Ингибиторы ns3 протеазы вируса гепатита с
Номер патента: 13331
Опубликовано: 30.04.2010
Авторы: Олсен Дэвид Б., Вакка Джозеф П., Ливертон Найджел Дж., Макинтайр Чарльз Дж., Холловэй М.Катарин, Радд Майкл Т., Макколи Джон А., Людмерер Стивен У.




Формула / Реферат
1. Соединение формулы I

или его фармацевтически приемлемая соль,
где р и q оба равны 1;
R1 является CONR10SO2R6;
R2 является C1-С6алкилом или С2-С6алкенилом, где указанный алкил или алкенил необязательно замещен 1-3 атомами галогена;
R3 является C1-С8алкилом или C3-С8циклоалкилом;
R5 является Н;
R6 является C3-С6циклоалкилом;
Y является С(=O);
Z является О;
М является С1-С12алкиленом или С2-С12алкениленом и
каждый R10 независимо является Н или C1-С6алкилом.
2. Соединение по п.1, где соединение имеет формулу III-а

или его фармацевтически приемлемая соль.
3. Соединение по п.2, где R1 является CONHSO2R6.
4. Соединение по п.3, где R6 является циклопропилом.
5. Соединение по п.4, где R2 является С1-С4алкилом или С2-С4алкенилом.
6. Соединение по п.5, где R3 является С5-С6циклоалкилом или С1-С4алкилом.
7. Соединение по п.6, где М является С4-С10алкиленом или С4-С10алкениленом.
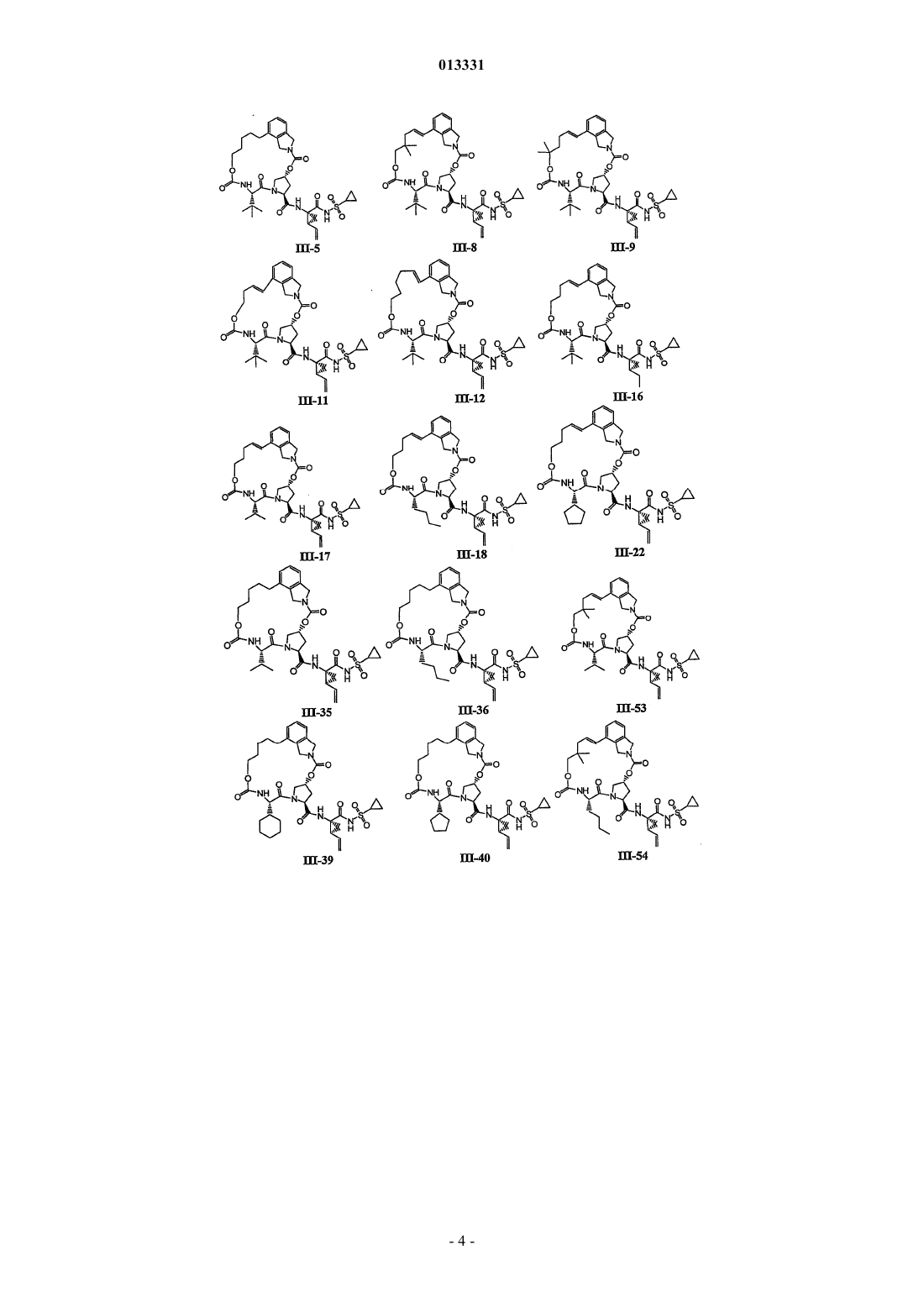
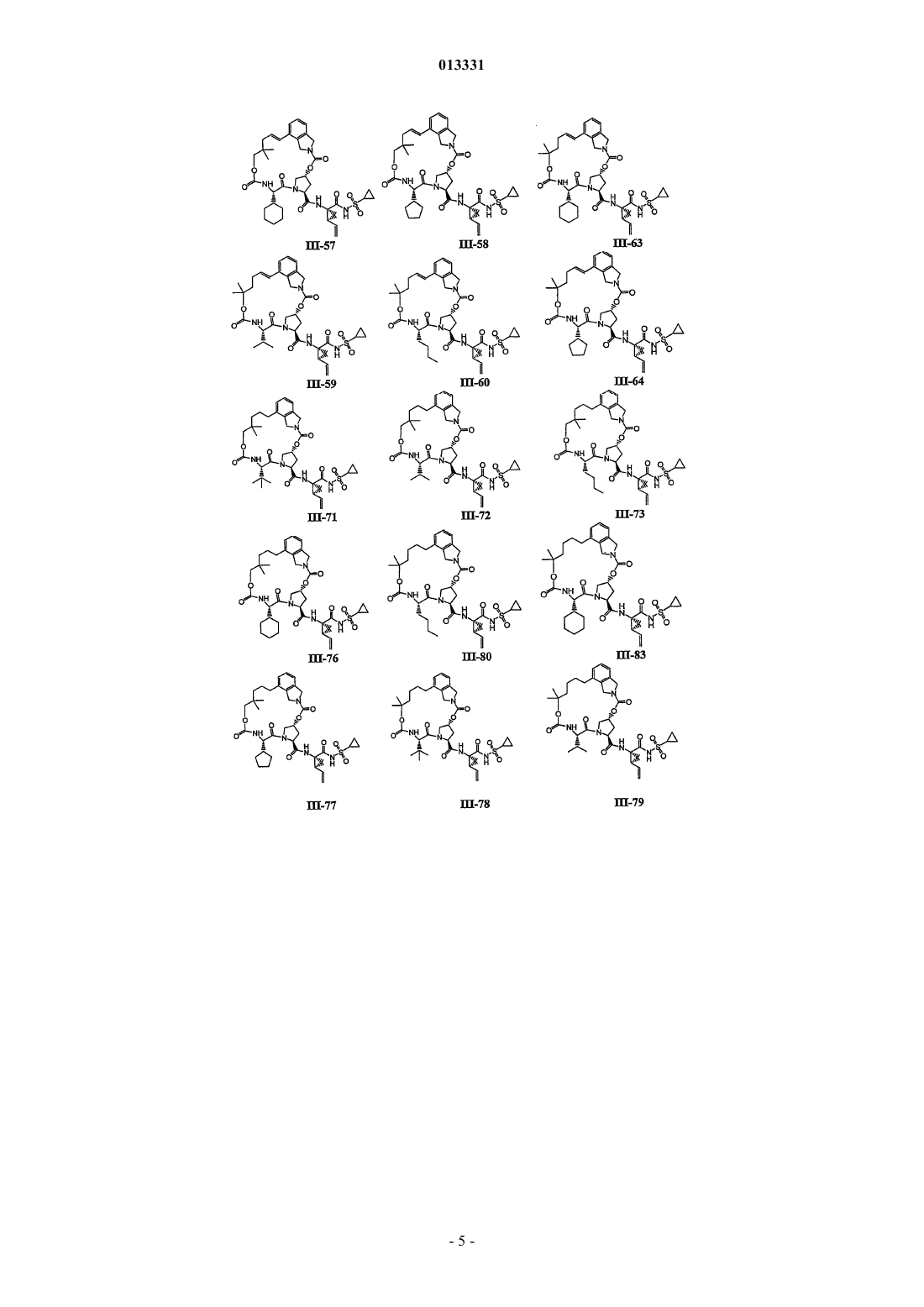
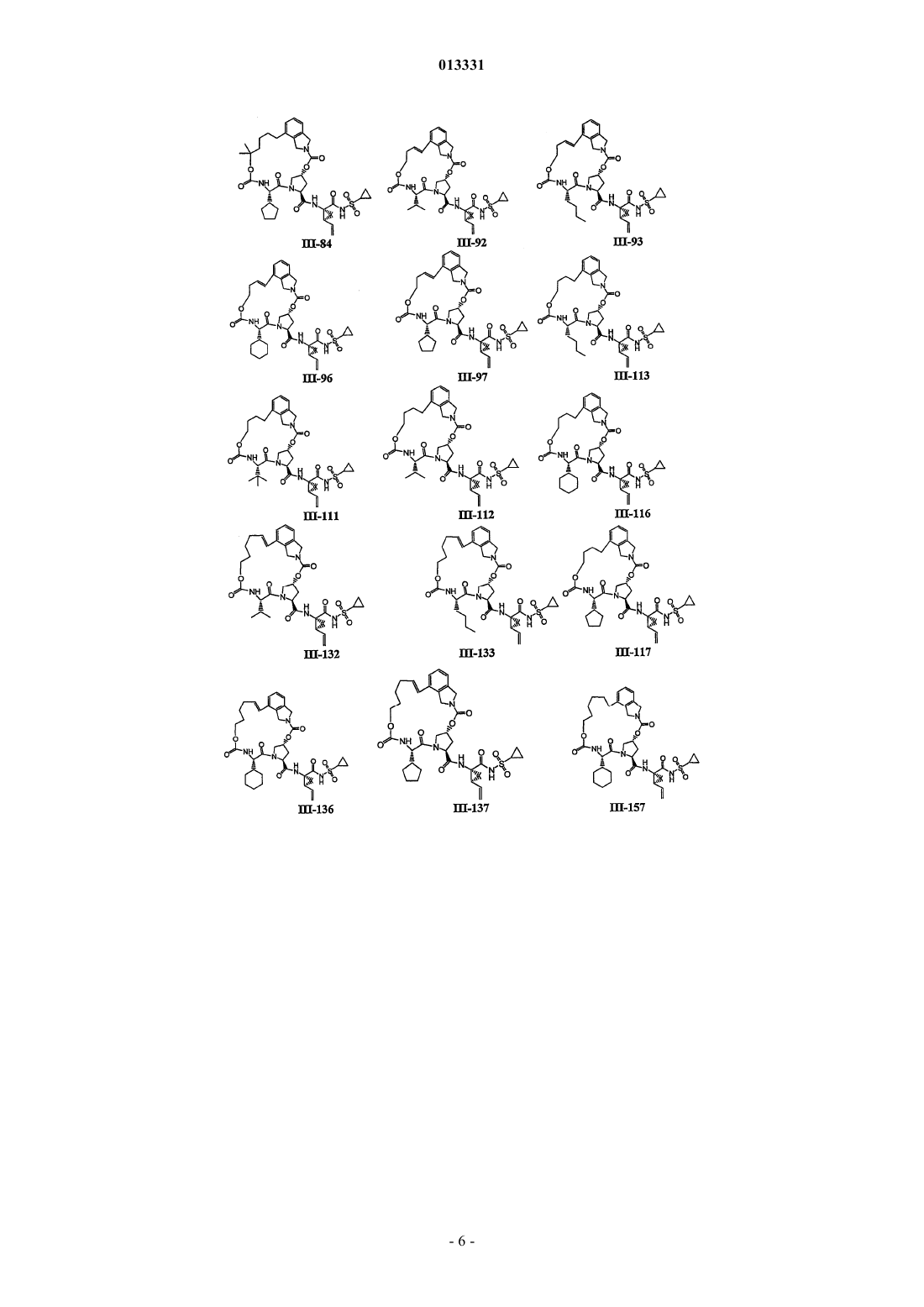
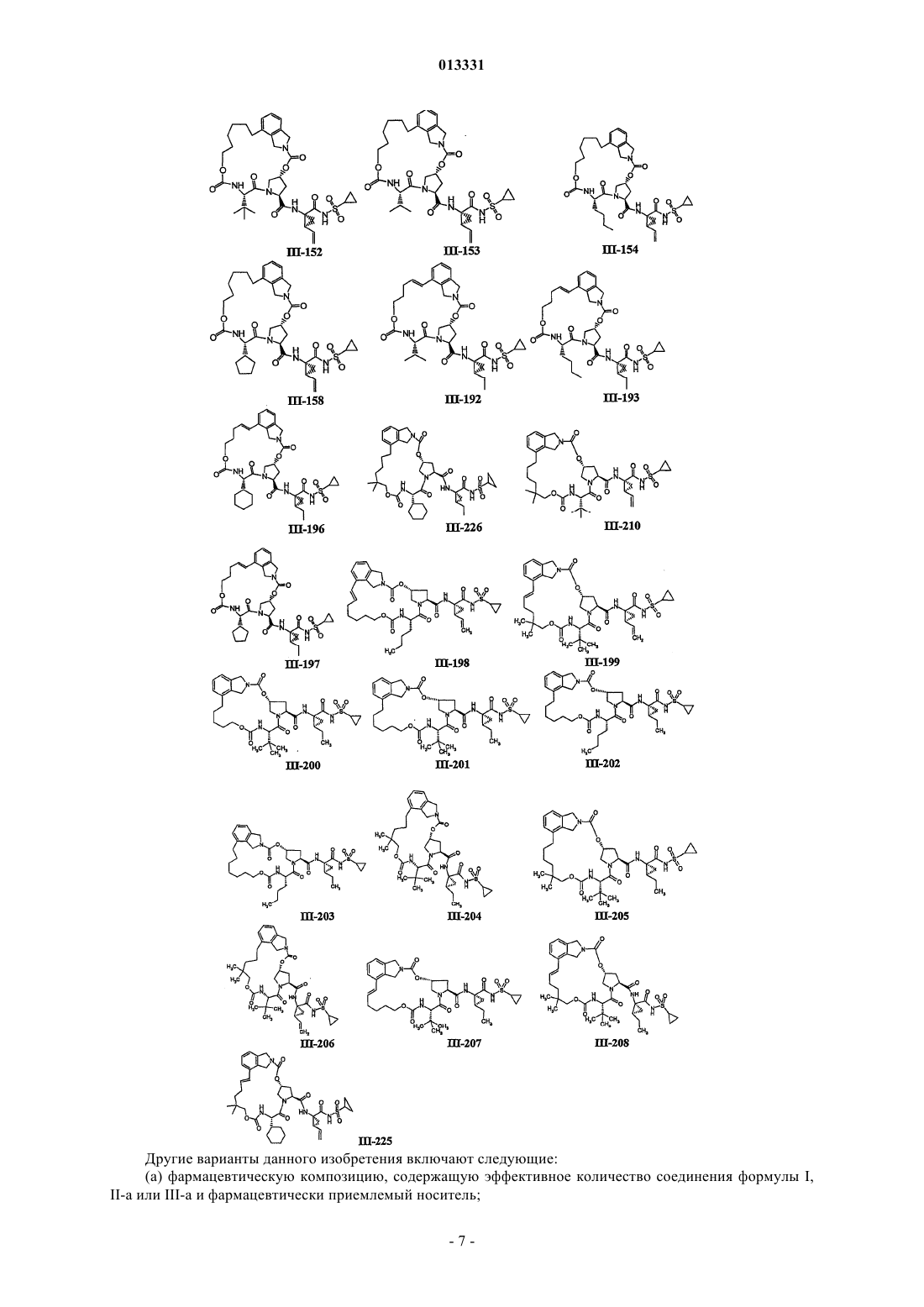
8. Соединение по п.1, где соединение выбирают из






или его фармацевтически приемлемая соль.
9. Фармацевтическая композиция, содержащая эффективное количество соединения по любому из пп.1-8 или его фармацевтически приемлемой соли и фармацевтически приемлемый носитель.
10. Фармацевтическая композиция по п.9, дополнительно содержащая второе терапевтическое средство, выбранное из группы, включающей средство против вируса гепатита С (HCV), иммуномодулятор и антибактериальное средство.
11. Фармацевтическая композиция по п.10, в которой средство против HCV представляет собой противовирусное средство, выбранное из группы, включающей ингибитор протеазы HCV и ингибитор HCV NS5B полимеразы.
12. Применение соединения по любому из пп.1-8 или его фармацевтически приемлемой соли для приготовления лекарственного средства для ингибирования активности HCV NS3 протеазы у пациента.
13. Применение соединения по любому из пп.1-8 или его фармацевтически приемлемой соли для приготовления лекарственного средства для предупреждения или лечения инфекции HCV у пациента.
14. Применение по п.13, где указанное лекарственное средство включает по меньшей мере одно дополнительное терапевтическое средство, выбранное из группы, включающей средство против HCV, иммуномодулятор и антибактериальное средство.
15. Применение по п.14, где средство против HCV представляет собой противовирусное средство, выбранное из группы, включающей ингибитор протеазы HCV и ингибитор HCV NS5B полимеразы.
16. Соединение по п.1, представляющее собой соединение формулы

или его фармацевтически приемлемую соль.
17. Фармацевтическая композиция, содержащая эффективное количество соединения по п.16 и фармацевтически приемлемый носитель.
18. Способ ингибирования активности HCV NS3 протеазы у пациента, где указанный способ включает введение пациенту эффективного количества соединения по п.16.
19. Способ предупреждения или лечения инфекции HCV у пациента, где указанный способ включает введение пациенту эффективного количества соединения по п.16.
Текст
013331 Область техники Данное изобретение относится к макроциклическим соединениям, которые применяются в качестве ингибиторов NS3 протеазы вируса гепатита С (HCV), их синтезу и их применению для лечения или профилактики инфекции HCV. Уровень техники Инфекция вирусом гепатита С (HCV) является основной проблемой здоровья, которая приводит к хроническому заболеванию печени, такому как цирроз и злокачественная гепатома, у значительного количества инфицированных людей, оцениваемого как 2-15% от всего мирового населения. По оценкам только в США насчитывается 3,9 млн инфицированных человек, что согласно данным Центра по контролю за заболеваниями США приблизительно в пять раз больше, чем человек, инфицированных вирусом иммунодефицита человека (ВИЧ). Согласно Всемирной организации здравоохранения в мире насчитывается более чем 170 млн инфицированных человек и по крайней мере от 3 до 4 млн человек инфицируется каждый год. При инфицировании около 20% людей устраняют вирус, но у остальных HCV остается на всю жизнь. У от 10 до 20% хронически инфицированных человек со временем развивается разрушающий печень цирроз или рак. Вирусное заболевание переносится парентерально через зараженную кровь и продукты крови, зараженные иглы или половым путем и по вертикали от зараженных матерей или матерей-носителей вируса к их потомству. Текущее лечение инфекции HCV, которое ограничено иммунотерапией с рекомбинантным интерфероном- отдельно или в сочетании с нуклеозидным аналогом рибавирином, оказывает ограниченную клиническую пользу. Кроме того, не создана вакцина от HCV. Следовательно, существует острая необходимость в улучшенных терапевтических агентах, которые эффективно борются с хронической инфекцией HCV. Текущее состояние известного уровня техники для лечения инфекции HCV обсуждается в следующих ссылках: Dymock В. et al. Novel approaches to the treatment of hepatitis С virus infection, Antiviral ChemistryChemotherapy, 11: 79-96, (2000); Rosen H. et al. Hepatitis С virus: current understanding andSpark Excitement about Hepatitis C, Science: 506-507, (2001). Несколько кодированных вирусами ферментов являются предполагаемыми целями для терапевтического вмешательства, включая металлопротеазу (NS2-3), серинпротеазу (NS3), геликазу (NS3) и РНК-зависимую РНК полимеразу (NS5B). NS3 протеаза расположена в N-концевом домене NS3 белка и считается первичной целью для лекарственных средств, так как она отвечает за внутримолекулярный распад в NS3/4A сайте и за нисходящий межмолекулярный процессинг на NS4A/4B, NS4B/5A иNS5A/5B сайтах. Предшествующие исследования определили классы пептидов, такие как гексапептиды,а также трипептиды, описанные в заявках на патент США US 2005/0020503, 2004/0229818 и 2004/00229776, показывающие степени активности при ингибировании NS3 протеазы. Задачей данного изобретения является предоставление других соединений, которые демонстрируют активность противNS3 протеазы HCV. Сущность изобретения Данное изобретение относится к новым макроциклическим соединениям формулы (I) и/или их фармацевтически приемлемым солям или гидратам. Эти соединения применяют для ингибирования NS3(неструктурной 3) протеазы HCV (вируса гепатита С) для профилактики или лечения одного или более из симптомов инфекции HCV, либо в виде соединений, либо в виде их фармацевтически приемлемых солей или гидратов (если они приемлемы), либо в виде ингредиентов фармацевтических композиций,отдельно или в сочетании с другими противо-HCV средствами, антибактериальными средствами, иммуномодуляторами, антибиотиками или вакцинами. Более конкретно, данное изобретение относится к соединению формулы I и/или его фармацевтически приемлемой соли или гидратуR2 является С 1-С 6 алкилом или С 2-С 6 алкенилом, где указанные алкил или алкенил необязательно за-1 013331 мещены от 1 до 3 атомами галогена;Z является О; М является С 1-С 12 алкиленом или С 2-С 12 алкениленом и каждый R10 независимо является Н или C1-С 6 алкилом. Данное изобретение также включает фармацевтические композиции, содержащие соединение в соответствии с данным изобретением и способы получения таких фармацевтических композиций. Данное изобретение также включает способы лечения или профилактики одного или более симптомов инфекцииHCV. Другие варианты, аспекты и характеристики данного изобретения либо описаны ниже, либо будут понятны из представленного ниже описания, примеров и формулы изобретения. Подробное описание изобретения Данное изобретение включает соединения формулы I выше и их фармацевтически приемлемые соли и/или гидраты. Эти соединения и их фармацевтически приемлемые соли и/или гидраты являются ингибиторами протеазы HCV (например, ингибиторами NS3 протеазы HCV). Данное изобретение также включает соединения формул II-а и III-а, где все переменные такие, как определены для формулы I. Первый вариант данного изобретения включает соединение формулы I, II-а или III-а или его фармацевтически приемлемую соль или гидрат, где R1 является CONHSO2R6 и все другие переменные такие,как определены выше (например, как определены в разделе Сущность изобретения). В первом аспекте первого варианта R1 является CONHSO2R6, где R6 является С 3-С 5 циклоалкилом; и все другие переменные имеют значения, определенные в первом варианте. В одном из вариантов первого аспекта первого варианта R1 является CONHSO2R6, где R6 является циклопропилом; и все другие переменные имеют значения, определенные в первом варианте. Второй вариант данного изобретения включает соединение формулы I, II-а или III-а или его фармацевтически приемлемую соль или гидрат, где R2 является С 1-С 6 алкилом или С 2-С 6 алкенилом; и все другие переменные такие, как определены выше, или такие, как определены в любом из предшествующих вариантов. В первом аспекте второго варианта R2 является С 1-С 4 алкилом или С 2-С 4 алкенилом и все другие переменные такие, как определены выше, или такие, как определены в любом из предшествующих вариантов. Во втором аспекте второго варианта R2 является С 2-С 4 алкенилом и все другие переменные такие, как определены выше, или такие, как определены в любом из предшествующих вариантов. В одном из вариантов второго аспекта второго варианта R2 является винилом и все другие переменные такие,как определены в третьем варианте, или такие, как определены в любом из предшествующих вариантов. В третьем аспекте второго варианта R2 является C1-С 4 алкилом и все другие переменные такие, как определены выше, или такие, как определены в любом из предшествующих вариантов. В одном из вариантов третьего аспекта третьего варианта R2 является этилом и все другие переменные такие, как определены в третьем варианте, или такие, как определены в любом из предшествующих вариантов. Третий вариант данного изобретения включает соединение формулы I, II-а или III-а или его фармацевтически приемлемую соль или гидрат, где R3 является С 3-С 8 циклоалкилом или C1-С 8 алкилом; и все другие переменные такие, как определены выше, или такие, как определены в любом из предшествующих вариантов. В первом аспекте третьего варианта R3 является С 5-С 7 циклоалкилом или С 1-С 8 алкилом и все другие переменные такие, как определены в третьем варианте, или такие, как определены в любом из предшествующих вариантов. Во втором аспекте третьего варианта R3 является С 5-С 6 циклоалкилом илиC1-С 8 алкилом и все другие переменные такие, как определены в третьем варианте, или такие, как определены в любом из предшествующих вариантов. В третьем аспекте третьего варианта R3 является пропилом или бутилом и все другие переменные такие, как определены в третьем варианте, или такие, как определены в любом из предшествующих вариантов. В одном из вариантов третьего аспекта третьего варианта R3 является изопропилом, н-пропилом или трет-бутилом и все другие переменные такие, как определены в третьем варианте, или такие, как определены в любом из предшествующих вариантов. В четвертом аспекте третьего варианта R3 является циклопентилом или циклогексилом и все другие перемен-2 013331 ные такие, как определены в третьем варианте, или такие, как определены в любом из предшествующих вариантов. Четвертый вариант данного изобретения включает соединение формулы I, II-а или III-а или его фармацевтически приемлемую соль или гидрат, где М является С 1-С 10 алкиленом или С 2-С 10 алкениленом(включая алкилен или алкенилен с линейной или разветвленной цепью); и все другие переменные такие,как определены выше, или такие, как определены в любом из предшествующих вариантов. В первом аспекте четвертого варианта М является С 1-С 8 алкиленом или С 2-С 8 алкениленом (включая алкилен или алкенилен с линейной или разветвленной цепью) и все другие переменные такие, как определены выше,или такие, как определены в любом из предшествующих вариантов. Во втором аспекте четвертого варианта М является С 4 алкиленом или С 4 алкениленом (включая алкилен или алкенилен с линейной или разветвленной цепью); и все другие переменные такие, как определены в четвертом варианте, или такие, как определены в любом из предшествующих вариантов. В третьем аспекте четвертого варианта М является С 5 алкиленом или С 5 алкениленом (включая алкилен или алкенилен с линейной или разветвленной цепью); и все другие переменные такие, как определены в четвертом варианте, или такие, как определены в любом из предшествующих вариантов. В четвертом аспекте четвертого варианта М является С 6 алкиленом или С 6 алкениленом (включая алкилен или алкенилен с линейной или разветвленной цепью) и все другие переменные такие, как определены в четвертом варианте, или такие, как определены в любом из предшествующих вариантов. В пятом аспекте четвертого варианта М является С 7 алкиленом или С 7 алкениленом (включая алкилен или алкенилен с линейной или разветвленной цепью) и все другие переменные такие, как определены в четвертом варианте, или такие, как определены в любом из предшествующих вариантов. В шестом аспекте четвертого варианта М является С 8 алкиленом или С 8 алкениленом(включая алкилен или алкенилен с линейной или разветвленной цепью) и все другие переменные такие,как определены в четвертом варианте, или такие, как определены в любом из предшествующих вариантов. В седьмом аспекте четвертого варианта М является С 9 алкиленом или С 9 алкениленом (включая алкилен или алкенилен с линейной или разветвленной цепью) и все другие переменные такие, как определены в четвертом варианте, или такие, как определены в любом из предшествующих вариантов. В восьмом аспекте четвертого варианта М является С 10 алкиленом или С 10 алкениленом (включая алкилен или алкенилен с линейной или разветвленной цепью) и все другие переменные такие, как определены в четвертом варианте, или такие, как определены в любом из предшествующих вариантов. В девятом аспекте четвертого варианта М выбирают из представленных ниже вариантов и все другие переменные такие, как определены в четвертом варианте, или такие, как определены в любом из предшествующих вариантов. Пятый вариант данного изобретения включает соединение или его фармацевтически приемлемую соль или гидрат, выбранное из группы, включающей следующие соединения. Другие варианты данного изобретения включают следующие:(a) фармацевтическую композицию, содержащую эффективное количество соединения формулы I,II-a или III-a и фармацевтически приемлемый носитель;(b) фармацевтическую композицию по пункту (а), дополнительно содержащую второе терапевтическое средство, выбранное из группы, включающей средство против HCV, иммуномодулятор и антибактериальное средство;(c) фармацевтическая композиция по пункту (b), где средством против HCV является противовирусное средство, выбранное из группы, включающей ингибитор протеазы HCV и ингибитор HCV NS5B полимеразы;(ii) второе терапевтическое средство, выбранное из группы, включающей противо-HCV средство, иммуномодулятор и антибактериальное средство; где каждое из соединения формулы I, II-а или III-а и второго терапевтического средства применяют в количестве, которое делает сочетание эффективным для ингибирования HCV NS3 протеазы или для лечения или профилактики инфекции HCV;(e) комбинацию по пункту (d), где средство против HCV представляет собой противовирусное средство, выбранное из группы, включающей ингибитор протеазы HCV и ингибитор NS5B полимеразы HCV;(f) способ ингибирования NS3 протеазы HCV у пациента, нуждающегося в таковом, который включает введение пациенту эффективного количества соединения формулы I, II-а или III-а;(g) способ предупреждения или лечения инфекции HCV у пациента, нуждающегося в таковом, который включает введение пациенту эффективного количества соединения формулы I, II-а или III-а;(h) способ по пункту (g), в котором соединение формулы I, II-а или III-а вводят в сочетании с эффективным количеством по крайней мере одного второго терапевтического средства, выбранного из группы, включающей противо-HCV средство, иммуномодулятор и антибактериальное средство;(i) способ по пункту (h), где противо-HCV средство представляет собой противовирусное средство,выбранное из группы, включающей ингибитор протеазы HCV и ингибитор NS5B полимеразы HCV;(j) способ ингибирования NS3 протеазы HCV у пациента, нуждающегося в таковом, который включает введение пациенту фармацевтической композиции по пунктам (а), (b) или (с) или комбинации по пунктам (d) или (е);(k) способ предупреждения или лечения инфекции HCV у пациента, нуждающегося в таковом, который включает введение пациенту фармацевтической композиции по пунктам (а), (b) или (с) или комбинации по пунктам (d) или (е). Данное изобретение также включает соединение в соответствии с данным изобретением (i) для применения в, (ii) для применения в качестве лекарственного средства для или (iii) для применения для получения лекарственного средства для (а) ингибирования NS3 протеазы HCV или (b) предупреждения или лечения инфекции HCV. В указанных областях применения соединения в соответствии с данным изобретением необязательно могут применяться в сочетании с одним или более терапевтическими средствами, выбранными из средств против HCV, антибактериальных средств и иммуномодуляторов. Дополнительные варианты данного изобретения включают фармацевтические композиции, комбинации и способы, перечисленные в пунктах (а)-(k) выше, и области применения, перечисленные в предшествующем параграфе, где применяемым соединением в соответствии с данным изобретением является соединение по одному или более варианту, аспекту, классу, подклассу или подварианту соединений,описанных выше. Во всех этих вариантах соединение необязательно может применяться в виде фармацевтически приемлемой соли или гидрата в случае необходимости. В данном описании термин алкил относится к любой линейной или разветвленной алкильной группе, имеющей количество атомов углерода в указанном интервале. Таким образом, например,C1-6 алкил (или C1-С 6 алкил) относится ко всем изомерам гексилалкила и пентилалкила, а также к н-,изо-, втор- и трет-бутилу, н- и изопропилу, этилу и метилу. В качестве другого примера С 1-4 алкил относится к н-, изо-, втор- и трет-бутилу, н- и изопропилу, этилу и метилу. Термин галоалкил относится к алкильной группе, в которой атом водорода замещен галогеном. Термин алкокси относится к алкил-O- группе. Термин алкилен относится к любой линейной или разветвленной алкиленовой группе, имеющей количество атомов углерода в указанном интервале. Таким образом, например, -C1-6 алкилен относится к любому из от C1 до С 6 линейных или разветвленных алкиленов. Класс алкиленов, которые представляют особенный интерес в свете данного изобретения, включает (CH2)1-6-, и особенно подклассы, представляющие особенный интерес, -(CH2)1-4-, -(CH2)1-3-, -(CH2)1-2- и -СН 2-. Также представляет интерес алкилен-СН(СН 3)-. Термин алкенилен относится к любой линейной или разветвленной двухвалентной алкениленовой группе, имеющей количество атомов углерода в указанном интервале. Термин циклоалкил относится к любому циклическому кольцу алкана или алкена, имеющему количество атомов углерода в указанном интервале. Таким образом, например, С 3-8 циклоалкил (или С 3-С 8 циклоалкил) относится к циклопропилу, циклобутилу, циклопентилу, циклогексилу, циклогептилу и циклооктилу. Термин циклоалкокси относится к циклоалкил-O- группе. Термин галоген (или гало) относится к фтору, хлору, брому и иоду (альтернативно, обозначен как фтор, хлор, бром и иод). Если не указано противоположное, все интервалы, представленные здесь, являются включающими.-8 013331 Например, гетероарильное кольцо, описанное как содержащее от 1 до 3 гетероатомов, означает, что кольцо может содержать 1, 2 или 3 гетероатома. Также должно быть понятно, что любой указанный здесь интервал включает в свой объем все подынтервалы в пределах данного интервала. Окисленные формы гетероатомов N и S также включены в объем данного изобретения. Если любая переменная (например, R10) возникает более одного раза в любом элементе или в формуле I, II-а или III-а или в любой другой формуле, изображенной и описывающей соединения в соответствии с данным изобретением, ее определение в каждом случае является независимым от ее определения в любом другом случае. Также сочетания заместителей и/или переменных допустимо, только если такое сочетание дает стабильные соединения. Если не указано обратное, замещение указанного заместителя разрешено на любом атоме в кольце(например, ариле, гетероароматическом кольце или насыщенном гетероциклическом кольце) при условии, что замещение кольца химически допустимо и дает стабильное соединение. Стабильным соединением является соединение, которое может быть получено и выделено и структура и свойства которого остаются или могут быть практически неизменными в течение периода времени, достаточного для применения соединения для описанных здесь целей (например, терапевтического или профилактического введения пациенту). В результате выбора заместителей и схем замещения определенные соединения в соответствии с данным изобретением могут иметь асимметрические центры и могут существовать в виде смесей стереоизомеров или в виде отдельных диастереомеров или энантиомеров. Все изомерные формы этих соединений, выделенные или в смесях, включены в объем данного изобретения. Как должно быть понятно специалисту в данной области техники, определенные соединения в соответствии с данным изобретением могут существовать в виде таутомеров. Для целей данного изобретения все ссылки на соединение формулы I, II-а или III-а являются ссылками на само соединение, или на любой из его таутомеров, или на смеси двух или более таутомеров. Соединения в соответствии с данным изобретением применяются для ингибирования протеазыHCV (например, NS3 протеазы HCV) и для предупреждения или лечения инфекции HCV. Например, соединения в соответствии с данным изобретением применяются для лечения инфекции HCV после предполагаемого заражения HCV через переливание крови, обмена жидкостями организма, укусов, случайного укола иглой или контакта с кровью пациента во время хирургической операции. Соединения в соответствии с данным изобретением применяются при получении и проведении скриннинговых исследований для противовирусных соединений. Например, для соединений в соответствии с данным изобретением применяются для выделения ферментных мутантов, которые являются превосходным средством для скрининга для более мощных противовирусных соединений. Более того, соединения в соответствии с данным изобретением применяются для установления или определения мест связывания других противовирусных средств с протеазой HCV, например, путем сравнительного ингибирования. Таким образом, соединения в соответствии с данным изобретением являются коммерческими продуктами, которые могут продаваться для указанных выше целей. Соединения в соответствии с данным изобретением могут вводиться в виде фармацевтически приемлемых солей. Термин фармацевтически приемлемая соль относится к соли, которая обладает эффективностью исходного соединения и которая не является биологически или каким-либо другим образом нежелательной (например, не является ни токсичной, ни вредной для пациента). Подходящие соли включают кислотно-аддитивные соли, которые могут, например, быть получены смешиванием раствора соединения в соответствии с данным изобретением с раствором фармацевтически приемлемой кислоты,такой как хлористо-водородная кислота, серная кислота, уксусная кислота, трифторуксусная кислота или бензойная кислота. Множество соединений в соответствии с данным изобретением имеют кислотную группу, в этом случае их фармацевтически приемлемые соли могут включать соли с щелочными металлами (например, соли натрия или калия), соли с щелочно-земельными металлами (например, соли кальция или магния) и соли, полученные с подходящими органическими лигандами, такими как соли четвертичного аммония. Также в случае присутствия кислоты (-СООН) или спиртовой группы фармацевтически приемлемые сложные эфиры могут применяться для изменения характеристик растворимости или гидролиза соединения. Термин введение и его варианты (например, вводить соединение), применяемый в отношении соединения в соответствии с данным изобретением, означает предоставление соединения или пролекарства соединения пациенту при необходимости лечения. Если соединение или пролекарство соединения дается в сочетании с одним или более активными агентами (например, противовирусными агентами,применяемыми для лечения инфекции HCV), каждый термин введение и его варианты понимаются как включающие одновременное и последовательное предоставление соединения или соли (или гидрата) и других агентов. В данном описании термин композиция охватывает продукт, содержащий определенные ингредиенты, а также любой продукт, который получают, прямо или косвенно при объединении определенных ингредиентов. Под фармацевтически приемлемым подразумевается, что ингредиенты фармацевтической компо-9 013331 зиции должны быть совместимы друг с другом и не причинять вред пациенту. Термин субъект (альтернативно обозначенный здесь как пациент) в данном описании относится к животному, предпочтительно млекопитающему, наиболее предпочтительно человеку, который является объектом лечения, наблюдения или эксперимента. Термин эффективное количество в данном описании означает количество активного соединения или фармацевтического агента, которое позволяет добиться биологической или медицинской реакции в ткани, системе у животного или человека, которая желательна для исследователя, ветеринара, врача или другого клинициста. В одном варианте эффективным количеством является терапевтически эффективное количество для облегчения симптомов лечимого заболевания или состояния. В другом варианте эффективным количеством является профилактически эффективное количество для профилактики симптомов лечимого заболевания или состояния. Термин также включает количество активного соединения, достаточное для ингибирования NS3 протеазы HCV, тем самым вызывая желаемую реакцию (например, эффективное для ингибирования количество). Если активное соединение (например, активный ингредиент) вводят в виде соли, ссылки на количество активного ингредиентаданы для свободной кислоты или свободного основания соединения. Для целей ингибирования NS3 протеазы HCV и предупреждения или лечения инфекции HCV соединения в соответствии с данным изобретением, необязательно в форме соли или гидрата, могут вводиться любыми способами, которые обеспечивают контакт активного агента с местом действия агента. Они могут вводиться любыми обычными способами, доступными для применения в сочетании с фармацевтическими средствами, либо в виде отдельных терапевтических агентов, либо в сочетании терапевтических агентов. Они могут вводиться отдельно, но обычно вводятся с фармацевтическим носителем, выбранным на основе применяемого способа введения и стандартной фармацевтической практики. Соединения в соответствии с данным изобретением, например, могут вводиться перорально, парентерально(включая подкожные инъекции, внутривенные, внутримышечные, надчревные инъекции или вливания),ингаляцией или ректально, в виде стандартной лекарственной формы фармацевтической композиции,содержащей эффективное количество соединения и подходящие нетоксичные фармацевтически приемлемые носители, адъюванты и наполнители. Жидкие композиции, подходящие для перорального введения (например, суспензии, сиропы, эликсиры и подобные), могут быть получены с применением методик, известных в данной области техники, и в них может применяться любая из обычных сред, такая как вода, гликоли, масла, спирты и подобные. Твердые препараты для перорального введения (например,порошки, пилюли, капсулы и таблетки) могут быть получены с применением методик, известных в данной области техники, и в них применяются такие твердые наполнители, как крахмалы, сахара, каолин,смазывающие агенты, связующие агенты, дезинтегрирующие агенты и подобные. Парентеральные композиции могут быть получены с применением методик, известных в данной области техники, и обычно в них применяется стерильная вода в качестве носителя и необязательно другие ингредиенты, такие как средства, улучшающие растворимость. Растворы для инъекций могут быть получены с применением методик, известных в данной области техники, где носитель включает физиологический раствор, раствор глюкозы или раствор, содержащий смесь физиологического раствора и глюкозы. Более подробное описание методик, подходящих для применения при получении фармацевтических композиций в соответствии с данным изобретением, и ингредиенты, подходящие для применения в указанных композициях,представлено в Remington's Pharmaceutical Sciences, 18th edition, edited by Gennaro A.R. Mack PublishingCo., 1990. Соединения в соответствии с данным изобретением могут вводиться перорально в дозе от 0,001 до 1000 мг/кг массы тела млекопитающего (например, человека) в сутки, одной дозой или многократными дозами. Предпочтительный интервал доз составляет от 0,01 до 500 мг/кг массы тела в сутки перорально,однократной дозой или многократными дозами. Другой предпочтительный интервал составляет от 0,1 до 100 мг/кг массы тела в сутки перорально, одной дозой или многократными дозами. Для перорального введения композиции могут иметь форму таблеток или капсул, содержащих от 1,0 до 500 мг активного ингредиента, особенно 1, 5, 10, 15, 20, 25, 50, 75, 100, 150, 200, 250, 300, 400 и 500 мг активного ингредиента для симптоматической корректировки дозы к пациенту. Конкретная дозировка и частота введения для каждого конкретного пациента может варьироваться и зависит от множества факторов, включая активность конкретного применяемого соединения, метаболическую стабильность и продолжительность действия соединения, возраст, массу тела, общее состояние здоровья, пол, режим питания, способ и время введения, скорость выведения, сочетание лекарственных средств, тяжесть конкретного состояния и пациента, который проходит терапию. Как отмечено выше, данное изобретение также относится к способу ингибирования HCV NS3 протеазы, ингибированию репликации HCV или предупреждению или лечению инфекции HCV с применением соединения в соответствии с данным изобретением в сочетании с одним или более терапевтическими средствами и фармацевтической композицией, содержащей соединение в соответствии с данным изобретением и одно или более терапевтических средств, выбранных из группы, включающей средство против HCV, иммуномодулятор и антибактериальное средство. Такие терапевтические средства, активные против HCV, включают, но не ограничены ими, рибавирин, левовирин, вирамидин, тимозин, альфа-1,- 10013331R7025 (улучшенный интерферон (Roche, интерферон-, интерферон-, пегилированный интерферон(пегинтерферон-), сочетание интерферона- и рибавирина, сочетание пегинтерферона- и рибавирина,сочетание интерферона- и левовирина и сочетание пегинтерферона- и левовирина. Интерферонвключает, но не ограничен ими, рекомбинантный интерферон-2 а (такой как интерферон Roferon отHoffmann-LaRoche, Nutley, NJ), пегилированный интерферон-2 а (Pegasys), интерферон-2b (такой как интерферон Intron-A от Schering Corp., Kenilworth, NJ), пегилированный интерферон-2b (PegIntron),рекомбинантный консенсусный интерферон (такой как интерферон альфакон-1), альбуферон (интерферон-, связанный с альбумином бычьей сыворотки (Human Genome Sciences и очищенный продукт интерферона-. Рекомбинантный консенсусный интерферон от Amgen имеет торговое наименование Infergen. Левовирин представляет собой L-энантиомер рибавирина, который демонстрирует иммуномодулирующее действие, схожее с рибавирином. Вирамидин представляет собой аналог рибавирина, описанный в WO 01/60379 (переуступленный ICN Pharmaceuticals). Согласно способу в соответствии с данным изобретением отдельные компоненты сочетания могут вводиться отдельно в различное время в течение курса терапии или одновременно в многократной или однократной форме сочетания. Для лечения инфекции HCV соединения в соответствии с данным изобретением также могут вводиться в сочетании с агентом, который является ингибитором NS3 серинпротеазы HCV. NS3 серинпротеаза HCV является жизненно важным вирусным ферментом и описана как превосходная цель для ингибирования репликации HCV. Ингибиторы NS3 протеазы HCV на основе субстратов и не содержащие субстратов описаны в WO 98/224 96, 98/46630, 99/07733, 99/07734, 99/38888, 99/50230, 99/64442,00/09543, 00/59929, GB-2337262, WO 02/48116, 02/48172 и патенте США 6323180. Рибавирин, левовирин и вирамидин могут проявлять их противо-HCV действие путем модулирования внутриклеточных пулов нуклеотидов гуанина путем ингибирования внутриклеточного фермента инозинмонофосфатдегидрогеназы (IMPDH). IMPDH является ограничивающим скорость ферментом в способе биосинтеза в de novo биосинтезе нуклеотида гуанина. Рибавирин легко фосфорилируется внутриклеточно, и производное монофосфата является ингибитором IMPDH. Таким образом, ингибированияIMPDH являются еще одной полезной целью для выявления ингибиторов репликации HCV. Поэтому соединения в соответствии с данным изобретением также могут вводиться в сочетании с ингибиторомIMPDH, таким как VX-497, который описан в WO 97/41211 и 01/00622 (переуступлены Vertex); другим ингибитором IMPDH, таким как описан в WO 00/25780 (переуступлен Bristol-Myers Squibb); или микофенолатом мофетилом [см. Allison А.С. and Eugui E.M. Agents Action, 44 (Suppl.): 165, (1993)]. Для лечения инфекции HCV соединения в соответствии с данным изобретением также могут вводиться в сочетании с противовирусным средством амантадином (1-аминоадамантан) [полное описание этого агента дано в Kirschbaum J. Anal. Profiles Drug Subs., 12: 1-36, (1983)]. Для лечения инфекции HCV соединения в соответствии с данным изобретением также могут вводиться в сочетании с противовирусным средством ингибитором полимеразы R7128 (Roche). Соединения в соответствии с данным изобретением также могут сочетаться для лечения инфекцииWO 04/003000 (8 января 2004) и международной публикации WO 04/002422 (8 января 2004); содержание которых включено сюда в качестве ссылок полностью. Такие 2'-С-разветвленные рибонуклеозиды включают, но не ограничены ими, 2'-С-метилцитидин, 2'-С-метилуридин, 2'-С-метиладенозин, 2'-С-метилгуанозин и 9-(2-С-метилD-рибофуранозил)-2,6-диаминопурин, и соответствующий сложный эфир аминокислоты С-2', С-3' и С-5' гидроксилов рибозы, и соответствующие необязательно замещенные циклические сложные эфиры 1,3-пропандиола производных 5'-фосфата. Соединения в соответствии с данным изобретением также могут быть объединены для лечения инфекции HCV с другими нуклеозидами, имеющими свойства против HCV, такими как описаны в(5 февраля 2004); 04/013300 (12 февраля 2004); US 2004/0063658 (1 апреля 2004) и WO 04/028481 (8 апреля 2004); содержание которых включено сюда в качестве ссылок полностью. Для лечения инфекции HCV соединения в соответствии с данным изобретением также могут вводиться в сочетании с агентом, который является ингибитором NS5B полимеразы HCV. Такие ингибито- 11013331 ры NS5B полимеразы HCV, которые могут применяться в комбинированной терапии, включают, но не ограничены ими, описаны в WO 02/057287, US 6777395, WO 02/057425, US 2004/0067901, WO 03/068244,2004/000858, 04/003138 и 2004/007512; содержание которых включено сюда в качестве ссылок полностью. Другие подобные ингибиторы полимеразы HCV включают, но не ограничены ими, валопицитабин(NM-283; Idenix) и 2'-F-2'-бета-метилцитидин (см. также WO 2005/003147, переуступленный Pharmasset,Ltd.). В одном варианте ингибиторы нуклеозида HCV NS5B полимеразы, которые применяются в сочетании с данными ингибиторами NS3 протеазы HCV, выбирают из следующих соединений: 4-амино-7-(2-СметилD-арабинофуранозил)-7 Н-пирроло[2,3-d]пиримидина; 4-амино-7-(2-С-метилD-рибофуранозил)-7 Н-пирроло[2,3-d]пиримидина; 4-метиламино-7-(2-С-метилD-рибофуранозил)-7 Н-пирроло[2,3d]пиримидина; 4-диметиламино-7-(2-С-метилD-рибофуранозил)-7 Н-пирроло[2,3-d]пиримидина; 4-амино-7-(2-С-ви 4-циклопропиламино-7-(2-С-метилD-рибофуранозил)-7 Н-пирроло[2,3-d]пиримидина; нилD-рибофуранозил)-7 Н-пирроло[2,3-d]пиримидина; 4-амино-7-(2-С-гидроксиметилD-рибофуранозил)-7 Н-пирроло[2,3-d]пиримидина; 4-амино-7-(2-С-фторметилD-рибофуранозил)-7 Н-пирроло[2,3-d]пиримидина; 4-амино-5-метил-7-(2-С-метилD-рибофуранозил)-7 Н-пирроло[2,3-d]пиримидина; 4-амино-7-(2-С-метилD-рибофуранозил)-7 Н-пирроло[2,3-d]пиримидин-5-карбоновой кислоты; 4 амино-5-бром-7-(2-С-метилD-рибофуранозил)-7 Н-пирроло[2,3-d]пиримидина; 4-амино-5-хлор-7-(2-СметилD-рибофуранозил)-7 Н-пирроло[2,3-d]пиримидина; 4-амино-5-фтор-7-(2-С-метилD-рибофуранозил)-7 Н-пирроло[2,3-d]пиримидина; 2,4-диамино-7-(2-С-метилD-рибофуранозил)-7 Н-пирроло[2,3d]пиримидина; 2-амино-7-(2-С-метилD-рибофуранозил)-7 Н-пирроло[2,3-d]пиримидина; 2-амино-4 циклопропиламино-7-(2-С-метилD-рибофуранозил)-7 Н-пирроло[2,3-d]пиримидина; 2-амино-7-(2-СметилD-рибофуранозил)-7 Н-пирроло[2,3-d]пиримидин-4-(3 Н)-она; 4-амино-7-(2-С-этилD-рибофуранозил)-7 Н-пирроло[2,3-d]пиримидина; 4-амино-7-(2-С,2-O-диметилD-рибофуранозил)-7 Н-пирроло[2,3-d]пиримидина; 7-(2-С-метилD-рибофуранозил)-7 Н-пирроло[2,3-d]пиримидин-4-(3 Н)-она; 2-амино-5 метил-7-(2-С,2-O-диметилD-рибофуранозил)-7 Н-пирроло[2,3-d]пиримидин-4-(3 Н)-она; 4-амино-7-(3-дезокси-2-С-метилD-рибофуранозил)-7 Н-пирроло[2,3-d]пиримидина; 4-амино-7-(3-дезокси-2-С-метилDарабинофуранозил)-7 Н-пирроло[2,3-d]пиримидина; 4-амино-2-фтор-7-(2-С-метилD-рибофуранозил)-7 Нпирроло[2,3-d]пиримидина; 4-амино-7-(3-С-метилD-рибофуранозил)-7 Н-пирроло[2,3-d]пиримидина; 4-амино-7-(3-С-метилD-ксилофуранозил)-7 Н-пирроло[2,3-d]пиримидина; 4-амино-7-(2,4-ди-С-метилDрибофуранозил)-7 Н-пирроло[2,3-d]пиримидина; 4-амино-7-(3-дезокси-3-фтор-2-С-метилD-рибофуранозил)7 Н-пирроло[2,3-d]пиримидина и соответствующих 5'-трифосфатов, или их фармацевтически приемлемых солей. Соединения в соответствии с данным изобретением также могут быть объединены для лечения инфекции HCV с ненуклеозидными ингибиторами полимеразы HCV, такими как описаны в WO 01/77091Istituto di Ricerche di Biologia Moleculare P. Angeletti S.P.A.; 02/20497 (3 марта 2002); 2005/016927 (в частности, JTK003), переуступленном Japan Tobacco, Inc.; содержание которых включено сюда в качестве ссылок полностью; и HCV-796 (Viropharma Inc.). В одном варианте не нуклеозидные ингибиторы HCV NS5B полимеразы, которые применяются в сочетании с данными ингибиторами HCV NS3 протеазы, выбирают из следующих соединений: 14 циклогексил-6-[2-(диметиламино)этил]-7-оксо-5,6,7,8-тетрагидроиндоло[2,1-а][2,5]бензодиазоцин-11-карбоновой кислоты; 14-циклогексил-6-(2-морфолин-4-илэтил)-5,6,7,8-тетрагидроиндоло[2,1-а][2,5]бензодиазоцин-11-карбоновой кислоты; 14-циклогексил-6-[2-(диметиламино)этил]-3-метокси-5,6,7,8-тетрагидроиндоло[2,1-а][2,5]бензодиазоцин-11-карбоновой кислоты; 14-циклогексил-3-метокси-6-метил-5,6,7,8-тетрагидроиндоло[2,1-а][2,5]бензодиазоцин-11-карбоновой кислоты; метил([(14-циклогексил-3-метокси-6 метил-5,6,7,8-тетрагидроиндоло[2,1-а][2,5]бензодиазоцин-11-ил)карбонил]аминосульфонил)ацетата;([(14-циклогексил-3-метокси-6-метил-5,6,7,8-тетрагидроиндоло[2,1-а][2,5]бензодиазоцин-11-ил)карбонил]аминосульфонил)уксусной кислоты; 14-циклогексил-N-[(диметиламино)сульфонил]-3-метокси-6 метил-5,6,7,8-тетрагидроиндоло[2,1-а][2,5]бензодиазоцин-11-карбоксамида; 3-хлор-14-циклогексил-6-[2(диметиламино)этил]-7-оксо-5,6,7,8-тетрагидроиндоло[2,1-а][2,5]бензодиазоцин-11-карбоновой кислоты; бис(трифторацетата) N'-(11-карбокси-14-циклогексил-7,8-дигидро-6 Н-индоло[1,2-е][1,5]бензоксазоцин 7-ил)-N,N-диметилэтан-1,2-диаминия; 14-циклогексил-7,8-дигидро-6 Н-индоло[2,1-е][1,5]бензоксазоцин 11-карбоновой кислоты; 14-циклогексил-6-метил-7-оксо-5,6,7,8-тетрагидроиндоло[2,1-а][2,5]бензодиазоцин-11-карбоновой кислоты; 14-циклогексил-3-метокси-6-метил-7-оксо-5,6,7,8-тетрагидроиндоло[2,1 а][2,5]бензодиазоцин-11-карбоновой кислоты; 14-циклогексил-6-[2-(диметиламино)этил]-3-метокси-7 оксо-5,6,7,8-тетрагидроиндоло[2,1-а][2,5]бензодиазоцин-11-карбоновой кислоты; 14-циклогексил-6-[3(диметиламино)пропил]-7-оксо-5,6,7,8-тетрагидроиндоло[2,1-а][2,5]бензодиазоцин-11-карбоновой кислоты; 14-циклогексил-7-оксо-6-(2-пиперидин-1-илэтил)-5,6,7,8-тетрагидроиндоло[2,1-а][2,5]бензодиазоцин-11-карбоновой кислоты; 14-циклогексил-6-(2-морфолин-4-илэтил)-7-оксо-5,6,7,8-тетрагидро- 12013331 индоло[2,1-а][2,5]бензодиазоцин-11-карбоновой кислоты; 14-циклогексил-6-[2-(диэтиламино)этил]-7 оксо-5,6,7,8-тетрагидроиндоло[2,1-а][2,5]бензодиазоцин-11-карбоновой кислоты; 14-циклогексил-6-(1 метилпиперидин-4-ил)-7-оксо-5,6,7,8-тетрагидроиндоло[2,1-а][2,5]бензодиазоцин-11-карбоновой кислоты; 14-циклогексил-N-[(диметиламино)сульфонил]-7-оксо-6-(2-пиперидин-1-илэтил)-5,6,7,8-тетрагидроиндоло[2,1-а][2,5]бензодиазоцин-11-карбоксамида; 14-циклогексил-6-[2-(диметиламино)этил]-N-[(диметиламино)сульфонил]-7-оксо-5,6,7,8-тетрагидроиндоло[2,1-а][2,5]бензодиазоцин-11-карбоксамида; 14-циклопентил-6-[2-(диметиламино)этил]-7-оксо-5,6,7,8-тетрагидроиндоло[2,1-а][2,5]бензодиазоцин-11-карбоновой кислоты; 14-циклогексил-5,6,7,8-тетрагидроиндоло[2,1-а][2,5]бензодиазоцин-11-карбоновой кислоты; 6-аллил-14-циклогексил-3-метокси-5,6,7,8-тетрагидроиндоло[2,1-а][2,5]бензодиазоцин-11-карбоновой кислоты; 14-циклопентил-6-[2-(диметиламино)этил]-5,6,7,8-тетрагидроиндоло[2,1-а][2,5]бензодиазоцин-11-карбоновой кислоты; 14-циклогексил-6-[2-(диметиламино)этил]-5,6,7,8-тетрагидроиндоло[2,1-а][2,5]бензодиазоцин-11 карбоновой кислоты; 13-циклогексил-5-метил-4,5,6,7-тетрагидрофуро[3',2':6,7][1,4]диазоцино[1,8-а]индол 10-карбоновой кислоты; 15-циклогексил-6-[2-(диметиламино)этил]-7-оксо-6,7,8,9-тетрагидро-5 Ниндоло[2,1-а][2,6]бензодиазонин-12-карбоновой кислоты; 15-циклогексил-8-оксо-6,7,8,9-тетрагидро-5 Н-индоло[2,1-а][2,6]бензодиазонин-12-карбоновой кислоты; 13-циклогексил-6-оксо-6,7-дигидро-5 Н-индоло[1,2d][1,4]бензодиазепин-10-карбоновой кислоты; и их фармацевтически приемлемых солей. Указанные выше ингибиторы NS5B на основе индола могут быть получены представленными ниже способами А-Е, где различные переменные могут быть выбраны в соответствии с конкретным получаемым тетрациклическим соединением индола Способ А Промежуточное соединение 2-броминдола (полученное, как описано в опубликованной заявке на международный патент WO 2004087714) функционализируют на атоме азота индола с введением предшествующей функциональной группы W'/X' на любом из двух или обоих элементах W/X связи. Методика Pd-медиированного перекрестного сочетания (например, Suzuki, Stille и т.д.) затем дает имеющую С 2 ароматическую группу предшествующую функциональную группу Z'/Y' на любом из двух или обоих элементах Z/Y связи. Обработка функциональной группы с последующим замыканием кольца дает тетрациклическую систему. Затем снятие защиты со сложного эфира дает целевые индольные карбоновые кислоты, в которых С 2 ароматическая группа связана с атомом азота индола. Способ В После сборки связей и получения подходящей 2-галогенароматической группы Pd-медиированное замыкание кольца дает конденсированную тетрациклическую систему. Затем снятие защиты со сложного эфира дает целевые индольные карбоновые кислоты, в которых С 2 ароматическая группа связана с атомами азота индола. С 2 ароматическую группу вводят на начальном этапе с применением методики Pd-медиированного перекрестного сочетания (Suzuki, Stille и т.д.). Затем образуют связь с циклизацией в атоме азота индола с замыканием кольца. Затем снятие защиты со сложного эфира дает целевые индольные карбоновые кислоты, в которых С 2 ароматическая группа связана с атомами азота индола. Способ D Конденсированные тетрациклические промежуточные соединения из способов А-С подвергают обработке функциональной группы в связи, затем снимают защиту со сложного эфира с получением целевых С 2-связанных индольных карбоновых кислот. Способ Е С 2-связанные индольные карбоновые кислоты, полученные в способах A-D, далее превращаются через обработку карбоксилатной функциональной группы с получением соединений, имеющих карбоксилатный заместитель или карбоксамид. Во время проведения указанных выше методов синтеза может быть необходимо и/или желательно защитить чувствительные или реакционноспособные группы в любых рассматриваемых молекулах. Это может быть достигнуто с применением обычных защитных групп,таких как описаны в Protective Groups in Organic Chemistry, ed. J.F.W. McOmie, Plenum Press, 1973 и T.W.GreeneP.G.M. Wuts, Protective Groups in Organic Synthesis, John WileySons, 3rd edition, 1999. Защитные группы могут быть удалены на удобной для этого последующей стадии замещения методами, известными в данной области техники. Ингибирующее действие на HCV NS3 протеазу соединений в соответствии с данным изобретением может быть протестировано с применением анализов, известных в данной области техники. Одним из таких анализов является анализ флуоресценции с временным разрешением (ФБР), описанный в примере 56. Другие примеры таких анализов описаны, например, в международной публикации патентаWO 2005/046712. Соединения, применяемые в качестве ингибиторов HCV NS3 протеазы, имеют Ki менее чем 50 мкМ, более предпочтительно менее чем 10 мкМ, даже более предпочтительно менее чем 100 нМ. Данное изобретение также включает способы получения соединений формулы I, II-а или III-а. Соединения в соответствии с данным изобретением могут быть легко получены согласно представленным ниже схемам реакции и примерам или с применением их модификаций, используя легко доступные исходные материалы, реагенты и обычные методики синтеза. В этих реакциях также возможно использовать варианты, которые сами по себе известны специалистам в данной области техники, но не указаны более детально. Более того, другие способы получения соединений в соответствии с данным изобретением будут очевидны специалисту в данной области техники в свете представленных ниже схем реакций и примеров. Если не указано иначе, все переменные такие, как определены выше. Представленные ниже схемы реакций и примеры служат только для иллюстрации данного изобретения и его практики. Примеры не должны считаться ограничивающими объем или суть данного изобретения.- 14013331 Общее описание синтеза Соединения в соответствии с данным изобретением могут быть синтезированы, как показано на общих схемах 1 и 2. Схема 1 На схеме 1 (n=0-9) изображен синтез характерной молекулы. Соответствующим образом защищенное производное 4-гидроксипролина (например, защищенный карбаматом азот и защищенная сложным эфиром кислота может быть подвергнута взаимодействию с карбонилдиимидазолом или эквивалентным реагентом и затем подвергнута взаимодействию с соответствующим образом защищенным изоиндолином или тетрагидроизохинолином). Алкенильная функциональная группа может быть введена на этой стадии или позднее путем катализируемой палладием реакции галогенидного заместителя, такого как хлорид, бромид и иодид, или другой функциональной группы, такой как трифлат, с металлорганическим реагентом, таким как винил или аллилтриалкилолово. Альтернативно, алкенильная функциональная группа может быть введена до реакции с защищенным пролинолом. На схеме 2 описан синтез олефина, содержащего аминокислотную группу. Аминокислота (либо коммерчески доступная, либо легко полученная методами, известными в данной области техники), в которой кислотная функциональная группа защищена в виде сложного эфира (например, R=метила), может быть превращена в амиды А сочетанием олефиновой карбоновой кислоты с применением широкого спектра пептидных связующих агентов, известных специалистам в данной области техники, таких как ДЦК, ЭДК, БОФ, ТБТУ и т.д. Получение сульфонамидов В может быть достигнуто взаимодействием с подходящим сульфонилхлоридом в органическом растворителе (например, ТГФ) с аминовым основанием в качестве поглотителя. Производные мочевины С могут быть получены взаимодействием аминоэфира с реагентом, таким как карбонилдиимидазол, с получением промежуточного изоцианата (Catalanoet al., WO 03/062192) с последующим добавлением второго олефинсодержащего амина. Альтернативно,фосген, дифосген или трифосген может применяться вместо карбонилдиимидазола. Производные цианогуанидина D могут быть получены взаимодействием сложного эфира аминокислоты с дифенил С-цианокарбонимидатом в органическом растворителе с последующим добавлением второго олефинсодержащего амина. Производные карбамата Е могут быть получены взаимодействием олефинсодержащего спирта с карбонилдиимидазолом (или фосгеном, трифосгеном или дифосгеном) в органическом растворителе с последующим добавлением аминоэфира. Схема 2 После функционализации амина сложный эфир может быть гидролизован с применением множест- 15013331 ва основных условий, известных специалистам в данной области техники (Theodora W. Greene, ProtectiveGroups in Organic Synthesis, Third Edition, John Wiley and Sons, 1999). Снятие карбаматной защитной группы на пролиновой группе может быть проведено множеством способов, известных специалистам в данной области техники (Theodora W. Greene, Protective Groups inOrganic Synthesis, Third Edition, John Wiley and Sons, 1999). Для завершения синтеза соединений в соответствии с данным изобретением производное аминокислоты может быть связано с производным пролина с применением множества пептидных связующих агентов, таких как ДЦК, ЭДК, БОФ, ТБТУ и т.д. (см. схему 1). Макроциклизация затем проводится путем олефиновой перестановки с применением множества катализаторов, которые описаны в литературе для этой цели. На этой стадии олефиновая связь, образованная во время перестановки с замыканием кольца,может быть необязательно гидрирована с получением насыщенной связи или функционализирована альтернативными методами, такими как циклопропанирование. Сложный эфир пролина затем гидролизуют в основных условиях и сочетают со сложным эфиром циклопропиламинокислоты (подходящая алкенильная или алкилциклопропановая группа молекулы может быть получена, как описано выше (LlinasBrunet et al., US 6323180 и подвергают дополнительному основному гидролизу с получением конечных соединений. Сложный эфир пролина также может быть гидролизован и непосредственно связан с имеющим подходящую функциональную группу сульфонамидом ацила циклопропиламинокислоты (который может быть получен согласно Wang X.A. et al., WO2003/099274) с получением конечных соединений. Катализаторы олефинового метатезиса включают следующие виды на основе рутения: F: MillerOrg. Lett. 1999, 1, 953; Hoveyda et al. US2002/0107138; K: Furstner et al. J. Org. Chem 1999, 64, 8275. Применение этих катализаторов при метатезисе с замыканием кольца хорошо известно в литературе (например, Trnka and Grubbs, Асе. Chem. Res. 2001, 34, 18). Список аббревиатур БОФ - гексафторфосфат бензотриазол-1-илокситрис(диметиламино)фосфония ДЦК - дициклогексилкарбодиимидHCl - хлористо-водородная кислота НОАс - уксусная кислота ГОАт - 1-гидрокси-7-азабензотриазолLiOH - гидроксид лития МеОН - метанолPPh3 - трифенилфосфин КТ - комнатная температура ТГФ - тетрагидрофуран ТБТУ - тетрафторборат O-бензотриазол-1-ил-N,N,N',N'-тетраметилурония Пример 1. (5R,7S,10S)-10-Бутил-N-1R,2S)-1-[(циклопропилсульфонил)амино]карбонил-2-винилциклопропил)-3,9,12-триоксо-1,6,7,9,10,11,12,14,15,16-декагидро-5 Н-2,22:5,8-диметано-4,13,2,8,11 бензодиоксатриазациклоикозин-7-карбоксамид (III-1). Смесь ангидрида 3-хлорфталевой кислоты (9 г, 49,2 ммоль) и формамида (100 мл) нагревают до температуры 125 С и перемешивают в течение 3 ч. Затем добавляют воду (300 мл) и смесь охлаждают до комнатной температуры. Смесь фильтруют и полученное белое твердое вещество промывают водой и сушат с получением 4-хлор-1 Н-изоиндол-1,3(2 Н)диона (7,7 г, выход 86%). К твердому 4-хлор-1 Н-изоиндол-1,3(2 Н)диону (4,0 г, 22,0 ммоль) добавляют комплекс боргидридТГФ (1 М/ТТФ, 88,1 мл, 88,1 ммоль) по каплям при перемешивании. После завершения добавления реакционную смесь нагревают до температуры кипения с обратным холодильником (80 С) и перемешивают в течение 6 ч. Затем реакционную смесь охлаждают до температуры 0 С, осторожно по каплям добавляют метанол (2,8 мл, 88,1 ммоль) и реакционную смесь нагревают до комнатной температуры. ДобавляютHCl (6 н.) до тех пор, пока смесь не станет кислой, и затем смесь концентрируют. Неочищенный продукт растворяют в 1 М HCl и дважды экстрагируют этиловым эфиром и дважды дихлорметаном. рН водного слоя доводят до рН 11 добавлением твердого NaOH и три раза экстрагируют этилацетатом. Объединенные этилацетатные экстракты сушат над Na2SO4, фильтруют и концентрируют с получением 4-хлоризоиндолина (1,8 г, выход 53%). МСНР (ИЭР) m/z 154 [(М+Н)+; рассчитано для C8H9ClN: 154]. Стадия 2. 1-трет-Бутил-2-метил (2S,4R)-4-[(4-хлор-1,3-дигидро-2 Н-изоиндол-2-ил)карбонил]оксипирролидин-1,2-дикарбоксилат. К раствору метилового эфира N-Boc пролина (2,87 г, 11,7 ммоль) в ДМФ (15 мл) при температуре 0 С добавляют карбонилдиимидазол (1,9 г, 11,7 ммоль). Реакционную смесь нагревают до комнатной температуры и перемешивают в течение 30 мин. Затем добавляют раствор 4-хлоризоиндолина (1,8 г,11,7 ммоль) в ДМФ (10 мл), реакционную смесь нагревают до температуры 50 С и перемешивают в течение 2 ч. Реакционную смесь выливают в этиловый эфир и 0,5 М HCl и слои разделяют. Органический слой промывают водой, сушат над Na2SO4, фильтруют и концентрируют. Неочищенный продукт очищают на силикагеле (градиентное элюирование от 10 до 90% этилацетат в гексане) с получением 1-третбутил-2-метил (2S,4R)-4-[(4-хлор-1,3-дигидро-2 Н-изоиндол-2-ил)карбонил]оксипирролидин-1,2-дикарбоксилата (3,3 г, выход 66%). МСНР (ИЭР) m/z 325 [(М+Н-Вос)+; рассчитано для C15H18ClN2O4: 325]. Раствор 1-трет-бутил-2-метил (2S,4R)-4-[(4-винил-1,3-дигидро-2 Н-изоиндол-2-ил)карбонил]оксипирролидин-1,2-дикарбоксилата (40 мг, 0,09 ммоль), винилтрибутилстаннана (36 мг, 0,11 ммоль) и фторида цезия (31 мг, 0,21 ммоль) в диоксане (0,5 мл) дегазируют с применением N2 в течение 15 мин. Затем добавляют бис(трибутилфосфин)палладий(0) (2 мг, 0,005 ммоль) и сосуд с реакционной смесью герметично закрывают и нагревают до температуры 100 С в течение 18 ч. После охлаждения реакционную смесь концентрируют и очищают хроматографией на силикагеле (от 10 до 90% этилацетата в гексане) с получением 1-трет-бутил-2-метил (2S,4R)-4-[(4-винил-1,3-дигидро-2 Н-изоиндол-2-ил)карбонил]оксипирролидин-1,2-дикарбоксилата (10 мг, выход 25%). МСНР (ИЭР) m/z 317 [(М+Н-Вос)+; рассчитано для В колбу, содержащую 1-трет-бутил-2-метил (2S,4R)-4-[(4-винил-1,3-дигидро-2 Н-изоиндол-2 ил)карбонил]оксипирролидин-1,2-дикарбоксилат (60 мг, 0,14 ммоль), добавляют 4 М раствор HCl в диоксане (2 мл). Через 1 ч ЖХ-МС анализ показывает полное поглощение исходного материала и образование желаемого Boc продукта. Летучие соединения затем удаляют в вакууме и неочищенный продукт помещают в ДМФ (2 мл). К этой смеси добавляют N-[(пент-4-ен-1-илокси)карбонил]-L-норлейцин (41 мг, 0,17 ммоль) (полученный по описанной ниже методике), ДИПЭА (0,076 мл, 0,43 ммоль), ЭДК (54 мг, 0,28 ммоль) и ГОАт(44 мг, 0,28 ммоль). После перемешивания при к.т. в течение 30 мин полное поглощение амина подтверждается ЖХ-МС. Затем реакционную смесь обрабатывают 0,5 н. HCl и EtOAc. Органический слой промывают насыщенным раствором соли и сушат над MgSO4. Затем растворитель удаляют в вакууме и неочищенный продукт очищают на двуокиси кремния (10-90% EtOAc/гексан) с получением 60 мг (выход 79%) метил N-[(пент-4-енилокси)карбонил]-L-норлейцил-(4R)-4-[(4-винил-1,3-дигидро-2 Н-изоиндол-2 ил)карбонил]окси-L-пролината. МСНР (ИЭР) m/z 542 [(М+Н)+; рассчитано для C29H40N3O7: 542]. Стадия 5. Метил (5R,7S,10S)-10-бутил-3,9,12-триоксо-1,6,7,9,10,11,12,14,15,16-декагидро-5 Н 2,22:5,8-диметано-4,13,2,8,11-бензодиоксатриазациклоикозин-7-карбоксилат. Раствор метил N-[(пент-4-енилокси)карбонил]-L-норлейцил-(4R)-4-[(4-винил-1,3-дигидро-2 Низоиндол-2-ил) карбонил]окси-L-пролината (60 мг, 0,11 ммоль) в ДХЭ (20 мл) дегазируют с применением N2 в течение 15 мин. Затем добавляют катализатор перестановки на основе рутения Zhan RC-301 (катализатор Zhan I (изображен как J на с.43), RC-301, Zannan Pharma Ltd.) (7 мг, 0,01 ммоль). Затем раствор нагревают до температуры 100 С в течение 1 ч. В это время анализы ЖХ-МС и ТСХ показали полное поглощение исходного материала и образование практически единственного продукта, который имеет желаемую массу. Затем растворитель удаляют в вакууме и неочищенный продукт очищают на двуокиси кремния (5-70% EtOAc/гексан) с получением 45 мг (выход 79%) метил (5R,7S,10S)-10-бутил-3,9,12 триоксо-1,6,7,9,10,11,12,14,15,16-декагидро-5 Н-2,22:5,8-диметано-4,13,2,8,11-бензодиоксатриазациклоикозин-7 карбоксилата. МСНР (ИЭР) m/z 514 [(М+Н)+; рассчитано для C27H36N3O7: 514]. Стадия 6.(2 мл), МеОН (0,5 мл) и воде (1 мл) добавляют LiOH (21 мг, 0,87 ммоль). Реакционную смесь нагревают до температуры 40 С и перемешивают в течение 1 ч, после чего полное поглощение исходного метилового эфира подтверждают ЖХ-МС. Затем смесь обрабатывают 0,5 н. HCl и EtOAc. Затем органический слой сушат над K2CO3 и растворитель удаляют в вакууме. Неочищенный продукт помещают в ДМФ(1 мл). К полученному выше раствору добавляют хлорид (1R,2S)-1-[(циклопропилсульфонил)амино]карбонил-2-винилциклопропанаминия (Llinas-Brunet et al. US 03/15755 и Wang et al. WO 03/099274)(32 мг, 0,12 ммоль), ТБТУ (51 мг, 0,16 ммоль) и ДИПЭА (0,071 мл, 0,40 ммоль) и реакционную смесь перемешивают при комнатной температуре в течение 2 ч. Реакционную смесь непосредственно очищают ВЭЖХ с обращенной фазой с получением (5R, 7S, 10S)-10-бутил-N-1R,2S)-1-[(циклопропилсульфонил)амино]карбонил-2-винилциклопропил)-3,9,12-триоксо-1,6,7,9,10,11,12,14,15,16-декагидро-5 Н-2,22:5,8-диметано 4,13,2,8,11-бензодиоксатриазациклоикозин-7-карбоксамида (27 мг, выход 47%). 1 Н ЯМР (500 МГц, ч./млн,CDCl3)10,01 (с, 1 Н), 7,27 (м, 2 Н), 7,12 (д, 1 Н), 7,04 (с, 1 Н), 6,40 (д, J=16,l Гц, 1 Н), 6,08 (м, 1 Н), 5,76 (м,1 Н), 5,44 (с, 1 Н), 5,36 (д, 1 Н), 5,25 (д, 1 Н), 5,14 (д, 1 Н) , 4,80-4,68 (м, 3 Н), 4,59 (д, 1 Н), 4,44 (м, 2 Н),4,38 (м, 1 Н), 4,28 (м, 1 Н), 3,95 (м, 1 Н), 3,77 (дд, 1 Н), 2,94 (м, 1 Н), 2,43 (м, 2 Н), 2,29 (д, 2 Н), 2,06 (м, 2 Н),1,94 (м, 1 Н), 1,78 (м, 4 Н), 1,45 (м, 1 Н), 1,38-1,06 (м, 5 Н), 1,04 (д, 2 Н), 0,92 (т, 3 Н) ч./млн. МСНР (ИЭР) m/z 712 [(М+Н)+; рассчитано для C35H46N5O9S: 712]. Пример 2. (5R,7S,10S)-10-трет-Бутил-N-1R,2S)-1-[(циклопропилсульфонил)амино]карбонил-2 винилциклопропил)-3,9,12-триоксо-1,6,7,9,10,11,12,14,15,16-декагидро-5 Н-2,22:5,8-диметано-4,13,2,8,11 бензодиоксатриазациклоикозин-7-карбоксамид (III-2) Пример 2 получают по методике, применяемой для примера 1, за исключением того, что 3-метил-N[(пент-4-енилокси)карбонил]-L-валин (получен по описанной ниже методике) применяют вместо Суспензию 3-бром-о-ксилола (196 г, 1,06 моль), N-бромсукцинимида (377 г, 2,15 моль) и перекиси бензоила (0,26 г, 1,0 ммоль) в четыреххлористом углероде (1800 мл) нагревают до температуры кипения с обратным холодильником в атмосфере азота в течение 15 ч. Содержимое реакционной колбы охлаждают, фильтруют и фильтрат выпаривают. Отгоняют неочищенный материал в высоком вакууме. Основные фракции отгоняют при температуре от 88 до 152 С. Восстанавливают 108 г чистого продукта. Восстанавливают 182 г слегка неочищенного материала, который может применяться в следующей реакции. 1 Н ЯМР (CDCl3)(ч./млн) 7,56 (д, J=8,0 Гц, 1 Н), 7,31 (д, J=8,0 Гц, 1 Н), 7,26 (с, 1 Н), 7,16 (т, J=8,0 Гц, 1 Н),4,84 (с, 2 Н), 4,64 (с, 2 Н). Стадия 2. 2-Бензил-4-бромизоиндолин. Бикарбонат калия (204 г, 2,04 моль) суспендируют в ацетонитриле (12 л) и смесь нагревают до тем- 19013331 пературы 80 С. Растворы 1-бром-2,3-бис(бромметил)бензола (280 г, 0,82 моль в 500 мл ацетонитрила) и бензиламина (87,5 г, 0,82 моль в 500 мл ацетонитрила) добавляют одновременно через капательные воронки в течение 1 ч. Реакционную смесь перемешивают при температуре 77 С в течение 16 ч. Содержимое реакционной колбы охлаждают, фильтруют и растворитель удаляют выпариванием. Реакционную смесь разделяют между 1 М K2CO3 и EtOAc. Органические фракции промывают насыщенным раствором соли, сушат над безводным Na2SO4, фильтруют и выпаривают. Флэш-хроматография на колонке (градиентное элюирование: гептан до 10% EtOAc в гептане) дает после выпаривания указанное в заголовке соединение в виде бледного масла. 1 Н ЯМР (CDCl3)(ч./млн) 7,41-7,39 (м, 2 Н), 7,37-7,34 (м, 2 Н), 7,32-7,27(м, 2 Н), 7,10-7,03 (м, 2 Н), 4,02 (с, 2 Н), 3,97 (с, 2 Н), 3,91 (с, 2 Н), МСНР (ИЭР) m/z 289 [(М+Н)+; рассчитано для C15H15BrN: 289]. Превращают соль HCl в HCl/МеОН. Добавляют МЭТБ и фильтруют твердое вещество с получением 188 г продукта в виде соли HCl. Стадия 3. 2-Бензил-4-винилизоиндолин. Раствор 2-бензил-4-бромизоиндолина (16,7 г, 58,0 ммоль) и трибутил(винил)олова (20,3 мл,69,6 ммоль) в толуоле (400 мл) дегазируют путем барботирования азота через раствор в течение 0,25 ч. Добавляют тетракис(трифенилфосфин)палладий(0) (1,30 г, 1,16 ммоль) и полученный раствор нагревают на масляной бане с температурой 100 С в атмосфере азота в течение 24 ч. Содержимое реакционной колбы охлаждают, выпаривают и подвергают флэш-хроматографии на колонке, элюируя гексаном/этилацетатом 95/5 с получением после выпаривания указанного в заголовке соединения в виде бледного масла, которое становится розовым при выстаивании. МСНР (ИЭР) m/z 236 [(М+Н)+; рассчитано для C17H18N: 236]. Стадия 4. 4-Винилизоиндолин. Раствор 2-бензил-4-винилизоиндолина (58 ммоль) в 1,2-дихлорэтане (150 мл) помещают в 1-литровую круглодонную колбу в атмосфере азота. К колбе присоединяют капательную воронку, содержащую раствор хлорформиата 1-хлорэтила (7,51 мл, 69,6 ммоль) в 1,2-дихлорэтане. Реакционную колбу охлаждают на ледяной бане и содержимое капательной воронки по каплям добавляют в течение 20 мин,сохраняя внутреннюю температуру 5 С. После завершения добавления реакционную колбу нагревают до комнатной температуры, затем нагревают до температуры кипения с обратным холодильником в течение 45 мин. Содержимое реакционной колбы охлаждают до комнатной температуры, затем растворитель удаляют выпариванием. Добавляют метанол (200 мл) и содержимое реакционной колбы нагревают до температуры кипения с обратным холодильником в течение 30 мин. Реакционную колбу охлаждают и растворитель удаляют выпариванием. Добавляют воду (200 мл) и полученную смесь промывают этилацетатом (2250 мл). Водный слой подщелачивают добавлением 2 N гидроксида натрия, затем экстрагируют метиленхлоридом (4250 мл). Объединенные органические экстракты сушат над безводным сульфатом натрия, фильтруют и фильтрат выпаривают. Остаток подвергают флэш-хроматографии на колонке, элюируя метиленхлоридом/метанолом/гидроксидом аммония от 97/3/0,3 до 95/5/0,5. Выпаривание фракций дает указанное в заголовке соединение в виде коричневого масла, 6,00 г (41,1 ммоль, выход 71% в две стадии). МСНР (ИЭР) m/z 146 [(М+Н)+; рассчитано для C10H12N: 146]. Стадия 5. 1-трет-Бутил 2-метил (2S,4R)-4-[(4-винил-1,3-дигидро-2 Н-изоиндол-2-ил)карбонил]оксипирролидин-1,2-дикарбоксилат. Раствор 1-трет-бутил 2-метил (2S,4R)-4-гидроксипирролидин-1,2-дикарбоксилата (10,1 г, 41,4 ммоль) в ДМФ (90 мл) в атмосфере азота охлаждают до температуры 0 С. К реакционной смеси добавляют твердый 1,1'-карбонилдиимидазол (6,70 г, 41,4 ммоль). Содержимое реакционной колбы нагревают до комнатной температуры и через 2 ч добавляют раствор 4-винилизоиндолина (6,00 г, 41,4 ммоль) в ДМФ(10 мл). Реакционную смесь нагревают на масляной бане с температурой 60 С в течение 2 ч, затем охлаждают и выливают в воду и 5% бисульфат калия. Полученную смесь экстрагируют этилацетатом(4250 мл). Объединенные органические экстракты промывают насыщенным раствором соли, сушат над безводным сульфатом натрия, фильтруют и выпаривают. Флэш-хроматография на колонке с элюированием гексаном/этилацетатом 70/30 дает указанное в заголовке соединение в виде белой пены, 13,9 г(2S,4R)-4-[(4-винил-1,3-дигидро-2 Н-изоиндол-2-ил)карбонил]оксипирролидин-1,2-дикарбоксилата (13,9 г, 33,4 ммоль) в этилацетате (700 мл), охлажденный на ледяной бане, насыщают газообразным хлороводородом. Реакционную колбу герметично закрывают и нагревают до комнатной температуры. Через 3,5 ч растворитель удаляют выпариванием с получением указанного в заголовке соединения в виде серого твердого вещества, 11,2 г, выход 95%. 1 Н ЯМР К раствору гидрохлорида (3R,5S)-5-(метоксикарбонил)пирролидин-3-ил 4-винил-1,3-дигидро-2 Низоиндол-2-карбоксилата (2,00 г, 5,67 ммоль) и N-[(2,2-диметилпент-4-енил)окси]карбонил-3-метил-Lвалина (1,54 г, 5,67 ммоль) в ДМФ (100 мл) добавляют ЭДК (1,41 г, 7,37 ммоль), ГОБт (1,00 г,7,37 ммоль) и ДИПЭА (3,16 мл, 22,8 ммоль). Реакционную смесь перемешивают при комнатной температуре в течение 18 ч и затем разбавляют этилацетатом и водным NaHCO3. Слои разделяют и органический слой промывают водой и насыщенным раствором соли, сушат над Na2SO4, фильтруют и концентрируют. Неочищенный остаток очищают на силикагеле (градиентное элюирование от 5 до 50% этилацетата в гексане) с получением метил N-[(2,2-диметилпент-4-енил)окси]карбонил-3-метил-L-валил-(4R)-4-[(4 винил-1,3-дигидро-2 Н-изоиндол-2-ил)карбонил]окси-L-пролината (2,75 г, 85% выход) в виде белой пены. МСНР (ИЭР) m/z 570 [(М+Н)+; рассчитано для C31H44N3O7: 570]. Стадия 8. Метил (5R,7S,10S)-10-трет-бутил-15,15-диметил-3,9,12-триоксо-1,6,7,9,10,11,12,14,15,16 декагидро-5 Н-2,22:5,8-диметано-4,13,2,8,11-бензодиоксатриазациклоикозин-7-карбоксилат. Раствор метил N-[(2,2-диметилпент-4-енил)окси]карбонил-3-метил-L-валил-(4R)-4-[(4-винил 1,3-дигидро-2 Н-изоиндол-2-ил)карбонил]окси-L-пролината (2,46 г, 4,32 ммоль) в безводном дихлорметане (450 мл) продувают азотом в течение 15 мин. Затем по каплям добавляют дихлорид бис(трициклогексилфосфин)-3-фенил-1 Н-инден-1-илиденрутения (катализатор Neolyst M1 от Strem)(0,40 г, 0,43 ммоль) в дегазированном безводном дихлорметане (50 мл) в течение более 30 мин. Реакционную смесь перемешивают при комнатной температуре, во время перемешивания добавляют по 0,2 г катализатора приблизительно каждые 8-12 ч. Прогресс реакции отслеживают с помощью ВЭЖХ до завершения реакции через 48 ч. Остаток очищают флэш-хроматографией на силикагеле, элюируя 10-70% К раствору метил (5R,7S,10S)-10-трет-бутил-15,15-диметил-3,9,12-триоксо-1,6,7,9,10,11,12,14,15,16 декагидро-5 Н-2,22:5,8-диметано-4,13,2,8,11-бензодиоксатриазациклоикозин-7-карбоксилата (0,9 г, 1,67 ммоль) в ТГФ:Н 2 О (2:1, 45 мл) добавляют LiOH (0,40, 16,7 ммоль). Реакционную смесь нагревают до температуры 40 С и перемешивают в течение 1 ч. Реакционную смесь разбавляют водной HCl и экстрагируют EtOAc. Объединенный слой EtOAc промывают водой, насыщенным раствором соли, сушат над Na2SO4, фильтруют и концентрируют. Продукт применяют без дальнейшей очистки. МСНР (ИЭР) m/z 528 [(М+Н)+; рассчитано для C28H38N3O7: 528]. Стадия 10. (5R,7S,10S)-10-трет-Бутил-N-1R,2S)-1-[(циклопропилсульфонил)амино]карбонил-2 винилциклопропил)-15,15-диметил-3,9,12-триоксо-1,6,7,9,10,11,12,14,15,16-декагидро-5 Н-2,22:5,8-диметано-4,13,2,8,11-бензодиоксатриазациклоикозин-7-карбоксамид. Раствор (5R,7S,10S)-10-трет-бутил-15,15-диметил-3,9,12-триоксо-1,6,7,9,10,11,12,14,15,16-декагидро-5 Н-2,22:5,8-диметано-4,13,2,8,11-бензодиоксатриазациклоикозин-7-карбоновой кислоты (100 мг,0,19 ммоль), хлорида (1R,2S)-1-[(циклопропилсульфонил)амино]карбонил-2-винилциклопропанаминия(Llinas-Brunet et al. US 03/15755 и Wang et al. WO 03/099274) (76 мг, 0,28 ммоль), фосфоргексафторида О-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония (ГАТУ, 108 мг, 0,28 ммоль), ДИПЭА (0,073 мл,0,42 ммоль) и 4-диметиламинопиридина (2 мг) в дихлорметане (5 мл) перемешивают при температуре 40 С в течение 1 ч. Реакционный раствор разбавляют водным насыщенным NaHCO3 и экстрагируют Указанное в заголовке соединение получают по методике, применяемой для примера 3, за исключением того, что 3-метил-N-[гекс-5-енилокси)карбонил]-L-валин (получен по описанной ниже методике) применяют вместо N-[(2,2-диметилпент-4-енил)окси]карбонил-3-метил-L-валина на стадии 7. 1 Н ЯМР Указанное в заголовке соединение получают по методике, применяемой для примера 3, за исключением того, что 3-метил-N-[гекс-5-енилокси)карбонил]-L-норлейцин (получен по описанной ниже методике) применяют вместо N-[(2,2-диметилпент-4-енил)окси]карбонил-3-метил-L-валина на стадии 7. 1 Н ЯМР(500 МГц, ч./млн, CD3OD)7,24 (т, 1 Н), 7,23 (д, 1 Н), 7,15 (д, 1 Н), 6,91 (д, 1 Н), 6,37 (д, J=16,1 Гц,1 Н), 6,07 (м, 1 Н), 5,75 (м, 1 Н), 5,39 (с, 1 Н), 5,29 (д, 1 Н), 5,12 (д, 1 Н), 4,77 (д, 1 Н), 4,66 (м, 3 Н), 4,57 (м,1 Н), 4,47 (кв, 1 Н), 4,39 (кв, 1 Н), 4,27 (д, 1 Н), 3,90 (дд, 1 Н), 3,77 (кв, 1 Н), 2,96 (м, 1 Н), 2,49 (кв, 1 Н), 2,29 Указанное в заголовке соединение получают по методике, применяемой для примера 3, за исключением того, что 3-метил-N-[гепт-6-ен-1-илокси)карбонил]-L-норлейцин (получен по описанной ниже методике) применяют вместо N-[(2,2-диметилпент-4-енил)окси]карбонил-3-метил-L-валина на стадии 7. 1 Н ЯМР (500 МГц, ч./млн., CD3OD)9,26 (с, 1 Н), 7,39 (д, 1 Н), 7,24 (т, 1 Н), 7,15 (д, 1 Н), 6,30 (д, J=15,9 Гц,1 Н), 6,20 (м, 1 Н), 5,75 (м, 1 Н), 5,53 (с, 1 Н), 5,31 (д, 1 Н), 5,12 (д, 1 Н), 4,70 (м, 4 Н), 4,43 (дд, 1 Н), 4,34 (м,2 Н), 4,27 (кв, 1 Н), 3,91 (дд, 1 Н), 3,79 (кв, 1 Н), 3,31 (м, 1 Н), 2,97 (м, 1 Н), 2,31 (м, 1 Н), 2,22 (м, 3 Н), 1,89 (дд,1 Н), 1,74 (м, 2 Н), 1,66 (м, 1 Н), 1,56 (м, 3 Н), 1,38 (м, 8 Н), 1,19 (м, 1 Н), 1,09 (м, 2 Н), 0,94 (т, 3 Н). МСНР Указанное в заголовке соединение получают по методике, применяемой для примера 3, за исключением того, что N-[(2,2-диметилгекс-5-енил)окси]карбонил-3-метил-L-валин (получен по описанной ниже методике) применяют вместо N-[(2,2-диметилпент-4-енил)окси]карбонил-3-метил-L-валина на стадии 7. 1 Н ЯМР (500 МГц, ч./млн, CD3OD)9,17 (с, 1 Н), 7,27 (т, J=7,5 Гц, 1 Н), 7,21 (т, J=7,5 Гц, 2 Н),7,16 (д, J=7,5 Гц, 1 Н), 6,38 (д, J=16 Гц, 1 Н), 6,03 (м, 1 Н), 5,79 (м, 1 Н), 5,32 (м, 2 Н), 5,13 (м, 1 Н), 4,82-4,77- 23013331 Продукт из примера 2 (0,32 мг, 0,45 ммоль) и палладий на угле (10 мас.%, 0,03 г) в EtOAc (10 мл) энергично перемешивают в атмосфере водорода в течение 1 ч. Реакционную смесь фильтруют и концентрируют. Остаток очищают ВЭЖХ с обращенной фазой (колонка DeltaPak C18), проливая 40-65%CH3CN в воде (с NH4OAc 1 г/л). Фракции концентрируют, разбавляют водным насыщенным NaHCO3 Указанное в заголовке соединение получают из продукта примера 4 по методике, описанной для примера 8. 1 Н ЯМР (500 МГц, ч./млн, CD3OD)7,23 (т, 1 Н), 7,14 (д, 1 Н), 7,10 (д, 1 Н), 7,02 (д, 1 Н), 5,36 (с,1 Н), 4,71 (м, 3 Н), 4,64 (т, 1 Н), 4,56 (м, 1 Н), 4,40 (м, 2 Н), 4,24 (д, 1 Н), 3,96 (дд, 1 Н), 3,72 (кв, 1 Н), 2,98 (м,1 Н), 2,58 (м, 1 Н), 2,49 (м, 2 Н), 2,15 (т, 1 Н), 1,69-1,19 (м, 15 Н), 1,09 (м, 1 Н), 1,06 (с, 9 Н), 0,98 (т, 3 Н). МСНР (ИЭР) m/z 730 [(М+Н)+; рассчитано для C36H52N5O9S: 730]. Пример 10. (5R,7S,10S)-10-Бутил-N-1R,2R)-1-[(циклопропилсульфонил)амино]карбонил-2 этилциклопропил)-3,9,12-триоксо-6,7,9,10,11,12,14,15,16,17,18,19-додекагидро-1 Н,5 Н-2,23:5,8-диметано 4,13,2,8,11-бензодиоксатриазациклогеникозин-7-карбоксамид (III-202). Указанное в заголовке соединение получают из продукта примера 5 по методике, описанной для примера 8. 1 Н ЯМР (500 МГц, ч./млн, CD3OD)7,23 (т, 1 Н), 7,14 (д, 1 Н), 7,09 (д, 1 Н), 6,99 (д, 1 Н), 5,39 (с,1 Н), 4,76-4,61 (м, 4 Н), 4,43 (м, 3 Н), 4,29 (д, 1 Н), 3,92 (дд, 1 Н), 3,69 (кв, 1 Н), 2,99 (м, 1 Н), 2,57 (м, 1 Н), 2,51 Указанное в заголовке соединение получают из продукта примера 6 по методике, описанной для примера 8. 1 Н ЯМР (500 МГц, ч./млн, CD3OD)7,25 (т, 1 Н), 7,15 (д, 1 Н), 7,11 (д, 1 Н), 5,55 (с, 1 Н), 4,70 Указанное в заголовке соединение получают из продукта примера 3 по методике, описанной для примера 8. 1 Н ЯМР (400 МГц, ч./млн, CD3OD)9,06 (с, 1 Н), 7,22 (дд, 1 Н), 7,13 (д, 1 Н), 7,07 (д, 1 Н), 5,51 Указанное в заголовке соединение получают из продукта примера 7 по методике, описанной для примера 8. 1 Н ЯМР (500 МГц, ч./млн, CD3OD)9,09 (с, 1 Н), 7,24 (т, J=7,5 Гц, 1 Н), 7,15 (д, J=7,5 Гц, 1 Н),7,10 (д, J=7,5 Гц, 1 Н), 5,53 (с, 1 Н), 4,75-4,59 (м, 4 Н), 4,44-4,37 (м, 3 Н), 4,20 (д, J=12 Гц, 1 Н), 3,95-3,91 (м,1 Н), 3,31 (м, 2 Н), 2,99-2,96 (м, 1 Н), 2,62-2,46 (м, 3 Н), 2,17-2,13 (м, 1 Н), 1,67-1,50 (м, 6 Н), 1,37-1,18 (м, 7 Н),1,15-0,96 (м, 16 Н), 0,80 (с, 3 Н). МСНР (ИЭР) m/z 758 [(М+Н)+; рассчитано для C38H56N5O9S: 758]. Альтернативное получение. Стадия 1. 1-Бром-2,3-бис(бромметил)бензол. К суспензии 3-бром-о-ксилола (999 г, 5,40 моль) в хлорбензоле (9 л) при комнатной температуре добавляют N-бромсукцинимид (1620 г, 9,1 моль) и перекись бензоила (2,6 г, 10,8 ммоль). Реакционную смесь нагревают до температуры 80 С и перемешивают в атмосфере азота в течение 18 ч. Реакционную смесь охлаждают до температуры 70 С и добавляют дополнительную порцию NBS (302 г, 1,7 моль). Реакционную смесь нагревают до температуры 80 С и перемешивают в атмосфере азота в течение 22 ч. Реакционную смесь охлаждают до комнатной температуры, разбавляют гептаном (6 л) и фильтруют. Фильтровальную лепешку промывают гептаном (4 л) и объединенные фильтраты выпаривают. Неочищенный продукт растворяют в гептане (2 л) и хлороформе (200 мл) и фильтруют через основную окись алюминия (500 г). Слой окиси алюминия промывают гептаном (4 л) и объединенные фильтраты выпаривают с получением 1-бром-2,3-бис(бромметил)бензола (1760 г вес неочищенного продукта), который применяют без дальнейшей очистки. 1 Н ЯМР (CDCl3)(ч./млн) 7,56 (д, J=8,0 Гц, 1 Н), 7,31 (д, J=8,0 Гц,1 Н), 7,26 (с, 1 Н), 7,16 (т, J=8,0 Гц, 1 Н), 4,84 (с, 2 Н), 4,64 (с, 2 Н). Стадия 2. Гидрохлорид 2-бензил-4-бромизоиндолин. Бикарбонат калия (657 г, 6,56 моль) суспендируют в MeCN (17 л) и смесь нагревают до температуры 80 С. Растворы неочищенного 1-бром-2,3-бис(бромметил)бензола (900 г, 2,63 моль в 1 л MeCN) и бензиламина (281 г, 2,63 моль в 1 л MeCN) одновременно добавляют через капательные воронки в течение более 2 ч. Реакционную смесь перемешивают при температуре 77 С в течение 2 ч и затем охлаждают- 25013331 до комнатной температуры и перемешивают в течение 16 ч. Содержимое реакционной колбы охлаждают,фильтруют и растворитель удаляют выпариванием. Реакционную смесь разделяют между водой (6 л) иEtOAc (2 л). рН доводят до 9 добавлением 1 М K2CO3, слои разделяют и водную фазу экстрагируют дополнительной порцией EtOAc (2 л). Объединенные органические фракции промывают насыщенным раствором соли, сушат над безводным Na2SO4, фильтруют и выпаривают. Неочищенное масло разбавляют EtOH (300 мл) и охлаждают до температуры 0 С. Добавляют метанольную HCl до тех пор, пока смесь не станет кислой, затем добавляют МЭТБ (700 мл) и смесь обрабатывают ультразвуком, затем перемешивают в течение 15 ч. Добавляют МЭТБ (1 л) и смесь фильтруют и промывают 20% EtOH в МЭТБ с последующим добавлением МЭТБ. Твердое вещество сушат на воздухе с получением гидрохлорида 2-бензил-4-бромизоиндолина (211 г). Дополнительную порцию продукта (86 г) выделяют концентрированием маточных растворов. МСНР (ИЭР) m/z 289 [(M+H)+; рассчитано для C15H15BrN: 289]. Стадия 3. 4-Бромизоиндолин. К раствору гидрохлорида 2-бензил-4-бромизоиндолина (11 г, 30,96 ммоль) в 200 мл EtOAc добавляют 1 М NaOH (100 мл) и смесь перемешивают в течение 30 мин. Органический слой разделяют, промывают насыщенным раствором соли, сушат над безводным Na2SO4 и растворитель выпаривают до масла, которое один раз подвергают азеотропной перегонке с толуолом (50 мл). Масло растворяют в хлорбензоле (50 мл) и в перемешиваемый раствор добавляют 4 молекулярные сита (5 г). Через 10 мин по каплям добавляют 1-хлорэтилхлорформиат (5,6 мл, 51 ммоль) в течение более 5 мин. Реакционную смесь затем нагревают до температуры 90 С в течение 2 ч, охлаждают до комнатной температуры и фильтруют. Твердые вещества промывают хлорбензолом (5 мл) и метанолом (40 мл). Фильтрат нагревают до температуры 70 С в течение 1 ч, охлаждают и перемешивают при комнатной температуре в течение ночи. Твердые вещества фильтруют, промывают хлорбензолом (2 мл) и гексаном и сушат с получением 6,84 г указанного в заголовке соединения. МСНР (ИЭР) m/z 198,1 [(М+Н)+; рассчитано для C8H9BrN: 198,0]. Стадия 4. 1-трет-Бутил 2-метил (2S,4R)-4-[(4-бром-1,3-дигидро-2 Н-изоиндол-2-ил)карбонил]оксипирролидин-1,2-дикарбоксилат. К раствору метилового эфира (2S,4R)-ВОС-4-гидроксипролина (126,3 г, 515 ммоль) в ДМФ (960 мл) при температуре 0 С добавляют N,N'-карбонилдиимидазол (83,51 г, 515 ммоль). Реакционную смесь перемешивают при комнатной температуре в течение 3 ч. Добавляют гидрохлорид 4-бромизоиндолина(120 г, 515 ммоль) и диизопропилэтиламин (96,3 мл, 540 ммоль) и реакционную смесь нагревают до температуры 50 С в течение 6 ч, затем охлаждают до комнатной температуры и перемешивают в течение ночи. Реакционную смесь разделяют между EtOAc (3 л) и 10% водным KHSO4 (6 л), водную фракцию повторно экстрагируют EtOAc (2 л) и объединенные органические фазы промывают 10% водным К раствору 1-трет-бутил 2-метил (2S,4R)-4-[(4-бром-1,3-дигидро-2 Н-изоиндол-2-ил)карбонил]оксипирролидин-1,2-дикарбоксилата (10,0 г, 21,3 ммоль) в этаноле (200 мл) добавляют винилтрифторборат калия (4,28 г, 32 ммоль) и триэтиламин (4,5 мл, 32 ммоль) с последующим добавлением продукта присоединения дихлорметана хлорида дихлор[1,1-бис(дифенилфосфино)ферроцен]палладия(II) (175 мг,0,21 ммоль). Реакционную смесь нагревают до температуры кипения с обратным холодильником в течение 6 ч, охлаждают до комнатной температуры, разбавляют 10% водным KHSO4 и этанол удаляют выпа- 26013331 риванием в вакууме. Водный остаток экстрагируют EtOAc и органическую фазу промывают насыщенным раствором соли, сушат над Na2SO4, растворитель выпаривают и неочищенный остаток очищают хроматографией на силикагеле, элюируя 40-60% EtOAc/гексаном с получением после выпаривания указанного в заголовке соединения (8,18 г). МСНР (ИЭР) m/z 411,2 [(М+Н)+; рассчитано для C22H29N2O6: 417,2]. Стадия 6. Гидрохлорид (3R,5S)-5-(метоксикарбонил)пирролидин-3-ил 4-винил-1,3-дигидро-2 Низоиндол-2-карбоксилат.(2S,4R)-4-[(4-винил-1,3-дигидро-2 Н-изоиндол-2 ил)карбонил]оксипирролидин-1,2-дикарбоксилата (18,0 г, 43,2 ммоль) и HCl/диоксана (4 М) (43,2 мл,173 ммоль) перемешивают при комнатной температуре в течение 2 ч. Реакционную смесь концентрируют для удаления диоксана, затем концентрируют из Et2O с получением гидрохлорида (3R,5S)-5(метоксикарбонил)пирролидин-3-ил 4-винил-1,3-дигидро-2 Н-изоиндол-2-карбоксилата в виде беловатого твердого вещества (15 г), которое применяют без дальнейшей очистки. МСНР (ИЭР) m/z 317 [(М+Н)+; рассчитано для C17H21N2O4: 317]. Стадия 7. Метил N-[(2,2-диметилгекс-5-ен-1-ил)окси]карбонил-3-метил-L-валил-(4R)-4-[(4 винил-1,3-дигидро-2 Н-изоиндол-2-ил)карбонил]окси-L-пролинат. К раствору гидрохлорида (3R,5S)-5-(метоксикарбонил)пирролидин-3-ил 4-винил-1,3-дигидро-2 Низоиндол-2-карбоксилата (5,0 г, 14,2 ммоль) и N-[(2,2-диметилгекс-5-енил)окси]карбонил-3-метил-Lвалина (4,0 г, 14,2 ммоль) в ДМФ (20 мл) при комнатной температуре добавляют ДИПЭА (2,5 мл,14,2 ммоль), ЭДК (5,5 г, 28,4 ммоль) и ГОАт (1,9 г, 14,2 ммоль). Через 18 ч реакционную смесь выливают в Et2O и экстрагируют 1 н. HCl. Водный слой экстрагируют EtOAc и объединенные органические слои промывают 1 н. HCl, водой, NaHCO3 и насыщенным раствором соли. Органический слой сушат надMgSO4 и растворитель удаляют в вакууме. Неочищенный продукт очищают на двуокиси кремния (30%Zhan 1B, RC-303, Zannan Pharma, Ltd.) (0,591 г, 0,805 ммоль). Затем смесь перемешивают при комнатной температуре в атмосфере N2. Через 19 ч реакцию завершают и добавляют ДМСО (57 мкл, 0,805 ммоль). Смесь перемешивают в течение 2 ч и смесь концентрируют в вакууме до 70 мл. Затем неочищенный продукт прямо очищают на двуокиси кремния (градиентное элюирование, 0-50% EtOAc в гексане) с получением 4,4 г указанного в заголовке соединения в виде масла. МСНР (ИЭР) m/z 556,3 [(М+Н)+; рассчитано для C30H42N3O7: 556,3]. Стадия 9. Метил (5R,7S,10S)-10-трет-бутил-15,15-диметил-3,9,12-триоксо-6,7,9,10,11,12,14,15,16,17,18,19 додекагидро-1 Н,5 Н-2,23:5,8-диметано-4,13,2,8,11-бензодиоксатриазациклогеникозин-7-карбоксилат.(4,4 г,7,92 ммоль) в EtOAc (79 мл) добавляют Pd/C (0,421 г, 0,396 ммоль). Затем баллон Н 2 помещают в реакционную колбу. Колбу быстро откачивают и наполняют Н 2. Через 17 ч реакция завершена по данным ЖХ-МС. Pd/C фильтруют через стеклянное волокно и неочищенный продукт очищают на двуокиси кремния (градиентное элюирование, 0-60% EtOAc в гексане) с получением 4,01 г указанного в заголовке соединения в виде белого порошка. МСНР (ИЭР) m/z 558,4 [(М+Н)+; рассчитано для C30H44N3O7: 558,3]. Стадия 10. К раствору метил (5R,7S,10S)-10-трет-бутил-15,15-диметил-3,9,12-триоксо-6,7,9,10,11,12,14,15,16,17,18,19 додекагидро-1 Н,5 Н-2,23:5,8-диметано-4,13,2,8,11-бензодиоксатриазациклогеникозин-7-карбоксилата (5,76 г,10,33 ммоль) в ТГФ (41,3 мл), МеОН (41,3 мл) и воды (20,7 мл) при комнатной температуре добавляютLiOH (4,33 г, 103 ммоль). После полного превращения (45 мин), подтвержденного ЖХ-МС, реакционную смесь обрабатывают путем разделения между Et2O и 1 н. HCl. Затем водный слой экстрагируют EtOAc. Объединенные органические слои сушат над MgSO4 и растворитель удаляют в вакууме с получением 5,53 г указанного в заголовке соединения, которое применяют без дальнейшей очистки. МСНР (ИЭР) m/z 544,4 [(М+Н)+; рассчитано для C29H42N3O7: 544,3]. Стадия 11. 5R,7S,10S)-10-трет-Бутил-N-1R,2R)-1-[(циклопропилсульфонил)амино]карбонил-2 этилциклопропил)-15,15-диметил-3,9,12-триоксо-6,7,9,10,11,12,14,15,16,17,18,19-додекагидро-1 Н,5 Н 2,23:5,8-диметано-4,13,2,8,11-бензодиоксатриазациклогеникозин-7-карбоксамид (III-205). К раствору(5,03 г, 13,22 ммоль). После полного превращения (1 ч) реакционную смесь разделяют между EtOAc и 1 н. HCl. Органический слой промывают насыщенным раствором соли три раза, сушат над MgSO4 и растворитель удаляют в вакууме. Затем неочищенный материал очищают на двуокиси кремния (градиентное элюирование, 20-80% EtOAc в гексане) с получением 5,8 г указанного в заголовке соединения в виде белого порошка. Пример 14. (5R,7S,10S)-10-трет-Бутил-N-1R,2S)-1-[(циклопропилсульфонил)амино]карбонил-2 винилциклопропил)-3,9,12-триоксо-1,6,7,9,10,11,12,14,15,16,17,18-додекагидро-5 Н-2,22:5,8-диметано 4,13,2,8,11-бензодиоксатриазациклоикозин-7-карбоксамид (III-5).(5R,7S,10S)-10-трет-бутил-3,9,12-триоксо-1,6,7,9,10,11,12,14,15,16-декагидро-5 Н-2,22:5,8 диметано-4,13,2,8,11-бензодиоксатриазациклоикозин-7-карбоксилат получают по методике, применяемой для получения метил (5R,7S,10S)-10-трет-бутил-15,15-диметил-3,9,12-триоксо-1,6,7,9,10,11,12,14,15,16-декагидро-5 Н-2,22:5,8-диметано-4,13,2,8,11-бензодиоксатриазациклоикозин-7-карбоксилата (пример 3, стадия 8),за исключением того, что 3-метил-N-[(пент-4-енилокси)карбонил]-L-валин (получен по описанной ниже методике) применяют вместо N-[(2,2-диметилпент-4-енил)окси]карбонил-3-метил-L-валина на стадии 7. МСНР (ИЭР) m/z 514 [(М+Н)+; рассчитано для C27H36N3O7: 514]. К раствору метил (5R,7S,10S)-10-трет-бутил-3,9,12-триоксо-1,6,7,9,10,11,12,14,15,16,17,18-додекагидро-5 Н-2,22:5,8-диметано-4,13,2,8,11-бензодиоксатриазациклоикозин-7-карбоксилата (0,10 г, 0,20 ммоль) в этилацетате (7 мл) добавляют 10% палладий на угле (0,01 г). Реакционную смесь перемешивают в атмосфере водорода в течение 5 ч при комнатной температуре. Содержимое реакционной колбы фильтруют через целит и фильтрат выпаривают. Неочищенный продукт применяют без дальнейшей очистки (0,09 г,выход 90%). МСНР (ИЭР) m/z 516 [(М+Н)+; рассчитано для C27H38N3O7 516]. Стадия 3. (5R,7S,10S)-10-трет-Бутил-N-1R,2S)-1-[(циклопропилсульфонил)амино]карбонил-2 винилциклопропил)-3,9,12-триоксо-1,6,7,9,10,11,12,14,15,16,17,18-додекагидро-5 Н-2,22:5,8-диметано 4,13,2,8,11-бензодиоксатриазациклоикозин-7-карбоксамид. К раствору метил(5R,7S,10S)-10-трет-бутил-3,9,12-триоксо-1,6,7,9,10,11,12,14,15,16,17,18 додекагидро-5 Н-2,22:5,8-диметано-4,13,2,8,11-бензодиоксатриазациклоикозин-7-карбоксилата (90 мг,0,18 ммоль) в ТГФ (2 мл) и МеОН (0,5 мл) добавляют LiOH (1 н. 1,75 мл, 1,75 ммоль). Реакционную смесь нагревают до температуры 40 С и перемешивают в течение 1 ч, после чего полное поглощение исходного метилового эфира подтверждают ЖХ-МС. Затем смесь обрабатывают 0,5 н. HCl и EtOAc. Органический слой затем сушат над K2CO3 и растворитель удаляют в вакууме. Неочищенный продукт помещают в ДМФ (1 мл). К полученному выше раствору добавляют хлорид(77 мг, 0,24 ммоль) и ДИПЭА (0,07 мл, 0,40 ммоль) и реакционную смесь перемешивают при комнатной температуре в течение 2 ч. Реакционную смесь непосредственно очищают ВЭЖХ с обращенной фазой с получением Указанное в заголовке соединение получают по методике, применяемой для получения примера 14(с применением стадий 2 и 3), за исключением того, что метил (5R,7S,10S)-10-трет-бутил-15,15-диметил 3,9,12-триоксо-1,6,7,9,10,11,12,14,15,16-декагидро-5 Н-2,22:5,8-диметано-4,13,2,8,11-бензодиоксатриазациклоикозин-7-карбоксилат (пример 3, стадия 1) применяют вместо метил (5R,7S,10S)-10-трет-бутил 3,9,12-триоксо-1,6,7,9,10,11,12,14,15,16-декагидро-5 Н-2,22:5,8-диметано-4,13,2,8,11-бензодиоксатриазациклоикозин-7-карбоксилата на стадии 2. 1 Н ЯМР (400 МГц, ч./млн, CDCl3)9,91 (с, 1 Н), 7,22 (т, 1 Н),7,09 (д, 2 Н), 7,05 (д, 1 Н), 5,77 (м, 1 Н), 5,60 (с, 1 Н), 5,45 (д, 1 Н), 5,29 (с, 1 Н), 5,15 (д, 1 Н), 4,72 (кв., 2 Н),4,40-4,55 (м, 4 Н), 4,30 (д, 1 Н), 4,25 (д, 1 Н), 3,78 (дд, 1 Н), 3,26 (д, 1 Н), 2,91 (м, 1 Н), 2,50 (м, 3 Н), 2,39 (м,3 Н), 2,11 (м, 1 Н), 1,98 (м, 2 Н), 1,51 (м, 2 Н), 1,38 (м, 4 Н), 1,18(м, 1 Н), 1,04 (с, 9 Н), 1,01 (т, 3 Н), 0,79 (с, 3 Н). МСНР (ИЭР) m/z 742 [(М+Н)+; рассчитано для C37H52N5O9S: 742].(60 мг, 0,12 ммоль) в ТГФ (1 мл) и МеОН (0,5 мл) добавляют LiOH (1 N 1,17 мл, 1,17 ммоль). Реакционную смесь нагревают до температуры 40 С и перемешивают в течение 1 ч, после чего полное поглощение исходного метилового эфира подтверждают ЖХ-МС. Затем смесь обрабатывают 0,5 н. HCl и EtOAc. Органический слой затем сушат над K2CO3 и растворитель удаляют в вакууме. Неочищенный продукт помещают в ДМФ (1 мл). К полученному выше раствору добавляют хлорид (1R,2R)-1-[(циклопропилсульфонил)амино]карбонил-2-этилциклопропанаминия (32 мг, 0,12 ммоль), ТБТУ (48 мг, 0,15 ммоль) и ДИПЭА (0,044 мл, 0,25 ммоль) и реакционную смесь перемешивают при комнатной температуре в течение 2 ч. Реакционную смесь непосредственно очищают ВЭЖХ с обращенной фазой с получением(5R,7S,10S)-10-трет-бутил-15,15-диметил-3,9,12-триоксо 1,6,7,9,10,11,12,14,15,16-декагидро-5 Н-2,22:5,8-диметано-4,13,2,8,11-бензодиоксатриазациклоикозин-7 карбоксилата (пример 3, стадия 8), за исключением того, что 3-метил-N-[(гекс-5-енилокси)карбонил]-Lвалин (получен по описанной ниже методике) применяют вместо N-[(2,2-диметилпент-4-енил)окси]карбонил-3-метил-L-валина на стадии 7. МСНР (ИЭР) m/z 528 [(М+Н)+; рассчитано для C28H38N3O7: 528]. Стадия 2. (5R,7S,10S)-10-трет-Бутил-N-1R,2R)-1-[(циклопропилсульфонил)амино]карбонил-2 этилциклопропил)-3,9,12-триоксо-6,7,9,10,11,12,14,15,16,17-декагидро-1 Н,5 Н-2,23:5,8-диметано-4,13,2,8,11 бензодиоксатриазациклогеникозин-7-карбоксамид. Пример 17 получают по методике, применяемой для получения примера 16, за исключением того,что метил (5R,7S,10S)-10-трет-бутил-3,9,12-триоксо-6,7,9,10,11,12,14,15,16,17-декагидро-1 Н,5 Н-2,23:5,8 диметано-4,13,2,8,11-бензодиоксатриазациклогеникозин-7-карбоксилат применяют вместо метил
МПК / Метки
МПК: C07K 5/08, A61P 31/14, A61K 38/06
Метки: ингибиторы, гепатита, вируса, протеазы
Код ссылки
<a href="https://eas.patents.su/30-13331-ingibitory-ns3-proteazy-virusa-gepatita-s.html" rel="bookmark" title="База патентов Евразийского Союза">Ингибиторы ns3 протеазы вируса гепатита с</a>
Ингибиторы серин-протеаз, в частности ns3 протеазы вируса гепатита c (hvc)

Номер патента: 1915
Опубликовано: 22.10.2001
Авторы: Мурко Марк А., Харбесон Скотт Л., Бхисетти Говинда Рао, Танг Роджер Д., Дейнинджер Дэвид Д, Фармер Люк Дж.
МПК: A61P 1/16, C07K 5/10, A61K 38/55...
Метки: ингибиторы, hvc, гепатита, вируса, серин-протеаз, протеазы, частности
Формула / Реферат:
1. Соединение структурной формулы (II) где W является m равно 0 или 1; каждый R2 представляет собой независимо водород, алкил, алкенил, арил, аралкил, аралкенил, циклоалкил, циклоалкилалкил, циклоалкенил, циклоалкенилалкил, гетероциклил, гетероциклилалкил, гетероциклилалкенил, гетероарил или гетероаралкил, или две R2 группы, которые связаны с одним и тем же атомом азота, образуют вместе с этим атомом азота 5-7-членную моноциклическую...
Трипептидные ингибиторы вируса гепатита с

Номер патента: 3906
Опубликовано: 30.10.2003
Авторы: Гудреу Натали, Тсантризо Иула С., Фоше Анн-Мари, Ллина-Брюне Монсе, Пупар Марк-Андре, Альмос Тедди, Гиро Элиза, Ранкур Жан, Верник Доминик М., Байли Мюррей Д., Симоно Брюно, Камерон Даль
МПК: A61K 38/06, A61P 31/12, C07C 229/48...
Метки: вируса, гепатита, ингибиторы, трипептидные
Формула / Реферат:
1. Соединение формулы (I), включая его рацематы, диастереоизомеры и оптические изомеры в которой B обозначает H, C6- или C10арил, C7-C16аралкил; Het или (низш.)алкил-Het, все необязательно замещенные C1-C6алкилом; C1-C6алкоксигруппу; C1-C6алканоил; гидроксигруппу; гидроксиалкил; галоген; галоалкил; нитрогруппу; цианогруппу; цианалкил; аминогруппу, необязательно замещенную C1-C6алкилом; амидогруппу; или (низш.)алкиламид; или B обозначает...
Пептидные ингибиторы вируса гепатита с

Номер патента: 4765
Опубликовано: 26.08.2004
Авторы: Пупар Марк-Андре, Байли Мюррей Д., Тсантризо Иула С., Ллина-Брюне Монсе, Гиро Элиза, Гудреу Натали, Ранкур Жан, Камерон Даль
МПК: C07K 14/81, A61K 38/55, A61P 31/14...
Метки: пептидные, вируса, гепатита, ингибиторы
Формула / Реферат:
1. Рацематы, диастериоизомеры и оптические изомеры соединения формулы (I) , в которой a равно 0 или 1, b обозначает 0 или 1, Y обозначает H или C1-C6алкил, B обозначает H, ацильное производное формулы R7-C(O)- или сульфонил формулы R7-SO2, где R7 обозначает C1-C10алкил, необязательно замещенный карбоксилом, C1-C6аланоилокси- или C1-C6алкоксигруппой, C6- или C10арил или C7-C16аралкил, необязательно замещенный C1-C6алкилом, гидрокси- или...
Ингибиторы рнк-зависимой рнк-полимеразы вируса гепатита с и композиции и способы лечения с их использованием

Номер патента: 12605
Опубликовано: 30.10.2009
Авторы: Гонсалес Хавьер, Линтон Анджелика, Тэтлок Джон Хоуард, Джуэлл Таня Мишель, Ли Гуи
МПК: A61K 31/366, A61P 31/14, A61K 31/351...
Метки: лечения, ингибиторы, рнк-полимеразы, композиции, способы, гепатита, рнк-зависимой, использованием, вируса
Формула / Реферат:
1. Соединение формулы (4) где R1 представляет собой циклопентил; R2 представляет собой -(CR6R7)n(5-6-членный гетероцикл), где указанная 5-6-членная гетероциклическая группа возможно замещена по меньшей мере одной группой R4; R3 представляет собой -(CR6R7)t(C6-C10-арил) или -(CR6R7)t(4-10-членный гетероцикл), где каждая из указанных группировок C6-C10-арил и 4-10-членный гетероцикл в указанных группах R3 возможно замещена по меньшей мере одной...
Синтез 1,3 – оксаселеноланнуклеозидов, обладающих активностью против вируса иммунодефицита человека и против вируса гепатита-b

Номер патента: 5097
Опубликовано: 28.10.2004
Авторы: Ду Джинфа, Шинази Раймонд Ф., Чу Чунг К.
МПК: A61P 31/18, C07D 329/00, A61K 31/513...
Метки: иммунодефицита, оксаселеноланнуклеозидов, обладающих, вируса, активностью, человека, гепатита-b, синтез, против
Формула / Реферат:
1. 1,3-Оксаселеноланлактон формулы где R обозначает водород, ацил, моно-, ди- или триэфир фосфорной кислоты, стабилизированный фосфат или остаток липида, или его фармацевтически приемлемая соль. 2. Способ получения 1,3-оксаселеноланлактона формулы где R обозначает водород, ацил, моно-, ди- или триэфир фосфорной кислоты, стабилизированный фосфат или остаток липида, или его фармацевтически приемлемой соли, включающий a) получение димера селенола...
Предыдущий патент: Бактериоцины и новые бактериальные штаммы
Следующий патент: Шланг с использованием высокопрочной проволоки некруглого сечения с повышенной усталостной выносливостью
Случайный патент: Фунгицидная смесь