Ингибиторы вируса гепатита с
Номер патента: 15827
Опубликовано: 30.12.2011
Авторы: Скола Пол Майкл, Д`андреа Стэнли, Чжао Цянь, Чжэн Барбара Чжичжэнь, Ван Алан Сяндун























Формула / Реферат
1. Соединение формулы (I)

или его фармацевтически приемлемая соль, где
n равно 1;
R1 выбран из гидрокси и -NHSO2R7;
R2 представляет собой С2алкил, С2алкенил или дифторметил;
R3 представляет собой 4-бифенил-4-ил;
R4 представляет собой -OR8;
R5 представляет собой С3-4алкил, С7алкенил или 4-трет-бутилбензил;
R6 представляет собой трет-бутоксикарбонил, возможно замещенный галогеном, трет-бутиламинокарбонил, адамантан-1-илоксикарбонил, адамантан-1-иламинокарбонил, фенил, возможно замещенный 1-2 заместителями, независимо выбираемыми из галогена и метокси;
R7 представляет собой циклопропил;
R8 представляет собой метокси или бензилокси.
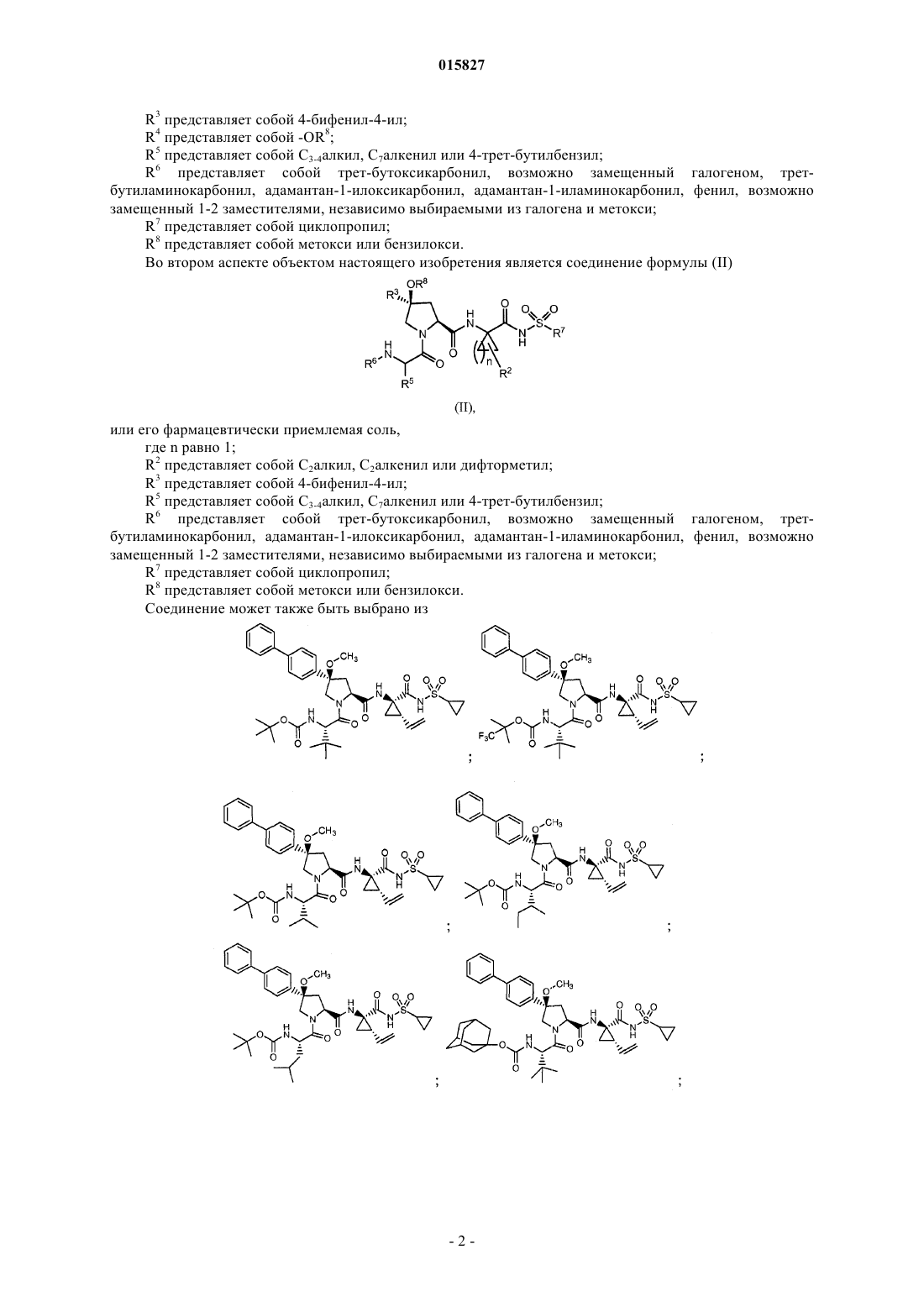
2. Соединение формулы (II)

или его фармацевтически приемлемая соль,
где n равно 1;
R2 представляет собой С2алкил, С2алкенил или дифторметил;
R3 представляет собой 4-бифенил-4-ил;
R5 представляет собой С3-4алкил, С7алкенил или 4-трет-бутилбензил;
R6 представляет собой трет-бутоксикарбонил, возможно замещенный галогеном, трет-бутиламинокарбонил, адамантан-1-илоксикарбонил, адамантан-1-иламинокарбонил, фенил, возможно замещенный 1-2 заместителями, независимо выбираемыми из галогена и метокси;
R7 представляет собой циклопропил;
R8 представляет собой метокси или бензилокси.
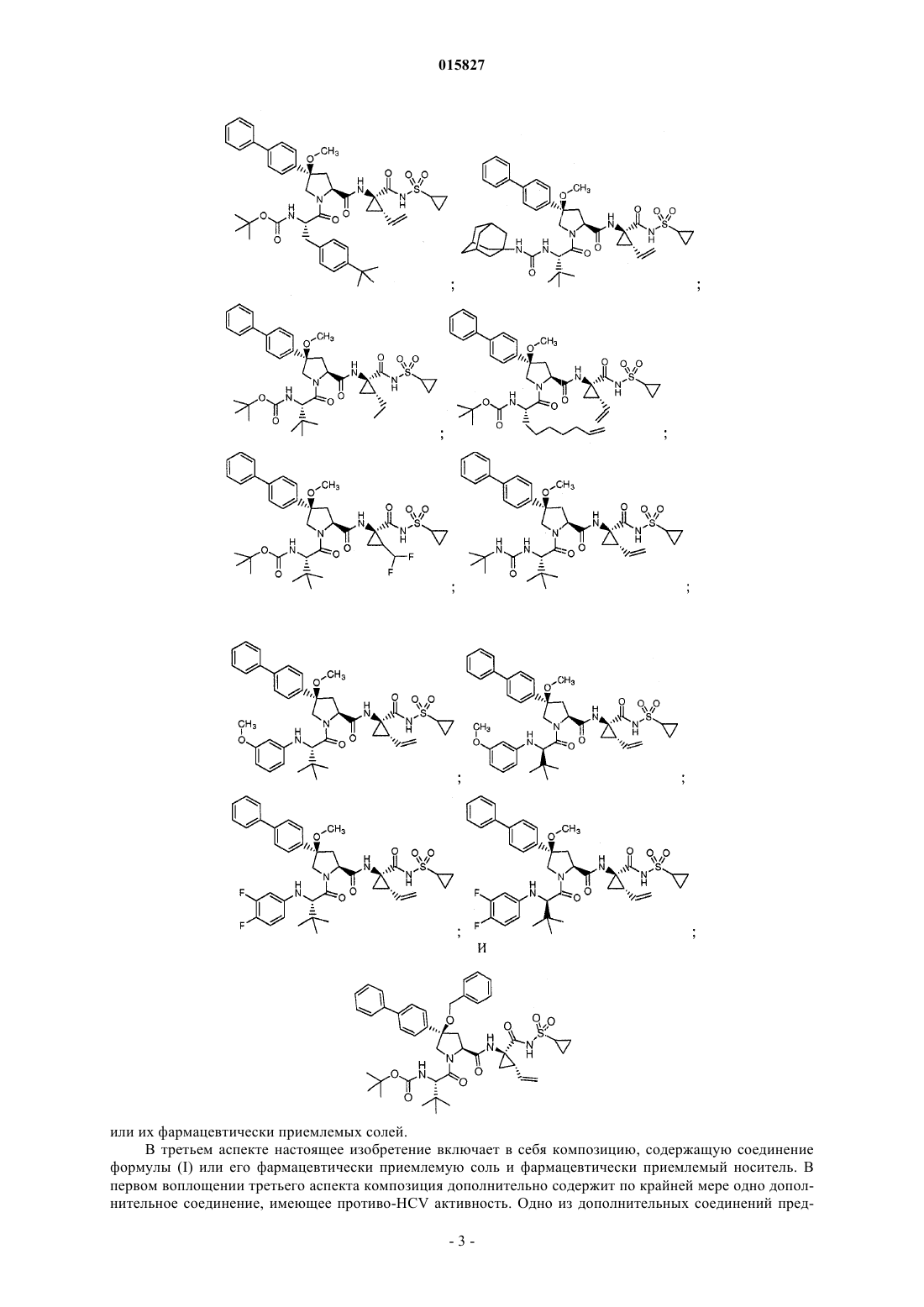
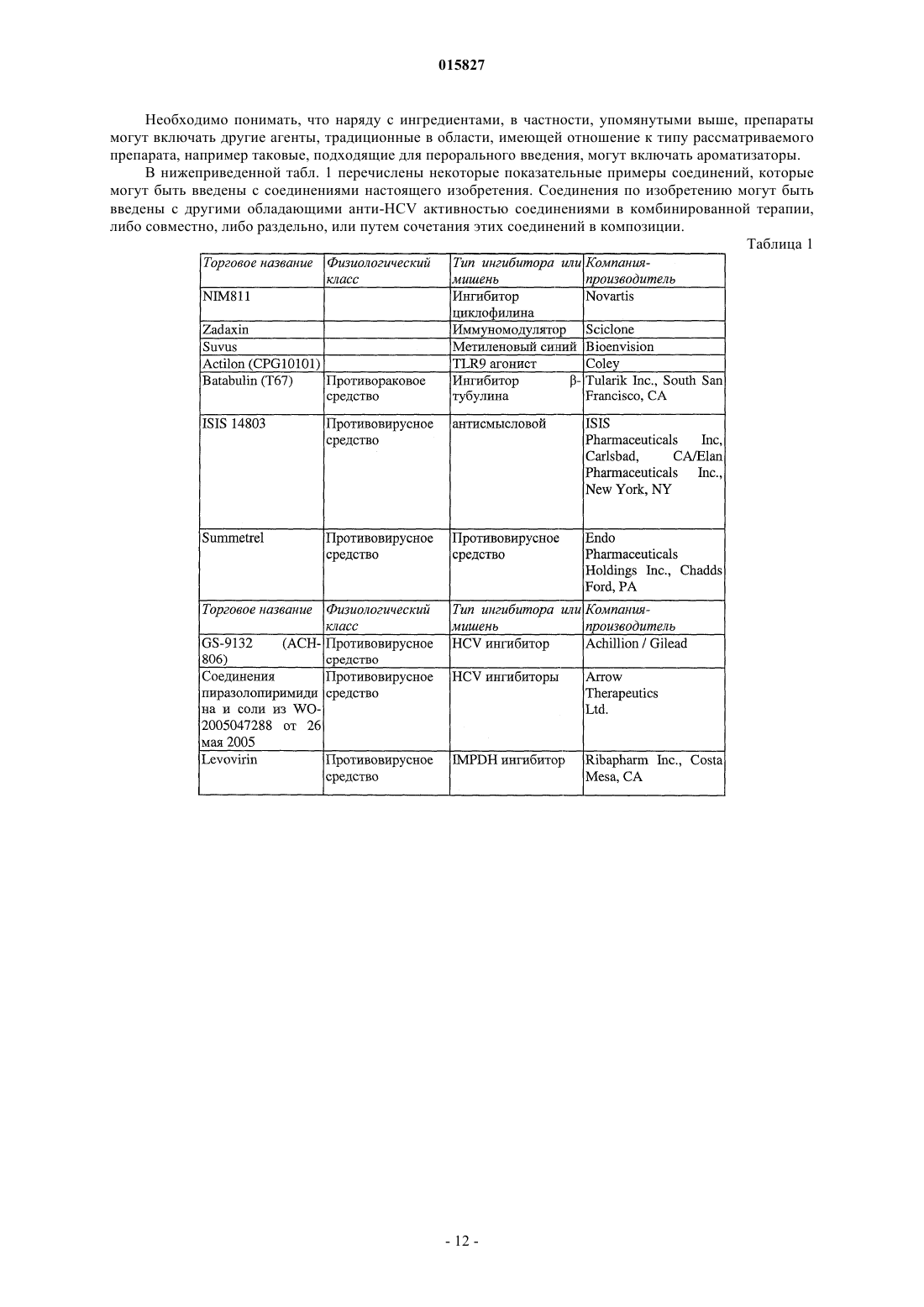
3. Соединение, выбранное из



или его фармацевтически приемлемая соль.
4. Фармацевтическая композиция, содержащая соединение по п.1 или его фармацевтически приемлемую соль и фармацевтически приемлемый носитель.
5. Фармацевтическая композиция по п.4, дополнительно содержащая по крайней мере одно дополнительное соединение, имеющее противо-HCV активность.
6. Фармацевтическая композиция по п.5, где по крайней мере одно из дополнительных соединений представляет собой интерферон или рибавирин.
7. Фармацевтическая композиция по п.6, где интерферон выбран из интерферона a-2В, пегилированного интерферона a, консенсусного интерферона, интерферона a-2А и лимфобластоидного интерферона t.
8. Фармацевтическая композиция по п.5, где по крайней мере одно из дополнительных соединений выбрано из интерлейкина 2, интерлейкина 6, интерлейкина 12, соединения, которое улучшает развитие реакции хелперных Т-клеток 1-го типа, интерферирующего РНК, антисмыслового РНК, имиквимода, рибавирина, ингибитора инозин 5'-монофосфат дегидрогеназы, амантадина и римантадина.
9. Фармацевтическая композиция по п.5, где по крайней мере одно из дополнительных соединений, эффективно ингибирующих функцию мишени, выбрано из HCV металлопротеазы, HCV сериновой протеазы, HCV полимеразы, HCV хеликазы, HCV NS4B белка, HCV входа, HCV сборки, HCV выхода, HCV NS5A белка и IMPDH для лечения HCV инфекции.
10. Способ лечения HCV инфекции, включающий введение пациенту терапевтически эффективного количества соединения по п.1 или его фармацевтически приемлемой соли.
11. Способ по п.10, дополнительно включающий введение по крайней мере одного дополнительного соединения, имеющего противо-HCV активность до, после или одновременно с соединением по п.1 или его фармацевтически приемлемой солью.
12. Способ по п.11, где по крайней мере одно из дополнительных соединений представляет собой интерферон или рибавирин.
13. Способ по п.12, где интерферон выбран из интерферона a-2В, пегилированного интерферона a, консенсусного интерферона, интерферона a-2А и лимфобластоидного интерферона t.
14. Способ по п.11, где по крайней мере одно из дополнительных соединений выбрано из интерлейкина 2, интерлейкина 6, интерлейкина 12, соединения, которое улучшает развитие реакции хелперных Т-клеток 1-го типа, интерферирующего РНК, антисмыслового РНК, имиквимода, рибавирина, ингибитора инозин 5'-монофосфат дегидрогеназы, амантадина и римантадина.
15. Способ по п.11, где по крайней мере одно из дополнительных соединений, эффективно ингибирующих функцию мишени, выбрано из HCV металлопротеазы, HCV сериновой протеазы, HCV полимеразы, HCV хеликазы, HCV NS4B белка, HCV входа, HCV сборки, HCV выхода, HCV NS5A белка и IMPDH для лечения HCV инфекции.
Текст
ОПИСАНИЕ ИЗОБРЕТЕНИЯ К ЕВРАЗИЙСКОМУ ПАТЕНТУ Дата публикации и выдачи патента ИНГИБИТОРЫ ВИРУСА ГЕПАТИТА С Раскрыты ингибиторы вируса гепатита С, имеющие общую формулу (I). Также раскрыты композиции, содержащие соединения и способы использования соединений для ингибирования Ван Алан Сяндун, Чжэн Барбара Чжичжэнь, ДАндреа Стэнли, Чжао Цянь, Скола Пол Майкл (US) Дементьев В.Н. (RU) 015827 Настоящее изобретение, главным образом, направлено на противовирусные соединения и, более конкретно, направлено на соединения, которые могут ингибировать функцию протеазы NS3 (также обозначаемой в настоящем изобретении как сериновая протеаза), кодируемой вирусом гепатита С (HCV),на композиции, содержащие такие соединения, и способы ингибирования функции протеазы NS3. Вирус гепатита С (HCV) является основным патогеном человека, инфицировавшим предположительно 170 млн человек по всему миру, что, грубо говоря, в пять раз больше, чем инфицированных вирусом иммунодефицита человека 1 типа. У значительного числа инфицированных HCV индивидуумов развивается серьезное прогрессирующее заболевание печени, включая цирроз и печеночно-клеточный рак. В настоящее время наиболее эффективная терапия HCV включает комбинацию -интерферона и рибавирина, которые приводят к устойчивому эффекту у 40% пациентов. Последние клинические результаты демонстрируют, что пегилированный -интерферон предпочтителен по сравнению с немодифицированным -интерфероном в виде монотерапии. Тем не менее, даже при экспериментальных терапевтических режимах, включающих комбинации пегилированного -интерферона и рибавирина, у существенной части пациентов не отмечается устойчивого снижения вирусной нагрузки. Таким образом, существует очевидная и важная необходимость развивать эффективные лекарственные средства для лечения HCV инфекции.HCV является положительно закрученным РНК вирусом. Основываясь на сравнении вычисленной последовательности аминокислот и значительном сходстве в 5'-нетранслируемой области, HCV классифицируется как отдельный род в семействе Flaviviridae. Все представители семейства Flaviviridae имеют оболочечные вирионы, которые содержат положительно закрученный РНК геном, кодирующий все известные вирус-специфические белки через трансляцию единичной непрерывной открытой рамки считывания. Существенная гетерогенность найдена в нуклеотидной и кодированной аминокислотной последовательности по всему геному HCV. Шесть основных генотипов и более чем 50 подтипов были описаны. Основные генотипы HCV отличаются по распространенности в мире и клиническое значение генетической гетерогенности HCV остается неясным, несмотря на множество исследований возможного действия генотипов на патогенез и терапию. Геном одноцепочечной РНК HCV составляет приблизительно 9500 нуклеотидов в длину и имеет одну открытую рамку считывания (ORF), кодирующую один большой полипротеин, состоящий примерно из 3000 аминокислот. В инфицированных клетках этот полипротеин расщепляется по многим сайтам клеточными и вирусными протеазами до получения структурных и неструктурных (NS) белков. В случаеHCV поколение зрелых неструктурных белков (NS2, NS3, NS4A, NS4B, NS5A и NS5B) подвергается воздействию двух вирусных протеаз. Первая расщепляет NS2-NS3 связь; вторая является сериновой протеазой, содержащей на N-конце NS3 (также обозначаемая как NS3 протеаза), и опосредует все последующие расщепления ниже по каскаду NS3 как в цис-положении, в сайте расщепления NS3-NS4A, так и в транс-положении, для остальных сайтов NS4A-NS4B, NS4B-NS5A, NS5A-NS5B. NS4A белок, как представляется, выполняет множество функций, действуя как кофактор для NS3 протеазы, и возможно способствует расположению в мембране NS3 и других компонентов вирусной репликазы. Образование комплекса NS3 белка и NS4A представляется необходимым для эффективного полибелкового процессинга,повышая протеолитическое расщепление во всех сайтах. Белок NS3 также проявляет активность нуклеозидной трифосфатазы и РНК хеликазы. NS5B (также обозначаемая как HCV полимераза) является РНКзависимой РНК полимеразой, которая вовлечена в репликацию HCV. Настоящее изобретение раскрывает пептидные соединения, которые могут ингибировать функционирование NS3 протеазы, например, в комбинации с NS4A протеазой. Кроме того, настоящее изобретение описывает введение пациенту комбинированной терапии, посредством чего соединение в соответствии с настоящим изобретением, которое является эффективным для ингибирования HCV NS3 протеазы,может быть введено с одним или двумя дополнительными соединениями, имеющими противо-HCV активность. В первом аспекте объектом настоящего изобретения является соединение формулы (I) или его фармацевтически приемлемая соль, гдеR1 выбран из гидрокси и -NHSO2R7;R6 представляет собой трет-бутоксикарбонил, возможно замещенный галогеном, третбутиламинокарбонил, адамантан-1-илоксикарбонил, адамантан-1-иламинокарбонил, фенил, возможно замещенный 1-2 заместителями, независимо выбираемыми из галогена и метокси;R8 представляет собой метокси или бензилокси. Во втором аспекте объектом настоящего изобретения является соединение формулы (II) или его фармацевтически приемлемая соль,где n равно 1;R6 представляет собой трет-бутоксикарбонил, возможно замещенный галогеном, третбутиламинокарбонил, адамантан-1-илоксикарбонил, адамантан-1-иламинокарбонил, фенил, возможно замещенный 1-2 заместителями, независимо выбираемыми из галогена и метокси;R8 представляет собой метокси или бензилокси. Соединение может также быть выбрано из или их фармацевтически приемлемых солей. В третьем аспекте настоящее изобретение включает в себя композицию, содержащую соединение формулы (I) или его фармацевтически приемлемую соль и фармацевтически приемлемый носитель. В первом воплощении третьего аспекта композиция дополнительно содержит по крайней мере одно дополнительное соединение, имеющее противо-HCV активность. Одно из дополнительных соединений пред-3 015827 ставляет собой интерферон или рибавирин. Интерферон может быть выбран из интерферона -2 В, пегилированного интерферона , консенсусного интерферона, интерферона -2 А и лимфобластоидного интерферона . Настоящее изобретение охватывает также композицию, содержащую соединение формулы (I) или его фармацевтически приемлемую соль, фармацевтически приемлемый носитель и по крайней мере одно дополнительное соединение, обладающее противо-HCV активностью, где по крайней мере одно из дополнительных соединений выбрано из интерлейкина 2, интерлейкина 6, интерлейкина 12, соединения,которое улучшает развитие реакции хелперных Т-клеток 1-го типа, интерферирующего РНК, антисмыслового РНК, имиквимода, рибавирина, ингибитора инозин 5'-монофосфат дегидрогеназы, амантадина и римантадина. Настоящее изобретение охватывает также композицию, содержащую соединение формулы (I) или его фармацевтически приемлемую соль, фармацевтически приемлемый носитель и по крайней мере одно дополнительное соединение, обладающее противо-HCV активностью, где по крайней мере одно из дополнительных соединений, эффективно ингибирующих функцию мишени, выбрано из HCV металлопротеазы, HCV сериновой протеазы, HCV полимеразы, HCV хеликазы, HCV NS4B белка, HCV входа, HCV сборки, HCV выхода, HCV NS5A белка и IMPDH для лечения HCV инфекции. В четвертом аспекте настоящее изобретение включает в себя способ лечения HCV инфекции у пациента, включающий введение пациенту терапевтически эффективного количества соединения формулы(I) или его фармацевтически приемлемой соли. В первом воплощении четвертого аспекта способ дополнительно включает введение одного дополнительного соединения, обладающего противо-HCV активностью, перед, после или одновременно с соединением формулы (I) или его фармацевтически приемлемой солью. В одном из воплощений четвертого аспекта изобретения по крайней мере одно из дополнительных соединений представляет собой интерферон или рибавирин. В другом его воплощении интерферон выбран из интерферона -2 В, пегилированного интерферона , консенсусного интерферона, интерферона -2 А и лимфобластоидного интерферона . В ещ одном воплощении четвертого аспекта настоящее изобретение включает в себя способ лечения HCV инфекции у пациента, включающий введение пациенту терапевтически эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли и по крайней мере одного дополнительного соединения, обладающего противо-HCV активностью, перед, после или одновременно с соединением формулы (I) или его фармацевтически приемлемой солью, где по крайней мере одно из дополнительных соединений выбрано из интерлейкина 2, интерлейкина 6, интерлейкина 12, соединения,которое улучшает развитие реакции хелперных Т-клеток 1-го типа, интерферирующего РНК, антисмыслового РНК, имиквимода, рибавирина, ингибитора инозин 5'-монофосфат дегидрогеназы, амантадина и римантадина. Настоящее изобретение включает также способ лечения HCV инфекции у пациента, включающий введение пациенту терапевтически эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли и по крайней мере одного дополнительного соединения, обладающего противо-HCV активностью, перед, после или одновременно с соединением формулы (I) или его фармацевтически приемлемой солью, где по крайней мере одно из дополнительных соединений, эффективно ингибирующих функцию мишени, выбрано из HCV металлопротеазы, HCV сериновой протеазы, HCV полимеразы, HCV хеликазы, HCV NS4B белка, HCV входа, HCV сборки, HCV выхода, HCV NS5A белка иIMPDH для лечения HCV инфекции. В пятом аспекте объектом настоящего изобретения является композиция, содержащая соединение формулы (I) или его фармацевтически приемлемую соль, один, два, три, четыре или пять дополнительных соединений, имеющих противо-HCV активность и фармацевтически приемлемый носитель. В первом воплощении пятого аспекта композиция содержит три или четыре дополнительных соединений,имеющих противо-HCV активность. Во втором воплощении пятого аспекта композиция содержит один или два дополнительных соединения, имеющих противо-HCV активность. В шестом аспекте объектом настоящего изобретения является способ лечения HCV инфекции у пациента, включающий введение пациенту терапевтически эффективного количества соединения формулы(I) или его фармацевтически приемлемой соли и один, два, три, четыре или пять дополнительных соединений, имеющих противо-HCV активность, перед, после или одновременно с соединением формулы (I) или его фармацевтически приемлемой солью. Способ может включать введение трех или четырех дополнительных соединений, имеющих противо-HCV активность, или же введение одного или двух дополнительных соединений, имеющих противо-HCV активность. Описание настоящего изобретения должно быть рассмотрено в соответствии с правилами и принципами образования химической связи. В некоторых случаях это может быть необходимо, чтобы удалить атом водорода для размещения заместителя на любом заданном месте при любой заданном положении. Должно быть понятно, что соединения, охваченные настоящим изобретением, являются теми, которые обеспечивают подходящую стабильность для применения в качестве фармацевтического агента. Все патенты, патентные заявки и опубликованные ссылки, процитированные в описании, при этом-4 015827 включены как ссылки в своей полноте. В случае несовместимости будет преобладать настоящее изобретение, включая определения. Как используют в настоящем описании, следующие термины имеют соответствующие значения. Термин алкенил, как используют в настоящем описании, относится к прямой или разветвленной цепи группы от двух до шести атомов углерода, содержащей по крайней мере одну углерод-углеродную двойную связь. Термин алкокси, как используют в настоящем описании, относится к алкильной группе, присоединенной к остатку исходной молекулы через атом кислорода. Термин алкоксиалкил, как используют в настоящем описании, относится к алкильной группе, замещенной одним, двумя или тремя алкоксигруппами. Термин алкоксикарбонил, как используют в настоящем описании, относится к алкоксигруппе,присоединенной к остатку исходной молекулы через карбонильную группу. Термин алкоксикарбонилалкил, как используют в настоящем описании, относится к алкильной группе, замещенной одним, двумя или тремя алкоксикарбонильными группами. Термин алкил, как используют в настоящем описании, относится к группе, производной от прямой или разветвленной цепи насыщенного углеводорода, содержащей от одного до десяти атомов углерода. Термин алкилкарбонил, как используют в настоящем описании, относится к алкильной группе,присоединенной к остатку исходной молекулы через карбонильную группу. Термин алкилсульфанил, как используют в настоящем описании, относится к алкильной группе,присоединенной к остатку исходной молекулы через атом серы. Термин алкилсульфонил, как используют в настоящем описании, относится к алкильной группе,присоединенной к остатку исходной молекулы через сульфонильную группу. Термин арил, как используют в настоящем описании, относится к фенильной группе или бициклической конденсированной кольцевой системе, где одно или оба кольца представляют собой фенильную группу. Бициклические конденсированные кольцевые системы состоят из фенильной группы, конденсированной с содержащим от четырех до шести членов ароматическим или неароматическим карбоциклическим кольцом. Арильные группы по настоящему изобретению могут быть присоединены к остатку исходной молекулы через любой замещаемый атом углерода в группе. Типичные примеры арильных групп включают, но без ограничения, инданил, инденил, нафтил, фенил и тетрагидронафтил. Арильные группы по настоящему изобретению могут быть необязательно замещены одним, двумя, тремя, четырьмя или пятью заместителями, независимо выбранными из алкокси, алкоксикарбонила, алкила, второй арильной группы, циано, галогена, галогеналкокси, галогеналкила, гетероциклила, гетероциклилалкила, гидрокси, гидроксиалкила, нитро, -NRxRy, (NRxRy)алкокси, (NRxRy)алкила, (NRxRy)карбонила и оксо; где вторая арильная группа, гетероциклил и гетероциклильная часть гетероциклилалкила может быть дополнительно необязательно замещена одним, двумя, тремя, четырьмя или пятью заместителями,независимо выбранными из алкокси, алкила, циано, галогена, галогеналкокси, галогеналкила, гидрокси и нитро. Термин арилалкил, как используют в настоящем описании, относится к алкильной группе, замещенной одним, двумя или тремя арильными группами. Термин карбонил, как используют в настоящем описании, относится к -С(О)-. Термин карбокси, как используют в настоящем описании, относится к -СО 2 Н. Термин карбоксиалкил, как используют в настоящем описании, относится к алкильной группе,замещенной одним, двумя или тремя карбоксигруппами. Термин циано, как используют в настоящем описании, относится к -CN. Термин цианоалкил, как используют в настоящем описании, относится к алкильной группе, замещенной одним, двумя или тремя цианогруппами. Термин циклоалкил, как используют в настоящем описании, относится к насыщенной моноциклической, бициклической или трициклической углеводородной кольцевой системе, имеющей от трех до четырнадцати атомов углерода и ноль гетероатомов. Типичные примеры циклоалкильных групп включают, но без ограничения, циклопропил, циклопентил, бицикло[3.1.1]гептил и адамантил. Циклоалкильные группы по настоящему изобретению могут быть необязательно замещены одним, двумя, тремя или четырьмя заместителями, независимо выбранными из алкенила, алкокси, алкоксиалкила, алкила, арилалкила, арилкарбонила, циано, циклоалкенила, (циклоалкил)алкила, галогена, галогеналкокси, галогеналкила и (NRjRk)карбонила; где Rj и Rk независимо выбраны из водорода, алкила, арила, арилалкила и гетероциклила; где арил, арильная часть арилалкила и гетероциклил необязательно замещены одним или двумя заместителями, независимо выбранными из алкокси, алкила и галогена. Термин(циклоалкил)алкил, как используют в настоящем описании, относится к алкильной группе, замещенной одним, двумя или тремя циклоалкильными группами. Термин циклоалкилкарбонил, как используют в настоящем описании, относится к циклоалкиль-5 015827 ной группе, присоединной к остатку исходной молекулы через карбонильную группу. Термин циклоалкилокси, как используют в настоящем описании, относится к циклоалкильной группе, присоединной к остатку исходной молекулы через атом кислорода. Термин циклоалкилоксикарбонил, как используют в настоящем описании, относится к циклоалкилоксигруппе, присоединенной к остатку исходной молекулы через карбонильную группу. Термины гало и галоген, как используют в настоящем описании, относится к F, Cl, Br и I. Термин галогеналкокси, как используют в настоящем описании, относится к галогеналкильной группе, присоединенной к остатку исходной молекулы через атом кислорода. Термин галогеналкоксиалкил, как используют в настоящем описании, относится к алкильной группе, замещенной одним, двумя или тремя галогеналкоксигруппами. Термин галогеналкоксикарбонил, как используют в настоящем описании, относится к галогеналкоксигруппе, присоединенной к остатку исходной молекулы через карбонильную группу. Термин галогеналкил, как используют в настоящем описании, относится к алкильной группе, замещенной одним, двумя, тремя или четырьмя атомами галогена. Термин галогеналкилкарбонил, как используют в настоящем описании, относится к галогеналкильной группе, присоединенной к остатку исходной молекулы через карбонильную группу. Термин гетероциклил, как используют в настоящем изобретении, относится к содержащему пять,шесть или семь членов кольцу, включающему один, два, три или четыре гетероатома, независимо выбранных из азота, кислорода и серы. Содержащее пять членов кольцо имеет от нуля до двух двойных связей, и содержащие шесть и семь членов кольца имеют от нуля до трех двойных связей. Термин гетероциклил также включает бициклические группы, в которых гетероциклильное кольцо является конденсированным еще с моноциклической гетероциклильной группой или содержащим от четырех до шести членов ароматическим или неароматическим карбоциклическим кольцом или другой моноциклической гетероциклильной группой. Гетероциклильные группы по настоящему изобретению могут быть присоединены к остатку исходной молекулы через атом углерода или атом азота в группе. Примеры гетероциклильных групп включают, но без ограничения, бензотиенил, фурил, имидазолил, индолинил, индолил, изотиазолил, изоксазолил, морфолинил, оксазолил, пиперазинил, пиперидинил, пиразолил, пиридинил, пирролидинил, пирролопиридинил, пирролил, тиазолил, тиенил и тиоморфолинил. Гетероциклильные группы по настоящему изобретению могут быть необязательно замещены одним, двумя, тремя,четырьмя или пятью заместителями, независимо выбранными из алкокси, алкоксикарбонила, алкила,арила, циано, галогена, галогеналкокси, галогеналкила, второй гетероциклильной группы, гетероциклилалкила, гидрокси, гидроксиалкила, нитро, -NRxRy, (NRxRy)алкокси, (NRxRy)алкила, (NRxRy)карбонила и оксо; где арил, вторая гетероциклильная группа и гетероциклильная часть гетероциклилалкила может быть дополнительно необязательно замещены одним, двумя, тремя, четырьмя или пятью заместителями,независимо выбранными из алкокси, алкила, циано, галогена, галогеналкокси, галогеналкила, гидрокси и нитро. Термин гетероциклилалкил, как используют в настоящем описании, относится к алкильной группе, замещенной одним, двумя или тремя гетероциклильными группами. Термин гидрокси, как используют в настоящем описании, относится к -ОН. Термин гидроксиалкил, как используют в настоящем описании, относится к алкильной группе,замещенной одним, двумя или тремя гидроксигруппами. Термин -NRaRb, как используют в настоящем описании, относится к двум группам Ra и Rb, которые присоединены к остатку исходной молекулы через атом азота. Ra и Rb, каждый, независимо выбран из водорода, алкокси, алкоксиалкила, алкила, арила, арилалкила, циклоалкила, галогеналкоксиалкила,галогеналкила, гетероциклила и гетероциклилалкила. Термин (NRaRb)алкил, как используют в настоящем описании, относится к алкильной группе, замещенной одним, двумя или тремя -NRaRb группами. Термин (NRaRb)карбонил, как используют в настоящем описании, относится к -NRaRb группе,присоединной к остатку исходной молекулы через карбонильную группу. Термин (NRaRb)карбонилалкил, как используют в настоящем описании, относится к алкильной группе, замещенной одним, двумя или тремя (NRaRb)карбонильными группами. Термин (NRaRb)сульфонил, как используют в настоящем описании, относится к -NRaRb группе,присоединной к остатку исходной молекулы через сульфонильную группу. Термин -NRcRd, как используют в настоящем описании, относится к двум группам Rc и Rd, которые присоединены к остатку исходной молекулы через атом азота. Rc и Rd независимо выбраны из алкокси, алкила, арила, арилалкила, циклоалкила, (циклоалкил)алкила, гетероциклила и гетероциклилалкила; или Rc и Rd вместе с атомом азота, с которым они связаны, образуют содержащее пять или шесть членов моноциклическое гетероциклическое кольцо. Термин -NReRf, как используют в настоящем описании, относится к двум группам Re и Rf, которые присоединены к остатку исходной молекулы через атом азота. Re и Rf независимо выбраны из водо-6 015827 рода, алкила и арилалкила; или Re и Rf вместе с атомом азота, с которым они связаны, образуют содержащее пять или шесть членов моноциклическое гетероциклическое кольцо, необязательно содержащее один дополнительный гетероатом, выбранный из О, NRx и S; где Rx выбран из водорода и алкила; и гдеR' выбран из водорода и алкила. Термин -NRgRh, как используют в настоящем описании, относится к двум группам Rg и Rh, которые присоединены к остатку исходной молекулы через атом азота. Rg и Rh независимо выбраны из водорода, алкоксиалкила, алкоксикарбонила, алкила, алкилкарбонила, арилалкила и галогеналкила. Термин -NRjRk, как используют в настоящем описании, относится к двум группам Rj и Rk, которые присоединены к остатку исходной молекулы через атом азота. Rj и Rk независимо выбраны из водорода, алкила, арила, арилалкила и гетероциклила; где арил, арильная часть арилалкила и гетероциклил необязательно замещены одним или двумя заместителями, независимо выбранными из алкокси, алкила и галогена. Термин (NRjRk)карбонил, как используют в настоящем описании, относится к -NRjRk группе, присоединной к остатку исходной молекулы через карбонильную группу. Термин (NRjRk)сульфонил, как используют в настоящем описании, относится к -NReRf группе,присоединной к остатку исходной молекулы через сульфонильную группу. Термин -NRxRy, как используют в настоящем описании, относится к двум группам Rx и Ry, которые присоединены к остатку исходной молекулы через атом азота. Rx и Ry независимо выбраны из водорода и алкила. Термин (NRxRy)алкокси, как используют в настоящем описании, относится к (NRxRy)алкильной группе, присоединной к остатку исходной молекулы через атом кислорода. Термин (NRxRy)алкил, как используют в настоящем описании, относится к алкильной группе, замещенной одним, двумя или тремя -NRxRy группами. Термин (NRxRy)карбонил, как используют в настоящем описании, относится к -NRxRy группе,присоединной к остатку исходной молекулы через карбонильную группу. Термин нитро, как используют в настоящем описании, относится к -NO2. Термин оксо, как используют в настоящем описании, относится к =О. Термин сульфонил, как используют в настоящем описании, относится к -SO2-. Соединения по настоящему изобретению могут существовать как пролекарства. Термин пролекарство, как используют в настоящем описании, представляет собой соединения, которые быстро преобразуются in vivo в исходные соединения путем гидролиза в крови. Пролекарства по настоящему изобретению включают сложные эфиры карбоксильных групп исходной молекулы, сложные эфиры гидроксильных групп исходной молекулы и амиды аминов исходной молекулы. Соединения по настоящему изобретению могут существовать в виде фармацевтически приемлемых солей. Термин фармацевтически приемлемая соль, как используют в настоящем изобретении, представляет собой соли или цвиттерионные формы соединений по настоящему изобретению, которые растворимы в воде или масле или диспергированы, которые в рамках указанного медицинского определения приемлемы для применения в контакте с тканями пациента без излишней токсичности, раздражения, аллергической реакции или другой проблемы или осложнения, соразмерных приемлемому соотношению польза/риск, и эффективны для их указанного определения. Соли могут быть получены при конечном выделении и очистке соединений или отдельно путем взаимодействия приемлемой основной функциональности с приемлемыми кислотами. Типичные кислотно-аддитивные соли включают ацетат, адипат,альгинат, цитрат, аспарат, бензоат, бензолсульфонат, бисульфат, бутират, камфорат, камфорсульфонат; диглюконат, глицерофосфат, гемисульфат, гептапоат, гексаноат, формиат, фумарат, гидрохлорид, гидробромид, гидроиодид, 2-гидроксиэтансульфонат, лактат, малеат, мезитиленсульфонат, метансульфонат,нафтиленсульфонат, никотинат, 2-нафталинсульфонат, оксалат, пальмоат, пектинат, персульфат, 3 фенилпропионат, пикрат, пивалат, пропионат, сукцинат, тартрат, трихлорацетат, трифторацетат, фосфат,глютамат, бикарбонат, паратолуолсульфонат и ундеканоат. Примеры кислот, которые могут быть применены, чтобы образовать фармацевтически приемлемые аддитивные соли, включают неорганические кислоты, такие как хлористо-водородная, бромисто-водородная, серная и фосфорная, и органические кислоты, такие как щавелевая, малеиновая, янтарная и лимонная. Основно-аддитивные соли могут быть получены при конечном выделении и очистке соединений путем взаимодействия кислотной группы с приемлемым основанием, таким как гидроксид, карбонат или бикарбонат катиона металла или с аммиаком, или с органическим первичным, вторичным или третичным амином. Катионы фармацевтически приемлемых солей включают литий, натрий, калий, кальций,магний и алюминий, так же как катионы нетоксичного четвертичного амина, такого как аммоний, тетраметиламмоний, тетраэтиламмоний, метиламин, диметиламин, триметиламин, триэтиламин, диэтиламин,этиламин, трибутиламин, пиридин, N,N-диметиланилин, N-метилпиперидин, N-метилморфолин, дициклогексиламин, прокаин, дибензиламин, N,N-дибензилфенэтиламин и N,N'-дибензилэтилендиамин. Другие типичные органические амины, пригодные для образования основно-аддитивных солей, включают этилендиамин, этаноламин, диэтаноламин, пиперидин и пиперазин.-7 015827 Как используют в настоящем описании, термин противо-HCV активность означает соединение,являющиеся эффективными для лечения HCV вируса. Термин соединения по настоящему изобретению и эквивалентные выражения предназначены,чтобы охватить соединения формулы (I) и их фармацевтически приемлемые энантиомеры, диастереомеры и соли. Подобным образом ссылки на промежуточные соединения предназначены, чтобы охватить их соли, если позволяет контекст. Термин пациент включает как человека, так и других млекопитающих. Термин фармацевтическая композиция означает композицию, содержащую соединение по изобретению в комбинации по крайней мере с одним дополнительным фармацевтическим носителем, то есть адъювант, наполнитель или связующее вещество, такое как разбавители, консерванты, наполнители,агенты регуляторы потока, агенты расщепления, агенты увлажнения, эмульгаторы, агенты суспендирования, подсластители, ароматизаторы, отдушки, противобактериальные агенты, противогрибковые агенты, смазывающие агенты и распределяющие агенты, в зависимости от сущности способа введения и формы дозирования. Могут быть использованы ингредиенты, перечисленные, например, в Remington'sPharmaceutical Sciences, 18th ed., Mack Publishing Company, Easton, PA (1999). Выражение фармацевтически приемлемый, используемое в данном описании, относится к тем соединениям, веществам, композициям и/или лекарственным формам, которые при взвешенном медицинском решении подходят для применения в контакте с тканями пациентов без избыточной токсичности,раздражения, аллергических реакций или других проблем или осложнений, соответствующих приемлемому отношению польза/риск, и являются эффективными в отношении их предполагаемого использования. Термин терапевтически эффективное количество означает общее количество каждого активного компонента, которое достаточно, чтобы продемонстрировать значительную пользу для пациента, например снижение вирусного воздействия. Когда используют индивидуальный активный ингредиент, вводимый сам по себе, термин сам по себе относится к одному ингредиенту. Когда используют комбинацию,термин относится к объединенному количеству активных ингредиентов, которые приводят к терапевтическому эффекту, или при введении в комбинации, периодически или одновременно. Термины лечить и лечение относятся к (i) профилактике заболевания, расстройства или патологического состояния, возникающих у пациента, который может быть предрасположен к заболеванию,расстройству и/или патологическому состоянию, которое еще не диагностировано у этого пациента; (ii) подавлению заболевания, расстройства или патологического состояния, т.е. торможение их развития; и(iii) облегчение заболевания, расстройства или патологического состояния, т.е. вызывание регрессии заболевания, расстройства и/или патологического состояния. Там, где используют при наименовании соединения по настоящему изобретению, обозначения Р 1',Р 1, Р 2, Р 2, Р 3 и Р 4, как используемые в настоящем описании, отображают относительные положения аминокислотных остатков ингибитора протеазы, связанных по отношению к связанному природному пептидному расщепленному субстрату. Расщепление происходит в природном субстрате между Р 1 и Р 1',где второстепенные положения обозначают аминокислоты, начиная с окончания С-конца пептидного природного расщепляемого сайта по направлению к N-концу; тогда как основные положения отсчитывают от окончания N-конца обозначеннного расщепляемого сайта и распространяются до С-конца. Например, Р 1' относится к первому положению от правого окончания С-конца расщепляемого сайта (т.е. исходному положению N-конца); тогда как Р 1 начинает нумерацию от левой стороны С-конца расщепляемого сайта, Р 2 - второе положение от С-конца и т.д. (см. Berger А.Schechter, Transactions of RoyalSociety London series (1970), B257, 249-264). Следующая фигура демонстрирует обозначения для соединений по настоящему изобретению. Асимметричные центры существуют в соединениях по настоящему изобретению. Например, соединения могут включать Р 1 циклопропильный элемент формулы где C1 и С 2, каждый, представляет собой асимметричный атом углерода в положениях 1 и 2 циклопропильного кольца. Понятно, что изобретение охватывает все стереохимические формы или их смеси, которые обладают способностями для ингибирования HCV протеазы. Определенные соединения по настоящему изобретению могут также существовать в различных стабильных конформационных формах, которые могут быть выделены. Спиральная асимметрия, благодаря ограниченной ротации около асимметричной простой связи, например, вследствие стерического несоответствия или деформации кольца, может допустить разделение различных конформеров. Настоящее изобретение включает каждый конформационный изомер этих соединений и их смесей. Определенные соединения по настоящему изобретению могут существовать в цвиттерионной форме, и настоящее изобретение включает каждую цвиттерионную форму этих соединений и их смесей. Когда возможно для использования в терапии, терапевтически эффективные количества соединения формулы (I), так же как его фармацевтически приемлемых солей, могут быть введены в виде сырого химического соединения, можно представить активный ингредиент в виде фармацевтической композиции. Таким образом, изобретение дополнительно обеспечивает фармацевтические композиции, которые включают терапевтически эффективные количества соединений формулы (I) или их фармацевтически приемлемые соли и один или большее количество фармацевтически приемлемых носителей, разбавителей или наполнителей. Соединения формулы (I) и их фармацевтически приемлемые соли являются теми,как описано выше. Носитель(и), разбавитель(и) или наполнитель(и) должны быть совместимы в смысле того, чтобы быть совместимыми с другими ингредиентами состава и не быть опасными для реципиента. В соответствии с другим аспектом настоящего изобретения также обеспечен способ получения фармацевтического состава, включая смешивание соединения формулы (I) или его фармацевтически приемлемой соли с одним или большим количеством фармацевтически приемлемых носителей, разбавителей или наполнителей. Фармацевтические составы могут быть представлены в дозированных лекарственных формах, содержащих заранее определенное количество активного ингредиента на единицу дозирования. Уровни дозировок соединений настоящего изобретения примерно от 0,01 до 250 мг на 1 кг массы тела в день,предпочтительно примерно от 0,05 до 100 мг/кг массы тела в день являются обычными для монотерапии с целью профилактики и лечения HCV-опосредованного заболевания. Обычно фармацевтические композиции настоящего изобретения вводят примерно от 1 до 5 раз в сутки или альтернативно в виде продолжающейся инфузии. Такое введение может применяться в качестве длительной или срочной терапии. Количество активного ингредиента, которое может быть комбинировано с веществами-носителями для получения единой лекарственной формы, будет варьироваться в зависимости от патологического состояния, по поводу которого проводится лечение, тяжести состояния, времени введения, пути введения, ско-9 015827 рости выведения используемого соединения, продолжительности лечения, возраста, пола, веса и состояния пациента. Предпочтительными дозированными лекарственными формами являются те, которые содержат суточную дозу или субдозу активного ингредиента, как было выше указано, или их подходящую часть. Как правило, лечение начинают малыми дозами, существенно меньшими, чем оптимальная доза соединения. После этого дозировку постепенно увеличивают до достижения оптимального эффекта в этих условиях. В общем, соединение наиболее желательно вводить при уровне концентрации, который обычно приводит к эффективным противовирусным результатам и не вызывает вредных или опасных побочных эффектов. Когда композиции настоящего изобретения включают комбинацию соединения по изобретению и одного или более дополнительного терапевтического или профилактического агента, оба соединения и дополнительный агент обычно находятся на уровне дозирования примерно от 10 до 150% и более предпочтительно от около 10 до 80% доз, обычно вводимых в режиме монотерапии. Фармацевтические составы могут быть адаптированы для введения посредством любого подходящего пути, например пероральным путем (включая защечный, подъязычный), ректальным, интраназальным, местным (включая защечный, подъязычный или чрескожный), вагинальным или парентеральным(включая подкожные, внутрикожные, внутримышечные, внутрисуставные, внутрисиновиальные, интрастернальные, интратекальные, внутриочаговые, внутривенные или внутрикожные инъекции или инфузии) путем. Такие препараты могут быть приготовлены с использованием любого из способов, известных в области фармацевтики, например путем сочетания активного ингредиента с носителем(ями) или наполнителем(ями). Фармацевтические составы, адаптированные для перорального введения, могут быть приготовлены как дискретные единицы, такие как капсулы или таблетки; порошки или гранулы; растворы или суспензии в водных или неводных жидкостях; пищевые пенки или взбитые массы; или жидкие эмульсии маслов-воде или эмульсии вода-в-масле. Например, для перорального введения в форме таблетки или капсулы активный лекарственный компонент может быть комбинирован с пероральным нетоксичным фармацевтически приемлемым инертным носителем, таким как этанол, глицерин, вода и т.п. Порошки готовят путем измельчения соединения до подходящего малого размера и смешивания с аналогично измельченным фармацевтическим носителем, таким как пищевой углевод, как, например, крахмал или маннитол. Ароматизатор, консервант, диспергирующий агент или краситель может также присутствовать. Капсулы готовят путем приготовления порошковой смеси, как описано выше, и заполнения ею формованного желатинового футляра. Глиданты и смазывающие вещества, такие как коллоидный кремний, тальк, стеарат магния, стеарат кальция, или твердый полиэтиленгликоль могут быть добавлены к порошковой смеси перед процедурой заполнения. Дезинтегрирующий или солюбилизирующий агент,такой как агар-агар, кальция карбонат или карбоната натрия, может быть добавлен для улучшения биодоступности лекарственного средства в случаях приема капсулы внутрь. Более того, по желанию или по необходимости подходящие связывающие вещества, смазывающие вещества, дезинтегрирующие агенты и красители могут также быть включены в эту смесь. Подходящие смазывающие вещества включают крахмал, желатин, природные сахара, такие как глюкоза или беталактоза, сахаристые вещества кукурузы, природные и синтетические камеди, такие как гуммиарабик,трагакант или натрия альгинат, карбоксиметилцеллюлоза, полиэтиленгликоль и т.п. Смызывающие вещества, используемые в этих лекарственных формах, включают натрия олеат, натрия хлорид и т.п. Дезинтегрирующие вещества включают, помимо прочего, крахмал, метилцеллюлозу, агар-агар, бетонит,ксантановую камедь и т.п. Таблетки изготавливают, например, путем приготовления порошковой смеси,гранулирования или комкования, добавления смазывающего вещества и дезинтегрирующего вещества и прессования в таблетки. Порошковую смесь готовят путем смешивания соединения, подходящим образом измельченного, с разбавителем или основой, как описано выше, и по необходимости со связывающим веществом, таким как карбоксиметилцеллюлоза, альгинат, желатин или поливинилпирролидон, замедлитель растворения, такой как парафин, ускоритель всасывания, такой как четвертичная соль и/или поглощающее вещество, такое как бетонит, каолин или дикальция фосфат. Порошковая смесь может быть приготовлена путем смачивания связующими веществами, такими как сироп, крахмальная паста,смесь гуммиарабика или полученный раствор целлюлозных или полимерных материалов и продавливания через сито. В качестве альтернативы гранулированию порошковая смесь может быть пропущена через таблеточную машину и результатом являются неправильной формы комки, разделенные на гранулы. Гранулы могут быть смазаны для предотвращения прилипания к таблетирующему прессу посредством добавления стеариновой кислоты, стеариновой соли, талька или минерального масла. Смазанную затем смесь прессуют в таблетки. Соединения настоящего изобретения могут быть также комбинированы с сыпучим инертным носителем и прессованы в таблетки напрямую без проведения этапов гранулирования или комкования. Прозрачная или непрозрачная оболочка, состоящая из герметичного слоя шеллака,оболочка из сахара или полимерного материала и гладкая оболочка из воска может быть предусмотрена. Красители могут быть добавлены к этим оболочкам, чтобы различать различные лекарственные формы. Жидкости для перорального применения, такие как раствор, сиропы и эликсиры, могут быть приго- 10015827 товлены в дозированных лекарственных формах, так что заданная доза содержит определенное количество соединения. Сиропы могут быть приготовлены путем растворения соединения в подходящем ароматизированном водном растворе, тогда как эликсиры готовят с помощью нетоксичного носителя. Солюбилизирующие средства и эмульгаторы, такие как этоксилированные изостеариловые спирты и эфиры полиоксиэтиленсорбитола, консерванты, ароматизирующие добавки, такие как перечное масло, или природные подсластители, или сахарин, или другие искусственные подсластители и т.п., могут быть добавлены. Если это необходимо, дозированные лекарственные средства для перорального введения могут быть микроинкапсулированными. Препарат также может быть приготовлен для продления или замедления высвобождения, например с помощью помещения состоящего из частиц материала в оболочку из полимеров, воска или т.п. Соединения формулы (I) и их фармацевтически приемлемые соли могут также быть введены в форме липосомальных систем доставки, таких как малые моноламеллярные везикулы, большие моноламеллярные везикулы и мультиламеллярные везикулы. Липосомы могут быть образованы из множества фосфолипидов, таких как холестерин, стеариламин или фосфатидилхолины. Соединения формулы (I) и их фармацевтически приемлемые соли могут быть также доставлены с помощью моноклональных антител в качестве индивидуальных носителей, к которым прикреплены молекулы соединения. Соединения могут также быть связаны с растворимыми полимерами, такими как носители нацеливаемых лекарств. Такие полимеры могут включать поливинилпирролидон, пиран сополимер, полигидроксипропилметакриламидфенол, полигидроксиэтиласпартамидфенол или полиэтиленоксидполилизин, замещенный остатками пальмитоила. Более того, соединения могут быть связаны с классом биоразлагаемых полимеров, применяемых для достижения контролируемого высвобождения лекарственного препарата, например полимолочная кислота, полиэпсилонкапролактон, полигидроксимасляная кислота, полиортоэфиры, полиацетали, полигидропираны, полицианоакрилаты и поперечно-сшитые или амфипатические блок-сополимеры гидрогелей. Фармацевтические составы, адаптированные для чрескожного введения, могут быть представлены в виде отдельных пластырей, предназначенных для пребывания в тесном контакте с эпидермисом реципиента в течение длительного периода времени. Например, активный ингредиент может поступать из пластыря посредством ионофореза, как в общем описано в Pharmaceutical Research 1986, 3(6), 318. Фармацевтические составы, адаптированные для местного введения, могут быть приготовлены в виде мазей, кремов, суспензий, лосьонов, порошков, растворов, паст, гелей, спреев, аэрозолей или масел. Для лечения глаз или других внешних тканей, например рта и кожи, составы предпочтительно применяют в виде местной мази или крема. Когда состав в виде мази, активный компонент может быть применен с или парафиновой, или смешанной с водой основой мази. Альтернативно, активный компонент может быть введен в крем вместе с основой крема масло-в-воде или с основой вода-в-масле. Фармацевтические составы, адаптированные для местного введения в глаза, включают глазные капли, в которых активный компонент растворен или суспендирован в подходящем носителе, в особенности водном растворителе. Фармацевтические составы, адаптированные для местного введения в рот, включают лепешки, пастилки и растворы для полоскания полости рта. Фармацевтические составы, адаптированные для ректального введения, могут быть приготовлены в виде суппозиториев или клизм. Фармацевтические составы, адаптированные для интраназального введения, в которых носитель является твердым веществом, включают сыпучий порошок, имеющий размер частиц, например, в диапазоне от 20 до 500 мкм, который вводится таким же образом, как и нюхательный порошок, т.е. путем быстрого вдыхания через носовой ход порошка из контейнера, который держат близко к носу. Подходящие составы в случае, когда носитель является жидким для введения в виде интраназального спрея или носовых капель, включают водные или масляные растворы активного ингредиента. Фармацевтические составы, адаптированные для введения путем ингаляции, включают мелкодисперсные пудры и аэрозоли, которые могут быть получены с помощью различных видов дозирующих аэрозолей, небулайзеров или инсуфляторов. Фармацевтические составы, адаптированные для вагинального введения, могут быть представлены в виде пессариев, тампонов, кремов, гелей, паст, пен или спреев. Фармацевтические составы, адаптированные для парентерального введения, включают водные и неводные стерильные растворы для инъекций, которые могут содержать антиоксиданты, буфера, бактериостатические средства, которые делают препарат изотоничным с кровью предполагаемого реципиента; и водные и неводные стерильные суспензии, которые могут включать суспендирующие агенты и загустители. Препараты могут быть представлены в однодозных или многодозных контейнерах, например запаянные ампулы и флаконы, и могут храниться в высушенном сублимацией (лиофилизированном) состоянии, требующем только добавления стерильного жидкого носителя, например воды для инъекций,непосредственно перед применением. Растворы для немедленного приготовления препарата для инъекции и суспензии могут быть приготовлены из стерильных порошков, гранул и таблеток.- 11015827 Необходимо понимать, что наряду с ингредиентами, в частности, упомянутыми выше, препараты могут включать другие агенты, традиционные в области, имеющей отношение к типу рассматриваемого препарата, например таковые, подходящие для перорального введения, могут включать ароматизаторы. В нижеприведенной табл. 1 перечислены некоторые показательные примеры соединений, которые могут быть введены с соединениями настоящего изобретения. Соединения по изобретению могут быть введены с другими обладающими анти-HCV активностью соединениями в комбинированной терапии,либо совместно, либо раздельно, или путем сочетания этих соединений в композиции. Таблица 1 Соединения по изобретению могут также быть использованы в качестве лабораторных реактивов. Соединения могут быть средством обеспечения исследовательских инструментов для проведения опытов по вирусной репликации, разработки опытных систем исследования животных и исследований структурной биологии для дальнейшего углубленного раскрытия механизмов заболевания HCV. Более того, со- 15015827 единения настоящего изобретения пригодны для установления или определения сайта связывания других противовирусных соединений, например, путем конкурентного ингибирования. Соединения по изобретению могут также быть пригодны для лечения или предотвращения вирусной контаминации материалов и, таким образом, снижения риска вирусной инфекции лабораторного или медицинского персонала или пациентов, которые контактируют с такими веществами, как, например,кровь, ткань, хирургические инструменты и костюмы, лабораторные инструменты и костюмы, а также аппараты и материалы для забора и переливания крови. Настоящее изобретение предназначено для охватывания соединений, имеющих формулу (I), полученных посредством способов синтеза или посредством метаболических способов, включая таковые,происходящие в организме человека или животного (in vivo), или способов, происходящих in vitro. Сокращения, используемые в настоящем описании, включая в особености включенные в иллюстративные схемы и последующие примеры, хорошо известны среднему специалисту. Некоторые из сокращений используют как следующие: CDI для 1,1-карбонилдиимидазола; ТГФ для тетрагидрофурана;DBU для 1,8-диазабицикло[5.4.0]ундек-7-ена; TFA для трифторуксусной кислоты; HATU для фосфата O(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония; РуВОР для гексафторфосфата бензотриазол-1-илокси-трис-пирролидинофосфония; MeI для метилиодида; Boc или ВОС для трет-бутоксикарбонила; OtBu для трет-бутокси; ТВМЕ для трет-бутилметилового эфира; Et3N для триэтиламина; DMSO для диметилсульфоксида; Оас для ацетата; DPPA для дифенилфосфорилазида; Me для метила; TBAF для фторида тетрабутиламмония; DMAP для 4-N,N-диметиламинопиридина; tBuLi для трет-бутиллития; LiHMDS для литий гексаметилдисилазид; Tle для трет-бутиллейцина, также ссылаются как трет-бутилглицин; 4BiphMgBr для 4-бифенилмагнийбромида; DCM для дихлорметана; МеО для метокси; EDAC или EDC для гидрохлорида 1-(3-диметиламинопропил)-3-этилкарбодиимида и HOBt для 1-гидроксибензотриазола. Исходные продукты, пригодные для синтеза соединений по настоящему изобретению, известны среднему специалисту и могут быть легко произведены или являются коммерчески доступными. Способы, представленные ниже, раскрыты с целью иллюстрации и не предназначены для ограничения объема заявленного изобретения. Понятно, что может быть необходимо получение такого соединения, в котором функциональная группа является защищенной, используя обычную защитную группу,затем удаляют защитную группу, чтобы обеспечить соединение по настоящему изобретению. Подробности, касающиеся применения защитных групп в соответствии с настоящим изобретением, известны среднему специалисту. Схема II демонстрирует общий способ, в котором соединения формулы (I) создают путем связывания трипептидкарбоновой кислоты (1) с сульфонамидом Р 1'. Указанная реакция связывания требует обработки карбоновой кислоты (1) реагентом связывания, таким как карбонилдиимидазол в растворителе,таком как ТГФ, который может быть нагрет до температуры кипения с обратным холодильником, с последующим добавлением образовавшегося производного соединения (1), с сульфонамидом Р 1', в растворителе, таком как ТГФ или дихлорметан, в присутствии основания, такого как DBU. Схема II Альтернативный способ получения соединений формулы (I) представлен на схеме III. Сульфонамидная частица Р 1' связывается с элементом Р 1, используя способ, примененный на схеме II. С полученного остатка Р 1-Р 1' может затем быть снята защита по его аминоконцам. В этом общем примере применяют Boc-защитную группу, но средним специалистом должно быть признано, что в этом способе может быть применен ряд подходящих аминозащитных групп. Boc защитная группа может быть удалена, используя кислоту, такую как трифторуксусная кислота, в растворителе, таком как дихлорэтан, чтобы обеспечить амин со снятой защитой в виде TFA соли. TFA соль амина может быть непосредственно применена в последующей реакции связывания или как альтернатива TFA соль амина может быть вначале превращена в HCl соль амина и эту HCl соль амина используют в указанной реакции связывания, как показано на схеме III. Связывание HCl соли амина (3) с карбоксильным концом промежуточного соединения Р 4-Р 3-Р 2 может быть выполнено, используя реагенты связывания, такие как HATU, в растворителях,таких как дихлорметан, чтобы обеспечить соединения формулы (I) (4). Альтернативный способ получения соединений формулы (I) представлен на схеме IV. В настоящем описании гидрохлоридную соль Р 1-Р 1' концевого амина (1) связывают со свободной карбоксильной группой элемента Р 2, используя агенты связывания, такие как РуВОР, в присутствии основания, такого как диизопропиламин, и в растворителе, таком как дихлорметан. Полученное промежуточное соединение Р 2-Р 1-Р 1' может быть превращено в соединения формулы (I) путем двухстадийного синтеза, где на первой стадии снимают защиту с аминного конца Р 2, используя кислоту, такую как TFA, в растворителе,таком как дихлорметан. Полученная соль трифторуксусной кислоты может быть связана с карбоксильным концом элемента Р 4-Р 3, используя типовые агенты связывания, такие как РуВОР, в присутствии основания, такого как диизопропиламин, и используя растворители, такие как дихлорметан, чтобы обеспечить соединения формулы (I) (4). Схема IV Промежуточное соединение Р 4-Р 3-Р 2, используемое в указанных выше схемах, может быть сформировано, как описано в последующем описании данного способа, показанного на общей схеме V. На указанной схеме свободный карбоксильный конец Р 4-Р 3 промежуточного соединения (1) может быть связан с аминоконцом элемента Р 2, чтобы обеспечить дипептид (2) Р 4-Р 3-Р 2. С карбоксильного конца промежуточного соединения Р 4-Р 3-Р 2 может быть снята защита путем омыления эфирной группы, чтобы обеспечить Р 4-Р 3-Р 2 в виде свободной карбоновой кислоты (3). Промежуточные соединения, подобные (3), могут быть превращены в соединения формулы (I), используя способы, описанные в настоящем описании. Соединения формулы (I) могут также быть превращены в другие соединения формулы (I), как описано в настоящем описании. Пример такого способа представлен на схеме VI, где соединение формулы(I) (1), которое несет Boc-группу по положению Р 4, преобразуют в соединение формулы (I) (3), где указанное соединение несет группу мочевины по положению Р 4. Превращение соединения (1) в соединение(3) может быть осуществлено путем двухстадийного синтеза, на первой стадии которого превращают соединение (1) в амин (2) путем обработки соединения (1) кислотой, такой как TFA, в растворителе, таком как дихлорметан. Полученная TFA соль амина может быть обработана изоцианатом, таким как третбутилизоцианат, в присутствии одного эквивалента основания, чтобы обеспечить соединение формулы(I) (3), где Р 3 остаток закрывается мочевиной. Как ранее отмечено, средним специалистом будет признано, что промежуточное соединение (2) может быть использовано в виде исходных продуктов для получения соединений формулы (I), где Р 3 группа закрывается амидом или сульфонамидом или тиомочевиной или сульфамидом. Создание указанных соединений формулы (I) может быть выполнено, используя типовые условия образования указанных Р 4 функциональностей из аминов. Схема VI При получении соединений формулы (I) конец Р 1' вводят в молекулу, используя один из общих способов, представленный выше и описанный более подробно ниже. В некоторых примерах элементы Р 1' представляют собой циклоалкил- или алкилсульфонамиды, которые являются коммерчески доступными или могут быть получены из соответствующих алкил- или циклоалкилсульфонилхлоридов путем обработки указанного сульфонилхлорида аммиаком. Альтернативно, эти сульфонамиды могут быть синтезированы, используя общий способ, представленный на схеме VII. На указанной схеме коммерчески доступный 3-хлорпропилсульфонилхлорид (1) преобразуют в соответствующий защищенный сульфонамид, как, например, путем обработки трет-бутиламином. Полученный сульфонамид (2) затем преобразуют в соответствующий циклоалкилсульфонамид путем обработки двумя эквивалентами основания, такого как бутиллитий, в растворителе, таком как ТГФ. С полученного циклоалкилсульфонамида может быть снята защита путем обработки кислотой, чтобы обеспечить требуемый незащищенный циклоалкилсульфонамид. Замещенные циклоалкилсульфонамиды могут также быть введены в соединения формулы (I), используя модификацию указанный выше методики. Например, промежуточное соединение 2 схемы VIII может быть обработано 2 экв. основания, такого как бутиллитий, и полученная реакционная смесь может быть обработана электрофилом, таким как метилиодид, чтобы обеспечить замещенный циклоалкилсульфонамид (3). С этого промежуточного соединения (3) может быть снята защита на N-конце, и полученное соединение (4) используют как промежуточное соединение при получении соединений формулы (I). Схема VIII Промежуточные соединения Р 1', применяемые для получения соединений формулы (I), являются в некоторых случаях производными от сульфамидных производных. В этих случаях промежуточные сульфамиды являются доступными с помощью ряда направлений синтеза, как, например, с помощью направления, представленного на схеме IX. Схема IX Сульфамоилхлорид (2) может быть получен in situ путем добавления воды (например, 1 экв.) к хлорсульфонилизоцианату 1 (например, 1 экв.) в растворителе, таком как ТГФ, пока поддерживается пониженная температура, такая как -20 С, и полученному раствору дают возможность нагреться до температуры 0 С. К этому раствору добавляют основание, такое как безводный триэтиламин (например, 1 экв.), затем добавляют амин (например, 1 экв.). Реакционную затем смесь нагревают до комнатной температуры, фильтруют и фильтрат концентрируют, чтобы получить требуемые сульфамиды (3). Сульфамиды могут быть введены в соединения формулы (I), следуя направлению синтеза, представленному на схеме X. На указанной схеме элемент (1) карбоновой кислоты Р 1 обрабатывают активирующим агентом, таким как CDI. В отдельной колбе к указанному выше описанному раствору сульфамида добавляют сильное основание и полученную реакционную смесь перемешивают в течение нескольких часов, после чего эту реакционную смесь добавляют в колбу, содержащую активированную карбоновую кислоту, чтобы обеспечить ацилсульфамидные производные (2). Промежуточные соединения, подобные 2, могут быть превращены в соединения формулы (I), как описано в настоящем описании. Схема X Следует отметить, что ацилсульфамидные производные могут также быть получены из трипептид- 19015827 карбоновых кислот на первой стадии способа, как определено на схеме XI. Схема XI Элементы Р 1, используемые для получения соединений формулы (I), являются в некоторых случаях коммерчески доступными, но в других случаях их синтезируют, используя способы, описанные в настоящем описании, и затем их вводят в соединения формулы (I), используя способы, описанные в настоящем описании. Замещенные Р 1 циклопропиламинокислоты могут быть синтезированы, следуя общему способу, представленному на схеме XII. Обработка коммерчески доступного или легко синтезируемого имина (1) 1,4-дигалогенбутеном (2) в присутствии полученного основания обеспечивает получение имина (3). Гидролиз кислоты (3) затем обеспечивает соединение (4) в виде основного продукта, которое имеет аллильный заместитель в synположении, к карбоксильной группе. Остаток амина (4) может быть защищен, используя Boc-группу,обеспечивает полностью защищенную аминокислоту (5). Это промежуточное соединение представляет собой рацемат, который может быть разделен с помощью энзиматического способа, в котором эфирный остаток (5) расщепляют с помощью протеазы, чтобы обеспечить соответствующую карбоновую кислоту. Не основываясь ни на какой отдельной теории, следует полагать, что эта реакция является селективной по той причине, что один из энантиомеров вступает в реакцию с более высокой скоростью, чем его зеркальное отображение, обеспечивая кинетическое разделение промежуточного рацемата. В воплощениях примеров, представленных в настоящем описании, стереоизомером для интегрирования в соединения формулы (I) является соединение (5 а), которое имеет (1R, 2S) стереохимию. В присутствии фермента этот энантиомер не подвергается эфирному расщеплению, и, таким образом, этот энантиомер (5 а) выделяют из реакционной смеси. Тем не менее, другой энантиомер (5b), который содержит (1S, 2R) стереохимию, подвергается эфирному расщеплению, т.е. гидролизу, чтобы обеспечить свободную кислоту (6). При завершении этой реакции сложный эфир (5 а) может быть отделен от кислотного продукта (6) с помощью обычных способов, таких как, например, способы водного экстрагирования или хроматография. Схема XII Неограничивающие методики получения промежуточных соединений Р 2 и соединений формулы (I)- 20015827 показаны на схемах ниже. Указанные промежуточные соединения, условия реакции и способы получения в конкретных примерах являются широкоприменимыми к соединениям с другими конфигурациями замещения. Например, синтез элементов Р 2, представленный в соединениях формулы (I) схемы XIII, может быть осуществлен, следуя определенному направлению синтеза. На указанной схеме легкосинтизируемый или коммерчески доступный N-Boc-4-оксо-L-пролин или N-Cbz-4-оксо-L-пролин обрабатывают металлоорганическим агентом, таким как реагент Гриньяра (или альтернативно алкил или ариллитиевые классы или альтернативно алкил или арилцинковые реагенты), чтобы обеспечить промежуточное соединение (2), в котором по С 4 положению пролина находится R3 заместитель и свободная третичная гидроксильная группа. Спиртовая группа промежуточного соединения (2) может затем функционировать так,чтобы обеспечить требуемую R8 функциональность. В этом способе промежуточный спирт (2) может быть включен в ряд общепринятых технологических процессов. Например, спирт (2) может быть ацилирован, чтобы обеспечить сложные эфиры, карбаматы или карбонаты; алкилирован, чтобы обеспечить эфиры, и фосфонилирован, чтобы обеспечить фосфаты. Для превращения промежуточного соединения(2) в промежуточное соединение (3), как показано на схемеы XIII, может быть необходимо вначале защитить карбоксильную группу (2). Химия на основе спиртовой функции описана для данного объекта в стандартных методиках, такой как Comprehensive Organic Transformations: A Guide to Functional Group Preparations, Второе издание, byRichard Larock. Этот текст издан Wiley and Sons. В нем выделены конкретные ссылки и обзоры, которые средним специалистом могут быть легко применены для превращения промежуточного соединения (2) по схеме XIII в промежуточное соединение (3). Например, условия и подходящие ссылки для образования эфиров из спиртов могут быть найдены на стр. 883-929 текста Ларока (Larock's). Более конкретно,условия и конкретные ссылки на стр. 890-894 являются более подходящими для практического осуществления настоящего изобретения. Также условия для превращения спиртов в соответствующие производные сложного эфира могут быть найдены на стр. 1952 и 1955 текста Ларока. Кроме того, химия, описанная в Journal of Organic Chemistry 2001, volume 66, page 8926 и содержащихся там подходящих цитируемых ссылках, является полезной для практического осуществления настоящего изобретения. Схема XIII Альтернативный подход к синтезу соединений формулы (I), как описано на схеме XIII, представлен на схеме XIV. Введиние функциональных групп по С 4 положению пролиновой группы происходит через синтез по Гриньяру с участием последнего промежуточного соединения (5), обеспечивает промежуточное соединение 6, которое может затем быть превращено в соединения формулы (I). Промежуточное соединение (5) является доступным через 4-стадийную последовательность, начинающуюся с коммерчески доступного промежуточного соединения (1), первая стадия которой касается связывания соединения(1) с промежуточными реагентами связывания Р 1-Р 1', установленными в технологии. Снятие защиты NBoc-группой в присутствии кислотного катализатора с промежуточного соединения (2) обеспечивает свободный промежуточный амин (3), который затем связывают с фрагментом Р 3-Р 4, чтобы обеспечить промежуточное соединение (4). Селективное окисление гидроксильной группы С 4 в промежуточном соединении (4), чтобы обеспечить промежуточное соединение (5), может быть выполнено, используя реагенты окисления, такие как реагент Десс-Мартина. Схема XIV Следует отметить, что присоединение металлоорганических агентов к кетоновому остатку пролинового производного 1 (схема XVI) хорошо известно в технологии. Например, Груби и коллеги (J. Org.Chem. 2001, 66, 3593) описали присоединение фенилмагнийбромида в промежуточные соединения общей структуры 1 (схема XVI). Эти находки являются доказательством того, что оптимальные выходы требуемых 1,2 продуктов присоединения (2, схемы XVI) получают, когда трет-бутиловую эфирную группу применяют как защитную группу карбоксильного остатка С 2. Кроме того, эта работа обеспечивает очевидное доказательство в виде данных кристаллографии, полученных с помощью рентгеновских- 22015827 лучей относительно стереохимического результата этой реакции присоединения. Конкретно, в результате вышеупомянутого присоединения реагента Гриньяра к кетону 1 получают единственный продукт, в котором гидроксильная группа С 4 и карбоксильная группа С 2 предполагают относительную synориентацию около пятичленного кольца. Из этого структурного определения лицо селективности при присоединении R3M к кетону 1 установлено как -положение в контексте структуры 1 схемы XVI. То есть металлоорганка селективно присоединяется к обратной стороне (нижняя часть) карбонила в соединениии 1, чтобы обеспечить соответствующие третичный спирт (2) с представленной стереохимией. Схема XVI Упомянутая выше работа Груби описывает присоединение конкретного реагента Гриньяра к производным соединения 1 (схема XVI). Тем не менее, присоединение различных реагентов Гриньяра в пролин 1 представлено в примерах настоящего изобретении. Основная литература, которая описывает введение металлоорганических агентов, включая реагенты Гриньяра, в кетоны, значительна и получена в итоге в общем обзоре в технологии, таком как Comprehensive Organic Functional Group Transformations.Volume 2: Synthesis: Carbon with one heteroatom attached by a single bond. Editor in Chief Alan. R. Katritzky,et al. 1995. Chapter 2.02, page 37. Этот класс реакций также описан в Comprehensive Organic Synthesis.Editor in Chief Barry M Trost, Volume 1: Additions to C-X pi-bonds (part 1). 1991. Последние исследования в технологии обеспечивают условия для дальнейшей оптимизации реагентов Гриньяра в реакциях присоединения к кетону, и эти работы могут быть полезны в настоящем изобретении. Например, Ишихара и коллеги (Org. Lett. 2005, Vol. 7, No. 4, 573) недавно описали образование и применимость магнийатных комплексов. Магнийатный комплекс R3MgLi представляет собой производную от реагентов Гриньяра и алкиллития. Как описано у Ишихара, эти комплексы обеспечивают превосходные выходы продуктов 1,2 в реакциях присоединения к кетонам. В отдельном исследовании Кнохель и коллеги (Angew. Chem Int. Ed. 2006, 45, 497) описали применение растворимых лантаноидных солей,таких как LnCl3, вместе с магнийорганическими реагентами. Присутствие этой лантаноидной соли приводит к улучшению эффективности реакции 1,2 присоединения к карбонильным соединениям. Эти рабо- 23015827 ты и ссылки, представленные в настоящем описании, отражают современное состояние уровня техники относительно оптимизации реакции Гриньяра с помощью простого присоединения к карбонильным соединениям и служат важным источником информации в настоящем изобретении. Должен также быть отмечен диапазон металлорганических реагентов, участвующих в реакциях присоединения к кетонам. Включенными в эту основную работу являются реагенты, такие как ариллитиевые, алкиллитиевые и гетероариллитивые реагенты, которые хорошо известны для введения 1,2 положения к карбонильным остаткам. Например, в недавнем исследовании Дондони и коллег (7. Org.Chem. 2005, 70, 9257) бензотиоазол литиируют, используя BuLi, и полученные С 2-литиевые группы присоединяют к 1,2 положениям лактона. В качестве аналогии литиирования бензотиазола следует ожидать присоединения по 1,2 положениям к кетону 1 схемы XVI, чтобы обеспечить промежуточное соединение,подобное 2 а. Средним специалистом должно быть признано, что металлоорганические реагенты, производные от гетероциклов, такие как оксазолы, тиазолы и имидазолы, могут также участвовать в 1,2 реакциях присоединения к кетону 1. Имеется значительное количество основной литературы, которая определяет уникальные условия, применяемые для каждый из этих гетероциклических систем, и эта информация является легко доступной среднему специалисту. Например, применение металлоорганических реагентов,производных от бензоксазола или оксазола, в реакциях присоединения к кетонам требует применение литиймагнезатов. Конкретно указанное исследование Байч и партнеров описано в J. Org. Chem., 2005, 70,5190. Введение бензоксазола в кетон 1 схемы XVI должен обеспечить доступ к промежуточным соединениям, подобным 2b. Имеется существенный литературный прецедент для присоединения к кетонам, используя широкий класс металлоорганических реагентов, производных от гетероциклов. Например, работа Бехинида и партнеров (Tet. Lett. 42, 2001, 647) описывает образование литиированного бензимидазола и его введение в простой лактон. По аналогии, применение этого литиированного бензимидазола в реакциях присоединения к кетону 1 схемы XVI должна обеспечить доступ к промежуточным соединениям, подобным 2 с. Кроме того, недавнее исследование Кавасаки и партнеров (Bioorganic и Medicinal Chem. Lett. 13, 2003,87) описывает образование ряда литиированных гетероароматических соединений и реакции их присоединения в активированным амидам. По аналогии, применение этих литиированных гетероароматических промежуточных соединений в реакциях присоединения к кетону 1 схемы XVI должно обеспечить доступ к промежуточным соединениям 2d-2k. Применение металлоорганических производных из биарильных или гетероариларильных систем в реакциях присоединения 1,2 к кетону 1 также имеет отношение к настоящему изобретению. Введение этого класса металлоорганических реагентов в кетон 1 должен обеспечить доступ к промежуточным соединениям, подобным 21 и 2m. Следует отметить, что в иллюстративных примерах по настоящему изобретению может быть необходим синтез металлоорганических биарилов или гетероарилов для последующего применения в реакциях присоединения к кетону 1 по схеме XVI. Средний специалист знаком с существенным количеством основной литературы, которая описывает получение металлоорганики этого класса и их предшественников. Например, недавняя статья Шиншилла и партнеров (Chem. Rev. 2004,104, 2667) описывает получение металлированных гетероциклов и их применение. Базовой химией для получения биарильных или гетероариларильных систем часто является реакция связывания, подобная Сузуки. Основная литература, издаваемая Грегори Фу, описывает современное состояние в таких реакциях связывания и подмножество этих ссылок включают следующиее: JACS 2004, 126, 1340; JACS,2002,124, 13662; Angew. Chem. Int. Ed. 2002, 41, No. 11, 1945; Angew. Chem. Int. Ed. 2002, 41, No.20, 3910;JACS 2002, 122, 4020; JACS 2001, 123, 10099; Org. Lett. 2001, Vol. 3, No. 26, 4295; Angew. Chem. Int. Ed. 1998, 37, No.24, 3387. В дополнение к этой основной работе легко доступными являются критические обзоры в данной области, такие как Rossi in Synthesis 2004, No. 15, 2419. Примеры Настоящее изобретение будет описано в связи с определенными воплощениями, которые не предназначены для ограничения его области. С другой стороны, настоящее изобретение охватывает все альтернативы, модификации и эквиваленты, которые могут быть включены в объем формулы изобретения. Таким образом, последующие примеры, которые включают конкретные воплощения, будут демонстрировать осуществление настоящего изобретения, при этом должно быть понятно, что примеры предназначены для иллюстративных целей определенных воплощений и представлены, чтобы обеспечить, как полагают, более полезное и легко понимаемое из описания воплощения приведенных методик и концептуальных аспектов. Процентное содержание раствора выражают отношением веса к объему и соотношения растворов выражают отношением объема 2 к объему, за исключением иным образом установленного. Спектр ядерного магнитного резонанса (ЯМР) регистрируют или на Bruker 300, 400 или 500 МГц спектрометре; химические сдвигивыражают в частях на миллион. Флеш-хроматографию проводят на силикагеле(SiO2) в соответствии с методикой флеш-хроматографии по Штилле (Still's) (J. Org. Chem. 1978, 43, 2923). Промежуточные соединения, описанные в примерах, представленные в настоящем описании, могут быть применены для синтеза соединений формулы I.- 24015827 Пример 1. Получение промежуточных соединений Р 1'. 1. Получение циклопропилсульфонамида. Способ 1: Стадия 1. трет-Бутиламин (3,0 моль, 315 мл) растворяют в ТГФ (2,5 л). Раствор охлаждают до температуры-20 С. Медленно добавляют 3-хлорпропансульфонилхлорид (1,5 моль, 182 мл). Реакционной смеси дают возможность нагреться до комнатной температуры и перемешивают в течение 24 ч. Смесь фильтруют и фильтрат концентрируют в вакууме. Остаток растворяют в CH2Cl2 (2,0 л). Полученный раствор промывают 1,0 М раствором HCl (1,0 л), водой (1,0 л) и рассолом (1,0 л), сушат над Na2SO4, фильтруют и концентрируют в вакууме, что дает твердое вещество слегка желтого цвета, который перекристаллизовывают из гексана, чтобы обеспечить продукт в виде твердого вещества белого цвета (316,0 г, 99%). 1(б, 1H). Стадия 2. К раствору продукта со стадии 1 (2,14 г, 10,0 ммоль) в ТГФ (100 мл) добавляют n-BuLi (2,5 М в гексане, 8,0 мл, 20,0 ммоль) при температуре -78 С. Реакционной смеси дают возможность нагреться до комнатной температуры в течение более часа и концентрируют в вакууме. Остаток распределяют между этилацетатом и водой (200 мл каждый). Отделенную органическую фазу промывают рассолом, сушат над Na2SO4, фильтруют и концентрируют в вакууме. Остаток повторно перекристаллизовывают из гексана, чтобы обеспечить требуемый продукт в виде твердого вещества белого цвета (1,0 г, 56%). 1 Н ЯМР (CDCl3)0.98-1.00 (м, 2 Н), 1.18-1.19 (м, 2 Н), 1.39 (с, 9 Н), 2.48-2.51 (м, 1H), 4.19 (б, 1H). Стадия 3. Раствор продукта со стадии 2 (110 г, 0,62 ммоль) в TFA (500 мл) перемешивают при комнатной температуре в течение 16 ч. Летучие вещества удаляют в вакууме. Остаток повторно перекристаллизовывают из смеси этилацетат/гексан (60 мл/240 мл), чтобы обеспечить требуемый продукт в виде твердого вещества белого цвета (68,5 г, 91%). 1 Н ЯМР (DMSO-d6)0.84-0.88 (м, 2 Н), 0.95-0.98 (м, 2 Н), 2.41-2.58 (м, 1H), 6.56 (б, 2 Н). Способ 2. В раствор 100 мл ТГФ, охлажденный до температуры 0 С, пробулькивают газообразный аммиак,пока не произойдет насыщение. К указанному раствору добавляют раствор 5 г (28,45 ммоль) циклопропилсульфонилхлорида (приобретенный от Array Biopharma) в 50 мл ТГФ. Раствор нагревают до комнатной температуры в течение ночи и перемешивают еще в течение дня. Смесь концентрируют, пока не останется 1-2 мл растворителя, и выливают на 30 г плотный слой из SiO2 (элюируют от 30 до 60% смесью этилацетат/гексан), чтобы обеспечить 3,45 г (100%) циклопропилсульфонамида в виде твердого вещества белого цвета. Раствор продукта со стадии 1 (4,3 г, 20 ммоль) растворяют в сухом ТГФ (100 мл) и охлаждают до температуры -78 С. К указанному раствору медленно добавляют н-бутиллития (17,6 мл, 44 ммоль, 2,5 М в гексане). Баню с сухим льдом удаляют и полученную реакционную смесь нагревают до комнатной температуры в течение более 1,5 ч. Указанную смесь охлаждают до температуры -78 С и добавляют раствор н-бутиллития (20 ммоль, 8 мл, 2,5 М в гексане). Реакционную смесь нагревают до комнатной температуры, охлаждают до температуры -78 С в течение более 2 ч и обрабатывают чистым раствором метилиодида (5,68 г, 40 ммоль). Реакционную смесь нагревают до комнатной температуры в течение ночи,затем гасят насыщенным раствором NH4Cl (100 мл) при комнатной температуре и экстрагируют этилацетатом (100 мл). Органическую фазу промывают рассолом (100 мл), сушат (MgSO4), фильтруют и концентрируют в вакууме, чтобы обеспечить масло желтого цвета, которое перекристаллизовывают из гексана,чтобы обеспечить требуемый продукт в виде слегка твердого вещества желтого цвета (3,1 г, 81%). 1 Н ЯМР (CDCl3)0.79 (м, 2 Н), 1.36 (с, 9 Н), 1.52 (м, 2 Н), 1.62 (с, 3 Н), 4.10 (широкий с, 1 Н). Стадия 3. Получение 1-метилциклопропилсульфонамида. Раствор продукта со стадии 2 (1,91 г, 10 ммоль) растворяют в TFA (30 мл) и полученную реакционную смесь перемешивают при комнатной температуре в течение 16 ч. Растворитель удаляют в вакууме,чтобы обеспечить масло желтого цвета, которое перекристаллизовывают из смеси этилацетат/гексан (1:4,40 мл), чтобы обеспечить требуемый продукт в виде твердого вещества белого цвета (1,25 г, 96%). 1 Н ЯМР (CDCl3)0.84 (м, 2 Н), 1.41 (м, 2 Н), 1.58 (с, 3 Н), 4.65 (широкий с, 2 Н). Вычислено для C4H9NO2S: С, 35.54; Н, 6.71; N, 10.36. Найдено: С, 35.67; Н, 6.80; N, 10.40. 2b. Получение 1-пропилциклопропилсульфонамида. Указанное соединение получают, используя методику, описанную для получения 1 метилциклопропилсульфонамида, заменяя метилиодид на пропилгалоид на второй стадии данной методики. 2 с. Получение 1-аллилциклопропилсульфонамида. Указанное соединение получают с 97% выходом в соответствии с методикой, описанной для синтеза N-трет-бутил-(1-метил)циклопропилсульфонамида, используя 1,25 экв. аллилбромида в качестве электрофила. Соединение используют на следующей стадии без дополнительной очистки. 1 Н ЯМР (CDCl3)0.83 (м, 2 Н), 1.34 (с, 9 Н), 1.37 (м, 2 Н), 2.64 (д, J = 7.3 Гц, 2 Н), 4.25 (широкий с,1H), 5.07-5.10 (м, 2 Н), 6.70-6.85 (м, 1 Н). Указанное соединение получают с 40% выходом из продукта со стадии 1 в соответствии с методикой, описанной для синтеза 1-метилциклопропилсульфонамида. Соединение очищают с помощью хроматографии на колонке с SiO2, используя 2% метанол в дихлорметане в виде элюента. 1 Н ЯМР (CDCl3)0.88 (м, 2 Н), 1.37 (м, 2 Н), 2.66 (д, J = 7.0 Гц, 2 Н), 4.80 (с, 2 Н), 5.16 (м, 2 Н), 5.82 (м,1H); 13 С ЯМР (CDCl3)11.2, 35.6, 40.7, 119.0, 133.6. 2d. Получение 1-бензилциклопропилсульфонамида. Указанное соединение получают с 60% выходом, используя методику, описанную для синтеза Nтрет-бутил-(1-метил)циклопропилсульфонамида, за исключением того, что используют 1,05 экв. бензилбромида, а затем растирают в порошок с 10% этилацетатом в гексане. 1 Н ЯМР (CDCl3)0.92 (м, 2 Н), 1.36 (м, 2 Н), 1.43 (с, 9 Н), 3.25 (с, 2 Н), 4.62 (широкий с, 1H), 7.29-7.36 Указанное соединение получают с 66% выходом из N-трет-бутил-(1-бензил)циклопропилсульфонамида, используя методику, описанную для синтеза 1-метилциклопропилсульфонамида, с последующей перекристаллизацией из минимального количества 10% этилацетата в гексане. 1 Указанное соединение получают с 84% выходом, используя методику, описанную для синтеза Nтрет-бутил-(1-метил)циклопропилсульфонамида, за исключением того, что используют 1,30 экв. циклогексанона с последующей перекристаллизацией из минимального количества 20% этилацетата в гексане. 1 Н ЯМР (CDCl3)1.05 (м, 4 Н), 1.26 (м, 2 Н), 1.37 (с, 9 Н), 1.57-1.59 (м, 6 Н), 1.97 (м, 2 Н), 2.87 (широкий с, 1H), 4.55 (широкий с, 1H). Стадия 2. Получение 1-(1-циклогексенил)циклопропилсульфонамида.N-трет-бутил-[1-(1-гидрокси)циклогексил]циклопропилсульфонамида, используя методику, описанную для синтеза 1-метилциклопропилсульфонамида, с последующей перекристаллизацией из минимального количества этилацетата и гексана. 1 Н ЯМР (DMSO-d6)0.82 (м, 2 Н), 1.28 (м, 2 Н), 1.51 (м, 2 Н), 1.55 (м, 2 Н), 2.01 (с, 2 Н), 2.16 (с, 2 Н),- 27015827 5.89 (с, 1H), 6.46 (с, 2 Н); 13 С ЯМР (DMSO-d6)11.6, 21.5, 22.3, 25.0, 27.2, 46.9, 131.6, 132.2; Указанное соединение получают с 66% выходом, используя методику, описанную для синтеза Nтрет-бутил-(1-метил)циклопропилсульфонамида, за исключением того, что 1,2 экв. метилбензоата используют в качестве электрофила. Соединение очищают с помощью хроматографии на колонке с SiO2,используя от 30 до 100% дихлорметана в гексане. 1 Н ЯМР (CDCl3)1.31 (с, 9 Н), 1.52 (м, 2 Н), 1.81 (м, 2 Н), 4.16 (широкий с, 1H), 7.46 (м, 2 Н), 7.57 (м,1H), 8.05 (д, J = 8.5 Гц, 2 Н). Стадия 2. Получение 1-бензоилциклопропилсульфонамида. Указанное соединение получают с 87% выходом из N-трет-бутил-(1-бензоил)циклопропилсульфонамида, используя способ, описанный для синтеза 1-метилциклопропилсульфонамида, с последующей перекристаллизацией из минимального количества этилацетата в гексане. 1 Н ЯМР (DMSO-d6)1.39 (м, 2 Н), 1.61 (м, 2 Н), 7.22 (с, 2 Н), 7.53 (т, J = 7.6 Гц, 2 Н), 7.65 (т, J = 7.6 Гц, 1 Н), 8.06 (д, J = 8.2 Гц, 2 Н); 13 С ЯМР (DMSO-d6)12.3, 48.4, 128.1, 130.0, 133.4, 135.3, 192.0. 2g. Получение N-трет-бутил-(1-фениламинокарбокси)циклопропилсульфонамида. Указанное соединение получают с 42% выходом, используя методику, описанную для синтеза Nтрет-бутил-(1-метил)циклопропилсульфонамида, используя 1 экв. фенилизоцианата с последующей перекристаллизацией из минимального количества этилацетата в гексане. 1 Н ЯМР (CDCl3)1.38 (с, 9 Н), 1.67-1.71 (м, 4 Н), 4.30 (широкий с, 1H), 7.10 (т, J = 7.5 Гц, 1H), 7.34(т, J = 7.5 Гц, 2 Н), 7.53 (т, J = 7.5 Гц, 2 Н). 3. Получение С 1-замещенных циклопропансульфонамидов. Применение N-Boc защитной группы. 3 а. Получение циклопропилсульфониламино-трет-бутилкарбамата, ключевого промежуточного соединения при получении С 1-замещенных циклопропилсульфонамидов. Раствор 3-хлорпропансульфонилхлорида (55 г, 310.7 ммоль) растворяют в ТГФ (200 мл) и добавляют по каплям в течение более 30 мин к раствору NH4OH (200 мл) при температуре 0 С. Реакционную смесь нагревают до комнатной температуры, перемешивают в течение 1 ч и водный слой экстрагируют дихлорметаном (4500 мл). Объединенные экстракты промывают 1N раствором HCl (150 мл), водой (150 мл), сушат над MgSO4, фильтруют и концентрируют в вакууме. Сырое твердое вещество повторно перекристаллизовывают из минимального количества дихлорметана в гексане, чтобы обеспечить требуемый продукт в виде твердого вещества белого цвета (45,3 г, 93%). 1 Н ЯМР (CDCl3)2.34 (м, 2 Н), 3.32 (т, J = 7.3 Гц, 2 Н), 3.70 (т, J = 6.2 Гц, 2 Н), 4.83 (с, 2 Н); 13 С ЯМР (CDCl3)27.10, 42.63, 52.57. Стадия 2. Получение 3-хлорпропилсульфониламин трет-бутилкарбамата. Раствор продукта со стадии 1 (30,2 г, 191,5 ммоль), триэтиламин (30,2 мл, 217,0 ммоль) и 4-DMAP(2,40 г, 19,6 ммоль) в дихлорметане (350 мл) при температуре 0 С обрабатывают по каплям раствором ди-трет-бутилдикарбоната (47,2 г, 216,9 ммоль) в дихлорметане (250 мл) в течение более 30 мин. Реакционную смесь нагревают до комнатной температуры, перемешивают еще 3 ч и промывают 1N растворомHCl (300 мл), водой (300 мл) и рассолом (300 мл), сушат над MgSO4, фильтруют и концентрируют в вакууме, чтобы обеспечить сырой продукт. Указанный продукт растирают в порошок с 70 мл 5% дихлорметана в гексане, чтобы обеспечить требуемый продукт в виде твердого вещества грязно-белого цвета Раствор н-бутиллития (74,7 мл, 119,5 ммоль, 1,6 М в гексане) растворяют в сухом ТГФ (105 мл) и охлаждают до температуры -78 С в атмосфере аргона. К указанному раствору добавляют по каплям раствор продукта со стадии 2 (14 г, 54,3 ммоль) в сухом ТГФ (105 мл) в течение более 20-30 мин. Баню с сухим льдом удаляют и реакционной смеси дают возможность нагреться до комнатной температуры в течение более 2 ч. Реакционную смесь гасят ледяной уксусной кислотой (3,4 мл), концентрируют в вакууме и распределяют между дихлорметаном (100 мл) и водой (100 мл). Органическую фазу промывают рассолом (100 мл), сушат (MgSO4), фильтруют и концентрируют в вакууме, чтобы обеспечить требуемый продукт в виде воскообразного твердого вещества грязно-белого цвета (12,08 г, 100%). 1 Н ЯМР (CDCl3)1.10 (м, 2 Н), 1.34 (м, 2 Н), 1.50 (с, 9 Н), 2.88 (м, 1H), 7.43 (с, 1H). 13 С ЯМР (CDCl3)6.21, 28.00, 31.13, 84.07, 149.82. 3b. Получение 1-метоксиметилциклопропилсульфонамида. Стадия 1. Получение 1-метоксиметилциклопропилсульфониламин трет-бутилкарбамата. К раствору циклопропилсульфониламин трет-бутилкарбамата (1,0 г, 4,5 ммоль), растворенному в ТГФ (30 мл), охлажденному до температуры -78 С, добавляют н-бутиллития (6,4 мл, 10,2 ммоль, 1,6 М в гексане) и полученную реакционную смесь перемешивают в течение 1 ч. К указанному раствору добавляют чистый раствор хлорметилметилового эфира (0-40 мл, 5,24 ммоль) и полученной смеси медленно дают возможность нагреться до комнатной температуры в течение ночи. Значение рН раствора доводят до 3, используя 1N водный раствор HCl, и затем экстрагируют этилацетатом (450 мл порции). Объединенные экстракты сушат (MgSO4), фильтруют и концентрируют, что дает 1-метоксиметилциклопропилсульфониламин трет-бутилкарбамат в виде воскообразного твердого вещества (1,20 г, 100%), которое переносят непосредственно на следующую стадию без дополнительной очистки. 1 Н ЯМР (CDCl3)1.03 (м, 2 Н), 1.52 (с, 9 Н), 1.66 (м, 2 Н), 3.38 (с, 3 Н), 3.68 (с, 2 Н), 7.54 (с, 1H); 13 С ЯМР (CDCl3)11.37, 28.29, 40.38, 58.94, 73.43, 83.61, 149.57. Стадия 2. Получение 1-метоксиметилциклопропилсульфонамида. Раствор 1-метоксиметилциклопропилсульфониламин трет-бутилкарбамата (1,14 г, 4,30 ммоль) растворяют в растворе 50% TFA/дихлорметан (30 мл) и перемешивают при комнатной температуре в течение 16 ч. Растворитель удаляют в вакууме и остаток хроматографируют над 80 г SiO2 (элюируя смесью от 0 до 60% этилацетат/гексан), что дает 1-метоксиметилциклопропилсульфонамид в виде твердого вещества белого цвета (0,55 г, 77% общий выход в течение двух стадий). 1 Н ЯМР (CDCl3)0.95 (м, 2 Н), 1.44 (м, 2 Н), 3.36 (с, 3 Н), 3.65 (с, 2 Н), 4.85 (с, 2 Н); 13 С ЯМР (CDCl3)11.17, 40.87, 59.23, 74.80;
МПК / Метки
МПК: A61P 31/00, A61K 31/401, C07K 5/08
Метки: вируса, гепатита, ингибиторы
Код ссылки
<a href="https://eas.patents.su/30-15827-ingibitory-virusa-gepatita-s.html" rel="bookmark" title="База патентов Евразийского Союза">Ингибиторы вируса гепатита с</a>
Ингибиторы вируса гепатита с (hcv)

Номер патента: 15415
Опубликовано: 31.08.2011
Авторы: Граупе Михаель, Линк Джон О., Венкатарамани Чандрэсикар
МПК: A61K 31/74, A61K 47/00, A61K 31/00...
Метки: вируса, гепатита, hcv, ингибиторы
Формула / Реферат:
1. Соединение формулы (I)где Е представляет собой -COCONHR6, где R6представляет собой водород, алкил, циклоалкил, аралкил или гетероаралкил, где ароматическое кольцо необязательно замещено одним или двумя галоидами;R1 представляет собой алкил, циклоалкилалкил, где алифатические или алициклические группы в R1необязательно замещены одним или двумя радикалами Rb, которые независимо выбирают из гидрокси, алкокси, арилокси, гетероарилокси, алкилтио,...
Ингибиторы вируса гепатита с

Номер патента: 15756
Опубликовано: 30.12.2011
Авторы: Бэчэнд Кэрол, Лавой Рико, Гуд Эндрю С., Ван Гань, Лопез Омар Д., Рудигер Эдвард Х., Джеймс Клинт А., Белема Маконен, Снайдер Лоуренс Б., Сен-Лоран Денис Р., Мартель Ален, Ромайн Джеффри Ли, Нгуен Ван Н., Лэнгли Дэвид Р., Минвелл Николас А., Гудрич Джейсон, Деон Дэниел Х., Ян Фукан, Хаманн Лоуренс Г.
МПК: A61K 31/4178, A61K 31/4025, A61P 31/12...
Метки: ингибиторы, гепатита, вируса
Формула / Реферат:
1. Соединение формулы (I)или его фармацевтически приемлемая соль, гдеm и n независимо имеют значения 0, 1 или 2;q и s независимо имеют значения 0, 1, 2, 3 или 4;u и v независимо имеют значения 0, 1, 2 или 3;X выбран из О, S, S(O), SO2, CH2, CHR5 и C(R5)2;при условии, что когда n равен 0, X выбран из СН2, CHR5 и C(R5)2;Y выбран из О, S, S(O), SO2, CH2, CHR6 и C(R6)2;при условии, что когда m равен 0, Y выбран из СН2, CHR6 и C(R6)2;каждый R1и R2...
Ингибиторы ns3 протеазы вируса гепатита с

Номер патента: 13331
Опубликовано: 30.04.2010
Авторы: Макколи Джон А., Радд Майкл Т., Вакка Джозеф П., Холловэй М.Катарин, Олсен Дэвид Б., Людмерер Стивен У., Ливертон Найджел Дж., Макинтайр Чарльз Дж.
МПК: A61K 38/06, C07K 5/08, A61P 31/14...
Метки: вируса, протеазы, гепатита, ингибиторы
Формула / Реферат:
1. Соединение формулы Iили его фармацевтически приемлемая соль,где р и q оба равны 1;R1 является CONR10SO2R6;R2 является C1-С6алкилом или С2-С6алкенилом, где указанный алкил или алкенил необязательно замещен 1-3 атомами галогена;R3 является C1-С8алкилом или C3-С8циклоалкилом;R5 является Н;R6 является C3-С6циклоалкилом;Y является С(=O);Z является О;М является С1-С12алкиленом или С2-С12алкениленом икаждый R10 независимо является Н или...
Макроциклические ингибиторы вируса гепатита с

Номер патента: 14188
Опубликовано: 29.10.2010
Авторы: Валльберг Ханс Кристиан, Ху Лили, Росенквист Оса Анника Кристина, Вехлинг Хорст Юрген, Сальберг Свен Кристер, Симмен Кеннет Алан, Самуэльссон Бенгт Бертил, Нильссон Карл Магнус, Классон Бьерн Олоф, Линдстрем Матс Стефан, Де Кок Херман Аугустинус, Линдквист Карин Карлотта, Канберг Пиа Сесилия, Бельфраге Анна Карин Гертруд Линнеа, Рабуассон Пьер Жан-Мари Бернар
МПК: A61K 31/517, C07D 239/72, A61P 31/12...
Метки: макроциклические, гепатита, ингибиторы, вируса
Формула / Реферат:
1. Соединение формулы (I)и его N-оксиды, соли и стереоизомеры,где А представляет собой OR1, NHS(=O)pR2; гдеR1 представляет собой водород, C1-C6-алкил;R2 представляет собой C3-C7-циклоалкил, причем указанный C3-C7-циклоалкил необязательно замещен 1-3 заместителями, независимо выбранными из C1-C6-алкила;р независимо равно 2;n равно 3, 4, 5 или 6;---- означает необязательную двойную связь;L представляет собой N или CRz;Rz представляет собой Н;Rq...
Макроциклические ингибиторы вируса гепатита с

Номер патента: 14646
Опубликовано: 30.12.2010
Авторы: Симмен Кеннет Алан, Вендевилль Сандрин Мари Элен, Ху Лили, Рабуассон Пьер Жан-Мари Бернар, Сюрлеро Доминик Луи Нестор Гилейн, Тахри Абделлах, Мак Гоуен Дэвид Крейг, Де Кок Херман Аугустинус, Ван Де Фрейкен Вим
МПК: A61K 38/05, C07D 487/04, A61P 31/14...
Метки: макроциклические, вируса, гепатита, ингибиторы
Формула / Реферат:
1. Соединение формулыего N-оксид, аддитивная соль и стереохимически изомерная форма, гдепунктирной линией обозначена необязательная двойная связь между атомами С7 и С8;R1 представляет собой частично ненасыщенную или полностью ненасыщенную 9-10-членную бициклическую гетероциклическую кольцевую систему, где указанная кольцевая система содержит один атом азота и где остальные члены кольца представляют собой атомы углерода; где указанная кольцевая...
Предыдущий патент: Смесительный аппарат
Следующий патент: Способ получения дулоксетина и новые ключевые интермедиаты для его применения
Случайный патент: Композиции на основе ганаксолона