Ингибиторы bace
Номер патента: 19892
Опубликовано: 30.07.2014
Авторы: Вот Грант Мэтьюс, Аудиа Джеймс Эдмунд, Ши Чуншэн Эрик, Мерготт Дастин Джеймс, Виннероски Леонард Лэрри, Уотсон Брайан Морган























Формула / Реферат
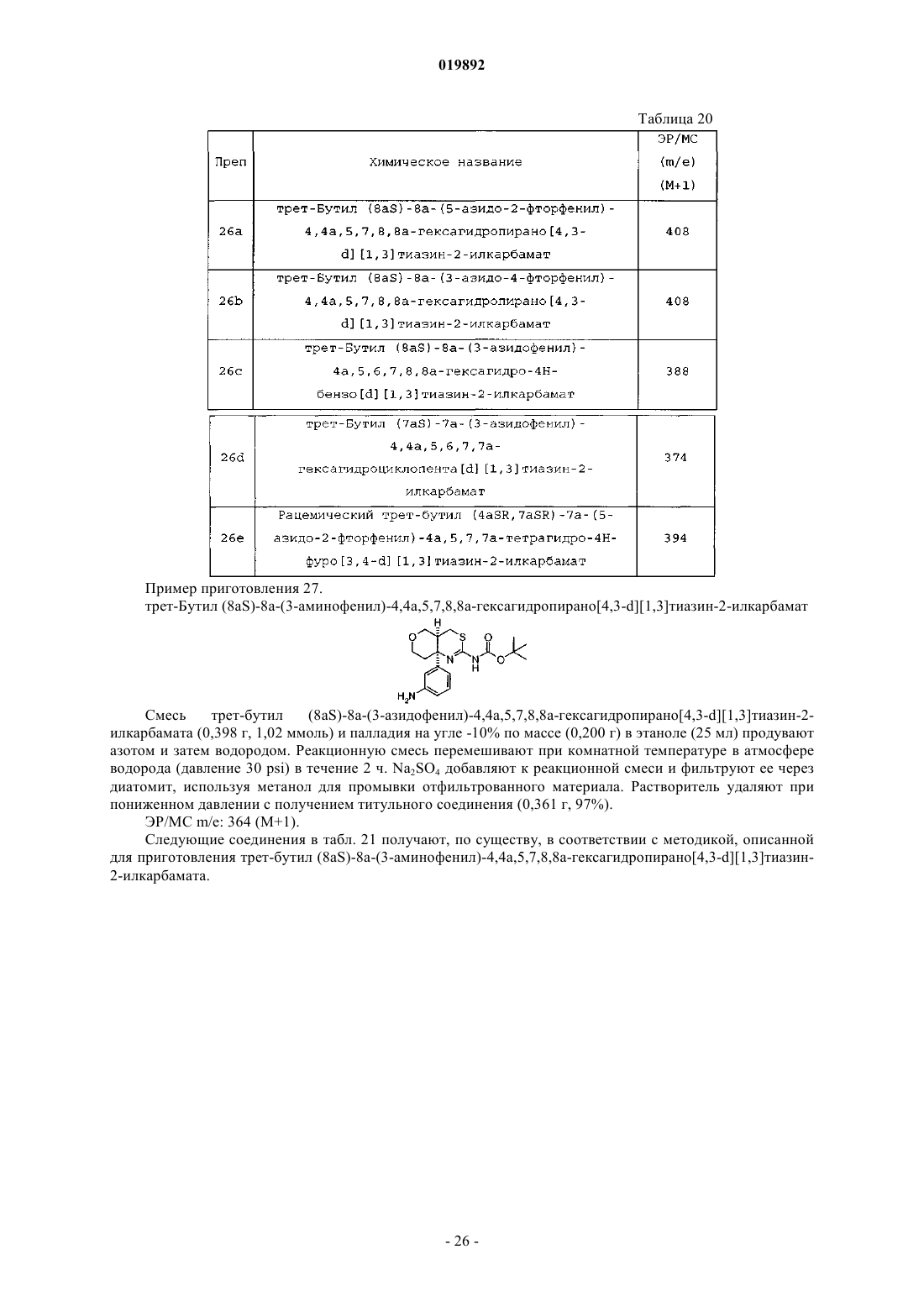
1. Соединение, которое представляет собой N-(3-((4aS,7aS)-2-амино-4а,5,7,7а-тетрагидро-4Н-фуро[3,4-d][1,3]тиазин-7а-ил)-4-фторфенил)-5-фторпиколинамид:

или его фармацевтически приемлемая соль.
2. Фармацевтическая композиция для лечения болезни Альцгеймера, содержащая эффективное количество соединения по п.1 или его фармацевтически приемлемую соль, в комбинации с фармацевтически приемлемым носителем, разбавителем или наполнителем.
3. Соединение, которое представляет собой N-(3-((4aS,7aS)-2-амино-4а,5,7,7а-тетрагидро-4Н-фуро[3,4-d][1,3]тиазин-7а-ил)-4-фторфенил)-5-фторпиколинамид:

4. Фармацевтическая композиция для лечения болезни Альцгеймера, содержащая эффективное количество соединения по п.3, в комбинации с фармацевтически приемлемым носителем, разбавителем или наполнителем.
Текст
Согласно изобретению предложен ингибитор BACE следующей формулы: или его фармацевтически приемлемая соль, а также фармацевтическая композиция для лечения болезни Альцгеймера, содержащая эффективное количество указанного соединения или его фармацевтически приемлемой соли.(71)(73) Заявитель и патентовладелец: ЭЛИ ЛИЛЛИ ЭНД КОМПАНИ (US) Настоящее изобретение относится к области лечения болезни Альцгеймера и других заболеваний и нарушений, связанных с амилоидным -пептидом (А-пептидом), пептидным сегментом белкапредшественника амилоида (APP), обладающим нейротоксичностью и высокой способностью к агрегации. Было показано, что полное или частичное ингибирование -секретазы или -сайта фермента расщепления белка-предшественника амилоида (BACE) оказывает значительное влияние на связанные с бляшками и зависящие от бляшек патологии у мышей, таким образом, это позволяет предположить, что даже небольшое снижение уровней А может привести к долгосрочному значительному снижению количества бляшек и уменьшению синаптического дефицита, обеспечивая тем самым значительный терапевтический эффект. Описанные к настоящему времени ингибиторы BACE представляют собой пептидомиметические аналоги переходного состояния, как правило, содержащие гидроксиэтильную часть. Несмотря на то что многие из этих соединений являются эффективными ингибиторами BACE, высокая молекулярная масса и низкая способность к проникновению через мембраны делают их плохими кандидатами в лекарственные препараты. См. Park и Lee, Journal of the American Chemical Society, 125(52), 16416-16422 (2003). Постепенно произошел переход от больших молекул пептидомиметиков к малым молекулам, например к разнообразным вариантам на основе гидроксиэтиламиновых или гетероциклических соединений. См.,например, Durham и Shepherd, Current Opinion in Drug DiscoveryDevelopment, 9(6), 776-791 (2006). Некоторые аминотиазольные соединения были описаны как ингибиторы BACE в WO 2007/049532,WO 2008/133273 и WO 2008/133274. Ингибиторы BACE, являющиеся сильными и более эффективными, необходимы для обеспечения лечения опосредуемых -пептидом заболеваний, таких как болезнь Альцгеймера. Согласно настоящему изобретению предложены новые сильные и эффективные ингибиторы BACE. Согласно настоящему изобретению предложены соединения формулы IR1 представляет собой -NHCOR4, пиримидинил, необязательно замещенный галогеном пиридинил или фенил, необязательно монозамещенный -C1-C3-алкокси;R4 представляет собой фенил, необязательно замещенный галогеном пиридинил, необязательно замещенный галогеном пиримидинил, пиризинил или тиазолил; или их фармацевтически приемлемые соли. Согласно настоящему изобретению также предложен способ лечения болезни Альцгеймера у пациентов, включающий введение пациентам, нуждающимся в таком лечении, эффективного количества соединения согласно настоящему изобретению. Согласно настоящему изобретению также предложен способ предотвращения прогрессирования легких когнитивных нарушений в болезнь Альцгеймера у пациентов, включающий введение пациентам,нуждающимся в таком лечении, эффективного количества соединения согласно настоящему изобретению или его фармацевтически приемлемой соли. Согласно настоящему изобретению также предложен способ предотвращения прогрессирования заболевания у пациентов с риском развития болезни Альцгеймера, включающий введение пациентам, нуждающимся в таком лечении, эффективного количества соединения согласно настоящему изобретению или его фармацевтически приемлемой соли. Согласно настоящему изобретению также предложен способ ингибирования BACE у пациентов,включающий введение млекопитающим, нуждающимся в таком лечении, эффективного количества соединения согласно настоящему изобретению или его фармацевтически приемлемой соли. Согласно настоящему изобретению также предложен способ ингибирования опосредуемого BACE расщепления белка-предшественника амилоида, включающий введение пациенту, нуждающемуся в таком лечении, эффективного количества соединения согласно настоящему изобретению или его фармацевтически приемлемой соли. Согласно настоящему изобретению также предложен способ ингибирования продуцирования пептида А, включающий введение пациенту, нуждающемуся в таком лечении, эффективного количества соединения согласно настоящему изобретению или его фармацевтически приемлемой соли. Согласно настоящему изобретению также предложена фармацевтическая композиция, содержащая соединение согласно настоящему изобретению или его фармацевтически приемлемую соль в комбинации с фармацевтически приемлемым носителем, разбавителем или наполнителем. В некоторых вариантах реализации композиция содержит дополнительно одно или несколько других терапевтических агентов. Кроме того, согласно настоящему изобретению предложен способ использования соединения согласно настоящему изобретению или его фармацевтически приемлемой соли в терапии, в частности для лечения болезни Альцгеймера или для профилактики прогрессирования легких когнитивных нарушений при болезни Альцгеймера. Далее согласно настоящему изобретению предложен способ использования соединения согласно настоящему изобретению или его фармацевтически приемлемой соли для изготовления лекарственного средства для лечения болезни Альцгеймера. Согласно настоящему изобретению также предложен способ использования соединения согласно настоящему изобретению или его фармацевтически приемлемой соли для изготовления лекарственного средства для профилактики прогрессирования легких когнитивных нарушений при болезни Альцгеймера. Также согласно настоящему изобретению предложен способ использования соединения согласно настоящему изобретению или его фармацевтически приемлемой соли для изготовления лекарственного средства для ингибирования BACE. Согласно настоящему изобретению также предложен способ использования соединения согласно настоящему изобретению или его фармацевтически приемлемой соли для изготовления лекарственного средства для ингибирования продуцирования А-пептида. Кроме того, согласно настоящему изобретению предложена фармацевтическая композиция, пригодная для лечения болезни Альцгеймера. Кроме того, согласно настоящему изобретению предложена фармацевтическая композиция, пригодная для профилактики прогрессирования легкого когнитивного расстройства при болезни Альцгеймера. Согласно настоящему изобретению также предложена фармацевтическая композиция, пригодная для ингибирования BACE. Кроме того, согласно настоящему изобретению предложена фармацевтическая композиция, пригодная для ингибирования опосредуемого BACE расщепления белка-предшественника амилоида. Согласно настоящему изобретению также предложена фармацевтическая композиция, пригодная для лечения состояний, возникающих при избыточных уровнях А-пептида, содержащая соединение согласно настоящему изобретению или его фармацевтически приемлемую соль в комбинации с одним или несколькими фармацевтически приемлемыми наполнителями, носителями или разбавителями. Общепринятые химические термины, используемые при описании вышеприведенных формул, употреблены в их обычном значении. Например, термин "-C1-C3-алкокси" обозначает -C1-C3-алкильную группу, связанную с атомом кислорода, и относится к метокси, этокси, пропокси и изопропокси. Термин "галоген" означает фтор и хлор. Термин "азотозащитная группа" означает группу, устойчивую к предполагаемым условиям реакции, которая, тем не менее, может быть селективно удалена при помощи реагентов и условий реакции,совместимых с регенерируемыми аминами. Такие группы хорошо известны специалистам в данной области и описаны в литературе. См., например, Greene and Wuts, Protective Groups in Organic Synthesis,Third Edition, Chapter 7, John WileySons Inc. (1999). Термин "ингибирование продуцирования A-пептида" обозначает уменьшение уровней А-пептида у млекопитающих in vivo. Термин "эффективное количество соединения формулы I" означает дозу/дозы соединения согласно настоящему изобретению, которые необходимы для обеспечения уровней ингибирования BACE, достаточных для уменьшения уровней А-пептида у млекопитающих in vivo. Легкое когнитивное расстройство определяется как потенциальная продромальная фаза развития деменции, связанной с болезнью Альцгеймера, определенная на основании клинической картины и данных о прогрессировании у пациентов с легкими когнитивными расстройствами деменции Альцгеймера с течением времени. (Morris, et al., Arch. Neurol., 58, 397-405 (2001); Petersen, et al., Arch. Neurol., 56, 303308 (1999. Термин "профилактика прогрессирования легкого когнитивного расстройства при болезни Альцгеймера" включает замедление, прекращение или обращение прогрессирования у пациента легкого когнитивного расстройства при болезни Альцгеймера. Для специалиста в данной области очевидно, что соединения согласно настоящему изобретению могут существовать в таутомерных формах, как показано в формуле (1). Любое указание на один из конкретных таутомеров соединений согласно настоящему изобретению в настоящем описании подразумевает обе таутомерные формы и все их смеси. Формула (1) Для специалиста в данной области очевидно, что соединения согласно настоящему изобретению включают ядро, содержащее не менее двух хиральных центров: Формула (2) Хотя настоящее изобретение включает все индивидуальные энантиомеры, а также смеси энантиомеров указанных соединений, включая рацематы, соединения с абсолютной конфигурацией атомов, обозначенных 1 и 2, показанные в формуле (2), являются предпочтительными вариантами осуществления соединений согласно настоящему изобретению. Кроме того, для специалиста в данной области очевидно, что при выборе определенных переменных дополнительные хиральные центры могут быть образованы в соединениях согласно настоящему изобретению. Настоящее изобретение включает все индивидуальные энантиомеры или диастереомеры, а также смеси указанных энантиомеров и диастереомеров указанных соединений, включая рацематы. Для специалиста в данной области будет очевидно, что в системе Кана-Ингольда-Прелога (R) или(S) обозначения для всех хиральных центров будут варьироваться в зависимости от состава заместителей конкретного соединения. Одиночные энантиомеры или диастереомеры могут быть получены на основе использования хиральных реагентов или с помощью стереоселективных или стереоспецифических методов синтеза. Кроме того, индивидуальные энантиомеры или диастереомеры могут быть выделены из смеси стандартными методами хиральной хроматографии или кристаллизацией в любой удобной точке синтеза соединений согласно настоящему изобретению. Одиночные энантиомеры и диастереоизомеры соединений согласно настоящему изобретению являются предпочтительным вариантом согласно настоящему изобретению. Соединения согласно настоящему изобретению представляют собой амины и, соответственно, реагируют с любой неорганической или органической кислотой с образованием фармацевтически приемлемых солей присоединения кислоты. Фармацевтически приемлемые соли и общая методология их получения хорошо известны в данной области техники. См., например, P. Stahl, et al. Handbook of Pharmaceutical Salts: Properties, Selection and Use, (VCHA/Wiley-VCH, 2002); S.M. Berge, et al., "PharmaceuticalSalts", Journal of Pharmaceutical Sciences, vol. 66, No. 1, January 1977. Фармацевтически приемлемые соли,образованные с присоединением соляной кислоты, являются предпочтительными вариантами осуществления. Хотя все соединения согласно настоящему изобретению способны ингибировать BACE, некоторые классы соединений являются предпочтительными вариантами осуществления. Ниже описываются такие предпочтительные классы:h) p должно быть равно 0, если X представляет собой -O-;n) R1 представляет собой пиримидинил; о) R1 представляет собой пиримидинил или необязательно замещенный галогеном пиридинил;ff) соединение согласно настоящему изобретению имеет цис-конфигурацию в хиральных центрах области сопряжения конденсированного аминотиазольного кольца;gg) соединение согласно настоящему изобретению представляет собой свободное основание;hh) соединение согласно настоящему изобретению представляет собой фармацевтически приемлемую соль;ii) соединение согласно настоящему изобретению представляет собой хлористо-водородную соль;jj) соединение согласно настоящему изобретению представляет собой дигидрохлорид;kk) соединение согласно настоящему изобретению представляет собой этансульфонат;ll) соединение согласно настоящему изобретению представляет собой соль п-толуолсульфоната. Предпочтительным вариантом осуществления соединения согласно настоящему изобретению являются такие соединения согласно настоящему изобретению, где X представляет собой -CH2 -или -O-; n равно 0 или 1; m равно 0, 1 или 2; р равно 0 или 1; р должно быть равно 0, если X представляет собой -O-;R2 представляет собой фтор; R3 представляет собой -C1-C3-алкокси или гидрокси; R4 представляет собой пиридинил, необязательно замещенный галогеном, пиримидинил, необязательно замещенный галогеном,пиризинил или тиазолил или их фармацевтически приемлемые соли. В указанном варианте галоген представляет собой хлор или фтор, если R4 представляет собой пиридинил; или хлор, если R4 представляет собой пиримидинил. Кроме того, для указанного варианта предпочтительнее соединения согласно настоящему изобретению, обладающие цис-конфигурацией в хиральных центрах области сопряжения конденсированного аминотиазольного кольца, или их фармацевтически приемлемые соли. Другим предпочтительным вариантом осуществления соединения согласно настоящему изобретению являются такие соединения формулы I, где X представляет собой -CH2- или -O-; n равно 0 или 1; m равно 0 или 1; р равно 1; р должно быть равно 0, если X представляет собой -O-; R1 представляет собой NHCOR4, фенил, необязательно замещенный -C1-C3-алкокси, пиримидинил или пиридинил, необязательно замещенный галогеном; R2 представляет собой фтор; R3 представляет собой -C1-C3-алкокси или гидрокси; R4 представляет собой необязательно замещенный галогеном пиридинил, необязательно замещенный галогеном пиримидинил или тиазолил или их фармацевтически приемлемые соли. В указанном варианте галоген представляет собой хлор или фтор, если R4 представляет собой пиридинил или пиримидинил, или их фармацевтически приемлемые соли. Кроме того, для указанного варианта предпочтительнее соединения, обладающие цис-конфигурацией в хиральных центрах области сопряжения конденсированного аминотиазольного кольца, или их фармацевтически приемлемые соли. Также предпочтительным вариантом осуществления соединения согласно настоящему изобретению являются такие соединения формулы I, где X представляет собой -CH2- или -O-; n равно 0 или 1; m равно 0, 1 или 2; р равно 0 или 1; р обязательно равно 0, если X представляет собой -O-; R1 представляет собой-NHCOR4 или пиримидинил; R2 представляет собой фтор; R3 представляет собой -C1-C3-алкокси или гидрокси; R4 представляет собой необязательно замещенный галогеном пиридинил, необязательно замещенный галогеном пиримидинил, пиризинил или тиазолил или их фармацевтически приемлемые соли. В указанном варианте галоген представляет собой хлор или фтор, если R4 представляет собой пиридинил или пиримидинил или их фармацевтически приемлемые соли. Кроме того, для указанного варианта предпочтительнее соединения, обладающие цис-конфигурацией в хиральных центрах области сопряжения конденсированного аминотиазольного кольца, или их фармацевтически приемлемые соли. Еще одним вариантом осуществления соединения согласно настоящему изобретению являются такие соединения формулы I, где X представляет собой -CH2- или -O-; n равно 0 или 1; m равно 0 или 1; р равно 0 или 1; р обязательно равно 0, если X представляет собой -O-; R1 представляет собой -NHCOR4,фенил, необязательно замещенный -C1-C3-алкокси или пиримидинил; R2 представляет собой фтор; R3 представляет собой -C1-C3-алкокси или гидрокси; R4 представляет собой необязательно замещенный галогеном пиридинил, необязательно замещенный галогеном пиримидинил, пиризинил или тиазолил или их фармацевтически приемлемые соли. В указанном варианте галоген представляет собой хлор или фтор,если R4 представляет собой пиридинил или пиримидинил. Кроме того, для указанного варианта предпочтительнее соединения, обладающие цис-конфигурацией в хиральных центрах области сопряжения конденсированного аминотиазольного кольца, или их фармацевтически приемлемые соли. Наиболее предпочтительным вариантом осуществления соединения согласно настоящему изобретению являются такие соединения формулы I, где X представляет собой -CH2- или -O-; n равно 0 или 1;m равно 0 или 1; р равно 0 или 1; р обязательно равно 0, если X представляет собой -O-; R1 представляет собой -NHCOR4 или пиримидинил; R2 представляет собой фтор; R3 представляет собой -C1-C3-алкокси или гидрокси; R4 представляет собой необязательно замещенный галогеном пиридинил или необязательно замещенный галогеном пиримидинил, или их фармацевтически приемлемые соли. В указанном варианте галоген представляет собой хлор или фтор, если R4 представляет собой пиридинил или пиримидинил, или их фармацевтически приемлемые соли. Кроме того, для указанного варианта предпочтительнее соединения, обладающие цис-конфигурацией в хиральных центрах области сопряжения конденсированного аминотиазольного кольца, или их фармацевтически приемлемые соли. Другим наиболее предпочтительным вариантом осуществления соединения согласно настоящему изобретению являются такие соединения формулы I, где X представляет собой -CH2- или -O-; n равно 0 или 1; m равно 0 или 1; р равно 0; R1 представляет собой -NHCOR4; R2 представляет собой фтор; R4 представляет собой пиридинил, необязательно замещенный галогеном, или их фармацевтически приемлемые соли. В указанном варианте галоген представляет собой хлор или фтор, если R4 представляет собой пиридинил или их фармацевтически приемлемые соли. Кроме того, для указанного варианта предпочтительнее соединения, обладающие цис-конфигурацией в хиральных центрах области сопряжения конденсированного аминотиазольного кольца, или их фармацевтически приемлемые соли. Наиболее предпочтительным вариантом осуществления соединения согласно настоящему изобретению являются такие соединения формулы I, где X представляет собой -O-; n равно 0; m равно 1; р равно 0; R1 представляет собой -NHCOR4; R2 представляет собой галоген; R4 представляет собой замещенный галогеном пиридинил или его фармацевтически приемлемую соль. Для указанного варианта предпочтительнее, если R2 представляет собой фтор или его фармацевтически приемлемую соль. Также для указанного варианта предпочтительнее соединения, обладающие цис-конфигурацией в хиральных центрах области сопряжения конденсированного аминотиазольного кольца, или их фармацевтически приемлемые соли. Еще одним наиболее предпочтительным вариантом осуществления соединения согласно настоящему изобретению является следующее соединение формулы I: или его фармацевтически приемлемая соль. Еще одним наиболее предпочтительным вариантом осуществления соединения согласно настоящему изобретению является следующее соединение формулы I: или его фармацевтически приемлемая соль. Соединения формулы I являются ингибиторами BACE. Таким образом, согласно настоящему изобретению также предложен способ ингибирования BACE у млекопитающих, включающий введение нуждающимся в таком лечении млекопитающим соединения формулы I в количествах, эффективных для ингибирования BACE. Под млекопитающим, получающим лечение посредством введения соединений формулы I, понимается преимущественно человек. Являясь ингибиторами BACE, соединения согласно настоящему изобретению пригодны для подавления продуцирования А-пептида и, следовательно, для лечения заболеваний, обусловленных избыточными уровнями А-пептида, возникающими из-за избыточного продуцирования и/или снижения клиренса А-пептида. Еще одним вариантом осуществления настоящего изобретения является использование соединения формулы I для изготовления лекарственного средства для лечения заболевания или состояния, которые могут быть облегчены или предотвращены путем ингибирования BACE. Таким образом,соединения формулы I предположительно могут быть пригодны для лечения или профилактики болезни Альцгеймера, легких когнитивных расстройств, синдрома Дауна, наследственной церебральной геморрагии с амилоидозом голландского типа, церебральной амилоидной ангиопатии, других дегенеративных деменций, таких как деменции смешанного сосудистого и дегенеративного происхождения, деменция,связанная с болезнью Паркинсона, деменция, связанная с прогрессирующим надъядерным параличом,деменция, связанная с корково-базальной дегенерацией, и диффузный вариант болезни Альцгеймера с тельцами Леви. Соединения согласно настоящему изобретению или их соли могут быть получены различными известными для данной области техники способами, некоторые из которых показаны ниже: на схемах и в разделах "Примеры приготовления" и "Примеры". Конкретные стадии синтеза для каждого из описанных способов можно комбинировать по-разному или сочетать со стадиями из других схем получения соединений формулы I или их солей. Продукты каждой стадии описанных ниже схем можно получить, используя обычные методы, в том числе экстрагирование, выпаривание, осаждение, хроматография, фильтрация, растирание и кристаллизация. На приведенных ниже схемах для наглядности не указаны некоторые стереохимические центры и не представлены некоторые заместители, что ни в коем случае не ограничивает информативности схем. Кроме того, отдельные изомеры, энантиомеры или диастереомеры могут быть отделены на любой удобной стадии синтеза соединений формулы I с использованием таких методов, как, например, хиральная хроматография. В дополнение к этому, промежуточные компоненты, описанные на приведенных ниже схемах, содержат определенное количество азотозащитных групп. Различные защитные группы могут быть одинаковыми или разными в каждом случае в зависимости от условий конкретной реакции и особенностей требуемых преобразований. Условия, обеспечивающие защиту и снятие защиты (депротекцию), хорошо известны специалистам в данной области и описаны в литературе. См., например, GreeneActa, vol. 17, No. 1, 1984. Другие сокращения соответствуют следующим терминам:IC50 означает "концентрация ингибитора, при которой максимальная активность фермента составляет 50% от исходной";"О.Е.Ф." означает "относительная единица флуоресценции". На приведенных ниже схемах все заместители, если не указано иное, соответствуют заместителям,определенным выше. Реагенты и исходные материалы, как правило, легкодоступны для специалистов в данной области. Также они могут быть изготовлены с помощью стандартных методов органической химии и химии гетероциклических соединений, аналогичных методам синтеза известных структурно подобных соединений, и методик, описанных в нижеследующих разделах "Примеры приготовления" и"Примеры", включая в том числе любые новые методики. Схема I На схеме I изображено ацилирование подходящего аминосодержащего соединения формулы (1) арилкарбоновой кислотой формулы (2) с образованием соединения формулы I после снятия защитных групп промежуточного соединения (3). "PG" представляет собой защитную группу, разработанную для аминогрупп, таких как карбаматы и амиды. Такие защитные группы хорошо известны и очевидны для специалистов в данной области. Соединение формулы (1) реагирует с соединением формулы (2) в условиях сочетания. Для специалиста в данной области очевидно существование ряда методик и реагентов для образования амида в результате реакции карбоновых кислот с аминами. Например, реакция подходящего соединения формулы(1) с подходящей кислотой формулы (2) при наличии связующего реагента и аминового основания, такого как DIPEA или триэтиламин, даст соединение формулы (3). Связующие реагенты включают карбодиимиды, такие как DCC, DIC, EDCI, и ароматические связующие реагенты, такие как HOBt и HOAt. Кроме того, вместо более традиционных связующих реагентов могут быть использованы соли урония или фосфония на основе не нуклеофильных анионов, такие как HBTU, HATU, PyBOP и PyBrOP. Для улучшения протекания реакции могут быть использованы добавки, такие как DMAP. Кроме того, соединения формулы I могут быть ацилированы с использованием замещенных бензоилхлоридов в присутствии оснований, например триэтиламина или пиридина. Защитная группа в промежуточном соединении (3) может быть удалена в кислотных или щелочных условиях с получением соединения формулы (1). Способы снятия защиты с таких соединений хорошо известны и очевидны для специалистов в данной области техники. Возможно, но не обязательно, получение фармацевтически приемлемой соли соединения формулы(I) путем реакции подходящего свободного основания формулы (I) с подходящей фармацевтически приемлемой кислотой в подходящем растворителе, при стандартных условиях. Кроме того, образование таких солей может происходить одновременно с депротекцией азотозащитной группы. Получение таких солей хорошо известно и очевидно для специалистов в данной области техники. Схема II На схеме II изображено алкилирование подходящего соединения формулы (4) арилбороновой кислотой (5) с получением соединения формулы I после снятия защиты с промежуточного соединения (6).Y представляет собой трифторметансульфонил или галоген, например Br или I. R1 представляет собой арильную группу, такую как фенил, или гетероарильную группу, такую как пиридинил. К примеру, подходящее соединение формулы (4) реагирует с подходящей бороновой кислотой (6) в позволяющих протекание реакции кросс-сочетания Судзуки-Мияуры условиях. Специалисту в данной области будет понятно, что существует целый ряд условий, обеспечивающих подобные реакции кросссочетания. Соответственно,подходящие палладийсодержащие реагенты включают бис(трифенилфосфин)палладия(II) хлорид, трис-(дибензилиденацетон)дипалладий(0) с трициклогексилфосфином, [1,1'-бис-(дифенилфосфино)ферроцен]палладия(II) хлорид, тетракис-(трифенилфосфин)палладий или палладия(II) ацетат. Подходящие основания включают карбонат цезия, карбонат натрия, карбонат калия или трехосновный калия фосфат моногидрат. Защитная группа может быть удалена в кислотных или щелочных условиях с получением биарильных соединений формулы I. Способы депротекции таких соединений хорошо известны и очевидны для специалистов в данной области техники. Схема III На схеме III изображены два варианта получения первичного амина (1) из подходящего арилбромида (7). В одном из вариантов проводится азидодегалогенирование подходящего арилбромида (7) в присутствии источника азида, например азида натрия. Подобные реакции азидодегалогенирования хорошо известны и очевидны для специалистов в данной области техники. На разложение результирующего азида(8) до первичного амина (1) могут повлиять некоторые восстанавливающие агенты, хорошо известные в данной области техники, такие как LiAlH4, NaBH4, PPh3, или гидрогенизирующие условия, хорошо известные и описанные в данной области техники. Кроме того, подходящий первичный амин (1) может быть получен непосредственно в ходе реакции подходящего арилбромида (7) с суррогатом аммонийной составляющей, например трифторацетамидом в присутствии катализатора, такого как йодид меди, основания, такого как карбонат калия, и лигандов,таких как (+/-)-транс-N,N-диметил-1,2-циклогександиамин. Подобные реакции хорошо известны и очевидны для специалистов в данной области техники. Очевидно, что соединения формулы (7) могут быть легко получены способами, подобными описанным здесь, при помощи методик, которые являются хорошо известными и общепринятыми в данной области техники. Понятно, что этапы получения соединений формулы I зависят от конкретного синтезируемого соединения, исходного соединения и относительной лабильности замещенных групп. Примеры приготовления и примеры Приведенные ниже примеры приготовления и соединений дополнительно иллюстрируют изобретение. Названия соединений согласно настоящему изобретению получены при помощи ChemDraw Ultra,версия 10,0. Масс-спектрометрические данные, если не указано иное, получены методом жидкостной хроматографии/масс-спектрометрии (LC/MS) с использованием колонки Phenomenex Gemini C18: (2,050 мкм,3,5 мкм) при температуре 5010 С при скорости потока 1 мл/мин. Элюирование в 5-100% градиентеACN с 0,1% гидроксида аммония в течение 7 мин с последующим выдерживанием в 100% ACN в течение 1 мин в сочетании с электрораспылительной ионизацией (диапазон сканирования 100-800 а.е.м., скорость сканирования 0,2 а.е.м.; делитель 80V; усиление 1,0; порог 80). Данные газовой хроматографии, если не указано иное, получены методом газовой хроматографии/масс-спектрометрии (GC/MS) с использованием газового хроматографа Agilent DB-5ms(0,25 мм 15 м 0,25 мкм) при температурной программе 60-280 С в течение 7,3 мин и последующем выдерживании при 280 С в течение 2 мин и отношением деления потока 20:1.(6,6 кг, 165 моль) в воде (14 л) и толуоле (14 л) при 20 С. Добавляют аллиловый спирт (801,5 г,13,8 моль) и смесь перемешивают при 20 С в течение 1 ч. Смесь остужают до 5 С и к ней при поддержании внутренней температуры на уровне ниже 15 С медленно добавляют трет-бутил 2-бромацетат (4 кг,20,5 моль). Реакционную смесь перемешивают при комнатной температуре в течение 16 ч. Смесь разводят водой (12 л) и гексанами (12 л) и органическую фазу отделяют. Водную фазу экстрагируют метилтрет-бутиловым эфиром (МТВЕ) (5 л). Объединенные органические фазы осушают над сульфатом магния, фильтруют и концентрируют с получением титульного соединения в виде бесцветного масла (2,6 кг,100%). ЭР/МС m/е: 173 (М+1). Пример приготовления 2. 2-(Аллилокси)уксусная кислота трет-Бутил 2-(аллилокси)ацетат (2,6 кг, 15,1 моль) добавляют к дихлорметану (14 л). 4 М HCl в диоксане (14 л) одной порцией и раствор перемешивают при 25 С в течение 16 ч. Растворитель удаляют при пониженном давлении и очищенный остаток сушат под вакуумом при комнатной температуре с получением титульного соединения (2,2 кг, 100%). ЭР/МС m/е: 117 (М+1). Пример приготовления 3. 2-(Аллилокси)-N-метокси-N-метилацетамид Тионилхлорид (1,5 л) добавляют одной порцией к раствору 2-(аллилокси)уксусной кислоты (2,2 кг,18,9 моль) в толуоле (3,0 л) и смесь нагревают при 65 С в атмосфере азота в течение 1 ч. Смесь охлаждают до комнатной температуры и добавляют к раствору N,O-диметилгидроксиламина гидрохлорида (2,1 кг, 21,5 моль) и N-метилморфолина (6,5 л, 59,2 моль) в дихлорметане (19 л) при 5 С. Реакционную смесь перемешивают при 25 С в течение 16 ч. К реакционной смеси добавляют воду и смесь экстрагируют дихлорметаном. Объединенные органические фазы собирают, промывают 1 М HCl (6 л), сушат над сульфатом магния и концентрируют при пониженном давлении. Остаток очищают хроматографией на силикагеле с элюированием этилацетатом в гексанах с получением титульного соединения (1,49 кг, 50%). ЭР/МС m/e: 160 (M+1). Пример приготовления 4. 2-(Аллилокси)-1-(5-бром-2-фторфенил)этанон К перемешиваемому раствору 4-бром-1-фтор-2-йодобензола (130,4 г, 433,5 ммоль) в тетрагидрофуране (722 мл) при температуре -72 С добавляют 2,5 М бутиллития в гексане (173,4 мл, 433,5 ммоль) в атмосфере азота за 40 мин. Реакционную смесь перемешивают в течение 30 мин при -72 С и в течение 35 мин по капле добавляют 2-(аллилокси)-N-метокси-N-метилацетамид (57,5 г, 361,2 ммоль) в тетрагидрофуране (115 мл). По истечении 45 мин при -72 С охлаждающую ванну убирают и смесь нагревают до 25 С. Реакцию останавливают насыщенным водным раствором NH4Cl (500 мл), разводят водой (300 мл) и трижды экстрагируют этилацетатом. Органические фракции объединяют, сушат над сульфатом магния,фильтруют и удаляют растворитель при пониженном давлении. Остаток очищают хроматографией на силикагеле в линейном градиенте 5-10% этилацетата в гексане с получением титульного соединения(63 г, 64%). ЭР/МС m/е (79Br/81Br): 273/275 (М+1). Следующие соединения в табл. 1 получают, по существу, в соответствии с методикой, описанной для приготовления 2-(аллилокси)-1-(5-бром-2-фторфенил)этанона.H ЯМР-спектр (400 МГц, CDCl3): 8,06 (т, J=1,6 Гц, 1 Н), 7,86-7,84 (м, 1 Н), 7,71-7,68 (м,1 Н), 7,34 (т, J=7,9 Гц, 1 Н), 5,98-5,91 (м, 1 Н), 5,34-5,23 (м, 2 Н), 4,69 (с, 2 Н), 4,14-4,11 (м,2 Н). 2 В качестве растворителя вместо ТГФ используется диэтиловый эфир. 3 В качестве растворителя вместо ТГФ используется толуол/гексан в пропорции 3:1. Соединения выделяют в виде смеси. К раствору 2-(аллилокси)-1-(5-бром-2-фторфенил)этанона (118 г, 432,1 ммоль) в этаноле (1,7 л) добавляют гидроксиламина гидрохлорид (34,5 г, 496,9 ммоль) и ацетат натрия (40,8 г, 496,9 ммоль) при 25 С. Реакционную смесь нагревают при 70 С в течение 1 ч. Реакционную смесь охлаждают и растворитель удаляют при пониженном давлении. Остаток промывают водой (1 л) и трижды экстрагируют дихлорметаном (3500 мл). Органическую фазу сушат над сульфатом магния, фильтруют и удаляют растворитель при пониженном давлении с получением титульного соединения в виде смеси двух возможных оксимов (120 г, 96%). ЭР/МС m/e (79Br/81Br): 288, 290 (М+1). Следующие соединения в табл. 2 получают, по существу, в соответствии с методикой, описанной для приготовления 2-(аллилокси)-1-(5-бром-2-фторфенил)этанон оксима. Таблица 2 Раствор 2-(аллилокси)-1-(5-бром-2-фторфенил)этанон оксима (120 г, 417 ммоль) в ксилоле (2 л) нагревают при 140 С в течение 6 ч. Реакционную смесь охлаждают и растворитель удаляют при пониженном давлении с получением твердой фазы. Твердую фазу очищают растиранием со смесью гексанов и метил-трет-бутилового эфира в пропорции 9:1 с получением титульного соединения (85 г, 72%). ЭР/МС m/е (79Br/81Br): 288, 290 (М+1). Следующие соединения в табл. 3 получают, по существу, в соответствии с методикой, описанной для приготовления рацемического Данная реакция протекает в толуоле в течение 18 ч при 150 С в герметично замкнутом сосуде. 6 Соединения выделяют в виде смеси. К рацемической смеси (3aSR,6aSR)-6 а-(5-бром-2-фторфенил)гексагидрофуро[3,4-с]изоксазола (84 г,290 ммоль) в уксусной кислоте (1,4 л) добавляют цинковую пыль (190 г, 2,91 моль) со скоростью, позволяющей поддерживать температуру ниже 30 С. Реакционную смесь нагревают при 40 С в течение 5 ч. Реакционную смесь охлаждают до комнатной температуры и фильтруют через диатомит, промывают уксусной кислотой и водой (200 мл). Растворитель удаляют при пониженном давлении. К остатку добавляют воду (500 мл), рН доводят до 10 добавлением 2 М водного гидроксида натрия. Основную водную суспензию трижды экстрагируют 15% изопропиловым спиртом в дихлорметане (3500 мл). Объединенный органический слой сушат над сульфатом магния и удаляют растворитель при пониженном давлении с получением титульного соединения в виде белой твердой фазы (73,0 г, 86%). ЭР/МС m/е (79Br/81Br): 290, 292 (М+1). Следующие соединения в табл. 4 получают, по существу, в соответствии с методикой, описанной для приготовления рацемического 3RS,4SR)-4-амино-4-(5-бром-2-фторфенил)тетрагидрофуран-3 ил)метанола. 20 экв. цинковой пыли используют для 0,06 М уксусной кислоты. Соединения выделяют в виде смеси. К раствору рацемического 3RS,4SR)-4-амино-4-(5-бром-2-фторфенил)тетрагидрофуран-3 ил)метанола (75 г, 259 ммоль) в тетрагидрофуране (1,3 л) при 25 С в атмосфере азота по капле добавляют бис-(триметилсилил)трифторацетамид (76,3 мл, 259 ммоль), поддерживая внутреннюю температуру не выше 30 С. Реакционную смесь перемешивают при 25 С в течение 30 мин. Добавляют бензоил изотиоцианат (38,4 мл, 284 ммоль) за 10 мин при поддержании внутренней температуры не выше 35 С и реакционную смесь перемешивают при 25 С в течение 30 мин. Реакционную смесь разводят этилацетатом (500 мл) и трижды промывают 1 н. HCl (3500 мл), а затем водой и солевым раствором. Раствор сушат над сульфатом магния и растворитель удаляют при пониженном давлении. Остаток очищают хроматографией на силикагеле в линейном градиенте 25-50% этилацетата в гексанах с получением титульного соединения (110 г, 94%). ЭР/МС m/е (79Br/81Br): 453, 455 (М+1). Следующие соединения в табл. 5 получают, по существу, в соответствии с методикой, описанной для приготовления рацемического(76,4 г, 291 ммоль) в тетрагидрофуране (970 мл) тремя частями за 10 мин добавляют ди-трет-бутил азодикарбоксилат (67,1 г, 291 ммоль), поддерживая внутреннюю температуру ниже 25 С. После добавления реакционную смесь перемешивают при 25 С в течение 1 ч. Растворитель удаляют при пониженном давлении и остаток очищают хроматографией на силикагеле в линейном градиенте 14-33% этилацетата в гексанах с получением титульного соединения (80 г, 76%). ЭР/МС m/е (79Br/81Br): 435, 437 (М+1). Следующие соединения в табл. 6 получают, по существу, в соответствии с методикой, описанной для приготовления рацемического N-4aSR,7aSR)-7a-(5-бром-2-фторфенил)-4 а,5,7,7 а-тетрагидро-4 Нфуро[3,4-d][1,3]тиазин-2-ил)бензамида. Очищенный радиальной хроматографией с элюированием в 10-15% этилацетата в гексане. 10 Соединения выделяют в виде смеси.N-4aSR,7aSR)-7a-(5-бром-2-фторфенил)-4a,5,7,7 а-тетрагидро-4 Н-фуро[3,4d][1,3]тиазин-2-ил)бензамид (108 г, 248 ммоль) очищают методом хиральной ВЭЖХ: колонка ChiralcelOJ-H (825 см); элюирование метанолом/ацетонитрилом (90:10) с 0,2% диметилэтиламина; скорость потока 300 мл/мин при УФ 254 нм. Выделяют второй элюирующий изомер с получением энантиомерно обогащенного титульного соединения (42,0 г, 40%). ЭР/МС m/е (79Br/81Br): 434,9/436,9 (М+1). Следующие соединения в табл. 7 получают, по существу, в соответствии с методикой, описанной для приготовления 5 н. водную соляную кислоту (158 мл) добавляют к N-(7 а-(5-бром-2-фторфенил)-4 а,5,7,7 атетрагидро-4 Н-фуро[3,4-d][1,3]тиазин-2-ил)бензамиду (16,35 г, 7,89 ммоль) и нагревают смесь до 90 С. Через 18 ч смеси дают остыть до комнатной температуры и промывают дихлорметаном. Органический слой экстрагируют однократно 5 н. водной соляной кислотой. рН водного слоя доводят до основных значений 50% водным гидрохлоридом натрия и экстрагируют дважды 10% изопропиловым спиртом в дихлорметане. Органический слой концентрируют при пониженном давлении. Полученный остаток очищают радиальной хроматографией, элюируя в 2-5% 7 н. аммиака в метаноле/дихлорметане с получением титульного соединения (2,23 г, 47%). ЭР/МС m/е (79Br/81Br): 331, 333 (М+1). Следующие соединения в табл. 8 получают, по существу, в соответствии с методикой, описанной для приготовления рацемического (4aSR,7aSR)-7 а-(5-бром-2-фторфенил)-4 а,5,7,7 а-тетрагидро-4 Нфуро[3,4-d][1,3]тиазин-2-амина. Таблица 8 В 2 л круглую колбу с плоским дном добавляют N-4aS,7aS)-7 а-(5-бром-2-фторфенил)-4 а,5,7,7 атетрагидро-4 Н-фуро[3,4-d][1,3]тиазин-2-ил)бензамид (35,0 г, 80,4 ммоль), трифторацетамид (16,2 г,143 ммоль), йодид меди(I) (2,66 г, 13,7 ммоль), йодид натрия (21,3 г, 141 ммоль) и углекислый калий(21,5 г, 153 ммоль). Колбу закрывают перегородкой, вакуумируют и затем заполняют азотом. Добавляют через канюлю 1,4-диоксан (731 мл) (предварительно дегазированный в вакууме, под азотом), затем добавляют (+/-)-транс-N,N-диметил-1,2-циклогександиамин (10,1 г, 70,8 ммоль). Смесь помещают в нагретую до 100 С масляную ванну и перемешивают при этой температуре в течение 19 ч. Перегородку заменяют обратным холодильником и добавляют через конденсатор смесь метанола (154 мл) и воды (154 мл). Смесь перемешивают при 100 С в течение 3,5 ч, охлаждают до 22 С и частично концентрируют при пониженном давлении (с получением объема 0,6 л). Добавляют водный гидроксид аммония (25%, 154 мл) и перемешивают в течение 10 мин. Смесь трижды экстрагируют этилацетатом (3500 мл) и растворитель удаляют при пониженном давлении. Получают остаток, который очищают флэш-хроматографией в линейном градиенте 50-75% этилацетата в гексане с получением титульного соединения (14,9 г, 47%). ЭР/МС m/е: 372 (М+1). Пример приготовления 13. Готовят 0,66 М раствор L-аскорбиновой кислоты, растворяя натриевую соль L-аскорбиновой кислоты (0,79 г, 2,0 ммоль) в воде (6 мл). N-4aS,7aS)-7 а-(3-Бромфенил)-4 а,5,7,7 а-тетрагидро-4 Н-фуро[3,4d][1,3]тиазин-2-ил)бензамид (1,40 г, 3,35 ммоль) и (+/-)-транс-N,N-диметил-1,2-циклогександиамин(162 мг, 1,11 ммоль) растворяют в этаноле (13,4 мл). Добавляют азид натрия (0,661 г, 10,1 ммоль). Добавляют 0,66 М водный раствор натриевой соли L-аскорбиновой кислоты (2,24 мл) и воду (2,58 мл). На реакционную колбу устанавливают обратный холодильник; смесь дегазируют и продувают азотом. Добавляют пентагидрат сульфата меди(II) (0,184 г, 0,738 ммоль), реакционную колбу нагревают до 80 С и содержимое перемешивают в течение 1,5 ч. Реакционную смесь охлаждают до комнатной температуры и добавляют ледяную воду. Реакционную смесь трижды экстрагируют этилацетатом. Объединенные органические фазы сушат над сульфатом магния и растворитель удаляют при пониженном давлении с получением остатка, который очищают на силикагеле 50% этилацетатом в гексане с получением титульного соединения (0,620 г, 49%). Дальнейшее элюирование флэш-колонки 100% этилацетатом позволяет получить больше титульного соединения (0,488 г, 41%). ЭР/МС m/е: 380 (М+1). Следующие соединения в табл. 9 получают, по существу, в соответствии с методикой, описанной для приготовления N-4aS,7aS)-7 а-(3-азидофенил)-4 а,5,7,7 а-тетрагидро-4 Н-фуро[3,4-d][1,3]тиазин-2 ил)бензамида. Таблица 9(0,62 г, 1,63 ммоль) разводят этанолом (10 мл) и Pd на угле (10%, влажный, 0,062 г). Смесь дегазируют и перемешивают при комнатной температуре под водородом (30 psi) до следующего утра. Смесь фильтруют через диатомит, используя для промывания этанол. Растворитель удаляют при пониженном давлении с получением титульного соединения (0,106 г, 18%). ЭР/МС m/е: 354 (М+1). Следующие соединения в табл. 10 получают, по существу, в соответствии с методикой, описанной для приготовления N-4aS,7aS)-7 а-(3-аминофенил)-4 а,5,7,7 а-тетрагидро-4 Н-фуро[3,4-d][1,3]тиазин-2 ил)бензамид. Таблица 10N-4aS,7aS)-7 а-(5-амино-2-фторфенил)-4 а,5,7,7 а-тетрагидро-4 Н-фуро[3,4-d][1,3]тиазин-2 ил)бензамид (20,4 г, 51,8 ммоль), 5-фторпиколиновой кислоты (8,77 г, 62,2 ммоль), гидрата 1-гидроксибензотриазола (10,3 г, 67,4 ммоль) и 1-(3-диметиламинопропил)-3-этил карбодиимида гидрохлорида (13,2 г, 67,4 ммоль) в смеси дихлорметана (345 мл) и ДМФ (6,5 мл) перемешивают при 22 С в течение 80 мин. Добавляют раствор 2 М гидроксида натрия (129,5 мл, 259 ммоль) и продолжают перемешивать в течение 10 мин. Смесь разделяют и водную фазу дважды экстрагируют дихлорметаном(2100 мл). Органический слой концентрируют при пониженном давлении и полученный остаток разводят этилацетатом (200 мл). Органический слой отмывают охлажденной водой (250 мл), солевым раствором (50 мл) и фильтруют через тонкий слой кварца с использованием 100% этилацетата с получением титульного соединения (23,8 г, 79%). ЭР/МС m/е: 495 (М+1). Следующие соединения в табл. 11 получают, по существу, в соответствии с методикой, описанной для приготовления N-(3-4aS,7aS)-2-бензамидо-4 а,5,7,7 а-тетрагидро-4 Н-фуро[3,4-d][1,3]тиазин-7 а-ил)-4 фторфенил)-5-фторпиколинамида. Таблица 11N-(3-4aS,7aS)-2-бензамидо-4 а,5,7,7 а-тетрагидро-4 Н-фуро[3,4-d][1,3]тиазин-7 а-ил)-4 фторфенил)-5-фторпиколинамида (23,7 г 40,8 ммоль), о-метилгидроксиламина гидрохлорида (34,4 г,412 ммоль) и пиридина (33,3 мл) в этаноле (735 мл) нагревают до 50 С в течение 4 ч. Смесь концентрируют. Остаток дважды промывают метил-трет-бутиловым эфиром (2250 мл) и выливают в насыщенный водный раствор бикарбоната натрия (453 мл). Суспензию взбалтывают в течение 5 мин и экстрагируют дихлорметаном (11 л и 20,5 л). Органический слой промывают водой (0,5 л), сушат над сульфатом магния и удаляют растворитель при пониженном давлении с получением твердой фазы. Дополнительное количество твердого вещества получают из водной фазы фильтрацией. Твердые фазы объединяют и растирают с водой (300 мл) в ультразвуковой ванне в течение 30 мин. Суспензию фильтруют, промывают водой и сушат под вакуумом с получением титульного соединения (17,3 г, 100%). ЭР/МС m/е: 391 (М+1). Порошок железа (16,49 г, 291 ммоль) добавляют к 1-бром-2,4-дифторбензолу (110 мл, 968 ммоль) в 1,2-дихлорэтане (968 мл) в трехгорлой колбе при комнатной температуре в потоке азота. Раствор брома(59,7 мл, 1,16 моль) в 1,2-дихлорэтане (968 мл) добавляют по капле за 1 ч и реакционную смесь перемешивают при температуре окружающей среды в течение 18 ч. Реакционную смесь охлаждают до 0 С, по частям добавляют насыщенный водный раствор бисульфита натрия (1,11 л, 533 ммоль) и смесь разделяют. Водную фазу экстрагируют дихлорметаном. Органический слой отмывают насыщенным водным раствором бикарбоната натрия, водой и солевым раствором. Органический слой сушат над сульфатом магния и растворитель удаляют при пониженном давлении с получением осадка, который очищают фильтрацией через слой кварца с использованием диэтилового эфира с получением титульного соединения (229 г, 76%). 1 Н ЯМР-спектр (400 МГц, CDCl3) : 7,70 (дд, J=4,6, 6,8 Гц, 1 Н), 6,95-6,92 (м, 1 Н). Пример приготовления 18. 4-(3-Бромфенил)тетрагидро-2 Н-пиран-4-ол К перемешиваемому (-78 С) раствору 1,3-дибромбензола (19,71 г, 81,05 ммоль) в ТГФ (150 мл) добавляют 1,6 М бутиллития в гексане (50,66 мл, 81,05 ммоль) и реакционную смесь перемешивают в течение 10 мин. Тетрагидро-4 Н-пиран-4-он (5,41 г, 54,04 ммоль) добавляют по капле и реакционную смесь перемешивают при -78 С в течение 2 ч. Реакцию останавливают добавлением насыщенного водного хлорида аммония (25 мл), затем разводят минимальным количеством воды и экстрагируют EtOAc. Органический слой сушат над Na2SO4 и растворитель удаляют при пониженном давлении с получением остатка,который очищают хроматографией на силикагеле с элюированием в линейном градиенте 5-100% EtOAc в гексане с получением титульного соединения (11,18 г, 76%). ГХ-МС (m/е) (79Br/81Br): 256, 258 (М-1). Следующие соединения в табл. 12 получают, по существу, в соответствии с методикой, описанной для приготовления 4-(3-бромфенил)тетрагидро-2 Н-пиран-4-ола. Таблица 12 В качестве растворителя в реакции используют толуол/гексан (2/1). В качестве растворителя в реакции используют диэтиловый эфир. 14 1 Н ЯМР-спектр (400 МГц, CDCl3) : 7,64 (т, J=2,0 Гц, 1 Н), 7,40-7,31 (м, 2 Н), 7,19 Смесь 4-(3-бромфенил)тетрагидро-2 Н-пиран-4-ола (11,17 г, 41,3 ммоль) и моногидрата п-толуолсульфоновой кислоты (0,797 г, 4,13 ммоль) в толуоле (100 мл) нагревают до температуры рефлюкса в течение 30 мин с использованием ловушки Дина-Старка для удаления воды. Реакционную смесь разбавляют водой и 5 н. NaOH и экстрагируют EtOAc. Органический слой сушат над Na2SO4 и растворитель удаляют при пониженном давлении с получением остатка, который очищают хроматографией на силикагеле с элюированием в линейном градиенте 5-100% EtOAc в гексане с получением титульного соединения (8,85 г, 90%). 1 Н ЯМР-спектр (400 МГц, CDCl3) : 2,45-2,50 (м, 2 Н), 3,92 (т, 2 Н, J=5,71 Гц), 4,31 (кв, 2 Н, J=3,07 Гц,J=5,71 Гц), 6,12-6,14 (м, 1 Н), 7,19 (т, 1 Н, J=7,91 Гц), 7,28-7,32 (м, 1 Н), 7,36-7,39 (м, 1 Н), 7,51 (т, 1 Н,J=1,76 Гц). Следующие соединения в табл. 13 получают, по существу, в соответствии с методикой, описанной для приготовления 4-(3-бромфенил)-3,6-дигидро-2 Н-пирана. Таблица 13 Раствор (0 С) 4-(3-бромфенил)-3,6-дигидро-2 Н-пирана (0,50 г, 2,09 ммоль) в CH2Cl2 (15 мл) обрабатывают параформальдегидом (0,208 г, 2,20 ммоль) и перемешивают в течение 5 мин при 0 С. 1 М раствор хлорида диметилалюминия в гексане (3,03 мл, 3,03 ммоль) по капле добавляют к суспензии. Реакционную смесь нагревают до комнатной температуры и перемешивают в течение 1 ч. Реакционную смесь охлаждают до 0 С и снова добавляют параформальдегид (0,208 г, 2,20 ммоль) и 1 М раствор хлорида диметилалюминия в гексане (3,03 мл, 3,03 ммоль). Реакционную смесь нагревают до комнатной температуры и перемешивают в течение ночи. Реакцию останавливают выливанием в смесь льда с 1 н.HCl и трижды экстрагируют EtOAc. Объединенные органические слои сушат над Na2SO4 и растворитель удаляют при пониженном давлении с получением остатка, который очищают хроматографией на силикагеле с элюированием в линейном градиенте 5-100% EtOAc в гексане с получением титульного соединения (0,315 г, 53%). 1 Н ЯМР-спектр (400 МГц, CDCl3) : 7,50 (т, J=2,0 Гц, 1 Н), 7,40 (дд, J=2,2, 7,9 Гц, 1 Н), 7,29-7,27 (м,1 Н), 7,20 (т, J=7,9 Гц, 1 Н), 6,11 (т, J=2,9 Гц, 1 Н), 4,32-4,29 (м, 2 Н), 4,28-4,25 (м, 1 Н), 3,76 (дд, J=3,1,11,4 Гц, 1 Н), 3,70-3,64 (м, 2 Н), 2,70 (д, J=2,2 Гц, 1 Н), 1,89 (дд, J=4,6, 6,4 Гц, 1 Н). Следующие соединения в табл. 14 получают, по существу, в соответствии с методикой, описанной для приготовления (4-(3-бромфенил)-3,6-дигидро-2 Н-пиран-3-ил)метанола. Таблица 14(10 мл) обрабатывают триэтиламином (0,218 г, 2,15 ммоль), затем метансульфонилхлоридом (0,148 г,1,29 ммоль) и реакционную смесь перемешивают при 0 С в течение 30 мин. Реакционную смесь разбавляют водой и экстрагируют CH2Cl2. Органический слой сушат над Na2SO4 и растворитель удаляют при пониженном давлении с получением титульного соединения (0,432 г, 97%). 1 Н ЯМР-спектр (400 МГц, CDCl3) : 7,51 (т, J=1,8 Гц, 1 Н), 7,44-7,41 (м, 1 Н), 7,31-7,29 (м, 1 Н), 7,257,21 (м, 1 Н), 6,18 (т, J=2,6 Гц, 1 Н), 4,33-4,31 (м, 2 Н), 4,20 (дд, J=1,5, 11,6 Гц, 2 Н), 4,14-4,08 (м, 1 Н), 3,723,68 (м, 1 Н), 3,01-2,98 (м, 1 Н), 2,95 (с, 3 Н). Следующие соединения в табл. 15 получают, по существу, в соответствии с методикой, описанной для приготовления (4-(3-бромфенил)-3,6-дигидро-2 Н-пиран-3-ил)метил метансульфоната.(м, 2 Н), 6,08-6,11 (м, 1 Н), 4,00-4,03 (м, 2 Н), 3,08-3,13 (ушир.д, 1 Н), 2,85 (с, 3 Н), 2,182,22 (м, 2 Н), 1,94-2,01 (м, 1 Н), 1,78-1,88 (м, 1 Н), 1,63-1,71 (м, 2 Н). 23 В этой реакции используют DMAP и триэтиламин. 24 1 Н ЯМР-спектр (400 МГц, CDCl3) : 7,53 (с, 1 Н), 7,37-7,29 (м, 2 Н), 7,19 (т, J=7,9 Гц,1 Н), 6,22 (с, 1 Н), 4,26 (дд, J=4,0, 10,1 Гц, 1 Н), 4,01 (дд, J=7,9, 9,7 Гц, 1 Н), 3,54-3,52 (м,1 Н), 2,86 (с, 3 Н), 2,59-2,51 (м, 2 Н), 2,28-2,19 (м, 1 Н), 2,04 (ддд, J=17,0, 7,8, 3,8 Гц, 1 Н). Смесь (4-(3-бромфенил)-3,6-дигидро-2 Н-пиран-3-ил)метил метансульфоната (4,82 г, 11,7 ммоль) и тиомочевины (1,78 г, 23,3 ммоль) в изопропиловом спирте (100 мл) нагревают до температуры рефлюкса в течение 24 ч. После того как реакционная смесь охладится, растворитель удаляют под частичным вакуумом с получением остатка, который объединяют с ацетонитрилом (30 мл) и гексаном (10 мл). Кристаллизируется твердая фаза и суспензию охлаждают до 0 С. Суспензию фильтруют, используя в качестве раствора для промывки ACN в гексане (3:1) (25 мл) с получением титульного соединения в форме мезилатной соли (3,45 г, 70%). ЭР/МС m/е (79Br/81Br): 327, 329 (М+1). Следующие соединения в табл. 16 получают, по существу, в соответствии с методикой, описанной для приготовления (4-(3-бромфенил)-3,6-дигидро-2 Н-пиран-3-ил)метил карбамимидотиоат метансульфоната.(3,41 г, 8,05 ммоль) в метансульфоновой кислоте (35 мл) нагревают при 50 С в течение 5 ч. Реакционную смесь охлаждают и выливают в ледяную воду. Смесь разводят EtOAc и рН доводят до основных значений, используя 5 н. NaOH. Основной водный слой трижды экстрагируется этилацетатом и органический слой сушат над Na2SO4. Растворитель удаляют при пониженном давлении. Полученный остаток растирают с CH2Cl2 с получением рацемического титульного соединения. Дополнительное количество рацемического продукта получают очищением фильтрата хроматографией на силикагеле с элюированием в линейном градиенте 1-10% 7 М NH3/MeOH в CH2Cl2 (1,99 г, 76%). ЭР/МС m/е (79Br/81Br): 327, 329 (М+1). Следующие соединения в табл. 17 получают, по существу, в соответствии с методикой, описанной для приготовления (4aSR,8aSR)-8 а-(3-бромфенил)-4,4 а,5,7,8,8 а-гексагидропирано[4,3-d][1,3]тиазин-2 амина. Реакционную смесь нагревают при 90 С в течение ночи. Реакционную смесь нагревают при 50 С в течение 5 ч. 27 Реакционную смесь перемешивают при комнатной температуре в течение 3 ч. 28 Реакционную смесь перемешивают при комнатной температуре в течение 17 ч. Продукт выделяют в форме соли. 26(60 мл) и насыщенный водный NaHCO3 (60 мл) перемешивают при комнатной температуре в течение 8 ч. Смесь разводят водой и трижды экстрагируют EtOAc. Объединенные органические слои сушат надNa2SO4 и растворитель удаляют при пониженном давлении с получением материала, который очищают хроматографией на силикагеле с элюированием в линейном градиенте 5-100% EtOAc в гексанах с получением титульного соединения (2,81 г, 100%). ЭР/МС m/е (79Br/81Br): 427, 429 (М+1). Следующие соединения в табл. 18 получают, по существу, в соответствии с методикой, описанной для приготовления рацемического трет-бутил(4aSR,8aSR)-8a-(3-бромфенил)-4,4 а,5,7,8,8 а-гексагидропирано[4,3d][1,3]тиазин-2-илкарбамат (2,80 г, 6,55 ммоль) очищают методом хиральной ВЭЖХ: Колонка: ChiralcelOJ 835 см; элюирование: метанол/ацетонитрил (75:25); поток: 400 мл/мин при УФ 260 нм. Выделяют второй элюирующий изомер с получением энантиомерно обогащенного титульного соединения (1,31 г,47%). ЭР/MC m/e (79Br/81Br): 427, 429 (М+1). Следующие соединения в табл. 19 получают, по существу, в соответствии с методикой, описанной для приготовления трет-бутил (8aS)-8a-(3-бромфенил)-4,4 а,5,7,8,8 а-гексагидропирано[4,3-d][1,3]тиазин 2-илкарбамата. Готовят 0,33 М раствор сульфата меди, растворяя пентагидрат сульфата меди(II) (1,0 г, 2,0 ммоль) в воде (12 мл). Готовят 0,66 М раствор L-аскорбиновой кислоты, растворяя натриевую сольL-аскорбиновой кислоты (1,58 г, 4,0 ммоль) в воде (12 мл). К раствору трет-бутил (8aS)-8 а-(3 бромфенил)-4,4 а,5,7,8,8 а-гексагидропирано[4,3-d][1,3]тиазин-2-илкарбамата (0,500 г, 1,20 ммоль) в этаноле (3,6 мл) добавляют азид натрия (0,228 г, 3,50 ммоль), (+/-) транс-N,N-диметил-1,2 циклогександиамин (0,0549 г, 0,386 ммоль), 0,66 М водную натриевую соль L-аскорбиновой кислоты(0,772 мл, 0,509 ммоль) и воду (0,71 мл) и смесь продувают азотом. Добавляют 0,33 М водный раствор пентагидрата сульфата меди(II) (0,773 мл, 0,255 ммоль) и реакционную смесь нагревают до 80 С в течение 12 мин. Реакционную смесь выливают в холодную воду с образованием смеси синего цвета, которую трижды экстрагируют EtOAc. Органический слой сушат над Na2SO4 с получением технического продукта, который очищают хроматографией на силикагеле с элюированием в линейном градиенте 5-100%EtOAc в гексане с получением титульного соединения (0,400 г, 88%). ЭР/МС m/е: 390 (М+1). Следующие соединения в табл. 20 получают, по существу, в соответствии с методикой, описанной для приготовления трет-бутил (8aS)-8 а-(3-азидофенил)-4,4 а,5,7,8,8 а-гексагидропирано[4,3-d][1,3]тиазин 2-илкарбамата.(8aS)-8 а-(3-азидофенил)-4,4 а,5,7,8,8 а-гексагидропирано[4,3-d][1,3]тиазин-2 илкарбамата (0,398 г, 1,02 ммоль) и палладия на угле -10% по массе (0,200 г) в этаноле (25 мл) продувают азотом и затем водородом. Реакционную смесь перемешивают при комнатной температуре в атмосфере водорода (давление 30 psi) в течение 2 ч. Na2SO4 добавляют к реакционной смеси и фильтруют ее через диатомит, используя метанол для промывки отфильтрованного материала. Растворитель удаляют при пониженном давлении с получением титульного соединения (0,361 г, 97%). ЭР/МС m/е: 364 (М+1). Следующие соединения в табл. 21 получают, по существу, в соответствии с методикой, описанной для приготовления трет-бутил (8aS)-8 а-(3-аминофенил)-4,4 а,5,7,8,8 а-гексагидропирано[4,3-d][1,3]тиазин 2-илкарбамата. К смеси трет-бутил (8aS)-8 а-(3-аминофенил)-4,4 а,5,7,8,8 а-гексагидропирано[4,3-d][1,3]тиазин-2 илкарбамат (0,15 г, 0,413 ммоль), 5-хлорпиридин-2-карбоновой кислоты (0,078 г, 0,495 ммоль) и 1-гидроксибензотриазола (0,073 г, 0,536 ммоль) в CH2Cl2 (2,75 мл) и ДМФ (0,3 мл) добавляют 1-(3-диметиламинопропил)-3-этил карбодиимида гидрохлорид (0,103 г, 0,536 ммоль). Реакционную смесь перемешивают в течение ночи при комнатной температуре. Реакционную смесь разбавляют водой,5 н. NaOH (0,5 мл) и трижды экстрагируют CH2Cl2. Объединенный органический слой сушат над Na2SO4 и неочищенный продукт очищают хроматографией на силикагеле с элюированием в линейном градиенте 5-100% EtOAc в гексане с получением титульного соединения (0,176 г, 85%). ЭР/МС m/e (35Cl/37Cl): 503, 505 (М+1). Следующие соединения в табл.22 получают, по существу, в соответствии с методикой, описанной для приготовления трет-бутил(4aSR,7aSR)-7a-(2-фтор-5-(5-фторпиколинамидо)фенил)-4 а,5,7,7 атетрагидро-4 Н-фуро[3,4-d][1,3]тиазин-2-илкарбамат (0,226 г, 0,460 ммоль) очищают хиральной ВЭЖХ: колонка Chiralcel OD-H 5 мкм, 2,125 см, 30% метанол/CO2, скорость потока: 70 мл/мин, УФ: 230 нм. Второй элюируемый изомер выделяют с получением энантиомерно обогащенного титульного соединения (0,092 г, 41%). ЭР/МС (m/е): 491 (М+1). Следующие соединения в табл. 23 получают, по существу, в соответствии с методикой, описанной для приготовления трет-бутил (4aS,7aS)-7a-(2-фтор-5-(5-фторпиколинамидо)фенил)-4 а,5,7,7 а-тетрагидро 4 Н-фуро[3,4-d][1,3]тиазин-2-илкарбамата. Таблица 23
МПК / Метки
МПК: C07D 279/08, C07D 498/04, C07D 513/02
Метки: ингибиторы
Код ссылки
<a href="https://eas.patents.su/30-19892-ingibitory-bace.html" rel="bookmark" title="База патентов Евразийского Союза">Ингибиторы bace</a>
Аминодигидротиазиновые производные в качестве ингибиторов bace для лечения болезни альцгеймера

Номер патента: 16955
Опубликовано: 30.08.2012
Авторы: Мерготт Дастин Джеймс, Аудиа Джеймс Эдмунд, Шихан Скотт Мартин, Уотсон Брайан Морган
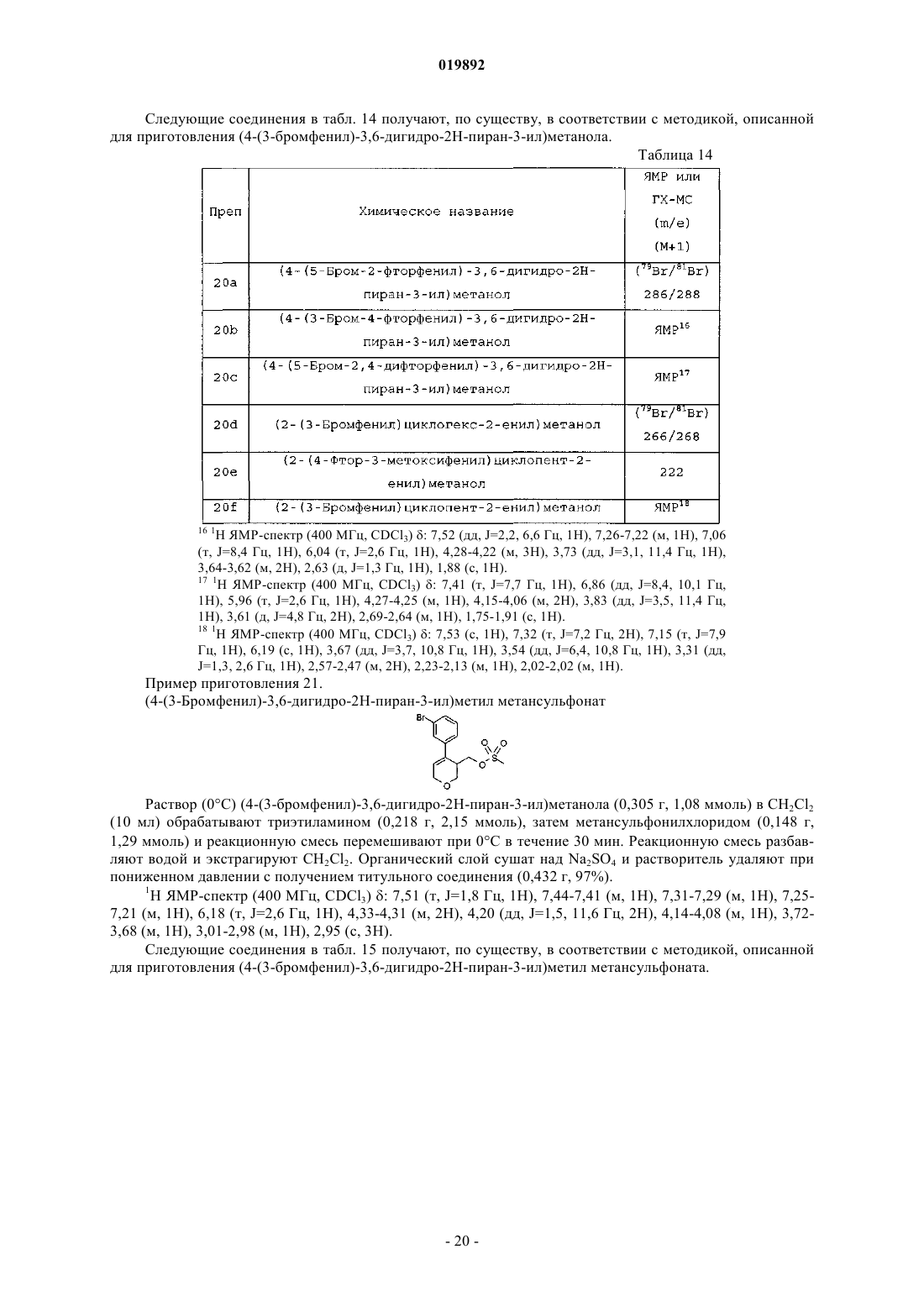
МПК: A61P 25/28, C07D 279/06, A61K 31/541...
Метки: лечения, болезни, ингибиторов, качестве, производные, альцгеймера, аминодигидротиазиновые
Формула / Реферат:
1. Соединение формулы Iгде n представляет собой 0, 1 или 2;R1 представляет собой пиримидинил, пиразинил, необязательно замещенный хлором или фтором, или пиридинил, необязательно замещенный одним или двумя заместителями, каждый из которых независимо выбран из хлора, фтора и C1-C3-алкокси;R2 в каждом случае независимо выбран из хлора или фтора;R3 представляет собой водород или C1-C4-алкил, необязательно замещенный гидроксилом;R4 представляет собой...
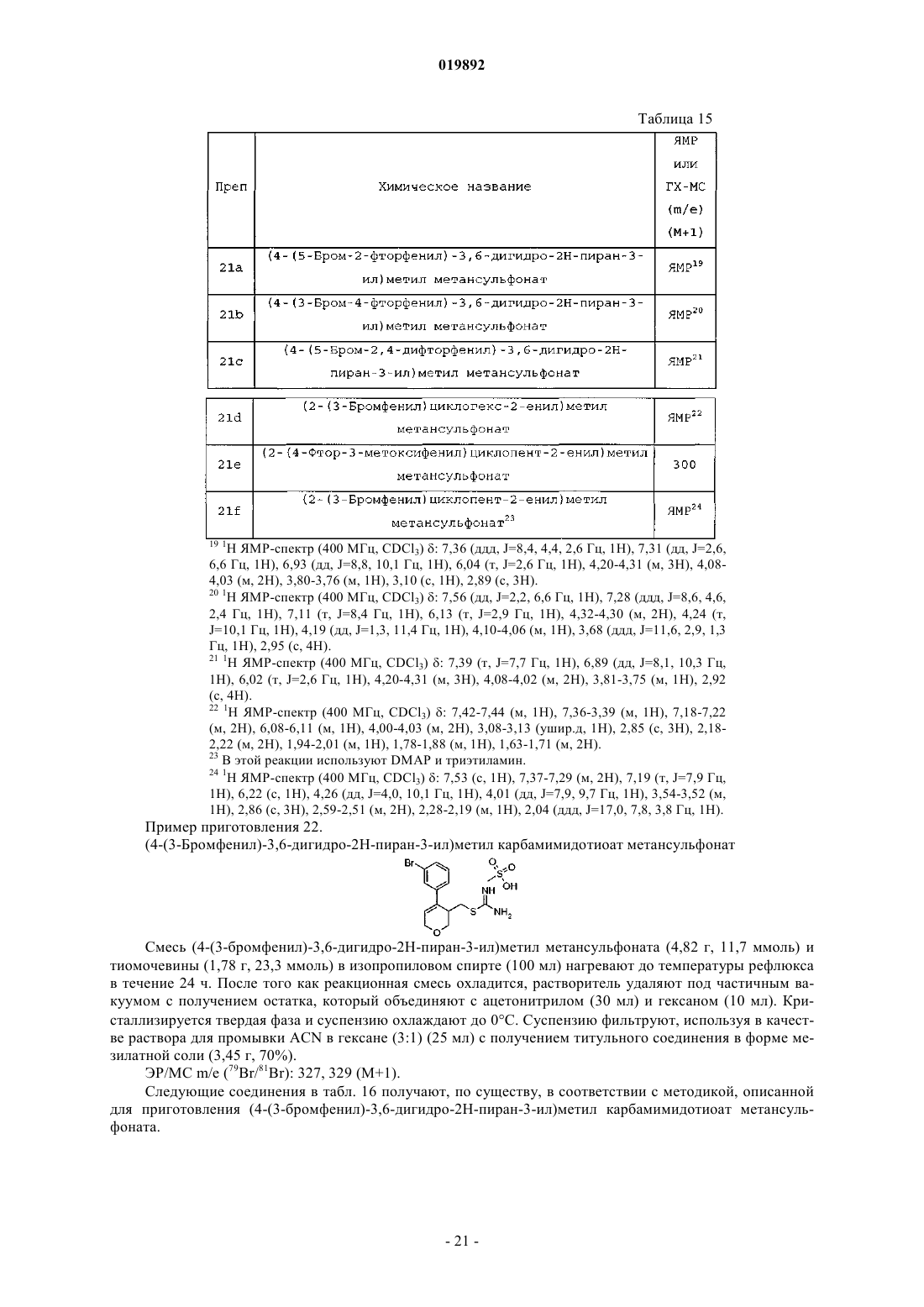
Ингибиторы p70 s6 киназы

Номер патента: 16445
Опубликовано: 30.05.2012
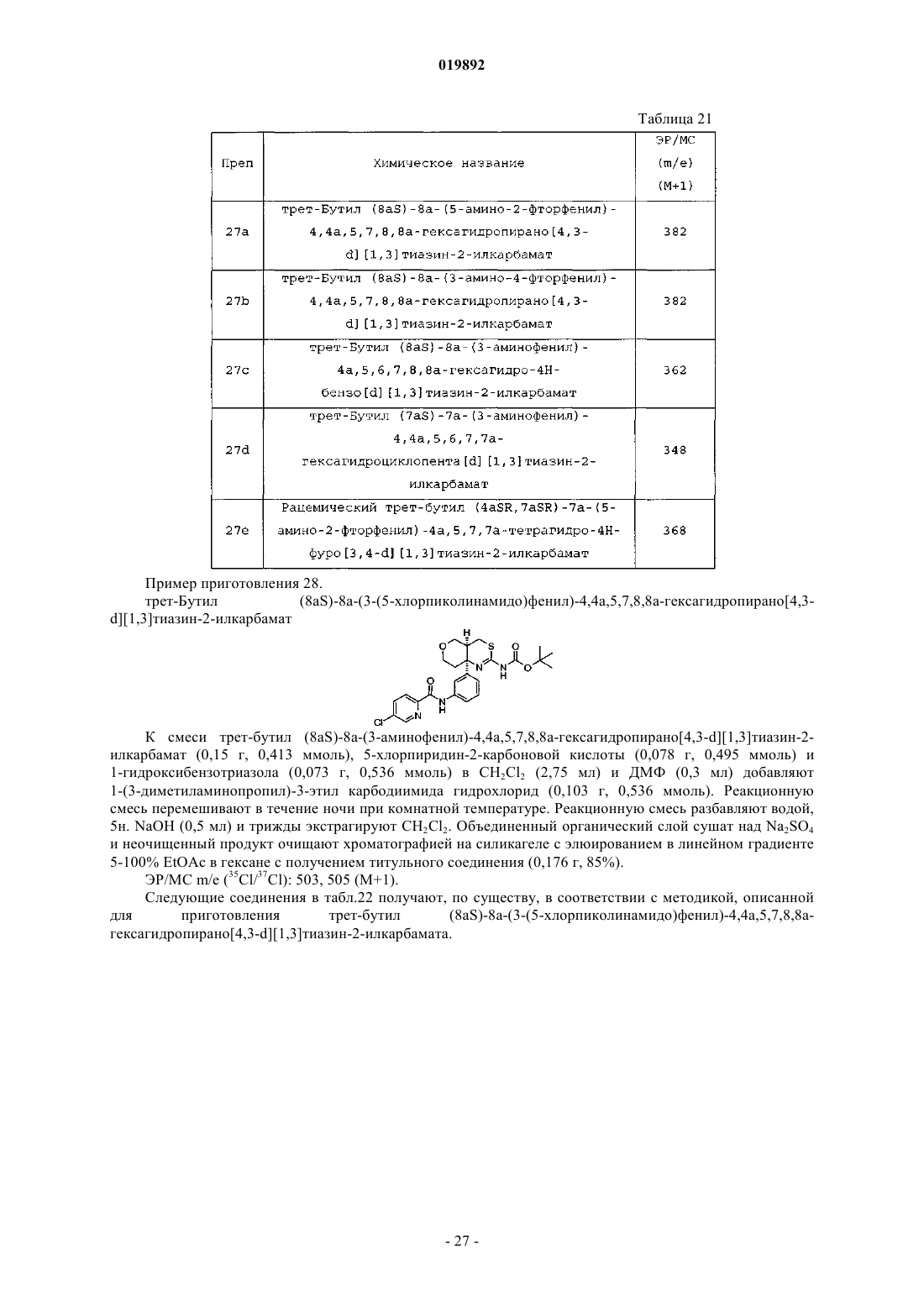
Авторы: Чжуан Цзяньпин, Хольст Кристиан Л., Шеферд Тимоти Алан, Джозеф Саджан, Дэлли Роберт Дин
МПК: C07D 487/04, A61K 31/437, A61P 35/00...
Метки: ингибиторы, киназы
Формула / Реферат:
1. Соединение формулыгде Y представляет собой CH;Z1 и Z2 представляют собой независимо CR3 или N при условии, что Z1 и Z2, оба, не являются N;R1 представляет собой H или CH3;R2 представляет собой фенил, содержащий первый заместитель, выбранный из галогена и трифторметила, и необязательно дополнительно содержащий второй заместитель, при этом указанный второй заместитель представляет собой галоген;R3 представляет собой водород, галоген,...
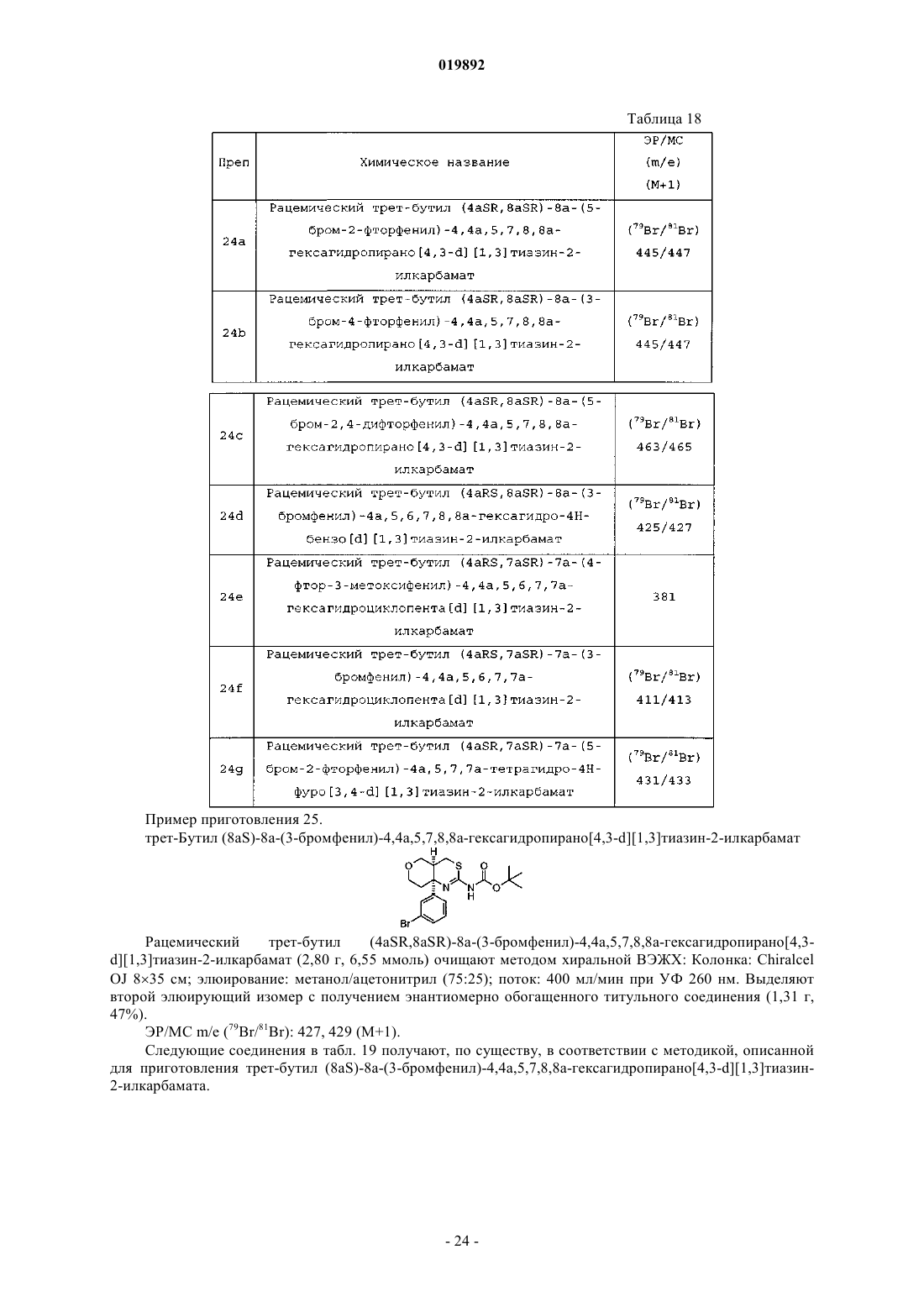
Ингибиторы киназы

Номер патента: 15189
Опубликовано: 30.06.2011
Авторы: Чжун Боюй, Майерс Майкл Рэй, Худзиак Кевин Джон, Бастиан Джолие Анне, Побанс Марк Эндрю, Лопес Де Уралде-Гармениа Беатрис, Блас Де Блас Хесус Андрес, Мэйдер Мэри Маргарет, Ли Течао, Де Дьес Альфонсо, Ших Чуан
МПК: A61K 31/44, A61K 31/497, C07D 401/14...
Метки: ингибиторы, киназы
Формула / Реферат:
1. Соединение формулы Iгде Z выбран из группы, которая включаетX выбран из группы, которая включаетR1 представляет собой С1-С7алкил, необязательно замещенный от одного до шести заместителями, выбранными из группы, которая включает галоген и C1-C4алкилгалоген; С3-С6циклоалкил, необязательно замещенный одним или двумя заместителями, выбранными из группы, которая включает C1-C4алкил и трифторметил; или триметилсилил;R2 представляет собой фенил,...
Ингибиторы тромбина

Номер патента: 2767
Опубликовано: 29.08.2002
Авторы: Зайтц Вернер, Мак Хельмут, Бём Ханс-Йоахим, Цирке Томас, Хорнбергер Вильфрид, Пфайффер Томас, Козер Штефан, Хёффкен Ханс Вольфганг
МПК: C07K 5/06, A61P 7/02, A61K 38/55...
Метки: тромбина, ингибиторы
Формула / Реферат:
1. Ингибиторы тромбина формулы (I) где R1 - алкил с 1-20 атомами углерода, фенил- или нафтилалкил с 1-10 атомами углерода в алкильной части, группа R2OOC-(CH2), где R2 означает водород или алкил с 1-10 атомами углерода, А - остаток a-аминокислоты формулы (II) где R3 - водород, алкил с 1-8 атомами углерода, циклоалкил с 3-7 атомами углерода, фенил и нафтил, незамещенные или замещенные алкилом или алкоксилом, каждый с 1-4 атомами углерода, или...
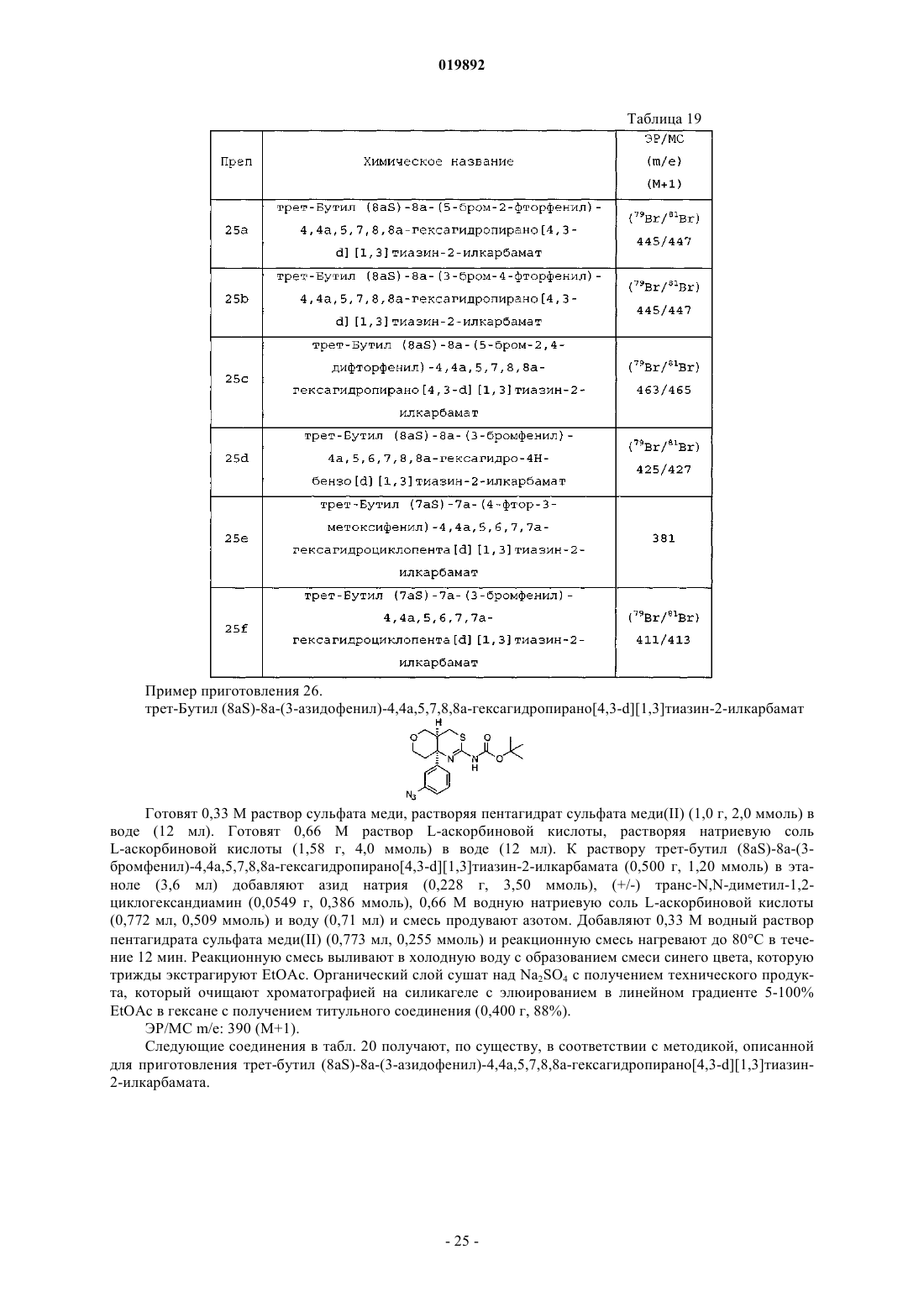
Ингибиторы акт и p70 s6-киназы

Номер патента: 18947
Опубликовано: 29.11.2013
Авторы: Дэлли Роберт Дин, Шеферд Тимоти Алан, Джозеф Саджан
МПК: A61P 35/00, C07D 473/00, A61K 31/52...
Метки: акт, s6-киназы, ингибиторы
Формула / Реферат:
1. Соединение формулыгде X представляет собой F, Cl, CF3, CN или Н;Y представляет собой F, Н или Cl;R1 и R2 независимо представляют собой Н, С1-С4алкил или СН2СН2ОН; или R1 и R2 вместе с атомом азота, к которому они присоединены, образуют пирролидиновое кольцо, необязательно замещенное гидроксиметилом в положении 2 или гидроксилом в положении 3, или азетидиновое кольцо, замещенное гидроксилом в положении 3;R3 представляет собой Н или ОН;R6...
Предыдущий патент: Способ приготовления хлеба
Следующий патент: Фармацевтическая композиция и способ лечения вич-инфекции
Случайный патент: Упругодемпфирующее устройство (варианты)