Бициклические пиримидиновые соединения
Номер патента: 25106
Опубликовано: 30.11.2016
Авторы: Норман Брайан Херст, Джонс Спенсер Брайан, Бочамп Томас Джеймс, Пфайфер Ланс Аллен, Дао Ен




























Формула / Реферат
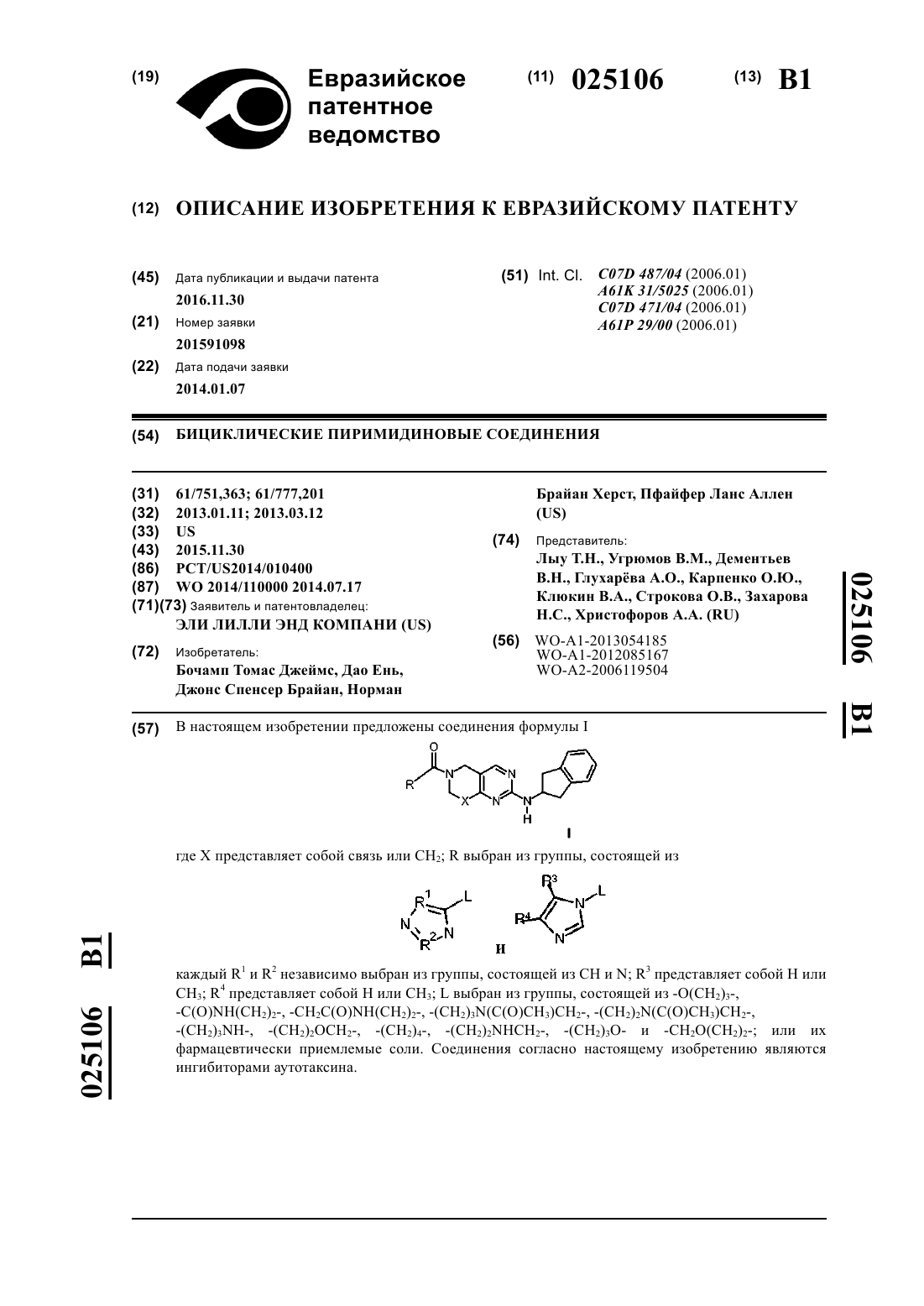
1. Соединение формулы I

где X представляет собой связь или СН2;
R выбран из группы, состоящей из

каждый R1 и R2 независимо выбран из группы, состоящей из СН и N;
R3 представляет собой Н или СН3;
R4 представляет собой Н или СН3;
L выбран из группы, состоящей из -О(СН2)3-, -C(O)NH(CH2)2-, -CH2C(O)NH(CH2)2-, -(CH2)3N(C(O)CH3)CH2-, -(CH2)2N(C(O)CH3)CH2-, -(CH2)3NH-, (CH2)2OCH2-, -(CH2)4-, -(CH2)2NHCH2-, -(CH2)3O- и -СН2О(СН2)2-;
или его фармацевтически приемлемая соль.
2. Соединение или соль по п.1, отличающееся тем, что R представляет собой

3. Соединение или соль по п.2, отличающееся тем, что R1 представляет собой СН.
4. Соединение или соль по любому из пп.2 или 3, отличающееся тем, что R2 представляет собой N.
5. Соединение или соль по любому из пп.1-4, отличающееся тем, что L выбран из группы, состоящей из -(СН2)4-, -О(СН2)3-,-(СН2)2ОСН2-, -(СН2)3О- и -СН2О(СН2)2-.
6. Соединение или соль по п.5, отличающееся тем, что L представляет собой -(СН2)2ОСН2-.
7. Соединение или соль по п.5, отличающееся тем, что L представляет собой -О(СН2)3-.
8. Соединение или соль по любому из пп.1-3, отличающееся тем, что L выбран из группы, состоящей из -(CH2)2N(C(O)CH3)CH2-, -(CH2)3N(C(O)CH3)CH2- и -CH2C(O)NH(CH2)2-.
9. Соединение или соль по любому из пп.1-8, отличающееся тем, что X представляет собой связь.
10. Соединение или соль по любому из пп.1-8, отличающееся тем, что X представляет собой СН2.
11. Соединение или соль по п.1, представляющее собой 1-[2-(2,3-дигидро-1H-инден-2-иламино)-7,8-дигидропиридо[4,3-d]пиримидин-6(5H)-ил]-2-[2-(1H-1,2,3-триазол-4-ил)этокси]этанон

12. Соединение или соль по п.1, представляющее собой 1-[2-(2,3-дигидро-1H-инден-2-иламино)-5,7-дигидро-6H-пирроло[3,4-d]пиримидин-6-ил]-2-[2-(1H-1,2,3-триазол-4-ил)этокси]этанон

13. Фармацевтическая композиция, содержащая соединение или его фармацевтически приемлемую соль по любому из пп.1-12 в комбинации с одним или более фармацевтически приемлемыми носителями, разбавителями или вспомогательными веществами.
14. Применение соединения или его фармацевтически приемлемой соли по любому из пп.1-12 в терапии.
15. Применение соединения или его фармацевтически приемлемой соли по любому из пп.1-12 для лечения боли.
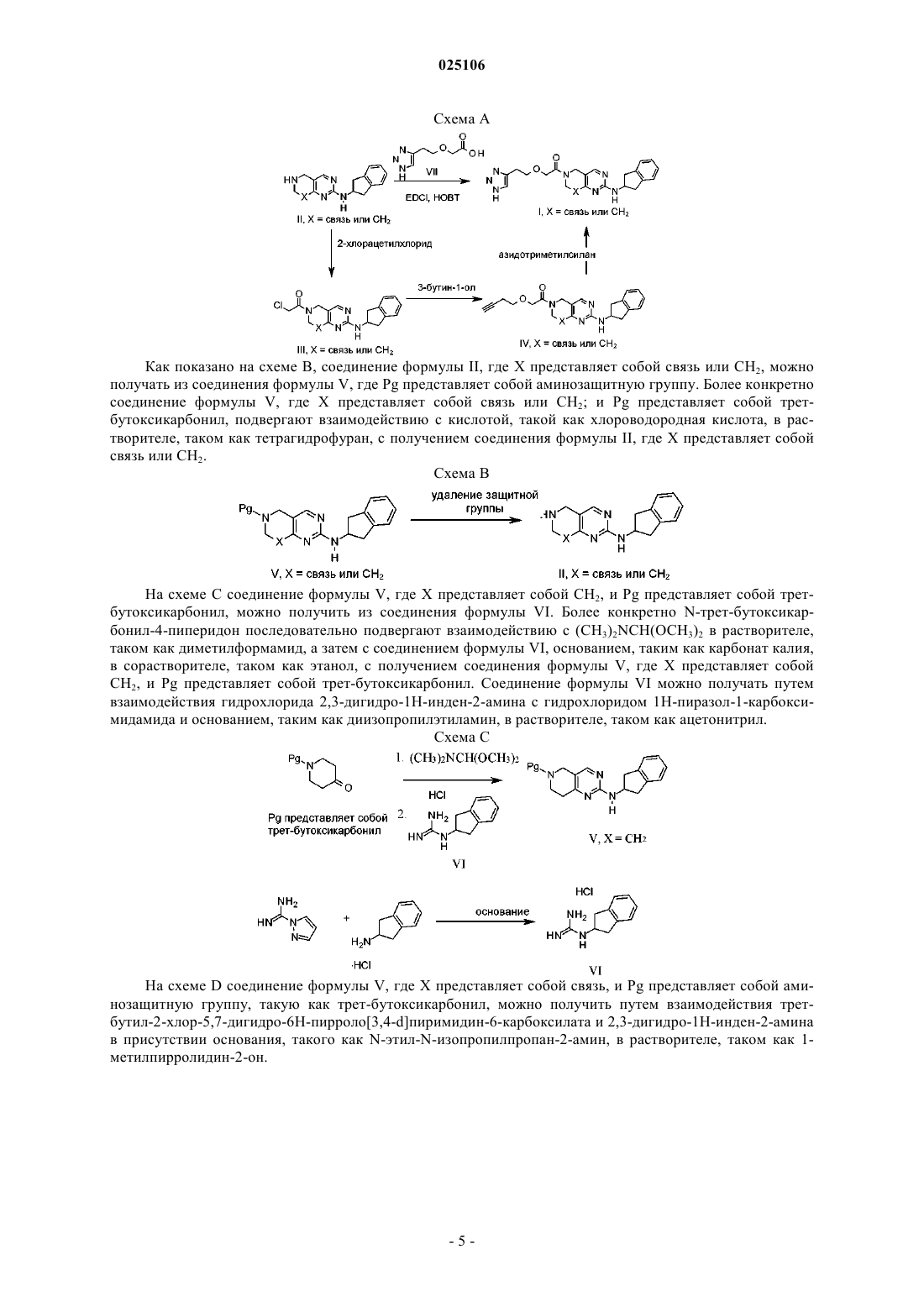
16. Промежуточное соединение формулы

17. Промежуточное соединение формулы

Текст
В настоящем изобретении предложены соединения формулы I где X представляет собой связь или СН 2; R выбран из группы, состоящей из каждый R1 и R2 независимо выбран из группы, состоящей из СН и N; R3 представляет собой Н или СН 3; R4 представляет собой Н или СН 3; L выбран из группы, состоящей из -О(СН 2)3-,-C(O)NH(CH2)2-, -CH2C(O)NH(CH2)2-, -(CH2)3N(C(O)CH3)CH2-, -(CH2)2N(C(O)CH3)CH2-,-(CH2)3NH-, -(CH2)2OCH2-, -(CH2)4-, -(CH2)2NHCH2-, -(CH2)3O- и -СН 2 О(СН 2)2-; или их фармацевтически приемлемые соли. Соединения согласно настоящему изобретению являются ингибиторами аутотаксина.(71)(73) Заявитель и патентовладелец: ЭЛИ ЛИЛЛИ ЭНД КОМПАНИ (US) Настоящее изобретение относится к бициклическим пиримидиновым соединениям или к их фармацевтически приемлемым солям и их терапевтическому применению. Соединения согласно настоящему изобретению являются ингибиторами аутотаксина. Аутотаксин представляет собой фермент, который, как сообщается, является источником лизофосфатидной кислоты (LPA), которая вызывает повышающую регуляцию белков, связанных с болью, посредством одного из родственных рецепторов, LPA1. LPA является внутриклеточным липидным медиатором, который влияет на множество биологических и биохимических процессов. Направленное ингибирование биосинтеза LPA, опосредованного аутотаксином, может обеспечивать новый механизм предотвращения невропатической боли, вызванной повреждением нервов. Для обеспечения возможного варианта лечения пациентов, нуждающихся в лечении боли, требуются соединения, ингибирующие аутотаксин. Известно, что боль, связанная с остеоартритом (ОА), является основным симптомом, приводящим к утрате функции нижних конечностей у пациентов с ОА. ОА, который является наиболее распространенным видом артропатии, диагностирован более чем у 20 миллионов американцев. Одобренные в настоящее время способы лечения боли, связанной с ОА, могут быть инвазивными, теряют эффективность при долгосрочном применении и подходят не для всех пациентов. Желательны дополнительные способы лечения пациентов, страдающих от боли, связанной с ОА. Соединения, ингибирующие аутотаксин, представляют собой другой возможный вариант лечения пациентов, страдающих от боли, связанной с ОА. В патенте США 7524852 ('852) предложены замещенные бициклические пиримидиновые производные в качестве противовоспалительных агентов. В заявке PCT/US 2011/048477 предложены индольные соединения в качестве ингибиторов аутотаксина. Существует потребность в новых соединениях, обеспечивающих ингибирование аутотаксина. В настоящем изобретении предложены новые соединения, которые являются ингибиторами аутотаксина. В настоящем изобретении предложены конкретные новые соединения, которые ингибируют выработкуLPA. Требуются соединения-ингибиторы аутотаксина для лечения состояний, опосредованных аутотаксином, таких как боль и боль, связанная с ОА. В настоящем изобретении предложены соединения формулы I где X представляет собой связь или СН 2; каждый R1 и R2 независимо выбран из группы, состоящей из СН и N;L выбран из группы, состоящей из -О(СН 2)3, -C(O)NH(CH2)2-, -CH2C(O)NH(CH2)2-,-(CH2)3N(C(O)CH3)CH2-, -(CH2)2N(C(O)CH3)CH2-, -(CH2)3NH-, -(CH2)2OCH2-, -(CH2)4-, -(CH2)2NHCH2-,-(CH2)3O- и -СН 2 О(СН 2)2-; или их фармацевтически приемлемые соли. В настоящем изобретении также предложен способ лечения боли у пациента, включающий введение пациенту, нуждающемуся в указанном лечении, эффективного количества соединения формулы I или его фармацевтически приемлемой соли. В настоящем изобретении также предложен способ лечения боли, связанной с остеоартритом, у пациента, включающий введение пациенту, нуждающемуся в указанном лечении, эффективного количества соединения формулы I или его фармацевтически приемлемой соли. В настоящем изобретении предложено соединение формулы I или его фармацевтически приемлемая соль для применения в терапии, в частности для лечения боли или для лечения боли, связанной с ОА. Кроме того, в настоящем изобретении предложено применение соединения формулы I или его фармацевтически приемлемой соли для получения лекарственного средства для лечения боли. В настоящем изобретении также предложено применение соединения формулы I или его фармацевтически приемлемой соли для получения лекарственного средства для лечения боли, связанной с ОА. В изобретении дополнительно предложена фармацевтическая композиция, содержащая соединение формулы I или его фармацевтически приемлемую соль в комбинации с одним или более фармацевтически приемлемыми носителями, разбавителями или вспомогательными веществами. Изобретение также охватывает новые промежуточные вещества и способы синтеза соединений формулы I. Термин "фармацевтически приемлемая соль" относится к соли соединения согласно настоящему изобретению, которую рассматривают как приемлемую для клинического и/или ветеринарного применения. Фармацевтически приемлемые соли и традиционная методика их получения хорошо известны в данной области техники (см., например, P. Stahl, et al., Handbook of Pharmaceutical Salts: Properties, Selection and Use, (VCHA/Wiley-VCH, 2002); S.M. Berge, et al., "Pharmaceutical Salts," Journal of PharmaceuticalSciences, Vol. 66, No. 1, January 1977). Термин "лечение" (или "лечить"), используемый в настоящем описании, относится к ограничению,замедлению, остановке или обращению вспять прогрессирования или степени тяжести существующего симптома, состояния или нарушения. Симптомы, состояния или нарушения могут быть выражены в виде"острых" или "хронических" явлений. При остром явлении соединение вводят при проявлении симптома,состояния или нарушения, введение заканчивают при прекращении явления. Хроническое явление подвергают лечению при течении нарушения или состояния, связанного с симптомом или явлением, при этом указанное хроническое лечение не зависит от конкретного проявления симптома или явления. Настоящее изобретение включает неотложное и хроническое лечение. Соединения согласно настоящему изобретению ингибируют аутотаксин и могут подходить для лечения заболевания или состояния, сопровождаемого увеличением уровня аутотаксина. Соединения согласно настоящему изобретению ингибируют выработку LPA и могут подходить для лечения заболевания или состояния, сопровождаемого увеличением уровня LPA. Соединения согласно настоящему изобретению могут ингибировать биосинтез LPA, опосредованный аутотаксином, по сравнению с другими липидными медиаторами LPA. Соединения согласно настоящему изобретению могут подходить для лечения заболевания или состояния, сопровождаемого увеличением связывания LPA1. Согласно настоящему описанию "пациент" относится к животному, нуждающемуся в лечении,предпочтительно, но не исключительно, к млекопитающему. В предпочтительном варианте реализации пациент представляет собой млекопитающее, которое предпочтительно представляет собой человека. В другом предпочтительном варианте реализации пациент представляет собой комнатное животное, такое как собака, кошка или домашняя птица. Согласно настоящему описанию термин "эффективное количество" относится к количеству или дозе соединения согласно настоящему изобретению или его фармацевтически приемлемой соли, которое при введении одной или нескольких доз пациенту обеспечивает желаемое действие у пациента, которому проводят диагностику или лечение. Следует понимать, что фактическое вводимое количество активного агента определяет лечащий врач с учетом важных факторов, включая состояние, подвергаемое лечению,выбранный способ введения, фактический вводимый активный агент, возраст, массу тела и ответ каждого пациента и тяжесть симптомов пациента, а также другие важные факторы. Соединение согласно настоящему изобретению предпочтительно вводят в состав фармацевтической композиции, которую вводят при помощи любого способа, обеспечивающего биодоступность соединения. Наиболее предпочтительно указанные композиции предназначены для перорального введения. Указанные фармацевтические композиции и способы их получения хорошо известны в данной области техники (см., например, Remington: The Science and Practice of Pharmacy (D.B. Troy, Editor, 21st Edition,Lippincott, WilliamsWilkins, 2006. Специалистам должно быть понятно, что структура охватывает все таутомерные формы. Для исключения неверного толкования следует отметить, что структура охватывает изомеры, которые легко превращаются друг в друга в результате таутомеризации. Структура охватывает, например, таутомеры Соединения формулы I особенно подходят для способов лечения согласно настоящему изобретению, но определенные группы, заместители и конфигурации являются предпочтительными для соединений формулы I. В следующих параграфах описаны указанные предпочтительные группы, заместители и конфигурации. Следует понимать, что указанные предпочтительные варианты относятся как к способам лечения, так и к новым соединениям согласно настоящему изобретению.R1 предпочтительно представляет собой СН.R2 предпочтительно представляет собой N. Предпочтительно, если R1 представляет собой СН, a R2 представляет собой N, то L представляет собой (СН 2)2OCH2 или О(СН 2)3. Предпочтительно, если R1 представляет собой,то каждый R3 и R4 представляет собой Н. Предпочтительно только один из R3 и R4 представляет собой СН 3.X предпочтительно представляет собой связь.X предпочтительно представляет собой СН 2. Особенно предпочтительно X представляет собой связь, a L представляет собой -(CH2)3NH-. Предпочтительно X представляет собой связь, R представляет собой Предпочтительно X представляет собой СН 2, R представляет собой(СН 2)2 ОСН 2 и соли указанных групп. Особенно предпочтительно X представляет собой СН 2, и L представляет собой -(СН 2)2 ОСН 2-, a R представляет собой Предпочтительные соединения представляют собой Н-3-[2-(2,3-дигидро-1H-инден-2-иламино)-7,8-дигидропиридо[4,3-d]пиримидин-6(5H)-ил]-3-оксопропил-2-(1H-имидазол-1-ил)ацетамид, 3-(4-метил-1H-имидазол-1-ил)пропил-2-(2,3-дигидро-1H-инден 2-иламино)-7,8-дигидропиридо[4,3-d]пиримидин-6(5H)-карбоксилат, 1-[2-(2,3-дигидро-1H-инден-2-иламино)-7,8-дигидропиридо[4,3-d]пиримидин-6(5H)-ил]-5-(1H-имидазол-1-ил)пентан-1-он, 3-(1H-имидазол-1-ил)пропил-2-(2,3-дигидро-1H-инден-2-иламино)-7,8-дигидропиридо[4,3-d]пиримидин-6(5H)-карбоксилат, 1-[2-(2,3-дигидро-1H-инден-2-иламино)-7,8-дигидропиридо[4,3-d]пиримидин-6(5H)-ил]-2-[2(1H-имидазол-1-ил)этокси]этанон и фармацевтически приемлемые соли указанных соединений. Предпочтительные соединения представляют собой 1-[2-(2,3-дигидро-1H-инден-2-иламино)-7,8-дигидропиридо[4,3-d]пиримидин-6(5H)-ил]-2-[2-(1 Н 1,2,3-триазол-4-ил)этокси]этанон, 1-[2-(2,3-дигидро-1H-инден-2-иламино)-7,8-дигидропиридо[4,3-d]пиримидин-6(5H)-ил]-5-(4H-1,2,4-триазол-3-ил)пентан-1-он,1-[2-(2,3-дигидро-1H-инден-2-иламино)-7,8 дигидропиридо[4,3-d]пиримидин-6(5H)-ил]-3-(1 Н-1,2,3-триазол-4-илметокси)пропан-1-он, 3-(1 Н-тетразол-5-ил)пропил-2-(2,3-дигидро-1H-инден-2-иламино)-7,8-дигидропиридо[4,3-d]пиримидин-6(5H)-карбоксилат, 1-[2-(2,3-дигидро-1H-инден-2-иламино)-7,8-дигидропиридо[4,3-d]пиримидин-6(5H)-ил]-2-[2(1H-1,2,3-триазол-4-ил)этил]аминоэтанон, 2-(2,3-дигидро-1H-инден-2-иламино)-N-[3-(1H-1,2,3-триазол 4-ил)пропил]-7,8-дигидропиридо[4,3-d]пиримидин-6(5H)-карбоксамид, N-2-[2-(2,3-дигидро-1H-инден 2-иламино)-7,8-дигидропиридо[4,3-d]пиримидин-6(5H)-ил]-2-оксоэтил-N-[3-(1H-1,2,3-триазол-4 ил)пропил]ацетамид, N-2-[2-(2,3-дигидро-1 Н-инден-2-иламино)-7,8-дигидропиридо[4,3-d]пиримидин 6(5H)-ил]-2-оксоэтил-N-[2-(1 Н-1,2,3-триазол-4-ил)этил]ацетамид,N-3-[2-(2,3-дигидро-1H-инден-2-3 025106 иламино)-7,8-дигидропиридо[4,3-d]пиримидин-6(5H)-ил]-3-оксопропил-2-(1H-1,2,3-триазол-5-ил)ацетамид, N-3-[2-(2,3-дигидро-1H-инден-2-иламино)-7,8-дигидропиридо[4,3-d]пиримидин-6(5H)-ил]-3-оксопропил-1 Н-1,2,3-триазол-4-карбоксамид и фармацевтически приемлемые соли указанных соединений. Предпочтительные соединения представляют собой 1-[2-(2,3-дигидро-1H-инден-2-иламино)-5,7-дигидро-6H-пирроло[3,4-d]пиримидин-6-ил]-5-(1 Н-тетразол-5-ил)пентан-1-он,1-[2-(2,3-дигидро-1H-инден-2-иламино)-5,7-дигидро-6H-пирроло[3,4-d]пиримидин-6-ил]-2-[2-(1H-1,2,3-триазол-5-ил)этил]аминоэтанон, 3-(1H-тетразол-5-ил)пропил-2-(2,3-дигидро-1H-инден-2-иламино)-5,7-дигидро-6H-пирроло[3,4-d]пиримидин-6-карбоксилат, 2-(2,3-дигидро-1Hинден-2-иламино)-N-[3-(1H-1,2,3-триазол-4-ил)пропил]-5,7-дигидро-6H-пирроло[3,4-d]пиримидин-6-карбоксамид,3-(1H-имидазол-5-ил)пропил-2-(2,3-дигидро-1H-инден-2-иламино)-5,7-дигидро-6H-пирроло[3,4-d]пиримидин-6-карбоксилат, 1-[2-(2,3-дигидро-1H-инден-2-иламино)-5,7-дигидро-6H-пирроло[3,4d]пиримидин-6-ил]-4-(1 Н-1,2,3-триазол-5-илокси)бутан-1-он,1-[2-(2,3-дигидро-1 Н-инден-2-иламино)5,7-дигидро-6H-пирроло[3,4-d]пиримидин-6-ил]-2-[2-(1H-1,2,3-триазол-4-ил)этокси]этанон и фармацевтически приемлемые соли указанных соединений. Особенно предпочтительные соединения представляют собой 1-[2-(2,3-дигидро-1H-инден-2-иламино)-5,7-дигидро-6H-пирроло[3,4-d]пиримидин-6-ил]-2-[2-(1 Н 1,2,3-триазол-4-ил)этокси]этанон и 1-[2-(2,3-дигидро-1H-инден-2-иламино)-7,8-дигидропиридо[4,3-d]пиримидин-6(5H)-ил]-2-[2-(1 Н-1,2,3-триазол-4-ил)этокси]этанон и фармацевтически приемлемые соли указанных соединений. Примеры получения и примеры Следующие примеры получения и примеры дополнительно иллюстрируют изобретение и представляют собой типовые способы синтеза соединения согласно настоящему изобретению. Следует понимать,что примеры получения и примеры приведены для иллюстрации, но не ограничения, и специалисты в данной области техники могут проводить различные их модификации. На схемах, приведенных ниже,все заместители, если конкретно не указано иное, являются такими, как определено выше. Для ясности на следующих схемах не указаны определенные стереохимические центры, также опущены определенные заместители, что никаким образом не ограничивает информацию, изображенную на схемах. Кроме того, специалисты в данной области техники могут выделять или разделять отдельные изомеры, энантиомеры или диастереомеры в любой подходящий момент синтеза соединений формулы I при помощи способов, таких как способы селективной кристаллизации или хиральной хроматографии (см., например,J. Jacques, et al., "Enantiomers, Racemates, and Resolutions", John Wiley and Sons, Inc., 1981, и E.L. Eliel andS.H. Wilen "Stereochemistry of Organic Compounds", Wiley-Interscience, 1994). Реагенты и исходные вещества в общем случае доступны специалистам в данной области техники, или же их можно получить при помощи традиционных способов органической и гетероциклической химии, аналогичных способам синтеза известных соединений со схожей структурой, а также способам, описанным в последующих примерах получения и примерах, включая любые новые способы. Если не указано иное, названия и нумерацию соединений, проиллюстрированных в настоящем описании, получали с использованием ACDLABS или Symyx Draw 3.2. В целом, соединение формулы I, где X представляет собой связь или СН 2, можно получать из соединения формулы II. Более конкретно, на схеме А соединение формулы II, где X представляет собой связь или СН 2, подвергают сочетанию с соединением формулы VII в присутствии карбодиимида, 1 гидроксибензотриазола и основания, такого как триэтиламин, с получением соединения формулы I, гдеX представляет собой связь или СН 2. Подходящие растворители включают дихлорметан. В качестве альтернативы на схеме А соединение формулы I, где X представляет собой связь или СН 2, можно получать из соединения формулы IV. Более конкретно соединение формулы IV, где X представляет собой связь или СН 2, подвергают взаимодействию с источником азида, таким как азидотриметилсилан, в присутствии сульфата меди (II) в растворителе, таком как диметилформамид/вода, с получением соединения формулы I, где X представляет собой связь или СН 2. Соединение формулы IV, где X представляет собой связь или СН 2, можно получать путем взаимодействия соединения формулы III с 3 бутин-1-олом и основанием, таким как гидрид натрия, в растворителе, таком как тетрагидрофуран. Соединение формулы III, где X представляет собой связь или СН 2, можно получать путем взаимодействия соединения формулы II с 2-хлорацетилхлоридом и основанием, таким как триэтиламин, в растворителе,таком как дихлорметан. Как показано на схеме В, соединение формулы II, где X представляет собой связь или СН 2, можно получать из соединения формулы V, где Pg представляет собой аминозащитную группу. Более конкретно соединение формулы V, где X представляет собой связь или СН 2; и Pg представляет собой третбутоксикарбонил, подвергают взаимодействию с кислотой, такой как хлороводородная кислота, в растворителе, таком как тетрагидрофуран, с получением соединения формулы II, где X представляет собой связь или СН 2. Схема В На схеме С соединение формулы V, где X представляет собой СН 2, и Pg представляет собой третбутоксикарбонил, можно получить из соединения формулы VI. Более конкретно N-трет-бутоксикарбонил-4-пиперидон последовательно подвергают взаимодействию с (CH3)2NCH(OCH3)2 в растворителе,таком как диметилформамид, а затем с соединением формулы VI, основанием, таким как карбонат калия,в сорастворителе, таком как этанол, с получением соединения формулы V, где X представляет собой СН 2, и Pg представляет собой трет-бутоксикарбонил. Соединение формулы VI можно получать путем взаимодействия гидрохлорида 2,3-дигидро-1H-инден-2-амина с гидрохлоридом 1H-пиразол-1-карбоксимидамида и основанием, таким как диизопропилэтиламин, в растворителе, таком как ацетонитрил. Схема С На схеме D соединение формулы V, где X представляет собой связь, и Pg представляет собой аминозащитную группу, такую как трет-бутоксикарбонил, можно получить путем взаимодействия третбутил-2-хлор-5,7-дигидро-6H-пирроло[3,4-d]пиримидин-6-карбоксилата и 2,3-дигидро-1H-инден-2-амина в присутствии основания, такого как N-этил-N-изопропилпропан-2-амин, в растворителе, таком как 1 метилпирролидин-2-он. В целом, соединение формулы VII можно получить из 3-бутин-1-ола с использованием нескольких стадий синтеза. Более конкретно, на схеме Е соединение формулы VIII, где Pg1 представляет собой защитную группу карбоновой кислоты, такую как трет-бутил, подвергают взаимодействию с кислотой,такой как трифторуксусная кислота, в растворителе, таком как дихлорметан, с получением соединения формулы VII. Соединение формулы VIII можно получить путем взаимодействия соединения формулыIX, где Pg1 представляет собой трет-бутил, с источником азида, таким как азидотриметилсилан, в присутствии йодида меди (I) в растворителе, таком как диметилформамид/метанол. Соединение формулыIX, где Pg1 представляет собой трет-бутил, можно получать путем взаимодействия 3-бутин-1-ола сYCH2C(O)OPg1, где Y представляет собой подходящую уходящую группу, такую как галоген, бром, aPg1 представляет собой трет-бутил. Взаимодействие эффективно проводят с использованием катализатора фазового переноса, такого как сульфат тетрабутиламмония, и основания, такого как водный гидроксид натрия, в растворителе, таком как дихлорметан. В качестве альтернативы согласно схеме Е соединение формулы VII можно получить путем взаимодействия соединения формулы X с водородом в присутствии катализатора на основе переходного металла, такого как хлорид палладия (II), в растворителе, таком как изопропиловый спирт/вода. Соединение формулы X можно получать путем взаимодействия соединения формулы XI с YCH2C(O)OPg1, где Y представляет собой подходящую уходящую группу, такую как галоген, бром, a Pg1 представляет собой трет-бутил. Взаимодействие эффективно проводят с использованием катализатора фазового переноса,такого как хлорид тетрабутиламмония, и основания, такого как водный гидроксид натрия, в растворителе, таком как дихлорметан. Полученное производное, содержащее трет-бутиловый эфир, подвергают взаимодействию с кислотой, такой как хлороводородная кислота, с получением соединения формулы X. Соединение формулы XI можно получить путем взаимодействия соединения формулы XII с хлоридом изопропилмагния и бензилазидом в растворителе, таком как тетрагидрофуран. Соединение формулы XII можно получить путем взаимодействия 3-бутин-1-ола с дигидропираном в присутствии органической кислоты, такой как п-толуолсульфокислота. Взаимодействие, как правило, проводят в растворителе, таком как дихлорметан. Схема Е Перемешивали раствор гидрохлорида 2,3-дигидро-1H-инден-2-амина (197 г; 1,08 экв.; 1,16 моль),гидрохлорида 1H-пиразол-1-карбоксимидамида (158 г; 1,00 экв.; 1,08 моль) и диизопропилэтиламина(400 г; 2,87 экв.; 3,09 моль; 539,74 мл) в ацетонитриле (2 л) при 62 С в течение 2 ч, за это время осаждалось белое твердое вещество. Охлаждали смесь до 25 С, затем фильтровали и промывали 300 мл ацето-6 025106 нитрила и 300 мл метил-трет-бутилового эфира. Сушили продукт на воздухе при 25 С в течение 1 ч с получением титульного соединения (200 г, 87%) в виде белого твердого вещества: МС (m/z): 176 (М+1). Пример получения 2 Синтез трет-бутил-2-(индан-2-иламино)-7,8-дигидро-5H-пиридо[4,3-d]пиримидин-6-карбоксилата Перемешивали раствор 1,1-диметокси-N,N-диметилметанамина (224 г; 2,15 экв.; 1,88 моль; 250,98 мл) и N-трет-бутоксикарбонил-4-пиперидона (250 г; 1,44 экв.; 1,25 моль) в диметилформамиде (1,2 л) при 109 С в атмосфере N2 в течение 4 ч. Охлаждали смесь до 25 С, а затем добавляли этанол (700 мл; 12,02 моль; 553,91 г). В смесь за один раз добавляли гидрохлорид 1-(2,3-дигидро-1H-инден-2-ил)гуанидина (185 г; 1,00 экв.; 873,90 ммоль) и карбонат калия (475 г; 3,44 моль) при 25 С с получением белой суспензии. Перемешивали смесь при 80-90 С в течение 24 ч, затем охлаждали до 25 С и выливали смесь в 5 л смеси лед/вода с получением желтой суспензии. Экстрагировали этилацетатом (33 л) и промывали органический слой 10% раствором хлорида лития (3 л), водой (3 л) и насыщенным раствором хлорида натрия (3 л). Сушили над безводным сульфатом натрия, фильтровали и концентрировали с получением примерно 300 мл красного раствора. Фильтровали раствор через небольшую колонку с силикагелем (высота 10 см, диаметр 5 см), а затем концентрировали досуха с получением титульного соединения в виде красного геля (320 г, 100%): МС (m/z): 367 (М+1). Пример получения 3 Синтез N-индан-2-ил-5,6,7,8-тетрагидропиридо[4,3-d]пиримидин-2-амина В раствор трет-бутил-2-(индан-2-иламино)-7,8-дигидро-5H-пиридо[4,3-d]пиримидин-6-карбоксилата (319 г; 1,00 экв.; 870,48 ммоль) в тетрагидрофуране (1,5 л) по частям добавляли хлороводородную кислоту (900 мл; 5 М в воде; 5,17 экв.; 4,50 моль; 1,08 кг). После завершения добавления перемешивали раствор при 50 С в течение 1 ч. Охлаждали смесь до 25 С, а затем добавляли 3 л метил-трет-бутилового эфира и 1 л воды. Оставляли раствор отстаиваться при 20 С на 16 ч. Разделяли фазы и экстрагировали водную фазу дихлорметаном (2 л). Отбрасывали органические экстракты и доводили рН водной фазы до 10 с использованием 4 М гидроксида натрия. Экстрагировали этилацетатом (33 л) и промывали объединенные органические экстракты насыщенным хлоридом натрия (2 л). Сушили над безводным сульфатом натрия, фильтровали и концентрировали досуха с получением красного геля. Перерастворяли вещество в этилацетате (300 мл) и петролейном эфире (200 мл) при 50 С и оставляли осаждаться на 24 ч. Фильтровали и сушили с получением титульного соединения (85 г, 37%). МС (m/z): 267 (М+1). Пример получения 4 Синтез 2-бут-3-инокситетрагидропирана Раствор 3-бутин-1-ола (50 г, 1 экв., 713 ммоль), моногидрата п-толуолсульфокислоты (1,36 г, 0,01 экв., 7,13 ммоль) и дигидропирана (71,7 мл, 1,10 экв., 785 ммоль) в дихлорметане (500 мл) перемешивали при 25 С в течение 24 ч. Затем медленно добавляли насыщенный раствор бикарбоната натрия (500 мл) и экстрагировали смесь дихлорметаном (500 мл). Органический экстракт сушили над безводным сульфатом натрия, фильтровали и концентрировали с получением неочищенного продукта, который очищали путем вакуумной перегонки с получением титульного соединения (90 г, 82%): Ткип: 65 С, 1 мм рт.ст. Пример получения 5 Синтез 2-(3-бензилтриазол-4-ил)этанола В раствор 2-бут-3-инокситетрагидропирана (113,50 г, 736,00 ммоль) в тетрагидрофуране (1,4 л) при 15 С по каплям добавляли хлорид изопропилмагния (368 мл, 736,00 ммоль), поддерживая внутреннюю температуру ниже 20 С. Удаляли баню лед-вода, оставляли нагреваться до 25 С и выдерживали при указанной температуре в течение 20 мин для обеспечения полноты выделения газа. Нагревали реакционную смесь до 30-35 С и в смесь по каплям добавляли бензилазид (70 г, 525,71 ммоль) в течение 2 ч, следя за тем, чтобы концентрация бензилазида в реакционной смеси не превышала приемлемый уровень, при помощи способа, такого как ЖХМС. После 4-часового перемешивания охлаждали реакционную смесь до 25 С и реакцию гасили насыщенным водным хлоридом аммония (400 мл), который добавляли по каплям. После 16-часового перемешивания разбавляли метил-трет-бутиловым эфиром (200 мл), разделяли слои и перемешивали органический слой с 3 М хлороводородной кислотой (600 мл) в течение 4 ч. При достижении конверсии более 98%, что определяли при помощи способа, такого как ЖХМС, повышали рН реакционной смеси до рН 10 путем добавления по каплям 6 М гидроксида натрия (примерно 350 мл). Экстрагировали полученную смесь метил-трет-бутиловым эфиром (3300 мл) и дихлорметаном (3300 мл). Концентрировали объединенные органические слои в вакууме и очищали полученный неочищенный продукт путем колоночной хроматографии на силикагеле, элюируя этилацетатом, с получением титульного соединения (53 г, 50%) в виде беловатого твердого вещества. МС (m/z): 204,1 (М+1). Пример получения 6 Синтез [2-(1-бензил-1H-1,2,3-триазол-5-ил)этокси]уксусной кислоты(6,84 г, 24,60 ммоль) в дихлорметане (400 мл) при 0 С помещали 400 мл 40% (мас./мас.) гидроксида натрия (5,73 моль). Добавляли этиловый эфир бром-1,1-диметилуксуной кислоты (62,38 г, 319,81 ммоль) и перемешивали в течение 16 ч, при этом температура повышалась до 25 С. Разделяли слои и экстрагировали водную фазу дихлорметаном (2100 мл). Концентрировали объединенные органические слои в вакууме с получением 75 г промежуточного трет-бутил-2-(2-(3-бензил-3H-1,2,3-триазол-4-ил)этокси)ацетата в виде желтой маслянистой жидкости. Разбавляли маслянистую жидкость 450 мл толуола и 150 мл 6 М хлороводородной кислоты и перемешивали при 25 С в течение 16 ч. Разделяли слои и доводили рН водного слоя примерно до рН 2,0. Отфильтровывали полученное белое твердое вещество, промывали водой (2200 мл) с получением титульного соединения (58 г, 90%) в виде белого твердого вещества. МС (m/z): 262,1 (М+1). Пример получения 7 Синтез 2-[2-(1H-триазол-5-ил)этокси]уксусной кислоты Нагнетали 1 атмосферу водорода (газ) в колбе, содержащей [2-(1-бензил-1 Н-1,2,3-триазол-5-ил)этокси]уксусную кислоту (10,1 г; 1,00 экв.; 38,66 ммоль) и хлорид палладия (II) (3 г; 16,92 ммоль; 3,00 г) в изопропиловом спирте (300 мл) и воде (60 мл). Выдерживали колбу в атмосфере водорода в течение 3 ч,затем фильтровали через Celite и концентрировали. Добавляли толуол (250 мл) и концентрировали с получением титульного соединения (7,96 г, 100%). 1 Н ЯМР (d6-ДМСО): 2,86 (t, J = 7 Гц, 2 Н), 3,65 (t, J = 7 Гц, 2 Н), 3,98 (s, 2 Н), 7,77 (s, 1H), 13,4-13,6 (шир.s, 2 Н). Пример 1 Синтез 1-[2-(2,3-дигидро-1H-инден-2-иламино)-7,8-дигидропиридо[4,3-d]пиримидин-6(5H)-ил]-2[2-(1 Н-1,2,3-триазол-4-ил)этокси]этанона В смесь 2-[2-(1H-триазол-5-ил)этокси]уксусной кислоты (2,7 г, 15,8 ммоль), 1-гидроксибензотриазола (3,20 г, 23,7 ммоль) и гидрохлорида (диметиламинопропил)-3-этилкарбодиимида (5,44 г, 28,4 ммоль) в дихлорметане (40 мл) при 25 С добавляли N-индан-2-ил-5,6,7,8-тетрагидропиридо[4,3-d]пиримидин-2-амин (4,2 г, 15,8 ммоль). В реакционную смесь добавляли триэтиламин (4,40 мл, 31,6 ммоль) и перемешивали в течение 16 ч. Промывали водой (250 мл) и концентрировали органический слой. Очищали путем колоночной хроматографии на силикагеле, элюируя смесью этилацетат/метанол, с получением титульного соединения (4,0 г, 60%) в виде твердого вещества. МС (m/z): 420 (М+Н). Пример получения 8 Синтез 2-хлор-1-[2-(2,3-дигидро-1H-инден-2-иламино)-7,8-дигидропиридо[4,3-d]пиримидин-6(5H)ил]этанона К N-индан-2-ил-5,6,7,8-тетрагидропиридо[4,3-d]пиримидин-2-амину (11,0 г, 41,3 ммоль) и триэтиламину (7,48 мл, 53,7 ммоль) в дихлорметане (200 мл) по каплям добавляли 2-хлорацетилхлорид (3,61 мл, 5,13 г, 45,4 ммоль) в течение 5 мин при 23 С. Перемешивали в течение 30 мин и выливали реакционную смесь в смесь 1:1 50% насыщенный водный бикарбонат натрия:дихлорметан (75 мл). Отделяли органический слой от водного слоя и дополнительно экстрагировали водный слой дихлорметаном (225 мл). Объединяли органические экстракты и сушили над безводным сульфатом натрия, фильтровали и концентрировали. Растворяли остаток в хлороформе (10 мл) и очищали путем колоночной хроматографии на силикагеле (элюировали с градиентом: от 25% этилацетата в гексанах до 100% этилацетата) с получением титульного соединения (9,75 г, 69%). 1 Н ЯМР (CDCl3,= неосновной амидный ротамер)2,77 (t, 2H), 2,84 (dd, 2H), 2,87 (t, 2H), 3,35 (dd, 2H), 3,76 (t, 2H), 3,85 (t, 2H), 4,12 (s, 2H), 4,52 (s, 2H),4,57 (s, 2H), 4,72-4,82 (m, 1H), 5,48-5,64 (m, 1H), 7,12-7,21 (m, 4H), 8,03-8,10 (m, 1H). Пример получения 9 Синтез 2-(бут-3-ин-1-илокси)-1-[2-(2,3-дигидро-1H-инден-2-иламино)-7,8-дигидропиридо[4,3-d]пиримидин-6(5H)-ил]этанона К гидриду натрия (60 мас.% в минеральном масле, 1,58 г, 39,6 ммоль) в тетрагидрофуране (50 мл) при 23 С по каплям добавляли 3-бутин-1-ол (7,93 г, 8,59 мл, 113,2 ммоль), затем перемешивали при 23 С в течение 20 мин. Добавляли полученный раствор к 2-хлор-1-[2-(2,3-дигидро-1H-инден-2-иламино)-7,8 дигидропиридо[4,3-d]пиримидин-6(5H)-ил]этанону (9,70 г, 28,3 ммоль) в тетрагидрофуране (150 мл) при 23 С и перемешивали в течение 1 ч. Выливали реакционную смесь в 50% насыщенный водный раствор бикарбоната натрия. Отделяли органический слой и дополнительно экстрагировали водный слой диэтиловым эфиром (2) и этилацетатом (2). Объединяли органические экстракты и промывали солевым раствором, затем сушили над безводным сульфатом натрия, фильтровали и концентрировали. Очищали полученный неочищенный продукт путем колоночной хроматографии на силикагеле (элюировали с градиентом: от 20% этилацетата в гексанах до 100% этилацетата) с получением титульного соединения (8,16 г,77%). МС (m/z): 377 (М+1). Пример 1 а Альтернативный способ синтеза 1-[2-(2,3-дигидро-1H-инден-2-иламино)-7,8-дигидропиридо[4,3d]пиримидин-6(5H)-ил]-2-[2-(1H-1,2,3-триазол-4-ил)этокси]этанона Раствор 2-(бут-3-ин-1-илокси)-1-[2-(2,3-дигидро-1H-инден-2-иламино)-7,8-дигидропиридо[4,3-d]пиримидин-6(5H)-ил]этанона (8,15 г, 21,7 ммоль) и натриевой соли L-аскорбиновой кислоты (8,58 г, 43,3 ммоль) в диметилформамиде (60 мл) и воде (60 мл) продували азотом в течение 10 мин, затем трижды проводили стадии вакуумирования и заполнения азотом. Добавляли пентагидрат сульфата меди (II) (1,08 г, 4,33 ммоль) и нагревали до 90 С, затем по каплям добавляли азидотриметилсилан (23,1 мл, 20,0 г, 173 ммоль) и перемешивали в течение 1 ч. Охлаждали реакционную смесь до 23 С и выливали в воду (50 мл). Экстрагировали полученную смесь этилацетатом (450 мл). Объединяли органические экстракты и промывали насыщенным водным хлоридом натрия, сушили над безводным сульфатом натрия, фильтровали и концентрировали. Очищали полученный неочищенный продукт путем колоночной хроматографии на силикагеле (элюировали с градиентом: от 0 до 10% метанола в этилацетате) с получением титульного соединения (3,60 г, 40%). МС (m/z): 420 (М+1). Пример получения 10 Синтез трет-бутил-2-(2,3-дигидро-1H-инден-2-иламино)-5,7-дигидро-6H-пирроло[3,4-d]пиримидин 6-карбоксилата В выдерживаемый при 15 С раствор трет-бутил-2-хлор-5,7-дигидро-6 Н-пирроло[3,4-d]пиримидин 6-карбоксилата (220 г, 860,37 ммоль) и 2,3-дигидро-1H-инден-2-амина (137,7 г, 1,03 моль) в 1-метилпирролидин-2-оне (3,6 л) помещали 450 мл (2,58 моль) N-этил-N-изопропилпропан-2-амина. Нагревали полученную смесь до 80 С в течение 16 ч, затем охлаждали до 30 С и переносили полученную смесь в 5 л воды при 25 С. Отфильтровывали полученное твердое вещество и промывали осадок водой (2300 мл). Повторно суспендировали твердое вещество в этилацетате (350 мл) в течение 45 мин при 15 С. Фильтро-9 025106 вали суспензию, промывали выдерживаемым при 15 С этилацетатом (2250 мл) и сушили с получением титульного соединения (226 г, 75%) в виде беловатого твердого вещества. 1 Н ЯМР (d6-ДМСО) 1,45 (s,9 Н), 2,87 (dd, J= 7,2, 15,8 Гц, 2 Н), 3,24 (dd, J= 7,2, 15,8 Гц, 2 Н), 4,36 (d, 10,4 Гц, 2 Н), 4,44 (d, J= 12,8 Гц,2 Н), 4,60 (m, 1H), 7,14 (m, 2H), 7,20 (m, 2H), 7,55 (d, J= 6,8 Гц, 1H), 8,27 (d, J= 7,2 Гц, 1 Н). Пример получения 11 Синтез гидрата дигидрохлорида N-(2,3-дигидро-1H-инден-2-ил)-6,7-дигидро-5H-пирроло[3,4-d]пиримидин-2-амина В раствор трет-бутил-2-(2,3-дигидро-1H-инден-2-иламино)-5,7-дигидро-6H-пирроло[3,4-d]пиримидин-6-карбоксилата (226 г, 641,25 ммоль) в тетрагидрофуране (2,0 л) при 17 С помещали 670 мл 5 М хлороводородной кислоты (3,35 моль), поддерживая при добавлении внутреннюю температуру ниже 26 С. Нагревали полученный раствор до 50 С в течение 16 ч, охлаждали до 25 С и разбавляли 500 мл воды и 500 мл метил-трет-бутилового эфира. Разделяли полученные слои и экстрагировали метил-третбутиловым эфиром (31 л). Концентрировали водную фазу до достижения объема реакционной смеси примерно 200 мл и фильтровали полученную суспензию. Промывали осадок метил-трет-бутиловым эфиром (2200 мл) и сушили с получением титульного продукта (177 г, 80%) в виде светло-коричневого твердого вещества. МС (m/z): 253,2 (М-2HCl-Н 2 О+1). Пример получения 12 Синтез трет-бутил-2-бут-3-иноксиацетата Перемешивали смесь бут-3-ин-1-ола (6,00 г; 85,60 ммоль), сульфата тетрабутиламмония (2,07 г; 8,54 ммоль) и гидроксида натрия (40% (мас./мас.); 150 мл) в дихлорметане (150 мл) при 0 С. По каплям добавляли трет-бутилбромацетат (19,34 мл; 128,40 ммоль) и перемешивали смесь в течение 2,5 ч при комнатной температуре. Разбавляли реакционную смесь дихлорметаном (200 мл) и водой (100 мл), разделяли слои и дополнительно экстрагировали водный слой дихлорметаном (2100 мл). Промывали объединенные органические слои солевым раствором (100 мл), сушили над безводным сульфатом натрия и концентрировали с получением неочищенного титульного соединения в виде коричневой маслянистой жидкости (11,93 г). Очищали маслянистую жидкость путем колоночной хроматографии на силикагеле,элюируя смесью гексан:этилацетат (смеси от 0 до 10%), с получением титульного соединения (11,35 г; 72%) в виде бесцветной маслянистой жидкости. Н ЯМР (CDCl3)1,48 (s, 9H), 2,00 (m, 1H), 2,52 (m, 2H),3,67 (m, 2H), 4,01 (шир.s, 2H). Пример получения 13 Синтез трет-бутил-2-[2-(1H-триазол-5-ил)этокси]ацетата Перемешивали трет-бутил-2-бут-3-иноксиацетат (11,34 г; 61,55 ммоль) и йодид меди (I) (584 мг; 3,07 ммоль) в смеси диметилформамида (56,70 мл) и метанола (11,34 мл) при 0 С. По каплям добавляли азидо(триметил)силан (12,33 мл; 86,47 ммоль) и грели смесь при 90 С в течение 18 ч. Во второй партии перемешивали трет-бутил-2-бут-3-иноксиацетат (4,38 г; 23,77 ммоль) и йодид меди (I) (226 мг; 1,19 ммоль) в смеси диметилформамида (22 мл) и метанола (6 мл) при 0 С. По каплям добавляли азидо(триметил)силан (4,8 мл; 33,66 ммоль) и грели смесь при 90 С в течение 18 ч. Охлаждали до комнатной температуры и объединяли неочищенные продукты из двух партий и концентрировали смесь с получением зеленоватого остатка. Очищали неочищенный продукт путем фильтрования через небольшую колонку с оксидом кремния, элюируя смесью дихлорметан:этилацетат(смеси от 75% до 100%), с получением титульного соединения (14,15 г, 73%) в виде бесцветной маслянистой жидкости. МС (m/z): 228,15 (М+1). Перемешивали смесь трет-бутил-2-[2-(1H-триазол-5-ил)этокси]ацетата (14,15 г; 62,26 ммоль) и трифторуксусной кислоты (70,75 мл, 935,69 ммоль) в дихлорметане (70,75 мл) в течение 2 ч при комнатной температуре. Концентрировали реакционную смесь при пониженном давлении с получением титульного соединения, дополнительно содержащего трифторуксусную кислоту, (20,22 г, 100%) в виде коричневого твердого вещества. МС (m/z): 172,05 (М+1). Пример 2 Синтез 1-[2-(2,3-дигидро-1H-инден-2-иламино)-5,7-дигидро-6H-пирроло[3,4-d]пиримидин-6-ил]-2[2-(1H-1,2,3-триазол-4-ил)этокси]этанона Перемешивали смесь соли 2,2,2-трифторуксусной кислоты и 2-[2-(1H-триазол-5-ил)этокси]уксусной кислоты (20,22 г; 70,90 ммоль), гидрата дигидрохлорида N-(2,3-дигидро-1H-инден-2-ил)-6,7 дигидро-5H-пирроло[3,4-d]пиримидин-2-амина (27,99 г; 81,54 ммоль) и триэтиламина (98,83 мл; 709,03 ммоль) в диметилформамиде (404,40 мл) при 0 С. Добавляли раствор циклического ангидрида 1-пропанфосфоновой кислоты (50% раствор в ДМФ; 51,89 мл; 81,54 ммоль) в течение 30 мин и перемешивали смесь при комнатной температуре в течение 18 ч. Концентрировали реакционную смесь при пониженном давлении с получением остатка. Добавляли воду (200 мл) и экстрагировали смесь этилацетатом (4250 мл) и дихлорметаном (4250 мл). Промывали объединенные органические слои насыщенным водным бикарбонатом натрия (2100 мл) и солевым раствором (100 мл), затем сушили над безводным сульфатом натрия. Фильтровали смесь и концентрировали раствор при пониженном давлении с получением красного твердого вещества (25,70 г), которое суспендировали в смеси этилацетат/метанол (смесь 9:1; 200 мл) в течение 2 ч при комнатной температуре. Фильтровали полученное твердое вещество и промывали холодным этилацетатом (50 мл) с получением твердого вещества (примерно 18,2 г), которое повторно суспендировали в этилацетате (200 мл) при температуре обратной конденсации в течение 1 ч. После охлаждения до комнатной температуры перемешивали смесь в течение 1 ч и отфильтровывали полученное светло-розовое твердое вещество. Суспендировали светло-розовое твердое вещество в смеси вода/метанол (смесь 1:1; 200 мл) и грели смесь при 50 С в течение 30 мин. Добавляли раствор гидроксида аммония (32%; 50 мл) и продолжали греть смесь при 50 С в течение 30 мин. После охлаждения до комнатной температуры дополнительно добавляли раствор гидроксида аммония (32%; 50 мл) и продолжали перемешивать в течение 1 ч при комнатной температуре. Отфильтровывали полученное светло-серое твердое вещество, сушили и снова суспендировали в этилацетате (200 мл) в течение 1 ч с получением светло-серого твердого вещества, которое отфильтровывали, промывали этилацетатом (25 мл) и сушили с получением титульного соединения (12,42 г; 43%) в виде серого твердого вещества. МС (m/z): 406 (М+1). Пример получения 15 Синтез 2-хлор-1-[2-(2,3-дигидро-1H-инден-2-иламино)-5,7-дигидро-6H-пирроло[3,4-d]пиримидин 6-ил]этанона Перемешивали суспензию гидрата дигидрохлорида N-(2,3-дигидро-1H-инден-2-ил)-6,7-дигидро-5Hпирроло[3,4-d]пиримидин-2-амина (14,4 г, 41,9 ммоль) и триэтиламина (14,3 г, 19,7 мл, 141,4 ммоль) в дихлорметане (200 мл) при 23 С в течение 10 мин, затем охлаждали до -30 С. Добавляли 2-хлорацетилхлорид (5,49 г, 3,86 мл, 48,6 ммоль) в течение 2 мин и нагревали до 23 С в течение 10 мин. Добавляли метанол (5 мл) и удаляли растворитель в вакууме. Суспендировали неочищенную реакционную смесь в метаноле (30 мл), добавляли 50 г силикагеля и удаляли растворитель в вакууме. Помещали полученный остаток в колонку и очищали путем колоночной хроматографии на силикагеле (элюировали с градиентом: от 50% этилацетата в гексанах до этилацетата и 10% метанола в этилацетате) с получением титульного соединения (11,5 г, 84%). МС (m/z): 329 (М+1). Пример получения 16 Синтез 2-(бут-3-ин-1-илокси)-1-[2-(2,3-дигидро-1H-инден-2-иламино)-5,7-дигидро-6H-пирроло[3,4d]пиримидин-6-ил]этанона К гидриду натрия (60 мас.% в минеральном масле, 2,06 г, 51,4 ммоль) в тетрагидрофуране (86 мл) при 0 С добавляли 3-бутин-1-ол (4,64 г, 5,03 мл, 64,3 ммоль), затем перемешивали при 23 С в течение 15 мин. Добавляли полученный раствор к 2-хлор-1-[2-(2,3-дигидро-1H-инден-2-иламино)-5,7-дигидро-6Hпирроло[3,4-d]пиримидин-6-ил]этанону (8,45 г, 25,7 ммоль) в тетрагидрофуране (86 мл) при 0 С и перемешивали в течение 5 мин. Выливали реакционную смесь в 50% насыщенный водный раствор бикарбоната натрия. Отделяли органический слой и дополнительно экстрагировали водный слой диэтиловым эфиром и этилацетатом (по 250 мл). Объединяли органические экстракты и промывали солевым раствором, затем сушили над безводным сульфатом натрия, фильтровали и концентрировали. Объединяли неочищенный продукт с неочищенным продуктом, полученным в результате второго взаимодействия(которое проводили в таких же условиях и с такой же стехиометрией с использованием 2-хлор-1-[2(индан-2-иламино)-5,7-дигидропирроло[3,4-d]пиримидин-6-ил]этанона (3,0 г, 9,1 ммоль, и очищали путем колоночной хроматографии на силикагеле (элюировали с градиентом: от 25% этилацетата в гексанах до 100% этилацетата) с получением титульного соединения (2,90 г, 23%). МС (m/z): 363 (М+1). Пример 2 а Альтернативный способ синтеза 1-[2-(2,3-дигидро-1H-инден-2-иламино)-5,7-дигидро-6H-пирроло[3,4-d]пиримидин-6-ил]-2-[2-(1H-1,2,3-триазол-4-ил)этокси]этанона В колбу, содержащую 2-(бут-3-ин-1-илокси)-1-[2-(2,3-дигидро-1H-инден-2-иламино)-5,7-дигидро 6H-пирроло[3,4-d]пиримидин-6-ил]этанон (2,90 г, 8,00 ммоль), добавляли диметилформамид (27 мл) и воду (27 мл). Добавляли пентагидрат сульфата меди (II) (400 мг, 1,60 ммоль) и натриевую соль Lаскорбиновой кислоты (3,17 г, 16,0 ммоль). Вакуумировали колбу и повторно заполняли азотом (2),затем добавляли азидотриметилсилан (7,37 г, 8,53 мл, 64,0 ммоль) и нагревали реакционную смесь до 90 С в течение 70 мин. Охлаждали реакционную смесь до 23 С и удаляли все растворители в вакууме. Суспендировали остаток в смеси метанол/дихлорметан, затем добавляли силикагель и удаляли растворитель в вакууме. Помещали полученное вещество в колонку и очищали путем колоночной хроматографии на силикагеле (элюировали с градиентом: 0-9% метанола в этилацетате) с получением титульного соединения (980 мг, 30%). МС (m/z): 406 (М+1). Схема F, стадия А. Продували хлороводород (газ) через выдерживаемый при 0 С раствор метилового эфира 5-цианопентановой кислоты (3,0 мл; 1,00 экв.; 22,02 ммоль) в метаноле (5 мл) и диэтиловом эфире (10 мл) в течение 1 ч. Оставляли раствор нагреваться до комнатной температуры, затем добавляли 50 мл диэтилового эфира, интенсивно перемешивали в течение 15 мин, затем фильтровали и собирали полученный осадок. Промывали твердое вещество диэтиловым эфиром с получением метил-6-имино-6 метоксигексаноата (3,82 г; 83%): 1H ЯМР (ДМСО)1,47-1,62 (m, 4 Н), 2,31 (t, 2H), 2,59 (t, 2H), 3,56 (s,3 Н), 4,03 (s, 3 Н). Пример получения 18 Синтез метил-5-(4H-1,2,4-триазол-3-ил)пентаноата Схема F, стадия В. В пробирку, содержащую метил-6-имино-6-метоксигексаноат (0,542 г; 1,00 экв.; 2,58 ммоль), добавляли метанол (1 мл) и триэтиламин (0,36 мл; 1,0 экв.; 2,58 ммоль), затем раствор формилгидразина (0,155 г; 1,0 экв.; 2,58 ммоль) в метаноле (3 мл). Перемешивали при комнатной температуре в течение 18 ч, затем нагревали до 70 С в течение 3 ч. Концентрировали смесь, затем разбавляли этилацетатом (30 мл) и фильтровали. Концентрировали фильтрат с получением метил-5-(4H-1,2,4-триазол-3 ил)пентаноата (0,498 г; 105%) в виде бесцветной маслянистой жидкости: МС (m/z): 184 (М+1). Пример получения 18 а Синтез 5-(4H-1,2,4-триазол-3-ил)пентановой кислоты Схема F, стадия С. В колбу, содержащую метил-5-(4H-1,2,4-триазол-3-ил)пентаноат (0,5 г; 1,0 экв.; 2,73 ммоль), добавляли метанол (3 мл), тетрагидрофуран (3 мл) и воду (2 мл). За один раз добавляли гидроксид лития (0,261 г; 4,0 экв.; 10,92 ммоль) и перемешивали при комнатной температуре в течение 90 мин. Охлаждали раствор до 0 С и медленно добавляли 5 н. хлороводород (2,18 мл; 4,0 экв.; 10,92 ммоль). Концентрировали смесь, а затем перегоняли остаток в виде азеотропа с толуолом (410 мл). Добавляли дихлорметан (30 мл), затем сульфат магния. Фильтровали и промывали осадок 100 мл 10% этанола в дихлорметане. Концентрировали с получением 5-(4H-1,2,4-триазол-3-ил)пентановой кислоты (1,6 г; 100%) в виде аддукта со спиртом: МС (m/z): 170 (М+1). Пример 3 Синтез 1-[2-(2,3-дигидро-1H-инден-2-иламино)-7,8-дигидропиридо[4,3-d]пиримидин-6(5 Н)-ил]-5(4 Н-1,2,4-триазол-3-ил)пентан-1-она Схема F, стадия D. В раствор, содержащий 5-(4H-1,2,4-триазол-3-ил)пентановую кислоту (0,40 г, 1,0 экв.; 0,71 ммоль), добавляли N-индан-2-ил-5,6,7,8-тетрагидропиридо[4,3-d]пиримидин-2-амин (0,189 г; 1,0 экв.; 0,71 ммоль), затем диметилформамид (2 мл). Перемешивали полученный раствор при комнатной температуре до достижения гомогенности. Добавляли N,N-диметил-4-пиридинамин (0,017 г; 0,2 экв.; 0,141 ммоль) и охлаждали раствор до 0 С. За один раз добавляли гидрохлорид 1-(3-диметиламинопропил)-3-этилкарбодиимида (0,204 г; 1,5 экв.; 1,06 ммоль). Оставляли смесь нагреваться до комнатной температуры и перемешивали в течение 30 мин. Добавляли дихлорметан (2 мл), нагревали до 40 С и перемешивали в течение 24 ч. Разбавляли 250 мл воды, а затем 50 мл этилацетата. Экстрагировали смесь этилацетатом (525 мл). Промывали объединенные органические экстракты солевым раствором (10 мл). Сушили органические слои над сульфатом магния, фильтровали, концентрировали и очищали путем колоночной хроматографии (от гексана до 12% метанола в этилацетате) с получением 1-[2-(2,3-дигидро 1H-инден-2-иламино)-7,8-дигидропиридо[4,3-d]пиримидин-6(5H)-ил]-5-(4H-1,2,4-триазол-3-ил)пентан-1 она (0,08 г; выход 27%) в виде белой пены: МС (m/z): 418 (М+1). Схема G(1,03 г; 1,2 экв.; 5,29 ммоль) в диметилформамиде (20 мл) добавляли гидрид натрия (0,212 г; 1,2 экв.; 5,29 ммоль). Перемешивали смесь в течение 18 ч, затем добавляли воду (15 мл). Экстрагировали смесь дихлорметаном и промывали солевым раствором (20 мл). Сушили органическую фазу над сульфатом натрия, фильтровали и концентрировали с получением неочищенного продукта, который очищали путем колоночной хроматографии (от 0 до 10% метанола в дихлорметане) с получением метил-5-имидазол-1 илпентаноата (0,655 г; 82%) в виде желтой маслянистой жидкости: МС (m/z): 183 (М+1). Пример получения 20 Синтез 5-имидазол-1-илпентановой кислоты Схема G, стадия В. Перемешивали раствор метил-5-имидазол-1-илпентаноата (0,60 г; 1,0 экв.; 3,29 ммоль) и гидроксида лития (0,553 г; 4 экв.; 13,17 ммоль) в тетрагидрофуране (20 мл) и воде (15 мл) в течение 18 ч. Добавляли 3 мл 5 н. хлороводородной кислоты, затем концентрировали смесь. Очищали неочищенный продукт путем колоночной хроматографии (от 0 до 15% метанола в дихлорметане) с получением 5-имидазол-1-илпентановой кислоты (0,72 г; 19%) в виде желтой маслянистой жидкости: МС (m/z): 169 (М+1). Пример 4 Синтез 1-[2-(2,3-дигидро-1H-инден-2-иламино)-7,8-дигидропиридо[4,3-d]пиримидин-6(5H)-ил]-5(1H-имидазол-1-ил)пентан-1-она(0,20 мг; 1,0 экв.; 0,75 ммоль), 5-имидазол-1-илпентановой кислоты (0,152 г; 1,2 экв.; 0,90 ммоль) и 1 гидроксибензотриазола (0,203 г; 2,0 экв.; 1,50 ммоль) в дихлорметане (20 мл) и триэтиламине (0,52 мл) добавляли гидрохлорид 1-(3-диметиламинопропил)-3-этилкарбодиимида (0,288 г; 2,0 экв.; 1,50 ммоль). Перемешивали смесь в течение 18 ч, затем концентрировали, добавляли воду (10 мл) и экстрагировали этилацетатом (320 мл). Сушили объединенные органические экстракты над безводным сульфатом натрия, фильтровали и концентрировали. Очищали неочищенный продукт путем колоночной хроматографии (от 0 до 10% метанола в дихлорметане) с получением 1-[2-(2,3-дигидро-1H-инден-2-иламино)-7,8 дигидропиридо[4,3-d]пиримидин-6(5H)-ил]-5-(1H-имидазол-1-ил)пентан-1-она (0,14 г; 45%) в виде бесцветной маслянистой жидкости: МС (m/z): 417 (М+1). Схема Н Схема Н, стадия А. В выдерживаемый при 0 С раствор N-индан-2-ил-5,6,7,8-тетрагидропиридо[4,3d]пиримидин-2-амина (0,781 г; 1,0 экв.; 2,93 ммоль) в дихлорметане (29 мл) по каплям добавляли триэти- 14025106 ламин (0,82 мл; 2,0 экв.; 5,86 ммоль), затем 2-пропеноилхлорид (0,24 мл; 1,0 экв.; 2,93 ммоль). Перемешивали раствор в течение 16 ч при температуре окружающей среды, затем разбавляли дихлорметаном,промывали солевым раствором, сушили над сульфатом натрия, фильтровали и концентрировали с получением оранжевой маслянистой жидкости. Очищали неочищенный продукт путем колоночной хроматографии с получением 1-[2-(индан-2-иламино)-7,8-дигидро-5H-пиридо[4,3-d]пиримидин-6-ил]проп-2-ен 1-она (0,66 г; 70%). МС (m/z): 321 (М+1). Пример получения 22 Синтез 1-[2-(2,3-дигидро-1H-инден-2-иламино)-7,8-дигидропиридо[4,3-d]пиримидин-6(5H)-ил]-3(проп-2-ин-1-илокси)пропан-1-она Схема Н, стадия В. В раствор 1-[2-(индан-2-иламино)-7,8-дигидро-5H-пиридо[4,3-d]пиримидин-6 ил]проп-2-ен-1-она (0,5 г; 1,0 экв.; 1,56 ммоль) и 2-пропин-1-ола (5,0 мл; 55 экв.; 85,86 ммоль) добавляли 1,8-диазабицикло[5.4.0]ундец-7-ен (0,035 мл; 0,15 экв.; 0,23 ммоль). Перемешивали реакционную смесь при температуре окружающей среды в течение 16 ч, затем нагревали до 50 С в течение 36 ч. Концентрировали раствор с получением коричневой маслянистой жидкости, затем очищали неочищенный продукт путем колоночной хроматографии (от 20 до 100% этилацетата в дихлорметане) с получением целевого 1[2-(2,3-дигидро-1H-инден-2-иламино)-7,8-дигидропиридо[4,3-d]пиримидин-6(5H)-ил]-3-(проп-2-ин-1 илокси)пропан-1-она (0,28 г; 48%): МС (m/z): 377 (М+1). Пример 5 Синтез 1-[2-(2,3-дигидро-1H-инден-2-иламино)-7,8-дигидропиридо[4,3-d]пиримидин-6(5H)-ил]-3(1 Н-1,2,3-триазол-4-илметокси)пропан-1-она Схема Н, стадия С. В раствор 1-[2-(2,3-дигидро-1H-инден-2-иламино)-7,8-дигидропиридо[4,3-d]пиримидин-6(5H)-ил]-3-(проп-2-ин-1-илокси)пропан-1-она (0,277 г; 1,0 экв.; 0,74 ммоль) в диметилформамиде (4 мл) и воде (4 мл) добавляли пентагидрат сульфата меди (II) (0,037 г; 0,2 экв.; 0,147 ммоль) и натриевую соль L-аскорбиновой кислоты (0,291 г; 2,0 экв.; 1,47 ммоль). Дважды вакуумировали систему и повторно заполняли азотом. Добавляли азидотриметилсилан (0,784 мл; 8,0 экв.; 5,89 ммоль) и нагревали гетерогенную смесь до 90 С в течение 1 ч. Разбавляли реакционную смесь водой (250 мл) и этилацетатом(50 мл) и экстрагировали этилацетатом (350 мл). Промывали объединенные органические экстракты солевым раствором (30 мл), сушили над сульфатом натрия, фильтровали и концентрировали в вакууме с получением желтой/оранжевой маслянистой жидкости. Очищали неочищенный продукт путем колоночной хроматографии (от этилацетата до 10% метанола в этилацетате) с получением целевого 1-[2-(2,3 дигидро-1H-инден-2-иламино)-7,8-дигидропиридо[4,3-d]пиримидин-6(5H)-ил]-3-(1H-1,2,3-триазол-4 илметокси)пропан-1-она (0,135 г; 44%): МС (m/z): 420 (М+1). Схема I Схема I, стадия А. В раствор 4-йод-1H-имидазола (0,388 г; 1,0 экв.; 2,00 ммоль), бензилпент-4 иноата (0,376 г; 1,0 экв.; 2,00 ммоль) и триэтиламина (0,84 мл; 3 экв.; 6,0 ммоль) в диметилформамиде (8 мл), из которого удаляли кислород, добавляли хлорид бис(трифенилфосфин)палладия (II) (0,14 г; 0,1 экв.; 0,20 ммоль) и йодид меди (I) (0,039 мг; 0,1 экв.; 0,20 ммоль). Нагревали смесь до 75 С в течение 16 ч, затем разбавляли смесь водой (30 мл) и экстрагировали 3 этилацетатом. Промывали объединенные органические экстракты солевым раствором, сушили над сульфатом магния, фильтровали и концентрировали с получением неочищенного продукта. Очищали неочищенный продукт путем колоночной хроматографии (от 0 до 10% метанола в дихлорметане) с получением бензил-5-(1H-имидазол-5-ил)пент-4 иноата (0,35 г; 69%) в виде бесцветной маслянистой жидкости: МС (m/z): 255 (М+1). Пример получения 24 Синтез трет-бутил-5-(5-бензилокси-5-оксопент-1-инил)имидазол-1-карбоксилата Схема I, стадия В. Перемешивали раствор бензил-5-(1H-имидазол-5-ил)пент-4-иноата (0,19 г; 1,00 экв.; 0,747 ммоль), ди-трет-бутилдикарбоната (0,210 г; 1,29 экв.; 0,96 ммоль), триэтиламина (0,21 мл; 2,0 экв.; 1,49 ммоль) и N,N-диметил-4-пиридинамина (9 мг; 0,1 экв.; 0,074 ммоль) в дихлорметане (10 мл) в течение 2 ч. Концентрировали раствор и очищали неочищенный продукт путем колоночной хроматографии (25% этилацетата в гексанах) с получением трет-бутил-5-(5-бензилокси-5-оксопент-1-инил)имидазол-1-карбоксилата (0,170 г; 64%) в виде белого твердого вещества: МС (m/z): 299 (M-t-Bu+1). Пример получения 25 Синтез 5-(3-трет-бутоксикарбонилимидазол-4-ил)пентановой кислоты Схема I, стадия С. Интенсивно перемешивали выдерживаемый при 60 С раствор трет-бутил-5-(5 бензилокси-5-оксопент-1-инил)имидазол-1-карбоксилата (0,17 г; 1,0 экв.; 0,48 ммоль) и палладия (10% на углеродной подложке; 0,030 г; 0,014 ммоль) в метаноле (13 мл) при 1 атмосфере водорода в течение 16 ч. Фильтровали и концентрировали с получением целевой 5-(3-трет-бутоксикарбонилимидазол-4-ил)пентановой кислоты (0,128 г; 99%) в виде белого твердого вещества: МС (m/z): 213 (M-t-Bu+1). Пример получения 26 Синтез трет-бутил-5-[5-[2-(индан-2-иламино)-7,8-дигидро-5H-пиридо[4,3-d]пиримидин-6-ил]-5-оксопентил]имидазол-1-карбоксилата Схема I, стадия D. В раствор 5-(3-трет-бутоксикарбонилимидазол-4-ил)пентановой кислоты (0,084 г; 1,0 экв.; 0,31 ммоль), N-индан-2-ил-5,6,7,8-тетрагидропиридо[4,3-d]пиримидин-2-амина (0,064 г; 0,77 экв.; 0,24 ммоль), 1-гидроксибензотриазола (0,065 г; 1,5 экв.; 0,48 ммоль) и триэтиламина (0,20 мл; 4,5 экв.; 1,44 ммоль) в дихлорметане (15 мл) добавляли гидрохлорид 1-(3-диметиламинопропил)-3-этилкарбодиимида (0,092 г; 1,5 экв.; 0,48 ммоль). Нагревали смесь до 40 С в течение 16 ч. Концентрировали смесь, а затем очищали неочищенный продукт путем колоночной хроматографии (от 0 до 10% метанола в дихлорметане) с получением трет-бутил-5-[5-[2-(индан-2-иламино)-7,8-дигидро-5H-пиридо[4,3-d]пиримидин-6-ил]-5-оксопентил]имидазол-1-карбоксилата (0,040 г; 25%) в виде белого твердого вещества: МС (m/z): 517 (М+1). Пример 6 Синтез 1-[2-(2,3-дигидро-1H-инден-2-иламино)-7,8-дигидропиридо[4,3-d]пиримидин-6(5H)-ил]-5(1H-имидазол-4-ил)пентан-1-онаd]пиримидин-6-ил]-5-оксопентил]имидазол-1-карбоксилата (0,030 г; 0,058 ммоль) в дихлорметане (8 мл) добавляли трифторуксусную кислоту (1,5 мл). Перемешивали реакционную смесь при комнатной температуре в течение 2 ч. Концентрировали смесь и добавляли насыщенный бикарбонат натрия (10 мл) и воду (20 мл), затем экстрагировали дихлорметаном (320 мл). Промывали объединенные органические экстракты солевым раствором, сушили над сульфатом натрия, фильтровали и концентрировали. Очищали неочищенный продукт путем колоночной хроматографии (от 0 до 10% метанола в дихлорметане) с получением 1-[2-(2,3-дигидро-1H-инден-2-иламино)-7,8-дигидропиридо[4,3-d]пиримидин-6(5H)-ил]-5-(1Hимидазол-4-ил)пентан-1-она (0,020 г; 83%) в виде белого твердого вещества: МС (m/z): 417 (М+1). Схема J Схема J, стадия А. В раствор 3-хлорпропил-2-(индан-2-иламино)-7,8-дигидро-5H-пиридо[4,3-d]пиримидин-6-карбоксилата (0,22 г; 1,0 экв.; 0,57 ммоль) и 1 Н-имидазола (0,082 г; 2,1 экв.; 1,20 ммоль) в диметилформамиде (2 мл) добавляли гидрид натрия (0,050 г; 2,2 экв.; 1,25 ммоль) и перемешивали в течение 18 ч. Разбавляли реакционную смесь дихлорметаном и водой. Разделяли слои и дополнительно экстрагировали водный слой дихлорметаном (2). Сушили объединенные органические экстракты сульфатом натрия, фильтровали и концентрировали. Очищали неочищенный продукт путем колоночной хроматографии (от 0 до 10% метанола в дихлорметане) с получением целевого 3-(1H-имидазол-1-ил)пропил 2-(2,3-дигидро-1H-инден-2-иламино)-7,8-дигидропиридо[4,3-d]пиримидин-6(5H)-карбоксилата (0,078 г; 33%) в виде белого твердого вещества: МС (m/z): 419 (М+1). Схема K до[4,3-d]пиримидин-6-карбоксилата (0,42 г; 1,0 экв.; 1,09 ммоль) и цианида натрия (0,085 г; 1,6 экв.; 1,73 ммоль) в диметилформамиде (3 мл) до 100 С в течение 2 ч. Охлаждали смесь до температуры окружающей среды, разбавляли дихлорметаном (20 мл) и промывали реакционную смесь водой (25 мл). Экстрагировали водный слой дихлорметаном (220 мл). Сушили объединенные органические экстракты над сульфатом натрия, фильтровали и концентрировали с получением неочищенного продукта. Объединяли неочищенный продукт и очищали путем колоночной хроматографии (от 0 до 10% метанола в дихлорметане) с получением 3-цианопропил-2-(2,3-дигидро-1H-инден-2-иламино)-7,8-дигидропиридо[4,3-d]пиримидин-6(5H)-карбоксилата в виде светло-коричневой маслянистой жидкости, который использовали на следующей стадии. МС (m/z): 378 (М+1). Пример 8 Пример получения 3-(1H-тетразол-5-ил)пропил-2-(2,3-дигидро-1H-инден-2-иламино)-7,8-дигидропиридо[4,3-d]пиримидин-6(5H)-карбоксилата(0,85 мл; 10 экв.; 6,38 ммоль) и дибутилоксостаннана (0,040 г; 0,25 экв.; 0,16 ммоль) в толуоле (5 мл) до 100 С в течение 24 ч, затем охлаждали до температуры окружающей среды и концентрировали. Очищали неочищенное вещество путем обращенно-фазовой хроматографии с получением продукта, содержащего примеси. Растворяли продукт, содержащий примеси, в 50 мл дихлорметана и промывали насыщенным бикарбонатом натрия и дополнительно экстрагировали водный слой 2 дихлорметаном. Сушили объединенные органические экстракты над сульфатом натрия, фильтровали и концентрировали с получением 3(1H-тетразол-5-ил)пропил-2-(2,3-дигидро-1H-инден-2-иламино)-7,8-дигидропиридо[4,3-d]пиримидин 6(5H)-карбоксилата (0,110 г; 41%) в виде желтого твердого вещества: МС (m/z): 421 (М+1). Схема L(0,205 мл; 1,2 экв.; 1,47 ммоль) в тетрагидрофуране (8 мл) быстро добавляли бут-3-ин-1-амин (2,71 г; 8 экв.; 9,80 ммоль). Через 15 мин нагревали раствор до 45 С в течение 2 ч, затем концентрировали смесь и перерастворяли остаток в дихлорметане (3 мл). Добавляли ди-трет-бутилдикарбонат (0,802 г; 3 экв.; 3,68 ммоль) и перемешивали при температуре окружающей среды в течение 1 ч. Концентрировали смесь и очищали неочищенный продукт путем колоночной хроматографии (от 0 до 100% этилацетат/гексаны) с получением трет-бутил-N-бут-3-инил-N-[2-[2-(индан-2-иламино)-7,8-дигидро-5H-пиридо[4,3-d]пиримидин-6-ил]-2-оксоэтил]карбамата (0,504 г; 87%) в виде белого твердого вещества: МС (m/z): 476 (М+1).(6 мл) и воде (3 мл) (содержащий толуол (0,05 мл) в качестве внутреннего стандарта), из которого удаляли кислород, добавляли пентагидрат сульфата меди (II) (0,026 г; 0,2 экв.; 0,106 ммоль) и натриевую сольL-аскорбиновой кислоты (0,21 г; 2,0 экв.; 1,06 ммоль). Нагревали реакционный сосуд до 90 С, а затем медленно добавляли азидотриметилсилан (0,565 мл; 8 экв.; 4,24 ммоль). Перемешивали при 90 С в течение 4 ч, затем разбавляли реакционную смесь водой (100 мл) и этилацетатом (50 мл) и экстрагировали 350 мл этилацетата. Промывали объединенные органические экстракты солевым раствором, сушили над сульфатом магния, фильтровали и концентрировали. Очищали неочищенный продукт путем колоночной хроматографии (от 0 до 10% метанола в этилацетате) с получением трет-бутил-2-[2-(2,3 дигидро-1H-инден-2-иламино)-7,8-дигидропиридо[4,3-d]пиримидин-6(5H)-ил]-2-оксоэтил[2-(1H-1,2,3 триазол-4-ил)этил]карбамата (0,174 г; 63%) в виде бесцветной пены: МС (m/z): 519 (М+1). Пример 9 Синтез 1-[2-(2,3-дигидро-1H-инден-2-иламино)-7,8-дигидропиридо[4,3-d]пиримидин-6(5H)-ил]-2[2-(1H-1,2,3-триазол-4-ил)этил]аминоэтанона(1 М в диэтиловом эфире; 2,55 мл; 8 экв.; 2,55 ммоль). После перемешивания в течение 1 ч при 0 С концентрировали смесь. Перерастворяли неочищенный продукт в смеси 10:1 дихлорметан: метанол и промывали насыщенным бикарбонатом натрия. Дополнительно экстрагировали водный слой 10:1 смесью дихлорметан:метанол (4). Сушили объединенные органические экстракты над сульфатом магния, фильтровали и концентрировали с получением 1-[2-(2,3-дигидро-1H-инден-2-иламино)-7,8-дигидропиридо[4,3-d]пиримидин-6(5H)-ил]-2-[2-(1H-1,2,3-триазол-4-ил)этил]аминоэтанона (0,136 г; 102%) в виде коричневой пены: МС (m/z): 419 (М+1). Схема М Схема М, стадия А. В выдерживаемый при 0 С раствор 1H-имидазол-1-этанола (2,01 г; 1,0 экв.; 17,93 ммоль) в тетрагидрофуране (90 мл) медленно добавляли гидрид натрия (0,79 г; 1,1 экв.; 19,75 ммоль). Перемешивали в течение 1 ч, затем по каплям добавляли 1,1-диметилэтиловый эфир бромуксусной кислоты (4,03 мл; 1,49 экв.; 26,76 ммоль) при 0 С. Нагревали раствор до температуры окружающей среды и перемешивали в течение 3 ч, затем добавляли 250 мл насыщенного водного раствора бикарбона- 19025106 та натрия. Экстрагировали смесь дихлорметаном (3150 мл). Промывали объединенные органические экстракты солевым раствором (100 мл), сушили над сульфатом натрия, фильтровали и концентрировали с получением неочищенного продукта. Очищали полученный остаток путем колоночной хроматографии Схема М, стадия В. В раствор трет-бутил-2-(2-имидазол-1-илэтокси)ацетата (0,375 г; 1,0 экв.; 1,66 ммоль) в дихлорметане (10 мл) по каплям добавляли трифторуксусную кислоту (2 мл). Перемешивали при комнатной температуре в течение 4 ч, а затем концентрировали смесь. Перерастворяли полученный остаток в диметилформамиде (5 мл) и добавляли диизопропилэтиламин (1,45 мл; 5 экв.; 8,31 ммоль), затем N-индан-2-ил-5,6,7,8-тетрагидропиридо[4,3-d]пиримидин-2-амин (0,44 г; 1,0 экв.; 1,65 ммоль). Добавляли гексафторфосфат О-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония (0,69 г; 1,1 экв.; 1,81 ммоль) и перемешивали при комнатной температуре в течение 16 ч. Выливали реакционную смесь в насыщенный водный раствор бикарбоната натрия (200 мл) и экстрагировали дихлорметаном (2200 мл). Промывали объединенные органические экстракты насыщенным водным раствором бикарбоната натрия(100 мл), сушили над сульфатом натрия, фильтровали и концентрировали с получением неочищенного продукта. Очищали полученный остаток путем колоночной хроматографии (от 0 до 20% метанола в дихлорметане) с получением 1-[2-(2,3-дигидро-1H-инден-2-иламино)-7,8-дигидропиридо[4,3-d]пиримидин 6(5H)-ил]-2-[2-(1H-имидазол-1-ил)этокси]этанона (0,152 г; 22%) в виде светло-коричневого твердого вещества: МС (m/z): 419 (М+1). Схема N Схема N, стадии А и В. В выдерживаемый при -78 С раствор 4-пентин-1-ола (0,50 мл; 5,39 ммоль) в дихлорметане (18 мл) медленно по каплям добавляли 1,1'-карбонилдиимидазол (0,936 г; 5,66 ммоль). Оставляли реакционную смесь нагреваться до температуры окружающей среды на 30 мин, затем половину полученного раствора добавляли в раствор N-индан-2-ил-5,6,7,8-тетрагидропиридо[4,3-d]пиримидин 2-амина (0,581 г; 1,05 экв.; 2,18 ммоль), N,N-диметил-4-пиридинамина (0,258 г; 1,0 экв.; 2,08 ммоль) и триэтиламина (0,58 мл; 2,0 экв.; 4,15 ммоль). Нагревали полученный бледно-желтый раствор до температуры окружающей среды и перемешивали в течение 16 ч, затем дополнительно нагревали до 40 С в течение 24 ч. Охлаждали реакционную смесь до температуры окружающей среды и непосредственно помещали в колонку с силикагелем. Очищали путем колоночной хроматографии (от 20 до 100% этилацетата в дихлорметане) с получением пент-4-ин-1-ил-2-(2,3-дигидро-1H-инден-2-иламино)-7,8-дигидропиридо[4,3-d]пиримидин-6(5H)-карбоксилата (0,76 г; 88%): МС (m/z): 377 (М+1). Пример 11 Синтез 3-(1 Н-1,2,3-триазол-4-ил)пропил-2-(2,3-дигидро-1H-инден-2-иламино)-7,8-дигидропиридо[4,3-d]пиримидин-6(5H)-карбоксилата Схема N, стадия С. В раствор пент-4-ин-1-ил-2-(2,3-дигидро-1H-инден-2-иламино)-7,8-дигидропиридо[4,3-d]пиримидин-6(5H)-карбоксилата (0,754 г; 1,0 экв.; 1,80 ммоль) в диметилформамиде (15 мл) и воде (15 мл), из которого удаляли кислород, добавляли пентагидрат сульфата меди (II) (0,090 г; 0,2 экв.; 0,36 ммоль) и натриевую соль L-аскорбиновой кислоты (0,714 г; 2,0 экв.; 3,61 ммоль). Добавляли азидотриметилсилан (1,92 мл; 8 экв.; 14,42 ммоль) и нагревали реакционную смесь до 90 С в течение 3 ч. Разбавляли реакционную смесь водой (250 мл) и этилацетатом Экстрагировали смесь этилацетатом (350 мл) Промывали объединенные органические экстракты водой (3) и солевым раствором, сушили над сульфатом натрия, фильтровали и концентрировали с получением коричневатой/оранжевой маслянистой жидкости Очищали неочищенный продукт путем колоночной хроматографии (от 0 до 5% метанола в дихлорметане) с получением 3-(1 Н-1,2,3-триазол-4-ил)пропил-2-(2,3-дигидро-1H-инден-2-иламино)-7,8 дигидропиридо[4,3-d]пиримидин-6(5H)-карбоксилата (0,26 г, 35%) МС (m/z) 420 (М+1). Схема О, стадия А В колбу, содержащую гидрат дигидрохлорида N-(2,3-дигидро-1 Н-инден-2-ил)6,7-дигидро-5H-пирроло [3,4-d] пиримидин-2-амина (1,96 г, 1,0 экв, 6,03 ммоль) и 5-бромвалериановую кислоту (1,4 г, 1,25 экв, 7,50 ммоль) в дихлорметане (25 мл), добавляли гидрохлорид 1-(3-диметиламинопропил)-3-этилкарбодиимида (1,33 г; 1,15 экв.; 6,94 ммоль). Перемешивали полученную реакционную смесь в течение 2 ч, затем непосредственно концентрировали смесь. Разделяли остаток в воде и дихлорметане и концентрировали органический экстракт. Очищали неочищенное вещество путем колоночной хроматографии (от 70 до 90% смеси этилацетат/гексаны) с получением промежуточного 5-бромвалерамида (0,66 г), который растворяли в диметилформамиде (10 мл). Добавляли цианид натрия (0,128 г; 2,48 ммоль) и нагревали полученную смесь до 100 С в течение 3 ч, затем охлаждали до температуры окружающей среды. Разделяли реакционную смесь в этилацетате и воде и отделяли слои. Экстрагировали водный слой этилацетатом (2100 мл) и промывали объединенные органические экстракты 1 н. хлороводородной кислотой, затем солевым раствором. Сушили объединенные органические экстракты над сульфатом натрия, фильтровали и концентрировали с получением неочищенного продукта. Очищали неочищенный продукт путем колоночной хроматографии (от 5 до 10% метанола в дихлорметане) с получением 6-[2-(индан-2-иламино)-5,7-дигидропиридо[3,4-d]пиримидин-6-ил]-6-оксогексаннитрила (0,0772 г; 4%): МС (m/z): 362 (М+1). Схема О, стадия В. В перемешиваемый раствор 6-[2-(индан-2-иламино)-5,7-дигидропиридо[3,4d]пиримидин-6-ил]-6-оксогексаннитрила (0,077 г; 1,0 экв.; 0,213 ммоль) в толуоле (6 мл) добавляли дибутилоксостаннан (0,071 г; 1,33 экв.; 0,29 ммоль) и азидотриметилсилан (1,6 мл; 56 экв.; 12,01 ммоль). Нагревали полученный раствор до 105 С в течение 16 ч. Охлаждали реакционную смесь до температуры окружающей среды и концентрировали. Растворяли остаток в метаноле (5 мл) и помещали раствор в колонку SCX (элюировали метанолом, затем 7 н. аммиаком в метаноле) с получением неочищенного продукта, который очищали путем обращенно-фазовой хроматографии с получением 1-[2-(2,3-дигидро-1Hинден-2-иламино)-5,7-дигидро-6H-пирроло[3,4-d]пиримидин-6-ил]-5-(1 Н-тетразол-5-ил)пентан-1-она Схема Р, стадия А. В раствор 3-хлорпропил-2-(индан-2-иламино)-5,7-дигидропирроло[3,4-d]пиримидин-6-карбоксилата (0,283 г; 1,0 экв.; 0,76 ммоль) и 1 Н-имидазола (0,105 г; 2,0 экв.; 1,54 ммоль) в диметилформамиде (2,4 мл) добавляли гидрид натрия (0,068 г; 2,25 экв.; 1,70 ммоль). Перемешивали реакционную смесь в течение 16 ч, затем разбавляли дихлорметаном и промывали водой. Дважды экстрагировали водный слой дихлорметаном. Концентрировали объединенные органические экстракты, а затем очищали неочищенный продукт путем колоночной хроматографии (от 0 до 10% метанола в дихлорметане) с получением 3-(1H-имидазол-1-ил)пропил-2-(2,3-дигидро-1H-инден-2-иламино)-5,7-дигидро-6Hпирроло[3,4-d]пиримидин-6-карбоксилата (0,0922 г; 30%) в виде желтого твердого вещества: МС (m/z): 405 (М+1). Схема Q, стадия А. В колбу, содержащую 2-хлор-1-[2-(2,3-дигидро-1H-инден-2-иламино)-5,7-дигидро-6H-пирроло[3,4-d]пиримидин-6-ил]этанон (0,184 г; 1,0 экв.; 0,56 ммоль) и триэтиламин (0,094 мл; 1,2 экв.; 0,67 ммоль) в тетрагидрофуране (5 мл), быстро добавляли бут-3-ин-1-амин (1,24 г; 8 экв.; 4,48 ммоль). Перемешивали при температуре окружающей среды в течение 15 мин, затем нагревали до 55 С в течение 1 ч и до 65 С в течение 30 мин. Концентрировали смесь, а затем растворяли остаток в дихлорметане (10 мл). Добавляли ди-трет-бутилдикарбонат (0,8 г; 6,5 экв.; 3,67 ммоль) и перемешивали при комнатной температуре в течение 30 мин. Помещали раствор непосредственно в колонку с силикагелем и очищали путем хроматографии (от 0 до 100% смеси этилацетат/гексаны) с получением трет-бутил-N-бут 3-инил-N-[2-[2-(индан-2-иламино)-5,7-дигидропирроло[3,4-d]пиримидин-6-ил]-2-оксоэтил]карбамата Схема Q, стадия В. В раствор трет-бутил-N-бут-3-инил-N-[2-[2-(индан-2-иламино)-5,7-дигидропирроло[3,4-d]пиримидин-6-ил]-2-оксоэтил]карбамата (0,204 г; 1,0 экв.; 0,44 ммоль) в диметилформамиде (6 мл) и воде (3 мл) (включающий толуол (0,05 мл) в качестве внутреннего стандарта), из которого удаляли кислород, добавляли пентагидрат сульфата меди (II) (0,022 г; 0,2 экв.; 0,088 ммоль) и натриевую соль Lаскорбиновой кислоты (0,175 г; 2,0 экв.; 0,88 ммоль). Нагревали до 90 С, а затем по каплям добавляли азидотриметилсилан (0,47 мл; 8 экв.; 3,54 ммоль) и продолжали перемешивать при 90 С. Перемешивали в течение 3 ч, затем разбавляли реакционную смесь водой (100 мл) и этилацетатом (50 мл). Экстрагировали этилацетатом (350 мл) и промывали объединенные органические экстракты солевым раствором. Сушили органические слои над сульфатом магния, фильтровали и концентрировали. Очищали неочищенный продукт путем колоночной хроматографии (от 0 до 10% метанола в этилацетате) с получением трет-бутил-2-[2-(2,3-дигидро-1H-инден-2-иламино)-5,7-дигидро-6H-пирроло[3,4-d]пиримидин-6-ил]-2 оксоэтил[2-(1H-1,2,3-триазол-5-ил)этил]карбамата (0,157 г; 70%) в виде желтоватой пены: МС (m/z): 505 (М+1). Пример 14 Синтез 1-[2-(2,3-дигидро-1H-инден-2-иламино)-5,7-дигидро-6H-пирроло[3,4-d]пиримидин-6-ил]-2[2-(1H-1,2,3-триазол-5-ил)этил]аминоэтанона Схема Q, стадия С. В выдерживаемый при 0 С раствор, содержащий трет-бутил-2-[2-(2,3-дигидро 1H-инден-2-иламино)-5,7-дигидро-6H-пирроло[3,4-d]пиримидин-6-ил]-2-оксоэтил[2-(1H-1,2,3-триазол 5-ил)этил]карбамат (0,15 г; 1,0 экв.; 0,297 ммоль) в дихлорметане (1 мл), медленно добавляли трифторуксусную кислоту (3 мл). Перемешивали в течение 2 ч, затем нагревали до температуры окружающей среды и перемешивали в течение 30 мин. Концентрировали смесь и растворяли остаток в дихлорметане (20 мл). Добавляли насыщенный бикарбонат натрия (20 мл), солевой раствор (20 мл) и воду (50 мл) и экстрагировали дихлорметаном (620 мл). Сушили объединенные органические экстракты на сульфатом магния, фильтровали и концентрировали с получением 1-[2-(2,3-дигидро-1H-инден-2-иламино)-5,7-дигидро 6H-пирроло[3,4-d]пиримидин-6-ил]-2-[2-(1H-1,2,3-триазол-5-ил)этил]аминоэтанона (0,093 г; 77%) в виде желтоватой пены: МС (m/z): 405 (М+1). Схема R, стадия А. В смесь гидрата дигидрохлорида N-(2,3-дигидро-1H-инден-2-ил)-6,7-дигидро 5H-пирроло[3,4-d]пиримидин-2-амина (1,0 г; 1,0 экв.; 3,07 ммоль) и триэтиламина (2,2 мл; 5,1 экв.; 15,78 ммоль) в тетрагидрофуране (21 мл) по каплям в течение 1 ч добавляли раствор 3-хлорпропилкарбонохлоридата (0,55 мл; 1,48 экв.; 4,54 ммоль) в тетрагидрофуране (21 мл). Перемешивали раствор в течение 18 ч, затем разбавляли дихлорметаном и промывали раствором бикарбоната натрия. Дополнительно экстрагировали водный слой дихлорметаном (2). Сушили объединенные органические экстракты над сульфатом натрия, фильтровали и концентрировали с получением 3-хлорпропил-2-(индан-2-иламино)5,7-дигидропирроло[3,4-d]пиримидин-6-карбоксилата (1,15 г; 100%) в виде коричневого твердого вещества: МС (m/z): 373 (М+1). Пример получения 36 Синтез 3-цианопропил-2-(индан-2-иламино)-5,7-дигидропирроло[3,4-d]пиримидин-6-карбоксилата Схема R, стадия В. В раствор, содержащий 3-хлорпропил-2-(индан-2-иламино)-5,7-дигидропирроло[3,4-d]пиримидин-6-карбоксилат (1,15 г; 1,0 экв.; 3,08 ммоль) в диметилформамиде (10 мл), добавляли цианид натрия (0,230 г; 1,5 экв.; 4,69 ммоль). Нагревали реакционную смесь до 100 С в течение 3 ч, затем охлаждали до температуры окружающей среды, разбавляли дихлорметаном, водой и 5% раствором хлорида лития. Разделяли слои и дополнительно экстрагировали водный слой дихлорметаном и этилацетатом. Сушили объединенные органические экстракты над сульфатом натрия, фильтровали и концентрировали с получением 3-цианопропил-2-(индан-2-иламино)-5,7-дигидропирроло[3,4-d]пиримидин-6 карбоксилата (0,98 г; 87%) в виде коричневого твердого вещества: МС (m/z): 364 (М+1). Пример 15 Синтез 3-(1H-тетразол-5-ил)пропил-2-(2,3-дигидро-1H-инден-2-иламино)-5,7-дигидро-6H-пирроло[3,4-d]пиримидин-6-карбоксилата(3,5 мл; 9,7 экв.; 26,27 ммоль) и дибутилоксостаннан (0,163 г; 0,25 экв.; 0,66 ммоль). Нагревали реакционную смесь до 100 С в течение 18 ч. Концентрировали раствор и очищали путем обращенно-фазовой хроматографии, затем путем хроматографии на силикагеле (от 0 до 15% метанола в хлороформе) с получением целевого 3-(1H-тетразол-5-ил)пропил-2-(2,3-дигидро-1H-инден-2-иламино)-5,7-дигидро-6H-пирроло[3,4-d]пиримидин-6-карбоксилата (0,097 г; 9%) в виде коричневого твердого вещества: МС (m/z): 407 (М+1).(0,265 г; 1,0 экв.; 1,50 ммоль) добавляли N-индан-2-ил-5,6,7,8-тетрагидропиридо[4,3-d]пиримидин-2 амин (0,418 г; 1,05 экв.; 1,57 ммоль), затем N,N-диметил-4-пиридинамин (0,185 г; 1,0 экв.; 1,50 ммоль) и триэтиламин (0,42 мл; 2,0 экв.; 2,99 ммоль). Нагревали полученный бледно-желтый раствор до комнатной температуры и перемешивали в течение 18 ч. Помещали реакционную смесь непосредственно в предколонку с оксидом кремния и очищали путем колоночной хроматографии (от 10 до 50% ацетона в гексанах) с получением 2-(2,3-дигидро-1H-инден-2-иламино)-N-(пент-4-ин-1-ил)-7,8-дигидропиридо[4,3d]пиримидин-6(5H)-карбоксамида (0,286 г; 51%) в виде белого твердого вещества: МС (m/z): 376 (М+1). Пример 16 Синтез 2-(2,3-дигидро-1H-инден-2-иламино)-N-[3-(1H-1,2,3-триазол-4-ил)пропил]-7,8-дигидропиридо[4,3-d]пиримидин-6(5H)-карбоксамида Схема S, стадия В. В раствор, содержащий 2-(2,3-дигидро-1H-инден-2-иламино)-N-(пент-4-ин-1 ил)-7,8-дигидропиридо[4,3-d]пиримидин-6(5H)-карбоксамид (0,275 г; 1,0 экв.; 0,73 ммоль) в диметилформамиде (2,44 мл) и воде (2,44 мл), из которого удаляли кислород, добавляли пентагидрат сульфата меди (II) (0,037 г; 0,2 экв.; 0,15 ммоль) и натриевую соль L-аскорбиновой кислоты (0,29 г; 2,0 экв.; 1,46 ммоль). Добавляли азидотриметилсилан (0,78 мл; 8 экв.; 5,86 ммоль) и нагревали до 90 С в течение 1 ч. Охлаждали раствор до температуры окружающей среды, разбавляли реакционную смесь 30 мл воды и экстрагировали этилацетатом (350 мл). Промывали объединенные органические экстракты водой и солевым раствором, сушили над сульфатом натрия, фильтровали и концентрировали в вакууме с получением желтой маслянистой жидкости. Очищали неочищенный продукт путем колоночной хроматографии(от 0 до 80% ацетона в дихлорметане) с получением 2-(2,3-дигидро-1H-инден-2-иламино)-N-[3-(1H-1,2,3 триазол-4-ил)пропил]-7,8-дигидропиридо[4,3-d]пиримидин-6(5H)-карбоксамида (0,132 г; 43%) в виде белого твердого вещества: МС (m/z): 419 (М+1). Схема Т, стадия А. В выдерживаемый при -78 С раствор 4-пентин-1-амина (0,258 мл; 1,0 экв.; 3,10 ммоль) в дихлорметане (10 мл) медленно по частям добавляли 1,1'-карбонилдиимидазол (0,539 мг; 1,05 экв.; 3,26 ммоль). Оставляли реакционную смесь нагреваться до температуры окружающей среды и перемешивали в течение 72 ч. Использовали раствор непосредственно на следующих стадиях химических превращений: МС (m/z): 178 (М+1). Пример получения 39 Синтез 2-(2,3-дигидро-1H-инден-2-иламино)-N-(пент-4-ин-1-ил)-5,7-дигидро-6H-пирроло[3,4-d]пиримидин-6-карбоксамида(0,275 г; 1,0 экв.; 1,55 ммоль) в дихлорметане (5 мл) добавляли гидрат дигидрохлорида N-(2,3-дигидро 1H-инден-2-ил)-6,7-дигидро-5H-пирроло[3,4-d]пиримидин-2-амина (0,266 г; 0,5 экв.; 0,78 ммоль), затем триэтиламин (1,08 мл; 5 экв.; 7,76 ммоль) и N,N-диметил-4-пиридинамин (0,029 г; 0,15 экв.; 0,23 ммоль). Оставляли светло-коричневый раствор нагреваться до температуры окружающей среды и перемешивали в течение 24 ч. Помещали реакционную смесь непосредственно в колонку с силикагелем и очищали путем колоночной хроматографии (от 0 до 30% этилацетата в гексанах) с получением 2-(2,3-дигидро-1Hинден-2-иламино)-N-(пент-4-ин-1-ил)-5,7-дигидро-6H-пирроло[3,4-d]пиримидин-6-карбоксамида (0,293 г; 63%) в виде белого твердого вещества: МС (m/z): 362 (М+1). Пример 17 Синтез 2-(2,3-дигидро-1H-инден-2-иламино)-N-[3-(1H-1,2,3-триазол-4-ил)пропил]-5,7-дигидро-6Hпирроло[3,4-d]пиримидин-6-карбоксамида Схема Т, стадия С. В раствор 2-(2,3-дигидро-1H-инден-2-иламино)-N-(пент-4-ин-1-ил)-5,7-дигидро 6H-пирроло[3,4-d]пиримидин-6-карбоксамида (0,354 г; 1,0 экв.; 0,98 ммоль) в диметилформамиде (3,26 мл) и воде (3,26 мл), из которого удаляли кислород, добавляли пентагидрат сульфата меди (II) (0,049 г; 0,2 экв.; 0,20 ммоль) и натриевую соль L-аскорбиновой кислоты (0,388 г; 2,0 экв.; 1,96 ммоль). Добавляли азидотриметилсилан (1,04 мл; 8 экв.; 7,84 ммоль) и нагревали смесь до 90 С в течение 1 ч, охлаждали до температуры окружающей среды и перемешивали в течение 16 ч. Концентрировали раствор досуха и очищали полученное оранжевое полутвердое вещество путем колоночной хроматографии (от 0 до 10% метанола в этилацетате) с получением 2-(2,3-дигидро-1H-инден-2-иламино)-N-[3-(1H-1,2,3-триазол-4 ил)пропил]-5,7-дигидро-6H-пирроло[3,4-d]пиримидин-6-карбоксамида (0,085 г; 21%) в виде твердого вещества: МС (m/z): 405 (М+1). Схема U, стадия А. В раствор 2-хлор-1-[2-(2,3-дигидро-1H-инден-2-иламино)-7,8-дигидропиридо[4,3-d]пиримидин-6(5H)-ил]этанона (0,534 г; 1,0 экв.; 1,56 ммоль) в тетрагидрофуране (5,19 мл) добавляли триэтиламин (0,434 мл; 2,0 экв.; 3,12 ммоль), затем 4-пентин-1-амин (0,196 мл; 1,50 экв.; 2,34 ммоль). Перемешивали в течение 48 ч при 55 С. Раствор непосредственно концентрировали с получением 1-[2-(индан-2-иламино)-7,8-дигидро-5H-пиридо[4,3-d]пиримидин-6-ил]-2-(пент-4-иниламино)этанона Схема U, стадия В. В раствор 1-[2-(индан-2-иламино)-7,8-дигидро-5H-пиридо[4,3-d]пиримидин-6 ил]-2-(пент-4-иниламино)этанона (0,606 г; 1,0 экв.; 1,56 ммоль) в дихлорметане (5,2 мл) добавляли ангидрид уксусной кислоты (0,176 мл; 1,20 экв.; 1,87 ммоль), затем триэтиламин (0,325 мл; 1,5 экв.; 2,33 ммоль). Разбавляли реакционную смесь водой и дихлорметаном. Разделяли слои и промывали органический экстракт солевым раствором, сушили над сульфатом натрия, фильтровали и концентрировали с получением оранжевой/коричневой маслянистой жидкости. Очищали неочищенный продукт путем колоночной хроматографии (от 0 до 2% метанола в этилацетате) с получением целевого N-2-[2-(2,3-дигидро 1H-инден-2-иламино)-7,8-дигидропиридо[4,3-d]пиримидин-6(5 Н)-ил]-2-оксоэтил-N-(пент-4-ин-1 ил)ацетамида (0,138 г; 21%): МС (m/z): 432 (М+1). Пример 18 Синтез N-2-[2-(2,3-дигидро-1H-инден-2-иламино)-7,8-дигидропиридо[4,3-d]пиримидин-6(5H)-ил]2-оксоэтил-N-[3-(1H-1,2,3-триазол-4-ил)пропил]ацетамида Схема U, стадия С. В раствор, содержащий N-2-[2-(2,3-дигидро-1H-инден-2-иламино)-7,8-дигидропиридо[4,3-d]пиримидин-6(5 Н)-ил]-2-оксоэтил-N-(пент-4-ин-1-ил)ацетамид (0,137 г; 1,0 экв.; 0,32 ммоль) в диметилформамиде (2,12 мл) и воде (1,06 мл), из которого удаляли кислород, добавляли пентагидрат сульфата меди (II) (0,016 г; 0,2 экв.; 0,063 ммоль) и натриевую соль L-аскорбиновой кислоты(0,126 г; 2,0 экв.; 0,64 ммоль). Добавляли азидотриметилсилан (0,34 мл; 8 экв.; 2,54 ммоль) и нагревали полученный раствор до 90 С в течение 1 ч. Концентрировали реакционную смесь с получением коричневой пасты и очищали неочищенный продукт путем колоночной хроматографии (от 0 до 10% метанола в этилацетате) с получением N-2-[2-(2,3-дигидро-1H-инден-2-иламино)-7,8-дигидропиридо[4,3-d]пиримидин-6(5H)-ил]-2-оксоэтил-N-[3-(1H-1,2,3-триазол-4-ил)пропил]ацетамида (0,036 г; 25%) в виде беловатого твердого вещества: МС (m/z): 475 (М+1). Схема V Схема V, стадия А. В смесь N-индан-2-ил-5,6,7,8-тетрагидропиридо[4,3-d]пиримидин-2-амина (1,00 г; 1,0 экв.; 3,75 ммоль) и триэтиламина (1,10 мл; 2,1 экв.; 7,88 ммоль) в тетрагидрофуране (25 мл) в течение 1 ч добавляли раствор 3-хлорпропил-карбонохлоридата (0,55 мл; 1,2 экв.; 4,51 ммоль) в тетрагидрофуране (25 мл). Перемешивали в течение 2 ч, затем разбавляли дихлорметаном и насыщенным бикарбонатом натрия и разделяли слои. Дополнительно экстрагировали водный слой дихлорметаном (2). Сушили объединенные органические экстракты над сульфатом натрия, фильтровали и концентрировали досуха с получением желтой маслянистой жидкости. Очищали неочищенный продукт путем колоночной хроматографии (10% метанола в дихлорметане) с получением 3-хлорпропил-2-(индан-2-иламино)-7,8 дигидро-5H-пиридо[4,3-d]пиримидин-6-карбоксилата (1,146 г; 79%): МС (m/z): 387 (М+1). Пример 19 Синтез 3-(5-метил-1H-имидазол-1-ил)пропил-2-(2,3-дигидро-1H-инден-2-иламино)-7,8-дигидропиридо[4,3-d]пиримидин-6(5H)-карбоксилата Схема V, стадия В. В раствор 4(5)-метилимидазола (0,180 г; 1,6 экв.; 2,19 ммоль) в тетрагидрофуране (15 мл) добавляли гидрид натрия (0,121 г; 2,2 экв.; 3,01 ммоль). Охлаждали реакционную смесь до 0 С и перемешивали в течение 15 мин, после чего добавляли 3-хлорпропил-2-(индан-2-иламино)-7,8 дигидро-5H-пиридо[4,3-d]пиримидин-6-карбоксилат (0,530 г; 1,0 экв.; 1,37 ммоль). Оставляли реакционную смесь перемешиваться при температуре окружающей среды на 24 ч. Разбавляли реакционную смесь дихлорметаном и водой и разделяли слои. Дополнительно экстрагировали водный слой дихлорметаном(2). Сушили объединенные органические экстракты над сульфатом натрия, фильтровали и концентрировали досуха с получением желтой жидкости. Очищали неочищенные продукты путем колоночной хроматографии (от 0 до 5% метанола в дихлорметане) с получением смеси двух региоизомерных продуктов. Дополнительная очистка с использованием хиральной подложки для хроматографии (Chiralcel OJ-H,5% ацетонитрила в метаноле, содержащем 0,2% изопропиламина) приводила к получению продукта 3-(5 метил-1H-имидазол-1-ил)пропил-2-(2,3-дигидро-1 Н-инден-2-иламино)-7,8-дигидропиридо[4,3-d]пиримидин-6(5H)-карбоксилата (0,055 г; 12%). МС (m/z): 433 (М+1). Пример 20 Синтез 3-(4-метил-1H-имидазол-1-ил)пропил-2-(2,3-дигидро-1H-инден-2-иламино)-7,8-дигидропиридо[4,3-d]пиримидин-6(5H)-карбоксилата Получали по существу согласно описанию примера 19. Дополнительная очистка с использованием хиральной подложки для хроматографии (Chiralcel OJ-H, 5% ацетонитрила в метаноле, содержащем 0,2% изопропиламина) приводила к получению продукта 3-(4-метил-1H-имидазол-1-ил)пропил-2-(2,3-дигидро-1H-инден-2-иламино)-7,8-дигидропиридо[4,3-d]пиримидин-6(5H)-карбоксилата (0,111 г; 25%). МС(2,0 г.; 1,0 экв.; 14,27 ммоль) в метаноле (18 мл) добавляли тионилхлорид (0,75 мл). Оставляли реакционную смесь нагреваться до температуры окружающей среды и перемешивали в течение 30 мин. В реакционную смесь добавляли насыщенный бикарбонат натрия (10 мл) и концентрировали. Суспендировали полученный остаток в метаноле (10 мл) и фильтровали, затем концентрировали с получением метил-3(1H-имидазол-5-ил)пропаноата (2,2 г; 100%) в виде белого твердого вещества: МС (m/z): 155 (М+1). Пример получения 44 Синтез 3-(1H-имидазол-5-ил)пропан-1-ола Схема W, стадия В. В выдерживаемый при 0 С раствор метил-3-(1H-имидазол-5-ил)пропаноата (2,2 г; 1,0 экв.; 8,52 ммоль) в тетрагидрофуране (85 мл) по каплям в течение 5 мин добавляли алюмогидрид лития (1 М в тетрагидрофуране; 17 мл; 2,0 экв.; 17,00 ммоль). Нагревали реакционную смесь до температуры окружающей среды и перемешивали в течение 16 ч. Последовательно добавляли воду (0,65 мл),15% гидроксид натрия (0,65 мл) и воду (1,95 мл), затем перемешивали в течение 30 мин. Отфильтровывали полученное твердое вещество и промывали ацетоном. Концентрировали фильтрат с получением 3(1H-имидазол-5-ил)пропан-1-ола (3,0 г; 61%) в виде белого твердого вещества. Для дополнительной очистки продукта суспендировали часть 3-(1H-имидазол-5-ил)пропан-1-ола (1,5 г) в воде (50 мл) и перемешивали при 100 С в течение 2 ч. Фильтровали смесь продуктов и концентрировали фильтрат с получением чистого 3-(1H-имидазол-5-ил)пропан-1-ола (0,5 г) в виде беловатого твердого вещества: МС (m/z): 127 (М+1). Пример 21 Синтез 3-(1H-имидазол-5-ил)пропил-2-(2,3-дигидро-1H-инден-2-иламино)-5,7-дигидро-6H-пирроло[3,4-d]пиримидин-6-карбоксилата Схема W, стадия С. В раствор 3-(1H-имидазол-5-ил)пропан-1-ола (0,50 г; 1,0 экв.; 2,62 ммоль) в диметилформамиде (8,5 мл) добавляли 1,Г-карбонилдиимидазол (0,51 г; 1,2 экв.; 3,15 ммоль) и перемешивали в течение 1 ч. В полученный выше раствор медленно добавляли гидрат дигидрохлорида N-(2,3 дигидро-1H-инден-2-ил)-6,7-дигидро-5H-пирроло[3,4-d]пиримидин-2-амина (0,94 г; 1,10 экв.; 2,89 ммоль). Перемешивали в течение 16 ч при температуре окружающей среды, а затем дополнительно нагревали до 60 С в течение 16 ч. Разделяли реакционную смесь в воде и хлороформе и отделяли слои. Дополнительно экстрагировали водный слой хлороформом (2). Сушили объединенные органические экстракты над сульфатом магния, фильтровали и концентрировали с получением вязкой жидкости. Очищали неочищенный продукт путем колоночной хроматографии (от 0 до 20% метанола в дихлорметане) с получением 3-(1H-имидазол-5-ил)пропил-2-(2,3-дигидро-1H-инден-2-иламино)-5,7-дигидро-6H-пирроло[3,4-d]пиримидин-6-карбоксилата (0,041 г; 4%) в виде коричневого твердого вещества: МС (m/z): 405
МПК / Метки
МПК: C07D 487/04, A61K 31/5025, A61P 29/00, C07D 471/04
Метки: соединения, бициклические, пиримидиновые
Код ссылки
<a href="https://eas.patents.su/30-25106-biciklicheskie-pirimidinovye-soedineniya.html" rel="bookmark" title="База патентов Евразийского Союза">Бициклические пиримидиновые соединения</a>
Пирроло[2,3-d]пиримидиновые соединения

Номер патента: 6034
Опубликовано: 25.08.2005
Авторы: Блуменкопф Тодд Эндрю, Браун Маттью Фрэнк, Фланаган Марк Эдуард, Чанджелиэн Пол Стивен
МПК: A61K 31/505, C07D 487/04, A61P 35/02...
Метки: пирроло[2,3-d]пиримидиновые, соединения
Формула / Реферат:
1. Соединение формулы или его фармацевтически приемлемая соль, где R1 представляет собой группу формулы где пунктирная линия обозначает возможные двойные связи; m равняется 0, 1 или 2; n равняется 1 или 2, X представляет собой кислород, NR6 или CR7R8; каждый из B и D представляет собой CR7R8; каждый из A и E представляет собой CR7R8; и R6 выбран из группы, состоящей из водорода, (C1-C6)алкила; каждый из R7 и R8 независимо выбран из группы,...
Пирроло[2,3-d]пиримидиновые соединения

Номер патента: 10377
Опубликовано: 29.08.2008
Авторы: Браун Маттью Фрэнк, Фланаган Марк Эдуард, Блуменкопф Тодд Эндрю, Чанджелиэн Пол Стивен
МПК: A61K 31/505, C07D 209/00, C07D 239/00...
Метки: соединения, пирроло[2,3-d]пиримидиновые
Формула / Реферат:
1. Фармацевтическая композиция для (а) лечения или предотвращения расстройства или состояния, выбранного из отторжения трансплантата органа, волчанки, рассеянного склероза, ревматоидного артрита, псориаза, диабета типа I и осложнений диабета, рака, астмы, атопического дерматита, аутоиммунных расстройств щитовидной железы, неспецифического язвенного колита, болезни Крона, болезни Альцгеймера, лейкоза и других аутоиммунных заболеваний, или (б)...
Пиримидиновые соединения, относящиеся к а2b – селективным антагонистам, их синтез и применение

Номер патента: 7468
Опубликовано: 27.10.2006
Авторы: Коллингтон Эрик Уилльям, Кастелано Арлиндо Л., Маккиббен Брайан, Штайниг Арно Г.
МПК: A61K 31/50, A61K 31/495, A61K 31/505...
Метки: относящиеся, синтез, антагонистам, соединения, пиримидиновые, применение, селективным
Формула / Реферат:
1. Соединение структуры где каждый R6 и R9 независимо представляет собой водород или алкил; R7 представляет собой водород, ОН, алкоксигруппу, сложный эфир, ацеталь, кеталь или CN; R8 представляет собой замещенный или незамещенный арил, арилоксигруппу или алкиларил; где заместители в алкильном фрагменте выбраны из галогена, гидроксила, алкилкарбонилоксигруппы, арилкарбонилоксигруппы, алкоксикарбонилоксигруппы, арилоксикарбонилоксигруппы,...
Пиримидиновые соединения, их применение в качестве а 2b-селективных антагонистов и способ их получения

Номер патента: 11809
Опубликовано: 30.06.2009
Авторы: Штайниг Арно Г., Маккиббен Брайан, Кастелано Арлиндо Л., Коллингтон Эрик Уилльям
МПК: A61K 31/50, A61K 31/505, A61K 31/495...
Метки: получения, способ, 2b-селективных, применение, пиримидиновые, качестве, соединения, антагонистов
Формула / Реферат:
1. Соединение структуры где R1 представляет собой фенил, незамещенный или замещенный галогеном; R2 представляет собой водород; R3 представляет собой водород или кислород, где пунктирная линия представляет собой вторую связь, которая может присутствовать или отсутствовать, и при ее отсутствии R3 представляет собой водород, и при ее наличии R3 представляет собой кислород; R4 и R5, каждый независимо, представляют собой замещенный или незамещенный...
Бициклические гетероароматические соединения в качестве ингибиторов протеин-тирозинкиназ

Номер патента: 2455
Опубликовано: 25.04.2002
Авторы: Смит Кэтрин Джейн, Картер Малком Клайв, Гантрип Стивен Барри, Лаки Карен Элизабет, Кокерилл Джордж Стюарт
МПК: C07D 471/04, A61P 35/00, A61K 31/519...
Метки: протеин-тирозинкиназ, бициклические, соединения, гетероароматические, качестве, ингибиторов
Формула / Реферат:
1. Соединение формулы (I) или его соль или сольват; где Y представляет собой CR1, а V представляет собой N; или Y представляет собой CR1, a V представляет собой CR2; R1 представляет собой группу СН3SO2СН2СН2NНСН2-Аr-, где -Аr- выбран из фурана и тиазола, каждый из которых может быть возможно замещен одной или двумя гало, С1-4алкильной или С1-4 алкоксигруппами; R2 выбран из группы, включающей в себя водород, гало, гидрокси, С1-4алкил,...
Предыдущий патент: Фармацевтическая композиция, содержащая мометазона фуроат и салициловую кислоту
Случайный патент: Способ идентификации товаров или услуг и система для его осуществления