Производные 6-циклобутил-1,5-дигидропиразоло[3,4-d]пиримидин-4-она и их применение в качестве ингибиторов pde9a
Номер патента: 23574
Опубликовано: 30.06.2016
Авторы: Хайне Никлас, Джованнини Риккардо, Феррара Марко




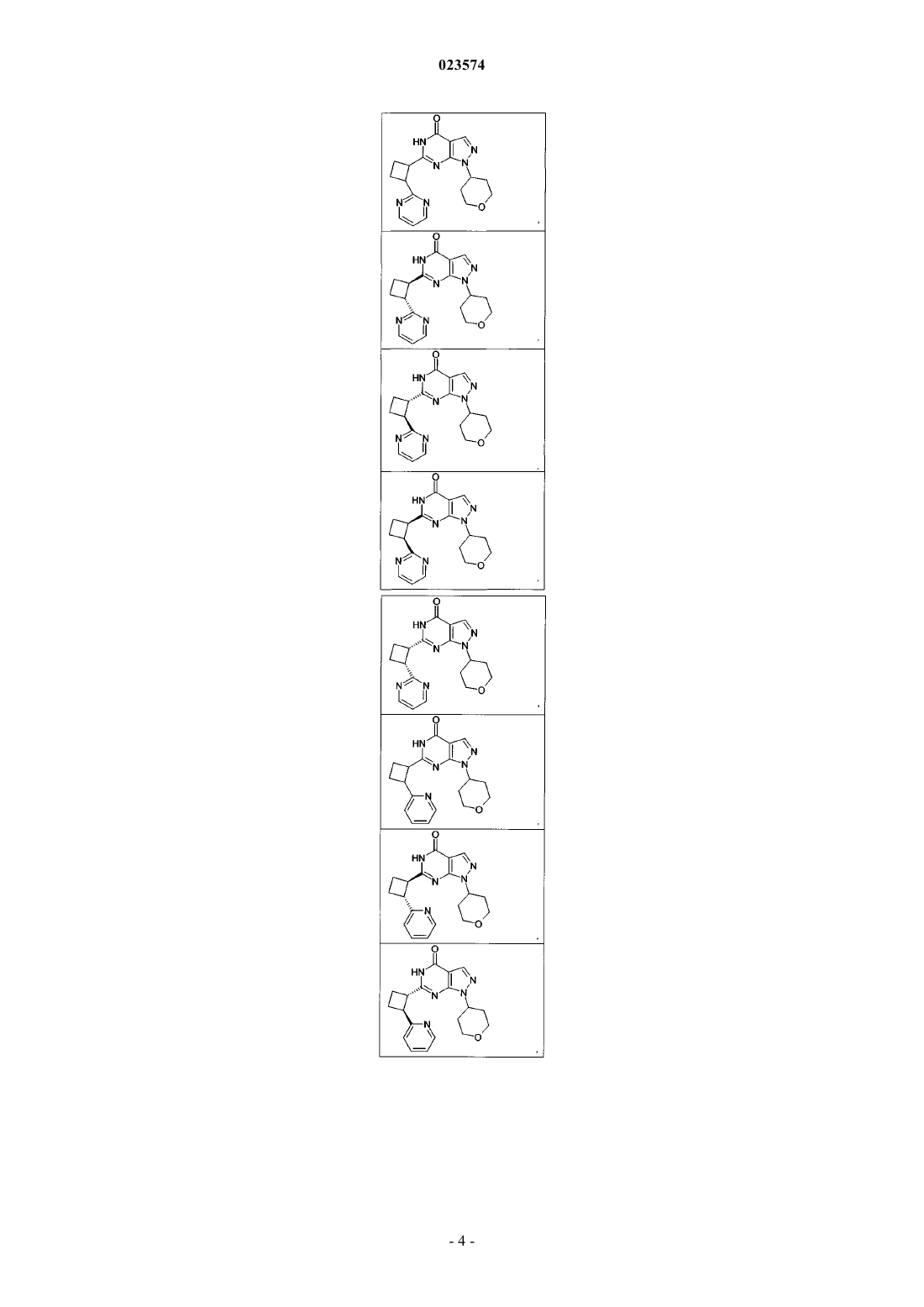
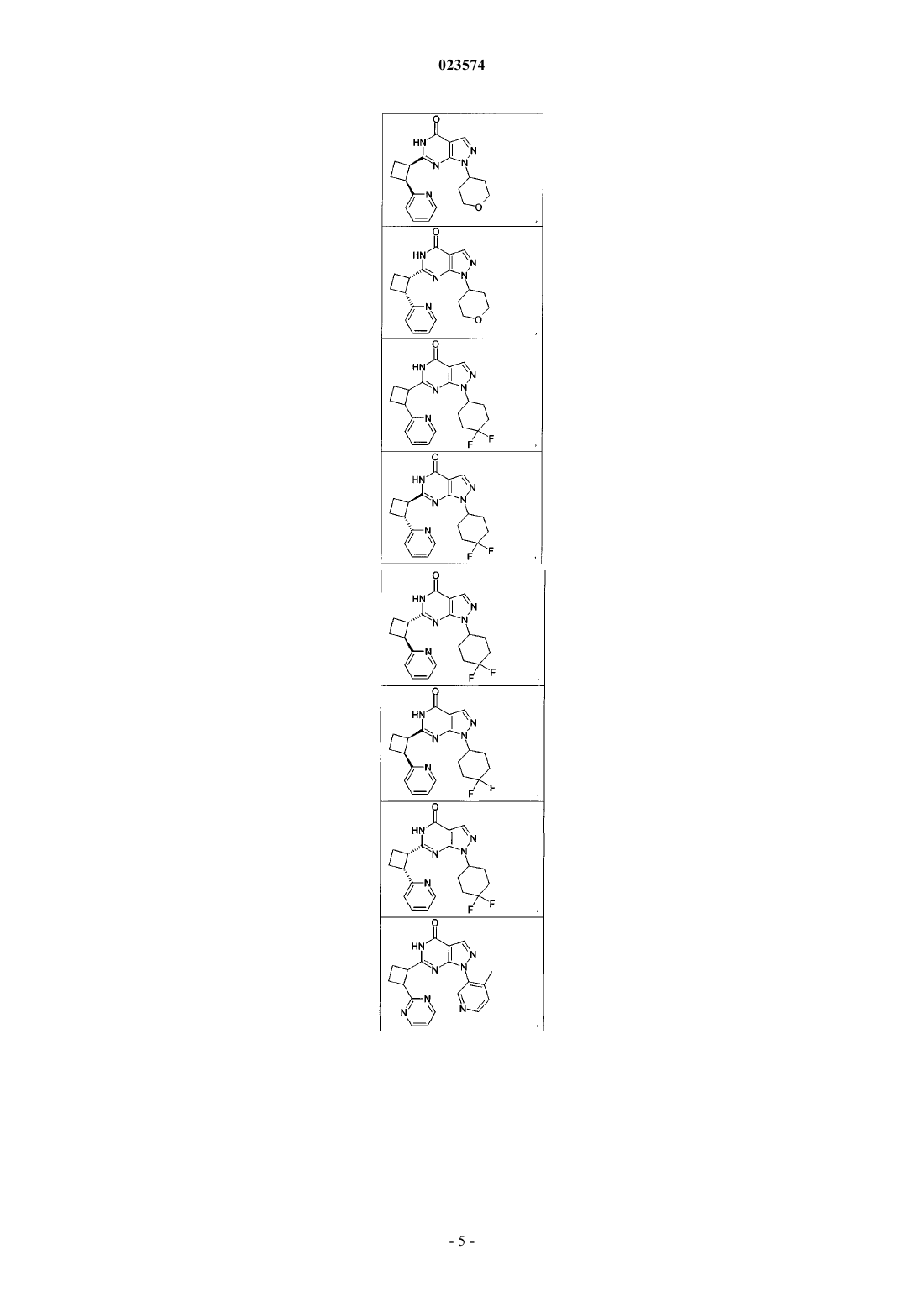



















Формула / Реферат
1. Соединение формулы (I)

где R1 и D такие, как указаны в нижеследующих соединениях:






и их фармацевтически приемлемые соли.
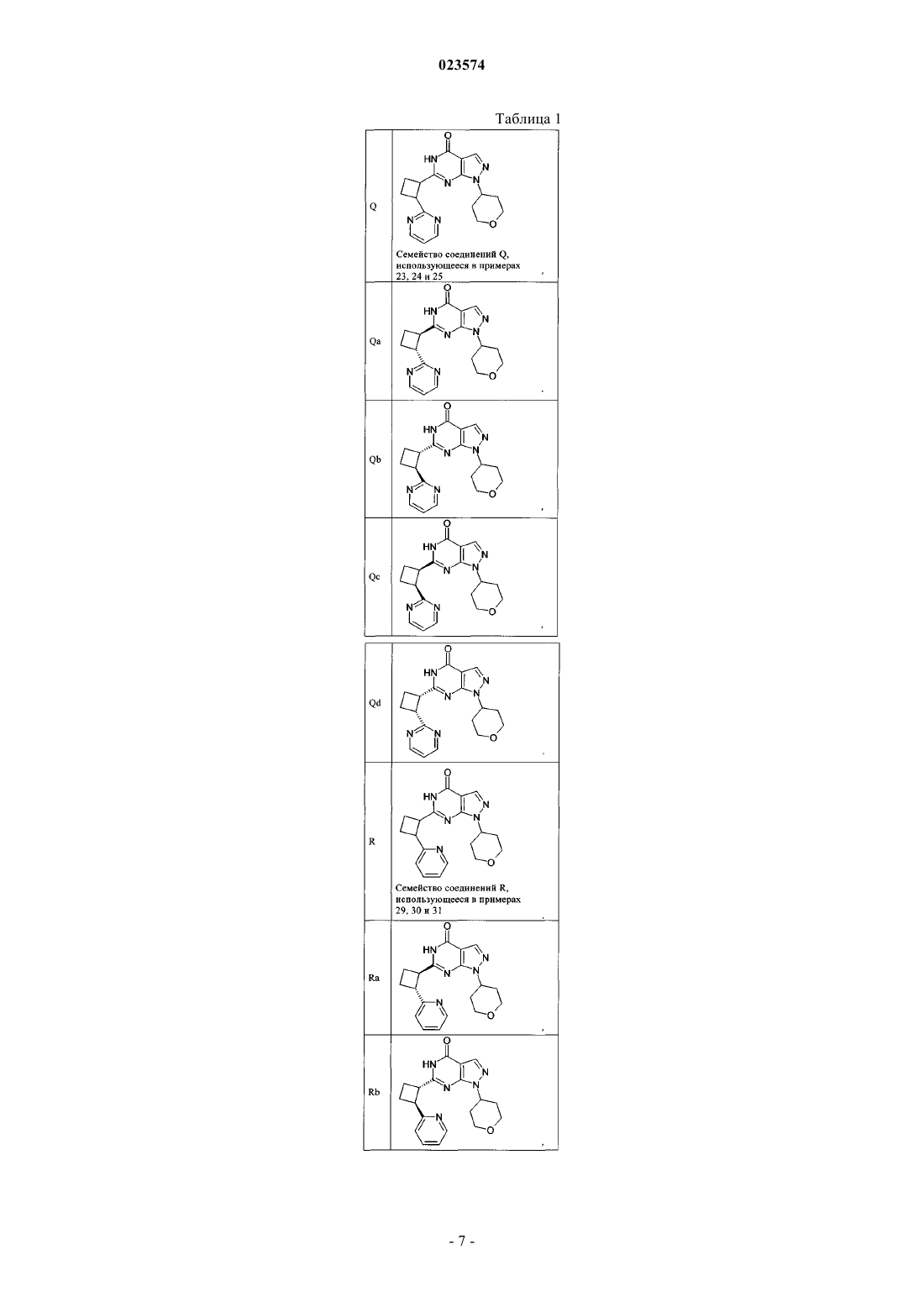
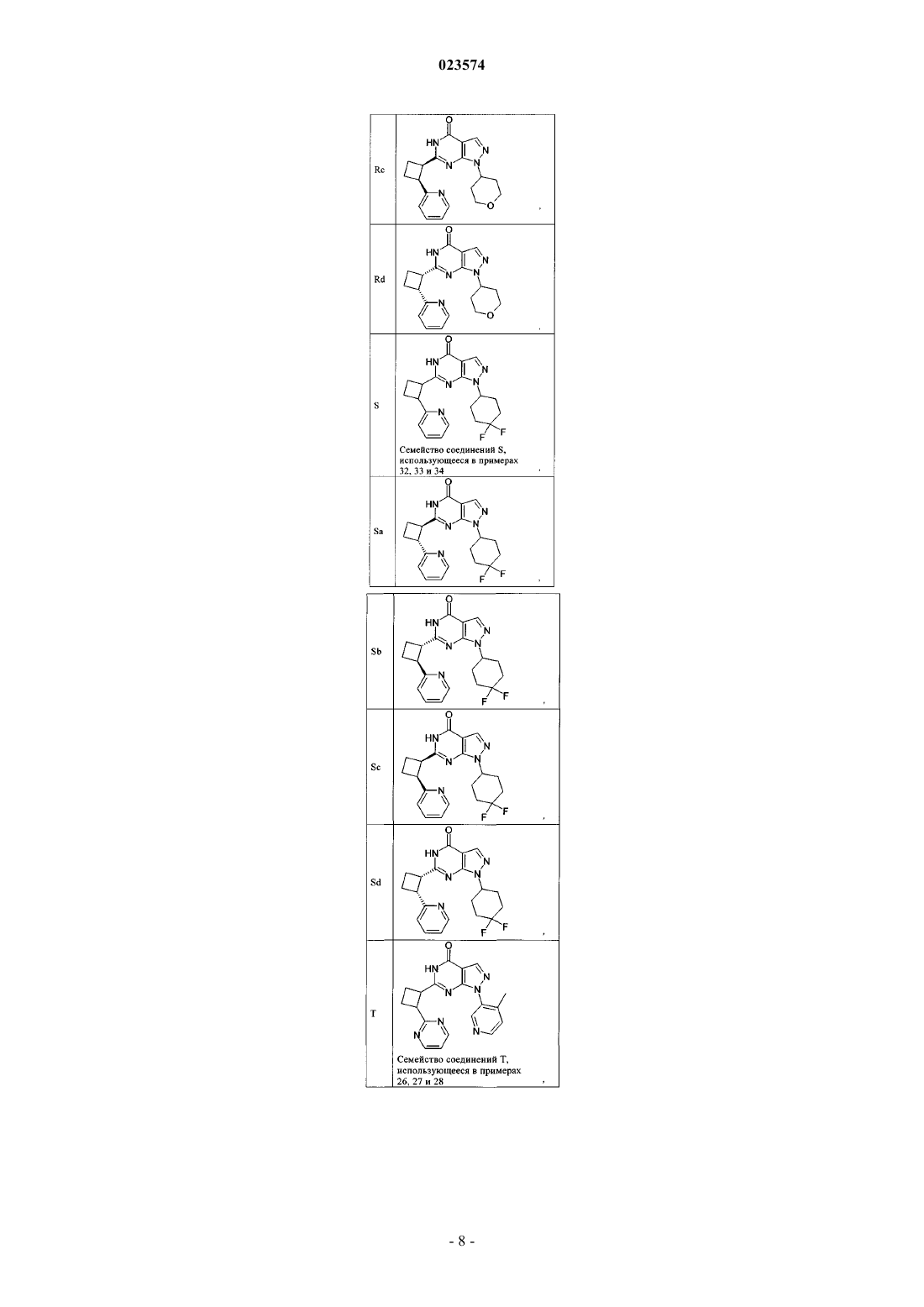
2. Соединение по п.1, где соединение выбрано из группы соединений формулы (IIa)

и их фармацевтически приемлемых солей.
3. Соединение по п.1, где соединение выбрано из группы соединений формулы (IIb)

и их фармацевтически приемлемых солей.
4. Соединение по п.1, где соединение выбрано из группы соединений формулы (IIc)

и их фармацевтически приемлемых солей.
5. Соединение по п.1, где соединение выбрано из группы соединений формулы (IId)

и их фармацевтически приемлемых солей.
6. Применение соединения по любому из пп.1-5 для приготовления лекарственного средства или для применения в терапевтическом способе.
7. Применение соединения по любому из пп.1-5 для приготовления лекарственного средства:
(a) для лечения заболевания центральной нервной системы;
(b) для лечения заболевания, лечение которого возможно путем ингибирования PDE9;
(c) для лечения или улучшения протекания или предупреждения, предпочтительно для лечения, патологического состояния, выбранного из группы, включающей нарушение познавательной способности, связанное с заболеванием или патологическим состоянием, выбранным из группы, включающей восприятие, сосредоточенность, познавательную способность, способность к обучению или память, где лечение или улучшение протекания или предупреждение нарушения познавательной способности предпочтительно относится к возрастным нарушениям способности к обучению и памяти, возрастной амнезии, мультиинфарктному слабоумию, черепно-мозговой травме, удару, слабоумию, возникшему после ударов (постинсультное слабоумие), посттравматическому слабоумию, общим нарушениям сосредоточенности, нарушениям сосредоточенности у детей, страдающих нарушениями способности к обучению и памяти, болезни Альцгеймера, слабоумию с тельцами Леви, слабоумию с дегенерацией лобных долей, включая синдром Пика, болезнь Паркинсона, прогрессирующий ядерный паралич, слабоумию с кортикобазальной дегенерацией, боковому амиотрофическому склерозу, болезни Гентингтона, рассеянному склерозу, дегенерации таламуса, слабоумию Крейтцфельдта-Якоба, слабоумию, связанному с ВИЧ (вирус иммунодефицита человека), эпилепсии, височной эпилепсии, шизофрении, шизофрении (со слабоумием), психозу Корсакова или нарушению познавательной способности, связанному с депрессией или биполярным нарушением;
(d) для лечения болезни Альцгеймера или нарушения познавательной способности, связанного с болезнью Альцгеймера;
(e) для лечения шизофрении или нарушения познавательной способности, связанного с шизофренией;
(f) для лечения эпилепсии или нарушения познавательной способности, связанного с эпилепсией;
(g) для лечения заболевания или патологического состояния, выбранного из группы, включающей нарушения сна, биполярное расстройство, метаболический синдром, ожирение, сахарный диабет, гипергликемию, дислипидемию, нарушенную переносимость глюкозы или заболевание яичек, головного мозга, тонкого кишечника, скелетных мышц, сердца, легких, вилочковой железы или селезенки.
8. Применение соединения по любому из пп.1-5 для приготовления лекарственного средства для лечения или профилактики нарушения познавательной способности.
9. Применение соединения по любому из пп.1-5 для приготовления лекарственного средства для лечения или профилактики нарушения познавательной способности, связанного с восприятием, сосредоточенностью, способностью к обучению или памятью.
10. Применение соединения по любому из пп.1-5 для приготовления лекарственного средства для лечения или профилактики нарушения познавательной способности в качестве симптомов иного основного подлежащего заболевания.
11. Применение соединения по любому из пп.1-5 для приготовления лекарственного средства для лечения или профилактики нарушения познавательной способности, связанного с восприятием, сосредоточенностью, способностью к обучению или памятью в качестве симптомов иного основного подлежащего заболевания.
12. Применение соединения по любому из пп.1-5 для приготовления лекарственного средства для улучшения навыков познавательной способности, связанных с восприятием, сосредоточенностью, способностью к обучению или памятью.
13. Фармацевтическая композиция, содержащая соединение, ингибирующее фосфодиэстеразу 9А, по любому из пп.1-5 и фармацевтический приемлемый носитель.
Текст
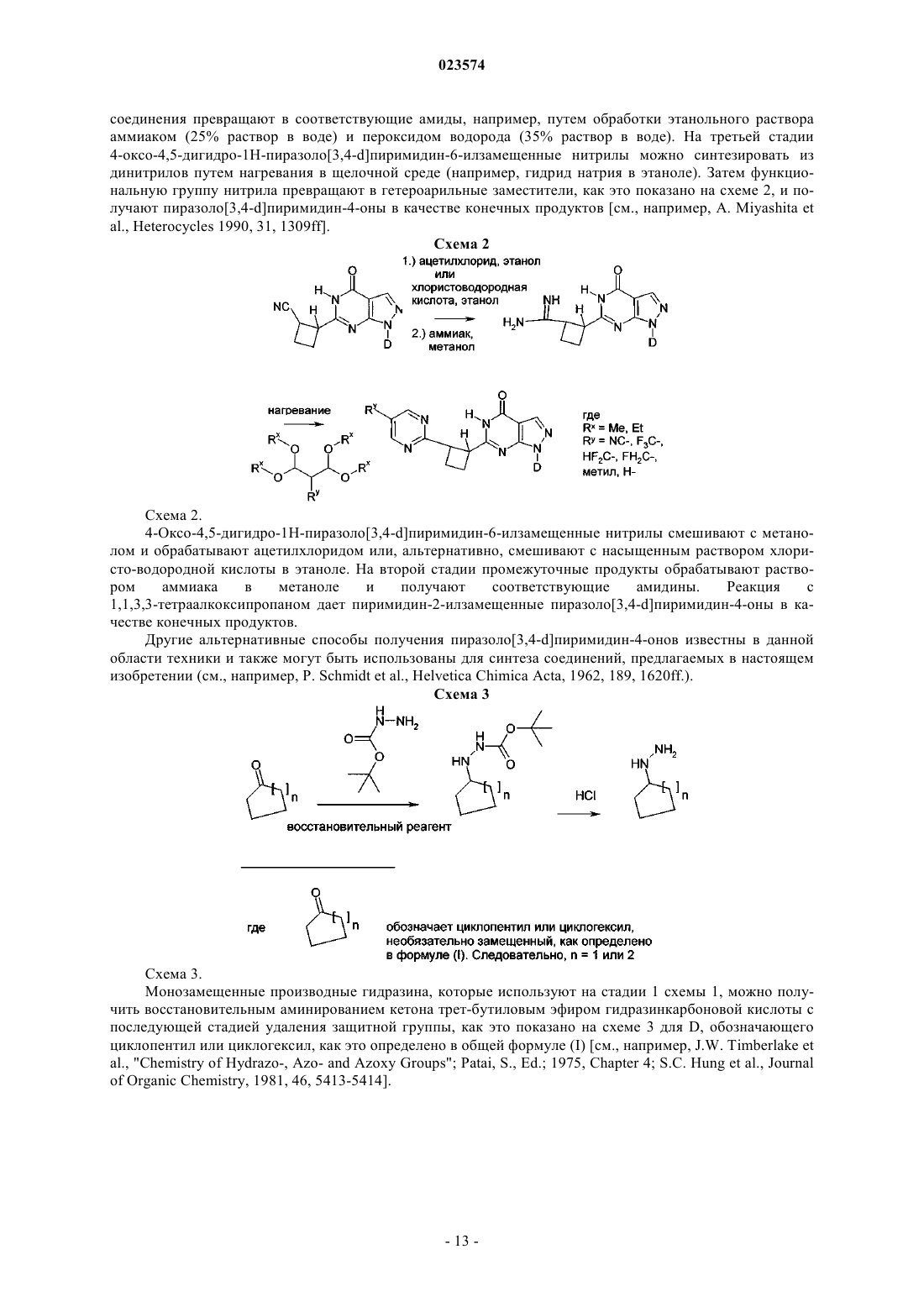
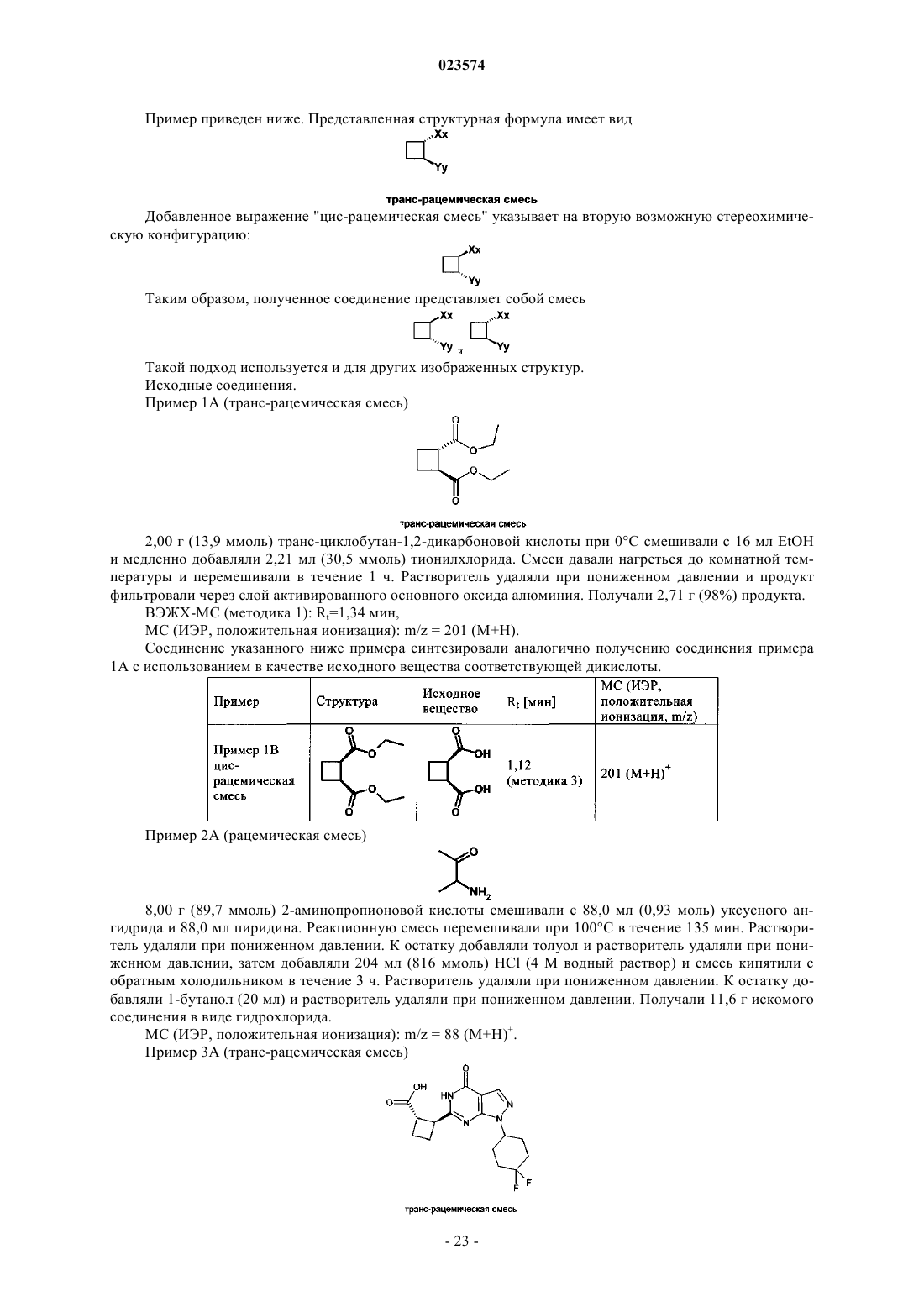
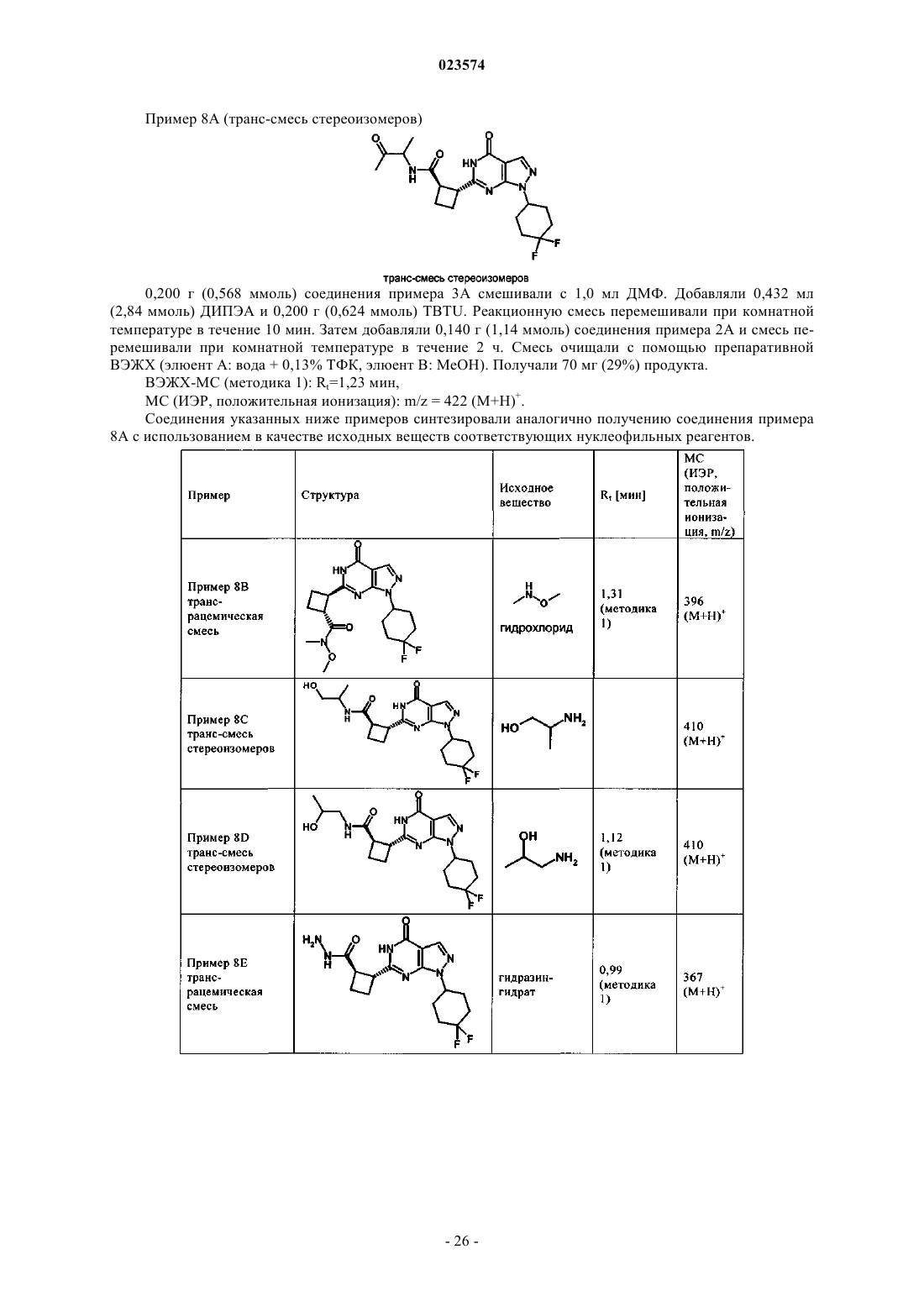
В изобретении описаны новые пиразолопиримидиноны формулы (I) в которой R1 обозначает пиридильную или пиримидинильную группу и D обозначает необязательно замещенный циклопентил, циклогексил, тетрагидрофуранил, тетрагидропиранил или 2-, 3- или 4-пиридил. Эти новые соединения предназначены для применения в качестве активного компонента лекарственных средств или для приготовления лекарственных средств соответственно, предпочтительно лекарственных средств, предназначенных для лечения патологических состояний, связанных с недостаточностью восприятия, сосредоточенности,способности к обучению или памяти. Такие патологические состояния могут быть связаны,например, с болезнью Альцгеймера, шизофренией и другими заболеваниями. Эти новые соединения также применимы, например, для приготовления лекарственных средств и/или для применения для лечения этих заболеваний, в частности нарушения познавательной способности,связанного с таким заболеванием. Описанные соединения обладают способностью ингибировать(71)(73) Заявитель и патентовладелец: БРИНГЕР ИНГЕЛЬХАЙМ ИНТЕРНАЦИОНАЛЬ ГМБХ (DE) Настоящее изобретение относится к новым пиразолопиримидинонам формулы (I) в которой R1 обозначает пиридильную или пиримидинильную группу иD обозначает необязательно замещенный циклопентил, циклогексил, тетрагидрофуранил, тетрагидропиранил или 2-, 3- или 4-пиридил. Эти новые соединения предназначены для применения в качестве активного компонента лекарственных средств или для приготовления лекарственных средств соответственно, предпочтительно лекарственных средств, предназначенных для лечения патологических состояний, связанных с недостаточностью восприятия, сосредоточенности, способности к обучению или памяти. Такие патологические состояния могут быть связаны, например, с болезнью Альцгеймера, шизофренией и другими заболеваниями. Эти новые соединения также применимы, например, для приготовления лекарственных средств и/или для применения для лечения этих заболеваний, в частности нарушения познавательной способности, связанного с таким заболеванием. Соединения, предлагаемые в настоящем изобретении, являются ингибиторами PDE9. Уровень техники Ингибирование фосфодиэстеразы 9 А (PDE9A) является одним из современных подходов к поиску новых путей лечения нарушений познавательной способности, вызванных нарушениями ЦНС (центральная нервная система), такими как болезнь Альцгеймера, шизофрения и другие заболевания, или вызванных любым другим нейродегенеративным процессом в головном мозге. В настоящем изобретении предложены новые соединения, которые соответствуют этому подходу. Фосфодиэстераза 9 А является одним представителем большого семейства фосфодиэстераз. Эти типы ферментов модулируют содержание циклических нуклеотидов циклического 5'-3'-аденозинмонофосфата (цАМФ) и циклического 5'-3'-гуанозинмонофосфата (цГМФ). Эти циклические нуклеотиды (цАМФ и цГМФ) являются важными вторыми мессенджерами и поэтому играют главную роль в каскадах передачи сигналов клеток. Каждый из них реактивирует, в частности, но не исключительно, протеинкиназы. Протеинкиназа, активированная с помощью цАМФ, называется протеинкиназой А (PKA), и протеинкиназа, активированная с помощью цГМФ, называется протеинкиназой G (PKG). В свою очередь, активированные PKA и PKG способны фосфорилировать целый ряд эффекторных клеточных белков (например, ионные каналы, связанные с белком G рецепторы, структурные белки, факторы транскрипции). Таким путем вторые мессенджеры цАМФ и цГМФ могут регулировать множество физиологических процессов во многих органах. Однако циклические нуклеотиды также могут непосредственно воздействовать на эффекторные молекулы. Так, например, известно, что цГМФ может непосредственно воздействовать на ионные каналы и тем самым может влиять на концентрацию иона в клетке(см. обзор: Wei et al., Prog. Neurobiol., 1998, 56, 37-64). Фосфодиэстеразы (PDE) являются средством регулирования активности цАМФ и цГМФ и, таким образом, регулируют соответствующие физиологические процессы. PDE гидролизуют циклические монофосфаты с образованием неактивных монофосфатов АМФ и ГМФ. В настоящее время на основании гомологии последовательностей соответствующих генов идентифицированы 11 семейств PDE. Отдельные гены PDE в семействе обозначают буквами (например,PDE1A и PDE1B). Если в гене также возникают разные варианты сплайсинга, то их указывают дополнительными цифрами после букв (например, PDE1 А 1).PDE9A человека клонировали и секвенировали в 1998 г. Совпадение аминокислотных последовательностей с последовательностями в других PDE не превышает 34% (PDE8A) и никогда не меньше 28%(PDE5A). При константе Михаэлиса-Ментена (Km), равной 170 нМ, PDE9A обладает высоким сродством к цГМФ. Кроме того, PDE9A селективна по отношению к цГМФ (Km для цАМФ = 230 мкмоль (мкМ.PDE9A не содержит домен связывания с цГМФ, и это показывает, что цГМФ не регулирует активность этого фермента. С помощью вестерн-блоттинга показано, что у людей PDE9A экспрессируется, в частности, в яичках, головном мозге, тонком кишечнике, скелетных мышцах, сердце, легких, вилочковой железе и селезенке. Наибольшее экспрессирование обнаружено в головном мозге, тонком кишечнике, почках,предстательной железе, толстой кишке и селезенке (Fisher et al., J. Biol. Chem., 1998, 273 (25), 1555915564; Wang et al., Gene, 2003, 314, 15-27). Ген PDE9A человека расположен в хромосоме 21q22.3 и содержит 21 экзон. Выявлены 4 альтернативных варианта сплайсинга PDE9A (Guipponi et al., Hum. Genet.,1998, 103, 386-392). Классические ингибиторы PDE не ингибируют PDE9A человека. Так, IBMX, дипиридамол, SKF94120, ролипрам и винпоцетин не ингибируют изолированный фермент при концентрациях, равных до 100 мкмоль (мкМ). Значение IC50 для запринаста найдено равным 35 мкМ (Fisher et al., J.PDE9A мышей клонировали и секвенировали в 1998 г. Soderling et al. (J. Biol. Chem., 1998, 273 (19),15553-15558). Она, как и PDE9A человека, обладает высоким сродством к цГМФ при Km, равной 70 нМ. Особенно высокое экспрессирование обнаружено в почках, головном мозге, легких и печени мы-1 023574 шей. IBMX не ингибирует PDE9A мышей при концентрациях, равных менее 200 мкМ; IC50 для запринаста равна 29 мкМ (Soderling et al., J. Biol. Chem., 1998, 273 (19), 15553-15558). Установлено, что PDE9A сильно экспрессируется в некоторых областях головного мозга крыс. К ним относятся обонятельные луковицы, гиппокамп, кора, базальное ядро и базальные отделы переднего мозга (Andreeva et al., J.Neurosci, 2001, 21 (22), 9068-9076). В частности, гиппокамп, кора и базальные отделы переднего мозга играют важную роль в процессах обучения и запоминания. Как уже указано выше, PDE9A отличается особенно высоким сродством к цГМФ. Поэтому PDE9A активна даже при низких физиологических концентрациях, в отличие от PDE2A (Km=10 мкМ; Martins et(Murashima et al., Biochemistry, 1990, 29, 5285-5292), каталитическая активность PDE9A не увеличивается под влиянием цГМФ, поскольку она не содержит домена GAF (домен связывания с цГМФ, с помощью которого аллостерически повышается активность PDE) (Beavo et al., Current Opinion in Cell Biology, 2000,12, 174-179). Поэтому ингибиторы PDE9A могут привести к повышению базовой концентрации цГМФ. Из этого общего описания следует, что PDE9A участвует в специфических физиологических процессах характерным и особым образом, что четко отличает роль PDE9A от роли любых других представителей семейства PDE. В WO 2004/099210 раскрыты 6-арилметилзамещенные пиразолопиримидиноны, которые являются ингибиторами PDE9. В WO 2004/099211 раскрыты 6-циклилметил- и 6-алкилметилзамещенные пиразолопиримидины и их применение для улучшения познавательной способности, сосредоточенности и т.п. В DE 10238722 раскрыто применение ингибиторов PDE9A для улучшения познавательной способности, сосредоточенности. В WO 2004/018474 раскрыты фенилзамещенные пиразолопиримидины и их применение для улучшения восприятия, сосредоточенности, способности к обучению и/или памяти. В WO 2004/026876 раскрыты алкилзамещенные пиразолопиримидины и их применение для улучшения восприятия, сосредоточенности, способности к обучению и/или работы памяти. В WO 2004/096811 раскрыты гетероциклические бициклические системы, как ингибиторы PDE9,предназначенные для лечения диабета, включая диабет типа 1 и типа 2, гипергликемии, дислипидемии,нарушенной переносимости глюкозы, метаболического синдрома и/или сердечно-сосудистого заболевания. В WO 2009/068617 раскрыты ингибирующие PDE9 соединения, полученные из пиразолопиримидинонов, с замещенной фенилметил- или пиридилметильной группой в положении 6. В WO 2010/112437 раскрыты ингибирующие PDE9 соединения, полученные из пиразолопиримидинонов, с фенил- или гетероарилзамещенной арилметил- или гетероарилметильной группой в положении 6. В WO 2009/121919 раскрыты ингибиторы PDE9, полученные из пиразолопиримидинонов, с неароматической гетероциклильной группой в положении 1, в число которых входит тетрагидропиранил. В WO 2010/026214 раскрыты ингибиторы PDE9, полученные из пиразолопиримидинонов, с циклоалкильной или циклоалкенильной группой в положении 1, в число которых входит 4,4-дифторциклогексил. Некоторые данные предшествующего уровня техники относятся к химически сходным производным нуклеозидов. В качестве примеров можно отметить WO 2002/057425, в которой раскрыты производные нуклеозидов, которые являются ингибиторами РНК-зависимой вирусной РНК полимеразы, илиWO 2001/060315, в которой раскрыты производные нуклеозидов, предназначенные для лечения инфицирования гепатитом С, или ЕР 679657, в которой раскрыты соединения, которые служат аналогами рибонуклеозидов, или US 2002058635, в которой раскрыты пуриновые L-нуклеозиды, в которых и пуриновые кольца, и углеводное кольцо (пентозное кольцо) модифицированы, снабжены функциональными группами или подвергнуты обеим операциям. Так, например, углеводное кольцо должно содержать по меньшей мере одну образовавшую сложный эфир группу OH. В WO 2005/051944 раскрыты содержащие оксетан нуклеозиды, предназначенные для лечения нарушений, связанных с аналогами нуклеозидов, таких как нарушения, включающие пролиферацию клеток и инфицирование. В WO 2006/084281 раскрыты ингибиторы активирующего фермента Е 1, которые содержат сульфонамидный фрагмент. В WO 1998/40384 раскрыты пиразолопиримидиноны, которые являются ингибиторами PDE1, 2 и 5,и могут использоваться для лечения сердечнососудистых и цереброваскулярных нарушений и нарушений мочеполовой системы. В СН 396924, СН 396925, СН 396926, СН 396927, DE 1147234, DE 1149013 описаны пиразолопиримидины, которые расширяют коронарные сосуды и которые можно использовать для лечения нарушений кровотока в миокарде. В US3732225 описаны пиразолопиримидины, которые обладают противовоспалительной способностью и способностью снижать содержание глюкозы в крови. В DE2408906 описаны стирилпиразолопиримидиноны, которые можно использовать в качестве противомикробных и противовоспалительных средств для лечения, например, отека. Задачи изобретения Изменения схемы замещения пиразолопиримидинонов приводят к представляющим интерес изменениям биологической активности и соответственно к изменениям сродства к различным ферментаммишеням. Поэтому задачей настоящего изобретения является получение соединений, описанных в настоящем изобретении, в частности в формуле изобретения, которые эффективно модулируют PDE9A, с целью разработки лекарственного средства, в частности, в связи с заболеваниями или патологическими состояниями, лечение которых возможно путем модулирования PDE9A. Другой задачей настоящего изобретения является получение соединений, которые применимы для приготовления лекарственного средства, предназначенного для лечения нарушений ЦНС. Еще одной задачей настоящего изобретения является получение соединений, которые обладают благоприятным профилем безопасности. Другой задачей настоящего изобретения является получение соединений, которые обладают благоприятным более селективным профилем ингибирования PDE9A по сравнению с профилем ингибирования других представителей семейства PDE и других фармакологических мишеней и которые вследствие этого могут обеспечить терапевтические преимущества. Еще одной задачей настоящего изобретения является получение такого лекарственного средства,которое применимо не только для лечения, но и для предупреждения или модификации соответствующего заболевания или патологического состояния. Настоящее изобретение также относится к фармацевтической композиции, содержащей соединение, описанное в настоящем изобретении, в частности в формуле изобретения, и фармацевтически приемлемый носитель. Настоящее изобретение также относится к способу лечения любого из патологических состояний,описанных в настоящем изобретении, у млекопитающего, нуждающегося в таком лечении, предпочтительно у человека, включающему введение млекопитающему соединения, описанного в настоящем изобретении, в частности в формуле изобретения, в терапевтически эффективном количестве. Настоящее изобретение также относится к соединению, описанному в настоящем изобретении, в частности, в формуле изобретения, предназначенному для применения в способе лечения организма человека или животного с помощью терапии. Подробное описание настоящего изобретения Вариант осуществления 1 настоящего изобретения. Соединение, предлагаемое в настоящем изобретении, которое характеризуется общей формулой (I) где соединение выбрано из группы, включающей и их соли, предпочтительно их фармацевтически приемлемые соли. В приведенной группе соединений R1 и D являются такими, как определено. Для всех приведенных в качестве примеров соединений конфигурация циклоалкильной группы в положении 6 пиразолопиримидиноновой группы по отношению к указанной пиразолопиримидиноновой группе и заместителю R1 может быть цис- или транс. В связи с этим соединения, предлагаемые в настоящем изобретении, могут обладать следующими конфигурациями: где R1 и D являются такими, как определено выше. Эти стереохимически определенные варианты осуществления являются другим объектом настоящего изобретения. В табл. 1 приведен обзор перечисленных выше соединений, предлагаемых в настоящем изобретении, причем соединения, обладающие одинаковыми определениями R1, R2, m, n и D, объединены в родственные группы, которые представляют собой группы соединений, обладающих одной и той же общей химической структурной формулой, если не рассматриваются стереохимические характеристики. Представители таких семейств соединений приведены в качестве примера в разделе "Типичные варианты осуществления". и их соли, предпочтительно их фармацевтически приемлемые соли, их сольваты и сольваты их указанных выше солей. В указанной группе соединений соединения, которые обладают транс-конфигурацией по отношению к заместителю циклобутильной группы, могут быть предпочтительнее соединений, обладающих цис-конфигурацией. Из возможных соединений, обладающих транс-конфигурацией, одно из них может быть предпочтительнее по эффективности. Чем эффективнее соединение, тем более предпочтительным оно является. Другим критерием, по которому могут различаться предпочтительные соединения, предлагаемые в настоящем изобретении, является баланс эффективности и безопасности, например, селективность по отношению к другим представителям семейства PDE, таким как PDE1 С. Для одной пары соединений, обладающих транс-конфигурацией, проведенный в экспериментальной части рентгеноструктурный анализ монокристаллов показал, что абсолютной стереохимической конфигурацией соединения, которое обладает наименьшей эффективностью, является его энантиомерR,R. Поэтому абсолютной стереохимической конфигурацией соединения, которое обладает наибольшей эффективностью, является S,S. Для указанного соединения S,S-конфигурация описывается следующей структурой, соответствующей общей формуле (IId) Аналогичным образом, можно предположить, что из соединений, предлагаемых в настоящем изобретении, такие соединения, которые обладают такой же абсолютной стереохимической конфигурацией,могут быть более активными, чем другие представители этого семейства соединений. В контексте настоящего изобретения для одного семейства соединений более активные соединения предпочтительнее менее активных соединений. Семейство соединений представляет собой группу соединений, химические структуры которых различаются только по стереохимическим характеристикам. Разные стереоизомеры относятся к отдельным вариантам осуществления настоящего изобретения. Дополнительные варианты осуществления настоящего изобретения. Вариант осуществления 2 настоящего изобретения относится к соединениям варианта осуществления 1 настоящего изобретения, которые обладают следующими стереохимическими характеристиками,указанными в формуле (IIa) Вариант осуществления 3 настоящего изобретения относится к соединению варианта осуществления 1 настоящего изобретения, где соединение обладает следующими стереохимическими характеристиками, указанными в формуле (IIb) Вариант осуществления 4 настоящего изобретения относится к соединению варианта осуществления 1 настоящего изобретения, где соединение обладает следующими стереохимическими характеристиками, указанными в формуле (IIc) Вариант осуществления 5 настоящего изобретения относится к соединению варианта осуществления 1 настоящего изобретения, где соединение обладает следующими стереохимическими характеристиками, указанными в формуле (IId) Термины и определения Терминам, специально не определенным в настоящем изобретении, следует придавать такие значения, которые им придал бы специалист в данной области техники с учетом раскрытия и контекста. Примеры включают конкретные заместители или атомы, обозначенные своими 1- или 2-буквенными обозначениями, такими как Н для водорода, N для азота, С для углерода, О для кислорода, S для серы и т.п. При использовании в настоящем описании, если не указано иное, приведенные ниже термины обладают указанными значениями и используются указанные ниже обозначения. Если ниже не указано иное, то во всех формулах и группах используются обычные определения терминов и обычные валентности атомов, соответствующие стабильным состояниям. В общем случае, если термины специально определены в определенном контексте, такие конкретные определения будут превалировать над более общими определениями, приведенными в этом разделе. Обычно в объем настоящего изобретения входят все "таутомерные формы и изомерные формы и смеси", а именно отдельные геометрические изомеры или оптические изомеры, или рацемические и нерацемические смеси изомеров химической структуры или соединения, если в названии или структуре соединения не указана конкретная стереохимическая конфигурация или изомерная форма. Конкретные определения являются превалирующими. Выражение "фармацевтически приемлемое" используется в настоящем изобретении для указания таких соединений, материалов, композиций и/или дозированных форм, которые в соответствии с основными положениями медицины являются подходящими для использования при соприкосновении с тканями людей или, как в случае животных, без проявления чрезмерной токсичности, раздражающего воздействия, аллергической реакции или других затруднений или осложнений при разумном соотношении польза/риск."Фармацевтически приемлемая соль (соли)" соединений, предлагаемых в настоящем изобретении,также являются объектом настоящего изобретения. Термин "фармацевтически приемлемая соль (соли)" означает производные раскрытых соединений, в которых исходное соединение изменено путем образо- 10023574 вания его солей с кислотой или основанием, предпочтительно солей присоединения. Примеры фармацевтически приемлемых солей включают, но не ограничиваются только ими, соли неорганических или органических кислот с основными остатками/фрагментами соединений, предлагаемых в настоящем изобретении, такими как аминогруппы; кислотные остатки/фрагменты соединений, предлагаемых в настоящем изобретении, могут образовывать соли со щелочами или органическими основаниями. Фармацевтически приемлемые соли включают обычные нетоксичные соли или четвертичные аммониевые соли исходного соединения, образованные, например, из нетоксичных неорганических или органических кислот. Например, такие обычные нетоксичные соли включают соли, образованные из неорганических кислот, таких как хлористо-водородная, бромисто-водородная, серная, сульфаминовая, фосфорная, азотная кислоты и т.п.; и соли, полученные из органических кислот, таких как уксусная, пропионовая, янтарная, гликолевая,стеариновая, молочная, яблочная, винная, лимонная, аскорбиновая, памоевая, малеиновая, гидроксималеиновая, фенилуксусная, глутаминовая, бензойная, салициловая, сульфаниловая, 2-ацетоксибензойная,фумаровая, толуолсульфоновая, метансульфоновая, этандисульфоновая, щавелевая, изэтионовая кислоты и т.п. Физиологически приемлемые соли с основаниями также могут включать соли с обычными основаниями, такие как, например и предпочтительно, соли щелочных металлов (например, соли натрия и калия), соли щелочноземельных металлов (например, соли кальция и магния) и аммония, соли с органическими аминами, содержащими от 1 до 16 атомов С, такие как, например и предпочтительно, этиламин,диэтиламин, триэтиламин, этилдиизопропиламин, моноэтаноламин, диэтаноламин, триэтаноламин, дихлоргексиламин, диметиламиноэтанол, прокаин, дибензиламин, N-метилморфолин, дегидроабиетиламин,аргинин, лизин, этилендиамин и метилпиперидин и т.п. Фармацевтически приемлемые соли, предлагаемые в настоящем изобретении, можно синтезировать из исходного соединения, которое обладает свойствами основания или кислоты, по обычным химическим методикам. Обычно такие соли можно получить по реакции этих соединений в форме свободной кислоты или основания со стехиометрическим количеством соответствующего основания или кислоты в воде или в органическом растворителе или в их смеси; обычно предпочтительными являются неводные среды, такие как эфир, этилацетат, этанол, изопропанол или ацетонитрил."Пролекарством" считают соединение, которое предназначено для высвобождения in vivo биологически активного соединения, предлагаемого в настоящем изобретении, когда такое пролекарство вводят млекопитающему. Пролекарства соединений, предлагаемых в настоящем изобретении, получают путем модификации функциональных групп, содержащихся в соединении, предлагаемом в настоящем изобретении, так что при физиологических условиях эти модифицированные группы превращаются в исходные функциональные группы. Следует понимать, что пролекарства соединений, предлагаемых в настоящем изобретении, также являются объектом настоящего изобретения."Метаболитами" считают производными соединений, предлагаемых в настоящем изобретении, которые образуются in vivo. Активными метаболитами являются такие метаболиты, которые оказывают фармацевтическое воздействие. Следует понимать, что метаболиты, в частности активные метаболиты соединений, предлагаемых в настоящем изобретении, также являются объектом настоящего изобретения. Некоторые из соединений могут образовывать "сольваты". Для задач настоящего изобретения термин "сольваты" означает такие формы соединений, которые в твердом или жидком состоянии образуют комплекс путем координации с молекулами растворителя. Гидраты являются особой формой сольватов,в которых координация происходит с водой. В контексте настоящего изобретения этот термин предпочтительно используется для твердых сольватов, таких как аморфные или более предпочтительно кристаллические сольваты."Ядро". Ядро соединений, предлагаемых в настоящем изобретении, описывается приведенной ниже основной структурой. Нумерация положений кольцевых атомов выполнена жирным шрифтом: Для специалиста в данной области техники должно быть очевидно, что это ядро можно описать его таутомерной "енольной" формой В контексте настоящего изобретения оба представления структуры ядра следует считать объектом настоящего изобретения, даже если приведено только одно из двух представлений. Без наложения какихлибо ограничений предполагается, что для большинства соединений при нормальных условиях окру- 11023574 жающей среды и при условиях, которые соответствуют условиям в фармацевтической композиции, содержащей указанные соединения, равновесие таутомерных форм смещено в сторону пиразолопиримидин-4-онового представления. Поэтому все варианты осуществления представлены в виде производных пиразолопиримидин-4-она или, точнее в виде производных пиразоло[3,4-d]пиримидин-4-она. Выражения "предупреждение", "профилактика", "профилактическое лечение" или "предупредительное лечение" при использовании в настоящем изобретении следует понимать, как синонимы и в том смысле, что риск развития патологического состояния, указанного выше в настоящем изобретении, снижается, особенно у пациента, для которого существует повышенная опасность возникновения указанных патологических состояний или соответствующий анамнез. Таким образом, выражение "предупреждение заболевания" при использовании в настоящем изобретении означает лечение и уход за индивидуумом,для которого существует опасность развития заболевания, до появления клинических симптомов заболевания. Целью предупреждения является борьба с развитием заболевания, патологического состояния или нарушения, и оно включает введение активных соединений для предупреждения или задержки появления симптомов или осложнений, или для предупреждения или задержки развития родственных заболеваний, патологических состояний или нарушений. Успех указанного предупредительного лечения отражен статистически в уменьшении частоты возникновения указанного патологического состояния в группе пациентов, для которых существует опасность возникновения этого патологического состояния, по сравнению с аналогичной группой пациентов, не подвергающихся предупредительному лечению. Выражение "лечение" или "терапия" предпочтительно означает лекарственное лечение пациентов(например, предпочтительно человека), у которых уже развилось одно или большее количество указанных патологических состояний в явной, острой или хронической форме, включая симптоматическое лечение, предназначенное для облегчения симптомов при конкретном показании, или этиотропное лечение, предназначенное для обращения или частичного обращения патологического состояния или для остановки или замедления прогрессирования заболевания настолько, насколько это возможно в зависимости от патологического состояния и его тяжести. Таким образом, выражение "лечение заболевания" при использовании в настоящем изобретении означает лечение и уход за пациентом, у которого развилось заболевание, патологическое состояние или нарушение. Целью лечения является борьба с заболеванием,патологическим состоянием или нарушением или с их симптомами. Лечение включает введение активных соединений для устранения заболевания, патологического состояния или нарушения или борьбы с ними, а также облегчение симптомов или осложнений, связанных с заболеванием, патологическим состоянием или нарушением. Приведенные ниже схемы в качестве примера в общем иллюстрируют способ получения соединений, предлагаемых в настоящем изобретении. Обозначенные аббревиатурами заместители могут быть такими, как определено для вариантов осуществления формулы (I), если в описаниях схем не приведено других определений: Схема 1 Схема 1. На первой стадии 2-этоксиметиленмалононитрил конденсируют с монозамещенными гидразинами путем нагревания в подходящем растворителе, таком как этанол, в присутствии основания (например,триэтиламина) и получают соответствующие 5-амино-1 Н-пиразол-4-карбонитрилы. На второй стадии эти соединения превращают в соответствующие амиды, например, путем обработки этанольного раствора аммиаком (25% раствор в воде) и пероксидом водорода (35% раствор в воде). На третьей стадии 4-оксо-4,5-дигидро-1 Н-пиразоло[3,4-d]пиримидин-6-илзамещенные нитрилы можно синтезировать из динитрилов путем нагревания в щелочной среде (например, гидрид натрия в этаноле). Затем функциональную группу нитрила превращают в гетероарильные заместители, как это показано на схеме 2, и получают пиразоло[3,4-d]пиримидин-4-оны в качестве конечных продуктов [см., например, A. Miyashita et Схема 2. 4-Оксо-4,5-дигидро-1 Н-пиразоло[3,4-d]пиримидин-6-илзамещенные нитрилы смешивают с метанолом и обрабатывают ацетилхлоридом или, альтернативно, смешивают с насыщенным раствором хлористо-водородной кислоты в этаноле. На второй стадии промежуточные продукты обрабатывают раствором аммиака в метаноле и получают соответствующие амидины. Реакция с 1,1,3,3-тетраалкоксипропаном дает пиримидин-2-илзамещенные пиразоло[3,4-d]пиримидин-4-оны в качестве конечных продуктов. Другие альтернативные способы получения пиразоло[3,4-d]пиримидин-4-онов известны в данной области техники и также могут быть использованы для синтеза соединений, предлагаемых в настоящем изобретении (см., например, Р. Schmidt et al., Helvetica Chimica Acta, 1962, 189, 1620ff.). Схема 3 Схема 3. Монозамещенные производные гидразина, которые используют на стадии 1 схемы 1, можно получить восстановительным аминированием кетона трет-бутиловым эфиром гидразинкарбоновой кислоты с последующей стадией удаления защитной группы, как это показано на схеме 3 для D, обозначающего циклопентил или циклогексил, как это определено в общей формуле (I) [см., например, J.W. Timberlake et Схема 4. Как показано на схеме 1, на первой стадии 2-этоксиметиленмалононитрил конденсируют с монозамещенными гидразинами путем нагревания в подходящем растворителе, таком как этанол, в присутствии основания (например, триэтиламина) с образованием соответствующих 5-амино-1 Н-пиразол-4 карбонитрилов. На второй стадии эти соединения превращают в соответствующие амиды, например,путем обработки этанольного раствора аммиаком (25% раствор в воде) и пероксидом водорода (35% раствор в воде). На третьей стадии нагревание с R1- и R2-замещенным эфиром циклобутил- или циклопентилкарбоновой кислоты в щелочной среде (например, гидрид натрия в этаноле) дает конечные пиразоло[3,4-d]пиримидин-4-оны в качестве конечных продуктов [см., например, A. Miyashita et al.,Heterocycles, 1990, 31, 1309ff]. Эта процедура для случая, когда R1 обозначает пиридил, подробнее описана в экспериментальном разделе (примеры 29-34). Дополнительная информация также приведена в WO 2004/099210 (в особенности от последнего абзаца на с. 9 до строки 8 с. 14, включена в настоящее изобретение в качестве ссылки); по общей методике получения соединений, в которых D означает тетрагидропиранил, дополнительная информация приведена в WO 2009/121919, в особенности с. 120-125, и в экспериментальной части этой заявки (включена в настоящее изобретение в качестве ссылки); для случая, когда D означает 4,4-дифторциклогексил, дополнительная информация приведена вWO 2010/026214, в особенности с. 59-63, и в экспериментальной части этой заявки (включена в настоящее изобретение в качестве ссылки); в экспериментальной части (типичные варианты осуществления) настоящего описания. В частности, в ней описано получение следующих двух структурных фрагментов: Способ лечения Настоящее изобретение относится к соединениям, которые считаются эффективными для лечения заболеваний. Соединения, предлагаемые в настоящем изобретении, являются эффективными и селективными ингибиторами фосфодиэстеразы 9 А и могут использоваться для разработки лекарственных средств. Такие лекарственные средства предпочтительно следует использовать для лечения заболеваний,при которых ингибирование PDE9A может привести к терапевтическому, профилактическому или модифицирующему заболевание эффекту. Лекарственные средства предпочтительно следует использовать для улучшения восприятия, сосредоточенности, познавательной способности, способности к обучению или памяти, таких как происходящих, в частности, при ситуациях/заболеваниях/синдромах, таких как слабое нарушение познавательной способности, возрастные нарушения способности к обучению и памяти, возрастная амнезия, мультиинфарктное слабоумие, черепно-мозговая травма, удар, слабоумие,возникшее после ударов (постинсультное слабоумие), посттравматическое слабоумие, общие нарушения сосредоточенности, нарушения сосредоточенности у детей, страдающих нарушениями способности к обучению и памяти, болезнь Альцгеймера, слабоумие с тельцами Леви, слабоумие с дегенерацией лобных долей, включая синдром Пика, болезнь Паркинсона, прогрессирующий ядерный паралич; слабоумие с кортикобазальной дегенерацией, боковой амиотрофический склероз (БАС), болезнь Гентингтона, рассеянный склероз, дегенерация таламуса, слабоумие Крейтцфельдта-Якоба, слабоумие,связанное с ВИЧ (вирус иммунодефицита человека), эпилепсия, височная эпилепсия, шизофрения, шизофрения (со слабоумием), психоз Корсакова или нарушение познавательной способности, связанное с депрессией или биполярным нарушением. Другим объектом настоящего изобретения является лечение заболевания, которое возможно путем модулирования PDE9A, в частности нарушений сна, таких как инсомния или нарколепсия, биполярного расстройства, метаболического синдрома, ожирения, сахарного диабета, включая диабет типа 1 или типа 2, гипергликемии, дислипидемии, нарушенной переносимости глюкозы или заболеваний яичек, головного мозга, тонкого кишечника, скелетных мышц, сердца, легких, вилочковой железы или селезенки. Таким образом, медицинский объект настоящего изобретения можно кратко описать так, что соединение формулы (I) или (II), определенное в настоящем изобретении, предпочтительно специально определенное соединение, применяется в качестве лекарственного средства. Такое лекарственное средство предпочтительно предназначено для применения в способе лечения заболевания ЦНС. В альтернативном применении лекарственное средство предназначено для применения в терапевтическом или профилактическом способе, предпочтительно в терапевтическом способе, для лечения заболевания ЦНС, лечение которого возможно путем ингибирования PDE9. В альтернативном применении лекарственное средство предназначено для применения в терапевтическом или профилактическом способе, предпочтительно в терапевтическом способе, для лечения заболевания, лечение которого возможно путем ингибирования PDE9, предпочтительно PDE9A. В наиболее предпочтительном альтернативном применении лекарственное средство предназначено для применения в терапевтическом или профилактическом способе, предпочтительно в терапевтическом способе, для лечения, улучшения протекания и/или предупреждения нарушения познавательной способности, связанного с восприятием, сосредоточенностью, познавательной способностью, способностью к обучению или памятью, предпочтительно, если такое нарушение познавательной способности связано с заболеванием или патологическим состоянием, описанным в этом разделе. В альтернативном применении лекарственное средство предназначено для применения в терапевтическом или профилактическом способе, предпочтительно в терапевтическом способе, для лечения или улучшения протекания, или предупреждения нарушения познавательной способности, связанного с возрастными нарушениями способности к обучению и памяти, возрастной амнезии, мультиинфарктного слабоумия, черепно-мозговой травмы, удара, слабоумия, возникшего после ударов (постинсультное слабоумие), посттравматического слабоумия, общих нарушений сосредоточенности, нарушений сосредоточенности у детей, страдающих нарушениями способности к обучению и памяти, болезни Альцгеймера,слабоумия с тельцами Леви, слабоумия с дегенерацией лобных долей, включая синдром Пика, болезни Паркинсона, прогрессирующего ядерного паралича, слабоумия с кортикобазальной дегенерацией, бокового амиотрофического склероза (БАС), болезни Гентингтона, рассеянного склероза, дегенерации таламуса, слабоумия Крейтцфельдта-Якоба, слабоумия, связанного с ВИЧ, эпилепсии, височной эпилепсии,шизофрении, шизофрении (со слабоумием), психоза Корсакова или нарушения познавательной способности, связанного с депрессией или биполярным нарушением. В альтернативном применении лекарственное средство предназначено для применения в терапевтическом или профилактическом способе, предпочтительно в терапевтическом способе, для лечения болезни Альцгеймера, шизофрении или нарушения познавательной способности, связанного с болезнью Альцгеймера или связанного с шизофренией. В альтернативном применении лекарственное средство предназначено для применения в терапевтическом или профилактическом способе, предпочтительно в терапевтическом способе, для лечения нарушений сна, биполярного расстройства, метаболического синдрома, ожирения, сахарного диабета, гипергликемии, дислипидемии, нарушенной переносимости глюкозы или заболеваний яичек, головного мозга, тонкого кишечника, скелетных мышц, сердца, легких, вилочковой железы или селезенки. В другом объекте настоящего изобретения настоящее изобретение относится к способу лечения или предупреждения патологического состояния или заболевания, выбранного из приведенных выше групп патологических состояний и заболеваний, при этом способ включает введение нуждающемуся в нем человеку соединения, предлагаемого в настоящем изобретении, в терапевтически эффективном количестве. Другим объектом настоящего изобретения являются соединения, предлагаемые в настоящем изобретении, предназначенные для применения в качестве лекарственного средства в терапевтическом или профилактическом способе, предпочтительно в терапевтическом способе. Если это показано, то терапевтический способ или лекарственное средство предпочтительно предназначено для лечения патологического состояния или заболевания, выбранного из приведенных группы патологических состояний или заболеваний, указанных выше в этом разделе "Способ лечения". Фармацевтические композиции Лекарственные средства для введения, которые также являются объектом настоящего изобретения,содержат соединение, предлагаемое в настоящем изобретении, или фармацевтически активный ингредиент в терапевтически эффективном количестве и фармацевтический носитель."Терапевтически эффективное количество" означает, что при введении лекарственного средства в соответствующем режиме, приспособленном к состоянию пациента, количество указанного соединения формулы (I) будет достаточно для эффективного лечения, предупреждения или замедления прогрессирования соответствующего заболевания или иного улучшения состояния пациента, страдающего от такого заболевания. Может оказаться, что "терапевтически эффективное количество", использующееся при монотерапии, отличается от "терапевтически эффективного количества", использующегося в комбинированной терапии вместе с другим лекарственным средством. Диапазон доз соединений общей формулы (I), вводимых в сутки, может составлять от 0,1 до 5000 мг, предпочтительно от 0,1 до 1000 мг, предпочтительно от 2 до 500 мг, более предпочтительно от 5 до 250 мг, наиболее предпочтительно от 10 до 100 мг. Дозированная форма (например, таблетка) предпочтительно может содержать от 2 до 250 мг, особенно предпочтительно от 10 до 100 мг соединений,предлагаемых в настоящем изобретении. Реальное фармацевтически эффективное количество или терапевтическая доза будет зависеть от факторов, известных специалистам в данной области техники, таких как возраст, масса тела, пол или другие характеристики пациента, путь введения, тяжесть заболевания и т.п. Соединения, предлагаемые в настоящем изобретении, можно вводить пероральным, парентеральным (внутривенным, внутримышечным и т.п.), назальным, сублингвальным, ингаляционным, внутриоболочечным, местным или ректальным путем. Подходящие препараты для введения соединений, предлагаемых в настоящем изобретении, включают, например, пластыри, таблетки, капсулы, пилюли, пеллеты, драже, порошки, лепешки, суппозитории, жидкие препараты, такие как растворы, суспензии, эмульсии, капли, сиропы, эликсиры или газообразные препараты, такие как аэрозоли, спреи и т.п. Содержание фармацевтически активного соединения (соединений) должно составлять от 0,05 до 90 мас.%, предпочтительно от 0,1 до 50 мас.% в пересчете на массу композиции в целом. Подходящие таблетки можно изготовить, например, путем смешивания активного вещества (веществ) с известными инертными наполнителями, например, инертными разбавителями, такими как карбонат кальция, фосфат кальция или лактоза, разрыхлителями, такими как кукурузный крахмал или альгиновая кислота, связующими, такими как крахмал или желатин, смазывающими веществами, такими как стеарат магния или тальк, и/или агентами для замедления высвобождения, такими как карбоксиметилцеллюлоза, ацетат-фталат целлюлозы или поливинилацетат. Таблетки также могут содержать несколько слоев. Таблетки с покрытием можно изготовить путем нанесения на ядра, полученные аналогично таблеткам, покрытия из вещества, обычно использующегося для нанесения на таблетки, например коллидона или шеллака, гуммиарабика, талька, диоксида титана или сахара. Для обеспечения замедленного высвобождения и предупреждения несовместимости ядро также может состоять из нескольких слоев. Аналогичным образом, покрытие таблетки может состоять из ряда слоев, обеспечивающих замедленное высвобождение, возможно, с включением инертных наполнителей, указанных выше для таблеток. Сиропы и эликсиры, содержащие активные вещества или их комбинации, предлагаемые в настоящем изобретении, могут дополнительно содержать подсластитель, такой как сахарин, цикламат, глицерин или сахар, и усилитель вкуса, например ароматизатор, такой как ванилин или апельсиновый экстракт. Они также могут содержать суспендирующие вспомогательные вещества или загустители, такие как натриевая соль карбоксиметилцеллюлозы, смачивающие агенты, такие как, например, продукты конденсации жирных спиртов с этиленоксидом, или консерванты, такие как п-гидроксибензоаты. Растворы готовят обычным образом, например, путем добавления изотонических агентов, консервантов, таких как п-гидроксибензоаты, или стабилизаторов, таких как соли щелочных металлов этилендиаминтетрауксусной кислоты, необязательно с использованием эмульгаторов и/или диспергирующих агентов, хотя если в качестве разбавителя используют воду, то в качестве солюбилизаторов или растворяющих средств необязательно можно использовать органические растворители и растворы можно помещать во флаконы или ампулы для инъекции или бутыли для вливания. Капсулы, содержащие одно или большее количество активных веществ или комбинации активных веществ, например, можно изготовить путем смешивания активных веществ с инертными носителями,такими как лактоза или сорбит, и их помещения в капсулы из желатина. Подходящие суппозитории, например, можно изготовить путем смешивания с носителями, предназначенным для этой цели, такими как нейтральные жиры или полиэтиленгликоль или его производные. Инертные наполнители, которые можно использовать, включают, например, воду, фармацевтически приемлемые органические растворители, такие как парафины (например, фракции нефти), растительные масла (например, арахисовое или кунжутное масло), одно- или многоатомные спирты (например, этанол или глицерин), носители, такие как, например, порошкообразные природные минералы (например, каолины, глины, тальк, мел), порошкообразные синтетические минералы (например, высокодисперсная кремниевая кислота и силикаты), сахара (например, тростниковый сахар, лактоза и глюкоза), эмульгаторы (например, лигнин, отработанные сульфитные щелоки, метилцеллюлоза, крахмал и поливинилпирролидон) и смазывающие вещества (например, стеарат магния, тальк, стеариновая кислота и лаурилсульфат натрия). Таблетки для перорального введения в дополнение к указанным носителям могут содержать добавки, такие как цитрат натрия, карбонат кальция и дикальций фосфат, вместе с различными дополнительными веществами, такими как крахмал, предпочтительно картофельный крахмал, желатин и т.п. Для приготовления таблеток также можно использовать смазывающие вещества, такие как стеарат магния,лаурилсульфат натрия и тальк. В случае водных суспензий в дополнение к указанным выше инертным наполнителям активные вещества можно объединить с различными средствами, усиливающими вкус,или красителями. Доза соединений, предлагаемых в настоящем изобретении, разумеется, сильно зависит от методики введения и подвергающегося лечению заболевания. Комбинации с другими активными веществами Другим объектом настоящего изобретения является комбинированная терапия, в которой соединение, предлагаемое в настоящем изобретении, вводят совместно с другим активным соединением. В соответствии с этим настоящее изобретение также относится к фармацевтическим препаратам, которые предоставляют собой такую комбинацию активных ингредиентов, в которой одним из этих соединений является соединение, предлагаемое в настоящем изобретении. Такие комбинации могут представлять собой фиксированные комбинации доз (фармацевтически активные ингредиенты, которые необходимо объединить, находятся в одном и том же фармацевтическом препарате) или нефиксированные комбинации доз (фармацевтически активные ингредиенты находятся в разных фармацевтических препаратах). Поэтому другим объектом настоящего изобретения является комбинация каждого из соединений,предлагаемых в настоящем изобретении, предпочтительно по меньшей мере одного соединения, предлагаемого в настоящем изобретении, с другим соединением, выбранным из группы, включающей, например, ингибиторы бета-секретазы; ингибиторы гамма-секретазы; модуляторы гамма-секретазы; ингибиторы агрегации амилоидов, такие как, например, альцгемед; нейропротективные вещества прямого или непрямого действия и/или модифицирующие заболевание вещества; антиоксиданты, такие как, например, витамин Е, гинкго билоба или гинколид; противовоспалительные вещества, такие как, например,ингибиторы Cox, НСПВС (нестероидные противовоспалительные средства), дополнительно или исключительно способные снижать содержание А (А-бета); ингибиторы HMG-СоА редуктазы, такие как статины; ингибиторы ацетилхолинэстеразы, такие как донепезил, ривастигмин, такрин, галантамин; антагонисты рецептора NMDA, такие как, например, мемантин; агонисты рецептора АМРА; позитивные модуляторы рецептора АМРА, АМРкины, ингибиторы переносчика 1 глицина; ингибиторы рецептора повторного захвата моноаминов; вещества, влияющие на концентрацию или высвобождение нейротрансмиттеров; вещества, вызывающие секрецию гормона роста, такие как ибутаморенмезилат и капроморелин; антагонисты или обратные агонисты рецептора СВ-1; антибиотики, такие как миноциклин или рифампицин; ингибиторы PDE1, PDE2, PDE4, PDE5 и/или PDE10, обратные агонисты рецептора GABAA; обратные агонисты рецептора GABAA альфа-5; антагонисты рецептора GABAA; агонисты или частичные агонисты, или позитивные модуляторы никотинового рецептора; агонисты или частичные агонисты,или позитивные модуляторы никотинового рецептора альфа-4-бета-2; агонисты или частичные агонисты никотинового рецептора альфа-7; антагонисты гистаминового рецептора Н 3; агонисты или частичные агонисты рецептора 5-НТ 4; антагонисты рецептора 5-НТ 6; антагонисты альфа-2-адренорецептора, антагонисты кальция; агонисты или частичные агонисты, или позитивные модуляторы мускаринового рецептора M1; антагонисты мускаринового рецептора М 2; антагонисты мускаринового рецептора М 4; позитивные аллостерические модуляторы метаботропного глутаматного рецептора 5; антагонисты метаботропного глутаматного рецептора 2, агонисты метаботропного глутаматного рецептора 2/3, позитивные аллостерические модуляторы метаботропного глутаматного рецептора 2 и другие вещества, которые модулируют рецепторы или ферменты таким образом, что повышается эффективность и/или безопасность соединений, предлагаемых в настоящем изобретении, или уменьшаются нежелательные побочные эффекты. Настоящее изобретение также относится к фармацевтическим композициям, содержащим одно или большее количество, предпочтительно одно активное вещество. По меньшей мере одно активное вещество выбрано из числа соединений, предлагаемых в настоящем изобретении, и/или их соответствующих солей. Композиция предпочтительно содержит только одно такое активное вещество. В случае,если содержится более одного активного вещества, другое может быть выбрано из указанной выше группы компонентов комбинации, включающей альцгемед, витамин Е, гинколид, донепезил, ривастигмин, такрин, галантамин, мемантин, ибутаморенмезилат, капроморелин, миноциклин и/или рифампицин. Композиция необязательно дополнительно содержит такие ингредиенты, как инертные носители и/или разбавители. Соединения, предлагаемые в настоящем изобретении, также можно использовать в комбинации с методиками иммунотерапии, такими как, например, активная иммунизация с помощью Abeta (амилоидные бета-пептиды) или ее части или пассивная иммунизация гуманизированными антителами к Abeta или фрагментами антител, для лечения указанных выше заболеваний и патологических состояний. Соединения, предлагаемые в настоящем изобретении, также можно объединять с димебоном. Соединения, предлагаемые в настоящем изобретении, также можно объединять с антидепрессантами, такими как амитриптилин, имипрамингидрохлорид (тофранил), имипраминмалеат (сурмонтил), лофепрамид, дезипрамин (норпрамин), доксепин (синекван, зоналон), тримипрамин (сурмонтил). Или соединения, предлагаемые в настоящем изобретении, также можно объединять с ингибиторами повторного всасывания серотонина (5-НТ), такими как алапроклат, циталопрам (целекса, ципрамил) эсциталопрам (лексапро, ципралекс), кломипрамин (анафранил), дулоксетин (цимбалта), феноксетин (малексил), фенфлурамин (пондимин), норфенфлурамин, флуоксетин (прозак), флувоксамин (лувокс), ин- 17023574(пристик), бразофензин и тезофензин. Компоненты комбинаций, предлагаемых в настоящем изобретении, могут находиться одновременно в одной дозированной форме, т.е. в виде комбинированного препарата, например два компонента могут быть включены в одну таблетку, например в разные слои указанной таблетки. Компоненты комбинаций также могут быть использоваться по отдельности в виде свободной комбинации, т.е. соединения,предлагаемые в настоящем изобретении, находятся в одной дозированной форме и один или большее количество указанных выше компонентов комбинации находятся в другой дозированной форме. Эти две дозированные формы могут представлять собой равноценные дозированные формы, например, при совместном введении двух таблеток, одна из которых содержит соединение, предлагаемое в настоящем изобретении, в терапевтически эффективном количестве, а вторая содержит указанный выше компонент комбинации в терапевтически эффективном количестве. При необходимости также можно комбинировать разные вводимые формы. Можно приготовить подходящие вводимые формы любого типа. Соединение, предлагаемое в настоящем изобретении, или его физиологически приемлемую соль в комбинации с другим активным веществом можно использовать одновременно или поочередно, но предпочтительно быстро одно за другим. При одновременном введении эти два активных вещества вводят пациенту совместно: при поочередном введении эти два активных вещества вводят пациенту последовательно с промежутком, меньшим или равным 12, предпочтительно меньшим или равным 6 ч. На дозированные или вводимые формы не налагаются ограничения, в контексте настоящего изобретения можно использовать любую подходящую дозированную форму. Типичные дозированные формы можно выбрать из числа твердых препаратов, таких как пластыри, таблетки, капсулы, пилюли, пеллеты, драже, порошки, лепешки, суппозитории, жидких препаратов, таких как растворы, суспензии, эмульсии, капли, сиропы, эликсиры, или газообразных препаратов, таких как аэрозоли, спреи и т.п. Дозированные формы с успехом готовят в виде дозированных единиц, каждая дозированная единица обеспечивает введение одной дозы каждого содержащегося активного компонента. Ингредиенты выбирают в соответствии с путем введения и дозированной формой. Дозировка компонентов указанной выше комбинаций предпочтительно составляет от 1/5 от обычно рекомендуемой минимальной дозы до 1/1 от обычно рекомендуемой дозы. Дозированные формы вводят пациенту, например, 1, 2, 3 или 4 раза в сутки в зависимости от типа препарата. В случае препаратов замедленного или пролонгированного высвобождения или других фармацевтических препаратов введение можно проводить по-другому (например, один раз в неделю или в месяц и т.п.). Соединения, предлагаемые в настоящем изобретении, предпочтительно вводить три раза в сутки или реже, более предпочтительно один или два раза в сутки. Примеры Фармацевтические композиции. Примеры могут иллюстрировать возможные фармацевтические препараты, которые не являются ограничивающими. Термин "активное вещество" означает одно или большее количество соединений, предлагаемых в настоящем изобретении, включая их соли. В случае, если одна из указанных выше комбинаций содержит одно или большее количество активных веществ, термин "активное вещество" также может включать дополнительные активные вещества. Пример А. Таблетки, содержащие 100 мг активного вещества. Препарат: таблетка. Пример С. Капсулы из твердого желатина, содержащие 150 мг активного вещества. Приготовление любого из указанных выше препаратов можно провести по стандартным методикам. Биологическое исследование. Воздействие in vitro соединений, предлагаемых в настоящем изобретении, можно продемонстрировать с помощью приведенных ниже биологических исследований. Протокол исследования PDE9A2. Исследование ферментативной активности PDE9A2 проводили с помощью сцинтилляционнопроксимального анализа (СПА) в целом в соответствии с протоколом изготовителя (GE Healthcare,ранее - Amersham Biosciences, product number: TRKQ 7100). В качестве источника фермента использовали лизат (ЗФФ (забуференный фосфатом физиологический раствор) с добавлением 1% Triton Х-100 и ингибиторов протеазы, продукт распада клеток удаляли центрифугированием при 13000 об/мин в течение 30 мин) клеток SF 9, экспрессирующих PDE9A2 человека. Полное количество белка, использованного при анализе, менялось в зависимости от степени заражения и эффективности продуцирования клетками SF9 и составляло 0,1-100 нг. Обычно условия проведения анализа были следующими: полный объем при анализе: 40 мкл; количество белка: 0,1-50 нг; концентрация субстрата (цГМФ): 20 нМ; 1 мкКи/л; длительность инкубации: 60 мин при комнатной температуре; конечная концентрация ДМСО: 0,2-1%. Анализы проводили в 384-луночных планшетах. Исследуемые реагенты, а также фермент и субстрат разводили в буфере для анализа. Буфер для анализа содержал 50 мМ Tris [трис(гидроксиметиламинометан)], 8,3 мМ MgCl2, 1,7 мМ ЭГТУ (этиленгликольтетрауксусная кислота), 0,1% БСА (бычий сывороточный альбумин), 0,05% Tween 20; значение рН буфера для анализа устанавливали равным 7,5. Реакцию останавливали путем введения избытка специфического ингибитора PDE9 (например, соединения, указанного в WO 04/099210 или WO 04/099211, такого как один из энантиомеров, приведенных в примере 37, например 1-(2-хлорфенил)-6-[(2R)-3,3,3-трифтор-2-метилпропил]-1,5-дигидро 4 Н-пиразоло[3,4-d]пиримидин-4-она). Литератураinhibitor BAY 73-6691 improves learning and memory in rodents. Neuropharmacology. 2008 Окт; 55(5):90818. Протокол исследования PDE1C. Анализ проводили аналогично анализу PDE9A2 с внесением следующих изменений: вместоPDE9A2 использовали PDE1C и буфер для анализа дополнительно содержал 50 нМ кальмодулина, 3 мМCaCl2. Реакцию можно остановить путем введения такого же ингибитора, как указанный вышеIC50 можно рассчитать с помощью GraphPadPrism или другого подходящего программного обеспечения с заданием значения, равного 100 для положительного контроля и равного 0 для отрицательного контроля. Для расчета значений IC50 разведения исследуемых соединений (субстратов) следует выбирать и исследовать по указанному выше протоколу. Данные. Приведенные ниже значения IC50 для ингибирования PDE9A2 [нМ] показывают, что соединения,предлагаемые в настоящем изобретении, ингибируют PDE9, в частности PDE9A2. Это свидетельствует о том, что соединения обладают полезными фармакологическими характеристиками. Примеры не являются ограничивающими. В таблице также приведены значения селективности (селективность), которые указывают на преимущество соединений при ингибировании PDE9A по сравнению с ингибированием PD1C. Селективность представляет собой отношение (IC50 для ингибирования PDE1C [нМ])/(IC50 для ингибированияPDE9A2 [нМ]). Номера примеров относится к конечным соединениям примеров, описанным в разделе "Типичные варианты осуществления" и определенным в приведенной выше таблице семейств соединений (табл. 1). Все значения получены по методике, описанной в настоящем изобретении. Определение энантиомер 1 или энантиомер 2 относится к порядку элюирования энантиомеров в хиральной СЖХ и хиральной ВЭЖХ. Таблица 2 Воздействие in vivo. Предполагается, что данные по благоприятной эффективности in vitro соединений, предлагаемых в настоящем изобретении, соответствуют благоприятной эффективности in vivo. Воздействие in vivo соединений, предлагаемых в настоящем изобретении, можно исследовать с помощью нового теста распознавания объектов по методике, описанной в публикации Prickaerts et al.(Neuroscience, 2002, 113, 351-361), с помощью теста социального распознавания или с помощью исследо- 20023574 вания самопроизвольных нарушений в Т-образном лабиринте по методикам, описанным в публикацииvan der Staay et al. (Neuropharmacology 2008, 55, 908-918). Дополнительная информация о биологическом исследовании также приведена в этих двух публикациях. Кроме способности ингибировать мишень PDE9, соединения, предлагаемые в настоящем изобретении, могут обладать другими полезными фармакокинетическими характеристиками. Например, соединения, предлагаемые в настоящем изобретении, могут обладать одной или большим количеством благоприятных характеристик, таких как безопасность, сбалансированный метаболизм, незначительная опасность лекарственных взаимодействий и/или сбалансированный клиренс. Соединения также могут обладать одной или большим количеством дополнительных или альтернативных благоприятных характеристик, таких как биологическая доступность, значительная степень всасывания, способность проходить через гематоэнцефалический барьер, подходящее (например, большое среднее) время удерживания, благоприятное воздействие на подвергающийся лечению участок и т.п. Получение соединений. Ниже представлены соединения и их методики получения, некоторые из них являются объектом настоящего изобретения, некоторые из них приведены для дополнительной иллюстрации. Соединения 23-34 являются объектами настоящего изобретения. Аббревиатуры: реагент Берджесса - (метоксикарбонилсульфамоил)триэтиламмоний-N-бетаин реагент Лавессона 2,4-бис-(4-метоксифенил)-[1,3,2,4]дитиадифосфетан-2,4-дисульфид; ХИАД - химическая ионизация при атмосферном давлении; АЦН - ацетонитрил; КДИ - 1,1'-карбонилдиимидазол; ДЭА - диэтиламин; ДИПЭА - диизопропилэтиламин; ДМЭ - 1,2-диметоксиэтан; ДМФ - диметилформамид; ИЭР - ионизация электрораспылением (в МС);EtOH - этанол; ч - час(ы); ГАТУ - О-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилуронийгексафторфосфат; ВЭЖХ - высокоэффективная жидкостная хроматография; ВЭЖХ-МС - связанная высокоэффективная жидкостная хроматография - масс-спектрометрия; М - молярная концентрация (моль/л);TBTU - О-(бензотриазол-1-ил)-N,N,N',N'-тетраметилуронийтетрафторборат; ТФК - трифторуксусная кислота; ТГФ - тетрагидрофуран; ТСХ - тонкослойная хроматография. Методики ЖХ-МС. Методика 1. Тип прибора для МС: Waters Micromass ZQ; Тип прибора для ВЭЖХ: Waters Alliance 2695, детектор с диодной матрицей Waters 2996; Колонка: Varian Microsorb 100 С 18, 304,6 мм, 3,0 мкм; Элюент А: вода + 0,13% ТФК, элюент В: АЦН; Градиентный режим: 0,0 мин 5% В 0,18 мин 5% В 2,0 мин 98% В 2,2 мин 98% В 2,3 мин 5% В 2,5 мин 5% В; Скорость потока: 3,5 мл/мин; УФ-детектирование: 210-380 нм. Методика 2. Тип прибора для МС: Waters Micromass ZQ; Тип прибора для ВЭЖХ: Waters Alliance 2695, детектор с диодной матрицей Waters 2996; Колонка: Varian Microsorb 100 С 18, 304,6 мм, 3,0 мкм; Элюент А: вода + 0,13% ТФК, элюент В: MeOH; Градиентный режим: 0,0 мин 5% В 0,35 мин 5% В 3,95 мин 100% В 4,45 мин 100% В 4,55 мин 5% В 4,9 мин 5% В; Скорость потока: 2,4 мл/мин; УФ-детектирование: 210-380 нм. Методика 3. Тип прибора для МС: Waters Micromass ZQ; Тип прибора для ВЭЖХ: Waters Alliance 2695, детектор с диодной матрицей Waters 2996; Колонка: Varian Microsorb С 18, 204,6 мм, 5,0 мкм; Элюент А: вода + 0,15% ТФК, элюент В: MeOH; Градиентный режим: 0,0 мин 5% В 0,25 мин 5% В 1,90 мин 100% В 2,05 мин 100% В 2,15 мин 5% В 2,25 мин 5% В; Скорость потока: 5,2 мл/мин; УФ-детектирование: 210-400 нм. Методика 1 Е hydro. Прибор: ЖХ/МС ThermoFinnigan. Hplc Surveyor DAD, квадрупольный MSQ; Колонка: Synergi Hydro-RP80A, 4 мкм, 4,60100 мм; Элюент А: 90% вода + 10% ацетонитрил +10 мМ формиат аммония; Элюент В = 90% АЦН +10% Н 2 О +10 мМ NH4COOH; Градиентный режим: А (100) в течение 1,5 мин, затем до В (100) за 10 мин и затем В (100) в течение 1,5 мин; Скорость потока: 1,2 мл/мин; УФ-детектирование: 254 нм; Источник ионов: ХИАД. Методики хиральной СЖХ: Методика 4. Тип прибора для СЖХ: Berger "Analytix"; Колонка: Daicel IC, 2504,6 мм, 5,0 мкм; Элюент: СО 2/25% MeOH/0,2% ДЭА (в изократическом режиме); Скорость потока: 4,0 мл/мин, 10 мин, температура: 40 С; УФ-детектирование: 210/220/254 нм. Методика 5. Тип прибора для СЖХ: Berger "Analytix"; Колонка: Daicel ADH, 2504,6 мм, 5,0 мкм; Элюент: СО 2/25% MeOH/0,2% ДЭА (в изократическом режиме); Скорость потока: 4,0 мл/мин, 10 мин, температура: 40 С; УФ-детектирование: 210/220/254 нм. Методики хиральной ВЭЖХ. Методика 6. Тип прибора для ВЭЖХ: Agilent 1100; Колонка: Daicel chiralcel OJ-H, 2504,6 мм, 5,0 мкм; Элюент: гексан/EtOH 80:20; Скорость потока: 1 мл/мин, температура: 25 С; УФ-детектирование: при переменной длине волны (200-500 нм). Методика 6.1. Тип прибора для ВЭЖХ: Agilent 1100; Колонка: Daicel chiralcel OJ-H, 2504,6 мм, 5,0 мкм; Элюент: гексан/EtOH 85:15; Скорость потока: 1 мл/мин, температура: 25 С; УФ-детектирование: при переменной длине волны (200-500 нм). Методика 7. Тип прибора для ВЭЖХ: Agilent 1100; Колонка: Chiralpak AD-H, 2504,6 мм, 5,0 мкм; Элюент: гексан/изопропанол 80:20; Скорость потока: 1 мл/мин, температура: 25 С; УФ-детектирование: при переменной длине волны (200-500 нм). Нагревание микроволновым излучением: Приборы Discover СЕМ, снабженные сосудами емкостью 10 и 35 мл;Biotage Initiator Sixty. Общие замечания о представлении структур. Соединения, содержащие стереогенный центр (центры). Структуры, представленные ниже в экспериментальном разделе, отображают только один, а необязательно все стереохимические варианты соединений. Однако в таких случаях после структуры приведено, например, выражение "транс-рацемическая смесь" или "цис-рацемическая смесь", чтобы отметить другие возможные стереохимические конфигурации. Пример приведен ниже. Представленная структурная формула имеет вид Добавленное выражение "цис-рацемическая смесь" указывает на вторую возможную стереохимическую конфигурацию: Таким образом, полученное соединение представляет собой смесь Такой подход используется и для других изображенных структур. Исходные соединения. Пример 1 А (транс-рацемическая смесь) 2,00 г (13,9 ммоль) транс-циклобутан-1,2-дикарбоновой кислоты при 0 С смешивали с 16 мл EtOH и медленно добавляли 2,21 мл (30,5 ммоль) тионилхлорида. Смеси давали нагреться до комнатной температуры и перемешивали в течение 1 ч. Растворитель удаляли при пониженном давлении и продукт фильтровали через слой активированного основного оксида алюминия. Получали 2,71 г (98%) продукта. ВЭЖХ-МС (методика 1): Rt=1,34 мин,МС (ИЭР, положительная ионизация): m/z = 201 (М+Н). Соединение указанного ниже примера синтезировали аналогично получению соединения примера 1 А с использованием в качестве исходного вещества соответствующей дикислоты. 8,00 г (89,7 ммоль) 2-аминопропионовой кислоты смешивали с 88,0 мл (0,93 моль) уксусного ангидрида и 88,0 мл пиридина. Реакционную смесь перемешивали при 100 С в течение 135 мин. Растворитель удаляли при пониженном давлении. К остатку добавляли толуол и растворитель удаляли при пониженном давлении, затем добавляли 204 мл (816 ммоль) HCl (4 М водный раствор) и смесь кипятили с обратным холодильником в течение 3 ч. Растворитель удаляли при пониженном давлении. К остатку добавляли 1-бутанол (20 мл) и растворитель удаляли при пониженном давлении. Получали 11,6 г искомого соединения в виде гидрохлорида. МС (ИЭР, положительная ионизация): m/z = 88 (М+Н)+. Пример 3 А (транс-рацемическая смесь)(см. заявку на патент РСТ WO 2010/026214, пример 8 А) смешивали с 15 мл безводного EtOH, добавляли 2,46 г (12,3 ммоль) соединения примера 1 А и 0,66 г (16,4 ммоль) гидрида натрия (60% суспензия в минеральном масле). Реакционную смесь нагревали в микроволновой печи при 140 С в течение 30 мин. Смесь охлаждали до комнатной температуры и добавляли раствор гидроксида натрия (4 М водный раствор). Растворитель удаляли при пониженном давлении. Остаток очищали с помощью препаративной ВЭЖХ(элюент А: вода + 0,13% ТФК, элюент В: MeOH). Получали 0,70 г (49%) продукта. ВЭЖХ-МС (методика 1): Rt=1,24 мин,МС (ИЭР, положительная ионизация): m/z = 353 (М+Н)+. Соединения указанных ниже примеров синтезировали аналогично получению соединения примера 3 А с использованием в качестве исходных веществ соответствующего амида и сложного эфира (исходные вещества описаны в публикации патента РСТ WO 2010/026214, WO 2009/121919 и WO 2004/09921). 0,200 г (0,568 ммоль) соединения примера 3 А смешивали с 0,157 мл (1,14 ммоль) триэтиламина и 5 мл ДМФ. К смеси добавляли 0,237 г (0,624 ммоль) ГАТУ, затем реакционную смесь перемешивали при комнатной температуре в течение 10 мин. К смеси добавляли 0,042 г (0,568 ммоль) гидразида уксусной кислоты и реакционную смесь перемешивали при комнатной температуре в течение 1 ч. Смесь очищали с помощью препаративной ВЭЖХ (элюент А: вода + 0,13% ТФК, элюент В: MeOH). Получали 30 мг продукта. ВЭЖХ-МС (методика 1): Rt=1,03 мин,МС (ИЭР, положительная ионизация): m/z = 409 (М+Н)+. Пример 5 А (транс-рацемическая смесь) 0,150 г (0,426 ммоль) соединения примера 3 А смешивали с 2 мл ТГФ. Смесь охлаждали до 0 С и добавляли 0,036 мл (0,426 ммоль) оксалилхлорида и одну каплю ДМФ. Реакционную смесь перемешивали при 0 С в течение 1 ч. К реакционной смеси добавляли 2 мл АЦН и 0,426 мл (0,851 ммоль) триметил- 24023574 силилдиазометана (2 М раствор в гексане). Смесь перемешивали в течение 2 ч, затем медленно добавляли 0,213 мл HCl (4 М раствор в диоксане). Реакционную смесь перемешивали в течение 3 ч. К смеси добавляли этилацетат и насыщенный водный раствор гидрокарбоната натрия. Органический слой промывали водой и рассолом и сушили над сульфатом натрия. Растворители частично выпаривали до объема,равного примерно 2 мл. Смесь использовали на следующей стадии без дополнительной очистки. ВЭЖХ-МС (методика 1): Rt=1,40 мин,МС (ИЭР, положительная ионизация): m/z = 385/387 (Cl). Соединение указанного ниже примера синтезировали аналогично получению соединения примера 5 А с использованием в качестве исходного вещества соответствующей кислоты. 0,200 г (0,628 ммоль) соединения примера ЗВ смешивали с 1 мл ДМФ. Добавляли 0,261 мл(1,89 ммоль) триэтиламина и 0,222 г (0,691 ммоль) TBTU. Реакционную смесь перемешивали при комнатной температуре в течение 10 мин. Затем добавляли 0,078 г (0,628 ммоль) соединения примера 2 А и смесь перемешивали при комнатной температуре в течение 1 ч. Смесь очищали с помощью препаративной ВЭЖХ (элюент А: вода + 0,13% ТФК, элюент В: MeOH). Получали 190 мг продукта. ВЭЖХ-МС (методика 3): Rt=1,03 мин,МС (ИЭР, положительная ионизация): m/z = 388 (М+Н). Пример 7 А (транс-рацемическая смесь)(1,26 ммоль) триэтиламина и 0,222 г (0,691 ммоль) TBTU. Реакционную смесь перемешивали при комнатной температуре в течение 10 мин. Затем добавляли 0,066 г (0,628 ммоль) 2,2-диметоксиэтиламина и смесь перемешивали при комнатной температуре в течение 1 ч. Затем добавляли HCl (2 М водный раствор) и смесь очищали с помощью препаративной ВЭЖХ (элюент А: вода + 0,13% ТФК, элюент В:MeOH). Остаток смешивали с 5 мл ацетона и 1 мл HCl (2 М водный раствор) и перемешивали в атмосфере азота в течение ночи. Затем смесь экстрагировали с помощью ДХМ (дихлорметан). Органический слой выпаривали и очищали с помощью препаративной ВЭЖХ (элюент А: вода + 0,13% ТФК, элюент В: MeOH). Получали 170 мг продукта. ВЭЖХ-МС (методика 3): Rt=1,01 мин,МС (ИЭР, положительная ионизация): m/z = 360 (М+Н)+.(2,84 ммоль) ДИПЭА и 0,200 г (0,624 ммоль) TBTU. Реакционную смесь перемешивали при комнатной температуре в течение 10 мин. Затем добавляли 0,140 г (1,14 ммоль) соединения примера 2 А и смесь перемешивали при комнатной температуре в течение 2 ч. Смесь очищали с помощью препаративной ВЭЖХ (элюент А: вода + 0,13% ТФК, элюент В: MeOH). Получали 70 мг (29%) продукта. ВЭЖХ-МС (методика 1): Rt=1,23 мин,МС (ИЭР, положительная ионизация): m/z = 422 (М+Н)+. Соединения указанных ниже примеров синтезировали аналогично получению соединения примера 8 А с использованием в качестве исходных веществ соответствующих нуклеофильных реагентов. 0,182 г (0,430 ммоль) перйодинана Десса-Мартина смешивали с 2,5 мл ДХМ. При комнатной температуре добавляли 0,160 г (0,391 ммоль) соединения примера 8D в 2,5 мл ДХМ. Реакционную смесь перемешивали при комнатной температуре в течение 30 мин и при 30 С в течение 30 мин. К смеси добавляли 10 мл раствора тиосульфата натрия (10% в воде) и 10 мл насыщенного раствора гидрокарбоната натрия и смесь перемешивали в течение 20 мин. Органический слой отделяли и водный слой экстрагировали с помощью ДХМ. Органический слой промывали насыщенным раствором гидрокарбоната натрия,сушили и выпаривали. Получали 93 мг (58%) продукта. ВЭЖХ-МС (методика 1): Rt=1,18 мин,МС (ИЭР, положительная ионизация): m/z = 408 (М+Н)+. Соединение указанного ниже примера синтезировали аналогично получению соединения примера 9 А с использованием в качестве исходного вещества соответствующего спирта. 0,450 г соединения примера 3 С смешивали с 3,5 мл ДМФ и 0,273 г (2,21 ммоль) соединения примера 2 А. Добавляли 1,00 мл (6,64 ммоль) ДИПЭА и 0,390 г (1,22 ммоль) TBTU и смесь перемешивали в течение 1 ч. Смесь очищали с помощью препаративной ВЭЖХ (элюент А: вода + 0,13% ТФК, элюент В:(см. WO 2010/026214, пример 8 А) в атмосфере азота смешивали с 4 мл безводного EtOH, 326 мг(3,07 ммоль) транс-циклобутан-1,2-дикарбонитрила и 0,197 г (4,91 ммоль) гидрида натрия (60% суспензия в минеральном масле). Реакционную смесь нагревали в микроволновой печи при 140 С в течение 45 мин. Растворитель удаляли при пониженном давлении. Остаток очищали с помощью препаративной ВЭЖХ (элюент А: вода + 0,13% ТФК, элюент В: MeOH). Получали 210 мг (51%) искомого соединения. ВЭЖХ-МС (методика 3): Rt=1,19 мин,МС (ИЭР, положительная ионизация): m/z = 334 (М+Н)+. К раствору 0,8 г (3,805 ммоль) амида 5-амино-1-(тетрагидропиран-4-ил)-1 Н-пиразол-4-карбоновой кислоты (см. заявку на патент РСТ WO 2010/026214) в 8 мл безводного EtOH при комнатной температуре в атмосфере азота добавляли 0,457 г (19,6 ммоль) гидрида натрия (60% суспензия в минеральном масле). Через 1 ч при перемешивании добавляли 1,2 г (11,42 ммоль) транс-циклобутан-1,2-дикарбонитрила и реакционную смесь нагревали в микроволновой печи при 140 С в течение 45 мин. Растворитель удаляли при пониженном давлении. Остаток растворяли в ДХМ, добавляли воду и фазы разделяли. Органические слои сушили над сульфатом натрия и выпаривали при пониженном давлении. Неочищенное вещество очищали с помощью флэш-хроматографии (Cy)/EtOAc от 80/20 до 100%) и получали искомое соединение в виде желтого твердого вещества (0,64 г, 55%). ВЭЖХ-МС (методика 1Eh):Rt=6,21 мин,МС (ХИАД): m/z = 300 (М+Н)+. Пример 11 С (транс-рацемическая смесь) К раствору 0,85 г (3,91 ммоль) амида 5-амино-1-(4-метилпиридин-3-ил)-1 Н-пиразол-4-карбоновой кислоты (см. заявку на патент РСТ WO 2004/09921) в 10 мл безводного EtOH в атмосфере азота при комнатной температуре добавляли 0,47 г (11,74 ммоль) гидрида натрия (60% суспензия в минеральном масле). Через 1 ч при перемешивании добавляли 1,28 г (11,74 ммоль) транс-циклобутан-1,2-дикарбонитрила и реакционную смесь нагревали в микроволновой печи при 140 С в течение 45 мин. Затем реакционную смесь загружали в картридж SCX, содержащие аммиак фракции собирали и выпаривали и остаток очищали с помощью флэш-хроматографии (ДХМ/MeOH 90:10) и получали искомое соединение в виде белого твердого вещества. (0,63 г, 52%). ВЭЖХ-МС (методика 1Eh): Rt=5,92 мин,МС (ХИАД, положительная ионизация): m/z = 307 (М+Н)+. Пример 12 А (транс-рацемическая смесь)(2,30 ммоль) безводного MeOH. При 0 С медленно добавляли 0,103 мл (1,45 ммоль) ацетилхлорида. Смесь перемешивали при комнатной температуре в течение 12 ч. Растворитель удаляли при пониженном давлении. К остатку добавляли 0,5 мл MeOH. Затем при 0 С добавляли 0,407 мл (2,85 ммоль) аммиака(7 М раствор в MeOH) и смеси давали нагреться до комнатной температуры. Через 30 мин реакционную смесь обрабатывали водой и значение рН доводили до рН 1 путем добавления ТФК. Смесь очищали с помощью препаративной ВЭЖХ (элюент А: вода + 0,13% ТФК, элюент В: MeOH) и получали 110 мг К смеси сухого EtOH (5 мл) и сухого CHCl3 (5 мл), охлажденной до 0 С, медленно добавляли ацетилхлорид (2,27 мл, 30,82 ммоль) и смесь перемешивали при 0 С в течение 20 мин. По каплям добавляли раствор соединения примера 11 В (0,410 г, 1,027 ммоль) в сухом CHCl3 (5 мл) и смесь перемешивали при комнатной температуре в течение ночи. Растворители выпаривали при пониженном давлении, остаток растворяли в сухом EtOH (5 мл) и добавляли 6,4 мл 7,0 М раствора аммиака в MeOH (30,82 ммоль). Смесь перемешивали при комнатной температуре в течение 12 ч. Растворитель удаляли при пониженном давлении. Конечный продукт получали в виде гидрохлорида и использовали на следующей стадии без дополнительной очистки (0,37 г, содержание 50%, определено с помощью ВЭЖХ-МС). ВЭЖХ-МС (методика 1Eh): Rt=5,38 мин,МС (ХИАД, положительная ионизация): m/z = 317 (М+Н)+. Пример 12 С (транс-рацемическая смесь) К смеси сухого EtOH (4 мл) и сухого CHCl3 (10 мл), охлажденной до 0 С, медленно добавляли ацетилхлорид (4,38 мл, 61,7 ммоль) и смесь перемешивали при 0 С в течение 20 мин. По каплям добавляли раствор соединения примера 11 С (0,63 г, 2,057 ммоль) в сухом CHCl3 (5 мл) и смесь перемешивали при комнатной температуре в течение ночи. Растворители выпаривали при пониженном давлении, остаток растворяли в сухом MeOH (10 мл) и добавляли 10,3 мл 7,0 М раствора аммиака в MeOH (72 ммоль). Смесь перемешивали при комнатной температуре в течение 12 ч. Растворитель удаляли при пониженном давлении. Конечный продукт, полученный в виде гидрохлорида, использовали на следующей стадии без дополнительной очистки (0,85 г, содержание 84%, определено с помощью 1 Н-ЯМР (ядерный магнитный резонанс. ВЭЖХ-МС (методика 1 Eh): Rt=5,15 мин,МС (ХИАД, положительная ионизация): m/z = 324 (М+Н)+. Пример 13 А (транс-рацемическая смесь) К раствору 1,6 г (10,24 ммоль) метилового эфира 2-ацетилциклобутанкарбоновой кислоты (получали, как это описано в публикации J. Med. Chem, 25, 109, 1982) в сухом EtOH (12 мл) добавляли пропаргиламин (1,4 мл, 20,4 ммоль), затем 0,122 г (0,307 ммоль) трихлорида натрия-золота. Реакционную смесь нагревали в микроволновой печи при 140 С в течение 45 мин, твердое вещество отфильтровывали и органические вещества выпаривали. Неочищенное вещество очищали с помощью флэш-хроматографии(Cy/EtOAc 70:30) и получали искомое соединение в виде желто-зеленого масла (0,18 г, 9,2%). ВЭЖХ-МС (методика 1Eh): Rt=0,87 мин,МС (ХИАД, положительная ионизация): m/z =192 (М+Н)+.
МПК / Метки
МПК: C07D 487/04, A61K 31/519, A61P 25/28
Метки: производные, 6-циклобутил-1,5-дигидропиразоло[3,4-d]пиримидин-4-она, применение, качестве, ингибиторов, pde9a
Код ссылки
<a href="https://eas.patents.su/30-23574-proizvodnye-6-ciklobutil-15-digidropirazolo34-dpirimidin-4-ona-i-ih-primenenie-v-kachestve-ingibitorov-pde9a.html" rel="bookmark" title="База патентов Евразийского Союза">Производные 6-циклобутил-1,5-дигидропиразоло[3,4-d]пиримидин-4-она и их применение в качестве ингибиторов pde9a</a>
Производные 1-гетероциклил-1,5-дигидропиразоло[3,4-d]пиримидин-4-она и их применение в качестве модуляторов pde9a

Номер патента: 21504
Опубликовано: 30.07.2015
Авторы: Шэнцле Герхард, Джованнини Риккардо, Фукс Клаус, Айкмайер Кристиан, Дорнер-Циоссек Корнелиа, Фиген Деннис, Хайне Никлас, Фокс Томас, Розенброк Хольгер
МПК: A61K 31/519, C07D 487/04
Метки: производные, модуляторов, качестве, 1-гетероциклил-1,5-дигидропиразоло[3,4-d]пиримидин-4-она, pde9a, применение
Формула / Реферат:
1. Соединение общей формулы (I)в которой Hc выбран из группы, включающей тетрагидропиранил, тетрагидрофуранил, пиперидинил и пирролидинил;R1 выбран из группы, включающей фенил, 2-, 3- и 4-пиридил-, пиримидинил, пиразолил, тиазолил, циклопропил, циклобутил, циклопентил, циклогептил, циклопентилметил, циклогексил, этил, пропил, 1- и 2-бутил-, 1-, 2- и 3-пентил-, тетрагидрофуранил и тетрагидропиранил, где эти группы необязательно могут содержать...
Производные 4,7-дигидропиразоло[1,5-a]пиразин-6-иламина, используемые в качестве ингибиторов бета-секретазы (васе)

Номер патента: 21723
Опубликовано: 31.08.2015
Авторы: Тресадерн Гэри Джон, Дельгадо-Хименес Франсиска, Трабанко-Суарес Андрес Авелино
МПК: A61P 25/28, C07D 487/04, A61K 31/4985...
Метки: используемые, качестве, 4,7-дигидропиразоло[1,5-a]пиразин-6-иламина, ингибиторов, производные, васе, бета-секретазы
Формула / Реферат:
1. Соединение формулы (I)или его таутомерная или стереоизомерная форма, у которогоR1 и R2 представляют собой водород;R3 выбран из группы, состоящей из C1-3алкила и С3-6циклоалкила;X1 и X3 представляют собой СН или F;X2 и X4 представляют собой СН;L представляет собой связь или -N(R5)CO-, где R5 представляет собой водород или C1-3алкил;R6 представляет собой водород или трифторметил;Ar представляет собой гомоарил или гетероарил;гомоарил...
Производные 6,7-дигидропиразоло[1,5-a]пиразин-4-иламина, пригодные в качестве ингибиторов бета-секретазы (bace)

Номер патента: 22649
Опубликовано: 29.02.2016
Авторы: Ван Гол Михиль Люк Мария, Вега-Рамиро Хуан Антонио, Трабанко-Суарес Андрес Авелино, Гейсен Хенрикус Якобус Мария, Дельгадо-Хименес Франсиска
МПК: A61P 25/28, C07D 487/04, A61K 31/4985...
Метки: производные, бета-секретазы, качестве, 6,7-дигидропиразоло[1,5-a]пиразин-4-иламина, bace, пригодные, ингибиторов
Формула / Реферат:
1. Соединение формулы (I)или его стереоизомерная форма,где R1 выбран из группы, состоящей из водорода, галогена;R2 выбран из группы, состоящей из водорода, циано, C1-3алкила, полигалоген-C1-3алкила;R3 выбран из группы, состоящей из C1-3алкила, полигалоген-C1-3алкила;X1 представляет собой C(R4), где R4 представляет собой водород, галоген;X2, X3, X4 представляют собой C(R4), где R4 представляет собой водород;L представляет собой -N(R5)CO-, где R5...
Производные 3-(3-пиримидин-2-илбензил)-1,2,4-триазоло[4,3-b]пиридазина в качестве ингибиторов met киназы

Номер патента: 19534
Опубликовано: 30.04.2014
Авторы: Блаукат Андрее, Штибер Франк, Дорш Дитер, Шадт Оливер
МПК: A61K 31/5025, A61P 35/02, C07D 487/04...
Метки: ингибиторов, киназы, качестве, 3-(3-пиримидин-2-илбензил)-1,2,4-триазоло[4,3-b]пиридазина, производные
Формула / Реферат:
1. Соединения формулы Iв которой R1 означает тиазолил, тиофенил, фуранил, пирролил, оксазолил, изоксазолил, оксадиазолил, пиразолил, имидазолил, тиадиазолил, пиридазинил, пиразинил, пиридинил или пиримидинил, где эти радикалы могут также быть моно-, ди- или тризамещенными Hal, [C(R5)2]nOR5 и/или А, или означает фенил, который является моно-, ди- или тризамещенным Hal и/или CN, или означает А;R2 означает O[C(R5)2]nOR5, Het, -[C(R5)2]nHet или...
Новые производные 6-триазолпиридинсульфанил-бензотиазола или -бензимидазола, способ их получения, применение их в качестве лекарственных средств, фармацевтические композиции и новое применение в качестве ингибиторов мет

Номер патента: 23465
Опубликовано: 30.06.2016
Авторы: Венслер Сильви, Альбер Эва, Бак Эрик, Юголини Антонио, Немесек Консепсьон
МПК: C07D 487/04, A61P 35/00, A61K 31/5025...
Метки: 6-триазолпиридинсульфанил-бензотиазола, качестве, производные, новое, лекарственных, ингибиторов, способ, средств, применение, бензимидазола, композиции, получения, мет, новые, фармацевтические
Формула / Реферат:
1. Производное 6-триазолпиридазинсульфанил-бензотиазола или -бензимидазола формулы (I)в которой ---- означает простую или двойную связь;Ra означает атом водорода; радикал С1-5алкокси, необязательно замещенный атомом хлора, гидроксильным радикалом или гетероциклоалкильным радикалом; ОС3-10циклоалкильный радикал; гетероарильный радикал, необязательно замещенный С1-6алкилом; фенильный радикал, необязательно замещенный атомом галогена; радикал...
Предыдущий патент: Дималеат 9-[4-(3-хлор-2-фторфениламино)-7-метоксихиназолин-6-илокси]-1,4-диазаспиро[5.5]ундекан-5-она, его применение в качестве лекарственного средства и его получение
Следующий патент: Способ изготовления агломератов, содержащих резину и воск, изготовленные агломераты и их применение в асфальтах или битумных массах
Случайный патент: Изомерные конденсированные пирролокарбазолы и изоиндолоны