Производные индола в качестве агонистов рецептора s1p1
Номер патента: 17406
Опубликовано: 28.12.2012
Авторы: Ахмед Махмуд, Риверс Дин Энрю, Нортон Дэвид, Майатт Джэймс






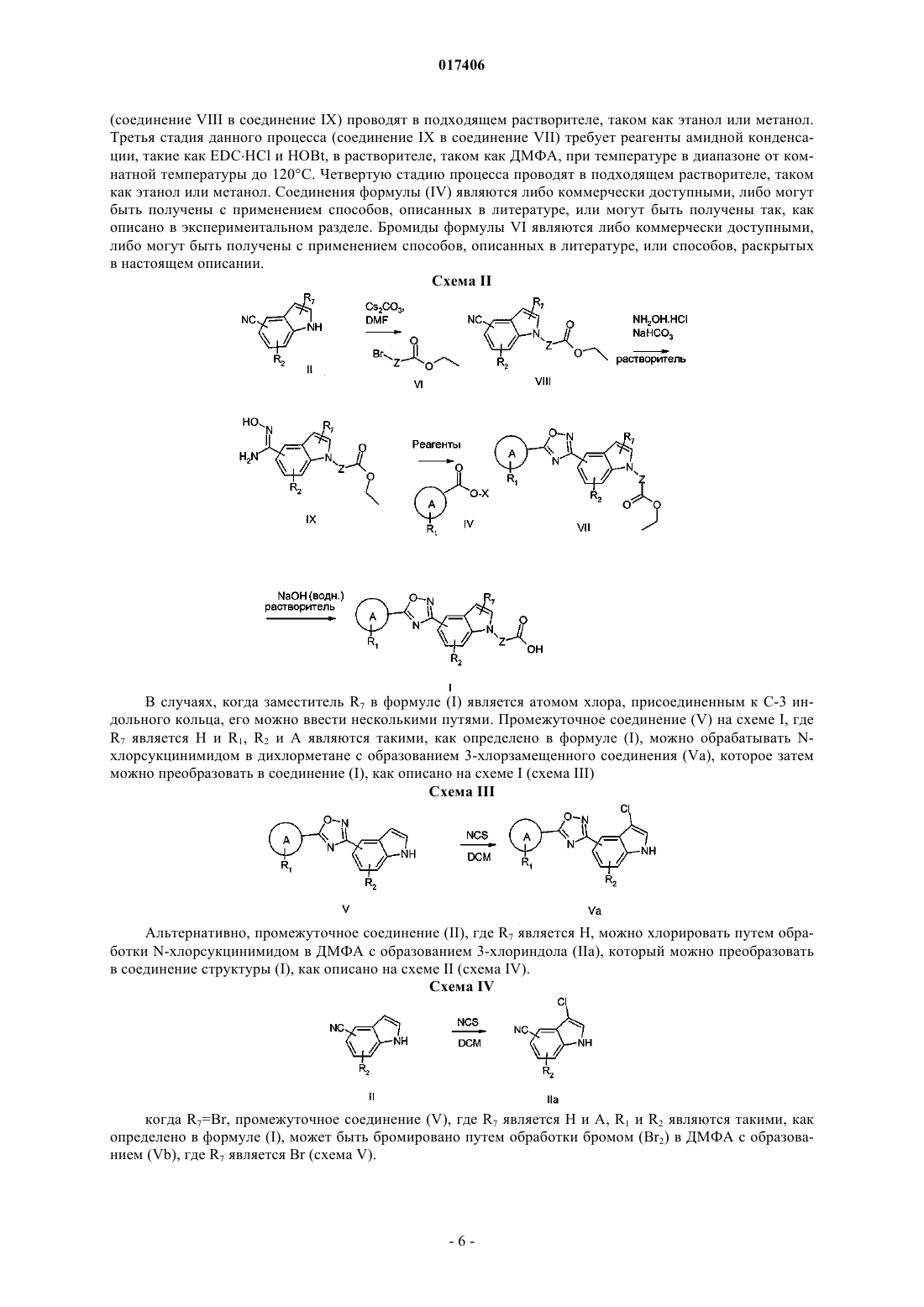






















Формула / Реферат
1. Соединение формулы (I) или его фармацевтически приемлемая соль

где один из R5 и R6 представляет собой водород и другой представляет собой группу (а)

А представляет собой фенил, тиенил или пиридил;
R1 представляет собой галоген, С(1-6)алкил, С(3-6)циклоалкил, С(1-6)алкоксигруппу, трифторметоксигруппу, трифторметил, цианогруппу, нитрогруппу, пирролидин или фенил, необязательно замещенный галогеном;
R2 представляет собой водород;
R7 представляет собой водород или галоген;
Z представляет собой С(1-4)алкил, который необязательно замещен галогеном или метилом.
2. Соединение, выбранное из:
3-(5-{5-[4-фенил-5-(трифторметил)-2-тиенил]-1,2,4-оксадиазол-3-ил}-1Н-индол-1-ил)пропановой кислоты;
3-[5-(5-{3-хлор-4-[(1-метилэтил)окси]фенил}-1,2,4-оксадиазол-3-ил)-1Н-индол-1-ил]пропановой кислоты;
3-[3-хлор-5-(5-{3-хлор-4-[(1-метилэтил)окси]фенил}-1,2,4-оксадиазол-3-ил)-1Н-индол-1-ил]пропановой кислоты;
3-(3-хлор-5-{5-[4-фенил-5-(трифторметил)-2-тиенил]-1,2,4-оксадиазол-3-ил}-1Н-индол-1-ил)пропановой кислоты;
3-(4-{5-[4-фенил-5-(трифторметил)-2-тиенил]-1,2,4-оксадиазол-3-ил}-1Н-индол-1-ил)пропановой кислоты;
3-[4-(5-{3-хлор-4-[(трифторметил)окси]фенил}-1,2,4-оксадиазол-3-ил)-1Н-индол-1-ил]пропановой кислоты;
3-[4-(5-{3-хлор-4-[(1-метилэтил)окси]фенил}-1,2,4-оксадиазол-3-ил)-1Н-индол-1-ил]пропановой кислоты;
3-[4-(5-{5-хлор-6-[(1-метилэтил)окси]-3-пиридинил}-1,2,4-оксадиазол-3-ил)-1Н-индол-1-ил]пропановой кислоты;
3-[5-(5-{3-хлор-4-[(1-метилэтил)окси]фенил}-1,2,4-оксадиазол-3-ил)-1Н-индол-1-ил]пропановой кислоты;
(5-{5-[4-фенил-5-(трифторметил)-2-тиенил]-1,2,4-оксадиазол-3-ил}-1Н-индол-1-ил)уксусной кислоты;
3-[3-бром-5-(5-{3-хлор-4-[(1-метилэтил)окси]фенил}-1,2,4-оксадиазол-3-ил)-1Н-индол-1-ил]пропановой кислоты;
5-[5-(5-{3-хлор-4-[(1-метилэтил)окси]фенил}-1,2,4-оксадиазол-3-ил)-1Н-индол-1-ил]пентановой кислоты;
4-[5-(5-{3-хлор-4-[(1-метилэтил)окси]фенил}-1,2,4-оксадиазол-3-ил)-1Н-индол-1-ил]бутановой кислоты;
(2S)-3-[5-(5-{3-хлор-4-[(1-метилэтил)окси]фенил}-1,2,4-оксадиазол-3-ил)-1Н-индол-1-ил]-2-метилпропановой кислоты;
2,2-диметил-3-(5-{5-[4-фенил-5-(трифторметил)-2-тиенил]-1,2,4-оксадиазол-3-ил}-1Н-индол-1-ил)пропановой кислоты;
3-[5-(5-{3-хлор-4-[(1-метилэтил)окси]фенил}-1,2,4-оксадиазол-3-ил)-1Н-индол-1-ил]-2,2,3-трифторпропановой кислоты;
4-[4-(5-{3-циано-4-[(1-метилэтил)окси]фенил}-1,2,4-оксадиазол-3-ил)-1Н-индол-1-ил]бутановой кислоты;
3-[5-(5-{3-циано-4-[(1-метилэтил)окси]фенил}-1,2,4-оксадиазол-3-ил)-1Н-индол-1-ил]пропановой кислоты;
3-[4-(5-{3-циано-4-[(1-метилэтил)окси]фенил}-1,2,4-оксадиазол-3-ил)-1Н-индол-1-ил]пропановой кислоты;
3-(4-{5-[2-(трифторметил)-4-бифенилил]-1,2,4-оксадиазол-3-ил}-1Н-индол-1-ил)пропановой кислоты;
3-(4-{5-[4-циклогексил-3-(трифторметил)фенил]-1,2,4-оксадиазол-3-ил}-1Н-индол-1-ил)пропановой кислоты;
3-(4-{5-[4-[(1-метилэтил)окси]-3-(трифторметил)фенил]-1,2,4-оксадиазол-3-ил}-1Н-индол-1-ил)пропановой кислоты;
[4-(5-{3-хлор-4-[(1-метилэтил)окси]фенил}-1,2,4-оксадиазол-3-ил)-1Н-индол-1-ил]уксусной кислоты;
[4-(5-{3-циано-4-[(1-метилэтил)окси]фенил}-1,2,4-оксадиазол-3-ил)-1Н-индол-1-ил]уксусной кислоты;
3-(4-{5-[2'-фтор-2-(трифторметил)-4-бифенилил]-1,2,4-оксадиазол-3-ил}-1Н-индол-1-ил)пропановой кислоты;
4-[4-(5-{3-хлор-4-[(1-метилэтил)окси]фенил}-1,2,4-оксадиазол-3-ил)-1Н-индол-1-ил]бутановой кислоты;
4-[4-(5-{3-хлор-4-[(трифторметил)окси]фенил}-1,2,4-оксадиазол-3-ил)-1Н-индол-1-ил]бутановой кислоты;
4-[4-(5-{5-хлор-6-[(1-метилэтил)окси]-3-пиридинил}-1,2,4-оксадиазол-3-ил)-1Н-индол-1-ил]бутановой кислоты;
4-(4-{5-[4-фенил-5-(трифторметил)-2-тиенил]-1,2,4-оксадиазол-3-ил}-1Н-индол-1-ил)бутановой кислоты;
4-(4-{5-[2-(трифторметил)-4-бифенилил]-1,2,4-оксадиазол-3-ил}-1Н-индол-1-ил)бутановой кислоты;
3-(4-{5-[4-(метилокси)-3-(трифторметил)фенил]-1,2,4-оксадиазол-3-ил}-1Н-индол-1-ил)пропановой кислоты;
4-{4-[5-(3-циано-4-{[(1R)-1-метилпропил]окси}фенил)-1,2,4-оксадиазол-3-ил]-1Н-индол-1-ил}бутановой кислоты;
4-{4-[5-(3-циано-4-{[(1S)-1-метилпропил]окси}фенил)-1,2,4-оксадиазол-3-ил]-1Н-индол-1-ил}бутановой кислоты;
3-(4-{5-[3-этил-4-(1-пиперидинил)фенил]-1,2,4-оксадиазол-3-ил}-1Н-индол-1-ил)пропановой кислоты;
3-{4-[5-(4-циклогексил-3-этилфенил)-1,2,4-оксадиазол-3-ил]-1Н-индол-1-ил}пропановой кислоты;
3-(4-{5-[5-хлор-6-(1-пирролидинил)-3-пиридинил]-1,2,4-оксадиазол-3-ил}-1Н-индол-1-ил)пропановой кислоты;
4-[4-(5-{3-бром-4-[(1-метилэтил)окси]фенил}-1,2,4-оксадиазол-3-ил)-1Н-индол-1-ил]бутановой кислоты;
4-(4-{5-[3-хлор-4-(2-метилпропил)фенил]-1,2,4-оксадиазол-3-ил}-1Н-индол-1-ил)бутановой кислоты;
3-(4-{5-[4-(2-метилпропил)-3-(трифторметил)фенил]-1,2,4-оксадиазол-3-ил}-1Н-индол-1-ил)пропановой кислоты;
4-(4-{5-[3-циано-4-(2-метилпропил)фенил]-1,2,4-оксадиазол-3-ил}-1Н-индол-1-ил)бутановой кислоты;
4-{4-[5-(2-циано-4-бифенилил)-1,2,4-оксадиазол-3-ил]-1Н-индол-1-ил}бутановой кислоты;
3-(3-хлор-4-{5-[4-фенил-5-(трифторметил)-2-тиенил]-1,2,4-оксадиазол-3-ил}-1Н-индол-1-ил)пропановой кислоты;
3-[3-хлор-5-(5-{3-хлор-4-[(трифторметил)окси]фенил}-1,2,4-оксадиазол-3-ил)-1Н-индол-1-ил]пропановой кислоты;
3-(3-хлор-5-{5-[3-хлор-4-(пропилокси)фенил]-1,2,4-оксадиазол-3-ил}-1Н-индол-1-ил)пропановой кислоты;
3-(3-хлор-5-{5-[3-хлор-4-(метилокси)фенил]-1,2,4-оксадиазол-3-ил}-1Н-индол-1-ил)пропановой кислоты;
3-(3-хлор-5-{5-[4-(метилокси)-3-(трифторметил)фенил]-1,2,4-оксадиазол-3-ил}-1Н-индол-1-ил)пропановой кислоты;
3-(3-хлор-5-{5-[3-хлор-4-(этилокси)фенил]-1,2,4-оксадиазол-3-ил}-1Н-индол-1-ил)пропановой кислоты;
3-[3-хлор-5-(5-{3-циано-4-[(1-метилэтил)окси]фенил}-1,2,4-оксадиазол-3-ил)-1Н-индол-1-ил]пропановой кислоты;
3-(3-хлор-5-{5-[4-нитро-3-(трифторметил)фенил]-1,2,4-оксадиазол-3-ил}-1Н-индол-1-ил)пропановой кислоты;
3-(3-хлор-5-{5-[6-(метилокси)-3-бифенилил]-1,2,4-оксадиазол-3-ил}-1Н-индол-1-ил)пропановой кислоты;
3-(5-{5-[6-(трифторметил)-3-бифенилил]-1,2,4-оксадиазол-3-ил}-1Н-индол-1-ил)пропановой кислоты и
их фармацевтически приемлемых солей.
3. Соединение по п.1, которое представляет собой 4-[4-(5-{5-хлор-6-[(1-метилэтил)окси]-3-пиридинил}-1,2,4-оксадиазол-3-ил)-1Н-индол-1-ил]бутановую кислоту.
4. Применение соединения по любому из пп.1-3 для лечения рассеянного склероза.
5. Применение соединения по любому из пп.1-3 для производства лекарственного средства для применения при лечении рассеянного склероза.
6. Фармацевтическая композиция, обладающая активностью агониста рецептора S1P1, содержащая соединение по любому из пп.1-3 и фармацевтически приемлемый носитель или эксципиент.
7. Способ лечения рассеянного склероза у млекопитающих, включая людей, который может быть опосредован рецепторами S1P1, включающий введение пациенту терапевтически безопасного и эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли.
Текст
ПРОИЗВОДНЫЕ ИНДОЛА В КАЧЕСТВЕ АГОНИСТОВ РЕЦЕПТОРА S1P1(71)(73) Заявитель и патентовладелец: ГЛЭКСО ГРУП ЛИМИТЕД (GB) Настоящее изобретение относится к соединениям формулы (I), где один из R5 и R6 представляет собой водород или R2 и другой представляет собой группу (а), к способам их получения,фармацевтическим композициям, содержащим их, и их применению для лечения состояний или расстройств, которые опосредуются рецептором S1P1. 017406 Настоящее изобретение относится к новым производным оксадиазола, обладающим фармакологической активностью, способу их получения, фармацевтическим композициям, содержащим их, и их применению для лечения различных расстройств. Сфингозин-1-фосфат (S1P) является биоактивным липидным медиатором, образованным фосфорилированием сфингозина сфингозинкиназами; его высокие уровни находят в крови. Он продуцируется и секретируется рядом типов клеток, включая клетки гемопоэтического происхождения, такие как тромбоциты и тучные клетки (Okamoto et al., 1998, J. Biol. Chem 273(42):27104; Sanchez and Hla, 2004, J. CellBiochem, 92:913). Он имеет широкий диапазон биологических эффектов, включая регуляцию пролиферации, дифференциации, подвижности клеток, регуляцию васкуляризации и активацию клеток очага воспаления и тромбоцитов (Pyne and Pyne 2000, Biochem J. 349:385). Описано пять подтипов рецепторов,чувствительных к S1P: S1P1 (Edg-1), S1P2 (Edg-5), S1P3 (Edg-3), S1P4 (Edg-6) и S1P5 (Edg-8), образующих часть семейства рецепторов гена эндотелиальной дифференциации, связанного с G-белком (Chun etal., 2002, Pharmacological Reviews 54:265, Sanchez and Hla 2004, J. Cellular Biochemistry, 92:913). Эти пять рецепторов показывают различную экспрессию мРНК; причем наиболее широко экспрессирован S1P1-3;S1P4 экспрессирован в лимфоидных и кроветворных тканях, и S1P5 экспрессирован, прежде всего в мозге и в меньшей степени в селезенке. Они передают сигналы через различные подгруппы G-белков, вызывая различные биологические реакции (Kluk and Hla 2002, Biochim et Biophysica Acta 1582:72, Sanchezand Hla 2004, J. Cellular Biochem 92:913). Предполагаемые роли для рецептора S1P1 включают миграцию лимфоцитов, индукцию/супрессию цитокинов и эффекты на эндотелиальные клетки (Rosen and Goetzl, 2005, Nat Rev Immunol. 5:560). Агонисты рецептора S1P1 применяли в ряде аутоиммунных и трансплантационных моделей животных,включая модели рассеянного склероза, основанные на экспериментальном аутоиммунном энцефаломиелите (ЕАЕ), для ослабления тяжести индуцированного заболевания (Brinkman et al., 2003, JBC 277:21453;Fujino et al., 2003, J. Pharmacol Exp Ther 305:70; Webb et al., 2004, J. Neuroimmunol 153:108; Rausch et al.,2004, J. Magn Reson Imaging 20:16). Сообщалось, что такая активность опосредуется эффектом агонистовS1P1 на циркуляцию лимфоцитов через систему лимфы. Обработка агонистами S1P1 приводит к секвестрации лимфоцитов во вторичных лимфоидных органах, таких как лимфатические узлы, вызывая обратимую периферическую лимфопению на моделях животных (Chiba et al., 1998, J. Immunology 160:5037,Forrest et al., 2004, J. Pharmacol Exp Ther 309:758; Sanna et al., 2004, JBC 279:13839). Опубликованные данные об агонистах свидетельствуют, что обработка данными соединениями вызывает исчезновение рецептора S1P1 с поверхности клеток посредством интернализации (Graler and Goetzl, 2004, FASEB J. 18:551; Matloubian et al., 2004, Nature 427:355; Jo et al., 2005 Chem Biol. 12:703) и что именно это уменьшение количества рецепторов S1P1 на иммунных клетках вносит свой вклад в уменьшение выхода Тклеток из лимфатических узлов обратно в кровоток. Делеция гена S1P1 вызывает гибель эмбриона. Эксперименты по исследованию роли рецептораS1P1 в миграции лимфоцитов и их транспорте включали адоптивный перенос меченых S1P1 дефицитных Т-клеток в облученных мышей дикого типа. Эти клетки показали пониженный выход из вторичных лимфоидных органов (Matloubian et al., 2004, Nature, 427:355). Рецептору S1P1 также приписывали роль в модулировании контактов эндотелиальных клеток(Allende et al., 2003, 102:3665, Blood; Singelton et al., 2005, FASEB J. 19:1646). В отношении такого эндотелиального действия сообщали, что агонисты S1P1 оказывают некоторый эффект на изолированные лимфатические узлы, который может вносить определенный вклад в их роль, модулирующую иммунные расстройства. Агонисты S1P1 вызывали закрытие эндотелиальных стромальных "ворот" лимфатических синусов, которые дренируют лимфатические узлы и предотвращают выход лимфоцитов (Wei et al., 2005,Nat. Immunology, 6:1228). Было показано, что иммуносупрессивное соединение FTY720 (JP1 1080026-А) снижает уровень циркулирующих лимфоцитов у животных и человека, обладает модулирующей активностью при заболеваниях на моделях иммунных расстройств у животных и снижают степень ремиссии при рецидивирующем перемежающемся рассеянном склерозе (Brinkman et al., 2002, JBC 277:21453, Mandala et al., 2002,Science 296:346, Fujino et al., 2003, J. Pharmacology and Experimental Therapeutics 305:45658, Brinkman etal., 2004, American J. Transplantation, 4:1019, Webb et al., 2004, J. Neuroimmunology 153:108, Morris et al.,2005, Eur J. Immunol, 35:3570, Chiba 2005, Pharmacology and Therapeutics, 108:308, Kahan et al., 2003,Transplantation 76: 1079, Kappos et al., 2006, New Eng J. Medicine 335:1124). Данное соединение является пролекарством, которое фосфорилируется in vivo сфингозинкиназами, образуя молекулу, которая обладает активностью агониста рецепторов S1P1, S1P3, S1P4 и S1P5. Клинические исследования продемонстрировали, что лечение соединением FTY720 приводит к брадикардии в течение первых 24 ч лечения(Kappos et al., 2006, New Eng J. Medicine 335:1124). Основываясь на ряде экспериментов, проведенных на клетках и с животными, считают, что брадикардия является следствием агонизма рецептора S1P3. Данные эксперименты включают использование животных с нокаутированным S1P3, которые в отличие от мышей дикого типа не демонстрировали брадикардии после введения FTY720, и применение S1P1 селективных соединений (Hale et al., 2004, Bioorganic and Medicinal Chemistry Letters 14:3501, Sanna et al.,2004, JBC 279:13839, Koyrakh et al., 2005, American J. Transplantation 5:529).-1 017406 Поэтому имеется потребность в соединениях, являющихся агонистами рецептора S1P1, селективность которых в отношении данного рецептора превышает их селективность в отношении рецептораS1P3, от которых можно было бы ожидать пониженной тенденции к индукции брадикардии. Следующие патентные заявки описывают производные оксадиазола в качестве агонистов S1P1:WO 03/105771, WO 05/058848, WO 06/047195, WO 06/100633, WO 06/115188, WO 06/131336,WO 07/024922 и WO 07/116866. Следующая патентная заявка описывает производные индолоксадиазола в качестве средств против пикорнавирусов: WO 96/009822. Следующие патентные заявки описывают производные индолкарбоновых кислот в качестве антагонистов рецепторов лейкотриенов, пестицидов и агрохимических фунгицидов соответственно:WO 06/090817, ЕР 0439785 и DE 3939238. В настоящее время найден структурно новый класс соединений, который предоставляет агонистов рецептора S1P1. Таким образом, настоящее изобретение предоставляет соединения (I) или их фармацевтически приемлемую соль где один из R5 и R6 представляет собой водород и другой представляет собой группу (а)R7 представляет собой водород или галоген;Z представляет собой С(1-4)алкил, который необязательно замещен галогеном или метилом. Термин "алкил" в качестве группы или части группы, например алкоксигруппы или гидроксиалкила, относится к прямой или разветвленной алкильной группе во всех изомерных формах. Термин"С(1-6)алкил" относится к алкильной группе, как определено выше, содержащей по меньшей мере 1 и по большей мере 6 атомов углерода. Примеры таких алкильных групп включают метил, этил, пропил, изопропил, н-бутил, изобутил, втор-бутил или трет-бутил. Примеры таких алкоксигрупп включают метоксигруппу, этоксигруппу, пропоксигруппу, изопропоксигруппу, бутоксигруппу, изобутоксигруппу, вторбутоксигруппу и трет-бутоксигруппу. Подходящие С(3-6)циклоалкильные группы включают циклопропил, циклобутил, циклопентил и циклогексил. Подходящие С(3-6)циклоалкилоксигруппы включают циклопропоксигруппу, циклобутоксигруппу,циклопентоксигруппу и циклогексилоксигруппу. Используемый в настоящем описании термин "галоген" относится к фтору (F), хлору (Cl), брому(Br) или йоду (I) и термин "галоид" относится к галогену (как заместителю): фтор- (-F), хлор- (-Cl), бром(-Br) и йод- (-I). Термин "гетероарил" представляет ненасыщенное кольцо, которое содержит один или более гетероатомов. Когда термин "гетероарил" представляет 5-членную группу, она содержит гетероатом, выбранный из О, N или S, и может необязательно содержать дополнительно от 1 до 3 атомов азота. Когда гетероарил представляет собой 6-членную группу, она содержит от 1 до 3 атомов азота. Примеры таких 5- или 6-членных гетероарильных колец включают пирролил, триазолил, тиадиазолил, тетразолил, имидазолил, пиразолил, изотиазолил, тиазолил, изоксазолил, оксазолил, оксадиазолил, фуразанил, фуранил,тиенил, пиридил, пиримидинил, пиразинил, пиридазинил и триазинил. В некоторых соединениях формулы (I) в зависимости от природы заместителя имеются хиральные атомы углерода, поэтому соединения формулы (I) могут существовать в виде стереоизомеров. Настоящее изобретение распространяется на все оптические изомеры, такие как стереоизомерные формы соединений формулы (I), включая энантиомеры, диастереомеры и их смеси, такие как рацематы. Различные стереоизомерные формы можно разделить или отделить одну от другой традиционными способами или данный изомер можно получить традиционным стереоселективным или асимметричным синтезом. Некоторые соединения согласно настоящему изобретению могут существовать в различных таутомерных формах и следует понимать, что настоящее изобретение охватывает все такие таутомерные формы.-2 017406 Подходящими соединениями согласно настоящему изобретению являются соединения, выбранные из: 3-(5-5-[4-фенил-5-(трифторметил)-2-тиенил]-1,2,4-оксадиазол-3-ил-1 Н-индол-1-ил)пропановой кислоты; 3-[5-(5-3-хлор-4-[(1-метилэтил)окси]фенил-1,2,4-оксадиазол-3-ил)-1 Н-индол-1-ил]пропановой кислоты; 3-[3-хлор-5-(5-3-хлор-4-[(1-метилэтил)окси]фенил-1,2,4-оксадиазол-3-ил)-1 Н-индол-1 ил]пропановой кислоты; 3-(3-хлор-5-5-[4-фенил-5-(трифторметил)-2-тиенил]-1,2,4-оксадиазол-3-ил-1 Н-индол-1 ил)пропановой кислоты; 3-(4-5-[4-фенил-5-(трифторметил)-2-тиенил]-1,2,4-оксадиазол-3-ил-1 Н-индол-1-ил)пропановой кислоты; 3-[4-(5-3-хлор-4-[(трифторметил)окси]фенил-1,2,4-оксадиазол-3-ил)-1 Н-индол-1-ил]пропановой кислоты; 3-[4-(5-3-хлор-4-[(1-метилэтил)окси]фенил-1,2,4-оксадиазол-3-ил)-1 Н-индол-1-ил]пропановой кислоты; 3-[4-(5-5-хлор-6-[(1-метилэтил)окси]-3-пиридинил-1,2,4-оксадиазол-3-ил)-1 Н-индол-1 ил]пропановой кислоты; 3-[5-(5-3-хлор-4-[(1-метилэтил)окси]фенил-1,2,4-оксадиазол-3-ил)-1 Н-индол-1-ил]пропановой кислоты;-3 017406 3-(4-5-[4-(метилокси)-3-(трифторметил)фенил]-1,2,4-оксадиазол-3-ил-1 Н-индол-1-ил)пропановой кислоты; 4-4-[5-(3-циано-4-[(1R)-1-метилпропил]оксифенил)-1,2,4-оксадиазол-3-ил]-1 Н-индол-1 илбутановой кислоты; 4-4-[5-(3-циано-4-[(1S)-1-метилпропил]оксифенил)-1,2,4-оксадиазол-3-ил]-1 Н-индол-1 илбутановой кислоты; 3-(4-5-[3-этил-4-(1-пиперидинил)фенил]-1,2,4-оксадиазол-3-ил-1 Н-индол-1-ил)пропановой кислоты; 3-4-[5-(4-циклогексил-3-этилфенил)-1,2,4-оксадиазол-3-ил]-1 Н-индол-1-илпропановой кислоты; 3-(4-5-[5-хлор-6-(1-пирролидинил)-3-пиридинил]-1,2,4-оксадиазол-3-ил-1 Н-индол-1 ил)пропановой кислоты; 4-[4-(5-3-бром-4-[(1-метилэтил)окси]фенил-1,2,4-оксадиазол-3-ил)-1 Н-индол-1-ил]бутановой кислоты; 4-(4-5-[3-хлор-4-(2-метилпропил)фенил]-1,2,4-оксадиазол-3-ил-1 Н-индол-1-ил)бутановой кислоты; 3-(4-5-[4-(2-метилпропил)-3-(трифторметил)фенил]-1,2,4-оксадиазол-3-ил-1 Н-индол-1 ил)пропановой кислоты; 4-(4-5-[3-циано-4-(2-метилпропил)фенил]-1,2,4-оксадиазол-3-ил-1 Н-индол-1-ил)бутановой кислоты; 4-4-[5-(2-циано-4-бифенилил)-1,2,4-оксадиазол-3-ил]-1 Н-индол-1-илбутановой кислоты; 3-(3-хлор-4-5-[4-фенил-5-(трифторметил)-2-тиенил]-1,2,4-оксадиазол-3-ил-1 Н-индол-1 ил)пропановой кислоты; 3-[3-хлор-5-(5-3-хлор-4-[(трифторметил)окси]фенил-1,2,4-оксадиазол-3-ил)-1 Н-индол-1 ил]пропановой кислоты; 3-(3-хлор-5-5-[3-хлор-4-(пропилокси)фенил]-1,2,4-оксадиазол-3-ил-1 Н-индол-1-ил)пропановой кислоты; 3-(3-хлор-5-5-[3-хлор-4-(метилокси)фенил]-1,2,4-оксадиазол-3-ил-1 Н-индол-1-ил)пропановой кислоты; 3-(3-хлор-5-5-[4-(метилокси)-3-(трифторметил)фенил]-1,2,4-оксадиазол-3-ил-1 Н-индол-1 ил)пропановой кислоты; 3-(3-хлор-5-5-[3-хлор-4-(этилокси)фенил]-1,2,4-оксадиазол-3-ил-1 Н-индол-1-ил)пропановой кислоты; 3-[3-хлор-5-(5-3-циано-4-[(1-метилэтил)окси]фенил-1,2,4-оксадиазол-3-ил)-1 Н-индол-1 ил]пропановой кислоты; 3-(3-хлор-5-5-[4-нитро-3-(трифторметил)фенил]-1,2,4-оксадиазол-3-ил-1 Н-индол-1 ил)пропановой кислоты; 3-(3-хлор-5-5-[6-(метилокси)-3-бифенилил]-1,2,4-оксадиазол-3-ил-1 Н-индол-1-ил)пропановой кислоты; 3-(5-5-[6-(трифторметил)-3-бифенилил]-1,2,4-оксадиазол-3-ил-1 Н-индол-1-ил)пропановой кислоты и их фармацевтически приемлемых солей. Особенно предпочтительным соединением согласно настоящему изобретению является 4-[4-(5-5 хлор-6-[(1-метилэтил)окси]-3-пиридинил-1,2,4-оксадиазол-3-ил)-1 Н-индол-1-ил]бутановая кислота. Фармацевтически приемлемые производные соединений формулы (I) включают любую фармацевтически приемлемую соль, сложный эфир или соль такого сложного эфира соединения формулы (I), которые после введения реципиенту способны предоставлять (прямо или косвенно) соединение формулы(I) или его активный метаболит или остаток. Соединения формулы (I) могут образовывать соли. Следует понимать, что для применения в медицине соли соединений формулы (I) должны быть фармацевтически приемлемыми. Подходящие фармацевтически приемлемые соли известны специалистам в данной области; они включают соли, описанные в J. Pharra. Sci, 1977, 66, 1-19, такие как кислотно-аддитивные соли, образованные неорганическими кислотами, например хлористо-водородной, бромисто-водородной, серной, азотной или фосфорной кислотой; и органическими кислотами, например янтарной, малеиновой, уксусной, фумаровой, лимонной,винной, бензойной п-толуолсульфоновой, метансульфоновой или нафталинсульфоновой кислотой. Некоторые соединения формулы (I) могут образовывать кислотно-аддитивные соли с одним или более эквивалентами кислоты. Настоящее изобретение включает в свой объем все возможные стехиометрические и нестехиометрические формы. Соли можно также получить из фармацевтически приемлемых оснований,включая неорганические основания и органические основания. Соли, являющиеся производными неорганических оснований, включают соли алюминия, аммония, кальция, меди, железа(III), железа(II), лития,магния, марганца, калия, натрия, цинка и т.п. Соли, являющиеся производными фармацевтически приемлемых органических оснований, включают соли первичных, вторичных и третичных аминов; замещенных аминов, включая природные замещенные амины; и циклические амины. Конкретные фармацевтиче-4 017406 ски приемлемые органические основания включают аргинин, бетаин, кофеин, холин, N,N'дибензилэтилендиамин, диэтиламин, 2-диэтиламиноэтанол, 2-диметиламиноэтил, этаноламин, этилендиамин, N-этилморфолин, N-этилпиперидин, глюкамин, глюкозамин, гистидин, гидрабамин, изопропиламин, лизин, метилглюкамин, морфолин, пиперазин, пиперидин, прокаин, пурины, теобромин, триметиламин, триметиламин, трипропиламин, трис(гидроксиметил)аминометан (TRIS, трометанол) и т.п. Соли также могут быть образованы из ионообменных смол основных ионов, например полиаминовых смол. Когда соединение согласно настоящему изобретению является основным, соли можно получить из фармацевтически приемлемых кислот, включая неорганические и органические кислоты. Такие кислоты включают уксусную, бензолсульфоновую, бензойную, камфорсульфоновую, лимонную, этансульфоновую, этандисульфоновую, фумаровую, глюконовую, глутамовую, бромисто-водородную, хлористоводородную, изетионовую, молочную, малеиновую, яблочную, миндальную, метансульфоновую, слизевую, памовую, пантотеновую, фосфорную, пропионовую, янтарную, серную, винную, птолуолсульфоновую кислоту и т.п. Соединения формулы (I) можно получить в кристаллической или некристаллической форме, и, если они являются кристаллическими, они необязательно могут быть гидратированными или сольватированными. Настоящее изобретение включает в свой объем стехиометрические гидраты или сольваты, а также соединения, содержащие переменные количества воды и/или растворителя. В следующем аспекте настоящее изобретение предоставляет способы получения соединения формулы (I). В одном аспекте соединение формулы (I) можно получить посредством способа, представленного на схеме I, где A, Z, R1, R2, R7 являются такими, как определено в формуле (I). Схема I Первую стадию процесса (соединение II в соединение III) проводят в подходящем растворителе, таком как метанол или этанол, и нагревают до такой температуры, как 50-80 С. На второй стадии процесса(соединение III в соединение V) подходящие реагенты включают EDCHCl и HOBt в растворителе, таком как ДМФА, при температуре в диапазоне от комнатной до 90 С или, альтернативно, с РуВОР в ДМФА. Альтернативно, соединение (III) можно преобразовать в соединение (V) обработкой сложным эфиром карбоновой кислоты соединения (IV) и этоксидом натрия в этаноле в микроволновом реакционном нагревателе при такой температуре, как 120 С. На третьей стадии (соединение V в соединение VII) используют основание, такое как карбонат цезия или, альтернативно, карбонат калия, и реакционную смесь нагревают либо традиционным образом, либо используя микроволновый реактор, до такой температуры,как 140 С. Четвертую стадию процесса (соединение VII в соединение I) проводят в подходящем растворителе, таком как метанол или, альтернативно, этанол; ее можно также проводить либо при комнатной температуре, либо при повышенной температуре, такой как 40 или 50 С. Соединения формулы (IV) являются либо коммерчески доступными, либо могут быть получены с применением способов, описанных в литературе, либо могут быть получены, как описано в экспериментальном разделе. Бромиды формулы(VI) являются либо коммерчески доступными, либо могут быть получены с применением способов, описанных в литературе, либо способами, раскрытыми в настоящем описании. В другом препаративном аспекте соединение формулы (I) может быть получено способом, представленным на схеме II, где A, Z, R1, R2, R7 являются такими, как определено в формуле (I). На первой стадии реакцию (соединение II в соединение VIII) можно нагревать до 80 С. Вторую стадию процесса(соединение VIII в соединение IX) проводят в подходящем растворителе, таком как этанол или метанол. Третья стадия данного процесса (соединение IX в соединение VII) требует реагенты амидной конденсации, такие как EDCHCl и HOBt, в растворителе, таком как ДМФА, при температуре в диапазоне от комнатной температуры до 120 С. Четвертую стадию процесса проводят в подходящем растворителе, таком как этанол или метанол. Соединения формулы (IV) являются либо коммерчески доступными, либо могут быть получены с применением способов, описанных в литературе, или могут быть получены так, как описано в экспериментальном разделе. Бромиды формулы VI являются либо коммерчески доступными,либо могут быть получены с применением способов, описанных в литературе, или способов, раскрытых в настоящем описании. Схема II В случаях, когда заместитель R7 в формуле (I) является атомом хлора, присоединенным к С-3 индольного кольца, его можно ввести несколькими путями. Промежуточное соединение (V) на схеме I, гдеR7 является Н и R1, R2 и А являются такими, как определено в формуле (I), можно обрабатывать Nхлорсукцинимидом в дихлорметане с образованием 3-хлорзамещенного соединения (Va), которое затем можно преобразовать в соединение (I), как описано на схеме I (схема III) Схема III Альтернативно, промежуточное соединение (II), где R7 является Н, можно хлорировать путем обработки N-хлорсукцинимидом в ДМФА с образованием 3-хлориндола (IIa), который можно преобразовать в соединение структуры (I), как описано на схеме II (схема IV). Схема IV когда R7=Br, промежуточное соединение (V), где R7 является Н и A, R1 и R2 являются такими, как определено в формуле (I), может быть бромировано путем обработки бромом (Br2) в ДМФА с образованием (Vb), где R7 является Br (схема V). В случаях, где R1 представляет собой фенильную группу, данную группу можно ввести по реакции перекрестного сочетания соединения структуры VII с образованием соединения VIIa (схема VI), где A, Z,R2 и R7 являются такими, как определено в формуле (I), и Ar представляет собой необязательно замещенный фенил, с последующим гидролизом до соединения I. В данном преобразовании М представляет собой группу, такую как В(ОН)2, что делает возможным осуществление реакции перекрестного сочетания, Y представляет собой группу, такую как бром, йод или трифторметансульфонат, и катализатор представляет собой палладиевый катализатор, такой как тетракистрифенилфосфинпалладий(0). Такие реакции обычно проводят при повышенной температуре. Схема VI В случаях, где R1 представляет собой алкоксигруппу, такую как О-этил или О-изопропил, алкильный заместитель можно ввести в соединение формулы VII, где R1 означает ОН, и A, Z, R7 и R2 являются такими, как определено в формуле (I), с образованием соединения формулы VIIb, где R1 означает Оалкил (схема VII). В этом случае Y представляет собой галоген, такой как йод. Реакцию можно проводить в полярном растворителе, таком как ДМФА, в присутствии основания, такого как карбонат калия. Схема VII В некоторых случаях можно алкилировать промежуточный индол (V), где R1, R2, R7 и А являются такими, как определено в формуле (I), непосредственно алкилбромидом, замещенным карбоновой кислотой, с образованием конечного соединения (I), не прибегая к стадии гидролиза (схема VIII). Подходящим основанием для такого преобразования является карбонат цезия. Схема VIII Фармацевтически приемлемые соли можно получить традиционным способом по реакции с соответствующей кислотой или производным кислоты. Силу и эффективность соединений согласно настоящему изобретению для рецептора S1P1 можно определить посредством GTPYS-теста, проводимого с клонированным человеческим рецептором, или посредством теста связывания дрожжей, также описанных в настоящем описании. Соединения формулы(I) продемонстрировали активность агонистов рецептора S1P1 в функциональных исследованиях, описанных в настоящем описании. Соединения формулы (I) и их фармацевтически приемлемые соли поэтому можно применять для лечения состояний или расстройств, опосредуемых рецептором S1P1. В частности, соединения формулы(I) и их фармацевтически приемлемые соли можно применять для лечения рассеянного склероза. Следует понимать, что используемый в настоящем описании термин "лечение" включает профилактику, а также ослабление установленных симптомов. Таким образом, настоящее изобретение также предоставляет соединение формулы (I) или его фармацевтически приемлемую соль для применения в качестве лекарственного средства, в частности для лечения состояний или расстройств, опосредуемых рецептором S1P1. В частности, настоящее изобретение предоставляет соединение формулы (I) или его фармацевтически приемлемую соль для применения-7 017406 для лечения рассеянного склероза. Кроме того, настоящее изобретение предоставляет способ лечения рассеянного склероза у млекопитающих, включая людей, который может быть опосредован рецепторами S1P1, включающий введение пациенту терапевтически безопасного и эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли. В другом аспекте настоящее изобретение предоставляет соединение формулы (I) или его фармацевтически приемлемую соль для применения при производстве лекарственного средства для применении при лечении рассеянного склероза. Для применения соединений формулы (I) и их фармацевтически приемлемых солей в терапии их обычно составляют в фармацевтические композиции согласно стандартной фармацевтической практике. Настоящее изобретение также представляет фармацевтическую композицию, которая содержит соединение формулы (I) или его фармацевтически приемлемую соль и фармацевтически приемлемый носитель или эксципиент. В следующем аспекте настоящее изобретение предоставляет способ получения фармацевтической композиции, включающий смешивание соединения формулы (I) или его фармацевтически приемлемой соли и фармацевтически приемлемого носителя или эксципиента. Фармацевтическая композиция согласно настоящему изобретению, которую можно получить смешиванием, как правило, при температуре окружающей среды и атмосферном давлении, обычно приспособлена для перорального, парентерального или ректального введения и, как таковая, может быть в форме таблеток, капсул, жидких препаратов для перорального введения, порошков, гранул, лепешек, восстанавливаемых порошков, инъекционных или инфузионных растворов или суспензий или суппозиториев. Как правило, предпочтительными являются композиции, вводимые перорально. Таблетки и капсулы для перорального введения могут быть в единичной дозированной форме и могут содержать традиционные эксципиенты, такие как связующие (например, предварительно желатинизированный кукурузный крахмал, поливинилпирролидон или гидроксипропилметилцеллюлоза); наполнители (например, лактоза, микрокристаллическая целлюлоза или гидрофосфат кальция); лубриканты для таблетирования (например, стеарат магния, тальк или кремнезем); дезинтегранты (например, картофельный крахмал или натриевая соль гликолята крахмала); и приемлемые увлажнители (например, лаурилсульфат натрия). Таблетки могут быть покрыты оболочкой согласно способам, хорошо известным в обычной фармацевтической практике. Жидкие препараты для перорального введения могут быть в форме, например, водной или масляной суспензии, растворов, эмульсий, сиропов или эликсиров или могут быть в форме сухого продукта для восстановления с водой или другим подходящим наполнителем непосредственно перед применением. Такие жидкие препараты могут содержать традиционные добавки, такие как суспендирующие средства (например, сироп сорбита, производные целлюлозы или гидрированные пищевые жиры), эмульгаторы (например, лецитин или камедь акации), неводные наполнители (которые могут включать пищевые масла, например, миндальное масло, сложные масляные эфиры, этиловый спирт или фракционированные растительные масла), консерванты (например, метил- или пропил-п-гидроксибензоаты или сорбиновую кислоту) и, если желательно, традиционные отдушки и красящие вещества, буферные соли и соответствующие подсластители. Препараты для перорального введения могут быть подходящим образом приготовлены для получения контролируемого высвобождения активного соединения. Для парентерального введения жидкие единичные дозированные формы получают, используя соединения согласно настоящему изобретению или их фармацевтически приемлемые соли и стерильный носитель. Составы для инъекций могут быть представлены в единичной дозированной форме, например в ампулах, или во множественных дозах, с применением соединения согласно настоящему изобретению или его фармацевтически приемлемых производных и стерильного носителя, необязательно с добавленным консервантом. Композиции могут принимать такие формы, как суспензии, растворы или эмульсии в масляных или водных носителях и могут содержать вспомогательные агенты, такие как суспендирующие, стабилизирующие и/или диспергирующие средства. Альтернативно, активный ингредиент может быть в форме порошка для восстановления подходящим носителем, например, стерильной апирогенной водой, непосредственно перед применением. Соединение в зависимости от носителя и используемой концентрации может быть либо суспендировано, либо растворено в носителе. При приготовлении растворов соединение может быть растворено для инъекций и стерилизовано фильтрованием в подходящий флакон или ампулу и герметизировано. Преимущественно в носителе растворяют вспомогательные средства, такие как местные анестетики, консерванты и буферные вещества. Для повышения стабильности,композицию можно замораживать после расфасовки по флаконам и удалять воду в вакууме. Парентеральные суспензии готовят существенно таким же образом, с тем исключением, что соединение суспендируют в носителе, а не растворяют в нем; кроме того, в этом случае невозможна стерилизация посредством фильтрования. Соединение можно стерилизовать воздействием этиленоксида перед суспендированием в стерильном носителе. Преимущественно для облегчения создания однородного распределения в композицию включают поверхностно-активное вещество и увлажнитель. Лосьоны можно получить на водной или масляной основе; обычно они также содержат не менее-8 017406 одного эмульгатора, стабилизаторов, диспергирующих средств, суспендирующих агентов, загустителей или окрашивающих средств. Капли можно получить на водной или неводной основе, также содержащей одно или более из диспергирующих средств, стабилизаторы, солюбилизаторы или суспендирующие средства. Они также могут содержать консервант. Соединение формулы (I) или его фармацевтически приемлемые соли могут быть получены в виде ректальных композиций, таких как суппозитории или удерживающие клизмы, например содержащие традиционные основы для суппозиториев, такие как масло какао или другие глицериды. Соединения формулы (I) или их фармацевтически приемлемые соли можно также получить в виде препаратов депо. Такие составы длительного действия можно вводить посредством имплантации (например, подкожно или внутримышечно) или посредством внутримышечной инъекции. Так, например,соединения согласно настоящему изобретению можно получить с подходящими полимерными или гидрофобными материалами (например, в виде эмульсии в приемлемом масле) или с ионообменными смолами или в виде труднорастворимых производных, например, в виде труднорастворимой соли. Для интраназального введения соединения формулы (I) или их фармацевтически приемлемые соли можно получить в виде растворов для введения с помощью соответствующего дозирующего устройства или устройства, предоставляющего единичные дозы, или, альтернативно, в виде порошкообразной смеси с соответствующим носителем для введения, используя соответствующее устройство для доставки. Таким образом, соединения формулы (I) или их фармацевтически приемлемые соли могут быть получены для перорального, буккального, парентерального, местного (включая офтальмологическое и интраназальное), депо или ректального введения или в форме, подходящей для введения посредством ингаляции или вдувания (через рот или нос). Соединения формулы (I) или их фармацевтически приемлемые соли могут быть получены для местного введения в форме мазей, кремов, гелей, лосьонов, пессариев, аэрозолей или капель (например,капель для глаз, ушей или для носа). Мази и кремы можно готовить, например на водной или масляной основе с добавлением подходящего загустителя и/или желирующих средств. Мази для введения в глаза можно готовить стерильно, используя стерилизованные компоненты. Композиция может содержать от 0,1 до 99 мас.%, предпочтительно от 10 до 60 мас.% активного ингредиента в зависимости от способа введения. Дозу соединения, применяемого для лечения вышеуказанных расстройств, будут менять обычным образом в зависимости от серьезности расстройств, массы тела пациента и других подобных факторов. Однако, как правило, подходящие единичные дозы могут составлять от 0,05 до 1000 мг, от 1,0 до 500 мг или от 1,0 до 200 мг и такие единичные дозы можно вводить более одного раза в день, например два или три раза в день. Соединения формулы (I) или их фармацевтически приемлемые соли можно применять в комбинированных препаратах. Например, соединения согласно настоящему изобретению можно применять в комбинации с циклоспорином А, метотрексатом, стероидами, рапамицином, ингибиторами воспалительных цитокинов, иммуномодуляторами, включая биологические или другие терапевтически активные соединения. Настоящее изобретение также включает изотопно-меченные соединения, которые идентичны соединениям, указанным в формуле (I) и последующих формулах, за исключением того, что один или более атомов заменены атомом, имеющим атомную массу или массовый номер, отличные от атомной массы или атомного номера, обычно встречающихся в природе. Примеры изотопов, которые можно вводить в соединения согласно настоящему изобретению, включают изотопы водорода, углерода, азота, кислорода, фосфора, фтора, йода и хлора, такие как 3 Н, 11C, 14C, 18F, 123I и 125I. Соединения согласно настоящему изобретению и фармацевтически приемлемые соли указанных соединений, которые содержат вышеуказанные изотопы и/или другие изотопы других атомов, входят в объем настоящего изобретения. Изотопно-меченные соединения согласно настоящему изобретению, например соединения, в которые введены радиоактивные изотопы, такие как 3 Н, 14 С, используют при исследованиях распределения лекарственного средства и/или субстрата по тканям. Особенно предпочтительны изотопы трития, т.е. 3 Н, и углерода-14, т.е. 14 С, поскольку они просты в получении и детектировании. Изотопы 11 С и 8F особенно полезны при PET (позитрон-эмиссионной томографии), и изотопы 125I особенно полезны при SPECT (однофотонной эмиссионной компьютерной томографии) для визуализации мозга. Кроме того, замена более тяжелыми изотопами, такими как дейтерий, т.е. 2 Н, может предоставить определенную терапевтическую пользу, вследствие большей метаболической стабильности, например, увеличения периода полужизни in vivo или снижения требуемых доз; поэтому в некоторых обстоятельствах они могут оказаться предпочтительными. Изотопно-меченные соединения формулы (I) и последующих формул согласно настоящему изобретению можно, как правило, получать, осуществляя методики, раскрытые ниже на схемах и/или примерах, заменяя легко доступным изотопно-меченным реагентом реагент, не меченный изотопом. Все публикации, включая патенты и заявки на патент, приведенные в настоящем описании, но, не ограничиваясь ими, включены в настоящее описание посредством ссылки, как если бы каждая индивидуальная публикация была конкретно и индивидуально указана как включенная в настоящее описание посредством ссылки.-9 017406 Следующие описания и примеры иллюстрируют получение соединений согласно настоящему изобретению. Условия, оборудование и программное обеспечение для аналитических систем ЖХМС Оборудование: градиентный насос Agilent 1100; автосэмплер Agilent 1100; детектор Agilent 1100 DAD; дегазатор Agilent 1100; термостат Agilent 1100; контроллер Agilent 1100; система подготовки растворителей Waters Acquity Binary Solvent Manager; система подготовки образцов Waters Acquity Sample Manager; матричный фотодиодный детектор Waters Acquity PDA; масс-спектрометр Waters ZQ;Waters MassLynx версии 4,0 SP2 или версии 4,1. Для 5-минутного способа. Колонка. Используют колонку Waters Atlantis с размерами 4,6 мм 50 мм. Размер частиц стационарной фазы 3 мкм. Растворители: А: водный растворитель=вода+0,05% муравьиной кислоты; В: органический растворитель=ацетонитрил+0,05% муравьиной кислоты. Методика. В стандартной методике используют 5-минутные хроматограммы. Скорость потока. В вышеуказанном способе используют скорость потока, равную 3 мл/мин. Для 2-минутного способа. Программное обеспечение:Waters MassLynx версии 4,1. Колонка. Используют колонку Waters Acquity ВЕН UPLC C18, с размерами 2,1 мм 50 мм. Размер частиц стационарной фазы 1,7 мкм. Растворители: А: водный растворитель=вода+0,05% муравьиной кислоты; В: органический растворитель=ацетонитрил+0,05% муравьиной кислоты; Слабая промывка=1:1 метанол:вода; Сильная промывка=вода. Методика. В стандартной методике используют 2-минутные хроматограммы. В вышеуказанном способе используют скорость потока, равную 1 мл/мин. В стандартной методике инъецируемый объем составляет 0,5 мкл. Температура колонки составляет 40 С. Диапазон детектирования в УФ-области составлял от 220 до 330 нм. Массовая направленная автопрепаративная система открытого доступа (MDAP). Оборудование. Набор инструментов для массовой направленной препаративной системы открытого доступа состоит из следующих компонентов: 1 градиентный насос Waters 600; 1 инжектор/коллектор Waters 2767;- 10017406 1 система для обработки реагентов Waters Reagent manager; 1 масс-спектрометр MicroMass ZQ; 1 коллектор сброса Gllson Aspec; 1 послефракционный УФ-детектор Gllson 115; 1 компьютерная система. Программное обеспечение:MicroMass MassLynx v. 4,0. Колонка. Обычно используют колонку Supelco LCABZ с внутренним диаметром 20 мм и длиной 100 мм. Размер частиц стационарной фазы составлял 5 мкм. Растворители: А: водный растворитель=вода+0,1% муравьиной кислоты; В: органический растворитель=MeCN/вода 95/5+0,05% муравьиной кислоты; Вспомогательный растворитель=МеОН/вода 80/20+50 мМ ацетат аммония; Растворитель для промывки иглы=МеОН/вода/ДМСО 80/10/10. Методики. В зависимости от аналитического времени удерживания представляющего интерес соединения можно применять одну из пяти методик. Все они выполняют 15-минутные хроматографические циклы, состоящие из 10-минутного градиента, с последующей 5-минутной промывкой колонки и стадией повторного уравновешивания.MDP 3,8-5,5=50-90% В. Скорость потока. Во всех вышеуказанных методиках скорость потока составляет 20 мл/мин. Альтернативная система. Оборудование: модуль формирования бинарного градиента Waters 2525; вспомогательный насос Waters 515; контрольный модуль для насосов Waters Pump Control Module; инжектор/коллектор Waters 2767; система управления потоком для колонок Waters Column Fluidics Manager; фотодиодный матричный детектор Waters 2996; масс-спектрометр Waters ZQ; коллектор фракций GIIson 202; коллектор сброса GIIson Aspec. Программное обеспечение:(крупный масштаб). Размер частиц стационарной фазы составлял 5 мкм. Растворители: А: водный растворитель=вода+0,1% муравьиной кислоты; В: органический растворитель=ацетонитрил+0,1% муравьиной кислоты; Вспомогательный растворитель=метанол:вода 80:20; Растворитель для промывки иглы=метанол. Методики. Имеется пять способов, применяемых в зависимости от аналитического времени удерживания представляющего интерес соединения. Они выполняют 13,5-минутные хроматографические циклы, состоящие из 10-минутного градиента, с последующей 3,5-минутной промывкой колонки и стадией повторного уравновешивания. Крупный/малый масштаб 1,0-1,5=5-30% В. Крупный/малый масштаб 1,5-2,2=15-55% В. Крупный/малый масштаб 2,2-2,9=30-85% В. Крупный/малый масштаб 2,9-3,6=50-99% В. Крупный/малый масштаб 3,6-5,0=80-99% В (спустя 6 мин за этим следует 7,5-минутная промывка и повторное уравновешивание). Скорость потока. Во всех вышеуказанных методиках скорость потока составляет 20 мл/мин (при малом масштабе) или 40 мл/мин (при крупном масштабе).Bruker DRX600. Программное обеспечение. Пользовательский интерфейс - NMR Kiosk. Управляющее программное обеспечение - XWin NMR версии 3,0. Хроматография. Если не указано иное, всю хроматографию проводили, используя колонки с диоксидом кремния. Сокращения: г - граммы; мг - миллиграммы; мл - миллилитры; мкл - микролитры;MDAP - массовая направленная автоматическая препаративная жидкостная хроматография; нас. - насыщенный.- 12017406 Общий химический раздел. Промежуточные соединения для получения соединений в примерах необязательно получали из конкретных описанных партий. Описание для D1. 5-Цианоиндол (1,00 г), гидроксиламинHCl (978 мг) и NaHCO3 (2,95 г) растворяли/суспендировали в МеОН (14 мл), нагревали до 50 С и перемешивали в течение ночи. ЖХМС-анализ показал, что после этого времени реакция не была завершена и реакционную температуру повышали до 80 С. Реакция завершилась через 4 ч. Реакционную смесь охлаждали до КТ и выпаривали досуха при пониженном давлении. Остаток обрабатывали 1 М водной HCl (50 мл) и экстрагировали EtOAc (250 мл). При этом не удавалось экстрагировать продукт из водного раствора, поэтому его обрабатывали 2 М водным NaOH, доводя рН около 7, после чего повторно экстрагировали EtOAc (350 мл). Объединенные органические растворы промывали насыщенным раствором соли (30 мл), сушили над MgSO4, фильтровали и выпаривали досуха, получая соединение, указанное в заголовке (1,36 г), в виде коричневого масла. Н (MeOD, 400 МГц): 6,50 (1 Н, с), 7,27 (1 Н, с), 7,36-7,45 (2 Н, м), 7,88 (1 Н, с). МС (ES): C9H8N3O вычислено 175; найдено 176 (МН+). Описание для D2. 5-5-[4-Фенил-5-(трифторметил)-2-тиенил]-1,2,4-оксадиазол-3-ил-1 Н-индол (D2)(286 мг), обрабатывали этоксидом натрия (21% по массе в EtOH, 411 мкл) и нагревали до 120 С в микроволновом реакторе в течение 30 мин. ЖХМС-анализ показал, что реакция не была завершена, поэтому микроволновое нагревание продолжали в течение следующих двух периодов по 30 мин. Затем реакционную смесь охлаждали до КТ, гасили Н 2 О (2 мл) и выпаривали досуха при пониженном давлении, получая неочищенный продукт (411 мг) в виде коричневого масла. Неочищенный остаток очищали на картридже 40+S Biotage, элюируя смесью 0-50% Et2O с петролейным эфиром. Получали соединение, указанное в заголовке (122 мг), в виде светло-серого твердого вещества. Н (CDCl3, 400 МГц): 6,68 (1 Н, с), 7,30 (1 Н, т), 7,41-7,55 (6 Н, м), 7,92 (1 Н, с), 7,99 (1 Н, д), 8,36 (1 Н,шир.с), 8,50 (1 Н, с). МС (ES): C21H12F3N3OS вычислено 411; найдено 410 (М-Н+). Описание для D3. Этил-3-(5-5-[4-фенил-5-(трифторметил)-2-тиенил]-1,2,4-оксадиазол-3-ил-1 Н-индол-1 ил)пропаноат (D3) Соединение D2 (100 мг) растворяли в ДМФА (1,2 мл), обрабатывали K2CO3 (50 мг), затем этил-3 бромпропионатом (90 мг) и нагревали при 130 С в течение ночи. После этого времени ЖХМС-анализ показал, что реакция не была завершена, поэтому дополнительно добавляли этил-3-бромпропионат(45 мг) и перемешивание продолжали при 130 С в течение 2 ч. ЖХМС-анализ показал отсутствие изменений, поэтому реакционную смесь выпаривали, затем распределяли между DCM и H2O. Органический слой удаляли, водный раствор экстрагировали DCM. Объединенные органические растворы сушили надMgSO4, фильтровали и выпаривали, получая неочищенный продукт (148 мг). Продукт очищали на картридже с диоксидом кремния (25+S), элюируя смесью 0-30% EtOAc с петролейным эфиром, с получением соединения MF105672-144A3, указанного в заголовке (38 мг), в виде белого твердого вещества. Н (CDCl3, 400 МГц): 1,21 (3 Н, т), 2,85 (2 Н, т), 4,12 (2 Н, кв), 4,50 (2 Н, т), 6,60 (1 Н, д), 7,21 (1 Н, д),7,42-7,52 (6 Н, м), 7,91 (1 Н, с), 8,00 (1 Н, д), 8,46 (1 Н, с). МС (ES+): C26H20F3N3O3S вычислено 511; найдено 512 (МН+). 5-5-[4-Фенил-5-(трифторметил)-2-тиенил]-1,2,4-оксадиазол-3-ил-1 Н-индол (D2) (600 мг), этил-3 бромпропаноат (374 мкл), карбонат цезия (950 мг) и ДМФА нагревали при 140 С в течение 1 ч в микроволновом реакторе. Добавляли дополнительно 1 экв. этил-3-бромпропаноата (187 мкл) и смесь нагревали в течение 30 мин. Реакционную смесь затем выпаривали, растворяли в DCM и фильтровали, получая соединение, указанное в заголовке (650 мг), в виде коричневого твердого вещества. Н (CDCl3, 400 МГц): 1,21 (3 Н, т), 2,85 (2 Н, т), 4,13 (2 Н, кв), 4,50 (2 Н, т), 6,61 (1 Н, д), 7,22 (1 Н, д),7,44-7,52 (6 Н, м), 7,89-7,92 (1 Н, м), 7,98-8,02 (1 Н, м), 8,45-8,46 (1 Н, м). МС (ES): C26H20F3N3O3S вычислено 511; найдено 512 (МН+). Описание для D4. 3-Хлор-4-[(1-метилэтил)окси]бензойная кислота (D4) Пропан-2-ол (2,45 мл) и PPh3 (1,18 г) растворяли в ТГФ (30 мл), охлаждали до 0 С, обрабатывали метил-3-хлор-4-гидроксибензоатом (6,00 г), затем по каплям добавляли DIAD (9,44 мл) и перемешивали при КТ в течение ночи. Реакционную смесь затем выпаривали и очищали на картриджах с диоксидом кремния (4100 г), элюируя смесью 0-40% EtOAc с пентаном, с получением неочищенного продукта(7,00 г) в виде бесцветного масла. Полученное масло растворяли в МеОН (30 мл) и 2 М водном NaOH(30 мл) и перемешивали при КТ в течение выходных дней. Реакционную смесь затем выпаривали и повторно растворяли в Н 2 О. Полученный раствор промывали Et2O, подкисляли до рН 1 и экстрагировалиEt2O. Последние экстракты сушили над MgSO4, фильтровали и выпаривали, получая соединение, указанное в заголовке (4,16 г), в виде белого твердого вещества. Н (MeOD, 400 МГц): 1,37 (6 Н, д), 4,77 (1 Н, септет), 7,12 (1 Н, д), 7,90 (1 Н, д), 7,98 (1 Н, с). МС (ES): C10H11ClO3 вычислено 214; найдено 215 (МН+). Альтернативный синтез. 3-Хлор-4-[(1-метилэтил)окси]бензойная кислота (D4)(19,9 г), затем изопропилбромидом (13,5 мл), полученную смесь нагревали до 70 С и перемешивали в течение ночи. Реакционную смесь затем охлаждали до КТ, выпаривали досуха, повторно растворяли вEtOH, фильтровали и выпаривали еще раз, что давало промежуточный сложный эфир (22,2 г) в виде белого твердого вещества. Полученное соединение было смесью этилового и метилового сложных эфиров; его использовали в неочищенном виде в следующей реакции. Неочищенное промежуточное соединение (22,2 г) растворяли в МеОН (75 мл), обрабатывали 2 М водным NaOH (75 мл), нагревали до 60 С и перемешивали в течение 2 ч. Реакционную смесь затем охлаждали до КТ, выпаривали МеОН и оставшийся водный раствор подкисляли 5 М водной HCl (30 мл). Осадок отфильтровывали и сушили, получая соединение, указанное в заголовке (15,1 г), в виде белого твердого вещества. Н (CDCl3, 400 МГц): 1,42 (6 Н, д), 4,70 (1 Н, септет), 6,97 (1 Н, д), 7,97 (1 Н, д), 8,12 (1 Н, с). МС (ES): C10H11ClO3 вычислено 214; найдено 213 (М-Н+). Описание для D5 MF105672-175A2. 5-(5-3-Хлор-4-[(1-метилэтил)окси]фенил-1,2,4-оксадиазол-3-ил)-1H-индол (D5) Соединение D1 (500 мг), D4 (611 мг) и РуВОР (1,66 г) растворяли в ДМФА и перемешивали в течение ночи. Реакционную смесь затем выпаривали и распределяли между EtOAc и Н 2 О. Органический слой промывали Н 2 О (2), затем насыщенным раствором соли, сушили над MgSO, фильтровали и выпаривали,получая неочищенный продукт. Продукт очищали на картридже с диоксидом кремния, элюируя смесью- 14017406 0-50% Et2O с гексаном, с получением соединения, указанного в заголовке (120 мг), в виде белого твердого вещества. Н (CDCl3, 400 МГц): 1,43 (6 Н, д), 4,69 (1 Н, септет), 6,92 (1 Н, с), 7,04 (1 Н, д), 7,25 (1 Н, с), 7,48 (1 Н,д), 8,00 (1 Н, д), 8,07 (1 Н, д), 8,25 (1 Н, с), 8,39 (1 Н, шир.с), 8,50 (1 Н, с). МС (ES): C19H16ClN3O2 вычислено 353; найдено 354 (МН+). Описание для D5 (альтернативная методика). 5-(5-3-Хлор-4-[(1-метилэтил)окси]фенил-1,2,4-оксадиазол-3-ил)-1H-индол (D5) Смесь 5-цианоиндола (5,00 г), NH2OHHCl (6,11 г) и NaHCO3 (14,77 г) в EtOH (176 мл) нагревали при 70 С в атмосфере Ar в течение ночи и затем при 80 С в течение 2,5 ч. Реакционную смесь затем фильтровали и выпаривали, получая желто-оранжевое твердое вещество (неочищенный продукт D1). Соединение D4 (7,55 г), НОВТ (5,23 г) и EDCl (7,42 г) растворяли в ДМФА (88 мл). Полученную смесь перемешивали в течение 10 мин и затем добавляли указанное выше желто-оранжевое твердое вещество (6,16 г), растворенное в ДМФА (88 мл). Реакционную смесь нагревали до 80 С в течение ночи,затем выпаривали и распределяли между EtOAc и Н 2 О. Разделяли фазы и водный раствор экстрагировали двумя дополнительными порциями EtOAc. Объединенные органические растворы сушили и выпаривали. Часть неочищенного остатка очищали на картридже 40+М Biotage, элюируя смесью 5-30% EtOAc с гексаном. Получали соединение, указанное в заголовке (1,45 г), в виде светло-серого твердого вещества. Н (CDCl3, 400 МГц): 1,45 (6 Н, д), 4,72 (1 Н, септет), 6,66-6,69 (1 Н, м), 7,06 (1 Н, д), 7,29 (1 Н, видимый триплет или дд), 7,50 (1 Н, д), 8,01 (1 Н, дд), 8,08 (1 Н, дд), 8,27 (1 Н, д), 8,49-8,52 (1 Н, м). МС (ES): C19H16ClN3O2 вычислено 353; найдено 354 (МН+). Оставшийся неочищенный остаток растирали с холодным МеОН с получением соединения, указанного в заголовке (3,54 г), в виде светло-серого твердого вещества. МС-данные являются такими, как указано выше. Описание для D6. Этил-3-[5-(5-3-хлор-4-[(1-метилэтил)окси]фенил-1,2,4-оксадиазол-3-ил)-1 Н-индол-1 ил]пропаноат (D6) Соединение D5 (100 мг) растворяли в ДМФА (1,5 мл). К полученному раствору добавляли K2CO3(58 мг) и этил-3-бромпропионат (72 мкл), смесь перемешивали и нагревали до 100 С. Спустя 1 ч с помощью ЖХМС установили, что преобразование прошло только на 5%, поэтому добавляли дополнительные порции K2CO3 (97 мг) и этил-3-бромпропионата (72 мкл). Через 3 ч реакционную смесь выпаривали, затем распределяли между DCM и H2O. Водный слой экстрагировали DCM, затем объединенные растворы в DCM промывали насыщенным раствором соли, сушили над MgSO4, фильтровали и выпаривали, получая неочищенный продукт. Продукт очищали на картридже с диоксидом кремния, элюируя смесью 050% Et2O с петролейным эфиром. Получали соединение, указанное в заголовке (40 мг), в виде белого твердого вещества. Соединение D5 (300 мг) и NCS (113 мг) растворяли в DCM (4,2 мл) и перемешивали в течение ночи при комнатной температуре. Реакционную смесь затем разбавляли DCM и промывали Н 2 О. Водный раствор экстрагировали двумя дополнительными порциями DCM и объединенные органические растворы выпаривали досуха. Неочищенный продукт растирали с метанолом, получая соединение, указанное в заголовке (42 мг), в виде коричневого твердого вещества. Метанол затем выпаривали и полученное коричневое твердое вещество растирали с DCM, получая вторую порцию соединения, указанного в заголовке (205 мг), в виде коричневого твердого вещества. Соединение D2 (200 мг) и NCS (65 мг) растворяли в DCM (5 мл) и перемешивали в течение ночи при комнатной температуре. Реакционную смесь затем распределяли между DCM и H2O. Раствор в DCM выпаривали досуха и очищали на картридже с диоксидом кремния Biotage, элюируя смесью 25-75% диэтилового эфира с гексаном. Получали соединение, указанное в заголовке (36 мг), в виде коричневого твердого вещества. Вторую порцию соединения, указанного в заголовке, также получали при данной очистке (86 мг) в виде коричневого твердого вещества. 4-Цианоиндол (850 мг) растворяли в EtOH (25 мл). К полученному раствору добавляли NaHCO3(2,51 г) и NH2OHHCl (831 мг). Смесь нагревали до 70 С и перемешивали в течение ночи. Реакция была незавершенной, поэтому нагревали при 80 С в течение дополнительных 4 ч. Реакционную смесь фильтровали и выпаривали, получая соединение, указанное в заголовке (980 мг), в виде желтого полутвердого вещества. Очистку не производили. Описание для D9 (альтернативная методика). Смесь 4-цианоиндола (5,0 г, 35,2 ммоль), гидрокарбоната натрия (8,9 г, 105,6 ммоль) и гидрохлорида гидроксиламина (4,9 г, 70,4 ммоль) в этаноле (200 мл) нагревали при 55 С в течение ночи. Добавляли гидрокарбонат натрия (5,9 г, 70 ммоль) и гидрохлорид гидроксиламина (4,9 г, 70,4 ммоль). Смесь нагревали в течение 4 дней, пока не осталось только очень малое количество исходного материла. Неорганические вещества отфильтровывали, тщательно промывая твердое вещество этанолом, и растворитель выпаривали. Остаток растирали с диэтиловым эфиром, получая 5,8 г светло-серого вещества. Н (400 МГц, метанол-d4): 6,76-6,78 (1 Н, м), 7,12 (1 Н, т), 7,24 (1 Н, дд), 7,29-7,33 (1 Н, м), 7,46 (1 Н,дд). Описание для D10. 4-5-[4-Фенил-5-(трифторметил)-2-тиенил]-1,2,4-оксадиазол-3-ил-1 Н-индол (D10)(242 мг) растворяли в ДМФА (3 мл) и перемешивали при комнатной температуре в течение 20 мин. Соединение D9 (200 мг) растворяли в ДМФА (3 мл) и добавляли к полученному выше раствору и перемешивание продолжали при комнатной температуре в течение 2 ч. Реакционную смесь затем нагревали до 90 С, охлаждали до КТ, оставляли стоять в течение ночи, повторно нагревали до 80 С и перемешивали в течение 3 ч, охлаждали до комнатной температуры и выпаривали досуха. Остаток повторно растворяли в Н 2 О, экстрагировали EtOAc (3) и объединенные органические экстракты выпаривали досуха. Остаток очищали флэш-хроматографией на диоксиде кремния, элюируя смесью 25-75% диэтилового эфира с гексаном, с получением соединения, указанного в заголовке (265 мг), в виде коричневого твердого вещества. Образец полученного соединения (100 мг) очищали MDAP, получая соединение, указанное в заголовке (62 мг), в виде светло-серого твердого вещества. 3-Бром-2,2-диметилпропановую кислоту (200 мг) растворяли в EtOH (5 мл) и обрабатывали концентрированной H2SO4 (0,4 мл). Полученную смесь нагревали при кипячении с обратным холодильником в течение ночи, затем выпаривали. Остаток экстрагировали EtOAc из Н 2 О (2) и объединенные органические растворы сушили и выпаривали, получая соединение, указанное в заголовке (316 мг), в виде прозрачного масла. 4-Амино-3-этилбензонитрил (3,0 г, 20,5 ммоль), 1,5-дибромпентан (11,1 мл, 82,1 ммоль), карбонат калия (5,67 г, 41,0 ммоль) и воду (39,6 мл) разделяли поровну по десяти микроволновым флаконам, каждый из которых нагревали при 160 С в течение 1 ч. Все реакционные смеси объединяли и дважды экстрагировали этилацетатом (40 мл), объединенные органические фракции сушили (с разделителем фаз) и концентрировали в вакууме. Добавляли дихлорметан и затем смесь фильтровали, фильтрат очищали хроматографией на диоксиде кремния, элюируя смесью 2-5% этилацетата с гексаном, с получением соединения, указанного в заголовке, в виде бесцветного масла (823 мг, 3,85 ммоль). Анализ показал, что полученное соединение содержало небольшое количество примеси дибромпентана. Н (метанол-d4, 400 МГц): 7,52 (1 Н, дд), 7,47 (1 Н, дд), 7,13 (1 Н, д), 2,89 (4 Н, дд), 2,71 (2 Н, кв), 1,761,71 (4 Н, м), 1,64-1,56 (2 Н, м), 1,25 (3 Н, т). МС (ES): C14H18N2 вычислено 214; найдено 215 (МН+). Описание для D13. 3-Этил-4-(1-пиперидинил)бензойная кислота (D13) 3-Этил-4-(1-пиперидинил)бензонитрил (D12) (817 мг, 3,82 ммоль) и гидроксид калия (2,14 г,38,2 ммоль) в этаноле (35 мл) и воду (8 мл) нагревали до 90 С (температура блока) в течение 9 ч. Дополнительно добавляли гидроксид калия (2,14 г, 38,2 ммоль) и воду (8 мл) и реакционную смесь нагревали в течение следующих 18 ч. Реакционной смеси давали возможность охладиться и нейтрализовали воднойHCl. Белое твердое вещество отделяли фильтрованием и предпринимали попытку очистить фильтрат,используя картридж SCX, но она оказалась неудачной. Объединяли твердое вещество и продукт, собранный с SCX, добавляли метанол и затем смесь подкисляли уксусной кислотой. Смесь фильтровали для получения фильтрата, который затем улавливали на картридже SCX, промывали метанолом и элюировали 2 М аммиаком в метаноле. В экспериментальном масштабе это давало соединение, указанное в заголовке, в виде белого твердого вещества (96 мг, 0,41 ммоль), и оставшийся продукт давал бесцветное масло (563 мг, 2,41 ммоль). Н (метанол-d4, 400 МГц): 7,85 (1 Н, д), 7,74 (1 Н, дд), 7,03 (1 Н, д), 2,85 (4 Н, дд), 2,73 (2 Н, кв), 1,72 Смесь этилового эфира 5,6-дихлорникотиновой кислоты (1,00 г, 4,57 ммоль), пирролидина (325 мг,4,57 ммоль), карбоната калия (632 мг, 4,57 ммоль) и порошка меди (34 мг) в ДМФА (6,8 мл) нагревали при 130 С в микроволновом нагревателе в течение 20 мин. Дополнительно добавляли пирролидин(163 мг, 2,29 ммоль) и реакционную смесь нагревали при 130 С в течение 20 мин. Добавляли воду (7 мл) и смесь экстрагировали этилацетатом (214 мл). Объединенные органические экстракты промывали водой (7 мл) и насыщенным раствором соли (7 мл), затем сушили (с разделителем фаз) и концентрировали в вакууме, получая соединение, указанное в заголовке, в виде масла оранжевого цвета (1,06 г,4,17 ммоль). Н (метанол-d4, 400 МГц): 8,45 (1 Н, д), 7,98 (1 Н, д), 4,31 (2 Н, кв), 3,82-3,75 (4 Н, м), 2,0-1,93 (4 Н, м),1,36 (3 Н, т) м.д. МС (ES): C12H15ClN2O2 вычислено 254, 256; найдено 255, 257 (МН+). Описание для D15. 5-Хлор-6-(1-пирролидинил)-3-пиридинкарбоновая кислота (D15) Этил-5-хлор-6-(1-пирролидинил)-3-пиридинкарбоксилат (D14) (1,06 г, 4,16 ммоль) в этаноле (20 мл) и водный гидроксид натрия (2 М, 2,08 мл, 4,16 ммоль) нагревали при 40 С в течение 18 ч. Реакционной смеси давали возможность охладиться и нейтрализовали 2 М HCl (водн.). Соединение, указанное в заголовке, образовывалось в виде белого твердого вещества, которое отфильтровывали и промывали метанолом, получая соединение SJ 108923-113A3, указанное в заголовке (243 мг, 1,08 ммоль). Фильтрат пропускали через колонку SCX, элюировали 2 М аммиаком в метаноле, получая дополнительное количество соединения, указанного в заголовке, в виде твердого вещества оранжевого цвета (467 мг, 2,07 ммоль). Н (метанол-d4, 400 МГц): 8,55 (1 Н, д), 8,03 (1 Н, д), 3,76-3,70 (4 Н, м), 1,96-1,90 (4 Н, м). МС (ES): C10H11ClN2O2 вычислено 226, 228; найдено 227, 229 (МН+). Описание для D16. 3-Этил-4-йодбензонитрил (D16) К 4-амино-3-этилбензонитрилу (2,50 г, 17,1 ммоль), перемешиваемому в воде (14 мл) при 0 С, по каплям добавляли концентрированную хлористо-водородную кислоту (7,80 мл, 257 ммоль), с последующим добавлением по каплям раствора нитрита натрия (1,24 г, 18,0 ммоль) в воде (3,43 мл). Полученную смесь перемешивали в течение 15 мин и затем в течение 15 мин добавляли раствор йодида калия (2,98 г,18,0 ммоль) в воде (6,0 мл) при 0 С. Смесь перемешивали при комнатной температуре в течение 2 ч. Смесь экстрагировали этилацетатом (3100 мл) и объединенные органические фракции промывали насыщенным раствором соли (100 мл), сушили (с разделителем фаз) и концентрировали в вакууме, получая соединение, указанное в заголовке, в виде коричневого твердого вещества (4,21 г, 16,4 ммоль). Н (метанол-d4, 400 МГц): 8,02 (1 Н, д), 7,61 (1 Н, д), 7,24 (1 Н, дд), 2,80 (2 Н, кв), 1,21 (3 Н, т).MC (ES): Ион с искомой массой не наблюдали. Описание для D17. 4-(1-Циклогексен-1-ил)-3-этилбензонитрил (D17)- 18017406 течение 10 мин в микроволновом нагревателе. Реакционную смесь распределяли между этилацетатом(40 мл) и водой (40 мл), затем органический слой дополнительно промывали водой (40 мл), сушили (с разделителем фаз) и концентрировали в вакууме. Неочищенный продукт очищали хроматографией на диоксиде кремния, элюируя смесью 0-5% EtOAc с гексаном в течение 30 мин, с получением соединения,указанного в заголовке, в виде желтого масла (824 мг, 3,91 ммоль). Н (метанол-d4, 400 МГц): 7,56 (1 Н, д), 7,46 (1 Н, дд), 7,19 (1 Н, д), 5,61-5,56 (1 Н, м), 2,68 (2 Н, квартет), 2,23-2,16 (4 Н, м), 1,85-1,68 (4 Н, м), 1,20 (3 Н, т). МС (ES): Ион с искомой массой не наблюдали. Описание для D18. 4-(1-Циклогексен-1-ил)-3-этилбензойная кислота (D18) 4-(1-Циклогексен-1-ил)-3-этилбензонитрил (D17) (824 мг, 3,91 ммоль) и гидроксид калия (2,19 г,39,1 ммоль) в этаноле (36 мл) и воду (8 мл) нагревали при 90 С (температура блока) в течение 20 ч. Реакционную смесь концентрировали в вакууме и остаток распределяли между этилацетатом (120 мл) и водной хлористо-водородной кислотой (2 М, 50 мл), затем органическую фазу дополнительно промывали хлористо-водородной кислотой (2 М, 50 мл), сушили (с разделителем фаз) и концентрировали в вакууме,получая соединение, указанное в заголовке, в виде желтого масла (808 мг, 3,51 ммоль). Н (метанол-d4, 400 МГц): 7,87 (1 Н д), 7,76 (1 Н дд), 7,11 (1 Н, д), 5,59-5,54 (1 Н, м), 2,68 (2 Н, кв),2,25-2,15 (4 Н, м), 1,84-1,67 (4 Н, м), 1,20 (3 Н, т). ЖХМС (ES): C15H18O2 вычислено 230; найдено 229 (М-Н+). Описание для D19. 4-Циклогексил-3-этилбензойная кислота (D19)(70 мл) и гидрировали в аппарате H-Cube, используя картридж с палладием на углероде. Раствор продукта концентрировали в вакууме, получая соединение, указанное в заголовке, в виде белого твердого вещества (792 мг, 3,41 ммоль). Н (метанол-d4, 400 МГц): 7,82-7,68 (2 Н, м), 7,33 (1 Н, д), 2,83 (1 Н, м), 2,73 (2 Н, кв), 1,87 (2 Н, м),1,85-1,70 (3 Н, м), 1,58-1,30 (5 Н, м), 1,22 (3 Н, т). ЖХМС (ES): Ион с искомой массой не наблюдали. Описание для D20. 1-Метилэтил-3-бром-4-[(1-метилэтил)окси]бензоат (D20) Смесь 3-бром-4-гидроксибензойной кислоты (2,00 г, 9,22 ммоль), 2-йодпропана (1,85 мл,18,4 ммоль) и карбоната калия (2,55 г, 18,4 ммоль) в ДМФА (175 мл) нагревали при кипении с обратным холодильником в течение 5 ч. Реакционной смеси давали возможность охладиться и смесь фильтровали. Фильтрат концентрировали в вакууме и остаток распределяли между этилацетатом (150 мл) и водой(150 мл), которую подщелачивали 2 М раствором NaOH. Органическую фазу сушили (с разделителем фаз) и концентрировали в вакууме, получая соединение, указанное в заголовке, в виде желтого масла(2,36 г, 7,84 ммоль). Н (метанол-d4, 400 МГц): 8,05 (1 Н, д), 7,90 (1 Н, дд), 7,25 (1 Н, д), 5,10 (1 Н, септет), 4,81 (1 Н, септет), 1,32 (6 Н, д), 1,31 (6 Н, д) м.д. МС (ES): Ион с искомой массой не наблюдали.(100 мл) и водный гидроксид натрия (2 М, 39 мл) нагревали при кипении с обратным холодильником в течение 5 ч. Реакционную смесь концентрировали в вакууме и распределяли между этилацетатом(125 мл) и водой (125 мл), последнюю подкисляли 2 М раствором HCl (40 мл). Водный слой экстрагировали дополнительным этилацетатом (70 мл), объединенные органические экстракты сушили (с разделителем фаз) и концентрировали в вакууме, получая соединение, указанное в заголовке, в виде светлосерого твердого вещества (1,83 г, 7,06 ммоль). Н (метанол-d4, 400 МГц): 8,05 (1 Н, д), 7,89 (1 Н, дд), 7,23 (1 Н, д), 4,79 (1 Н, септет), 1,32 (6 Н, д). К суспензии 4-бром-3-хлорбензойной кислоты (5,00 г, 21,2 ммоль) в этаноле (50 мл) добавляли серную кислоту (5 мл) и полученную смесь нагревали при кипении с обратным холодильником в течение 60 ч. Реакционную смесь распределяли между этилацетатом (50 мл) и водой (50 мл). Водный слой экстрагировали дополнительным этилацетатом, объединенные органические фракции сушили (с разделителем фаз) и концентрировали в вакууме, получая соединение, указанное в заголовке, в виде коричневого масла/твердого вещества (5,09 г, 19,3 ммоль). Н (d6-ДМСО, 400 МГц): 8,06 (1 Н, д), 7,96 (1 Н, д), 7,80 (1 Н, дд), 4,33 (2 Н, кв), 1,33 (3 Н, т). МС (ES): Ион с искомой массой не наблюдали. Описание для D23. Этил-3-хлор-4-(2-метилпропил)бензоат (D23) Раствор бромида изобутилцинка в ТГФ (0,5 М, 30 мл, 15,0 ммоль) добавляли в атмосфере аргона к этил-4-бром-3-хлорбензоату (D22) (2,00 г, 7,60 ммоль) и затем добавляли комплекс 1,1'-бис(дифенилфосфино)ферроценпалладий(II)дихлорида с дихлорметаном (930 мг, 1,14 ммоль). Реакционную смесь нагревали при кипении с обратным холодильником в течение 4,5 ч. Смесь концентрировали в вакууме и остаток распределяли между этилацетатом (125 мл) и водой (125 мл). Образовывалось твердое вещество, которое отфильтровывали и отбрасывали. Органический слой промывали водой (100 мл), сушили (с разделителем фаз) и концентрировали в вакууме. Неочищенный продукт очищали хроматографией на диоксиде кремния, элюируя 0-5% EtOAc в гексане в течение 30 мин, с получением соединения,указанного в заголовке, в виде бесцветного масла (1,76 г, 7,33 ммоль). Н (d6-ДМСО, 400 МГц): 7,91 (1 Н, д), 7,80 (1 Н, дд), 7,46 (1 Н, д), 4,30 (2 Н, кв), 2,66 (2 Н, д), 1,88-2,01(1 Н, м), 1,32 (3 Н, т), 0,89 (6 Н, д). МС (ES): Ион с искомой массой не наблюдали. Описание для D24. 3-Хлор-4-(2-метилпропил)бензойная кислота (D24)- 20017406 натрия (2 М, 3,70 мл, 7,4 ммоль) в этаноле (30 мл) нагревали при 40 С в течение 3 ч. Реакционную смесь концентрировали в вакууме и остаток распределяли между этилацетатом (100 мл) и водой (100 мл), которую подкисляли 2 М HCl (4 мл). Водный слой экстрагировали этилацетатом (100 мл), объединенные органические экстракты сушили (с разделителем фаз) и концентрировали в вакууме, получая соединение,указанное в заголовке, в виде белого твердого вещества (1,35 г, 6,36 ммоль). Н (d6-ДМСО, 400 МГц): 13,20 (1 Н, шир.с), 7,89 (1 Н, д), 7,82 (1 Н, дд), 7,44 (1 Н, д), 2,64 (2 Н, д), 1,94 К раствору метил-3-циано-4-гидроксибензоата (3 г, 16,93 ммоль) и триэтиламина (3,54 мл,25,4 ммоль) в сухом дихлорметане (60 мл) при 0 С в токе аргона медленно по каплям добавляли трифторметансульфоновый ангидрид (3,15 мл, 18,63 ммоль). Реакционной смеси давали возможность нагреться до комнатной температуры и полученную смесь перемешивали в течение 1 ч. Реакционную смесь промывали 10% водным карбонатом калия (250 мл) и затем водной HCl (2 М, 250 мл), затем органическую фазу сушили (с разделителем фаз) и растворитель удаляли в вакууме, получая соединение,указанное в заголовке, в виде темно-коричневого масла (5,165 г, 16,70 ммоль). Н (CDCl3, 400 МГц): 8,44 (1 Н, д), 8,38 (1 Н, дд), 7,60 (1 Н, д), 3,99 (3 Н, с). МС (ES): Ион с искомой массой не наблюдали. Описание для D26. Метил 2-циано-4-бифенилкарбоксилат (D26) Следующую реакционную смесь разделяли на две порции с половинными количествами: метил-3 циано-4-[(трифторметил)сульфонил]оксибензоат (D25) (1,5 г, 4,85 ммоль), фенилбороновую кислоту(0,561 г, 0,485 ммоль) растворяли в ДМФА (24 мл) и смесь нагревали в микроволновом нагревателе в течение 30 мин при 150 С. Объединяли две реакционные смеси, разбавляли этилацетатом (50 мл) и смесь фильтровали через кизельгур для удаления остатков палладия. Фильтрат концентрировали в вакууме для уменьшения количества ДМФА и затем остаток распределяли между насыщенным водным бикарбонатом натрия (50 мл) и этилацетатом (50 мл). Органическую фазу промывали дополнительным бикарбонатом натрия (50 мл) и затем водой (50 мл), затем сушили (MgSO4), фильтровали и растворитель удаляли в вакууме. Коричневое твердое вещество очищали хроматографией на диоксиде кремния, элюируя 0-25%EtOAc в изогексане в течение 35 мин, с получением соединения, указанного в заголовке, в виде белого твердого вещества (935 мг, 3,94 ммоль). Н (d6-ДМСО, 400 МГц): 8,42 (1 Н, д), 8,29 (1 Н, дд), 7,81 (1 Н, д), 7,65 (2 Н, м), 7,60-7,50 (3 Н, м), 3,92(3 Н, с). МС (ES): Ион с искомой массой не наблюдали. Описание для D27. 2-Циано-4-бифенилкарбоновая кислота (D27) К метил-2-циано-4-бифенилкарбоксилату (D26) (935 мг, 3,94 ммоль) добавляли этанол (18 мл), но растворение не происходило, поэтому добавляли дихлорметан (10 мл). Затем добавляли гидроксид на- 21017406 трия (2 мл, 4,00 ммоль) и реакционную смесь перемешивали в течение 2 ч. К смеси добавляли дихлорметан (20 мл) и 2 М водную HCl (10 мл). Разделяли слои и водный слой экстрагировали дополнительным дихлорметаном (20 мл). Объединенную органическую фазу сушили (с разделителем фаз) и растворитель удаляли в вакууме,получая белое твердое вещество, которое растворяли в метаноле (30 мл), и добавляли водный гидроксид натрия (2 М, 3 мл). Реакционную смесь перемешивали при комнатной температуре в течение 1 ч, затем добавляли воду (20 мл). Реакционную смесь перемешивали в течение одного следующего часа. Добавляли дихлорметан (60 мл), смесь встряхивали и разделяли слои. Водную фазу экстрагировали дополнительным дихлорметаном (50 мл), затем объединенную органическую фазу сушили (с разделителем фаз) и растворитель удаляли в вакууме, получая соединение, указанное в заголовке, в виде белого твердого вещества (849 мг, 3,80 ммоль). Н (d6-ДМСО, 400 МГц): 13,60 (1 Н, шир.с), 8,38 (1 Н, д), 8,28 (1 Н, дд), 7,78 (1 Н, д), 7,63 (2 Н, м),7,60-7,50 (3 Н, м). 4-Хлор-3-(трифторметил)бензойную кислоту (1 г, 4,45 ммоль) растворяли в этаноле (3 мл) и добавляли концентрированную серную кислоту (0,15 мл). Смесь нагревали в микроволновом нагревателе при 100 С в течение 5 мин и затем при 120 С в течение 15 мин. Растворитель удаляли в вакууме и остаток распределяли между насыщенным водным бикарбонатом натрия (50 мл) и этилацетатом (50 мл). Водный слой экстрагировали дополнительным EtOAc (50 мл), органические фазы объединяли, сушили с разделителем фаз и концентрировали в вакууме, получая соединение, указанное в заголовке (1,026 г) (DN108121148A3), в виде бесцветного масла. Н (метанол-d4, 400 МГц): 1,40 (3 Н, т), 4,41 (2 Н, кв), 7,76 (1 Н, д), 8,21 (1 Н, дд), 8,33 (1 Н, д). МС (ES): Ион с искомой массой не наблюдали. Описание для D29. 2-(Трифторметил)-4-бифенилкарбоновая кислота (D29) Реакционную смесь разделяли на 4 части, используя четверть реагентов в каждой из них: к смеси 4 бром-3-(трифторметил)бензонитрила (4 г, 16,00 ммоль), фенилбороновой кислоты (3,90 г, 32,0 ммоль) и карбоната калия (6,63 г, 48,0 ммоль) в N,N-диметилформамиде (ДМФА) (64 мл) добавляли тетракистрифенилфосфинпалладий(0) (1,849 г, 1,600 ммоль). Каждую реакционную смесь нагревали в микроволновом нагревателе при 150 С в течение 30 мин. Объединенные реакционные смеси фильтровали через целит, промывали этилацетатом и растворитель удаляли в вакууме. Остаток распределяли между этилацетатом (100 мл) и водой (100 мл) и органическую фазу промывали раствором бикарбоната натрия(100 мл). Органическую фазу сушили (MgSO4), фильтровали и растворитель удаляли в вакууме. Коричневое масло растирали с дихлорметаном и фильтровали, получая бледно-желтое твердое вещество, 2(трифторметил)-4-бифенилкарбоксамид (2,47 г), который использовали без дальнейшей очистки. К 2(трифторметил)-4-бифенилкарбоксамиду (2 г, 7,54 ммоль) в этаноле (80 мл) добавляли гидроксид калия(4,23 г, 75 ммоль) и воду и смесь нагревали до 90 С в течение 18 ч. Реакционную смесь концентрировали в вакууме и остаток распределяли между дихлорметаном (100 мл) и 2 М HCl (100 мл). Органическую фазу отделяли, сушили (с разделителем фаз) и растворитель удаляли в вакууме, получая неочищенный продукт. Очистку проводили, используя картридж с обращенной фазой Biotage Horizon, элюируя 5-100%(689 мг, 11,9 ммоль) в ТГФ (8 мл) нагревали в микроволновом нагревателе при 120 С в общей сложности в течение 40 мин. Порция В. Смесь D28 (500 мг, 1,98 ммоль), фенилбороновой кислоты (290 мг, 2,38 ммоль), ацетата палладия (2,2 мг), (дициклогексилфосфино)бифенила (7 мг) и фторида калия (344 мг, 5,8 ммоль) в ТГФ(4 мл) нагревали в микроволновом нагревателе при 120 С в течение 20 мин. Реакционные смеси из порций А и В объединяли, фильтровали и фильтрат концентрировали в вакууме. Остаток очищали флэш-хроматографией (0-5% EtOAc в гексане), получая смесь исходного продукта и продукта конденсации. Полученный продукт растворяли в этаноле (10 мл) и 2 М NaOH (водн.)(5 мл) и затем нагревали при кипении с обратным холодильником в течение 3 ч. Растворитель удаляли в вакууме и остаток распределяли между DCM и 2 М водн. HCl. Водную фазу экстрагировали дополнительным DCM. Органические фазы объединяли и концентрировали в вакууме. Неочищенный продукт очищали хроматографией с обращенной фазой на картридже Horizon, элюируя 5-100% MeCN в воде, с получением соединения, указанного в заголовке, в виде белого твердого вещества (367 мг). Указанное соединение получали, применяя способ, подобно описанному для соединения D29, используя (2-фторфенил)бороновую кислоту и соединение D98, с тем исключением, что проводили только одну реакцию конденсации, подобно описанной для порции А, и нагревали в течение 20 мин. МС (ES): C14H8F4O2 вычислено 284; найдено 283 (М-Н+). Описание для D31. Метил-3-циано-4-(2-метилпропил)бензоат (D31) К метил-3-циано-4-[(трифторметил)сульфонил]оксибензоату (D25) (1,5 г, 4,85 ммоль) добавляли бром(2-метилпропил)цинк (48,5 мл, 24,25 ммоль) в тетрагидрофуране (50 мл) в атмосфере аргона. К раствору затем добавляли комплекс 1,1'-бис-(дифенилфосфино)ферроцендихлорпалладия(II) и дихлорметана (0,355 г, 0,485 ммоль) и реакционную смесь нагревали при кипении с обратным холодильником в течение 6 ч. Смесь гасили водой (2 мл) и затем фильтровали через целит, промывая этилацетатом. Растворитель удаляли в вакууме. Остаток распределяли между этилацетатом (50 мл) и водой (50 мл), органическую фазу сушили (с разделителем фаз) и растворитель удаляли в вакууме. Остаток очищали хроматографией на диоксиде кремния, элюируя 0-15% EtOAc в изогексане в течение 40 мин. Собирали две порции, одна из которых была соединением, указанным в заголовке, в виде бесцветного масла (233 мг,1,072 ммоль). Н (CDCl3, 400 МГц): 8,28 (1 Н, д), 8,15 (1 Н, дд), 7,38 (1 Н, д), 3,94 (3 Н, с), 2,78 (2 Н, д), 2,02 (1 Н, м),0,96 (6 Н, д). Метил-3-циано-4-(2-метилпропил)бензоат (D31) (233 мг, 1,072 ммоль) растворяли в этаноле (4 мл) и добавляли 2 М водный гидроксид натрия (1 мл, 2 ммоль). Реакционную смесь перемешивали в течение 1 ч. Добавляли 2 М водную HCl (10 мл) и смесь экстрагировали дихлорметаном (20 мл+10 мл). Органические фазы отделяли, сушили разделителем фаз и объединяли, затем растворитель удаляли в вакууме,получая соединение, указанное в заголовке, в виде белого твердого вещества (203 мг, 0,999 ммоль). Н (d6-ДМСО, 400 МГц) 13,43 (1 Н, шир.с), 8,29 (1 Н, д), 8,14 (1 Н, дд), 7,59 (1 Н, д), 2,74 (2 Н, д), 1,96(25 ммоль) в ТГФ (50 мл, 25 ммоль) в атмосфере аргона добавляли комплекс 1,1'-бис(дифенилфосфино)ферроцендихлорпалладия(II) и дихлорметана (612 мг, 0,75 ммоль) и реакционную смесь нагревали при кипячении с обратным холодильником в течение 5 ч. Смесь концентрировали в вакууме и остаток распределяли между этилацетатом (80 мл) и водой (80 мл). Образовавшееся твердое вещество отфильтровывали и отбрасывали. Органический слой промывали водой (80 мл), затем сушили (с разделителем фаз) и концентрировали в вакууме, получая неочищенное соединение, указанное в заголовке, в виде черного масла. Его использовали непосредственно на следующей стадии (1,35 г). Описание для D34. 4-(2-Метилпропил)-3-(трифторметил)бензойная кислота (D34) 4-(2-Метилпропил)-3-(трифторметил)бензамид (D33) (1,35 г, 5,50 ммоль) растворяли вместе с гидроксидом калия (3,09 г, 55,0 ммоль) в этаноле (40 мл) и воде (10,0 мл) и раствор нагревали при кипении с обратным холодильником в течение 18 ч. Реакционную смесь концентрировали в вакууме и смесь распределяли между EtOAc (150 мл) и водным гидроксидом натрия (2 М, 150 мл). Разделяли слои и органическую фазу экстрагировали дополнительным раствором гидроксида натрия (200 мл). ЖХМС-анализ показал наличие продукта в обеих фазах. Поэтому водную фазу подкисляли до рН 1 добавлением HCl(5 м) и обратно экстрагировали в EtOAc (2150 мл), полученные органические фазы объединяли с исходной органической фазой. Растворитель удаляли в вакууме и остаток очищали хроматографией с обращенной фазой, элюируя 5-100% MeCN в H2O объемом 2000 мл; растворитель удаляли в вакууме, с получением коричневого твердого вещества (690 мг, 2,410 ммоль). Полученное твердое вещество растирали с гексаном, получая соединение, указанное в заголовке, в виде темно-желтого твердого вещества(135 мг, 0,548 ммоль), и фильтрат очищали MDAP, получая дополнительное количество соединения, указанного в заголовке, в виде белого твердого вещества (102 мг, 0,414 ммоль).(2S)-2-Бутанол (0,99 г, 0,013 моль) растворяли в ДМФА (50 мл) и раствор охлаждали до 0 С. К полученному раствору порциями добавляли гидрид натрия (60%-ная дисперсия в минеральном масле,1,54 г, 0,036 моль), смесь перемешивали при 0 С в течение 10 мин после завершения добавления. Затем добавляли 2-фтор-5-формилбензонитрил (2,0 г, 0,013 моль) и реакционной смеси давали возможность нагреваться до комнатной температуры (медленно, на ледяной бане) и реакционную смесь перемешивали в течение ночи при комнатной температуре. Затем реакционную смесь охлаждали до 0 С, гасили насыщенным раствором соли и разбавляли EtOAc (25 мл). Смесь разделяли и органическую фракцию экстрагировали водой (30 мл); объединенные органические фракции сушили путем пропускания через фазоделительный картридж и затем выпаривали досуха при пониженном давлении, получая неочищенный продукт. Неочищенный остаток очищали на картридже 40+М Biotage, элюируя смесью 20-50% EtOAc в гексане. Получали соединение, указанное в заголовке (220 мг), в виде белого твердого вещества. Н (d6-ДМСО, 400 МГц): 9,88 (1 Н, с), 8,30 (1 Н, с), 8,15 (1 Н, д), 7,49 (1 Н, д), 4,73-4,81 (1 Н, м), 1,631,79 (2 Н, м), 1,33 (3 Н, д), 0,95 (3 Н, т). К раствору 5-формил-2-[(1S)-1-метилпропил]оксибензонитрила (D35) (220 мг, 1,08 ммоль) в уксусной кислоте (20 мл) добавляли тетрагидрат пербората натрия (334 мг, 2,17 ммоль), реакционную смесь нагревали при 50 С в течение выходных дней. Реакционную смесь концентрировали в вакууме. Добавляли воду (50 мл), добавляли EtOAc (30 мл) и разделяли слои; водный слой еще два раза экстрагировали EtOAc (30 мл) и объединенные органические фазы выпаривали досуха при пониженном давлении, получая соединение, указанное в заголовке (245 мг), в виде светло-серого твердого вещества. Н (d6-ДМСО, 400 МГц): 8,17 (2 Н, видимый д), 7,39 (1 Н, с), 4,68-4,74 (1 Н, м), 1,55-1,76 (2 Н, м), 1,31(2R)-2-Бутанол (0,99 г, 0,013 моль) растворяли в ДМФА (50 мл) и раствор охлаждали до 0 С. К полученному раствору порциями добавляли гидрид натрия, 60%-ную дисперсию в минеральном масле(1,54 г, 0,036 моль), смесь перемешивали при 0 С в течение 10 мин после завершения добавления. Затем добавляли 2-фтор-5-формилбензонитрил (2,0 г, 0,013 моль) и реакционной смеси давали возможность нагреваться до комнатной температуры (медленно, на ледяной бане), реакционную смесь перемешивали в течение ночи при комнатной температуре. Затем реакционную смесь охлаждали до 0 С, гасили насыщенным раствором соли и разбавляли EtOAc (25 мл). Смесь разделяли и органическую фракцию экстрагировали водой (30 мл); объединенные органические фракции сушили путем пропускания через фазоделительный картридж и выпаривали досуха при пониженном давлении, получая неочищенный продукт. Неочищенный остаток очищали на картридже 40+М Biotage, элюируя смесью 20-50% EtOAc с гексаном. Получали соединение, указанное в заголовке (310 мг), в виде желтого масла. Н (d6-ДМСО, 400 МГц): 9,88 (1 Н, с), 8,30 (1 Н, с), 8,15 (1 Н, д), 7,49 (1 Н, д), 4,73-4,81 (1 Н, м), 1,631,79 (2 Н, м), 1,33 (3 Н, д), 0,95 (3 Н, т) м.д. Описание для D38. 3-Циано-4-[(1R)-1-метилпропил]оксибензойная кислота (D38) К раствору 5-формил-2-[(1R)-1-метилпропил]оксибензонитрила (D37) (310 мг, 1,53 ммоль) в ук- 25017406 сусной кислоте (30 мл) добавляли тетрагидрат пербората натрия (471 мг, 3,05 ммоль), реакционную смесь нагревали при 50 С в течение выходных дней. Реакционную смесь концентрировали в вакууме и добавляли воду (50 мл), добавляли EtOAc (30 мл) и разделяли слои; водный слой еще два раза экстрагировали EtOAc (30 мл) и объединенные органические фазы выпаривали досуха при пониженном давлении, получая соединение, указанное в заголовке (315 мг), в виде светло-серого твердого вещества. Н (d6-ДМСО, 400 МГц): 8,07-8,24 (2 Н, м), 7,38 (1 Н, д), 4,63-4,77 (1 Н, м), 1,55-1,83 (2 Н, м), 1,31 (3 Н,д), 0,95 (3 Н, т). МС (ES+): C12H13NO2 вычислено 219; найдено 220 (МН+). Описание для D39. Метил-6-(метилокси)-3-бифенилкарбоксилат (D39)DME с 2 н. Na2CO3 (2:1, 18 мл) и затем добавляли фенилбороновую кислоту (244 мг) и тетракистрифенилфосфинпалладий(0) (58 мг). Реакционную смесь нагревали до 80 С и затем оставляли охлаждаться в течение выходных дней. Добавляли EtOAc и воду, отделяли органическую фазу, сушили и выпаривали,получая черное смолообразное вещество. Очистка флэш-хроматографией давала соединение, указанное в заголовке (194 мг), в виде смолообразного вещества. Н (d6-ДМСО, 400 МГц): 3,83 (3 Н, с), 3,85 (3 Н, с), 7,24 (1 Н, д), 7,33-7,50 (5 Н, м), 7,83 (1 Н, д), 7,97NaOH (3 мл) и метаноле (3 мл). Перемешивали при комнатной температуре в течение ночи и затем органический растворитель выпаривали в вакууме. Добавляли EtOAc с водой, разделяли, затем подкисляли водный слой и повторно экстрагировали. Органические экстракты сушили и выпаривали, получая 202 мг соединения, указанного в заголовке, в виде белого твердого вещества. Н (6-ДМСО, 400 МГц): 3,84 (3 Н, с), 7,22 (1 Н, д), 7,33-7,49 (5 Н, м), 7,82 (1 Н, д), 7,95 (1 Н, дд). МС (ES+): C14H12O3 вычислено 228; найдено 229 (М+Н+). Описание для D41. 3-Бром-5-(5-3-хлор-4-[(1-метилэтил)окси]фенил-1,2,4-оксадиазол-3-ил)-1 Н-индол (D41) К 5-(5-3-хлор-4-[(1-метилэтил)окси]фенил-1,2,4-оксадиазол-3-ил)-1 Н-индолу (D5) (450 мг, 1,27 ммоль), растворенному в ДМФА (12 мл), добавляли по каплям бром (213 мг, 1,35 ммоль). Перемешивали в течение 15 мин, выпаривали ДМФА, добавляли диэтиловый эфир (70 мл) и промывали водой(270 мл). Сушили над MgSO4 и выпаривали растворитель. Остаток кристаллизовали из смеси диэтилового эфира с гексаном, получая 160 мг соединения, указанного в заголовке, в виде белого твердого вещества.(1 Н, дд), 8,13-8,32 (3 Н, м). МС (ES) C19H1579BrClN3O2 вычислено 431; найдено 432 (МН+). Следующие сложные эфиры получали способом, подобно описанному в предыдущих примерах (таких как для соединения D6), используя соответствующий индол и алкилирующий агент. Алкилгалогениды доступны коммерчески (кроме соединения D11, используемого для получения соединения D48). Если не указано иное, реакции проводили в ДМФА. В некоторых случаях реакционные смеси промывали водными растворами, в остальных случаях неочищенный продукт использовали непосредственно на стадии гидролиза после выпаривания реакционного растворителя.(367 мг, 2,67 ммоль) и раствор оставляли стоять в течение 30 мин. Добавляли N-гидрокси-1 Н-индол-4 карбоксимидамид (D9) (427 мг, 2,44 ммоль) и оставляли стоять в течение 1 ч. К раствору добавляли(100 мг, 0,29 ммоль), этил-4-бромбутирата (85 мг, 0,44 ммоль) и карбоната цезия (189 мг, 0,58 ммоль) в ДМФА (2 мл) нагревали при 80 С в течение 1 ч. Добавляли этил-4-бромбутират (85 мг, 0,44 ммоль) и нагревали в течение ночи при 80 С. Добавляли этил-4-бромбутират (85 мг, 0,44 ммоль) и карбонат цезия(189 мг, 0,58 ммоль) и нагревали в течение 24 ч. Добавляли этил-4-бромбутират (85 мг, 0,44 ммоль) и нагревали в течение 24 ч. Добавляли этил-4-бромбутират (85 мг, 0,44 ммоль) и нагревали в течение 6 ч. Добавляли EtOAc (20 мл) и промывали водой (20 мл). Сушили над MgSO4 и выпаривали растворитель. Остаток кристаллизовали из этанола, получая соединение, указанное в заголовке (55 мг), в виде белого твердого вещества. МС (ES): C26H26N4O4 вычислено 458; найдено 459 (МН+). Описание 51. Этил-3-(5-циано-1 Н-индол-1-ил)пропаноат (D51) Смесь 1 Н-индол-5-карбонитрила (1,42 г, 10 ммоль), этил-3-бромпропаноата (1,92 мл, 15 ммоль) и карбоната цезия (6,5 г, 20 ммоль) в ДМФА (50 мл) нагревали при 80 С в течение 4 ч. Раствор охлаждали,добавляли диэтиловый эфир (300 мл) и промывали водой (3300 мл). Сушили над MgSO4 и выпаривали растворитель, получая 2,4 г масла бледно-оранжевого цвета. Неочищенный продукт использовали на следующей стадии (получение D52). Описание для D52. Этил-3-5-[(гидроксиамино)(имино)метил]-1H-индол-1-илпропаноат(1,0 г, 14,4 ммоль) и гидрокарбонат натрия (2,42 г, 28,9 ммоль) суспендировали в этаноле (100 мл) и перемешивали при 50 С в течение 3 дней. Образовывался единственный продукт, но оставалось 15% исходного вещества. Смесь охлаждали, отфильтровывали неорганические вещества и выпаривали растворитель. Продукт кристаллизовали из смеси EtOAc, диэтилового эфира и гексана, получая 1,9 г соединения, указанного в заголовке, в виде белого твердого вещества. МС (ES): C14H17N3O3 вычислено 275; найдено 276 (МН+). Описание для D53. Этил-3-[5-(5-3-циано-4-[(1-метилэтил)окси]фенил-1,2,4-оксадиазол-3-ил)-1 Н-индол-1 ил]пропаноат (D53)WO 2005/58848) (215 мг, 1,05 ммоль), EDAC (219 мг, 1,14 ммоль) и HOBt (156 мг, 1,14 ммоль) в сухом ДМФА (10 мл) перемешивали при КТ в течение 10 мин. Добавляли этил-3-5[(гидроксиамино)(имино)метил]-1 Н-индол-1-илпропаноат (D52) (288 мг, 1,05 ммоль) и перемешивали в течение 1 ч при КТ. Нагревали при 80 С в течение 7 ч. Раствор охлаждали и добавляли EtOAc (50 мл). Промывали водой (50 мл), насыщенным гидрокарбонатом натрия (50 мл) и водой (50 мл). Сушили надMgSO4 и выпаривали растворитель. Остаток кристаллизовали из эфира, получая 200 мг соединения, указанного в заголовке, в виде бледно-розового твердого вещества. МС (ES): C25H24N4O4 вычислено 444; найдено 445 (МН+)
МПК / Метки
МПК: A61P 37/00, C07D 413/14, A61K 31/405, C07D 413/04
Метки: производные, рецептора, индола, качестве, агонистов
Код ссылки
<a href="https://eas.patents.su/30-17406-proizvodnye-indola-v-kachestve-agonistov-receptora-s1p1.html" rel="bookmark" title="База патентов Евразийского Союза">Производные индола в качестве агонистов рецептора s1p1</a>
Производные индола, полезные в качестве антагонистов рецептора эндотелина.

Номер патента: 1471
Опубликовано: 23.04.2001
Авторы: Дэк Кевин Нил, Росон Дейвид Джеймс, Дикинсон Роджер Питер, Джеймс Ким
МПК: A61P 9/10, A61K 31/405, C07D 209/18...
Метки: производные, эндотелина, антагонистов, качестве, индола, полезные, рецептора
Формула / Реферат:
1. Соединение формулы I где R1 и R2 являются возможными заместителями и независимо представляют собой C1-6алкил, С3-6циклоалкил, С2-6алкенил [возможно замещенные СО2Н, СO2(С1-6алкилом) или СО2(С3-6циклоалкилом)], С2-6алкинил, галоген, С1-3перфторалкил, С3перфторциклоалкил, (CH2)mAr1, (СН2)mНеt1, (CH2)mCONR7R8, (CH2)mCO2R8, O(CH2)qCO2R8, (CH2)mCOR8, (CH2)mOR8, O(CH2)pOR8, (CH2)mNR7R8, CO2(CH2)qNR7R8, (CH2)mCN, S(O)nR8, SO2NR7R8, CONH(CH2)mAr1...
Производные оксииндола в качестве агонистов 5-ht4 рецептора

Номер патента: 12615
Опубликовано: 30.10.2009
Авторы: Кавамура Кийоси, Соне Хироки, Утида Тикара
МПК: A61P 1/04, A61K 31/454, C07D 401/12...
Метки: производные, агонистов, качестве, 5-ht4, оксииндола, рецептора
Формула / Реферат:
1. Соединение формулы (I) где А представляет собой C1-C2-алкиленовую группу, указанная алкиленовая группа является незамещенной или замещенной 1-4 заместителями, независимо выбираемыми из группы, состоящей из C1-C4-алкильной группы, гидроксизамещенной C1-C4-алкильной группой и C1-C2-алкоксизамещенной C1-C4-алкильной группой, где 2 из указанных заместителей могут необязательно образовывать мостиковую связь, образуя 3-6-членное кольцо; R1...
Производные феноксиуксусных кислот, применимые в качестве двойных агонистов активируемого пероксисомным пролифератором рецептора ( ppar )

Номер патента: 9804
Опубликовано: 28.04.2008
Авторы: Ван Айхуа, Шэнь Лань, Чжан Янь, Ко Джи-Хун
МПК: C07D 285/08, A61P 31/10, A61K 31/433...
Метки: активируемого, качестве, применимые, феноксиуксусных, пероксисомным, агонистов, производные, пролифератором, рецептора, кислот, двойных
Формула / Реферат:
1. Соединение формулы (I) где m равно 1, 2 или 3; n равно 0 или 1; X означает S или О; Y означает S, СН2 или О; R1 и R2 независимо выбраны из Н, C1-4алкила, C1-3алкокси, галогена и -NRaRb, где каждый из Ra и Rb независимо выбран из Н и C1-4 алкила; каждый из R3 и R4 независимо выбран из Н, галогена, циано, C1-4алкила, C1-3алкокси и NRcRd, где каждый из Rc и Rd независимо выбран из Н и C1-4алкила и где по меньшей мере один из R3 и R4 не Н; и...
Производные индола как антагонисты рецептора 5-нт

Номер патента: 304
Опубликовано: 29.04.1999
Авторы: Дакворт Дэвид Малькольм, Вайман Пол Эдриан, Дэвис Дэвид Томас, Форбес Ян Томсон, Гэстер Лэрэми Мэри, Джоунз Грэхэм Элджин, Малхоллэнд Кит Раймонд
МПК: C07D 401/12, A61K 31/44
Метки: индола, рецептора, 5-нт, антагонисты, производные
Формула / Реферат:
1. Соединение формулы (I) или его соль где Р1 и Р2 независимо представляет собой фенил, ароматические или частично насыщенные моноциклические или бициклические гетероциклические кольца, содержащие до трех гетероатомов, выбранных из азота, кислорода или серы; А представляет собой связь, цепь из 1-5 атомов, необязательно замещенных С1-6алкилом, или А представляет собой необязательно замещенный фенил или необязательно замещенное 5-7-членное...
Соединения пиперазинилпиразинов в качестве агонистов или антагонистов серотонин 5-ht2 рецептора

Номер патента: 6552
Опубликовано: 24.02.2006
Автор: Нильссон Бьерн М.
МПК: A61K 31/445, A61K 31/4427, A61K 31/497...
Метки: качестве, рецептора, 5-ht2, серотонин, антагонистов, пиперазинилпиразинов, агонистов, соединения
Формула / Реферат:
1. Соединение общей формулы (I) в которой: (i) X и Y оба являются азотом и Z является CH, образуя производное пиразина, или (ii) X и Z оба являются CH и Y является азотом, образуя производное пиридина, или (iii) X является C-CF3, Z является CH и Y является азотом, образуя производное 4-трифторметилпиридина, или (iv) Y и Z оба являются азотом и X является CH, образуя производное пиримидина, и где R1 и R2, каждый независимо, выбирают из группы A,...
Предыдущий патент: Соединения и композиции в качестве ингибиторов протеинкиназы
Следующий патент: Новое терапевтическое применение 4-[2-(4-метилфенилсульфанил)фенил]пиперидина
Случайный патент: Катализатор дегидрирования и способ получения носителя катализатора дегидрирования