной для замедленного выделения, эффективного для достижения необходимых результатов. В состав стандартных лекарственных форм, предназначенных для перорального введения, например таблеток либо капсул, может входить от приблизительно 0,1 до приблизительно 100 мг бензотиепинового соединения, в предпочтительном варианте от приблизительно 1 до приблизительно 75 мг соединения, в более предпочтительном варианте от приблизительно 10 до приблизительно 50 мг соединения. В случае фармацевтически приемлемых солей, вышеупомянутая масса относится к массе бензотиепинового иона, полученного из упомянутой соли. Пероральная доставка ингибитора перемещения желчных кислот в подвздошной кишке, соответствующего настоящему изобретению, может осуществляться с помощью лекарственных форм, которые хорошо известны в данной отрасли, способных обеспечить длительное либо замедленное введение лекарственного препарата в желудочно-кишечный тракт посредством целого ряда различных механизмов. К их числу относятся, но ими не ограничиваются, реагирующее на уровень рН выделение из стандартной лекарственной формы, основанное на изменении рН тонкого кишечника, медленное эродирование таблетки либо капсулы,задержка в желудке, обусловленная физическими свойствами лекарственной формы, биоадгезия стандартной лекарственной формы к слизистой выстилке кишечника либо ферментное выделение активного лекарственного препарата из упомянутой стандартной дозированной формы. Предполагаемый эффект заключается в продлении периода времени, в течение которого молекула активного лекарственного препарата доставляется к месту действия (подвздошная кишка) посредством манипулирования упомянутой стандартной лекарственной формой. Так, в объем настоящего изобретения входят лекарственные формы с энтеросолюбильным покрытием и лекарственные формы с энтеросолюбильным покрытием и регулируемым выделением. К 29 числу подходящих энтеросолюбильных покрытий относятся ацетофталат целлюлозы, поливинилацетофталат, гидроксипропилметилфталат целлюлозы и анионные полимеры метакриловой кислоты и метилового эфира метакриловой кислоты. В случае внутривенного введения, упомянутая доза, например, может находиться в пределах от приблизительно 0,1 до приблизительно 1,0 мг/кг массы тела, в предпочтительном варианте от приблизительно 0,25 до приблизительно 0,75 мг/кг массы тела, в более предпочтительном варианте от приблизительно 0,4 до приблизительно 0,6 мг/кг массы тела. Эта доза может быть подходящим образом введена посредством вливания от приблизительно 10 до приблизительно 100 нг/кг массы тела в минуту. Жидкости для вливания, пригодные для этой цели,могут содержать, например, от приблизительно 0,1 до приблизительно 10 нг, в предпочтительном варианте от приблизительно 1 до приблизительно 10 нг на миллилитр. Стандартные дозы могут содержать, например, от приблизительно 1 мг до приблизительно 10 г упомянутого соединения, соответствующего настоящему изобретению. Таким образом, ампулы для инъекций могут содержать, например, от приблизительно 1 до приблизительно 100 мг. К числу фармацевтических композиций,соответствующих настоящему изобретению,могут относиться композиции, пригодные для перорального, ректального, местного, трансбуккального (например, подъязычного) и парентерального (например, подкожного, внутримышечного, внутрикожного либо внутривенного) ведения, хотя наиболее приемлемый путь в любом данном случае будет зависеть от природы и тяжести состояния, подвергаемого лечению, и от природы конкретного используемого соединения. В большинстве случаев, предпочтительным путем введения является пероральный. Фармацевтические композиции, пригодные для перорального введения, могут быть представлены в виде дискретных единиц, например капсул, каше, лепешек либо таблеток,каждая из которых содержит предопределенное количество как минимум одного соединения,соответствующего настоящему изобретению; в виде порошков или гранул; в виде раствора или суспензии в водной либо неводной жидкости; либо в виде эмульсии масла в воде или водомасляной эмульсии. Как указывалось, такие композиции могут изготовляться с помощью любого пригодного фармацевтического способа,который включает этап объединения активного соединения (соединений) и носителя (который может включать один либо несколько дополнительных ингредиентов). В общем, композиции получают посредством однородного и тщательного смешивания активного соединения с жидкостью, либо тонко измельченным твердым носителем, либо с тем и другим, с последующим, в 30 случае необходимости, приданием продукту необходимой формы. Например, таблетка может быть получена посредством прессования либо формования порошка либо гранул упомянутого соединения факультативно с одним либо несколькими дополнительными ингредиентами. Прессованные таблетки могут быть получены посредством прессования, в соответствующей машине, соединения в сыпучем состоянии, например в виде порошка либо гранул, факультативно смешанного со связующим, смягчающим компонентом, инертным разбавителем и/или поверхностно-активным/диспергирующим веществом(ами). Формованные таблетки могут изготовляться посредством формования, в подходящей машине, порошкообразного соединения, увлажненного инертным жидким разбавителем. Фармацевтическими композициями, пригодными для трансбуккального (подъязычного) введения, являются лепешки, в состав которых входит соединение, соответствующее настоящему изобретению, в ароматизированной основе, как правило, сахарозе, и аравийской камеди либо трагаканте, и пастилки, включающие упомянутое соединение, в инертной основе, например желатине и глицерине или сахарозе и аравийской камеди. К числу фармацевтических композиций,пригодных для парентерального введения, традиционно относят стерильные водные препараты соединения, соответствующего настоящему изобретению. Эти препараты в предпочтительном варианте вводятся внутривенно, хотя введение может осуществляться также посредством подкожного, внутримышечного либо внутрикожного впрыскивания. Такие препараты могут быть подходящим образом получены посредством смешивания упомянутого соединения с водой, с последующим превращением полученного раствора в стерильный и изотонический с кровью. В состав композиций для инъекций,соответствующих настоящему изобретению,входит, как правило, от 0,1 до 5% (в массовом отношении) соединения, раскрываемого в настоящем описании. Фармацевтические композиции, пригодные для ректального введения, представлены в предпочтительном варианте в виде стандартных суппозиториев. Они могут быть получены посредством смешивания соединения, соответствующего настоящему изобретению, с одним либо несколькими подходящими твердыми носителями, например маслом какао, с последующим приданием полученной смеси соответствующей формы. Фармацевтическим композициям, пригодным для местного нанесения на кожу, в предпочтительном варианте придается форма мази,крема, лосьона, пасты, геля, спрея, аэрозоля либо масла. В число носителей, которые могут использоваться, входят вазелин, ланолин, поли 31 этиленгликоли, спирты и комбинации двух или более упомянутых веществ. Активное соединение представляется, как правило, в концентрации от 0,1 до 15% (в массовом отношении) от упомянутой композиции, например от 0,5 до 2%. Возможно также чрескожное введение. Фармацевтические композиции, пригодные для чрескожного введения, могут представляться в виде дискретных пластырей, приспособленных для нахождения в тесном соприкосновении с кожей реципиента в течение длительного промежутка времени. В состав таких пластырей,как правило, входит соединение, соответствующее настоящему изобретению, в факультативно забуференном водном растворе, растворенное и/или диспергированное в клейком веществе либо диспергированное в полимере. Подходящая концентрация упомянутого активного соединения составляет приблизительно от 1 до 35%, в предпочтительном варианте от приблизительно 3 до 15%. Как один из конкретных возможных вариантов, упомянутое соединение может доставляться из пластыря посредством, например,электропереноса либо электрофореза, как описано в Pharmaceutical Research, 3 (6), 318 (1986). В любом случае, количество активного ингредиента, которое может комбинироваться с материалами-носителями для получения стандартной дозированной формы для введения, будет изменяться в зависимости от пациента, подвергаемого лечению, и конкретного способа введения. В состав твердых стандартных дозированных лекарственных форм для перорального введения, к которым относятся упоминавшиеся ранее капсулы, таблетки, пилюли, порошки и гранулы, входит одно либо несколько соединений, соответствующих настоящему изобретению, смешанных как минимум с одним инертным разбавителем, например сахарозой, лактозой либо крахмалом. В состав таких стандартных дозированных лекарственных форм, при обычном практическом применении, могут входить также, кроме инертных разбавителей, дополнительные вещества, например смягчающие компоненты, например стеарат магния. В случае капсул, таблеток и пилюль, в состав упомянутых стандартных дозированных лекарственных форм могут входить также буферные вещества. Наряду с этим, таблетки и пилюли могут изготовляться с энтеросолюбильными покрытиями. Жидкие стандартные дозированные лекарственные формы для перорального введения могут включать фармацевтически приемлемые эмульсии, растворы, суспензии, сиропы и эликсиры, в состав которых входят инертные разбавители, традиционно используемые в данной отрасли, например вода. Такие композиции могут включать также адъюванты, например увлажняющие вещества, эмульгирующие и суспендирующие вещества, а также подслащивающие вещества, отдушки и корригенты. 32 Препараты для впрыскивания, например стерильные водные либо маслянистые суспензии для впрыскивания, могут готовиться по рецептам, известным в данной отрасли, с использованием подходящих диспергирующих или стабилизирующих и суспендирующих веществ. Стерильным препаратом для впрыскивания может также быть стерильный раствор или суспензия для впрыскивания в нетоксическом парентерально приемлемом разбавителе или растворителе, например в виде раствора в 1,3 бутандиоле. К числу приемлемых носителей и растворителей, которые могут быть использованы, относятся вода, раствор Рингера-Локка и изотонический раствор хлорида натрия. Наряду с этим, в качестве растворителя либо суспендирующей среды традиционно используются стерильные нелетучие масла. С этой целью может быть использовано любое слаболетучее масло,например синтетические моно- или диглицериды. Наряду с этим, для получения растворов для впрыскивания могут использоваться жирные кислоты, например олеиновая кислота. К числу фармацевтически приемлемых носителей относятся все вышеупомянутые и подобные. Лечебная схема Схема приема лекарственного средства для профилактики, облегчения либо улучшения болезненного состояния, составным элементом которого является гиперлипидемия, например атеросклероза, либо для защиты или дополнительного лечения соединениями и/либо композициями, соответствующими настоящему изобретению, с целью снижения высоких концентраций холестерина в плазме или крови, выбирается в соответствии с различными факторами. К их числу относятся тип, возраст, масса, пол,рацион и состояние здоровья пациента, тяжесть заболевания, путь введения, фармакологические соображения, такие как активность, эффективность, фармакокинетические и токсикологические профили конкретного примененного соединения, применяется ли система доставки лекарственного препарата или же соединение вводится как часть комбинации лекарственных препаратов. Таким образом, фактически применяемая схема приема лекарственного средства может колебаться в широких пределах и, таким образом,отклоняться от предпочтительной схемы приема лекарственного средства, изложенной ранее. Предварительное лечение пациента, страдающего от гиперлипидемического состояния,может начинаться с введения вышеуказанных доз. Лечение, как правило, должно продолжаться, при необходимости, в течение периода времени от нескольких недель до нескольких месяцев либо лет до тех пор, пока упомянутое гиперлипидемическое болезненное состояние не окажется под контролем либо не будет ликвидировано. Состояние пациентов, подвергаемых лечению с помощью соединений либо композиций, раскрываемых в настоящем описании, мо 33 жет постоянно контролироваться посредством,например, измерения уровней содержания холестерина в сыворотке с помощью любого хорошо известного в данной отрасли метода с целью определения эффективности лечения. Постоянный анализ таких данных позволяет вносить изменения в лечебную схему в течение процесса лечения, благодаря чему в любое время вводятся оптимальные эффективные количества соединений, соответствующих настоящему изобретению, и благодаря чему может быть определена продолжительность лечения. Таким образом, в процессе лечения лечебная схема/схема применения лекарственного средства может подвергаться рациональному изменению, что обеспечивает введение минимального количества ингибитора переноса желчных кислот в подвздошной кишке, соответствующего настоящему изобретению, которое обеспечивает удовлетворительную эффективность, и благодаря чему введение продолжается не дольше периода времени, необходимого для успешного излечения гиперлипидемического состояния. Приведенные далее неограничивающие примеры служат иллюстрацией различных аспектов настоящего изобретения. Примеры способов синтезирования Препарат 1. 2-Этил-2-(мезилоксиметил) гексанал (1).(0,13 моль) триэтиламина добавляли капля за каплей 15,8 г 2-этил-2-(гидроксиметил)гексанала,полученного с помощью способа, описание которого было приведено в Chem. Ber. 98, 728-734(1965), с поддержанием температуры реакции ниже 30 С. Полученную реакционную смесь перемешивали при комнатной температуре в течение 18 ч, быстро прекращали реакцию посредством смешивания с холодной разбавленной НСl и экстрагировали с помощью метиленхлорида. Полученный метиленхлоридный экстракт сушили над MgSO4 и концентрировали in vacuo с получением 24,4 г масла коричневого цвета. Препарат 2. 2-2-Бензоилфенилтио)метил)2-этилгексанал (2). Смесь 31 г (0,144 моль) 2-меркаптобензофенона, полученного по способу, описание которого было приведено в WO 93/16055, 24,4 г(1), 14,8 г (0,146 моль) триэтиламина и 80 мл 2 метоксиэтилового эфира выдерживали с нагреванием в колбе с обратным холодильником при температуре перегонки в течение 24 ч. Полученную реакционную смесь выливали в 3N НСl и экстрагировали с помощью 300 мл метиленхлорида. Полученный метиленхлоридный слой промывали 300 мл 10% NaOH, сушили надMgSO4 и концентрировали in vacuo для удаления 2-метоксиэтилового эфира. Полученный остаток очищали посредством высокоэффективного жидкостного хроматографирования 35 Общая схема 10. Посредством нуклеофильного замещения соответствующим образом замещенного 2 фторбензальдегида с помощью сульфида лития либо другого нуклеофильного сульфидного аниона в полярном растворителе (например,диметилформамиде,N,N-диметилацетамиде,диметилсульфоксиде и т.п.), с последующим добавлением альдегида диалкилмезилата (X),был получен диальдегид диалкилбензола Y. Посредством восстановления диальдегида при низкой температуре с помощью диизобутиадипината лития был получен моноальдегид бензилового спирта Z. Посредством превращения бензилового спирта в бензилбромид, с последующим окислением сульфида до сульфона, был получен ключевой промежуточный продукт W. Соединения, соответствующие настоящему изобретению, также могут быть синтезированы с использованием циклического сульфата(XL, далее) в качестве реактива, как показано на представленных далее схемах 11 и 12. В приведенных далее примерах представлено описание способа использования циклического сульфата в качестве реактива. Схема 11 Схема 11 иллюстрирует еще один способ получения бензотиепин-1,1-диоксидов, в частности аналогов 3,3-диалкила, начиная с тиофенола XVIIIA. Тиофенол XVIIIA может вступать в реакцию с циклическим сульфатом XL с образованием спирта XLI, который оксидируется с получением альдегида XLII. Сам альдегид XLII может подвергаться дополнительному оксидированию с получением сульфона XLIII, который может циклизоваться с получением стереоизомерной смеси бензотиепина XLIVa и XLIVb. 36 Тиофенол XVIIIA может быть получен в соответствии со cхемой 3, как обсуждалось ранее, и имеет представленную далее формулу где R5, Rx и q соответствуют определению, которое было представлено ранее для соединений формулы I. Циклический сульфат XL может быть получен в соответствии со способами синтезирования, известными в данной отрасли, и имеет представленную далее формулу где R1 и R2 соответствуют определению, которое было представлено ранее для соединений формулы I. В предпочтительном варианте R1 иR2 представляют собой алкил; в более предпочтительном варианте они выбираются из группы,включающей метил, этил, пропил, изопропил, nбутил, изобутил, втор-бутил, трет-бутил и пентил; в еще более предпочтительном варианте R1 и R2 представляют собой n-бутил. В способе, представленном на cхеме 11,тиофенол XVIIIA вначале реагирует с циклическим сульфатом XL. Эта реакция в предпочтительном варианте осуществляется в апротонном растворителе, таком как метоксиэтиловый эфир. Несмотря на то, что условия прохождения упомянутой реакции, например температура и время, не являются жестко определенными, упомянутая реакция может осуществляться при приблизительно комнатной температуре в течение приблизительно 2 ч. Для осуществления упомянутой реакции в предпочтительном варианте используют приблизительно стехиометрическое соотношение исходных материалов, причем предпочтение отдается использованию незначительного избытка циклического сульфата XL. Время прохождения реакции и выход могут быть улучшены путем использования приблизительно от 1,01 до 1,3 эквивалента циклического сульфата XL на каждый присутствующий эквивалент тиофенола XVIIIA. В более предпочтительном варианте упомянутое соотношение составляет приблизительно 1,1 эквивалента циклического сульфата XL на каждый присутствующий эквивалент тиофенола XVIIIA. В процессе осуществления способа, соответствующего настоящему изобретению, тиофенол XVIIIA подвергают также обработке с помощью отщепляющего вещества. Упомянутое отщепляющее вещество может добавляться к растворителю, содержащему тиофенол XVIIIA,перед, одновременно либо после добавления циклического сульфата XL. He придерживаясь 37 конкретной теории, полагают, что упомянутое отщепляющее вещество отщепляет атом водорода от меркаптановой группы, присоединенной к бензольному кольцу тиофенола XVIIIA. После этого полученный серный анион тиофенола реагирует с циклическим сульфатом XL с разрывом сульфатного кольца. После этого серный анион тиофенола связывается с концевым атомом углерода разорванного сульфатного кольца. Концевой группой на несвязанном конце разорванного сульфатного кольца является сульфатная группа. Упомянутым отщепляющим веществом,как правило, является основание, имеющее рН более приблизительно 10. В предпочтительном варианте упомянутым основанием является гидрид щелочного металла, например гидрид натрия, гидрид лития либо гидрид калия; в более предпочтительном варианте упомянутым основанием является гидрид натрия. Отдается предпочтение незначительному избытку отщепляющего вещества относительно тиофенолаXVIIIA. Время прохождения реакции и выход улучшаются в случае использования от приблизительно 1,0 до приблизительно 1,1 эквивалента отщепляющего вещества на каждый присутствующий эквивалент тиофенола XVIIIA. В более предпочтительном варианте упомянутое соотношение составляет приблизительно 1,1 эквивалента отщепляющего вещества на каждый присутствующий эквивалент тиофенола XVIIIA. После этого удаляется сульфатная группа упомянутого промежуточного продукта реакции тиофенола XVIIIA с циклическим сульфатомXL, в предпочтительном варианте посредством гидролиза, с получением спирта XLI. Подходящими гидролизующими веществами являются минеральные кислоты, в частности хлористоводородная кислота и серная кислота. Несколько реакций, вовлекающих тиофенол XVIIIA, циклический сульфат XL, отщепляющее вещество и гидролизующее вещество,могут осуществлять in situ без необходимости выделения каких-либо образовавшихся промежуточных продуктов. После этого спирт XLI отделяют посредством традиционных способов (например, путем экстрагирования с помощью водного раствора метилсалицилата) и оксидируют с помощью стандартных оксидантов с получением альдегида XLII. В предпочтительном варианте упомянутым оксидантом является триоксид серы либо хлорохромат пиридиния, а в более предпочтительном варианте им является хлорохромат пиридиния. Упомянутую реакцию осуществляют в подходящем органическом растворителе, например метиленхлориде либо хлороформе. После этого альдегид XLII отделяют посредством традиционных способов и подвергают дополнительному оксидированию посредством стандартных оксидантов с получением альдегида сульфокислоты XLIII. В предпочтитель 004650 38 ном варианте упомянутым оксидантом является метахлорпербензойная кислота. Подобным же образом традиционными способами отделяют альдегид сульфокислотыXLIII с последующей циклизацией с получением стереоизомерных бензотиепинов XLIVa иXLIVb. Упомянутым агентом циклизации в предпочтительном варианте является основание, имеющее рН в промежутке от приблизительно 8 до приблизительно 9. В более предпочтительном варианте упомянутым основанием является алкоксидное основание, и в еще более предпочтительном варианте упомянутым основанием является трет-бутоксид калия. Два этапа оксидирования cхемы 11 могут реверсироваться без отрицательного воздействия на общий результат реакции. Вначале может быть оксидирован спирт XLI с получением сульфонового спирта, который, в последующем,оксидируется с получением альдегида сульфокислоты. Схема 12 Схема 12 иллюстрирует еще один способ получения бензотиепин-1,1-диоксидов, в частности аналогов 3,3-диалкила, начиная с галогенбензола L. Галогенбензол L может вступать в реакцию с циклическим сульфатом XL, раскрытым ранее, с образованием спирта LI, который может оксидироваться с получением сульфонового спирта LII. Сам сульфоновый спиртLII может подвергаться дополнительному оксидированию с получением альдегида сульфокислоты LIII, который может циклизоваться с получением стереоизомерной смеси бензотиепинаLIVa и LIVb. Галогенбензол L (который коммерчески доступен либо может быть синтезирован из коммерчески доступных галогенбензолов специалистом в данной отрасли) имеет представленную далее формулу где R5, Rx и q соответствуют определению, которое было представлено ранее для соединений 39 формулы I; Rh представляет собой галоген, например хлор, бром, фтор или йод; и Re представляет собой группу акцептирования электронной пары в орто- либо пара-положении галогенбензола и в предпочтительном варианте представляет собой p-нитро- либо о-нитрогруппу. Циклический сульфат XL может быть получен, как показано на cхеме 11, и может иметь представленную далее формулу где R1 и R2 соответствуют определению, которое было представлено ранее для соединений формулы I. В предпочтительном варианте R1 иR2 представляют собой алкил; в более предпочтительном варианте они выбираются из группы,включающей метил, этил, пропил, изопропил, nбутил, изобутил, втор-бутил, трет-бутил и пентил; в еще более предпочтительном варианте R1 и R2 представляют собой n-бутил. В способе, представленном на cхеме XII,галогенбензол L вначале реагирует с циклическим сульфатом XL. Эта реакция в предпочтительном варианте осуществляется в апротонном растворителе, например диметилформамиде илиN,N-диметилацетамиде и в более предпочтительном варианте в диметилформамиде. Несмотря на то, что условия прохождения упомянутой реакции, например температура и время,не являются жестко определенными, упомянутая реакция может осуществляться в пределах от приблизительно 70 до приблизительно 90 С в течение приблизительно 8-12 ч. В более предпочтительном варианте реакционная температура поддерживается на уровне приблизительно 80 С. Для осуществления упомянутой реакции в предпочтительном варианте используют приблизительно стехиометрическое соотношение исходных материалов, причем предпочтение отдается использованию незначительного избытка циклического сульфата XL. Время прохождения реакции и выход могут быть улучшены путем использования приблизительно от 1,1 до 1,3 экв. циклического сульфата XL на каждый присутствующий эквивалент галогенбензола L. В более предпочтительном варианте упомянутое соотношение составляет приблизительно 1,1 экв. циклического сульфата XL на каждый присутствующий эквивалент галогенбензола L. В процессе осуществления способа, соответствующего настоящему изобретению, галогенбензол L подвергают также обработке с помощью отщепляющего вещества. Упомянутое отщепляющее вещество может добавляться к растворителю, содержащему галогенбензол L,перед, одновременно либо после добавления циклического сульфата XL. He придерживаясь 40 конкретной теории, полагают, что упомянутое отщепляющее вещество отщепляет атом галогена, присоединенный к бензольному кольцу галогенбензола L, и заменяет этот атом двухвалентным атомом серы. После этого полученный серный анион реагирует с циклическим сульфатом XL с разрывом сульфатного кольца. После этого серный анион галогенбензола связывается с концевым атомом углерода разорванного сульфатного кольца. Концевой группой на несвязанном конце разорванного сульфатного кольца является сульфатная группа. Упомянутым отщепляющим веществом, как правило,является дисульфид щелочного металла, и в предпочтительном варианте им является дисульфид лития. Отдается предпочтение незначительному избытку отщепляющего вещества относительно галогенбензола L. Время прохождения реакции и выход улучшаются в случае использования от приблизительно 1,01 до приблизительно 1,3 экв. отщепляющего вещества на каждый присутствующий эквивалент галогенбензола L. В более предпочтительном варианте упомянутое соотношение составляет приблизительно 1,05 эквивалента отщепляющего вещества на каждый присутствующий эквивалент галогенбензола L. После этого удаляется сульфатная группа упомянутого продукта реакции тиофенолаXVIIIA с циклическим сульфатом XL, в предпочтительном варианте посредством гидролиза,с получением смеси эфира и спирта LI. Подходящими гидролизующими веществами являются минеральные кислоты, в частности хлористоводородная кислота и серная кислота. После этого упомянутый эфир превращается в спиртLI посредством обработки гидроксидом щелочного металла, в предпочтительном варианте гидроксидом натрия. Несколько реакций, вовлекающих галогенбензол L, циклический сульфат XL, отщепляющее вещество и гидролизующее вещество,могут осуществлять in situ без необходимости выделения каких-либо образовавшихся промежуточных продуктов. После этого спирт LI отделяют посредством традиционных способов (например, путем экстрагирования с помощью водного раствора метилсалицилата) и оксидируют с помощью стандартных оксидантов с получением сульфонового спирта LII. В предпочтительном варианте упомянутым оксидантом является метахлорпербензойная кислота. Упомянутую реакцию осуществляют в подходящем органическом растворителе, например метиленхлориде либо хлороформе. После этого сульфоновый спирт LII отделяют посредством традиционных способов и подвергают дополнительному оксидированию посредством стандартных оксидантов с получением альдегида сульфокислоты LIII. В предпочтительном варианте упомянутым оксидантом 41 является триоксид серы либо хлорохромат пиридиния, и в более предпочтительном варианте им является хлорохромат пиридиния. Упомянутую реакцию осуществляют в подходящем органическом растворителе, например метиленхлориде либо хлороформе. После этого альдегид сульфокислотыXLIII превращают в необходимые бензотиепин 1,1-диоксиды в соответствии с процедурой, ранее представленной на схеме XI. Два этапа оксидирования могут реверсироваться без отрицательного воздействия на общий результат реакции. Вначале может быть оксидирован спирт XLI с получением альдегида, который затем оксидируется с получением альдегида сульфокислоты. Использование циклического сульфатного реактива вместо мезилатного реактива на схемах 11 и 12 улучшает общий выход и позволяет избежать множества трудностей, с которыми сталкиваются в процессе очистки, по сравнению с реакционными схемами, в которых используется мезилатный промежуточный продукт. Общий выход значительно повышается в случае использования циклического сульфата вместо мезилатного реактива. В дополнение к этому,отпадает необходимость хроматографического разделения промежуточного продукта этапа соединения циклического сульфата этой реакции. Например, промежуточным продуктом на схемах 11 и 12 является водорастворимая соль щелочного металла и примеси могут удаляться посредством экстрагирования эфиром. После этого упомянутый промежуточный продукт гидролизуется до необходимого спирта. Пример, соответствующий схеме 11. Этап 1. Получение 2,2-дибутил-1,3-пропандиола. Алюмогидрид лития (662 мл, 1,2 экв., 0,66 моль) в 662 мл 1 М тетрагидрофурана добавляли капля за каплей к перемешиваемому раствору дибутилдиэтилмалоната (150 г, 0,55 моль) (компания Aldrich) в сухом тетрагидрофуране (700 мл) с поддержанием температуры полученной реакционной смеси в пределах от приблизительно -20 до приблизительно 0 С, с помощью ванны со смесью ацетона/твердой углекислоты. После этого реакционную смесь перемешивали при комнатной температуре в течение ночи. Полученную реакционную смесь охлаждали до температуры -20 С и к ней последовательно добавляли капля за каплей 40 мл воды, 80 мл 10% NaOH и 80 мл воды. Полученную суспензию фильтровали. Фильтрат сушили над суль 004650 42 фатом натрия и концентрировали под вакуумом с получением 98,4 г (выход 95%) диола в виде масла. Данные протонного ЯМР, углеродного ЯМР и масс-спектроскопии подтвердили идентичность полученного продукта. Этап 2. Циклический дибутилсульфит. Раствор дибутилдиола этапа 1 (103 г,0,5478 моль) в безводном метиленхлориде (500 мл) и триэтиламине (221 г, 4 экв., 2,19 моль) перемешивали при температуре 0 С в атмосфере азота. К полученной смеси добавляли капля за каплей тионилхлорид (97,78 г, 0,82 моль). Через 5 мин полученный раствор приобрел желтый цвет; после завершения добавления, т.е. приблизительно через полчаса, упомянутый раствор окрасился в черный цвет. Реакция завершилась в течение 3 ч (данные газовой хроматографии подтвердили отсутствие исходного материала). Полученную смесь дважды промывали смесью воды со льдом и дважды рассолом. Полученную органическую фазу сушили над сульфатом магния и концентрировали под вакуумом с получением 128 г (выход 100%) циклического дибутилсульфата в виде масла черного цвета. Данные ЯМР и масс-спектроскопии подтвердили идентичность полученного продукта. Этап 3. Циклический дибутилсульфат. К раствору циклического дибутилсульфита этапа 2 (127,5 г, 0,54 моль) в 600 мл ацетонитрила и 500 мл воды, охлажденного в ванне со льдом в атмосфере азота, добавляли рутения(III) хлорид (1 г) и натрия периодат (233 г, 1,08 моль). Полученную реакционную смесь перемешивали в течение ночи; раствор стал черным. Данные газовой хроматографии подтвердили отсутствие исходного материала. Полученную смесь экстрагировали 1 раз с помощью 300 мл эфира и трижды с помощью рассола. Полученную органическую фазу сушили над сульфатом магния и пропускали через слой целита. Полученный фильтрат концентрировали под вакуумом и получили 133 г (выход 97,8%) циклического дибутилсульфата в виде масла. Данные протонного ЯМР, углеродного ЯМР и массспектроскопии подтвердили идентичность полученного продукта. 60% дисперсию гидрида натрия (0,27 г,6,68 ммоль) в масле промывали гексаном. Гексан сливали, к промытому гидриду натрия добавляли 20 мл метоксиэтилового эфира и охлаждали в ванне со льдом. В течение 15 мин добавляли капля за каплей смесь дифенилметантиофенола (1,55 г, 6,68 ммоль) в 10 мл метоксиэтилового эфира. После этого добавляли смесь циклического дибутилсульфата этапа 3 (2,17 г,8,66 ммоль) в 10 мл метоксиэтилового эфира. Полученную смесь перемешивали в течение 30 мин при температуре 0 С и в течение 1 ч при комнатной температуре в атмосфере азота. Данные газовой хроматографии подтвердили отсутствие тиола. Упомянутый растворитель выпаривали и дважды промывали водой и эфиром. Полученный водной слой отделяли и добавляли 20 мл 10% NaOH. Полученную водную смесь кипятили в течение 30 мин, охлаждали, подкисляли 6N НСl и кипятили в течение 10 мин. Полученную смесь охлаждали и экстрагировали с помощью эфира. Полученный органический слой последовательно промывали водой и рассолом, сушили над сульфатом магния и концентрировали под вакуумом с получением 2,47 г(выход 92,5%) гексанола в виде масла. Данные протонного ЯМР, С 13-ЯМР и масс-спектроскопии подтвердили идентичность полученного продукта. Этап 5. 2-[(2-4'-Фторбензил-4-метилфенилтио)метил]-2-бутилгексанол. К раствору гексанола этапа 4 (2 г, 4,9 ммоль) в 40 мл метиленхлорида, охлажденному в ванне со льдом в атмосфере азота, добавляли пиридиния хлорхромат (2,18 г, 9,9 ммоль). Полученную реакционную смесь перемешивали в течение 3 ч и фильтровали через слой силикагеля. Полученный фильтрат концентрировали под вакуумом с получением 1,39 г (выход 70%) гексанола в виде масла. Данные протонного ЯМР, углеродного ЯМР и масс-спектроскопии подтвердили идентичность полученного продукта. К раствору гексанола этапа 5 (0,44 г, 1,1 ммоль) в 20 мл метиленхлорида, охлажденному в ванне со льдом в атмосфере азота, добавляли 70% метахлорпербензойную кислоту (0,54 г, 2,2 ммоль). Полученную реакционную смесь перемешивали в течение 18 ч и фильтровали. Полученный фильтрат последовательно промывали 10% NaOH (3X), водой и рассолом, сушили над сульфатом магния и концентрировали под вакуумом с получением 0,42 г (выход 90%) гексанола в виде масла. Данные протонного ЯМР,углеродного ЯМР и масс-спектроскопии подтвердили идентичность полученного продукта. Этап 7. цис-3,3-Дибутил-7-метил-5-(4'-фторфенил)-2,3,4,5-тетрагидробензотиепин-1,1-диоксид. Смесь гексанола этапа 6 (0,37 г, 0,85 ммоль) в 30 мл безводного тетрагидрофурана перемешивали в ванне со льдом при температуре приблизительно 0 С. После этого добавляли третбутоксид калия (102 мг, 0,85 ммоль). Через 3 ч средствами тонкослойной хроматографии было подтверждено присутствие продукта и небольшого количества исходного материала. Неочищенную реакционную смесь подкисляли 10% НСl, экстрагировали эфиром, последовательно промывали водой и рассолом, сушили надMgSO4 и концентрировали под вакуумом, полученный концентрат очищали средствами высокоэффективного жидкостного хроматографирования (10% смесь EtOAc-гексана). Первая полученная фракция представляла собой 0,1 г исходного материала в виде масла. Вторая фракция представляла собой 0,27 г (75% выход) необходимого бензотиепина в виде твердого вещества белого цвета. Данные протонного ЯМР,углеродного ЯМР и масс-спектроскопии подтвердили идентичность полученного продукта(1,75 г, 1,05 экв.). Полученный раствор приобрел красный цвет. Реакционную смесь нагревали при температуре 80 С в течение ночи. Раствор охлаждали до температуры 0 С, добавляли циклический дибутилсульфат (9,9 г; полученный в соответствии с описанием, приведенным на этапе 3 примеров к схеме XI) в 10 мл диметилформамида и перемешивали при комнатной температуре в течение ночи. Растворитель выпаривали и последовательно промывали водой и эфиром (трижды). Полученный водный слой отделяли, добавляли 40 мл концентрированной серной кислоты, и полученную реакционную смесь кипятили в течение ночи. Смесь охлаждали и экстрагировали с помощью этилацетата. Полученный органический слой последовательно промывали водой и рассолом, сушили над сульфатом магния и концентрировали под вакуумом, полученный продукт кипятили с 3 МNaOH в течение 1 ч. Полученную смесь охлаждали и экстрагировали с помощью этилацетата. Полученный органический слой последовательно промывали водой и рассолом, сушили над сульфатом магния и концентрировали под вакуумом. Полученный концентрат растворяли в метиленхлориде, фильтровали через слой силикагеля, элюировали 20% смесью этилацетата и гексана и концентрировали под вакуумом с получением 11,9 г (выход 74%) гексанола в виде масла. Данные протонного ЯМР, С 13-ЯМР и масс-спектроскопии подтвердили идентичность полученного продукта. Этап 2. 2-[(2-4'-Метоксибензил-4-нитрофенилтио)метил]-2-бутилгексанол. К раствору гексанола этапа 1 (6 г, 13 ммоль) в 50 мл метиленхлорида, охлажденному в ванне со льдом в атмосфере азота, добавляли 70% метахлорпербензойную кислоту (8,261 г, 33 ммоль). Полученную реакционную смесь перемешивали в течение 18 ч при комнатной температуре и фильтровали. Полученный фильтрат последовательно промывали 10% NaOH (3 х),водой и рассолом, сушили над сульфатом магния и концентрировали под вакуумом. Полу 004650 46 ченный концентрат растворяли в метиленхлориде, фильтровали через слой силикагеля,элюировали 20% смесью этилацетата и гексана и концентрировали под вакуумом с получением 5 г (выход 77,7%) гексанола в виде твердого вещества белого цвета, температура плавления 58-60 С. Данные протонного ЯМР, С 13-ЯМР и масс-спектроскопии подтвердили идентичность полученного продукта. Пример 1398. Этап 1. Получение соединения 2. К раствору 6,0 г диальдегида дибутил 4 фторбензола примера 1395 (14,3 ммоль) в 72 мл толуола и 54 мл этанола добавляли 4,7 г 3-нитробензолбороновой кислоты (28,6 ммоль), 0,8 г тетракис(трифенилфосфин)палладия(0) (0,7 ммоль) и 45 мл 2 М раствора карбоната натрия в воде. Эту гетерогенную смесь нагревали в колбе с обратным холодильником при температуре перегонки в течение 3 ч, затем охлаждали до температуры окружающей среды и распределяли между этилацетатом и водой. Полученный органический слой сушили над MgSО 4 и концентрировали in vacuo. В результате очистки посредством гель-хроматографирования (WatersPrep-2000) с использованием смеси этилацетата/ гексанов (25/75) получали 4,8 г (73%) целевого соединения в виде твердого вещества желтого цвета. 1 Н ЯМР (СDСl3)0,88 (t, J=7,45 Гц, 6 Н),0,99-1,38 (m, 8H), 1,62-1,75 (m, 2H), 1,85-2,00 Раствор 4,8 г (10,4 ммоль) соединения 2 в 500 мл тетрагидрофурана охлаждали до температуры 0 С в ванне со льдом. Медленно добавляли 20 мл 1 М раствора t-бутоксида калия, поддерживая температуру 5 С. Перемешивание продолжали в течение 30 мин, после чего реакцию быстро прекращали посредством добавления 100 мл холодного насыщенного раствора хлорида аммония. Полученную смесь распределяли между этилацетатом и водой; полученный органический слой промывали рассолом, затем сушили (MgSO4) и концентрировали in vacuo. В результате очистки посредством гель-хроматографирования через 100 мл пробку с использованием СН 2 Сl2 в качестве элюента получали 4,3 47 г (90%) соединения 3 в виде пены бледножелтого цвета. 1H ЯМР (CDCl3)0,93 (t, J=7,25 Гц, 6 Н), 1,00-1,55 (m, 8H), 1,59-1,74 (m, 3H),2,15-2,95 (m, 1H), 3,16 (qAB, JAB=15,0 Гц,V=33,2 Гц, 2 Н), 4,17 (d, J=6,0 Гц, 1 Н), 5,67 (s,1H), 6,34 (dd, J=9,6 и 3,0 Гц, 1H), 7,08 (dt, J=8,5 и 2,9 Гц, 1H), 7,64 (t, J=8,1 Гц, 1H), 7,81 (d, J=8,7 Гц, 1H), 8,13 (dd, J=9,9 и 3,6 Гц, 1H), 8,23-8,30 К охлажденному (температура 0 С) раствору 4,3 г (9,3 ммоль) соединения 3 в 30 мл тетрагидрофурана, находившемуся в реакционном сосуде из нержавеющей стали, добавляли 8,2 г диметиламина (182 ммоль). Упомянутый реакционный сосуд герметизировали и нагревали при температуре 110 С в течение 16 ч. Упомянутый реакционный сосуд охлаждали до температуры окружающей среды и содержимое концентрировали in vacuo. В результате очистки посредством гель-хроматографирования (WatersPrep-2000) с использованием градиентной смеси этилацетата/гексанов (10-40% этилацетата) получали 4,0 г (88%) соединения 4 в виде твердого вещества желтого цвета. 1 Н ЯМР (СDСl3)0,800,95 (m, 6 Н), 0,96-1,53 (m, 8H), 1,60-1,69 (m,3 Н), 2,11-2,28 (m, 1H), 2,79 (s, 6H), 3,09 (qАВ,JAB=15,0 Гц, V=45,6 Гц, 2 Н), 4,90 (d, J=9,0 Гц,1H), 5,65 (s, 1H), 5,75 (d, J=2,1 Гц, 1H), 6,52 (dd,J=9,6 и 2,7 Гц, 1H), 7,59 (t, J=8,4 Гц, 1H), 7,85 (d,J=7,80 Гц, 1H), 7,89 (d, J=9,0 Гц, 1H), 8,20 (dd,J=8,4 и 1,2 Гц, 1H), 8,43 (s, 1H). MS (FABH+) m/e К суспензии 1,0 г (2,1 ммоль) соединения 4 в 100 мл этанола в реакторе Парра из нержавеющей стали добавляли 1 г 10% палладия на углероде. Упомянутый реакционный сосуд герметизировали, дважды продували Н 2, после чего заполняли Н 2 под давлением (100 фунтов/дюйм 2(7,031 кг/см 2 и нагревали до температуры 45 С в течение 6 ч. Упомянутый реакционный сосуд охлаждали до температуры окружающей среды и содержимое фильтровали для удаления ката 004650 48 лизатора. Полученный фильтрат концентрировали in vacuo с получением 0,9 г (96%) соединения 5. 1H ЯМР (CDCl3)0,80-0,98 (m, 6H), 1,001,52 (m, 10 Н), 1,52-1,69 (m, 1H), 2,15-2,29 (m,1H), 2,83 (s, 6H), 3,07 (qAB, JAB=15,1 Гц,V=44,2 Гц, 2 Н), 3,70 (s, 2 Н), 4,14 (s, 1H), 5,43 (s,1H), 6,09 (d, J=2,4 Гц, 1H), 6,52 (dd, J=12,2 и 2,6 Гц, 1H), 6,65 (dd, J=7,8 и 1,8 Гц, 1H), 6,83 (s,1H), 6,93 (d, J=7,50 Гц, 1H), 7,19 (t, J=7,6 Гц,1H), 7,89 (d, J=8,9 Гц, 1 Н). MS (FABH+) m/e (относительная интенсивность) 459,7 (100). HRMS вычислено для М+Н 459,2681. Установлено: 459,2670. Этап 6. Получение соединения 6. К раствору 914 мг (2,0 ммоль) соединения 5 в 50 мл тетрагидрофурана добавляли 800 мг(4,0 ммоль) 5-бромвалероилхлорида. После этого добавляли 4 г (39,6 ммоль) трифторуксусной кислоты. Полученную реакционную смесь перемешивали в течение 10 мин, затем распределяли между этилацетатом и рассолом. Полученный органический слой сушили (MgSO4) и концентрировали in vacuo. В результате очистки посредством гель-хроматографирования через 70 мл колонку (жидкостная хроматография среднего давления) с использованием градиентной смеси этилацетата (20-50%) в гексане в качестве элюента получали 0,9 г (73%) соединения 6 в виде масла бледно-желтого цвета. 1 Н ЯМР (СDСl3)0,84-0,95 (m, 6 Н), 1,02-1,53 (m,10 Н), 1,53-1,68 (m, 1H), 1,80-2,00 (m, 4H), 2,122,26 (m, 4H), 2,38 (t, J=6,9 Гц, 2 Н), 2,80 (s, 6H),3,07 (qAB, JАВ=15,6 Гц, V=40,4 Гц, 2 Н), 3,43 (t,J=6,9 Гц, 2 Н), 4,10 (s, 1H), 5,51 (s, 1H), 5,95 (d,J=2,4 Гц, 1H), 6,51 (dd, J=9,3 и 2,7 Гц, 1H), 7,28 К раствору 0,9 г (1,45 ммоль) соединения 6 в 25 мл ацетонитрила добавляли 18 г (178 ммоль) триэтаноламина. Нагревали при температуре 55 С в течение 16 ч. Полученную реакционную смесь охлаждали до температуры окружающей среды и концентрировали in vacuo. В результате очистки посредством гель-хроматографирования с обращенной фазой (Waters Delta Prep 3000) с использованием градиентной смеси ацетонитрила/воды, содержащей 0,05% трифторуксусной кислоты (20-65% ацетонитрила), получали 0,8 г (73%) соединения 7 в виде пены белого цвета. 1 Н ЯМР (CDCl3)0,80-0,96 (m, 6H),0,99-1,54 (m, 19H), 1,59-1,84 (m, 3 Н), 2,09-2,24 В инертной атмосфере отвесить 68,3 г пентахлорида фосфора (0,328 моль Aldrich 15, 7775) в 2-горлую 500 мл круглодонную колбу. Установить на колбу переходный ниппель для подвода N2 и воздухонепроницаемое уплотнение. Извлечь из инертной атмосферы и приступить к продувке N2. С помощью шприца добавить к PCl5 50 мл безводного хлорбензола (Aldrich 28, 451-3) и начать перемешивание с помощью магнитной мешалки. Отвесить 60 г 2-хлор-5-нитробензойной кислоты (0,298 моль Aldrich 12, 511-3). Медленно добавить к раствору хлорбензола с одновременной продувкой N2. Перемешивать при комнатной температуре в течение ночи. После перемешивания при комнатной температуре в течение 20 ч поместить на масляную ванну и нагревать при температуре 50 С в течение 1 ч. Удалить хлорбензол под высоким вакуумом. Промыть остаток безводным гексаном. Высушить с помощью хлорангидрида, масса=61,95 г. Хранить в инертной и сухой атмосфере. В инертной атмосфере растворить хлорангидрид 105 мл безводного анизола (0,97 моль,Aldrich 29, 629-5). Поместить раствор в 2 горлую 500 мл круглодонную колбу. Отвесить 45,1 г хлорида алюминия (0,34 моль, Aldrich 29, 471-3) и поместить в воронку для добавления сыпучих веществ. Установить на реакционной колбе воронку и переходный ниппель для подвода N2. Извлечь из инертной атмосферы. Охладить реакционный раствор в ледяной бане и приступить к продувке N2. К охлажденному раствору медленно добавить АlСl3. После завершения добавления выдержать полученный раствор до нагревания до комнатной температуры. Перемешивать в течение ночи. Быстро остановить реакцию, поместив реакционную смесь в холодный раствор 300 мл 1NHCl и лед. Перемешивать в течение 15 мин. Дважды экстрагировать с помощью эфира. Смешать органические слои и дважды экстрагировать с помощью 2% NaOH, затем дважды с помощью деионизированной Н 2 О. Высушить с помощью MgSO4, профильтровать и высушить на роторном испарителе до сухости. Удалить анизол под высоким вакуумом. Кристаллизовать продукт из 90% этанола-10% этилацетата. Растворить 38,10 г (0,131 моль) бензофенона этапа 1 в 250 мл безводного метиленхлорида. Перенести в 3 л колбу, снабженную переходным ниппелем для подвода N2, капельной воронкой и пробкой. Перемешать с помощью магнитной мешалки. Охладить раствор на ледяной бане. Получить раствор 39,32 г трифторметансульфоновой кислоты (0,262 моль Aldrich 15,853-4) и 170 мл безводного метиленхлорида. Поместить в капельную воронку и добавлять капля за каплей к охлажденному раствору в атмосфере N2. После завершения добавления, перемешивать в течение 5 мин. Получить раствор 22,85 г триэтилсилана(0,197 моль Aldrich 23, 019-7) и 170 мл безводного метиленхлорида. Поместить в капельную воронку и добавлять капля за каплей к охлажденному раствору в атмосфере N2. После завершения добавления, перемешивать в течение 5 мин. Получить второй раствор 39,32 г трифторметансульфоновой кислоты и 170 мл безводного метиленхлорида. Поместить в капельную воронку и добавлять капля за каплей к охлажденному раствору в атмосфере N2. После завершения добавления перемешивать в течение 5 мин. Получить второй раствор 22,85 г триэтилсилана и 170 мл безводного метиленхлорида. Поместить в капельную воронку и добавлять капля за каплей к охлажденному раствору в атмосфере N2. После завершения всех добавлений выдержать с медленным нагреванием до комнатной температуры в течение ночи. Перемешивать в атмосфере N2 в течение ночи. Получить 1300 мл насыщенного раствораNaHCO3 в 4 л химическом стакане. Охладить на ледяной бане. Медленно добавить реакционную смесь с энергичным перемешиванием. Перемешивать при температуре охлаждения в течение 30 мин. Вылить в делительную воронку и произвести разделение. Удалить органический слой и дважды экстрагировать водный слой с помощью метиленхлорида. Высушить органические слои с помощью MgSO4. Кристаллизовать из этанола. Высушить под вакуумом. Сухая масса=28,8 г. Подтвердить идентичность продукта путем осуществления ЯМР- и масс-спектроскопирования (m/z=278). Растворить 10,12 г (0,036 моль) продукта 2 в 200 мл безводного диметилсульфоксида. Внести в 500 мл круглодонную колбу с магнитной мешалкой. Установить на колбу водяной конденсатор, переходный ниппель для подвода N2 и пробку. Добавить 1,84 г Li2S (0,040 моль Aldrich 21, 324-1). Поместить колбу на масляную ванну и нагревать при температуре 75 С в атмосфереN2 в течение ночи, затем охладить до комнатной температуры. Отвесить 10,59 г дибутилмезилата (0,040 моль). Растворить безводным диметилсульфоксидом и добавить к реакционному раствору. Тщательно продуть N2, нагревать в течение ночи при температуре 80 С. Охладить до комнатной температуры. Приготовить 500 мл 5% уксусной кислоты в 2 л химическом стакане. Медленно, с перемешиванием, добавить реакционную смесь. Перемешивать в течение 30 мин. Трижды экстрагировать с помощью эфира. Объединить органические слои и экстрагировать водой и насыщеннымNaCl. Высушить органический слой с помощьюMgSO4, профильтровать и высушить на роторном испарителе до сухости. Высушить масло под вакуумом. Получить чистый продукт посредством хроматографирования на колонках с использованием 95% гексана и 5% этилацетата в качестве подвижной фазы. Сухая масса=7,8 г. Выполнить ЯМР- и масс-спектроскопирование Растворить 9,33 г (0,021 моль) продукта 3 в 120 мл безводного метиленхлорида. Перенести в 250 мл круглодонную колбу с магнитной мешалкой. Установить на колбу переходный ниппель для подвода N2 и пробку. Охладить раствор на ледяной бане с продувкой N2. Медленно добавить 11,54 г 3-хлорпербензойной кислоты(0,0435 моль, Fluka 25800, приблизительно 65%). После завершения добавления нагреть до ком 004650 52 натной температуры и контролировать прохождение реакции посредством тонкослойного хроматографирования. Результатом непродолжительной реакции является получение сульфоксидного промежуточного продукта, однако,продолжительность реакции до получения сульфона составляет 8 ч. Охладить раствор в морозильной камере в течение ночи. Отфильтровать твердое вещество, экстрагировать фильтрат посредством 10% К 2 СО 3. Дважды экстрагировать водный слой с помощью метиленхлорида. Объединить органические слои и высушить с помощью MgSO4. Профильтровать и высушить на роторном испарителе до сухости. Получить чистый продукт посредством кристаллизации из этанола либо выделить посредством хроматографирования на колонках. Выполнить ЯМР- и масс-спектроскопирование (m/z=476). Этап 5. Реакцию осуществляют в 300 мл миниреакторе Парра из нержавеющей стали. Внести 9,68 г (0,0204 моль) продукта 4 в реактор. Добавить 160 мл этанола. Следующие два соединения, по соображениям безопасности, добавляют в защитном мешке с манипуляционными перчатками в атмосфере N2. Добавить в защитном мешке с манипуляционными перчатками 15,3 мл формальдегида (0,204 моль, Aldrich 25, 2549, около 37 маc.% в воде) и 1,45 г 10% Pd на углероде (Aldrich 20, 569-9). Реактор герметизировать перед выемкой из защитного мешка. Реактор трижды продуть H2. Нагреть до температуры 55 С в атмосфере Н 2. Поддерживать в реакторе избыточное давление 200 фунтов/дюйм 2(14,062 кг/см 2), температуру 55 С и частоту перемешивания 250 об./мин. Поддерживать указанные условия в реакторе в течение ночи. Охладить реактор и выпустить Н 2 в атмосферу. Продуть реактор N2. Проверить прохождение реакции посредством тонкослойного хроматографирования. Реакционная смесь представляет собой смесь необходимого и промежуточного продукта. Профильтровать реакционную смесь через слой целита с тщательной промывкой эфиром. Высушить на роторном испарителе и повторно растворить в эфире. Экстрагировать с помощью воды. Высушить органический продукт с помощью MgSO4, профильтровать и высушить на роторном испарителе до сухости. Высушить под вакуумом. 53 Вновь загрузить в реактор такое же количество исходного материала, загерметизировать и осуществить реакцию в таких же условиях в течение ночи. После завершения второй процедуры весь материал оказывается превращенным в необходимый продукт. Охладить и выпустить Н 2 в атмосферу. Продуть N2. Профильтровать через слой целита с тщательной промывкой эфиром. Высушить на роторном испарителе до сухости. Растворить в эфире и экстрагировать с помощью воды. Высушить органический слой с помощью MgSO4, профильтровать и высушить на роторном испарителе до сухости. Высушить под вакуумом. Выполнить ЯМР- и массспектроскопирование (m/z=474). Этап 6. Растворить 8,97 г (0,0189 моль) продукта 5 в 135 мл безводного тетрагидрофурана. Перенести в 250 мл круглодонную колбу с магнитной мешалкой. Установить на колбу переходный ниппель для подвода N2 и пробку. Охладить раствор в ванне со смесью льда и соли с продувкой N2. Медленно добавить 2,55 г tбутоксида калия (0,227 моль, Aldrich 15, 667-1). После завершения добавления продолжать перемешивание при температуре около 10 С с контролированием прохождения реакции посредством тонкослойного хроматографирования. После завершения реакции быстро охладить реакционную смесь добавлением 135 мл холодной 10% НСl с перемешиванием в течение 10 мин. Трижды экстрагировать с помощью эфира. Высушить органический слой с помощью MgSO4, профильтровать и высушить на роторном испарителе до сухости. Кристаллизовать из эфира. Выполнить ЯМР- и массспектроскопирование (m/z=474). Этап 7. Растворить 4,67 г (0,01 моль) продукта 6 в 100 мл безводного хлороформа. Перенести в 250 мл круглодонную колбу с магнитной мешалкой. Установить на колбу переходный ниппель для подвода N2 и воздухонепроницаемое уплотне 004650 54 ние. Охладить раствор в ванне со смесью твердой углекислоты/ацетона с продувкой N2. С помощью шприца медленно добавить 2,84 мл трибромида бора (0,03 моль, Aldrich 20, 220-7). Перемешивать при температуре охлаждения в течение 15 мин после завершения добавления,затем выдержать с нагреванием до комнатной температуры. Контролировать прохождение реакции посредством тонкослойного хроматографирования. Реакция завершается, как правило, в течение 3 ч. Охладить раствор на ледяной бане. Быстро прекратить реакцию посредством добавления 100 мл холодного 10% раствора К 2 СО 3 с быстрым перемешиванием. Перемешивать в течение 10 мин, затем перенести в делительную воронку и произвести разделение. Удалить водный слой. Экстрагировать органический слой 1 раз с помощью 10% НСl, 1 раз с помощью Н 2 О и 1 раз с помощью насыщенного раствора NaCl. Высушить органический слой с помощью MgSO4,профильтровать и высушить на роторном испарителе до сухости. Кристаллизовать продукт из эфира. Выполнить ЯМР- и масс-спектроскопирование (m/z=460). Этап 8. Отвесить 0,38 г NaH (9,57 ммоль, Aldrich 19, 923-0, 60% дисперсия в минеральном масле) в 250 мл круглодонную колбу с магнитной мешалкой. Установить на колбу переходный ниппель для подвода N2 и пробку. Охладить NaH на ледяной бане и начать продувку N2. Растворить 4,0 г (8,7 ммоль) продукта 7 в 60 мл безводного диметилформамида. Добавить к холодному NaH. Перемешивать при температуре охлаждения в течение 30 мин. Добавить 1,33 г К 2 СО 3 (9,57 ммоль, Fischer P-208). Растворить 16,1 г 1,2-бис-(2-йодоэтокси) этана (43,5 ммоль Aldrich 33, 343-3) в 60 мл безводного диметилформамида. Добавить к холодной реакционной смеси. Нагреть до комнатной температуры, затем нагревать до температуры 40 С в течение ночи в атмосфере N2. Очистить посредством разбавления эфиром и последовательного экстрагирования 5%NaOH, H2O и насыщенного раствора NaCl. Высушить органический слой с помощью MgSO4,профильтровать и высушить. Получить чистый продукт посредством хроматографирования на колонках с использованием смеси 75% гексана 25% этилацетата в качестве подвижной фазы. Растворить 1,0 г (1,43 ммоль) продукта 8 в 10 мл безводного ацетонитрила. Перенести в реактор высокого давления Фишера-Портера(28,6 ммоль, Aldrich 23, 962-3), растворенного в 10 мл безводного ацетонитрила. Тщательно продуть N2, затем закрыть систему. Нагревать при температуре 45 С. Контролировать прохождение реакции посредством тонкослойного хроматографирования. Реакция завершается,как правило, в течение 48 ч. Осуществить очистку посредством удаления ацетонитрила под вакуумом. Повторно растворить в безводном хлороформе и осадить соль четвертичного аммония эфиром. Повторить несколько раз. Высушить с получением кристаллического продукта. Выполнить ЯМР- и массспектроскопирование (m/z=675). Пример 1399. Этап 1. Получение соединения 1. К раствору 144 г КОН (2560 ммоль) в 1,1 л диметилсульфоксида медленно через капельную воронку добавили 120 г 2-бромбензилового спирта (641 ммоль). Затем через капельную воронку добавили 182 г метилйодида (80 мл, 1282 ммоль). Перемешали при температуре окружающей среды в течение 15 мин. Вылили полученную реакционную смесь в 1,0 л воды и трижды экстрагировали с помощью этилацетата. Полученный органический слой сушили надMgSO4 и концентрировали in vacuo. Осуществили очистку посредством гель-хроматографирования через 200 мл пробку с использованием гексанов (100%) в качестве элюента с получением 103,2 г (80%) соединения 1 в виде прозрачной бесцветной жидкости. 1H ЯМР (СDСl3)3,39 (s, 3 Н), 4,42 (s, 2H), 7,18-7,27 (m, 2 Н), 7,12 56 К охлажденному (температура -78 С) раствору 95 г (472 ммоль) соединения 1 в 1,5 л тетрагидрофурана добавляли 240 мл 2,5 М nбутиллития (576 ммоль). Полученную смесь перемешивали в течение 1 ч, после этого к ней добавляли 180 г йодида цинка (566 ммоль), растворенного в 500 мл тетрагидрофурана. Полученную смесь перемешивали в течение 30 мин,выдерживали с нагреванием до температуры 5 С, охлаждали до температуры -10 С, после чего к ней добавляли 6 г Рd(РРh3)4 (5,2 ммоль) и 125 г 2,5-дифторбензоилхлорида (708 ммоль). Полученную смесь перемешивали при температуре окружающей среды в течение 18 ч, после чего охлаждали до температуры 10 С, быстро останавливали реакцию посредством смешивания с холодной водой, распределяли между этилацетатом и водой и промывали органический слой 1N HCl и 1N NaOH. Полученный органический слой сушили над MgSO4 и концентрировали in vacuo. В результате очистки посредством гель-хроматографирования (WatersPrep-500) с использованием 5% смеси ацетата/гексанов в качестве элюента получали 53,6 г(43%) соединения 2 в виде масла оранжевого цвета. 1H ЯМР (CDCl3)3,40 (s, 3H), 4,51 (s,2H), 7,12-7,26 (m, 3H), 7,47 (t, J=7,50, 1H), 7,57 Раствор 53 г (202,3 ммоль) соединения 2 и 11,2 г Li2S (242,8 ммоль) в 250 мл диметилформамида нагревали до температуры 100 С в течение 18 ч. Полученную реакционную смесь охлаждали (температура 0 С) и добавляли 60,7 гX' (циклическое сульфатное соединение примера 1397) (242,8 ммоль) в 50 мл диметилформамида. Перемешивали при температуре окружающей среды в течение 18 ч, затем конденсировали in vacuo. К органическому остатку добавляли 1 л воды и дважды экстрагировали с помощью диэтилового эфира. Водный слой подкисляли (рН 1) и нагревали в колбе с обратным холодильником при температуре перегонки в течение 2 дней. Охлаждали до температуры окружающей среды и экстрагировали с помощью метиленхлорида, высушивали органический слой над MgSO4 и конденсировали invacuo. В результате очистки посредством гельхроматографирования (Waters Prep-500) с использованием 10% смеси ацетата/гексанов в качестве элюента получали 42,9 г (48%) соединения 3 в виде масла желтого цвета. 1H ЯМР К охлажденному (температура около 40 С) раствору 42,9 г (96,2 ммоль) соединения 3 в 200 мл метиленхлорида добавляли 21,6 г трифторметансульфоновой кислоты (12,8 мл, 144 ммоль), с последующим добавлением 22,4 г триэтилсилана (30,7 мл, 192,4 ммоль). Перемешивали при температуре около 20 С в течение 2 ч, быстро охлаждали посредством смешивания с холодной водой и нагревали до температуры окружающей среды. Распределяли между метиленхлоридом и водой, сушили полученный органический слой над MgSO4 и конденсировалиin vacuo. В результате очистки посредством гель-хроматографирования (Waters Prep-500) с использованием 10% смеси этилацетата/гексанов в качестве элюента получали 24,2 г (60%) соединения 4 в виде масла. 1H ЯМР (CDCl3)0,89 (t,J=7,05 Гц, 6 Н), 1,17-1,40 (m, 12H), 1,46 (t, J=5,84 Гц, 1 Н), 2,81 (s, 2H), 3,38 (s, 3H), 3,43 (d, J=5,23 Гц,2 Н), 4,16 (s, 2H), 4,42 (s, 2H), 6,80 (d, J=9,67 Гц,1H), 6,90 (t, J=8,46 Гц, 1 Н), 7,09 (d, J=7,45 Гц, 1H),7,15=7,21 (m, 2H), 7,25-7,32 (m, 2H), 7,42 (m, 1H). Этап 5. Получение соединения 5.(55,8 ммоль) соединения 4 в 100 мл диметилсульфоксида добавляли 31,2 г комплекса триоксида серы-пиридина (195 ммоль). Перемешивали при температуре окружающей среды в течение 30 мин. Выливали в холодную воду и трижды экстрагировали с помощью этилацетата. Полученный органический слой промывали 5% НСl (300 мл), затем рассолом (300 мл), сушили над MgSO4 и конденсировали in vacuo с получением 23,1 г (96%) соединения 5 в виде масла светло-коричневого цвета. 1H ЯМР (СDСl3)0,87 58 К охлажденному (температура 0 С) раствору 23,1 г (53,6 ммоль) соединения 5 в 200 мл метиленхлорида добавляли 28,6 г m-хлорпероксибензойной кислоты (112,6 ммоль). Перемешивали при температуре окружающей среды в течение 24 ч. Быстро охлаждали посредством смешивания со 100 мл холодного 10% Na2SO3, распределяли между водой и метиленхлоридом. Полученный органический слой сушили над MgSO4 и конденсировали in vacuo с получением 24,5 г (98%) соединения 6 в виде масла светло-желтого цвета. 1 К раствору 24,5 г (52,9 ммоль) соединения 6 в 20 мл тетрагидрофурана, находившегося в реакционном сосуде из нержавеющей стали,добавляли 100 мл 2,0 М раствора диметиламина и 20 мл неразбавленного диметиламина. Реакционный сосуд герметизировали и нагревали до температуры 110 С в течение 16 ч. Упомянутый реакционный сосуд охлаждали до температуры окружающей среды и содержимое концентрировали in vacuo. В результате очистки посредством гель-хроматографирования (Waters Prep500) с использованием 15% смеси этилацетата/гексанов получали 21,8 г (84%) соединения 7 в виде прозрачного бесцветного масла. 1H ЯМР Раствор 21,8 г (44,8 ммоль) соединения 7 в 600 мл тетрагидрофурана охлаждали до температуры 0 С. Медленно добавляли 58,2 мл 1 М раствора t-бутоксида калия, поддерживая температуру 5 С. Перемешивали в течение 30 мин,затем реакцию быстро прекращали посредством смешивания с 50 мл холодного насыщенного раствора хлорида аммония. Полученный органический слой распределяли между этилацетатом и водой, сушили над MgSO4 и концентрировали in vacuo. В результате очистки посредством рекристаллизации из приблизительно 10% 59 смеси этилацетата/гексанов получали 15,1 г соединения 8 в виде твердого вещества белого цвета. Маточную жидкость очищали посредством гельхроматографирования (Waters Prep-500) с использованием 30% смеси этилацетата/гексанов в качестве элюента с получением 3,0 г соединения 8 в виде твердого вещества белого цвета. Жидкостная вторично-ионная масс-спектрометрия при бомбардировке ускоренными атомами (MS (FABLi+m/e 494,6. HRMS по методу ионизации под действием электронного удара (EI+) вычислено для М+Н 487,2756. Установлено: 487,2746. Этап 9. Получение соединения 9. Раствор 2,0 г (4,1 ммоль) соединения 8 в 20 мл метиленхлорида охлаждали до температуры 60 С. Добавляли 4,1 мл 1 М раствора трибромида бора. Перемешивали при температуре окружающей среды в течение 30 мин. Реакционную смесь охлаждали до температуры приблизительно 10 С и реакцию быстро останавливали посредством смешивания с 50 мл холодной воды. Полученный органический слой распределяли между метиленхлоридом и водой, сушили над MgSO4 и концентрировали in vacuo. В результате очистки посредством рекристаллизации из 50% смеси этилацетата/метиленхлорида получали 1,95 г (89%) соединения 9 в виде твердого вещества белого цвета. MS методом бомбардировки быстрыми атомами (FABH+) m/е 537. HRMS (FAB) вычислено для М 536,1834. Установлено: 536,1822. Этап 10. Получение соединения 10. Раствор 1,09 г (2,0 ммоль) соединения 9 и 4,9 г (62 ммоль) пиридина в 30 мл ацетонитрила перемешивали при температуре окружающей среды в течение 18 ч. Полученную реакционную смесь концентрировали in vacuo. В результате очистки посредством рекристаллизации из смеси метанола/диэтилового эфира получали 1,19 г(96%) соединения 10 в виде твердого вещества не совсем белого цвета. MS (FAB+) m/e 535,5. Пример 1400. Этап 1. 60 На 12 л 4-горлую круглодонную колбу устанавливали парциальный конденсатор горячего орошения, переходный ниппель для подвода N2,механическую мешалку и капельную воронку. Полученную систему продували N2. Добавляли суспензию гидрида натрия (126,0 г/4,988 моль) в толуоле (2,5 л) и полученную смесь охлаждали до температуры 6 С. Через капельную воронку в течение 2,5 ч добавляли раствор 4-фторфенола(560,5 г/5,000 моль) в толуоле (2,5 л). Полученную реакционную смесь нагревали в колбе с обратным холодильником при температуре перегонки (100 С) в течение 1 ч. Через капельную воронку добавляли раствор 3-метоксибензилхлорида (783,0 г/5,000 моль) в толуоле (750 мл) с поддержанием температуры перегонки. После нагревания в колбе с обратным холодильником при температуре перегонки в течение 15 ч полученную смесь охлаждали до комнатной температуры и выливали в Н 2 О (2,5 л). После перемешивания в течение 20 мин полученные слои разделяли, полученный органический слой экстрагировали с помощью раствора гидроксида калия (720 г) в МеОН (2,5 л). Полученный слой МеОН добавляли к 20% водному раствору гидроксида калия и полученную смесь перемешивали в течение 30 мин. После этого полученную смесь 5 раз промывали толуолом. Полученные толуоловые смывы экстрагировали посредством 20% водного раствора КОН. Все 20% водные растворы КОН смешивали и подкисляли концентрированной НСl. Полученный кислотный раствор трижды экстрагировали этиловым эфиром, сушили (MgSO4), фильтровали и концентрировали in vacuo. Полученный неочищенный продукт очищали посредством дистилляции через трубку с шаровым расширением с получением прозрачного бесцветного масла (449,0 г/39% выход). b.р. (температура кипения): 120130 С/50 мторр Hg. Данные 1H ЯМР и массспектроскопии [(М+Н=233)] подтвердили необходимую структуру. Этап 2. На 12 л 3-горлую круглодонную колбу устанавливали механическую мешалку и переходный ниппель для подвода N2. Полученную систему продували N2. Добавляли 4-фтор-2-(3 метоксибензил)фенол (455,5 г/1,961 моль) и диметилформамид. Полученный раствор охлаждали до температуры 6 С и медленно добавляли гидрид натрия (55,5 г/2,197 моль). После нагревания до комнатной температуры добавляли диметилтиокарбамоилхлорид (242,4 г/1,961 моль).
МПК / Метки
МПК: C07D 337/08, C07F 9/6553, A61K 31/38, C07K 5/06, C07C 323/18
Метки: перемещения, кислот, ингибиторов, активностью, желчных, поглощения, обладающие, кишке, бензотиепины, таурохолата, подвздошной
Код ссылки
<a href="https://eas.patents.su/30-4650-benzotiepiny-obladayushhie-aktivnostyu-ingibitorov-peremeshheniya-zhelchnyh-kislot-v-podvzdoshnojj-kishke-i-pogloshheniya-tauroholata.html" rel="bookmark" title="База патентов Евразийского Союза">Бензотиепины, обладающие активностью ингибиторов перемещения желчных кислот в подвздошной кишке и поглощения таурохолата</a>
Производные желчных кислот

Номер патента: 3392
Опубликовано: 24.04.2003
Автор: undefined
МПК: A61P 1/16, A61K 31/575, C07J 41/00...
Метки: производные, желчных, кислот
Формула / Реферат:
1. Конъюгаты (соединения) желчной кислоты или желчной соли с жирной кислотой общей формулы II W-X-G в которой G представляет собой радикал желчной кислоты или желчной соли, которая, при желании, соединена в положении 24 с подходящей аминокислотой, W представляет собой один или два радикала жирных кислот, имеющих 6-26 атомов углерода, и X представляет собой NH-связь между радикалом указанной желчной кислоты или желчной соли и жирной...
Производные хиноксалин-2,3-диона и их фармацевтически приемлемые соли, фармацевтические композиции, обладающие антагонистической активностью в отношении рецептора глютамата и противосудорожной активностью, способы лечения пациентов, страдающих от удара или заболеваний, при помощи этих соединений.

Номер патента: 762
Опубликовано: 24.04.2000
Авторы: Рафферти Майкл Фрэнсис, Корнберг Брайэн Эдвард, Никам Шам
МПК: C07D 241/44
Метки: этих, фармацевтически, отношении, рецептора, способы, помощи, пациентов, фармацевтические, хиноксалин-2,3-диона, обладающие, глютамата, страдающих, противосудорожной, активностью, приемлемые, композиции, лечения, заболеваний, соединений, удара, производные, соли, антагонистической
Формула / Реферат:
1. Производные хиноксалин-2,3-диона общей формулы I где R - группа - NR4R5, где R4 и R5 независимо друг от друга означают водород, низший алкил, незамещенный или замещенный низшим алкилом, низший циклоалкил, который может содержать 1 или 2 атома кислорода в качестве гетероатома, R1 и R2 независимо друг от друга означают водород или нитрогруппу, R3 - низший алкил, при этом радикалы R3 и R-CH2- могут каждый находиться в положениях 5 или...
Соединения, обладающие антивирусной активностью

Номер патента: 1316
Опубликовано: 26.02.2001
Авторы: Теббе Марк Дж., Шеферд Тимоти А., Юнгхейм Луи Н., Спитцер Вейн А.
МПК: A61P 31/12, A61K 31/4184, C07D 235/04...
Метки: обладающие, соединения, антивирусной, активностью
Формула / Реферат:
1. Соединение формулы I где а равно 0, 1, 2 или 3; каждый R независимо обозначает водород, галоген, циано, амино, галоген (C1-С6) алкил, ди (C1-С4) алкиламино, азидо, C1-С6-алкил, карбамоил, карбамоилокси, карбамоиламино, C1-С4-алкокси, C1-С4-алкилтио, C1-С4-алкилсульфинил, C1-С4-алкилсульфонил, пирролидино, пиперидино или морфолино; R0 обозначает водород, галоген, C1-С4-алкил или C1-С4-алкокси; R1 обозначает галоген, циано-, гидрокси,...
Соединения, обладающие антивирусной активностью

Номер патента: 1442
Опубликовано: 23.04.2001
Авторы: Виктор Франц, Миллер Шон К., Юнгхейм Луи Н., Теббе Марк Дж., Спитцер Вейн А.
МПК: A61P 31/12, C07D 235/30, A61K 31/4184...
Метки: обладающие, соединения, активностью, антивирусной
Формула / Реферат:
1. Соединение формулы I где а равно 0, 1, 2 или 3; каждый R независимо обозначает водород, галоген, циано, амино, галоген(С1-С4)алкил, ди(С1-С4алкиламино, азидо, С1-С6-алкил, карбамоил, карбамоилокси, карбамоиламино, С1-С4-алкокси, С1-С4-алкилтио, С1-С4-алкилсульфинил, С1-С4-алкилсульфонил, пирролидино, пиперидино или морфолино; R0 обозначает водород, галоген, С1-С4-алкил или С1-С4-алкокси; R1 обозначает галоген, циано-, гидрокси, метил,...
Производные камптотецина, обладающие противоопухолевой активностью

Номер патента: 3605
Опубликовано: 26.06.2003
Авторы: Карминати Паоло, Мерлини Лючио, Цунино Франко, Пенко Серджо
МПК: A61K 31/435, C07D 491/22
Метки: производные, обладающие, камптотецина, активностью, противоопухолевой
Формула / Реферат:
1. Соединения формулы I в которой R1 обозначает группу -C(R5)=N-O(n)R4, в которой R4 обозначает нормальный или разветвленный C1-C8алкил, нормальную или разветвленную C1-C8алкенильную группу, C3-C10циклоалкил, (C3-C10)циклоалкилнормальный или разветвленный (C1-C8)алкил, (C6-C14)арил, (C6-C14)арилнормальный или разветвленный (C1-C6)алкил, гетероциклическую группу или гетероциклонормальный или разветвленный-(C1-C8)алкил, где гетероциклическая...
Предыдущий патент: Фармацевтические композиции, содержащие производные 3-аминоазетидина, новые производные и способ их получения
Следующий патент: Способ получения изотретиноина
Случайный патент: Упаковка тонких слоев для остекления